Embed Size (px)
Citation preview

Nuclear magnetic resonance in magnetic systems and metals
Denis Arčon
University of Ljubljana, Faculty of mathematics and physics, Slovenia
Institute “Jozef Stefan”, Ljubljana, Slovenia
1

Motivation
2
Solid-state physics: - Crystal structure and lattice dynamics, - Electronic properties, - Magnetism, (anti)ferroelectrics, multiferroics - Superconductors
Local probes that would be simultaneously sensitive to lattice as well to electrons
Experimentalists toys: Scattering techniques: - XRD - Neutron diffraction Measurements of macroscopic properties: - Dielectric spectroscopy, - Magnetization measurements
(SQUID, … ) - Specific heat, thermal
conductivity, thermal dilatometry, …
Vibrational spectroscopies

So, why NMR?
3
Nuclear magnetic resonance (NMR) A TOOL to study condensed matter systems o Local, microscopic, site‐specific probe o Virtually all elements are NMR active o study electronic spin structure, lattice
structure
o Non‐invasive – no current, no contacts on the sample
o ωNMR ≈ 0 (μeV), gives partial q information (cf. neutron diffraction)
o can be combined with other techniques: transport, magnetization, dielectric, optical, …
o IJS laboratory: extreme conditions: low temperatures, high pressures, different magnetic fields up to 9.34 T
Elvis is alive and well, working as an NMR spectroscopist in Ljubljana.

Magnetic resonance - basics
4
History: Purcell (NMR in paraffin), Bloch (NMR in H2O), Zavoisky (EPR) Magnetic resonance phenomena is found in systems that poses magnetic moments and we are “in tune” with a natural frequency of the magnetic system. Magnetic resonance technique offers a high resolution experimental probe for the study of static and dynamics properties of local magnetic fields. Nuclear magnetic resonance (NMR): typically in the 10-500 MHz range Electron paramagnetic resonance (EPR): typically in the 1-100 GHz range NMR is utilized widely not only in physics and/or chemistry but also in medical diagnostics (MRI) and so on. ・Physics Condensed matter physics ・Chemical Analysis and/or identification of material ・Biophysics Analysis of Protein structure ・Medical MRI (Magnetic Resonance Imaging)
„It‘s lupus, do the MRI!“

Outline
5
Lesson Subject
1 Introduction to magnetic resonance; Bloch equations, relaxation times, dynamics susceptibility
2 The basic spin Hamiltonian for NMR
3 Hyperfine coupling interaction, Knight shift
4 The Moriya theory of spin-lattice relaxation
5 NMR in the superconducting state (just briefly)
6 NMR in the magnetically ordered state
7 Examples: fullerides, pnictides and quasi-1D magnetoelectric system

Magnetic resonance - basics
6
Nuclear magnetic moments: A system such as nucleus may consist of many particles coupled together so that for a given state the nucleus possesses a total magnetic moment m related to a total angular momentum G Here g is a scalar called gyromagnetic ratio and is typical for nuclei or electrons. Simple model: m = iS = ipr2 ; G=mvr=m(2pr/T)r ; i=e/T g = e/2m g thus decreases with increasing particle mass
Γ
gm =
γ [MHz/T ] ν [MHz]
proton 2.675102 42.58 B0 [T]
electron 1.759105 27990 B0 [T]
QM: II Nˆˆˆˆ gm ==G

Magnetic resonance - basics
7
Nucleus γ/2π
(MHz/T)
1H 42.576
2H 6.53566
3He -32.434
7Li 16.546
13C 10.705
14N 3.0766
15N -4.3156
17O -5.7716
23Na 11.262
63Cu 11.284
65Cu 12.109
51V 11.193
31P 17.235
129Xe -11.777

Magnetic moment in the external magnetic field
• Torque on the magnetic moment:
• Change of the angular momentum
8
BT
= m
Bdt
dB
dt
Γd
== mgm
m
Larmor precesion m
0BNL g =
Larmor frequency Typically in the rf range up several houndred MHz

Effect of rf field • But, in order to see precession, we first need to shift
magnetization away from the z-axis. This is the job of rf pulses.
• In the rotating frame, that rotates with Larmor frequency L, m is static Beff=B0- L/g=0.
• rf field B10cos(Wt) = BR+BL
• Rotation around the xR axis
9
0=R
effB
m
xR rfB
90o pulse tWgB10=p/2
yR
B10 of the order of 10 mT

Magnetic resonance - basics
10
Quantum mechanics: we have to treat m and G as operators.
IIΓ nn
ˆˆˆˆ
gm ==
In an applied field, Example: I = ½ (case of 1H or 13C for instance)
0ˆ
a B=B z → , 1, , 1,Im I I I I=
ZZ IBBH ˆˆˆ0
gm ==
mBBE Zm 00 gm ==
In the MR we attempt to detect the splitting of these energy levels by appropriate perturbation. The interaction must be such, that the angular frequency matches the splitting: LARMOR FREQUENCY
0BEL g ==
Which perturbation will trigger the transitions between these levels? R.f. irradiation perpendicular to B0 will do the job!
tIBH Lxrfrf g cosˆ=

Magnetic resonance - basics
11
Time dependent Schrödinger equation has the general solution in the form We can now calculate the expectation value for mz, for instance
ZHt
i ˆ=
=m
tiE
mImmeuc/
,
)()(
ˆ'
)(ˆ)(*
22
21
2/1
*
2/12/1
*
2/1212/1
*
',
/)(*
''
bacccc
cmc
mImecc
dtt
I
m
mm
mm
z
tEEi
mm
zz
mm
==
=
=
=
=
gg
g
g
tmm
Normalised WF: 2
2/1
*
2/1
2
2/1
*
2/1 & bccacc ==

Magnetic resonance - basics
12
The calculation for the expectation value for mx, is slightly more complicated, but follows the same steps By recalling we can derive
=
=
',
/)(*
'ˆ'
)(ˆ)(*
'
mm
x
tEEi
mm
xx
mImecc
dtt
mm g
tmm
1,)1()1(,ˆ
1,)1()1(,ˆ
=
=
mImmIImII
mImmIImII
gm
gm
=
=
tab
tab
y
x
0
0
sin
cos
)( 22
21 baz = gm

Magnetic resonance – basics spin-lattice relaxation
13
N-
N+
= NWNW
dt
dN
Simple rate equations
Fermi golden rule: p
= bbab EEaVbP2
ˆ2
WWWbVaaVb ==
22
ˆˆ
If we introduce n=N+ -N- and N=N+ +N- then N+=½(N+n) and N-=½(N-n) then we get
WtentnWndt
dn 2
0)(2 ==Initial difference in energy population will exponential decay to zero!

