Embed Size (px)
Citation preview

1521-0103/354/3/269–278$25.00 http://dx.doi.org/10.1124/jpet.115.224816THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 354:269–278, September 2015Copyright ª 2015 by The American Society for Pharmacology and Experimental Therapeutics
Orally Administered Mucolytic Drug L-Carbocisteine InhibitsAngiogenesis and Tumor Growth in Mice s
Tomohiro Shinya, Tsubasa Yokota, Shiori Nakayama, Sayuri Oki, Junpei Mutoh,Satoru Takahashi, and Keizo SatoDepartment of Clinical Biochemistry (T.S.,T.Y., S.N., S.O., K.S.) and Second Department of Pharmacology (J.M.), School ofPharmaceutical Science, Kyushu University of Health and Welfare, Nobeoka, Miyazaki, Japan; and Department ofImmunobiology, School of Pharmacy and Pharmaceutical Science, Mukogawa Women’s University, Nishinomiya, Hyogo, Japan(S.T.)
Received April 1, 2015; accepted June 29, 2015
ABSTRACTAngiogenesis, the formation of new blood vessels from pre-existing vessels, is essential for the growth and metastasis oftumors. In this study, we found that L-carbocisteine, a widelyused expectorant, potently inhibits angiogenesis in vitro and invivo. An in vivo Matrigel plug assay revealed that L-carbocisteine(2.5 mg/kg i.p. twice daily) significantly inhibited vascular endo-thelial growth factor (VEGF)–induced angiogenesis. L-Carbocisteinealso suppressed VEGF-stimulated proliferation, migration, andformation of capillary-like structures of human umbilical veinendothelial cells (HUVECs). We examined the signaling path-ways affected in VEGF-stimulated HUVECs, and found thatL-carbocisteine significantly inhibited VEGF-induced phosphorylation
of phospholipase C (PLC) g, protein kinase C (PKC) m, andextracellular signal-related kinases (ERK) 1/2, which havebeen shown to be essential for angiogenesis. However, theseinhibitory effects of L-carbocisteine were not observed in theHeLa human cervical cancer cell line. An in vivo study of Colon-26tumor-bearing mice found that tumor volumes were significantlysmaller in mice treated with L-carbocisteine (150 mg/kg adminis-tered orally twice daily) in comparison with vehicle-treated mice.However, L-carbocisteine had no direct effect on Colon-26 cellproliferation or ERK activation. Collectively, our results suggestthat L-carbocisteine inhibits tumor angiogenesis by suppressingPLCg/PKC/ERK signaling.
IntroductionAngiogenesis plays an important role in tumor growth
(Thairu et al., 2011) because blood vessels generated via thispathophysiological process supply oxygen and nutrients tocancer cells and subsequently remove carbon dioxide andmetabolites, both of which are indispensable to the pro-liferation and survival of cells (McMahon, 2000; Bhat andSingh, 2008; Claesson-Welsh, 2012). Considerable evidenceshows that appropriate suppression of tumor angiogenesis canattenuate tumor growth (Bhat and Singh, 2008; Claesson-Welsh, 2012). Vascular endothelial growth factor (VEGF)-A isa key regulator of angiogenesis. Angiogenesis-related VEGFsignaling is mediated primarily by VEGF receptor 2 (VEGFR2/KDR) activation (Nagy et al., 2007; Takahashi, 2011; Shibuya,2014), which activates various cell-signalingmolecules, such asphosphoinositide 3-kinase/Akt, Cdc42/p38 mitogen-activatedprotein (MAP) kinase, focal adhesion kinase (FAK), Src family
kinase, phospholipase C (PLC)/protein kinase C (PKC), andmitogen extracellular kinase (MEK)/extracellular signal-related kinase (ERK) (Zachary and Gliki, 2001).
L-Carbocisteine (S-carboxymethylcysteine) is used widely asan expectorant (Rhinathiol, Mucodyne) because it normalizessialic acid and fucose contents inmucins through the regulationof glycosyltransferase activity, and its use is not associatedwith serious side effects. L-Carbocisteine removes phlegm, andindications for its use include inflammation of the upperrespiratory tract, acute bronchitis, bronchial asthma, chronicbronchitis, bronchiectasis, pulmonary tuberculosis, and chronicsinusitis (Hooper and Calvert, 2008). In recent years, novelbiologic activities of L-carbocisteine have been reported in thecontext of inhibition of inflammation associated with influenzavirus infection and chronic obstructive pulmonary disease(Yasuda et al., 2006; Zheng at al., 2008; Yamaya et al., 2010;Asada et al., 2012). Another report showed that L-carbocisteinepossessed free radical–scavenging properties in vitro (Nogawaet al., 2009). Various inflammatory cells, including neutrophils,mast cells, natural killer cells, macrophages, and dendriticcells, are involved in the induction and promotion of angiogen-esis (Noonan et al., 2008; Kim et al., 2013). Moreover, generationof reactive oxygen species (ROS) is a primary function of
This work was supported by Kyorin Pharmaceutical Co.dx.doi.org/10.1124/jpet.115.224816.s This article has supplemental material available at jpet.aspetjournals.
org.
ABBREVIATIONS: DMEM, Dulbecco’s modified Eagle’s medium; EGF, epidermal growth factor; ERK, extracellular signal-related kinase; HUVEC,human umbilical vein endothelial cell; JNK, c-Jun N-terminal qwkinase; MAP kinase, mitogen-activated protein kinase; MEK, mitogen extracellularkinase; NAC, N-acetylcysteine; PBS, phosphate-buffered saline; PKC, protein kinase C; PLC, phospholipase C; ROS, reactive oxygen species;VEGF, vascular endothelial growth factor.
269
http://jpet.aspetjournals.org/content/suppl/2015/07/13/jpet.115.224816.DC1Supplemental material to this article can be found at:
at ASPE
T Journals on M
ay 15, 2018jpet.aspetjournals.org
Dow
nloaded from

activated inflammatory cells, which serve as important stimulifor angiogenic signaling (Reuter et al., 2010; Grote et al., 2011;Kim et al., 2013). However, the effects of L-carbocisteine onangiogenesis have not been reported.We hypothesized that L-carbocisteine produces antiangio-
genic activity, and tested this hypothesis in vitro and in vivo,because an understanding of the molecular mechanisms andtargets of established drugs is essential for safe drug use andthe development of novel indications.
Materials and MethodsAntibodies and Reagents. L-Carbocisteine was a gift from
Kyorin Pharmaceutical Co. (Tokyo, Japan). L-2-Aminoadipic acid wasobtained from TCI (Tokyo, Japan). Human recombinant VEGF165 andepidermal growth factor (EGF) were purchased from PeproTech (RockyHill, NJ). Anti-phospho-Akt (Ser473), anti-Akt, anti-phospho ERK1/2(Thr202/Tyr204), anti-ERK1/2, anti–phospho-stress-activated protein kinase(SAPK)/c-Jun N-terminal kinase (JNK) (Thr183/Tyr185), anti-SAPK/JNK,anti-MEK1/2, anti–phospho-PLCg (Tyr783), anti-PLCg, anti–phospho-PKCm/PKD (Ser744/748), anti-PKCm/PKD, anti–phospho-VEGFR2 (Tyr1175),anti-VEGFR2, and horseradish peroxidase–conjugated anti-rabbit/mouse IgG antibodies were obtained from Cell Signaling Technology(Beverly, MA). Anti-CD31 antibodies were purchased from eBioscience(San Diego, CA). Anti–phospho-p38 MAP kinase (Thr180/Tyr182) anti-bodies, anti–p38 MAP kinase antibodies, anti-ERK1 antibodies, andgrowth factor-reduced Matrigel basement membrane matrix wereobtained from BD Biosciences (Lexington, KY). Protein G Sepharose
was obtained from GE Healthcare (Pittsburgh, PA). Cellmatrix typesI-A and I-C and reconstitution buffer were obtained fromNitta Gelatin,Inc. (Osaka, Japan). Dulbecco’s modified Eagle’s medium (DMEM) andRPMI-1640 medium were obtained from Nissui Pharmaceutical Co.,Ltd. (Tokyo, Japan).
