Embed Size (px)
Citation preview

Original Articles
Analysis of Dystrophin Deletion Mutations Predicts Age ofCardiomyopathy Onset in Becker Muscular DystrophyRita Wen Kaspar, PhD, RN; Hugh D. Allen, MD; Will C. Ray, PhD; Carlos E. Alvarez, PhD;
John T. Kissel, MD; Alan Pestronk, MD; Robert B. Weiss, PhD; Kevin M. Flanigan, MD;Jerry R. Mendell, MD; Federica Montanaro, PhD
Background—Becker muscular dystrophy (BMD) and X-linked dilated cardiomyopathy often result from deletionmutations in the dystrophin gene that may lead to expression of an altered dystrophin protein in cardiac muscle. Cardiacinvolvement is present in �70% of BMD and all X-linked dilated cardiomyopathy cases. To date, the timing ofcardiomyopathy development remains unpredictable. We analyzed 78 BMD and X-linked dilated cardiomyopathypatients with common deletion mutations predicted to alter the dystrophin protein and correlated their mutations tocardiomyopathy age of onset. This approach was chosen to connect dystrophin structure with function in the heart.
Methods and Results—Detailed cardiac information was collected for BMD and X-linked dilated cardiomyopathy patientswith defined dystrophin gene deletion mutations. Patients were grouped based on the dystrophin protein domain affectedby the deletion. Deletions affecting the amino-terminal domain are associated with early-onset dilated cardiomyopathy(DCM; mid-20s), whereas deletions removing part of the rod domain and hinge 3 have a later-onset DCM (mid-40s).Further, we modeled the effects of the most common mutations occurring in the rod domain on the overall structure ofthe dystrophin protein. By combining genetic and protein information, this analysis revealed a strong correlationbetween specific protein structural modifications and DCM age of onset.
Conclusions—We identified specific regions of the dystrophin gene that when mutated predispose BMD patients toearly-onset DCM. In addition, we propose that some mutations lead to early-onset DCM by specific alterations in proteinfolding. These findings have potential implications for early intervention in the cardiac care of BMD patients and fortherapeutic approaches that target the heart in dystrophinopathies. (Circ Cardiovasc Genet. 2009;2:544-551.)
Key Words: cardiomyopathy � genetics � risk factors � muscular dystrophy � dystrophin
The dystrophin gene, located on the X-chromosome, is thelargest known human gene (2.4 Mb, 79 exons), resulting
in a high rate of spontaneous disease-causing mutations (30%of cases) with deletions forming the majority (�60%).Dystrophin plays an essential structural role in both cardiacand skeletal muscle, protecting the sarcolemma from mechan-ical stresses of muscle contraction. Complete loss of dystro-phin leads to Duchenne muscular dystrophy (DMD), the mostcommon severe form of childhood muscular dystrophy,complicated by skeletal muscle degeneration and dilatedcardiomyopathy (DCM).
Clinical Perspective on p 551In contrast to the well-defined clinical course of DMD,
mutations that do not disrupt the reading frame can result in
expression of an altered dystrophin protein, leading to a morevariable clinical presentation. This includes Becker musculardystrophy (BMD) that presents primarily with progressiveskeletal muscle degeneration with variable age of onset andseverity, and X-linked DCM (XLDCM) that typically has nodetectable skeletal muscle signs accompanying the cardiacinvolvement. Although BMD is historically diagnosed basedon skeletal muscle manifestations, the primary cause of deathis heart failure.1 Indeed, �70% of BMD patients developDCM,1–4 and a recent longitudinal study demonstrated anonset of cardiac involvement in the early teens in some BMDpatients.5 Similar to DMD and XLDCM, the severity and ageof onset of cardiac involvement in BMD show no correlationto skeletal muscle involvement.2,3 Furthermore, DCM is often
Received March 25, 2009; accepted September 21, 2009.From the Center for Gene Therapy (R.W.K., J.R.M., F.M.), The Research Institute at Nationwide Children’s Hospital; College of Nursing (R.W.K.),
The Ohio State University; Division of Pediatric Cardiology (H.D.A.), The Ohio State University College of Medicine, Nationwide Children’s Hospital,Heart Center; Battelle Center for Mathematical Medicine (W.C.R.), The Research Institute at Nationwide Children’s Hospital; Biophysics GraduateProgram (W.C.R.), The Ohio State University; Center for Molecular and Human Genetics (C.E.A.), The Research Institute at Nationwide Children’sHospital; Departments of Pediatrics (C.E.A., J.R.M., F.M.) and Neurology (J.T.K., J.R.M.), The Ohio State University College of Medicine, Columbus,Ohio; Department of Neurology (A.P.), Washington University, St. Louis, Mo; and Departments of Genetics (R.B.W.) and Pediatrics (K.M.F.), Universityof Utah School of Medicine, Salt Lake City, Utah.
The online-only Data Supplement is available at http://circgenetics.ahajournals.org/cgi/content/full/CIRCGENETICS.109.867242.Correspondence to Federica Montanaro, PhD, The Research Institute at Nationwide Children’s Hospital, Center for Gene Therapy, 700 Children’s
Drive, WA3020, Columbus, OH 43205. E-mail [email protected]© 2009 American Heart Association, Inc.
Circ Cardiovasc Genet is available at http://circgenetics.ahajournals.org DOI: 10.1161/CIRCGENETICS.109.867242
544
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
by guest on M
ay 27, 2018http://circgenetics.ahajournals.org/
Dow
nloaded from

diagnosed after cardiac symptoms manifest, diminishing theefficacy of cardioprotective drugs. Therefore, the identifica-tion of parameters for cardiac risk assessment before symp-tom manifestation bears undeniable relevance for the clinicalcare of BMD patients and would assist in patient stratificationin clinical trials testing the efficacy of cardiac treatments. It iswith this in mind that we extensively examined a large cohortof BMD and XLDCM patients with deletion mutations toexplore the hypothesis that loss of specific domains of thedystrophin protein predispose to early-onset cardiomyopathyby potentially affecting protein expression levels, function,and/or structure. This hypothesis was born from previousstudies exploring correlation between genotype and the pres-ence of DCM in BMD patients2–4 as well as reports on theassociation of deletions in specific dystrophin domains withseverity of skeletal muscle symptoms.6–8
Materials and MethodsPatient Sources and Data CollectionThis is a cross-sectional retrospective study of patient informationobtained from 3 major sources: (1) a database from the MuscularDystrophy Association clinics at Nationwide Children’s Hospitaland The Ohio State University Medical Center; (2) the UnitedDystrophinopathy Project database (PI, Kevin Flanigan, Co-PIs JerryR Mendell and Alan Pestronk); and (3) published studies. Informa-tion was obtained related to age of onset and severity of skeletal andcardiac muscle manifestations, description of cardiac evaluation andskeletal muscle biopsy, the gene mutation, family history, and serumcreatine kinase. Echocardiograms at Nationwide Children’s Hospitalwere interpreted by the same cardiologist (H.D.A.). The InstitutionalReview Board at Nationwide Children’s Hospital and The Ohio StateUniversity approved all protocols.
Inclusion and Exclusion CriteriaInclusion criteria included (1) a diagnosis of BMD or XLDCM orboth, (2) cardiac evaluation, and (3) a confirmed exon deletionmutation spanning up to 11 exons. The 11 exon limit was set toexclude large deletions potentially affecting multiple functionaldomains of dystrophin, while including most patients who werefound to have deletions affecting 1 to 8 exons. Patients with exon 45to 55 deletions known to be associated with mild, late-onset skeletalmuscle involvement9,10 were also included. More stringent criteriapotentially compromised statistical power.
Exclusion criteria included (1) reported or suspected cardiac viralinfections, (2) cardiac biopsy without dystrophin expression, (3)deletions restricted to noncoding regions of the dystrophin genepotentially containing ill-defined regulatory elements, (4) subjectsyounger than 12 years without proven family history of BMD, and(5) wheelchair-dependent patients by age 12 years carrying adiagnosis of BMD (preferably considered a severe form of dystro-phinopathy11). Deletions selectively affecting myocardial expressionof dystrophin have been excluded from this study primarily becauseof low patient numbers, precluding meaningful statistical analysis.Supplemental Table I lists subjects excluded based on these criteria.Of 320 subjects initially screened for inclusion in this study, 118satisfied the selection criteria.
Definition of Cardiomyopathy and Disease OnsetCardiomyopathy was defined as follows: ejection fraction �55% orshortening fraction �32% or both. The ejection fraction cutoffagrees with previous studies based on the natural history of thedisease5,12 and with timing of cardiac drug intervention common inclinical practice for BMD patients. When available, additionalparameters were considered to support cardiac dilation: E-point
septal separation above 5 mm, left ventricular (LV) end-diastolicdiameter above 58 mm or above 2 z scores when indexed to bodysurface area, or cardiomegaly consistent with cardiomyopathy bychest radiograph. Electrocardiograms were not used to define car-diomyopathy.13 In this cross-sectional study, the time at whichabnormal cardiac findings were first reported defines the “onset” ofcardiomyopathy. The age of onset of DCM represents the youngestreported age at which cardiac parameters met the definition ofcardiomyopathy.
For analyses involving noncardiomyopathic patients, age corre-sponds to the oldest reported age at which cardiac findings werenormal.
EchocardiographyFor United Dystrophinopathy Project and Muscular DystrophyAssociation clinic patients, images from 2D and Doppler ultrasoundstudies were evaluated by standard techniques. Measurements in-cluded LV diameter, shortening fraction (LV diastolic diameterminus LV systolic diameter divided by LV diastolic diameter), andejection fraction (Simpsons’ formula applied to planimeterized diastolicand systolic LV cavity images derived from the apex view). Forpublished cases, deviations from this methodology can be found inthe original articles (supplemental Tables II and III).
Patient GroupingPatients were categorized into 3 groups based on the affectedfunctional domain of the dystrophin protein. Group 1: subjects withdeletions affecting any portion of the actin-binding amino-terminaldomain of dystrophin (exons 2 to 9). Hinge 3 (a specific proteinsequence joining 2 segments of dystrophin that allows flexiblemovement accounting for intrinsic protein folding) has been impli-cated in skeletal muscle involvement6 and thus served to divideBMD subjects into 2 additional groups. Group 2: subjects withdeletions preserving hinge 3 and affecting exons 45 to 49 (spectrinrepeats 17 up to 19). Group 3: subjects with deletions affecting exon50 or 51 or both, removing or disrupting hinge 3.
Dystrophin Protein ModelingRod-region spectrin repeats were modeled based on the publishedstructure of repeats 15 and 16 of chicken brain �-spectrin (PDB1U5P).14 The structure was manually extended by replication andRMS alignment of corresponding terminal residues, using PyMol(http://www.pymol.org/). The structure was briefly minimized usingVMD/NAMD (http://www.ks.uiuc.edu/Research/namd/) to removesignificant bad contacts. Hinge region and out-of-phase deletionmutation structures were constructed by manual deletion of structur-ally equivalent residues from the extended spectrin repeat andstructural realignment of the resulting fragments. A brief minimiza-tion using VMD/NAMD was used to correct any significant mis-placement of fragment ends.
Statistical AnalysisNonparametric Kruskal-Wallis test was performed for cross-groupage comparisons (a priori P�0.05), followed by Mann–Whitney Utest post hoc comparisons among groups (Bonferroni adjustment wasused to achieve overall significance of P�0.05). Mann–Whitney Utest was used to compare age of cardiomyopathy onset betweenin-phase and out-of-phase mutations in group 2 patients. For bloodrelatives, only 1 patient was randomly selected for inclusion instatistical analyses. Three sibling pairs were identified within car-diomyopathic patients: 224 and 225, 251 and 3, AH11 and MJ13(supplemental Table II). Concordance in the age of cardiomyopathyonset was observed among siblings. All data were analyzed in SPSSversion 15 (SPSS, Chicago).
Kaspar et al Dystrophin Mutations Predict Cardiomyopathy Onset 545
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from

ResultsPatient Selection and DescriptionA total of 118 BMD and XLDCM patients (supplementalTables II and III) were enrolled. Table 1 shows the break-down of patients based on diagnosis, source, and whetherthey were categorized as cardiomyopathic or noncardiomyo-pathic. Only subjects with cardiomyopathy (n�78) wererequired to test our hypothesis that the age of DCM manifes-tation is associated with deletion of specific dystrophinprotein domains. However, we did analyze noncardiomyo-pathic patients for evidence of a cardioprotective effect ofsome deletion mutations. We found that the noncardiomyo-pathic BMD patients were significantly younger than cardio-
myopathic BMD patients (P�0.001) and that their deletionmutations overlap with those of cardiomyopathic patients(supplemental Figure 1). This suggests that noncardiomyo-pathic patients were too young to manifest cardiac involve-ment and will require follow-up studies to further test thehypothesis under consideration.
Distribution of Mutations Relative to DystrophinProtein Structure and DiagnosisTo determine whether all cardiomyopathic patients can becombined for maximum statistical power, we first testedwhether the source of patient information (published versusUnited Dystrophinopathy Project) or the diagnosis (BMD
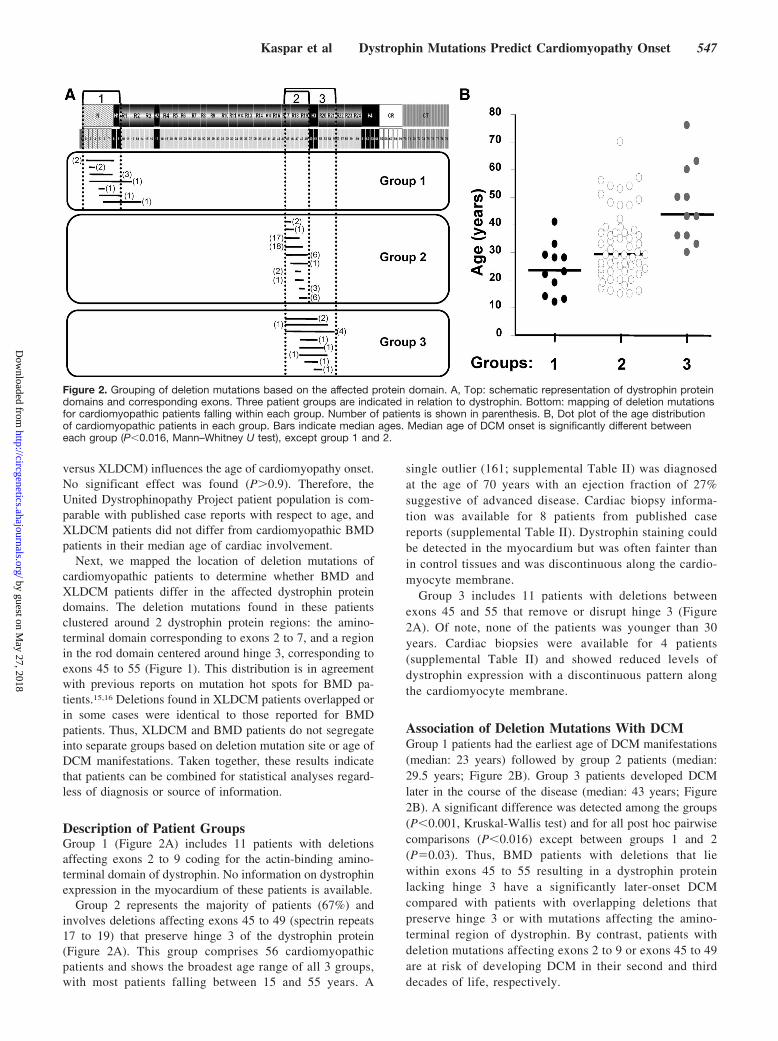
Figure 1. Mapping of deletion mutations in cardiomyopathic patients. Dystrophin protein domains and corresponding exons are sche-matically represented at the top. Deleted regions are indicated below by a line. The number of patients with any given deletion muta-tion is indicated in parenthesis. Patients are grouped based on diagnosis and source. Dystrophin domains: N indicates amino terminus(diagonal stripes); R, spectrin repeats 1 through 24 (gray); H, hinges 1 through 4 (black); CR, cysteine-rich domain (white); CT, carboxylterminus (vertical stripes).
Table 1. Patient Distribution According to Diagnosis, Source of Information, andCategorization as Affected or Not With DCM
Diagnosis Source n
Cardiomyopathic Noncardiomyopathic
Median Age* N Median Age† n
XLDCM Publication 17 30 17 … …
BMD Publication 59 29.5 38 18 21
BMD UDP 42 33 23 13 19
The median age in years of the patients in each category is indicated as well as the No. of patients (n).*Youngest age when fulfilling criteria for a diagnosis of cardiomyopathy.†Oldest age at which cardiac function was found to be within normal parameters.
546 Circ Cardiovasc Genet December 2009
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
versus XLDCM) influences the age of cardiomyopathy onset.No significant effect was found (P�0.9). Therefore, theUnited Dystrophinopathy Project patient population is com-parable with published case reports with respect to age, andXLDCM patients did not differ from cardiomyopathic BMDpatients in their median age of cardiac involvement.
Next, we mapped the location of deletion mutations ofcardiomyopathic patients to determine whether BMD andXLDCM patients differ in the affected dystrophin proteindomains. The deletion mutations found in these patientsclustered around 2 dystrophin protein regions: the amino-terminal domain corresponding to exons 2 to 7, and a regionin the rod domain centered around hinge 3, corresponding toexons 45 to 55 (Figure 1). This distribution is in agreementwith previous reports on mutation hot spots for BMD pa-tients.15,16 Deletions found in XLDCM patients overlapped orin some cases were identical to those reported for BMDpatients. Thus, XLDCM and BMD patients do not segregateinto separate groups based on deletion mutation site or age ofDCM manifestations. Taken together, these results indicatethat patients can be combined for statistical analyses regard-less of diagnosis or source of information.
Description of Patient GroupsGroup 1 (Figure 2A) includes 11 patients with deletionsaffecting exons 2 to 9 coding for the actin-binding amino-terminal domain of dystrophin. No information on dystrophinexpression in the myocardium of these patients is available.
Group 2 represents the majority of patients (67%) andinvolves deletions affecting exons 45 to 49 (spectrin repeats17 to 19) that preserve hinge 3 of the dystrophin protein(Figure 2A). This group comprises 56 cardiomyopathicpatients and shows the broadest age range of all 3 groups,with most patients falling between 15 and 55 years. A
single outlier (161; supplemental Table II) was diagnosedat the age of 70 years with an ejection fraction of 27%suggestive of advanced disease. Cardiac biopsy informa-tion was available for 8 patients from published casereports (supplemental Table II). Dystrophin staining couldbe detected in the myocardium but was often fainter thanin control tissues and was discontinuous along the cardio-myocyte membrane.
Group 3 includes 11 patients with deletions betweenexons 45 and 55 that remove or disrupt hinge 3 (Figure2A). Of note, none of the patients was younger than 30years. Cardiac biopsies were available for 4 patients(supplemental Table II) and showed reduced levels ofdystrophin expression with a discontinuous pattern alongthe cardiomyocyte membrane.
Association of Deletion Mutations With DCMGroup 1 patients had the earliest age of DCM manifestations(median: 23 years) followed by group 2 patients (median:29.5 years; Figure 2B). Group 3 patients developed DCMlater in the course of the disease (median: 43 years; Figure2B). A significant difference was detected among the groups(P�0.001, Kruskal-Wallis test) and for all post hoc pairwisecomparisons (P�0.016) except between groups 1 and 2(P�0.03). Thus, BMD patients with deletions that liewithin exons 45 to 55 resulting in a dystrophin proteinlacking hinge 3 have a significantly later-onset DCMcompared with patients with overlapping deletions thatpreserve hinge 3 or with mutations affecting the amino-terminal region of dystrophin. By contrast, patients withdeletion mutations affecting exons 2 to 9 or exons 45 to 49are at risk of developing DCM in their second and thirddecades of life, respectively.
Figure 2. Grouping of deletion mutations based on the affected protein domain. A, Top: schematic representation of dystrophin proteindomains and corresponding exons. Three patient groups are indicated in relation to dystrophin. Bottom: mapping of deletion mutationsfor cardiomyopathic patients falling within each group. Number of patients is shown in parenthesis. B, Dot plot of the age distributionof cardiomyopathic patients in each group. Bars indicate median ages. Median age of DCM onset is significantly different betweeneach group (P�0.016, Mann–Whitney U test), except group 1 and 2.
