Embed Size (px)
Citation preview

1
Organometallic Chemistry of Transition
Metal – Group 13 Complexes and
Metal-Organic Precursor Synthesis for
ALD Cobalt Oxide Thin Films
Cumulative Dissertation
Jiyeon Kim

Part I
Organometallic Synthesis of Transition
Metal – Group 13 Complexes
Part II
Precursor Synthesis and Atomic Layer
Deposition of Cobalt Oxide
Dissertation
Faculty of Chemistry and Biochemistry
Ruhr-University Bochum
Submitted by
Jiyeon Kim, M.Sc.
February 2017

This work has been performed in the time between May 2013 and January 2017 under
supervision of Prof. Roland A. Fischer and Prof. Anjana Devi at the Chair of Inorganic
Chemistry II – Organomatallics & Materials Chemistry of Ruhr-University Bochum, Germany.
1st referee: Prof. Dr. Roland A. Fischer
2nd referee: Prof. Dr. Anjana Devi
Herein I declare that I have written the corresponding cumulative thesis myself independently
without any other help or source which are not specifically and explicitly marked or authorized
in this dissertation. Furthermore, I declare that I have not applied to any other review procedures
or submitted this cumulative thesis in the same, similar or different form to another faculty or
university as a dissertation.
Jiyeon Kim, February 2017

Acknowledgement
I am deeply grateful to many people who supported me with every meaningful way to manage
this work. Amongst, I sincerely appreciate my supervisor Prof. Roland A. Fischer for his
support and generosity for giving the great opportunity to be at AC II. With no doubt, it was a
dream to work in organometallics regardless of the frustrating outcome.
I wish to express my gratitude to Prof. Anjana Devi who is the supervisor of the ALD part of
my thesis. I appreciate your efforts for giving me guidance from totally different field to another.
I was given the inspiration as well as the motivation of learning about materials science in very
productive environment.
I wish to thank to Christian Gemel for his supervision also scientific support.
I am thankful to Prof. Markku Leskelä from University of Helsinki in Finland for providing me
the opportunity to learn atomic layer deposition from one of the pioneering group in the ALD
field.
I would like to thank Dr, Mariusz Molon, Dr. Arik Puls, Hung Banh who provided me a
guidance to establish and settle down at OM group at the beginning.

I am thankful to Dr. Andreas Schneemann, Suttipong Wannapaiboon, Dr. Clarissa Kroll, Dr.
Sunja Kim for welcoming me and giving the introduction of Ruhrgebiet at the beginning.
I would like to thank Dr. Markus Halbherr, Dr. Arik Puls, Dr. Mariusz Molon, Dr. Kerstin
Freitag, Hung Banh, Jana Weßing and Manuela Winter for the introduction and guidance of
single crystal X-ray diffraction and synthetic skills in the lab.
I am very thankful to Tomi Iivonen from University of Helsinki in Finland who offered me an
excellent supervision and fun while learning the ALD process including the maintenance of F-
120.
I would like to thank the OM group and the CVD group for the scientific support and advise as
well as to the whole ACII including the former members for providing me the great and
comfortable working environment. Especially, thanks to all the office member at NC 2/27 who
always brought interesting conversation topic to drag me out of work.
I would like to thank Prof. Radim Beranek for successfully instructing the 4G Photocat EU
project as a leader and organizing the interesting meetings in different countries which allowed
me to broaden my knowledge on different field. Also, his contribution of photoelectrochemical
catalysis expertise on my work.
I would like to thank the New York and Boston crew, Dr. Christian Wiktor, Dr. Andreas
Schneemann, Dr. Wenhua Zhang, Jana Weßing, Inke Hante who joined the ACS fall meeting

on 2015, especially Wenhua Zhang and Inke Hante for having girls’ night out in Boston and
Jana Weßing and Andreas Schneemann for having together a wonderful time during the round
trip in the US and Canada afterwards.
I thank Dr. Clarissa Kroll who joined me to the ICOMC 2014 in Sapporo, Japan and during the
travel to Tokyo and Kyoto afterwards.
I would like to thank Dr. Mariusz Molon, Dr. Arik Benjamin Puls and Hung Banh for their
guidance at the beginning to help me settle down in the group and learn quickly to handle the
air sensitive reactions.
I would like to thank Inke Hante or Schwedler for her friendship and having an awesome chat
in the chair as well as outside of the chair. Thanks to Jana Weßing for hanging out together at
scientific and non-scientific meetings accompanied with beer. I am thankful to Sarah Karle
having a short break together every day at 02 in front of NC. Thanks to Stefan Cwik for his
guidance in CVD as well as common humor. Thanks to Alexander Sadlo for introducing
Bouldering which I still cannot manage to do well but it was fun. I would like to thank all the
CVD group for their help and welcoming me as a CVD group.
I would like to thank Dr. Andreas Schneemann and Richard O’Donoghue, Dr. Steven Hughes,
Kathryn Poole for generously proof reading my English during my study.
I would like to thank Sabine Pankau who is a wonderful colleague always being supportive
with non-scientific issues even though it was not her task.

I would like to thank Niklas Stegmann for his assistance on the synthesis in the lab during the
in-depth practical.
I express my special thanks to my friends Meike Stiepel and Christian Groß and Andreas
Schneemann for their friendship and spending plenty of time together that I could avoid feeling
home sick. The chemistry what we did together was the most fun during my PhD while drinking
and brewing Panda Gom Pale Ale.
Finally, I wish to thank from the bottom of my heart to Dr. Andreas Schneemann for his
unconditional love and support and made it possible for in many ways.

“I am one of those who think, like Nobel, that humanity will draw more good than evil from
new discoveries.”
Marie Curie

Contents
Part 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
1 Motivation .......................................................................................................................................1
List of Abbreviation .............................................................................................................................2
2 Introduction .....................................................................................................................................3
2.1 Organometallic Chemistry .........................................................................................................3
2.2 Historic Development and Significance of Organometallic Chemistry.........................................3
2.3 Discovery of the Low-Valent Group 13 Organyls and Chronological Development of Their
Synthesis ....................................................................................................................................... 10
2.4 Coordination chemistry of ECp* toward transition metals ....................................................... 12
2.5 Redox Chemistry of Transition Metal Group 13 Complexes...................................................... 16
3. References ................................................................................................................................ 19
3 Oxidation of Ni-ECp* Complexes: Stable Open-Shell NiI Cations [Ni(ECp*)n(PPh3)4−n]+ (n = 2, 4; E = Al,
Ga).................................................................................................................................................... 24
3.1 Abstract ...................................................................................................................................... 24
3.2 Introduction ............................................................................................................................ 24
3.3 Result and Discussion .............................................................................................................. 26
3.4 Conclusion .............................................................................................................................. 29
3.6 Experimental Section .................................................................................................................. 33
3.6.1 General Comments............................................................................................................... 33
3.6.2 Synthetic Procedures ............................................................................................................ 34
3.6.3 Spectroscopic and crystallographic data ............................................................................... 36
4 References ................................................................................................................................. 47
4. Organoaluminum Palladium Complex via Reductive Elimination ................................................... 49
4.1 Abstract .................................................................................................................................. 49
4.2 Introduction ............................................................................................................................ 49
4.3 Result and Discussion .............................................................................................................. 50
4.4 Conclusion .............................................................................................................................. 53
4.5 Experimental Section............................................................................................................... 55
4.5.1 General Comments ........................................................................................................... 55
4.5.2 Synthetic procedure ......................................................................................................... 55
4.5.3 Spectroscopic, Spectrometric, Crystallographic Data......................................................... 56
5. References ................................................................................................................................ 62

List of Abbreviation ........................................................................................................................... 64
5 General Introduction ...................................................................................................................... 67
5.1 General Introduction ............................................................................................................... 67
Physical Vapor Deposition ......................................................................................................... 68
Chemical Vapor Deposition ....................................................................................................... 68
5.2 Atomic Layer Deposition ......................................................................................................... 69
5.3 ALD of metal oxides thin films ................................................................................................. 72
5.3.1 Precursors ........................................................................................................................ 72
5.3.2 Transition metal oxides and cobalt ................................................................................... 80
6. References ................................................................................................................................ 82
6. Low temperature atomic layer deposition of cobalt oxide as an effective catalyst layer for
photoelectrochemical water splitting devices ................................................................................... 88
6.1 Abstract .................................................................................................................................. 88
6.2 Introduction ............................................................................................................................ 89
6.3 Results and Discussion............................................................................................................. 91
6.3.3 Film Characterization............................................................................................................ 95
6.4 Conclusion and Outlook ........................................................................................................ 102
6.5 Appindex ............................................................................................................................... 102
6.5 Experimental Section............................................................................................................. 107
6.5.1 Precursor synthesis......................................................................................................... 108
6.5.2 Film deposition ............................................................................................................... 109
6.5.3 Film characterization ...................................................................................................... 109
6.5.4 Photoelectrochemistry ................................................................................................... 111
6.6 References ............................................................................................................................ 112
7. Homoleptic all nitrogen coordinated bis-guanidinato cobalt (II) complex: Volatile and thermally
stable precursors suitable for ALD/CVD ........................................................................................... 136
7.1 Abstract ................................................................................................................................ 136
7.2 Introduction .......................................................................................................................... 136
7.3 Result and Discussion ............................................................................................................ 139
7.4 Conclusion ............................................................................................................................ 148
7.5 Experimental Section............................................................................................................. 150
7.5.1 General Comments ......................................................................................................... 150
7.5.2 Synthetic Procedure ....................................................................................................... 150
7.5.3 Crystallographic data .......................................................................................................... 152
8. References .............................................................................................................................. 153
Curriculum Vitae ............................................................................................................................. 155

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
1
1 Motivation
This cumulative thesis consists of two parts whereas Part 1 deals with the synthesis and characterization
of novel organometallic complexes. On the other hand, Part 2 describes the synthesis of new cobalt
precursors for the use in thin film deposition techniques (Part 2 chapter XY) as well as the development
of a ALD process for the preparation of efficient cobalt-oxide thin film photocatalysts.
The driving force for pursuing the first project based on pure organometallic chemistry, was the
preparation of new organometallic complexes with unusual bonding situation and structural features. In
Part 1 chapter 4 the use of reductive elimination as a tool for the preparation of new building blocks and
precursors for metal-rich clusters is explored. These clusters bear potential for a wide range of
applications including catalysis and medical application. In chapter 3 of Part 1 the focus lays on the
preparation of stable open shell NiI species, which are extremely rare and the exploration of these
compounds and in particular the documentation of reliable synthesis conditions can be of extreme
usefulness for future studies.
The second part of the thesis is much closer to application and was embedded in the Photocat-4G
Network. The overall goal of this project was the preparation of thin film devices useful for the
photocatalytic degradation of pollutants in water. As part of my thesis, new precursors were developed
for this project and in close collaboration with the University of Helsinki, we worked on new atomic
layer deposition processes for the preparation of spinel-cobaltoxide thin films, which is are promising
photocatalytic materials.
My role within this project was on the one hand the preparation of known (commercially unavailable)
and novel precursor molecules for the thin film depositions, and additionally, I was during two academic
stays at the university of Helsinki deeply involved in the film depositions as well as characterizations.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
1
PART 1 Organometallic Synthesis of Transition Metal –
Group 13 Complexes

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
2
List of Abbreviation
BAr4F B[C6H3(CF3)2]4
DDP 2-Diisopropylphenylamino-4-diisopropylphenylimino-2-pentene
dvds 1,3-Divinyl-1,1,3,3-tetramethyldisiloxane
COD 1,5-Cyclooctadiene
COT Cyclooctatetraene
Cp Cyclopentadienylanion
CpH 1,3-Cyclopentadiene
Cp* Pentamethylcyclopentadienylanion
Cp*H 1,3-Pentamethylcyclopentadiene
E Group 13 metals
EA Elemental Analysis
Et Ethyl
EIR Monovalent group 13 organyls
Et2O Diethylether
iPr iso-propyl
LIFDI Liquid Injection Field Desorption Ionization
Me Methyl
NHC N-heteocyclic-carbene
Ph Phenyl
Rt Room temperature
TM Transition metals
tBu tert-butyl
tmeda N,N,N’,N’-Tetramethylethylen-1,2-diamine

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
3
2 Introduction
2.1 Organometallic Chemistry
Organometallic Chemistry describes the chemistry of compounds, which contain at least one
direct carbon to metal bond. This includes all transition metals, earth alkaline and alkaline as
well as lanthanides, actinides, post transition metals and metalloids. The field of organometallic
chemistry has a huge impact on the day-to-day life of millions of people, since organometallic
complexes are used for a wide range of large-scale homogeneously catalyzed reactions as well
as precursors for the preparation of thin films in the semiconductor industry. The influence of
this field was honoured with several Nobel Prizes in chemistry. For instance, for the discovery
of the Grignard Reagent and Metal Catalyzed Hydrogenation for Victor Grignard and Paul
Sabatier in 1912 and the discovery of the Metallocenes by E.O. Fischer and G. Wilkinson in
1973. Also, several Nobel Prizes were awarded for the development of groundbreaking
catalysts based on organometallic compounds (e.g. 1963 for Ziegler-Natta Catalysis, 2001 for
assymetric hydrogenation, 2005 for metal-catalyzed metathesis and 2010 for palladium
catalyzed cross-couplings).
2.2 Historic Development and Significance of Organometallic Chemistry
Figure 2.1: Depiction of the synthesis of one of the first organometallic compounds cacodyl by
Louis Claude Cadet de Gassicourt in 1760.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
4
As in many cases in the history of new fields in chemistry, the discovery of organometallic
compounds was a mere serendipity. The first reported man-made organometallic compound is
most likely (As2(CH3)4), or Cacodyl, which was a component of “Cadet’s Fuming Liquid”. This
liquid was prepared in 1760 by Louis Claude Cadet de Gassicourt by the reaction of AsO3 and
potassium acetate. A side product of this reaction is cacodyl oxide. The presence of methyl
radicals, who are bound to the Arsen was later postulated by Robert Bunsen.
Figure 2.2: Schematic of the preparation of Zeise Salt by the reaction of K2PtCl4 in boiling
ethanol.
In 1827 William Zeise discovered the first Olefin Transition Metal-Complex, the so called Zeise
Salt which has the formuly [K[PtCl3(C2H4)]·H2O. This compound was prepared by reacting
PtCl4 in boiling ethanol. In this comlex the platinum is fourfold coordinated, with each chlorine
taking up one coordination site and the C2H4 coordinating to the fourth site with a η2 bond.
However, the real composition and structure remained for a long time under controversial
discussion. The chemist at that time very influential chemist Justus Liebig highly criticized the
assumption by Zeisse that Ethylene is bound within the structure. The ultimate proof for the
presence of Ethylene in this structure was finally given by the chemist Birnbaum in 1868, who
prepared the same compound using Ethylene gas. During the second half of the 19th century the

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
5
constitution of this compound remained uncertain and was finally resolved in the 1920 with the
development of X-ray diffraction techniques.
Figure 2.3: Depiction of the synthetic route of Edward Frankland towards diethylzinc.
Edward Frankland discovered another milestone compound of historical significance, the very
pyrophoric compound diethyl zinc (Zn(Et)2). It was prepared by the reaction of zinc with
ethyliodide. Due to its reactive nature it has become a very important reactant in organic
chemistry.
Ludwig Mond discovered the first carbonyl compounds in 1890. Ni(CO)4 is an intermediate in
the so called Mond process, which is used for the preparation of highly pure Nickel.
Nickeloxide, NiO reacts with Syngas (H2 and CO), forming impure Ni and H2O. At
temperatures of roughly 50-60 °C the NiCO4 is formed from the prepared impure Nickel.
NiCO4 is a highly volatile gaseous compound and can be deposited as ultrathin pure Nickel
films on substrates or pallets at around 220 °C. Among the carbonyl preparation reactions, this
is the one occurring under the mildest conditions.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
6
Figure 2.4: Illustration of the three steps occuring during the Mond process for the preparation
of ultra-pure Nickel
The chemistry of carbonyls was promoted later on by the likes of Walter Hieber and Walter
Reppe. Walter Hieber did a lot of groundbreaking research at the Technical University Munich
since the 1920s and developed among other things the first routes towards Metal Carbonyl
Hydrides, for instance Fe(CO)4H2 or Mn(CO)H5. These hydrides, particularly Co(CO)4H and
Rh(CO)4H are still today important intermediates of the catalysts for the hydroformulation,
which is used for the preparation of Aldehydes from Olefins (Figure 5). Walter Repp on the
other hand worked for the BASF (Badische Soda und Anillin Fabrik) and developed a manifold
of catalytic processes using Ni(CO)4 or Co(CO)4 as catalysts for the hydrocarboxylation of
Olefins and Alkynes using CO as a reagent. The carbonyl chemistry however is not only limited
to large scale industrial reactions, also in nature, so far three active centers of enzymes have
been identified to be metal-carbonyls.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
7
Figure 2.5: Scheme of the reaction process of hydroformulation using one of the metal carbonyl
hydrides discovered by Walter Hieber.
A further class of important organometallic compounds are the so called sandwhich complexes.
This class of molecules features usually two arenes bound to a metal center on opposite sites
by a haptic covalent bond. The most famous subclass of these materials are the metallocenes,
which feature two cyclopentadienyl units bound to a metal center. The first metallocene
reported was Ferrocene, which was independently discovered by Kealy and Pauson, as well as
Miller et al. in 1951. Kealy and Pauson were actually trying to prepare Fulvalene from a
cylopentadienyl salt using anhydrous ironchloride as an electron acceptor. The reaction of
Miller et al. featured Cyclopentadiene, iron as well as the oxides of either aluminum, potassium
or molybdenum. Both reactions yielded a product with the sum formula C10H10Fe.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
8
Figure 2.6: Depiction of the two different synthetic pathways of Pauson and Kealey (a) and
Miller (b) towards Ferrocene.
In the following year Fischer and Wilkinson determined the structure of the compound and
concentrated on the further development of the metallocenes. Their works demonstrated that
the electrons located on the cyclopentadienyl ring equally contributed to the bonding of the
metal-center between the two aromatic rings, leading to a η5 bond. Fischer and coworkers
furthermore developed analogues compounds for Nickel and Cobalt. Metallocenes and their
derivatives are of significance for polymerization reactions, and are used as Ziegler Natta
Catalysts.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
9
Figure 2.7: Depiction of the Singlet-Fischer-Carbenes as well as the Triplet-Schrock-Carbenes
including an example for both classes.
Apart from his research on metallocenes, E.O. Fischer also helped establishing the class of
metal-carbenes, with R. Schrock being another important figure in this field of organometallic
chemistry. In the honor of these two scientists two classes of metallcarbenes are named Fischer
and Schrock Carbenes. The Fischer Carbenes feature high oxidation state metal-centers, usually
use middle and late transition metals and are π-acceptor ligands, while the Schrock Carbenes
consist of low oxidation state metal centers of early transition metals and are usually π-donor
ligands. Apart from these two classes, N-heterocyclic carbenes (NHC) are much more
commonly used, since they are purely spectator ligands, that do not influence the electronic
structure of the metal as heavily as the afore mentioned Fischer and Schrock Carbenes. These
NHC Ligands are of high relevance, in the research towards new catalysts and have also been
established in the chemical industry, e.g. as a part of Grubbs catalysts used for the olefin
methathesis (Figure 8).

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
10
Figure 2.8: Depiction of the first and second generation Grubbs Catalysts, which both employ
carbenes in their structure.
2.3 Discovery of the Low-Valent Group 13 Organyls and Chronological
Development of Their Synthesis
The exotic chemistry of low valent group 13 organyls EIR (E = Al, Ga, In, Tl) has caught
enormous attention by the organometallic community due to their stability and availability as
ligands in coordination chemistry. The first synthesized low valent group 13 compounds were
InCp and TlCp reported already in 1957 by E. O. Fischer and H. Meister.1-2 Since the synthetic
route of those heavier element containing EIR group 13 organyls is not feasible for the lighter
elements such as aluminum and gallium, the syntheses of AlIR and GaIR were published 34
years later. Schnöckel et al. successfully synthesized [AlCp*]4 in 1991. Reaction of AlCl and
[MgCp2] led to tetrameric [AlCp*]4.3 Two years later in 1993, Schnöckel et al. achieved to
synthesized the related gallium (I) organyls including [GaCp*]6 and its Cp derivatives using the
same synthetic pathway.4 (Scheme 1)

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
11
Scheme 2.1 Synthesis of ECp*
Thereafter, Jutzi et al. reported an alternative synthetic method which provided more
convenient access to EIR.5-6 The former methods required metastable AlCl and GaCl generated
at harsh conditions e.g. heating to 1000 °C, while the routes provided by Jutzi comprises
reductive elimination of salt in Cp*AlCl2 and Cp*GaI2 employing elemental potassium.
(Scheme 2)
Scheme 2.2 Synthesis of ECp*
Nowadays the most commonly used synthetic method to prepare GaCp* is using metal stable
GaI generated by ultra-sonication in benzene which is a modified procedure of a report by Green
et al. who synthesized in 1990 a different, unrelated GaI species.7 (Scheme 3)
Scheme 2.3 Synthesis of GaCp* (most commomly adopted procedure)

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
12
In 2012, Ganesamoorthy et al. developed a new large scale synthetic route towards AlCp* via
thermally induced reductive elimination of Cp*H from the organo-aluminum hydride species
Cp*2AlH in an exceptionaly high yield (93 %).8 The reaction of AlCl3 with LiAlH4 in 3:1 molar
ratio leads to Cl2AlH which reacts further with KCp* in Et2O to afford Cp*2AlH. Cp*2AlH
reductively eliminates Cp*H in toluene at 110 °C. (Scheme 4)
Scheme 2.4 Synthesis of AlCp* (improved procedure for large scale synthesis)
Due to the stability as well as the isolobal structure to CO or phosphines, EIR species are
interesting ligands for transition metals in coordination chemistry. This topic will be discussed
more in detail in the next section. (See 2.4)
2.4 Coordination chemistry of ECp* toward transition metals
EIR species are an isolobal analogs of CO or phosphine, they possess two vacant p orbitals
perpendicular E−R axis and one lone pair electron in the orbital. (Figure 9)

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
13
Figure 2.9 Isolobality of EIR species and CO
Therefore, the interaction between the EIR species and transition metals follows as shown in
Figure 2.
Figure 2.10 Bonding situation in transiton metal-EIR complexes.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
14
The synthetic strategy of ECp* transition metal compounds is frequently found in the form of
ligand substitution reactions with labile ligands. The first attempt for synthesizing TM-EIR
complexes was performed using homoleptic TM−carbonyls. However, the strong -donating
properties of ECp* increase the -backdonating properties of carbonyls which prohibits the
further substitution of ligands. Accordingly, labile ligands with weaker -accepting properties
such as olefins were adopted to access homoleptic ECp*−TM complexes. (Scheme 5) In the
case of palladium, the redox reaction takes place at the palladium center transforming one
equivalent of GaICp* to Me2GaIIICp* as a byproduct.
Scheme 2.5 Synthesis of homoleptic TM−ECp* complexes
The homoleptic [M(ECp*)4] complexes are used as molecular building blocks for multi-nuclear
clusters. T. Steinke et al. synthesized homoleptic dimeric clusters including [Pt2(2-
AlCp*)(AlCp*)2] and [Pd2(2-GaCp*)(PPh3)2] by treatment of [M(GaCp*)4] with [M(cod)2]
or [Pd(dvds)3] (cod = 1,5-Cyclooctadiene, dvds = 1,3-Divinyl-1,1,3,3-tetramethyldisiloxane)
and subsequent addition of ECp*.9 (Scheme 6) Heteroleptic dimeric clusters are accessed by
the treatment of [Pd2(dvds)3] or [Pt(cod)2] by 3.0 equiv. of GaCp* and 2.0 equiv. of PMe3.
(Scheme 7)

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
15
Scheme 2.6 [M(GaCp*)4] as building block for dimeric clusters of the type [MPt(GaCp*)2(-
GaCp*)3].9 Reprinted with permission from Organometallics, 2003, 22 (13), pp 2705–2710.
Copyright (2003) American Chemical Society.
Scheme 2.7 Synthesis of heteroleptic dimeric clusters of the type [M2(GaCp*)3(PMe3)2].10
Reprinted with permission from Dalton Trans., 2014, 43, 3114–3120. Copyright (2014) Royal
Society of Chemistry.
In a similar manner, trimeric homoleptic clusters can be synthesized by controlling the
stoichiometry of ECp*.10-11
Recently, it has been discovered a novel pathway to approach heteroloptic dimeric cluster via
reductive elimination unlike the conventional method by substitution of labile ligand with ECp*.
This will be discussed in chapter 4 as a major discussion of this thesis.
The ECp* containing transition metal complexes are used as well in synthesis of metal-rich
complexes. M. Molon et al synthesized organozinc-rich palladium and platinum clusters.10
(Scheme 8)

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
16
Treatment of 6.0 equivalents of ZnMe2 and excess of PMe3 with [M(2-GaCp*)3(PMe3)] (M =
Pd or Pt) leads to mononuclear compounds [M(ZnCp*)2(ZnMe2)(PMe3)2].
Scheme 2.8 Synthesis of zinc rich transition metal clusters.10 Reprinted with permission from
Dalton Trans., 2014, 43, 3114–3120. Copyright (2014) Royal Society of Chemistry.
2.5 Redox Chemistry of Transition Metal Group 13 Complexes
The reaction of cationic complexes such as [Ga2Cp*][BAr4F] and [FeCp2][BAr4
F] with ECp*
containing transition metal complexes gives further insight into the redox chemistry of ECp*
species. Treatment of homoleptic complex [M(GaCp*)4] with one equivalent of
[Ga2Cp*][BAr4F] or [FeCp2][BAr4
F] led to the product [MGa(GaCp*)4][BAr4F] (M = Ni or Pt)
whereas 1/3 equiv. of [FeCp2][BAr4F] yielded [(2-Ga)Pt3(GaCp*)6][BAr4
F].12-13

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
17
Scheme 2.9 Reaction of [M(GaCp*)4] (M = Ni or Pt)with [FeCp2][BAr4F] leading to an
oxidative cleavage of Cp* to form “naked” Ga+ containing complexes.[Buchin 2006, Halbherr
2010] Reprinted with permission from Angew. Chem. Int. Ed. 2006, 45, 5207 –5210 and Angew.
Chem. Int. Ed. 2010, 49, 1878 –1881 Copyright (2006, 2010) Wiley.
Reaction behavior of heteroleptic complex of the type [Ni(ECp*)2(PPh3)2] with [FeCp2][BAr4F]
will be discussed as a major topic of this thesis in chapter 3 in detail.
Aforementioned new synthetic route from aluminum (III) hydride species to AlICp* developed
by C. Ganesamoorthy et. al.8 is an inspiration of redox chemistry of group 13 complexes. Indeed,
hydrides of main group metals are playing a key role in organic synthesis as Lewis acid
catalysis14-21 and hydrogen storage materials22-23 as well as in material chemistry due to its
volatility in chemical vapor deposition techniques 24-27 Moreover, the comprehensive
elucidation of chemical reactivity of hydrides in terms of addition, elimination and redox
reactions has been a goal for inorganic chemical society.28-31 Previous studies of main group
hydride compounds show the unusual stabilization of aluminum with +1 oxidation state via

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
18
reductive dehalogenation from the aluminum halide species5, 28, 30, 32 and ECp* is remains
arguably the excellent example of those studies due to their exotic chemistry and strong
donating and accepting properties towards transition metal complexes.6
C. Ganesamoorthy et al reported a novel pathway to AlICp* via reductive elimination of Cp*H
from Cp*2AlH while Cp*AlH2 reductively eliminates Cp*H and forms metallic Al0. Both
results provided very important information in terms of not only the final product but also the
type of chemical bonding. Cp*2AlH and Cp*AlH2 lead to the formation of metal-metal bonds
where the aluminum metal is clearly containg metal-metal bonds as well as AlCp* known as
tetrameric complex [AlCp*]4.8 This work was an inspiration of preparing intermetallic
complexes including bonds between transition metals and group 13 metals via intramolecular
reductive elimination. The reaction with aluminum hydride towards transition metal complexes
leading to the formation of TM-Al will be discussed in detail in chapter 4.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
19
3. References
1. Fischer, E. O.; Hofmann, H. P., Metall-cyclopentadienyle des Indiums. Angew. Chem.
1957, 69, 639-640.
2. Meister, H., Cyclopentadienyl-thallium. Angew. Chem. 1957, 69, 533-534.
3. Dohmeier, C.; Robl, C.; Tacke, M.; Schnöckel, H., The Tetrameric Aluminum(I)
Compound [{Al(η5-C5Me5)}4]. Angew. Chem., Int. Ed. 1991, 30, 564-565.
4. Loos, D.; Schnöckel, H., (Cyclopentadienyl)-Gallium(I)-Verbindungen. J. Organomet.
Chem. 1993, 463, 37-40.
5. Schulz, S.; Roesky, H. W.; Koch, H. J.; Sheldrick, G. M.; Stalke, D.; Kuhn, A., A Simple
Synthesis of [(Cp*Al)4] and Its Conversion to the Heterocubanes [(Cp*AlSe)4] and
[(Cp*AlTe)4] (Cp* = η5-C5(CH3)5). Angew. Chem., Int. Ed. 1993, 32, 1729-1731.
6. Jutzi, P.; Neumann, B.; Reumann, G.; Schebaum, L. O.; Stammler, H.-G.,
Pentamethylcyclopentadienylindium (Cp*In) as Terminal Ligand in the Chemistry of
Chromium. Organometallics 1999, 18, 2550-2552.
7. Green, M. L. H.; Mountford, P.; Smout, G. J.; Speel, S. R., New synthetic pathways into
the organometallic chemistry of gallium. Polyhedron 1990, 9, 2763-2765.
8. Ganesamoorthy, C.; Loerke, S.; Gemel, C.; Jerabek, P.; Winter, M.; Frenking, G.;
Fischer, R. A., Reductive elimination: a pathway to low-valent aluminium species. Chem.
Commun. 2013, 49, 2858-2860.
9. Gemel, C.; Steinke, T.; Weiss, D.; Cokoja, M.; Winter, M.; Fischer, R. A., [M(GaCp*)4]
(M = Pd, Pt) as Building Blocks for Dimeric Homoleptic Cluster Compounds of the Type
[MPt(GaCp*)5]. Organometallics 2003, 22, 2705-2710.
10. Molon, M.; Gemel, C.; Fischer, R. A., Organogallium- and organozinc-rich palladium
and platinum clusters. Dalton Trans. 2014, 43, 3114-3120.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
20
11. Steinke, T.; Gemel, C.; Winter, M.; Fischer, R. A., The Clusters [Ma(ECp*)b] (M=Pd,
Pt; E=Al, Ga, In): Structures, Fluxionality, and Ligand Exchange Reactions. Chem. Eur. J.
2005, 11, 1636-1646.
12. Buchin, B.; Gemel, C.; Cadenbach, T.; Fernández, I.; Frenking, G.; Fischer, R. A.,
“Naked” Ga+ and In+ as Pure Acceptor Ligands: Structure and Bonding of
[GaPt(GaCp*)4][BArF]. Angew. Chem., Int. Ed. 2006, 45, 5207-5210.
13. Halbherr, M.; Bollermann, T.; Gemel, C.; Fischer, R. A., Selective Oxidative Cleavage
of Cp* from Coordinated GaCp*: Naked Ga+ in [GaNi(GaCp*)4]+ and [(μ2-
Ga)nM3(GaCp*)6]n+ Angew. Chem., Int. Ed. 2010, 49, 1878-1881.
14. Bulychev, B. M., A new stage in the development of transition metal alumohydrides.
Polyhedron 1990, 9, 387-408.
15. Duchateau, R.; Meetsma, A.; Teuben, J. H., Sterically crowded monomeric neutral
bis(benzamidinato) compounds of aluminium, [PhC(NSiMe3)2]2AlX (X = Cl, H); X-ray
crystal structure of [PhC(NSiMe3)2]2AlH. Chem. Commun. 1996, 223-224.
16. Natta, G.; Pino, P.; Mazzanti, G.; Longi, P.; Bernardini, F., The Reaction between
Styrene and Triisobutylaluminum. J. Am. Chem. Soc. 1959, 81, 2561-2563.
17. Spielmann, J.; Buch, F.; Harder, S., Early Main-Group Metal Catalysts for the
Hydrogenation of Alkenes with H2. Angew. Chem., Int. Ed. 2008, 47, 9434-9438.
18. Harder, S., Molecular early main group metal hydrides: synthetic challenge, structures
and applications. Chem. Commun. 2012, 48, 11165-11177.
19. Mandal, S. K.; Roesky, H. W., Group 14 Hydrides with Low Valent Elements for
Activation of Small Molecules. Acc. Chem. Res. 2012, 45, 298-307.
20. Harder, S., Molecular early main group metal hydrides: synthetic challenge, structures
and applications. Chem. Commun. (Cambridge, U. K.) 2012, 48, 11165-11177.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
21
21. Spielmann, J.; Buch, F.; Harder, S., Early main-group metal catalysts for the
hydrogenation of alkenes with H2. Angew. Chem., Int. Ed. 2008, 47, 9434-9438.
22. Zidan, R.; Garcia-Diaz, B. L.; Fewox, C. S.; Stowe, A. C.; Gray, J. R.; Harter, A. G.,
Aluminium hydride: a reversible material for hydrogen storage. Chem. Commun. (Cambridge,
U. K.) 2009, 3717-3719.
23. Orimo, S.-i.; Nakamori, Y.; Eliseo, J. R.; Zuettel, A.; Jensen, C. M., Complex Hydrides
for Hydrogen Storage. Chem. Rev. (Washington, DC, U. S.) 2007, 107, 4111-4132.
24. Choi, H.; Hwang, S., Volatile Amidoalane Compounds for Chemical Vapor Deposition
of Aluminum. Chem. Mater. 1998, 10, 2323-2325.
25. Xenidou, T. C.; Prud'homme, N.; Vahlas, C.; Markatos, N. C.; Boudouvis, A. G.,
Reaction and Transport Interplay in Al MOCVD Investigated Through Experiments and
Computational Fluid Dynamic Analysis. J. Electrochem. Soc. 2010, 157, D633-D641.
26. Hanabusa, M.; Ikeda, M., Wavelength dependence in photochemical vapor deposition
of aluminum film using dimethylaluminum hydride. Appl. Organomet. Chem. 1991, 5, 289-93.
27. Baum, T. H.; Larson, C. E.; Jackson, R. L., Laser-induced chemical vapor deposition of
aluminum. Appl. Phys. Lett. 1989, 55, 1264-6.
28. Aldridge, S.; Downs, A. J., Hydrides of the main-group metals. New variations on an
old theme. Chem. Rev. (Washington, D. C.) 2001, 101, 3305-3365.
29. Uhl, W.; Matar, M., Hydroalumination of nitriles and isonitriles. Z. Naturforsch., B:
Chem. Sci. 2004, 59, 1214-1222.
30. Jones, C.; Rose, R. P., Synthesis and characterisation of tetramethylpiperidinyloxide
(TEMPO) complexes of group 13 metal hydrides. New J. Chem. 2007, 31, 1484-1487.
31. Dagorne, S.; Atwood, D. A., Synthesis, Characterization, and Applications of Group 13
Cationic Compounds. Chem. Rev. (Washington, DC, U. S.) 2008, 108, 4037-4071.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
22
32. Roesky, H. W., Hydroalumination reactions in organic chemistry. Aldrichimica Acta
2004, 37, 103-108.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
23
3.
Oxidation of Ni-ECp* Complexes: Stable Open-Shell NiI Cations
[Ni(ECp*)n(PPh3)4−n]+ (n = 2, 4; E = Al, Ga)
Jiyeon Kim, Markus Halbherr, Christian Gemel, and Roland A. Fischer

