Embed Size (px)
Citation preview

NOTCH1 Regulates Osteoclastogenesis Directly in OsteoclastPrecursors and Indirectly via Osteoblast Lineage Cells*
Received for publication, August 21, 2007, and in revised form, December 18, 2007 Published, JBC Papers in Press, December 22, 2007, DOI 10.1074/jbc.M707000200
Shuting Bai‡, Raphael Kopan§, Wei Zou‡, Matthew J. Hilton¶, Chin-tong Ong§, Fanxin Long¶, F. Patrick Ross‡,and Steven L. Teitelbaum‡1
From the Departments of ‡Pathology and Immunology, §Molecular Biology and Pharmacology, and ¶Internal Medicine,Washington University School of Medicine, St. Louis, Missouri 63110
NOTCH signaling is a key regulator of cell fate decisions inprenatal skeletal development and is active during adult tissuerenewal. In addition, its associationwith neoplasia suggests thatit is a candidate therapeutic target. We find that attenuatedNOTCH signaling enhances osteoclastogenesis and boneresorption in vitro and in vivo by a combination of molecularmechanisms. First, deletion of Notch1–3 in bone marrow mac-rophages directly promotes their commitment to the osteoclastphenotype.Theseosteoclast precursors proliferatemore rapidlythan the wild type in response to macrophage colony-stimulat-ing factor and are sensitized toRANKLandmacrophage colony-stimulating factor, undergoing enhanced differentiation inresponse to lowdoses of either cytokine. Conformingwith a rolefor NOTCH in this process, presentation of the NOTCH ligandJAGGED1 blunts the capacity of wild-type bonemarrowmacro-phages to become osteoclasts. Combined, these data establishthat NOTCH suppresses osteoclastogenesis via ligand-medi-ated receptor activation. AlthoughNOTCH1 andNOTCH3 col-laborate in regulating osteoclast formation, NOTCH1 is thedominant paralog. In addition, NOTCH1 deficiency promotesosteoclastogenesis indirectly by enhancing the ability of osteo-blast lineage cells to stimulate osteoclastogenesis. This isachieved by decreasing the osteoprotegerin/RANKL expressionratio. Thus, NOTCH1 acts as a net inhibitor of bone resorption,exerting its effect both directly in osteoclast precursors andindirectly via osteoblast lineage cells. These observations raisecaution that therapeutic inhibition of NOTCH signaling mayadversely accelerate bone loss in humans.
NOTCH signaling is an evolutionarily conserved pathwaythat profoundly impacts mammalian development by regulat-ing survival, proliferation, and cell fate decision in a context-de-pendent manner. It contributes to tissue maintenance and/orrenewal in the adult intestine (1), skin (2), hematopoietic sys-tem (3), mammary epithelium (4), and central nervous system(5) and can either promote (6–9) or suppress (10) cancer.
There are fourNOTCH receptors (NOTCH1–4) and at leastseven NOTCH ligands (JAGGED1, JAGGED2, DLL1 (Delta-like1), DLL3, DLL4, and DNER (11) and contactin/F3/NB-3(12)) inmice and humans. The receptors and ligands are single-pass transmembrane proteins expressed on the surface of adja-cent cells. Activation of NOTCH signaling requires cell/cellcontact because ligand binding to specific epidermal growthfactor-like repeats in the extracellular domain of NOTCHreceptors must induce a conformational change (13), mostlikely by trans-endocytosis (14), to expose the juxtamembraneregion to cleavage by ADAMmetalloproteases. The exposed Nterminus is recognized by �-secretase (15), which cleavesNOTCH again within its transmembrane domain. This cleav-age releases the NOTCH intracellular domain (NICD),2 whichtranslocates to the nucleus, where it associates with the DNA-binding protein CSL and other transcriptional coactivators.This complex is responsible for the transcription of NOTCHtarget genes, including those of HES (hairy and enhancer ofsplit) and the HES-related transcription factor HEY (16, 17).NOTCH signaling plays a critical role in somitogenesis (18).
For example, mice deficient in presinilin-1, the catalytic com-ponent of �-secretase, develop deformed vertebrae (19). More-over, deletion of the NOTCH ligand Jagged2 results in cleftpalate and syndactyly in mice (20). More important, patientsbearing mutations of the NOTCH ligand DLL3, the modifierFng, or the targetMesp2 also have severe deformities of the axialskeleton (18, 21).Due to the contribution of NOTCH signaling to somitogen-
esis and fetal skeletal development, attentionhas turned to skel-etal cells (22–26). Regarding the impact of NOTCH on osteo-blast differentiation, these efforts have yielded disparate results.Whereas some investigators report that NOTCH signalingdampens osteoblastogenesis (22, 24), others describe the oppo-site (25, 26). NOTCH signaling has also been reported to neg-atively impact osteoclastogenesis in vitro via a mechanisminvolving osteoclast precursors per se as well as regulatory stro-mal cells (23).Although these efforts have been informative, genetic assess-
ment of the role of NOTCH signaling in osteoblast and oste-oclast formation and function is not in hand. Most important,
* This work was supported by National Institutes of Health Grants HD044056(to R. K.), DK065789 (to F. L.), AR46852 and AR48812 (to F. P. R.), andAR32788, AR46523, and AR48853 (to S. L. T.). The costs of publication ofthis article were defrayed in part by the payment of page charges. Thisarticle must therefore be hereby marked “advertisement” in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
1 To whom correspondence should be addressed: Dept. of Pathology andImmunology, Washington University School of Medicine, Campus Box8118, 660 South Euclid Ave., St. Louis, MO 63110. Tel.: 314-454-8463; Fax:314-454-5505; E-mail: [email protected].
2 The abbreviations used are: NICD, Notch intracellular domain; BMMs, bonemarrow macrophages; M-CSF, macrophage colony-stimulating factor;OPG, osteoprotegerin; HA, hemagglutinin; TRAP, tartrate-resistant acidphosphatase; PBS, phosphate-buffered saline; BrdUrd, bromodeoxyuri-dine; CTx, C-terminal cross-linking telopeptide of bone collagen; PTH, par-athyroid hormone; WT, wild-type.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 283, NO. 10, pp. 6509 –6518, March 7, 2008© 2008 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
MARCH 7, 2008 • VOLUME 283 • NUMBER 10 JOURNAL OF BIOLOGICAL CHEMISTRY 6509
by guest on July 17, 2018http://w
ww
.jbc.org/D
ownloaded from

no information exists regarding the role of NOTCH in main-taining the mature skeleton, a highly plastic tissue, in vivo andhow it would respond to inhibition of NOTCH signaling in theadult.Osteoclasts differentiate from bone marrow macrophages
(BMMs) in response to RANKL (receptor activator of NF-�Bligand) and macrophage colony-stimulating factor (M-CSF)(27). Alternatively, recruitment of these resorptive polykaryonsis physiologically restricted by osteoprotegerin (OPG) (27).Molecules that modulate the resorptive process often do so byregulating expression of these osteoblast-produced cytokines.Alternatively, osteoclastogenesis is also governed by direct tar-geting of osteoclast precursors by agents such as tumor necrosisfactor-� (28).In this study, we focused on mice with individual and com-
bined deletions of Notch1–3 and found that the selectiveabsence ofNotch1 in either osteoclast or osteoblast lineage cellsenhances osteoclastogenesis by different mechanisms, result-ing in stimulated bone resorption. Thus, NOTCH1 signalinglimits postnatal bone degradation. These observations identifyosteoporosis as a potential complication of therapeutic inhibi-tion of NOTCH activity in humans.
