Embed Size (px)
Citation preview

Molecular and Cellular Pathobiology
Overexpression of the Transcription Factor MEF2D inHepatocellular Carcinoma Sustains Malignant Character bySuppressing G2–M Transition Genes
Leina Ma1, Jia Liu1, Limei Liu1, Guangjie Duan1, Qingliang Wang1, Yanmin Xu1, Feng Xia2, Juanjuan Shan1,Junjie Shen1, Zhi Yang1, Ping Bie2, Youhong Cui1, Xiu-Wu Bian1, Jesus Prieto3,4, Matías A. Avila3,4, andCheng Qian1
AbstractThe underlying molecular pathogenesis in hepatocellular carcinoma remains poorly understood. The tran-
scription factor MEF2D promotes survival in various cell types and it seems to function as an oncogene inleukemia. However, its potential contributions to solid cancers have not been explored. In this study, weinvestigated MEF2D expression and function in hepatocellular carcinoma, finding that MEF2D elevation inhepatocellular carcinoma clinical specimens was associated with poor prognosis. MEF2D-positive primaryhepatocellular carcinoma cells displayed a faster proliferation rate compared with MEF2D-negative cells, andsilencing or promoting MEF2D expression in these settings limited or accelerated cell proliferation, respectively.Notably, MEF2D-silencing abolished hepatocellular carcinoma tumorigenicity in mouse xenograft models.Mechanistic investigations revealed that MEF2D-silencing triggered G2–M arrest in a manner associated withdirect downregulation of the cell-cycle regulatory genes RPRM, GADD45A, GADD45B, and CDKN1A. Furthermore,we identified MEF2D as an authentic target of miR-122, the reduced expression of which in hepatocellularcarcinoma may be responsible for MEF2D upregulation. Together, our results identify MEF2D as a candidateoncogene in hepatocellular carcinoma and a potential target for hepatocellular carcinoma therapy. CancerRes; 74(5); 1452–62. �2014 AACR.
IntroductionHepatocellular carcinoma is a highly lethal cancer, with
increasing worldwide incidence (1). Lack of effective treatmentof hepatocellular carcinoma is due to relatively poor under-standing on molecular mechanisms underlying pathogenesisof hepatocellular carcinoma (2). Numerous studies have beenfocused on identification of hepatocellular carcinoma associ-ated genes (3, 4). However, current knowledge aboutmolecularpathogenesis of hepatocellular carcinoma is far from completeelucidation. Therefore, identification of new genes participat-ing in hepatocarcinogenesis is critical for the development of
novel targeted therapeutic strategies in hepatocellular carci-noma (5).
The MEF2 family of transcription factors comprises fourmembers in mammals, MEF2A, 2B, 2C, and 2D. They wereoriginally identified as major transcriptional activators formuscle differentiation (6, 7). Subsequently, MEF2 factors werefound to participate in diverse gene regulatory programs,including muscle and neural differentiation, cardiac morpho-genesis, blood vessel formation, and growth factor responsive-ness (8–11). In addition to their effect on development, MEF2family members behave as survival factors in different types ofcells (12–15). Cyclic AMP–dependent protein kinase A signal-ing promotes apoptosis by regulating negatively MEF2D func-tion in primary hippocampal neurons (16). And a small mol-ecule, bis(3)-cognitin, acts as a potent neuroprotective agent inParkinson disease neurons against toxic stress by upregulationof MEF2D (17).
However, expression and function of MEF2 is poorly under-stood in human tumors. Studies on leukemia showed thatMEF2D/DAZAP1 and DAZAP1/MEF2D fusion proteins pro-duced by t(1;19)(q23;p13.3) chromosome translocation main-tained the malignant phenotype of acute lymphoblastic leu-kemia cells (ALL; refs. 18, 19). Integrated transcript andgenome analysis demonstrated that ectopically activatedMEF2C served as an oncogene in human ALL (20, 21). Con-sistent with this, large-scale retrovirus-mediated insertionmutagenesis identified the mouse MEF2D gene as a potential
Authors' Affiliations: 1Institute of Pathology and Southwest CancerCenter; 2Institute of Hepatobiliary Surgery, Southwest Hospital, ThirdMilitary Medical University, Chongqing, China; 3Division of Hepatologyand Gene Therapy, CIMA, University of Navarra, Pamplona; and 4CIBER-ehd. Instituto de Salud Carlos III, Madrid, Spain
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
L. Ma and J. Liu contributed equally to this work.
CorrespondingAuthor:ChengQian, Institute ofPathology andSouthwestCancer Center, Southwest Hospital, Third Military Medical University,Chongqing, China. Phone: 86-23-6876-5957; Fax: 86-23-6875-2247;E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-13-2171
�2014 American Association for Cancer Research.
CancerResearch
Cancer Res; 74(5) March 1, 20141452
on June 23, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 3, 2014; DOI: 10.1158/0008-5472.CAN-13-2171

oncogene in the development of both myeloid and lymphoidtumors (22, 23). Therefore, MEF2 transcription factors mayplay an important role in progression of leukemia. It has beenreported that MEF2D was overexpressed in nasopharyngealcarcinoma (24). However, the biologic function of MEF2 familymembers in solid cancers is not known.In liver, expression of MEF2A, MEF2C, and MEF2D
increased in the activated hepatic stellate cell (HSC) andenhancing MEF2 significantly increased the expression ofa-smooth muscle actin (a-SMA), activated collagen I pro-moter activity, and stimulated HSC proliferation, suggestingthat MEF2 plays a critical role in regulating multiple keyaspects of HSC activation and fibrotic response (25, 26). Ithas been shown that retinoic acid could inhibit the expres-sion of MEF2D in murine hepatocytes (27). Retinoic acid hasa key function in the control of cell proliferation, differen-tiation, and apoptosis and retinoic acid may play a role inthe prevention and treatment of tumors (28). The closeassociation between liver fibrosis and hepatic cancer andregulation of MEF2D by retinoic acid suggest MEF2 familymembers might participate in hepatocarcinogenesis.In this study, we investigated MEF2 expression and func-
tions in hepatocellular carcinoma. Our data showed thatMEF2D was overexpressed in hepatocellular carcinoma andhigh level of MEF2D expression was correlated with a poorprognosis in patients with hepatocellular carcinoma. MEF2Dparticipated in tumorigenecity of hepatocellular carcinoma bytranscriptional regulation of G2–M transition-retarding genes.
