Embed Size (px)
Citation preview

Neuroprotective effects of bcl-2 overexpression in hippocampal
cultures: interactions with pathways of oxidative damage
Sarah Howard,* Clement Bottino,* Sheila Brooke,* Elise Cheng,* Rona G. Giffard�,�and Robert Sapolsky*,�,§
*Department of Biological Sciences, Stanford University, Stanford, California, USA
Departments of �Anesthesiology, �Neurosurgery and §Neurology and Neurological Sciences, Stanford University School of Medicine,
Stanford, California, USA
Abstract
Overexpression of bcl-2 protects neurons from numerous
necrotic insults, both in vitro and in vivo. While the bulk of such
protection is thought to arise from Bcl-2 blocking cytochrome c
release from mitochondria, thereby blocking apoptosis, the
protein can target other steps in apoptosis, and can protect
against necrotic cell death as well. There is evidence that these
additional actions may be antioxidant in nature, in that Bcl-2
has been reported to protect against generators of reactive
oxygen species (ROS), to increase antioxidant defenses and to
decrease levels of ROS and of oxidative damage. Despite this,
there are also reports arguing against either the occurrence,
or the importance of these antioxidant actions. We have
examined these issues in neuron-enriched primary hippo-
campal cultures, with overexpression of bcl-2 driven by a her-
pes simplex virus amplicon: (i) Bcl-2 protected strongly against
glutamate, whose toxicity is at least partially ROS-dependent.
Such protection involved reduction in mitochondrially derived
superoxide. Despite that, Bcl-2 had no effect on levels of lipid
peroxidation, which is thought to be the primary locus of glu-
tamate-induced oxidative damage; (ii) Bcl-2 was also mildly
protective against the pro-oxidant adriamycin. However, it did
so without reducing levels of superoxide, hydrogen peroxide or
lipid peroxidation; (iii) Bcl-2 protected against permanent
anoxia, an insult likely to involve little to no ROS generation.
These findings suggest that Bcl-2 can have antioxidant actions
that may nonetheless not be central to its protective effects,
can protect against an ROS generator without targeting steps
specific to oxidative biochemistry, and can protect in the
absence of ROS generation. Thus, the antioxidant actions of
Bcl-2 are neither necessary nor sufficient to explain its pro-
tective actions against these insults in hippocampal neurons.
Keywords: apoptosis, bcl-2, necrosis, neurotoxicity, oxygen
radicals, reactive oxygen species.
J. Neurochem. (2002) 83, 914–923.
An extensive work demonstrates the capacity of Bcl-2 to block
cell death in numerous cell types, including both neurons and
glia (Green and Reed 1998). The protein is thought to
heterodimerize with pro-apoptotic proteins such as BAX,
thereby impeding the release of cytochrome c from mitochon-
dria. Release of cytochrome c is critical to the activation of
caspases and the execution of programmed cell death.
However, Bcl-2 has a variety of other actions within cells
and, as the most explicit example of this, Bcl-2 can also block
apoptosis following cytochrome c release (Rosse et al. 1998).
These other actions include Bcl-2 enhancing mitochondrial
calcium uptake (Murphy et al. 1996) and decreasing nuclear
calcium accumulation (Marin et al. 1996), forming ion
channels in mitochondria (Green and Reed 1998), and causing
the translocation of kinases to mitochondria (Wang et al.
1996). Moreover, Bcl-2 is capable of blocking instances of
necrotic, as well as apoptotic cell death (Kane et al. 1993, in
neural cell lines; Yang et al. 1998, in the substantia nigra;
Papadopoulos et al. 1998, in cortical astrocytes).
Thus, Bcl-2 appears to have salutary effects other than
preventing cytochrome c release. It has long been postulated
Resubmitted manuscript received August 13, 2002; accepted August 19,
2002.
Address correspondence and reprint requests to Robert Sapolsky,
Department of Biological Sciences, Gilbert Laboratory MC 5020,
Stanford University, Stanford, CA 94305–5020, USA.
E-mail: [email protected]
Abbreviations used: ABTS, 2,3-azino-bis(ethylbezothiazoline-6-sulf-
onic) acid; DCF, dichlorodihydrofluorescein diacetate; DMSO, dimethyl
sulphoxide; HE, dihydroethidium; MEM, a modified minimum essential
medium; MOI, multiplicity of infection; PBS, phosphate-buffered saline;
ROS, reactive oxygen species.
Journal of Neurochemistry, 2002, 83, 914–923
914 � 2002 International Society for Neurochemistry, Journal of Neurochemistry, 83, 914–923

that some of these additional effects occur in the oxidative
realm. Such protection could involve Bcl-2 acting as a
classical antioxidant [i.e. quenching reactive oxygen species
(ROS)]. However, there is little evidence for this (Lee et al.
2001). In addition, Bcl-2 may decrease ROS generation,
increase the level/activity of antioxidants and/or protecting
targets of oxidative damage. Overexpression of bcl-2
attenuates the cell death caused by numerous insults whose
toxicities depend heavily upon ROS generation (such as
adriamycin, paraquat, hydrogen peroxide, 6-OHDA, MPTP;
see below). Moreover, many of these instances of protection
are accompanied by a reduction in ROS accumulation (Kane
et al. 1993, insult of glutathione depletion in neural cell
lines; Lawrence et al. 1996, adriamycin in primary hippo-
campal cultures; Papadopoulos et al. 1998, aglycemia in
cortical astrocyte cultures). In addition, Bcl-2 decreases
oxidative damage (Kane et al. 1993, glutathione depletion in
neural cell lines; Myers et al. 1995, cyanide/aglycemia in
hypothalamic tumor lines; Bruce-Keller et al. 1998, hydro-
gen peroxide and amyloid b-peptide in PC12 cells; Giardinoet al. 1996, hyperglycemia in peripheral tissue; Lee et al.
2001, hydrogen peroxide in teratocarcinoma and neurobla-
stoma cell lines). Studies also report that Bcl-2 can increase
the activity or levels of antioxidants (Kane et al. 1993, in
neural cell line; Ellerby et al. 1996, in a hypothalamic cell
line; Papadopoulos et al. 1998, in cultured cortical astro-
cytes; Steinman 1995, in peripheral tissue; Voehringer et al.