Cell Culture. Human umbilical vein endothelial cells (HUVECs)were obtained from Lonza (Basel, Switzerland) and maintained inendothelial basement medium-2 supplemented with EGM-2 BulletKit(Lonza). HeLa human cervical cancer cells were cultured in DMEMsupplementedwith 10% fetal bovine serum (FBS; Cell Culture Bioscience/Nichirei Biosciences, Inc., Tokyo, Japan). Colon-26 murine coloncarcinoma cells were obtained from Riken BioResource Center (Ibaraki,Japan) and maintained in RPMI-1640 medium supplemented with 10%FBS. Cells were cultured in a humidified atmosphere of 5% CO2 at 37°C.
Animals. Specific pathogen-free inbred C57BL6/JJms mice (weigh-ing 19–21 g) and BALB/cCrmice (weighing 20–22 g) for use in this studywere obtained from Japan SLC, Inc. (Shizuoka, Japan) and housed ina laminar airflow room with a 12-hour light-dark cycle under specificpathogen-free conditions. All animals were allowed to acclimatize totheir new environment for 1 week before experimentation. The animalexperiments were performed according to the guidelines of the KyushuUniversity of Health and Welfare (Nobeoka, Japan), which compliedwith the “Law Concerning the Protection and Control of Animals” and“Standards relating to the care and management, etc. of experimentalanimals” (Office of the Prime Minister of Japan; http://law.e-gov.go.jp).
In Vivo Angiogenesis Assay. The in vivo antiangiogenic activityof L-carbocisteine was assessed with a Matrigel plug assay asdescribed elsewhere (Suehiro et al., 2010). Matrigel was mixed withvehicle or 30 ng/ml of VEGF and injected subcutaneously in a 500-ml
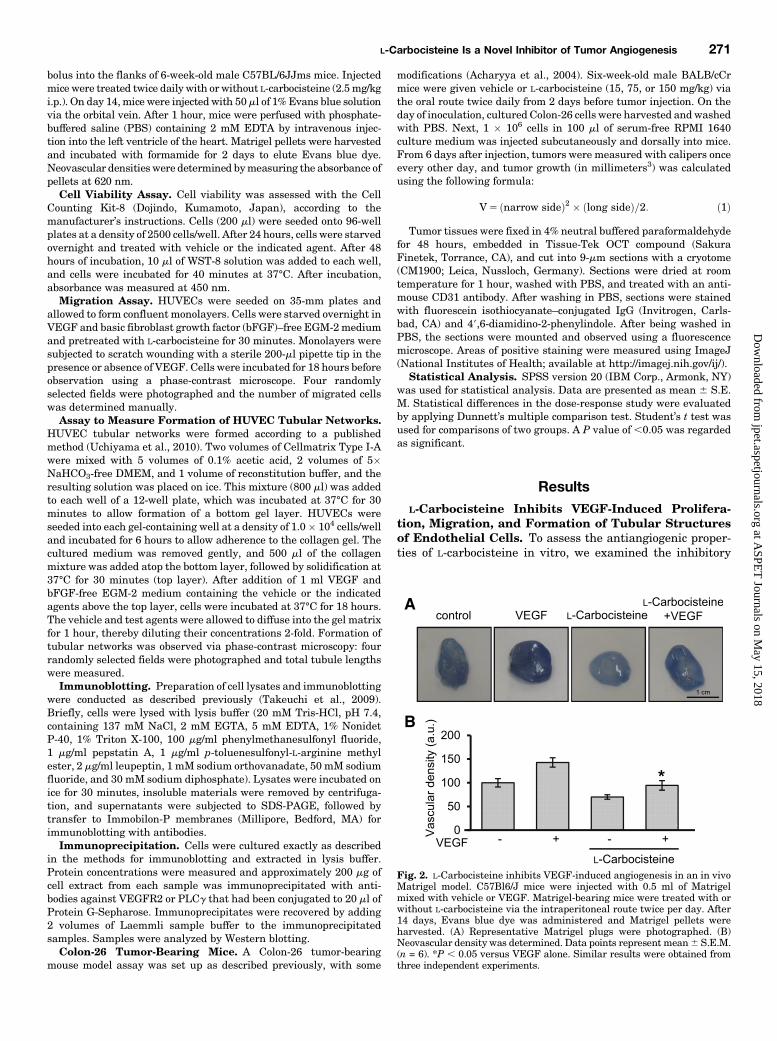
Fig. 1. L-Carbocisteine inhibits VEGF-induced changes in viability, migration, and capillary-structure formation in endothelial cells. (A) HUVECs werepretreated with various concentrations of L-carbocisteine and incubated with VEGF. After 2 days of incubation, cell viability was quantified using a CellCounting Kit-8. (B) HUVECs were pretreated with 100 mM L-carbocisteine for 30 minutes and then incubated with 30 ng/ml VEGF. Migrated cells werequantified by manual counting. (C) HUVECs were pretreated with 100 mM L-carbocisteine and incubated with 30 ng/ml VEGF. After 18 hours ofincubation, total tubule length was assayed using a phase-contrast microscope (100� magnification). Values are mean 6 S.E.M. *P , 0.05; **P , 0.01versus the VEGF-treated group. Similar results were obtained from three independent experiments.
270 Shinya et al.
at ASPE
T Journals on M
ay 15, 2018jpet.aspetjournals.org
Dow
nloaded from
bolus into the flanks of 6-week-old male C57BL/6JJms mice. Injectedmice were treated twice daily with or without L-carbocisteine (2.5mg/kgi.p.). On day 14, mice were injected with 50ml of 1% Evans blue solutionvia the orbital vein. After 1 hour, mice were perfused with phosphate-buffered saline (PBS) containing 2 mM EDTA by intravenous injec-tion into the left ventricle of the heart. Matrigel pellets were harvestedand incubated with formamide for 2 days to elute Evans blue dye.Neovascular densities were determined bymeasuring the absorbance ofpellets at 620 nm.
Cell Viability Assay. Cell viability was assessed with the CellCounting Kit-8 (Dojindo, Kumamoto, Japan), according to themanufacturer’s instructions. Cells (200 ml) were seeded onto 96-wellplates at a density of 2500 cells/well. After 24 hours, cells were starvedovernight and treated with vehicle or the indicated agent. After 48hours of incubation, 10 ml of WST-8 solution was added to each well,and cells were incubated for 40 minutes at 37°C. After incubation,absorbance was measured at 450 nm.