EXPERIMENTAL PROCEDURES
Mice—The generation of Notch1flox/flox, Notch2flox/flox, andNotch1flox/flox Notch2flox/flox mice has been described (29).Notch3�/� B6.129P2-Lyzstm1(cre)Ifo/J (also known as LysMcre)were purchased fromThe Jackson Laboratory. Mice transgenicfor Col1-cre, which expresses Cre under the control of a 2.3-kbpromoter sequence of the murineCol�1(I) gene and which wasshown previously to function effectively in more committedosteoblasts, were provided by Dr. Henry Kronenberg (Massa-chusetts General Hospital) (30). All mice were housed in theanimal care unit of the Department of Pathology, WashingtonUniversity School ofMedicine, and weremaintained accordingto guidelines of the Association for Assessment and Accredita-tion of Laboratory Animal Care. All animal experimentationwas approved by theAnimal StudiesCommittee ofWashingtonUniversity School of Medicine. 6–8-week-old mice were usedin all circumstances.Reagents—Recombinant murine M-CSF was obtained from
R&D Systems (Minneapolis, MN). Glutathione S-transferase-RANKL was expressed in our laboratory as described (31).Anti-hemagglutinin (HA) antibodywas obtained fromCovanceResearch Products (Princeton, NJ). All other chemicals wereobtained from Sigma. The plasmid transfection reagentFuGENE 6 was purchased from Roche Applied Science.Plasmids and Transfection—Turbo-cre was cloned into the
pMX vector to generate retroviruses. NICD1 was amplifiedfrom a mouse macrophage cDNA library by PCR and clonedinto the pMX vector linked to a HA tag at the C terminus.MutantNicd1 (referred to asM2) (32) was cloned from amousefull-length Notch1 plasmid by PCR into the pMX vector. Fortransfection, cells were plated into 100-mm tissue culturedishes for 24 h, and 8 �g of plasmid was transfected accordingto the protocol of Roche Applied Science.Western Blotting—HA-tagged Nicd1 and M2 in the pMX
vector were transfected into Plat-E cells to generate retrovi-
ruses. BMMs from C57B/6 mice were infected with the virusesand selected by blasticidin. Anti-HA antibody was used todetect the expression levels of these proteins. Anti-NOTCH1and anti-NOTCH3 antibodies were obtained from Abcam(Cambridge, MA).Osteoclast Lineage Cells—Macrophages/osteoclast precur-
sors and osteoclasts were generated from bonemarrow precur-sors as described (33).Characterization of Osteoclasts—BMMs were cultured in
96-well cell culture dishes in the presence ofM-CSF (10 ng/ml)and RANKL (100 ng/ml or the doses detailed in the figure leg-ends). Media were changed on day 3. On day 5, osteoclast-likecells were characterized by staining for tartrate-resistant acidphosphatase (TRAP) activity. The number of osteoclasts wascounted as described previously (33).Primary Osteoblast and Macrophage Co-culture Assay—Pri-
mary osteoblasts were extracted from 3–5-day-old neonatalcalvariae.Macrophageswere extracted frombonemarrow. Thecells (5 � 104 of each type/well) were mixed and cultured in�-10 cell culturemedium in 24-well plates with 1,25-dihydroxyvi-tamin D (10�8 M) for 7 days. The osteoblasts were lifted by colla-genase, and the remaining cells were stained for TRAP activity.Bone Resorption Assay in Vitro—Osteoclasts were generated
on whale dentin slices from BMMs in the presence of M-CSFand RANKL. On day 7, the cells were fixed in 4% paraformal-dehyde and stained for TRAP activity. To visualize resorptionlacunae, cells were removed from the dentin slices with 0.5 Mammonium hydroxide and mechanical agitation. Dentin slideswere stained with toluidine blue.Cell Stimulation and Immunoblotting—For M-CSF and
RANKL/RANK signaling experiments, BMMs were starved for6 h, following which RANKL (100 ng/ml) or M-CSF (50 ng/ml)was added to the media. The cells were lysed over time, and thelysate was subjected to immunoblotting.Mineralized Bone Nodule Formation—Osteoblasts were cul-
tured in osteoblast differentiationmedium for 14 days (34). Thecells were washed with phosphate-buffered saline (PBS) andfixed in ice-cold 70% ethanol for 1 h at 4 °C. The cells werewashed with and incubated in 0.4% alizarin red S solution atroom temperature for 10 min. The cells were then washed,dehydrated in ethanol, and air-dried.Reverse Transcription-PCR—Total RNA was isolated using
RNeasy kits (Qiagen, Valencia, CA). First-strand cDNA wasgenerated from 1 �g of total RNA using the SuperScript first-strand synthesis system for reverse transcription-PCR (Invitro-gen) as recommended by the manufacturer. PCR was per-formed with 1 �l of cDNA reaction mixture using Taqpolymerase and appropriate primers in a volume of 50 �l. Thegenes to be tested were amplified in a PCR Express ThermalCycler (ThermoHybaid, Ulm,Germany). The cDNAwas dena-tured at 94 °C for 5 min and subsequently subjected to variousamplification cycles consisting of 94 °C for 30 s, 60 °C for 30 s,and 72 °C for 30 s. The primers used were as follows: glyceral-dehyde-3-phosphate dehydrogenase, 5�-ACTTTGTCAAGC-TCATTTCC-3� and 5�-TGCAGCGAACTTTATTGATG-3�;�-actin, 5�-ATGGATGACGATATCGCTG-3� and 5�-ATGA-GGTAGTCTGTCAGGT-3�; M-CSF, 5�-GACTTCATGCCA-GATTGCC-3� and 5�-GGTGGCTTTAGGGTACAGG-3�;
NOTCH1 Regulates Osteoclastogenesis
6510 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 10 • MARCH 7, 2008
by guest on July 17, 2018http://w
ww
.jbc.org/D
ownloaded from

OPG, 5�-CCACTCTTATACGGACAGCT-3� and 5�-TCTCG-GCATTCACTTTGGTC-3�; RANKL, 5�-CAGGTTTGCAGG-ACTCGAC-3� and 5�-AGCAGGGAAGGGTTGGACA-3�;TLR2, 5�-GGAGACTCTGGAAGCAGGCG-3� and 5�-GGCT-TCCTCTTGGCCTGGAG-3�; c-Fms, 5�-TGAATGGCTCTG-ATGTCCTG-3� and 5�-TTCTCATTGTAGGGCAACTG-3�;and RANK, 5�-CCAGTCAGCAAGAAGTGTGT-3� and5�-TCTGTTGAGTTGGAGCTGAC-3�.Proliferation Assay—BMMs (plated in 96-well dishes at a
density of 5000 cells/well) were maintained for 2 days in �-10medium with 20 ng/ml M-CSF and labeled with bromode-oxyuridine (BrdUrd) for the last 4 h of culture. BrdUrd incor-poration was determined using the Biotrak ELISA system(Amersham Biosciences).Media C-terminal Cross-linking Telopeptide of Bone Colla-
gen (CTx) Assay—BMMs were cultured on dentin in 96-wellplates with RANKL and M-CSF for 4 days. �-10 medium waschanged 1 day before harvesting. ThemediaCTx concentrationwas determined using a CrossLaps for Culture ELISA kit (Nor-dic Bioscience Diagnostics A/S, Herlev, Denmark).Serum CTx Assay—RANKL (100 �g/mouse/day) or PBS
was injected subcutaneously daily for 7 days. 2 days after thefinal injection, serum was collected. The CTx concentrationwas determined by using a RatLaps ELISA kit (Nordic Bio-science Diagnostics A/S). For parathyroid hormone (PTH)-based experiments, 10 �g of synthetic human PTH-(1–34)(Bachem California Inc., Torrance, CA) in 25 �l of vehicle (1mM HCl and 0.1% bovine serum albumin) or vehicle alonewas injected four times/day for 4 days into supracalvarialsubcutaneous tissue.RANKL-stimulated Bone Loss in Vivo—RANKL (100 �g/
mouse/day) orPBSwas injected subcutaneously onceperday for 7days. 2 days after the final injection, the mice were killed, and thetibias (devoid of soft tissue) were used to determine trabecularbone volume bymicro-computerized tomography.Statistics—Data are expressed as themeans� S.D. Statistical
significance was determined by two-tailed Student’s t test. Eachexperiment was performed at least twice, and representativedata are presented.