1998, in peripheral cell lines; Lee et al. 2001, in a peripheral
and a neuroblastoma cell line). In one scenario revolving
around the putative antioxidant effects of Bcl-2, the protein is
thought to protect mitochondrial membranes from peroxida-
tive damage (Bruce-Keller et al. 1998). Another study
focuses on the capacity of Bcl-2 to maintain mitochondrial
potential and decrease mitochondrial ROS production (Green
and Reed 1998).
Thus, it is clearly that Bcl-2 can exert significant
antioxidant actions. However, there are findings which
suggest that this is not always the case:
1. In a number of cases in peripheral tissue, Bcl-2 prevents
cell death under circumstances which are highly unlikely to
involve ROS (for example, growth factor deprivation under
anaerobic conditions; (Jacobson and Raff 1995; Shimizu
et al. 1995).
2. In at least some reports, Bcl-2 does not prevent the cell
death induced by the pro-oxidant 6-OHDA (Oh et al. 1995,
1998, in a dopaminergic cell line; Yamada et al. 1999, in the
substantia nigra for example of protection against 6-OHDA).
3. In one instance where Bcl-2 reduced both the ROS
accumulation and cell death caused by an insult, the
reduction in ROS accumulation was shown to be irrelevant
to the sparing from death (Gardner et al. 1997, in fibro-
blasts), and could only account for part of the protection
observed (Papadopoulos et al. 1998).
4. Finally, there has been the suggestion that Bcl-2 can act
as a pro-oxidant. One report involving bcl-2 overexpression
in Escherichia coli, and in a B-cell line (Steinman 1995), and
one involving cultured cortical astrocytes (Papadopoulos
et al. 1998) showed that a primary effect of Bcl-2 was to
increase ROS levels, and the increase in antioxidant levels
could be viewed as a secondary compensation.
Relatively few of the reports in this confusing literature are
derived from studies of the nervous system, which is
particularly vulnerable to ROS and in which Bcl-2 can be
highly protective from various insults. Because of this, we
have examined these issues in primary hippocampal cultures
overexpressing bcl-2. We observe that: (i) Bcl-2 can reduce
the neurotoxicity of an excitotoxic insult while having
inconsistent effects upon indices of oxidative stress; (ii)
Bcl-2 can reduce the neurotoxicity of a pro-oxidant while
having no effect on indices of oxidative stress; (iii) Bcl-2 can
spare neurons from an insult whose toxicity is not likely to
involve the generation of ROS. Collectively, these findings
argue against the importance of an antioxidant role for Bcl-2 in
its neuroprotective actions in cultured hippocampal neurons.
Materials and methods
Hippocampal cell cultures
Tissue culture methods were described previously (Brooke et al.
1997) Briefly, hippocampus from 18-day-old fetal rats were
removed, dissociated with papain, filtered through an 80-lm cell
strainer, and resuspended in a modified minimum essential medium
(MEM; UCSF Tissue Culture Facility, San Francisco, CA, USA)
containing 25 mM glucose and 10% horse serum (Hyclone, Logan,
UT, USA). Cells were plated at a density of 30 000/cm2 on poly-D-
lysine-treated 96-well plates for the toxicity and superoxide studies,
on 24-well plates for the lipid peroxidation studies, and on 48-well
plates for the anoxia and anoxia/aglycemia studies. Cells were used
after 10–12 days. Cells used in glutamate experiments were treated
with 10 lM cytosine arabinoside on day 3 in culture, which
increased the percentage of neurons to approximately 70–80%.
Bcl-2 overexpression
A modified herpes simplex virus was used to deliver plasmids to
hippocampal neurons in culture. A bipromoter plasmid, pa22bgala4bcl-2, containing bcl-2 and the lacZ reporter gene under a4and a22 promoters, respectively, was used to overexpress bcl-2. Thecontrol plasmid, pa4bgal, contained the a4 promoter and lac Z
reporter gene. Both plasmids also contained the oriS and a
sequences required for replication and packaging. Construction of
plasmids was described previously (Lawrence et al. 1996).
Vectors were generated by transfection of plasmids into E5 cells
using lipofectamine (Gibco-BRL, Gaithersburg, MD, USA) and
superinfecting 24 h later with the helper virus d120 at a multiplicity
of infection (MOI) of 0.03 (Ho 1994). E5 cells stably transformed
with the a4 gene allowed d120, which lacks a4, propagation(DeLuca et al. 1985). Amplicons and d120 helper virus were titered
Neuroprotective effects of bcl-2 overexpression 915
� 2002 International Society for Neurochemistry, Journal of Neurochemistry, 83, 914–923

on Vero and E5 cells, respectively, and both were in the range of
0.5–3 · 107 infectious particles/mL. In all experiments, hippocam-pal cultures were infected approximately 12 h prior to experimental
treatment. Under these conditions, 63% of neurons are infected, as
are 11% of glia.
Solutions
For all experiments, working solutions were prepared in MEM
Eagle media (UCSF Tissue Culture Facility). A 1-M glutamate stock
solution in water was prepared prior to each experiment and further
diluted in MEM Eagle media. A 2-mM adriamycin (also known as
doxorubicin hydrochloride; Sigma, St Louis, MO, USA) stock
solution was kept at 4�C and diluted in MEM Eagle media for use.
EDTA (Sigma) was diluted from a 100-mM stock for use.
Toxicity studies
Glutamate or adriamycin was added directly to tissue culture wells
to the final concentrations of 5, 10, 15, 20 lM and 10, 30, 40, 50 and60 lM, respectively. After 24 h, cells were fixed with cold methanoland analyzed according to a published method (Brooke et al. 1997).
