Embed Size (px)
Citation preview

,1
*Center for Molecular Neurobiology, Department of Molecular and Cellular Biochemistry, The Ohio State University College of
Medicine, Columbus, Ohio, USA
�Department of Statistics, The Ohio State University, Columbus, Ohio, USA
Frontotemporal lobar degeneration (FTLD) is a clinically andpathologically heterogeneous group of diseases associatedwith cognitive decline and degeneration of prefrontal andanterior temporal neocortex (Neary et al. 2005). FTLD isoften familial, with �20% of inherited cases caused bymutations in the MAPT gene located on chromosome 17(Rademakers et al. 2004). MAPT encodes the microtubule-associated protein tau, which exists in human brain as amixture of six isoforms owing to alternative splicing of exons2, 3, and 10 from MAPT transcripts (Fig. 1a). Tau isoformsnormally bind to and function in conjunction with themicrotubule cytoskeleton, but they aggregate to formfilamentous inclusions in FTLD. To date more than 40pathogenic MAPT mutations that lead to tau lesion formationand FTLD have been identified in 127 families worldwide(Rademakers et al. 2004), 34 of which change tau primarystructure at one of 27 different amino-acid residues (Fig. 1a).These residues are found predominantly in the C-terminalhalf of tau isoforms (i.e. those encoded by exons 9–12),where the 31–32 residue imperfect repeats that mediate both
microtubule binding (Goode et al. 2000) and tau fibrilliza-tion (Novak et al. 1993) are located. However, pathologicalmutations have been discovered in exon 1 as well, leading toamino acid substitutions at residue five near the N-terminus(Hayashi et al. 2002; Poorkaj et al. 2002), whereas othershave been found in exon 13, which encodes the segment C-terminal to the repeat region. Each of these mutations couldaffect lesion formation through different mechanisms. Forexample, they could promote tau lesion formation directly byincreasing the aggregation propensity of tau isoforms, or
Received July 23, 2008; revised manuscript received September 4, 2008;accepted September 9, 2008.Address correspondence and reprint requests to Jeff Kuret, OSU
Center for Molecular Neurobiology, 1060 Carmack Rd, Columbus, OH43210, USA. E-mail: [email protected] present address of Haishan Yin is the Department of Pharmacology,Emory University School of Medicine, Atlanta, GA 30322, USA.Abbreviations used: 3R, three-repeat tau isoforms; 4R, four-repeat tau
isoforms; FTLD, frontotemporal lobar degeneration; PHF, paired helicalfilament; WT, wild-type tau.
Abstract
Mutations in the MAPT gene encoding tau protein lead to
neurofibrillary lesion formation, neurodegeneration, and cog-
nitive decline associated with frontotemporal lobar degenera-
tion. While some pathogenic mutations affect MAPT introns,
resulting in abnormal splicing patterns, the majority occur in
the tau coding sequence leading to single amino acid changes
in tau primary structure. Depending on their location within the
polypeptide chain, tau missense mutations have been re-
ported to augment aggregation propensity. To determine the
mechanisms underlying mutation-associated changes in
aggregation behavior, the fibrillization of recombinant patho-
genic mutants R5L, G272V, P301L, V337M, and R406W
prepared in a full-length four-repeat human tau background
was examined in vitro as a function of time and submicromolar
tau concentrations using electron microscopy assay methods.
Kinetic constants for nucleation and extension phases of
aggregation were then estimated by direct measurement and
mathematical simulation. Results indicated that the mutants
differ from each other and from wild-type tau in their aggre-
gation propensity. G272V and P301L mutations increased the
rates of both filament nucleation and extension reactions,
whereas R5L and V337M increased only the nucleation
phase. R406W did not differ from wild-type in any kinetic
parameter. The results show that missense mutations can
directly promote tau filament formation at different stages of
the aggregation pathway.
Keywords: aggregation reaction mechanism, frontotemporal
lobar degeneration, neurofibrillary tangle, tau.
J. Neurochem. (2008) 107, 1113–1123.
JOURNAL OF NEUROCHEMISTRY | 2008 | 107 | 1113–1123 doi: 10.1111/j.1471-4159.2008.05692.x
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1113–1123 1113

indirectly by altering tau-microtubule binding equilibria,levels of tau post-translational modification, or levels of tauexpression. Because sequences encoded by exons 1, 9, 11,
12, and 13 affect protein segments common to all tauisoforms, mutations in these regions may affect all six tausplice variants. In contrast, mutations that change tauresidues encoded by exon 10, which encodes a fourthimperfect repeat sequence, are expressed only in four-repeat(4R) tau isoforms.
Previous studies support a direct effect of mutation on tauaggregation propensity, with certain tau mutants supportingincreased rate [P301L (Nacharaju et al. 1999; Gamblin et al.2000)] and/or extent [I260V (Grover et al. 2003); L266V(Hogg et al. 2003); P301L and P301S (Barghorn et al. 2000;Gamblin et al. 2000; Spina et al. 2007); and Q336R(Pickering-Brown et al. 2004)] of aggregation in vitro. Somemutations have been reported to support an increase in thenumbers of filaments formed [R5H (Hayashi et al. 2002);K257T (Rizzini et al. 2000); G272V (Goedert et al. 1999);P301L and P301S (Goedert et al. 1999); and K369I (Neu-mann et al. 2001)], suggesting that tau structure mayinfluence the rate-limiting step in fibrillization. Many ofthese studies were only semiquantitative and did not addressthe mechanism of aggregation enhancement. In addition, tauaggregation in vitro requires addition of an aggregationinducer, typically in the form of an anionic substance, whichadds another layer of complexity to analysis of the reaction(Kuret et al. 2005). A more systematic study of the kineticsof the tau aggregation reaction is needed to address intrinsictau mutant aggregation propensity and the mechanismthrough which the aggregation reaction may be affected.
Recently, we showed that small-molecule inducers such asThiazine red drive aggregation of full-length tau at submi-cromolar concentrations in the absence of macromolecularinducers such as heparin (Chirita et al. 2005). The reactionapproximates a homogeneous nucleation scheme character-ized by initial formation of an unstable dimeric nucleus,followed by filament elongation through monomer addition(Congdon et al. 2008). Under these conditions, it is possibleto fit the reaction to a mathematical model and estimate theelementary rate constants for tau aggregation. Thus, theinherent aggregation propensity for mutant tau may bequantified and mechanistically localized under near physio-logical tau protein concentrations.
Here, we examine the effects of tauopathy mutants R5L,G272V, P301L, V337M, and R406W on aggregationpropensity. The results suggest that some but not alltauopathy mutants increase tau aggregation propensity, andthat they can do so at both the nucleation and elongationreaction steps of the reaction.