Materials and MethodsCell cultureHuman hepatocellular carcinoma cell lines Huh7, PLC/PRF/
5, SMMC-7721, BEL-7404, MHCC97-H, MHCC97-L, Hep3B, andHepG2, human uterine cervix cancer line HeLa, were pur-chased from the Shanghai Cell Collection. HEK293 andHEK293FT cell lines were obtained fromMicrobix Biosystems.The cells were authenticated by short tandem repeat profilingand cultured according to the manufacturer's specificationsfor less than 6 months. The patient-derived primary hepato-cellular carcinoma cultures of hepatocellular carcinoma cellswere obtained from fresh tumor specimens from patients withhepatocellular carcinoma described previously (29). In brief,the single-cell suspension was obtained from tumors by mech-anical manipulation. The primary culture was establishedinitially in Dulbecco's Modified Eagle Medium (DMEM) sup-plemented with 15% FBS and maintained in DMEM supple-mented with 10% FBS. The cells were verified for expression ofa-fetoprotein (AFP), albumin, and a-SMA by immunofluores-cence staining. All cells expressed AFP and albumin, but didnot express a-SMA. The primary cultures were named asT1216, T0127, T0408, T0420, and T0421.
Tissue arraysThe samples (n ¼ 145) were randomly collected from
patients with hepatocellular carcinoma who underwent cura-tive resection in the Institute of Hepatobiliary Surgery inSouthwest Hospital (Chongqing, China). No antitumor treat-ment was performed before hepatectomy. Tissue array blocks
containing hepatocellular carcinoma tissues and their corre-sponding nonhepatocellular carcinoma tissues were generatedwith a tissue microarrayer (Leica).
The procedure of human sample collection and use ofhuman samples for primary culture and gene expression wereapproved by the Ethical Committee of the Third MilitaryMedical University (Chongqing, China).
ImmunohistochemistryImmunohistochemical staining was performed on tissue
array slides and formalin-fixed, paraffin-embedded tissue sec-tions using the streptavidin–biotin–peroxidase complexmeth-od. The antigen retrieval procedure was performed by heatingthe samples in Dako antigen retrieval solution containing 10mmol/L EDTA (pH 8.0) with a pressure cooker. Rabbit anti-human MEF2D antibody (HPA004807; Sigma-Aldrich; 1:350)was used to detect MEF2D expression. Slides were counter-stained with hematoxylin (Sigma). Expression of MEF2D wasevaluated using graded semiquantitatively scoring system. Theintensity of staining was classified into none (0), weak (1),strong (2), or very strong (3), and the staining patterns wereclassified into negative (0:�10%), sporadic (1:11% to 25%), focal(2:26% to 50%), or diffuse(3:�51%). An overall expression scorewas calculated by multiplying the intensity and positivityscores: 0 score (negative), 1 to 4 score (moderate), and 5 to9 score (strong). In the analysis of clinical significance andprognosis, malignant samples with strongly and moderatelypositive MEF2D staining were merged as MEF2Dþ, whereasMEF2D� indicated no MEF2D expression.
Animal experimentsAll procedures for animal experiments were approved by the
Committee on the Use and Care on Animals (The ThirdMilitaryMedical University, Chongqing, China) and performedin accordance with the institution guidelines. After infectionwith indicated lentiviral vectors, Huh7 tumor xenografts wereestablished by subcutaneously inoculating 5� 105 cells into theboth flanks of 6-week-old BALB/c nude mice (the Lv-scram-bled–infected group, n ¼ 9; the Lv-shMef2d-1–infected group,n ¼ 9; and the Lv-shMef2d-2–infected group, n ¼ 9). Twenty-one days later, animals were sacrificed to weight the estab-lished tumors. All animals received humane care according tothe criteria outlined in the "Guide for the Care and Use ofLaboratory Animals" prepared by the National Academy.
Statistical analysisThe statistical significance of correlation between MEF2D
expression and survival was estimated by the log-rank test. Tostudy the relationship between MEF2D expression and othervariables, we used either the independent sample t test or thenonparametric Mann–Whitney test for continuous variables.We used the Spearman rank test to analyze correlationsbetween variables. The values of quantitative real-time PCR(qRT-PCR), cell growth rate, and colony formation wereexpressed as means� SD, and compared at a given time pointby a two-tailed independent sample t test. Data were consid-ered to be statistically significant (�, P < 0.05; ��, P < 0.01).
Other methodologies are detailed in Supplementary Data.
Promotion of Cancer Cell Growth by MEF2D in HCC
www.aacrjournals.org Cancer Res; 74(5) March 1, 2014 1453
on June 23, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 3, 2014; DOI: 10.1158/0008-5472.CAN-13-2171

ResultsAberrant expression of MEF2D in hepatocellularcarcinoma samples
To investigate expression of MEF2 in hepatocellular carci-noma specimens, we measured mRNA levels ofMEF2A, 2B, 2C,and 2D in eight fresh hepatocellular carcinoma samples andtheir corresponding nonhepatocellular carcinoma tissues. Ourdata showed that no significant differences in mRNA level ofMEF2A and MEF2C were found between tumors and theircorresponding noncancerous tissues (Fig. 1A and B) and therewas no detectableMEF2B expression in these tissues (data notshown). However, mRNA abundance of MEF2D was signifi-cantly higher in hepatocellular carcinoma tissue than non-hepatocellular carcinoma tissues (Fig. 1C). We confirmed theelevated MEF2D expression in hepatocellular carcinoma sam-
ples at protein level by immunoblotting and MEF2D proteinwas not detectable in normal livers (Fig. 1D). Immunohisto-chemical staining showed an increased expression ofMEF2D incancerous tissue. MEF2D protein was mainly localized in thenuclei of cancer cells. No obvious staining was observed innoncancerous liver tissues nor in normal livers (Fig. 1E). Thesefindings reveal a marked upregulation of MEF2D in hepato-cellular carcinoma.
Overexpression of MEF2D in cancer cells correlates withpoor prognosis of patients with hepatocellularcarcinoma
Subsequently, we investigated whether aberrant expressionof MEF2D predicts prognosis in patients with hepatocellularcarcinoma. We generated a tissue array containing 145
Figure 1. Aberrant expressionprofile of MEF2D in hepatocellularcarcinoma tissue samples. A–C,mRNA levels of MEF2A, 2B,2C, and 2D were quantified inhepatocellular carcinoma samplesand their correspondingnoncancerous liver tissues as wellas normal liver tissues by qRT-PCR(n ¼ 8). MEF2 expression wasnormalized by glyceraldehyde-3-phosphate dehydrogenase(GAPDH) expression and eachnoncancerous liver tissue wasused as a control. Data, mean �SD. T, tumors; N, noncancerousliver tissues. D, protein level ofMEF2D was determined byimmunoblotting. GAPDHexpression was used asendogenous reference. E,immunohistochemical analysis onMEF2D expression was performedin hepatocellular carcinomasamples and their correspondingnoncancerous liver tissues, as wellas in normal liver tissues. Therepresentative microphotographsof MEF2D expression aredisplayed.
Ma et al.
