Embed Size (px)
Citation preview

Orbital dependent correlation potentialsin ab initio density functional theory
noniterative - one step - calculations
Orbital dependent correlation potentialsin ab initio density functional theory
noniterative - one step - calculations
Ireneusz GrabowskiInstitute of Physics
Nicolaus Copernicus UniversityToruń, Poland
OEP Workshop, Berlin March 2005

Collaborators:Collaborators:
Quantum Theory ProjectUniversity of Florida,
Gainesville, FL
• So Hirata• Stanislav Ivanov• Victor Lotrich• Igor Schweigert• Rod Bartlett

Two independent theories of the electronic structure of atoms, molecules and solids.
WFT DFT
Two independent theories of the electronic structure of atoms, molecules and solids.
WFT DFT
• Expensive, but provides results which are quaranteed to converge to the solution of the Schrödinger equation as electron correlation and basis set is extended.
• MBPT(2) < CCD < CCSD <CCSD(T) < CCSDTQ < FCI
• Its one particle structure make it possible to treat much larger systems than WFT.
• Almost all unknown informations are contained in an exchange –correlation functional Exc and its associated potential Vxc.
• In standard formulation ‐parameter dependent

• Whereas WFT is a „constructive” theory that provides a prescription for obtaining increasingly more accurate solutions of Schrödinger equation, DFT provides the existence of Exc, but does not provide the energy functional (or even theoretical prescription) , nor systematic converging series of approximations to it.
• Standard LDA, GGA, or hybrid functionals works well in some cases but usually results are unpredictable and it is difficult (or impossible) to improve the functional approximation systematically.

Method which could define a rigorious exchange-correlation functional, potential and orbitals in context ofthe Kohn-Sham theory:
The exploiting in DFT orbital-dependentfunctionals and potentials – OEP method.
Ab initio density functional theory• From coupled-cluster theory and many-body
perturbation theory we derived the local exchange-correlation potential of DFT in an orbital dependentform.
• Parameter free• It guarantees to converge to the right answer in the
correlation and basis set limit, just as does ab initioWFT.
• Specyfying initially to second-order terms –Optimized Effective Potential Method with correlationincluded - OEP-MBPT(2)-KS, OEP-MBPT(2)-f,...

Two different ways to obtain OEP methodswith correlation included
Two different ways to obtain OEP methodswith correlation included
• Functional derivative path– taking functional derivativewith respect to density oforbital dependent energyfunctional (from ab initio WFT) to get exchange‐correlation potential in KS theory.
MBPT(2) levelProblems with extending to higher orders
• Density condition pathGeneral theoreticalframework based on thedensity condition in KohnSham theory involvingcoupled cluster method, many body perturbationtheory, and technicallydiagramatic manipulation.

Basic formalism – functional derivative pathBasic formalism – functional derivative pathThe spin densities ρσ (r) and KS orbitals {φpσ(r)} are obtained by a self consistently solving the KS equation:
The local exchange-correlation potential is formally defined as
the functional derivative of the exchange-correlation energy

In the Optimized Effective Potential (OEP) method, the Exc
OEP[{φpσ}] is an explicit functional of spinorbitals,and the spinorbitals are the solutions of the KS equationwith a local effective OEP potential, which is determinedby the condition that its orbitals be ones that minimizethe energy functional:
The resulting integral OEP equation have to be solved for the VXC in each KS(OEP) iteration,

Formally we can represent Vxc as
Where Xsσ-1 is the inverse of the static KS linear response
function of a system of noninteracting particles
In the LCAO-OEP procedure, the potential and response
function and it inverse are represented in the AO basis.

