Embed Size (px)
Citation preview

Outcome of Liver Transplantation for FamilialAmyloidotic Polyneuropathy
Pratima Sharma,* Roman E. Perri,† Joseph E. Sirven,‡ Steven R. Zeldenrust,§
David J. Brandhagen,† Charles B. Rosen,� David D. Douglas,* David C. Mulligan,¶
Jorge Rakela,* Russell H. Wiesner,† and Vijayan Balan*
Familial amyloidotic polyneuropathy (FAP) is an autosomaldominant disorder caused by mutation in the transthyretingene. The most common mutation is substitution of valinefor methionine at position 30 (MET30). Liver transplanta-tion (LT) is the preferred treatment. After LT, althoughmany patients show stabilization or improvement in the dis-ease, adverse outcomes have been reported in those who havemalnutrition, long-standing disease, and non-MET (NMET)mutations at position 30. Our aim is to compare survival andoutcome of symptoms associated with FAP after LT inpatients with MET30 and NMET30 mutations. Medicalrecords of all patients who underwent LT for amyloidosis atour institution were reviewed to obtain demographic infor-mation and clinical features, such as severity of neuropathy,diarrhea, orthostatic hypotension, and posterior wall or ven-tricle septal thickness before and after LT. Fifteen patientsunderwent LT for amyloidosis at our institution between1990 and 2000 (MET30, n � 5; NMET30, n � 7; hereditaryamyloidosis, n � 2; primary amyloidosis, AL type, n � 1).Patients with hereditary and primary amyloidosis wereexcluded from analysis. One- and 3-year survival rates afterLT in MET30 patients were 100%. Before LT, five of fivepatients had sensorimotor neuropathy; five of five patientshad diarrhea, and four of five patients had orthostatic hypo-tension. After LT, improvement or stabilization of neuropa-thy was seen in two of five patients; of diarrheal symptoms, inthree of five patients; and of orthostatic hypotension, in threeof four patients. One- and 3-year survival rates after LT inNMET30 patients were 100% and 85.7%, respectively.Before LT, six of seven patients had sensorimotor neuropa-thy, six of seven patients had diarrhea, and five of sevenpatients had orthostatic hypotension. After LT in this group,improvement or stabilization of neuropathy was seen in twoof six patients; of diarrhea, in six of six patients; and oforthostatic hypotension, in five of five patients. Before LT,posterior wall and/or ventricle septal thickness was increasedin two of five MET patients and seven of seven NMETpatients. Five of seven NMET30 patients (71.4%) whoreceived a combined liver and heart transplant had stabiliza-tion, and two patients in the NMET group and one patient inthe MET group had progression of heart disease. Outcomesfor LT for patients with FAP with MET or NMET mutationswere similar. Earlier LT for patients with FAP with MET30or NMET30 mutation would improve outcomes after LT.(Liver Transpl 2003;9:1273-1280.)
Familial amyloidotic polyneuropathy (FAP) type 1is the Portuguese type autosomal dominant hered-
itary disorder caused by methionine at position 30mutation (MET30) in the transthyretin (TTR) gene.1,2
The first symptoms usually occur during the third
decade of life, subsequently followed by progressivesensorimotor neuropathy with autonomic dysfunction,weight loss, and involvement of several other organs,such as the heart, kidneys, gastrointestinal tract, andeyes.3-5 The clinical course of the disease is progressiveand usually fatal within a mean of 10.8 years afteronset.3,5
TTR, formerly called prealbumin, is a normal tet-rameric serum protein involved in the transport ofserum thyroxine and retinol-binding protein.1 A singlegene located on chromosome 18 encodes it. More than70 autosomal dominant inherited point mutationsoccurring at 51 different sites have been described;among these, substitution of valine by methionine atposition 30 is by far the most frequent and most widelydisseminated form.1,6-8 In other mutations, cardiomy-opathy may be the most important symptom. Becausemost TTR (wild-type and variant TTR) is synthesizedby hepatocytes, liver transplantation (LT) has been thetreatment for FAP.9-13
According to the FAP World Transplant Registry,more than 350 LTs have been performed in patientswith FAP type 1.14 Various investigators have reportedstabilization or improvement of FAP disease after LT inpatients with MET30 mutations.10,14,15 However,adverse outcomes also have been observed in patientswith FAP who have malnutrition, long-standing diseasebefore LT, or non-MET30 (NMET30) mutations.14,16
Thus, the purpose of this study is to compare survivaland outcome of MET30 and NMET30 patients in theareas of polyneuropathy, orthostasis, diarrhea, and pro-gression of heart disease.
From the *Divisions of Transplant Medicine and ¶TransplantationSurgery and ‡Department of Neurology, Mayo Clinic, Scottsdale, AZ;and Divisions of †Gastroenterology and Hepatology, §Hematology, and�Transplantation Surgery, Mayo Clinic, Rochester, MN.
