Embed Size (px)
Citation preview

Paediatric Extracranial Solid TumoursFRACP Teaching 2017
Dr. Andy Wood, FRACPPaediatric Haematologist‐OncologistUniversity of Auckland
Cancer Most Common Cause of Death From a Disease Age 1 – 19 Years
1
Neoplasms29%
Diseases of the nervous system
17%Congenital anomalies
Diseases of the respiratory system
10%
Diseases of the circulatory system
9%
Infectious and parasitic disease
7%
Endocrine, nutritional and
metabolic diseases6%
Other11%
MORTALITY BY MEDICAL CAUSE OF DEATH, AGE 1‐19 YEARS, NEW ZEALAND, 2010‐2014
2
1/3 Paediatric Cancers are Solid + Outside Brain 75 Minutes To Cover
1. Introduction 2. Practical Principles3. Major solid tumours ‐Wilms, Sarcomas, NB, Liver,
GCT, Melanoma briefly4. Key Cancer Predispositions (depending on time)5. MCQ with full explanations will be available on pdf if
run out of time.6. RED = important.
3

Definitions
• Uncontrolled cellular proliferation mass / “tumour”• A tumour is a “cancer” if it has: a) spread, b )potential to spread
• Process of metastasis produces metastases = metastatic cancer
• In clinical usage cancer may be used to refer to some non‐inflammatory tumours with deleterious local effects, e.g. compression, obstruction, damage.
• Solid tumours arise from solid organs• Blood cells can form masses too – lymphoma and leukaemia can form chloroma
Genome Malfunction Cancer
• The normal human genome• 3 billion DNA bases from both your mum and dad • = 6 billion base pairs in total• Arranged into 23 pieces called chromosomes• Genes encode ~ 23,000 proteins but each protein has large number of variations
• Several thousand proteins expressed in any given cell • Protein expression specific to developmental stage, tissue and cell, environmental and physiological context
• Significant variation from person to person
Mutations Inherent Risk of DNA• Single cell to adult human with 37 trillion cells so replication / repair reliable
• Controlled alterations to the genetic code occur in limited and very specific circumstances,
• Immune repertoire• Meiosis sperm and eggs
• DNA replication and repair is imperfect, and mechanisms of healthy genetic alterations may be misappropriated with the consequence of mutations
Dinosaur Bone Cancer
Some Mutations Cause Cancer, Most Don’t.Major types of cancer or “driver” mutations:1. Oncogenes, 2. Tumour suppressors, and 3. DNA repair genes
• Mutations at conception = inherited or de novo
• Mutations acquired after conception in a subset of cells = somatic. Most cancer mutations are somatic.
Types of Cancer Mutations
• Oncogenes – proto‐oncogene that usually promotes healthy cell growth and division is inappropriately over‐activated.
• Tumour suppressor genes – suppress excess growth. Cancers damages or suppresses, e.g. TP53 activation tanning vs. skin peeling after sunburn kill cells with UV induced DNA damage and malignant potential.
• DNA repair genes ‐ safe guard genetic information, and repair environmental and stoichastic DNA replication errors.
Somatic Mutations are Acquired After Conception• Typical situation
• Born with no cancer mutations• Acquire them over lifespan• Acquire enough bad ones in the same cell cancer• Becomes like a snowball• Genomic instability may beget genome instability
$$ https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3749880/
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer Genome Landscapes. Science (New York, NY). 2013;339(6127):1546‐1558. doi:10.1126/science.1235122.
Cancer Formation: Stepwise Process Cancer Predisposition
• From “Germline” / “Constitutional” DNA
• Inherit from parents. “Heritable” as mutation was present in parent’s germ cells, hence the term “germline genome”.
• de novo. Acquired during meiosis, conception, or very early embryonic development

Inherit APC Mutation, and You’ve Already Taken 1 Step Towards Cancer. ‐ Germline APC mutations predispose to hepatoblastoma as well, so parent with polyps, Colon Ca age < 40 years.
Cancer Predisposition: Effect Size • Some genetic mutations produce a large increase in risk and so are clinically relevant
• Trisomy 21. ~3% get leukaemia by age 30 years. • Of 30% with TMD (transient myeloproliferative disorder) 25% Myeloid Leukaemia of Down’s Syndrome (ML‐DS) = MDS or acute megakaryoblasticleukaemia.
• RR = 500x for AMKL, RR = 20x for ALL versus general population.• Age < 5 years, general population ALL:AML = 4:1, in DS 1:1
• Single nucleotide polymorphism (SNP) produce a very small increase in risk and not clinically relevant (yet)
• Theory = thousands of subtle cancer predispositions add up to create vulnerability
• other end of spectrum is 9/10 smokers don’t get lung cancer
Adult Cancer Paed cancerFrequency 1 in 2 1 in 500
Cell of Origin Mature differentiated cells Developing cells
Oncogenesis SLOW. Over decades FASTER. Months to Years
Mutations MANY. FEWER
Proven CausativeEnvironmental
Aetiology
MAJOR, 30‐50% preventable by lifestyle modification
NOT REALLY KNOWN / RAREtreatment related – radiation therapy, alkylators e.g. cyclophosphamide
Screening YES NO
Metastatic Solid Tumours VAST MAJORITY INCURABLE OFTEN CURABLE
14
Paediatric Cancer is Normal Development Gone Wrong
• Embryonal cancers arise from embryonic remnants• Embryonal cancers end in ”blastoma” • A blast is an immature• May be healthy and maturing, or cancerous • A cancerous blast in blood is leukaemia• Lumps of blasts = blastomas, ‐oma means tumour• Neuroblastoma, Nephroblastoma (Wilms), Hepatoblastoma
• Adolescent growth spurt is associated bone sarcoma • Germ Cell Tumours arise from primordial germ cells destined to produce sperm or eggs, but 2/3 migration problem, 1/3 aberrant in gonad
15