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
24
3 Oxidation of Ni-ECp* Complexes: Stable Open-Shell NiI Cations
[Ni(ECp*)n(PPh3)4−n]+ (n = 2, 4; E = Al, Ga)1
3.1 Abstract
Uncommon NiI cationic complexes were synthesized by treating [Ni(ECp*)2(PPh3)2] (E = Al
or Ga, Cp* = pentamethylcyclopentadienyl) with one equivalent of [FeCp2][BAr4F]. All
compounds have been prepared readily in a high yield. The paramagnetic compounds were
characterized by single crystal X-ray crystallography, Mass spectroscopy, elemental analysis,
magnetic susceptibility and EPR spectroscopy.
3.2 Introduction
During the past two decades the coordination chemistry of low-valent EIR (E = Al, Ga) ligands
to transition metals has been thoroughly investigated. In the case of Cp*, so far every reported
monomeric homoleptic [M(Ecp*)] complex is a saturated species containing 18 valence
electrons (VE). This feature is independent of the used transition metal M. However, for GaR
species containing bulkier R groups, i.e. β-diketiminates (e.g. Ga(ddp) with ddp =
CH(CMeNC6H3(C6H3Pri2-2,6))2) or guanidinates (e.g. Ga(guan) with guan = (C6H3Pri
2-
2,6)NC(NCy2)N(C6H3Pri2-2,6) unsaturated species including [(1,3-cod)Pt(Ga(ddp))2]
4 (cod =
cyclooctadiene) or [Pt(Ga(guan))3] have been reported. The very strong and polar M-E bond
is responsible for the reactivity of these class of mixed-metal complexes. In these compounds
1 The results presented in this chapter have been published in a peer reviewed journal. Reprinted
with permission from Inorg. Chem., 2015, 54 (20), pp 9675–9677. Copyright (2015) American
Chemical Society.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
25
the ligand dissociation is prevented by the close vicinity of the nucleophilic M and electrophilic
E. Strong and weakly polar bond such as C-H, Si-H or C-C bonds were activated by compounds
with this interesting electronic settings. To further elucidate this correlation, recent studies
deployed by our group focused on investigating the behavior of [M(ECp*)n] complexes towards
strong one-electron oxidizing agents, for example ferrocenium salts. Suprisingly, neither for
[M(GaCp*)4] (M = Ni, Pt) nor for [Pd3(GaCp*)8] a reaction on the metal-site was observed but
on the organic ligand sphere, leading to the removal of decamethylfulvalene as a dimerization
product of the cp* radical. The reaction products were [Ni(GaCp*)4(Ga)]+ as well as the
triangular clusters [M3(GaCp*)6(Ga)n]n+ (M = Pd, Pt; n = 1, 2), which contained “naked” Ga+
ions as terminal or bridging ligands, respectively. Whithin this chapter, the oxidation of
[Ni(ECp*)n(PPh3)(4−n)] complexes (E = Al, Ga; n = 2, 4) by [FeCp2][OTf] and [FeCp2][BAr4F]
(OTf = trifluoromethanesulfonate; Cp = cyclpentadienyl; BAr4F = B[3,5-(CF3)2C6H3]4) will be
discussed. Interestingly, [Ni(ECp*)n(PPh3)(4−n)]+ cations are formed by oxidation of the
transition-metal center, which reveal the oxidation state NiI. Outstaningly, these products are
the first mixed d-block metal/main-group metal open-shell complexes ever reported and
represent new examples for only a handful known organometallic monomeric open-shell d9
complexes.8
In general the oxidation state 1+ for nickel is uncommon, however, in fact, a few stable
compounds have been reported in the literature. For example the tetraazamacrocycle [Ni(tmc)]+
(tmc = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) is an important model for
enzymatic processes e.g. Acetyl-CoA synthesis. This structure is also related to the methyl
coenzyme M Reductase F430 which generates in metabolic reactions in the Achea methane
from thiomethyl compounds. Other stable NiI complexes include [NiCl(NHC)L] [NHC = 1,3-
bis(2,6-diisopropylphenyl)imidazol-2-ylidene; L = NHC, PPh3], which is used for the catalysis
of cross-coupling and amination reactions.10 A number of low coordinate and neutral NiI

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
26
complexes were investigated by Laskowski and Hillhouse In the recent literature NiI complexes
which were supported by NHC and Cp ligands have been explored. Furthermore, a small
number of cationic compounds have been discovered, such as the tricoordinated and
tetracordinated complexes [NiI(PPh3)3][BF4]13 and [NiI(PMe3)4][BPh4], respectively.14 For the
latter compound a disproportion into Ni0 and Ni was observed, as it is not stable in solution.
3.3 Result and Discussion
The oxidation the Ni(0) centers to Ni(1) is achieved by the addition of one equivalent of
[FeCp2][BAr4F] in fluorobenzene at room temperature to the complexes [Ni(ECp*)2(L)2] (L =
AlCp*, PPh3; E = Al, Ga). The E(I) ligands and the Cp* remain unchanged during this reaction.
Isolation of the complexes [NiI(AlCp*)4][Cp*Al(OTf)3] (1), [NiI(AlCp*)2(PPh3)2]-[BAr4F] (2)
and [NiI(GaCp*)2(PPh3)2][BAr4F] (3) was possible. Noticeably 1 is thermally highly unstable
and decomposes during workup, hence only few crystals were isolated and were used for
characterization. On the other hand, compounds 2 and 3 were stable at inert conditions at room
temperature either in the solid state as well as in solution.
A quite suprising result is the finding that the trasition metal center is oxidized instead of the
ECp* ligand. In particular, because for the analogous compound [Ni(GaCp*)4] the oxidation
Scheme 3.3.1 Synthesis of NiI Compounds 1-3

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
27
by [FeCp2][BAr4F] resulted in the formation of [NiGa(GaCp*)4]
+ containing Ga+ moieties.
Additionally, in the case of analogous palladium and platinum complexes a similar oxidation
accompanied by the formation of decamethylfulvalene.
The compounds 1-3 are crystallized from a mixture of fluorobenzene and hexane. In Figure
3.3.1 depiction of the molecular structures of all three compounds is displayed and in Table
3.6.1 all important structural paramters are summarized. An overview of the important
interatomic distances is given in Table 3.6.2. No complete single crystal diffraction datasets of
compound 1 could be obtained, because of the high instability of this material. During the
experiment the compound slowly decomposed. However, an overall completeness of 80 % with
a final data to parameter ratio of 9:1 could be achieved and also a sufficiently good R value
leads to high confidence in the interpretation of the diffraction data. More or less strong
distortions of the tetrahedral coordination environment of the Ni center was observed in all
three compounds. Particularly obvious was this finding for the homoleptic complex 1, where
the Al-Ni-Al angles amount to 103.23(15) ° and 132.00(17) °. In the parent, neutral [Ni(AlCp*)4]
species the ideal tetrahedral angles of 109.5° are present. The Jahn-Teller distortion is held
accountable for this flattening of the structure with the prototypical [CuCl4]2- as an example.
For the similar homoleptic cation [Ni(PMe3)4]+ similar distortions were observed (104.6(1)° to
119.9(1)°). Even larger distortion have been found in the phosphine substituted complex 2 and
3 with angles between 98.07(4)° and 131.24(4)°) and 3 (L-Ni-L angles between 98.75(2)° and
134.35(3)°, respectively. This is caused most probably by the steric repulsion of the very bulky
PPh3 ligands. Notably, no indication for hydride species were found using spectroscopic
methods (NMR). Additionally, the compounds are all paramagnetic, which was confirmed by
EPR spectroscopy.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
28
The Ni-E bond distances are only slightly affected by the oxidation. The mean Ni-Al distance
amounts to 2.17 Å in [Ni(AlCp*)4]+ and is the same in the parent neutral compound
[Ni(AlCp*)4]. In the phosphine substituted species a slight increase is observed in the Ni-Al
bond upon oxidation, from 2.21 Å to 2-27 Å and from 2.25 Å to 2.32 Å in compounds 2 and 3,
respectively. Moreover, a decrease of the E-Cp*centroid distance was found, most likely because
of the increase of the E-Cp* bonds polarity. This is induced by the over-all positive charge of
the complex.
Figure 3.3.1 Molecular structure (POV-Ray Plot) of the cationic parts of the salts 1-3, namely
[Ni(AlCp*)4]+ (1), [Ni(AlCp*)2(PPh3)2]
+ (2) and [Ni(GaCp*)2(PPh3)2]+ (3) in the solid state as
determined by single crystal X-ray diffraction; displacement ellipsoids are shown at the 50%
probability level, hydrogen atoms are omitted for clarity. Selected interatomic distances and angles are
summarized in Table 3.6.2.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
29
Broad signals in the 1H-NMR spectra of compounds 2 and 3 indicate their paramagnetic nature.
Nonetheless, in the 31P-NMR unusually sharp signals were observed. Via Evans NMR method
at room temperature the magnetic susceptibility (eff) of these complexes were determined.
Values for eff of 1.09 BM and 1.15 BM were calculated in fluorobenzene (BM = Bohr
Magnetron). These values are slightly lower than the theoretically calculated values for NiI in
tetrahedral geometry (1.73 BM). These two compounds showed also EPR activity, with average
g-factors of 2.11 and 2.15 for 2 and 3 respectively. (Figure 3.6.3) These findings are in
accordance with other NiI complexes. The recorded spectra are highly complex and thus the
hyperfine couplings of the EPR signals could not be simulated. The cation peaks of these two
complexes were also revealed using liquid injection field desorption ionization mass
spectrometry (LIFDI-MS), with the cation peaks at m/z [a.u] of 906 and 993 for compounds 2
and 3, respectively.
3.4 Conclusion
Using the more electrophilic metal ligator Al, both the homoleptic [Ni(AlCp*)4] and the
phosphine containing analogue [Ni(AlCp*)2(PPh3)2] yield cationic 17 VE complexes (1 and 2).
For the metal Ligator Ga, only the heteroleptic complex [Ni(GaCp*)2(PPh3)2] is reacting in the
same fashion, yielding 3. The homoleptic [Ni(GaCp*)4] complexe yields after oxidation the
cation [Ni(GaCp*)4(Ga)]+ featuring naked Ga. This is because the cleavage of a C5Me5 radical
occurs easier on the less polar E-Cp* bond, showcased by the homoleptic GaCp* substituted
Nickel complex. Furthermore, the substitution of ECp* by PPh3 forms more electron deficient
metal centers increasing the polarity of the E-Cp* bond.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
30
3.5 References
(1) (a) Asay, M.; Jones, C.; Driess, M. Chem. Rev. 2011, 111, 354-396; (b) Molon, M.;
Bollermann, T.; Gemel, C.; Schaumann, J.; Fischer, R. A. Dalton Trans. 2011, 40, 10769-10774;
(c) Steinke, T.; Cokoja, M.; Gemel, C.; Kempter, A.; Krapp, A.; Frenking, G.; Zenneck, U.;
Fischer, R. A. Angew. Chem., Int. Ed. 2005, 44, 2943-2946; (d) Steinke, T.; Gemel, C.; Cokoja,
M.; Winter, M.; Fischer, R. A. Angew. Chem., Int. Ed. 2004, 43, 2299-2302; (e) Steinke, T.;
Gemel, C.; Winter, M.; Fischer, R. A. Chem. Eur. J. 2005, 11, 1636-1646; (f) Bollermann, T.;
Cadenbach, T.; Gemel, C.; von Hopffgarten, M.; Frenking, G.; Fischer, R. A. Chem. Eur. J.
2010, 16, 13372-13384.
(2) Hardman, N. J.; Eichler, B. E.; Power, P. P. Chem. Commun. 2000, 1991-1992.
(3) Jones, C.; Junk, P. C.; Platts, J. A.; Stasch, A. J. Am. Chem. Soc. 2006, 128, 2206-2207.
(4) Kempter, A.; Gemel, C.; Fischer, R. A. Chem. Eur. J. 2007, 13, 2990-3000.
(5) Green, S. P.; Jones, C.; Stasch, A. Inorg. Chem. 2007, 46, 11-13.
(6) Cadenbach, T.; Gemel, C.; Schmid, R.; Block, S.; Fischer, R. A. Dalton Trans. 2004, 3171-
3172.
(7) Halbherr, M.; Bollermann, T.; Gemel, C.; Fischer, R. A. Angew. Chem., Int. Ed. 2010, 49,
1878-1881.
(8) Margulieux, G. W.; Weidemann, N.; Lacy, D. C.; Moore, C. E.; Rheingold, A. L.; Figueroa,
J. S. J. Am. Chem. Soc. 2010, 132, 5033-5035.
(9) (a) Ram, M. S.; Riordan, C. G.; Ostrander, R.; Rheingold, A. L. Inorg. Chem. 1995, 34,
5884-5892; (b) Ram, M. S.; Yap, G. P. A.; Liable-Sands, L.; Rheingold, A. L.; Marchaj, A.;

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
31
Norton, J. R. J. Am. Chem. Soc. 1997, 119, 1648-1655; (c) Lafrance, M.; Gorelsky, S. I.; Fagnou,
K. J. Am. Chem. Soc. 2007, 129, 14570-14571; (d) Telser, J. J. Braz. Chem. Soc. 2010, 21,
1139-1157.
(10) Nagao, S.; Matsumoto, T.; Koga, Y.; Matsubara, K. Chem. Lett. 2011, 40, 1036-1038.
(11) (a) Laskowski, C. A.; Hillhouse, G. L. J. Am. Chem. Soc. 2008, 130, 13846-13847; (b)
Anderson, J. S.; Iluc, V. M.; Hillhouse, G. L. Inorg. Chem. 2010, 49, 10203-10207; (c) Iluc, V.
M.; Hillhouse, G. L. J. Am. Chem. Soc. 2010, 132, 15148-15150; (d) Lipschutz, M. I.; Yang,
X.; Chatterjee, R.; Tilley, T. D. J. Am. Chem. Soc. 2013, 135, 15298-15301.
(12) (a) Wu, J.; Nova, A.; Balcells, D.; Brudvig, G. W.; Dai, W.; Guard, L. M.; Hazari, N.; Lin,
P.-H.; Pokhrel, R.; Takase, M. K. Chem. Eur. J. 2014, 20, 5327-5337; (b) Pelties, S.; Herrmann,
D.; de Bruin, B.; Hartl, F.; Wolf, R. Chem. Commun. 2014, 50, 7014-7016.
(13) Saraev, V. V.; Kraikivskii, P. B.; Svoboda, I.; Kuzakov, A. S.; Jordan, R. F. J. Phys. Chem.
A 2008, 112, 12449-12455.
(14) Gleizes, A.; Dartiguenave, M.; Dartiguenave, Y.; Galy, J.; Klein, H. F. J. Am. Chem. Soc.
1977, 99, 5187-5189.
(15) (a) Helmholz, L.; Kruh, R. F. J. Am. Chem. Soc. 1952, 74, 1176-1181; (b) Willett, R. D.;
Larsen, M. L. Inorg. Chim. Acta 1971, 5, 175-179.
(16) (a) Hüttner, W.; Flygare, W. H. J. Chem. Phys. 1969, 50, 2863-2868; (b) de Luca, G.;
Russo, N.; Sicilia, E.; Toscano, M. J. Chem. Phys. 1996, 105, 3206-3210; (c) Bloor, J. E.;
Maksić, Z. B. Mol. Phys. 1973, 26, 397-408; (d) Hoppe, J. I. J. Chem. Educ. 1972, 49, 505; (e)
Bain, G. A.; Berry, J. F. J. Chem. Educ. 2008, 85, 532; (f) Flygare, W. H.; Benson, R. C. Mol.
Phys. 1971, 20, 225-250; (g) Piguet, C. J. Chem. Educ. 1997, 74, 815; (h) Sur, S. K. J. Magn.
Reson. 1989, 82, 169-173.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
32
(17) (a) Kotani, M. Progr. Theor. Exp. Phys. Supp. 1961, 17, 4-13; (b) Petuker, A.; Merten, C.;
Apfel, U.-P. Eur. J. Inorg. Chem. 2015, 2015, 2139-2144.
(18) Eckert, N. A.; Dinescu, A.; Cundari, T. R.; Holland, P. L. Inorg. Chem. 2005, 44, 7702-
7704

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
33
3.6 Experimental Section
3.6.1 General Comments
Material and Methods. All manipulations were carried out in an atmosphere of purified argon
using standard Schlenk and glove-box techniques. n-Hexane was dried using an MBraun
Solvent Purification System and fluorobenzene and d6-benzene were dried by passing through
a column of dried activated neutral Al2O3. The final H2O content in all solvents was checked
by Karl Fischer titration and did not exceed 5 ppm. [AlCp*]1, [GaCp*]2, [Ni(AlCp*)4]3,
[Ni(AlCp*)2(PPh3)2]4, [Ni(GaCp*)2(PPh3)2]
4, [FeCp2][BAr4F]5 and [FeCp2][OTf]6 were
prepared as previously described in the literature. Elemental analyses were performed by the
Microanalytical Laboratory (Rubiospek) of the Ruhr-University Bochum. The LIFDI mass
spectra were measured with a Jeol AccuTOF GCv spectrometer with a Linden-CMS.de LIFDI
source. NMR spectra of paramagnetic substances were recorded on a Bruker Avance DPX-250
spectrometer in fluorobenzene/benzene (10:1 v/v ratio) solution at 298 K. Effective magnetic
moment calculated by Evan’s method was conducted in fluorobenzene and dichloromethane
with chemical shifts given relative to TMS and were referenced to the residual solvent. The
chemical shifts are described in parts per million (ppm), downfield shifted from TMS and are
consecutively reported as position (δ H, δ P, δ B), relative integral, multiplicity (s = singlet, m
= multiplet), coupling constant (J in Hz) and assignment. EPR spectra were measured with an
X-band spectrometer in fluorobezene at 16 K (Bruker ELEXSYS500 X-band spectrometer).
FT-IR spectra were measured on a Bruker Alpha FT-IR spectrometer. The molecular structures
were measured on an Agilent Technologies SuperNova diffractometer with an Atlas CCD

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
34
detector, using CuK radiation (λ = 1.541 84 Å) from multilayer X-ray optics. The crystals were
coated with a perflusotopolyether, picked up with a cryo loop, and immediately mounted in the
nitrogen cold gas stream of the diffractometer. The data were processes with CrysAlisPro.7 The
crystal structure was solved by direct methods using SHELXS-97 and refined with SHELXL-
2013.19 Two of the CF3 groups of the BAr4F anion show rotational disorder.8
3.6.2 Synthetic Procedures
[Ni(AlCp*)4][Cp*Al(OTf)3] (1) [Ni(AlCp*)4] (100 mg, 0.141 mmol) was dissolved in
fluorobenzene (5 ml) and [FeCp2][OTf] (47 mg, 0.141 mmol) was suspended in fluorobenzene
(5 ml) in a different schlenk tube and both cooled with an iso-propanol/dry-ice bath to -35°C.
The latter suspension of [FeCp2][OTf] was transferred slowly via cannula to [Ni(AlCp*)4]
giving a deep red solution. Storage at -30°C for 3 months gave few crystals suitable for single
crystal x-ray diffraction. Because of very low stability of the compound 1, further
characterization could not be done.
[Ni(AlCp*)2(PPh3)2][BAr4F] (2) [Ni(AlCp*)2(PPh3)2] (100 mg, 0.110 mmol) and
[FeCp2][BAr4F] (116 mg, 0.110 mmol) were dissolved in fluorobenzene (5 ml) and stirred for
1 h during which the color changed from deep red to dark green. Crystals suitable for single
crystal x-ray diffraction were obtained by layering the fluorobenzene solution with n-hexane.
Anal. Calcd. (found) for C88H72BF24Al2NiP2: C 59.68% (59.04%); H 4.1% (4.11%). LIFDI-MS:
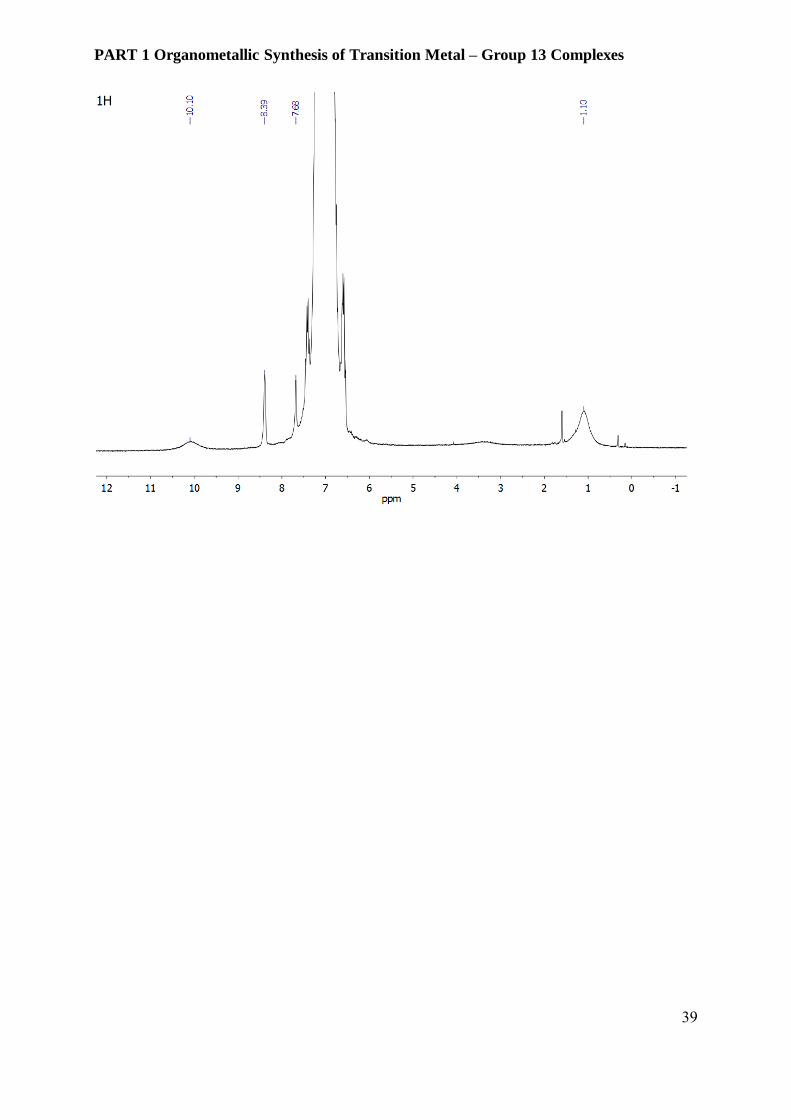
m/z = 906.4 [M] +, for the cation [Ni(AlCp*)2(PPh3)2]+. 1H-NMR (C6H5F/10% C6D6): δ 10.10
ppm, 1.10 ppm. 31P-NMR : 53.16 ppm.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
35
[Ni(GaCp*)2(PPh3)2][BAr4F] (3) [Ni(GaCp*)2(PPh3)2] (100 mg, 0.101 mmol) and
[FeCp2][BAr4F] (106 mg, 0.101 mmol) were dissolved in fluorobenzene (5 ml) and stirred for
1 h during which the color changed from deep red to light red. Crystals suitable for single crystal
x-ray diffraction were obtained by layering the fluorebenzene solution with n-hexane. Anal.
Calcd. (found) for C88H72BF24Ga2NiP2: C 56.98 % (56.53 %); H 3.92 % (4.01 %). LIFDI-MS:
m/z = 993.0 [M] + for the cation [Ni(GaCp*)2(PPh3)2]+. 1H-NMR (C6H5F/10% C6D6): δ 11.32
ppm, 3.77 ppm. 31P-NMR : 48.84 ppm.
Comment to the x-ray diffraction experiment of 1:
The high chemical instability and sensitivity of 1 also at low temperatures leads to slow
decomposition of the single crystal during the x-ray diffraction experiment and no complete
dataset was available. However, the overall completeness of 80%, a final data:parameter ratio
of 9:1 and sufficiently good R values (R1=0.0485) allow a consistent interpretation of the
data, and therefore a structural discussion of this compound has been included in the
manuscript.
PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
36
3.6.3 Spectroscopic and crystallographic data
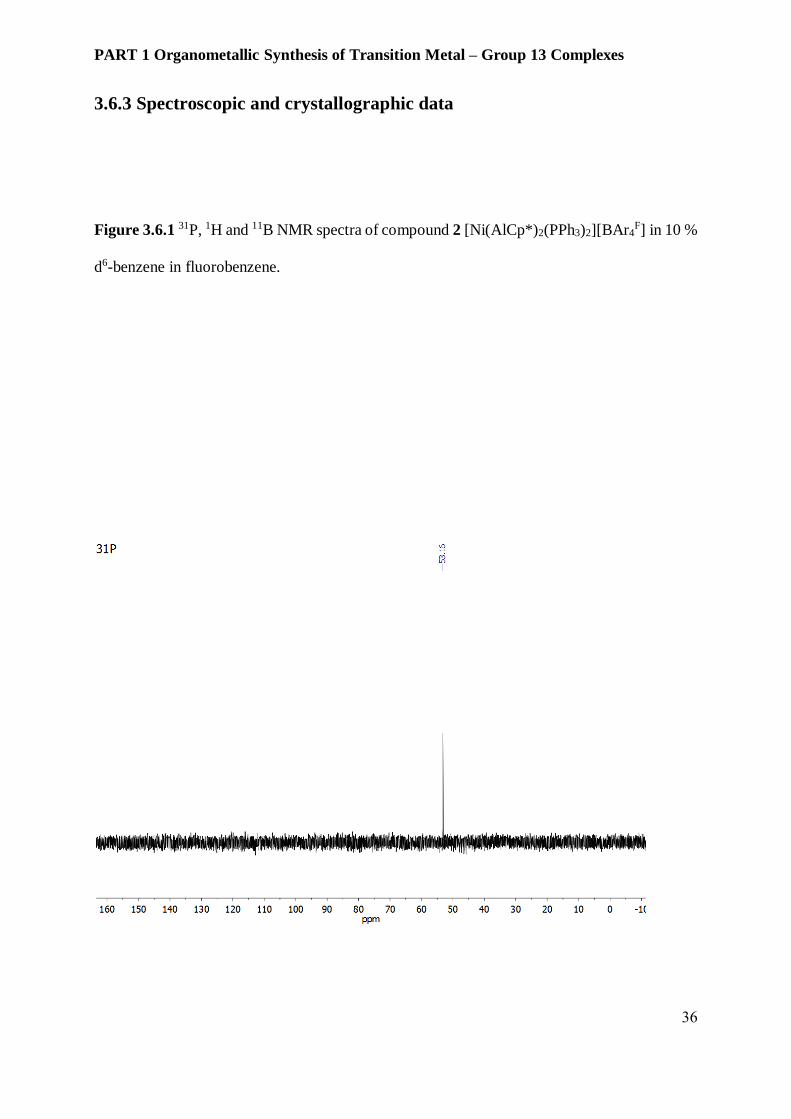
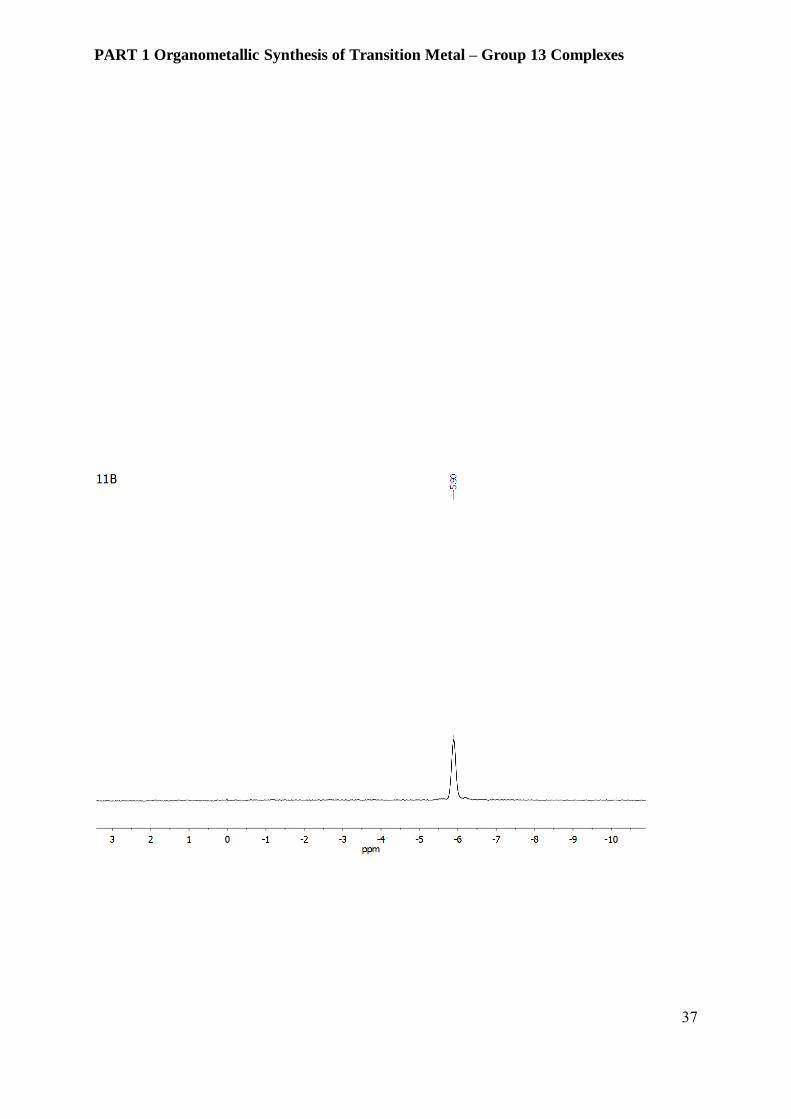
Figure 3.6.1 31P, 1H and 11B NMR spectra of compound 2 [Ni(AlCp*)2(PPh3)2][BAr4F] in 10 %
d6-benzene in fluorobenzene.
PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
37
PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
38
PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
39
PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
40
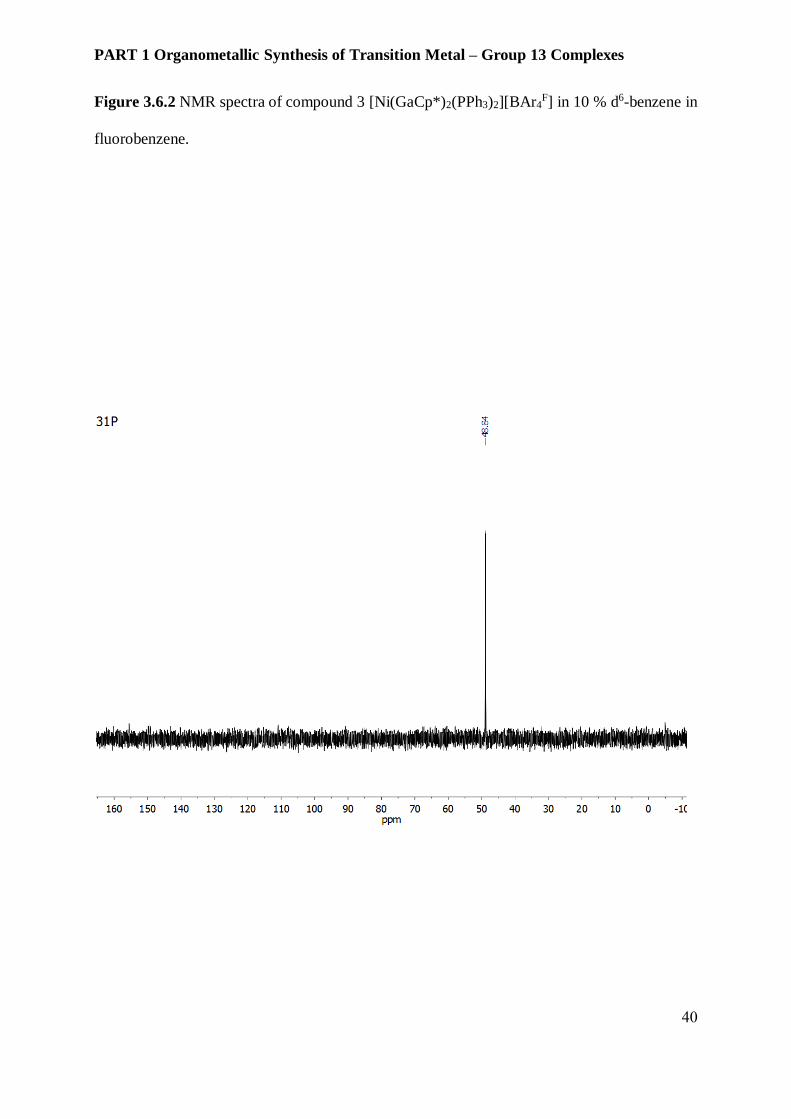
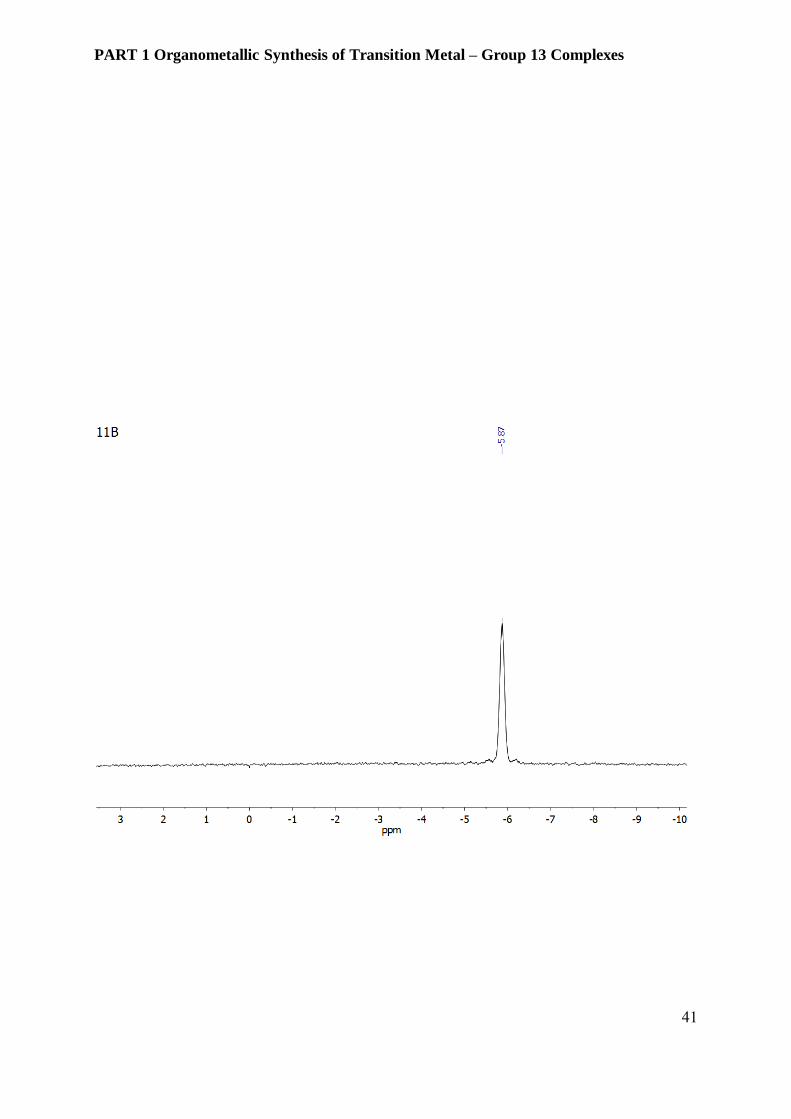
Figure 3.6.2 NMR spectra of compound 3 [Ni(GaCp*)2(PPh3)2][BAr4F] in 10 % d6-benzene in
fluorobenzene.
PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
41