Cancer Res; 74(5) March 1, 2014 Cancer Research1454
on June 23, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 3, 2014; DOI: 10.1158/0008-5472.CAN-13-2171

hepatocellular carcinoma samples and their correspondingnoncancerous liver tissues to determineMEF2D expression levelby immunohistochemical staining. The representative images ofhepatocellular carcinoma samples with strong, moderate, or noMEF2D expression andof nonhepatocellular carcinoma sampleswith moderate or noMEF2D expression were shown in Fig. 2A.Of note, 59% of hepatocellular carcinoma samples had strongMEF2D expression, whereas percentages of moderate and noMEF2D expression were 28% and 13%, respectively (Fig. 2B). Incontrast,MEF2D expression was relatively low in noncancerousliver tissues. Percentages of moderate and no expression ofMEF2D protein were 71% and 29%, respectively (Fig. 2C).Clinicopathologic analysis showed that overexpression ofMEF2D in hepatocellular carcinoma was correlated with earlierhepatocellular carcinoma recurrence (P¼ 0.0158) and increasedincidences of death (P ¼ 0.0352; Supplementary Table S1 andS2). Importantly, MEF2D-positive patients had shorter totalsurvival than MEF2D-negative patients (P ¼ 0.033; Fig. 2D).
Expression of MEF2D in hepatocellular carcinoma celllines and patient-derived primary hepatocellularcarcinoma cultures of tumor cellsMEF2D expression pattern was evaluated in hepatocellular
carcinoma cell lines and in patient-derived primary hepatocel-lular carcinoma cultures of tumor cells at mRNA and protein
levels.HeLacell linewasused as apositive control because it hasbeen validated to expressMEF2D (30). High expression level ofMEF2D mRNA was detected in Huh7, PLC/PRF/5, BEL-7404,SMMC-7721, and Hep3B cell lines as well as T1216, T0420, andT0421 patient-derived primary hepatocellular carcinoma cul-tures of tumor cells, but not in MHCC97-L, MHCC97-H, andHepG2 as well as T0127 and T0408 patient-derived primaryhepatocellular carcinoma cultures of tumor cells (Supplemen-tary Fig. S1A). Immunoblotting analysis showed that expressionof MEF2D protein paralleled mRNA level in the tested cells(Supplementary Fig. S1D). The location of MEF2D protein wasalso studied by immunofluorescence in MEF2D-positive hepa-tocellular carcinoma cell lines. Similar to HeLa cells, nuclearaccumulation ofMEF2Dwas observed extensively in the major-ity of Huh7, PLC/PRF/5, and SMMC-7721 cells. No MEF2Dstaining was detected in the cytoplasm or on the membraneof hepatocellular carcinoma cells (Supplementary Fig. S1E).
MEF2D promotes the growth of hepatocellularcarcinoma cells
Our data showed that theMEF2D-positive cells (T1216, T0420,and T0421) had increased proliferation rates than MEF2D-negative cells (T0127 and T0408; Fig. 3A). Correlation analysisshowed an inverse correlation between MEF2D mRNA abun-dance and doubling time of proliferation in these primary
Figure 2. MEF2D overexpression incancer cells correlates with poorprognosis of patients withhepatocellular carcinoma. A, therepresentative microphotographsshowed strong, moderate, and noMEF2D expression in hepatocellularcarcinoma tissues and theircorresponding noncancerous livertissues by immunohistochemistry intissuearray.BandC,percentagesofstrong, moderate, and no MEF2Dexpression in hepatocellularcarcinoma samples (B) andcorresponding noncancerous livertissues (C) are shown in the piecharts. D, Kaplan–Meier analysis ofoverall survival of patients withhepatocellular carcinoma accordingto the expression level of MEF2Dprotein in hepatocellular carcinomatissues [MEF2D� (n ¼ 19) andMEF2Dþ (n ¼ 126)].
Promotion of Cancer Cell Growth by MEF2D in HCC
www.aacrjournals.org Cancer Res; 74(5) March 1, 2014 1455
on June 23, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 3, 2014; DOI: 10.1158/0008-5472.CAN-13-2171

hepatocellular carcinoma cells (Fig. 3A). These results suggesteda proliferation-promoting role of MEF2D in hepatocellular car-cinoma cells. To confirm this notion, we constructed a lentiviralvector carrying short hairpin RNA that specifically knockeddown MEF2D expression (Lv-shMEF2D-1). Our data showedthat Lv-shMEF2D-1 inhibited the expression ofMEF2D, but notMEF2A and MEF2C (Fig. 3B and Supplementary Fig. S2A–S2C).We found that silencing MEF2D inhibited the growth of Huh7andPLC/PRF/5cell lines aswell asT0420primaryhepatocellularcarcinoma cells (Fig. 3C). Moreover, the growth-suppressing
effect seemed to depend on the extent of MEF2D reduction,because infection of cells with Lv-shMEF2D-1 at multiplicity ofinfection (MOI) of 10 inhibited MEF2D expression and cellgrowth more efficiently, as compared with infection of cellswithLv-shMEF2D-1atMOIof1 (Fig. 3BandC). Similardatawereobtained when a second MEF2D-silencing lentiviral vector (Lv-shMEF2D-2) was used to reduce MEF2D expression in hepato-cellular carcinomacells (SupplementaryFig. S2D). Thus,weusedanMOI of 10 at the following experiments. In addition, we foundthat the efficiency of colony formation was also decreased when
Figure 3. Downregulation of MEF2D expression results in the inhibition of hepatocellular carcinoma cell growth. A, growth rates of patient-derived primaryhepatocellular carcinoma cultures of tumor cells with various levels ofMEF2D expression as determined by cell proliferation assay. Data, means � SD fromthree independent experiments. A highly significant inverse correlation is shown between MEF2D mRNA levels and cell doubling time for these primaryhepatocellular carcinoma cells. B, hepatocellular carcinoma cell lines were infected with lentiviral vectors (Lv-scrambled and Lv-shMEF2D-1) at MOI of1 or 10. MEF2D protein was determined by immunoblotting at 72 hours of infection. GAPDH served as endogenous reference protein. C, growth rates ofhepatocellular carcinoma cell lines and primary hepatocellular carcinoma cells in which MEF2D expression was suppressed were determined. Data,means � SD from three independent experiments. D, colony formation ability was performed on Huh7 cells at 72 hours after infection with Lv-scrambledand Lv-shMEF2D-1 atMOI of 10. The number of Huh7 cells colonies is shown asmeans� SD from three independent experiments (��,P < 0.01). E, cell-cycleanalysis was performed on Huh7 and SMMC-7721 cells at 72 hours after infection with Lv-scrambled and Lv-shMEF2D-1 at MOI of 10. The averagevalues of population percentages at G0–G1, S, and G2–M phases are shown as mean � SD from three independent experiments (��, P < 0.01).
Ma et al.