Exc=Ex+Ec
Ex - HF exchange energy functional in terms of KS orbitals
Vx – orbital dependent exchage-only OEP potential

Orbital dependent OEP–DFT correlation functional
Energy expression for the second order RS PerturbationTheory
OEP-MBPT(2)-KS method
I. Grabowski, S. Hirata, S. Ivanov, R. J. Bartlett Ab-initio density functional theory: OEP-MBPT(2) – a new orbital-dependent correlation functional.,J. Chem. Phys. 116, 4415 (2002


Density condition in KS theory & Coupled Cluster theory
The KS density by construction is an exact density, then any corrections to the converged KS densityintroduced by changes in φi(r) have to vanish.
ρ(r)= ρKS(r)+δρ(r) and δρ(r)=0
The total density from Coupled Cluster (CC) density matrix

We can represent CC density using antisymmetrized diagrams


Equivalence with the OEP-MBPT(2) correlation potentialderived fromfunctional derivativepath.

17

For defining our perturbation at the second order level, we have several different choices for the partitioning of theHamiltonian.
• OEP‐MBPT(2)‐KS
H0=
• OEP‐MBPT(2)‐f
H0=
• OEP‐MBPT(2)‐sc
H0=
}{ ppp
p+∑ε
}{ ppfp
pp+∑
}{}{}{ bafjifppfba
abji
ijp
pp+
≠
+
≠
+ ∑∑∑ ++

19
ResultsResults
• We are NOT doing CC calculations or morecomplicated MBPT(2) !
• We are doing KS DFT‐OEP iterations withcorrectly defined exchange‐correlation potentials(orbital dependent)
• In each KS DFT iteration, using one‐ and two‐electron integrals we calculate VXC,
• Going back with VXC to the KS equation we obtain new set of orbitals and then we can repeatour procedure until self consistency is achieved.

Correlation potential of helium
-0,10
-0,08
-0,06
-0,04
-0,02
0,00
0,02
0,04
0 1 2 3 4 5 6 7
R / a.u.
Cor
rela
tion
pote
ntia
l / a
.u.
Exact (Umrigar & Gonze)
Vosko-Wilk-Nusair correlation potential
Lee-Yang-Parr correlation potential
KLICS correlation potential
OEP-MBPT(2)-KS
OEP-MBPT(2)-f