Microscope: Embryonal Cancer Resemble Disorganised Version of Healthy Embryonic Counterpart.
stroma
Mimic renal tubule
blastema
Wilms = Nephroblastoma. Small round blue cells on H&E stains = large nucleus, small cytoplasm + disorganised looking tubule/glomeruli
0
50
100
150
200
250
1 8 15 22 29 36 43 50 57 64 71 78 85 92 99 106 113 120 127 134 141 148 155 162 169 176Age in Months
Num
ber
of Cas
es NEUROBLASTOMA INCIDENCE
• Median age diagnosis 19 months• Most common cancer in infancy• 95% of cases before age 5
London JCO 2005
‐ Embryonic Remnants Usually Resolve With Age ‐ Breeding Ground of Embryonic Cancer Dries Up ‐ Incidence of Embryonal Cancers Declines with Age,
17
Practical Principles
18
Symptoms and Signs to Identify Primary Site
• What things are missed? When people don’t consider cancer as a possibility by the third presentation / third week
• If you think anatomically and physiologically most times you can identify the potential primary site
• As with any illness localising signs are less specific and harder to elicit in younger children, e.g. limp 2ry pelvic met in HR NBL
• A mass that is stable or improves only slightly on antibiotics may be cancer, e.g. cervical adenopathy
• Abdo can hide big tumours. When I’ve seen big masses missed usually the abdominal contour is abnormal through T‐shirt
19

Cancer TESTABLE GENETICS
Ewings t(11:22) EWSR1:FLI1, EWSR1 = 22q11
Rhabdomyosarcoma (Alveolar only; not embryonal)
t(1;13) or t(2;13) PAX-FKHR (worse prognosis if present, incurable if widely metastatic)
Neuroblastoma
MYCN amplification = poorly prognosticALK mutation* also a drug target for ALK inhibitorsPHOX2 if breathing problemLOH 1p or 11q worse prognosis intermediate risk
Retinoblastoma RB1: 1 gene lost = predisposition; 2 lost = cancer
Rhabdoid brain or kidney (ATRT) Loss INI1= SMARCB1, germline 35%. Poor prognosis.
Anaplastic large cell lymphoma t(2;5) NPM-ALK *drug target for ALK inhibitors
Li Fraumeni Syndrome
TP53 in germline. Germline loss of TP53 is autosomal dominant predisposition solid tumours & leukaemia. Screening exam, bloods and whole body MRI, USS, etc decreased mortality. May test family.
20
Translocations Help Diagnosis, Guide Rx• Pathognomonic, e.g. alveolar Rhabdo t(1;13) and t(2;13).
• Alveolar rhabdomyosarcoma worse prognosis than embryonal RMS.
• Not pathognonomic but still very helpful: • t(8;14) may be Burkitts lymphoma or B‐ALL• 22q12 EWSR1 in sarcomas, many translocation partners.
• EWSR1 22q12 with FLI1, t(11;22) in bone is Ewings• Intraperitoneal Desmoplastic Small Round Cell Tumor (EWSR1/WT1), • AFH (EWSR1/CREB1), • Clear cell sarcoma joints/tendons (EWSR1/ATF1)
• Pathognomonic based on age and location, e.g. t(12;15) ETV6‐NTRK3 is infantile fibrosarcoma, or adult secretory breast carcinoma. Tyrosine kinase inhibitors (TKI) of NTRK3 have spared children from mutilating surgeries. Best activity of TKI in sarcoma.
21
Solid Tumours Can Spread to Bone Marrow
Abnormal FBC do not always mean leukaemia
Cytopenias may be due to non‐malignant (viral, AIHA, occult bleeding) or malignant ‐ leukaemia, or solid tumour infiltration
Paediatric Small Round Blue Cell Tumours can invade the marrow and cause bone marrow failure
• Ewings, Neuroblastoma, Rhabdomyosarcoma, Medulloblastoma, Wilms, Retinoblastoma, Anaplastic Hepatoblastic (not fetal or embryonal HBL subtypes)
• H&E blue means nucleus. Big nucleus because rapidly dividing. Small cytoplasm. Not specific.
22
Chemo May Cure Metastases in Paeds
Chemotherapy improves survival in localised osteosarcoma, mainly because the chemo kills metastases that are undetectable
• Micro‐metastases undetectable• Local disease resected but die
from untreated metastases.
• Metastases seen at diagnosis on imaging and bone marrow may disappear forever
• Cure metastatic disease you can make solid organ transplant a possibility
• Unresectable hepatoblastoma• Bilateral Wilms‐kidney transplant if bilateral nephrectomy (anephric) bridge with dialysis, and 1‐2 year remissionJCO
1987
EFS
Days 23
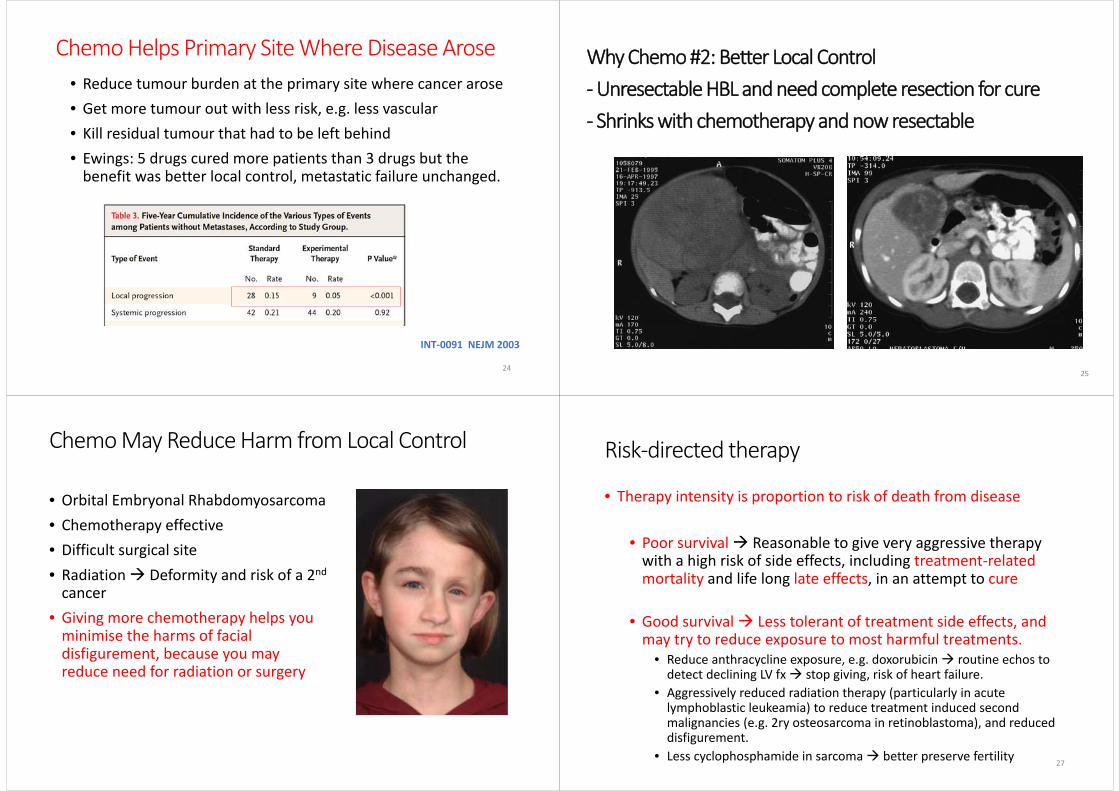
Chemo Helps Primary Site Where Disease Arose
INT‐0091 NEJM 2003
• Reduce tumour burden at the primary site where cancer arose• Get more tumour out with less risk, e.g. less vascular• Kill residual tumour that had to be left behind• Ewings: 5 drugs cured more patients than 3 drugs but the benefit was better local control, metastatic failure unchanged.
24
Hepatoblastoma at diagnosisHepatoblastoma at diagnosis
Why Chemo #2: Better Local Control ‐Unresectable HBL and need complete resection for cure‐ Shrinks with chemotherapy and now resectable
25
Chemo May Reduce Harm from Local Control
• Orbital Embryonal Rhabdomyosarcoma • Chemotherapy effective• Difficult surgical site• Radiation Deformity and risk of a 2ndcancer
• Giving more chemotherapy helps you minimise the harms of facial disfigurement, because you may reduce need for radiation or surgery
Risk‐directed therapy
• Therapy intensity is proportion to risk of death from disease
• Poor survival Reasonable to give very aggressive therapy with a high risk of side effects, including treatment‐related mortality and life long late effects, in an attempt to cure
• Good survival Less tolerant of treatment side effects, and may try to reduce exposure to most harmful treatments.
• Reduce anthracycline exposure, e.g. doxorubicin routine echos to detect declining LV fx stop giving, risk of heart failure.
• Aggressively reduced radiation therapy (particularly in acute lymphoblastic leukeamia) to reduce treatment induced second malignancies (e.g. 2ry osteosarcoma in retinoblastoma), and reduced disfigurement.
• Less cyclophosphamide in sarcoma better preserve fertility27