Address reprint requests to Vijayan Balan, MD, Division of Trans-plantation Medicine, Mayo Clinic Hospital, 5777 E Mayo Blvd, Phoe-nix, AZ 85054. Telephone: 480-342-0516; FAX: 480-342-2324;E-mail: [email protected]
Copyright © 2003 by the American Association for the Study ofLiver Diseases
1527-6465/03/0912-0006$30.00/0doi:10.1016/j.lts.2003.09.016
1273Liver Transplantation, Vol 9, No 12 (December), 2003: pp 1273-1280

Methods
Patients
We retrospectively reviewed all patients with the diagnosis ofFAP who underwent LT between 1990 and 2000 at ourinstitution after obtaining approval from the Mayo Founda-tion Institutional Review Board. Demographic information,albumin level, height, weight, date of first symptom, severityof neuropathy, orthostatic hypotension, diarrhea, thickness ofthe posterior wall and ventricular septum before and after LT,and last date of follow-up were collected for the study.
Clinical Findings
The diagnosis of FAP was reviewed for the presence of symp-toms, electromyographic findings of peripheral axonal neu-ropathy, and genetic testing. Symptoms of peripheral andautonomic neuropathy were graded according to previouslypublished parameters.17-19 A modified polyneuropathy dis-ability (PND) score was used to evaluate peripheral sensoryand motor disturbances as follows: score I, sensory distur-bances in limbs without motor impairment; score II, diffi-culty walking without the need of a walking aid; score IIIa,one stick or one crutch required for walking; score IIIb, twosticks or two crutches needed; and score IV, wheelchairrequired or patient confined to bed.
Routine nerve conduction studies (NCSs) and electro-myography (EMG) were performed before and after LT.Severity of diarrhea and orthostatic hypotension was assessedbefore and after LT by using the scoring of autonomic dys-function by Tashima et al18,19 (Table 1). All patients had anechocardiogram performed to assess cardiac involvement.Patients who underwent simultaneous heart transplantationhad an endomyocardial biopsy specimen obtained before theoperation. Nutritional status was assessed by determiningmodified body mass index (mBMI). mBMI was calculatedusing the following formula: (weight [kg]/length [m]2)/serumalbumin (g/L). A person with an mBMI less than 700 isconsidered malnourished. Duration of disease is defined astime in years from the approximate date of appearance of thefirst symptom to the day of LT surgery. Follow-up time afterLT is defined as time in years from date of LT to date of lastfollow-up or death. Time elapsed from the last pre-LT NCSand EMG study and LT was calculated in months. Survival
and outcome of polyneuropathy, orthostasis, and diarrheaand progression of heart disease after LT were evaluated byreviewing follow-up notes, PND scoring, EMG, autonomictesting, and echocardiography.
Liver Transplantation
All patients received cadaveric liver transplants. Five patientsreceived heart and liver transplants, and one patient received aheart, liver, and kidney transplant. Patients receiving bothliver and heart transplants were administered cyclosporine-based triple-drug immunosuppression, whereas patientsreceiving a combined liver and kidney transplant or livertransplant alone were administered tacrolimus-based immu-nosuppression.
Statistical Analysis
All statistical analyses were performed using SPSS statisticalsoftware for Windows, version 11 (SPSS Inc, Chicago, IL).Results are expressed as mean � SD. Duration of disease isexpressed as median. Pearson’s correlation was calculated forcontinuous variables with normal distribution.
Results
Fifteen patients underwent LT for amyloidosis at ourinstitution between 1990 and 2000. Of the 15 patients,5 patients had MET30 mutation, 7 patients hadNMET30 mutation, 2 patients had hereditary amy-loidosis with fibrinogen mutation, and 1 patient hadliver AL-type primary amyloidosis. Patients with hered-itary and primary amyloidosis were excluded from theoutcome analysis. Characteristics of patients withhereditary amyloidosis with fibrinogen mutation weredescribed previously from our institution.20 All patientsincluded in the analysis were unrelated. All except 3 ofthe patients analyzed had an amyloid deposit in one ofthe following biopsy specimens: sural nerve, gastroin-testinal tract, or subcutaneous fat.
MET30 Patients
Demographics, duration of symptoms, mBMI, and fol-low-up time for the five MET30 patients are listed inTable 2. Table 3 shows the comparison betweenMET30 and NMET30 patient characteristics. Neurop-athy, diarrhea, orthostasis, and cardiac outcomes arelisted in Table 4.