RESULTS
Notch1–3 Deletion Promotes Osteoclast Differentiation—Toavoid ambiguity due to possible redundancy among NOTCHparalogs and to circumvent embryonic lethality associated withloss of Notch1 or Notch2 (35–38), we generated compoundmice lacking all three receptors in osteoclast lineage cells. Thus,we mated Notch1flox/flox Notch2flox/flox Notch3�/� mice withanimals transgenic for Cre recombinase driven by themyeloid-specific lyzs promoter, producing offspring bearing theLysMcre(�)Notch1flox/� Notch2flox/� Notch3�/� genotype.These animals served as controls in all experiments relatingto the absence of Notch1–3 in osteoclasts. They were alsobackcrossed with Notch1flox/flox Notch2flox/flox Notch3�/�
mice. One product of this mating, viz. LysMcre(�)Notch1flox/flox Notch2flox/flox Notch3�/�, were then bred withNotch1flox/flox Notch2flox/flox Notch3�/� animals to generateLysMcre(�)Notch1flox/flox Notch2flox/flox Notch3�/� mice
whose osteoclast lineage cells were deleted of all three Notchgenes (hereafter referred to as Notch1,2,3OC�/�).
Using these compoundmice, we investigated whether loss ofNOTCH signaling in osteoclast lineage cells impacts theircapacity to differentiate into mature resorptive polykaryons.Osteoclast precursors in the form of BMMs isolated fromNotch1,2,3OC�/� and control mice were cultured with increas-ing amounts of M-CSF and/or RANKL. After 4 days, the cellswere stained for TRAP activity as a marker of osteoclast differ-entiation. Compared with control cells, Notch1,2,3OC�/�
BMMs differentiated into more and larger osteoclasts inresponse to cytokines (Fig. 1). The enhancement of osteoclastnumber was particularly evident at lower concentrations ofM-CSF or RANKL, indicating that global Notch deficiency sen-sitizes BMMs to osteoclastogenic cytokines.
FIGURE 1. NOTCH suppresses M-CSF- or RANKL-stimulated osteoclast dif-ferentiation in vitro. Notch1,2,3OC�/� and control BMMs were treated with25 or 100 ng/ml M-CSF in the presence of low dose RANKL (25 ng/ml) (A) orwith 25 or 100 ng/ml RANKL in the presence of low dose M-CSF (10 ng/ml) (B).After 5 days, the cultures were stained for TRAP activity. The number of oste-oclasts/well were counted (C). *, p � 0.05; ***, p � 0.001.
NOTCH1 Regulates Osteoclastogenesis
MARCH 7, 2008 • VOLUME 283 • NUMBER 10 JOURNAL OF BIOLOGICAL CHEMISTRY 6511
by guest on July 17, 2018http://w
ww
.jbc.org/D
ownloaded from
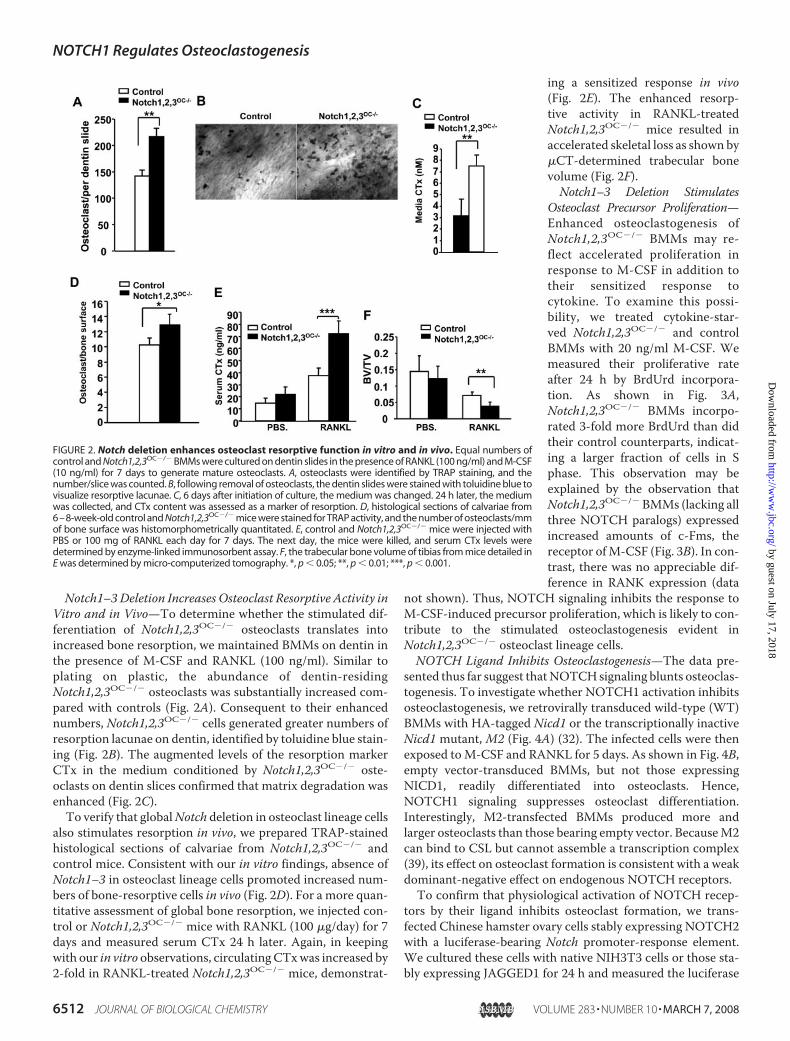
Notch1–3Deletion Increases Osteoclast Resorptive Activity inVitro and in Vivo—To determine whether the stimulated dif-ferentiation of Notch1,2,3OC�/� osteoclasts translates intoincreased bone resorption, we maintained BMMs on dentin inthe presence of M-CSF and RANKL (100 ng/ml). Similar toplating on plastic, the abundance of dentin-residingNotch1,2,3OC�/� osteoclasts was substantially increased com-pared with controls (Fig. 2A). Consequent to their enhancednumbers, Notch1,2,3OC�/� cells generated greater numbers ofresorption lacunae on dentin, identified by toluidine blue stain-ing (Fig. 2B). The augmented levels of the resorption markerCTx in the medium conditioned by Notch1,2,3OC�/� oste-oclasts on dentin slices confirmed that matrix degradation wasenhanced (Fig. 2C).To verify that globalNotch deletion in osteoclast lineage cells
also stimulates resorption in vivo, we prepared TRAP-stainedhistological sections of calvariae from Notch1,2,3OC�/� andcontrol mice. Consistent with our in vitro findings, absence ofNotch1–3 in osteoclast lineage cells promoted increased num-bers of bone-resorptive cells in vivo (Fig. 2D). For a more quan-titative assessment of global bone resorption, we injected con-trol or Notch1,2,3OC�/� mice with RANKL (100 �g/day) for 7days and measured serum CTx 24 h later. Again, in keepingwith our in vitro observations, circulatingCTxwas increased by2-fold in RANKL-treated Notch1,2,3OC�/� mice, demonstrat-
ing a sensitized response in vivo(Fig. 2E). The enhanced resorp-tive activity in RANKL-treatedNotch1,2,3OC�/� mice resulted inaccelerated skeletal loss as shown by�CT-determined trabecular bonevolume (Fig. 2F).Notch1–3 Deletion Stimulates
Osteoclast Precursor Proliferation—Enhanced osteoclastogenesis ofNotch1,2,3OC�/� BMMs may re-flect accelerated proliferation inresponse to M-CSF in addition totheir sensitized response tocytokine. To examine this possi-bility, we treated cytokine-star-ved Notch1,2,3OC�/� and controlBMMs with 20 ng/ml M-CSF. Wemeasured their proliferative rateafter 24 h by BrdUrd incorpora-tion. As shown in Fig. 3A,Notch1,2,3OC�/� BMMs incorpo-rated 3-fold more BrdUrd than didtheir control counterparts, indicat-ing a larger fraction of cells in Sphase. This observation may beexplained by the observation thatNotch1,2,3OC�/�BMMs (lacking allthree NOTCH paralogs) expressedincreased amounts of c-Fms, thereceptor ofM-CSF (Fig. 3B). In con-trast, there was no appreciable dif-ference in RANK expression (data
not shown). Thus, NOTCH signaling inhibits the response toM-CSF-induced precursor proliferation, which is likely to con-tribute to the stimulated osteoclastogenesis evident inNotch1,2,3OC�/� osteoclast lineage cells.NOTCH Ligand Inhibits Osteoclastogenesis—The data pre-