Local Control
• Surgery• Typically need to resect or at least debulk primary to cure• May be a role to resect metastases, e.g. osteosarcoma
• Radiation therapy• Sometimes used instead of surgery if primary site is inoperable or surgery is undesirable due to morbidity
• If surgery used, but want to tidy up around the edges in case there’s viable residual disease, often microscopic
• Sometimes used to target individual metastases
28
Major Solid Tumours individually
29
Wilms Tumour = Nephroblastoma• 1899 named by surgeon Max Wilms
• Surgeon alone early 1900s 25% perioperative mortality
• Routine radiation (RT) ~1950s 50% survival • Lots of really nasty radiation side effects ‐ Scoliosis, 2nd cancer, infertility• Trend to use less radiation• If lung mets respond to chemo quickly, don’t need whole lung irradiation
• Dactinomycin + Vincristine ~ 1960s 70‐80% survival
• Doxorubicin cardiomyopathy risk justified only if advanced disease
• Anaplastic histological subtype needs more chemo and RT
• 90% + survival with less late effects
30
Wilms Tumor Genetics
• At least 10 genes / syndromes implicated in predisposition to Wilms tumor
• What do you need to know? Surveillance can catch disease early save lives.
• Screening q3mth USS until age 8 years• Risk of bilateral Wilms, so screen s/p nephrectomy
• How do you recognize them?• Problems with kidneys and genital malformation• WT1 deletions (11p13) = WAGR syndrome
• Wilms 50%, Aniridia 100% (PAX6 del), GU, Retardation• WT1missense mutation = Denys‐Drash syndrome
• Nephropathy and intersex• FWT1 (17q12‐q21) and FWT2 (19q13) with the familial form• Beckwith Wiedeman, 11p15, Wilms > Hepatoblastoma. (More later)
ANIRIDIA

Bilateral Wilms Tumor 5%
• Synchronus is both kidneys, same time.• Metachronus – years later • 20‐yr ESRF risk: 1% unilateral; 12% bilateral• Risks dialysis, and need for transplant • Try to avoid this by giving more aggressive chemo. • If complete response, no surgery. Partial response partial nephrectomy = nephron sparing surgery = try to leave healthy kidney behind.
• If remove both kidneys, dialysis 1‐2 years, then renal transplant if remission• Principle: Maximising cure means preserving renal function at the price of slightly “under‐treating” versus unilateral cancer
• Bottom lines: treatable, if you look after a patient cured of Wilms Tumour they may have an elevated risk of contralateral Wilms Tumour
32
Chemo and Radiation can Kill over Decades Radiation was reduced. Doxorubicin that damages hearts was
reduced reduced late treatment‐related mortality
Unrelated to cancer, e.g. road accidents
Late treatment‐related mortality:‐ Second cancer 15x‐ Heart 7x
Death from original cancer progression
or recurrence
JCO 2009
33
Kidney Tumour + Mets Outside Lung May Not be Wilms
• Wilms LUNGS• Rhabdoid BRAIN (infants, rare, nasty, loss of INI1/SMARC)
• Clear Cell Sarcoma SKELETON• Renal Cell Carcinoma (age > 10 years)
34
Sarcomas• Sarc (Greek): flesh or meat
• sarcomere• sarcophagus• sarcasm
• ‐oma (Greek): tumor• Chemotherapy‐sensitive
• Rhabdomyosarcoma• Osteosarcoma• Ewing sarcoma/peripheral PNET (can be bone or soft tissue
• Chemotherapy‐resistant = Non‐rhabdo soft tissue sarcomas (NRSTS) treated with surgery and radiation
• Radiation often used, except osteosarcoma.35