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
42

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
43
Figure 3.6.3 EPR spectra of compound 2 [Ni(AlCp*)2(PPh3)2][BAr4F] (left) and 3
[Ni(GaCp*)2(PPh3)2][BAr4F] (right). The average g-factors for 2 (g = 2.11) and 3 (g = 2.15)
were obtained. The measurements were reproduced at 6 K on a different instrument (Varian
E112 EPR- Spectrometer (X-Band)) with identical results. The interpretation of the hyperfine
couplings of the highly complicated EPR signals is not possible.
1000 2000 3000 4000 5000 6000
B (G)
1000 2000 3000 4000 5000
B (G)

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
44
Figure 3.6.4 UV-Vis (black) and Photoluminescence (PL, blue) spectra of compound 2 (left)
and 3 (right).The UV-Vis and PL were measured in fluorobenzene solution. The spectra of UV-
Vis/PL of compound 2 and 3 show absorbance band and emission peak at abs = 350 nm, em =
404, 428 nm and abs = 355, 444 nm, em = 404, 428 nm.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
45
Crystallographic Data
Table 3.6.1 Important crystallographic data of compounds 1, 2 and 3
1 2 3
Empirical formula C59H75Al5F10NiO9S3 C88H72Al2BF24NiP2 C88H72BF24Ga2NiP2
Formula weight 1407.98 1770.88 1856.36
Temperature (K) 108.15 104.15 110.15
Crystal system monoclinic monoclinic monoclinic
Space group Cc P21/n P21/n
a/Å 16.7480(8) 20.4713(5) 20.5202(3)
b/Å 15.5917(9) 20.1109(4) 20.1408(2)
c/Å 26.5642(15) 20.5530(5) 20.5787(3)
α/° 90 90 90
β/° 95.151(5) 101.560(2) 101.965(1)
γ/° 90 90 90
Volume/Å3 6908.7(6) 8290.0(3) 8320.26(19)
Z 4 4 4
ρcalcg/cm3 1.354 1.419 1.482
μ/mm-1 0.512 0.393 1.004
F(000) 2928 3620 3764
Crystal size/mm3 0.15 × 0.12 × 0.09 0.14 × 0.12 × 0.10 0.17 × 0.14 × 0.09
Radiation MoKα (λ = 0.71073) MoKα (λ = 0.71073) MoKα (λ = 0.71073)
2Θ range for data collection/° 5.666 to 48.498 5.72 to 58 5.72 to 55.12
Index ranges -17 ≤ h ≤ 17 -26 ≤ h ≤ 27 -26 ≤ h ≤ 26
-11 ≤ k ≤ 17 -26 ≤ k ≤ 26 -26 ≤ k ≤ 26
-28 ≤ l ≤ 27 -26 ≤ l ≤ 27 -25 ≤ l ≤ 26
Reflections collected 11368 157220 152416
Data/restraints/parameters 6835/56/784 20038/0/1063 19082/0/1073
Goodness-of-fit on F2 0.789 1.025 1.02
Final R indexes [I>=2σ (I)] R1 = 0.0485 R1 = 0.0704 R1 = 0.0460
wR2 = 0.0668 wR2 = 0.1517 wR2 = 0.1016
Final R indexes [all data] R1 = 0.1000 R1 = 0.1343 R1 = 0.0736
wR2 = 0.0746 wR2 = 0.1792 wR2 = 0.1136
Largest diff. peak/hole / e Å-3 0.48/-0.30 1.39/-0.88 1.29/-0.85
PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
46
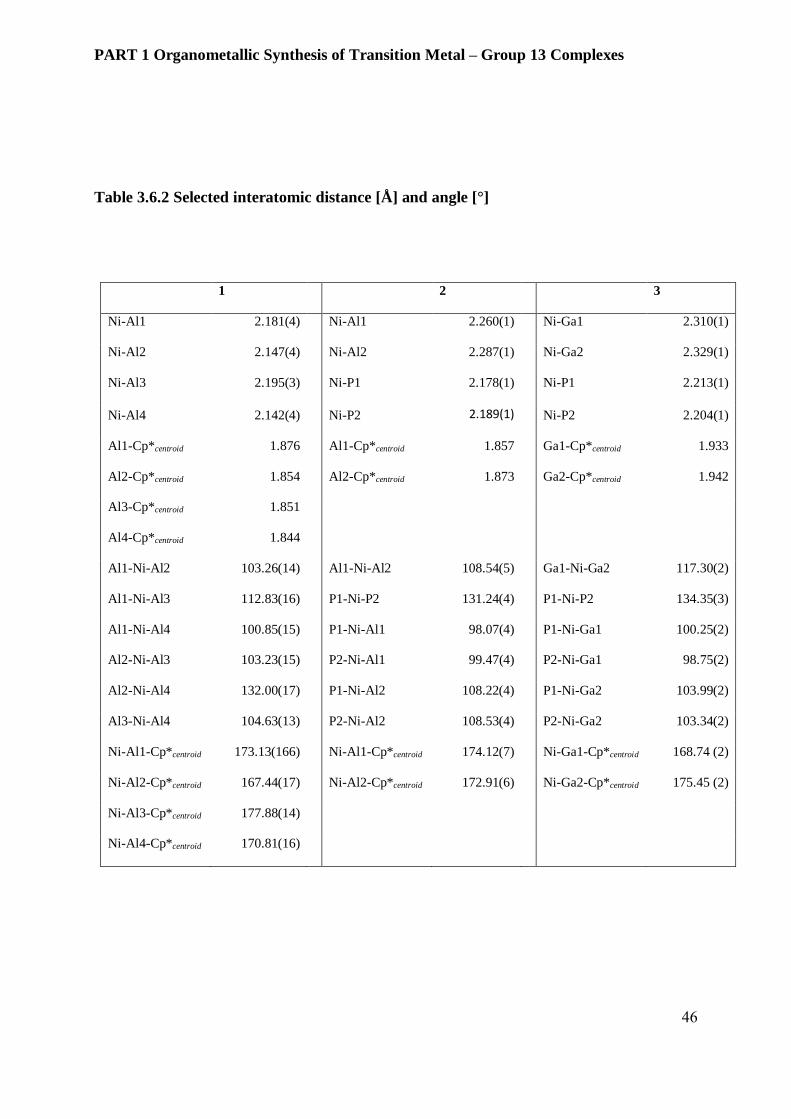
Table 3.6.2 Selected interatomic distance [Å] and angle [°]
1 2 3
Ni-Al1 2.181(4) Ni-Al1 2.260(1) Ni-Ga1 2.310(1)
Ni-Al2 2.147(4) Ni-Al2 2.287(1) Ni-Ga2 2.329(1)
Ni-Al3 2.195(3) Ni-P1 2.178(1) Ni-P1 2.213(1)
Ni-Al4 2.142(4) Ni-P2 2.189(1) Ni-P2 2.204(1)
Al1-Cp*centroid 1.876 Al1-Cp*centroid 1.857 Ga1-Cp*centroid 1.933
Al2-Cp*centroid 1.854 Al2-Cp*centroid 1.873 Ga2-Cp*centroid 1.942
Al3-Cp*centroid 1.851
Al4-Cp*centroid 1.844
Al1-Ni-Al2 103.26(14) Al1-Ni-Al2 108.54(5) Ga1-Ni-Ga2 117.30(2)
Al1-Ni-Al3 112.83(16) P1-Ni-P2 131.24(4) P1-Ni-P2 134.35(3)
Al1-Ni-Al4 100.85(15) P1-Ni-Al1 98.07(4) P1-Ni-Ga1 100.25(2)
Al2-Ni-Al3 103.23(15) P2-Ni-Al1 99.47(4) P2-Ni-Ga1 98.75(2)
Al2-Ni-Al4 132.00(17) P1-Ni-Al2 108.22(4) P1-Ni-Ga2 103.99(2)
Al3-Ni-Al4 104.63(13) P2-Ni-Al2 108.53(4) P2-Ni-Ga2 103.34(2)
Ni-Al1-Cp*centroid 173.13(166) Ni-Al1-Cp*centroid 174.12(7) Ni-Ga1-Cp*centroid 168.74 (2)
Ni-Al2-Cp*centroid 167.44(17) Ni-Al2-Cp*centroid 172.91(6) Ni-Ga2-Cp*centroid 175.45 (2)
Ni-Al3-Cp*centroid 177.88(14)
Ni-Al4-Cp*centroid 170.81(16)

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
47
4 References
(1) S. Schulz, H. W. Roesky, H. J. Koch, G. M. Sheldrick, D. Stalke and A. Kuhn, Angew.
Chem. Int. Ed., 1993, 32, 1729–1731.
(2) P. Jutzi, B. Neumann, G. Reumann and H.-G. Stammler, Organometallics, 1998, 17, 1305–
1314.
(3) Steinke, T.; Gemel, C.; Cokoja, M.; Winter, M.; Fischer, R. A Angew. Chem. Int. Ed, 2004,
43, 2299–2302.
(4) M. Molon, T. Bollermann, C. Gemel, J. Schaumann, R. A. Fischer, Dalton Trans, 2011.
(5) J. Le Bras, H. Jiao, W. E. Meyer, F. Hampel and J. A. Gladysz, J. Organomet. Chem., 2000,
616, 54–66.
(6) Z.-W. Li, A. Yeh and H. Taube, Inorg. Chem., 1994, 33, 2874–2881.
(7) CrysAlisPro Software System, version 1.171.36.20; AgilentTechnologies UK Ltd.: Oxford,
U.K., 2012.
(8) Sheldrick, G. M. Acta Crystallogr. 2008, A64, 112−122.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
48
4. Organoaluminum Palladium Complex via Reductive
Elimination
Jiyeon Kim, Hung Banh, Mathies Evers, Christian Gemel, Roland A. Fischer

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
49
4. Organoaluminum Palladium Complex via Reductive
Elimination
4.1 Abstract
The reaction of AlIII hydride species and palladium phosphine complexes is described in this
chapter. The reaction product [(PCy3)Pd-2-AlCp*]2 was obtained via reductive elimination of
hydrogen and characterized by NMR spectroscopy, LIFDI-MS, single crystal X-ray diffraction.
4.2 Introduction
There has been a significant amount of efforts from fundamental organometallic chemists to
access heterometallic complexes containing direct metal-metal bonds. Although, several
classical methods are known, such as oxidative addition etc., metal-metal coupling still remains
a challenge. The coordination chemistry of low valent EIR species towards transition metals
opened new doors in intermetallic chemistry. Since EIR is an isolobal analog of CO and
phosphine, many group 6-12 transition metal complexes containing EIR with the fomular
[M(EIR)n] have been investigated.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
50
In 2012, Ganesamoorthy et al. reported a new synthetic pathway for synthesizing [AlICp*]4
(Cp* = pentamethylcyclopentadienyl) via reductive elimination.1 This procedure suggested a
route with a mechanism distinct from the literature known procedure (Schnöckel)2. Thermally
induced reaction of AlIIICp*2H leads to the formation of [AlCp*]4 which includes the formation
of a direct Al-Al bond via reductive elimination of Cp*H. This work was an inspiration of
preparing intermetallic complexes including bonds between transition metals and group 13
metals via intramolecular reductive elimination. Even though, reductive elimination in
transition metal-complexes is commonly observed particularly in small molecule activations
(C-H or C-C coupling), reports on intramolecular reductive elimination resulting in direct
metal-metal bonds remain scarce. Herein, we report the formation of Pd-Al bonds via reductive
elimination of H2.
4.3 Result and Discussion
The stoichiometric reaction of Pd(PCy3)2 (Cy = cyclohexyl) and AlCp*H2 in benzene
reductively eliminates H2 thermally at 70 °C. (Scheme 4.1) Additionally, the loss of one
equivalent of free PCy3 affords [(PCy3)Pd-2-AlCp*]2 (4). Reaction completeness was
monitored by in-situ 1H-NMR spectroscopy. A strong gas evolution was already observed as
soon as benzene was added into a J-Young NMR tube before sealing. Although, in-situ 1H
NMR measurements show a weak but clear evidence of H2 evolution at 4.46 ppm from the
H2 cleavage in AlCp*H2. (Figure 4.4) Compound 4 decomposes in air and is moisture sensitive
and readily dissolve in non-polar solvents such as hexane or toluene. The chemical shift of
bridging AlCp* from compound 4 was observed at 2.14 ppm whereas AlCp* and AlCp*H2
protons appear at 1.89 and 1.91 ppm. Protons in the phosphine ligand are displayed as broad

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
51
peaks at 1.22 and 1.72 ppm, whereas the protons of free phosphine appear in the area of 1.26
– 2.24. The 31P NMR spectrum of compound 4 shows a resonance at 57.09 ppm with trace
amounts of free PCy3 at 9.97 ppm. the reactant Pd(PCy3)2 has the phosphine resonance peak
at 39.18 ppm and cannot be identified in the final product. Compound 4 could not be
completely isolated from free PCy3 by recrystallization due to the indistinct solubility
differences. Liquid injection field desorption ionization mass spectrometry (LIFDI-MS) of 4
clearly identifies molecular ion peak at m/z 1098.17. This observation indicates there is no
hydride takes place in the molecule particularly at the two palladium atoms. Moreover, the
transition metal hydrides exhibit the IR vibration near 2000 cm-1 where no peak has been
observed in this area. (Figure 4.7)
Scheme 4.1 Reaction of Pd(PCy3)2 with AlCp*H2
Crystals suitable for single crystal X-ray diffraction are obtained in pentane at -30 °C. Figure
4 shows a depiction of the molecular structure of 4. Crystallographic data and refinement details
are shown in Table 4.1 whereas important bond distance and angles are shown in Table 4.2.
The solvate free compound 4 crystallizes in the monoclinic space group C2/c with two
fragments in the asymmetric unit. The molecular structure of the type [Pd2Al2P2] has two
bridging AlCp* and two cis-terminal phosphine ligands with a geometry described best as two
edge-sharing trigonal planar units. (Figure 4.2) The two aluminum atoms are shared within
both units. The two bridging AlCp*s are bent to lopsided with an angle of Pd−Pd axis and two
aluminum atoms found 130.7°. Interestingly, the two terminal phosphines are bent to opposite

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
52
direction of AlCp* ligands resulting in a P−Pd−Pd angle of 152.7 °. (Figure 4.1) These effects
explain the biased structure of bridging AlCp* of compound 4. In comparison to previous
observations by Fischer et al. where the compounds [Pd2(GaCp*)3(PMe3)2]3-4 [Mariusz Dalton
2014] and [Pd2(GaCp*)3(PPh3)2] [Steinke 2005 chemeurj] with a similar core building unit but
with three bridging ligands of the type [Pd2Ga3P2] was reported, quite different results regarding
the bonding situation were found. The angle of P-Pd-Pd in those structures, the tree atoms are
in linear positions in contrast to compound 4. Also, the steric bulk cyclohexyl groups in
phosphine are inclined to the opened site of Pd−Pd bond which consequently stabilize the
structure (Figure 2). The Pd1-Pd2 bond length of 2.592 Å in compound 4 is slightly shortened
than as found for the Pd0-Pd0 complexes [Pd2(AlCp*)5]4 [Steinke 2005 chemeurj] and
[Pd2(GaCp*)3(PMe3)2]3 [Mariusz Dalton 2014] (2.633(1); and 2.623(1)). The bridging Pd−Al
bond length of 4 (a 2.424 Å) is in the same range as found in [Pd2AlCp*5] (a 2.448 Å). [Steinke
chemeurj 2005]
Figure 4.1 Povray plot of the molecular structure of 1 in the solid state as determined by single
crystal X-ray diffraction (the asymmetric unit contains two molecules; thermal ellipsoids are
shown 50 % probability; hydrogen atoms are omitted for clarity). Selected atomic distances (Å)
as well as angles (°): Pd1−Pd2: 2.5914(6), Al1−Pd1: 2.4284(15), Pd1−Al2: 2.4102(14),
Pd2−Al2: 2.4201(15), Pd2−Al1: 2.4377(15), Pd1−P1: 2.2807 (13), Pd2−P2: 2.2936(13),

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
53
Al−Cp*: a1.951, Pd1−Al1−Pd2: 100.25(5), Pd1−Al2−Pd2: 99.71(5), Cp* values are calculated
from the centroid of the Cp* moiety.
Figure 4.2 Illustration of coordination geometry of compound 1 (M2Al2P2) and the
comparison of molecular type M2Ga3P2.
4.4 Conclusion
Within this chapter a facile route to a dinuclear Pd core structure with heteroleptic ligand
coordination sphere was presented. Reductive elimination was used as a tool to prepare this
new structure. Regarding the state of the art, this is the first described synthesis of a transition
metal−group 13 complex via this mechanistic route. It is highly expected that this synthetic
approach can help to extend the possibilities in constructing compounds containing metal-metal

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
54
bonds and will help open up new doors in the field of organomettalic chemistry. Furthermore,
we showed a new Pd2Al2P2 core with aluminum ligands which have been directed by the steric
bulk of the R groups on the P ligands. The found geometry is very uncommon and the
assymetric palladium dimer with relatively unshielded metal site might be of interest as a
building unit to access intermetallic clusters.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
55
4.5 Experimental Section
4.5.1 General Comments
All manipulations were carried out in an atmosphere of purified argon using standard Schlenk
and glove-box techniques. n-Hexane and n-pentane were dried using an MBraun Solvent
Purification System. Benzene was dried by passing through a column of dried activated neutral
Al2O3. d6-Benzene was degassed and stored under the molecular sieves (4 Å). All the
instruments for characterization were identically used described in section 3.6. [AlCp*H2] and
[Pd(PCy3)2] were prepared as previously described in the literature. [Ganesh + a]
4.5.2 Synthetic procedure
[(Pd(AlCp*)(PCy3))2] Pd(PCy3)2 (200 mg, 0.300 mmol) and AlCp*H2 (50 mg, 0.305 mmol)
were dissolved in benzene (5 ml), immediate colour change to dark red and gas evolution were
observed. The reaction mixture was stirred for 1 h at 80 °C. All the volatiles were removed
under vacuum and the residue was extracted with cold (- 60 °C) pentane. Dark-red crystals
suitable for single crystal x-ray diffraction were obtained at -30 °C in the concentrated pentane
solution. 1H-NMR (C6D6): 2.14 ppm (Cp*Al), 1.77-1.72 ppm (Cy3P). 31P-NMR (C6D6):
57.09 ppm (P-Pd). LIFDI-MS: m/z = 1098.2 [M] +, for the cation [(Pd(AlCp*)(PCy3))2]+.
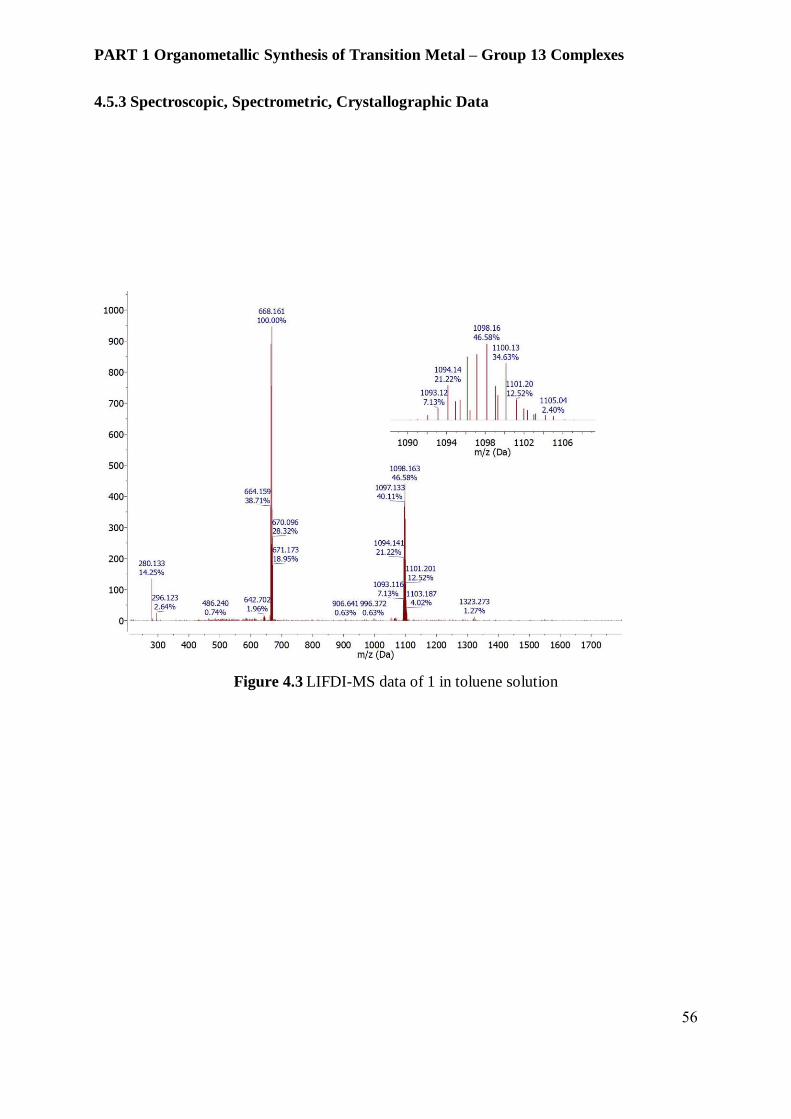
PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
56
4.5.3 Spectroscopic, Spectrometric, Crystallographic Data
Figure 4.3 LIFDI-MS data of 1 in toluene solution
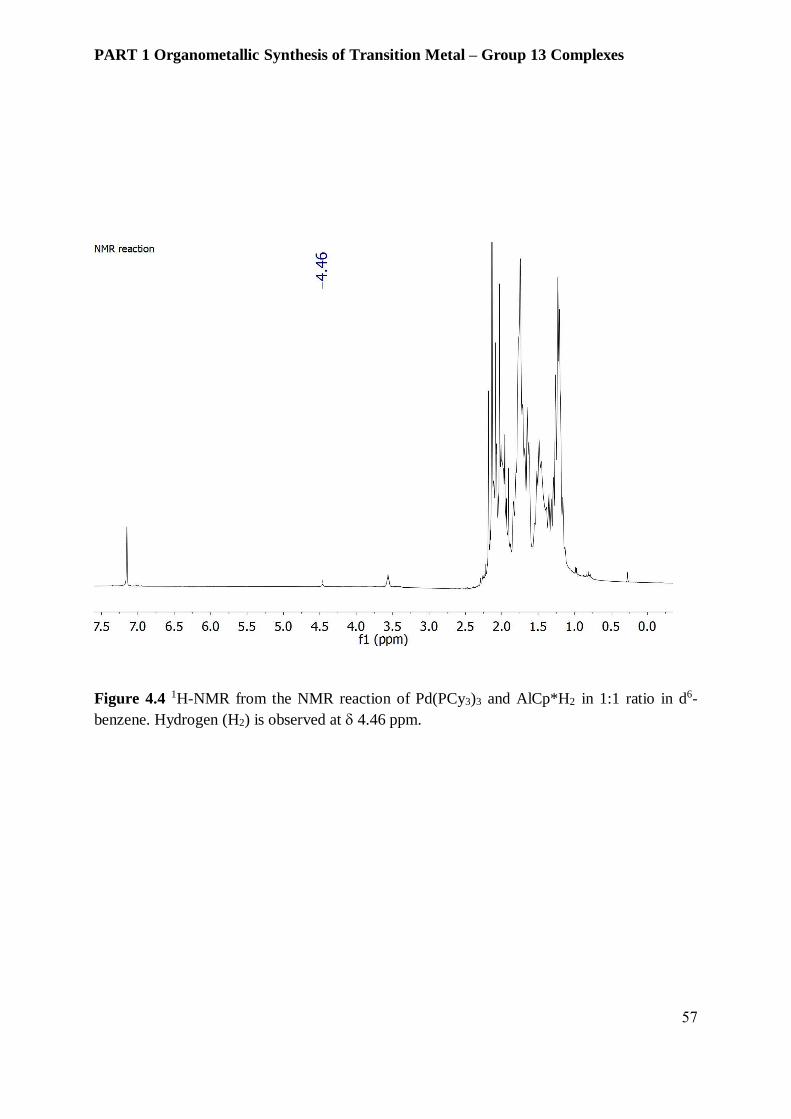
PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
57
Figure 4.4 1H-NMR from the NMR reaction of Pd(PCy3)3 and AlCp*H2 in 1:1 ratio in d6-
benzene. Hydrogen (H2) is observed at 4.46 ppm.
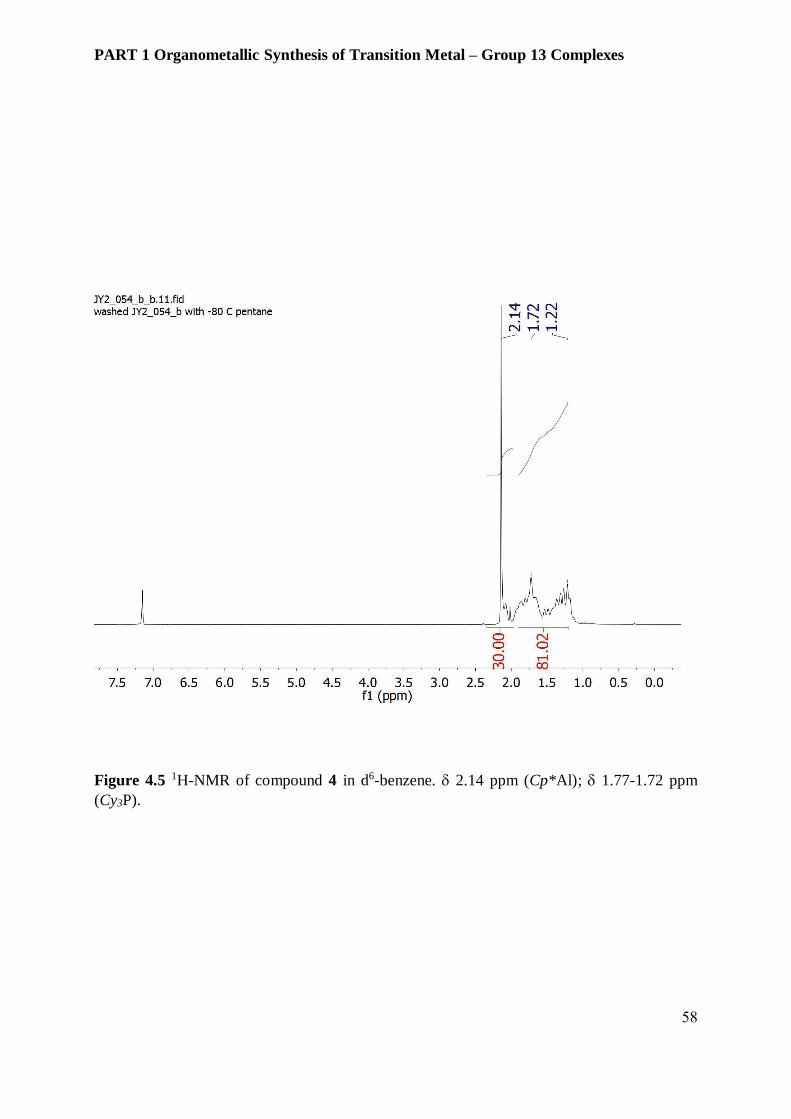
PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
58
Figure 4.5 1H-NMR of compound 4 in d6-benzene. 2.14 ppm (Cp*Al); 1.77-1.72 ppm
(Cy3P).
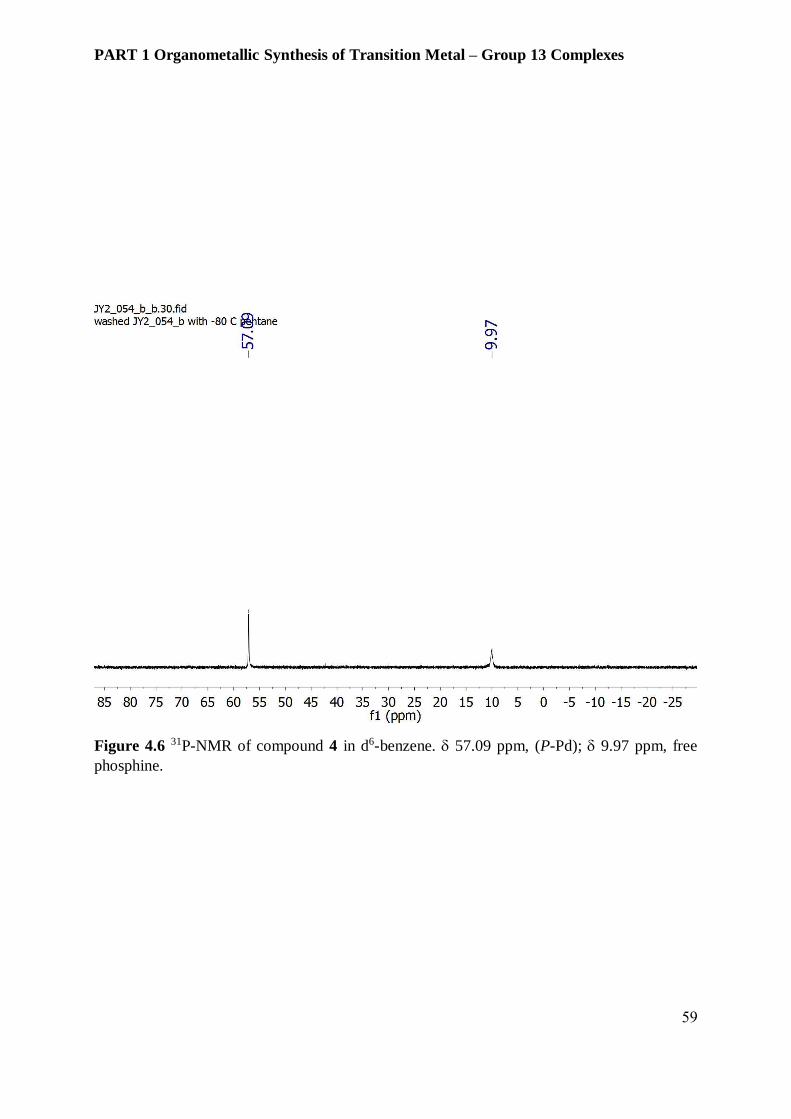
PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
59
Figure 4.6 31P-NMR of compound 4 in d6-benzene. 57.09 ppm, (P-Pd); 9.97 ppm, free
phosphine.
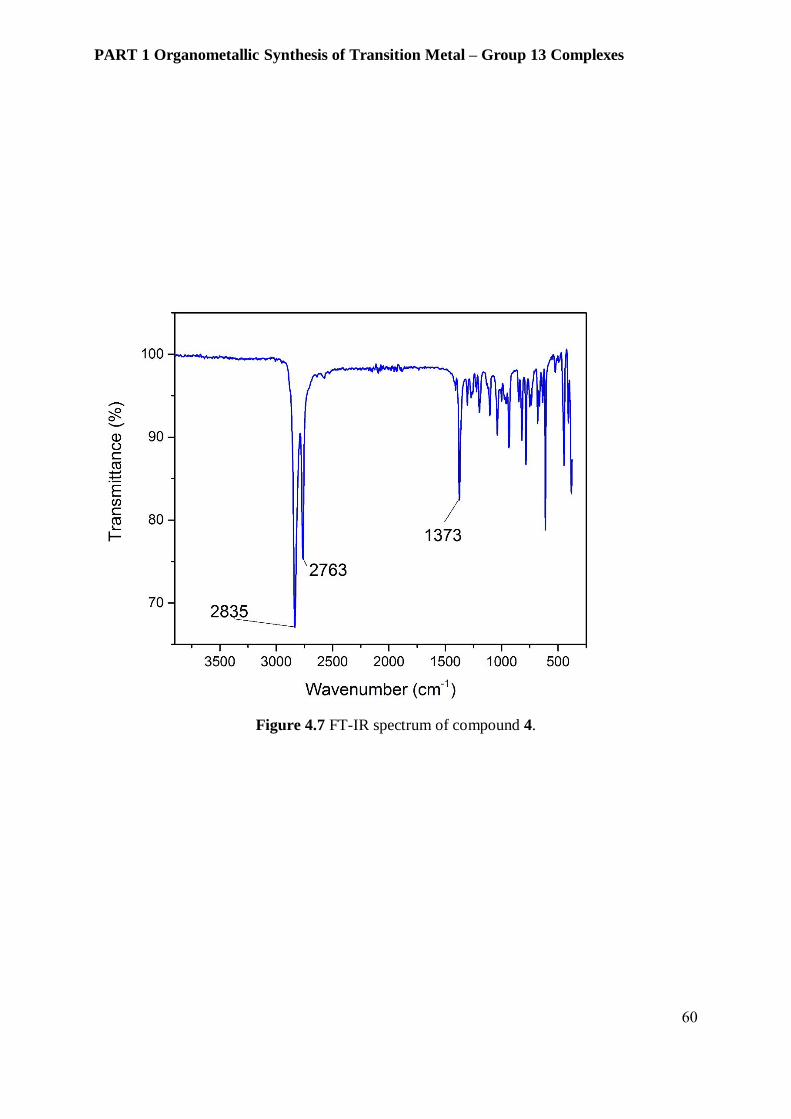
PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
60
Figure 4.7 FT-IR spectrum of compound 4.