Experimental procedures
MaterialsRecombinant polyhistidine-tagged human 2N4R tau, and mutants
G272V, P301L, V337M, and R406W in 2N4R background, were
(a)
(b) (c)
(d) (e)
(f) (g)
Fig. 1 Filament morphology. (a) Distribution of 27 amino acid resi-
dues affected by pathological missense tau mutations currently tabu-
lated at http://www.molgen.ua.ac.be/FTDMutations (Rademakers
et al. 2004) and depicted on isoform 2N4R. This isoform contains
alternatively spliced exons 2 and 3 (E2 and E3), each of which en-
codes an acidic 29 residue segment and exon 10 (E10), which en-
codes an additional microtubule binding repeat sequence. Mutants
R5L, G272V, P301L, V337M, and R406W modeled herein span the
tau molecule and are distinguished graphically with raised hollow
symbols. (b–g) Full-length wild-type 2N4R tau (b) and mutants R5L
(c), G272V (d), P301L (e), V337M (f), and R406W (g) were incubated
(1 lM concentration) without agitation in the presence of 100 lM
Thiazine red (24 h at 37�C), spotted onto copper Formvar-carbon
mesh grids, stained with 2% uranyl acetate, and viewed by trans-
mission electron microscopy. All tau species produced unbranched
filaments �16 nm in diameter with no obvious differences in mor-
phology or length distribution. Scale bar, 200 nm.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1113–1123� 2008 The Authors
1114 | E. Chang et al.

prepared as described previously (Carmel et al. 1996; Gamblin
et al. 2000). R5L was generated by site-directed mutagenesis
(Stratagene QuikChange XL; Stratagene, La Jolla, CA, USA) of the
pT7C-htau40 vector using the following primers: forward (5¢-CTGAGCCCCTCCAGGAGTTCGAAGTGATG-3¢) and reverse
(5¢-CATCACTTCGAACTCCTGGAGGGGCTCAG-3¢). Successfulmutagenesis was confirmed by DNA sequence analysis. R5L mutant
tau was then expressed and purified as described for wild-type 2N4R
tau (Carmel et al. 1996).Aggregation inducer Thiazine red (Chemical Abstract Service
registry number 2150-33-6) was obtained from TCI America
(Portland, OR, USA). Formvar/carbon-coated copper grids (300
mesh), glutaraldehyde, and uranyl acetate were obtained from
Electron Microscopy Sciences (Fort Washington, PA, USA).
Tau fibrillization assayPurified tau preparations were incubated at 37�C without agitation in
assembly buffer (10 mM HEPES, pH 7.4, 100 mM NaCl, and
5 mM dithiothreitol) containing aggregation inducer Thiazine red
(100 lM final concentration) for various times up to 24 h. Reactions
were then fixed with 2% glutaraldehyde, adsorbed onto Formvar/
carbon-coated copper grids, stained with 2% uranyl acetate, and
examined by transmission electron microscopy as described
previously (Necula and Kuret 2004b). At least three fields were
captured for each reaction condition with a Tecnai G2 Spirit
BioTWIN transmission electron microscope (FEI, Hillsboro, OR,
USA) operated at 80 kV and 23 000–49 000· magnification, and
tau filaments >10 nm in length were quantified with IMAGEJ software
(National Institutes of Health, Bethesda, MD, USA). Total filament
length is defined as the sum of the lengths of all resolved filaments
per field and is reported ± SD.
Critical concentration determinationKcrit values were determined from linear regression analysis of the
tau concentration dependence of aggregation by inverse prediction
of abscissa intercepts (Congdon et al. 2008). The accompanying
standard error of the estimate, Sx, was calculated as:
Sx ¼CI
2ðt0:975; n�2 Þ; ð1Þ
where CI is the length of the Fieller 95% confidence interval of
each regression and t0.975, n)2 is the Student’s t distribution
percentage at 1)a = 0.975 and n)2 degrees of freedom.
Dissociation ratesTau filaments prepared as described above for 24 h were diluted
10-fold into assembly buffer containing 100 lM Thiazine red and
incubated at 37�C. Aliquots were withdrawn as a function of time
up to 5 h post-dilution and then assayed for filament length. The
resultant disaggregation time series was fit to an exponential
decay function to obtain kapp, the pseudo-first-order rate constant
describing the time-dependent decrease in filament length, and L0,the total filament length at time zero. The rate constant ke) was
estimated from kapp, L0, and the number of filaments at time zero
as described previously (Kristofferson et al. 1980; Necula
and Kuret 2005). The association rate constant for elongation,
ke+, was then obtained from the relationship (Congdon et al.2008):
Kcrit ¼ ke�=keþ ð2Þ
Aggregation time seriesAliquots of tau aggregation reactions prepared as described above
were removed as a function of time and assayed for filament
formation by electron microscopy. Aggregation lag times, defined as
the time when the tangent to the point of maximum aggregation rate
intersects the abscissa of the sigmoidal curve (Evans et al. 1995),were obtained ± SE from each time series by Gompertz regression
as described in Necula and Kuret (2004a). To determine the
nucleation dissociation equilibrium constant, Kn, filament length
data were converted to protomer concentration, cp*, by assuming
that all protein above the critical concentration formed filaments
(Congdon et al. 2008), and that the resultant filaments were
composed of two tau protomers per b-sheet spacing (Congdon
et al. 2008). Data were then fitted to the simplified homogeneous
nucleation scheme of Wegner and Engel (1975) assuming a dimeric
nucleus (Congdon et al. 2008):
c1 ¼ ctotal � c�p ð3Þ
dcpdt¼ knþðkeþc1 � ke�Þc21
kn� þ keþc1 � ke�ð4Þ
dc�pdt¼ ðkeþ c1 � ke�Þcp; ð5Þ
where ctotal, c1, and cp represent bulk tau, tau monomer, and tau
filament concentrations, respectively. Parameter estimates were
obtained by fitting experimentally determined values of ctotal, cp*,ke), and ke+ to eqns 3–5 in JACOBIAN� modeling software
(Numerica Technology, LLC, Cambridge, MA, USA). The simula-
tion yielded estimates of forward and reverse nucleation rate
constants kn+ and kn), with the ratio kn)/kn+ recorded as Kn.
Statistical analysisThe probability (p) of obtaining the observed results, assuming the
null hypothesis, was assessed by z-test:
z ¼ x1 � x2ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiðSx1 Þ
2 þ ðSx2Þ2
q ; ð6Þ
where x1 ± Sx1 and x2 ± Sx2 are the pair of estimates ± SE being
compared, z is the 1)a point of the standard normal distribution,
and p is 2a. All statistical analyses were carried out using JMP 7.0
(SAS Institute, Cary, NC, USA).
Results
Initial characterizationMissense tau mutants R5L, G272V, P301L, V337M, andR406W were selected for analysis because their associationwith FTLD is well established and because all except R5Lhave been shown to promote neurofibrillary lesion formationin transgenic mice (Denk and Wade-Martins 2008). Further-more, the mutations span the length of the tau molecule(Fig. 1a) to include the N-terminal projection domain (R5L),the first, second, and third microtubule-binding repeats
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1113–1123
Aggregation propensity of tauopathy mutants | 1115
(G272V, P301L, and V337M), and the C-terminal tail(R406W). Because P301L can be accommodated in 4R butnot three-repeat (3R) tau isoforms, all mutants were preparedin a 2N4R background to facilitate direct comparison to eachother and also wild-type 2N4R prepared by identicalmethods.