Cancer Res; 74(5) March 1, 2014 Cancer Research1456
on June 23, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 3, 2014; DOI: 10.1158/0008-5472.CAN-13-2171

Huh7 cellswere infectedwithLv-shMEF2D-1 (Fig. 3D). Cell-cycleanalysis revealed that silencingMEF2D expression in Huh7 andSMMC-7721 cells caused moderate G2–M arrest (Fig. 3E andSupplementary Fig. S5). However, MEF2D knockdown did notinduce cell apoptosis (data not shown).On the other hand, we infectedMEF2D-negative MHCC97-H
and HepG2 cells with lentiviral vector expressing MEF2D (Lv-MEF2D) to evaluate its growth-promoting effect on hepato-cellular carcinoma cells. After infection of MHCC97-H andHepG2 cells with Lv-MEF2D, MHCC97-H, and HepG2 cellsexpressed high level of exogenous MEF2D (Fig. 4A and B). Lv-MEF2D–infectedMHCC97-H andHepG2 cells exhibited higherproliferation rates, as compared with control vector Lv-GFP–infected cells (Fig. 4C). Consistently, Lv-MEF2D–infectedMHCC97-H and HepG2 cells also displayed the increasedcolony forming capacity in comparison with Lv-GFP–infectedcounterparts (Fig. 4D). Overexpression ofMEF2D in MHCC97-H and HepG2 cells resulted in the accelerated G2–M transition(Fig. 4E and Supplementary Fig. S5).
Downregulation of MEF2D abolished tumorigenecity ofhepatocellular carcinoma cellsThe role of MEF2D in tumor formation of hepatocellular
carcinoma cells was also investigated in the animal model.Lv-shMEF2D-1- and Lv-shMEF2D-2–infectedHuh7 cells formed
small tumors in only 22% and 11% of nudemice, respectively. Incontrast, Lv-scrambled–infected cells formed tumors in 89% ofnude mice (Fig. 5A). The average weight of tumors was signif-icantly lower in Lv-shMEF2D–infected groups than that in theLv-scrambled–infected group (Fig. 5B). Immunohistochemicalstaining analysis revealed extensive expression of MEF2D intumors from the Lv-scrambled–infected group, whereasMEF2Dexpression was not detected in the formed tumors from Lv-shMEF2D-1- and Lv-shMEF2D-2–infected groups (Fig. 5C).These data show thatMEF2D targeting blocks tumor formationin vivo.
MEF2D regulates the G2–M transition of cell cycle inhepatocellular carcinoma cells
To elucidate the mechanisms by which MEF2D promotesproliferation of hepatocellular carcinoma cells, we examinedglobal gene expression profiles inHuh7 cells after infectionwithLv-shMEF2D-1 and Lv-scrambled as well as after transfectionwith siRNA againstMEF2D and control siRNA by cDNA micro-array. By an analysis of the combined data from Lv-shMEF2D-1infection and siRNA MEF2D transfection, we found that therewere 1,397 genes with 2-fold or higher change in their expres-sion when MEF2D was knocked down. The Shanghai Biotech-nology CorporationAnalysis Systemanalysis showed that thesegenes enriched in the categories of cell processes, including
Figure 4. Overexpression ofMEF2Dresults in increased growth ofhepatocellular carcinoma cells. A,MHCC97-H and HepG2 cells wereinfected with Lv-GFP or Lv-MEF2Dlentiviral vectors at MOI of 10.MEF2D protein level wasdetermined by immunoblotting at72 hours of infection. GAPDHserved as endogenous reference.B, MHCC97-H and HepG2 cellswere infected with Lv-GFP and Lv-MEF2D at MOI of 10. Seventy-twohours later MEF2D protein wasdetermined by immunofluorescentstaining (original magnification,�200). C, growth rates of MEF2D-expressing MHCC97-H andHepG2 cells and control GFP-expressing cells were determined.Data, means � SD from threeindependent experiments. D,colony formation ability of controland MEF2D-expressing MHCC97-HandHepG2cells. The numbers ofcolonies are shown asmeans�SDfrom three independentexperiments (�, P < 0.05). E, cell-cycle analysis was performed onMHCC97-H and HepG2 cells 72hours after infection with Lv-GFPand Lv-MEF2D. The averagevalues of population percentagesat G0–G1, S, and G2–M phasesare shown as mean � SD fromthree independent experiments(�, P < 0.05).
Promotion of Cancer Cell Growth by MEF2D in HCC
www.aacrjournals.org Cancer Res; 74(5) March 1, 2014 1457
on June 23, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 3, 2014; DOI: 10.1158/0008-5472.CAN-13-2171

nicotinate and nicotinamide metabolism, mitogen-activatedprotein kinase (MAPK) signaling pathway, TGF-b signalingpathway, Janus-activated kinase–STAT signaling pathway, andcell cycle (Supplementary Fig. S3). Further analysis indicated ashift toward G2–M arrest in the cells with reduced MEF2Dexpression. The genes that inhibit G2–M transition were foundto be expressed at higher levels in the MEF2D-downregulatedgroup, as compared with the control group (Fig. 6A). Mean-while, mRNA abundance of G2–M transition-promoting genes,except CDC2 and CDC25C, was reduced when MEF2D expres-sion was depressed in Huh7 cells (Fig. 6A).
MEF2D suppresses the transcription of RPRM, CDKN1A,GADD45A, and GADD45B in hepatocellular carcinomacells
To know whether MEF2D directly regulates the transcrip-tion of G2–M transition-related genes, we analyzed putativeMEF2 recognition elements (MRE) in the upstream region of
transcription starting points of these genes by bioinformat-ics approach. We found multiple putative MREs in regula-tory regions of RPRM, CDKN1A, GADD45A, and GADD45B,providing grounds for the ability of MEF2D to modulate thetranscription of these genes (Fig. 6B and SupplementaryTable S3). Furthermore, our data confirmed that silencingMEF2D expression in Huh7 cells lead to increased expressionof RPRM, CDKN1A, GADD45A, and GADD45B, whereas over-expression of MEF2D resulted in decreased expression ofRPRM, CDKN1A, GADD45A, and GADD45B in MHCC97-Hcells by RT-PCR (Fig. 6C and D).
To further confirm the binding of MEF2D to the putativeMREs located in the upstream regions of RPRM, CDKN1A,GADD45A, and GADD45B promoters, we performed chromatinimmunoprecipitation (ChIP) assay on Huh7 cells. The resultsshowed that anti-MEF2D antibody coprecipitated all the DNAfragments containing the predicted MREs in the regulatoryregions of the four genes (Fig. 6E), indicating that MEF2Ddirectly binds the MREs in Huh7 cells. Subsequently, wegenerated a series of constructs in which luciferase expressionwas driven by regulatory regions of RPRM, CDKN1A,GADD45A,and GADD45B (Supplementary Fig. S4). We found thatMEF2Doverexpression significantly suppressed luciferase activitydriven by the four gene promoters. Consistently, knockingdown endogenous MEF2D levels resulted in increased pro-moter-driven luciferase activity (Fig. 6F). These data indicatedthat MEF2D directly suppressed transcription of RPRM,CDKN1A, GADD45A, and GADD45B genes, which promotedG2–M transition in hepatocellular carcinoma cells.