sented thus far suggest thatNOTCHsignaling blunts osteoclas-togenesis. To investigate whether NOTCH1 activation inhibitsosteoclastogenesis, we retrovirally transduced wild-type (WT)BMMs with HA-tagged Nicd1 or the transcriptionally inactiveNicd1 mutant, M2 (Fig. 4A) (32). The infected cells were thenexposed toM-CSF and RANKL for 5 days. As shown in Fig. 4B,empty vector-transduced BMMs, but not those expressingNICD1, readily differentiated into osteoclasts. Hence,NOTCH1 signaling suppresses osteoclast differentiation.Interestingly, M2-transfected BMMs produced more andlarger osteoclasts than those bearing empty vector. BecauseM2can bind to CSL but cannot assemble a transcription complex(39), its effect on osteoclast formation is consistent with a weakdominant-negative effect on endogenous NOTCH receptors.To confirm that physiological activation of NOTCH recep-
tors by their ligand inhibits osteoclast formation, we trans-fected Chinese hamster ovary cells stably expressing NOTCH2with a luciferase-bearing Notch promoter-response element.We cultured these cells with native NIH3T3 cells or those sta-bly expressing JAGGED1 for 24 h and measured the luciferase
FIGURE 2. Notch deletion enhances osteoclast resorptive function in vitro and in vivo. Equal numbers ofcontrol and Notch1,2,3OC�/� BMMs were cultured on dentin slides in the presence of RANKL (100 ng/ml) and M-CSF(10 ng/ml) for 7 days to generate mature osteoclasts. A, osteoclasts were identified by TRAP staining, and thenumber/slice was counted. B, following removal of osteoclasts, the dentin slides were stained with toluidine blue tovisualize resorptive lacunae. C, 6 days after initiation of culture, the medium was changed. 24 h later, the mediumwas collected, and CTx content was assessed as a marker of resorption. D, histological sections of calvariae from6–8-week-old control and Notch1,2,3OC�/� mice were stained for TRAP activity, and the number of osteoclasts/mmof bone surface was histomorphometrically quantitated. E, control and Notch1,2,3OC�/� mice were injected withPBS or 100 mg of RANKL each day for 7 days. The next day, the mice were killed, and serum CTx levels weredetermined by enzyme-linked immunosorbent assay. F, the trabecular bone volume of tibias from mice detailed inE was determined by micro-computerized tomography. *, p � 0.05; **, p � 0.01; ***, p � 0.001.
NOTCH1 Regulates Osteoclastogenesis
6512 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 10 • MARCH 7, 2008
by guest on July 17, 2018http://w
ww
.jbc.org/D
ownloaded from

activity. Relative to parental NIH3T3 cells, co-culture withthose expressing JAGGED1 activated the NOTCH reporterconstruct in N2-CHO cells by �7-fold (Fig. 4C). Next, we co-cultured WT BMMs for 5 days with parental NIH3T3 cells orthose overexpressing JAGGED1 in the presence of M-CSF andRANKL. JAGGED1-expressing NIH3T3 cells markedlyreduced the differentiation of WT BMMs into osteoclasts (Fig.4D), establishing that ligand-mediatedNOTCHactivation sup-presses osteoclastogenesis.NOTCH1 and NOTCH3 Inhibit Osteoclastogenesis—Having
established that NOTCH signaling inhibits osteoclastogenesis,we turned to the specific role of individual NOTCH receptorsin the differentiation of osteoclast lineage cells. First, we meas-ured the abundance of each paralog throughout osteoclasto-genesis. Immunoblot analysis showed that NOTCH1 andNOTCH3 proteins were expressed in BMMs and throughoutthe osteoclast differentiation process (Fig. 5, A and B). On theother hand, NOTCH2 protein was undetectable in osteoclastsand their precursors (data not shown).We next transduced Notch1flox/flox BMMs in vitro with Cre
recombinase-expressing, blasticidin-selectable retrovirus orwith empty vector controls. The infected cells were selectedwith blasticidin in the presence of M-CSF. These Notch1-defi-cient BMMs, their empty vector-containing counterparts, orthose obtained from Notch3�/� or WT mice were placed in
M-CSF and RANKL for 5 days to determine their osteoclast-forming potential. Because NOTCH2 was not detected in oste-oclast lineage cells, Cre recombinase-treated Notch2flox/floxBMMs served as an additional control. Loss of NOTCH1 inosteoclast lineage cells greatly enhanced osteoclastogenesis(Fig. 5C). Notch3 deletion also modestly promoted osteoclastformation. As expected, removal of Notch2 had no impact ondifferentiation of the resorptive cell. Thus, both NOTCH1 andNOTCH3were required to fully inhibit osteoclast formation inthese assays, most likely by mutual inhibition, as osteoclasto-genic macrophages express JAGGED1 and JAGGED2 (40). Weconclude that NOTCH1 in BMMs plays a dominant role insuppressing their differentiation into osteoclasts.
FIGURE 3. Notch deletion accelerates BMM proliferation. A, control andNotch1,2,3OC�/� BMMs were starved for 6 h and then exposed to M-CSF (20mg/ml) for 4 h. Proliferation was determined as a function of BrdUrd incorpo-ration. B, control (Con) and Notch1,2,3OC�/� BMMs were assayed for c-Fmsexpression (arrowheads) by immunoblotting of total cell lysate. Actin servedas a loading control. **, p � 0.01.