When incubated in the presence of Thiazine red aggre-gation inducer under near physiological conditions of pH,reducing conditions, and ionic strength, submicromolarconcentrations of wild-type 2N4R form twisted ribbons(Chirita et al. 2005) with a mass-per-unit length similar toauthentic brain-derived paired helical filament (PHFs)(Congdon et al. 2008). When the missense mutants wereincubated under identical conditions up to 1 lM concen-tration, all formed filaments of similar width and morphol-ogy as wild-type 2N4R (Fig. 1b–g). Filament lengthdistributions were also similar to wild-type 2N4R tau (datanot shown). These data indicate that all five missensemutations retain the fundamental aggregation properties ofwild-type tau and can be characterized at physiological bulktau concentrations.
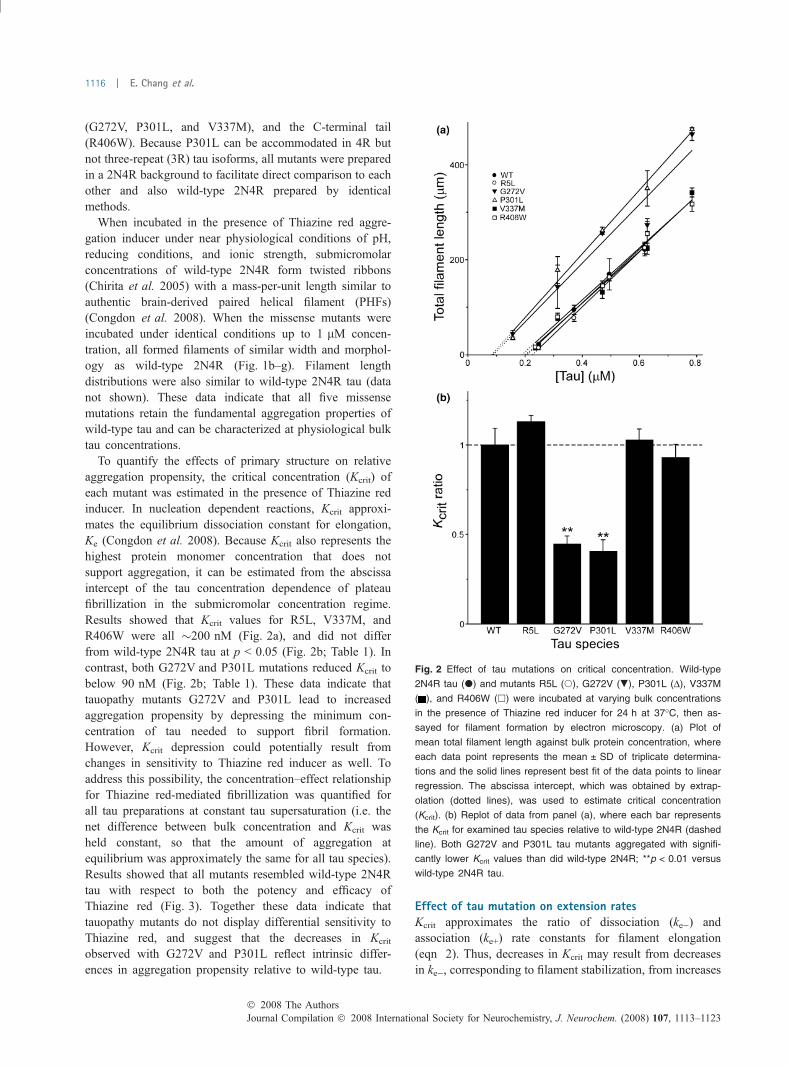
To quantify the effects of primary structure on relativeaggregation propensity, the critical concentration (Kcrit) ofeach mutant was estimated in the presence of Thiazine redinducer. In nucleation dependent reactions, Kcrit approxi-mates the equilibrium dissociation constant for elongation,Ke (Congdon et al. 2008). Because Kcrit also represents thehighest protein monomer concentration that does notsupport aggregation, it can be estimated from the abscissaintercept of the tau concentration dependence of plateaufibrillization in the submicromolar concentration regime.Results showed that Kcrit values for R5L, V337M, andR406W were all �200 nM (Fig. 2a), and did not differfrom wild-type 2N4R tau at p < 0.05 (Fig. 2b; Table 1). Incontrast, both G272V and P301L mutations reduced Kcrit tobelow 90 nM (Fig. 2b; Table 1). These data indicate thattauopathy mutants G272V and P301L lead to increasedaggregation propensity by depressing the minimum con-centration of tau needed to support fibril formation.However, Kcrit depression could potentially result fromchanges in sensitivity to Thiazine red inducer as well. Toaddress this possibility, the concentration–effect relationshipfor Thiazine red-mediated fibrillization was quantified forall tau preparations at constant tau supersaturation (i.e. thenet difference between bulk concentration and Kcrit washeld constant, so that the amount of aggregation atequilibrium was approximately the same for all tau species).Results showed that all mutants resembled wild-type 2N4Rtau with respect to both the potency and efficacy ofThiazine red (Fig. 3). Together these data indicate thattauopathy mutants do not display differential sensitivity toThiazine red, and suggest that the decreases in Kcrit
observed with G272V and P301L reflect intrinsic differ-ences in aggregation propensity relative to wild-type tau.
Effect of tau mutation on extension ratesKcrit approximates the ratio of dissociation (ke)) andassociation (ke+) rate constants for filament elongation(eqn 2). Thus, decreases in Kcrit may result from decreasesin ke), corresponding to filament stabilization, from increases
Fig. 2 Effect of tau mutations on critical concentration. Wild-type
2N4R tau (d) and mutants R5L (s), G272V (.), P301L (D), V337M
( ), and R406W (h) were incubated at varying bulk concentrations
in the presence of Thiazine red inducer for 24 h at 37�C, then as-
sayed for filament formation by electron microscopy. (a) Plot of
mean total filament length against bulk protein concentration, where
each data point represents the mean ± SD of triplicate determina-
tions and the solid lines represent best fit of the data points to linear
regression. The abscissa intercept, which was obtained by extrap-
olation (dotted lines), was used to estimate critical concentration
(Kcrit). (b) Replot of data from panel (a), where each bar represents
the Kcrit for examined tau species relative to wild-type 2N4R (dashed
line). Both G272V and P301L tau mutants aggregated with signifi-
cantly lower Kcrit values than did wild-type 2N4R; **p < 0.01 versus
wild-type 2N4R tau.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1113–1123� 2008 The Authors
1116 | E. Chang et al.