Magnetic resonance – basics spin-lattice relaxation
14
Problems: 1. Absorbed power dE/dt=N+WħωL-N-WħωL=WħωLn(t) will also vanish after some time
(not supported by the experiments). 2. What if W = 0 (no perturbation) and we change magnetic field (MZ should change,
what is not predicted by this equation)! Therefore, there MUST be a process that allows the system to accept (or release) the
magnetic energy. It is the coupling to the lattice spin-lattice relaxation Thermal equilibrium: difference in the population given by the energy difference
TkBTkE BB eeN
N0
0
0g
==
= NWNWdt
dN
In thermal equilibrium d/dt=0 TkB BeWW 0/g
=

Magnetic resonance – basics spin-lattice relaxation
15
Why now WW? Rate equations are solved by introducing n and N and we get In terms of magnetization, this actually reads as
Spin Lattice
)(
)()(
1
)()(
0
1
1
0
=
==
WW
WWNnWW
T
T
nnWWnWWN
dt
dn
0
1
zzM MdM
dt T
=

Magnetic resonance – basics spin-lattice relaxation
16
Spin-lattice relaxation also solves the problem of absorption of r.f. energy. Namely, if we take into account external perturbation (described by W) and spin-lattice relaxation together, then we derive In the steady state And the absorbed power will be
1
02T
nnWn
dt
dn =
1
021
1
WTnn
=
1
021 WT
WnnWdt
dE
==
0 2 4 6 8 10
0.0
0.2
0.4
0.6
0.8
1.0
P/P
S
2WT1

Magnetic resonance – basics Bloch equations
17
Classical description of the motion of magnetic moments in external magnetic field Torque due to the action of B0
This torque will change the angular momentum In addition we have also relaxation phenomena to return back to equilibrium. We define two relaxation times T1 – spin-lattice relaxation time and T2 – spin-spin relaxation time to allow longitudinal and transverse magnetizations to come back
0B
m
000 BMdt
MdB
dt
dB
dt
d
===G
gmgm
m
0
2
x xy
dM MB M
dt Tg=
0
2
y y
x
dM MB M
dt Tg=
0
1
zzM MdM
dt T
=
Please note that changes in Mx and My do not change the magnetic energy

Magnetic resonance – basics Bloch equations
18
Bloch equations in compact form
==
1
2
2
00
/100
0/10
00/1
T
T
T
RMMRBMdt
Md
g
With initial conditions
2/
0cost T
xM t m e t=
00 , 0 0,x y zM m M M M= = =
we have solutions in the form
2/
0sint T
yM t m e t=
1/
0 1t T
zM t M e
=
0 5 10 15
-2
0
2
Mx, M
y (
arb
. u
nits )
time
Mx
My

Magnetic resonance – basics Bloch equations
19
The application of rf field perpendicular to the static magnetic field will thus lead to an extra transverse magnetization. In a typical experiment, the rf field will be linearly polarized (along the x-axis)
xlab xrot
yrot ylab
t
The result of previous calculations is that, we can write Mx component of the magnetization in the laboratory frame as We thus defined a complex r.f. susceptibility with components
1sin''cos'sincos)( BtttMtMtM rot
y
rot
xx ==
''' i=
2
2
2
0
200
2
2
2
0
2020
0
1
1
2''
12'
TT
T
TT
=
=
Rotating frame with field
Stat
ion
ary
solu
tio
ns
dM
/dt=
0

Magnetic resonance – basics Bloch equations
20
When the detection coil is filled with measured material, its inductance changes by
L=L0(1+) The complex impedance of the coil thus also changes as Z=R+iL=R-L0’’+iL0’ The real part of Z changes by R/R=L0’’/R=Q’’ In unperturbed coil, the relation between the magnetic energy and the current producing that magnetic field is ½L0i0
2= ½B12V/m0
Because of change in the impedance, the average dissipated power is In MR experiments we are thus measuring ’’. But ’ is related to ’’ through Kramers-Kronig relations, so we in principle know both of them. Also, please note that P is proportional to 0 so it can provide a quantitative information about the static magnetic susceptibility of the samples!
''2
1
2
1 2
1
0
2
0 m
VBRiP ==

Magnetic resonance – basics Method of moments
21
The experimentalists thus measure the absorption ’’(). The theorists tend to be more familiar with the Green functions or time-correlation functions of spin operators. In a linear response theory, developed for MR by Kubo and Tomita, ’’() is expressed as A time evolution of Mx operator is calculated in the interaction representation Lets define the spectral lineshape as If we perform inverse FT of the above expression, we can derive
dteMtMTk
V ti
xx
B
= 0ˆˆ2
''
/ˆ/ˆ ˆˆ tHi
x
tHi
x eMetM =
dteMtM
Tk
Vf ti
xx
B
== 0ˆˆ2
'')(
p
defMtMTk
V ti
xx
B
= )(2
10ˆˆ
2

Magnetic resonance – basics Method of moments
22
If we look at the above expression at time t=0, then we notice the following Therefore <Mx
2> is a measure for the area under the resonance curve. But, if we take the time derivative and then take its value at t=0, we can make even a step further The n-th time derivative will be thus
p
dfMMTk
Vxx
B
= )(2
10ˆ0ˆ
2
p
dfiMtMdt
d
Tk
Vtxx
B
= = )(2
10ˆˆ
20
p
dfi
MtMdt
d
Tk
V n
n
txxn
n
B
= = )(2
0ˆˆ2
0
3
)1(ˆˆˆˆ
ˆˆ
222
312
,
2
,
2
,
,
====
=
IIiziyixi
i
xix
gmmmm
mm

Magnetic resonance – basics Method of moments
23
We can thus define n-th moment of the resonance as Let us calculate the second moment of the the line Taking the above expression at t=0 and slightly rearranging terms, we finally derive
2
0
ˆ
0ˆˆ
)(
)(
x
txxn
n
n
n
n
M
MtMdt
d
i
df
df=
==
0ˆˆ,,1
0ˆ)ˆˆ(0ˆ0ˆ
/ˆ/ˆ
2
/ˆ/ˆ/ˆ/ˆ
2
2
x
tHi
x
tHi
x
tHi
xx
tHi
x
tHi
x
tHi
MeMHHe
MeHMMHedt
diMeMe
dt
d
=
=
2
2
2
2
2
ˆ
ˆ,1
)(
)(
x
x
M
MH
df
df
==
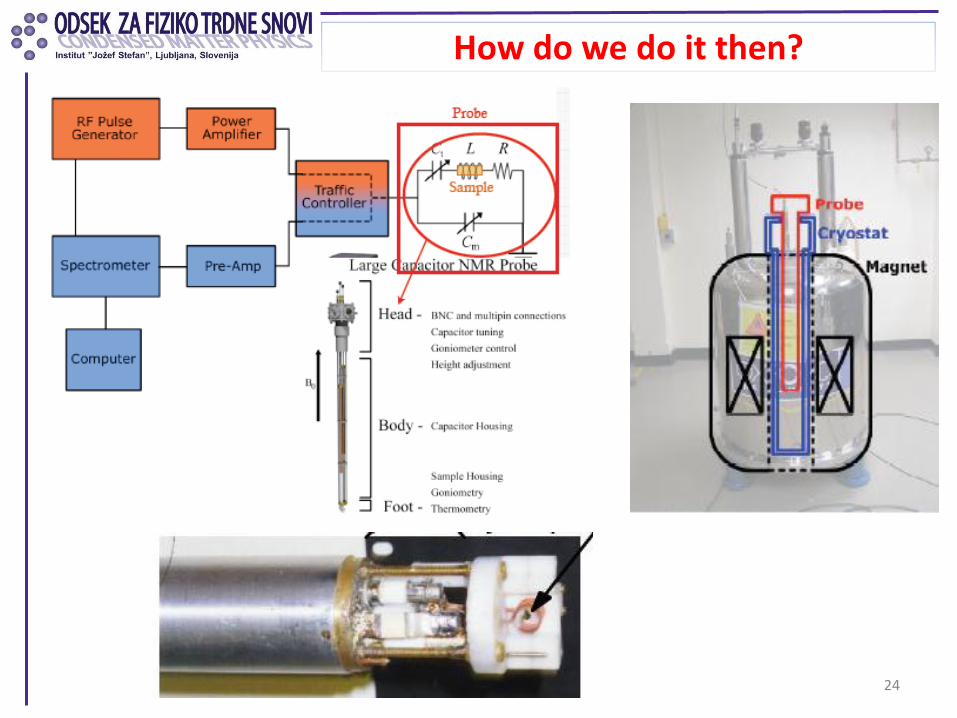
How do we do it then?
24