Kaspar et al Dystrophin Mutations Predict Cardiomyopathy Onset 547
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
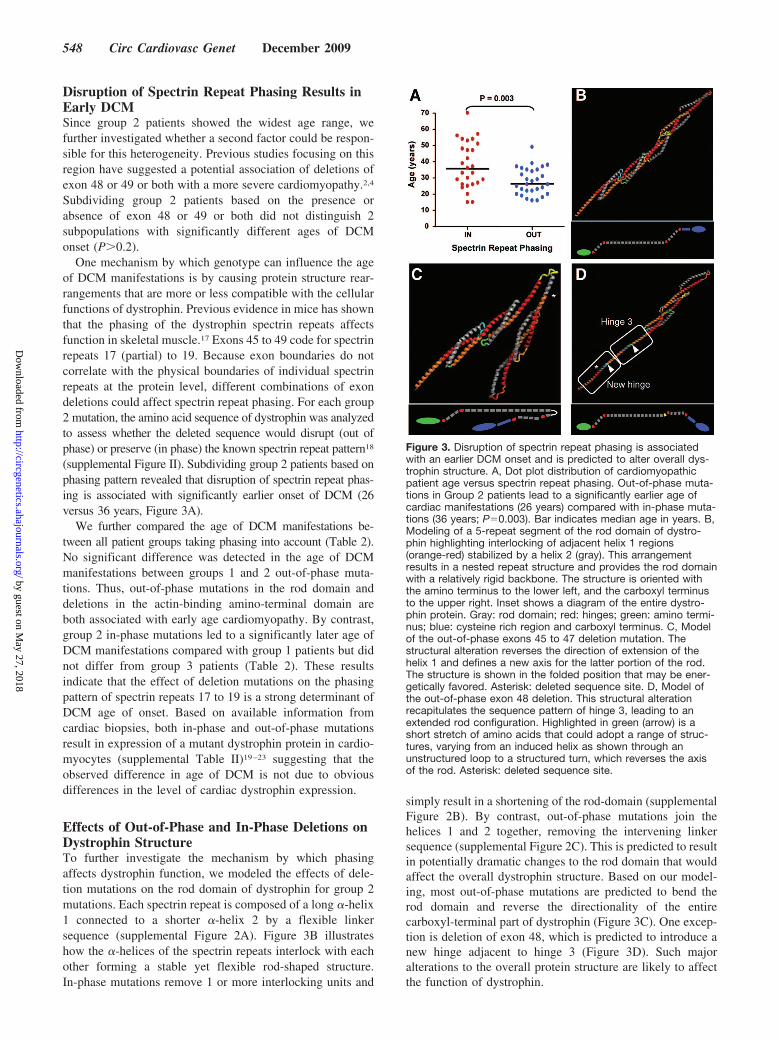
Disruption of Spectrin Repeat Phasing Results inEarly DCMSince group 2 patients showed the widest age range, wefurther investigated whether a second factor could be respon-sible for this heterogeneity. Previous studies focusing on thisregion have suggested a potential association of deletions ofexon 48 or 49 or both with a more severe cardiomyopathy.2,4
Subdividing group 2 patients based on the presence orabsence of exon 48 or 49 or both did not distinguish 2subpopulations with significantly different ages of DCMonset (P�0.2).
One mechanism by which genotype can influence the ageof DCM manifestations is by causing protein structure rear-rangements that are more or less compatible with the cellularfunctions of dystrophin. Previous evidence in mice has shownthat the phasing of the dystrophin spectrin repeats affectsfunction in skeletal muscle.17 Exons 45 to 49 code for spectrinrepeats 17 (partial) to 19. Because exon boundaries do notcorrelate with the physical boundaries of individual spectrinrepeats at the protein level, different combinations of exondeletions could affect spectrin repeat phasing. For each group2 mutation, the amino acid sequence of dystrophin was analyzedto assess whether the deleted sequence would disrupt (out ofphase) or preserve (in phase) the known spectrin repeat pattern18
(supplemental Figure II). Subdividing group 2 patients based onphasing pattern revealed that disruption of spectrin repeat phas-ing is associated with significantly earlier onset of DCM (26versus 36 years, Figure 3A).
We further compared the age of DCM manifestations be-tween all patient groups taking phasing into account (Table 2).No significant difference was detected in the age of DCMmanifestations between groups 1 and 2 out-of-phase muta-tions. Thus, out-of-phase mutations in the rod domain anddeletions in the actin-binding amino-terminal domain areboth associated with early age cardiomyopathy. By contrast,group 2 in-phase mutations led to a significantly later age ofDCM manifestations compared with group 1 patients but didnot differ from group 3 patients (Table 2). These resultsindicate that the effect of deletion mutations on the phasingpattern of spectrin repeats 17 to 19 is a strong determinant ofDCM age of onset. Based on available information fromcardiac biopsies, both in-phase and out-of-phase mutationsresult in expression of a mutant dystrophin protein in cardio-myocytes (supplemental Table II)19–23 suggesting that theobserved difference in age of DCM is not due to obviousdifferences in the level of cardiac dystrophin expression.
Effects of Out-of-Phase and In-Phase Deletions onDystrophin StructureTo further investigate the mechanism by which phasingaffects dystrophin function, we modeled the effects of dele-tion mutations on the rod domain of dystrophin for group 2mutations. Each spectrin repeat is composed of a long �-helix1 connected to a shorter �-helix 2 by a flexible linkersequence (supplemental Figure 2A). Figure 3B illustrateshow the �-helices of the spectrin repeats interlock with eachother forming a stable yet flexible rod-shaped structure.In-phase mutations remove 1 or more interlocking units and
simply result in a shortening of the rod-domain (supplementalFigure 2B). By contrast, out-of-phase mutations join thehelices 1 and 2 together, removing the intervening linkersequence (supplemental Figure 2C). This is predicted to resultin potentially dramatic changes to the rod domain that wouldaffect the overall dystrophin structure. Based on our model-ing, most out-of-phase mutations are predicted to bend therod domain and reverse the directionality of the entirecarboxyl-terminal part of dystrophin (Figure 3C). One excep-tion is deletion of exon 48, which is predicted to introduce anew hinge adjacent to hinge 3 (Figure 3D). Such majoralterations to the overall protein structure are likely to affectthe function of dystrophin.
Figure 3. Disruption of spectrin repeat phasing is associatedwith an earlier DCM onset and is predicted to alter overall dys-trophin structure. A, Dot plot distribution of cardiomyopathicpatient age versus spectrin repeat phasing. Out-of-phase muta-tions in Group 2 patients lead to a significantly earlier age ofcardiac manifestations (26 years) compared with in-phase muta-tions (36 years; P�0.003). Bar indicates median age in years. B,Modeling of a 5-repeat segment of the rod domain of dystro-phin highlighting interlocking of adjacent helix 1 regions(orange-red) stabilized by a helix 2 (gray). This arrangementresults in a nested repeat structure and provides the rod domainwith a relatively rigid backbone. The structure is oriented withthe amino terminus to the lower left, and the carboxyl terminusto the upper right. Inset shows a diagram of the entire dystro-phin protein. Gray: rod domain; red: hinges; green: amino termi-nus; blue: cysteine rich region and carboxyl terminus. C, Modelof the out-of-phase exons 45 to 47 deletion mutation. Thestructural alteration reverses the direction of extension of thehelix 1 and defines a new axis for the latter portion of the rod.The structure is shown in the folded position that may be ener-getically favored. Asterisk: deleted sequence site. D, Model ofthe out-of-phase exon 48 deletion. This structural alterationrecapitulates the sequence pattern of hinge 3, leading to anextended rod configuration. Highlighted in green (arrow) is ashort stretch of amino acids that could adopt a range of struc-tures, varying from an induced helix as shown through anunstructured loop to a structured turn, which reverses the axisof the rod. Asterisk: deleted sequence site.
548 Circ Cardiovasc Genet December 2009
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
DiscussionAn association of genotype with DCM has long been sus-pected but has not been clearly established.2–4,24 In this study,we demonstrated that genotype is a determinant of the age ofDCM manifestations in BMD and XLDCM patients. Thelarge sample size (78 patients) and stringent selection criteriaincluding only patients with small deletions (11 exons)enabled us to capture informative dystrophin protein do-mains. This allowed patient grouping based on dystrophinmutations affecting a single rather than multiple adjacentfunctional protein domains, thus increasing the statisticalpower of the study. We also provided novel evidence of astrong association of cardiomyopathy with specific structuralalterations of the dystrophin rod domain. Previous studiesreported contradictory findings on the potential link ofdeletions including exon 48 or 49 or both with severity andoccurrence of DCM.2–4,24 Analysis of our 56 patients in group2 showed that DCM in this region is more sensitive to alteredphasing of the spectrin repeats rather than absence or pres-ence of any individual exon. Our analysis of the effects ofspecific deletions on the 3D structure of dystrophin and theircorrelation with cardiac phenotype highlights the importanceof integrating protein structure information in genotype-phe-notype studies.
Our results also indicate that the rod domain of dystrophinmay not be as permissive to alterations as previously thought.This study suggests that preservation of phasing delays theonset of DCM by about a decade. This is likely not due to adifference in expression of cardiac dystrophin protein be-cause Arbustini et al20 reported similar amount and distribu-tion of dystrophin in cardiac biopsy samples from BMDpatients with either in-phase or out-of-phase mutations.Rather, our modeling suggests that the alterations caused byout-of-phase mutations extend beyond the spectrin repeat unitand may lead to a severely altered configuration of the roddomain, ultimately affecting the entire dystrophin protein.This major structural change is likely a main determinant ofearly-onset DCM. Interestingly, most group 2 BMD patients
have late-onset skeletal muscle symptoms and mild diseaseprogression, irrespective of the effects of their mutation onphasing. This is in agreement with studies in dystrophin-nullmdx mice expressing a minidystrophin construct that lacksthe exons 45 to 49 region but has an intact hinge 3 domain. Inthese mice, only a partial restoration of cardiac function wasachieved despite a complete rescue of the skeletal musclepathology.25 Thus, cardiac dystrophin may be particularlysensitive to structural disruptions of the exons 45 to 49 regioncompared with skeletal muscle dystrophin. The reasons forthis disparity are currently unknown but highlight the impor-tance of mapping domains of dystrophin essential for cardiacfunction to improve on current treatment approaches relyingon exon skipping or gene replacement with mini-/microdys-trophin constructs.
This study provides expected median ages of onset ofDCM associated with 3 distinct regions of the dystrophinprotein and with specific rearrangements of its rod domain.The deletion mutations studied here are among the mostfrequent, rendering our findings relevant to most BMD andXLDCM patients. Of interest, within groups, XLDCM pa-tients did not have an earlier age of cardiomyopathy com-pared with BMD patients. Instead, the earliest age of DCM isassociated with mutations affecting the amino-terminal do-main of the protein (early 20s), and out-of-phase mutations inthe exons 45 to 49 region of the dystrophin rod domain(mid-20s). Although cardiac expression of dystrophin hasbeen confirmed in several out-of-phase group 2 patients, suchinformation is not available for group 1 patients. The beststudied mutations affecting the 5� region of the dystrophingene (including the muscle promoter, exon 1, or intronicregions that alter exon splicing26–28) lead to a selective lack ofcardiac dystrophin. Although none of our group 1 patientshad mutations affecting noncoding regions or exon 1, wecannot exclude the possibility that the early DCM onset ingroup 1 patients reflects a selective absence of cardiacdystrophin. Of note, the median age of XLDCM patients withlack of cardiac dystrophin (24 years for 6 independentfamilies, supplemental Table I) is very similar to that of group1 patients (23 years). Further studies are needed to determinethe mechanism(s) by which deletion mutations in the amino-terminal region lead to earlier onset of cardiomyopathycompared with mutations affecting other regions. A greaterawareness of the value of cardiac tissue sampling at the timeof cardiac transplantation and the design of transgenic mdxmice mimicking human mutations could yield importantinformation on cardiac-specific mechanisms regulating thisregion of dystrophin at a transcriptional and protein level.
Significantly later DCM onset is associated with group 2in-phase (mid-1930s) and group 3 mutations (mid-1940s).The cardio-protective effect of hinge 3 deletion seen in group3 patients mirrors findings reported for skeletal muscle inboth mice and humans.6,17 However, although loss of hinge 3delays onset of cardiomyopathy, it correlated with slowerdisease progression in skeletal muscle but had no effect onage of onset.6 Because of the small number of group 3patients with identical deletions, we could not determinewhether this partially protective effect is associated with aspecific structural alteration of the dystrophin protein back-
Table 2. Pairwise Comparison of Cardiomyopathic PatientGroups Based on the Location of the Deletion Mutation and theEffects on Spectrin Repeat Phasing
Groups n Median Age, y P
Group 2 (in phase) vs: 27 36
Group 2 (out of phase) 29 26 0.003
Group 1 11 23 0.003
Group 3 11 43 0.107
Group 2 (out of phase) vs: 29 26
Group 1 11 23 0.24
Group 3 11 43 �0.001
Group 1 vs: 11 23
Group 3 11 43 �0.001
The Mann–Whitney U test was used for post hoc statistical comparisonsamong all 4 groups. Significance was set a priori at P�0.0125 (Bonferroniadjustment for overall significance of P�0.05). Significant P values areitalicized. The median age of cardiac manifestations is indicated for eachgroup.
Kaspar et al Dystrophin Mutations Predict Cardiomyopathy Onset 549
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from

bone. Further studies are needed to explain the significantcardio-protective effect conferred by the loss of hinge 3.
The median age of cardiac involvement for each patientgroup reported here is currently the best approximationavailable for this patient population. This information isvaluable because cardiac involvement in BMD patients isoften asymptomatic in its initial stages and can therefore beunderestimated. Because genotyping has become a morecommon practice, the median ages reported here may provevaluable for individualized risk assessment and for timelycardiac evaluation and intervention. An important next step isto conduct a large-scale longitudinal study to further refinethe age of DCM onset associated with the dystrophin domainsidentified here. This information underscores the importanceof genotype information in the cardiac care of BMD patientsand bears relevance to the design of therapies aimed at themyocardium in BMD, XLDCM, and DMD patients.
AcknowledgmentsWe acknowledge the input of the United Dystrophinopathy ProjectConsortium including the following individuals: Brenda Wong atCincinnati Children’s Hospital Medical Center, Richard Finkel,Carsten Bonnemann, and Livje Medne at Children’s Hospital ofPhiladelphia, Julaine Florence and Anne Connolly, WashingtonUniversity, Katherine Mathews, University of Iowa, Jacinda Samp-son, Mark Bromberg, and Kathryn J. Swoboda, University of Utah,and John W. Day, University of Minnesota. We thank Dr XiomaraRosales for her diagrams of the alignment of dystrophin exons withprotein domains and Brent Yetter for assistance in the identificationof patients seen at Muscular Dystrophy Association clinics who weresuitable for this study. We also thank Drs Carlos Miranda, JenniferThomas-Ahner, and Christopher Pierson for editing assistance andfor mentorship and support to R.W.K. from Dr Donna McCarthy,Professor of Nursing at The Ohio State University. We are indebtedto Dr Christopher Holloman from the College of Mathematical andPhysical Sciences at The Ohio State University for assistance withstatistical analysis.
Sources of FundingSupported by a grant from the NIH Roadmap Training Program inClinical Research (T32-RR023260-03, to R.W.K.). The UnitedDystrophinopathy Project is supported by grants from the NationalInstitute of Neurological Diseases and Stroke (R01 NS043264) andthe National Center for Research Resources (M01-RR00064, to theUniversity of Utah, Dr L. Betz, P.I.).
DisclosuresNone.
References1. Bushby K, Muntoni F, Bourke JP. 107th ENMC international
workshop: the management of cardiac involvement in muscular dys-trophy and myotonic dystrophy. 7th-9th June 2002, Naarden, theNetherlands. Neuromuscul Disord. 2003;13:166 –172.
2. Nigro G, Politano L, Nigro V, Petretta VR, Comi LI. Mutation ofdystrophin gene and cardiomyopathy. Neuromuscul Disord. 1994;4:371–379.
3. Melacini P, Fanin M, Danieli GA, Villanova C, Martinello F, Miorin M,Freda MP, Miorelli M, Mostacciuolo ML, Fasoli G, Angelini C, DallaVolta S. Myocardial involvement is very frequent among patients affectedwith subclinical Becker’s muscular dystrophy. Circulation. 1996;94:3168–3175.
4. Melacini P, Fanin M, Danieli GA, Fasoli G, Villanova C, Angelini C,Vitiello L, Miorelli M, Buja GF, Mostacciuolo ML, Pegoraro E, Della
Volta S. Cardiac involvement in Becker muscular dystrophy. J Am CollCardiol. 1993;22:1927–1934.
5. Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD,Neish SR, Smith EOB, Towbin JA. Genetic predictors and remodeling ofdilated cardiomyopathy in muscular dystrophy. Circulation. 2005;112:2799–2804.
6. Carsana A, Frisso G, Tremolaterra MR, Lanzillo R, Vitale DF, Santoro L,Salvatore F. Analysis of dystrophin gene deletions indicates that the hingeIII region of the protein correlates with disease severity. Ann Hum Genet.2005;69:253–259.
7. Beggs AH, Hoffman EP, Snyder JR, Arahata K, Specht L, Shapiro F,Angelini C, Sugita H, Kunkel LM. Exploring the molecular basis forvariability among patients with Becker muscular dystrophy: dystrophingene and protein studies. Am J Hum Genet. 1991;49:54–67.
8. Comi GP, Prelle A, Bresolin N, Moggio M, Bardoni A, Gallanti A, VitaG, Toscano A, Ferro MT, Bordoni A, Fortunato F, Ciscato P, Felisari G,Tedeschi S, Castelli E, Garghentino R, Turconi A, Fraschini P, Marchi E,Negretto GG, Adobbati L, Meola G, Tonin P, Papadimitriou D, ScarlatoG. Clinical variability in Becker muscular dystrophy. Genetic, bio-chemical and immunohistochemical correlates. Brain. 1994;117(pt1):1–14.
9. Yazaki M, Yoshida K, Nakamura A, Koyama J, Nanba T, Ohori N, IkedaS. Clinical characteristics of aged Becker muscular dystrophy patientswith onset after 30 years. Eur Neurol. 1999;42:145–149.
10. Nakamura A, Yoshida K, Fukushima K, Ueda H, Urasawa N, Koyama J,Yazaki Y, Yazaki M, Sakai T, Haruta S, Takeda S, Ikeda S. Follow-up ofthree patients with a large in-frame deletion of exons 45–55 in theDuchenne muscular dystrophy (DMD) gene. J Clin Neurosci. 2008;15:757–763.
11. Brooke MH, Fenichel GM, Griggs RC, Mendell JR, Moxley R, Miller JP,Province MA. Clinical investigation in Duchenne dystrophy. II. Deter-mination of the “power” of therapeutic trials based on the natural history.Muscle Nerve. 1983;6:91–103.
12. Duboc D, Meune C, Lerebours G, Devaux JY, Vaksmann G, Becane HM.Effect of perindopril on the onset and progression of left ventriculardysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol. 2005;45:855–857.
13. Thrush PT, Allen HD, Viollet L, Mendell JR. Re-examination of the elec-trocardiogram in boys with Duchenne muscular dystrophy and correlationwith its dilated cardiomyopathy. Am J Cardiol. 2009;103:262–265.
14. Kusunoki H, Minasov G, Macdonald RI, Mondragon A. Independentmovement, dimerization and stability of tandem repeats of chicken brainalpha-spectrin. J Mol Biol. 2004;344:495–511.
15. Worton RG, Thompson MW. Genetics of Duchenne muscular dystrophy.Annu Rev Genet. 1988;22:601–629.
16. Worton RG, Molnar MJ, Brais B, Karpati G. The muscular dystrophies.In: Scriver CR, Beaudet A, Sly WS, Valle D, eds. The Metabolic andMolecular Bases of Inherited Disease: New York: McGraw Hill; 2001:5493–5523.
17. Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, PhelpsSF, Harper HA, Robinson AS, Engelhardt JF, Brooks SV, ChamberlainJS. Modular flexibility of dystrophin: implications for gene therapy ofDuchenne muscular dystrophy. Nat Med. 2002;8:253–261.