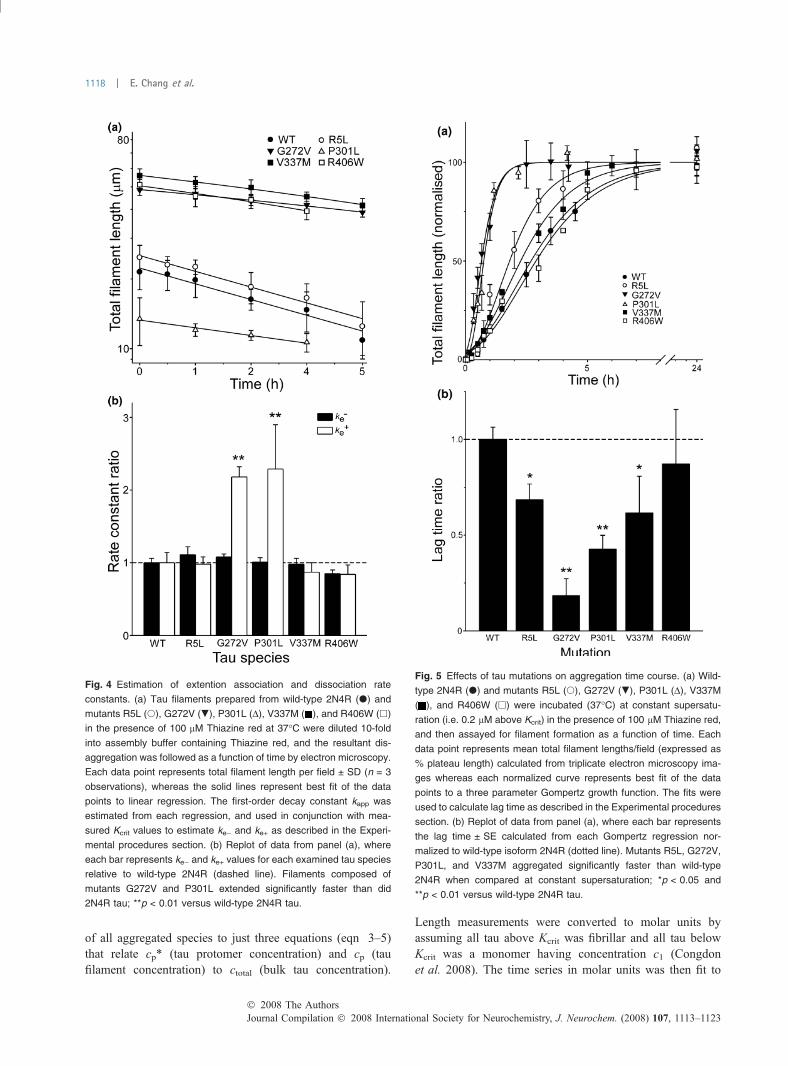
in ke+, reflecting a more efficient association reaction, or fromboth. To determine the mechanism of Kcrit depression, ke)was estimated for each tau preparation by diluting preas-sembled filaments in assembly buffer containing Thiazine redbut no tau protein and following the time-dependent loss offilament length by electron microscopy. Loss of filamentlength was first order as predicted for endwise depolymer-ization from a Poisson-like length distribution (Kristoffersonet al. 1980) (Fig. 4a). On the basis of the relationshipbetween tau mass and filament length established for wild-type 2N4R tau (Congdon et al. 2008), the dissociationelongation constant ke) was derived from the disaggregationrate of each mutant (Table 1). Rate constant ke+ was then
calculated from estimates of ke) and Kcrit for each mutantthrough eqn 2 (Table 1). Pairwise comparison of any mutantwith wild-type 2N4R tau failed to show a significantdifference in ke) at p < 0.05 (Fig. 4b). In contrast, bothG272V and P301L aggregated with significantly greater ke+values than wild-type tau. These data indicate that G272Vand P301L mutations depress Kcrit by increasing theefficiency of monomer addition to nascent filament ends.
Effect of tau mutation on nucleation ratesIn the presence of Thiazine red inducer, tau aggregationapproximates an equilibrium nucleation-elongation reaction,where assembly competent monomer rapidly equilibrateswith a thermodynamically unstable species termed thenucleus (Ferrone 1999). Once the critical nucleus clustersize is reached, subsequent additions to the nascent filamentends are energetically favorable and elongation proceedsefficiently. As a result, aggregation rate depends not only onthe rate of filament elongation, but on the efficiency of thenucleation step as well. To assess the effects of primarystructure on nucleation rate, the time course of aggregationwas quantified for each tau construct at constant supersat-uration. Under these conditions, differences in reaction ratesprimarily reflect differences in rates of nucleation and proteinconcentrations (Fesce et al. 1992). All resultant reactionprogress curves were sigmoidal with lag, exponential growth,and equilibrium phases (Fig. 5a). Lag times, which varyinversely with nucleation rate (Evans et al. 1995), wereobtained after fitting the data to a 3-parameter Gompertzgrowth function as described in Experimental procedures.The values are summarized in Table 1 and comparedgraphically with wild-type 2N4R tau in Fig. 5b. The datashow that all tauopathy mutants except R406W aggregatedwith significantly shorter lag times than wild-type 2N4R tau,suggesting that R5L, G272V, P301L, and V337M mutationscan directly accelerate the nucleation phase of the aggrega-tion reaction.
To quantify the nucleation reaction, all reaction progresscurves were fit to the approximation of Wegner and Engel(1975) as described previously for wild-type isoform 2N4R(Congdon et al. 2008). This approach simplifies the familyof differential equations describing nucleation and extension
Table 1 Summary of aggregation parameters
aKcrit (nM) ake) (per seconds) ake+ (per mM/s) Lag time (h) Kn (mM) bDGr (kcal/mol)
2N4R 200 ± 15 0.019 ± 0.001 94.8 ± 12.8 0.54 ± 0.09 15.0 6.9
R5L 226 ± 7 0.021 ± 0.002 92.9 ± 9.9 0.37 ± 0.12* 6.5 6.3
G272V 89 ± 8** 0.020 ± 0.001 229.0 ± 22.0** 0.10 ± 0.05** 2.2 6.2
P301L 80 ± 12** 0.019 ± 0.001 240.0 ± 38.0** 0.23 ± 0.04** 3.0 6.5
V337M 205 ± 12 0.019 ± 0.002 90.5 ± 9.3 0.33 ± 0.10* 8.6 6.6
R406W 186 ± 14 0.016 ± 0.001 87.1 ± 8.5 0.47 ± 0.15 11.8 6.8
aOverall constants reflecting events at both filament ends; bDGr = )RT lnr, where r = Kcrit/Kn. *p < 0.05 and **p < 0.01 versus 2N4R tau.
Fig. 3 Tau mutants share a common sensitivity to aggregation in-
ducer Thiazine red. Wild-type 2N4R (d) and mutants R5L (s), G272V
(.), P301L (D), V337M ( ), and R406W (h) were incubated (24 h at
37�C) at constant supersaturation (i.e. 0.5 lM above Kcrit) in the
presence of varying concentrations of Thiazine red, and then assayed
for filament formation by electron microscopy. Each data point repre-
sents mean total filament lengths/field from triplicate determinations,
whereas the solid curve is drawn solely to aid visualization. Under
these conditions, the concentration effect relationship for Thiazine red
was similar for all tau species.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1113–1123
Aggregation propensity of tauopathy mutants | 1117
of all aggregated species to just three equations (eqn 3–5)that relate cp* (tau protomer concentration) and cp (taufilament concentration) to ctotal (bulk tau concentration).
Length measurements were converted to molar units byassuming all tau above Kcrit was fibrillar and all tau belowKcrit was a monomer having concentration c1 (Congdonet al. 2008). The time series in molar units was then fit to
Fig. 4 Estimation of extention association and dissociation rate
constants. (a) Tau filaments prepared from wild-type 2N4R (d) and
mutants R5L (s), G272V (.), P301L (D), V337M ( ), and R406W (h)
in the presence of 100 lM Thiazine red at 37�C were diluted 10-fold
into assembly buffer containing Thiazine red, and the resultant dis-
aggregation was followed as a function of time by electron microscopy.