Current Piston-cylinder cell for NMR
• for pressures up to 20 kbar
• large sample volumes (< 200 mm3)
• easy to manufacture
• inexpensive materials
• dangerous
clamp/screw (Be-Cu)
bottom piston (Be-Cu)
body (Ni-Cr-Al)
upper piston (Ni-Cr-Al)

Magnetic resonance – basics pulses
26
If we can neglect the effect of relaxation times (typically this is justfied when the duration of rf fields is short compared to T1 and T2), then one can also show that the effect of rf field will be to rotate the magnetization
After p/2 pulse we observe FID
Small current induced in the coil is amplified and then analyzed

Magnetic resonance – spin echo
27
When FID is to short to be observed then we may want to try with spin echo

Measurements of relaxation times
28

Magnetic resonance – what we have learnt so far?
29
1. Classical Bloch equations (torque + T1 + T2) 2. T1 processes must result in a transfer of energy since it involves magnetic
dipoles reorienting in a magnetic field. Quantum mechanically, it is a change of populations between spin-down states to spin-up states which are nondegenerate in a magnetic field. Since the energy is typically gained by the lattice, T1 is termed as the lattice or longitudinal spin relaxation time.
3. QM treatment in the Schrödineger and Heisenberg picture 4. In magnetic resonance we are measuring the imaginary part of the r.f. spin
susceptiblity ”(). 5. Method of moments:
1. O-th moment M0=f()d is proportional to the static spin susceptibility 2. Higher moments are in fact given by the commuators [H’, Ix]. In
particular we emphasized the first moment M1= f()d / M0, which is just the center of the resonance and the second moment M2= (-M1)2 f()d / M0 which is measure for the linewidth.

Magnetic resonance – a brief summary after first lectures
30
Bloch equations in compact form Magnetic resonance measures Method of moments
==
1
2
2
00
/100
0/10
00/1
T
T
T
RMMRBMdt
Md
g0 5 10 15
-2
0
2
Mx, M
y (
arb
. u
nits )
time
Mx
My
dteMtMTk
V ti
xx
B
= 0ˆˆ2
''
B0
Brf
2
22
0
2
0
2
0
0
ˆ
ˆ,'1
)(
)(
ˆ
ˆˆ,'1
)(
)(
x
x
x
xx
M
MH
df
df
M
MMH
df
df
=
=
=
=

NMR observables
31
NMR = local, real-space probe where the behaviour of nuclear spins can be monitored on a site-to-site basis. Types of NMR observables: 1. NMR spectrum is fundamental and major element of sample
characterization • Width & distribution
Sample quality Crystallographic inequivalent sites Local site disorder
• NMR shifts, which are measured as a frequency shift proportional to the applied field is a fundamental measure of the various terms in spin Hamiltonian. In magnetic systems is a measure of local spin susceptibilities. Various sources: s-contact shift (in metals = Knight shift) Core-polarization shift Dipolar shift Chemical shift
2. NMR dynamics as represented by the spin-lattice relaxation time T1. T1 is linked to “(q,) via the fluctuation-dissipation theorem.
1100
==
g
resres
B

NMR – basic Hamiltonian
32
Nuclear magnetic moments in solids constitute nearly perfect example of an ensemble that is weakly coupled to its neighborhood. In NMR we attempt to explore this weak coupling to measure sample’s static and dynamic properties. General spin Hamiltonian 1. Zeeman term
NMR: detect the transitions between these levels. It is usually the strongest term and in most experiments it will range between 10-500 MHz.
neQdipZ HHHHH =
ZZ IBH ˆ0g=

NMR – basic Hamiltonian
33
2. Dipolar term It is much weaker than Zeeman term as it is at most in the several 10 kHz range. It will
give rise to a Gaussian type of broadening of resonances with a second moment Calculated from the definition of the second moment
=
ji ij
ijjiji
ij
ji
dipr
rIrI
r
IIH
,53
22 ˆˆ3ˆˆ
2
g
=
ji ij
ij
rNII
,6
22
242)1cos3(1
)1(4
3 g
2
2
2
2
0
2
ˆ
ˆ,1
)(
)(
x
xdip
M
MH
df
df
=
=

NMR – dipolar interaction; example
34
2. Dipolar term Effect of molecular motions: we need to Take a time average over the dipolar term
=
ji ij
ij
rNII
,6
22
242)1cos3(1
)1(4
3 g
)1cos3( 2 ij

Nuclei of spin I 1 have electric quadrupole moment.
2
,
3
6 2 1 2Q
eQH V I I I I I
I I
=
Q > 0
Q > 0 for convex (egg shaped) charge distribution.
,
1
3!QH V Q
= 23 k k k
k protons
Q e x x r
=
Wigner –Eckart Theorem:
2
0
VV
x x
=
=
r
Quadrupole term: zzeq V=
223 1
4 2 1Q
e qQE m I I
I I =
Ref: C.P.Slichter, “Principles of Magnetic Resonance”, 2nd ed., Chap 9.
zzeQ I I Q I I=
Q in field gradient
=
2222
2)1(ˆ3
)12(4IIIII
II
qQeH ZQ
Axial symmetry: =(Vxx-Vyy)/Vzz
Quadrupole Interaction

NMR – basic Hamiltonian
36
Some comments about the Quadrupole term: 1. It is frequently the leading (perturbation) term in NMR when I1/2. It is not
unusual to find it in the several 10 MHz range. In fact in some experiments we deliberately switch off magnetic field and observe only the transitions between the quadrupole split levels (Nuclear Quadrupole Resonance).
2. For I=1/2 the nuclear quadrupole moment Q vanishes identically according to the Wigner-Eckart theorem.
3. The choice made in the above equation is such that the principal axes fo the EFG tensor are chosen in such a way that |Vxx|<|Vzz|<|Vzz|. That is, |Vzz| is the largest principal value.
4. The symmetry plays an important role. For instance in cubic symmetries because of V=0 we see that Vxx=Vzz=Vzz=0. In other words, in this case HQ=0! However, even in cubic structures, strains can play a role as they effectively give rise to some distribution of Q. Then due to the first order broadening only the central transition -1/2<->1/2 will be resolved.
5. In tetragonal or trigonal symmetries we find that Vxx=Vyy or =0! We have an axially symmetric EFG.