FIGURE 4. NOTCH signaling inhibits osteoclastogenesis. A, WT BMMs wereretrovirally transduced with the pMX vector, HA-Nicd1, or HA-M2. Theinfected cells were selected by 3-day exposure to blasticidin, after which HA-NICD1 and HA-M2 expression was determined by immunoblotting. Actinserved as a loading control. B, pMX-, Nicd1-, and M2-transduced BMMsexposed to RANKL and M-CSF for 5 days were stained for TRAP activity.C, NOTCH2-expressing NIH3T3 cells transfected with NOTCH-response ele-ment-luciferase reporter plasmid were co-cultured with native NIH3T3 cells(3T3) or those overexpressing JAGGED1 (3T3-J1). Luciferase activity wasmeasured 24 h later. **, p � 0.01. D, NIH3T3 and JAGGED1-expressing NIH3T3cells were co-cultured with WT BMMs in the presence of M-CSF and RANKL. 5days later, the NIH3T3 and JAGGED1-expressing NIH3T3 cells were removedwith trypsin, and osteoclasts were visualized by TRAP staining.
NOTCH1 Regulates Osteoclastogenesis
MARCH 7, 2008 • VOLUME 283 • NUMBER 10 JOURNAL OF BIOLOGICAL CHEMISTRY 6513
by guest on July 17, 2018http://w
ww
.jbc.org/D
ownloaded from

Absence of NOTCH1 in Osteoblasts Enhances OsteoclastFormation—Having established that NOTCH activation atten-uates osteoclast differentiation and bone resorption, we
assessed the impact of NOTCH signaling on the osteoblast, acell that regulates osteoclast differentiation by expression of thepositive regulators RANKL and M-CSF or the repressor OPG.We first addressed this issue in vitro by infecting calvarial
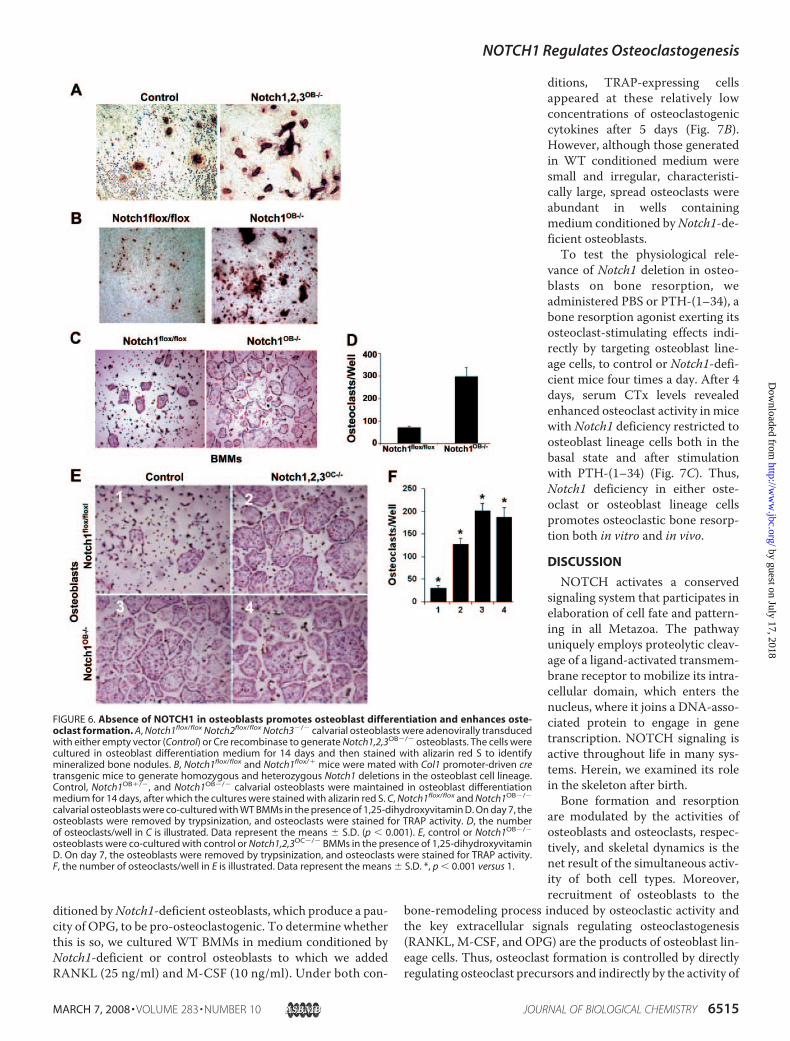
osteoblasts (isolated from 3–5-day-old Notch1flox/floxNotch2flox/flox Notch3�/� mice) with empty vector or Cre-bear-ing adenovirus. Green fluorescent protein-tagged adenovirus,serving as efficiency control, demonstrated that �90% of thecells were infected (data not shown). We examined the effectsof NOTCH loss on osteoblast function by culturing the Creadenovirus-infected cells (Notch1,2,3OB�/�) in osteoblasto-genicmediumand after 14 days assessedmineralized bonenod-ule formation. Similar to the effects on osteoclasts, the absenceof the three NOTCH paralogs enhanced the osteogenic capac-ity of osteoblasts (Fig. 6A). To determine whether Notch1 is afunctional receptor in osteoblast differentiation, we gener-ated Col1-cre;Notch1flox/flox mice to yield animals lackingNotch1 in osteoblast lineage cells (Notch1OB�/�). Similar toNotch1,2,3OC�/� osteoblasts, calvarial cells lacking Notch1underwent enhanced mineralized bone nodule formation(Fig. 6B), confirming a role for NOTCH1 in delaying osteo-blast differentiation.We next investigated whether deletion of Notch1 in osteo-
blasts impacts osteoclastogenesis. To this end, we culturedNotch1flox/flox or Notch1OB�/� osteoblasts with WT BMMs for 5days in the presence of 1,25-dihydroxyvitaminD,which promotesosteoclast differentiation in this co-culture system. The osteo-blasts were then removed with collagenase, and osteoclasts wereidentified by TRAP staining. More mature osteoclasts were pres-ent in co-cultures containing Notch1-deficient osteoblasts com-pared with control osteoblasts (Fig. 6,C andD).The data presented thus far have established thatNotch dele-
tion in either BMMs or osteoblast lineage cells enhances oste-oclastogenesis. To establish whether osteoclast recruitmentcan be enhanced further with Notch deficiency in both celltypes, we cultured various combinations of WT and Notch-deficient BMMs and osteoblasts in 1,25-dihydroxyvitamin D.Cultures containing osteoblasts and BMMs (both lackingNOTCH) were substantially more osteoclastogenic (Fig. 6E,panel 4) than those in which both cells wereWT (panel 1). Theabsence of the receptor in only BMMs (panel 2) or osteoblastlineage cells (panel 3) yielded osteoclast cultures indistinguish-able from those in which both cell types lacked Notch (Fig. 6F).Hence,Notch deficiencies in either osteoclast or osteoblast lin-eage cells optimally promote osteoclast formation.Absence of Notch1 in Osteoblasts Suppresses OPG Expression
and Enhances Osteoclast Resorptive Activity—To understandhow Notch-deficient osteoblasts promote osteoclastogenesis,we measured the expression of RANKL and M-CSF mRNAs,the principal cytokines with which osteoblasts promote oste-oclastogenesis, and that of OPG, a RANKL decoy receptor thatsuppresses osteoclast formation. Although the quantity ofM-CSF mRNA was essentially unaltered in Notch1-deficientosteoblasts, RANKLmRNAwas enhanced, and therewas a pro-found decrease in OPG mRNA (Fig. 7A), resulting in a pro-nounced pro-osteoclastogenic shift in the cytokines produced.Unlike osteoblast-produced RANKL, which is membrane-
bound, OPG is secreted. Thus, one would expect medium con-
FIGURE 5. NOTCH1 and NOTCH3 inhibit osteoclastogenesis. A, control andNotch1,2,3OC�/� BMMs were cultured with RANKL and M-CSF over time. TheNOTCH1 levels in total cell lysate were assessed by immunoblotting at days 0,3, and 5. �-Actin served as a loading control. B, cells were treated as describedfor A, following which NOTCH3 expression was assessed by immunoblotting.c-Src and �3 integrin (�3) expression served as a marker of osteoclast differ-entiation. C, Notch1flox/flox and Notch2flox/flox BMMs were retrovirally trans-duced with either cre or the pMX vector. Blasticidin-selected cells were geno-typed to assure Notch1 or Notch2 deletion. The selected cells and Notch3�/�
BMMs were exposed to RANKL and M-CSF to induce osteoclastogenesis. 5days later, the cells were stained for TRAP activity, and the number of oste-oclasts was counted. KO, knock-out. **, p � 0.01; ***, p � 0.001.