Neuropathy
All five MET30 patients had sensorimotor neuropathy,evidenced by findings on EMG and NCS. Two patientshad a PND score of I; two patients, a score of II; and onepatient, a score of IIIa. After transplantation, three of
Table 1. Scale for Familial Amyloidotic Polyneuropathy
Scoring for diarrhea2 Alternating constipation and diarrhea3 Regular diarrhea6 Severe diarrhea
Scoring for orthostatic hypotension2 Systolic blood pressure (0-20 mm Hg decrease)4 Systolic blood pressure (�20 mm Hg decrease)6 Severe with faintness
1274 Sharma et al
five patients showed progression in neuropathy, evi-denced by findings on follow-up EMG and NCS. Pre-transplantation PND scores were IIIa, II, and I. Thepatient with the IIIa score had absent sensory responsesand low-amplitude or absent motor responses with rel-atively preserved conduction velocities and distal laten-cies on nerve conduction studies pretransplantation.Blink reflexes were normal.
One-year after LT, repeated EMG and NCSsshowed progression of neuropathy. This patient’s PNDscore deteriorated to IIIb. At the 1-year post-LT follow-
up, two patients had either improvement or stabiliza-tion (one patient with PND score II before LT hadstabilization, and one patient with PND score I hadimprovement) in neuropathy. The patient withimprovement in neuropathy showed improvement inthe modified sigma compound muscle action potentialfrom 6.1 to 8.4 mV (change � 1.0 is significant) 1 yearafter LT. Modified sigma sensory nerve action potentialimproved from 23.0 to 28.3 �V. Improvement evi-denced by EMG and NCS findings was observed inonly 20% of patients (one of five patients) after trans-plantation, and stabilization of neuropathy wasobserved in only 20% of patients (one of five patients).Median time elapsed between LT and last pre-LTEMG/NCS study was 5 months.
Diarrhea
All five patients had diarrhea. Two of five patients hadalternating constipation and diarrhea, two patients hadmoderate diarrhea with more than 10 loose bowelmovements per day, and one patient had severe diarrheawith more than 20 loose bowel movements per day. Nopatient in this group showed improvement in gastroin-testinal symptoms after LT in the follow-up period.Furthermore, progression of diarrhea was noted in twoof five patients (40%), and malnutrition caused bychronic diarrhea developed in one of these two patients.Three of five patients (60%) showed neither improve-
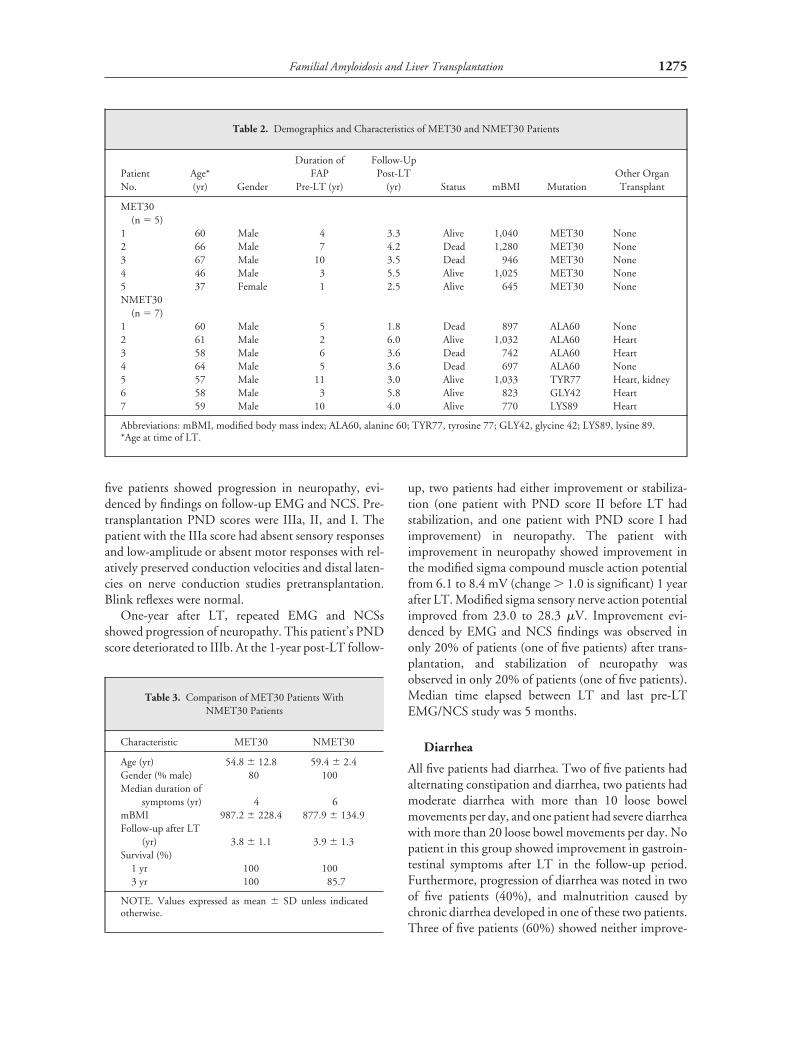
Table 2. Demographics and Characteristics of MET30 and NMET30 Patients
PatientNo.