BONE CANCER EWINGS OSTEOSARCOMA
AGE Teenager Teenager
LOCATION
BONE + SOFT TISSUE primary
Ewing's-Diaphysis = MiddleE" is closer to "D"
Also pelvic bones
JUST BONE
Osteosarcoma-Metaphysis"O" is closer to M"
Knee and proximal humerus
XRAY Lytic, onion skin Sunburst, sclerotic
NON-METASTATIC ( 70% ) 70% survival 70% survival
METASTATIC doworse( 30% )
~30% survival
Lungs, bone, marrow
~ 30% survival
Lung and bones
SYMPTOMS Pain, Fx, Fever, Anaemia, Unwell Pain, Fx and pathological #
PREDISPOSITION* t(11:22) in 90% Retinoblastoma, TP53, prior RT
TREATMENT Surgery, Chemo, may get Radiation Therapy* Surgery, Chemo, no RT
36Radiopaedia.org
Sunburst = osteoid bone formation like rays of light
Diaphysis is more typical location of EWS
Lytic = black hole
Growth plates and joints
Onionlayer
37
EWINGS SARCOMA OF BONE OSTEOSARCOMA
Endoprosthetic Reconstruction ‐ may spare limbs and prevent amputation, still get disability and prolonged rehab
Sarcomas Spread to Lungs
Lung Bone Marrow Nodes Other
Osteo + +Ewing + + + ~ERMS + ~ ~ +ARMS + + + + +NRSTS + + +
39
I don’t think you need to know anything else other than to recognise Ewings and Alveolar Rhabdomyosarcoma can spread to bone marrow

Rhabdomyosarcoma of soft tissueshas 2 major types
Embryonal Better Alveolar Worse
Age younger older
Location central peripheral
Invasiveness low‐moderate High
Metastases lung everywhere
Prognosis 40‐90+% EFS 0‐60% EFS
Local disease Needs more therapy
Metastatic disease Curable *Incurable => palliativechemo
Translocation No Yes t(1;13) or t(2;13)40
*Alveolar Rhabdomyosarcoma t(1;13) or t(2;13)Embryonal do not have translocations
PAX‐3 or PAX‐7
FOXO1 (FKHR)
Questions tend to check if you understand cancer translocations, e.g. Fusion oncoprotein expressed “Frankenstein” protein, in this case a mutant transcription factor cancer
Regulatory Domain – on and off switch
DNA binding doman– controls target gene
41
Mixture of 2 proteins
NEUROBLASTOMA
42
Neuroblastoma Encompasses Diseases with Very Different Natural History in Pelvis, Abdo, Chest and Neck:
“The Syphilis of Paediatric Oncology”
Asymptomatic mass• neck• thorax• abdomen• pelvis
Horner’s Syndrome Spinal Cord Compression (medical
emergency)• back pain• neurologic deficits
“Raccoon eyes” Hepatomegaly Marrow failure: bruising, petechiae,
pallor
• Systemic symptoms– weight loss– irritability– fever– hypertension– intractable diarrhea (VIP)– opsoclonus/myoclonus =
jumping eyes• Bone pain
– Limp but abdo primary or pelvic metastases.
– refusal to walk• Pallor• Subcutaneous nodules• MS syndrome 43

Neuroblastoma metastases have a predilection for cortical bone. In the orbits bleeding metastases produce bilateral black eyes called raccoon eyes. NBL is a ddx in NAI work‐up. Exclude NBL in neck, chest, abdo. Consider urine catecholamines and MIBG (both ~90% reliable) and whole body MRI.
44
New ptosis 4 weeks + cervical adenopathy 1 week that grew rapidly despite first line antibiotics USS and diagnosis. New onset ptosis is abnormal. Earlier diagnosis may have made local control easier.
Rule: Cervical adenopathy or mass + ptosis = consider NBL
45
3 Types of Neuroblastoma RiskHigh-Risk MALIGNANT and AGGRESSIVE ~ 70% survive• MYCN AMPLIFICATION. Intense Rx lasts 13- 18 months• Kitchen sink: intense chemo, high dose chemotherapy with
autologous stem cell rescue, surgery, radiation, +/- MIBG radioactive isotope, immune therapy with ch14.18, differentiation therapy with retinoic acid.
• Local disease + “pitbull” biology still needs max therapy
Intermediate-risk local or metastatic < 18 mth – 95% survive• 2 to 8 cycles of moderate chemo + surgery. • No transplant, no immune therapy, rarely radiation
Low-Risk is LOCALISED and STAY THAT WAY – 97% - 99% survive• Small ones in babies usually resolve spontaneously• Larger ones cured with resection. Don’t need any chemo.
46
Neuroblastoma Risk Uses Big Complex Tables
KEY POINT:
Combine clinical features (stage: metastatic vs local, extent = unresectable big ugly primary versus easy surgery, older generally does worse, cut‐off at 18 months.
And lab tests (histology, genetics = MYCN amplification, 11q missing are bad).
47

A. Normal MYCN FISH B. MYCN Amplification: DMs
A FISH probe (a DNA sequence) binds to a copy of a target gene and has a fluorescent colour attached. Each pink spot is 1 copy of MYCN. Healthy cells and low risk neuroblastoma have 2 MYCN copies.High‐risk neuroblastoma may have MYCN amplification = 8 to 200 copies
MYCN Amplification Indicates An Aggressive Nasty Neuroblastoma even if Local
48
Ch14.18 Monoclonal Antibody Targeting “GD2” Glycophospholipid Improved Survival in HR‐NBL
NEJM 2010
• Antibody‐dependent cellular cytotoxicity = ADCC • Attacks healthy nerves need morphine +/‐ ketamine, • Physiological instability, fever, oedema
49
Neuroblastoma 4S/MS can remit spontaneous or kill rapidly• MS= “Metastatic Special” age < 18 months
• adrenal mass and metastases limited to:• Liver (expanding liver ‐‐> squash lungs rapid death without Rx)• Bone marrow < 10%, but not cortical bone• Skin nodules
• Highest rate of spontaneous resolution of any cancer• Can also rapidly grow.• Blueberry nodules on baby + rapidly expanding liver respiratory embarrassment death
50
Malignant Benign
1. Hepatoblastoma Hemangioendothelioma
2. Hepatocellular Carcinoma Hemangioma
Embryonal Sarcoma Mesenchymal HamartomaRhabdomyosarcoma Simple Cyst
Angiosarcoma Adenoma
Carcinoid (APUDoma)
Leiomyosarcoma
Malignant Teratoma
Primary Lymphoma
Rhabdoid Tumor
Paediatric Liver Cancer – Big 2
51
Don’t freak patients out just because you see a mass or cyst in the paediatric liver