MiR-122 regulates MEF2D expression in hepatocellularcarcinoma cells
Bioinformatic analysis using multiple algorithms showedthat MEF2D is a predictive target of miR-122. Thus, we exper-imentally verified whether miR-122 can modulate MEF2Dexpression in hepatocellular carcinoma cells. In the sametumors from patients with hepatocellular carcinoma, whichhad increased expression level ofMEF2D (Fig. 1C),miR-122wasfound to be strongly downregulated (Fig. 7A). Expression levelsof miR-122 and MEF2D were inversely correlated in thesehepatocellular carcinoma samples (Fig. 7A). Also, there wasan inverse correlation between MEF2D and miR-122 levels inhepatocellular carcinoma cell lines and patient-derived pri-mary hepatocellular carcinoma cultures of tumor cells (Sup-plementary Fig. S1B and S1C). Next, we determined MEF2Dexpression in hepatocellular carcinoma cells by our previouslyconstructed adenoviral vector expressing exogenous miR-122(Ad-miR122; 31). Infection of hepatocellular carcinoma cellswith Ad-miR122 resulted in the reduced MEF2D expressionboth at the mRNA and protein levels (Fig. 7B and C). With thehelp of a series of online databases, we predicted thatmiR-122–specific binding site was located within the 30 untranslatedregion (UTR) ofMEF2DmRNA (Fig. 7D).We then constructed avector to investigate whether miR-122 could directly targetMEF2D 30UTR. We found that miR-122 markedly inhibitedluciferase activity when MEF2D 30UTR was inserted down-stream of luciferase cDNA in our reporter vector (pMIR-MEF2D3UTR). In contrast, no significant suppressive effect
Figure 5. Downregulation of MEF2D expression abolishes the in vivotumorigenic capacity of hepatocellular carcinoma cells. A, Huh7 cellswere subcutaneously inoculated in BALB/c nudemice after infectionwithLv-scrambled, Lv-shMEF2D-1, or Lv-shMEF2D-2 at MOI of 10 (n¼ 9 foreach group). Twenty-one days later tumors were removed. B, the weightof established tumors was measured and is shown in a scatter plot.Horizontal lines, average values. C, immunohistochemical analysis ofMEF2D expression was performed on Huh7 tumor xenografts. Therepresentative images are shown (original magnification, �200).
Ma et al.
Cancer Res; 74(5) March 1, 2014 Cancer Research1458
on June 23, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 3, 2014; DOI: 10.1158/0008-5472.CAN-13-2171

on luciferase activity was observed in cells transfected with acontrol vector with mutant MEF2D 30UTR (MIR-MEF2-D3UTRm) when miR-122 expression was elevated (Fig. 7E).These data indicate that downregulation of miR-122 could beresponsible for elevated expression ofMEF2D in hepatocellularcarcinoma cells.
DiscussionIn this study, we found elevated MEF2D expression in
hepatocellular carcinoma tissues, compared with noncancer-ous tissue and normal liver. Importantly, we observed thatoverexpression of MEF2D in hepatocellular carcinoma wascorrelated with more frequent tumor recurrence and shorter
Figure 6. MEF2D regulates the G2–Mcell-cycle transition in hepatocellular carcinoma cells. A, Huh7 cells were infected with Lv-scrambled or Lv-shMEF2D-1lentiviruses, or transfected with MEF2D-specific siRNAs and control siRNAs. Forty-eight hours later global gene expression profiles of these cells weredetermined using expressionmicroarrays. The changes in the expression ofG2–Mtransition involved genes of both lentivirus-infected and siRNA-transfectedcells and are shown as means � SD. B, bioinformatic analysis for putative MEF2D-binding sites in the regulatory regions of RPRM, CDKN1A, GADD45A,andGADD45Bgenes are shown.CandD, expression ofRPRM,CDKN1A,GADD45A, andGADD45Bat themRNA level in both Lv-shMEF2D-1–infectedHuh7cells (C) and Lv-MEF2D–infected MHCC97H cells (D) were determined by RT-PCR. mRNA levels were normalized by GAPDH expression and controlvector-infected cells were used as a controls. Data,means�SD from three independent experiments (�,P <0.05; ��,P < 0.01). E, ChIP assayswere performedto detect the binding of MEF2D to the potential MREs identified in the promoter regions mentioned above. The IgG-incubated and blank groups wereconsidered as negative controls, whereas the input fraction was the positive control. F, luciferase expression driven by the regulatory regions ofRPRM, CDKN1A, GADD45A, and GADD45B was tested in Lv-MEF2D–infected MHCC97-H cells and Lv-shMEF2D-1–infected Huh7 cells. The foldchanges of relative luciferase activity in Lv-MEF2D- and Lv-shMEF2D-1–infected cells were normalized to Lv-GFP- and Lv-scrambled–infected cells,respectively. Data, means � SD from three independent experiments (�, P < 0.05; ��, P < 0.01).