Each data point represents total filament length per field ± SD (n = 3
observations), whereas the solid lines represent best fit of the data
points to linear regression. The first-order decay constant kapp was
estimated from each regression, and used in conjunction with mea-
sured Kcrit values to estimate ke) and ke+ as described in the Experi-
mental procedures section. (b) Replot of data from panel (a), where
each bar represents ke) and ke+ values for each examined tau species
relative to wild-type 2N4R (dashed line). Filaments composed of
mutants G272V and P301L extended significantly faster than did
2N4R tau; **p < 0.01 versus wild-type 2N4R tau.
Fig. 5 Effects of tau mutations on aggregation time course. (a) Wild-
type 2N4R (d) and mutants R5L (s), G272V (.), P301L (D), V337M
( ), and R406W (h) were incubated (37�C) at constant supersatu-
ration (i.e. 0.2 lM above Kcrit) in the presence of 100 lM Thiazine red,
and then assayed for filament formation as a function of time. Each
data point represents mean total filament lengths/field (expressed as
% plateau length) calculated from triplicate electron microscopy ima-
ges whereas each normalized curve represents best fit of the data
points to a three parameter Gompertz growth function. The fits were
used to calculate lag time as described in the Experimental procedures
section. (b) Replot of data from panel (a), where each bar represents
the lag time ± SE calculated from each Gompertz regression nor-
malized to wild-type isoform 2N4R (dotted line). Mutants R5L, G272V,
P301L, and V337M aggregated significantly faster than wild-type
2N4R when compared at constant supersaturation; *p < 0.05 and
**p < 0.01 versus wild-type 2N4R tau.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1113–1123� 2008 The Authors
1118 | E. Chang et al.

eqns 3–5 assuming a dimeric nucleus (Congdon et al.2008) and constrained by the elongation parameters sum-marized in Table 1. The results confirm that tauopathymutants R5L, G272V, P301L, and V337M increase the
efficiency of the nucleation reaction, and indicate that theydo so by decreasing the dissociation equilibrium constantrelative to wild-type 2N4R (Table 1). In the cases of G272Vand P301L, the decreases in Kn were as much as five- tosevenfold.
Equations 4 and 5 predict that filament number concen-tration (cp) should be a function of tau concentration and thefour rate constants that govern the nucleation and extensionphases of the aggregation reaction. Therefore, filamentnumber is not a direct measure of any kinetic parameter,including nucleation rate. However, because both R5L andV337M display decreased Kn relative to wild-type 2N4Rwhile their elongation rate constants remain unchanged, theywould be predicted to yield more filaments relative to wild-type 2N4R tau when compared at identical tau concentra-tions. A simulation of this hypothetical reaction at constantsupersaturation (i.e. 0.2 lM above Kcrit) using the kineticconstants summarized in Table 1 is shown in Fig. 6a. R5Land V337M were predicted to produce �1.4- to 1.6-foldincreases in filament number concentration relative to wild-type 2N4R at reaction plateau. To test this prediction, R5L,V337M, and wild-type 2N4R were aggregated under theseconditions and then subjected to transmission electronmicroscopy to quantify filament number. The results showedthat R5L and V337M yielded 1.5 ± 0.2- and 1.7 ± 0.1-foldincreases in filament number relative to wild-type 2N4R tau,respectively (Fig. 6b). These data are consistent with theincreases in nucleation rates predicted by mathematicalsimulation, and support the hypothesis that missense tauop-athy mutants R5L and V337M act in part to accelerate thenucleation phase of the aggregation reaction.
The ratio of elongation and nucleation equilibriumconstants, termed r, provides an index of reaction cooper-ativity (Zhao and Moore 2003). Expressed in terms of freeenergy, DGr represents the difference in energy that accom-panies contacts formed in the elongation step relative to thenucleation step. DGr varied over the narrow range of 6.2–6.9 kcal/mol for wild-type and mutant tau preparations(Table 1), indicating that the free energy of nucleation wasconsistently less favorable than that for elongation. Overallthese data indicate that tauopathy mutants can strongly anddirectly increase aggregation propensity at nucleation andextension steps while retaining the cooperative aggregationmechanism of wild-type tau.
Discussion
Certain tau missense mutations cause FTLD, indicating thattau dysfunction alone can induce neurofibrillary lesionformation, neurodegeneration, and cognitive decline. Thefindings here, along with those from previous studies, areconsistent with the disease promoting activity of somemissense mutations being related to augmentation of tauaggregate formation. In the case of the R5L, G272V, P301L,
Fig. 6 Mutants R5L and V337M yield increased filament number
concentration. (a) Mathematical simulation of reaction time course
for R5L, V337M, and wild-type 2N4R tau at constant supersaturation
(i.e. 0.2 lM above Kcrit) using eqns 3–5 and the kinetic parameters
summarized in Table 1. Each curve represents the predicted evo-
lution of filament number concentration (cp) as function of time. The
simulations predict that the decreases in Kn associated with R5L
and V337M should yield increases in filament number concentration
relative to 2N4R tau under these conditions. (b) R5L, V337M, and
wild-type 2N4R were incubated (37�C) under identical conditions as
in panel (a) and then subjected to electron microscopy analysis at
24 h. Each column represents mean total filament number/
field ± SD (normalized to wild-type 2N4R tau) calculated from qua-
druplicate electron microscopy images. Consistent with mathemati-
cal simulation, R5L and V337M yielded significantly more tau
filaments relative to wild-type 2N4R tau at reaction plateau;
**p < 0.01 and n.sp > 0.05.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1113–1123
Aggregation propensity of tauopathy mutants | 1119

and V337M, aggregation was promoted directly at the levelof intrinsic aggregation propensity. The largest effects onaggregation were observed with G272V and P301L muta-tions, which are located in exons 9 and 10 regions thatencode the first and second binding repeats, respectively. Asexon 10 is alternatively spliced, P301L only affects four-repeat isoforms of tau whereas G272Vaffects both three- andfour-repeat isoforms. Both mutants promote filament nucle-ation by decreasing the dissociation equilibrium constant fordimerization and support aggregation at low tau concentra-tions by lowering the dissociation equilibrium constant forfilament extension. For both mutants, effects on the extensionreaction are driven primarily by an increase in the associationrate constant, ke+, governing association of tau monomerwith filament ends. Therefore, the aggregation of G272V andP301L tau is predicted to be highly sensitive to changes infree cytosolic tau concentrations. Although bulk tau levelsare reportedly low micromolar (Drubin et al. 1985; Khatoonet al. 1994), normal free cytosolic concentrations are wellbelow this owing to high-affinity microtubule binding andsequestration (Ackmann et al. 2000; Makrides et al. 2004).Thus, the ability of G272V and P301L to support rapid tauaggregation at submicromolar concentrations is likely to beespecially important in the early stages of disease when taudissociation from microtubules is incomplete.