Quadrupole interaction
37
Case of =0
)12(2
3)1(
2
1 2
312
0
==IhI
qQeIImhmBE QQm g
Quadrupole frequency
L+Q
L
L-Q L+Q L-Q L
First order satelites
)1cos3( 2
21 = QL nFor q 0:

Quadrupole interaction – powder lineshape
38
In case of powder samples with uniform distribution of q over the unit sphere, the first order transitions give rise to an intensity distribution between L+nQ and L-½nQ with a square root singularity at L-½nQ
)1cos3( 2
21 = QL n
m is no longer strickly a good quantum number so there are a second order frequency shifts. Interestingly they cancel out for the satelite transition, but they give a second order shift
)1cos9(sin4
3)1(
16
22
2
)2(
21
=
II
L
Q
L
I = 3/2

Quadrupole interacation - example
39
bcc lattice: only a single intercalation site
fcc lattice: Octahedral (O) and tetrahedral (T) sites
133Cs I=7/2 7/25/2, 5/23/2, …, -5/2-7/2)
fcc: site symmetries for the two Cs sites are 23 and m-3 => EFG=0
bcc: site symmetry (-4m.2) => =0
-5000 0 5000 52.3 52.4 52.5
SIM
bcc phase
133Cs
Inte
nsi
ty (
arb
. u
nit
s)
- ref
(ppm)
fcc phase
133Cs
Inte
nsi
ty (
arb
. u
nit
s)
(MHz)
O
T
T'

Electron-nuclear interaction
40
The coupling between the nuclear and electron moments can be divided into three contributions
hf
dip
nelne HHHH =
Chemical shift:
3/ rlIgH Bl
= mg … is the interaction to the electron orbital moment
and is usually contained in the so-called chemical shift in materials where the angular momentum is quenched. If this is the case, then the extra magnetic field at the nuclear site, which is a result of the orbital moment of the electrons, is expressed as
0BIHCS
= g
=
zzzyzx
yzyyyx
xzxyxx
Dimensionless second-rank tensor
0
0
=
Shift:

Chemical Shift
41
The range of component is somehere in the ppm range, so it s really a small perturbation to the Zeeman hamiltonian. For this reason we will need only zz component of the tensor.
Chemical shift is widely explored in chemistry
for recognising chemical groups and thus for the reconstruction of the chemical components. The calculation of is however very tedious job and we shall leave it to quantum chemists. For us it will be a parameter, which is determined directly from the experiments.

Chemical Shift – powder spectrum
42
Zeeman + Chemical shift Hamiltonians ij<<1 Principal axes system
0)1( BIH
= g
)1()1(0 zzLzzB g ==
=
zz
yy
xx
00
00
00
'
x’
y,y’
z’ x’
q
q
Axial symmetry xx=yy
1' = yy RR

Chemical Shift – powder spectrum
43
The zz component, which we are looking for is The resonance frequency this reads To calculate the spectrum
2
|| cos =zz
)cos1( 2
|| = L
)]1(),1([)1(2
.
cos2
.
cos
.
cos.
||
||
||
=
==
=WW=
LL
LL
L
constf
const
d
d
constf
dconstdgdf
Presence of motions: can average the CS anisotropy even in powders
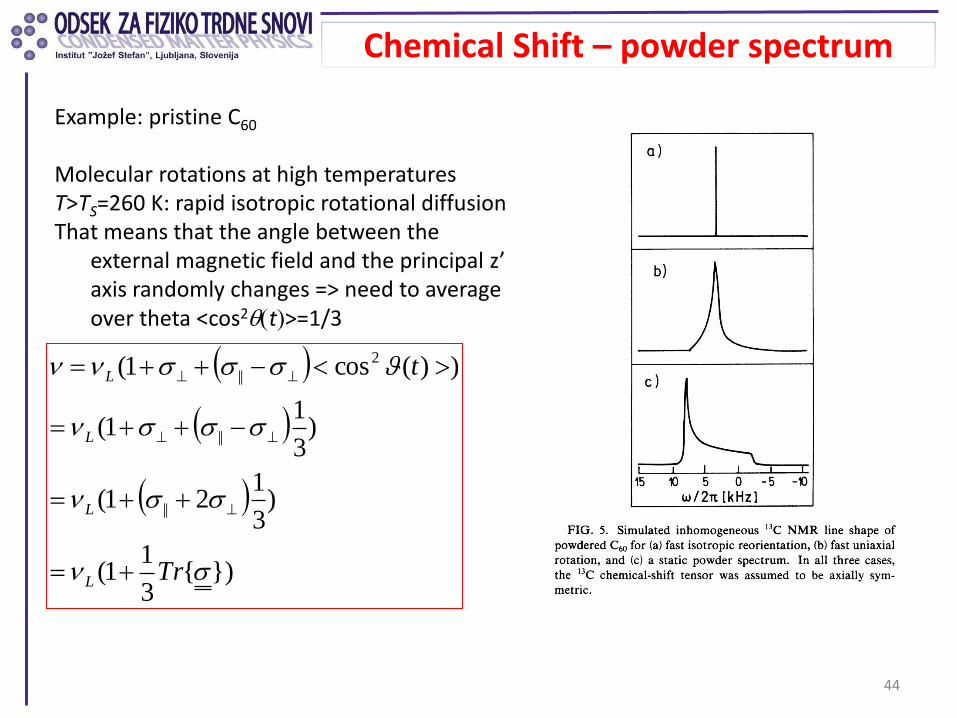
Chemical Shift – powder spectrum
44
Example: pristine C60 Molecular rotations at high temperatures T>TS=260 K: rapid isotropic rotational diffusion That means that the angle between the
external magnetic field and the principal z’ axis randomly changes => need to average over theta <cos2qt>=1/3
}){3
11(
)3
121(
)3
11(
))(cos1(
||
||
2
||
Tr
t
L
L
L
L
=
=
=
=

Chemical Shift – powder spectrum
45
Example: pristine C60

Electron-nuclear interactions cont.
46
We mentioned that the angular momentum interaction will give rise to a temperature independent chemical shift. However, in paramagnetic solids we need to take into account also the interaction between the electron and nuclear spins. When they are separated in space, then we can always count on the el-nuclear dipolar interactions
This will hold well also for p- and d-state electrons. For them we can in fact rewrite the
above equation in terms of a coupling traceless tensor T For p-electrons the spatial averaging gives the components
=
ji ij
ji
ij
ijjiji
B
ne
dipr
IS
r
rIrSgH
,35
2ˆˆˆˆ3
mg
0
2 ˆBTIgH B
ne
dip
= mg
SSr
Tr
T 33||
1
5
21
5
4==

Electron-nuclear interactions cont.
47
The difficulties arise for the s-state electrons. There the dipolar approximation breaks down since we have a non-zero spin density at the nuclear site. We will treat this case within a simple first-order approximation that is reasonably accurate at high magnetic fields. A more rigorous treatment utilises the Dirac equation (see for instance C.P. Slichter, Principles of Magnetic resonance)
S-orbitals have spherical symmetry and non-zero spin density at the nuclear site. Therefore hf interactions involving s-orbitals are large and isotropic!
The simple model for this interaction is a current loop representing the magnetic moment of the nucleus.
Biot-Savart law: MF at the center of the loop is The average field inside a sphere of radius r is
e
n 2/ˆ rinSi NN pmm ==
NNrr
rldiB m
p
m
p
m
3
0
3
0
24=
=
2
0
320
3
2
3
40
)(
eNeN
Nav
rB
volumedensityyprobabilitBB
mmp ==
==