NOTCH1 Regulates Osteoclastogenesis
6514 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 10 • MARCH 7, 2008
by guest on July 17, 2018http://w
ww
.jbc.org/D
ownloaded from
ditioned byNotch1-deficient osteoblasts, which produce a pau-city of OPG, to be pro-osteoclastogenic. To determine whetherthis is so, we cultured WT BMMs in medium conditioned byNotch1-deficient or control osteoblasts to which we addedRANKL (25 ng/ml) and M-CSF (10 ng/ml). Under both con-
ditions, TRAP-expressing cellsappeared at these relatively lowconcentrations of osteoclastogeniccytokines after 5 days (Fig. 7B).However, although those generatedin WT conditioned medium weresmall and irregular, characteristi-cally large, spread osteoclasts wereabundant in wells containingmedium conditioned byNotch1-de-ficient osteoblasts.To test the physiological rele-
vance of Notch1 deletion in osteo-blasts on bone resorption, weadministered PBS or PTH-(1–34), abone resorption agonist exerting itsosteoclast-stimulating effects indi-rectly by targeting osteoblast line-age cells, to control or Notch1-defi-cient mice four times a day. After 4days, serum CTx levels revealedenhanced osteoclast activity inmicewith Notch1 deficiency restricted toosteoblast lineage cells both in thebasal state and after stimulationwith PTH-(1–34) (Fig. 7C). Thus,Notch1 deficiency in either oste-oclast or osteoblast lineage cellspromotes osteoclastic bone resorp-tion both in vitro and in vivo.
DISCUSSION
NOTCH activates a conservedsignaling system that participates inelaboration of cell fate and pattern-ing in all Metazoa. The pathwayuniquely employs proteolytic cleav-age of a ligand-activated transmem-brane receptor to mobilize its intra-cellular domain, which enters thenucleus, where it joins a DNA-asso-ciated protein to engage in genetranscription. NOTCH signaling isactive throughout life in many sys-tems. Herein, we examined its rolein the skeleton after birth.Bone formation and resorption
are modulated by the activities ofosteoblasts and osteoclasts, respec-tively, and skeletal dynamics is thenet result of the simultaneous activ-ity of both cell types. Moreover,recruitment of osteoblasts to the
bone-remodeling process induced by osteoclastic activity andthe key extracellular signals regulating osteoclastogenesis(RANKL, M-CSF, and OPG) are the products of osteoblast lin-eage cells. Thus, osteoclast formation is controlled by directlyregulating osteoclast precursors and indirectly by the activity of
FIGURE 6. Absence of NOTCH1 in osteoblasts promotes osteoblast differentiation and enhances oste-oclast formation. A, Notch1flox/flox Notch2flox/flox Notch3�/� calvarial osteoblasts were adenovirally transducedwith either empty vector (Control) or Cre recombinase to generate Notch1,2,3OB�/� osteoblasts. The cells werecultured in osteoblast differentiation medium for 14 days and then stained with alizarin red S to identifymineralized bone nodules. B, Notch1flox/flox and Notch1flox/� mice were mated with Col1 promoter-driven cretransgenic mice to generate homozygous and heterozygous Notch1 deletions in the osteoblast cell lineage.Control, Notch1OB�/�, and Notch1OB�/� calvarial osteoblasts were maintained in osteoblast differentiationmedium for 14 days, after which the cultures were stained with alizarin red S. C, Notch1flox/flox and Notch1OB�/�
calvarial osteoblasts were co-cultured with WT BMMs in the presence of 1,25-dihydroxyvitamin D. On day 7, theosteoblasts were removed by trypsinization, and osteoclasts were stained for TRAP activity. D, the numberof osteoclasts/well in C is illustrated. Data represent the means � S.D. (p � 0.001). E, control or Notch1OB�/�
osteoblasts were co-cultured with control or Notch1,2,3OC�/� BMMs in the presence of 1,25-dihydroxyvitaminD. On day 7, the osteoblasts were removed by trypsinization, and osteoclasts were stained for TRAP activity.F, the number of osteoclasts/well in E is illustrated. Data represent the means � S.D. *, p � 0.001 versus 1.
NOTCH1 Regulates Osteoclastogenesis
MARCH 7, 2008 • VOLUME 283 • NUMBER 10 JOURNAL OF BIOLOGICAL CHEMISTRY 6515
by guest on July 17, 2018http://w
ww
.jbc.org/D
ownloaded from

the osteoblast.We have demonstrated, for example, that tumornecrosis factor-� exerts its osteoclastogenic effects by stimulat-ing both cell types (28). Hence, we investigated whetherNOTCH signaling impacts osteoclast formation in vitro and invivo and dissected the osteoclast-autonomous requirements aswell as the role of NOTCH signaling in the supportive osteo-blast lineage cells. Both contribute to bone resorption.NOTCH1 and NOTCH3 are constitutively expressed
throughout osteoclastogenesis, whereas NOTCH2 is undetect-able. To determine whether NOTCH receptors impact differ-entiation of osteoclast precursors, we produced single or com-
pound mutant mice lacking either one or all three NOTCHparalogs in this lineage. To avoid lethality associatedwith globaldeletion of Notch1 or Notch2, we generated Notch3�/� micewith conditional alleles forNotch1 andNotch2 andmated themwith transgenicmice expressing Cre recombinase driven by themyeloid-specific lysozyme M-cre promoter. Our rationale fordeleting theNotch2 gene in our compound knock-outmice wasto avoid the possibility of compensatory NOTCH2 expressionin the absence of the other two paralogs. Using this strategy, wediscovered that although NOTCH1 is the dominant NOTCHreceptor mediating osteoclastogenesis, NOTCH3 is also activein this regard.M-CSF andRANKLare the key regulators of bone resorption
and act directly on osteoclast precursors. We found that theabsence of Notch1–3 in these cells lowers the threshold foreithermolecule, eventuating in enhanced bone resorption bothin vitro and in vivo. This finding suggests that NOTCH nega-tively controls osteoclastogenesis at low doses of RANKL orM-CSF. We confirmed this hypothesis by demonstrating thatJAGGED1-expressing cells inhibit differentiation of BMMs tothe osteoclast phenotype in the presence of RANKL.NOTCH signaling requires trans-endocytosis of receptor
bound to ligand. We propose that NOTCH-mediated regula-tion of osteoclastogenesis involves physical interactionbetween BMMs or their interaction with ligand-expressingstromata. Because osteoclastogenic macrophages expressNOTCH ligand and utilize NOTCH to regulate their differen-tiation, our data suggest that osteoclastogenesis is modulatedby lateral or mutual inhibitory interactions between osteoclastprecursors.Osteoclast formation also reflects the number of progenitors,
and others have shown that NOTCH activation prompts pro-grammed death of osteoclast lineage cells (23, 41). Similarly, wefound that the absence of NOTCH potentiates the proliferativeresponse of BMMs to M-CSF most likely caused by enhancedexpression of c-Fms, the cytokine receptor, by Notch-deficientosteoclast precursors.NOTCH paralogs are similar in structure and ligand recog-
nition, but eachmay regulate particular events in a cell-specificmanner (42). NOTCH1 and NOTCH3 are constitutivelyexpressed during osteoclastogenesis, and Notch1 deletion dra-matically promotes osteoclast formation. However, NOTCH1does not completely regulate osteoclastogenesis, as eliminatingonly NOTCH3 increases the number of bone-resorptivepolykaryons. Although both paralogs have been considered bysome to be antagonistic (43), this is the first example of a proc-ess controlled by each in a parallel fashion. Consistent with ourfailure to detect NOTCH2 protein in BMMs, Cre-mediateddeletion of its gene in osteoclast lineage cells does not alterRANKL- and M-CSF-induced differentiation. In a separatestudy, however, we found that NOTCH2 plays a predominantrole in bone marrow mesenchymal progenitors to controlosteoblastogenesis.3Having established that NOTCH signaling regulates oste-
oclastogenesis cell-autonomously, we investigated whether
3 Hilton, M. J., Tu, X., Bai, S., Zhao, J., Kobayashi, T., Kronenberg, H. M., Teitel-baum, S. L., Ross, F. P., Kopan, R., and Long, F. (2008) Nat. Med., in press.