With respect to structure, both mutations affect PGGGsegments located in the repeat region of several microtubule-associated proteins (Lewis et al. 1988; Olson et al. 1995);G272Valters the segment in the first repeat to 270PGVG273,whereas P301L alters the segment in the second repeat to301LGGG304. Each segment lies adjacent to hexapeptidemotifs involved in transition of the repeat region to cross-b-sheet structure (von Bergen et al. 2000). Both mutationsincrease local hydrophobicity, which is a major determinantof aggregation propensity (Pawar et al. 2005; Rojas Quijanoet al. 2006; Tartaglia et al. 2008), and a hypothetical modellinking the conformation of PGGG segments to localhexapeptide b-sheet formation has been proposed (vonBergen et al. 2001). The similarity in aggregation propensityprofile – reduced Kcrit, increased ke+, and reduced lag timeand Kn – are consistent with these two mutations targetinghomologous segments of the repeat region.
In contrast, the R5L mutation led to increased nucleationrate but no change in extension kinetics relative to wild-typetau. The effects on nucleation are consistent with the abilityof another missense mutation at the same site, R5H, toincrease numbers of filaments formed at high concentrationsin vitro relative to wild-type tau (Hayashi et al. 2002). As thecore of the PHF is mainly composed of the repeat region oftau (Novak et al. 1993), it is perhaps surprising that an N-terminal mutation can result in increased aggregationpropensity. However, conformation-sensitive antibody bind-ing studies suggest that the N-terminus is in proximity to orinfluenced by the microtubule binding repeat region (Carmel
et al. 1996). In fact, the N-terminus of tau may contact therepeat domain in tau monomers (Jeganathan et al. 2006), andelimination of this interaction through truncation results indecreased aggregation rates in vitro (Gamblin et al. 2003).The change of Arg to Leu eliminates a positive charge andagain introduces a hydrophobic residue, both of which arepredicted to increase aggregation propensity (Pawar et al.2005; Rojas Quijano et al. 2006; Tartaglia et al. 2008).
Mutation V337M is found in the third repeat, whichcomposes part of the PHF core (Novak et al. 1993).Nonetheless, this region is not essential for arachidonic acidinduced fibrillization in vitro (Abraha et al. 2000). V337Mresembles R5L in terms of aggregation propensity, displayingan increased nucleation rate without change in Kcrit, ke), orke+. Consistent with these observations, a previous studyshowed an increase in aggregation rate but not in plateaulevels in the presence of heparin (Nacharaju et al. 1999).These data are consistent with V337M favoring fasternucleation rate with little effect on the extension reaction. Asimilar spectrum of activity may accompany K257T, whichreportedly increases filament number (Rizzini et al. 2000) butnot total filament mass at reaction plateau (Grover et al.2003).
R406W is located in the C-terminal segment flanking therepeat region. Although truncation of this region reportedlyincreases extent of aggregation (Abraha et al. 2000), here itwas found that R406W did not differ from wild-type tau withrespect to aggregation propensity. Therefore, the biologicaleffects of R406Won aggregation are most likely indirect, anddepend on differential interaction with cellular factors, suchas phosphotransferases (Goedert et al. 2000; Alonso et al.2004).
Interplay of tau mutations with cellular factorsTau aggregation in cells is a complex process, where theintrinsic aggregation propensity of tau proteins is modulatedby interactions with other factors. We have proposed that thepathway involves four principal steps that must be overcomefor filamentous aggregates to accumulate in disease (Cong-don et al. 2008) (Fig. 7). First, tau must dissociate frommicrotubules so that its free cytosolic concentration exceedsthe minimal tau concentration necessary to support aggrega-tion. Although marked decreases in microtubule assemblykinetics have been observed for many missense mutations,including G272V, P301L, V337M, and R406W (Hasegawaet al. 1998; Barghorn et al. 2000; DeTure et al. 2000;Poorkaj et al. 2002), their binding affinity for microtubules,which may more closely reflect normal tau function (Qianget al. 2006), is only weakly affected in vitro and remainshigh affinity (Barghorn et al. 2000; DeTure et al. 2000). Solong as the dissociation equilibrium constant for bindingremains lower than the concentration of available tau bindingsites on microtubules, small changes in affinity may notappreciably affect free tau concentrations. In Michigan
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1113–1123� 2008 The Authors
1120 | E. Chang et al.

Cancer Foundation 7 (MCF7) cells, for example, mutant tau(i.e. G272V, P301L, or R406W) microinjected at physiolog-ical concentrations colocalizes with microtubules much likewild-type tau (Bunker et al. 2006). Similarly, bulk tau levelsraised by increased expression or decreased degradation oftau may have limited effects on free tau levels. R5L isassociated with approximately twofold bulk tau over-expres-sion (Poorkaj et al. 2002), whereas V337M and R406Wstabilize tau against proteolytic degradation by calpain I (EC3.4.22.52) (Yen et al. 1999). Although the magnitude ofthese changes may be insufficient to overcome the largeexcess of tubulin in cells (Drubin et al. 1985), both increasedexpression and decreased turnover could make more tauavailable for aggregation once liberated from microtubulesby other means. Binding affinity is modulated by post-translational modifications such as phosphorylation (Biernatet al. 1993; Bramblett et al. 1993), which may be theprimary gatekeeper controlling the amount of free tauavailable for aggregation. The missense tauopathy mutantsexamined here (R5L, G272V, P301L, V337M, and R406W)all have been reported to be better protein kinase substratesin vitro, whereas G272V, P301L, V337M, and R406W areweaker phosphoprotein phosphatase 2A (EC 3.1.3.16) bind-ing partners than wild-type tau isoforms (Goedert et al. 2000;Alonso et al. 2004). Both of these interactions would beexpected to increase occupancy of phosphorylation sitesmediating tau-microtubule affinity, and to increase amountsof cytosolic tau available for aggregation (regardless of anydirect affects of phosphorylation on the aggregation reac-tion). Overall, changes in microtubule binding affinity,expression levels, and degradation may synergize with theeffects of post-translational modifications on tau-microtubuleequilibria to increase the amount of free tau available for
aggregation. These interactions may be especially importantfor aggregation of R406W which, as shown here, does notdirectly modify intrinsic aggregation propensity.
The second step involves a conformational change to anassembly competent state (Fig. 7). This step is proposed tobe a barrier to aggregation because high concentrations (i.e.up to 100 lM) of free tau alone are insufficient to supportaggregation or seeding reactions in vitro (Ko et al. 2002). Incontrast, tau proteins readily aggregate in the presence ofanionic inducers such as Thiazine red. The effects ofmissense mutations on this step are not clear, but on thebasis of infrared spectroscopy, neither G272V, P301L,V337M, nor R406W increases secondary structure contentof tau monomers relative to wild-type constructs (von Bergenet al. 2001).
Once aggregation-competent conformations are adopted,the rate-limiting step in filament formation becomes dimer-ization, which is energetically disfavored at physiological tauconcentrations, and therefore a third key point of control(Fig. 7). As shown here, missense mutations can directlymodulate this step. Because of its dimeric nature, nucleusstructure may be a key determinant of whether 3R or 4Rforms of tau predominate in aggregates. For example,disulfide bond formation can favor nucleation of 3R isoformswhile inhibiting nucleation of 4R isoforms (Schweers et al.1995).