Migration Assay. HUVECs were seeded on 35-mm plates andallowed to form confluent monolayers. Cells were starved overnight inVEGF and basic fibroblast growth factor (bFGF)–free EGM-2mediumand pretreated with L-carbocisteine for 30 minutes. Monolayers weresubjected to scratch wounding with a sterile 200-ml pipette tip in thepresence or absence of VEGF. Cells were incubated for 18 hours beforeobservation using a phase-contrast microscope. Four randomlyselected fields were photographed and the number of migrated cellswas determined manually.
Assay to Measure Formation of HUVEC Tubular Networks.HUVEC tubular networks were formed according to a publishedmethod (Uchiyama et al., 2010). Two volumes of Cellmatrix Type I-Awere mixed with 5 volumes of 0.1% acetic acid, 2 volumes of 5�NaHCO3-free DMEM, and 1 volume of reconstitution buffer, and theresulting solution was placed on ice. This mixture (800 ml) was addedto each well of a 12-well plate, which was incubated at 37°C for 30minutes to allow formation of a bottom gel layer. HUVECs wereseeded into each gel-containing well at a density of 1.0� 104 cells/welland incubated for 6 hours to allow adherence to the collagen gel. Thecultured medium was removed gently, and 500 ml of the collagenmixture was added atop the bottom layer, followed by solidification at37°C for 30 minutes (top layer). After addition of 1 ml VEGF andbFGF-free EGM-2 medium containing the vehicle or the indicatedagents above the top layer, cells were incubated at 37°C for 18 hours.The vehicle and test agents were allowed to diffuse into the gel matrixfor 1 hour, thereby diluting their concentrations 2-fold. Formation oftubular networks was observed via phase-contrast microscopy: fourrandomly selected fields were photographed and total tubule lengthswere measured.
Immunoblotting. Preparation of cell lysates and immunoblottingwere conducted as described previously (Takeuchi et al., 2009).Briefly, cells were lysed with lysis buffer (20 mM Tris-HCl, pH 7.4,containing 137 mM NaCl, 2 mM EGTA, 5 mM EDTA, 1% NonidetP-40, 1% Triton X-100, 100 mg/ml phenylmethanesulfonyl fluoride,1 mg/ml pepstatin A, 1 mg/ml p-toluenesulfonyl-L-arginine methylester, 2 mg/ml leupeptin, 1 mM sodium orthovanadate, 50 mM sodiumfluoride, and 30 mM sodium diphosphate). Lysates were incubated onice for 30 minutes, insoluble materials were removed by centrifuga-tion, and supernatants were subjected to SDS-PAGE, followed bytransfer to Immobilon-P membranes (Millipore, Bedford, MA) forimmunoblotting with antibodies.
Immunoprecipitation. Cells were cultured exactly as describedin the methods for immunoblotting and extracted in lysis buffer.Protein concentrations were measured and approximately 200 mg ofcell extract from each sample was immunoprecipitated with anti-bodies against VEGFR2 or PLCg that had been conjugated to 20 ml ofProtein G-Sepharose. Immunoprecipitates were recovered by adding2 volumes of Laemmli sample buffer to the immunoprecipitatedsamples. Samples were analyzed by Western blotting.
Colon-26 Tumor-Bearing Mice. A Colon-26 tumor-bearingmouse model assay was set up as described previously, with some
modifications (Acharyya et al., 2004). Six-week-old male BALB/cCrmice were given vehicle or L-carbocisteine (15, 75, or 150 mg/kg) viathe oral route twice daily from 2 days before tumor injection. On theday of inoculation, cultured Colon-26 cells were harvested andwashedwith PBS. Next, 1 � 106 cells in 100 ml of serum-free RPMI 1640culture medium was injected subcutaneously and dorsally into mice.From 6 days after injection, tumors were measured with calipers onceevery other day, and tumor growth (in millimeters3) was calculatedusing the following formula:
V5 ðnarrow sideÞ2 � ðlong sideÞ=2: ð1Þ
Tumor tissues were fixed in 4% neutral buffered paraformaldehydefor 48 hours, embedded in Tissue-Tek OCT compound (SakuraFinetek, Torrance, CA), and cut into 9-mm sections with a cryotome(CM1900; Leica, Nussloch, Germany). Sections were dried at roomtemperature for 1 hour, washed with PBS, and treated with an anti-mouse CD31 antibody. After washing in PBS, sections were stainedwith fluorescein isothiocyanate–conjugated IgG (Invitrogen, Carls-bad, CA) and 49,6-diamidino-2-phenylindole. After being washed inPBS, the sections were mounted and observed using a fluorescencemicroscope. Areas of positive staining were measured using ImageJ(National Institutes of Health; available at http://imagej.nih.gov/ij/).
Statistical Analysis. SPSS version 20 (IBM Corp., Armonk, NY)was used for statistical analysis. Data are presented as mean 6 S.E.M. Statistical differences in the dose-response study were evaluatedby applying Dunnett’s multiple comparison test. Student’s t test wasused for comparisons of two groups. A P value of ,0.05 was regardedas significant.
ResultsL-Carbocisteine Inhibits VEGF-Induced Prolifera-
tion, Migration, and Formation of Tubular Structuresof Endothelial Cells. To assess the antiangiogenic proper-ties of L-carbocisteine in vitro, we examined the inhibitory
Fig. 2. L-Carbocisteine inhibits VEGF-induced angiogenesis in an in vivoMatrigel model. C57Bl6/J mice were injected with 0.5 ml of Matrigelmixed with vehicle or VEGF. Matrigel-bearing mice were treated with orwithout L-carbocisteine via the intraperitoneal route twice per day. After14 days, Evans blue dye was administered and Matrigel pellets wereharvested. (A) Representative Matrigel plugs were photographed. (B)Neovascular density was determined. Data points represent mean6 S.E.M.(n = 6). *P , 0.05 versus VEGF alone. Similar results were obtained fromthree independent experiments.
L-Carbocisteine Is a Novel Inhibitor of Tumor Angiogenesis 271
at ASPE
T Journals on M
ay 15, 2018jpet.aspetjournals.org
Dow
nloaded from

effects of L-carbocisteine onHUVEC proliferation. L-Carbocisteineattenuated VEGF-induced proliferation in a concentration-dependent manner and exerted a significant inhibitory effectat concentrations greater than 100 mM (Fig. 1A). The effectsof L-carbocisteine on chemotactic motility were examined inawound-healingmigration assay. Treatment with L-carbocisteine(100 mM) significantly inhibited VEGF-induced HUVEC migra-tion (Fig. 1B). We examined the potential effects of L-carbocisteine on the formation of tubular structures usinga collagen gel matrix assay and found that HUVECs formedan extended network of tubular structures in response toVEGF. Treatment with L-carbocisteine significantly abrogatedVEGF-stimulated formation of tubular networks in endothelialcells (Fig. 1C).
L-Carbocisteine Inhibits VEGF-Induced Angiogene-sis In Vivo. To ascertain the effects of L-carbocisteine onangiogenesis in vivo, we conducted a Matrigel plug assay.VEGF-loaded Matrigel (30 ng/ml) was stained positively withEvans blue, suggesting that new blood vessels formed within
the Matrigel via VEGF-induced angiogenesis (Fig. 2A). Incontrast, treatment with 2.5 mg/kg L-carbocisteine almost com-pletely abolished angiogenesis, as evidenced by the remarkablyreduced level of Evans blue staining in the L-carbocisteine–treated group (Fig. 2B), suggesting that L-carbocisteine effectivelyinhibited angiogenesis in vivo.