18. Cross RA, Stewart M, Kendrick-Jones J. Structural predictions for thecentral domain of dystrophin. FEBS Lett. 1990;262:87–92.
19. Maeda M, Nakao S, Miyazato H, Setoguchi M, Arima S, Higuchi I,Osame M, Taira A, Nomoto K, Toda H, Tahara M, Atsuchi Y, Tanaka H.Cardiac dystrophin abnormalities in Becker muscular dystrophy assessedby endomyocardial biopsy. Am Heart J. 1995;129:702–707.
20. Arbustini E, Diegoli M, Morbini P, Dal Bello B, Banchieri N, Pilotto A,Magani F, Grasso M, Narula J, Gavazzi A, Vigano M, Tavazzi L.Prevalence and characteristics of dystrophin defects in adult male patientswith dilated cardiomyopathy. J Am Coll Cardiol. 2000;35:1760–1768.
21. Politano L, Passamano L, Petretta VR, Nigro V, Papparella S, Nigro G,Santangelo L, Esposito MG, Come LI, Nigro G. Familial dilated cardio-myopathy associated with the typical dystrophin BMD mutation: reporton two additional cases. Acta Myol. 1999;3:3329–3336.
22. Muntoni F, Di Lenarda A, Porcu M, Sinagra G, Mateddu A, Marrosu G,Ferlini A, Cau M, Milasin J, Melis MA, Marrosu MG, Cianchetti C,Sanna A, Falaschi A, Camerini F, Giacca M, Mestroni L. Dystrophin geneabnormalities in two patients with idiopathic dilated cardiomyopathy.Heart. 1997;78:608–612.
23. Fanin M, Melacini P, Angelini C, Danieli GA. Could utrophin rescue themyocardium of patients with dystrophin gene mutations? J Mol CellCardiol. 1999;31:1501–1508.
550 Circ Cardiovasc Genet December 2009
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from

24. Politano L, Colonna-Romano S, Esposito MG, Nigro V, Comi LI, Pas-samano L, Nigro G. Genotype-phenotype correlation in patients withdeletions of Duchenne/Becker gene. Acta Cardiomyologica. 1991;3:239–244.
25. Bostick B, Yue Y, Long C, Marschalk N, Fine DM, Chen J, Duan D.Cardiac Expression of a mini-dystrophin that normalizes skeletal muscleforce only partially restores heart function in aged Mdx mice. Mol Ther.2009;17:253–261.
26. Milasin J, Muntoni F, Severini GM, Bartoloni L, Vatta M, Krajinovic M,Mateddu A, Angelini C, Camerini F, Falaschi A, Mestroni L, Giacca M. A
point mutation in the 5� splice site of the dystrophin gene first intron responsiblefor X-linked dilated cardiomyopathy. Hum Mol Genet. 1996;5:73–79.
27. Muntoni F, Cau M, Ganau A, Congiu R, Arvedi G, Mateddu A, MarrosuMG, Cianchetti C, Realdi G, Cao A, Melis MA. Brief report: deletion ofthe dystrophin muscle-promoter region associated with X-linked dilatedcardiomyopathy. N Engl J Med. 1993;329:921–925.
28. Muntoni F, Wilson L, Marrosu G, Marrosu MG, Cianchetti C, MestroniL, Ganau A, Dubowitz V, Sewry C. A mutation in the dystrophin geneselectively affecting dystrophin expression in the heart. J Clin Invest.1995;96:693–699.
CLINICAL PERSPECTIVEExon deletions of the dystrophin gene lead to Becker muscular dystrophy and X-linked dilated cardiomyopathy. Bothconditions are associated with cardiomyopathy with variable onset between the second and sixth decade of life. Betterunderstanding of the predictive pathogenic factors influencing time of onset and severity of cardiac involvement wouldenable clinicians to begin early intervention, and potentially prevent premature death. In this study, insight into theevolution of cardiomyopathy was gained from analyzing a large patient population with the most prevalent exon deletionsaffecting discrete dystrophin protein domains. Four patient groups emerged from our study. Their expected ages ofcardiomyopathy onset seem to be associated with the location of the exon deletion mutation and the effects on dystrophinprotein structure. The complexity of our findings illustrates that dystrophin exon deletions must be correlated with proteinstructural alterations to predict outcomes. Prospective testing of these relationships potentially will empower clinicians touse genotype information to intervene more effectively in the treatment of patients with Becker muscular dystrophy orX-linked dilated cardiomyopathy. In addition, the findings pave the way for improvements on current therapeuticapproaches targeting the heart in dystrophinopathies and may be valuable for patient stratification in clinical trials.
Kaspar et al Dystrophin Mutations Predict Cardiomyopathy Onset 551
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from

Pestronk, Robert B. Weiss, Kevin M. Flanigan, Jerry R. Mendell and Federica MontanaroRita Wen Kaspar, Hugh D. Allen, Will C. Ray, Carlos E. Alvarez, John T. Kissel, Alan
Becker Muscular DystrophyAnalysis of Dystrophin Deletion Mutations Predicts Age of Cardiomyopathy Onset in
Print ISSN: 1942-325X. Online ISSN: 1942-3268 Copyright © 2009 American Heart Association, Inc. All rights reserved.
Dallas, TX 75231is published by the American Heart Association, 7272 Greenville Avenue,Circulation: Cardiovascular Genetics
doi: 10.1161/CIRCGENETICS.109.8672422009;2:544-551; originally published online September 30, 2009;Circ Cardiovasc Genet.
http://circgenetics.ahajournals.org/content/2/6/544World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circgenetics.ahajournals.org/content/suppl/2009/09/30/CIRCGENETICS.109.867242.DC1Data Supplement (unedited) at:
http://circgenetics.ahajournals.org//subscriptions/
is online at: Circulation: Cardiovascular Genetics Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer information about this process is available in the
requested is located, click Request Permissions in the middle column of the Web page under Services. FurtherCenter, not the Editorial Office. Once the online version of the published article for which permission is being
can be obtained via RightsLink, a service of the Copyright ClearanceCirculation: Cardiovascular Geneticsin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on May 27, 2018
http://circgenetics.ahajournals.org/D
ownloaded from
Wen Kaspar et al.
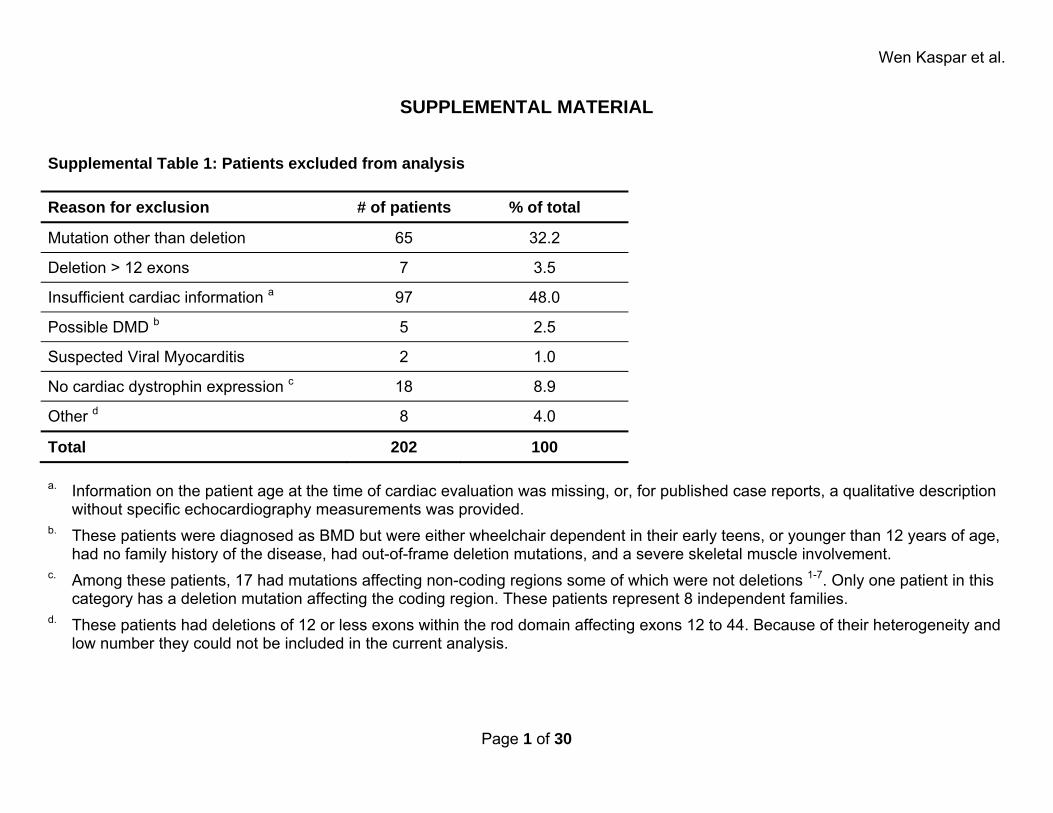
SUPPLEMENTAL MATERIAL Supplemental Table 1: Patients excluded from analysis Reason for exclusion # of patients % of total
Mutation other than deletion 65 32.2
Deletion > 12 exons 7 3.5
Insufficient cardiac information a 97 48.0
Possible DMD b 5 2.5
Suspected Viral Myocarditis 2 1.0
No cardiac dystrophin expression c 18 8.9
Other d 8 4.0
Total 202 100 a. Information on the patient age at the time of cardiac evaluation was missing, or, for published case reports, a qualitative description
without specific echocardiography measurements was provided. b. These patients were diagnosed as BMD but were either wheelchair dependent in their early teens, or younger than 12 years of age,
had no family history of the disease, had out-of-frame deletion mutations, and a severe skeletal muscle involvement. c. Among these patients, 17 had mutations affecting non-coding regions some of which were not deletions 1-7. Only one patient in this
category has a deletion mutation affecting the coding region. These patients represent 8 independent families. d. These patients had deletions of 12 or less exons within the rod domain affecting exons 12 to 44. Because of their heterogeneity and
low number they could not be included in the current analysis.
Page 1 of 30

Wen Kaspar et al.
References: 1. Ferlini A, Galie N, Merlini L, Sewry C, Branzi A, Muntoni F. A novel Alu-like element rearranged in the dystrophin gene causes a
splicing mutation in a family with X-linked dilated cardiomyopathy. Am J Hum Genet. 1998;63(2):436-446. 2. Bies RD, Maeda M, Roberds SL, Holder E, Bohlmeyer T, Young JB, Campbell KP. A 5' dystrophin duplication mutation causes
membrane deficiency of alpha-dystroglycan in a family with X-linked cardiomyopathy. J Mol Cell Cardiol. 1997;29(12):3175-3188.
3. Saotome M, Yoshitomi Y, Kojima S, Kuramochi M. Dilated cardiomyopathy of Becker-type muscular dystrophy with exon 4 deletion--a case report. Angiology. 2001;52(5):343-347.
4. Yoshida K, Ikeda S, Nakamura A, Kagoshima M, Takeda S, Shoji S, Yanagisawa N. Molecular analysis of the Duchenne muscular dystrophy gene in patients with Becker muscular dystrophy presenting with dilated cardiomyopathy. Muscle Nerve. 1993;16(11):1161-1166.
5. Muntoni F, Wilson L, Marrosu G, Marrosu MG, Cianchetti C, Mestroni L, Ganau A, Dubowitz V, Sewry C. A mutation in the dystrophin gene selectively affecting dystrophin expression in the heart. J Clin Invest. 1995;96(2):693-699.
6. Muntoni F, Cau M, Ganau A, Congiu R, Arvedi G, Mateddu A, Marrosu MG, Cianchetti C, Realdi G, Cao A, Melis MA. Brief report: deletion of the dystrophin muscle-promoter region associated with X-linked dilated cardiomyopathy. N Engl J Med. 1993;329(13):921-925.
7. Milasin J, Muntoni F, Severini GM, Bartoloni L, Vatta M, Krajinovic M, Mateddu A, Angelini C, Camerini F, Falaschi A, Mestroni L, Giacca M. A point mutation in the 5' splice site of the dystrophin gene first intron responsible for X-linked dilated cardiomyopathy. Hum Mol Genet. 1996;5(1):73-79.
Page 2 of 30
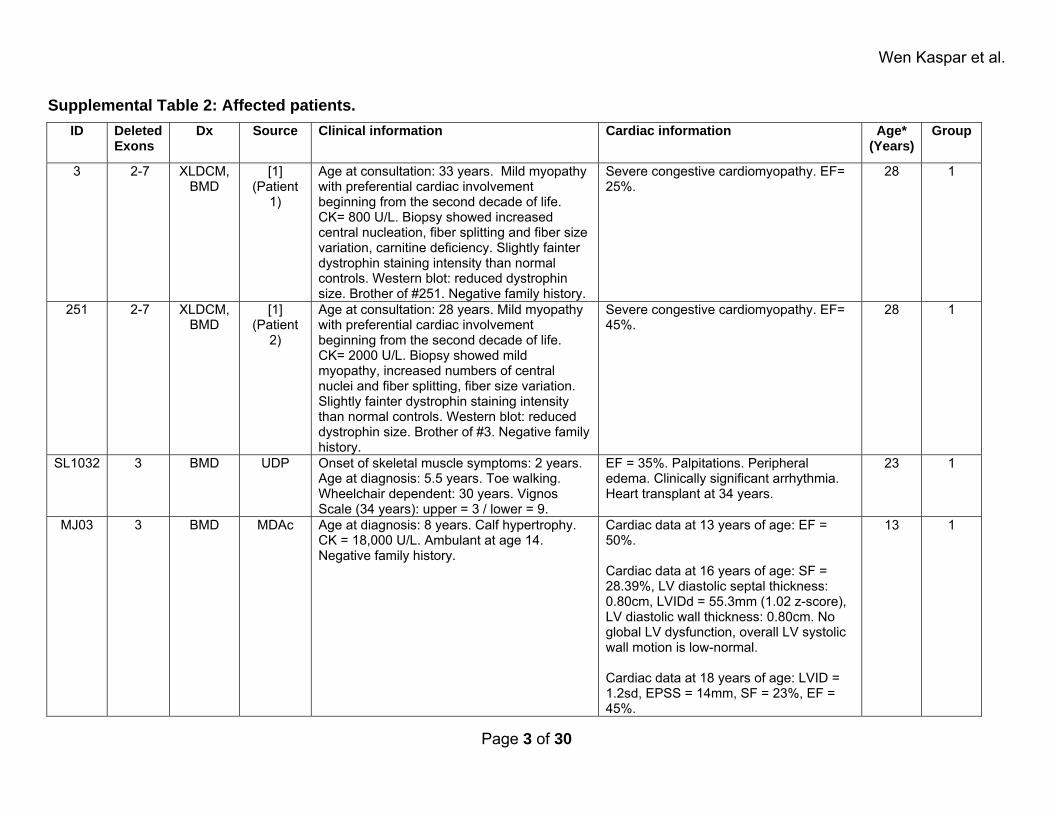
Wen Kaspar et al.
Supplemental Table 2: Affected patients. ID Deleted
Exons Dx Source Clinical information Cardiac information Age*
(Years)Group
3 2-7 XLDCM, BMD
[1] (Patient
1)
Age at consultation: 33 years. Mild myopathy with preferential cardiac involvement beginning from the second decade of life. CK= 800 U/L. Biopsy showed increased central nucleation, fiber splitting and fiber size variation, carnitine deficiency. Slightly fainter dystrophin staining intensity than normal controls. Western blot: reduced dystrophin size. Brother of #251. Negative family history.
Severe congestive cardiomyopathy. EF= 25%.
28 1
251 2-7 XLDCM, BMD
[1] (Patient
2)
Age at consultation: 28 years. Mild myopathy with preferential cardiac involvement beginning from the second decade of life. CK= 2000 U/L. Biopsy showed mild myopathy, increased numbers of central nuclei and fiber splitting, fiber size variation. Slightly fainter dystrophin staining intensity than normal controls. Western blot: reduced dystrophin size. Brother of #3. Negative family history.
Severe congestive cardiomyopathy. EF= 45%.
28 1
SL1032 3 BMD UDP Onset of skeletal muscle symptoms: 2 years. Age at diagnosis: 5.5 years. Toe walking. Wheelchair dependent: 30 years. Vignos Scale (34 years): upper = 3 / lower = 9.
EF = 35%. Palpitations. Peripheral edema. Clinically significant arrhythmia. Heart transplant at 34 years.
23 1
MJ03 3 BMD MDAc Age at diagnosis: 8 years. Calf hypertrophy. CK = 18,000 U/L. Ambulant at age 14. Negative family history.
Cardiac data at 13 years of age: EF = 50%. Cardiac data at 16 years of age: SF = 28.39%, LV diastolic septal thickness: 0.80cm, LVIDd = 55.3mm (1.02 z-score), LV diastolic wall thickness: 0.80cm. No global LV dysfunction, overall LV systolic wall motion is low-normal. Cardiac data at 18 years of age: LVID = 1.2sd, EPSS = 14mm, SF = 23%, EF = 45%.
13 1
Page 3 of 30
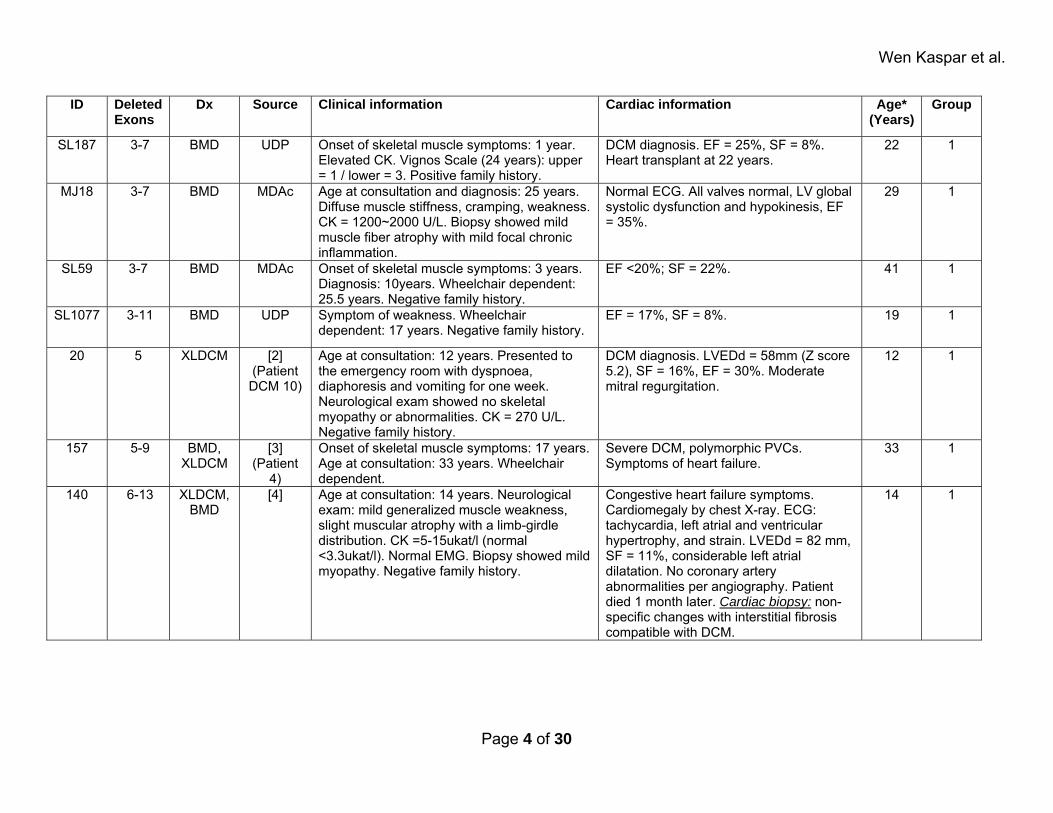
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
SL187 3-7 BMD UDP Onset of skeletal muscle symptoms: 1 year. Elevated CK. Vignos Scale (24 years): upper = 1 / lower = 3. Positive family history.
DCM diagnosis. EF = 25%, SF = 8%. Heart transplant at 22 years.