FIGURE 7. NOTCH deficiency modulates OPG and RANKL expression.A, RANKL, OPG, M-CSF, and NOTCH1 mRNA levels in Notch1flox/flox andNotch1OB�/� osteoblasts were measured by reverse transcription-PCR. Glyc-eraldehyde-3-phosphate dehydrogenase (GAPDH) served as a loading con-trol. B, WT BMMs were cultured in medium conditioned for 1 day byNotch1flox/flox or Notch1OB�/� osteoblasts in the presence of low dose RANKL(25 ng/ml) and M-CSF (10 ng/ml). 5 days later, the cells were stained for TRAPactivity. C, Notch1flox/flox and Notch1OB�/� mice were injected with PBS orPTH-(1–34) four times/day for 4 days, after which serum CTx levels were deter-mined by enzyme-linked immunosorbent assay. *, p � 0.05; **, p � 0.01.
NOTCH1 Regulates Osteoclastogenesis
6516 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 10 • MARCH 7, 2008
by guest on July 17, 2018http://w
ww
.jbc.org/D
ownloaded from

NOTCH activity in accessory cells, viz. those of the osteoblastlineage, contributes to regulation of osteoclastogenesis. Weherein report that Col1�-cre-mediated inactivation of only theNotch1 gene in osteoblasts enhances osteoclastogenesis in vitroand osteoclast activity in vivo.We favor the hypothesis that thisis due to an increased ratio of RANKL/OPG expression becausewe observed a reduced ability of medium conditioned byNotch1-deficient osteoblasts to block osteoclastogenesis inresponse to low concentrations of M-CSF or RANKL. Thismost likely represents deficiency of the RANKLdecoy receptor,given that RANKL is typically membrane-residing and OPG issecreted. This increase in both the osteogenic and osteoclasto-genic capacity of Notch1-deficient osteoblasts provides a likelyexplanation for the normal skeletal mass of 2-month-oldNotch1OB�/� mice. On the other hand, mice lacking bothNotch1 and Notch2 in osteoblasts (by Col1-Cre) have a lowerbonemass compared with controls by 5months of age, indicat-ing that suppressed bone formation ultimately dominates theskeletal phenotype.4
The increased bone resorption in mice lacking Notch1, spe-cifically in osteoblast lineage cells, occurs under basal condi-tions and in mice treated with PTH-(1–34), a resorption-en-hancing hormone that targets the osteoblast to promote itsexpression of RANKL and to inhibit that of OPG. On the otherhand, Calvi et al. (44, 45) have shown that PTH-(1–34)increases the number of hematopoietic stem cells by inducingJAGGED1 expression in osteoblasts. Given that PTH-(1–34) isproresorptive and that osteoclasts are of hematopoietic origin,these data are seemingly at odds with ours, which have estab-lished that the population of osteoclasts diminishes in the faceof JAGGED1-induced NOTCH activation. However, becausethe increase in hematopoietic stem cell abundance does notnecessarily translate to a change in the number of BMMs, ourdata, together with those of Calvi et al., indicate that NOTCHsignaling dampens osteoclast recruitment not by targetingprimitive precursors, but by targeting those committed tomac-rophage differentiation.Finally, these results have significant therapeutic implica-
tions. Because NOTCH signaling is implicated in a variety ofhematological and solid malignancies, it is an attractive thera-peutic target (46, 47). Our findings demonstrate that NOTCHinhibition in the adult stimulates bone resorption and raise cau-tion that drugs inhibiting this pathwaymay accelerate bone lossand predispose patients to fracture.
REFERENCES1. van Es, J. H., van Gijn, M. E., Riccio, O., van den Born, M., Vooijs, M.,
Begthel, H., Cozijnsen, M., Robine, S., Winton, D. J., Radtke, F., and Clev-ers, H. (2005) Nature 435, 959–963
2. Lee, J., Basak, J. M., Demehri, S., and Kopan, R. (2007) Development(Camb.) 134, 2795–2806
3. Maillard, I., He, Y., and Pear, W. S. (2003) Immunity 18, 587–5894. Callahan, R., and Egan, S. E. (2004) J. Mammary Gland Biol. Neoplasia 9,
145–1635. Costa, R. M., Honjo, T., and Silva, A. J. (2003) Curr. Biol. 13, 1348–13546. Ball, D. W., and Leach, S. D. (2003) Cancer Treat. Res. 115, 95–1217. Ellisen, L. W., Bird, J., West, D. C., Soreng, A. L., Reynolds, T. C., Smith,
S. D., and Sklar, J. (1991) Cell 66, 649–6618. Maillard, I., and Pear, W. S. (2007) Science 316, 840–8429. Nam, Y., Aster, J. C., and Blacklow, S. C. (2002) Curr. Opin. Chem. Biol. 6,
501–50910. Nicolas,M.,Wolfer, A., Raj, K., Kummer, J. A.,Mill, P., vanNoort,M., Hui,
C. C., Clevers, H., Dotto, G. P., and Radtke, F. (2003) Nat. Genet. 33,416–421
11. Eiraku, M., Tohgo, A., Ono, K., Kaneko, M., Fujishima, K., Hirano, T., andKengaku, M. (2005) Nat. Neurosci. 8, 873–880
12. Hu, Q. D., Ang, B. T., Karsak, M., Hu, W. P., Cui, X. Y., Duka, T., Takeda,Y., Chia, W., Sankar, N., Ng, Y. K., Ling, E. A., Maciag, T., Small, D.,Trifonova, R., Kopan, R., Okano, H., Nakafuku, M., Chiba, S., Hirai, H.,Aster, J. C., Schachner, M., Pallen, C. J., Watanabe, K., and Xiao, Z. C.(2003) Cell 115, 163–175
13. Gordon, W. R., Vardar-Ulu, D., Histen, G., Sanchez-Irizarry, C., Aster,J. C., and Blacklow, S. C. (2007) Nat. Struct. Mol. Biol. 14, 295–300
14. Parks, A. L., Klueg, K. M., Stout, J. R., and Muskavitch, M. A. (2000)Development (Camb.) 127, 1373–1385