Briefly, cells were blocked with 5% milk in phosphate-buffered
saline (PBS), followed by immunocytochemistry with a neuron
specific primary antibody against MAP-2 (Sigma) at a dilution of
1 : 1000 in 5% milk in PBS. Following incubation with a rat-
adsorbed biotinylated secondary anti-IgG antibody (Vector, Burlin-
game, CA, USA), cells were treated with avidin-bound horseradish
peroxidase (ABC reagent, Vector). Finally, 2,3-azino-bis(ethylbezo-
thiazoline-6-sulfonic) acid (ABTS) was added according to manu-
facturer’s instructions (Vector), producing a color change in
proportion to amount of MAP-2 present. Absorbance at 405 nM
wavelength was read on a plate reader. During analysis, blanks
(wells treated with cold media to kill neurons), were subtracted from
all values and data were expressed as percentage survival according
to comparison with control wells that received no insult.
Hydroethidine studies
Generation of intracellular superoxide was determined according to
fluorescence of ethidium as a result of oxidation of hydroethidine
[also known as dihydroethidium (HE); Molecular Probes, Eugene,
OR, USA; Lagrange et al. 1994]. HE, 10 lg/lL in dimethyl
sulphoxide (DMSO), was stored under nitrogen at )80�C. Experi-mental treatments were added in 10–20 lL aliquots directly to tissueculture wells. Final concentrations of treatments were 40 lMadriamycin with and without 1.5 mM EDTA, and 10 and 20 lMglutamate. HE (16 lM) or DMSO vehicle was added at the same
time. Thirty minutes later, media were aspirated and replaced with
PBS containing 1% Triton-X. Fluorescence with excitation 480 nm
and emission 590 nm was read on a fluorescence plate reader.
Blanks without HE were subtracted from readings. A Pierce assay
was done in order to standardize for the amount of protein. Data
were expressed as percentage of control on each plate. For the
rotenone experiments, cells were pretreated with a final concentra-
tion of 10 lM rotenone (Sigma) in MEM, 40 min prior to
experimental treatment.
DCF studies
Dichlorodihydrofluorescein diacetate (DCF; Molecular Probes)
fluoresces upon oxidation by hydrogen peroxide (Lebel et al.
1990). DCF was stored at 4 mM in DMSO at – 80�C. Experimentaltreatments were added in 10–20 lL aliquots with final concentra-
tions of 40 lM adriamycin with and without 1.5 mM EDTA. DCF
(20 lM) or DMSO vehicle was added at the same time. Fifteen
minutes later, media were aspirated and replaced with PBS
containing 1% Triton-X. Fluorescence was read on a plate reader
with excitation 480 nm and emission 520 nm. Blanks, without DCF,
were subtracted from readings. A Pierce assay was used to
determine amounts of protein for standardization. Data were
expressed as a percentage of control on each plate. Because DCF
is pH-sensitive and glutamate decreases intracellular pH, attempts to
detect glutamate-induced increases in hydrogen peroxide were not
useful.
Lipid peroxidation studies
Lipid peroxidation was determined by measuring the loss of
fluorescence due to peroxidation of the naturally fluorescent fatty
acid, cis-parinaric acid (Molecular Probes; Kuypers et al. 1987).
Hippocampal cultures were incubated with 10 lM cis-parinaric
acid or 90% EtOH vehicle for 1 h prior to addition of 10 lLinsult or vehicle. Final conditions were 10 lM glutamate or 40 lMadriamycin with and without 1.5 mM EDTA. Two hours later,
cells were scraped from tissue culture wells and suspended in
PBS bubbled with nitrogen. Fluorescence was measured on a
Perkin-Elmer LS50B spectrometer (Perkin-Elmer, Foster City,
CA, USA) with excitation 312 nm and emission 414 nm. Blanks
containing no cis-parinaric acid were subtracted from readings
and data were expressed as percent of control. Because cis-
parinaric acid is light sensitive, all manipulations were performed
in the dark.
Anoxia studies
Experimental and control cells received two media changes with a
balanced salt solution. The experimental solution contained NaCl
116 mM, CaCl2 1.8 mM, MgSO4 0.8 mM, KCl 5.4 mM, NaH2PO41 mM, glucose 5.5 mM, NaHCO3 14.7 mM, and HEPES 10 mM. The
control solution was the same except it lacked HEPES and had a
higher concentration of NaHCO3 (27 mM). Experimental cells were
transferred to an anoxia chamber (Sheldon Manufacturing, Cornel-
ius, OR, USA) in an atmosphere of 5% CO2, 5% H2 and 90% N2,
where the media was changed twice to the deoxygenated salt
solution lacking glucose. Control plates received the same treatment
with solution incubated in the 37�C incubator with 5% CO2.
Experimental plates were incubated at 37�C in the anoxia chamber(with N2 gas). After 5–6 h, survival was assessed by the ABTS
assay described.
Data analysis and statistics
For all studies, data were expressed as a percentage increase above
Bcl-2 or bgal control after comparison by t-test to determine that
Bcl-2 and bgal controls were not significantly different from each
other. For toxicity studies, two-way ANOVA followed by Tukey post-
hoc test was used to determine difference between bgal and Bcl-2groups. For cPnA, HE and DCF studies, t-test was used to compare
insult group with Bcl-2 versus insult group with bgal. In the HEstudy with two concentrations of glutamate, a 2-way ANOVA
followed by Tukey’s post-hoc test was used to compare the bgalgroup versus the Bcl-2 group. For the anoxia and anoxia/aglycemia
916 S. Howard et al.
� 2002 International Society for Neurochemistry, Journal of Neurochemistry, 83, 914–923

studies, percentage survival with bgal was compared with
percentage survival with Bcl-2 by t-test. For all statistics, signifi-
cance was set at p < 0.05, and data are presented as mean ± SE.
Results
As would be expected, glutamate was neurotoxic to neuron-
enriched hippocampal cultures in a dose-dependent manner
[Fig. 1; the extent of toxicity did not differ from mock-
infected cultures exposed to equivalent amounts of glutamate
(data not shown)]. In support of the picture of excitotoxin-
induced neuron death involving ROS accumulation and
damage, glutamate also caused a significant accumulation of
superoxide (Fig. 2) and a significant amount of lipid
peroxidation (Fig. 3). Such superoxide appeared to be
derived from mitochondria. As evidence, treatment of
cultures with rotenone, which blocks mitochondrial super-
oxide production (Sensi et al. 1999; Saybasili et al. 2001),
blocked glutamate-induced superoxide accumulation (super-
oxide accumulation above baseline induced by 10 lMglutamate: 35% ± 9; p < 0.01 by t-test, compared with
0 glutamate; superoxide accumulation above baseline
induced by 10 lM glutamate plus 10 lM rotenone:
15% ± 6; n.s. as compared with 0 glutamate plus rotenone).