L-Carbocisteine Inhibits VEGF-Induced Phosphory-lation of ERK1/2 in HUVECs. To evaluate the molecularmechanisms associated with L-carbocisteine–induced inhibi-tion of VEGF-dependent angiogenesis, we measured byWestern blotting the phosphorylation of key proteins down-stream of VEGFR2 activation: Akt, ERK1/2, JNK, and p38MAP kinase (Wu et al., 2006; Dellinger and Brekken, 2011;Song et al., 2012;). L-Carbocisteine (100 mM) potently sup-pressed VEGF-induced ERK1/2 activation in HUVECs buthad no effect on activation of Akt, JNK, or p38 MAP kinase(Fig. 3, A–D).To determine whether L-carbocisteine inhibits ERK1/2
phosphorylation in nonendothelial cells, we examined the
Fig. 3. L-Carbocisteine attenuated VEGF-induced ERK1/2 phosphorylation in endothelial cells. (A–D) HUVECs were pretreated with L-carbocisteinefor 30minutes and stimulated with 30 ng/ml VEGF for the indicated periods, and cellular lysates were analyzed by SDS-PAGE and immunoblotting withphosphorylation site–specific antibodies, after which the membranes were reprobed with antibodies against unmodified proteins. Protein levels of p-Akt(A), p-ERK (B), p-JNK (C), and p-p38 MAPK (D) were determined. **P , 0.01 versus VEGF (5-minute)-treated group.
272 Shinya et al.
at ASPE
T Journals on M
ay 15, 2018jpet.aspetjournals.org
Dow
nloaded from

effect of L-carbocisteine on ERK1/2 activation induced by100 ng/ml EGF in HeLa cells. However, L-carbocisteine didnot affect ERK1/2 activation in epidermal cells (Fig. 4).
L-Carbocisteine Inhibits Activation of VEGFR2/PLCg/PKC/MEK Signaling in Endothelial Cells. Toclarify the mechanisms underlying L-carbocisteine–mediatedinhibition of the activation of ERK, we examined the effects ofL-carbocisteine on phosphorylation of VEGFR2, PLCg, PKC,and MEK. VEGFR2 phosphorylation in VEGF-stimulatedHUVECs was not suppressed by L-carbocisteine. In contrast,pretreatment with L-carbocisteine significantly suppressedthe phosphorylation of PLCg and PKCm (Fig. 5, A–C). Inaddition, L-carbocisteine inhibited MEK1/2 phosphorylationafter VEGF treatment (Fig. 5D).
L-Carbocisteine Attenuates the Association of PLCgwith VEGFR2. To determine whether L-carbocisteine sup-presses the formation of the PLCg/VEGFR2 complex, celllysates were immunoprecipitated with antibodies againstVEGFR2 or PLCg and immunoblotted with reciprocal anti-bodies. VEGF stimulated complex formation in HUVECs,whereas pretreatment with L-carbocisteine prevented com-plex formation (Fig. 6). L-Carbocisteine suppressed signals forangiogenesis by inhibiting VEGF-induced formation of thePLCg/VEGFR2 complex (Fig. 7).
L-Carbocisteine Suppresses Tumor Growth and An-giogenesis. To determine the effects of L-carbocisteine ontumor growth and angiogenesis in vivo, we evaluated theeffect of L-carbocisteine in Colon-26 tumor-bearing mice. Forthis purpose, we injected Colon-26 tumor cells into maleBALB/c mice, following which they were orally administeredvarious concentrations of L-carbocisteine (experimentalgroup) or vehicle (control group) daily for 26 days (Fig. 8A).On day 22, mice treated orally twice daily with 150 mg/kgL-carbocisteine presented with considerably smaller tumorsthan those observed in control mice (Fig. 8B). At 15 mg/kg and75 mg/kg doses, L-carbocisteine was associated with slightretardation of tumor growth in comparison with the controltreatment. From 10 days after inoculation with tumor cells,
tumor volumes were significantly smaller in mice treatedwith 150 mg/kg L-carbocisteine in comparison with the controlgroup (Fig. 8C). No apparent toxic effects were observed in anyof the treatment groups. Capillary density in the peritumoralregion was determined by staining sections with anti-CD31antibodies. Treatment with 150 mg/kg L-carbocisteine signifi-cantly reduced the number of capillary microvessels (Fig. 8D).From 12 days after the injection of tumor cells, tumor volumewas significantly smaller in mice treated intraperitoneallytwice daily with 10 mg/kg L-carbocisteine in comparison withthe control group (Supplemental Fig. 1, A and B).To determine whether L-carbocisteine directly induces
apoptosis in tumor cells, we tested the effect of L-carbocisteineon Colon-26 cell viability. We found that treatment with a highconcentration (approximately 500 mM) of L-carbocisteine hadno effect on Colon-26 cell proliferation (Supplemental Fig. 2A).Subsequently, we used Western blot analysis to examine theeffect of L-carbocisteine on growth factor–induced phosphory-lation of ERK1/2 in tumor cells, and found that L-carbocisteinedid not suppress EGF-induced activation of ERK1/2 in Colon-26cells (Supplemental Fig. 2B).
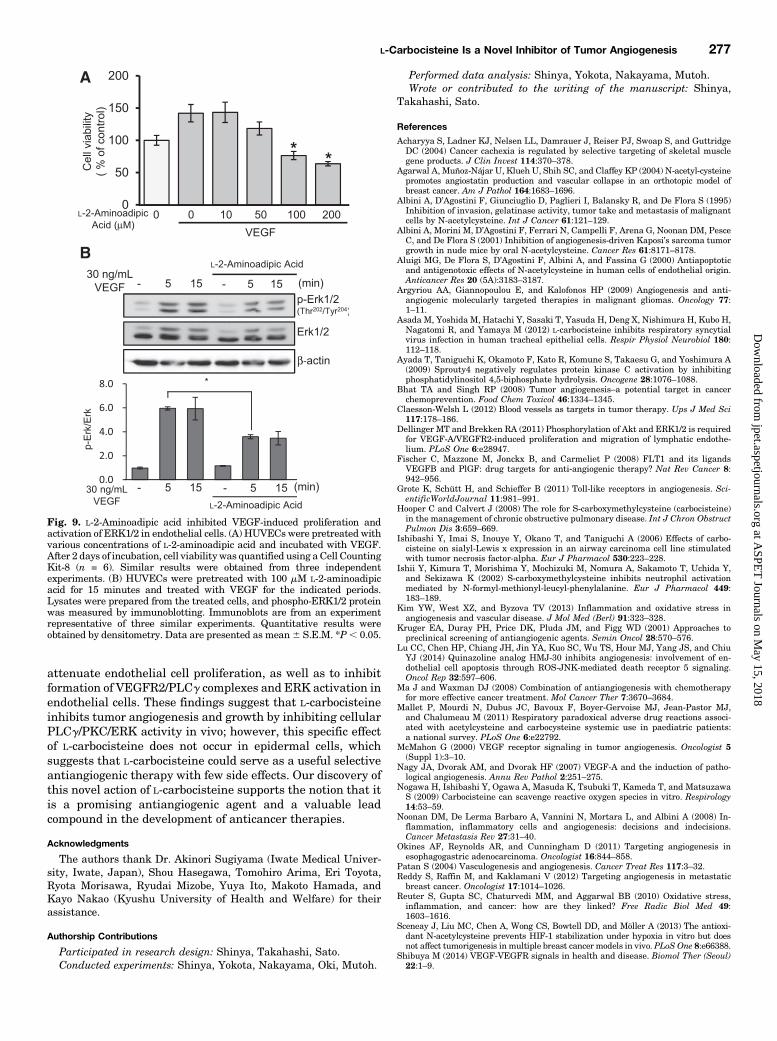
L-2-Aminoadipic Acid Inhibits VEGF-Induced Pro-liferation and Activation of ERK1/2 in EndothelialCells. L-2-Aminoadipic acid is a substitution product ofL-carbocisteine (sulfur to carbon). To confirm whether sulfuris important for the antiangiogenic effect of L-carbocisteine,we evaluated the effects of L-2-aminoadipic acid in VEGF-stimulated endothelial cells. L-2-Aminoadipic acid suppressedVEGF-induced proliferation in a concentration-dependentmanner and suppressed VEGF-induced ERK1/2 activationin HUVECs (Fig. 9, A and B).