Age*(yr) Gender
Duration ofFAP
Pre-LT (yr)
Follow-UpPost-LT
(yr) Status mBMI MutationOther OrganTransplant
MET30(n � 5)
1 60 Male 4 3.3 Alive 1,040 MET30 None2 66 Male 7 4.2 Dead 1,280 MET30 None3 67 Male 10 3.5 Dead 946 MET30 None4 46 Male 3 5.5 Alive 1,025 MET30 None5 37 Female 1 2.5 Alive 645 MET30 NoneNMET30
(n � 7)1 60 Male 5 1.8 Dead 897 ALA60 None2 61 Male 2 6.0 Alive 1,032 ALA60 Heart3 58 Male 6 3.6 Dead 742 ALA60 Heart4 64 Male 5 3.6 Dead 697 ALA60 None5 57 Male 11 3.0 Alive 1,033 TYR77 Heart, kidney6 58 Male 3 5.8 Alive 823 GLY42 Heart7 59 Male 10 4.0 Alive 770 LYS89 Heart
Abbreviations: mBMI, modified body mass index; ALA60, alanine 60; TYR77, tyrosine 77; GLY42, glycine 42; LYS89, lysine 89.*Age at time of LT.
Table 3. Comparison of MET30 Patients WithNMET30 Patients
Characteristic MET30 NMET30
Age (yr) 54.8 � 12.8 59.4 � 2.4Gender (% male) 80 100Median duration of
symptoms (yr) 4 6mBMI 987.2 � 228.4 877.9 � 134.9Follow-up after LT
(yr) 3.8 � 1.1 3.9 � 1.3Survival (%)
1 yr 100 1003 yr 100 85.7
NOTE. Values expressed as mean � SD unless indicatedotherwise.
1275Familial Amyloidosis and Liver Transplantation

ment nor progression. They were treated symptomati-cally with antidiarrheal medication.
Orthostatic Hypotension
Four of five patients had orthostatic hypotension. Oneof four patients had worsening of symptoms, and theother three patients had no change in severity of hypo-tension.
Cardiac Involvement
There was increased thickness in the posterior wall andventricular septal wall in three of five patients. Informa-tion on the other two patients was not available. Ejec-tion fraction was normal in all three patients. Conduc-tion defects were present in two of three patients. Nopatient underwent simultaneous heart transplantation.Worsening of septal and posterior wall thickness of theleft ventricle was observed in one patient (19/16 mmpre-LT v 23/21 mm post-LT), and this patient died 4.2years after LT from progression of heart disease.
One patient had no change and one patient had adecrease in posterior wall and septal wall thickness ofthe left ventricle (13/12 mm pre-LT v 8/9 mm post LT)on serial echocardiography post-LT.
Survival
One- and 3-year survival rates after LT were 100%.Two patients died 3.5 and 4.2 years after LT of progres-sion of disease.
NMET30 Patients
Demographics, mutation, duration of symptoms,mBMI, and follow-up time of the nine NMET30patients are listed in Table 2. Outcomes of neuropathy,diarrhea, orthostasis, and cardiac involvement are listedin Table 5. Outcome according to individual NMET30mutations is discussed next.
ALA60 Patients
Four of seven NMET30 patients had the ALA60 muta-tion.
Neuropathy. Three of four patients (75%) had sensori-motor neuropathy, and one of four patients had no evi-dence of neuropathy. Pre-LT PND scores were IIIb, IIIa,and I. Patients with IIIa and IIIb scores deteriorated to IIIband IV scores, whereas the patient with a PND score of Ihad stabilization of neuropathy. NCS and EMG was notavailable for three patients, and one patient with a PNDscore of I underwent the study 2 months before LT.
Diarrhea and orthostatic hypotension. Three of fourpatients with this mutation had moderate to severediarrhea that showed stabilization after LT. Three offour patients had moderate to severe orthostasis that gotbetter in one patient and stabilized in two patients.
Cardiac involvement. Echocardiographic findings ofthickened posterior wall and/or ventricular septal wallwere seen in four of four patients. However, only two offour patients had evidence of low ejection fractions of30% and 43% at the time of pre-LT evaluation, which
Table 4. Outcome of Neuropathy, Orthostasis, and Posterior Wall and Ventricular Septal Thickness in the MET30 Group
Patient No.