In agreement with a prior report (Lawrence et al. 1996),
overexpression of bcl-2 decreased the neurotoxicity of
glutamate, causing an approximate doubling of the LD50(Fig. 1). We then explored whether bcl-2 overexpression
attenuated the ROS-related effects of glutamate. We observed
that Bcl-2 completely blocked the superoxide accumulation
induced by 10 lM glutamate, a concentration at which Bcl-2also decreased the neurotoxicity (Fig. 2; superoxide produc-
tion was maximal at this time point). In addition, Bcl-2
caused a trend towards decreased superoxide accumulation at
20 lM glutamate, a concentration at which Bcl-2 was not
protective. Surprisingly, despite this effect, bcl-2 overexpres-
sion had no effect on the extent of lipid peroxidation induced
by 10 lM glutamate (Fig. 3).
We then examined the effects of bcl-2 overexpression on
the actions of the ROS generator, adriamycin. Bcl-2 signi-
ficantly decreased adriamycin-induced neurotoxicity, causing
an approximate 35% increase in the LD50 (Fig. 4). As would
be expected, adriamycin markedly increased superoxide
accumulation (Fig. 5; note that the scale on the x-axis differs
from Fig. 2), hydrogen peroxide accumulation (Fig. 6; as
Fig. 1 Glutamate-induced neurotoxicity in neuron-enriched hippo-
campal cultures treated with either control vector (m) or Bcl-2 (d).
Bcl-2 caused a significant decrease in neurotoxicity (p < 0.02,
F ¼ 6.04, d.f. ¼ 1/113, two-way ANOVA). n ¼ averaged 10/data point,
derived from three different weekly culture preparations.
Fig. 2 Effects of glutamate and of differing viral vectors on superoxide
accumulation in neuron-enriched hippocampal cultures. Cultures were
exposed to indicated concentrations of glutamate and either the con-
trol (bgal) or bcl-2-expressing vector. Increasing glutamate concen-
trations produced increasing superoxide accumulation in control
cultures (p < 0.02 by one-way ANOVA), but not in Bcl-2-treated cultures
(n.s). When exposed to 10 lM glutamate, Bcl-2-treated cultures had
significantly less accumulation than did control cultures (*p < 0.05,
Tukey test following two-way ANOVA). n ¼ 22–23/group, derived from
five different weekly culture preparations.
Fig. 3 Effects of glutamate and of differing viral vectors on lipid per-
oxidation in neuron-enriched hippocampal cultures. Cultures were
exposed to 10 lM glutamate and were infected with either control or
bcl-2-expressing vector. Glutamate caused a highly significant
increase in lipid peroxidation, regardless of vector treatment
(*p < 0.001, comparing bgal/0 glutamate with bgal/10 glutamate, or
Bcl-2/0 glutamate with Bcl-2/10 glutamate; Tukey’s post-hoc test fol-
lowing two-way ANOVA). Viral vector treatment, however, had no effect
on the extent of lipid peroxidation (n.s., by two-way ANOVA). n ¼ 23–28/
data point, taken from five different weekly culture preparations.
Neuroprotective effects of bcl-2 overexpression 917
� 2002 International Society for Neurochemistry, Journal of Neurochemistry, 83, 914–923

noted in the Methods section, similar measures were not
carried out with glutamate, because the DCF assay is
disrupted by the pH changes caused by the excitotoxin),
and lipid peroxidation (Fig. 7).
Despite the protective effects of Bcl-2 against adriamycin
neurotoxicity, we observed no effects of overexpression on
these ROS-related endpoints. Bcl-2 did not decrease adria-
mycin-induced superoxide accumulation (Fig. 5); as a pos-
itive control, under those same conditions, such
accumulation was significantly blunted by the calcium-
chelating effects of EDTA. Of note, the positive effects of
Bcl-2 on glutamate-induced superoxide accumulation were
demonstrable at a single time point (Fig. 2). The failure of
Bcl-2 to decrease adriamycin-induced superoxide accumula-
tion was not due to having chosen the wrong single time
point, as this was demonstrable over a range of times
(Fig. 5b). Similarly, Bcl-2 had no effect on adriamycin-
induced hydrogen peroxide accumulation, under conditions
where EDTA was effective (Fig. 6). Moreover, overexpres-
sion had no effect on the extent of lipid peroxidation under
conditions where EDTA was protective (Fig. 7).
We then examined whether Bcl-2 could protect against a
necrotic insult whose damaging effects were unlikely to
involve the generation of ROS. Specifically, we tested a
model of permanent anoxia. Five to six hours of anoxia was
significantly damaging to neurons in cultures transfected
with control vector (Fig. 8). In contrast, Bcl-2 overexpres-
sion provided complete protection.
Discussion
As discussed, the role of Bcl-2 in preventing neuron death is
more complicated than the protein solely preventing apop-
tosis by antagonizing the actions of BAX. The interactions of
Bcl-2 with the mitochondrial membrane are likely to be
pivotal to its larger role. While this appears to be central to
Fig. 4 Adriamycin-induced neurotoxicity in hippocampal cultures
treated with either control vector (m) or Bcl-2 (d). Bcl-2 caused a
significant decrease in neurotoxicity (p < 0.001, F ¼ 22.68,
d.f. ¼ 1/76, two-way ANOVA). n ¼ averaged 6/data point, taken from
two different weekly culture preparations.