Promotion of Cancer Cell Growth by MEF2D in HCC
www.aacrjournals.org Cancer Res; 74(5) March 1, 2014 1459
on June 23, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 3, 2014; DOI: 10.1158/0008-5472.CAN-13-2171
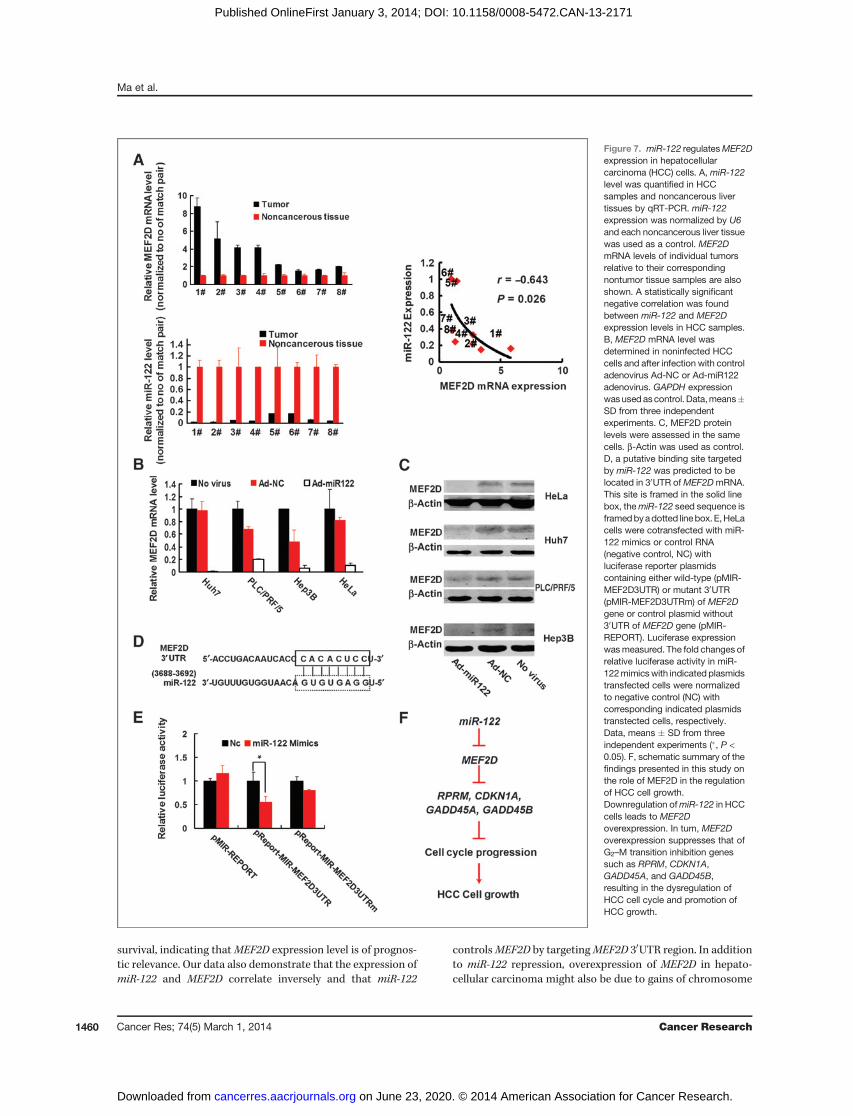
survival, indicating thatMEF2D expression level is of prognos-tic relevance. Our data also demonstrate that the expression ofmiR-122 and MEF2D correlate inversely and that miR-122
controlsMEF2D by targetingMEF2D 30UTR region. In additionto miR-122 repression, overexpression of MEF2D in hepato-cellular carcinoma might also be due to gains of chromosome
Figure 7. miR-122 regulatesMEF2Dexpression in hepatocellularcarcinoma (HCC) cells. A, miR-122level was quantified in HCCsamples and noncancerous livertissues by qRT-PCR. miR-122expression was normalized by U6and each noncancerous liver tissuewas used as a control. MEF2DmRNA levels of individual tumorsrelative to their correspondingnontumor tissue samples are alsoshown. A statistically significantnegative correlation was foundbetween miR-122 and MEF2Dexpression levels in HCC samples.B, MEF2D mRNA level wasdetermined in noninfected HCCcells and after infection with controladenovirus Ad-NC or Ad-miR122adenovirus. GAPDH expressionwasused ascontrol. Data,means�SD from three independentexperiments. C, MEF2D proteinlevels were assessed in the samecells. b-Actin was used as control.D, a putative binding site targetedby miR-122 was predicted to belocated in 30UTR ofMEF2DmRNA.This site is framed in the solid linebox, themiR-122 seed sequence isframedbyadotted linebox.E,HeLacells were cotransfected with miR-122 mimics or control RNA(negative control, NC) withluciferase reporter plasmidscontaining either wild-type (pMIR-MEF2D3UTR) or mutant 30UTR(pMIR-MEF2D3UTRm) of MEF2Dgene or control plasmid without30UTR of MEF2D gene (pMIR-REPORT). Luciferase expressionwasmeasured. The fold changes ofrelative luciferase activity in miR-122mimicswith indicated plasmidstransfected cells were normalizedto negative control (NC) withcorresponding indicated plasmidstranstected cells, respectively.Data, means � SD from threeindependent experiments (�, P <0.05). F, schematic summary of thefindings presented in this study onthe role of MEF2D in the regulationof HCC cell growth.Downregulation ofmiR-122 in HCCcells leads to MEF2Doverexpression. In turn, MEF2Doverexpression suppresses that ofG2–M transition inhibition genessuch as RPRM, CDKN1A,GADD45A, and GADD45B,resulting in the dysregulation ofHCC cell cycle and promotion ofHCC growth.
Ma et al.
Cancer Res; 74(5) March 1, 2014 Cancer Research1460
on June 23, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 3, 2014; DOI: 10.1158/0008-5472.CAN-13-2171

1q12-q23 frequently detected in hepatocellular carcinomacells, in which MEF2D gene is located (32, 33).Interestingly, we observed that there was a correlation
between MEF2D expression level and cell growth rate, sug-gesting that MEF2D may contribute to cancer cell prolifer-ation. In fact, knocking down MEF2D expression in MEF2D-positive hepatocellular carcinoma cells suppressed cancercell growth, whereas overexpression of MEF2D in MEF2D-negative hepatocellular carcinoma cells accelerated theirproliferation. Most importantly, downregulation of MEF2Din hepatocellular carcinoma cells could abolish their tumor-igenecity, when they were implanted into animals. In coin-cidence with our findings, MEF2D also plays an importantrole in cell growth in normal tissues. Zhao and colleaguesdemonstrated that MEF2D is required for p38- and BMK1MAPKs-induced proliferation of vascular smooth musclecells (34). This function of MEF2D was also reported duringstress-dependent cardiac growth (35).The critical role ofMEF2D in proliferation of hepatocellular
carcinoma cells was further supported by the finding thatknocking down MEF2D expression in hepatocellular carcino-ma cells blocked cell cycle at theG2–Mcheckpoints. Analysis ofglobal expression microarray revealed that suppression ofMEF2D resulted in downregulation of G2–M transition-pro-moting genes and upregulation of G2–M transition-inhibitinggenes. Bioinformatics analysis identified multiply putativeMREs in regulatory regions of some G2–M checkpoint genes,including RPRM, CDKN1A, GADD45A, and GADD45B, suggest-ing that MEF2D protein maybe bound these regions to mod-ulate their transcription. Our data further confirmed thatexpression of RPRM, CDKN1A, GADD45A, and GADD45B wasupregulated when MEF2D expression was decreased. Consis-tently, expression of RPRM, CDKN1A,GADD45A, andGADD45Bwas downregulated when MEF2D expression level wasincreased. Previous studies have shown that hypermethylationofRPRM promoter with reduced expression is a common eventin many human cancers and that RPRM overexpression medi-ated by an adenoviral vector induced a strong G2–M arrest inHeLa cells (36, 37). CDKN1A (p21, Cip1) upregulation is alsorequired for G2–M arrest induced by a variety of drugs (38).GADD45A is known to allow HepG2 cells to undergo G2–Marrest (39), whereas decreased GADD45B expression in humanhepatocellular carcinoma tissues is significantly associatedwith histologic grading of tumors (40). Collectively, our dataindicated that MEF2D promoted cell growth by downregulat-ing G2–M transition-inhibiting genes. The pathways involvedinMEF2D-mediated pathogenesis of hepatocellular carcinomaare outlined in Fig. 7F.Our findings also confirmed that MEF2D protein directly
binds to the putative MREs located in the upstream region
from the transcription sites of RPRM, CDKN1A, GADD45A andGADD45B, andMEF2D regulated activity of these promoters. Ithas been demonstrated that MEF2D regulated the transcrip-tion of target genes by recruiting the necessary corepressors,such as histonemodifiers SIRT1,HDAC4, andHDAC9. However,our preliminary data showed that some histone modifiers(SIRT1, HDAC4, and HDAC9) that are reported to form com-plexes with MEF2 family members did not interact withMEF2D in Huh7 cells (data not shown), suggesting that othercorepressors may partner with MEF2D to inhibit the expres-sion of G2–M checkpoint genes in hepatocellular carcinomacells. The detailed mechanisms need further studies.