• Environment• Prematurity/LBW
– LBW < 1000 gm = RR 38x– Many theories but not modifiable
• Genetic predisposition (~15%)– Beckwith‐Wiedemann syndrome
• IGF2 (11p15.5) overexpression• Relative risk 2,280• HBL 2nd most common
– Familial adenomatous polyposis• APC mutations• 1/250 lifetime risk of HB• ~1/20 with HB have familial polyps in colon
– Li‐Fraumeni– Trisomy 18– NF‐1– Ataxia‐telangiectasia– Fanconi anemia– Tuberous sclerosis
Hepatoblastoma Has Many Predispositions
52
HBL HCCMolecular spectrum
Age < 3yr Age > 3yraFP (fetal ALB is not specific) Elevated in HBL, HCC, Germ Cell Tumour
Chronic liver injuryHBV, HCV, ETOH,
Metabolic disease
Predispositions = things you’re born with‐ Prematurity‐ Genetic predisposition 15%
Complete resection = key to cure
• CisPLATIN, CarboPLATIN can cure• Minority get no chemo
Metastatic HCC is incurable
53
Liver Transplantation
• Increasingly performed for hepatoblastoma• <2% of liver transplants from 1989‐1993• 7.5% of liver transplants in 2010
• Persistance of viable extrahepatic disease after chemotherapy (not amenable to surgical resection) is the only absolute contraindication to liver transplantation
• Emerging evidence supports post‐transplant chemotherapy (Browne, J Ped Surg 2008)
54
STS protects against CisPLATINinduced hearing loss
• Sodium thiosulfate STS (antidote for cyanide poisoning) • STS infusion soon after cisplatin infusion protects against induced sensorineural hearing loss without compromising anti‐tumour efficacy.
• Phase 3 RCT x 2 (ACCL0431 = NBL, HBL, MB, GCT, OS) • SIOPEL 6 = SR HBL
55

Germ Cell Tumour (GCT)
A type of tumor that begins in the cells that give rise to sperm or eggs. Germ cell tumors can occur almost anywhere in the body and can be either benign or malignant. A germ cell tumour in the brain is still classified as a germ cell tumour.
Germ Cell Tumours
• Germ cell are precursor cells that make egg and sperm, and the egg and sperm
• Primordial germ cells migrate to gonads • GCT: 1/3 gonadal, 2/3 extragonadal from aberrant migration• GCT appearance may mimic any cell in embryo, or extra‐embryonic, e.g. yolk sac or chorion.
57
Increasing malignant potential• Some GCT have a mixture histopathology• Malignant (spreading) potential defined by most aggressive element
Benign so no chemo
AFP normal values decline with age
59

Diverse Primary Site
Klinefelter47, XXY
Dysgenetic gonads = aneuploidy of sex chrCryptorchid testis (5% risk)
60
Teeth in an Ovarian Mature Teratoma
Teeth are ectodermal derivatives
• Tumour components resemble normal tissues
• Tissue components need to be derived from more than one germ layer
• Implies cell of origin was totipotent germ cell so turn resemble anything
GCT Treatment
•Surgery• Chemotherapy – platinum based• Radiation therapy – least favourite
Checkpoint Inhibitors Activate T‐cells Prolongs Survival in Melanoma
• Why aren’t the T-cells killing cancer?• T-cells have an “off-switch” • Cancers Turn T-cell off
• Checkpoint Blockade turns the T-cells back on
• Ipilimumab inhibit CTLA-4 receptor• Nivolumab & Keytruda inhibit PD1
receptor – PDL1/2 ligand
63

Germline Predisposition to Paediatric Solid Tumours
64
8% of Paed Cancer may have Germline Genetic Predisposition
• 8.5% of children with cancer have pathogenic or probably pathogenic protein coding mutations
• 1.1% of healthy person have comparable mutations. • Family history is unreliable, even when performed by genetic counsellors who spend 30 min on cancer history
• Osteosarcoma, 3 – 10% have a germline mutation in TP53 consistent with Li Fraumeni Syndrome
65Zhang J et al. N Engl J Med 2015. DOI: 10.1056/NEJMoa1508054
Beckwith‐Wiedemann Syndrome and Hemihypertrophy
• Macrosomia• Macroglossia• Visceromegaly• Omphalocele• Neonatal hypoglycemia• Ear creases and pits• Adrenocortical cytomegaly• Renal abnormalities
~5 to 10% get cancer
1. Wilms tumor2. Hepatoblastoma3. Adrenocortical carcinoma4. Neuroblastoma5. Rhabdomyosarcoma
66BECKWITH WIEDEMAN
Beckwith Wiedeman Cancer Screening• AFP q3mth for 4 years to detect hepatoblastoma• AFP may be elevated in first year of life so refer to normal values.
• Abdominal USS q3mth until age 8 years.• Exact risk of cancer depends on exact mutations, and rates have varied significantly
• Most don’t get cancer
67

Li‐Fraumeni Syndrome = TP53 MutationsIncreased risk for Rhabdomyosarcoma in age < 5 years,
Osteosarcoma age < 30 years = 3‐ 10%
ALL hypodiploid
Significance?
Child has increased risk of second cancer
Parents and siblings may wish to have genetic testing and counselling and may with to start screening, e.g. whole body MRIs several times per year
68
• PTEN Hamartoma Tumor Syndrome (AKA Cowden syndrome, Riley‐Ruvalcaba syndrome, Proteus syndrome)
• Macrocephaly, autism/LD, skin lesions, lipomas, AVMs
• Thyroid, breast and endometrial cancer; also colon cancer, melanoma, renal cell carcinoma
• Thyroid cancer screening starting at 10‐12 yo; breast and colon cancer screening at 25‐30 yo.
PTEN Syndrome and Surveillance
69
Recent Cancer Predisposition Syndromes (own reference)DICER1 Syndrome
• Mutations in DICER1 gene
• Tumors include PPB, cystic nephroma, Sertoli‐Leydig cell tumors, thyroid goiter, cervical RMS, CNS tumors (pineal, pituitary, ciliary body medulloepithelioma)
• Penetrance low, most tumors in 1st& 2nd decades
• ~50% have a germline mutation
• No standard surveillance guidelines
Rhabdoid Tumor Syndrome
• Mutations in SMARCB1/INI1 gene
• Atypical teratoid/rhabdoid tumors (AT/RT) – CNS
• Malignant rhabdoid tumors (renal and extra‐renal)
• Incomplete penetrance, most rhabdoid tumors occur <5 years
• Adults can develop schwannomatosis
• 35% have a germline mutation
• No standard surveillance guidelines
70
MCQs
71