22 1
MJ18 3-7 BMD MDAc Age at consultation and diagnosis: 25 years. Diffuse muscle stiffness, cramping, weakness. CK = 1200~2000 U/L. Biopsy showed mild muscle fiber atrophy with mild focal chronic inflammation.
Normal ECG. All valves normal, LV global systolic dysfunction and hypokinesis, EF = 35%.
29 1
SL59 3-7 BMD MDAc Onset of skeletal muscle symptoms: 3 years. Diagnosis: 10years. Wheelchair dependent: 25.5 years. Negative family history.
EF <20%; SF = 22%. 41 1
SL1077 3-11 BMD UDP Symptom of weakness. Wheelchair dependent: 17 years. Negative family history.
EF = 17%, SF = 8%. 19 1
20 5 XLDCM [2] (Patient DCM 10)
Age at consultation: 12 years. Presented to the emergency room with dyspnoea, diaphoresis and vomiting for one week. Neurological exam showed no skeletal myopathy or abnormalities. CK = 270 U/L. Negative family history.
DCM diagnosis. LVEDd = 58mm (Z score 5.2), SF = 16%, EF = 30%. Moderate mitral regurgitation.
12 1
157 5-9 BMD, XLDCM
[3] (Patient
4)
Onset of skeletal muscle symptoms: 17 years. Age at consultation: 33 years. Wheelchair dependent.
Severe DCM, polymorphic PVCs. Symptoms of heart failure.
33 1
140 6-13 XLDCM, BMD
[4] Age at consultation: 14 years. Neurological exam: mild generalized muscle weakness, slight muscular atrophy with a limb-girdle distribution. CK =5-15ukat/l (normal <3.3ukat/l). Normal EMG. Biopsy showed mild myopathy. Negative family history.
Congestive heart failure symptoms. Cardiomegaly by chest X-ray. ECG: tachycardia, left atrial and ventricular hypertrophy, and strain. LVEDd = 82 mm, SF = 11%, considerable left atrial dilatation. No coronary artery abnormalities per angiography. Patient died 1 month later. Cardiac biopsy: non-specific changes with interstitial fibrosis compatible with DCM.
14 1
Page 4 of 30
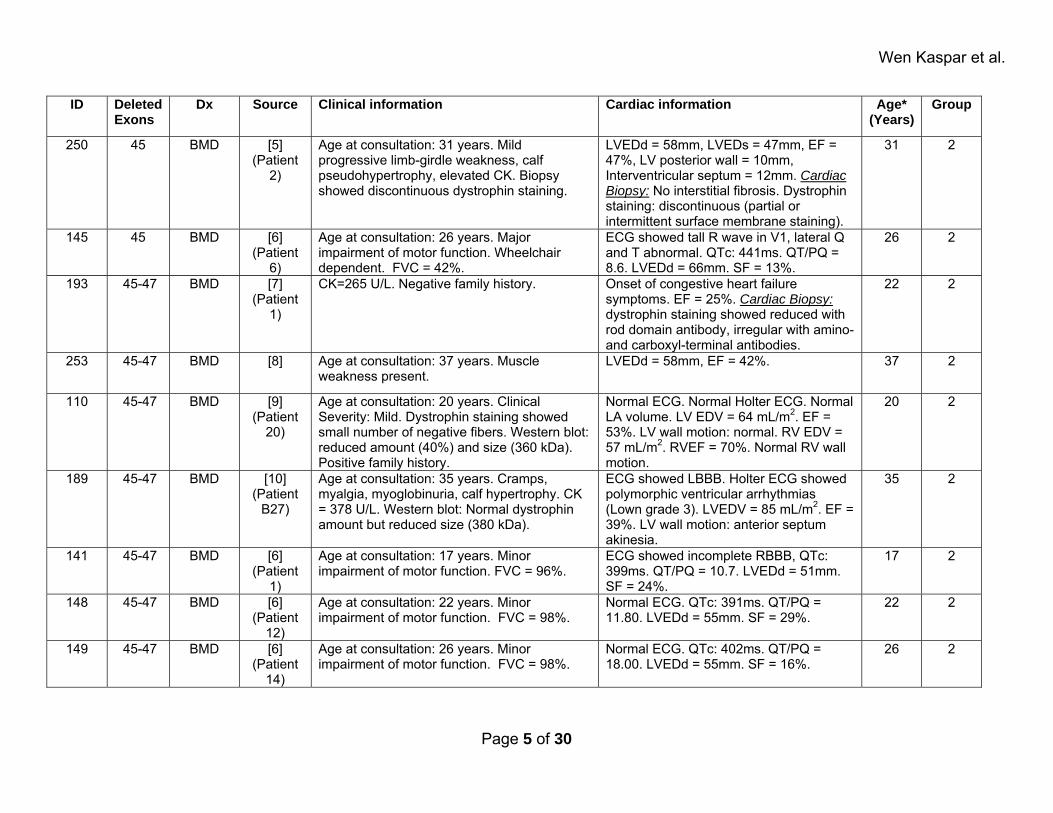
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
250 45 BMD [5] (Patient
2)
Age at consultation: 31 years. Mild progressive limb-girdle weakness, calf pseudohypertrophy, elevated CK. Biopsy showed discontinuous dystrophin staining.
LVEDd = 58mm, LVEDs = 47mm, EF = 47%, LV posterior wall = 10mm, Interventricular septum = 12mm. Cardiac Biopsy: No interstitial fibrosis. Dystrophin staining: discontinuous (partial or intermittent surface membrane staining).
31 2
145 45 BMD [6] (Patient
6)
Age at consultation: 26 years. Major impairment of motor function. Wheelchair dependent. FVC = 42%.
ECG showed tall R wave in V1, lateral Q and T abnormal. QTc: 441ms. QT/PQ = 8.6. LVEDd = 66mm. SF = 13%.
26 2
193 45-47 BMD [7] (Patient
1)
CK=265 U/L. Negative family history. Onset of congestive heart failure symptoms. EF = 25%. Cardiac Biopsy: dystrophin staining showed reduced with rod domain antibody, irregular with amino- and carboxyl-terminal antibodies.
22 2
253 45-47 BMD [8] Age at consultation: 37 years. Muscle weakness present.
LVEDd = 58mm, EF = 42%. 37 2
110 45-47 BMD [9] (Patient
20)
Age at consultation: 20 years. Clinical Severity: Mild. Dystrophin staining showed small number of negative fibers. Western blot: reduced amount (40%) and size (360 kDa). Positive family history.
Normal ECG. Normal Holter ECG. Normal LA volume. LV EDV = 64 mL/m2. EF = 53%. LV wall motion: normal. RV EDV = 57 mL/m2. RVEF = 70%. Normal RV wall motion.
20 2
189 45-47 BMD [10] (Patient
B27)
Age at consultation: 35 years. Cramps, myalgia, myoglobinuria, calf hypertrophy. CK = 378 U/L. Western blot: Normal dystrophin amount but reduced size (380 kDa).
ECG showed LBBB. Holter ECG showed polymorphic ventricular arrhythmias (Lown grade 3). LVEDV = 85 mL/m2. EF = 39%. LV wall motion: anterior septum akinesia.
35 2
141 45-47 BMD [6] (Patient
1)
Age at consultation: 17 years. Minor impairment of motor function. FVC = 96%.
ECG showed incomplete RBBB, QTc: 399ms. QT/PQ = 10.7. LVEDd = 51mm. SF = 24%.
17 2
148 45-47 BMD [6] (Patient
12)
Age at consultation: 22 years. Minor impairment of motor function. FVC = 98%.
Normal ECG. QTc: 391ms. QT/PQ = 11.80. LVEDd = 55mm. SF = 29%.
22 2
149 45-47 BMD [6] (Patient
14)
Age at consultation: 26 years. Minor impairment of motor function. FVC = 98%.
Normal ECG. QTc: 402ms. QT/PQ = 18.00. LVEDd = 55mm. SF = 16%.
26 2
Page 5 of 30
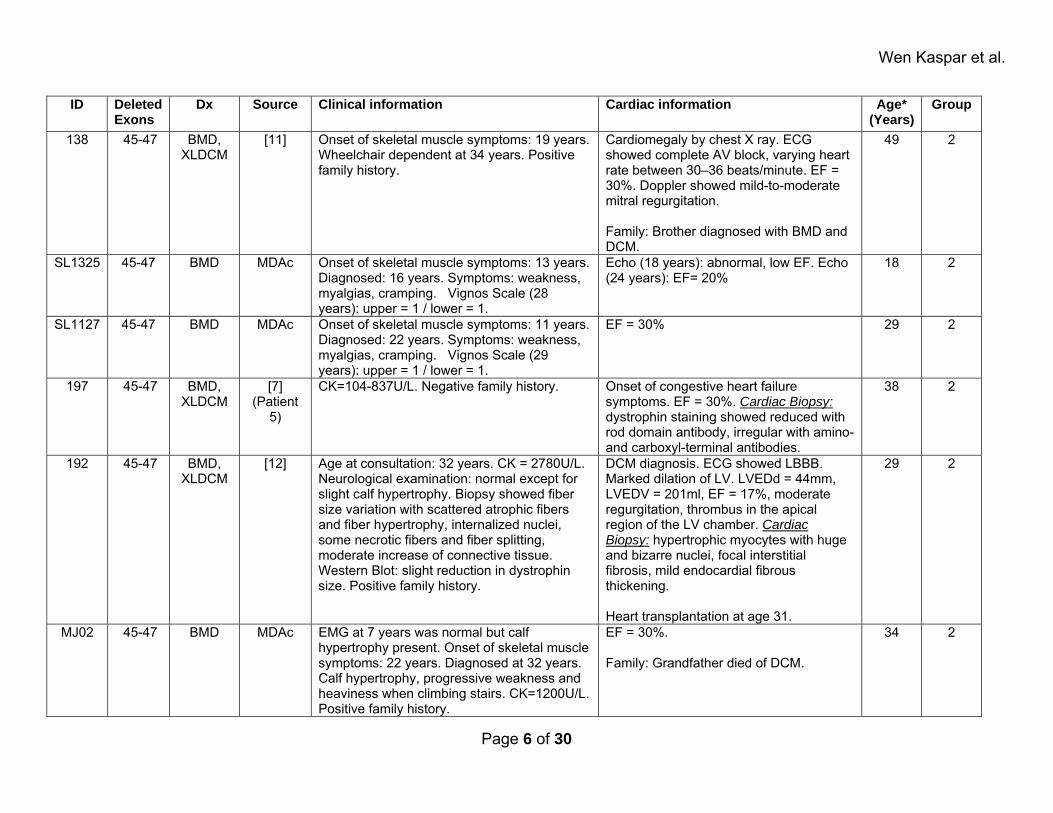
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
138 45-47 BMD, XLDCM
[11] Onset of skeletal muscle symptoms: 19 years. Wheelchair dependent at 34 years. Positive family history.
Cardiomegaly by chest X ray. ECG showed complete AV block, varying heart rate between 30–36 beats/minute. EF = 30%. Doppler showed mild-to-moderate mitral regurgitation. Family: Brother diagnosed with BMD and DCM.
49 2
SL1325 45-47 BMD MDAc Onset of skeletal muscle symptoms: 13 years. Diagnosed: 16 years. Symptoms: weakness, myalgias, cramping. Vignos Scale (28 years): upper = 1 / lower = 1.
Echo (18 years): abnormal, low EF. Echo (24 years): EF= 20%
18 2
SL1127 45-47 BMD MDAc Onset of skeletal muscle symptoms: 11 years. Diagnosed: 22 years. Symptoms: weakness, myalgias, cramping. Vignos Scale (29 years): upper = 1 / lower = 1.
EF = 30% 29 2
197 45-47 BMD, XLDCM
[7] (Patient
5)
CK=104-837U/L. Negative family history. Onset of congestive heart failure symptoms. EF = 30%. Cardiac Biopsy: dystrophin staining showed reduced with rod domain antibody, irregular with amino- and carboxyl-terminal antibodies.
38 2
192 45-47 BMD, XLDCM
[12] Age at consultation: 32 years. CK = 2780U/L. Neurological examination: normal except for slight calf hypertrophy. Biopsy showed fiber size variation with scattered atrophic fibers and fiber hypertrophy, internalized nuclei, some necrotic fibers and fiber splitting, moderate increase of connective tissue. Western Blot: slight reduction in dystrophin size. Positive family history.
DCM diagnosis. ECG showed LBBB. Marked dilation of LV. LVEDd = 44mm, LVEDV = 201ml, EF = 17%, moderate regurgitation, thrombus in the apical region of the LV chamber. Cardiac Biopsy: hypertrophic myocytes with huge and bizarre nuclei, focal interstitial fibrosis, mild endocardial fibrous thickening. Heart transplantation at age 31.
29 2
MJ02 45-47 BMD MDAc EMG at 7 years was normal but calf hypertrophy present. Onset of skeletal muscle symptoms: 22 years. Diagnosed at 32 years. Calf hypertrophy, progressive weakness and heaviness when climbing stairs. CK=1200U/L. Positive family history.
EF = 30%. Family: Grandfather died of DCM.
34 2
Page 6 of 30
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
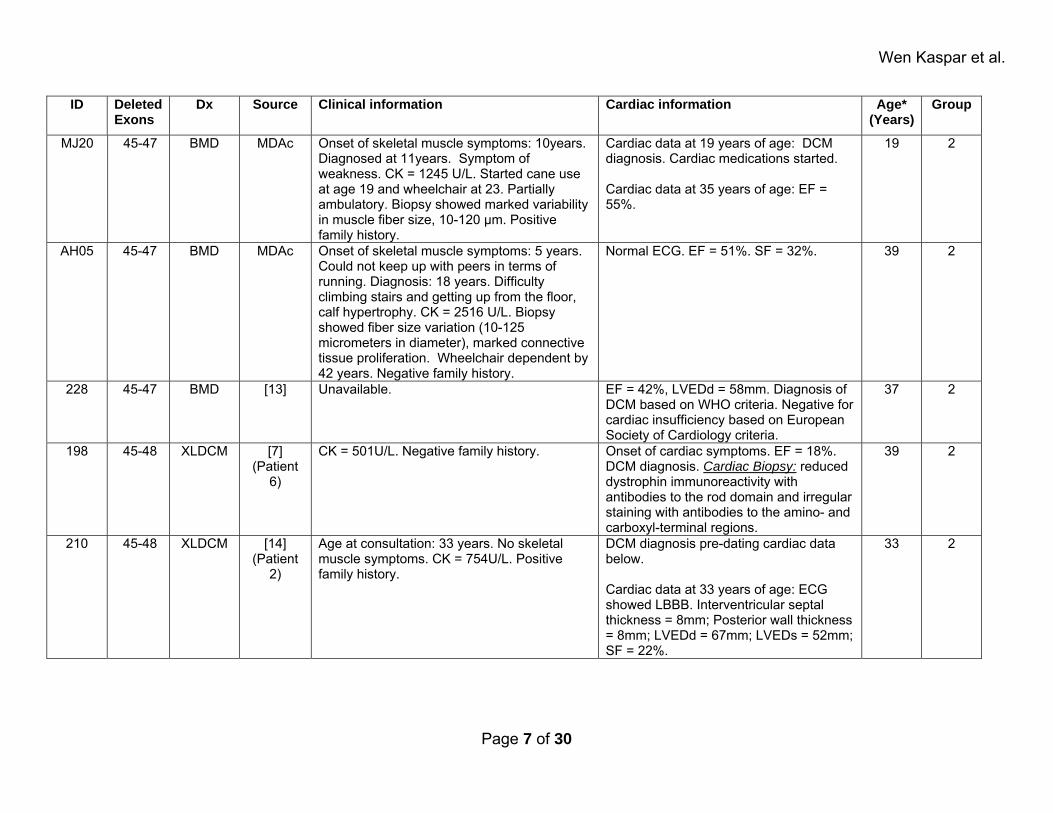
MJ20 45-47 BMD MDAc Onset of skeletal muscle symptoms: 10years. Diagnosed at 11years. Symptom of weakness. CK = 1245 U/L. Started cane use at age 19 and wheelchair at 23. Partially ambulatory. Biopsy showed marked variability in muscle fiber size, 10-120 μm. Positive family history.
Cardiac data at 19 years of age: DCM diagnosis. Cardiac medications started. Cardiac data at 35 years of age: EF = 55%.
19 2
AH05 45-47 BMD MDAc Onset of skeletal muscle symptoms: 5 years. Could not keep up with peers in terms of running. Diagnosis: 18 years. Difficulty climbing stairs and getting up from the floor, calf hypertrophy. CK = 2516 U/L. Biopsy showed fiber size variation (10-125 micrometers in diameter), marked connective tissue proliferation. Wheelchair dependent by 42 years. Negative family history.
Normal ECG. EF = 51%. SF = 32%. 39 2
228 45-47 BMD [13] Unavailable. EF = 42%, LVEDd = 58mm. Diagnosis of DCM based on WHO criteria. Negative for cardiac insufficiency based on European Society of Cardiology criteria.
37 2
198 45-48 XLDCM [7] (Patient
6)
CK = 501U/L. Negative family history. Onset of cardiac symptoms. EF = 18%. DCM diagnosis. Cardiac Biopsy: reduced dystrophin immunoreactivity with antibodies to the rod domain and irregular staining with antibodies to the amino- and carboxyl-terminal regions.
39 2
210 45-48 XLDCM [14] (Patient
2)
Age at consultation: 33 years. No skeletal muscle symptoms. CK = 754U/L. Positive family history.
DCM diagnosis pre-dating cardiac data below. Cardiac data at 33 years of age: ECG showed LBBB. Interventricular septal thickness = 8mm; Posterior wall thickness = 8mm; LVEDd = 67mm; LVEDs = 52mm; SF = 22%.
33 2
Page 7 of 30
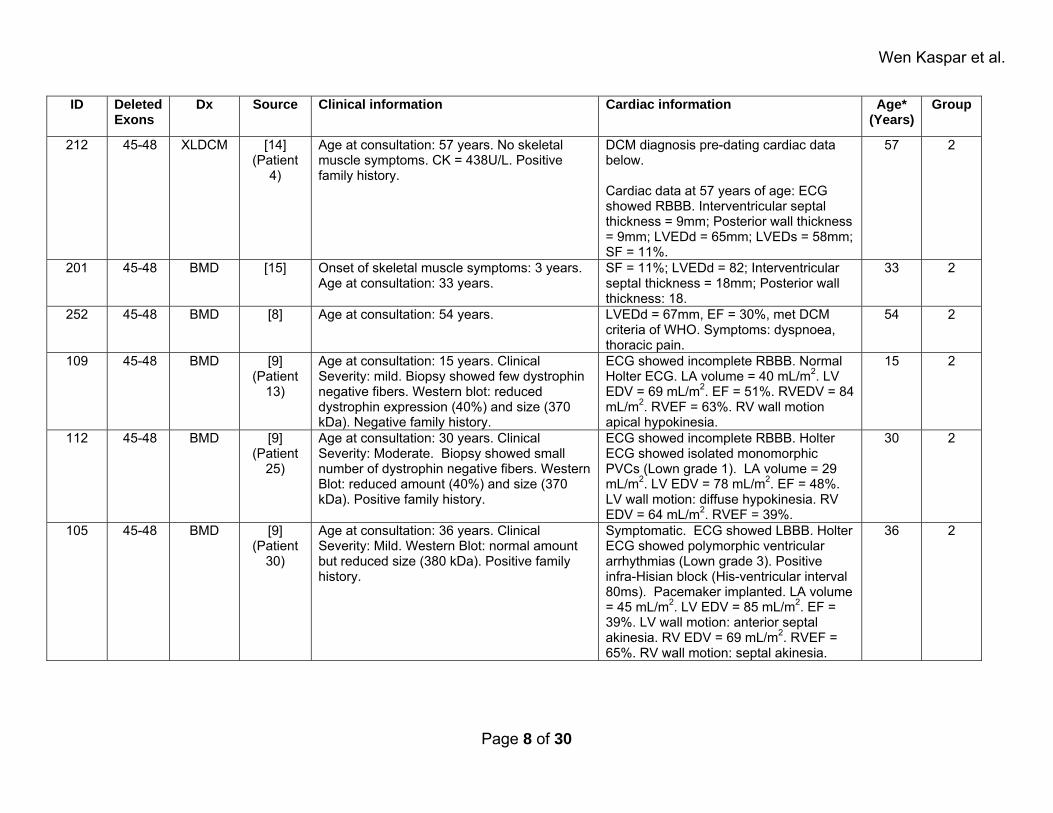
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
212 45-48 XLDCM [14] (Patient
4)
Age at consultation: 57 years. No skeletal muscle symptoms. CK = 438U/L. Positive family history.
