Embed Size (px)
Citation preview

Course: Nutrition and Metabolism
Part (3): Amino Acids & Protein Metabolism
Lecture (4): Biosynthesis of Non-essential Amino Acids
Dr. Nuha AminMobile: +249910050800

Classification of Amino Acids
According to the diet, amino acids can be classified into
• Essential amino acids -our body cannot
synthesize them.
• Nonessential amino acids-our body can
synthesize them

Essential Amino Acids in Humans10 amino acids are essential and must be provided by
the diet
– Phenylalanine
– Valine
– Threonine
– Tryptophan PVT TIM HALL
– Isoleucine
– Methionine
– Histidine *
– Arginine *
– Leucine
– Lysine
*Required to some degree in young growing children.

Non-essential amino acids in humans
Amino acids that can be synthesized in the body.
Alanine
Asparagine
Aspartate
Glutamate
Glutamine
Glycine
Proline
Serine
Cysteine (from Met*)
Tyrosine (from Phe*)
* Essential amino acids

SYNTHESIS OF NON-ESSENTIAL AMINO
ACIDS
• All ,except tyrosine, are synthesized from
common intermediates of the TCA cycle
and glycolysis.
– PYRUVATE
– OXALOACETATE
– -KETOGLUTARATE
– 3-PHOSPHOGLYCERATE

Synthesis of Amino Acids
• In humans, transamination of compounds from glycolysis or the citric acid cycle produces nonessential amino acids.

Biosynthesis of amino acids: transamination reactions
amino acid1 +-keto acid2 amino acid2 +a-keto acid1

From pyruvate
• alanine

CH3 C C-OH
O O
Glutamate -ketoglutarate
CH3 CH C-OH
NH2 O
Pyruvate Alanine
Alanine
transaminase
PLP
1. Alanine Biosynthesis

From oxaloacetate
• Aspartate
• Asparagine

O
Glutamate -ketoglutarate
NH2
Oxaloacetate Aspartate
Aspartate
transaminase
PLPHO-C-CH2-CH-C-OH
O O
HO-C-CH2-C-C-OH
O O
2. Aspartate Biosynthesis

Chapter 17
3.Asparagine Biosynthesis


Chapter 17
4.Glutamate synthesis
• Addition of an amino group to -ketoglutarate
produces glutamate.
• The enzyme is called glutamate dehydrogenase

5.Glutamine synthesis
• Glutamine is synthesized by adding another amino
group to glutamate using glutamine synthetase.

Chapter 17
6. Proline and Arginine Biosynthesis


7.Serine Biosynthesis

8. Glycine Biosynthesis
NH2 C C-OH
CH2 O
THFmethylene THF
NH2 CH C-OH
H O
Serine Glycine
Serine hydroxymethyltransferase
OH

The sulfur for cysteine synthesis comes from the essential
amino acid methionine.
SAM serves as a precurosor for numerous methyl transfer reactions (e.g. the
conversion of norepinephrine to epinenephrine).
9.Cysteine Biosynthesis
Condensation of ATP and
methionine yield S-
adenosylmethionine (SAM)
SAM

1. Conversion of
SAM to
homocysteine.
2. Condensation of
homocysteine
with serine to
cystathione.
3. Cystathione is
cleavaged to
cysteine.
*

Methionine is catabolised through cysteine synthesis
Propionyl
CoA
Succinyl CoA

10. Tyrosine Biosynthesis

INBORN ERRORS OF METABOLISM
Genetic diseases!

What is an inborn error disease?
A
D
B Csubstrate
excess
Toxic compound
no product

26
– Absence of phenylalanine hydroxylase
enzyme
1.Phenylketonuria

Phenylketonuria (PKU) Disease
CH2CCO2-
O
Phe
Tyr
Transamination
Phenylpyruvate
(urine)
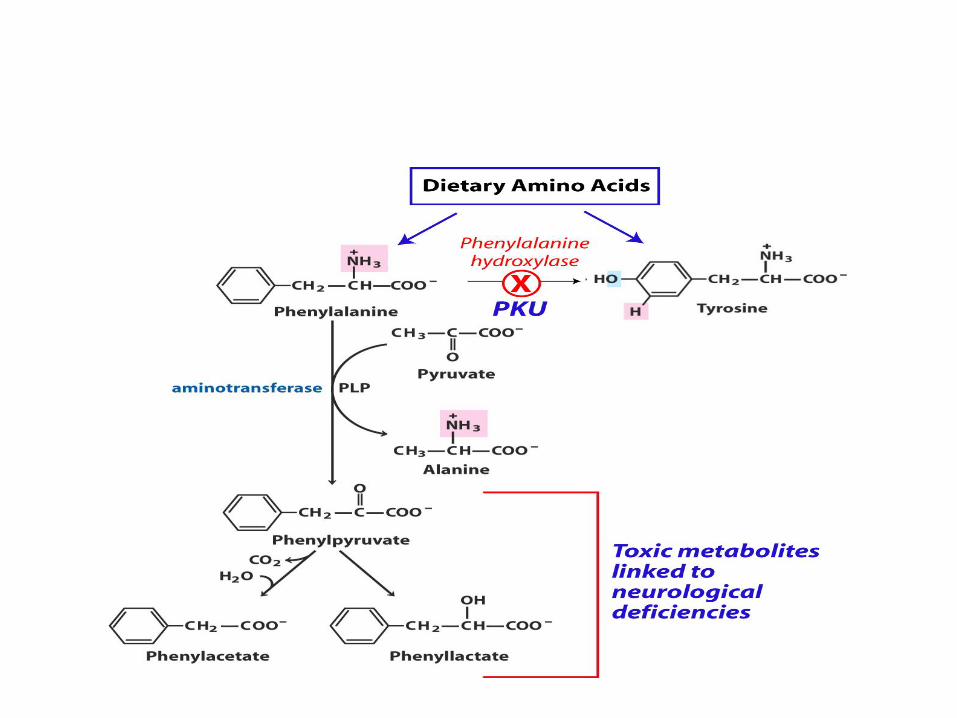