Regarding the aetiology of paediatric cancers which of the following is correct?
A. The majority of childhood cancers have proven links to environmental factors
B. The majority of children with Beckwith‐Wiedeman Syndrome develop hepatoblastoma or nephroblastoma
C. Proto‐oncogenes are genes that normally restrict cell growth and cell division
D. All translocations are pathognomonic E. Nephrogenic rests are embryonic remnants and may be pre‐
malignant
72
Regarding the aetiology of paediatric cancers which of the following is correct?
A. The majority of childhood cancers have proven links to environmental factors
B. The majority of children with Beckwith‐Wiedeman Syndrome develop hepatoblastoma or nephroblastoma
C. Proto‐oncogenes are genes that normally restrict cell growth and cell division
D. All translocations are pathognomonic E. Nephrogenic rests are embryonic remnants and may be pre‐malignant
Embryonal cancers develop from embryonic remnants. In the kidney these ”nephrogenic rests” may develop into nephroblastoma, aka Wilms Tumour.
Etoposide and alkylating chemotherapy and radiation predispose to treatment related second malignant neoplasms, but otherwise you need nuclear disasters or toxic waste spells. Infection exposure may play a role in ALL. EBV plays a role in nasopharyngeal carcinoma. Some predisposition syndromes are more dangerous than others, and there is often wide variation between kindreds, and within kindreds. 90% of BWS patients do not get cancer. Some translocations are pathognomonic but some are not. The translocations in fusion positive Alveolar Rhabdomyosarcoma t(1;13) or t(2;13) are pathognomonic ,and when metastatic there is zero chance of long‐term survival.
73
A 16-year old male presents to the doctor with chest pain and shortness of breath. Medical records identify a spiral tibial fracture aged 2 years. Examination reveals bilateral breast development, small gonads, and a systolic murmur. Imaging reveals an abnormal mass behind the sternum. DNA testing of the lymphocytes from the patient’s blood is most likely to show:
A. MYC amplification
B. A mutation in the WT1 gene
C. A normal karyotype
D. 47, XXY
E. 22q11.2 deletion
74
A 16-year old male presents to the doctor with chest pain and shortness of breath for 3 weeks. The medical record records a spiral tibial fracture aged 2 years. Examination reveals bilateral breast development, small gonads, and a systolic murmur. Imaging reveals an abnormal mass behind the sternum. DNA testing of the mononuclear cells from the patient’s blood is most likely to show:
A. MYC amplification
B. A mutation in the WT1 gene
C. A normal karyotype
D. 47, XXY
E. 22q11.2 deletion
Klinefelters (47, XXY) phenotype varies but individuals often have small for age testes and produce insufficient testosterone that may lead to delayed or incomplete puberty and gynaecomastia. People with klinefelters have an increased risk of mediastinal GCT and breast cancer.
MYC amplification implies somatic acquisition, and the DNA was obtained from lymphocytes in the peripheral blood implying the blood test is measuring constitutional germline DNA. Leukaemia and lymphoma cells are mostly mononuclear and can cause solid masses in the anterior mediastinum however there is no clear increased risk of either in klinefelters. MYC is not the same as MYCN (N for neural / neuroblastoma), they are different genes. MYC is also called MYC-C. MYC-C is translocated in most cases of Burkitts which can present anywhere, but it is not typically amplified.
75

An 8 year old girl presented with variable hip pain of 4 months duration. The pain progressed and she presented after being unable to climb out of the bath. MRI shows a mass arising from the neck of femur and lesser trochanter. Interventional radiology performed a biopsy. A fluorescence in situ hybridisation (FISH) breakapart probe at 22q12‐q13 was consistent with a translocation.
76
Which of the following is most correct:a. Germline TP53 mutations are found in 3‐10% of this histologyb. This histology is radiation sensitive.c. The location and number of metastases is not prognostic.d. This histology is largely resistant to radiation therapye. Treatment includes high dose methotrexate.
An 8 year old girl presented with variable hip pain of 4 months duration. The pain progressed and she presented after being unable to climb out of the bath. MRI shows a mass arising from the neck of femur and lesser trochanter. Interventional radiology performed a biopsy. A fluorescence in situ hybridisation (FISH) breakapart probe at 22q12‐q13 was consistent with a translocation.
77
Which of the following is most correct:a. Germline TP53 mutations are found in 3‐10% of this histologyb. This histology is radiation sensitive.c. The location and number of metastases is not prognostic.d. This histology is largely resistant to radiation therapye. Treatment includes high dose methotrexate.
The question is Ewings versus Osteosarcoma. Despite the location which suggests OS, only Ewings is associated with translocation of the EWSR1 gene that mostly commonly produces a t(11;22) translocation. Radiation therapy is regularly used in Ewings , not OS. A greater burden of metastatic disease is intuitively bad and studies confirm this for most solid tumours, so C is incorrect. A, D, and E are correct for osteosarcoma. Take home: Ewings has a translocation and OS does not. Ewings is also much more sensitive to radiation than osteosarcoma, an observation that helped James Ewings differentiate Ewings Sarcoma from osteosarcoma in 1972.
Regarding treatment of germ cell tumours, select the answer that is most correctA. Germ cell tumours have very poor survivalB. When necessary, chemotherapy regimens containing platinum
are most effectiveC. Chemotherapy is the standard of care for all localised germ cell
tumours to prevent death from micrometastasesD. Radiation therapy is first line treatmentE. Surgery alone is always curative for extra‐gonadal disease
78
Regarding treatment of extra‐cranial germ cell tumours, select the answer that is most correct:
A. Germ cell tumours have very poor survival
B. When necessary, chemotherapy regimens containing platinum are most effective
C. Chemotherapy is the standard of care for all localised germ cell tumours to prevent death frommicrometastases
D. Radiation therapy is first line treatment
E. Surgery alone is always curative for extra‐gonadal disease.
Most GCT are curable, many localised GCT are cured with surgery alone (e.g. Ovarian teratoma), and chemotherapy is used for some localised disease with metastatic potential or disease with overt metastases. Cisplatin and Carboplatin are typically effective at treating GCT.
The Germ Cell Tumour (GCT) cell of origin is aberrant germ cells, or primordial germ cells that were meant to end up making sperm and eggs but something went wrong. Survival is typically high because paediatric extracranial disease is often resectable,and/or chemosensitive. Normal migration can lead to gonadal GCT (1/3) and abnormal migration can lead to extra‐gonadal GCT (2/3) at virtually any site. The tissue components of teratomas need to be derived from more than one germ layer (e.g. Need 2out of 3: endoderm, mesoderm, ectoderm). Teratoma is divided into immature that resembles developing tissues under the microscope, and mature resembles full differentiated tissues, e.g. Teeth. Paediatric teratoma that is localised and completely resected is typically cured by surgery alone. GCT histologies can mix so one tumour has a mixture of non‐malignant (teratoma) and malignant histologies. But if there is pure teratoma there is no metastatic potential and therefore no need to treat micro‐metastatic disease with chemotherapy. By contrast some GCT do have metastatic potential. Therefore, even a localised and completely resected GCT may already metastased, and micrometastases may be missed by imaging. Germinomas have confusing terminology. But you need to know some are highly malignant.
79