Electron-nuclear interactions cont.
48
The energy of the electron magnetic moment in this field will be Lets take 1s WF to get The more general treatment would actually give the famous Fermi contact interaction
SIaSIgBE eBave
===
2
0 03
2gmmm
Brr
B
e er
r/
3
1 =
p
rB=5.29·10-11 m
T5076.003
2/
2
01 == eBs ga gmm
rSIgH Bhf
32
3
8gm
p=
More formalistic calucaltion: see J.D. Jackson, Classical Electrodynamics, Ch. 5

Core-polarization hf coupling
49
d-electrons: Do we expect Fermi contact hf interaction (zero spin density)? They will frequently lead to the anisotropic hf interactions, which can be generally written as We may thus define the Knight-shift tensor with component
The result may be at first sight surprising since d-WF vanishes at the nuclear site. However, d-electrons can interact indirectly by polarizing core electrons. The Coulomb repulsion drives core s-states (fully occupied) closer to the nucleus. However, because of the Pauli principle “down-spin” s-orbitals will shrink more than “up-spin” s-orbitals. This will create a total negative hf field at the nuclear site. The associated fields can be in fact quite substantial sometimes. For Mn2+ in MnF2 it can be as large as -127 kOe/mB.
==
=zyxi
ii
B
i
zyxi
iii BIg
aSIaH,,,, m
i
B
iii
g
aK
gm 2=

Hyperfine interaction
50
What will Fermi contact inetraction do in magnetic systems? Since we expect that the electron spin dynamics will be fast on the time-scale of NMR
experiments (over 10 ms) we can take average of e- spin operator ||B0
rSIgH Bhf
32
3
8gm
p=
0BgN
AI
gAISAISIA
BAn
SZn
B
ZZZZ
mg
g
m
m==
hfnlocnn BBB == 0gg
=i
iihf SAB Hyperfine tensor: - on-site: Fermi contact, A= very strong and
known - Transferred: A can be anything - Dipole: A can be calculated, usually very weak; it
vanished in sites having cubic symmetry
S
BAn gN
AK
mg =
Units of A found in the literature: - A/ [s-1] - 10-3 A/gn [kG/spin] - 10-3 A/gng [kG/mB] - A/1.60210-12 [eV]

Hyperfine interaction
51
How do we extract hyperfine tensor?
Temperature (K)
0 50 100 150 200 250 300
Ma
gn
etic s
usce
ptib
ility
, (1
0-3
em
u m
ol-1
)
0.0
0.5
1.0
1.5
2.0
2.5
3.0
x = 0
x = 0.35
x = 0.5
x = 0.75
x = 1
0 50 100 150 200 250 300
0
-100
-200
-300
-400
-500
-600
13
3K
, 8
7K
(ppm
)
Temperature (K)
O-site
T-site
0.8 1.0 1.2 1.4
0
-100
-200
-300
-400
-500
-600
13
3K
O,T (
ppm
)
105 K
105 K
300 K
30 K
SQUID
(10-3 emu/mol)
300 K
133KO,T=133CS,O,T+(133aO,T/NAmB)
133CS,O = 15(8), 133CS,T = 148(15)
133aO = -2.28 kOe/mB, 133aT = -1.35 kOe/mB

Hyperfine interaction
52
But, be carefull! NMR is still a local real-space probe! Example, doping of Haldane chain system
89Y NMR Y2BaNi0.95Mg0.05O5
Tii
i THTH /1
1
1e1
=
valence bond solid with a single valence bond connecting every neighboring pair of sites
the ends of the chain behave like spin S=1/2 moments even though the system consists of S= 1 only
F. Tedoldi et al., PRL 1999

Knight shift
53
C60 versus K3C60
In metals: -The shift is always (almost) positive -The shift is very nearly temperature independent -The shift in principle increases with the nuclear charge Z.
The fact that metals have a weak spin-paramagnetism suggest that the shift may simply represent the pulling of the magnetic flux into the metal. However, the susceptibilities are to small to account for such effect. Knight postulated that the shift arises because of the interaction of nuclear spins with the conduction electrons through the s-state hf interaction.

Knight shift
54
What will Fermi contact inetraction do in metals? Since we expect that the electron spin dynamics will be fast on the time-scale of NMR
experiments we can In order to calculate the spin density at the nuclear site, we start with the Bloch WF We need to consider only states at the Fermi level Putting everything together we derive With a Knight shift
rSIgH Bhf
32
3
8gm
p=
0Bg
Ig
ISISIB
molZ
B
ZZZZ
m
m
m==
2
rerurke
rki
kk
==
00
20
3
8BKIBIuH ZZEkhf
F
ggp
=
p 2
03
8
FEkuK = Measurement of spin-only susceptibility → N(EF)

METALLIC/SC STATE TMIT , TSC dependence pressure/doping
55
700 720 740 760 780 8000
40
80
120
160
200
RbxCs
3-xC
60
13
C Tc
133
Cs Tc
13
C TMIT
133
Cs TMIT
____________
susc. TMIT
XRD TMIT
SUPERCONDUCTOR
T (
K)
Cs3C
60
13
C Tc
133
Cs Tc
13
C TMIT
133
Cs TMIT
V / C3-
60 (A
3)
AFI
METAL
INSULATOR
MIT line is not vertical
Thinking about CC explanation dp/dT=S/V If the magnetic entropy (calculated per C60) is S = kB ln [S(S+1)] = kB ln 2. Structural study: V = 4 Å3 per C60
We thus estimate dp/dT = kB ln 2/V = 1.4·106 Pa/K Experimental value: dp/dT = 0.6 GPa/150 K = 4·106 Pa/K.
0 2 4 6 8 10 12 14 160
40
80
120
160
200
T (
K)
Cs3C
60
13
C Tc
133
Cs Tc
13
C TMIT
133
Cs TMIT
p (kbar)
NMR TMIT vs p diagram

Superconducting state
56
0 50 100 150 200 250 300120
140
160
180
200
220
M1 (
pp
m )
T (K)
15.5 kbarTc
chemical shift
aiso
S
Superconducting state
0
SCS χB
B
N
a
BA
=
m
demagnetization due to the Meissner screening currents and is in general not known
SCS χδδ A=
After subtraction of two shifts
CSCSCS δδ =
BANaaA m /
=
1.0 1.5 2.0 2.5 3.00.01
0.1
1 13
C - 87
Rb
13
C - 133
CsO
133
CsO -
87Rb
No
rma
lize
d S
pin
Su
sce
ptib
ility
Tc/T
2/kBTc=4.7(3)

Summary of interactions
57
neQdipZ HHHHH =
ZZ IBH ˆ0g=
=
ji ij
ijjiji
ij
ji
dipr
rIrI
r
IIH
,53
22 ˆˆ3ˆˆ
2
g
=
2222
2)1(ˆ3
)12(4IIIII
II
qQeH ZQ
0BIHCS
= g
0
2 ˆBTIgH B
ne
dip
= mg
rSIgH Bhf
32
3
8gm
p= i
B
iii
g
aK
gm 2=