Fig. 5 (a) Left: Effects of adriamycin and of differing viral vectors on
superoxide accumulation in hippocampal cultures. Cultures were
exposed to 40 lM adriamycin and either the control or bcl-2-expressing
vector. Adriamycin caused a significant increase in superoxide accu-
mulation, regardless of vector treatment (***p < 0.001, comparing bgal/
0 adriamycin with bgal/40 adriamycin, or Bcl-2/0 adriamycin with Bcl-2/
40 adriamycin; Tukey’s post-hoc test following two-way ANOVA). Viral
vector treatment, however, had no effect on superoxide accumulation
(n.s., by two-way ANOVA). Right: Effects of adriamycin and of EDTA on
superoxide accumulation in hippocampal cultures. Cultures were
exposed to 40 lM adriamycin with or without 1.5 mM EDTA. Adriamycin
caused a significant accumulation of superoxide generation in the
absence of EDTA (p < 0.01, when compared with control; Tukey’s
post-hoc test), while treatment with EDTA prevented such adriamycin-
induced accumulation (*p < 0.05, Tuvkey’s post-hoc test). For
unknown reasons, adriamycin was not as effective at increasing
superoxide generation in the EDTA experiment as in the vector
experiment; (p < 0.05, when compared with 40 lM adriamycin, bgal).
n ¼ 12–13/group, from two weekly culture preparations. (b) The effects
of adriamycin and indicated vectors on superoxide accumulation at 10,
20, 30 and 40 min Adriamycin significantly increased accumulation
regardless of vector, but vector treatment did not alter accumulation
(n.s). n ¼ 12–15/data point. d, bgal; s, bgal + 40 lM adriamycin; .,
bcl-2; ,, bcl-2 + 40 lM adriamycin.
918 S. Howard et al.
� 2002 International Society for Neurochemistry, Journal of Neurochemistry, 83, 914–923

Bcl-2 preventing BAX-induced release of cytochrome c from
the mitochondria, the maintenance of mitochondrial function
is also likely to help explain the protein’s capacity to protect
against necrotic cell death (Kane et al. 1993; Papadopoulos
et al. 1998; Yang et al. 1998). This involvement of Bcl-2 in
mitochondrial function has also prompted explorations of its
Fig. 7 Left: Effects of adriamycin and of differing viral vectors on
lipid peroxidation in hippocampal cultures. Cultures were exposed to
40 lM adriamycin and either the control or bcl-2-expressing vector.
Adriamycin caused a significant increase in lipid peroxidation,
regardless of vector treatment (***p < 0.001, comparing bgal/0
adriamycin with bgal/40 adriamycin, or Bcl-2/0 adriamycin with Bcl-2/
40 adriamycin; Tukey’s post-hoc test following two-way ANOVA). Viral
vector treatment, however, had no effect on superoxide accumula-
tion (n.s., by two-way ANOVA). Right: Effects of adriamycin and of
EDTA on lipid peroxidation in hippocampal cultures. Cultures were
exposed to 40 lM adriamycin with or without 1.5 mM EDTA. Adria-
mycin caused significant lipid peroxidation in the absence of EDTA
(**p < 0.01, when compared with control; Tukey’s post-hoc test),
while treatment with EDTA caused a significant diminution of this
adriamycin effect (*p < 0.05). n ¼ 18–22, from five different weekly
culture preparations.
Fig. 6 Left: Effects of adriamycin and of differing viral vectors on
hydrogen peroxide generation accumulation (as measured with DCF
fluorescence) in hippocampal cultures. Cultures were exposed to 40 lM
adriamycin and either the control or Bcl-2-expressing vector. Adria-
mycin caused a significant increase in DCF fluorescence, regardless of
vector treatment **p < 0.01, ***p < 0.001, comparing bgal/0 adriamycin
with bgal/40 adriamycin, or Bcl-2/0 adriamycin with Bcl-2/40 adriamy-
cin, respectively; Tukey’s post-hoc test following two-way ANOVA). Viral
vector treatment, however, had no effect on superoxide accumulation
(n.s., by two-way ANOVA). Right: Effects of adriamycin and of EDTA on
hydrogen peroxide generation in hippocampal cultures. Cultures were
exposed to 40 lM adriamycin with or without 1.5 mM EDTA. Adriamycin
caused significant generation in the absence of EDTA (**p < 0.01,
when compared to control; Tukey’s post-hoc test), while treatment with
EDTA prevented such adriamycin-induced accumulation. n ¼ 6–19/
group, from three different weekly cultures.
Neuroprotective effects of bcl-2 overexpression 919
� 2002 International Society for Neurochemistry, Journal of Neurochemistry, 83, 914–923
potential role as an antioxidant. In an early version of this
view, the proximity of Bcl-2 to the mitochondria suggested
that it would be preferentially poised to quench ROS released
from mitochondria (Hockenberry et al. 1993; this view pre-
dated the discovery of Bcl–2 interactions with BAX). More
recently, there has emerged the view that Bcl-2, while not
necessarily functioning as a classical ROS quencher, may
decrease ROS production by mitochondria, secondary to its
capacity to preserve both mitochondrial potential and
function of the electron transport chain. Furthermore, Bcl-2
can decrease peroxidative damage to mitochondrial mem-
brane (Bruce-Keller et al. 1998).
As reviewed, the evidence of Bcl-2 attenuating oxidative
damage, and for such actions being critical to its overall
protective effects, is equivocal, especially in the nervous
system. Our data suggest that Bcl-2’s protective effects in
hippocampal neurons need not be heavily dependent on its
capacity to protect against the generation and/or proximal
consequences of ROS, in contrast to our prior findings in
astrocytes (Papadopoulos et al. 1998).
Glutamate neurotoxicity
We initially observed Bcl-2 to protect against glutamatergic
excitotoxicity in hippocampal cultures. This protection
involved an approximate doubling of the LD50, such that at
its most efficacious, Bcl-2 decreased neuron death more than
50% (at 10 lM glutamate). Given that herpes viral vectors
have a strong preference for infecting neurons over glia in
primary cultures (Ho et al. 1995) and that these studies utilized
neuron-enriched cultures, the protective effects of Bcl-2 were
overwhelmingly likely to be due to direct actions within
infected neurons, rather than secondary to glial effects. This
should be contrasted with our previous observation that
selective bcl-2 overexpression in astrocytes cocultured with
wild-type neurons does afford protection from combined
oxygen glucose deprivation, an injury largely dependent on
the activation of glutamate receptors (Xu et al. 1999).