Treatment of solid tumours uses systemic therapy and local therapy. Which of the following answers is correct?
A. Chemotherapy does not influence disease control at the primary site
B. Surgical resection is limited to the primary siteC. Chemotherapy improves survival in metastatic osteosarcoma,
but does not improve survival in local osteosarcoma with a complete resection and clear margins.
D. The primary function of checkpoint inhibitors is activation of B‐cells and dendritic cells.
E. In solid tumours radiation therapy is a form of local control that may be used to treat the primary tumour, and potentially metastatic disease.
80
Treatment of solid tumours uses systemic therapy and local therapy. Which of the following answers is correct?
A. Chemotherapy does not influence disease control at the primary site B. Surgical resection is limited to the primary siteC. Chemotherapy improves survival in metastatic osteosarcoma, but does not improve
survival in local osteosarcoma with a complete resection and clear margins.D. The primary function of checkpoint inhibitors is activation of B‐cells and dendritic cells.E. In solid tumours radiation therapy is a form of local control that may be used to treat the
primary tumour, and potentially metastatic disease.
Surgery and radiation are local control modalities. Both can be used at the primary site and are sometimes used to control metastatic disease. Generally far prefer to use surgery over radiation because radiation has bad long term side effects like cancer, deformity, etc.
Chemotherapy and immune therapy are systemic therapies that circulate around the entire body and penetrate into many tissues. Chemotherapy can shrink cancer and make it less vascular to facilitate a more complete and safer resection. Pre‐surgical chemotherapy can turn an unresectable cancer into a resectable cancer. Systemic therapies also target metastatic disease that may be detectable or undetectable. In apparently localised Ewings Sarcoma, Osteosarcoma, and biologically aggresive localised neuroblastoma, the majority of patients died from metastatic disease that was not detected at diagnosis despite complete resection of the primary tumour. Checkpoint inhibitors primarily re‐activate T cells by interfering with signals from cancer cells that make them dormant.
81
The following may be associated with an inherited cancerpredisposition to germ cell tumour except:
A. Trisomy 21
B. Polymorphisms associated with KITLG on 12q22
C. Somatic mutations in the Wilms Tumour 1 gene
D. Undescended testis in a 7-year old male
E. 46, XY/45,X mosaic
82
The following may be associated with an inherited cancer predisposition to germcell tumour except:A. Trisomy 21
B. Polymorphisms associated with KITLG on 12q22
C. Somatic mutations in the Wilms Tumour 1 gene
D. Undescended testis in a 7-year old male
E. 46, XY/45,X mosaic
Somatic mutations are acquired after conception, do not occur in egg or sperm, are not inherited and do not predispose patients to cancer, so C is incorrect. Germline mutations are DNA alterations in the reproductive cells (sperm or egg) that become incorporated in the DNA of every cell in the body of the offspring. Germline mutations are passed from parents to offspring and are also called hereditary mutations.
Germline genetic polymorphisms (KITLG 12q22), chromosomal abnormalities (Trisomy 21, Turners), and developmental anomalies (cryptorchidism) can predispose to paediatric cancer and may require anticipatory surveillance or referral.
WT1-related Wilms Tumour syndromes are a group of hereditary disorders caused by alterations in the WT1 gene but the genetic alteration is germline / hereditary, e.g. WAGR, Denys-Drash, Frasier Syndrome, Genitourinary anomalies. Frasier syndrome has been associated with an increased risk of GCT.
People with genetic mosaicism (one fertilised zygote produces subpopulations of cells with different genotypes) or chimerism (two fertilized zygotes merge, e.g. absorb cells from miscarried sibling in utero) may incur an increased cancer risk if a cancer predisposition in contained in only one cell population. 83