DiscussionAngiogenesis plays a crucial role in the tumor growth and
metastasis (Fischer et al., 2008; Zetter, 2008). Therefore,inhibition of tumor angiogenesis has become an importantstrategy for cancer treatment. Several inhibitors of tumorangiogenesis have been shown to prevent the growth andmetastasis of solid tumors (Argyriou et al., 2009), and suchfindings have spurred efforts to discover novel angiogenicinhibitors. HUVECs are derived from the endothelium of largeveins in the umbilical cord and are used as a model system forangiogenesis studies (Wang et al., 2015). L-Carbocisteine wassynthesized in the 1930s and was first used as amucoregulatoryagent (Rhinathiol, Mucodyne) in the treatment of respiratorydiseases in the 1960s (Hooper and Calvert, 2008). In recentyears, novel biologic activities of L-carbocisteine have beenreported. L-Carbocisteine inhibits inflammation associatedwith influenza virus infection and chronic obstructive pul-monary disease (Yasuda et al., 2006; Zheng et al., 2008;Yamaya et al., 2010; Asada et al., 2012), suppresses oxaliplatin-induced hepatocyte toxicity by inhibiting oxaliplatin-induceddecreases in the Bcl2/Bim ratio, and inhibits oxaliplatin-induced apoptosis in vitro (Zhai et al., 2012). Moreover,L-carbocisteine possesses free radical–scavenging and anti-inflammatory properties in vitro (Zheng et al., 2008; Nogawaet al., 2009).On the basis of recent studies showing that L-carbocisteine
inhibits multiple steps of VEGF-induced angiogenesis, wehypothesized that it is a promising novel anticancer agent.This is the first report to demonstrate comprehensively that
Fig. 4. L-Carbocisteine had no effect on EGF-induced ERK activation inepithelial cells. HeLa cells were pretreated with L-carbocisteine for 30minutes and incubated with EGF for the indicated periods. The cells wereharvested and equal aliquots of protein were analyzed for anti–phospho-ERK1/2 by immunoblotting. Results are from an experiment representativeof three independent experiments. Data are presented as mean 6 S.E.M.
L-Carbocisteine Is a Novel Inhibitor of Tumor Angiogenesis 273
at ASPE
T Journals on M
ay 15, 2018jpet.aspetjournals.org
Dow
nloaded from

L-carbocisteine inhibits angiogenesis and tumor growth.Unlike conventional anticancer agents, the uses of whichare complicated by various side effects and/or severe cytotox-icity, L-carbocisteine produces exceptional antiangiogenicactivity without cytotoxicity or side effects.Angiogenesis is a complex, multistep process that involves
the proliferation, migration, and tubular-network formation ofendothelial cells (Patan, 2004), and inhibition of any step of thisprocess has been shown to prevent formation of new bloodvessels (Tournaire et al., 2004). In this study, we showed thatL-carbocisteine significantly inhibits endothelial cell prolifera-tion in a concentration-dependent manner (Fig. 1A). Moreover,L-carbocisteine inhibits VEGF-induced angiogenic responsessuch as cellmigration and formation of capillary-like structures(Fig. 1, B and C). Furthermore, L-carbocisteine inhibitedangiogenesis in a Matrigel plug assay in mice (Fig. 2), showingthat L-carbocisteine inhibits angiogenesis in vitro and in vivo.VEGFR2-mediated activation of Akt, ERK, JNK, and p38
MAP kinase contributes to VEGF-induced survival, prolifera-tion, migration, and tubular-network formation of endothelialcells (Zachary and Gliki, 2001; Wu et al., 2006; Dellinger andBrekken, 2011; Song et al., 2012). Our data showed thatL-carbocisteine significantly abrogated ERK activation
specifically in endothelial cells; no effect was observed inepidermal cells (Figs. 3B and 4). Reports have noted that,unlike other representative growth factor–receptor tyrosinekinases, VEGFR2 forms a complex with and subsequentlyphosphorylates PLCg, which is critical for ERK activation(Takahashi and Shibuya 1997; Wu et al., 2000; Takahashiet al., 2001). In contrast, Ras is weakly activated by VEGF(Takahashi et al., 1999). VEGF stimulates activation of PKCm(PKD) via the VEGFR2/PLCg/PKC pathway. PKCm in endo-thelial cells is rapidly phosphorylated at Ser744/Ser748 inresponse to VEGF, and PKCm is involved in VEGF-inducedERK signaling and endothelial cell proliferation (Wong andJin, 2005). In the present study, L-carbocisteine had no effect onVEGFR2 phosphorylation. However, L-carbocisteine signifi-cantly attenuated VEGF-induced phosphorylation of ERK andPLCg, as well as upstream formation of VEGFR2/PLCgcomplexes (Figs. 5 and 6). Taken together, our data suggestthat L-carbocisteine affects formation of VEGFR2 and PLCgcomplexes without inhibiting VEGFR2 phosphorylation, whichsubsequently affects signaling cascades in a manner that maybe responsible for the antiangiogenic effects of L-carbocisteine.VEGF-induced VEGFR2/PLCg complex formation and activa-tion of PLCg evoke Ca21 mobilization, phosphatidylinositol
Fig. 5. L-Carbocisteine inhibits VEGF-induced PLCg/PKC/ERK signaling in HUVECs. (A–D) HUVECs were pretreated with L-carbocisteine andstimulated with VEGF for the indicated periods. Lysates were subjected to SDS-PAGE and the membranes were hybridized with phospho-specificantibodies, after which the membranes were reprobed. Protein levels of p-VEGFR2 (A), p-PLCg (B), p-PKCm (C), and p-MEK1/2 (D) were determined.Quantitative results were obtained by densitometry. Data are presented as mean 6 S.E.M. from three independent experiments. *P , 0.05.