15. Ilagan, M. X., and Kopan, R. (2007) Cell 128, 1246.e1–1246.e216. Fischer, A., and Gessler, M. (2003) Trends Cardiovasc. Med. 13, 221–22617. Iso, T., Kedes, L., and Hamamori, Y. (2003) J. Cell. Physiol. 194, 237–25518. Gridley, T. (2006) Dev. Dyn. 235, 2330–233619. Shen, J., Bronson, R. T., Chen, D. F., Xia, W., Selkoe, D. J., and Tonegawa,
S. (1997) Cell 89, 629–63920. Jiang, R., Lan, Y., Chapman, H. D., Shawber, C., Norton, C. R., Serreze,
D. V., Weinmaster, G., and Gridley, T. (1998) Genes Dev. 12, 1046–105721. Bulman, M. P., Kusumi, K., Frayling, T. M., McKeown, C., Garrett, C.,
Lander, E. S., Krumlauf, R., Hattersley, A. T., Ellard, S., and Turnpenny,P. D. (2000) Nat. Genet. 24, 438–441
22. Zamurovic, N., Cappellen, D., Rohner, D., and Susa, M. (2004) J. Biol.Chem. 279, 37704–37715
23. Yamada, T., Yamazaki, H., Yamane, T., Yoshino, M., Okuyama, H.,Tsuneto, M., Kurino, T., Hayashi, S., and Sakano, S. (2003) Blood 101,2227–2234
24. Deregowski, V., Gazzerro, E., Priest, L., Rydziel, S., and Canalis, E. (2006)J. Biol. Chem. 281, 6203–6210
25. Tezuka, K., Yasuda, M., Watanabe, N., Morimura, N., Kuroda, K., Miya-tani, S., and Hozumi, N. (2002) J. Bone Miner. Res. 17, 231–239
26. Nobta, M., Tsukazaki, T., Shibata, Y., Xin, C., Moriishi, T., Sakano, S.,Shindo, H., and Yamaguchi, A. (2005) J. Biol. Chem. 280, 15842–15848
27. Teitelbaum, S. L. (2007) Am. J. Pathol. 170, 427–43528. Kitaura, H., Zhou, P., Kim,H. J., Novack, D. V., Ross, F. P., andTeitelbaum,
S. L. (2005) J. Clin. Investig. 115, 3418–342729. Pan, Y., Lin,M.-H., Tian, X., Cheng,H.-T., Gridley, T., Shen, J., andKopan,
R. (2004) Dev. Cell 7, 731–74330. Miao, D., He, B., Jiang, Y., Kobayashi, T., Soroceanu,M. A., Zhao, J., Su, H.,
Tong, X., Amizuka, N., Gupta, A., Genant, H. K., Kronenberg, H. M.,Goltzman, D., and Karaplis, A. C. (2005) J. Clin. Investig. 115, 2402–2411
31. McHugh, K. P., Hodivala-Dilke, K., Zheng, M. H., Namba, N., Lam, J.,Novack, D., Feng, X., Ross, F. P., Hynes, R. O., and Teitelbaum, S. L. (2000)J. Clin. Investig. 105, 433–440
32. Kopan, R., Nye, J. S., andWeintraub, H. (1994)Development (Camb.) 120,2385–2396
33. Wang,M.W.,Wei, S., Faccio, R., Takeshita, S., Tebas, P., Powderly,W. G.,Teitelbaum, S. L., and Ross, F. P. (2004) J. Clin. Investig. 114, 206–213
34. Zou, W., Kitaura, H., Reeve, J., Long, F., Tybulewicz, V. L., Shattil, S. J.,Ginsberg, M. H., Ross, F. P., and Teitelbaum, S. L. (2007) J. Cell Biol. 176,877–888
35. Huppert, S. S., Le, A., Schroeter, E. H.,Mumm, J. S., Saxena,M. T.,Milner,L. A., and Kopan, R. (2000) Nature 405, 966–970
36. Hamada, Y., Kadokawa, Y., Okabe, M., Ikawa, M., Coleman, J. R., andTsujimoto, Y. (1999) Development (Camb.) 126, 3415–3424
37. Conlon, R. A., Reaume, A. G., and Rossant, J. (1995)Development (Camb.)121, 1533–1545
38. Swiatek, P. J., Lindsell, C. E., del Amo, F. F., Weinmaster, G., and Gridley,T. (1994) Genes Dev. 8, 707–719
39. Lubman,O. Y., Korolev, S. V., andKopan, R. (2004)Mol. Cell 13, 619–62640. Monsalve, E., Perez, M. A., Rubio, A., Ruiz-Hidalgo, M. J., Baladron, V.,
4 M. J. Hilton, X. Tu, S. Bai, J. Zhao, T. Kobayashi, H. M. Kronenberg, S. L. Teitel-baum, F. P. Ross, R. Kopan, and F. Long, unpublished data.
NOTCH1 Regulates Osteoclastogenesis
MARCH 7, 2008 • VOLUME 283 • NUMBER 10 JOURNAL OF BIOLOGICAL CHEMISTRY 6517
by guest on July 17, 2018http://w
ww
.jbc.org/D
ownloaded from

Garcia-Ramirez, J. J., Gomez, J. C., Laborda, J., and Diaz-Guerra, M. J.(2006) J. Immunol. 176, 5362–5373
41. Ohishi, K., Varnum-Finney, B., Serda, R. E., Anasetti, C., and Bernstein,I. D. (2001) Blood 98, 1402–1407
42. Cheng, H.-T., Kim, M., Valerius, M. T., Surendran, K., Schuster-Gossler,K., Gossler, A., McMahon, A. P., and Kopan, R. (2007) Development(Camb.) 134, 801–811
43. Apelqvist, A., Li, H., Sommer, L., Beatus, P., Anderson, D. J., Honjo, T.,Hrabe de Angelis, M., Lendahl, U., and Edlund, H. (1999) Nature 400,877–881
44. Calvi, L. M., Adams, G. B., Weibrecht, K.W.,Weber, J. M., Olson, D. P.,Knight, M. C., Martin, R. P., Schipani, E., Divieti, P., Bringhurst, F. R.,Milner, L. A., Kronenberg, H. M., and Scadden, D. T. (2003) Nature425, 841–846
45. Weber, J.M., Forsythe, S. R., Christianson, C.A., Frisch, B. J., Gigliotti, B. J.,Jordan, C. T., Milner, L. A., Guzman, M. L., and Calvi, L. M. (2006) Bone39, 485–493
46. Bolos, V., Grego-Bessa, J., and de la Pompa, J. L. (2007) Endocr. Rev. 28,339–363
47. van Es, J. H., and Clevers, H. (2005) Trends Mol. Med. 11, 496–502
NOTCH1 Regulates Osteoclastogenesis
6518 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 283 • NUMBER 10 • MARCH 7, 2008
by guest on July 17, 2018http://w
ww
.jbc.org/D
ownloaded from

Long, F. Patrick Ross and Steven L. TeitelbaumShuting Bai, Raphael Kopan, Wei Zou, Matthew J. Hilton, Chin-tong Ong, Fanxin
Indirectly via Osteoblast Lineage CellsNOTCH1 Regulates Osteoclastogenesis Directly in Osteoclast Precursors and
doi: 10.1074/jbc.M707000200 originally published online December 22, 20072008, 283:6509-6518.J. Biol. Chem.
10.1074/jbc.M707000200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/283/10/6509.full.html#ref-list-1
This article cites 47 references, 16 of which can be accessed free at
by guest on July 17, 2018http://w
ww
.jbc.org/D
ownloaded from




