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
61
Table 4.1 Crystallographic data and refinement details of compound 4.
Table S1 Selected Bond Length (Å) and Angles (°) for compound 4.
Pd(1)−Pd(2) 2.5914(6) Pd(1)−Al(1)−Pd(2) 100.25(5)
Pd(1)−Al(1) 2.4284(15) Pd(1)−Al(2)−Pd(2) 99.71(5)
Pd(1)−Al(2) 2.4102(14) P(1)−Pd(1)−Pd(2) 153.70(4)
Pd(2)−Al(2) 2.4201(15) P(1)−Pd(2)−Pd(1) 151.74(4)
Pd(2)−Al(1) 2.4377(15) Al(1)−Pdaxis−Al(2) 130.7
Pd(1)−P(1) 2.2807 (13)
Pd(2)−P(2) 2.2936(13)
Al−Cp*centroid a1.951
a means value
4
Chemical formula C56H96Al2P2Pd2
Formula weight 1098.02
Crystal system Monoclinic
Space group C2/c
a (Å) 71.238(5)
b (Å) 10.2931(3)
c (Å) 45.665(3)
(°) 136.980(12)
Volume (Å3) 22845(4)
Z 16
Dcalc (g × cm-3) 1.277
/mm-1 6.142
F(000) 9280.0
2θ range for data collection (°) 7.276 to 139.998
Reflections collected/unique 42875/20848 [Rint = 0.0564]
Data/restraints/parameters 20848/0/1137
goodness-of-fit on F2 1.006
Final R indices [I > 2(I)] R1 = 0.0469, wR2 = 0.1028
R indices (all data) R1 = 0.0717, wR2 = 0.1503

PART 1 Organometallic Synthesis of Transition Metal – Group 13 Complexes
62
5. References
1. Ganesamoorthy, C.; Loerke, S.; Gemel, C.; Jerabek, P.; Winter, M.; Frenking, G.;
Fischer, R. A., Reductive elimination: a pathway to low-valent aluminium species. Chem.
Commun. 2013, 49, 2858-2860.
2. Dohmeier, C.; Robl, C.; Tacke, M.; Schnöckel, H., The Tetrameric Aluminum(I)
Compound [{Al(η5-C5Me5)}4]. Angew. Chem., Int. Ed. 1991, 30, 564-565.
3. Molon, M.; Gemel, C.; Fischer, R. A., Organogallium- and organozinc-rich palladium
and platinum clusters. Dalton Trans. 2014, 43, 3114-3120.
4. Steinke, T.; Gemel, C.; Winter, M.; Fischer, R. A., The Clusters [Ma(ECp*)b] (M=Pd,
Pt; E=Al, Ga, In): Structures, Fluxionality, and Ligand Exchange Reactions. Chem. Eur. J.
2005, 11, 1636-1646.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
63
PART 2 Precursor Synthesis and Atomic Layer Deposition
of Cobalt Oxide

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
64
List of Abbreviation
AFM Atomic Force Microscopy
ALD Atomic Layer Deposition
AMD Amidinate
CDI Carbodiimide
Cp Cyclopentadiene
CVD Chemical Vapor Deposition
Cy Cyclohexyl
DTA Differential Thermal Analysis
DSC Differential Scanning Calorimetry
EA Elemental Analysis (CHN)
EDX Energy Dispersive X-ray
EI-MS Electron Impact Mass Spectrometry
Et2O Diethylether
tBu2DAD 1,4-di-tert-butyl-1,3-diazadiene
GI Grazing incidence
GPC Growth per cycle
Guan Guanidinate
Hacac Acetylacetone
HTXRD High Temperature X-ray Diffraction
IR Infra-Red
Me Methyl

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
65
MOCVD Metalorganic Chemical Vapor Deposition
NMR Nuclear Magnetic Resonance
iPr Isopropyl
PEALD Plasma Enhanced ALD
PVD Physical Vapor Depsoition
RBS Rutherford Backscattering Spectrometry
Rms Root mean square
RT Room Temperature
SEM Scanning Electron Microscopy
tBu tert-butyl
TEM Transmission Electron Microscopy
TGA Thermogravimetric Analysis
Thd 2,2,6,6,-tetramethyl-3,5-heptadione, C11H20O2
THF Tetrahydrofuran
ToF-ERDA Time of Flight Elastic Recoil Detection Analysis
UV Ultraviolet
XPS X-ray Photoelectron Spectroscopy
XRD X-ray Diffraction

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
66
5. General Introduction

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
67
5 General Introduction
5.1 General Introduction
Already many examples for thin films possessing functional properties exist in nature, for
instance the brilliant colors of the feathers of a peacock or the wings of butterflies caused by
the interference of light with extremely thin films covering these. Another example for thin
films in daily life are simple household mirrors. Mirrors are nowadays produced by covering a
suitable substrate with a reflective coating. The first attempts on making very crude mirrors in
this fashion were already reported 2000 years ago in Pliny the Elders book on Natural History.
In Sidon, which is located in nowadays Lebanon, circular cuts of brown glass were coated with
molten lead. Since these early reports on the preparation of functional films a lot happened and
many methods have been developed to coat substrate materials in order to improve their
properties for certain useful applications and alter their mechanic, optical, chemical or
electronic functions.
Rusting of steel and iron parts was a big issue in former times, which reduced the stability and
longevity of tools and parts in i.e. machinery and buildings drastically. By applying a thin film
of zinc to the used parts the iron was protected from outer influences and the reaction to iron
oxide (rust) is inhibited. Even in the case of scratching or partly destruction of the zinc layer,
still a protective effect is occurring, as the zinc acts as a sacrificial anode, which reacts with
corrosive reagents from the surroundings. A method to apply these films was developed by Paul
Jacques Malouin, who coated iron with zinc by dipping it into molten zinc in 1742.
For the preparation of thin films nowadays, two general concepts are of outstanding importance
and will be introduced in the following, (1) physical vapor deposition and (2) chemical vapor

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
68
deposition. These methods have a plethora of sub-methods and variations and the following
description of methods is by no means complete. Both methods are very powerful to prepare
thin films and are conducted at vacuum conditions, and the deposited thin film is transported
through the gas phase and deposited on a substrate.
Physical Vapor Deposition
The most common methods of physical vapor deposition are sputtering and evaporation
techniques. In sputtering techniques, atoms from a metal target are ejected by collisions of high
energetic particles, most commonly gas atoms. The ejected metal atoms are travelling from the
target to the substrate where they are condensing, forming a thin film. In physical evaporation
techniques, the target is heated to very high temperatures at very low pressures, leading to the
evaporation of atoms from the target. The evaporated atoms can then react with the substrate
and deposit on its surface forming a thin film. The advantages of these physical deposition
techniques are the ability to use nearly any kind of target and substrate. The biggest
disadvantage is the so called “line-of-sight-coverage” which makes the coverage of complex
samples with advanced geometrical features, e.g. protrusions unfeasible
Chemical Vapor Deposition
Chemical Vapor deposition summarizes all methods in which a chemical precursor is
decomposed and deposited on a surface. Many variations of this method exist, but the general
advantages of this method is the uniform coverage of complex shaped substrates. A big
drawback of this method is the limitation of precursors, not for every kind of coating suitable
precursor molecules have been developed. A very useful variation of this method, which will
be the focal point in the following is atomic layer deposition, or short ALD.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
69
5.2 Atomic Layer Deposition
The fabrication of highly conformal thin films of metal or metal oxides are of eminent
importance for the production of modern devices in electronics and nanotechnology. There are
various methods to prepare thin films, the most common used techniques are either physical or
chemical methods. For instance, The most common methods to prepare cobalt oxide thin films
are for example sol-gel processes,1-3 spray pyrolysis,4-7 electrodeposition and physical vapor
deposition (PVD).8-12 The precision of these methods is still limited and hence the development
of new deposition techniques for these films is necessary. Up to date, the most promising
methods, which outperform the afore mentioned by means of film thickness and phase purity
are chemical vapor deposition (CVD) and atomic layer deposition (ALD).13 Among these
techniques, specifically ALD is a very powerful method to deposit films which possess
exceptional conformality and distinct morphology and phase control on the nanometer scale is
possible.9-10 Furthermore, the deposition of thin films on complex-shaped substrates is possible
by this method.
ALD also called ALE (atomic layer epitaxy) was developed in the 1970s by T. Suntola and J.
Antson to deposit thin films from the chemical vapor phase. The ALD process allowed
producing high quality, uniform and conformal thin films over large and complex-shaped areas
with precise control of thickness. These characteristic function of ALD has caught an enormous
attention from the microdevice and semiconductor industries. Fundamental studies as well as
todays application fields of ALD have been excellently reviewed in several reports by T.
Suntola, L. Niinistö, M. Leskelä, M. Ritala.14-20
ALD is considered as an advanced version CVD, because ALD shows two unique features
compared to CVD processes. ALD is a thin film deposition technique, which relies on
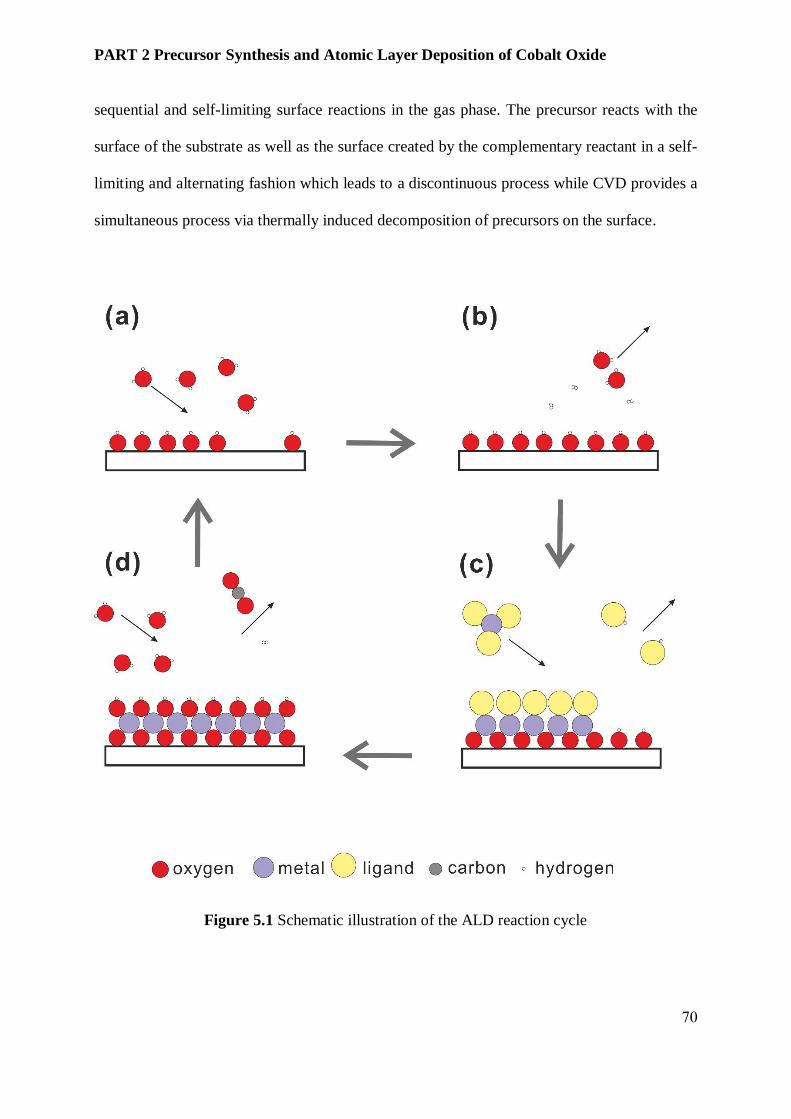
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
70
sequential and self-limiting surface reactions in the gas phase. The precursor reacts with the
surface of the substrate as well as the surface created by the complementary reactant in a self-
limiting and alternating fashion which leads to a discontinuous process while CVD provides a
simultaneous process via thermally induced decomposition of precursors on the surface.
Figure 5.1 Schematic illustration of the ALD reaction cycle

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
71
Unlike precursors used for CVD growth, an ALD precursor should be thermally stable at
deposition temperature to guarantee the self-limiting saturation of the surface. (Criteria of ALD
precursors will be discussed in detail in chapter 5.3) Full ALD cycles consist of metal
containing precursor and the complementary reactant. In the first step, the surface (most likely
OH groups on the surface) is exposed to a pulse of gaseous metal-containing precursor and
saturated by the precursor via chemisorption which inhibits the further growth (Figure 5.1a).
Excess precursor and by-products are purged out with an inert gas (e.g. nitrogen) (Figure 5.1b).
The resulting chemisorbed layer from the first step, is then exposed to the second
complementary reactant (e.g. H2O, O2, O3 H2, NH3 etc) (Figure 5.1c). This leads to the
formation of the desired material (for instance metals, metal oxides or metal nitrides depending
on the second reactant) and the excess of reactants and reaction by-products are subsequently
purged by inert gas. (Figure 5.1d) Repetition of these steps initiates the film growth restrained
by saturative and self-limiting surface reactions leading to uniform, conformal and precisely
controlled thickness (simply tuning the applied number of cycles controls film thickness).

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
72
Figure 5.2 Possible film growth behavior of ALD growth per cycles versus deposition
temperature.
To obtain an ideal ALD grown thin film, an optimized deposition temperature (Tdep) is required.
The temperature range where a constant growth rate is achieved is the so-called ALD window
(Figure 5.2).21 At temperatures below the ALD window the precursor either condenses on the
surface, or the surface reaction remains incomplete, due to the low temperature. Above the ALD
temperatures window, the precursors can decompose on the surface, which will lead to a CVD
like film growth or the surface species desorbs again.
The presence of an ALD window and the linearity of the thickness vs number of applied cycles
are typical features of the self-limiting character, however, it does not necessarily guarantee the
self-limiting growth process will actually take place. Only saturation studies with the growth
rate as a function of the precursor pulses finally demonstrate the self-limiting process.
5.3 ALD of metal oxides thin films
5.3.1 Precursors
Although atomic layer deposition techniques are widely used to provide very uniform and
conformal thin films, suitable and effective precursors remain scarce. Precursors employed for
ALD require several conditions. The criteria of ALD precursors are as follow. 1) Highly
reactive towards the functionalities on the surface as well as the surface prepared by the
complementary reactant. Furthermore, a complete reaction is desired. 2) Volatile (vapor
pressure > 0.1 Torr;)22-23 and thermally stable at growth (> seconds) and sublimation
temperatures (> months). Evaporation should occur in a single step without thermal

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
73
decomposition. 3) Non-reactive and non-corrosive byproduct formation is strongly preferred.
As is often the case, the reactive by-products can damage the film materials by etching or
reabsorption onto the reactive sites of the surface, thereby inducing non-uniformity and low
growth rates.23
ALD precursors can occur in the solid, liquid or gaseous state, but solid precursors own certain
drawbacks as unwanted particles might be transported by the carrier gas and incorporated into
the films. This in turn might cause defects in the films. Thus, the liquid or gaseous precursors
are preferred particularly in industrial scale application, if those are accessible.
Chronological development of precursors including metal halides, cyclopentadienyls (Cps),
alkoxides, -ketonates, alkylamides, trimethylsilanes (TMS), heteroleptic compounds of
mixture of theses ligands.
Figure 5.3 shows a list of the common ligands used for ALD precursors. In the following
chapters, the ALD precursors properties in detail will be discussed.
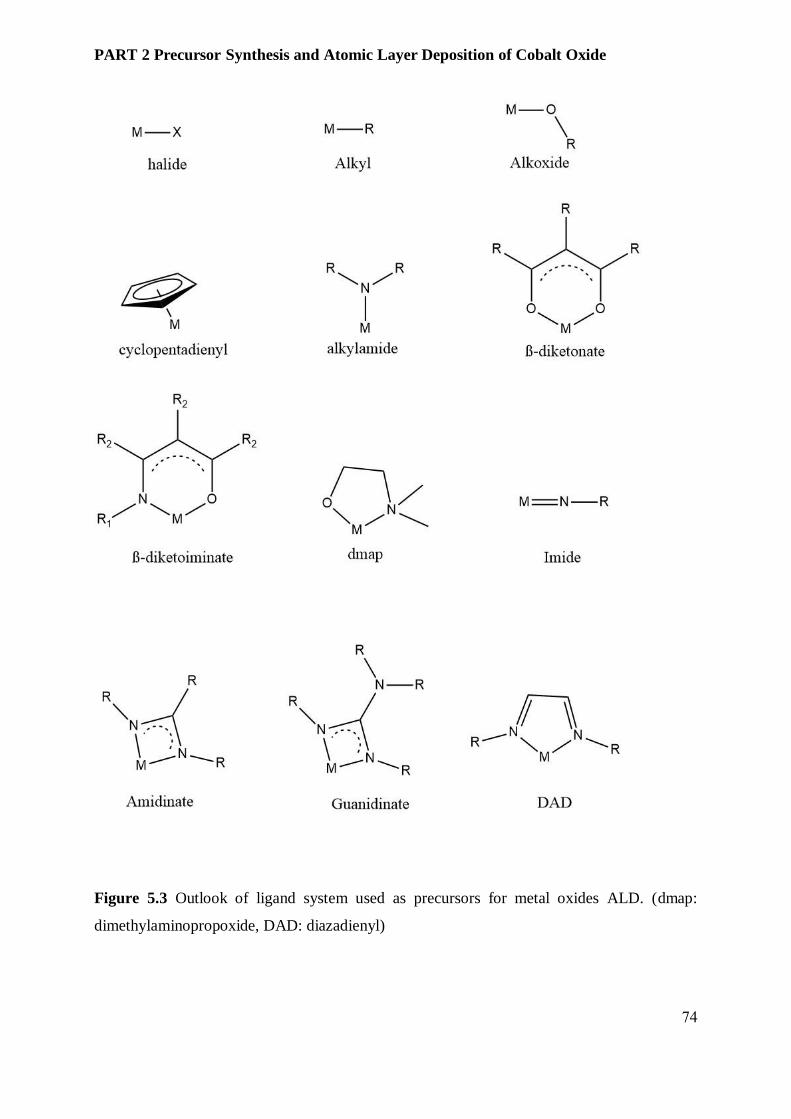
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
74
Figure 5.3 Outlook of ligand system used as precursors for metal oxides ALD. (dmap:
dimethylaminopropoxide, DAD: diazadienyl)

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
75
A. Metal halides
The metal halides with the general formula MXn (X = F, Al, Br or I; n = depends on the
oxidation state of metals) have the general advantage of stability at a broad range of
temperatures and a high reactivity. However, metal halides produce HX which is a strong acid
can damage or etch the surface of the metal/metal oxide thin films and this cause the
inhomogeneous film thickness. Moreover, metal halides precursors are not preferred in many
occasions in particular in our group due to the resulting halogen contamination.
B. ß-diketonates
Metal ß-diketonates with the general formula of M(thd)n (thd = 2,2,6,6-tetramethyl-3,5-
heptanedionyl) are one of the most common precursors used for gas phase depositions and
nearly every corresponding transition metal complexes across the periodic table as well as rare
earth metals compounds have been explored.24-25 The synthesis is relatively easy and straight
forward and very often they are commercially available. Owing to four strong M−O bonds in
the structure (low basicity of the ß-diketonate ligand), metal ß-diketonates possess low
reactivity towards water. Thus, metal ß-diketonates are rather developed for CVD precursor or
ALD with powerful oxidant such as ozone to combust the ligands. Metal ß-diketonates have
relatively low volatility and evaporation late but they have very high thermal stability (>
300 °C). Although metal ß-diketonates are the most intensively used precursor, they cause the
major problems concerning the low growth rates (0.2 – 0.4 Å/cycle), high quantity of residue
related to the high evaporation temperature due to the high thermal stability.26 [Andrian 167,168]

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
76
C. Amidinates
The bidendate chelating effects of amidinates improve the thermal stability which is the major
drawback of many monodendate alkylamides. Moreover, easily tunable steric crowding in the
ligand sphere of the system prevents the oligomerization, which has a huge impact on the
volatility of the compounds. In fact, the presence of two substituents (NR2) at the nitrogen atom
readily leads to a distinct increase of steric hindrance compared to oxygen based systems
(OR).27-28 Also amidinates have good reactivity towards oxidizing reagent in the ALD process
such as water and ozone. Amidinates were intensively investigated which diminish the
drawbacks of conventional precursors. Although numerous amidinates have been reported, only
a few ALD process are known for the preparation of metal oxides.29 Details of amnidinates
synthetic route and their electronic structures will be dicussed in the following chapter in
comparison to the guanidinates. (See 1.2.1.7 Guanidinates)
D. Guanidinates
Guanidinates are closely related to the amidinates regarding the tunability of their steric
hindrance, sturcture and bidendate chelating effect, however guanidinates possess an extended
electronic flexibility since the lone pair electrons of the amido-nitrogen is delocalized in the
ligand -system.30-32 Additionally, guanidinate ligands are stronger bases in comparison to the

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
77
amidinates, which increases the reactivity of the corresponding guanidinato metal complexes.
As an ALD precursor, these features give rise to advantages, in particular from the increase of
reactivity towards hydroxyl groups on the surface leading to a high surface coverage and high
growth rates.
Amidinate and guanidinate compounds, particularly Group 13 and transition metals are
investigated intensively. Synthetic strategies to prepare metal guanidinate complexes include
several different routes, most common approaches are summarized in the following.30, 33
(a) Salt metathesis reaction
The principle of salt metathesis reaction is the chemical process of the exchange of cations
between a metal halide and an alkali metal (mostly Li or Na) guanidinates. This approach
includes a two-step reaction, where an alkali metal guanidinate [Li(NR’}2CNR2]x is created by
insertion of carbodiimide into the alkyl lithium amide. Normally this step is a quantitative
reaction, [Li(NR’)2CNR2]n is not separated but its solution (most of the case Et2O) is used
directly for the next step. In the second step, reaction with metal halides give the metal
guanidinates with the general formula of [Mn(NR’)2CNR2]n (n is related to the oxidation state
of the metals) and LiCl as a byproduct.30, 34-35 (Scheme 1) This synthetic route is most common
way to synthesize the metal guanidinates. Reaction of dialkylamide and N,N’-dialkyl-
carbodiimide gives almost quantitative yield of lithiated guanidinato complex, normally metal
halides are added in-situ without isolation of lithiated guanidinate.
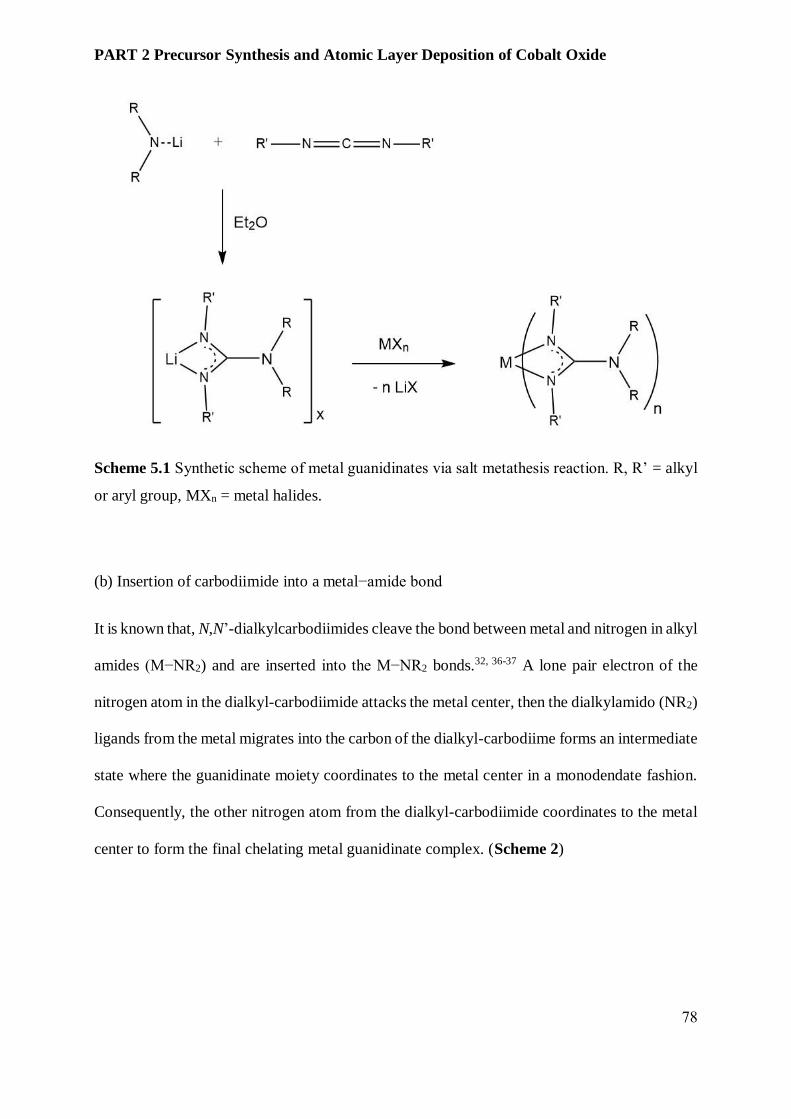
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
78
Scheme 5.1 Synthetic scheme of metal guanidinates via salt metathesis reaction. R, R’ = alkyl
or aryl group, MXn = metal halides.
(b) Insertion of carbodiimide into a metal−amide bond
It is known that, N,N’-dialkylcarbodiimides cleave the bond between metal and nitrogen in alkyl
amides (M−NR2) and are inserted into the M−NR2 bonds.32, 36-37 A lone pair electron of the
nitrogen atom in the dialkyl-carbodiimide attacks the metal center, then the dialkylamido (NR2)
ligands from the metal migrates into the carbon of the dialkyl-carbodiime forms an intermediate
state where the guanidinate moiety coordinates to the metal center in a monodendate fashion.
Consequently, the other nitrogen atom from the dialkyl-carbodiimide coordinates to the metal
center to form the final chelating metal guanidinate complex. (Scheme 2)
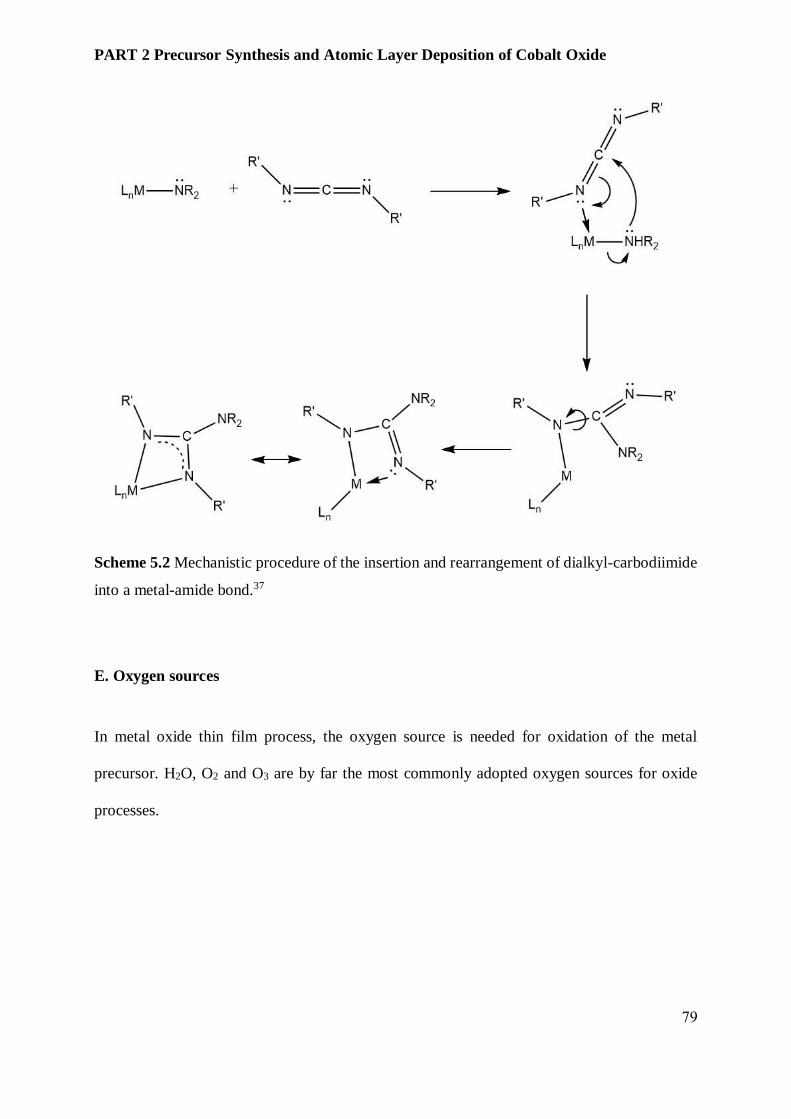
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
79
Scheme 5.2 Mechanistic procedure of the insertion and rearrangement of dialkyl-carbodiimide
into a metal-amide bond.37
E. Oxygen sources
In metal oxide thin film process, the oxygen source is needed for oxidation of the metal
precursor. H2O, O2 and O3 are by far the most commonly adopted oxygen sources for oxide
processes.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
80
5.3.2 Transition metal oxides and cobalt
Transition metal oxides are known to exhibit distinctive physical/chemical properties including
electric/optical and catalytic/semiconductive properties due to the concurrence of delocalized
and localized d-electrons. Notwithstanding, the performance of those properties are highly
related to the morphology, structure and impurities of the materials. These properties can be
reasoned by the band structure of transition metal oxides. The bulk band structure of transition
metal oxides consists of overlapping p-orbitals from the oxygen atoms. These orbitals are highly
populated and of comparatively low energy, forming the valence band. Additionally, the d-
orbitals of the metals are also overlapping, although they are only sparsely populated thus
forming the conduction band. This band is of higher energy than the valence band and the
energy difference is called band gap. This band gap prevents the recombination of electron-hole
pairs which are separated into the valence and conduction band. Exactly this separation allows
the charge transfer onto adsorbed surface species, a requirement for using transition metal
oxides in a manifold of catalytic processes.
Owing to its peculiar and versatile physical/chemical properties of cobalt oxides such as
electrochemical capacitors, high energy density as Li ion batteries, oxygen evolution reaction
(OER) catalyst, optical gas sensors as well as their abundacy in earth, they have attracted
substantial attention from the material scientists. In general, cobalt oxide exists in three different
forms, CoIIO, CoIII2O3 and CoII,III
3O4 and the latest stoichiometry is predominantly studied for
the applications due to its chemical stability and electrochemical properties.6 The focus in this
thesis will lay on the description of cobalt-oxide materials in particular the mixed valent CoII
CoIII containing spinel Co3O4. Co3O4 is studied in the field of water splitting system as an

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
81
effective photoelectrocatalyst. The water splitting system consists two half reactions: namely
hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) and the latter reaction
is the rate-determining step because it requires the coupling of four electrons and four proton
transfers and formation of O−O bond which normally demands high overpotential to initiate
the reaction.38 The Co3O4 unit cell has a spinel structure and direct optical band gap of 1.4-2
eV in the bulk solid state. However, nanostructured Co3O4 shows considerably larger band gap
(ca 2.5 eV).39 A prerequisite for the high activity in OER is the presence of two distinct cobalt
sites in spinel Co3O4. There are tetrahedrally coordinated Co2+Td sites and octahedrally
coordinated Co3+Oh sites.40 It was shown in different studies, that the OER reaction is highly
dependent on the nature of the exposed surface sites, which in turn are interacting with adsorbed
species. The importance of both sites remains controversially discussed in the literature, even
though, both sites are important as they are building up the geometrical environment for the
catalytically active site. In a study by Wang et al. the dependence of the OER on both metal
sites was probed using an ion substitution technique. They used CoAl2O4 only containing
Co2+Td sites as well as ZnCo2O4, which only contains Co3+
Oh sites. From their study using in
operando spectroscopic methods it was revealed that the Co2+Td site is forming CoOOH species
during the reaction which is an important intermediate in the OER reaction, whilst the Co3+Oh
containing species remained catalytically inactive.40
Photocatalytic reactions often require a harsh alkali environment which result in a corrosion of
the layers, limiting the catalysis operation time.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
82
6. References
1. Barrera, E.; Viveros, T.; Avila, A.; Quintana, P.; Morales, M.; Batina, N., Cobalt oxide
films grown by a dipping sol-gel process. Thin Solid Films 1999, 346, 138-144.
2. Armelao, L.; Barreca, D.; Gross, S.; Martucci, A.; Tieto, M.; Tondello, E., Cobalt oxide-
based films: sol–gel synthesis and characterization. J. Non-Cryst. Solids 2001, 293–295, 477-
482.
3. Quinlan, F. T.; Vidu, R.; Predoana, L.; Zaharescu, M.; Gartrner, M.; Groza, J.; Stroeve,
P., Lithium Cobalt Oxide (LiCoO2) Nanocoatings by Sol−Gel Methods. Ind. Eng. Chem. Res.
2004, 43, 2468-2477.
4. Athey, P. R.; Urban, F. K.; Tabet, M. F.; McGahan, W. A., Optical properties of cobalt
oxide films deposited by spray pyrolysis. J. Vac. Sci. Technol., A 1996, 14, 685-692.
5. Shinde, V. R.; Mahadik, S. B.; Gujar, T. P.; Lokhande, C. D., Supercapacitive cobalt
oxide (Co3O4) thin films by spray pyrolysis. Appl. Surf. Sci. 2006, 252, 7487-7492.
6. Louardi, A.; Rmili, A.; Ouachtari, F.; Bouaoud, A.; Elidrissi, B.; Erguig, H.,
Characterization of cobalt oxide thin films prepared by a facile spray pyrolysis technique using
perfume atomizer. J. Alloy. Compd. 2011, 509, 9183-9189.
7. Abbas, T. A.-H.; Slewa, L. H.; Khizir, H. A.; Kakil, S. A., Synthesis of cobalt oxide
(Co3O4) thin films by electrostatic spray pyrolysis technique (ESP). J. Mater. Sci. Mater.
Electron. 2016, 1-7.
8. Meyer, W.; Biedermann, K.; Gubo, M.; Hammer, L.; Heinz, K., Surface structure of
polar Co3O4 (111) films grown epitaxially on Ir(100)-(1 × 1). J. Phys. Condens. Matter 2008,
20, 265011.
9. Ngo, T. Q.; Posadas, A.; Seo, H.; Hoang, S.; McDaniel, M. D.; Utess, D.; Triyoso, D.
H.; Buddie Mullins, C.; Demkov, A. A.; Ekerdt, J. G., Atomic layer deposition of photoactive

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
83
CoO/SrTiO3 and CoO/TiO2 on Si(001) for visible light driven photoelectrochemical water
oxidation. J. Appl. Phys. 2013, 114, 084901.
10. Johnson, R. W.; Hultqvist, A.; Bent, S. F., A brief review of atomic layer deposition:
from fundamentals to applications. Mater. Today 2014, 17, 236-246.
11. Mazzi, A.; Bazzanella, N.; Orlandi, M.; Edla, R.; Patel, N.; Fernandes, R.; Miotello, A.,
Physical vapor deposition of mixed-metal oxides based on Fe, Co and Ni as water oxidation
catalysts. Mater. Sci. Semicond. Process. 2016, 42, Part 1, 155-158.
12. Xu, T.; Schwarz, M.; Werner, K.; Mohr, S.; Amende, M.; Libuda, J., Structure-
Dependent Anchoring of Organic Molecules to Atomically Defined Oxide Surfaces: Phthalic
Acid on Co3O4(111), CoO(100), and CoO(111). Chem. Eur. J. 2016, 22, 5384-5396.
13. Klepper, K. B.; Nilsen, O.; Fjellvåg, H., Growth of thin films of Co3O4 by atomic layer
deposition. Thin Solid Films 2007, 515, 7772-7781.
14. Leskelä, M.; Ritala, M., Atomic Layer Deposition Chemistry: Recent Developments
and Future Challenges. Angew. Chem. Int. Ed. 2003, 42, 5548-5554.
15. Leskelä, M.; Ritala, M., Rare-earth oxide thin films as gate oxides in MOSFET
transistors. J. Solid State Chem. 171, 170-174.
16. Tuomo Suntola, J. H., Atomic Layer Epitaxy. Annual Review of Materials Science 1985,
15, 177-195.
17. Suntola, T., Atomic layer epitaxy. Materials Science Reports 1989, 4, 261-312.
18. Suntola, T., Surface chemistry of materials deposition at atomic layer level. Appl. Surf.
Sci. 1996, 100, 391-398.
19. Niinistö, L., Atomic layer epitaxy. Current Opinion in Solid State and Materials Science
1998, 3, 147-152.
20. Niinistö, L.; Päiväsaari, J.; Niinistö, J.; Putkonen, M.; Nieminen, M., Feature Article:
Advanced electronic and optoelectronic materials by Atomic Layer Deposition: An overview