274 Shinya et al.
at ASPE
T Journals on M
ay 15, 2018jpet.aspetjournals.org
Dow
nloaded from

4,5-biphosphate breakdown, and inositol 1,4,5-triphosphateproduction, which are signaling events upstream of PKC(Ayada et al., 2009). Therefore, the results of our study suggestthat L-carbocisteine suppressed Ca21 mobilization, 4,5-biphos-phate breakdown, and 1,4,5-triphosphate production.Inhibition of tumor angiogenesis represents a novel thera-
peutic modality for controlling tumor metastasis (Kruger et al.,2001; Yi et al., 2008). In this report, we elucidated somemechanisms underlying the inhibitory effect of L-carbocisteineon VEGF-induced angiogenesis by using Matrigel containing
VEGF. However, we also studied the effects of L-carbocisteinein a tumor-bearing mouse model, because malignant cellsrelease a wide range of growth factors in addition to VEGF. Inour in vivo Colon-26 tumor-bearing mouse model, wedemonstrated the effectiveness of oral administration of 150mg/kg L-carbocisteine as a tumor suppressor (Fig. 8, B and C).Related immunohistochemical analyses further revealed thatexpression of the endothelial marker CD31 was reducedmarkedly in tumor sections from L-carbocisteine–treatedmice (Fig. 8D). Furthermore, we determined that 500 mML-carbocisteine did not directly induce apoptosis or inhibitproliferation of Colon-26 cells (Supplemental Fig. 2A). Theseresults suggest that L-carbocisteine inhibits tumor growthindirectly by inhibiting tumor angiogenesis.It has been reported that L-carbocisteine suppresses tumor
necrosis factor (TNF)-a–induced activation of phosphatidylinositol-specific phospholipase C in NCH-H292 epithelial cells (Ishibashiet al., 2006). L-Carbocisteine has also been shown to at-tenuate N-formyl-Met-Leu-Phe (FMLP)–stimulated neutro-phil activation by inhibiting phosphatidylinositol-specificphospholipase C–mediated signal transduction (Ishii et al.,2002). In this study, we demonstrated for the first time thatL-carbocisteine directly inhibits formation of VEGFR2 andPLCg complexes in endothelial cells.Aswith L-carbocisteine,N-acetylcysteine (NAC) is a cysteine-
derivative mucolytic drug that acts by breaking disulfidebridges between macromolecules (Mallet et al., 2011). At thecellular level, NAC inhibits endothelial cell invasion andangiogenesis, probably by inhibiting metalloproteinase activi-ties (Albini et al., 1995). NAC has also been shown to exertdirect cytoprotective and antigenotoxic effects on endothelialcells (Aluigi et al., 2000). Given the possible associationbetween NAC treatment and reduced tumor-dependent angio-genesis, a reported and potentially important aspect of theeffectiveness of NAC is its ability to limit VEGF expression(Albini et al., 2001; Agarwal et al., 2004), and this effect may berelated to its suppression of ROS and hypoxia-inducedtranscription via hypoxia inducible factor-1 (Albini et al.,1995; Agarwal et al., 2004; Sceneay et al., 2013). Therefore,the antiangiogenic effects of NAC are attributable to its
Fig. 6. L-Carbocisteine attenuated VEGF-induced formation of PLCg/VEGFR2 complexes. HUVECs were pretreated with L-carbocisteine andstimulated with VEGF for the indicated periods. The cells were harvestedand equal aliquots of protein extracts were immunoprecipitated withantibodies against VEGFR2 or PLCg. Immunoprecipitates were subjectedto SDS-PAGE and blotted with antibodies against PLCg or VEGFR2 asindicated. Total cell extracts were prepared and subjected to SDS-PAGEfor detection of VEGFR2 and PLCg. The blot was reprobed with beta-actinantibodies as a loading control. Data are presented as mean6 S.E.M. fromthree independent experiments. *P , 0.05.
Fig. 7. Schematic representation of the mechanism by which L-carbocis-teine inhibits VEGF-stimulated angiogenesis. VEGF stimulates forma-tion of complexes between VEGFR2 and PLCg, and this phenomenoninduces angiogenesis. Conversely, L-carbocisteine suppresses VEGFR2/PLCg complex formation and downstream signaling.
L-Carbocisteine Is a Novel Inhibitor of Tumor Angiogenesis 275
at ASPE
T Journals on M
ay 15, 2018jpet.aspetjournals.org
Dow
nloaded from

antioxidant activity and are distinct from the antiangiogeniceffects of L-carbocisteine reported in the present study.VEGF stimulates ROS production (Ushio-Fukai, 2007) and
ROS play a critical role in stimulation of angiogenic signaling,including ERK and JNK signaling (Lu et al., 2014). Becausesulfur compounds have strong anti-ROS activity, we consid-ered whether the inhibitory effect of L-carbocisteine on VEGF-induced ERK activation was the result of anti-ROS activity.We showed that L-2-aminoadipic acid inhibited proliferationand activation of ERK1/2 in VEGF-stimulated endothelialcells (Fig. 9), indicating that the antiangiogenic effect ofL-carbocisteine is not conferred by its constituent sulfur. Webelieve that steric effects associated with L-carbocisteine andL-2-aminoadipic acid are important to their inhibitory effects.Additionally, we expect that addition of another carboxy-methyl or amino group to L-carbocisteine could enhance itsinterference with VEGFR2 and suppression of ERK activationin endothelial cells. The effect of L-carbocisteine does not seemto be stronger than other available antiangiogenic agents,such as bevacizumab, sunitinib, and sorafenib, which atten-uate VEGFR2- and VEGFR2-mediated phosphorylation andactivation of ERK, Akt, JNK, and p38 MAP kinase (Okines
et al., 2011; Reddy et al., 2012). In the present study, we foundthat L-carbocisteine suppressed VEGF-induced ERK1/2 acti-vation but had no effect on activation of Akt, JNK, or p38MAPkinase (Fig. 3, A–D). Furthermore, the usual oral dose ofL-carbocisteine prescribed to adults is 500mg of L-carbocisteine(3 times daily). In our study, L-carbocisteine inhibited angio-genesis but did so at a dose about 10 times greater than thenormally prescribed dose. One of the reasons why a higherconcentration of L-carbocisteine was required is its shortbiologic half-life (t1/2) (about 2 hours; from a medical packageinsert of Mucodyne). However, antiangiogenic effects might beproduced with lower doses of L-carbocisteine by reducing thedosing interval. Currently used antiangiogenic drugs such asthe anti-VEGF antibody bevacizumab can induce transientfunctional normalization of tumor vasculature that canpotentiate the activity of coadministered chemoradiotherapeu-tics (Ma et al., 2008). We believe that the combination ofL-carbocisteine with conventional chemotherapeutic agentsmight increase their efficacy.To our knowledge, this is the first report to demonstrate
that the mucolytic drug L-carbocisteine inhibits angiogenesisin vitro and in vivo. Moreover, L-carbocisteine was found to
Fig. 8. L-Carbocisteine inhibits tumor growth and angiogenesis in Colon 26-bearing mice. (A) Experimental schedule of in vivo tumor growth(schematic). (B) Typical example of tumor-bearing mice from the groups treated with vehicle or 150mg/kg L-carbocisteine on day 6 and day 22. (C) Tumorgrowth was measured with calipers once every other day and calculated in cubic millimeters. All data are presented as mean tumor volume 6 S.E.M.(n = 8 animals per group). (D) Representative photomicrographs of CD31 capillaries in tumor sections stained with antibodies against CD31 (greenfluorescence), an endothelial marker. Nuclei were counterstained with 49,6-diamidino-2-phenylindole (DAPI; blue). The area of CD31-stained capillarieswas measured using ImageJ software. Data are the mean 6 S.E.M. of four experiments. *P , 0.05 versus the vehicle group.