Finally, we confirmed thatmiR-122 inhibitedMEF2D expres-sion by targeting its mRNA 30UTR. Gramantieri and colleagueshave reported that miR-122 was able to inhibit cell-cycleprogression in hepatocellular carcinoma cells (41). Our previ-ous study also showed that overexpression of miR-122 medi-ated by adenoviral vector induced a G2–M arrest in hepato-cellular carcinoma cells and rendered hepatocellular carcino-ma cells sensitive to chemotherapy (31, 42). Therefore, wehypothesized that MEF2D suppression is responsible, at leastin part, for the inhibitory effect of miR-122 on cell-cycleprogression.
In conclusion, this study provides evidence identifyingMEF2D as a tumor-promoting gene for human hepatocellularcarcinoma. Our data also suggest thatMEF2Dmay be a usefulprognostic marker and a potential therapeutic target inpatients with primary liver cancer.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: L. Ma, J. Liu, P. Bie, X.-W. Bian, M.A. Avila, C. QianDevelopment of methodology: L. Ma, J. Liu, J. Shan, J. Shen, Z. YangAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): L. Ma, J. Liu, L. Liu, G. Duan, Q. Wang, X.-W. BianAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): L. Ma, J. Liu, L. Liu, G. Duan, X.-W. BianWriting, review, and/or revision of the manuscript: L. Ma, J. Liu, Y. Cui, J.Prieto, M.A. Avila, C. QianAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructingdatabases): L.Ma, J. Liu, L. Liu, G. Duan, Q.Wang, Y.Xu, F. Xia, X.-W. BianStudy supervision: F. Xia, P. Bie, X.-W. Bian, C. Qian
Grant SupportThis work was supported by funds from National Natural Sciences Founda-
tion of China (nos. 81090423 and 81020108026 to C. Qian; 81001104 to L. Ma;81000966 to J. Shen; and 81101630 to J. Shan), National Basic Research Program ofChina (973 program, no. 2010CB529406; C. Qian), grant "UTE project CIMA," andgrants RTICC-RD06 00200061 and PI10/00038 from Instituto de Salud Carlos III,Spain (M.A. Avila).
Received July 30, 2013; revised December 2, 2013; accepted December 2, 2013;published OnlineFirst January 3, 2014.
References1. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer
J Clin 2010;60:277–300.2. Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in
HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer2013;13:123–35.
3. Ranzani M, Cesana D, Bartholomae CC, Sanvito F, Pala M, Benedi-centi F, et al. Lentiviral vector-based insertional mutagenesis identifiesgenes associated with liver cancer. Nat Methods 2013;10:155–61.
4. Cleary SP, Jeck WR, Zhao X, Chen K, Selitsky SR, Savich GL,et al. Identification of driver genes in hepatocellular carcinoma by
Promotion of Cancer Cell Growth by MEF2D in HCC
www.aacrjournals.org Cancer Res; 74(5) March 1, 2014 1461
on June 23, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 3, 2014; DOI: 10.1158/0008-5472.CAN-13-2171

exome sequencing. Hepatology. 2013 May 31. [Epub ahead ofprint].
5. Feng GS. Conflicting roles of molecules in hepatocarcinogenesis:paradigm or paradox. Cancer Cell 2012;21:150–4.
6. Molkentin JD,OlsonEN.Combinatorial control ofmuscle developmentby basic helix-loop-helix and MADS-box transcription factors. ProcNatl Acad Sci U S A 1996;93:9366–73.
7. McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependentregulator of cell division, differentiation and death. Trends BiochemSci 2002;27:40–7.
8. Potthoff MJ, Olson EN. MEF2: a central regulator of diverse develop-mental programs. Development 2007;134:4131–40.
9. Lin Q, Schwarz J, Bucana C, Olson EN. Control of mouse cardiacmorphogenesis and myogenesis by transcription factor MEF2C. Sci-ence 1997;276:1404–7.
10. ArnoldMA, Kim Y, Czubryt MP, Phan D,McAnally J, Qi X, et al. MEF2Ctranscription factor controls chondrocytehypertrophy andbonedevel-opment. Dev Cell 2007;12:377–89.
11. Hayashi M, Kim SW, Imanaka-Yoshida K, Yoshida T, Abel ED, EliceiriB, et al. Targeted deletion of BMK1/ERK5 in adult mice perturbsvascular integrity and leads to endothelial failure. J Clin Invest 2004;113:1138–48.
12. Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronalactivity–dependent cell survival mediated by transcription factorMEF2. Science 1999;286:785–90.
13. Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, et al.Activity-dependent regulation of MEF2 transcription factors sup-presses excitatory synapse number. Science 2006;311:1008–12.
14. Gaudilliere B, Shi Y, Bonni A. RNA interference reveals a requirementfor myocyte enhancer factor 2A in activity-dependent neuronal sur-vival. J Biol Chem 2002;277:46442–6.
15. YangQ, SheH, GearingM, Colla E, LeeM, Shacka JJ, et al. Regulationof neuronal survival factorMEF2Dby chaperone-mediated autophagy.Science 2009;323:124–7.
16. Salma J, McDermott JC. Suppression of a MEF2-KLF6 survival path-way by PKA signaling promotes apoptosis in embryonic hippocampalneurons. J Neurosci 2012;32:2790–803.
17. Yao L, Li W, She H, Dou J, Jia L, He Y, et al. Activation of transcriptionfactor MEF2D by bis(3)-cognitin protects dopaminergic neurons andameliorates Parkinsonian motor defects. J Biol Chem 2012;287:34246–55.
18. Prima V, Hunger SP. Cooperative transformation by MEF2D/DAZAP1andDAZAP1/MEF2D fusion proteins generated by the variant t(1;19) inacute lymphoblastic leukemia. Leukemia 2007;21:2470–5.