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
84
with special emphasis on recent progress in processing of high-k dielectrics and other oxide
materials. physica status solidi (a) 2004, 201, 1375-1375.
21. Adelmann, C.; Delabie, A.; Schepers, B.; Rodriguez, L. N. J.; Franquet, A.; Conard, T.;
Opsomer, K.; Vaesen, I.; Moussa, A.; Pourtois, G.; Pierloot, K.; Caymax, M.; Van Elshocht,
S., Atomic Layer Deposition of Tantalum Oxide and Tantalum Silicate from Chloride
Precursors. Chem. Vap. Deposition 2012, 18, 225-238.
22. Mikko Ritala, M. L., Handbook of Thin Films Materials: Deposition and Processing of
Thin Films. In Handbook of Thin Films, Nalwa, H. S., Ed. Academic Press: Burlington, 2002;
Vol. 1.
23. Ritala, M.; Niinisto, J., Chapter 4 Atomic Layer Deposition. In Chemical Vapour
Deposition: Precursors, Processes and Applications, The Royal Society of Chemistry: 2009;
pp 158-206.
24. Tiitta, M.; Niinistou, L., Volatile Metal β-Diketonates: ALE and CVD precursors for
electroluminescent device thin films. Chem. Vap. Deposition 1997, 3, 167-182.
25. Binnemans, K., Chapter 225 - Rare-earth beta-diketonates. In Handbook on the Physics
and Chemistry of Rare Earths, Karl A. Gschneidner, J.-C. G. B.; Vitalij, K. P., Eds. Elsevier:
2005; Vol. Volume 35, pp 107-272.
26. Luten, H. A.; Rees, W. S.; Goedken, V. L., Preparation and structural characterization
of, and chemical vapor deposition studies with, certain yttrium tris(β-diketonate) compounds.
Chem. Vap. Deposition 1996, 2, 149-161.
27. Lim, B. S.; Rahtu, A.; Park, J.-S.; Gordon, R. G., Synthesis and Characterization of
Volatile, Thermally Stable, Reactive Transition Metal Amidinates. Inorg. Chem. 2003, 42,
7951-7958.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
85
28. Milanov, A. P.; Thiede, T. B.; Devi, A.; Fischer, R. A., Homoleptic Gadolinium
Guanidinate: A Single Source Precursor for Metal−Organic Chemical Vapor Deposition of
Gadolinium Nitride Thin Films. J. Am. Chem. Soc. 2009, 131, 17062-17063.
29. Lim, B. S.; Rahtu, A.; Gordon, R. G., Atomic layer deposition of transition metals. Nat.
Mater. 2003, 2, 749-754.
30. Bailey, P. J.; Pace, S., The coordination chemistry of guanidines and guanidinates.
Coord. Chem. Rev. 2001, 214, 91-141.
31. Coyle, J. P.; Johnson, P. A.; DiLabio, G. A.; Barry, S. T.; Müller, J., Gas-Phase
Thermolysis of a Guanidinate Precursor of Copper Studied by Matrix Isolation, Time-of-Flight
Mass Spectrometry, and Computational Chemistry. Inorg. Chem. 2010, 49, 2844-2850.
32. Barry, S. T., Amidinates, guanidinates and iminopyrrolidinates: Understanding
precursor thermolysis to design a better ligand. Coord. Chem. Rev. 2013, 257, 3192-3201.
33. Musashi, Y.; Sakaki, S., Insertion of carbon dioxide into a rhodium(III)-hydride bond:
a theoretical study [dagger]. Journal of the Chemical Society, Dalton Transactions 1998, 577-
584.
34. Edelmann, F. T., Chapter 3 - Advances in the Coordination Chemistry of Amidinate and
Guanidinate Ligands. In Adv. Organomet. Chem., Anthony, F. H.; Mark, J. F., Eds. Academic
Press: 2008; Vol. Volume 57, pp 183-352.
35. Edelmann, F. T., Lanthanide amidinates and guanidinates: from laboratory curiosities
to efficient homogeneous catalysts and precursors for rare-earth oxide thin films. Chem. Soc.
Rev. 2009, 38, 2253-2268.
36. Chang, C.-C.; Hsiung, C.-S.; Su, H.-L.; Srinivas, B.; Chiang, M. Y.; Lee, G.-H.; Wang,
Y., Carbodiimide Insertion into Organoaluminum Compounds and Thermal Rearrangement of
the Products. Organometallics 1998, 17, 1595-1601.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
86
37. Rowley, C. N.; DiLabio, G. A.; Barry, S. T., Theoretical and Synthetic Investigations
of Carbodiimide Insertions into Al−CH3 and Al−N(CH3)2 Bonds. Inorg. Chem. 2005, 44,
1983-1991.
38. Koper, M. T. M., Thermodynamic theory of multi-electron transfer reactions:
Implications for electrocatalysis. J. Electroanal. Chem. 2011, 660, 254-260.
39. Ramakrishnan, V.; Kim, H.; Park, J.; Yang, B., Cobalt oxide nanoparticles on TiO2
nanorod/FTO as a photoanode with enhanced visible light sensitization. RSC Adv. 2016, 6,
9789-9795.
40. Wang, H.-Y.; Hung, S.-F.; Chen, H.-Y.; Chan, T.-S.; Chen, H. M.; Liu, B., In Operando
Identification of Geometrical-Site-Dependent Water Oxidation Activity of Spinel Co3O4. J.
Am. Chem. Soc. 2016, 138, 36-39.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
87
6.
Low temperature atomic layer deposition of cobalt oxide
as an effective catalyst layer for photoelectrochemical
water splitting devices
Jiyeon Kim, Tomi Iivonen, Jani Hämäläinen, Marianna Kemell, Kristoffer Meinander,
Kenichiro Mizohata, Lidong Wang, Radim Beranek, Markku Leskelä, Anjana Devi
All contents of this paper have been published in Chem. Mater., 2017, 29 (14), 5796–5805.
Reprinted with permissions, Copyright © 2017 American Chemical Society.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
88
6. Low temperature atomic layer deposition of cobalt oxide as an
effective catalyst layer for photoelectrochemical water splitting
devices2
6.1 Abstract
We have developed a low-temperature atomic layer deposition (ALD) process for depositing
crystalline and phase pure spinel cobalt oxide (Co3O4) films at 120 °C using Co(tBu2DAD)2 and
ozone as co-reagent. X-ray diffraction, UV-Vis spectroscopy, atomic force microscopy, field
emission scanning electron microscopy, X-ray photoelectron spectroscopy and time-of-flight
elastic recoil detection analysis were performed to characterize the structure and properties of
the films. Full characterization of the film demonstrates that as-deposited Co3O4 films are
crystalline with low amount of impurities (<5 % hydrogen) despite low deposition temperatures.
Deposition of ultra-thin Co3O4 (< 1.1 nm) layers onto thin TiO2 films (100 nm) resulted in 30 %
improvement of photocurrent compared to pristine TiO2 films, demonstrating the applicability
of the developed ALD process for deposition of effective, conformal electrocatalytic layers in
photoelectrochemical water-splitting devices.
2 This chapter has been in part submitted for publication and is currently under revision.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
89
6.2 Introduction
The interest in nanostructured cobal(II,III) oxide has been growing enormously over the past
few years, due to its gigantic potential for a variety of applications, in such areas as
electrochemistry, catalysis, sensing, batteries and energy storage.1-5 Co3O4 proved to be an
excellent electrocatalyst for water oxidation and is hence of outstanding importance, since the
water oxidation reaction remains the kinetic bottleneck in water splitting systems. Because of
that, CO3O4 catalysts are among the most favorable materials in this field, which is directly
connected to a sustainable and low-cost energy supply. 6-13 Electrocatalyst activity and their use
in thin-film devices (e.g. for chemical water splitting) generally depends on a variety of factors,
such as composition, morphology, thickness and optical properties.8, 14-17 The most common
methods to prepare cobalt oxide thin films are for example sol-gel processes,18-20 spray
pyrolysis,21-24 electrodeposition14 and physical vapor deposition (PVD).25-28 The precision of
these methods is still limited and hence the development of new deposition techniques for these
films is necessary. Up to date, the most promising methods, which outperform the afore
mentioned by means of film thickness and phase purity are chemical vapor deposition (CVD)
and atomic layer deposition (ALD).29 Among these techniques, specifically ALD is a very
powerful method to deposit films which possess exceptional conformality and distinct
morphology and phase control on the nanometer scale is possible. 26, 30 Furthermore, the
deposition of thin films on complex-shaped substrates is possible by this method. The before
mentioned benefits of ALD are in particular in the light of applicability of Co3O4 in devices of
marked importance. 31-33 These applications include for example the deposition of light
adsorbers, protective layers or catalysts film for photoelectrochemical water splitting devices.
34-39 Noticeably, only very few reports on the preparation of cobalt oxide exist, eventhough for
the deposition of other metal oxides ALD processes are quite common. Among the few

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
90
described ALD based routes towards cobalt oxides are for instance the deposition of cobalt
oxide from the cobalt amidinates precursor Co(iPrNCMeNiPr)2 in a water assisted ALD
process.30, 40 Other examples are the use of CoCp2 (Cp = cyclopentadienyl),11, 41 CCTBA
(dicobalthexacarbonyl-tert-butylacetylene),42 Co(thd)2 (thd = 2,2,6,6-tetramethylheptan-3,5-
dionate)29 and Co(acac)2 (acac = acetylacetonate)32, 43 combined with either O2 plasma or O3 as
the oxygen source. Drawbacks of these processes are however the high deposition temperatures
in combination with low film growth rates and impure thin films. 29, 32 Therefore, the
development of new cobalt precursors made from bidentate chelating ligands, for example
amidinates was an interesting alternative.44 By the bidentate binding of the amidinate
counteracts effects observed in monodentate compounds (i.e. amido or imido compounds),
particularly low thermal stability. At the same time, the all nitrogen coordinated amidinates
possess low deposition temperatures caused by the lower bond strength of M-N compared to
M-O.45 Remarkably, in the group of Winter, a amidinate build up from redox non-innocent
linkers was prepared, namely bis(1,4-di-tert-butyl-1,3-diazadiene) cobalt (II) Co(tBu2DAD)2.46
This kind of compound bear much higher redox activity under mild conditions. In a recent
report, a low temperature ALD process for the preparation of metallic Co0 films using formic
acid as a reducing agent and the before mentioned Co(tBu2DAD)2 was described. 5

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
91
6.3 Results and Discussion
2.3.1 Thermal properties of precursor [Co(tBu2DAD)2]
This chapter describes the development of a low temperature ALD process used for the
fabrication of phase pure Co3O4 thin films. The process employs [Co(tBu2DAD)2] as cobalt and
ozone as oxygen sources respectively. Additionally, it will be demonstrated that the prepared
films can be effectively used for the preparation of Co3O4/TiO2 electrocatalysts. TiO2 thin film
have gained massive attention and are utilized as protective layers in novel water splitting
devices.47-50
Scheme 6.1: Synthetic route towards [Co(tBu2DAD)2]
Scheme 6.1 depicts the synthesis route towards [Co(tBu2DAD)2], which was prepared in analogy
to the known procedure from the literature (Details Supporting Information). The precursor
shows very promising thermal properties, TGA/DTA and isothermal TG studies are presented
in Figure 6.1. Thermogravimetric studies of [Co(tBu2DAD)2] show a single step weight loss
with an onset at 160 °C. The residual mass is very low and accounts to 4 % of the initial mass.
Differential Thermal Analysis (DTA) reveals a melting point of 175 °C, which is in line with
the previously reported value (174-175 °C).46 The conducted isothermal studies show a linear
weight loss, and constant evaporation rate over a time range of 180 minutes. This makes them
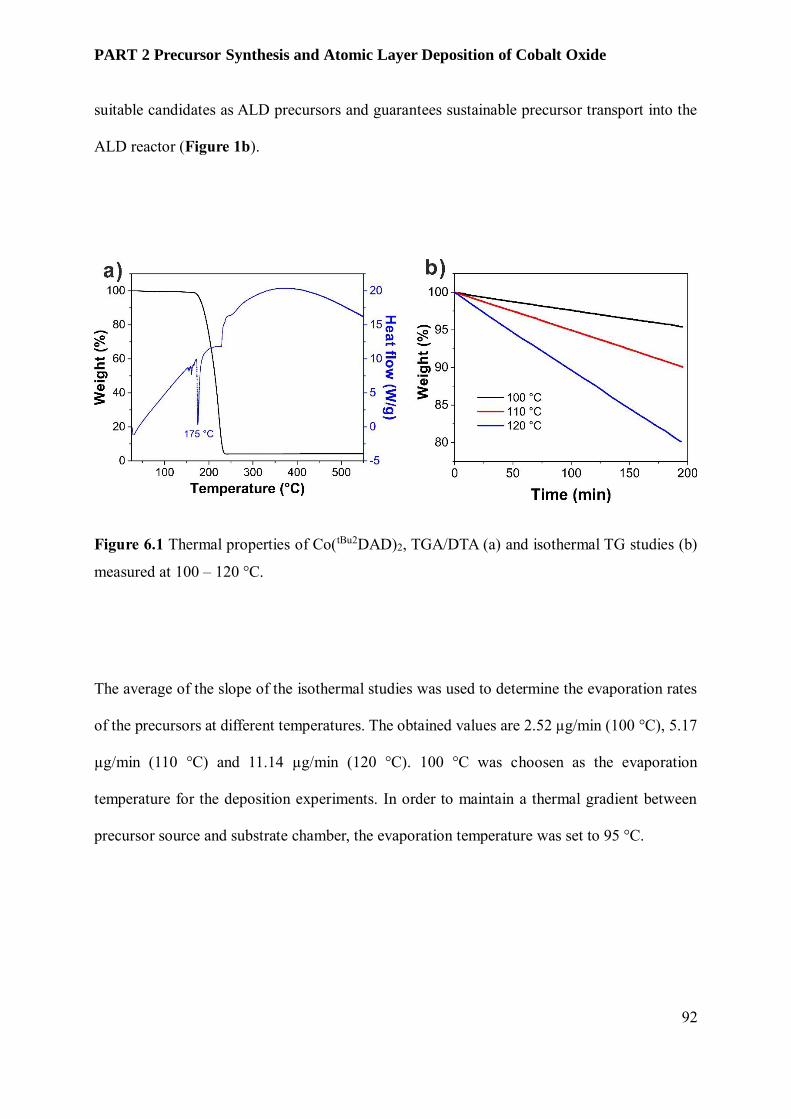
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
92
suitable candidates as ALD precursors and guarantees sustainable precursor transport into the
ALD reactor (Figure 1b).
Figure 6.1 Thermal properties of Co(tBu2DAD)2, TGA/DTA (a) and isothermal TG studies (b)
measured at 100 – 120 °C.
The average of the slope of the isothermal studies was used to determine the evaporation rates
of the precursors at different temperatures. The obtained values are 2.52 µg/min (100 °C), 5.17
µg/min (110 °C) and 11.14 µg/min (120 °C). 100 °C was choosen as the evaporation
temperature for the deposition experiments. In order to maintain a thermal gradient between
precursor source and substrate chamber, the evaporation temperature was set to 95 °C.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
93
6.3.2 Film Deposition
In order to verify the ALD growth of Co3O4, the deposition temperature, the precursor pulse
lengths of Co(tBu2DAD)2/O3 and the number of deposition cycles were varied (Figure 6.1). In
Figure 6.1a the growth rate is depicted as a function of temperature in the range 100 – 150 °C.
The temperature window with a constant growth rate is very narrow (110 − 120°C), which has
been observed for other ozone assisted metal oxide ALD proceses at high temperatures, due to
decomposition of ozone on the metal oxide surface.51-52 At deposition temperatures higher than
120 °C full coverage was not feasible over the whole substrate, due to the configuration of the
used F-120 reactor. Using 40 mm Si trenches, fully conformal Co3O4 deposition was also not
achieved, although 6 s pulses and 10 s purge times were used for both precursors. An
explanation for not reaching conformal depositions in the trenches are the ozone decomposition
of Co3O4 and the large surface area of the trench substrate. Hence, 120 °C were chosen as the
ideal deposition temperature for all further experiments (See Appendix, Figure 6.9).
After 2 second pulses of Co precursors followed by 2 sec nitrogen purges saturation of film
growth was observed (Figure 6.2b). For O3 saturation was also found to occur after 2 second
ozone purges. These findings are proof for the self-limiting character of the deposition at a
temperature of 120 °C. Noticeably, the linear dependence of the film thickness on the number
of deposition cycles further underlines the occurrence of a typical ALD process. Astonishingly,
the determined growth rate of 1.1 Å/cycle is five times higher than the previously reported
Co3O4 ALD deposition rate (0.25 Å/cycle, CoCp2/O3 ALD Window 110-300 °C).29This rate is
also slightly higher as recently found results, which used a different cobalt precursor
(diazadienyl Co precursor) and O3 as well as O2 as oxygen sources.53
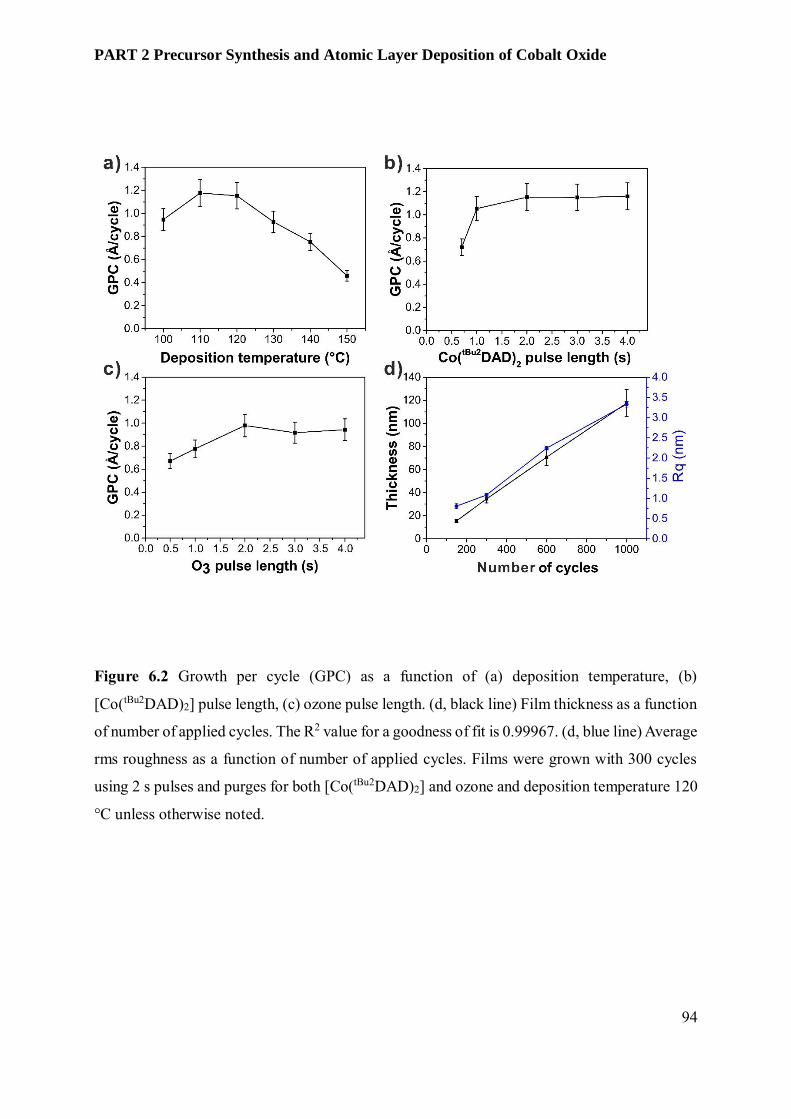
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
94
Figure 6.2 Growth per cycle (GPC) as a function of (a) deposition temperature, (b)
[Co(tBu2DAD)2] pulse length, (c) ozone pulse length. (d, black line) Film thickness as a function
of number of applied cycles. The R2 value for a goodness of fit is 0.99967. (d, blue line) Average
rms roughness as a function of number of applied cycles. Films were grown with 300 cycles
using 2 s pulses and purges for both [Co(tBu2DAD)2] and ozone and deposition temperature 120
°C unless otherwise noted.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
95
6.3.3 Film Characterization
A. Structure and Morphology
In order to determine the structure and the composition of the prepared Cobalt oxide thin films,
X-ray diffraction measurements, FESEM and atomic force microscopy measurements were
performed.
The X-ray diffraction measurements were performed in grazing incidence mode and are shown
in Figure 3. The films deposited at 120 °C are of polycrystalline nature and the reflections
found can be attributed to spinel cobalt lattice planes, namely the (111), (220), (311), (222),
(400), (511) and (440) planes.54 Already for the as-deposited films high intensities indicating
high crystallinity can be observed, which is in strong contrast to previously reported, much less
crystalline Co3O4 thin films prepared by ALD. 29, 53, 55 High Temperature XRD showed in air
and nitrogen atmosphere an increase in crystallinity in the range from room temperature to 700
°C. However, in oxygen free environment a reduction of the prepared Co3O4 thin films to CoO
was observed. This was indicated by new Bragg reflections fitting to the (111), (200) and (220)
lattice planes of periclase CoO. 56

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
96
Figure 6.3 HTXRD patterns of Co3O4 films deposited over 1000 cycles at 120 °C in a) air b)
nitrogen. The measurement temperature is shown under each diffractogram.
Via FSEM and AFM the surface morphology of the as-deposited films was analyzed. In Figure
6.4a and b the SEM and AFM images of the Co3O4 films deposited at 120 °C over 1000 cycles
are displayed. It is visible in both figures that homogeneous, well defined nano crystallites were
deposited without the necessity of any post annealing processes. The grain size of the nano
crystallites were in a range of 75 – 90 nm. In Figure 6.4c roughness studies with increasing
number of deposition cycles are depicted. Noticeably, the Surface average Rq values rise
linearly with the number of applied deposition cycles, with values of 0.81 nm, 1.1nm, 2.2 nm
and 3.3 nm for 150, 300, 600 and 1000 deposition cycles respectively (dependency of roughness
on number of cycles depicted in Figure 6.4c).

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
97
Figure 6.4 (a) Plane-view SEM and (b) representative AFM images of Co3O4 thin film
deposited over 1000 cycles at 120 °C. (c) AFM topographs of Co3O4 thin films deposited over
different cycles at 120 °C and the corresponding Rq values.
B. Film Composition
To confirm the chemical composition and the surface properties of the Co3O4 films prepared
at 120 °C over 1000 cycles, XPS and ToF-ERDA measurements were conducted. The XPS
measurements were performed in order to analyze the chemical states of surface cobalt and
oxygen in the prepared samples. In Figure 6.5 the survey spectrum of CO3O4 deposited on
Si(100) is depicted. Figure 6.5a shows the core level spectrum of Co 2p, which indicates spin
orbit splitting into 2p1/2 and 2p3/2. The plateau shake-up peaks confirm the presence of a
mixed Co2+/3+ oxidation state. From the satellite line tit is possible to make a distinction
between Co2+ and Co3+ states. Pure Co2+ shows a projecting shake-up peak within the areas of
786 and 790 eV. For Co3+ this shake-up peak can be found at 790 eV. 57 Because the 2p1/2 and
2p3/2 components contain identical chemical information, the 2p3/2 peak was chosen for curve
fitting and qualitative analysis, due to higher intensity. The binding energy peaks at 779.6 and
781.1 eV strongly suggest the presence of Co3+ and Co2+ species within the sample and the
shake up peaks belonging to these peaks can be found at 786.0 and 789.7 eV. 57-59 At 782.7
eV a peak that can be ascribed to Co(OH)2 is identified, which suggests the presence of this
species at the surface. 57 The O1s area also shows two peaks, one located at 531.3 eV and
529.6 eV. The first one is attributed to the lattice oxygen present in spinel Co3O4 and the latter
arises from hydroxyl groups present in Co(OH)2.
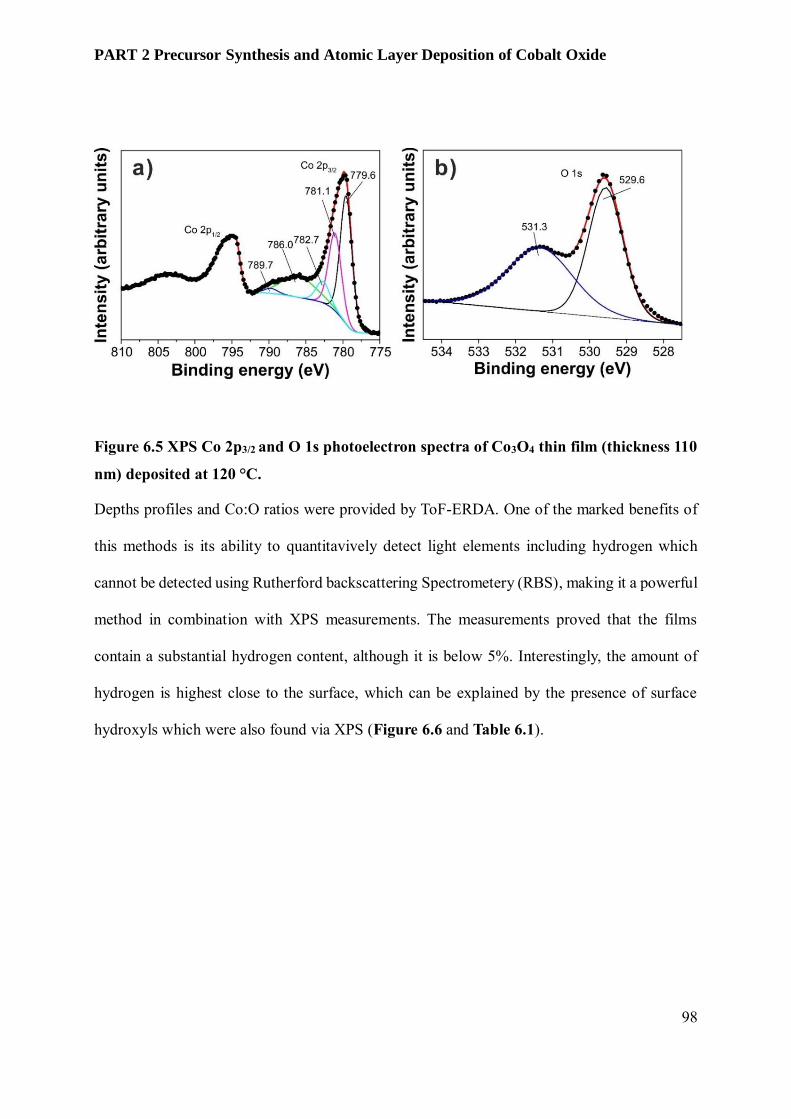
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
98
Figure 6.5 XPS Co 2p3/2 and O 1s photoelectron spectra of Co3O4 thin film (thickness 110
nm) deposited at 120 °C.
Depths profiles and Co:O ratios were provided by ToF-ERDA. One of the marked benefits of
this methods is its ability to quantitavively detect light elements including hydrogen which
cannot be detected using Rutherford backscattering Spectrometery (RBS), making it a powerful
method in combination with XPS measurements. The measurements proved that the films
contain a substantial hydrogen content, although it is below 5%. Interestingly, the amount of
hydrogen is highest close to the surface, which can be explained by the presence of surface
hydroxyls which were also found via XPS (Figure 6.6 and Table 6.1).
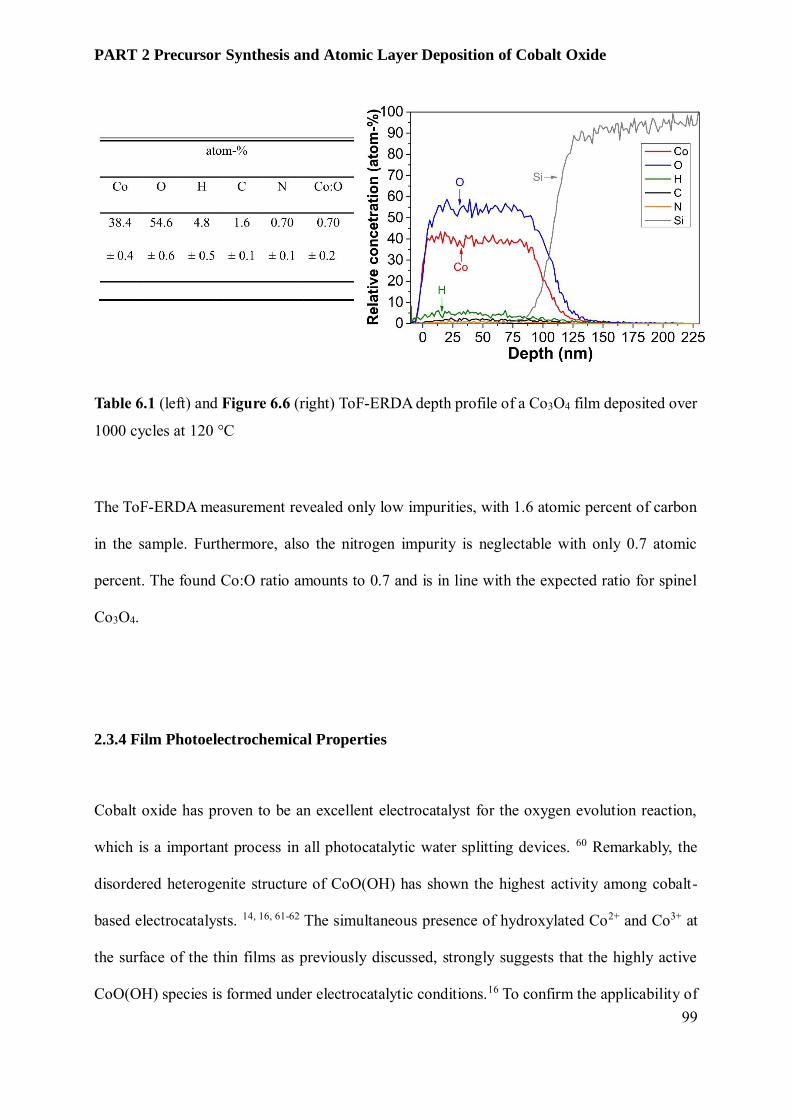
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
99
Table 6.1 (left) and Figure 6.6 (right) ToF-ERDA depth profile of a Co3O4 film deposited over
1000 cycles at 120 °C
The ToF-ERDA measurement revealed only low impurities, with 1.6 atomic percent of carbon
in the sample. Furthermore, also the nitrogen impurity is neglectable with only 0.7 atomic
percent. The found Co:O ratio amounts to 0.7 and is in line with the expected ratio for spinel
Co3O4.
2.3.4 Film Photoelectrochemical Properties
Cobalt oxide has proven to be an excellent electrocatalyst for the oxygen evolution reaction,
which is a important process in all photocatalytic water splitting devices. 60 Remarkably, the
disordered heterogenite structure of CoO(OH) has shown the highest activity among cobalt-
based electrocatalysts. 14, 16, 61-62 The simultaneous presence of hydroxylated Co2+ and Co3+ at
the surface of the thin films as previously discussed, strongly suggests that the highly active
CoO(OH) species is formed under electrocatalytic conditions.16 To confirm the applicability of

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
100
the prepared cobalt oxide thin films for water splitting, ultrathin films (1.1 nm) of the material
were deposited on 100 nm thick TiO2 layers on FTO glass substrate. For future
photoelectrochemical water splitting systems these composite TiO2/Co3O4 composites are of
very high interest. This is because currently TiO2 film are in the focus of research as highly
effective protection layers for low-gap semiconductors including Si, GaAs, Gap and CdTe.
These semiconductors usually undergo severe photocorrosion.47-50
The photoaction spectra (Figure 6.7) as well as the spectra under simulated solar light (Figure
6.8) show a striking increase of the photocurrent response of 30% in comparison to pristine
TiO2. Notably, the improvement excludes the presence of any harmful surface defect states in
TiO2 caused by the ALD-grown Co3O4. These effects could lead to enhanced surface
recombination. It is noticeable that the charge separation is enhanced, because the Co3O4
effectively extracts the holes from TiO2. This finally leads to the observed dramatic increase of
photocurrent. 63 Additionally, another advantage of Co3O4 is its role in the passivation of surface
states in TiO2, which has been recently reported for some photoanodes based on Fe2O3 or
BiVO4. 64-66 The photoholes in TiO2 have a highly positive nature and the presence of a co-
catalyst to induce full water oxidation under photoelectrochemical conditions is not necessary.
Therefore, the increased oxygen evolution rate can be compared to an enhancement of
photocurrent without a severe change in the faradaic oxygen evolution efficiency. Anyhow,
these results showcase that the developed low temperature ALD process is useful for the
preparation of electrocatalytic cobalt oxide layers on top of TiO2 layers. These TiO2 layers are
of crucial interest for the preparation of photoanodes for tandem water-splitting devices, which
are currently very susceptible towards photocorossion.47-50
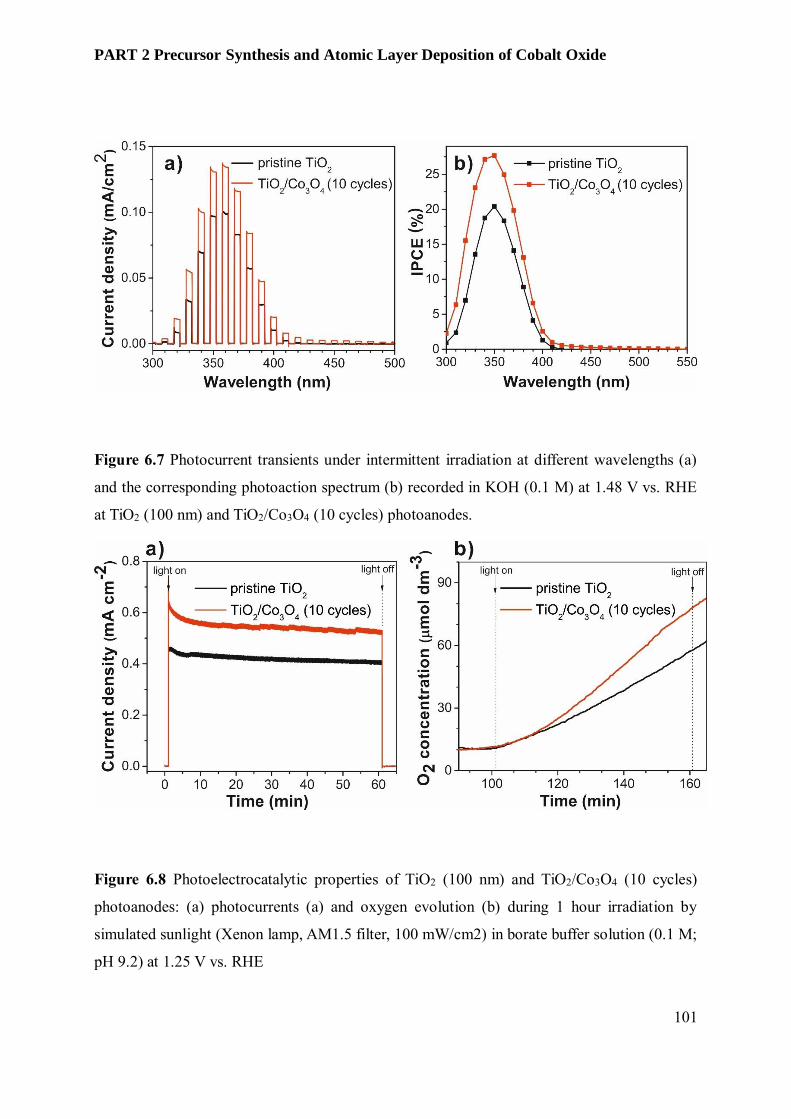
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
101
Figure 6.7 Photocurrent transients under intermittent irradiation at different wavelengths (a)
and the corresponding photoaction spectrum (b) recorded in KOH (0.1 M) at 1.48 V vs. RHE
at TiO2 (100 nm) and TiO2/Co3O4 (10 cycles) photoanodes.
Figure 6.8 Photoelectrocatalytic properties of TiO2 (100 nm) and TiO2/Co3O4 (10 cycles)
photoanodes: (a) photocurrents (a) and oxygen evolution (b) during 1 hour irradiation by
simulated sunlight (Xenon lamp, AM1.5 filter, 100 mW/cm2) in borate buffer solution (0.1 M;
pH 9.2) at 1.25 V vs. RHE