276 Shinya et al.
at ASPE
T Journals on M
ay 15, 2018jpet.aspetjournals.org
Dow
nloaded from
attenuate endothelial cell proliferation, as well as to inhibitformation of VEGFR2/PLCg complexes and ERK activation inendothelial cells. These findings suggest that L-carbocisteineinhibits tumor angiogenesis and growth by inhibiting cellularPLCg/PKC/ERK activity in vivo; however, this specific effectof L-carbocisteine does not occur in epidermal cells, whichsuggests that L-carbocisteine could serve as a useful selectiveantiangiogenic therapy with few side effects. Our discovery ofthis novel action of L-carbocisteine supports the notion that itis a promising antiangiogenic agent and a valuable leadcompound in the development of anticancer therapies.
Acknowledgments
The authors thank Dr. Akinori Sugiyama (Iwate Medical Univer-sity, Iwate, Japan), Shou Hasegawa, Tomohiro Arima, Eri Toyota,Ryota Morisawa, Ryudai Mizobe, Yuya Ito, Makoto Hamada, andKayo Nakao (Kyushu University of Health and Welfare) for theirassistance.
Authorship Contributions
Participated in research design: Shinya, Takahashi, Sato.Conducted experiments: Shinya, Yokota, Nakayama, Oki, Mutoh.
Performed data analysis: Shinya, Yokota, Nakayama, Mutoh.Wrote or contributed to the writing of the manuscript: Shinya,
Takahashi, Sato.
References
Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S, and GuttridgeDC (2004) Cancer cachexia is regulated by selective targeting of skeletal musclegene products. J Clin Invest 114:370–378.
Agarwal A, Muñoz-Nájar U, Klueh U, Shih SC, and Claffey KP (2004) N-acetyl-cysteinepromotes angiostatin production and vascular collapse in an orthotopic model ofbreast cancer. Am J Pathol 164:1683–1696.
Albini A, D’Agostini F, Giunciuglio D, Paglieri I, Balansky R, and De Flora S (1995)Inhibition of invasion, gelatinase activity, tumor take and metastasis of malignantcells by N-acetylcysteine. Int J Cancer 61:121–129.
Albini A, Morini M, D’Agostini F, Ferrari N, Campelli F, Arena G, Noonan DM, PesceC, and De Flora S (2001) Inhibition of angiogenesis-driven Kaposi’s sarcoma tumorgrowth in nude mice by oral N-acetylcysteine. Cancer Res 61:8171–8178.
Aluigi MG, De Flora S, D’Agostini F, Albini A, and Fassina G (2000) Antiapoptoticand antigenotoxic effects of N-acetylcysteine in human cells of endothelial origin.Anticancer Res 20 (5A):3183–3187.
Argyriou AA, Giannopoulou E, and Kalofonos HP (2009) Angiogenesis and anti-angiogenic molecularly targeted therapies in malignant gliomas. Oncology 77:1–11.
Asada M, Yoshida M, Hatachi Y, Sasaki T, Yasuda H, Deng X, Nishimura H, Kubo H,Nagatomi R, and Yamaya M (2012) L-carbocisteine inhibits respiratory syncytialvirus infection in human tracheal epithelial cells. Respir Physiol Neurobiol 180:112–118.
Ayada T, Taniguchi K, Okamoto F, Kato R, Komune S, Takaesu G, and Yoshimura A(2009) Sprouty4 negatively regulates protein kinase C activation by inhibitingphosphatidylinositol 4,5-biphosphate hydrolysis. Oncogene 28:1076–1088.
Bhat TA and Singh RP (2008) Tumor angiogenesis–a potential target in cancerchemoprevention. Food Chem Toxicol 46:1334–1345.
Claesson-Welsh L (2012) Blood vessels as targets in tumor therapy. Ups J Med Sci117:178–186.
Dellinger MT and Brekken RA (2011) Phosphorylation of Akt and ERK1/2 is requiredfor VEGF-A/VEGFR2-induced proliferation and migration of lymphatic endothe-lium. PLoS One 6:e28947.
Fischer C, Mazzone M, Jonckx B, and Carmeliet P (2008) FLT1 and its ligandsVEGFB and PlGF: drug targets for anti-angiogenic therapy? Nat Rev Cancer 8:942–956.
Grote K, Schütt H, and Schieffer B (2011) Toll-like receptors in angiogenesis. Sci-entificWorldJournal 11:981–991.
Hooper C and Calvert J (2008) The role for S-carboxymethylcysteine (carbocisteine)in the management of chronic obstructive pulmonary disease. Int J Chron ObstructPulmon Dis 3:659–669.
Ishibashi Y, Imai S, Inouye Y, Okano T, and Taniguchi A (2006) Effects of carbo-cisteine on sialyl-Lewis x expression in an airway carcinoma cell line stimulatedwith tumor necrosis factor-alpha. Eur J Pharmacol 530:223–228.
Ishii Y, Kimura T, Morishima Y, Mochizuki M, Nomura A, Sakamoto T, Uchida Y,and Sekizawa K (2002) S-carboxymethylcysteine inhibits neutrophil activationmediated by N-formyl-methionyl-leucyl-phenylalanine. Eur J Pharmacol 449:183–189.
Kim YW, West XZ, and Byzova TV (2013) Inflammation and oxidative stress inangiogenesis and vascular disease. J Mol Med (Berl) 91:323–328.
Kruger EA, Duray PH, Price DK, Pluda JM, and Figg WD (2001) Approaches topreclinical screening of antiangiogenic agents. Semin Oncol 28:570–576.
Lu CC, Chen HP, Chiang JH, Jin YA, Kuo SC, Wu TS, Hour MJ, Yang JS, and ChiuYJ (2014) Quinazoline analog HMJ-30 inhibits angiogenesis: involvement of en-dothelial cell apoptosis through ROS-JNK-mediated death receptor 5 signaling.Oncol Rep 32:597–606.
Ma J and Waxman DJ (2008) Combination of antiangiogenesis with chemotherapyfor more effective cancer treatment. Mol Cancer Ther 7:3670–3684.
Mallet P, Mourdi N, Dubus JC, Bavoux F, Boyer-Gervoise MJ, Jean-Pastor MJ,and Chalumeau M (2011) Respiratory paradoxical adverse drug reactions associ-ated with acetylcysteine and carbocysteine systemic use in paediatric patients:a national survey. PLoS One 6:e22792.
McMahon G (2000) VEGF receptor signaling in tumor angiogenesis. Oncologist 5(Suppl 1):3–10.
Nagy JA, Dvorak AM, and Dvorak HF (2007) VEGF-A and the induction of patho-logical angiogenesis. Annu Rev Pathol 2:251–275.
Nogawa H, Ishibashi Y, Ogawa A, Masuda K, Tsubuki T, Kameda T, and MatsuzawaS (2009) Carbocisteine can scavenge reactive oxygen species in vitro. Respirology14:53–59.
Noonan DM, De Lerma Barbaro A, Vannini N, Mortara L, and Albini A (2008) In-flammation, inflammatory cells and angiogenesis: decisions and indecisions.Cancer Metastasis Rev 27:31–40.
Okines AF, Reynolds AR, and Cunningham D (2011) Targeting angiogenesis inesophagogastric adenocarcinoma. Oncologist 16:844–858.
Patan S (2004) Vasculogenesis and angiogenesis. Cancer Treat Res 117:3–32.Reddy S, Raffin M, and Kaklamani V (2012) Targeting angiogenesis in metastaticbreast cancer. Oncologist 17:1014–1026.