DCM diagnosis pre-dating cardiac data below. Cardiac data at 57 years of age: ECG showed RBBB. Interventricular septal thickness = 9mm; Posterior wall thickness = 9mm; LVEDd = 65mm; LVEDs = 58mm; SF = 11%.
57 2
201 45-48 BMD [15] Onset of skeletal muscle symptoms: 3 years. Age at consultation: 33 years.
SF = 11%; LVEDd = 82; Interventricular septal thickness = 18mm; Posterior wall thickness: 18.
33 2
252 45-48 BMD [8] Age at consultation: 54 years. LVEDd = 67mm, EF = 30%, met DCM criteria of WHO. Symptoms: dyspnoea, thoracic pain.
54 2
109 45-48 BMD [9] (Patient
13)
Age at consultation: 15 years. Clinical Severity: mild. Biopsy showed few dystrophin negative fibers. Western blot: reduced dystrophin expression (40%) and size (370 kDa). Negative family history.
ECG showed incomplete RBBB. Normal Holter ECG. LA volume = 40 mL/m2. LV EDV = 69 mL/m2. EF = 51%. RVEDV = 84 mL/m2. RVEF = 63%. RV wall motion apical hypokinesia.
15 2
112 45-48 BMD [9] (Patient
25)
Age at consultation: 30 years. Clinical Severity: Moderate. Biopsy showed small number of dystrophin negative fibers. Western Blot: reduced amount (40%) and size (370 kDa). Positive family history.
ECG showed incomplete RBBB. Holter ECG showed isolated monomorphic PVCs (Lown grade 1). LA volume = 29 mL/m2. LV EDV = 78 mL/m2. EF = 48%. LV wall motion: diffuse hypokinesia. RV EDV = 64 mL/m2. RVEF = 39%.
30 2
105 45-48 BMD [9] (Patient
30)
Age at consultation: 36 years. Clinical Severity: Mild. Western Blot: normal amount but reduced size (380 kDa). Positive family history.
Symptomatic. ECG showed LBBB. Holter ECG showed polymorphic ventricular arrhythmias (Lown grade 3). Positive infra-Hisian block (His-ventricular interval 80ms). Pacemaker implanted. LA volume = 45 mL/m2. LV EDV = 85 mL/m2. EF = 39%. LV wall motion: anterior septal akinesia. RV EDV = 69 mL/m2. RVEF = 65%. RV wall motion: septal akinesia.
36 2
Page 8 of 30
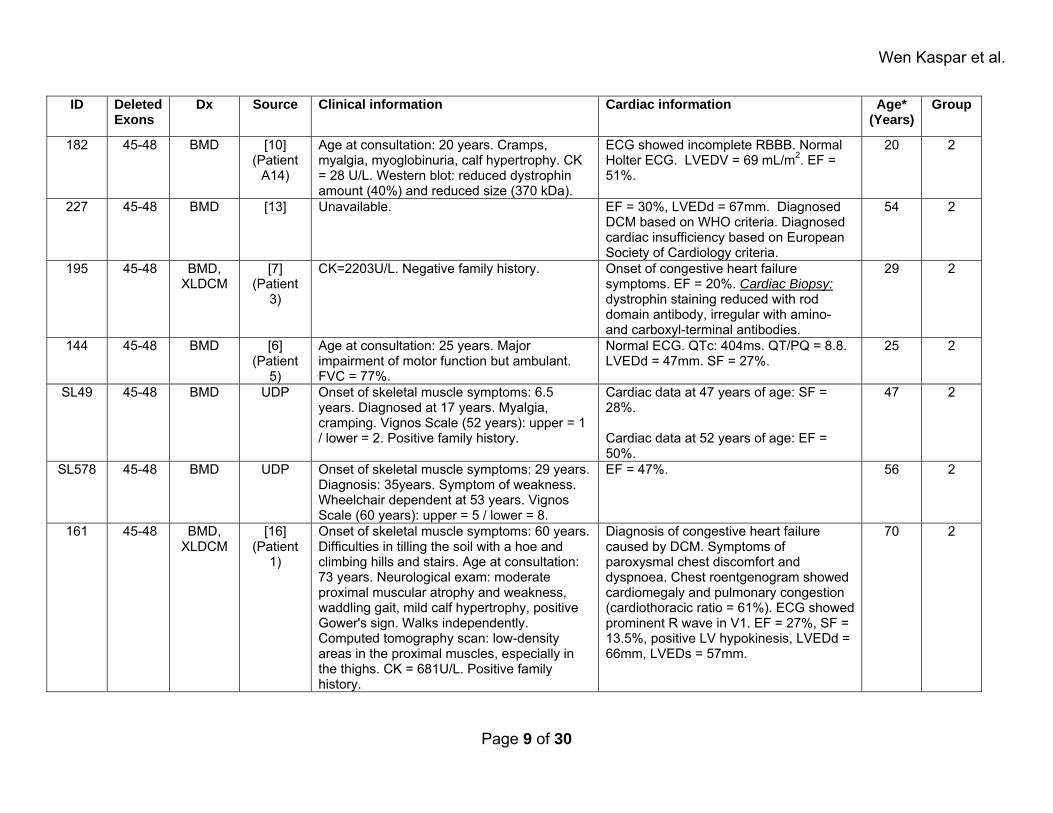
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
182 45-48 BMD [10] (Patient
A14)
Age at consultation: 20 years. Cramps, myalgia, myoglobinuria, calf hypertrophy. CK = 28 U/L. Western blot: reduced dystrophin amount (40%) and reduced size (370 kDa).
ECG showed incomplete RBBB. Normal Holter ECG. LVEDV = 69 mL/m2. EF = 51%.
20 2
227 45-48 BMD [13] Unavailable. EF = 30%, LVEDd = 67mm. Diagnosed DCM based on WHO criteria. Diagnosed cardiac insufficiency based on European Society of Cardiology criteria.
54 2
195 45-48 BMD, XLDCM
[7] (Patient
3)
CK=2203U/L. Negative family history. Onset of congestive heart failure symptoms. EF = 20%. Cardiac Biopsy: dystrophin staining reduced with rod domain antibody, irregular with amino- and carboxyl-terminal antibodies.
29 2
144 45-48 BMD [6] (Patient
5)
Age at consultation: 25 years. Major impairment of motor function but ambulant. FVC = 77%.
Normal ECG. QTc: 404ms. QT/PQ = 8.8. LVEDd = 47mm. SF = 27%.
25 2
SL49 45-48 BMD UDP Onset of skeletal muscle symptoms: 6.5 years. Diagnosed at 17 years. Myalgia, cramping. Vignos Scale (52 years): upper = 1 / lower = 2. Positive family history.
Cardiac data at 47 years of age: SF = 28%. Cardiac data at 52 years of age: EF = 50%.
47 2
SL578 45-48 BMD UDP Onset of skeletal muscle symptoms: 29 years. Diagnosis: 35years. Symptom of weakness. Wheelchair dependent at 53 years. Vignos Scale (60 years): upper = 5 / lower = 8.
EF = 47%. 56 2
161 45-48 BMD, XLDCM
[16] (Patient
1)
Onset of skeletal muscle symptoms: 60 years. Difficulties in tilling the soil with a hoe and climbing hills and stairs. Age at consultation: 73 years. Neurological exam: moderate proximal muscular atrophy and weakness, waddling gait, mild calf hypertrophy, positive Gower's sign. Walks independently. Computed tomography scan: low-density areas in the proximal muscles, especially in the thighs. CK = 681U/L. Positive family history.
Diagnosis of congestive heart failure caused by DCM. Symptoms of paroxysmal chest discomfort and dyspnoea. Chest roentgenogram showed cardiomegaly and pulmonary congestion (cardiothoracic ratio = 61%). ECG showed prominent R wave in V1. EF = 27%, SF = 13.5%, positive LV hypokinesis, LVEDd = 66mm, LVEDs = 57mm.
70 2
Page 9 of 30
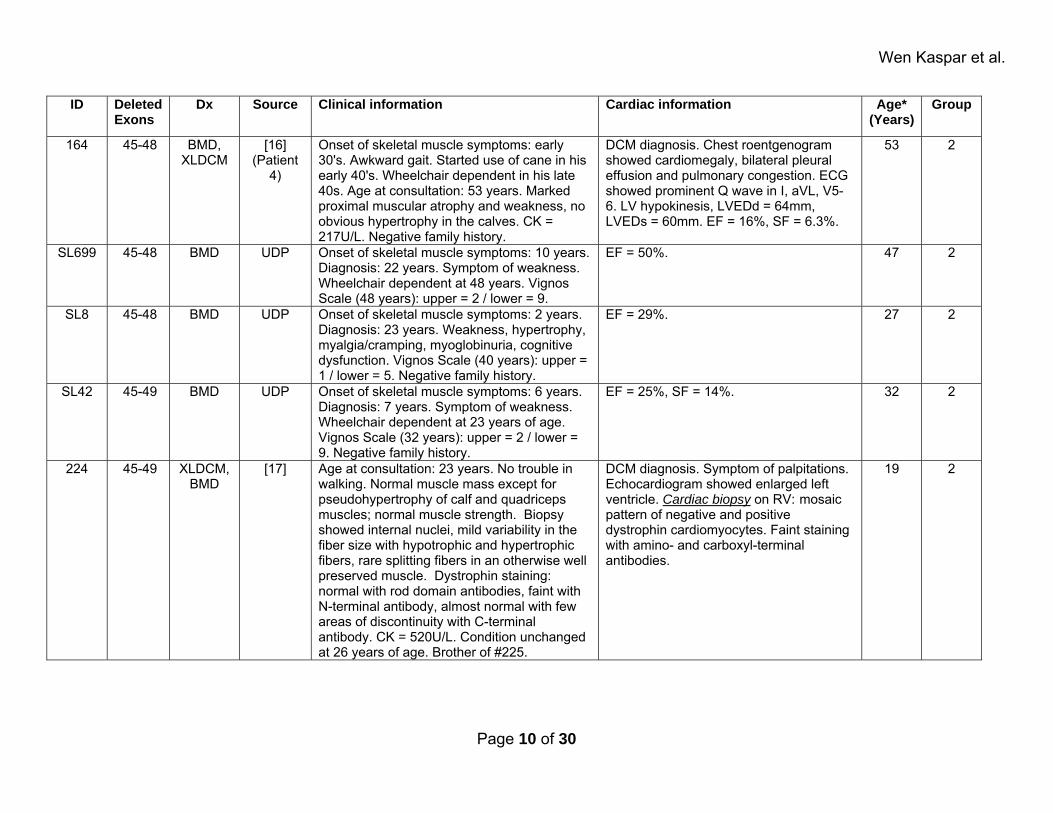
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
164 45-48 BMD, XLDCM
[16] (Patient
4)
Onset of skeletal muscle symptoms: early 30's. Awkward gait. Started use of cane in his early 40's. Wheelchair dependent in his late 40s. Age at consultation: 53 years. Marked proximal muscular atrophy and weakness, no obvious hypertrophy in the calves. CK = 217U/L. Negative family history.
DCM diagnosis. Chest roentgenogram showed cardiomegaly, bilateral pleural effusion and pulmonary congestion. ECG showed prominent Q wave in I, aVL, V5-6. LV hypokinesis, LVEDd = 64mm, LVEDs = 60mm. EF = 16%, SF = 6.3%.
53 2
SL699 45-48 BMD UDP Onset of skeletal muscle symptoms: 10 years. Diagnosis: 22 years. Symptom of weakness. Wheelchair dependent at 48 years. Vignos Scale (48 years): upper = 2 / lower = 9.
EF = 50%. 47 2
SL8 45-48 BMD UDP Onset of skeletal muscle symptoms: 2 years. Diagnosis: 23 years. Weakness, hypertrophy, myalgia/cramping, myoglobinuria, cognitive dysfunction. Vignos Scale (40 years): upper = 1 / lower = 5. Negative family history.
EF = 29%. 27 2
SL42 45-49 BMD UDP Onset of skeletal muscle symptoms: 6 years. Diagnosis: 7 years. Symptom of weakness. Wheelchair dependent at 23 years of age. Vignos Scale (32 years): upper = 2 / lower = 9. Negative family history.
EF = 25%, SF = 14%. 32 2
224 45-49 XLDCM, BMD
[17] Age at consultation: 23 years. No trouble in walking. Normal muscle mass except for pseudohypertrophy of calf and quadriceps muscles; normal muscle strength. Biopsy showed internal nuclei, mild variability in the fiber size with hypotrophic and hypertrophic fibers, rare splitting fibers in an otherwise well preserved muscle. Dystrophin staining: normal with rod domain antibodies, faint with N-terminal antibody, almost normal with few areas of discontinuity with C-terminal antibody. CK = 520U/L. Condition unchanged at 26 years of age. Brother of #225.
DCM diagnosis. Symptom of palpitations. Echocardiogram showed enlarged left ventricle. Cardiac biopsy on RV: mosaic pattern of negative and positive dystrophin cardiomyocytes. Faint staining with amino- and carboxyl-terminal antibodies.
19 2
Page 10 of 30
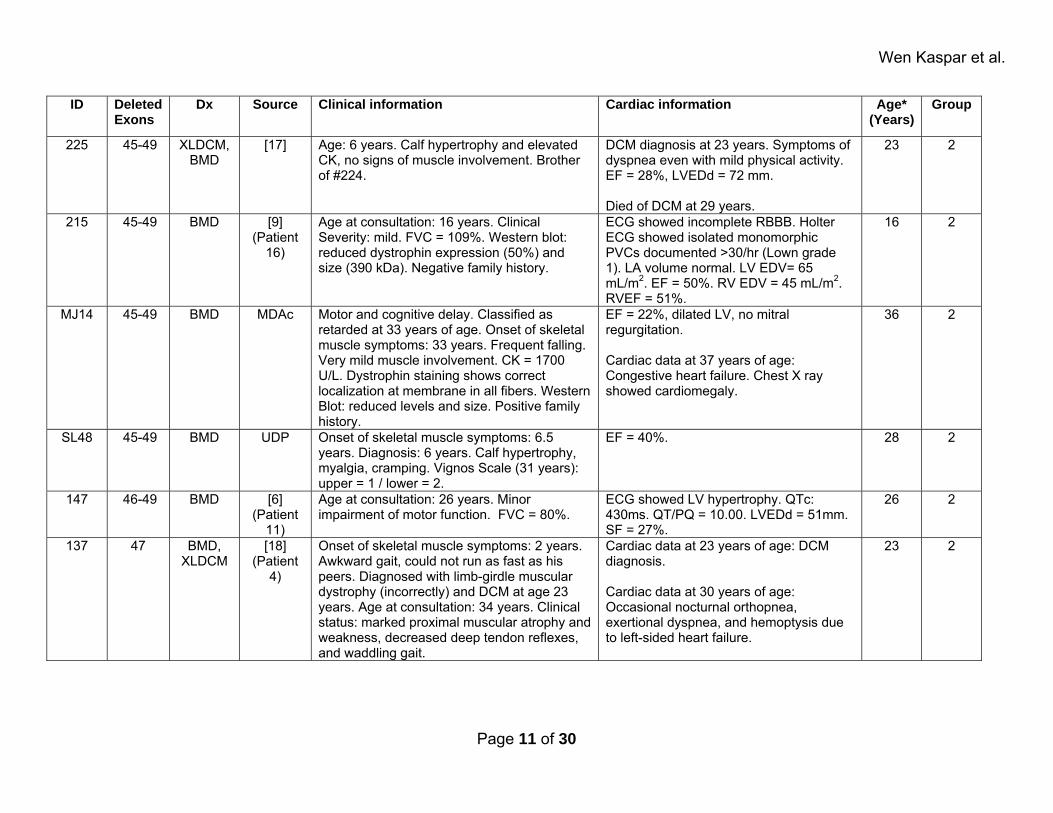
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
225 45-49 XLDCM, BMD
[17] Age: 6 years. Calf hypertrophy and elevated CK, no signs of muscle involvement. Brother of #224.
DCM diagnosis at 23 years. Symptoms of dyspnea even with mild physical activity. EF = 28%, LVEDd = 72 mm. Died of DCM at 29 years.
23 2
215 45-49 BMD [9] (Patient
16)
Age at consultation: 16 years. Clinical Severity: mild. FVC = 109%. Western blot: reduced dystrophin expression (50%) and size (390 kDa). Negative family history.
ECG showed incomplete RBBB. Holter ECG showed isolated monomorphic PVCs documented >30/hr (Lown grade 1). LA volume normal. LV EDV= 65 mL/m2. EF = 50%. RV EDV = 45 mL/m2. RVEF = 51%.
16 2
MJ14 45-49 BMD MDAc Motor and cognitive delay. Classified as retarded at 33 years of age. Onset of skeletal muscle symptoms: 33 years. Frequent falling. Very mild muscle involvement. CK = 1700 U/L. Dystrophin staining shows correct localization at membrane in all fibers. Western Blot: reduced levels and size. Positive family history.
EF = 22%, dilated LV, no mitral regurgitation. Cardiac data at 37 years of age: Congestive heart failure. Chest X ray showed cardiomegaly.
36 2
SL48 45-49 BMD UDP Onset of skeletal muscle symptoms: 6.5 years. Diagnosis: 6 years. Calf hypertrophy, myalgia, cramping. Vignos Scale (31 years): upper = 1 / lower = 2.
EF = 40%. 28 2
147 46-49 BMD [6] (Patient
11)
Age at consultation: 26 years. Minor impairment of motor function. FVC = 80%.
ECG showed LV hypertrophy. QTc: 430ms. QT/PQ = 10.00. LVEDd = 51mm. SF = 27%.
26 2
137 47 BMD, XLDCM
[18] (Patient
4)
Onset of skeletal muscle symptoms: 2 years. Awkward gait, could not run as fast as his peers. Diagnosed with limb-girdle muscular dystrophy (incorrectly) and DCM at age 23 years. Age at consultation: 34 years. Clinical status: marked proximal muscular atrophy and weakness, decreased deep tendon reflexes, and waddling gait.
Cardiac data at 23 years of age: DCM diagnosis. Cardiac data at 30 years of age: Occasional nocturnal orthopnea, exertional dyspnea, and hemoptysis due to left-sided heart failure.
23 2
Page 11 of 30
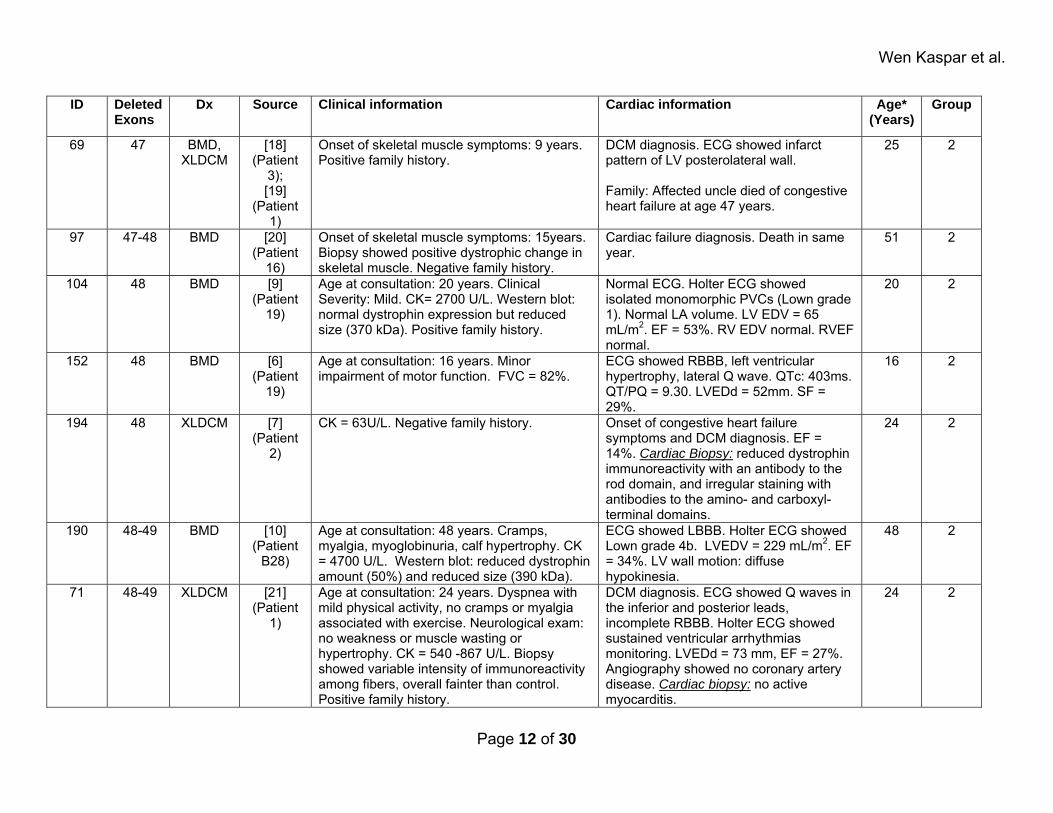
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
69 47 BMD, XLDCM
[18] (Patient
3); [19]
(Patient 1)
Onset of skeletal muscle symptoms: 9 years. Positive family history.