These protective Bcl-2 effects agree with prior reports
showing neuroprotection by Bcl-2 against excitotoxins,
hypoglycemia and adriamycin in primary cultures derived
from a number of brain regions (Jia et al. 1996; Lawrence
et al. 1996;McLaughlin et al. 2000; Tamatani et al. 2000) and
against in vivo models of excitotoxicity, hypoxia–ischemia,
ROS generators, or mechanical trauma (Linnik et al. 1995;
Lawrence et al. 1996, 1997; Antonawich et al. 1999; Yamada
et al. 1999; Phillips et al. 2000; Shimazaki et al. 2000).
Moreover, we observed that such protection was accom-
panied by a complete block of glutamate-induced superoxide
accumulation at the time point where such accumulation is
maximal post-glutamate. Mitochondria appear to be a major
source of such accumulation during excitotoxic insults
(Dugan et al. 1995), probably secondary to the disruption
of mitochondrial potential; supporting this, we observed that
rotenone, which blocks mitochondrial superoxide produc-
tion, blocked the effects of glutamate on this endpoint. Thus,
the Bcl-2 effect is commensurate with the broadly protective
array of effects of Bcl-2 in mitochondria [nonetheless, in this
particular culture system, we observe that Bcl-2 does not
alter glutamate-induced declines in mitochondrial potential
(manuscript in preparation)]. As an alternative or additional
mechanism, Bcl-2 can increase the activity or levels of
antioxidants, or optimize their subcellular distribution (Kane
et al. 1993; Steinman 1995; Ellerby et al. 1996; Papadopo-
ulos et al. 1998; Voehringer et al. 1998; Lee et al. 2001);
insofar as this involves an increase in SOD activity, this
should lead to a reduction in superoxide accumulation.
Finally, the effects of Bcl-2 on availability of substrates such
as GSH or GSSG are unknown, although the glutamatergic
excitotoxin kainic acid does not alter levels of either in this
culture system (Patel et al. 2002).
Glutamate-induced superoxide production appears to be an
important contributor to the subsequent neuron death (Dugan
et al. 1995) and administration or overexpression of a variety
of antioxidants (SOD, vitamin E, 21-amino steroids, oxy-
purinol, glutathione peroxidase) can decrease glutamatergic
injury in the brain (Acosta et al. 1987; Monyer et al. 1990;
Chan et al. 1991; Lin and Phillis 1991; Lafon-Cazal et al.
1993). Therefore, the decreased accumulation could help
explain the neuroprotective actions of Bcl-2. Thus, while there
is little evidence that Bcl-2 is acting in this case as a classical
quencher of ROS, some of its protective functionsmay revolve
around it indirectly decreasing ROS accumulation.
Our data also indicate that the glutamate-induced super-
oxide accumulation does not necessarily lead to peroxidative
damage to lipid membranes, as Bcl-2 reduced the former
without altering the latter. Excitotoxin-induced peroxidative
damage to cell membranes probably reflects ROS generation
in the cytosol and membrane itself. This would typically be
secondary to calcium-induced activation of xanthine oxidase,
phospholipase A2, and NOS, and the generation of hydrogen
peroxide, hydroxyl radicals, and peroxynitrites. In addition,
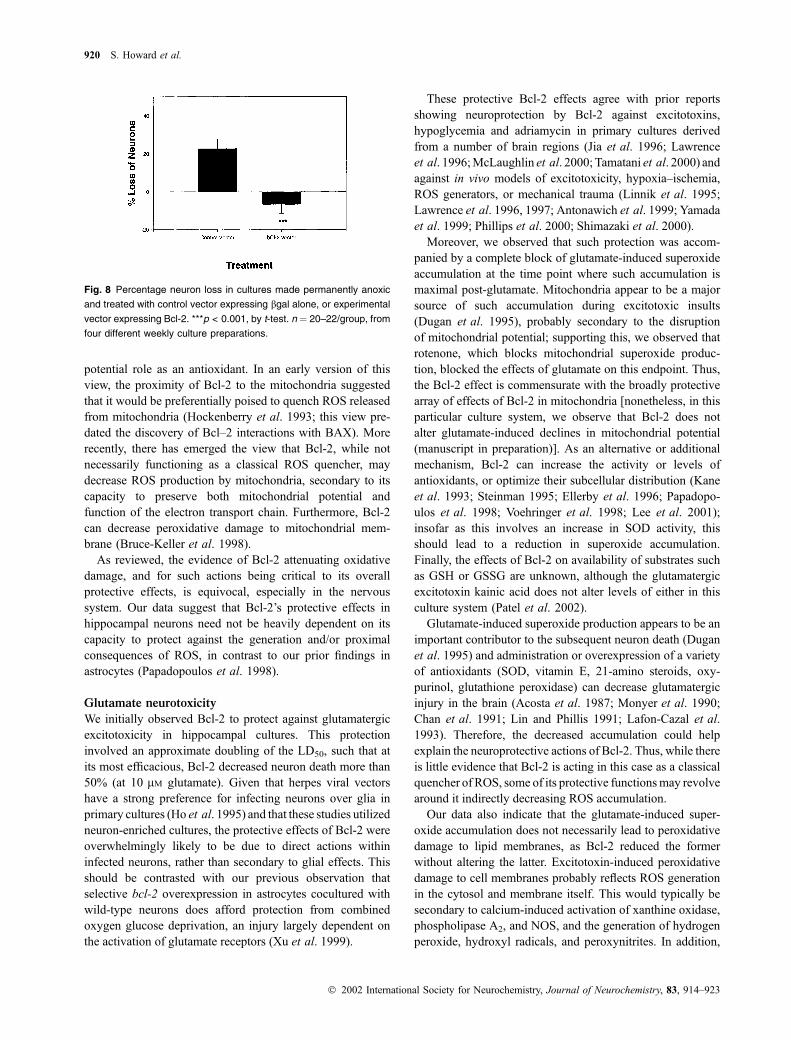
Fig. 8 Percentage neuron loss in cultures made permanently anoxic
and treated with control vector expressing bgal alone, or experimental
vector expressing Bcl-2. ***p < 0.001, by t-test. n ¼ 20–22/group, from
four different weekly culture preparations.