Reuter S, Gupta SC, Chaturvedi MM, and Aggarwal BB (2010) Oxidative stress,inflammation, and cancer: how are they linked? Free Radic Biol Med 49:1603–1616.
Sceneay J, Liu MC, Chen A, Wong CS, Bowtell DD, and Möller A (2013) The antioxi-dant N-acetylcysteine prevents HIF-1 stabilization under hypoxia in vitro but doesnot affect tumorigenesis inmultiple breast cancer models in vivo. PLoS One 8:e66388.
Shibuya M (2014) VEGF-VEGFR signals in health and disease. Biomol Ther (Seoul)22:1–9.
Fig. 9. L-2-Aminoadipic acid inhibited VEGF-induced proliferation andactivation of ERK1/2 in endothelial cells. (A) HUVECs were pretreated withvarious concentrations of L-2-aminoadipic acid and incubated with VEGF.After 2 days of incubation, cell viability was quantified using aCell CountingKit-8 (n = 6). Similar results were obtained from three independentexperiments. (B) HUVECs were pretreated with 100 mM L-2-aminoadipicacid for 15 minutes and treated with VEGF for the indicated periods.Lysates were prepared from the treated cells, and phospho-ERK1/2 proteinwas measured by immunoblotting. Immunoblots are from an experimentrepresentative of three similar experiments. Quantitative results wereobtained by densitometry. Data are presented as mean6 S.E.M. *P, 0.05.
L-Carbocisteine Is a Novel Inhibitor of Tumor Angiogenesis 277
at ASPE
T Journals on M
ay 15, 2018jpet.aspetjournals.org
Dow
nloaded from

Song Y, Dai F, Zhai D, Dong Y, Zhang J, Lu B, Luo J, Liu M, and Yi Z (2012) Usnicacid inhibits breast tumor angiogenesis and growth by suppressing VEGFR2-mediated AKT and ERK1/2 signaling pathways. Angiogenesis 15:421–432.
Suehiro J, Hamakubo T, Kodama T, Aird WC, and Minami T (2010) Vascular en-dothelial growth factor activation of endothelial cells is mediated by early growthresponse-3. Blood 115:2520–2532.
Takahashi S (2011) Vascular endothelial growth factor (VEGF), VEGF receptors andtheir inhibitors for antiangiogenic tumor therapy. Biol Pharm Bull 34:1785–1788.
Takahashi T and Shibuya M (1997) The 230 kDa mature form of KDR/Flk-1 (VEGFreceptor-2) activates the PLC-gamma pathway and partially induces mitotic sig-nals in NIH3T3 fibroblasts. Oncogene 14:2079–2089.
Takahashi T, Ueno H, and Shibuya M (1999) VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA syn-thesis in primary endothelial cells. Oncogene 18:2221–2230.
Takahashi T, Yamaguchi S, Chida K, and Shibuya M (2001) A single autophos-phorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation ofPLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J 20:2768–2778.
Takeuchi K, Shin-ya T, Nishio K, and Ito F (2009) Mitogen-activated protein kinasephosphatase-1 modulated JNK activation is critical for apoptosis induced by in-hibitor of epidermal growth factor receptor-tyrosine kinase. FEBS J 276:1255–1265.
Thairu N, Kiriakidis S, Dawson P, and Paleolog E (2011) Angiogenesis as a thera-peutic target in arthritis in 2011: learning the lessons of the colorectal cancerexperience. Angiogenesis 14:223–234.
Tournaire R, Simon MP, le Noble F, Eichmann A, England P, and Pouysségur J(2004) A short synthetic peptide inhibits signal transduction, migration and an-giogenesis mediated by Tie2 receptor. EMBO Rep 5:262–267.
Uchiyama T, Toda K, and Takahashi S (2010) Resveratrol inhibits angiogenic re-sponse of cultured endothelial F-2 cells to vascular endothelial growth factor, butnot to basic fibroblast growth factor. Biol Pharm Bull 33:1095–1100.
Ushio-Fukai M (2007) VEGF signaling through NADPH oxidase-derived ROS.Antioxid Redox Signal 9:731–739.
Wang B, Yu W, Guo J, Jiang X, Lu W, Liu M, and Pang X (2015) The antiparasiticdrug, potassium antimony tartrate, inhibits tumor angiogenesis and tumor growthin nonsmall-cell lung cancer. J Pharmacol Exp Ther 352:129–138.
Wong C and Jin ZG (2005) Protein kinase C-dependent protein kinase D activationmodulates ERK signal pathway and endothelial cell proliferation by vascular en-dothelial growth factor. J Biol Chem 280:33262–33269.
Wu G, Luo J, Rana JS, Laham R, Sellke FW, and Li J (2006) Involvement of COX-2 inVEGF-induced angiogenesis via P38 and JNK pathways in vascular endothelialcells. Cardiovasc Res 69:512–519.
Wu LW, Mayo LD, Dunbar JD, Kessler KM, Baerwald MR, Jaffe EA, Wang D,Warren RS, and Donner DB (2000) Utilization of distinct signaling pathways byreceptors for vascular endothelial cell growth factor and other mitogens in theinduction of endothelial cell proliferation. J Biol Chem 275:5096–5103.
Yamaya M, Nishimura H, Shinya K, Hatachi Y, Sasaki T, Yasuda H, Yoshida M,Asada M, Fujino N, Suzuki T, et al. (2010) Inhibitory effects of carbocisteine ontype A seasonal influenza virus infection in human airway epithelial cells. Am JPhysiol Lung Cell Mol Physiol 299:L160–L168.
Yasuda H, Yamaya M, Sasaki T, Inoue D, Nakayama K, Yamada M, Asada M,Yoshida M, Suzuki T, and Nishimura H, et al. (2006) Carbocisteine inhibits rhi-novirus infection in human tracheal epithelial cells. Eur Respir J 28:51–58.
Yi T, Cho SG, Yi Z, Pang X, Rodriguez M, Wang Y, Sethi G, Aggarwal BB, and Liu M(2008) Thymoquinone inhibits tumor angiogenesis and tumor growth throughsuppressing AKT and extracellular signal-regulated kinase signaling pathways.Mol Cancer Ther 7:1789–1796.
Zachary I and Gliki G (2001) Signaling transduction mechanisms mediating biologicalactions of the vascular endothelial growth factor family. Cardiovasc Res 49:568–581.
Zetter BR (2008) The scientific contributions of M. Judah Folkman to cancer re-search. Nat Rev Cancer 8:647–654.
Zhai Q, Bian XL, Lu SR, Zhu B, and Yu B (2012) Carbocisteine reduces the cyto-toxicity of oxaliplatin. Z Naturforsch C 67:215–221.
Zheng JP, Kang J, Huang SG, Chen P, Yao WZ, Yang L, Bai CX, Wang CZ, Wang C,Chen BY, et al. (2008) Effect of carbocisteine on acute exacerbation of chronicobstructive pulmonary disease (PEACE Study): a randomised placebo-controlledstudy. Lancet 371:2013–2018.
Address correspondence to: Dr. Keizo Sato, Kyushu University of Healthand Welfare, 1714-1 Yoshino-machi, Nobeoka-shi, Miyazaki 882-8508, Japan.E-mail: [email protected]
278 Shinya et al.
at ASPE
T Journals on M
ay 15, 2018jpet.aspetjournals.org
Dow
nloaded from





