DCM diagnosis. ECG showed infarct pattern of LV posterolateral wall. Family: Affected uncle died of congestive heart failure at age 47 years.
25 2
97 47-48 BMD [20] (Patient
16)
Onset of skeletal muscle symptoms: 15years. Biopsy showed positive dystrophic change in skeletal muscle. Negative family history.
Cardiac failure diagnosis. Death in same year.
51 2
104 48 BMD [9] (Patient
19)
Age at consultation: 20 years. Clinical Severity: Mild. CK= 2700 U/L. Western blot: normal dystrophin expression but reduced size (370 kDa). Positive family history.
Normal ECG. Holter ECG showed isolated monomorphic PVCs (Lown grade 1). Normal LA volume. LV EDV = 65 mL/m2. EF = 53%. RV EDV normal. RVEF normal.
20 2
152 48 BMD [6] (Patient
19)
Age at consultation: 16 years. Minor impairment of motor function. FVC = 82%.
ECG showed RBBB, left ventricular hypertrophy, lateral Q wave. QTc: 403ms. QT/PQ = 9.30. LVEDd = 52mm. SF = 29%.
16 2
194 48 XLDCM [7] (Patient
2)
CK = 63U/L. Negative family history. Onset of congestive heart failure symptoms and DCM diagnosis. EF = 14%. Cardiac Biopsy: reduced dystrophin immunoreactivity with an antibody to the rod domain, and irregular staining with antibodies to the amino- and carboxyl- terminal domains.
24 2
190 48-49 BMD [10] (Patient
B28)
Age at consultation: 48 years. Cramps, myalgia, myoglobinuria, calf hypertrophy. CK = 4700 U/L. Western blot: reduced dystrophin amount (50%) and reduced size (390 kDa).
ECG showed LBBB. Holter ECG showed Lown grade 4b. LVEDV = 229 mL/m2. EF = 34%. LV wall motion: diffuse hypokinesia.
48 2
71 48-49 XLDCM [21] (Patient
1)
Age at consultation: 24 years. Dyspnea with mild physical activity, no cramps or myalgia associated with exercise. Neurological exam: no weakness or muscle wasting or hypertrophy. CK = 540 -867 U/L. Biopsy showed variable intensity of immunoreactivity among fibers, overall fainter than control. Positive family history.
DCM diagnosis. ECG showed Q waves in the inferior and posterior leads, incomplete RBBB. Holter ECG showed sustained ventricular arrhythmias monitoring. LVEDd = 73 mm, EF = 27%. Angiography showed no coronary artery disease. Cardiac biopsy: no active myocarditis.
24 2
Page 12 of 30
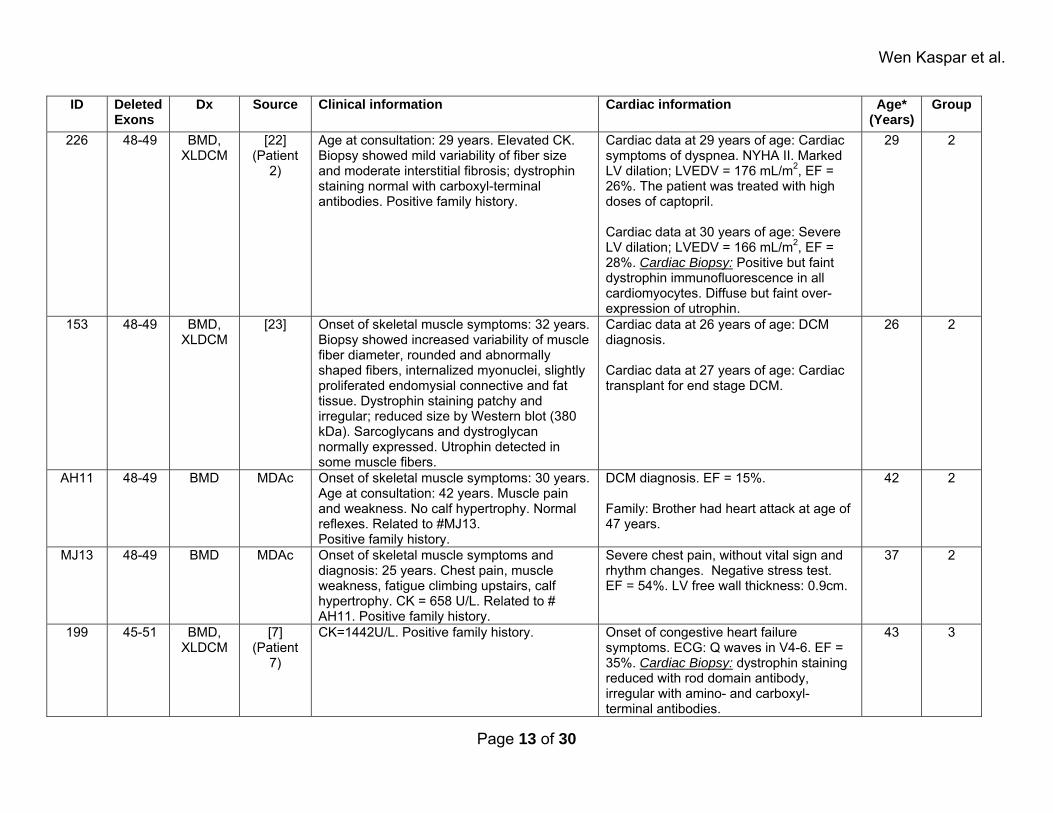
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
226 48-49 BMD, XLDCM
[22] (Patient
2)
Age at consultation: 29 years. Elevated CK. Biopsy showed mild variability of fiber size and moderate interstitial fibrosis; dystrophin staining normal with carboxyl-terminal antibodies. Positive family history.
Cardiac data at 29 years of age: Cardiac symptoms of dyspnea. NYHA II. Marked LV dilation; LVEDV = 176 mL/m2, EF = 26%. The patient was treated with high doses of captopril. Cardiac data at 30 years of age: Severe LV dilation; LVEDV = 166 mL/m2, EF = 28%. Cardiac Biopsy: Positive but faint dystrophin immunofluorescence in all cardiomyocytes. Diffuse but faint over-expression of utrophin.
29 2
153 48-49 BMD, XLDCM
[23] Onset of skeletal muscle symptoms: 32 years. Biopsy showed increased variability of muscle fiber diameter, rounded and abnormally shaped fibers, internalized myonuclei, slightly proliferated endomysial connective and fat tissue. Dystrophin staining patchy and irregular; reduced size by Western blot (380 kDa). Sarcoglycans and dystroglycan normally expressed. Utrophin detected in some muscle fibers.
Cardiac data at 26 years of age: DCM diagnosis. Cardiac data at 27 years of age: Cardiac transplant for end stage DCM.
26 2
AH11 48-49 BMD MDAc Onset of skeletal muscle symptoms: 30 years. Age at consultation: 42 years. Muscle pain and weakness. No calf hypertrophy. Normal reflexes. Related to #MJ13. Positive family history.
DCM diagnosis. EF = 15%. Family: Brother had heart attack at age of 47 years.
42 2
MJ13 48-49 BMD MDAc Onset of skeletal muscle symptoms and diagnosis: 25 years. Chest pain, muscle weakness, fatigue climbing upstairs, calf hypertrophy. CK = 658 U/L. Related to # AH11. Positive family history.
Severe chest pain, without vital sign and rhythm changes. Negative stress test. EF = 54%. LV free wall thickness: 0.9cm.
37 2
199 45-51 BMD, XLDCM
[7] (Patient
7)
CK=1442U/L. Positive family history. Onset of congestive heart failure symptoms. ECG: Q waves in V4-6. EF = 35%. Cardiac Biopsy: dystrophin staining reduced with rod domain antibody, irregular with amino- and carboxyl-terminal antibodies.
43 3
Page 13 of 30
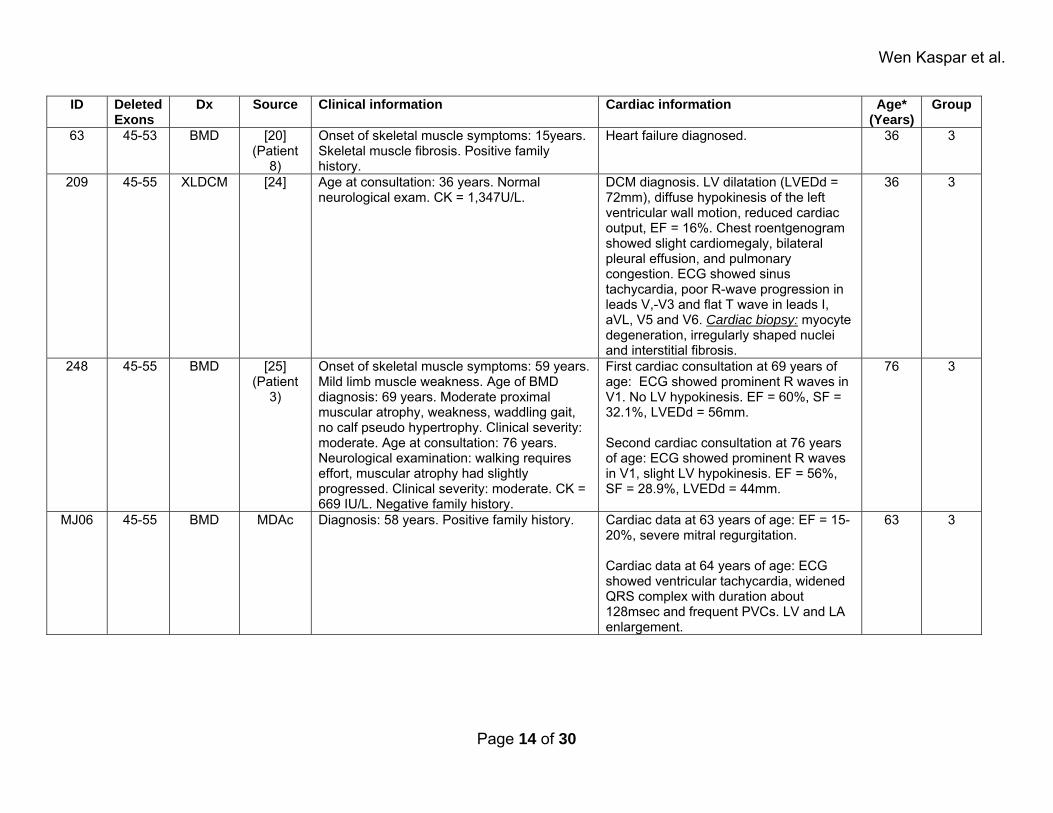
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
63 45-53 BMD [20] (Patient
8)
Onset of skeletal muscle symptoms: 15years. Skeletal muscle fibrosis. Positive family history.
Heart failure diagnosed. 36 3
209 45-55 XLDCM [24] Age at consultation: 36 years. Normal neurological exam. CK = 1,347U/L.
DCM diagnosis. LV dilatation (LVEDd = 72mm), diffuse hypokinesis of the left ventricular wall motion, reduced cardiac output, EF = 16%. Chest roentgenogram showed slight cardiomegaly, bilateral pleural effusion, and pulmonary congestion. ECG showed sinus tachycardia, poor R-wave progression in leads V,-V3 and flat T wave in leads I, aVL, V5 and V6. Cardiac biopsy: myocyte degeneration, irregularly shaped nuclei and interstitial fibrosis.
36 3
248 45-55 BMD [25] (Patient
3)
Onset of skeletal muscle symptoms: 59 years. Mild limb muscle weakness. Age of BMD diagnosis: 69 years. Moderate proximal muscular atrophy, weakness, waddling gait, no calf pseudo hypertrophy. Clinical severity: moderate. Age at consultation: 76 years. Neurological examination: walking requires effort, muscular atrophy had slightly progressed. Clinical severity: moderate. CK = 669 IU/L. Negative family history.
First cardiac consultation at 69 years of age: ECG showed prominent R waves in V1. No LV hypokinesis. EF = 60%, SF = 32.1%, LVEDd = 56mm. Second cardiac consultation at 76 years of age: ECG showed prominent R waves in V1, slight LV hypokinesis. EF = 56%, SF = 28.9%, LVEDd = 44mm.
76 3
MJ06 45-55 BMD MDAc Diagnosis: 58 years. Positive family history. Cardiac data at 63 years of age: EF = 15-20%, severe mitral regurgitation. Cardiac data at 64 years of age: ECG showed ventricular tachycardia, widened QRS complex with duration about 128msec and frequent PVCs. LV and LA enlargement.
63 3
Page 14 of 30
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
196 48-51 XLDCM [7] (Patient
4)
CK = 442-515U/L. Negative family history. Onset of cardiac symptoms. Congestive heart failure diagnosed. EF = 30%. Cardiac Biopsy: reduced dystrophin immunoreactivity with an antibody to the rod domain, and irregular staining with antibodies to the amino- and carboxyl- terminal domains.
30 3
203 48-51 XLDCM [26] Age at consultation: 65 years. Investigated following incidental finding of elevated CK in his 5-year-old grandson. Normal muscle strength, no trophic changes. Normal CK. Normal EMG. Biopsy was normal apart from occasional small fibers and some internal nuclei. Dystrophin immunostaining: normal in distribution, slightly reduced in amount. Western blot: reduced size of dystrophin. Alpha-sarcoglycan, beta-sarcoglycan, gamma-sarcoglycan, spectrin and merosin showed normal distribution.
Cardiac data at 60 years of age: DCM diagnosis. Severe LV dilation with reduced EF and mitral and aortic regurgitation. Coronary arteriography showed very mild atherosclerosis without significant obstructive lesions. Died suddenly of cardiac arrest at age 68. Family: “Cardiomyopathy” caused the death of his mother and two brothers in their sixth decade.
60 3
211 48-52 XLDCM [14] (Patient
3)
Age at consultation: 43 years. No skeletal muscle symptoms. CK = 96U/L. Positive family history.
DCM diagnosis pre-dating cardiac data below. Cardiac data at 43 years of age: ECG showed LBBB. Interventricular septal thickness = 9mm; Posterior wall thickness = 9mm; LVEDd = 72mm; LVEDs = 69mm; SF = 4%.
43 3
200 48-53 XLDCM [7] (Patient
8)
CK below 200U/L. Negative family history. Onset of cardiac symptoms. Congestive heart failure diagnosed. EF = 30%. Cardiac Biopsy: reduced dystrophin immunoreactivity with an antibody to the rod domain, and irregular staining with antibodies to the amino- and carboxyl- terminal domains.
50 3
Page 15 of 30
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
208 49-51 XLDCM [21] (Patient
2)
Age at consultation: 52 years. No muscle atrophy or pseudohypertrophy, normal muscle strength. CK= 84 U/L.
Cardiac data at 50 years of age: Onset of congestive heart failure symptoms. Cardiac data at 52 years of age: Dyspnoeic at rest, systolic murmur present. ECG showed negative T waves in leads V5-V6. Dilated LV (LVEDd = 70mm), EF = 20%, dilated RV and both atria. Moderate mitral and tricuspidal valve regurgitation. Cardiac biopsy: significant fibrosis, separating individual cardiomyocytes in some areas, gross variability of fiber size mainly due to hypertrophic cardiomyocytes; strong and continuous dystrophin staining with antibodies to amino-terminus, mid rod domain, and carboxyl-terminus. Slight reduction in amount by Western blot.
50 3
MJ01 51-52 BMD MDAc Onset of skeletal muscle symptoms: birth. Diagnosis: 5 years. Clinical findings: scoliosis since the teenage years, limited mobility at 29 years. Wheelchair dependent at 40 years of age.
Cardiac data at 33 years of age: Congestive heart failure. Pacemaker implanted. Cardiac data at 35 years of age: Severe LV dilation with global LV dysfunction, EF = 34%.
33 3
* Age refers to the youngest age in years at which the patient fulfills our criteria for classification as affected with cardiomyopathy. Abbreviations: AV: atrioventricular; BMD: Becker muscular dystrophy; CK: creatine kinase; DCM: dilated cardiomyopathy; Dx: diagnosis; ECG: electrocardiogram; EDV: end diastole volume; EF: ejection fraction (left ventricular unless otherwise specified); EPSS: E-point septal separation; FVC: forced vital capacity; LA: left atrium; LBBB: left branch bundle block; LV: left ventricle; LVEDd: left ventricular end-diastolic diameter; LVEDs: left ventricular end systolic diameter; MDAc: muscular dystrophy clinics of Nationwide Children’s Hospital and The Ohio State University Medical Center; NSI: no specific information; PVC: premature ventricular contraction; RA: right atrium;
Page 16 of 30

Wen Kaspar et al.
RBBB: right branch bundle block; RV: right ventricle; SF: shortening fraction; UDP: United Dystrophinopathy Project; WHO: World Health Organization. Normal values for measured parameters: CK: 200 U/L (unless otherwise stated); EF: above or equal to 55%; EPSS: below 5 mm; FVC: 80%-120% predicted; Intraventricular Septal Thickness: below 12mm; LVEDd: below 58 mm, 2 z scores, or 2 SD; Posterior Wall Thickness: below 12mm; QTc: below 440ms; SF: above or equal to 32%; Vignos Scale: described in JAMA (1963), 184:89-96. Published Sources: 1. Gold R, Kress W, Meurers B, Meng G, Reichmann H, Muller CR: Becker muscular dystrophy: detection of unusual disease courses by combined approach to dystrophin analysis. Muscle Nerve 1992, 15:214-218.
2. Feng J, Yan J, Buzin CH, Towbin JA, Sommer SS: Mutations in the dystrophin gene are associated with sporadic dilated cardiomyopathy. Mol Genet Metab 2002, 77:119-126.
3. Novakovic I, Bojic D, Todorovic S, Apostolski S, Lukovic L, Stefanovic D, Milasin J: Proximal dystrophin gene deletions and protein alterations in becker muscular dystrophy. Ann N Y Acad Sci 2005, 1048:406-410.
4. Oldfors A, Eriksson BO, Kyllerman M, Martinsson T, Wahlstrom J: Dilated cardiomyopathy and the dystrophin gene: an illustrated review. Br Heart J 1994, 72:344-348.
5. Maeda M, Nakao S, Miyazato H, Setoguchi M, Arima S, Higuchi I, Osame M, Taira A, Nomoto K, Toda H, et al.: Cardiac dystrophin abnormalities in Becker muscular dystrophy assessed by endomyocardial biopsy. Am Heart J 1995, 129:702-707.
6. Steare SE, Dubowitz V, Benatar A: Subclinical cardiomyopathy in Becker muscular dystrophy. Br Heart J 1992, 68:304-308.
7. Arbustini E, Diegoli M, Morbini P, Dal Bello B, Banchieri N, Pilotto A, Magani F, Grasso M, Narula J, Gavazzi A, et al.: Prevalence and characteristics of dystrophin defects in adult male patients with dilated cardiomyopathy. J Am Coll Cardiol 2000, 35:1760-1768.