Example - Pnictides
(La3+O2–)+(FeAs)–
Ba2+(Fe2As2)2– Li+(FeAs)–
Se atom has one extra electron compared to an As atom, Se2+: (4s)2(4p)6 As3-: (4s)2(4p)6
Chu et al., Nat. Phys. 5, 788 (2009)

Phase diagrams
59
Chu et al., Nat. Phys. 5, 787 (2009) SC: superconducting AFI: AFM insulating SDW: spin density wave
1. AFI phase next to SC is a hallmark of electron correlations. Correlations are strong and onsite.
2. No symmetry change through MIT! 3. Charge versus pressure controle of SC!
4. No disorder caused by the doping (off stoichiometries ...) 5. 3D vs 2D! 6. Importance of cubic symmetry!
Also see our papers Science 323, 1585 (2009) Nature 466, 221-225 (2010) Phys. Rev. B80, 195424 (2009)

Example: NMR in magnetically ordered state SDW in pnictides (1111 phase)
Cruz et al., Nature 453, 899 (2008)
Yildirim, arXiv:0804.2252
Fully frustrated
The frustration is lifted by a structural distortion
•SDW order with Q=(p,p) for 2a x 2a due to the interband nesting between the hole - and electron -bands •magnetic moments ~ 0.3 mB

75As coupling to Fe moments i
i
iAsB m
= =
4
1
A
=
=
=
=
zzyzxz
yzyyxy
xzxyxx
zzyzxz
yzyyxy
xzxyxx
zzyzxz
yzyyxy
xzxyxx
zzyzxz
yzyyxy
xzxyxx
aaa
aaa
aaa
aaa
aaa
aaa
aaa
aaa
aaa
aaa
aaa
aaa
43
21
;
;
AA
AA75As
a
b
3
1
4
2
Fe1 Fe2
Fe3 Fe4
Symmetry considerations: mirror reflection with respect to bc plane
AF phase – taken from LaOFeAs data Q = (1, 0, 1) Fe magnetic moments are aligned along the a−axis μFe ~ 0.3μB
cFexzAs eaB ˆ4 = m
cos1 0int
2
0int0 BBBBBBeff =
ONLY OFF-DIAGONAL TERMS!!!!
PM phase: 0HM
=
04 Baxx =
ONLY ISOTROPIC PART!!!!
Example: NMR in magnetically ordered state SDW in pnictides (1111 phase)

Example: NMR in magnetically ordered state
-10 -5 0 5 10
Inte
nsi
ty (
arb.
un
its)
80 K
180 K
-ref
(MHz)
NdOFeAs T phase: As reside on the 2c (1/4, 1/4, z) position, which is axially symmetric. = (Vxx-Vyy)/Vzz = 0 Q = 3 eVzzQ/ 2I(2I−1) =11.8 MHz NdFeAsO0.85F0.15
Q = 12.8 MHz
comparison (LaO0.9F0.1FeAs: Q = 11 MHz)
Low-T orthorhombic phase: As is on 4g (0, 1/4, z) position, =0.102
ONSET OF LOCAL MAGNETIC FIELDS
PHYSICAL REVIEW B 79, 094515 2009
EFG mainly originates from As 4p Electrons with a prolate p electron distribution in agreement with the negative Vzz.
g ,, )2()1(
1 QQeffAsmm B =
Axz=10.5 kOe/mB
large anisotropic hyperfine fields → experimental evidence for the Fe 3d and As 4p hybridizations

‘111’ family
LiFeAs
M.J. Pitcher et al., Chemical Communications 5918 - 5920 (2008).
NaFeAs
D.R. Parker et al., PRL 104, 057007 (2010).
Bulk superconductor
SDW below TSDW = 45 K
They are isostructural
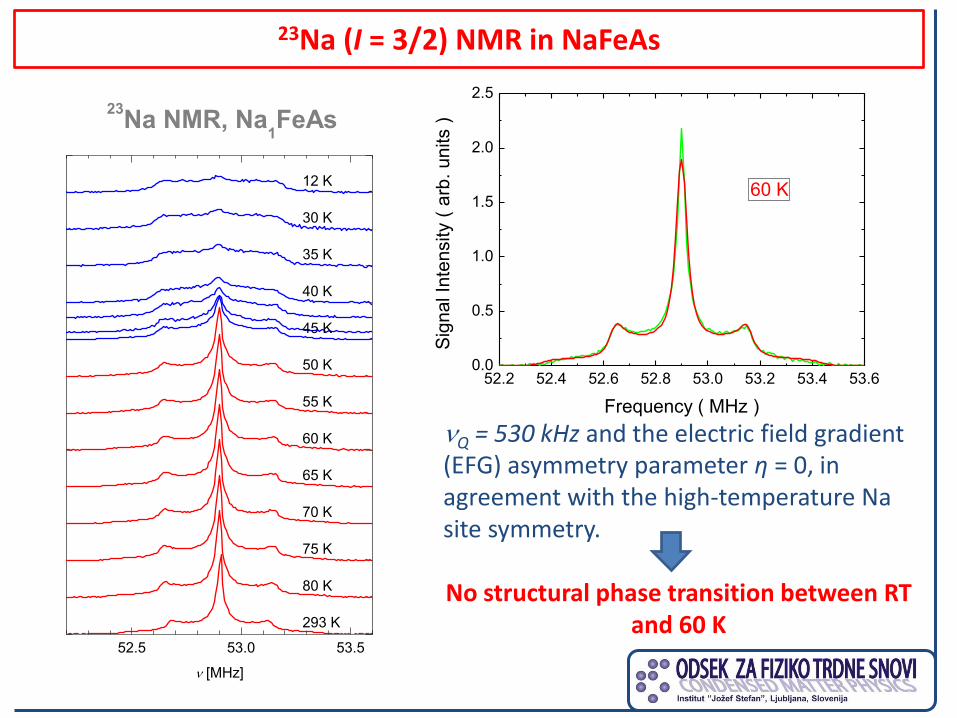
23Na (I = 3/2) NMR in NaFeAs
52.5 53.0 53.5
293 K
80 K
75 K
70 K
65 K
60 K
55 K
50 K
45 K
40 K
35 K
30 K
12 K
23Na NMR, Na
1FeAs
[MHz]
52.2 52.4 52.6 52.8 53.0 53.2 53.4 53.60.0
0.5
1.0
1.5
2.0
2.5
Sig
nal In
tensity (
arb
. u
nits )
Frequency ( MHz )
60 K
Q = 530 kHz and the electric field gradient (EFG) asymmetry parameter η = 0, in agreement with the high-temperature Na site symmetry. No structural phase transition between RT
and 60 K

23Na (I = 3/2) NMR in NaxFeAs
52.6 52.8 53.0 53.2
52.5 53.0 53.5 0 100 200 3000
50
100
150
300 K
(MHz)
Na1FeAs
Na0.9
FeAs
Na0.8
FeAs
(b)
Na1FeAs
Inte
nsity (
arb
. u
nits)
(MHz)
60 K
30 K
(c)
(a)
Na1FeAs
Na0.9
FeAs
Na0.8
FeAs
Wid
th (
kH
z)
T (K)
They are isostructural! Na vacancies in Na-deficient samples are expected to result in the local disorder and thus broadened NMR lines, which is in contrast to the measured 23Na central transition linewidth in the temperature range 50 − 300K Na+ migration??
Lineshape broadening is a clear indication of SDW transition. Variation of SDW order paramater with x. Still single line so NO phase segregation in this case.
Klanjšek et al., PHYSICAL REVIEW B 84, 054528 (2011)