19. Prima V, Gore L, Caires A, Boomer T, Yoshinari M, Imaizumi M, et al.Cloning and functional characterization of MEF2D/DAZAP1 andDAZAP1/MEF2D fusion proteins created by a variant t(1;19)(q23;p13.3) in acute lymphoblastic leukemia. Leukemia 2005;19:806–13.
20. Homminga I, Pieters R, Langerak AW, deRooi JJ, Stubbs A, VerstegenM, et al. Integrated transcript andgenomeanalyses reveal NKX2–1 andMEF2C as potential oncogenes in T cell acute lymphoblastic leukemia.Cancer Cell 2011;19:484–97.
21. Schwieger M, Schuler A, Forster M, Engelmann A, Arnold MA, DelwelR, et al. Homing and invasiveness of MLL/ENL leukemic cells isregulated by MEF2C. Blood 2009;114:2476–88.
22. Lund AH, Turner G, Trubetskoy A, Verhoeven E, Wientjens E, HulsmanD, et al.Genome-wide retroviral insertional taggingof genes involved incancer in Cdkn2a-deficient mice. Nat Genet 2002;32:160–5.
23. Suzuki T, Shen H, Akagi K, Morse HC, Malley JD, Naiman DQ, et al.New genes involved in cancer identified by retroviral tagging. NatGenet 2002;32:166–74.
24. Li Y, Zhang L, Nong J, Bian S, Zhao Z, Ren Y, et al. [Expression ofMEF2D on nasopharyngeal carcinoma tissues and its influence of
prognostic]. Lin Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi2011;25:840–2.
25. Wang X, Tang X, Gong X, Albanis E, Friedman SL, Mao Z. Regu-lation of hepatic stellate cell activation and growth by transcriptionfactor myocyte enhancer factor 2. Gastroenterology 2004;127:1174–88.
26. Moreira RK. Hepatic stellate cells and liver fibrosis. Arch Pathol LabMed 2007;131:1728–34.
27. Mamoon A, Ventura-Holman T, Maher JF, Subauste JS. Retinoic acidresponsive genes in the murine hepatocyte cell line AML 12. Gene2008;408:95–103.
28. Kojima S, OkunoM, Matsushima-Nishiwaki R, Friedman SL, MoriwakiH. Acyclic retinoid in the chemoprevention of hepatocellular carcinoma(review). Int J Oncol 2004;24:797–805.
29. Shan J, Shen J, Liu L, Xia F, Xu C, Duan G, et al. Nanog regulates self-renewal of cancer stem cells through the insulin-like growth factorpathway in human hepatocellular carcinoma. Hepatology 2012;56:1004–14.
30. Ornatsky OI, McDermott JC. MEF2 protein expression, DNAbinding specificity and complex composition, and transcriptionalactivity in muscle and non-muscle cells. J Biol Chem 1996;271:24927–33.
31. Ma L, Liu J, Shen J, Liu L, Wu J, Li W, et al. Expression of miR-122mediated by adenoviral vector induces apoptosis and cell-cycle arrestof cancer cells. Cancer Biol Ther 2010;9:554–61.
32. Wong N, Lai P, Pang E, Leung TW, Lau JW, Johnson PJ. A compre-hensive karyotypic study on human hepatocellular carcinoma byspectral karyotyping. Hepatology 2000;32:1060–8.
33. Nishimura T, Nishida N, Itoh T, Komeda T, Fukuda Y, Ikai I, et al.Discrete breakpoint mapping and shortest region of overlap of chro-mosome arm 1q gain and 1p loss in human hepatocellular carcinomadetected by semiquantitative microsatellite analysis. Genes Chromo-somes Cancer 2005;42:34–43.
34. Zhao M, Liu Y, Bao M, Kato Y, Han J, Eaton JW. Vascular smoothmuscle cell proliferation requires both p38 and BMK1 MAP kinases.Arch Biochem Biophys 2002;400:199–207.
35. Kim Y, Phan D, van Rooij E, Wang DZ, McAnally J, Qi X, et al. TheMEF2D transcription factor mediates stress-dependent cardiac remo-deling in mice. J Clin Invest 2008;118:124–32.
36. Takahashi T, Suzuki M, Shigematsu H, Shivapurkar N, Echebiri C,Nomura M, et al. Aberrant methylation of Reprimo in human malig-nancies. Int J Cancer 2005;115:503–10.
37. Ohki R, Nemoto J, Murasawa H, Oda E, Inazawa J, Tanaka N, et al.Reprimo, a new candidate mediator of the p53-mediated cell-cyclearrest at the G2 phase. J Biol Chem 2000;275:22627–30.
38. Wu Q, Qin SK, Teng FM, Chen CJ, Wang R. Lobaplatin arrests cell-cycle progression in human hepatocellular carcinoma cells. J HematolOncol 2010;3:43.
39. Zhu N, Shao Y, Xu L, Yu L, Sun L. Gadd45-alpha and Gadd45-gammautilize p38 and JNK signaling pathways to induce cell-cycle G2–Marrest in Hep-G2 hepatoma cells. Mol Biol Rep 2009;36:2075–85.
40. Qiu W, David D, Zhou B, Chu PG, Zhang B, Wu M, et al. Down-regulation of growth arrest DNA damage-inducible gene 45betaexpression is associated with human hepatocellular carcinoma. AmJ Pathol 2003;162:1961–74.
41. Gramantieri L, Ferracin M, Fornari F, Veronese A, Sabbioni S, Liu CG,et al. Cyclin G1 is a target of miR-122a, a microRNA frequentlydownregulated in human hepatocellular carcinoma. Cancer Res 2007;67:6092–9.
42. Xu Y, Xia F, Ma L, Shan J, Shen J, Yang Z, et al. MicroRNA-122sensitizes HCC cancer cells to adriamycin and vincristine throughmodulating expression of MDR and inducing cell-cycle arrest. CancerLett 2011;310:160–9.
Ma et al.
Cancer Res; 74(5) March 1, 2014 Cancer Research1462
on June 23, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 3, 2014; DOI: 10.1158/0008-5472.CAN-13-2171

2014;74:1452-1462. Published OnlineFirst January 3, 2014.Cancer Res Leina Ma, Jia Liu, Limei Liu, et al.
M Transition Genes−2Suppressing GHepatocellular Carcinoma Sustains Malignant Character by Overexpression of the Transcription Factor MEF2D in
Updated version
10.1158/0008-5472.CAN-13-2171doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2014/01/06/0008-5472.CAN-13-2171.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/74/5/1452.full#ref-list-1
This article cites 41 articles, 13 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/74/5/1452.full#related-urls
This article has been cited by 4 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/74/5/1452To request permission to re-use all or part of this article, use this link
on June 23, 2020. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 3, 2014; DOI: 10.1158/0008-5472.CAN-13-2171





