8. Martins E, Silva-Cardoso J, Silveira F, Nadais G, Goncalves FR: Left ventricular function in adults with muscular dystrophies: genotype-phenotype correlations. Rev Port Cardiol 2005, 24:23-35.
Page 17 of 30

Wen Kaspar et al.
9. Melacini P, Fanin M, Danieli GA, Fasoli G, Villanova C, Angelini C, Vitiello L, Miorelli M, Buja GF, Mostacciuolo ML, et al.: Cardiac involvement in Becker muscular dystrophy. J Am Coll Cardiol 1993, 22:1927-1934.
10. Melacini P, Fanin M, Danieli GA, Villanova C, Martinello F, Miorin M, Freda MP, Miorelli M, Mostacciuolo ML, Fasoli G, et al.: Myocardial involvement is very frequent among patients affected with subclinical Becker's muscular dystrophy. Circulation 1996, 94:3168-3175.
11. Akdemir R, Ozhan H, Gunduz H, Yazici M, Erbilen E, Uyan C, Imirzalioglu N: Complete atrioventricular block in Becker muscular dystrophy. N Z Med J 2004, 117:U895.
12. Piccolo G, Azan G, Tonin P, Arbustini E, Gavazzi A, Banfi P, Mora M, Morandi L, Tedeschi S: Dilated cardiomyopathy requiring cardiac transplantation as initial manifestation of Xp21 Becker type muscular dystrophy. Neuromuscul Disord 1994, 4:143-146.
13. Sousa RC, Silva P, Pais F, Fortuna A, Relvas S, Simoes L, Miranda O, Gama V: [Dilated cardiomyopathy in a patient with Becker's muscular dystrophy. A clinical case report]. Rev Port Cardiol 1993, 12:563-570, 511.
14. Shimizu M, Ino H, Yasuda T, Fujino N, Uchiyama K, Mabuchi T, Konno T, Kaneda T, Fujita T, Masuta E, et al.: Gene mutations in adult Japanese patients with dilated cardiomyopathy. Circ J 2005, 69:150-153.
15. Finsterer J, Stollberger C, Blazek G, Kunafer M, Prager E: Cardiac involvement over 10 years in myotonic and Becker muscular dystrophy and mitochondrial disorder. Int J Cardiol 2007, 119:176-184.
16. Yazaki M, Yoshida K, Nakamura A, Koyama J, Nanba T, Ohori N, Ikeda S: Clinical characteristics of aged Becker muscular dystrophy patients with onset after 30 years. Eur Neurol 1999, 42:145-149.
17. Politano L, Passamano L, Petretta VR, Nigro V, Papparella S, Nigro G, Santangelo L, M.G.Esposito, Come LI, Nigro G: Familial Dilated Cardiomyopathy Associated with the Typical Dystrophin BMD Mutation: Report on Two Additional Cases. Acta Myol 1999:3329-3336.
18. Yoshida K, Ikeda S, Nakamura A, Kagoshima M, Takeda S, Shoji S, Yanagisawa N: Molecular analysis of the Duchenne muscular dystrophy gene in patients with Becker muscular dystrophy presenting with dilated cardiomyopathy. Muscle Nerve 1993, 16:1161-1166.
19. Yazawa M, Ikeda S, Owa M, Haruta S, Yanagisawa N, Tanaka E, Watanabe M: A family of Becker's progressive muscular dystrophy with severe cardiomyopathy. Eur Neurol 1987, 27:13-19.
Page 18 of 30

Wen Kaspar et al.
20. Saito M, Kawai H, Akaike M, Adachi K, Nishida Y, Saito S: Cardiac dysfunction with Becker muscular dystrophy. Am Heart J 1996, 132:642-647.
21. Muntoni F, Di Lenarda A, Porcu M, Sinagra G, Mateddu A, Marrosu G, Ferlini A, Cau M, Milasin J, Melis MA, et al.: Dystrophin gene abnormalities in two patients with idiopathic dilated cardiomyopathy. Heart 1997, 78:608-612.
22. Fanin M, Melacini P, Angelini C, Danieli GA: Could utrophin rescue the myocardium of patients with dystrophin gene mutations? J Mol Cell Cardiol 1999, 31:1501-1508.
23. Finsterer J, Bittner RE, Grimm M: Cardiac involvement in Becker's muscular dystrophy, necessitating heart transplantation, 6 years before apparent skeletal muscle involvement. Neuromuscul Disord 1999, 9:598-600.
24. Tasaki N, Yoshida K, Haruta SI, Kouno H, Ichinose H, Fujimoto Y, Urasawa N, Kawakami T, Taniguchi M, Kurushima S, et al.: X-linked dilated cardiomyopathy with a large hot-spot deletion in the dystrophin gene. Intern Med 2001, 40:1215-1221.
25. Nakamura A, Yoshida K, Fukushima K, Ueda H, Urasawa N, Koyama J, Yazaki Y, Yazaki M, Sakai T, Haruta S, et al.: Follow-up of three patients with a large in-frame deletion of exons 45-55 in the Duchenne muscular dystrophy (DMD) gene. J Clin Neurosci 2008, 15:757-763.
26. Palmucci L, Mongini T, Chiado-Piat L, Doriguzzi C, Fubini A: Dystrophinopathy expressing as either cardiomyopathy or Becker dystrophy in the same family. Neurology 2000, 54:529-530.
Page 19 of 30
Wen Kaspar et al.
Supplemental Table 3: Non-affected patients.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
216 3-4 BMD [1] (Patient 22)
Age at consultation: 24 years. Clinical Severity: severe. Myoglobinuria. FVC = 92%. Positive family history.
ECG showed incomplete RBBB. Holter ECG showed isolated monomorphic PVCs (Lown grade 1). Normal LA volume. LV EDV = 50 mL/m2. EF = 67%. Normal LV wall motion. Normal RV EDV. Normal RVEF.
24 1
218 3-4 BMD [1] (Patient 29)
Age at consultation: 35 years. Clinical Severity: Severe. FVC = 75% (mild restrictive respiratory insufficiency). Positive family history.
Normal ECG. Holter ECG showed isolated monomorphic PVCs (Lown grade 1). Normal LA volume. LV EDV = 62 mL/ m2. EF = 61%. Normal LV wall motion. Normal RV EDV. Normal RVEF. Normal RV motion.
35 1
AH15 3-4 BMD MDAc Ambulatory at 9 years of age. EF = 55%, SF = 34%, EPSS = 5mm. 9 1
SL459 3-7 BMD UDP Vignos Scale (22 years): upper = 1 / lower = 2.
EF = 55% 25 1
99 3-9 BMD [1] (Patient 1)
Age at consultation: 6 years. Clinical Severity: Mild. Skeletal muscle symptoms are present. CK = 1770 U/L. Western blot: reduced dystrophin expression (60%) and size (370 kDa). Negative family history.
Normal ECG. Normal LA volume. LV EDV = 37 mL/ m2. EF = 60%. Normal LV wall motion. RV EDV=41 mL/ m2. RVEF = 68%. Normal RV wall motion.
6 1
AH14 5 BMD MDAc Onset of skeletal muscle symptoms: 7 years. Age at consultation: 13 years. Clinical Severity: Mild. Skeletal muscle symptoms are present. Elevated CK. Biopsy: dystrophic changes; Dys2 staining positive but Dys 1 and Dys 3 severely reduced; Utrophin is upregulated. Western Blot: severely reduced dystrophin.
EF = 58%, SF = 31%, EPSS = 5mm, no mitral valve regurgitation, no sign of LV dilation.
13 1
Page 20 of 30
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
AH02 5 BMD MDAc Onset of skeletal muscle symptoms: 10 years. Frequent falling, difficulty climbing stairs, calf hypertrophy, positive Gower's sign. CK = 7651 U/L. Negative family history.
LVEDd = 0.5sd, EF = 60%, SF = 40%. 14 1
SL188 45 BMD UDP Onset of skeletal muscle symptoms: 3 years. Diagnosis: 6 years. Weakness, calf hypertrophy and Gower’s sign. Vignos Scale (14 years): upper = 1 / lower = 2.
EF = 63%, SF = 35%. 15 2
213 45-47 BMD [1] (Patient 2)
Age at consultation: 6 years. Clinical Severity: Mild. CK = 5000 U/L. FVC = 56% (moderate restrictive respiratory insufficiency).
Normal ECG. Normal LA volume. LV EDV = 67 mL/ m2. EF = 64%. Normal LV wall motion. RV EDV = 46 mL/ m2. RVEF = 57%. Normal RV motion.
6 2
217 45-47 BMD [1] (Patient 24)
Age at consultation: 28 years. Clinical Severity: Moderate.
ECG showed T wave changes. Normal Holter ECG. Normal LA volume. LV EDV = 69 mL/ m2. EF = 55%. Normal LV wall motion. RV EDV = 66 mL/ m2. RVEF = 64%.
28 2
106 45-47 BMD [1] (Patient 28)
Age at consultation: 33 years. Clinical Severity: Moderate. Biopsy showed small number of dystrophin negative fibers. Western Blot: reduced amount (50%) and size (380 kDa). Positive family history.
Normal ECG. Holter ECG showed isolated monomorphic PVCs (Lown grade 1). LA volume = 29 mL/ m2. LV EDV = 44 mL/ m2. EF = 55%. Normal LV wall motion. RV EDV = 52 mL/ m2. RVEF = 52%.
33 2
KJ03 45-47 BMD MDAc Onset of skeletal muscle symptoms: childhood. Diagnosis: 37 years. Age at consultation: 47 years. Patient is ambulant with wide-based gait with exaggerated lordosis and some waddle.
Echocardiography and stress test negative for cardiac dilation.
45 2
SL382 45-47 BMD UDP Onset of skeletal muscle symptoms: 1.5 years. Diagnosis: 9 years. Toe walking, myalgia, cramping. Vignos Scale (19 years): upper = 1 / lower = 1. Negative family history.
EF=57%, SF= 32%. 19 2
Page 21 of 30
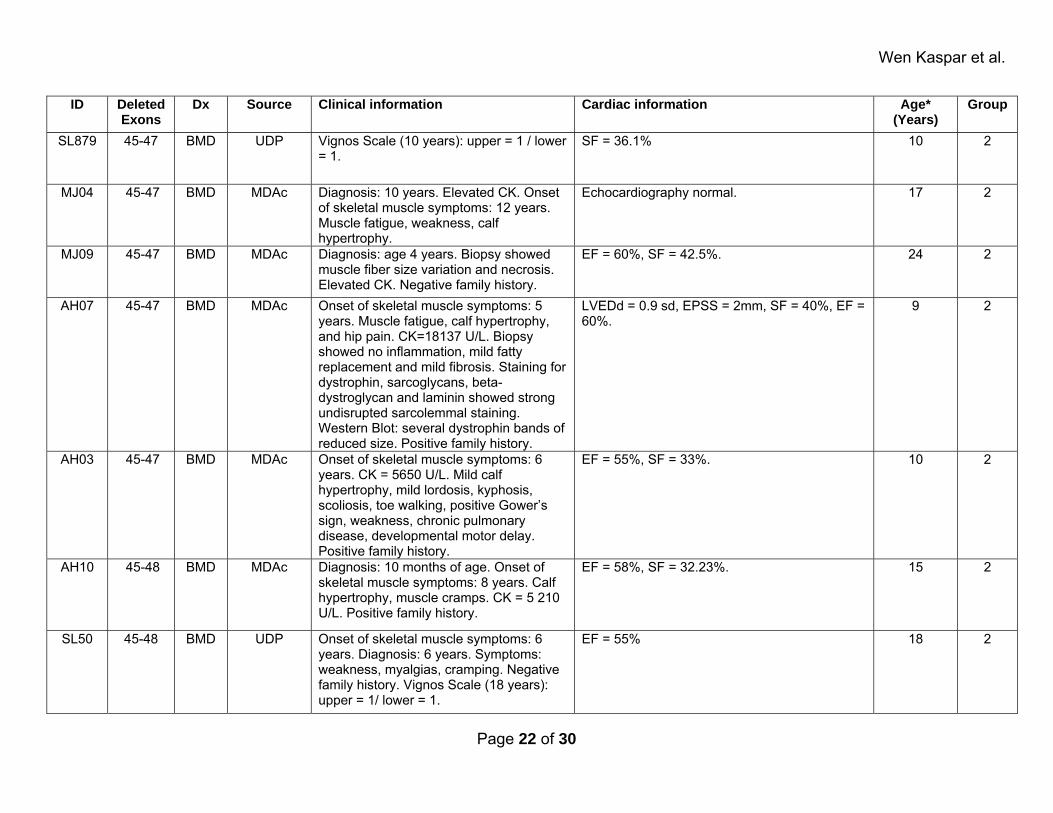
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
SL879 45-47 BMD UDP Vignos Scale (10 years): upper = 1 / lower = 1.
SF = 36.1% 10 2
MJ04 45-47 BMD MDAc Diagnosis: 10 years. Elevated CK. Onset of skeletal muscle symptoms: 12 years. Muscle fatigue, weakness, calf hypertrophy.
Echocardiography normal. 17 2
MJ09 45-47 BMD MDAc Diagnosis: age 4 years. Biopsy showed muscle fiber size variation and necrosis. Elevated CK. Negative family history.
EF = 60%, SF = 42.5%. 24 2
AH07 45-47 BMD MDAc Onset of skeletal muscle symptoms: 5 years. Muscle fatigue, calf hypertrophy, and hip pain. CK=18137 U/L. Biopsy showed no inflammation, mild fatty replacement and mild fibrosis. Staining for dystrophin, sarcoglycans, beta-dystroglycan and laminin showed strong undisrupted sarcolemmal staining. Western Blot: several dystrophin bands of reduced size. Positive family history.
LVEDd = 0.9 sd, EPSS = 2mm, SF = 40%, EF = 60%.
9 2
AH03 45-47 BMD MDAc Onset of skeletal muscle symptoms: 6 years. CK = 5650 U/L. Mild calf hypertrophy, mild lordosis, kyphosis, scoliosis, toe walking, positive Gower’s sign, weakness, chronic pulmonary disease, developmental motor delay. Positive family history.
EF = 55%, SF = 33%. 10 2
AH10 45-48 BMD MDAc Diagnosis: 10 months of age. Onset of skeletal muscle symptoms: 8 years. Calf hypertrophy, muscle cramps. CK = 5 210 U/L. Positive family history.
EF = 58%, SF = 32.23%. 15 2
SL50 45-48 BMD UDP Onset of skeletal muscle symptoms: 6 years. Diagnosis: 6 years. Symptoms: weakness, myalgias, cramping. Negative family history. Vignos Scale (18 years): upper = 1/ lower = 1.
EF = 55% 18 2
Page 22 of 30
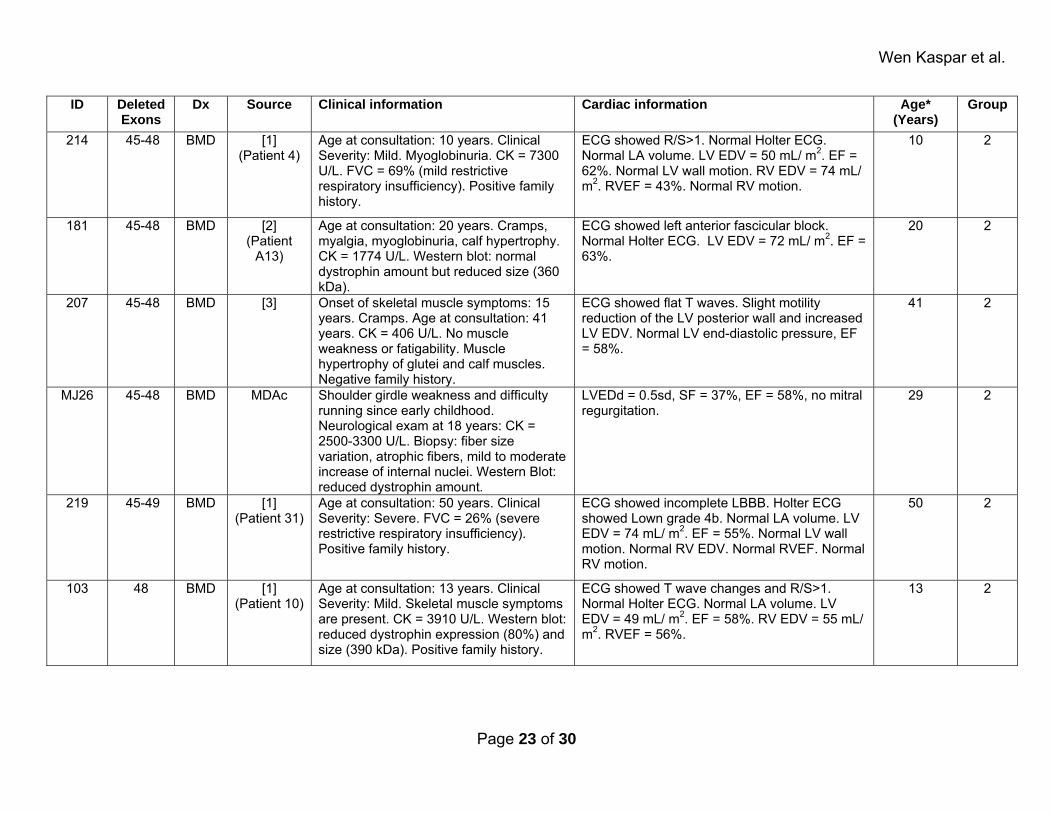
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
214 45-48 BMD [1] (Patient 4)
Age at consultation: 10 years. Clinical Severity: Mild. Myoglobinuria. CK = 7300 U/L. FVC = 69% (mild restrictive respiratory insufficiency). Positive family history.
ECG showed R/S>1. Normal Holter ECG. Normal LA volume. LV EDV = 50 mL/ m2. EF = 62%. Normal LV wall motion. RV EDV = 74 mL/ m2. RVEF = 43%. Normal RV motion.
10 2
181 45-48 BMD [2] (Patient
A13)
Age at consultation: 20 years. Cramps, myalgia, myoglobinuria, calf hypertrophy. CK = 1774 U/L. Western blot: normal dystrophin amount but reduced size (360 kDa).
ECG showed left anterior fascicular block. Normal Holter ECG. LV EDV = 72 mL/ m2. EF = 63%.
20 2
207 45-48 BMD [3] Onset of skeletal muscle symptoms: 15 years. Cramps. Age at consultation: 41 years. CK = 406 U/L. No muscle weakness or fatigability. Muscle hypertrophy of glutei and calf muscles. Negative family history.
ECG showed flat T waves. Slight motility reduction of the LV posterior wall and increased LV EDV. Normal LV end-diastolic pressure, EF = 58%.
41 2
MJ26 45-48 BMD MDAc Shoulder girdle weakness and difficulty running since early childhood. Neurological exam at 18 years: CK = 2500-3300 U/L. Biopsy: fiber size variation, atrophic fibers, mild to moderate increase of internal nuclei. Western Blot: reduced dystrophin amount.
LVEDd = 0.5sd, SF = 37%, EF = 58%, no mitral regurgitation.
29 2
219 45-49 BMD [1] (Patient 31)
Age at consultation: 50 years. Clinical Severity: Severe. FVC = 26% (severe restrictive respiratory insufficiency). Positive family history.
ECG showed incomplete LBBB. Holter ECG showed Lown grade 4b. Normal LA volume. LV EDV = 74 mL/ m2. EF = 55%. Normal LV wall motion. Normal RV EDV. Normal RVEF. Normal RV motion.
50 2
103 48 BMD [1] (Patient 10)
Age at consultation: 13 years. Clinical Severity: Mild. Skeletal muscle symptoms are present. CK = 3910 U/L. Western blot: reduced dystrophin expression (80%) and size (390 kDa). Positive family history.
ECG showed T wave changes and R/S>1. Normal Holter ECG. Normal LA volume. LV EDV = 49 mL/ m2. EF = 58%. RV EDV = 55 mL/ m2. RVEF = 56%.