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
102
6.4 Conclusion and Outlook
This chapter nicely demonstrates the possibility to deposit spinel cobalt oxide via ALD by using
an ozone assisted process with [Co(tBu2DAD)2]as a precursor. The process was conducted at
low temperatures (120 °C) and the obtained films are conformal, crystalline, phase pure and
only contain extremely low carbon and hydrogen contamination. A Post annealing treatment to
increase these films properties is not necessary. Furthermore, by the ALD process ultrathin
Co3O4 electrocatalyst films were prepared on top of TiO2 films, which lead to a striking
improvement of photoconversion efficiency. Due to the conformal nature of the ALD grown
films and the importance of TiO2 protective layers for photoelectrodes these finding will be of
very high relevance for the fabrication of a variety of solar water splitting devices.
6.5 Appindex
Thickness(nm) Top Middle Bottom
114 101 84
122 93 85
111 93 77
119 93 71

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
103
130 89 77
138 92 71
80
Average
thickness (nm) 122 94 78
Figure 6.9 SEM image of Si trench with an aspect ratio of 20:1 (depth 40 m, width 2 m).
Co3O4 film has been deposited over 1000 cycles at 120 °C using pulse lengths of 6 seconds
for both Co(tBu2DAD)2 and ozone. The purge length for both precursors was 10 seconds.
Fully conformal Co3O4 films could not be deposited on 40 m Si trenches (aspect ratio of
1:20) despite using 6 s pulse and 10 s purge times of both precursors. A conceivable
explanation of for not achieving full conformality in the trench structure is the combined
effect of ozone decomposing effect of Co3O4 and high surface area of the substrate.
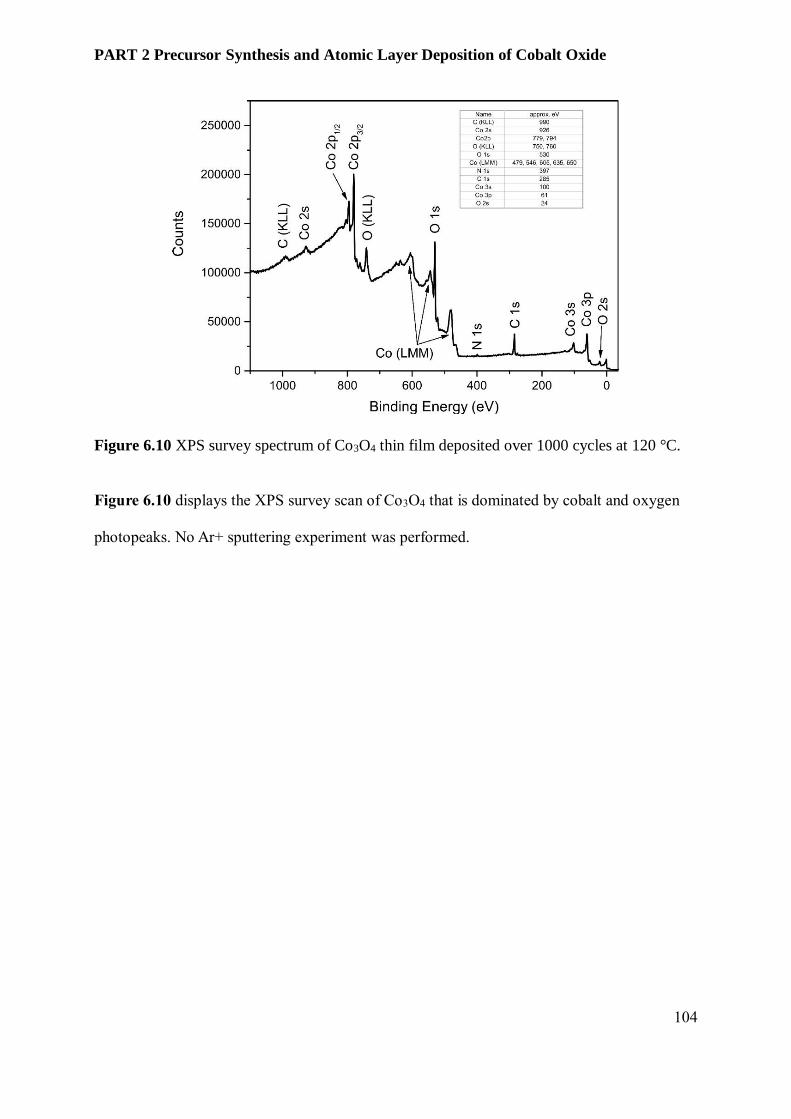
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
104
Figure 6.10 XPS survey spectrum of Co3O4 thin film deposited over 1000 cycles at 120 °C.
Figure 6.10 displays the XPS survey scan of Co3O4 that is dominated by cobalt and oxygen
photopeaks. No Ar+ sputtering experiment was performed.
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
105
Figure 6.10 UV-Vis transmission spectrum and corresponding Tauc plot of Co3O4 thin film
deposited over 1000 cycles at 120 °C.
UV-Vis spectroscopy was performed on the as-deposited Co3O4 film grown on soda-lime
glass to determine the optical bandgap of Co3O4 thin films via Tauc plot. Obtained data
indicated a direct band gap value of 2.02 eV.
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
106
Figure 6.11 UV-Vis transmission spectra of TiO2 and Co3O4
UV-Vis spectroscopy was performed on pristine TiO2 (black line) and TiO2/Co3O4 (red line)
grown on FTO glass. As can be seen in Figure S5, the shift between pristine TiO2 and
TiO2/Co3O4 was not observed.
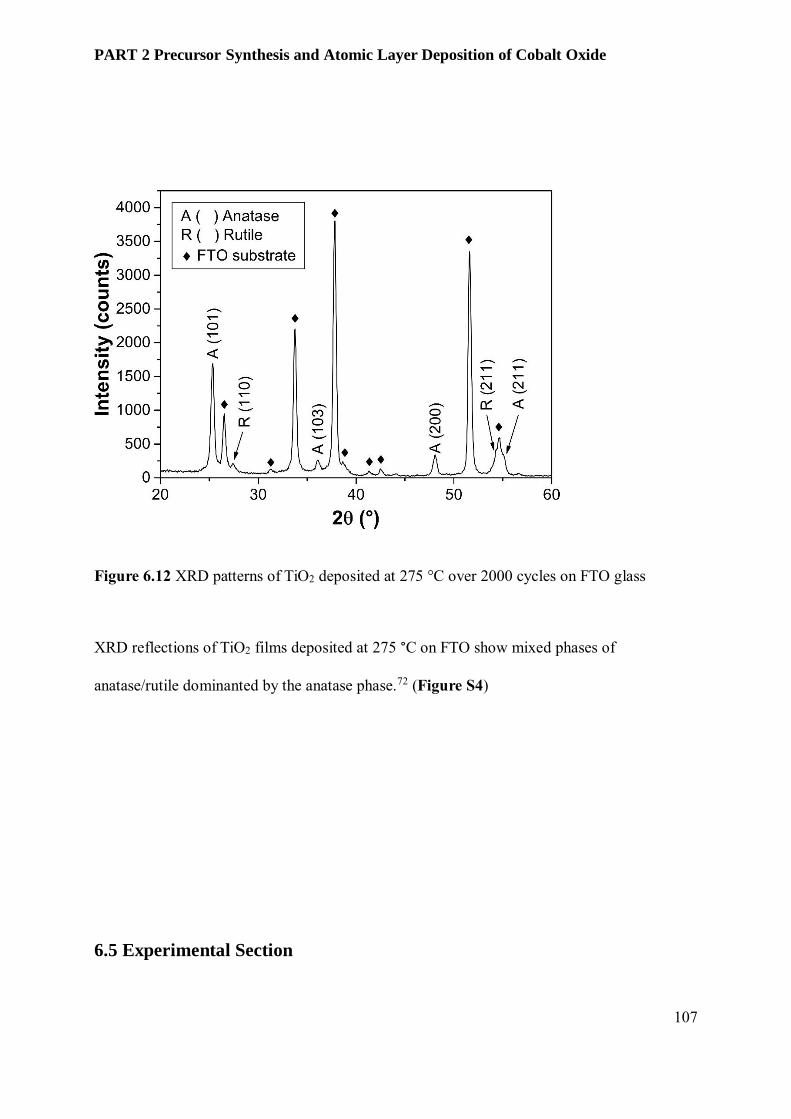
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
107
Figure 6.12 XRD patterns of TiO2 deposited at 275 °C over 2000 cycles on FTO glass
XRD reflections of TiO2 films deposited at 275 °C on FTO show mixed phases of
anatase/rutile dominanted by the anatase phase.72 (Figure S4)
6.5 Experimental Section

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
108
6.5.1 Precursor synthesis
[Co(tBu2DAD)2] was synthesized modifying the literature reported procedure.46 All
manipulations were carried out in an atmosphere of argon purified by Cu catalysts and
molecular sieves using standard Schlenk and glove-box techniques. All solvents were dried
using an MBraun Solvent Purification System. The crude mixture after the synthesis was
sublimed at 60 °C in vacuo (10-3 mbar), then the temperature was gradually increased up to 90
°C to remove excess amount of free ligands. Deep dark- green crystals were collected on a
cooling finger by slow sublimation (100 °C, 10-3 mbar). The purity and the thermal properties
were analyzed using elemental analysis, mass spectrometry and thermogravimetric analysis
(TGA, EXSTAR 6000 TG/DTA 6200, Seiko Instruments Inc.). The measurements were
performed under inert gas atmosphere using nitrogen (99.999 %) at ambient pressure.
Approximately 10 mg of samples were filled in aluminum crucibles with a circular opening.
The heating rate was 5 °C/min and a nitrogen flow of 300 ml/min was used. For isothermal TG
studies, the sample was heated with a rate of 10 °C/min until the desired temperature was
reached, then mass loss was measured for 180 min at constant temperature.
EI-MS [m/z]: 395.3 [M]+, 380.2 [M]+-CH3, 338.2 [M]+-C(CH3)3, 227.1 [M]+-L, 212.1 [M]+-L-
CH3

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
109
6.5.2 Film deposition
Cobalt oxide thin films were deposited using a hot-wall flow-type ALD reactor (ASM
Microchemistry F-120) operated under a nitrogen atmosphere of approximately 5 mbar.
Nitrogen (99.999 %) was used as both carrier and purging gas. Si (100) and soda lime glass cut
to 5 × 5 cm2 pieces were used as substrates. Co(tBuDAD)2 was sublimed from an open glass
boat held inside the reactor at 95 − 100 °C. Ozone was produced from oxygen (99.999 %) using
a Weeco Ozomatic Modulator 4 HC Lab Ozone generator (ozone concentration of
approximately 100 g/Nm3) and introduced to the reactor using needle and solenoid valves. The
deposition temperature was varied from 100 – 150 °C. Thin films for photoelectrochemical
studies were prepared as follows: 100 nm TiO2 thin films were deposited on 1.5 x 2.0 cm2 FTO
substrates (Solems TEC7) by applying the TiOMe4 / H2O ALD process at 275 °C.67 The
TiO2/Co3O4 structure was obtained by applying 10 deposition cycles of the Co3O4 process at a
temperature of 120 °C.
6.5.3 Film characterization
All characterizations were conducted using films deposited over 300 cycles (film thickness of
approximately 30 nm) on Si (100) substrates unless otherwise noted. All thickness
measurements related to saturation studies and the effect of deposition temperature were done
on Si (100) with an in situ aluminum oxide layer deposited over 100 cycles using
trimethylaluminum (TMA) and ozone (Al2O3 film thickness of approximately 10 nm). The
purpose of the Al2O3 layer was to twofold: I) To produce a reproducible, hydroxylated starting
surface68 for each cobalt oxide film deposition and II) to cover and thus passivate cobalt oxide
that would grow on the ALD reactor walls and eventually cause ozone decomposition.52

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
110
Film thickness was measured with energy dispersive X-ray spectrometry (EDX). The EDX
spectra were collected using an Oxford INCA 350 microanalysis system connected to a Hitachi
S-4800 field emission scanning electron microscope (FESEM). Film thicknesses were
calculated from the EDX spectra using the GMRfilm software and assuming bulk density values
of 6.1 g cm-3 for the Co3O4 films.69
All X-ray diffraction measurements were performed in the grazing incidence (GI) geometry
using an incident angle of 1 °. Film crystal structure was identified GI-(XRD) patterns obtained
with a PANalytical X’Pert Pro MPD diffractometer. The high temperature XRD (HTXRD)
measurements were done using an Anton-Paar HTK1200N oven in both air and nitrogen
atmospheres. The nitrogen gas (99.999%) was purified in situ prior to the experiment in an
Entegris 35KF-I-4R gas purification system.
Film morphology was studied using both FESEM and atomic force microscopy (AFM). AFM
images were captured in tapping mode using a Veeco Multimode V tool equipped with a
Nanoscope V controller. Film roughness was calculated as average root mean square (Rq)
values from 2 x 2 µm2 images obtained using Si probes (RTESP, Bruker).
X-ray photoelectron spectra (XPS) were obtained using an Omicron ARGUS spectrometer
operated at a pass energy of 20 eV. Samples were illuminated with X-rays emitted from a
standard Mg source (K alpha line) at a photon energy of 1253.6 eV. No sputtering was done on
the films studied. Binding energies were calibrated using the C 1s peak of ambient hydrocarbons
(284.8 eV). Peak fitting was done using the CasaXPS software package.
The time-of-flight elastic recoil detection analysis (ToF-ERDA) measurements were done on
films deposited on Si (100) over 1000 cycles using a 50 MeV 127I7+ ion beam in a setup
described in full elsewhere.70

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
111
UV-Vis transmittance measurements were performed on films deposited on soda lime glass
substrates using a Hitachi U2000 spectrophotometer.
6.5.4 Photoelectrochemistry
Photocurrent measurements were carried out using a three electrode setup with a platinum
counter electrode and a Ag/AgCl (3M KCl) reference electrode using a SP-300 BioLogic
potentiostat. The photoelectrodes were pressed against an O-ring of the cell leaving an
irradiated area of 0.5 cm2. The electrodes were irradiated from the back side (through the FTO
glass). The monochromatic wavelength-
resolved photocurrent measurements were performed using a tunable monochromatic light
source (Instytut Fotonowy) provided with a 150 W Xenon lamp and a grating monochromator
with a bandwidth of 10 nm and SP-300 BioLogic potentiostat. The monochromatic intensities
between 300 nm and 800 nm were in the range of 0.16–2.48 mW/cm2. Appropriate cut-off filters
were used in order to eliminate the second-order diffraction radiation. The oxygen evolution
was monitored by an OxySense 325i oxygen analyzer in a two-compartment cell, and the
electrodes were irradiated by a 150 W Xenon lamp (LOT Oriel) equipped with a KG-3 (Schott)
heat-absorbing filter and an AM 1.5 filter. Prior to experiments the electrolyte solutions were
purged with argon for 30 minutes.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
112
6.6 References
1. Švegl, F.; Orel, B.; Hutchins, M. G.; Kalcher, K., Structural and Spectroelectrochemical
Investigations of Sol‐Gel Derived Electrochromic Spinel Co3O4 Films. J. Electrochem. Soc.
1996, 143, 1532-1539.
2. Ando, M.; Kobayashi, T.; Iijima, S.; Haruta, M., Optical recognition of CO and H2 by
use of gas-sensitive Au-Co3O4 composite films. J. Mater. Chem. 1997, 7, 1779-1783.
3. Li, W. Y.; Xu, L. N.; Chen, J., Co3O4 Nanomaterials in Lithium-Ion Batteries and Gas
Sensors. Adv. Funct. Mater. 2005, 15, 851-857.
4. Bekermann, D.; Gasparotto, A.; Barreca, D.; Maccato, C.; Comini, E.; Sada, C.;
Sberveglieri, G.; Devi, A.; Fischer, R. A., Co3O4/ZnO Nanocomposites: From Plasma Synthesis
to Gas Sensing Applications. ACS Appl. Mater. Interfaces 2012, 4, 928-934.
5. Klesko, J. P.; Kerrigan, M. M.; Winter, C. H., Low Temperature Thermal Atomic Layer
Deposition of Cobalt Metal Films. Chem. Mater. 2016, 28, 700-703.
6. Lewis, N. S.; Nocera, D. G., Powering the planet: Chemical challenges in solar energy
utilization. Proc. Natl. Acad. Sci. 2006, 103, 15729-15735.
7. Dau, H.; Limberg, C.; Reier, T.; Risch, M.; Roggan, S.; Strasser, P., The Mechanism of
Water Oxidation: From Electrolysis via Homogeneous to Biological Catalysis. ChemCatChem
2010, 2, 724-761.
8. Trotochaud, L.; Mills, T. J.; Boettcher, S. W., An Optocatalytic Model for
Semiconductor–Catalyst Water-Splitting Photoelectrodes Based on In Situ Optical
Measurements on Operational Catalysts. J. Phys. Chem. Lett. 2013, 4, 931-935.
9. Yang, J.; Wang, D.; Han, H.; Li, C., Roles of Cocatalysts in Photocatalysis and
Photoelectrocatalysis. Acc. Chem. Res. 2013, 46, 1900-1909.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
113
10. Peter, L. M., Photoelectrochemical Water Splitting. A Status Assessment.
Electroanalysis 2015, 27, 864-871.
11. Ager, J. W.; Shaner, M. R.; Walczak, K. A.; Sharp, I. D.; Ardo, S., Experimental
demonstrations of spontaneous, solar-driven photoelectrochemical water splitting. Energy
Environ. Sci. 2015, 8, 2811-2824.
12. Zhou, X.; Shen, X.; Xia, Z.; Zhang, Z.; Li, J.; Ma, Y.; Qu, Y., Hollow Fluffy Co3O4
Cages as Efficient Electroactive Materials for Supercapacitors and Oxygen Evolution Reaction.
ACS Appl. Mater. Interfaces 2015, 7, 20322-20331.
13. Chua, C. S.; Ansovini, D.; Lee, C. J. J.; Teng, Y. T.; Ong, L. T.; Chi, D.; Hor, T. S. A.;
Raja, R.; Lim, Y.-F., The effect of crystallinity on photocatalytic performance of Co3O4 water-
splitting cocatalysts. Phys. Chem. Chem. Phys. 2016, 18, 5172-5178.
14. Kanan, M. W.; Nocera, D. G., In Situ Formation of an Oxygen-Evolving Catalyst in
Neutral Water Containing Phosphate and Co2+. Science 2008, 321, 1072-1075.
15. Lin, F.; Boettcher, S. W., Adaptive semiconductor/electrocatalyst junctions in water-
splitting photoanodes. Nat. Mater. 2014, 13, 81-86.
16. Bergmann, A.; Martinez-Moreno, E.; Teschner, D.; Chernev, P.; Gliech, M.; de Araújo,
J. F.; Reier, T.; Dau, H.; Strasser, P., Reversible amorphization and the catalytically active state
of crystalline Co3O4 during oxygen evolution. Nat. Commun. 2015, 6, 8625.
17. Nellist, M. R.; Laskowski, F. A. L.; Lin, F.; Mills, T. J.; Boettcher, S. W.,
Semiconductor–Electrocatalyst Interfaces: Theory, Experiment, and Applications in
Photoelectrochemical Water Splitting. Acc. Chem. Res. 2016, 49, 733-740.
18. Barrera, E.; Viveros, T.; Avila, A.; Quintana, P.; Morales, M.; Batina, N., Cobalt oxide
films grown by a dipping sol-gel process. Thin Solid Films 1999, 346, 138-144.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
114
19. Armelao, L.; Barreca, D.; Gross, S.; Martucci, A.; Tieto, M.; Tondello, E., Cobalt oxide-
based films: sol–gel synthesis and characterization. J. Non-Cryst. Solids 2001, 293–295, 477-
482.
20. Quinlan, F. T.; Vidu, R.; Predoana, L.; Zaharescu, M.; Gartrner, M.; Groza, J.; Stroeve,
P., Lithium Cobalt Oxide (LiCoO2) Nanocoatings by Sol−Gel Methods. Ind. Eng. Chem. Res.
2004, 43, 2468-2477.
21. Athey, P. R.; Urban, F. K.; Tabet, M. F.; McGahan, W. A., Optical properties of cobalt
oxide films deposited by spray pyrolysis. J. Vac. Sci. Technol., A 1996, 14, 685-692.
22. Shinde, V. R.; Mahadik, S. B.; Gujar, T. P.; Lokhande, C. D., Supercapacitive cobalt
oxide (Co3O4) thin films by spray pyrolysis. Appl. Surf. Sci. 2006, 252, 7487-7492.
23. Louardi, A.; Rmili, A.; Ouachtari, F.; Bouaoud, A.; Elidrissi, B.; Erguig, H.,
Characterization of cobalt oxide thin films prepared by a facile spray pyrolysis technique using
perfume atomizer. J. Alloy. Compd. 2011, 509, 9183-9189.
24. Abbas, T. A.-H.; Slewa, L. H.; Khizir, H. A.; Kakil, S. A., Synthesis of cobalt oxide
(Co3O4) thin films by electrostatic spray pyrolysis technique (ESP). J. Mater. Sci. Mater.
Electron. 2016, 1-7.
25. Meyer, W.; Biedermann, K.; Gubo, M.; Hammer, L.; Heinz, K., Surface structure of
polar Co3O4 (111) films grown epitaxially on Ir(100)-(1 × 1). J. Phys. Condens. Matter 2008,
20, 265011.
26. Johnson, R. W.; Hultqvist, A.; Bent, S. F., A brief review of atomic layer deposition:
from fundamentals to applications. Mater. Today 2014, 17, 236-246.
27. Mazzi, A.; Bazzanella, N.; Orlandi, M.; Edla, R.; Patel, N.; Fernandes, R.; Miotello, A.,
Physical vapor deposition of mixed-metal oxides based on Fe, Co and Ni as water oxidation
catalysts. Mater. Sci. Semicond. Process. 2016, 42, Part 1, 155-158.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
115
28. Xu, T.; Schwarz, M.; Werner, K.; Mohr, S.; Amende, M.; Libuda, J., Structure-
Dependent Anchoring of Organic Molecules to Atomically Defined Oxide Surfaces: Phthalic
Acid on Co3O4(111), CoO(100), and CoO(111). Chem. Eur. J. 2016, 22, 5384-5396.
29. Klepper, K. B.; Nilsen, O.; Fjellvåg, H., Growth of thin films of Co3O4 by atomic layer
deposition. Thin Solid Films 2007, 515, 7772-7781.
30. Ngo, T. Q.; Posadas, A.; Seo, H.; Hoang, S.; McDaniel, M. D.; Utess, D.; Triyoso, D.
H.; Buddie Mullins, C.; Demkov, A. A.; Ekerdt, J. G., Atomic layer deposition of photoactive
CoO/SrTiO3 and CoO/TiO2 on Si(001) for visible light driven photoelectrochemical water
oxidation. J. Appl. Phys. 2013, 114, 084901.
31. Leskelä, M.; Ritala, M., Atomic Layer Deposition Chemistry: Recent Developments
and Future Challenges. Angew. Chem. Int. Ed. 2003, 42, 5548-5554.
32. Backman, L. B.; Rautiainen, A.; Lindblad, M.; Krause, A. O. I., The interaction of cobalt
species with alumina on Co/Al2O3 catalysts prepared by atomic layer deposition. Appl. Catal.,
A 2009, 360, 183-191.
33. Barreca, D.; Devi, A.; Fischer, R. A.; Bekermann, D.; Gasparotto, A.; Gavagnin, M.;
Maccato, C.; Tondello, E.; Bontempi, E.; Depero, L. E.; Sada, C., Strongly oriented Co3O4 thin
films on MgO(100) and MgAl2O4(100) substrates by PE-CVD. CrystEngComm 2011, 13,
3670-3673.
34. Liu, R.; Lin, Y.; Chou, L.-Y.; Sheehan, S. W.; He, W.; Zhang, F.; Hou, H. J. M.; Wang,
D., Water Splitting by Tungsten Oxide Prepared by Atomic Layer Deposition and Decorated
with an Oxygen-Evolving Catalyst. Angew. Chem. Int. Ed. 2011, 50, 499-502.
35. Paracchino, A.; Laporte, V.; Sivula, K.; Grätzel, M.; Thimsen, E., Highly active oxide
photocathode for photoelectrochemical water reduction. Nat. Mater. 2011, 10, 456-461.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
116
36. Morales-Guio, C. G.; Tilley, S. D.; Vrubel, H.; Grätzel, M.; Hu, X., Hydrogen evolution
from a copper(I) oxide photocathode coated with an amorphous molybdenum sulphide catalyst.
Nat. Commun. 2014, 5, 3059.
37. Young, K. M. H.; Hamann, T. W., Enhanced photocatalytic water oxidation efficiency
with Ni(OH)2 catalysts deposited on [small alpha]-Fe2O3 via ALD. Chem. Commun. 2014, 50,
8727-8730.
38. Steier, L.; Luo, J.; Schreier, M.; Mayer, M. T.; Sajavaara, T.; Grätzel, M., Low-
Temperature Atomic Layer Deposition of Crystalline and Photoactive Ultrathin Hematite Films
for Solar Water Splitting. ACS Nano 2015, 9, 11775-11783.
39. Hajibabaei, H.; Zandi, O.; Hamann, T. W., Tantalum nitride films integrated with
transparent conductive oxide substrates via atomic layer deposition for photoelectrochemical
water splitting. Chem. Sci. 2016, 7, 6760-6767.
40. Lim, B. S.; Rahtu, A.; Gordon, R. G., Atomic layer deposition of transition metals. Nat.
Mater. 2003, 2, 749-754.
41. Lichterman, M. F.; Shaner, M. R.; Handler, S. G.; Brunschwig, B. S.; Gray, H. B.;
Lewis, N. S.; Spurgeon, J. M., Enhanced Stability and Activity for Water Oxidation in Alkaline
Media with Bismuth Vanadate Photoelectrodes Modified with a Cobalt Oxide Catalytic Layer
Produced by Atomic Layer Deposition. J. Phys. Chem. Lett. 2013, 4, 4188-4191.
42. Han, B.; Choi, K. H.; Park, K.; Han, W. S.; Lee, W.-J., Low-Temperature Atomic Layer
Deposition of Cobalt Oxide Thin Films Using Dicobalt Hexacarbonyl tert-Butylacetylene and
Ozone. Electrochem. Solid State Lett. 2011, 15, D14-D17.
43. Taheri Najafabadi, A.; Khodadadi, A. A.; Parnian, M. J.; Mortazavi, Y., Atomic layer
deposited Co/γ-Al2O3 catalyst with enhanced cobalt dispersion and Fischer–Tropsch synthesis
activity and selectivity. Appl. Catal., A 2016, 511, 31-46.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
117
44. Lim, B. S.; Rahtu, A.; Park, J.-S.; Gordon, R. G., Synthesis and Characterization of
Volatile, Thermally Stable, Reactive Transition Metal Amidinates. Inorg. Chem. 2003, 42,
7951-7958.
45. Barry, S. T., Amidinates, guanidinates and iminopyrrolidinates: Understanding
precursor thermolysis to design a better ligand. Coord. Chem. Rev. 2013, 257, 3192-3201.
46. Knisley, T. J.; Saly, M. J.; Heeg, M. J.; Roberts, J. L.; Winter, C. H., Volatility and High
Thermal Stability in Mid- to Late-First-Row Transition-Metal Diazadienyl Complexes.
Organometallics 2011, 30, 5010-5017.
47. Chen, Y. W.; Prange, J. D.; Duehnen, S.; Park, Y.; Gunji, M.; Chidsey, C. E. D.;
McIntyre, P. C., Atomic layer-deposited tunnel oxide stabilizes silicon photoanodes for water
oxidation. Nat. Mater. 2011, 10, 539-544.
48. Scheuermann, A. G.; Prange, J. D.; Gunji, M.; Chidsey, C. E. D.; McIntyre, P. C.,
Effects of catalyst material and atomic layer deposited TiO2 oxide thickness on the water
oxidation performance of metal-insulator-silicon anodes. Energy Environ. Sci. 2013, 6, 2487-
2496.
49. Hu, S.; Shaner, M. R.; Beardslee, J. A.; Lichterman, M.; Brunschwig, B. S.; Lewis, N.
S., Amorphous TiO2 coatings stabilize Si, GaAs, and GaP photoanodes for efficient water
oxidation. Science 2014, 344, 1005-1009.
50. Lichterman, M. F.; Carim, A. I.; McDowell, M. T.; Hu, S.; Gray, H. B.; Brunschwig, B.
S.; Lewis, N. S., Stabilization of n-cadmium telluride photoanodes for water oxidation to O2(g)
in aqueous alkaline electrolytes using amorphous TiO2 films formed by atomic-layer
deposition. Energy Environ. Sci. 2014, 7, 3334-3337.
51. Li, W.; Gibbs, G. V.; Oyama, S. T., Mechanism of Ozone Decomposition on a
Manganese Oxide Catalyst. 1. In Situ Raman Spectroscopy and Ab Initio Molecular Orbital
Calculations. J. Am. Chem. Soc. 1998, 120, 9041-9046.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
118
52. Knoops, H. C. M.; Elam, J. W.; Libera, J. A.; Kessels, W. M. M., Surface Loss in Ozone-
Based Atomic Layer Deposition Processes. Chem. Mater. 2011, 23, 2381-2387.
53. Han, B.; Park, J.-M.; Choi, K. H.; Lim, W.-K.; Mayangsari, T. R.; Koh, W.; Lee, W.-J.,
Atomic layer deposition of stoichiometric Co3O4 films using bis(1,4-di-iso-propyl-1,4-
diazabutadiene) cobalt. Thin Solid Films 2015, 589, 718-722.
54. Will, G.; Masciocchi, N.; Parrish, W.; Hart, M., Refinement of simple crystal structures
from synchrotron radiation powder diffraction data. J. Appl. Crystallogr. 1987, 20, 394-401.
55. Nandi, D. K.; Manna, J.; Dhara, A.; Sharma, P.; Sarkar, S. K., Atomic layer deposited
cobalt oxide: An efficient catalyst for NaBH4 hydrolysis. J. Vac. Sci. Technol., A 2016, 34,
01A115.
56. Liu, J. F.; Yin, S.; Wu, H. P.; Zeng, Y. W.; Hu, X. R.; Wang, Y. W.; Lv, G. L.; Jiang, J.
Z., Wurtzite-to-Rocksalt Structural Transformation in Nanocrystalline CoO. J. Phys. Chem. B
2006, 110, 21588-21592.
57. Yang, J.; Liu, H.; Martens, W. N.; Frost, R. L., Synthesis and Characterization of Cobalt
Hydroxide, Cobalt Oxyhydroxide, and Cobalt Oxide Nanodiscs. J. Phys. Chem. C 2010, 114,
111-119.
58. Biesinger, M. C.; Payne, B. P.; Grosvenor, A. P.; Lau, L. W. M.; Gerson, A. R.; Smart,
R. S. C., Resolving surface chemical states in XPS analysis of first row transition metals, oxides
and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717-2730.
59. Li, J.; Lu, G.; Wu, G.; Mao, D.; Guo, Y.; Wang, Y.; Guo, Y., Effect of TiO2 crystal
structure on the catalytic performance of Co3O4/TiO2 catalyst for low-temperature CO
oxidation. Catal. Sci. Tech. 2014, 4, 1268-1275.
60. Harriman, A.; Pickering, I. J.; Thomas, J. M.; Christensen, P. A., Metal oxides as
heterogeneous catalysts for oxygen evolution under photochemical conditions. J. Chem. Soc.
Faraday Trans. 1988, 84, 2795-2806.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
119
61. Kanan, M. W.; Yano, J.; Surendranath, Y.; Dincă, M.; Yachandra, V. K.; Nocera, D. G.,
Structure and Valency of a Cobalt−Phosphate Water Oxidation Catalyst Determined by in Situ
X-ray Spectroscopy. J. Am. Chem. Soc. 2010, 132, 13692-13701.
62. Risch, M.; Ringleb, F.; Kohlhoff, M.; Bogdanoff, P.; Chernev, P.; Zaharieva, I.; Dau,
H., Water oxidation by amorphous cobalt-based oxides: in situ tracking of redox transitions and
mode of catalysis. Energy Environ. Sci. 2015, 8, 661-674.
63. Ramakrishnan, V.; Kim, H.; Park, J.; Yang, B., Cobalt oxide nanoparticles on TiO2
nanorod/FTO as a photoanode with enhanced visible light sensitization. RSC Adv. 2016, 6,
9789-9795.
64. Barroso, M.; Cowan, A. J.; Pendlebury, S. R.; Gratzel, M.; Klug, D. R.; Durrant, J. R.,
The Role of Cobalt Phosphate in Enhancing the Photocatalytic Activity of α-Fe2O3 toward
Water Oxidation. J. Am. Chem. Soc. 2011, 133, 14868-14871.
65. Cummings, C. Y.; Marken, F.; Peter, L. M.; Tahir, A. A.; Wijayantha, K. G. U., Kinetics
and mechanism of light-driven oxygen evolution at thin film α-Fe2O3 electrodes. Chem.
Commun. 2012, 48, 2027-2029.
66. Ma, Y.; Le Formal, F.; Kafizas, A.; Pendlebury, S. R.; Durrant, J. R., Efficient
suppression of back electron/hole recombination in cobalt phosphate surface-modified undoped
bismuth vanadate photoanodes. J. Mater. Chem. A 2015, 3, 20649-20657.
67. Pore, V.; Rahtu, A.; Leskelä, M.; Ritala, M.; Sajavaara, T.; Keinonen, J., Atomic Layer
Deposition of Photocatalytic TiO2 Thin Films from Titanium Tetramethoxide and Water. Chem.
Vap. Deposition 2004, 10, 143-148.
68. Goldstein, D. N.; McCormick, J. A.; George, S. M., Al2O3 Atomic Layer Deposition
with Trimethylaluminum and Ozone Studied by in Situ Transmission FTIR Spectroscopy and
Quadrupole Mass Spectrometry. J. Phys. Chem. C 2008, 112, 19530-19539.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
120
69. Waldo, R. A., An iteration procedure to calculate film compositions and thicknesses in
electron-probe microanalysis. Microbeam Anal. 1988, 23rd, 310-14.
70. Jokinen, J.; Keinonen, J.; Tikkanen, P.; Kuronen, A.; Ahlgren, T.; Nordlund, K.,
Comparison of TOF-ERDA and nuclear resonance reaction techniques for range profile
measurements of keV energy implants. Nucl. Instr. Meth. Phys. Res. 1996, 119, 533-542.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
121
Supporting Information
Jiyeon Kim,a Tomi Iivonen,b Jani Hämäläinen,b Marianna Kemell,b Kristoffer Meinander,c Kenichiro Mizohata,c Lidong Wang,a Jyrki Räisänen,c Radim Beranek,a,d Markku Leskelä,b Anjana Devia*
a) Inorganic Materials Chemistry, Chair of Inorganic Chemistry II, Ruhr-University Bochum, Universitätstr. 150, 44801 Bochum, Germany b) Laboratory of Inorganic Chemistry, Department of Chemistry, University of Helsinki, P.O. Box 55, FI-00014 Helsinki, Finland c) Division of Materials Physics, Department of Physics, University of Helsinki, P.O. Box 43, FI-00014 Helsinki, Finland d) Institute of Electrochemistry, Ulm University, Albert-Einstein-Allee 47, 89081 Ulm, Germany
Table of Contents
Figure S1 EI-MS fragmentation pattern of [Co(tBu2DAD)2].
Figure S2 Schematic depiction of the reaction chamber of F-120 ALD reactor
Figure S3 Co3O4 film thickness vs distance from leading edge of the 5 x 5 cm2 silicon
substrate depending on the deposition temperature.
Figure S4 The photograph of Co3O4 thin film on Si and soda lime glass deposited at 120 °C
over 1000 cycles.
Figure S5 Plane view SEM of Co3O4 thin film deposited over 1000 cycles at 120 °C.
Figure S6 SEM image of Si trench with an aspect ratio of 20:1 (depth 40 m, width 2 m).
Figure S7X-ray Photoelectron Spectroscopy survey spectrum of Co3O4 thin films
Figure S8 UV-Vis transmission spectrum and corresponding Tauc plot of Co3O4 thin film
deposited over 1000 cycles at 120 °C.
Figure S9 UV-Vis transmittance plots of a 100 nm TiO2 film (pristine TiO2) modified by the
depositing series of Co3O4 (10, 50, 100, 300, 500, and 10000 cycles) on the TiO2 films.
Figure S10 Photocurrent transients of TiO2/Co3O4 structures with depositing series of Co3O4
catalyst (10 – 1000 ALD cycles) on FTO glass under irradiation at different wavelengths (a),