21
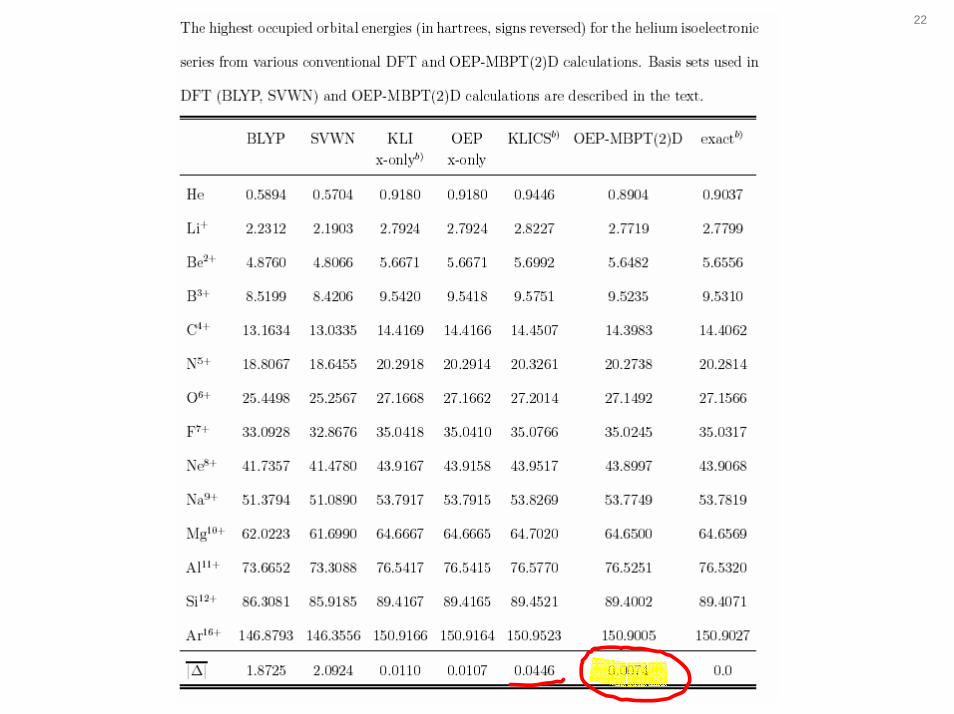
22
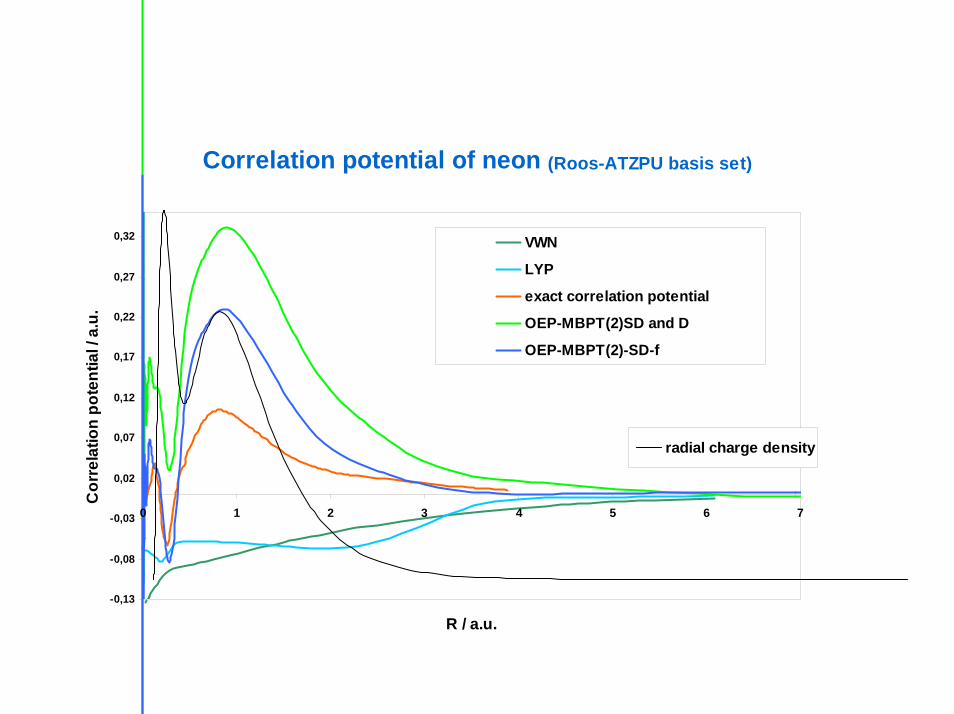
Correlation potential of neon (Roos-ATZPU basis set)
-0,13
-0,08
-0,03
0,02
0,07
0,12
0,17
0,22
0,27
0,32
0 1 2 3 4 5 6 7
R / a.u.
Cor
rela
tion
pote
ntia
l / a
.u.
VWN
LYP
exact correlation potential
OEP-MBPT(2)SD and D
OEP-MBPT(2)-SD-f
radial charge density

Exchange-correlation potential of neon (Roos-ATZPU)one step calculations
-9
-8
-7
-6
-5
-4
-3
-2
-1
00 1 2 3 4 5 6
R / a.u.
Pote
ntia
l / a
.u.
exchange-correlation OEP-MBPt(2)-f and OEP-MBPT(2)`exchange-only' OEP
SVWN
BLYP
OEP-MBPT(2)-f-1shot-HF


Correlation potentials of Be atom
-0,50
-0,40
-0,30
-0,20
-0,10
0,00
0,10
0 1 2 3 4 5 6 7
R / a.u.
Cor
rela
tion
pote
ntia
l / a
.u.
OEP-MBPT(2)-f
vc exact
OEP-MBPT(2)-KS - (non converged)
LYP
VWN