Incommensurate SDW vs. other solutions
Commensurate SDW Commensurate SDW order with large local amplitude variations in the vicinity of the dopant Curro et al (2010).
INCOMMENSURATE SDW
-1 0 1 2 3 -1 0 1 2 3
a x = 0
Inte
nsity (
arb
. u
nits)
-ref
(MHz)
300 K
170 K
140 K
130 K
120 K
BA
36 K
38 K
41 K
43 K
50 K
120 K
130 K
140 K
300 K
170 K
x = 0.15b
-ref
(MHz)
NdOFeAs; P. Jeglič, PRB 2009
-5 -4 -3 -2 -1 0 1 2 3 4 5
Frequency ( MHz )
Bint = B0 sin α with α ∈ [0, 2π] and B0 = 0.45 T ordering vector (1/2− ϵ,0,1/2) for ϵ → 0
-6 -4 -2 0 2 4 6
Na1FeAs
- ref
(MHz)
Inte
nsi
ty (
arb
. u
nits
)
75As NMR
IC SDW amplitude of x=1 0.28μB x=0.9 0.04μB
x=0.8 0.13μB

75As (I = 3/2) NMR in NaxFeAs for T<TSDW: Nature of the order?
-6 -4 -2 0 2 4 6 4 5 6
c-SDW
dis-SDW
inc-SDW
= 0
0
Na1FeAs
- ref
(MHz)
In
ten
sity (
arb
. u
nits)
40 K
50 K
70 K
- ref
(MHz)
Still no structural phase transition, but some distribution due to local inhomogeneities
Structural phase with low-temperature tetragonal phase between 60 K and TSDW
THE MAGNETIC ORDER IS INCOMMENSURATE!!!
Klanjšek et al., PHYSICAL REVIEW B 84, 054528 (2011)

LiFeAs – 75As NMR
-2 -1 0 1 2
0 100 200 300
20.821.021.221.4
1.0 1.5
Inte
nsi
ty (
arb
. u
nit
s)
20 K
(a)
sim.
200 K
- ref
(MHz)
Q (
MH
z)
T (K)
(b)9 K
10 K
12 K
100 K
16 K
200 K
300 K
Inte
nsi
ty (
arb
. u
nit
s)
- ref
(MHz)
•Axially symetric tensor in agreement with the 75As site symmetry •Weakly temperature dependent Q
•No annomalies that would indicate SDW transition •Below Tc 16 K (9 T) – wipeout effect => onset of SC
Jeglič et al., PHYSICAL REVIEW B 81, 140511R (2010)

Spin-lattice relaxation time
69
Lets recall that
In order to calculate spin-lattice relaxation, we should look at the terms in the spin Hamiltonian that produce transitions between energy levels => we need transverse time-dependent magnetic field
)()(2
)(' tBItBItBIH nn ==
gg

Spin-lattice relaxation time
70
Fermi golden rule We next use the definition of Dirac’s delta function And plug it into the above expression Spin-lattice relaxation time is then given by
gp
p
=
=
EEtBmImP
EEaHbP
nmm
baab
'
22
1
2
)('12
2
'2
dteEE
tEEi
=
/
''
2
1
p
=
==
dteBtBmImI
dteBeBemImP
tin
tBeBe
titiEtiEnmm
tiHtiH
g
g
)0()()1)((2
)0()0('12
2
)()0(
//2
2
1
//
'
)1)((
1 11
1
=
mImI
PP
T
mmmm

Spin-lattice relaxation time – some simple considerations
71
Lets assume that the local magnetic field jumps between two sites with a typical correlation time t. Therefore, the probability for B to change is given by ½t with =1/t.
S(t)S(0)=(1- ½t ) S2 + ½t (-S2)=S2(1- t) ½t 1-½t
S(Nt)S(0)= S2(1- t)N
<S(t)S(0)>= S2e-t = S2e-t/t
FT
22
2/2
1
2cos
t
t
pt
=
SdtteS t1/T1 maximum when t=1 BPP dependence
= tdtStSa
Tcos)0(),(
2
12
2
1

Spin-lattice relaxation time
72
Lets now assume, that the fluctuation field comes from the electronic field through the hyperfine coupling intercation Expressing B we finally get for T1
After Fourier transformation Finally we fluctuation-dissipation theorem, which states To get T1 in the high-T expansion
== BItSaIH nii
g)('
= tdtStSa
Tcos)0(),(
2
12
2
1
=i
rq
iqieSS
[A,B]=(AB+BA)/2
=q
qqqq tdtStSAAT
cos)0(),(2
11
1
kT
e
qqe
qtdtStS
/21
),(''2cos)0(),(
2
1g
=
=q
e
n qAA
kT
T
g
g ),(''212
2
1

Spin-lattice relaxation in metals
73
rSIgH Bhf
32
3
8gm
p=
The perturbing interaction is the s-contact interaction Transitions from state |k m> to |k m+1> where m is the initial nuclear quantum number
kkhfmkmkEEmkHmkP =
'
2
1'1'
2
p
)1)(()0()0(3
41'
2
'
22
= mImIuumkHmk kkenhf gg
p
We take Bloch WFs and calculate the corresponding matrix elements We make an approximation that all states in the vicinity of Fermi surface have approximately the same probability density at the nucleus. Next we need to sum over all k and k’ states then we can finaly write
=
',
'
222
1
)0(3
441
kk
kkken EEuT
ggpp

Spin-lattice relaxation in metals
74
It is important to bear in mind, that the sum over k,k’ is restricted to states fully occupied at k and empty at k’. The sum over k can be replaced by the integration over energy with introducing the density of states n(E), i.e. n(E) dE. The occupation restriction is eforced with the Fermi occupation function f(E)=[exp((E-EF)/kT)+1]-1. The summation then becomes
2
222
1
)()0(3
441Fken
B EnuTk
T
=
gg
pp
'))'(1)(()'()(' EEEfEfEnEndEdE
FBB EETkE
fTkEfEf =
= ))'(1)((
Remember Knight shift:
p 2
03
8
FEkuK =
22
1
41
e
Bk
TKT g
p
=
Famous Korringa relation The final result depends only on elementary constants and not on materials; this hold only for simple metals, where correlations can be neglected

Korringa relation - examples
75
22
1
41
e
Bk
TKT g
p
=
In practice, simple Korringa relation is not satisfied. Let’s check some simple alkali metals
metal KS(%) T1T (exp) T1T (calc) ratio
7Li 0.0263 45 Ks 26 Ks 0.58
23Na 0.112 4.8 Ks 3.1 Ks 0.65
63Cu 0.232 0.7 Ks 0.7 Ks 0.78
Various corrections can be made to Korringa relation. In principle electron-electron interaction potential can enhance the spin susceptibility thus correcting K.R. in the right way. However, exchange fluctutations would also enhance spin-lattice relaxation. We will come to this point latter, but for now we just introduce Korringa factor , which should be <1 (AFM) fluctutaions, >1 (FM) fluctuations
g
p22
1
41
e
Bk
TKT =
13C NMR in K3C60
Yoshinari et al. (1993)