1 2 3 4 5
NeuropathyPND score pre-LT IIIa II II I IPND score post-LT IIIb IIIa II II I
Diarrhea*Pre-LT 4 2 4 2 6Post-LT 6 2 4 2 �6†
Orthostasis*Pre-LT 4 2 4 0 2Post-LT 6 2 4 0 2
Posterior wall/ventricular septal thickness (mm)Pre-LT — 19/16 15/15 13/12 —Post-LT — 23/21 15/15 8/9 —
Abbreviation: PND, polyneuropathy disability.*Grades of diarrhea and orthostasis are based on patient symptoms, as listed in Table 1.†Grade greater than 6 indicates severe diarrhea with malnutrition.
1276 Sharma et al

decreased further with overt symptoms of congestiveheart failure. These two patients received combinedheart and liver transplants.
Survival. Three of four patients died. Two patients(who did not receive a heart transplant) died of progres-sion of heart disease. One of three patients who receivedcombined liver and heart transplants died of progres-sion of neuropathy.
TYR77 Patient
One of the seven NMET30 patients had the TYR77mutation.
Neuropathy, diarrhea, and orthostatic hypotension. Thispatient had mild neuropathy (PND score I) that pro-gressed to PND score II after LT. Diarrhea and ortho-static hypotension stabilized after LT. Pre-LT NCS andEMG were performed 15 months before LT.
Cardiac involvement. A pre-LT echocardiogramshowed posterior wall and ventricle septal thickness of 26mm (normal, 9 to 12 mm), posterior wall thickness of 25mm (normal, 9 to 12 mm), and left ventricular ejectionfraction of 20% to 25%, with moderate mitral and tricus-pid regurgitation. This patient also had conduction abnor-malities and a dual-chamber pacemaker. The patientunderwent combined liver, heart, and kidney transplanta-tion. One year posttransplantation, an echocardiogramshowed normal wall thickness and systolic function.
Renal involvement. This patient also had biopsy-proven renal involvement, with a low creatinine clear-ance and proteinuria, and underwent combined three-organ (heart, liver, and kidney) transplantation.
Survival. The patient is alive after 3 years of follow-up.
GLY42 Patient
One of seven NMET30 patients had the GLY42 muta-tion.
Neuropathy, diarrhea, and orthostatic hypotension. Thispatient had peripheral neuropathy with a PND score ofII and mild diarrhea. NCS and EMG and autonomictesting were not available. Neuropathy and diarrhealsymptoms are stable after transplantation.
Cardiac involvement. This patient also had a familyhistory of FAP and biopsy-proven cardiac amyloidosis.Initial symptoms were fatigue and dyspnea 3 yearsbefore transplantation. The pretransplantation echo-cardiogram showed a thickened ventricular septum andposterior wall (19 and 16 mm) and left ventricularejection fraction of 35%, which deteriorated to 18%during the waiting time. The patient also had a ventric-ular pacemaker. This patient underwent combined liverand heart transplantation.
Survival. This patient is alive after 5.8 years of fol-low-up.
LYS89 Patient
One of seven NMET30 patients had the LYS89 muta-tion.
Neuropathy, diarrhea, and orthostatic hypotension. A pre-transplantation PND score of II progressed to IIIa aftertransplantation. One-year posttransplantation EMG
Table 5. Outcome of Neuropathy, Orthostasis, and Posterior Wall and Ventricular Septal Thickness in the NMET30 Group
Patient No. (Mutation)
1(ALA60)
2(ALA60)
3(ALA60)
4(ALA60)
5(TYR77)
6(GLY42)
7(LYS89)
NeuropathyPND scores pre-LT IIIb None IIIa I I II IIPND scores post-LT IV None IIIb I II II IIIa
Diarrhea*Pre-LT 6 0 4 4 4 2 6Post-LT 6 0 4 4 4 2 4
Orthostasis*Pre-LT 6 0 4 4 4 Not available 4Post-LT 4 0 4 4 2 Not available 2
Posterior wall/ventricular septal thickness (mm)Pre-LT 17/14 17/16 20/20 17/12 26/25 19/16 18/18Post-LT 18/16 HT HT 19/19 HT HT HT
Abbreviations: ALA60, alanine 60; TYR77, tyrosine 77; GLY42, glycine 42; LYS89, lysine 89; PND, polyneuropathy disability; HT,heart transplant.*Grades of diarrhea and orthostasis are based on patient symptoms as listed in Table 1.
1277Familial Amyloidosis and Liver Transplantation

showed progression of sensorimotor neuropathy. Thepretransplantation NCS and EMG study was per-formed 14 months before transplantation. Diarrhealsymptoms were severe in intensity, but improved aftertransplantation. Orthostatic hypotension also im-proved after transplantation.
Cardiac involvement. This patient had biopsy andechocardiographic evidence of cardiac involvement,with a thickened posterior wall and ventricular septumand low left ventricular ejection fraction. He underwentcombined liver and heart transplantation and is doingwell.
Survival. This patient is alive after 4 years of follow-up.