The final step in filament formation is extension. Althoughnot rate-limiting, equilibria at filament ends dictate theminimal concentration of tau required to support aggregation.In addition to promoting microtubule dissociation, thenegative charge associated with phosphorylation can enhancethe extension reaction by inhibiting filament dissociation(Necula and Kuret 2004c, 2005). This reaction may synergize
Fig. 7 Effect of FTLD mutations on the fibrillization pathway. Normal
tau binds tightly to microtubules, but dissociates upon phosphorylation
to form free tau, which exists as a natively disordered, assembly
incompetent monomer (Ux). A conformational change to an
assembly competent state accelerates polymerization (Uc). Once
assembly competent species form, the rate-limiting step in tau
fibrillization is formation of dimer, which represents the thermodynamic
nucleus (N). Following nucleation, extension occurs through further
addition of assembly competent monomers to the filament (F) ends.
Tau mutations promote fibrillization at multiple points in the path-
way. Reactions characterized herein are shown in green lettering,
whereas those reported in the literature are shown in red lettering. See
text for details.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1113–1123
Aggregation propensity of tauopathy mutants | 1121

with the effects of P301L and G272Von extension to furtheraugment the rate of monomer association with filament ends.
Overall, the findings here are consistent with a growingliterature suggesting that missense tau mutations promote tauaggregation by acting at multiple steps along a singlepathway. For some mutations, these include direct effects onthe rate and/or extent of aggregation. The differential activityof missense mutations on individual steps in the pathwaymay influence how tau misfunction leads to clinically andhistopathologically distinct diseases.
Acknowledgements
We thank Ranjan Batra and Lauren Crissman for technical
assistance. This work was supported by Grants from the National
Institutes of Health (AG14452), the Alzheimer’s Association (IIRG-
05-14288), and NIH CTSA Grant UL1RR025755-01 awarded to
The Ohio State University.
References
Abraha A., Ghoshal N., Gamblin T. C., Cryns V., Berry R. W., KuretJ. and Binder L. I. (2000) C-terminal inhibition of tau assemblyin vitro and in Alzheimer’s disease. J. Cell Sci. 113, 3737–3745.
Ackmann M., Wiech H. and Mandelkow E. (2000) Nonsaturable bindingindicates clustering of tau on the microtubule surface in a pairedhelical filament-like conformation. J. Biol. Chem. 275, 30335–30343.
Alonso A., Mederlyova A., Novak M., Grundke-Iqbal I. and Iqbal K.(2004) Promotion of hyperphosphorylation by frontotemporaldementia tau mutations. J. Biol. Chem. 279, 34873–34881.
Barghorn S., Zheng-Fischhofer Q., Ackmann M., Biernat J., von BergenM., Mandelkow E. M. and Mandelkow E. (2000) Structure,microtubule interactions, and paired helical filament aggregationby tau mutants of frontotemporal dementias. Biochemistry 39,11714–11721.
von Bergen M., Friedhoff P., Biernat J., Heberle J., Mandelkow E. M.and Mandelkow E. (2000) Assembly of tau protein into Alzheimerpaired helical filaments depends on a local sequence motif(306VQIVYK311) forming b structure. Proc. Natl Acad. Sci. USA97, 5129–5134.
von Bergen M., Barghorn S., Li L., Marx A., Biernat J., MandelkowE. M. and Mandelkow E. (2001) Mutations of tau protein infrontotemporal dementia promote aggregation of paired helicalfilaments by enhancing local beta-structure. J. Biol. Chem. 276,48165–48174.
Biernat J., Gustke N., Drewes G., Mandelkow E. M. and Mandelkow E.(1993) Phosphorylation of Ser262 strongly reduces binding of tauto microtubules: distinction between PHF-like immunoreactivityand microtubule binding. Neuron 11, 153–163.
Bramblett G. T., Goedert M., Jakes R., Merrick S. E., TrojanowskiJ. Q. and Lee V. M. (1993) Abnormal tau phosphorylation atSer396 in Alzheimer’s disease recapitulates development andcontributes to reduced microtubule binding. Neuron 10, 1089–1099.
Bunker J. M., Kamath K., Wilson L., Jordan M. A. and Feinstein S. C.(2006) FTDP-17 mutations compromise the ability of tau to reg-ulate microtubule dynamics in cells. J. Biol. Chem. 281, 11856–11863.
Carmel G., Mager E. M., Binder L. I. and Kuret J. (1996) The structuralbasis of monoclonal antibody Alz50’s selectivity for Alzheimer’sdisease pathology. J. Biol. Chem. 271, 32789–32795.
Chirita C. N., Congdon E. E., Yin H. and Kuret J. (2005) Triggers offull-length tau aggregation: a role for partially folded intermediates.Biochemistry 44, 5862–5872.
Congdon E. E., Kim S., Bonchak J., Songrug T., Matzavinos A. andKuret J. (2008) Nucleation-dependent tau filament formation: theimportance of dimerization and an estimation of elementary rateconstants. J. Biol. Chem. 283, 13806–13816.
Denk F. and Wade-Martins R. (2008) Knock-out and transgenic mousemodels of tauopathies. Neurobiol. Aging (In press).
DeTure M., Ko L. W., Yen S., Nacharaju P., Easson C., Lewis J., vanSlegtenhorst M., Hutton M. and Yen S. H. (2000) Missense taumutations identified in FTDP-17 have a small effect on tau-microtubule interactions. Brain Res. 853, 5–14.
Drubin D. G., Feinstein S. C., Shooter E. M. and Kirschner M. W. (1985)Nerve growth factor-induced neurite outgrowth in PC12 cells in-volves the coordinate induction of microtubule assembly andassembly-promoting factors. J. Cell Biol. 101, 1799–1807.
Evans K. C., Berger E. P., Cho C. G., Weisgraber K. H. and LansburyP. T. Jr (1995) Apolipoprotein E is a kinetic but not a thermody-namic inhibitor of amyloid formation: implications for the patho-genesis and treatment of Alzheimer disease. Proc. Natl Acad. Sci.USA 92, 763–767.
Ferrone F. (1999) Analysis of protein aggregation kinetics. MethodsEnzymol. 309, 256–274.
Fesce R., Benfenati F., Greengard P. and Valtorta F. (1992) Effects of theneuronal phosphoprotein synapsin I on actin polymerization. II.Analytical interpretation of kinetic curves. J. Biol. Chem. 267,11289–11299.
Gamblin T. C., King M. E., Dawson H., Vitek M. P., Kuret J., BerryR. W. and Binder L. I. (2000) In vitro polymerization of tau proteinmonitored by laser light scattering: method and application to thestudy of FTDP-17 mutants. Biochemistry 39, 6136–6144.
Gamblin T. C., Berry R. W. and Binder L. I. (2003) Tau polymerization:role of the amino terminus. Biochemistry 42, 2252–2257.
Goedert M., Jakes R. and Crowther R. A. (1999) Effects of frontotem-poral dementia FTDP-17 mutations on heparin-induced assemblyof tau filaments. FEBS Lett. 450, 306–311.
Goedert M., Satumtira S., Jakes R., Smith M. J., Kamibayashi C., WhiteC. L. III and Sontag E. (2000) Reduced binding of protein phos-phatase 2A to tau protein with frontotemporal dementia and par-kinsonism linked to chromosome 17 mutations. J. Neurochem. 75,2155–2162.