920 S. Howard et al.
� 2002 International Society for Neurochemistry, Journal of Neurochemistry, 83, 914–923

superoxide can be generated in the cytosol by oxygenases,
and thus could readily contribute to oxidative damage to the
cell membrane. However, our rotenone data suggest that the
superoxide generated by glutamate in our insult model is
predominately mitochondrial in origin. Thus, a change in
superoxide generation from mitochondria is not likely to
impact the endpoint of lipid peroxidation in the cell
membrane. It has been speculated that Bcl-2 can protect
mitochondrial membrane from lipid peroxidation (Bruce-
Keller et al. 1998). Such peroxidation could be generated by
the superoxide and be diminished by Bcl-2 in the present
case; however, any such peroxidation would be in amounts
below the level of detection in our assay.
Our data also demonstrate that in this model, glutamate-
induced lipid peroxidation is not likely to be playing an
obligatory role in the neurotoxicity, insofar as bcl-2
overexpression reduced neurotoxicity without altering the
extent of peroxidation. This is in contrast to the situation in
ischemic brain injury, in which lipid peroxidation is thought
to play a more central role in the neuron death (Traystman
et al. 1991; Chan 1996; Liu et al. 1998). However, it is
quite plausible that the peroxidative damage could impair
functional recovery in surviving neurons. In support of this,
under a number of circumstances, bcl-2 overexpression can
spare neurons from insult-induced death, but not from
insult-induced dysfunction (McLaughlin et al. 2000; Dumas
et al. 2000).
Adriamycin neurotoxicity
Bcl-2 can decrease the toxicity of a number of insults that are
heavily or entirely oxidative in nature, such as adriamycin,
paraquat, hydrogen peroxide, or 6-OHDA (Oh et al. 1995;
Lawrence et al. 1996; Lezoualc’h et al. l996; Marin et al.
1996; Bruce-Keller et al. 1998; ; Hochman et al. 1998; Yang
et al. 1998Yamada et al. 1999; Luc Cadet et al. 2000). We
examined whether Bcl-2 could protect against an insult that
is overwhelmingly oxidative in nature, and if any such
protection arose as a result of reducing ROS accumulation or
oxidative damage. We utilized adriamycin (doxorubicin), a
potent pro-oxidant commonly used in the treatment of
malignant tumors, which is toxic to cultured neurons in the
lM range. As expected, the neurotoxin generated a consid-
erable oxidative challenge; at its approximate LD50 (40 lM),adriamycin caused a 200% increase in superoxide accumu-
lation (in contrast, glutamate, at its LD50 of 10 lM, caused a15% increase).
We then observed that bcl-2 overexpression decreased
adriamycin neurotoxicity, although to a lesser extent than
against glutamate. Despite these neuroprotective effects,
Bcl-2 had no effect on the accumulation of superoxide,
hydrogen peroxide at a range of time points, or on the extent
of lipid peroxidation. This dissociation between protecting
from ROS while not decreasing a measure of oxidative
damage is reminiscent of the finding that in a peripheral cell
line, Bcl-2 decreased hydrogen peroxide toxicity without
decreasing oxidative damage to lipids, DNA or protein (Lee
et al. 2001). As discussed, the ability of Bcl-2 to lessen
glutamate-induced superoxide accumulation is, most parsi-
moniously, a consequence of the protein’s actions at
mitochondria. Commensurate with this, we observe that
Bcl-2 also blocks cytochrome c release from mitochondria in
this insult model (manuscript in preparation). The lack of an
effect of Bcl-2 against the far greater superoxide accumula-
tion induced by adriamycin suggests either that (i) the
superoxide is derived from mitochondria but exceeds Bcl-2’s
capacity to constrain such accumulation, and/or (ii) the
superoxide is predominately derived from non-mitochondrial
sites not subject to Bcl-2’s effects.
Recent work has emphasized the potential role of nitro-
sylative rather than oxidative damage in cell death. Along
these lines, in a case where Bcl-2 protected against hydrogen
peroxide without decreasing oxidative damage, it decreased
3-nitrotyrosine levels (Lee et al. 2001). Thus, the same may
hold in the present study.
ROS accumulation is thought to be one of the signals
initiating injury-induced apoptosis (e.g. the translocation of
BAX to the mitochondria). Our data suggest that Bcl-2
reduces adriamycin neurotoxicity by one of two routes. First,
it may protect downstream of the oxidative realm, with
blocking of cytochrome c release being the most implicated,
but not sole, possible mechanism. Second, it is currently not
known whether adriamycin causes nitrosylative damage and
whether such damage can also initiate apoptosis. If so, Bcl-2
might be blocking the nitrosylation pathway. Thus, while
protecting against an ROS generator, such protection may not
be centered in the oxidative realm, a point emphasized
previously (Oh et al. 1995).
Anoxic neurotoxicity
Glutamatergic excitotoxicity represents a model in which
ROS generation is likely to contribute at least somewhat to
damage, while adriamycin toxicity is overwhelmingly oxi-
dative in nature. Anoxia, in contrast, represents an insult in
which ROS play a minimal role, if any, in the neuron death.
As noted, Bcl-2 protects against insults under anaerobic
conditions in peripheral cell types (Jacobson and Raff 1995;
Shimizu et al. 1995), and this has been strongly interpreted
as evidence against Bcl-2’s protective actions being solely
antioxidant in nature. We observed that Bcl-2 overexpression
blocked the toxicity induced by permanent anoxia in these
cultures. Broadly, this suggests the same non-oxidative facets
of Bcl-2 actions within the CNS.
In conclusion, these data suggest a mixed picture
concerning the antioxidant actions of Bcl-2. The glutamate
data suggest that, while Bcl-2 can have antioxidant actions,
they may not impact one of the major endpoints of
oxidative damage. The adriamycin data, moreover, suggest
that, while Bcl-2 can protect against a classical ROS
Neuroprotective effects of bcl-2 overexpression 921
� 2002 International Society for Neurochemistry, Journal of Neurochemistry, 83, 914–923

generator, such protection may arise from actions either
lateral to, or downstream of, specific oxidative events.