Correlation of Clinical Features
There was a negative correlation between survival afterLT and pre-LT severity of diarrhea (r � �0.770; P �.003) and orthostatic hypotension (r � �0.687; P �.028). Increased severity of diarrhea and orthostatichypotension before LT was associated with worse sur-vival after LT. Furthermore, severity of diarrhea beforeLT correlated with pre-LT severity of orthostatic hypo-tension (r � 0.791; P � .006).
Complications of LT
One patient in each group had acute hepatic arterythrombosis and had to undergo a second LT. Two of 12patients developed a biliary leak. One patient had acuterenal failure during the postoperative period that wasmanaged conservatively.
Discussion
FAP, first described by Andrade,4 is a debilitating dis-ease that causes progressive organ dysfunction and ulti-mately leads to death. The genetic defect leads to abnor-mal accumulation of amyloidogenic TTR protein intissues, resulting in their deterioration. Previously,treatment was limited to palliative care of organ dys-function (e.g., pacemakers for conduction abnormali-ties of the heart, hemodialysis therapy for renal failure,and parenteral nutrition for gastrointestinal dysfunc-tion), without producing improvement in overall sur-vival. Both wild-type and variant TTR are synthesizedmainly by hepatocytes. Thus, LT is the definitive treat-ment of the disease.
This is the largest series of NMET30 patients fromthe United States to report on their outcome after LT.Less than 50% of the 12 patients with MET30 or
NMET30 mutations showed either improvement orstabilization in the EMG and NCS study and PNDscore after LT. However, diarrhea and orthostatichypotension improved or stabilized after LT in mostMET and NMET patients.
Cardiac involvement was observed more commonlyin NMET patients. Three of our four ALA60 patientsdied after LT of progression of heart disease and neu-ropathy. Four ALA60 patients are described in the lit-erature.21 Two of these patients died after LT of pro-gression of heart disease. Kotani et al22 recentlydescribed a first non–Caucasian patient harboring theALA60 TTR mutation who presented with severe late-onset restrictive cardiomyopathy, as well as sensorimo-tor and autonomic polyneuropathy. In our study, threeof four patients with the ALA60 TTR mutation died;thus, taken together, ALA60 appears to be a mutationof bleak prognosis, and it appears these patients need acombined liver and heart transplant.
Dubrey et al16 reported that some patients who hadFAP showed continued left ventricular wall thickeningafter LT, with a concomitant trend to deterioration inventricular function, which was observed more fre-quently in NMET30 patients. Dubrey et al16 also sug-gested that preexisting amyloid fibrils could act as anidus for nonhepatic sources of mutant TTR or for thedeposition of normal TTR produced by the trans-planted liver. They reported progression of left ventric-ular wall thickening after LT in the GLY42 TTR vari-ant.16 Our patient with the GLY42 mutation who hadrestrictive cardiomyopathy with a low ejection fractionunderwent combined liver and heart transplantationand had no posttransplantation evidence of cardiacamyloid.
There are three described cases in the literature withthe TYR77 mutation. One of these patients had pro-gression of cardiomyopathy and neuropathy after LT.23
The other two patients with the TYR77 mutation werereported to undergo combined heart and liver trans-plantation, and none of these patients had amyloiddeposit on routine posttransplantation endomyocardialbiopsy.21,24 Our patient with the TYR77 TTR variantreceived a liver, heart, and kidney transplant. This is thefirst patient with this mutation to undergo three-organtransplantation. This patient with the TYR77 mutationhad more predominant cardiac involvement and wouldbenefit from combined heart and liver transplantation.Stangou et al21 reported very depressing results regard-ing the proline 52 (PRO52) mutation. Thus, type ofmutation probably is of paramount importance for out-come; some mutations do well and others encounterserious problems and may need combined liver and
1278 Sharma et al

heart transplants.21 Some mutations, such as PRO52and cysteine 114, may not be helped by transplantationat all. Because a relatively small number of patients withNMET30 mutations undergo transplantation, reportsof outcome and problems with each of these mutationsare important.
Recently, the FAP World Transplant Registry col-lected data for several centers and reported an overall5-year survival rate of 75%.25 Adverse outcomes areassociated with poor nutritional status, long-standingdisease of more than 7 years’ duration before LT, andTTR variants other than MET30.25 In our series of 12patients, because of the small sample size, none of thesefactors showed a correlation with outcome. At 1 year,the survival rate was 100%, and at 3 years, there wasonly one death in the NMET30 group.
Previous reports suggested that patients withNMET30 compared with MET30 mutations are asso-ciated with a poor outcome after LT.16,26-29 Based onthese previous findings, we expected stabilization orimprovement in neuropathy, diarrhea, orthostatichypotension, and heart disease in most patients withMET30 mutations compared with those withNMET30 mutations. However, we found that bothgroups had similar presentations for neuropathy, gas-trointestinal involvement, and orthostasis, and alsosimilar outcomes.