The clinical symptoms of PKU are
caused by
• the accumulation of phenylalanine in the blood
that is 30-50 times higher than normal.
• This high level of phenylalanine leads to the
production of phenylalanine metabolites such as
phenylpyruvate, phenylacetate and
phenyllactate
• these lead to the neurological and developmental
problems.

Disease symptoms of PKU include
• severe mental retardation,
• stunted growth

Treatment of Phenylketonuria (PKU)
Decreasing dietary PHE prevents brain damage
in children with PKU.
PHE is less damaging once brain has developed.
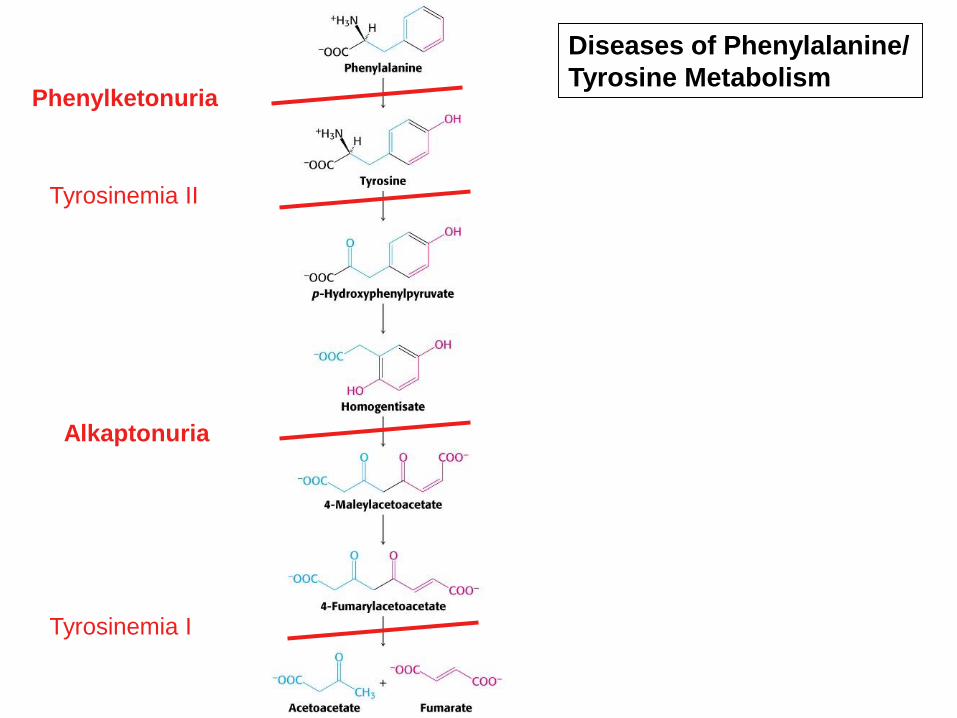
Phenylketonuria
Alkaptonuria
Tyrosinemia II
Tyrosinemia I
Diseases of Phenylalanine/
Tyrosine Metabolism

33
– Absence of homogentisate oxidase activity
2.Alkaptonuria
Dark
urine

Alkaptonuria - Clinical signs
• The pigmentation is called ochronosis.
– Note the pigmentation of the ear and eyes.
• The arthritis is associated with calcification of joints.