Energy surface of He2 (17s10p2d)
-0,004
-0,003
-0,002
-0,001
0,001
0,002
3 4 5 6 7 8 9 10 11 12
r / au
E-E ∞
/ e
V
MBPT(2)
CCSDT
OEP-MBPT(2)
PBE

Ne2 dimer potential energy - AUG-CC-PVTZ basis set
-200
-150
-100
-50
0
50
100
150
200
250
300
5 5,5 6 6,5 7 7,5 8
r [a.u]
Ener
gy[c
m-1
]
MP2
CCSD
CCSD(T)
OEP_MBPT(2)-fsc

Approximated one step calculationsApproximated one step calculations
• Using exchange-only OEP orbitals we can generatecorrelation potentials using one step procedure by simply inserting orbitals into an orbital-dependentexpression for the correlation potential.
• We can even do the same one step procedure usingHF orbitals, and then generate correlation andexchange correlation potential, without doing anyOEP & KS self interaction procedure.

Correlation potential of helium 1 step calculations
-0,10
-0,08
-0,06
-0,04
-0,02
0,00
0,02
0,04
0 1 2 3 4 5 6 7
R / a.u.
Cor
rela
tion
pote
ntia
l / a
.u.
Exact (Umrigar & Gonze)Vosko-Wilk-Nusair correlation potentialLee-Yang-Parr correlation potentialKLICS correlation potentialOEP-MBPT(2)SD-fOEP-MBPT(2)-f-SD-1 shotOEP-MBPT(2)-SD-f-1shot-HF
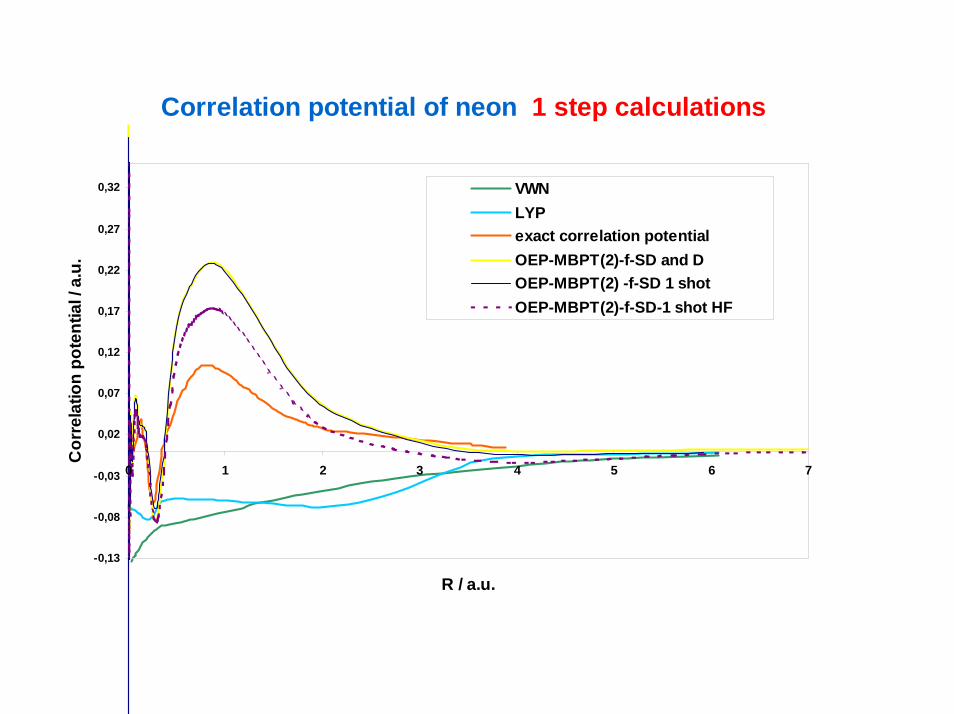
Correlation potential of neon 1 step calculations
-0,13
-0,08
-0,03
0,02
0,07
0,12
0,17
0,22
0,27
0,32
0 1 2 3 4 5 6 7
R / a.u.
Cor
rela
tion
pote
ntia
l / a
.u.
VWNLYPexact correlation potentialOEP-MBPT(2)-f-SD and DOEP-MBPT(2) -f-SD 1 shot OEP-MBPT(2)-f-SD-1 shot HF