Our study also identified diminishing gastrointesti-nal symptoms reported by earlier investigators.19,30 Thestabilization of diarrhea observed in most patients inboth groups may have been a result of LT or may haveoccurred because a majority were treated symptomati-cally with antidiarrheal agents. Long-term administra-tion of steroids and immunosuppressive agents31 andthe use of midodrine for symptomatic treatment oforthostatic hypotension could partially explainimprovement of this symptom. However, autonomicimprovement could be attributed to regeneration ofsmall unmyelinated fibers in peripheral nerves,32 incontrast to the large myelinated nerve fibers in whichregeneration is more prolonged.
Most of our patients with FAP who underwent LTwere older and referred late in the course of the disease.Only one patient had an mBMI less than 700 pretrans-plantation. No previously known preoperative prog-nostic factors (e.g., disease duration, NMET30 muta-tion, and mBMI)25 showed a correlation with pooroutcome after LT in our series of patients.
In conclusion, outcomes of LT in MET30 andNMET30 patients are similar with regard to outcomesof neuropathy, diarrhea, and orthostatic hypotension inour cohort of patients. Cardiac involvement is common
in the NMET group, and these patients should be eval-uated for combined liver and heart transplantation,when appropriate. Earlier LT for patients with FAPwith MET30 or NMET30 mutation would improveoutcome after LT. Last, combined liver and heart trans-plantation should be considered in patients withNMET30 mutation with a significant increase in leftventricular posterior wall and septal wall thickness andlow ejection fraction.
Acknowledgment
The authors thank J. Hernandez, Section of Biostatistics,Mayo Clinic, Scottsdale, AZ, for assistance with statisticalanalysis.
References1. Murakami T, Uchino M, Ando M. Genetic abnormalities and
pathogenesis of familial amyloidotic polyneuropathy. Pathol Int1995;45:1-9.
2. Saraiva MJ. Recent advances in the molecular pathology of famil-ial amyloid polyneuropathy. Neuromuscul Disord 1991;1:3-6.
3. Coutinho P, Martin da Silva A, Lopes Lima J, Resende BarbosaA. Forty years of experience with type I amyloid neuropathy.Review of 483 cases. In: Glenner GG, Pe CP, Falcao De FreitasA (eds). Amyloid and amyloidosis. Amsterdam: Experta Medica,1980:88-98.
4. Andrade C. A peculiar form of peripheral neuropathy. Brain1952;75:408-427.
5. Anderrson R. Familial amyloidosis with polyneuropathy: A clin-ical study based on patients living in northern Sweden. Acta MedScand 1976;(suppl S590);198:1-64.
6. Sousa A, Coelho T, Barros J, Sequeiros J. Genetic epidemiologyof familial amyloidotic polyneuropathy (FAP)-type I in Povoa doVarzim and Vila do Conde (north of Portugal). Am J Med Genet1995;60:512-521.
7. Holmgren G, Costa PM, Andersson C, Asplund K, Steen L,Beckman L, et al. Geographical distribution of TTR met30carriers in northern Sweden: Discrepancy between carrier fre-quency and prevalence rate. J Med Genet 1994;31:351-354.
8. Palacios SA, Bittencourt PL, Cancado EL, Farias AQ, MassarolloPCB, Mies S, et al. Familial amyloidotic polyneuropathy type Iin Brazil is associated with Val30MET variant. Amyloid 1999;6:289-291.
9. Suhr OB, Holmgren G, Steen L, Wikstrom L, Norden G, Fri-man S, et al. Liver transplantation in familial amyloidotic poly-neuropathy. Follow-up of the first 20 Swedish patients. Trans-plantation 1995;60:933-938.
10. Pomfret EA, Lewis WD, Jenkins RL, Bergethon P, Dubrey SW,Reisinger J, et al. Effect of orthotopic liver transplantation on theprogression of familial amyloidotic polyneuropathy. Transplan-tation 1998;65:918-925.
11. Parrilla P, Lopez-Andreu FR, Ramirez P, Bueno FS, Robles R,Miras M, et al. Familial amyloidotic polyneuropathy type I(Andrade’s disease): A new indication for liver transplant. Trans-plantation 1994;57:473-475.
12. Holmgren G, Ericzon BG, Groth CG, Steen L, Suhr O,Andersen O, et al. Clinical improvement and amyloid regression
1279Familial Amyloidosis and Liver Transplantation

after liver transplantation in hereditary transthyretin amyloid-osis. Lancet 1993;341:1113-1116.
13. Holmgren G, Steen L, Ekstedt J, Groth CG, Ericzon BG, Eriks-son S, et al. Biochemical effect of liver transplantation in twoSwedish patients with familial amyloidotic polyneuropathy(FAP-met30). Clin Genet 1991;40:242-246.