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
122
selected area (b, c) and the corresponding photoaction spectra (d) measured in KOH (0.1 M)
at 1.48 V vs RHE at TiO2 and TiO2/Co3O4 photoanodes.
Figure S11 AFM image of Co3O4 deposited over 10 cycles at 120 °C on Si (100) substrate.
Figure S12 XPS spectra of Co3O4 thin films deposited over 10 cycles on Si and TiO2 at 120
°C.
Figure S13 XRD patterns of TiO2 deposited at 275 °C over 2000 cycles on FTO glass
Table S1 XPS surface distribution of Co, O, C, Ti and Si on samples deposited over 10 and 50 cycles
on a 100 nm thick TiO2 film and Si (100).
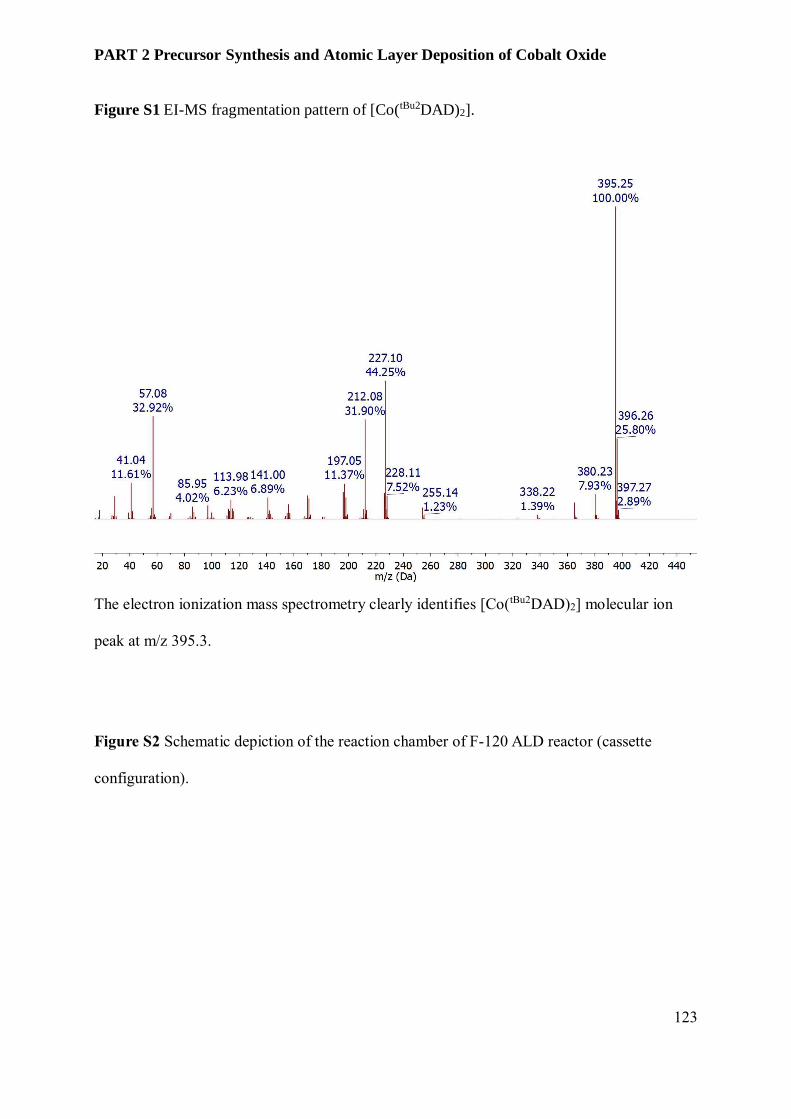
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
123
Figure S1 EI-MS fragmentation pattern of [Co(tBu2DAD)2].
The electron ionization mass spectrometry clearly identifies [Co(tBu2DAD)2] molecular ion
peak at m/z 395.3.
Figure S2 Schematic depiction of the reaction chamber of F-120 ALD reactor (cassette
configuration).
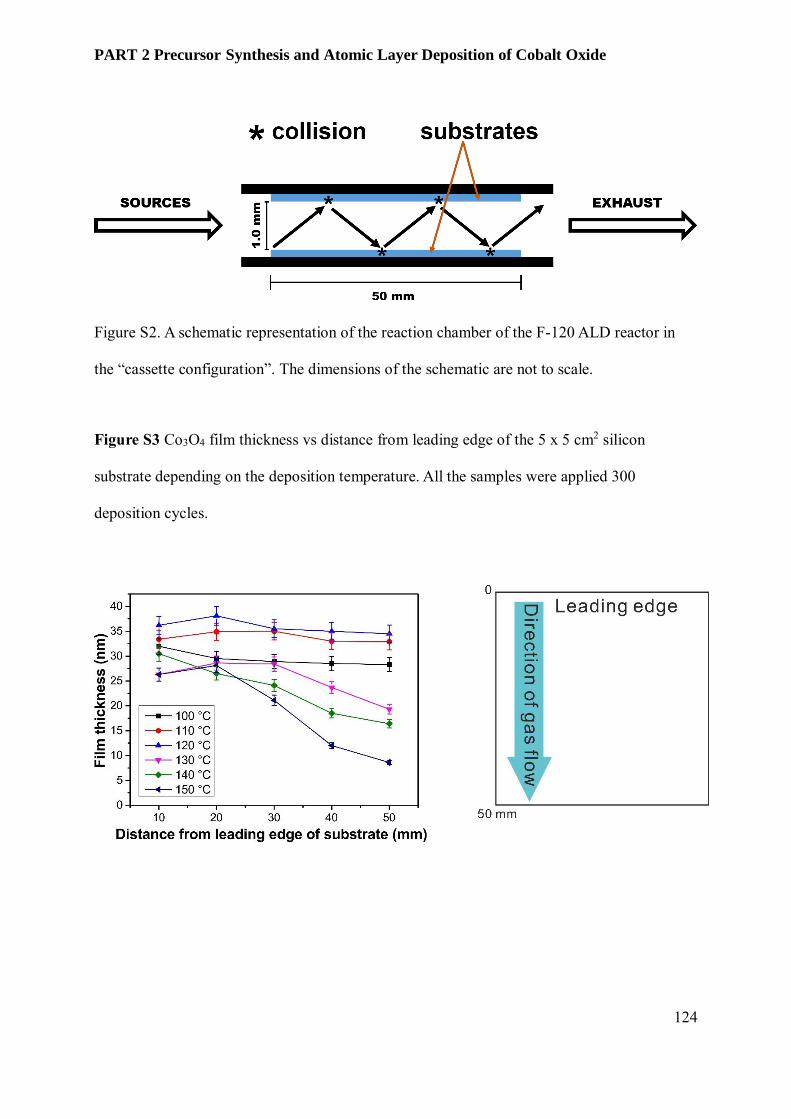
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
124
Figure S2. A schematic representation of the reaction chamber of the F-120 ALD reactor in
the “cassette configuration”. The dimensions of the schematic are not to scale.
Figure S3 Co3O4 film thickness vs distance from leading edge of the 5 x 5 cm2 silicon
substrate depending on the deposition temperature. All the samples were applied 300
deposition cycles.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
125
Figure S4 The photograph of Co3O4 thin film on Si and soda lime glass deposited at 120 °C
over 300 cycles.
Co3O4 on Si (100) Co3O4 on soda lime glass Soda lime glass
Figure S5 Plane view SEM of Co3O4 thin film deposited over 1000 cycles at 120 °C.
500 nm 1.00 µm

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
126
Figure S6 SEM image of Si trench with an aspect ratio of 20:1 (depth 40 m, width 2 m).
Co3O4 film has been deposited over 1000 cycles at 120 °C using pulse lengths of 6 seconds
for both Co(tBu2DAD)2 and ozone. The purge length for both precursors was 10 seconds.
Thickness(nm) Top Middle Bottom
114 101 84
122 93 85
111 93 77
119 93 71
130 89 77
138 92 71
80
Average
thickness (nm) 122 94 78
Fully conformal Co3O4 films could not be deposited on 40 m Si trenches (aspect ratio of
1:20) despite using 6 s pulse and 10 s purge times of both precursors. A conceivable
explanation of for not achieving full conformality in the trench structure is the combined
effect of ozone decomposing effect of Co3O4 and high surface area of the substrate.
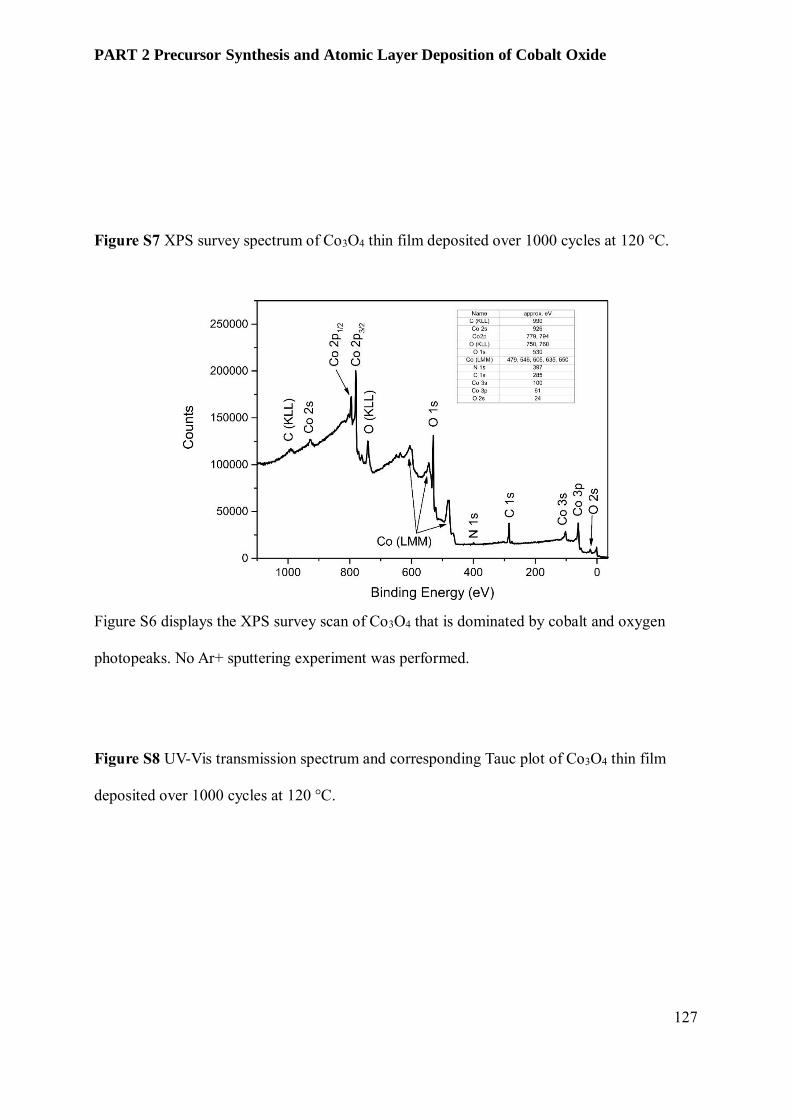
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
127
Figure S7 XPS survey spectrum of Co3O4 thin film deposited over 1000 cycles at 120 °C.
Figure S6 displays the XPS survey scan of Co3O4 that is dominated by cobalt and oxygen
photopeaks. No Ar+ sputtering experiment was performed.
Figure S8 UV-Vis transmission spectrum and corresponding Tauc plot of Co3O4 thin film
deposited over 1000 cycles at 120 °C.
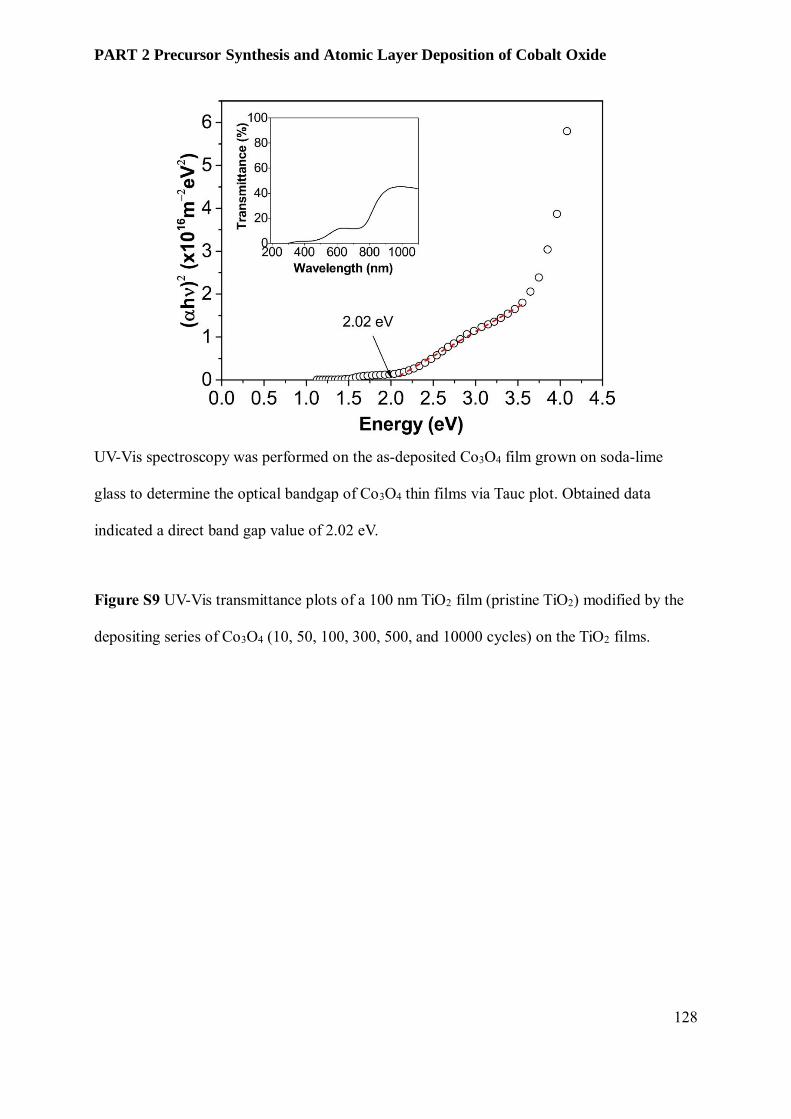
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
128
UV-Vis spectroscopy was performed on the as-deposited Co3O4 film grown on soda-lime
glass to determine the optical bandgap of Co3O4 thin films via Tauc plot. Obtained data
indicated a direct band gap value of 2.02 eV.
Figure S9 UV-Vis transmittance plots of a 100 nm TiO2 film (pristine TiO2) modified by the
depositing series of Co3O4 (10, 50, 100, 300, 500, and 10000 cycles) on the TiO2 films.
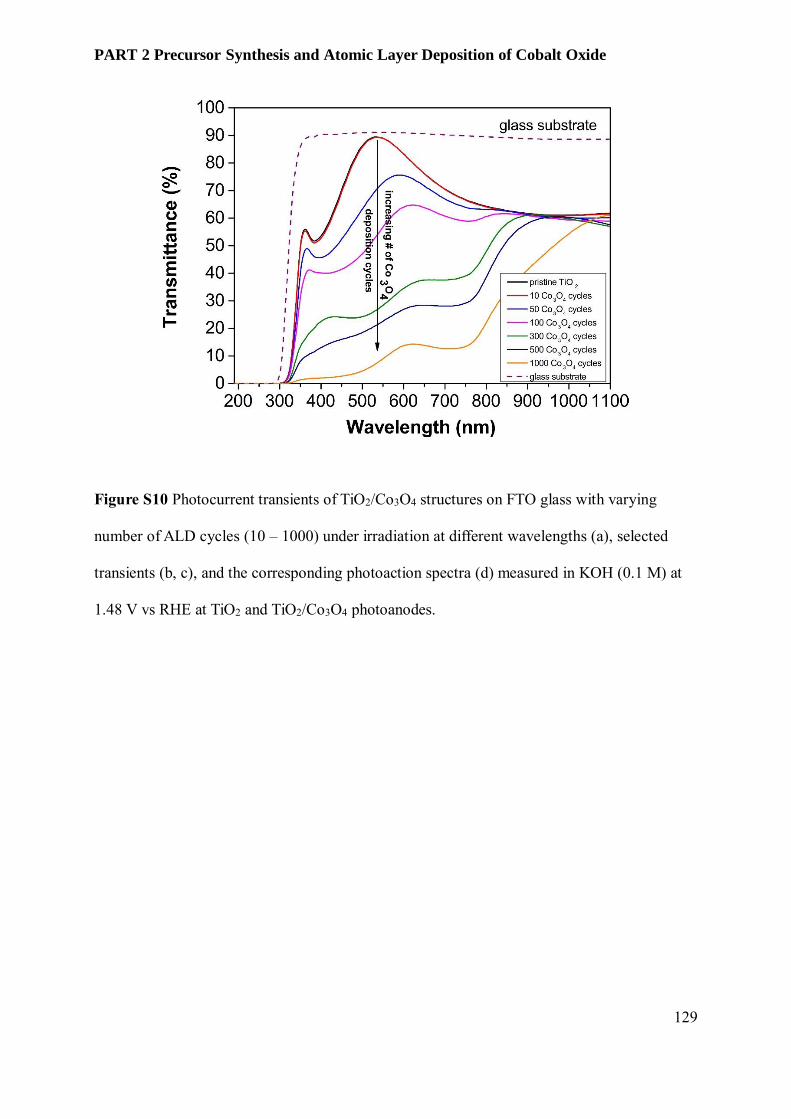
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
129
Figure S10 Photocurrent transients of TiO2/Co3O4 structures on FTO glass with varying
number of ALD cycles (10 – 1000) under irradiation at different wavelengths (a), selected
transients (b, c), and the corresponding photoaction spectra (d) measured in KOH (0.1 M) at
1.48 V vs RHE at TiO2 and TiO2/Co3O4 photoanodes.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
130

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
131
Figure S11 AFM images of Co3O4 deposited over 10 cycles (a,b) and 50 cycles (c,d) at 120
°C on Si (100) substrate. After 10 deposition cycles, island-like nanoparticles are formed
instead of a closed film. After 50 deposition cycles, the island-like features are no longer
present and a homogeneous and smooth surface is observed, indicating that a closed film has
been formed. Images (a) and (b) were captured from different positions of a sample deposited
over 10 cycles. Images (c) and (d) were captured from different positions of a sample
deposited over 50 cycles.
Figure S12 XPS spectra of cobalt oxide deposited over 10 and 50 cycles on a 100 nm thick
TiO2 layer and on Si (100) substrate at 120 °C.
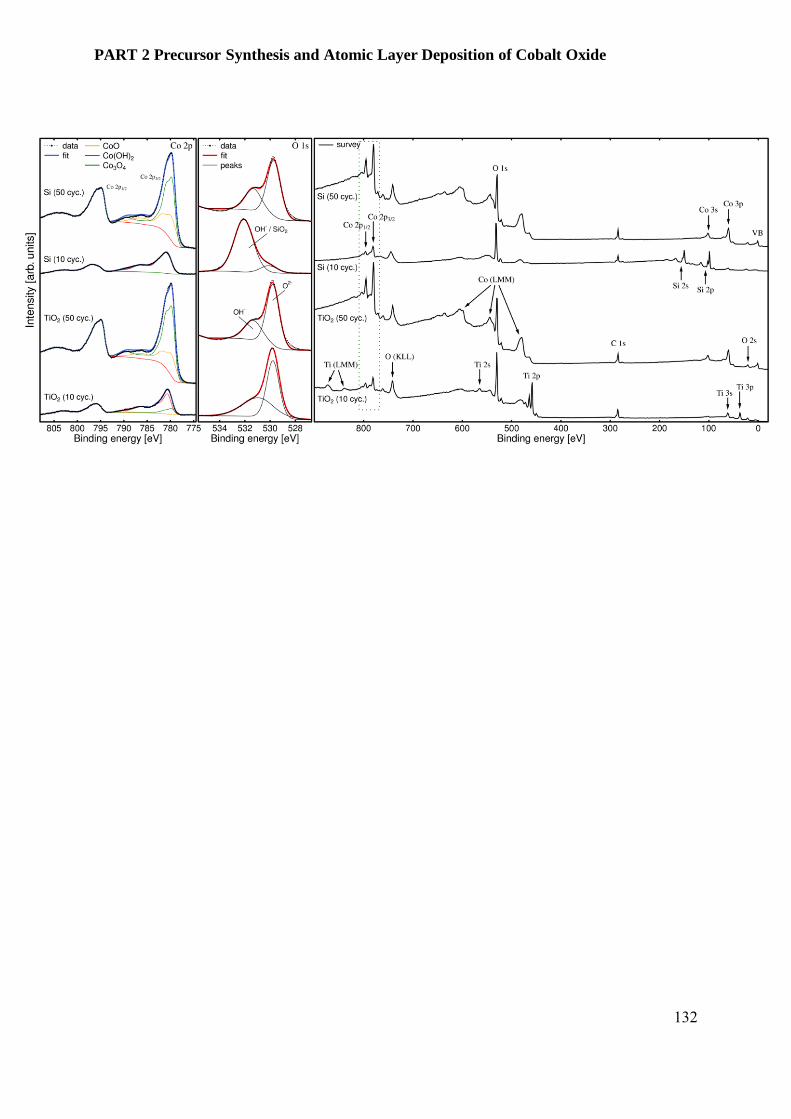
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
132
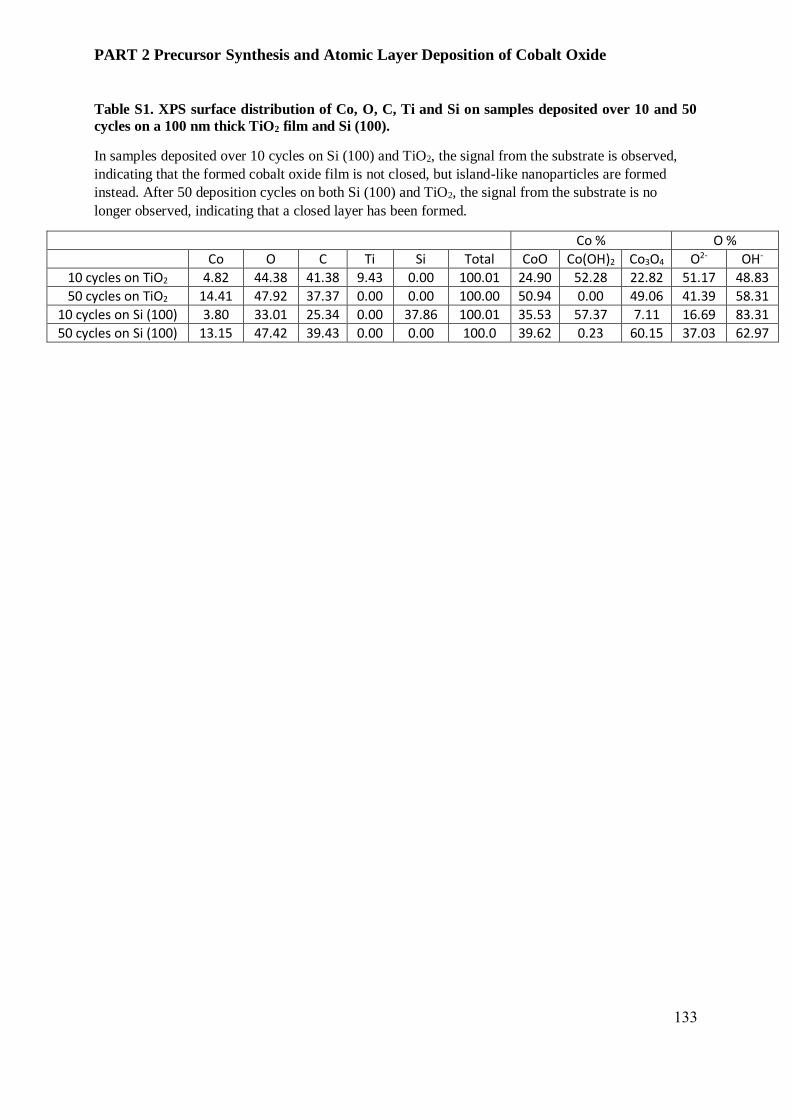
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
133
Table S1. XPS surface distribution of Co, O, C, Ti and Si on samples deposited over 10 and 50
cycles on a 100 nm thick TiO2 film and Si (100).
In samples deposited over 10 cycles on Si (100) and TiO2, the signal from the substrate is observed,
indicating that the formed cobalt oxide film is not closed, but island-like nanoparticles are formed
instead. After 50 deposition cycles on both Si (100) and TiO2, the signal from the substrate is no
longer observed, indicating that a closed layer has been formed.
Co % O %
Co O C Ti Si Total CoO Co(OH)2 Co3O4 O2- OH-
10 cycles on TiO2 4.82 44.38 41.38 9.43 0.00 100.01 24.90 52.28 22.82 51.17 48.83
50 cycles on TiO2 14.41 47.92 37.37 0.00 0.00 100.00 50.94 0.00 49.06 41.39 58.31
10 cycles on Si (100) 3.80 33.01 25.34 0.00 37.86 100.01 35.53 57.37 7.11 16.69 83.31
50 cycles on Si (100) 13.15 47.42 39.43 0.00 0.00 100.0 39.62 0.23 60.15 37.03 62.97

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
134
Figure S13 XRD patterns of TiO2 deposited at 275 °C over 2000 cycles on FTO glass
GI-XRD reflections of TiO2 films deposited at 275 °C on FTO show mixed phases of
anatase/rutile dominated by the anatase phase.1
References:

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
135
7.
Homoleptic all nitrogen coordinated bis-guanidinato
cobalt (II) complex: Volatile and thermally stable
precursors suitable for ALD/CVD

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
136
7. Homoleptic all nitrogen coordinated bis-guanidinato cobalt (II)
complex: Volatile and thermally stable precursors suitable for
ALD/CVD
7.1 Abstract
This work describes the synthesis of novel homoleptic all nitrogen coordinated bis-guanidinato
cobalt (II) complexes. The used guanidinato ligands feature different side groups attached to
the guanidinates, namely isopropyl and cycohexyl. Interestingly, after reaction of the ligand
precursors with the cobalt (II) source a carbodiimide insertion occurs in the case of the isopropyl
containing guanidinato ligands, which is not observed in the case of cyclohexyl. In this work
the chemistry of steric hindrance effect of cobalt guanidinate complexes will be discussed. The
cobalt (II) complexes were fully characterized using NMR spectroscopy, single crystal X-ray
diffraction and TGA-DSC.
7.2 Introduction
Highly conformal thin films of metal or metal oxides are highly desired for their applications
in worldwide field including microelectronics, optics, protection against corrosion and
oxidation, diffusion barriers, sensor for gas or liquid chemicals, photo sensitive coatings and so
on. Although chemical vapor deposition techniques including atomic layer depositions are
widely used to provide very uniform and conformal thin films, suitable and effective precursors
remained scarce. The criteria of ALD precursors are as follow. 1) Highly reactive towards the
functionalities on the surface as well as surface prepared by complementary reactant. 2) Volatile
and thermally stable at growth temperatures (> seconds) and sublimation temperatures (>
months). 3) Non-reactive and non-corrosive byproduct formation is preferred. Chronological

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
137
development of precursors including metal halides, Cps, alkoxides, b-ketonates, alkylamides,
TMS, heteroleptic compounds of mixture of theses ligands meet several limits. Metal halides
produce highly corrosive byproducts and precursors involving M¬C bonds lead to carbon
incorporation into the resulting films.2-3
Precursors containing metals for chemical vapor depositions were investigated across the across
the periodic table for the last century. While oxygen based precursors has reached the limit,
nitrogen based ligand system has caught an attention due to its ease of tunable steric bulk and
electronic saturation. In fact, the presence of two substituents (NR2) at the nitrogen atom readily
leads to increase of steric hinderance compared to oxygen based system (OR). Consequently, a
higher steric crowding surround the electro positive metal atom prevents the oligomerization
which influence the volatility of the compounds. Amidinates were intensively investigated
which diminish the drawbacks of conventional precursors.3 Our interest to develop reactive,
volatile and thermally stable all nitrogen coordinated cobalt complexes as CVD/ALD
precursors, we were inspired by the guanidinates as versatile ligand system closely related to
amidinates. Guanidinates have caught attention as ligand for transition metals, main group3-4
and lanthanides. Rare earth guanidinate are not so common still can be found in several
literatures.5
Guanidinates are in similarity of amidinates in terms of steric hinderance, however guanidinates
possess extended electronic flexibility where the lone pair electrons of the amido-nitrogen are
delocalized in the ligand -system.2 Additionally, guanidinate ligand is stronger base than the
amidinate which will prompt the reactivity of corresponding guanidinato metal complexes.
Synthetic strategy of guanidinato complex include several routes,2, 6 however we will focus on
salt metathesis approach in the paper.2, 7-8

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
138
We have synthesized homoleptic all nitrogen coordinated bis-guanidinato cobalt (II) complexes
with the general formula of [M((NR’)2CNR2)2]and the steric bulk effect to stabilize the bis-
guanidinato complexes depends on R’ substituent.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
139
7.3 Result and Discussion
Synthetic strategy of guanidinato complex include several routes,6 however we will focus on
salt metathesis approach in this work.7-8 The salt metathesis contains two step reaction.
Stoichiometric reaction of a carbodiimide (CDI) with lithium amide generates a lithium
guanidinate [Li((NR’)2CNR2)]. An in-situ reaction with metal halide affords the desired
guanidinate complex along with LiCl as a byproduct.
Stoichiometric reaction of iPrCDI and LiNMe2 results in Li(iPr2Guan) (iPr2Guan =
(NiPr)2CNMe2). The reaction of two equivalents of Li(iPr2Guan) with CoCl2 was expected to
result in Co(iPr2Guan)2. Unexpectedly, heteroleptic compound [CoII((NiPr)2CNMe2)8
(NiPr)2CNMe2(iPrNHCNiPr))] (5) was obtained whereas iPrCDI insertion occurred on the
guanidinate ligand (Scheme 7.1 and Figure 7.1). In fact, homoleptic six-fold coordinated
guanidinate compounds (tris-guanidinates) are already known for second/third row transition
metals, metalloid such as antimony and rare earth metals. Meanwhile guanidinates (e.g. 2,6-
diisopropylphenyl, 2,4,6-mesityl) with high streric bulk ligands were used to stabilize the low
coordination first row transition metal (I) complexes9 but two guanidinates coordination
complexes were not investigated but still bis-guanidinate compounds are not investigated.
One molecule of iPrCDI cleaves the cobalt-N bond and lead to a wider N−M−N angel to stabilize
the compound 5. Interestingly, the structurally similar ligand system iPrAMD is known that it
leads to a homoleptic cobalt amidinate compound.10 This can be explained by the difference of
the electronic situation between the amidinates and guanidinates. Richer electron density and
electronical flexibility on the iPrguan ligand discourage the iPr substituent to be not bulky
enough for stabilization of the Co(II) metal center. On the other hand, our attempt of
reproducing Co(iPrAMD)2 afforded contrasting results.10 According to our observation,
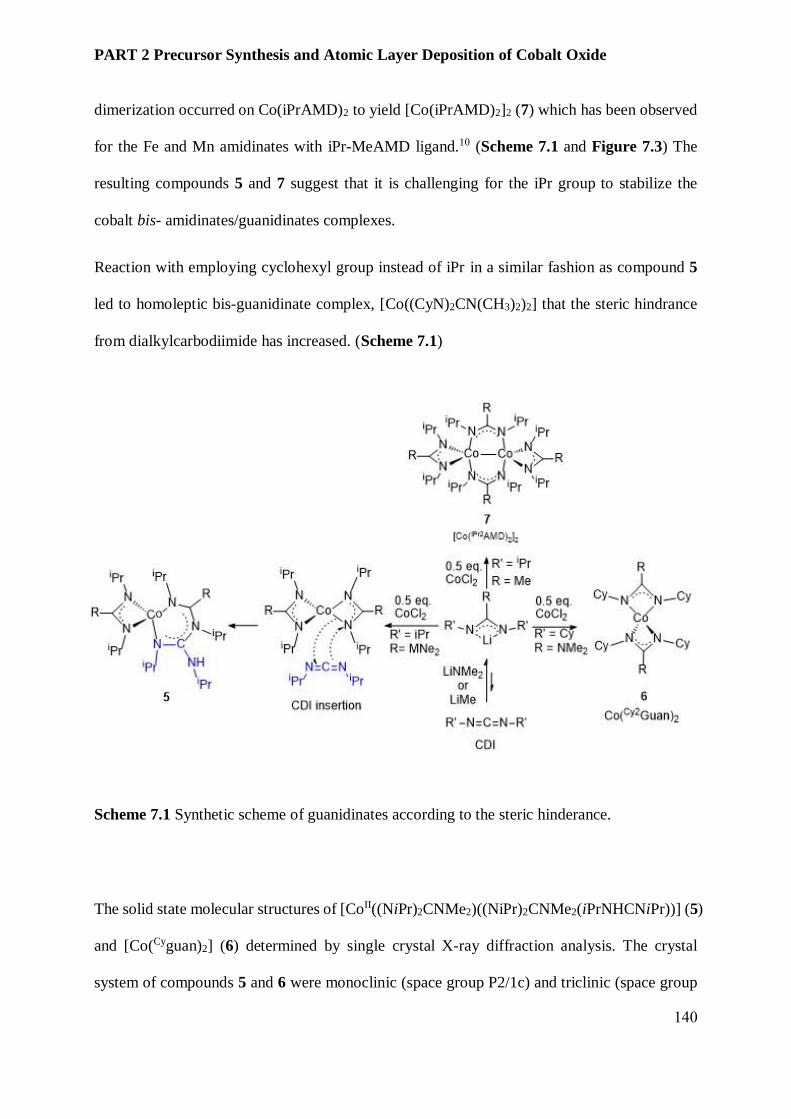
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
140
dimerization occurred on Co(iPrAMD)2 to yield [Co(iPrAMD)2]2 (7) which has been observed
for the Fe and Mn amidinates with iPr-MeAMD ligand.10 (Scheme 7.1 and Figure 7.3) The
resulting compounds 5 and 7 suggest that it is challenging for the iPr group to stabilize the
cobalt bis- amidinates/guanidinates complexes.
Reaction with employing cyclohexyl group instead of iPr in a similar fashion as compound 5
led to homoleptic bis-guanidinate complex, [Co((CyN)2CN(CH3)2)2] that the steric hindrance
from dialkylcarbodiimide has increased. (Scheme 7.1)
Scheme 7.1 Synthetic scheme of guanidinates according to the steric hinderance.
The solid state molecular structures of [CoII((NiPr)2CNMe2)((NiPr)2CNMe2(iPrNHCNiPr))] (5)
and [Co(Cyguan)2] (6) determined by single crystal X-ray diffraction analysis. The crystal
system of compounds 5 and 6 were monoclinic (space group P2/1c) and triclinic (space group