Correlation potential of magnesium (Roos-ATZPU basis set)1 step calculations
-0,17
-0,12
-0,07
-0,02
0,03
0,08
0,13
0 0,5 1 1,5 2 2,5 3 3,5 4 4,5
r / a.u.
Corr
elat
ion
pote
ntia
l / a
.u.
VWN
LYP
OEP-MBPT(2)SD-f
OEP-MBPT(2)-f-1 shot
OEP-MBPT(2)-f-1 shot - HF

SummarySummary
Starting from a general theoretical framework based on thedensity condition in Kohn-Sham theory and coupled clustertheory, we have defined a rigorious exchange-correlationfunctionals, potentials and orbitals.
We have performed an ab initio correlated dft calculationsemploying the OEP-MBPT(2) exchange and correlation potentials.
We show the interconnections between the CC & MBPT approach and DFT.
The calculations are fully self-consistent. (Except approximated “one step” calculations)

The OEP-MBPT(2) correlation potentials and the exact correlationpotentials are in the excellent agreement with each other, while thestandard approximate DFT correlation potentials have a qualitativelywrong behavior.
The total energies, and highest occupied orbital energies calculated byOEP-MBPT(2) method are very accurate (CCSD(T) accuracy).
With OEP-MBPT(2) method we can treat with almost CCSD(T)accuracy “week interactions” systems.
Our ab initio dft correlation potentials will be instrumental indeveloping accurate and systematically improvable exchange-correlation functionals and potentials.
The non expensive “one step” procedure which do not need selfconsistent process can be very useful in obtaining approximatecorrelation potentials

36
Some negative aspects
Exc and Vxc are orbital dependent
Strong basis set dependency of the LCAO-OEP results
Slow convergence in some cases
Numerical cost scales as Nit nocc2 nvirt
3
Exchange and correlation potentials are very complicated –they reflects the shell structure, changes in number ofparticles

Ab initio dft (OEP) papersAb initio dft (OEP) papers
• S. Hirata, , S. Ivanov, I. Grabowski, R.J. Bartlett, K. Burke and J. Talman Is an OEP potential determined uniquely? J. Chem. Phys. 115 ,1635 ,(2001)
• I. Grabowski, S. Hirata, S. Ivanov, R. J. Bartlett Ab‐initio density functional theory: OEP‐MBPT(2) – a new orbital‐dependent correlation functional.,J. Chem. Phys. 116, 4415 (2002)
• S. Hirata, S. Ivanov, I. Grabowski, R. J. Bartlett Time‐dependent density functional theory employing optimized effective potentials, J. Chem. Phys. 116, 6468, (2002)
• S. Ivanov, S. Hirata, I. Grabowski, R. J. Bartlett Connections between Second‐Order Gorling‐Levy and Many Body perturbation Approaches in Density Functional Theory. J. Chem. Phys. 118, 461 (2003)
• R. J. Bartlett , I. Grabowski, S. Hirata, S. Ivanov, The Exchange‐Correlation Potential in ab initio Density Functional Theory. J. Chem. Phys. 122, 034104 (2005)
• V. Lotrich, I.Grabowski, R.J. Bartlett Intermolecular potential energy surfaces ofweakly bound dimers computed from ab initio dft: the right answer for the rightreason. Chem. Phys. Lett. xxx, (2004)
• I. Grabowski, V. Lotrich Acurate orbital‐dependent correlation and exchange‐correlation potentials from noniterative ab initio dft calculations. Mol. Phys. xxx (2005)
• S. Hirata, S. Ivanov, R. J. Bartlett , I. Grabowski Exact‐Exchange time dependent density functional theory for frequency‐dependent polarizabilities. ,Phys. Rev. A 71, 1, (2005)

38





