14. Suhr OB, Herlenius G, Friman S, Ericzon BG. Liver transplan-tation for hereditary transthyretin amyloidosis. Liver Transpl2000;6:263-276.
15. Adams D, Samuel D, Goulon-Goeau C, Nakazato M, CostaPM, Feray C, et al. The course and prognostic factors of familialamyloid polyneuropathy after liver transplantation. Brain 2000;123:1495-1504.
16. Dubrey SW, Davidoff R, Skinner M, Bergethon P, Lewis D,Falk RH. Progression of ventricular wall thickening after livertransplantation for familial amyloidosis. Transplantation 1997;64:74-80.
17. Suhr OB, Ericzon BG, Friman S. Long-term follow-up of sur-vival of liver transplant recipients with familial amyloid polyneu-ropathy (Portuguese type). Liver Transpl 2002;8:787-794.
18. Tashima KAY, Ando E, Tanaka Y, Ando M, Uchino M. Heter-ogeneity of clinical symptoms in patients with familial amyloid-otic polyneuropathy (FAP TTR Met30) amyloid. Int J Exp ClinInvest 1996;4:108-111.
19. Tashima K, Ando Y, Terazaki H, Yoshimatsu S, Suhr OB, Oba-yashi K, et al. Outcome of liver transplantation for transthyretinamyloidosis: Follow-up of Japanese familial amyloidotic poly-neuropathy patients. J Neurol Sci 1999;171:19-23.
20. Zeldenrust S, Gertz M, Uemichi T, Bjornsson J, Wiesner R,Schwab T, et al. Orthotopic liver transplantation for hereditaryfibrinogen amyloidosis. Transplantation 2003;75:560-561.
21. Stangou AJ, Hawkins PN, Heaton ND, Rela M, Monaghan M,Nihoyannopoulos P, et al. Progressive cardiac amyloidosis fol-lowing liver transplantation for familial amyloid polyneurop-athy: Implication for amyloid fibrillogenesis. Transplantation1998;66;229-233.
22. Kotani N, Hattori T, Yamagata S, Tokuda T, Shirasawa A,Yamaguchi S, et al. Transthyretin Thr60Ala Appalachian type
mutation in a Japanese family with familial amyloidotic polyneu-ropathy. Amyloid J 2002;9:31-34.
23. Garcia-Herola A, Prieto M, Pascual S, Berenguer M, Lopez-Viedma B, Mir J, et al. Progression of cardiomyopathy andneuropathy after liver transplantation in a patient with familialamyloidotic polyneuropathy caused by tyrosine 77 transthyere-tin variant. Liver Transpl 1999;5:246-248.
24. Ruygrok PN, Gane EJ, McCall JL, Chen XZ, Haydock DA,Munn SR. Combined heart and liver transplantation for familialamyloidosis. Int Med J 2001;31:66-67.
25. Herlenius G, Larsson M, Ericzon BG. Results from the familialamyloidotic polyneuropathy world transplant registry. Trans-plant Proc 2001;33:2454.
26. Ericzon BG, Suhr O, Broome U, Holmgren G, Duraj F, EleborgL, et al. Liver transplantation halts the progress of familial amy-loidotic polyneuropathy. Transplant Proc 1995;27:1233.
27. Monteiro E, Perdigoto R, Furtado AL. Liver transplantation forfamilial amyloid polyneuropathy. Hepatogastroenterology1998;45:1375-1380.
28. Bittencourt PL, Couto CA, Farias AQ, Marchiori P, Bosco Mas-sarollo PC, Mies S. Results of liver transplantation for familialamyloid polyneuropathy type I in Brazil. Liver Transpl 2002;8:34-39.
29. Munitiz V, Ramirez P, Munar M, Andreu F, Robles R, BuenoSF, et al. Reversibility of the neurologic alterations in familialamyloidotic polyneuropathy type I after liver transplantation (22cases). Transplant Proc 2002;34:310-311.
30. Parrilla P, Ramirez P, Andreu LF, Bueno SF, Robles R, Miras M,et al. Long-term results of liver transplantation in familial amy-loidotic polyneuropathy type I. Transplantation 1997;64:646-649.
31. Winkler M, Brinkmann C, Jost U, Oldhafer K, Ringe B, Pichl-mayr R. Long-term side effects of cyclosporine-based immuno-suppression in patients after liver transplantation. TransplantProc 1994;26:2679-2682.
32. Ando Y, Tanaka Y, Ando E, Yamashita T, Nishida Y, Tashima K,et al. Effect of liver transplantation on autonomic dysfunction infamilial amyloidotic polyneuropathy type I. Lancet 1995;345:195-196.
1280 Sharma et al





