Goode B. L., Chau M., Denis P. E. and Feinstein S. C. (2000) Struc-tural and functional differences between 3-repeat and 4-repeattau isoforms. Implications for normal tau function and theonset of neurodegenetative disease. J. Biol. Chem. 275, 38182–38189.
Grover A., England E., Baker M. et al. (2003) A novel tau mutation inexon 9 (1260V) causes a four-repeat tauopathy. Exp. Neurol. 184,131–140.
Hasegawa M., Smith M. J. and Goedert M. (1998) Tau proteins withFTDP-17 mutations have a reduced ability to promote microtubuleassembly. FEBS Lett. 437, 207–210.
Hayashi S., Toyoshima Y., Hasegawa M., Umeda Y., Wakabayashi K.,Tokiguchi S., Iwatsubo T. and Takahashi H. (2002) Late-onsetfrontotemporal dementia with a novel exon 1 (Arg5His) tau genemutation. Ann. Neurol. 51, 525–530.
Hogg M., Grujic Z. M., Baker M. et al. (2003) The L266V tau mutationis associated with frontotemporal dementia and Pick-like 3R and4R tauopathy. Acta Neuropathol. (Berl.) 106, 323–336.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1113–1123� 2008 The Authors
1122 | E. Chang et al.

Jeganathan S., von Bergen M., Brutlach H., Steinhoff H. J. andMandelkow E. (2006) Global hairpin folding of tau in solution.Biochemistry 45, 2283–2293.
Khatoon S., Grundke-Iqbal I. and Iqbal K. (1994) Levels of normal andabnormally phosphorylated tau in different cellular and regionalcompartments of Alzheimer disease and control brains. FEBS Lett.351, 80–84.
Ko L. W., DeTure M., Sahara N., Chihab R. and Yen S. H. (2002)Cellular models for tau filament assembly. J. Mol. Neurosci. 19,311–316.
Kristofferson D., Karr T. L. and Purich D. L. (1980) Dynamics of linearprotein polymer disassembly. J. Biol. Chem. 255, 8567–8572.
Kuret J., Congdon E. E., Li G., Yin H., Yu X. and Zhong Q. (2005)Evaluating triggers and enhancers of tau fibrillization. Microsc.Res. Tech. 67, 141–155.
Lewis S. A., Wang D. H. and Cowan N. J. (1988) Microtubule-associ-ated protein MAP2 shares a microtubule binding motif with tauprotein. Science 242, 936–939.
Makrides V., Massie M. R., Feinstein S. C. and Lew J. (2004) Evidencefor two distinct binding sites for tau on microtubules. Proc. NatlAcad. Sci. USA 101, 6746–6751.
Nacharaju P., Lewis J., Easson C., Yen S., Hackett J., Hutton M. and YenS. H. (1999) Accelerated filament formation from tau protein withspecific FTDP-17 missense mutations. FEBS Lett. 447, 195–199.
Neary D., Snowden J. and Mann D. (2005) Frontotemporal dementia.Lancet Neurol. 4, 771–780.
Necula M. and Kuret J. (2004a) A static laser light scattering assay forsurfactant-induced tau fibrillization. Anal. Biochem. 333, 205–215.
Necula M. and Kuret J. (2004b) Electron microscopy as a quantitativemethod for investigating tau fibrillization. Anal. Biochem. 329,238–246.
Necula M. and Kuret J. (2004c) Pseudophosphorylation and glycation oftau protein enhance but do not trigger fibrillization in vitro. J. Biol.Chem. 279, 49694–49703.
Necula M. and Kuret J. (2005) Site-specific pseudophosphorylationmodulates the rate of tau filament dissociation. FEBS Lett. 579,1453–1457.
Neumann M., Schulz-Schaeffer W., Crowther R. A., Smith M. J.,Spillantini M. G., Goedert M. and Kretzschmar H. A. (2001) Pick’sdisease associated with the novel Tau gene mutation K369I. Ann.Neurol. 50, 503–513.
Novak M., Kabat J. and Wischik C. M. (1993) Molecular characteriza-tion of the minimal protease resistant tau unit of the Alzheimer’sdisease paired helical filament. EMBO J. 12, 365–370.
Olson K. R., McIntosh J. R. and Olmsted J. B. (1995) Analysis of MAP4 function in living cells using green fluorescent protein (GFP)chimeras. J. Cell Biol. 130, 639–650.
Pawar A. P., Dubay K. F., Zurdo J., Chiti F., Vendruscolo M. and DobsonC. M. (2005) Prediction of ‘‘aggregation-prone’’ and ‘‘aggregation-susceptible’’ regions in proteins associated with neurodegenerativediseases. J. Mol. Biol. 350, 379–392.
Pickering-Brown S. M., Baker M., Nonaka T. et al. (2004) Frontotem-poral dementia with Pick-type histology associated with Q336Rmutation in the tau gene. Brain 127, 1415–1426.
Poorkaj P., Muma N. A., Zhukareva V. et al. (2002) An R5L tau mutationin a subject with a progressive supranuclear palsy phenotype. Ann.Neurol. 52, 511–516.
Qiang L., Yu W., Andreadis A., Luo M. and Baas P. W. (2006) Tauprotects microtubules in the axon from severing by katanin.J. Neurosci. 26, 3120–3129.
Rademakers R., Cruts M. and van Broeckhoven C. (2004) The role oftau (MAPT) in frontotemporal dementia and related tauopathies.Hum. Mutat. 24, 277–295.
Rizzini C., Goedert M., Hodges J. R., Smith M. J., Jakes R., Hills R.,Xuereb J. H., Crowther R. A. and Spillantini M. G. (2000) Taugene mutation K257T causes a tauopathy similar to Pick’s disease.J. Neuropathol. Exp. Neurol. 59, 990–1001.
Rojas Quijano F. A., Morrow D., Wise B. M., Brancia F. L. and Goux W.J. (2006) Prediction of nucleating sequences from amyloidogenicpropensities of tau-related peptides. Biochemistry 45, 4638–4652.
Schweers O., Mandelkow E. M., Biernat J. and Mandelkow E. (1995)Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein tau controls the in vitro assembly of pairedhelical filaments. Proc. Natl Acad. Sci. USA 92, 8463–8467.
Spina S., Murrell J. R., Yoshida H. et al. (2007) The novel Tau mutationG335S: clinical, neuropathological and molecular characterization.Acta Neuropathol. 113, 461–470.
Tartaglia G. G., Pawar A. P., Campioni S., Dobson C. M., Chiti F. andVendruscolo M. (2008) Prediction of aggregation-prone regions instructured proteins. J. Mol. Biol. 380, 425–436.
Wegner A. and Engel J. (1975) Kinetics of the cooperative association ofactin to actin filaments. Biophys. Chem. 3, 215–225.
Yen S., Easson C., Nacharaju P., Hutton M. and Yen S. H. (1999) FTDP-17 tau mutations decrease the susceptibility of tau to calpain Idigestion. FEBS Lett. 461, 91–95.
Zhao D. and Moore J. S. (2003) Nucleation-elongation: a mechanism forcooperative supramolecular polymerization. Org. Biomol. Chem. 1,3471–3491.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 107, 1113–1123
Aggregation propensity of tauopathy mutants | 1123





