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
141
P-1). Crystallographic refinement data of the compounds 5-7 can be found section in 7.5.3. All
the Co−N bond lengths of heteroleptic compound 5 show in the same range (2.022 Å). The
bond angle of N4−Co−N5 from the ligand where the CDI insertion took place displays much
wider angle (96.16(12) °) than N1−Co−N2 in the guanidinate ligand (65.97(12) °). (Figure 7.1
and Table 7.1) Predictably, in compound 6 the bond lengths of Co−N and the angles of
N−Co−N are in the same range (2.016 Å and 66.62 °) Both structures show more or less strong
distortion of tetrahedron geometry, however the distortion observed in 6 was more prominent
than in 5. This is ascribed to the cyclohexyl group which is much higher sterically demanding
compared to iso-propyl group, enables the nitrogen atoms from each guanidinates ligands are
more distanced. (Figure 7.2 and Table 7.2)
The solid state X-ray crystal structure of [Co(iPrAMD)2]2 (7) shows the dimerized structure
which has not been reported where only the monomeric Co(iPrAMD)2 is reported. [Gordon] We
postulate the both monomeric and dimeric structures are in the equilibrium in the solution so
that the crystallization behavior can differ dependent on the applied crystallization condition.
The structure of dimeric compound 7 is shown in Figure 7.3, and selected bond distances and
angles are contained in Table 7.3. Crystallographic data can be found in Table 7.4 in section
7.5.3. Each cobalt atoms in dimeric compound 7 are bridged by two 1-amidinate ligands and
one chelated by one 2-amidinate ligand. The Co−N bond lengths of (2.0880(12)-2.0731(12)
Å) in the terminal chelating ligands are slightly enlongated than the Co−N distances in the
bridging ligand (2.0450(12)-2.430(12) Å), but are in the same range. The compound 7 is not of
direct goal and interest in this work, further characterization of 7 was not performed.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
142
Figure 7.1 Povray plot of the molecular structure of compound 5 in the solid state as
determined by single crystal X-ray diffraction. Thermal ellipsoids are shown at the 50 %
probability level and the hydrogen atoms are omitted for clarity.
Table 7.1 Selected Bond Length (Å) and Angles (°) for
[CoII((NiPr)2CNMe2)((NiPr)2CNMe2(iPrNHCNiPr))] (5)
Co−N(1) 2.074(3) N(1)−Co−N(2) 65.97(12)
Co−N(2) 2.017(3) N(4)−Co−N(5) 96.16(12)
Co−N(4) 1.969(3) N(1)−Co−N(5) 115.87(12)
Co−N(5) 2.029(3) N(2)−Co−N(4) 128.34(13)
N(1)−C(1) 1.328(5) Co−N(4)−C(10) 110.6(2)
N(2)−C(1) 1.340(5) Co−N(5)−C(20) 116.3(2)
N(4)−C(10) 1.360(5) N(4)−C(10)−N(6) 112.2(3)
N(5)−C(20) 1.316(5) N(5)−C(20)−N(6) 119.5(3)
N(6)−C(10) 1.478(5) C(10)−N(6)−C(20) 119.3(3)
N(6)−C(20) 1.376(5)

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
143
N(7)−C(10) 1.267(5)
Figure 7.2 Povray plot of the molecular structure of [Co(Cyguan)2] (6) (one of the two whole
fragments in the asymmetric unit are shown) in the solid state as determined by single crystal
X-ray diffraction. Thermal ellipsoids are shown at the 50 % probability level and the
hydrogen atoms are omitted for clarity. Structural disorder is confined to the cyclohexyl
groups in the other half of fragments.
Table 7.2 Selected Bond Length (Å) and Angles (°) for [Co(CyGuan)2] (6)
Co−N(1) 2.023(3) N(1)−Co−N(2) 66.53(13)
Co−N(2) 2.012(3) N(4)−Co−N(5) 66.70(13)
Co−N(4) 2.009(3) N(1)−Co−N(4) 129.77(14)
Co−N(5) 2.021(3) N(2)−Co−N(5) 132.40(14)
N(1)−C(1) 1.336(5)
N(2)−C(1) 1.345(5)
N(4)−C(16) 1.348(5)
N(5)−C(16) 1.348(5)

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
144
N(3)−C(1) 1.374(5)
N(6)−C(16) 1.363(5)
Figure 7.3 Molecular structure (POV-ray plot) of [Co(iPrAMD)2]2 as determined by single-
crystal X-ray diffraction. Displacement ellipsoids are shown at the 50% probability level, and
hydrogen atoms are omitted for clarity.
Table 7.3 Selected Bond Length (Å) and Angles (°) for [Co(iPrAMD)2]2 (7)
Co−N(1) 2.0880(12) N(1)−Co−N(2) 64.50(5)
Co−N(2) 2.0731(12) N(3)−Co−N(4) 132.45(5) Co−N(3) 2.0450(12) N(1)−Co−N(3) 113.42(5)
Co−N(4) 2.0430(12) N(1)−Co−N(4) 106.97(5)
N(1)−C(1) 1.333(2) N(2)−Co−N(3) 105.80(5) N(2)−C(1) 1.325(2) N(2)−Co−N(4) 113.70(5)
N(3)−C(9) 1.329(2) N−C−Nchelating 113.28(13)
N(4)−C(9) 1.332 N−C−Nbridging 116.01(13)

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
145
A crude oil of compound 5 as synthesized without crystallization was measured TGA to study
thermal behavior. (Figure 7.4) TGA curve shows onset of volatilization (1 % weight loss) at
73 °C and a shouldered curve can be seen at ca. 220 °C that is a phenomenon named CDI
deinsertion of guanidinate demonstrated in previous report.11 The final residual mass show ca.
10 %. Isothermal studies at ambient pressure exhibit non-linear weight loss which is attributed
to a partial decomposition of ligand (CDI deinsertion from ((NiPr)2CNMe2(iPrNHCNiPr))).
TGA studies were repeated on crystallized compound 5 in combination with DSC (Differential
Scanning Calorimetry) to screen the solid-liquid phase transition. (Figure 7.5) In the solid state
TGA, a single step weight loss was observed due to the higher purity and less contact to air
during the sample loading. The compound 5 is in the liquid state at room temperature and
crystallizes at ca. -30 °C. Notably, it is observed that after warming up the crystals to room
temperature, the compound remains in the solid state. DSC studies confirm this behavior. After
cooling, a phase transition is observed in the DSC trace, however, on the heating branch the
phase transition to solid occurs at substantially higher temperature (75 °C). This result indicates
a good agreement with the melting point found in DTA. Both measurements TGA/DTA and
DSC were using a heating rate of 5 K/min which was apparently insufficient to trigger the phase
transition in the used temperature range.
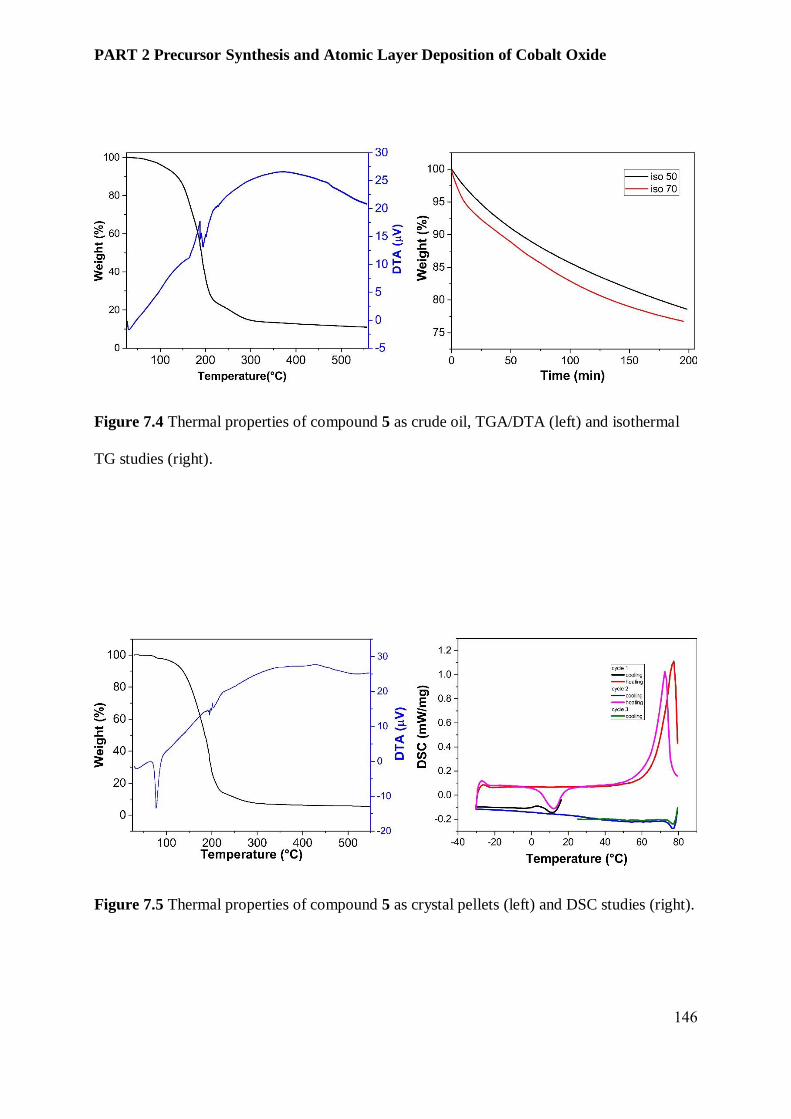
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
146
Figure 7.4 Thermal properties of compound 5 as crude oil, TGA/DTA (left) and isothermal
TG studies (right).
Figure 7.5 Thermal properties of compound 5 as crystal pellets (left) and DSC studies (right).
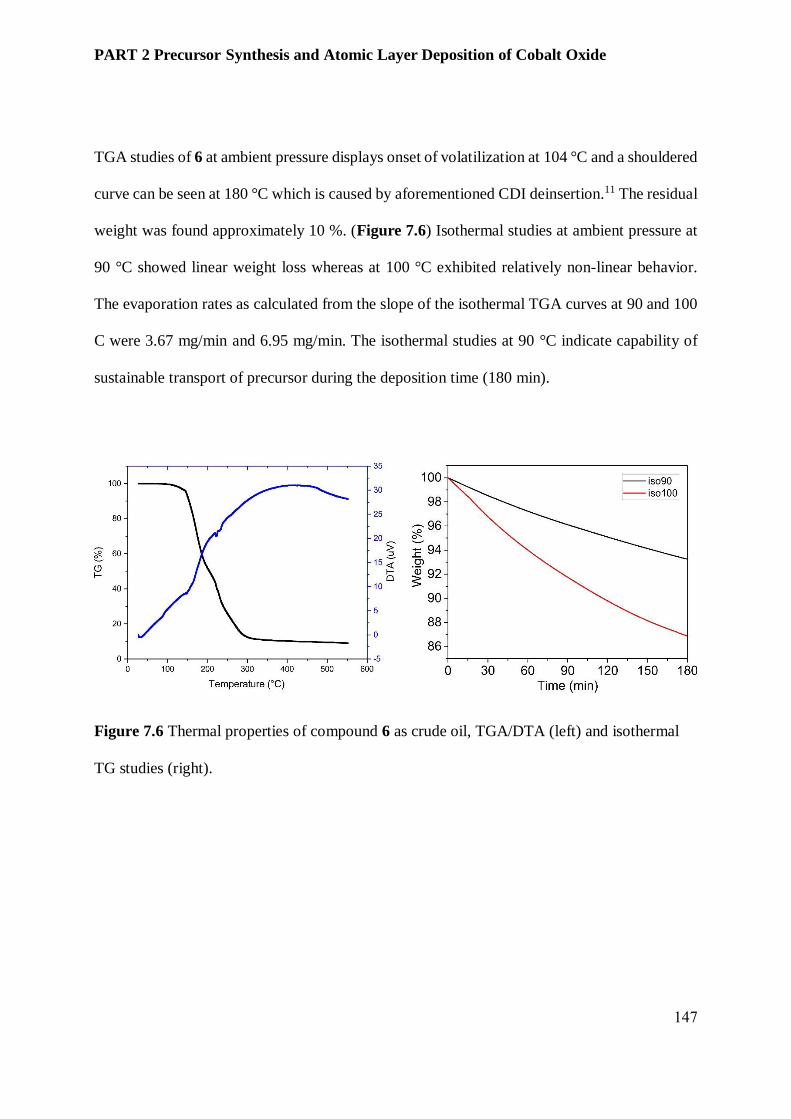
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
147
TGA studies of 6 at ambient pressure displays onset of volatilization at 104 °C and a shouldered
curve can be seen at 180 °C which is caused by aforementioned CDI deinsertion.11 The residual
weight was found approximately 10 %. (Figure 7.6) Isothermal studies at ambient pressure at
90 °C showed linear weight loss whereas at 100 °C exhibited relatively non-linear behavior.
The evaporation rates as calculated from the slope of the isothermal TGA curves at 90 and 100
C were 3.67 mg/min and 6.95 mg/min. The isothermal studies at 90 °C indicate capability of
sustainable transport of precursor during the deposition time (180 min).
Figure 7.6 Thermal properties of compound 6 as crude oil, TGA/DTA (left) and isothermal
TG studies (right).
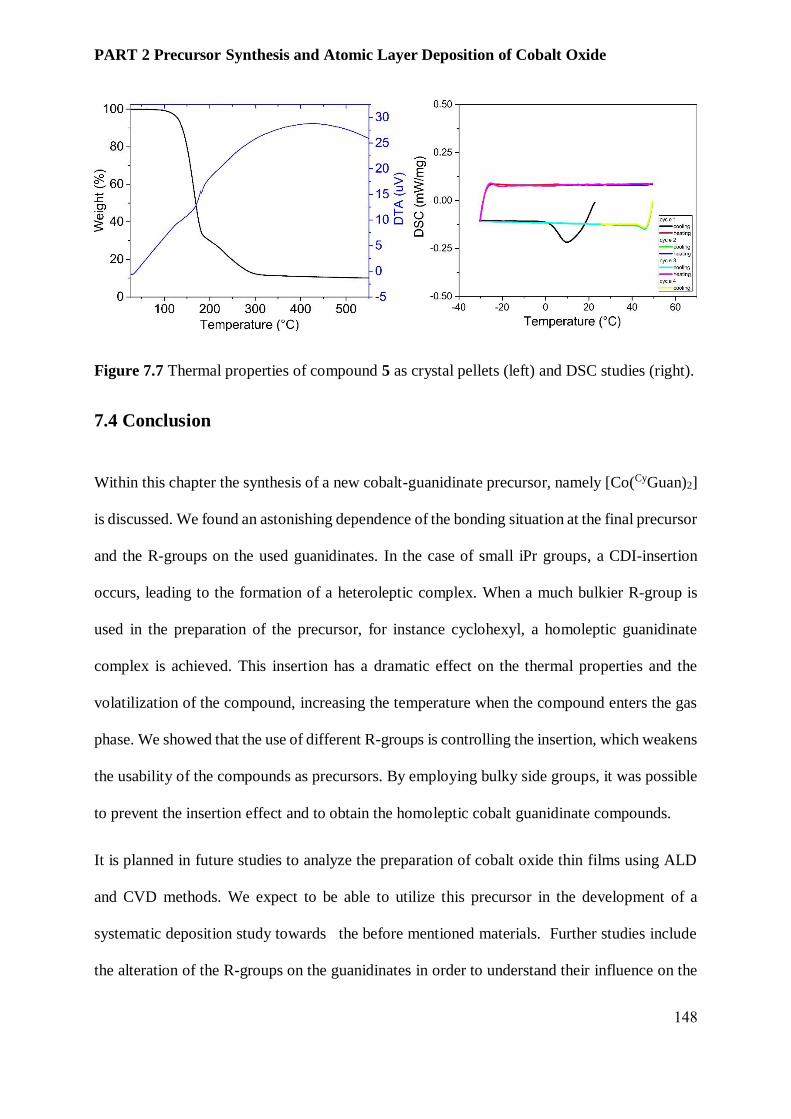
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
148
Figure 7.7 Thermal properties of compound 5 as crystal pellets (left) and DSC studies (right).
7.4 Conclusion
Within this chapter the synthesis of a new cobalt-guanidinate precursor, namely [Co(CyGuan)2]
is discussed. We found an astonishing dependence of the bonding situation at the final precursor
and the R-groups on the used guanidinates. In the case of small iPr groups, a CDI-insertion
occurs, leading to the formation of a heteroleptic complex. When a much bulkier R-group is
used in the preparation of the precursor, for instance cyclohexyl, a homoleptic guanidinate
complex is achieved. This insertion has a dramatic effect on the thermal properties and the
volatilization of the compound, increasing the temperature when the compound enters the gas
phase. We showed that the use of different R-groups is controlling the insertion, which weakens
the usability of the compounds as precursors. By employing bulky side groups, it was possible
to prevent the insertion effect and to obtain the homoleptic cobalt guanidinate compounds.
It is planned in future studies to analyze the preparation of cobalt oxide thin films using ALD
and CVD methods. We expect to be able to utilize this precursor in the development of a
systematic deposition study towards the before mentioned materials. Further studies include
the alteration of the R-groups on the guanidinates in order to understand their influence on the

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
149
inclusion reaction and to demonstrate the tunability of this class of ligands by slight alterations
on the ligand backbone.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
150
7.5 Experimental Section
7.5.1 General Comments
Material and Methods. All manipulations were carried out in an atmosphere of purified argon
using standard Schlenk and glove-box techniques. n-Hexane, n-pentane, toluene, diethylether
were dried using an MBraun Solvent Purification System. Tetrahydrofuran and d6-benzene
were dried by passing through a column of dried activated neutral Al2O3. The final H2O content
in all solvents was checked by Karl Fischer titration and did not exceed 5 ppm. All
characterization and instrumental details are identical described in chapter 3.6 and 6.5. Lithium
guanidinate species, [Li(NR)2CNMe2] were prepared by stoichiometric reaction of LiNMe2 and
corresponding dialkylcarbodiimide in diethylether in prior to the salt metathesis reaction and
used without any further isolation or purification.
7.5.2 Synthetic Procedure
[CoII((NiPr)2CNMe2)((NiPr)2CNMe2(
iPrNHCNiPr))] (5) Freshly prepared [Li(iPr)2CNMe2]
etherate solution was added into a CoCl2 (0.5 equivalent) etherate solution at 0 °C. The reaction
mixture was stirred overnight at room temperature. All the volatiles were removed under
vacuum and the residue was extracted with n-hexane. The residual solution was filtered through
the celite pad in a shlenk frit to remove LiCl salts. The filterate was concentrated to a quarter
of initial volume, then recrystallized at -30 °C. Blue crystals suitable for single crystal X-ray
diffraction were obtained.

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
151
[Co(CyGuan)2] (6) Freshly prepared [Li(Cy)2CNMe2] etherate solution was added into a CoCl2
(0.5 equivalent) etherate solution at 0 °C. The reaction mixture was stirred overnight at room
temperature. All the volatiles were removed under vacuum and the residue was extracted with
n-hexane. The residual solution was filtered through the celite pad in a shlenk frit to remove
LiCl salts. The filterate was concentrated to a quarter of initial volume, then recrystallized at -
30 °C. Blue-violate crystals suitable for single crystal X-ray diffraction were obtained.
[Co(iPrAMD)2]2 (7) was prepared by a modified procedure described in the literature.10 A
solution of methyllithium (1.6 M in Et2O) was added dropwise to a etherate solution of 1,3-
diisopropylcarbodiimide in 1:1 molar ratio at -30 °C. The reaction mixture was stirred for
overnight at room temperature. The formed Li(iPrAMD) solution was added into an Et2O
solution of CoCl2 (0.5 equivalent) by means of cannula at 0 °C. The reaction mixture was stirred
overnight at room temperature. All the volatiles were removed under vacuum and the residue
was extracted with n-hexane. The extract was filtered through a celite pad on a schlenk frit to
afford dark blue solution. The filtrate was concentrated to 10-15 mL and recrystallized at -30 °C.
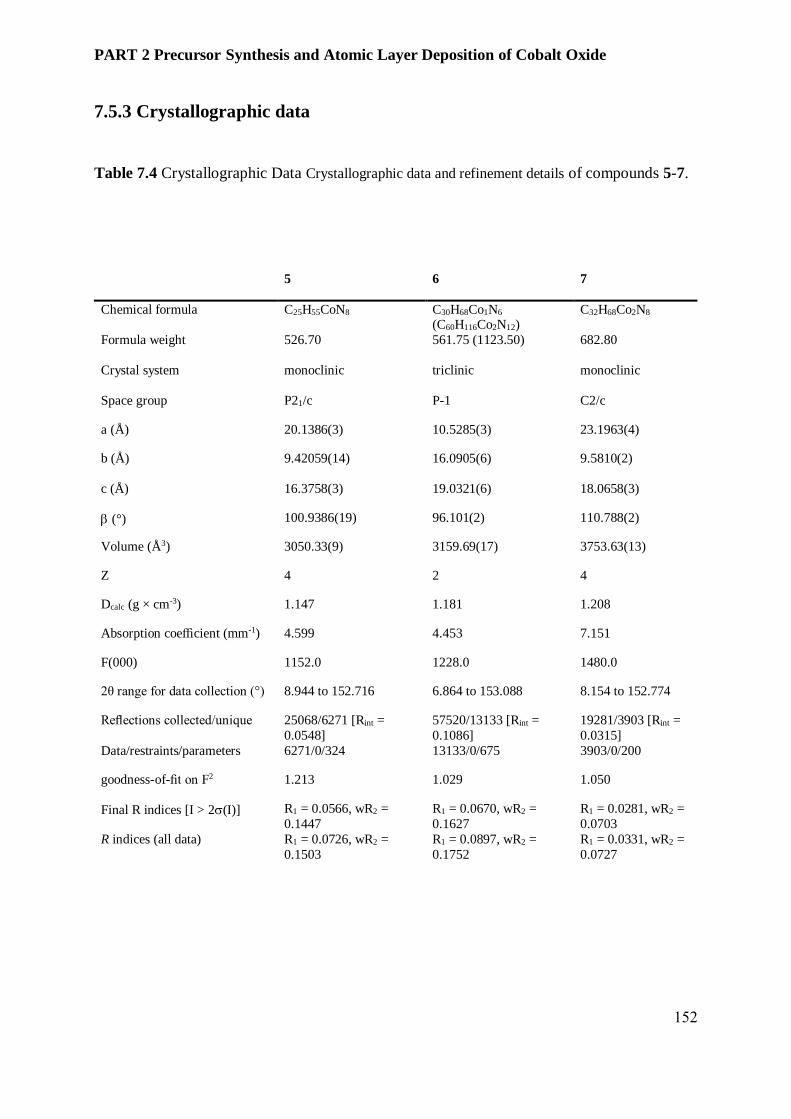
PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
152
7.5.3 Crystallographic data
Table 7.4 Crystallographic Data Crystallographic data and refinement details of compounds 5-7.
5 6 7
Chemical formula C25H55CoN8 C30H68Co1N6
(C60H116Co2N12) C32H68Co2N8
Formula weight 526.70 561.75 (1123.50) 682.80
Crystal system monoclinic triclinic monoclinic
Space group P21/c P-1 C2/c
a (Å) 20.1386(3) 10.5285(3) 23.1963(4)
b (Å) 9.42059(14) 16.0905(6) 9.5810(2)
c (Å) 16.3758(3) 19.0321(6) 18.0658(3)
(°) 100.9386(19) 96.101(2) 110.788(2)
Volume (Å3) 3050.33(9) 3159.69(17) 3753.63(13)
Z 4 2 4
Dcalc (g × cm-3) 1.147 1.181 1.208
Absorption coefficient (mm-1) 4.599 4.453 7.151
F(000) 1152.0 1228.0 1480.0
2θ range for data collection (°) 8.944 to 152.716 6.864 to 153.088 8.154 to 152.774
Reflections collected/unique 25068/6271 [Rint =
0.0548]
57520/13133 [Rint =
0.1086]
19281/3903 [Rint =
0.0315]
Data/restraints/parameters 6271/0/324 13133/0/675 3903/0/200
goodness-of-fit on F2 1.213 1.029 1.050
Final R indices [I > 2(I)] R1 = 0.0566, wR2 =
0.1447
R1 = 0.0670, wR2 =
0.1627
R1 = 0.0281, wR2 =
0.0703
R indices (all data) R1 = 0.0726, wR2 =
0.1503
R1 = 0.0897, wR2 =
0.1752
R1 = 0.0331, wR2 =
0.0727

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
153
8. References
1. Ballirano, P.; Caminiti, R., Rietveld refinements on laboratory energy dispersive X-ray
diffraction (EDXD) data. J. Appl. Crystallogr. 2001, 34, 757-762.
2. Bailey, P. J.; Pace, S., The coordination chemistry of guanidines and guanidinates.
Coord. Chem. Rev. 2001, 214, 91-141.
3. Barry, S. T., Amidinates, guanidinates and iminopyrrolidinates: Understanding
precursor thermolysis to design a better ligand. Coord. Chem. Rev. 2013, 257, 3192-3201.
4. Coyle, J. P.; Johnson, P. A.; DiLabio, G. A.; Barry, S. T.; Müller, J., Gas-Phase
Thermolysis of a Guanidinate Precursor of Copper Studied by Matrix Isolation, Time-of-Flight
Mass Spectrometry, and Computational Chemistry. Inorg. Chem. 2010, 49, 2844-2850.
5. Milanov, A. P. MOCVD and ALD of Rare Earth Containing Multifunctional Materials:
From Precursor Chemistry to Thin Film Deposition and Application. Dissertation, Ruhr-
University Bochum, Bochum, Germany, 2010.
6. Musashi, Y.; Sakaki, S., Insertion of carbon dioxide into a rhodium(III)-hydride bond:
a theoretical study [dagger]. Journal of the Chemical Society, Dalton Transactions 1998, 577-
584.
7. Edelmann, F. T., Chapter 3 - Advances in the Coordination Chemistry of Amidinate and
Guanidinate Ligands. In Adv. Organomet. Chem., Anthony, F. H.; Mark, J. F., Eds. Academic
Press: 2008; Vol. Volume 57, pp 183-352.
8. Edelmann, F. T., Lanthanide amidinates and guanidinates: from laboratory curiosities
to efficient homogeneous catalysts and precursors for rare-earth oxide thin films. Chem. Soc.
Rev. 2009, 38, 2253-2268.
9. Jones, C.; Schulten, C.; Rose, R. P.; Stasch, A.; Aldridge, S.; Woodul, W. D.; Murray,
K. S.; Moubaraki, B.; Brynda, M.; La Macchia, G.; Gagliardi, L., Amidinato– and Guanidinato–

PART 2 Precursor Synthesis and Atomic Layer Deposition of Cobalt Oxide
154
Cobalt(I) Complexes: Characterization of Exceptionally Short Co–Co Interactions. Angew.
Chem., Int. Ed. 2009, 48, 7406-7410.
10. Lim, B. S.; Rahtu, A.; Park, J.-S.; Gordon, R. G., Synthesis and Characterization of
Volatile, Thermally Stable, Reactive Transition Metal Amidinates. Inorg. Chem. 2003, 42,
7951-7958.
11. Kenney, A. P.; Yap, G. P. A.; Richeson, D. S.; Barry, S. T., The Insertion of
Carbodiimides into Al and Ga Amido Linkages. Guanidinates and Mixed Amido Guanidinates
of Aluminum and Gallium. Inorg. Chem. 2005, 44, 2926-2933.
Curriculum Vitae
155
Curriculum Vitae
Jiyeon Kim
E-mail: [email protected]
PROFESSIONAL EXPERIENCE
1/2017 6/2017 Scientific Researcher Technical University Munich, Chair of Inorganic and Metal Organic
Chemistry
Synthesis of Rhenium Sulfide nanoparticles for anti-skin cancer application
12/2012 – 12/2016 Scientific Researcher Ruhr-University Bochum, Chair of Inorganic Chemistry II
Atomic Layer Deposition (ALD) of Transition Metal Oxides Thin Film and Precursor Chemistry
Synthesis of Organometallic Complexes Expertized in Handling Extremely Air Sensitive Materials
Analytic Techniques; Single Crystal X-ray Diffraction, TG, XRD, XPS, AFM, SEM, NMR, IR, EI-MS etc.
ALD Process Development
Teaching Assistant in Advanced Inorganic Chemistry (Lab Course, 4 semesters in total) and Supervision of Bachelor and Master Students
09/2012 – 11/2012 Research Assistant Kookmin University, Laboratory of Inorganic Chemistry
Synthesis of Metal/Metal Oxide Nanoparticles and Their Surface Modification via Ligand Exchange
03/2012 – 05/2012 Visiting Researcher Ruhr-University Bochum, Chair of Inorganic Chemistry II
Metal Organic Framework coated Silver Nanowires
03/2010 – 02/2012 Research Assistant
Kookmin University, Laboratory of Inorganic Chemistry
Photo-luminescent Ceramic Nano Materials
Analysis of Fluorescent Materials (NMR, UV-Vis, PL)
01/2009 – 02/2010 Undergraduate Research Opportunities Program (UROP)
Kookmin University, LABORATORY of Inorganic Chemistry

Curriculum Vitae
156
EDUCATION
12/2012 – 4/2017 Doctor of Philosophy (Ph.D.) Inorganic Chemistry Bochum, Germany Chair of Inorganic Chemistry II, Ruhr-University Bochum
Prof. Dr. Anjana Devi and Prof. Dr. Roland A. Fischer
“Precursor Chemistry for Atomic Layer Deposition of Metal Oxides”
Development of novel cobalt precursor for ALD and process aiming
for photoelectrocatalytic water splitting application.
“Synthesis of Heteronuclear Metal Complexes Containing Direct Metal-Metal Bonds” Metal-Metal bond formation via Reductive Elimination
03/2010 – 02/2012 Mater of Science (M.Sc.) Chemistry
Seoul, Republic of Korea Inorganic Chemistry, Kookmin University, Prof. Dr. Sungho Yoon GPA 4.36 / 4.5
“Surface Modification of Silica Nanoparticles with Zn(II) Complexes Mimicking the Active Site of Carbonic Anhydrase”
03/2006 – 02/2010 Bachelor of Science (B.Sc.) Chemistry Seoul, Republic of Korea Inorganic Chemistry, Kookmin University, Prof. Dr. Sungho Yoon
GPA 3.26 / 4.5
“Investigating the Gelation Properties of Mixed Ag(I) Carboxylates”
INTERNATIONAL EXPERIENCE
03/2016 and 06/2015 Research Stay Helsinki, Finland University of Helsinki, Helsinki, Finland
Prof. Dr. Markku Leskelä “Atomic Layer Deposition of Cobalt Oxide”
Development of Ozone Assisted ALD Process Resulting in Co3O4 Thin Films for Photoelectrocatalysis
01/2013 – 12/2015 Research Contribution 4G-Photocat EU Project (FP7-309636) Objectives: Development of Highly Active Composite Photocatalysts Based on Cheap and Abundant Elements, and Their Deposition onto Various Surfaces.
Lab Visits for Scientific Presentation and Discussion during the Project in Czech Republic, Finland, Vietnam, Poland, Malaysia
07/2014 Research Visit Sapporo, Japan Hokkaido University, Sapporo, Japan
Prof. Dr. Hajime Ito Organometallic Chemistry and Catalysis (C-H Activation)

Curriculum Vitae
157
FELLOWSHIPS & HONOURS
05/2013 – 12/2015 4G-Photocat EU Project Ph.D. Fellowship
07/2014 – 07/2016 Two Ruhr-University Bochum Research School Plus Grants for
International Projects
02/2012 Excellent Researcher Scholarship from Kookmin University
LANGUAGE SKILLS
Korean Native English Fluent in Spoken and Written Form
2.5 Years Elementary School in USA (Los Angeles County, CA) TOFEL Score of 91 in 2012 Official Language during Ph.D. Project
German Basic in Spoken and Written Form
SOFTWARE SKILLS
Microsoft Office (Word, Power Point, Excel), Origin
Photoshop, CorelDraw, ChemDraw, POV-Ray
Shelxl, Olex
Jiyeon Kim July, 20th, 2017

Curriculum Vitae
158
PUBLICATIONS
Homoleptic all nitrogen coordinated bis-guanidinato cobalt (II) complex: Volatile and
thermally stable precursor suitable for ALD/CVD Jiyeon Kim, Niklas Stegmann, Stefan Cwik and Anjana Devi*
Manuscript in progress 2017
Low temperature atomic layer deposition of cobalt oxide as an effective catalyst layer for
photoelectrochemical water splitting devices Jiyeon Kim, Tomi Iivonen, Jani Hämäläinen, Marianna Kemell, Kristoffer Meinander,
Kenichiro Mizohata, Radim Beranek, Markku Leskelä and Anjana Devi*
Accepted in Chemistry of Materials (ACS) 2017 (DOI: 10.1021/acs.chemmater.6b05346)
Oxidation of Nickel-ECp* Complexes: Stable Open-Shell NiI Cations [Ni(ECp*)n(PPh3)4-n]+ (n =
2, 4; E = Al, Ga) Jiyeon Kim, Markus Halbherr, Christian Gemel and Roland A. Fischer*
Inorganic Chemistry, 2015, 54, 9675
Comparative Synthesis of Cu and Cu2O Nanoparticles from Different Copper Precursors in an
Ionic Liquid or Propylene Carbonate Raquel Marcos Esteban, Hajo Meyer, Jiyeon Kim, Christian Gemel, Roland A. Fischer* and Christoph
Janiak*
Eur. J. Inorg. Chem. 2016, 2016, 2106
Low-temperature atomic layer deposition of copper(II) oxide thin films Tomi Iivonen*, Jani Hämäläinen, Benoît Marchand, Kenichiro Mizohata, Miika Mattinen, Georgi Popov,
Jiyeon Kim, Roland A. Fischer, and Markku Leskelä Journal of Vacuum Science & Technology A, 2016, 34, 01A109
A fluorescent ammonia sensor based on a porphyrin cobalt(II)–dansyl complex Jiyeon Kim, Si Hyung Lim, Yeoil Yoon, T. Daniel Thangadurai and Sungho Yoon*
Tetrahedron Letters, 2011, 52, 2645
CONFERENCE
7/2016 Low Temperature Ozone Assisted ALD of Co3O4
Dublin, Ireland Jiyeon Kim, Tomi Iivonen, Radim Beranek, Markku Leskelä and Anjana Devi Oral 16th Atomic Layer Deposition Conference, Dublin, Ireland
8/2015 Low Valent Group 13 Organyls at Transition Metals
Boston, USA Jiyeon Kim, Christian Gemel and Roland A. Fischer Oral + Poster
Precursor Synthesis and Atomic Layer Deposition of CoOx Thin Films Jiyeon Kim, Tomi Iivonen, Roland A. Fischer, Markku Leskelä and Anjana Devi
250th National Meeting of the American Chemical Society
7/2014 Transition Metal-mediated Elimination of H2 from (Organo-)Al Hydrides
Sapporo, Japan Jiyeon Kim, Christian Gemel and Roland A. Fischer Poster XXVI International Conference on Organometallic Chemistry
Oral The Coordination Chemistry of Low Valent Group 13 Species at Transition
Metal Complexes Jiyeon Kim, Christian Gemel and Roland A. Fischer
International Workshop of the Japanese Society of Coordination Chemistry

Curriculum Vitae
159





