Korringa relation - corrections
76
Various corrections can be made to Korringa relation. In principle electron-electron interaction potential can enhance the spin susceptibility thus correcting K.R. in the right way. However, exchange fluctutations would also enhance spin-lattice relaxation. We will come to this point latter, but for now we just introduce Korringa factor , which should be <1 (AFM) fluctutaions, >1 (FM) fluctuations
g
p22
1
41
e
Bk
TKT =
Stoner enhancement factor
0
0
1
=
=<(1-q)2/(1-0)2>EF

Spin-lattice relaxation time – some simple considerations
77
To a reasonable appoximation we may try to write Then Where ex=[2zkB
2J2S(S+1)/3ħ2]1/2
e
n
ne
ne qqq
)0,()0,(),(''
22
=
=
q e
e
n qAA
kT
T
g
g )0,(212
2
1

C
Example: YBa2Cu3O7-y
Moriya expression
=
j
jj
n
n
iCA
ATT
)exp()(
),()(
1 2
1
rqq
q
CuO2 layer with Cu2+ localized moments
63Cu
17O
Mila & Rice, Physica C 157, 561 (1990)
B
A
2
222
17
2263
17
63
cos4)(
coscos2)(
2
)4(
aq
yx
s
s
xCA
aqaqBAA
CK
BAK
=
=
=
=
q
q
A: on-site coupling B,C: transferred couplings
Millis, Monien & Pines PRB 42, 167 (1990)
a Millis, Monien and Pines assumed that the spin dynamics is described by
G=
2
22
4
0
0 11),(lim
p
AFMqq
q
0)(
4)(
,
217
2263
=
=
=
AFM
AFM
aaAFM
A
BAA
q
q
q pp
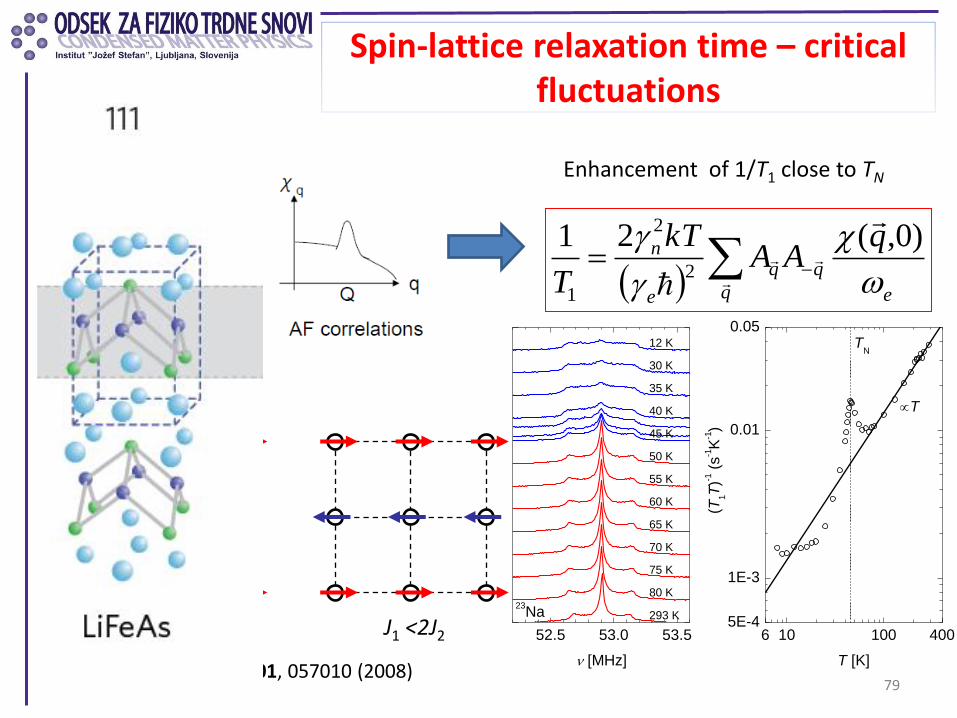
Spin-lattice relaxation time – critical fluctuations
79
Enhancement of 1/T1 close to TN
Yildirim, PRL 101, 057010 (2008)
Checkerboard AFM
J1 J2
J1 >2J2 J1 <2J2 6 10 100 4005E-4
1E-3
0.01
0.05
52.5 53.0 53.5
293 K
80 K
75 K
70 K
65 K
60 K
55 K
50 K
45 K
40 K
35 K
30 K
12 K
23Na
T
TN
(T1T
)-1 (
s-1K
-1)
T [K]
[MHz]
=
q e
e
n qAA
kT
T
g
g )0,(212
2
1

Example: LiFeAs
Localized Fe2+ spins Itinerant Fe 3d electrons
57Fe
75As
C
B
A
2
2
2
222
75
75
coscos16)(
4
aqaq
s
yxCA
CK
=
=
q
a
22
75
75
)( cA
cK s
=
=
q
For noninteracting spins can be taken out of the summation. In this limit we can calculate 0 and 0’ for the localized spins and itinerant scenario. If
n
nATT
),()(
1 2
1
q
),( n q
= Cc 44
coscosdd16C
dd16C
)(
2
2
2
22
2
2
2
0
0
==
==
aqaq
yx
yx
yxqq
A
c
q
q
q
Supported by density-functional calculations: - Katrin Koch & Helge Rosner, MPI-CPfS - Calculated electric-field gradients correctly reproduce the experimental values for both 75As and 7Li sites.
Jeglič et al., ArXiv: 0912.0692
A B

Example: AFM fluctuations in LiFeAs
How strong are AFM fluctuations in LiFeAs? 1. We cannot unambiguously discriminate between the on-site Fermi contact (itinerant) and the transferred coupling mechanism (localized moment at the Fe sites). 2. Cross-terms between different bands in the LiFeAs multiband structure can modify Korringa relation.
Korringa factor , measured for LiFeAs (green circles) and SrFe2As2 (red circles). Horizontal dashed lines indicate expected values for noninteracting electrons. If the transferred coupling is active, 1/T1T is enhanced for a factor of 15±5!
0 100 200 3000.0
0.2
0.4
0.6
0 100 200 3000
1
2
3
4
0.0
0.1
0.2
0.3
T (K)
1/(
T1T
) (s
-1K
-1)
T (K)
0'
0
(c)
(b)
(a)
LiFeAs
SrFe2As
2
Ks (
%)
Jeglič et al., ArXiv: 0912.0692

Summary
NMR = local, real-space probe where the behaviour of nuclear spins can be monitored on a site-to-site basis. Observables: - NMR spectrum - Relaxation rates
Hyperfine interaction (Fermi contact, transferred hf, dipolar interaction) Knight shift, shift in the superconducting state Spin-lattice relaxation rate, Korringa relation
=q
e
n qAA
kT
T
g
g ),(''212
2
1

Acknowledgements
83
Ljubljana: Peter Jeglič, Martin Klanjšek, Andrej Zorko, Anton Potočnik, Kristjan Anderle
and Andraž Kranjc
www.lemsuper.eu





