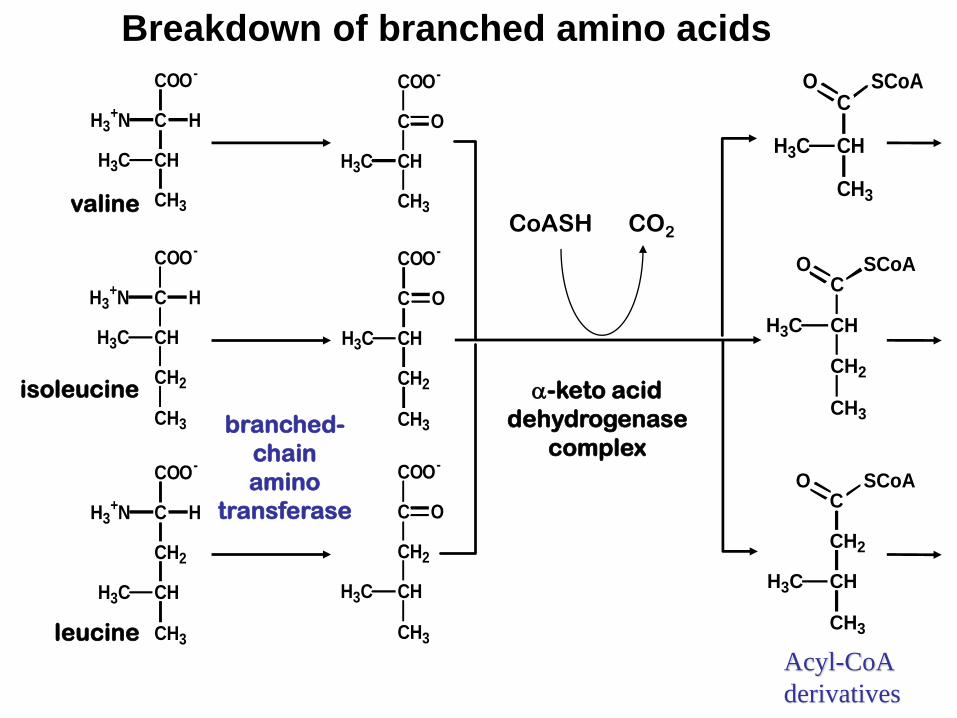
3.Maple syrup urine disease
Genetic disease caused by deficiency of Branched Chain alpha-Keto Acid Dehydrogenase
Alpha keto amino acids, valine, isolucine and
leucine will accumulated in the blood and excreted
in the urine (smells like maple syrup).
If untreated, mental retardation and early death.
COO-
CH3+N
CHH3C
CH3
H
COO-
CH3+N
CHH3C
CH2
H
CH3
COO-
CH3+N
CH2
CH
H
CH3
H3C
COO-
C
CHH3C
CH3
O
COO-
C
CHH3C
CH2
O
CH3
COO-
C
CH2
CH
O
CH3
H3C
C
CHH3C
CH3
O SCoA
C
CHH3C
CH2
CH3
O SCoA
C
CH2
CH
CH3
H3C
O SCoA
CoASH CO2
-keto acid
dehydrogenase
complexbranched-
chain
amino
transferase
valine
isoleucine
leucine
Acyl-CoA
derivatives
Breakdown of branched amino acids
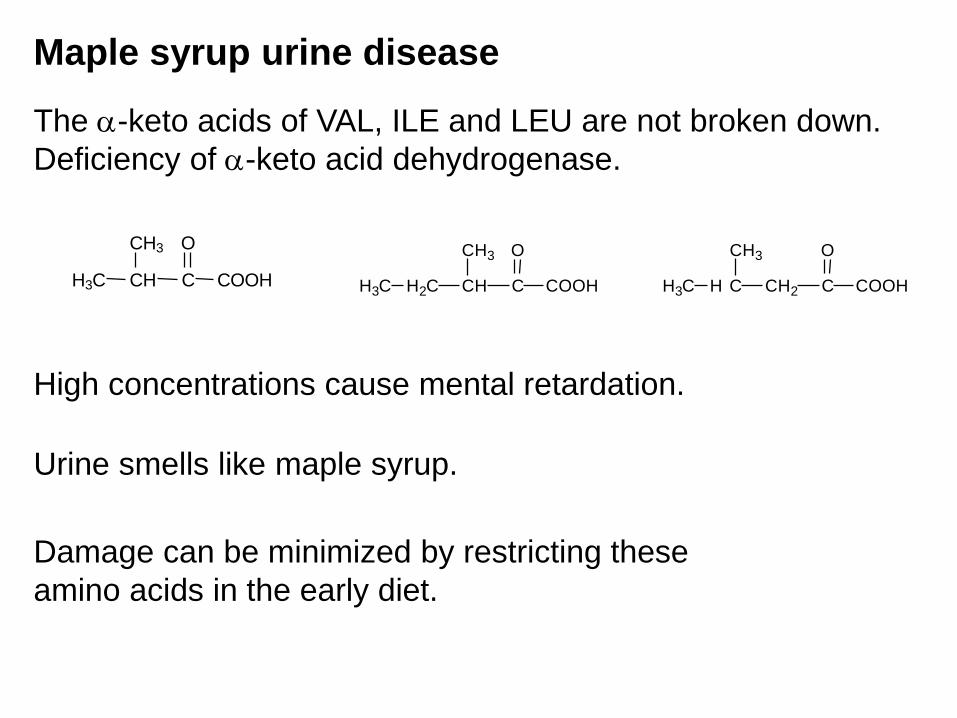
Maple syrup urine disease
The -keto acids of VAL, ILE and LEU are not broken down.
Deficiency of -keto acid dehydrogenase.
Urine smells like maple syrup.
High concentrations cause mental retardation.
Damage can be minimized by restricting these
amino acids in the early diet.
H2C CH C
CH3
COOH
O
H3CH3C CH C
CH3
COOH
O
H C CH2 C COOH
O
H3C
CH3
