A 4 year old with widely disseminated neuroblastoma of the left adrenal gland has completed induction chemotherapy, surgery, and high dose chemotherapy with autologous stem cell rescue. The parents ask you about the next phase of therapy that involves immune therapy. Regarding immune therapy with the ch14.18 monoclonal antibody, all of the following are correct except:
A. The target is GD1.B. The antibody binds to neuroblastoma cells and induces antibody‐dependent
cell‐mediated cytotoxicity.C. Severe pain requiring morphine infusion is frequent.D. Administration is associated with swelling and hypersensitivity reactions.E. Ch14.18 improves survival in high‐risk neuroblastoma.
84
A 4 year old with widely disseminated neuroblastoma of the left adrenal gland has completed induction chemotherapy, surgery, and high dose chemotherapy with autologous stem cell rescue. The parents ask you about the next phase of therapy that involves immune therapy. Regarding immune therapy with the ch14.18 monoclonal antibody, all of the following are correct except:
A. The target is GD1.
B. The antibody binds to neuroblastoma cells and induces antibody‐dependent cell‐mediated cytotoxicity.
C. Severe pain requiring morphine infusion is frequent.
D. Administration is associated with swelling and hypersensitivity reactions.
E. Ch14.18 improves survival in high‐risk neuroblastoma.
The target of ch14.18 immune therapy is disialoganglioside GD2 and this was associated with improved event free (20% better) and overall survival (9% better). The GD2 antigen is expressed on neuroblastoma but is also expressed on nerve cells. Treatment leads to significant pain although slower infusions may mitigate this. In the USA the antibody is given with IL‐2 and GM‐CSF but in Europe there is variable practice. GM‐CSF and IL‐2 are associated with capillary leak and hypersensitivity, but these side effects are recognized with all monoclonal antibodies even when given without immune modulating agents.
85
A 2 year old presents with lethargy and abdominal distension to his GP. You identify a large epigastric and LUQ mass. FBC shows haemoglobin 76 g/L (105‐140), White Cell Count 9.75 (5‐14.5 x 109/L), and Platelets 1050 (150‐500 x 109/L). Imaging identifies a heterogenous mass of the left liver lobe involving the portal triad and widespread metastasis. Alpha fetoprotein > 60,000 ug/L. Which answer is false?
A. Metastatic hepatoblastoma at diagnosis is not an absolute contraindication to possible liver transplantation.
B. High dose chemotherapy with allogeneic stem cell support is appropriate to prevent hepatoblastoma contaminating autologous marrow.
C. Cisplatin chemotherapy is highly active
D. A family history of colonic polyposis is relevant.
E. Elevated Alpha fetoprotein is not specific to hepatoblastoma
86
A 2 year old presents with lethargy and abdominal distension to his GP. You identify a large epigastric and LUQ mass. FBC shows haemoglobin 76 g/L (105‐140), White Cell Count 9.75 (5‐14.5 x 109/L), and Platelets 1050 (150‐500 x 109/L). Imaging identifies a heterogenous mass of the left liver lobe involving the portal triad and widespread metastasis. Alpha fetoprotein > 60,000 ug/L. Which answer is incorrect?
A. Metastatic hepatoblastoma at diagnosis is not an absolute contraindication to possible liver transplantation.
B. High dose chemotherapy with allogeneic stem cell support is appropriate to prevent hepatoblastoma contaminating autologous marrow.
C. Cisplatin chemotherapy is highly active
D. A family history of colonic polyposis is relevant.
E. Elevated Alpha fetoprotein is not specific to hepatoblastoma
87
Allogeneic haematopoietic stem cell transplantation is only used for leukaemia and lymphoma. In paediatric solid tumours there is no graft vs. disease so the risk of life threatening side effects like GVHD, delayed engraftment, and engraftment failure are not worth it. Autologous transplant is used in high‐risk neuroblastoma, some centres use for metastatic or large volume Ewings, and/or refractory Wilms. AFP is increased in HBL, HCC, and germ cell tumours most notably yolk sac or endodermal sinus tumour. If hepatoblastoma metastases resolve with cisplatin based therapy, and partial hepatectomy is not feasible, then total hepatectomy and liver transplantation may be curative. Detailed and focused family history has identified new kindreds with hereditary cancer predispositions including APC and early detection of Dukes A . Large liver + thrombocytosis = HBL and is not specific for haemorrhage or marrow infiltration.

A 17 year old hockey player presents to the ED at 10 pm for the third time with recent onset lower back pain. Pain persists despite paracetamol and NSAID. There is no history of trauma or strain, no difficulty passing urine or passing bowel motions. He has not noticed alterations in sensation or strength. Afebrile. No tenderness. Normal power. Sensation is subjectively altered in the perianal area. Plain Xray, FBC, chemistry are normal. ESR 24 (0‐22 mm/hr). Orthopaedics aren’t answering their pager. What further management is most appropriate:
A. ESR indicates infection. Confirm with a CRP and send blood Cx.
B. MRI T and L‐spine in the morning. Prolapsed disc is most likely.
C. Urgent MRI L‐S spine, if that is not available urgent CT.
D. You’ve got great indemnity insurance. Discharge and follow up with GP is appropriate.
E. Don’t bother neurosurgery. Nobody likes to be seen as over‐excited.
88
A 17 year old hockey player presents to the ED at 10 pm for the third time with recent onset lower back pain. Pain persists despite paracetamol and NSAID. There is no history of trauma or strain, no difficulty passing urine or passing bowel motions. He has not noticed alterations in sensation or strength. Afebrile. No tenderness. Normal power. Sensation is subjectively altered in the perianal area. Plain Xray, FBC, chemistry are normal. ESR 24 (0‐22 mm/hr). Orthopaedics aren’t answering their pager. What further management is most appropriate:
A. ESR indicates infection. Confirm with a CRP and send blood Cx.
B. MRI T and L‐spine in the morning. Prolapsed disc is most likely.
C. Urgent MRI L‐S spine, if that is not available urgent CT.
D. You’ve got great indemnity insurance. Discharge and follow up with GP is appropriate.
E. Don’t bother neurosurgery. Nobody likes to be seen as over‐excited.
Alterations in perianal sensation / saddle region often proceeds complete loss of touch sensation, bladder incontinence, and bowel incontinence. The findings may be consistent with cauda equina that requires urgent MRI
A potentially farcical question. Yet I have seen several cases where a subtle but potentially critical examination finding is elicited but the physician hesitates to believe or act on their own findings with ideal degree of urgency. Or the critical aspect of the examination was omitted (like sensation), or a false conclusion was drawn (you can’t reliably exclude sensory deficits in pre/non‐verbal children). Cauda equina and cord compression may progress and/or complete in under 24 hours, and deficits are typically irreversible. Early signs are frequently vague but prompt imaging, initiation of high dose steroids, and surgical decompression may be appropriate, and may prevent a life time of morbidity. If imaging or consultation is not available it may be appropriate to discuss starting starting steroids as precaution and make the patient NBM in case urgent intervention is needed. 89
MRI Spine – There’s a Reason!
X‐Ray CT scan MRIAnalogous Situations





