13 2
Page 23 of 30
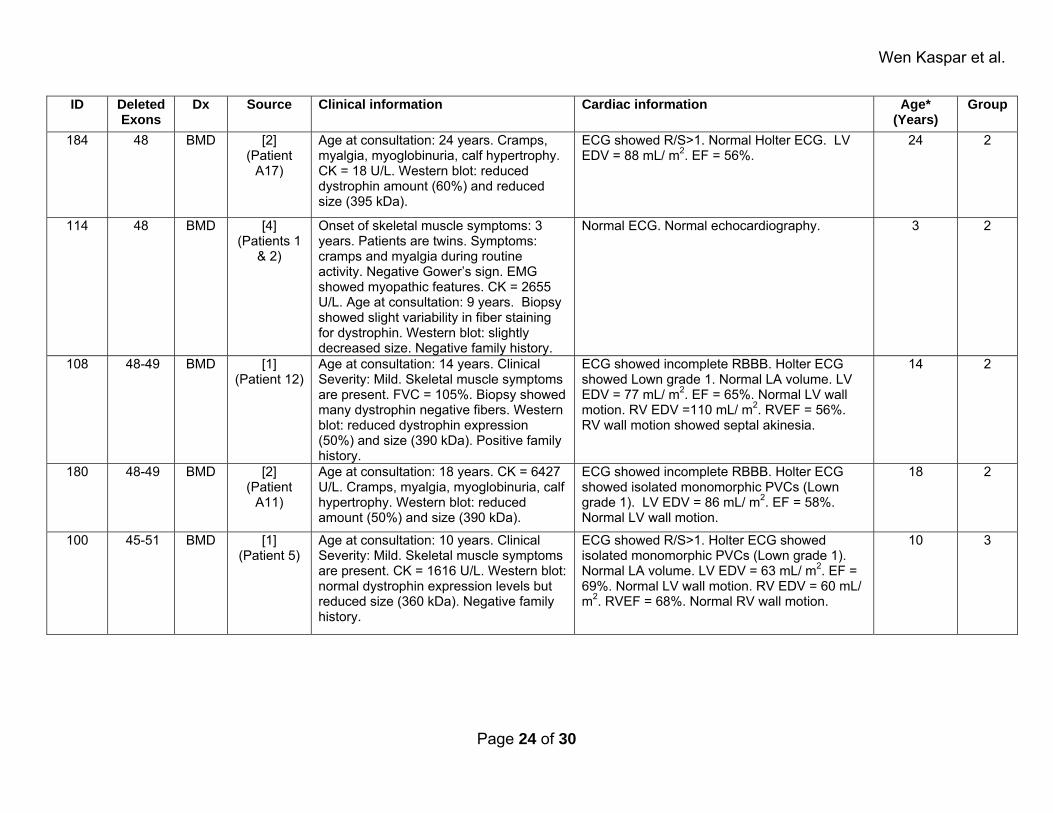
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
184 48 BMD [2] (Patient
A17)
Age at consultation: 24 years. Cramps, myalgia, myoglobinuria, calf hypertrophy. CK = 18 U/L. Western blot: reduced dystrophin amount (60%) and reduced size (395 kDa).
ECG showed R/S>1. Normal Holter ECG. LV EDV = 88 mL/ m2. EF = 56%.
24 2
114 48 BMD [4] (Patients 1
& 2)
Onset of skeletal muscle symptoms: 3 years. Patients are twins. Symptoms: cramps and myalgia during routine activity. Negative Gower’s sign. EMG showed myopathic features. CK = 2655 U/L. Age at consultation: 9 years. Biopsy showed slight variability in fiber staining for dystrophin. Western blot: slightly decreased size. Negative family history.
Normal ECG. Normal echocardiography. 3 2
108 48-49 BMD [1] (Patient 12)
Age at consultation: 14 years. Clinical Severity: Mild. Skeletal muscle symptoms are present. FVC = 105%. Biopsy showed many dystrophin negative fibers. Western blot: reduced dystrophin expression (50%) and size (390 kDa). Positive family history.
ECG showed incomplete RBBB. Holter ECG showed Lown grade 1. Normal LA volume. LV EDV = 77 mL/ m2. EF = 65%. Normal LV wall motion. RV EDV =110 mL/ m2. RVEF = 56%. RV wall motion showed septal akinesia.
14 2
180 48-49 BMD [2] (Patient
A11)
Age at consultation: 18 years. CK = 6427 U/L. Cramps, myalgia, myoglobinuria, calf hypertrophy. Western blot: reduced amount (50%) and size (390 kDa).
ECG showed incomplete RBBB. Holter ECG showed isolated monomorphic PVCs (Lown grade 1). LV EDV = 86 mL/ m2. EF = 58%. Normal LV wall motion.
18 2
100 45-51 BMD [1] (Patient 5)
Age at consultation: 10 years. Clinical Severity: Mild. Skeletal muscle symptoms are present. CK = 1616 U/L. Western blot: normal dystrophin expression levels but reduced size (360 kDa). Negative family history.
ECG showed R/S>1. Holter ECG showed isolated monomorphic PVCs (Lown grade 1). Normal LA volume. LV EDV = 63 mL/ m2. EF = 69%. Normal LV wall motion. RV EDV = 60 mL/ m2. RVEF = 68%. Normal RV wall motion.
10 3
Page 24 of 30
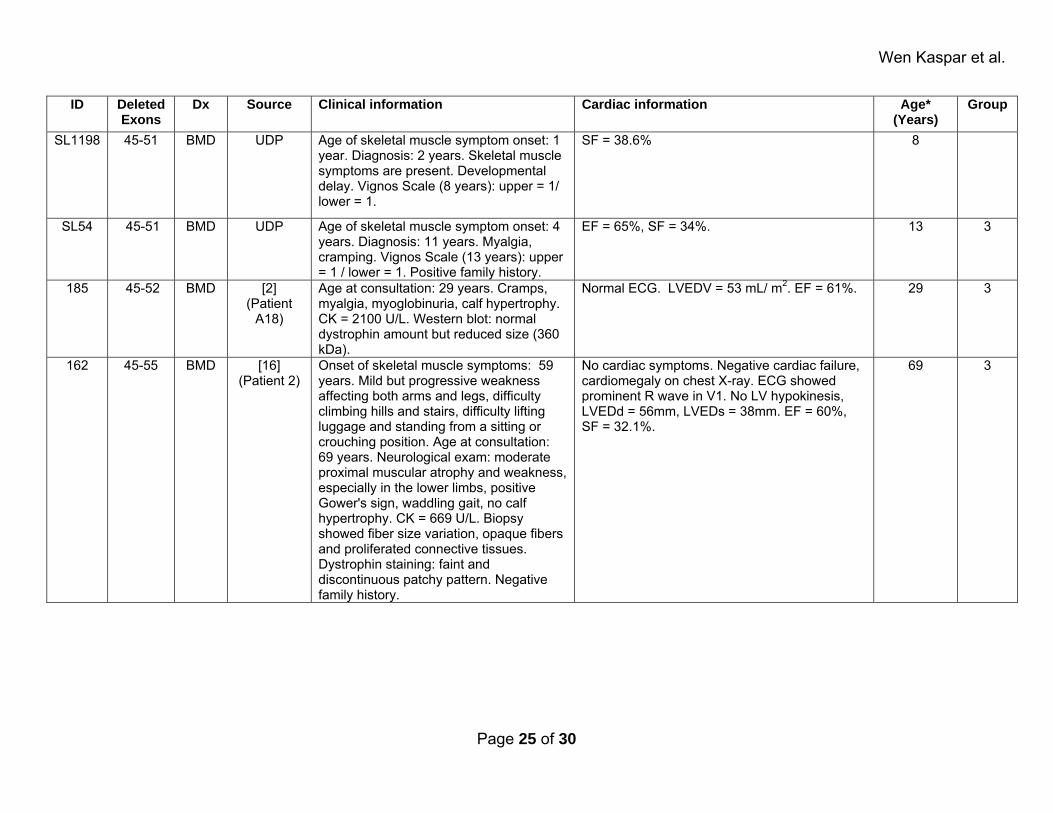
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
SL1198 45-51 BMD UDP Age of skeletal muscle symptom onset: 1 year. Diagnosis: 2 years. Skeletal muscle symptoms are present. Developmental delay. Vignos Scale (8 years): upper = 1/ lower = 1.
SF = 38.6% 8
SL54 45-51 BMD UDP Age of skeletal muscle symptom onset: 4 years. Diagnosis: 11 years. Myalgia, cramping. Vignos Scale (13 years): upper = 1 / lower = 1. Positive family history.
EF = 65%, SF = 34%. 13 3
185 45-52 BMD [2] (Patient
A18)
Age at consultation: 29 years. Cramps, myalgia, myoglobinuria, calf hypertrophy. CK = 2100 U/L. Western blot: normal dystrophin amount but reduced size (360 kDa).
Normal ECG. LVEDV = 53 mL/ m2. EF = 61%. 29 3
162 45-55 BMD [16] (Patient 2)
Onset of skeletal muscle symptoms: 59 years. Mild but progressive weakness affecting both arms and legs, difficulty climbing hills and stairs, difficulty lifting luggage and standing from a sitting or crouching position. Age at consultation: 69 years. Neurological exam: moderate proximal muscular atrophy and weakness, especially in the lower limbs, positive Gower's sign, waddling gait, no calf hypertrophy. CK = 669 U/L. Biopsy showed fiber size variation, opaque fibers and proliferated connective tissues. Dystrophin staining: faint and discontinuous patchy pattern. Negative family history.
No cardiac symptoms. Negative cardiac failure, cardiomegaly on chest X-ray. ECG showed prominent R wave in V1. No LV hypokinesis, LVEDd = 56mm, LVEDs = 38mm. EF = 60%, SF = 32.1%.
69 3
Page 25 of 30
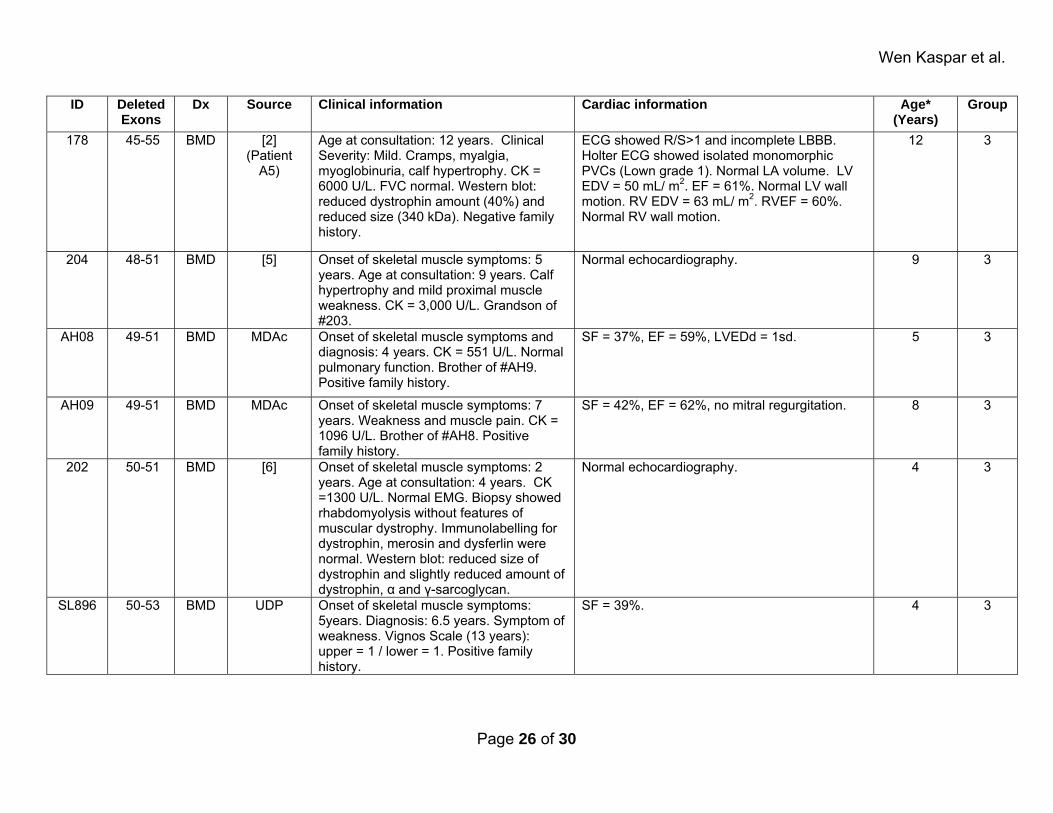
Wen Kaspar et al.
ID Deleted Exons
Dx Source Clinical information Cardiac information Age* (Years)
Group
178 45-55 BMD [2] (Patient
A5)
Age at consultation: 12 years. Clinical Severity: Mild. Cramps, myalgia, myoglobinuria, calf hypertrophy. CK = 6000 U/L. FVC normal. Western blot: reduced dystrophin amount (40%) and reduced size (340 kDa). Negative family history.
ECG showed R/S>1 and incomplete LBBB. Holter ECG showed isolated monomorphic PVCs (Lown grade 1). Normal LA volume. LV EDV = 50 mL/ m2. EF = 61%. Normal LV wall motion. RV EDV = 63 mL/ m2. RVEF = 60%. Normal RV wall motion.
12 3
204 48-51 BMD [5] Onset of skeletal muscle symptoms: 5 years. Age at consultation: 9 years. Calf hypertrophy and mild proximal muscle weakness. CK = 3,000 U/L. Grandson of #203.
Normal echocardiography. 9 3
AH08 49-51 BMD MDAc Onset of skeletal muscle symptoms and diagnosis: 4 years. CK = 551 U/L. Normal pulmonary function. Brother of #AH9. Positive family history.
SF = 37%, EF = 59%, LVEDd = 1sd. 5 3
AH09 49-51 BMD MDAc Onset of skeletal muscle symptoms: 7 years. Weakness and muscle pain. CK = 1096 U/L. Brother of #AH8. Positive family history.
SF = 42%, EF = 62%, no mitral regurgitation. 8 3
202 50-51 BMD [6] Onset of skeletal muscle symptoms: 2 years. Age at consultation: 4 years. CK =1300 U/L. Normal EMG. Biopsy showed rhabdomyolysis without features of muscular dystrophy. Immunolabelling for dystrophin, merosin and dysferlin were normal. Western blot: reduced size of dystrophin and slightly reduced amount of dystrophin, α and γ-sarcoglycan.
Normal echocardiography. 4 3
SL896 50-53 BMD UDP Onset of skeletal muscle symptoms: 5years. Diagnosis: 6.5 years. Symptom of weakness. Vignos Scale (13 years): upper = 1 / lower = 1. Positive family history.
SF = 39%. 4 3
Page 26 of 30

Wen Kaspar et al.
* Age refers to the age in years of the patient at the last reported cardiac evaluation with cardiac findings that fulfill our criteria for a classification of non-affected with cardiomyopathy. Abbreviations: BMD: Becker muscular dystrophy; CK: creatine kinase; Dx: diagnosis; ECG: electrocardiogram; EDV: end diastole volume; EF: ejection fraction (left ventricular unless otherwise specified); EMG: electromyography; EPSS: E-point septal separation; FVC: forced vital capacity; LA: left atrium; LBBB: left branch bundle block; LV: left ventricle; LVEDd: left ventricular end-diastolic diameter; MDAc: muscular dystrophy clinics of Nationwide Children’s Hospital and The Ohio State University Medical Center; PVC: premature ventricular contraction; RBBB: right branch bundle block; RV: right ventricle; SF: shortening fraction; UDP: United Dystrophinopathy Project. Normal values for measured parameters: CK: 200 U/L (unless otherwise stated); EF: above or equal to 55%; EPSS: below 5 mm; FVC: 80%-120% predicted; LVEDd: below 58 mm, 2 z scores, or 2 SD; SF: above or equal to 32%; Vignos Scale: described in JAMA (1963), 184:89-96. Published sources: 1. Melacini P, Fanin M, Danieli GA, Fasoli G, Villanova C, Angelini C, Vitiello L, Miorelli M, Buja GF, Mostacciuolo ML, et al.: Cardiac involvement in Becker muscular dystrophy. J Am Coll Cardiol 1993, 22:1927-1934.
2. Melacini P, Fanin M, Danieli GA, Villanova C, Martinello F, Miorin M, Freda MP, Miorelli M, Mostacciuolo ML, Fasoli G, et al.: Myocardial involvement is very frequent among patients affected with subclinical Becker's muscular dystrophy. Circulation 1996, 94:3168-3175.
3. Siciliano G, Fanin M, Angelini C, Pollina LE, Miorin M, Saad FA, Freda MP, Muratorio A: Prevalent cardiac involvement in dystrophin Becker type mutation. Neuromuscul Disord 1994, 4:381-386.
4. Ramelli GP, Joncourt F, Luetschg J, Weis J, Tolnay M, Burgunder JM: Becker muscular dystrophy with marked divergence between clinical and molecular genetic findings: case series. Swiss Med Wkly 2006, 136:189-193.
5. Palmucci L, Mongini T, Chiado-Piat L, Doriguzzi C, Fubini A: Dystrophinopathy expressing as either cardiomyopathy or Becker dystrophy in the same family. Neurology 2000, 54:529-530.
Page 27 of 30

Wen Kaspar et al.
6. Lesca G, Testard H, Streichenberger N, Pelissier JF, Lestra C, Burel E, Jonveaux P, Michel-Calemard L: [Family study allows more optimistic prognosis and genetic counselling in a child with a deletion of exons 50-51 of the dystrophin gene]. Arch Pediatr 2007, 14:262-265.
Page 28 of 30
Wen Kaspar et al.
Page 29 of 30
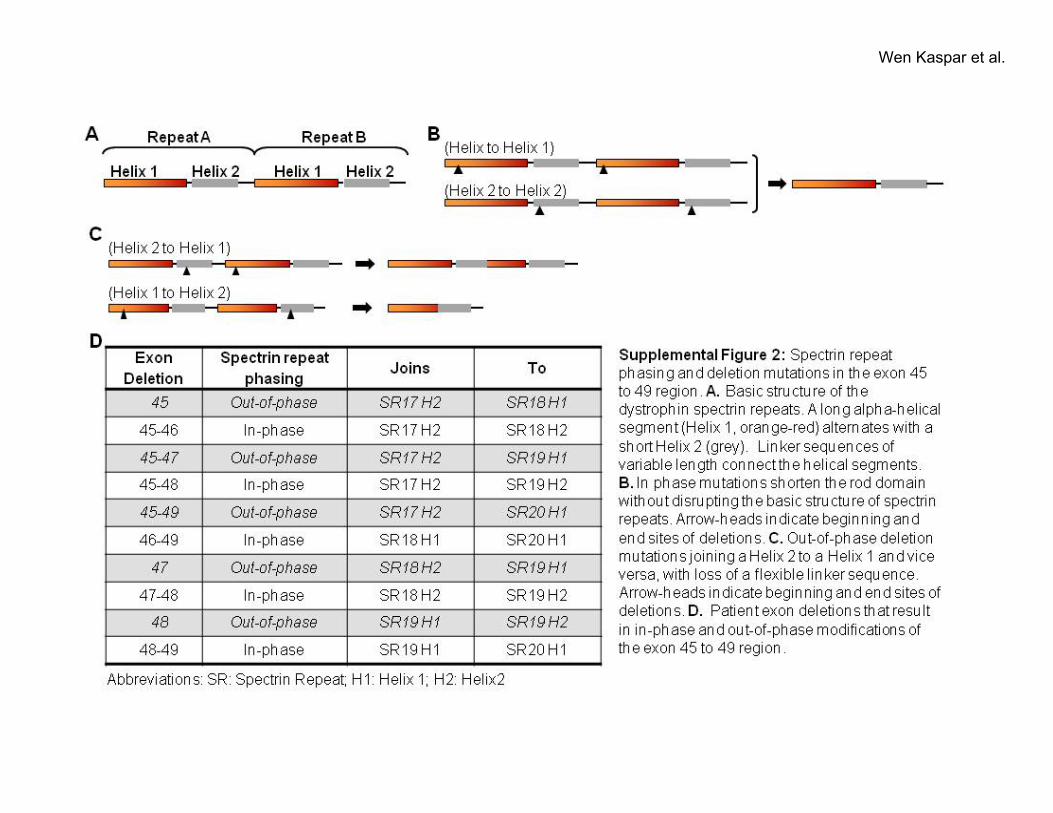
Wen Kaspar et al.
Page 30 of 30





