Finally, the anoxia data show that Bcl-2 can protect against
an insult likely to have little or no elements of ROS
generation. Thus, in this model system, the antioxidant
actions of Bcl-2 may not be either necessary or sufficient to
explain its protective actions.
Acknowledgements
Funding was provided by NIH PO1 NS27520 (RG and RS), a
TRDRP State of California grant (RS), the International Anesthesia
Research Society (RG) and a URO Grant (CB). Technical
assistance was provided by Martin Brown, Nick Denko, Adrian
Dunn, Pedram Ghafourifar, Mark Mattson, Stefano Sensi and John
Weiss.
References
Acosta D., Kass I. and Cottrell J. (1987) Effect of a-tocopherol and freeradicals on anoxic damage in the rat hippocampal slice. Exp.
Neurol. 97, 607–614.
Antonawich F., Federoff H. and Davis J. (1999) Bcl-2 transduction,
using a HSV ampolicon, protects hippocampal neurons from
transient global ischemia. Exp. Neurol. 156, 130–136.
Brooke S. M., Chan R., Howard S. and Sapolsky R. (1997) Endocrine
modulation of the neurotoxicity of gp120: implications for AIDS-
related dementia complex. Proc. Natl Acad. Sci. USA 94, 9457–
9461.
Bruce-Keller A., Begley J., Fu W., Butterfield D., Bredesen D., Hutchins
J., Hensley K. and Mattson M. (1998) Bcl-2 protects isolated
plasma and mitochondrial membranes against lipid peroxidation
induced by hydrogen peroxide and amyloid b-peptide. J. Neuro-
chem. 70, 31–38.
Chan P. (1996) Role of oxidants in ischemic brain damage. Stroke 27,
1124–1129.
Chan P., Yang G., Chen S., Carlson E. and Epstein C. (1991) Cold-
induced brain edema and infarction are reduced in transgenic mice
overexpressing CuZn-superoxide dismutase. Ann. Neurol. 29, 482–
489.
DeLuca N., McCarthy A. and Schaffer P. (1985) Isolation and charac-
terization of deletion mutants of herpes simplex virus type I in the
gene encoding immediate-early regulatory protein ICP4. J. Virol.
56, 558–570.
Dugan L., Sensi S. and Choi D. (1995) Mitochondrial production of
reactive oxygen species in cortical neuron sfollowing exposure to
N-methyl-D-aspartate. J. Neurosci. 15, 6377–6388.
Dumas T., McLaughlin J., Ho D., Lawrence M. and Sapolsky R. (2000)
Gene therapies that enhance hippocampal neuron survival after an
excitotoxic insult are not equivalent in their ability to maintain
synaptic transmission. Exp. Neurol. 166, 180–188.
Ellerby L., Ellerby H., Park S., Hlleran A., Murphy a Fiskum G., Kane
D., Testa M., Kayalar C. and Bredesen D. (1996) Shift of the
cellular oxidation-reduction potential in neural cells expressing
Bcl-2. J. Neurochem. 67, 1259–1266.
Gardner A., Xy F., Fady C., Sarafian T., Tu Y. and Lichtenstein A.
(1997) Evidence against the hypothesis that BCL-2 inhibits
apoptosis thorugh an anti-oxidant effect. Cell Death Different. 4,
487–494.
Giardino I., Edelstein D. and Brownlee M. (1996) BCL-2 expression or
antioxidants prevent hyperglycemia-induced formation of intra-
cellular advanced glycation endproducts in bovine endothelial
cells. J. Clin. Invest. 97, 1422–1428.
Green D. and Reed J. (1998) Mitochondria and apoptosis. Science 281,
1309–1313.
Ho D. Y. (1994) Amplicon-based herpes simplex virus vectors. Meth.
Cell Biol. 43, 191–210.
Ho D. Y., Fink S., Lawrence M., Meier T., Saydam T., Dash R. and
Sapolsky R. (1995) Herpes simplex virus vector system: analysis
of its in vivo and in vitro cytopathic effects. J. Neurosci. Meth. 57,
205–216.
Hochman A., Sternin H., Gorodin S., Korsmeyer S., Ziv I., Melamed E.
and Offen D. (1998) Enhanced oxidative stress and altered anti-
oxidants in brains of Bcl-2-deficient mice. J. Neurochem. 71, 741–
748.
Hockenberry D., Oltvai Z., Yin X., Milliman C. and Korsmeyer S.
(1993) Bcl-2 functions in an antioxidant pathway to prevent
apoptosis. Cell 75, 241–248.
Jacobson M. and Raff M. (1995) Programmed cell death and Bcl-2
protection in very low oxygen. Nature 374, 814–816.
Jia W., Wang Y., Qiang D., Tufaro F., Remington R. and Cynader M.
(1996) A bcl-2 expressing viral vector protects cortical neurons
from excitotoxictiy even when administered several hours after the
toxic insult. Mol Brain Res. 42, 350–356.
Kane D., Sarafian T., Anton R., Hahn H., Gralla E., Valentine J., Ord
T. and Bredesen D. (1993) Bcl-2 inhibition of neural death:
decreased generation of reactive oxygen species. Science 262,
1274–1277.
Kuypers F., van den Berg J., Schalwijk B., Roelofsen B. and Op den
Kamp J. (1987) Parinaric acid as a sensitive fluorescent probe for
the determination of lipid peroxidation. Biochim. Biophys. Acta
921, 266–274.
Lafon-Cazal M., Pietri S., Culcasi M. and Bockaert J. (1993) NMDA-
dependent superoxide production and neurotoxicity. Nature 364,
535–537.
Lagrange P., Livertoux H., Grassiot M. and Minn A. (1994) Superoxide
anion production during monoelectronic reduction of xenobiotics
by preparations of rat brain cortex, microvessels, and choroid
plexus. Free Rad. Biol. Med. 17, 355–359.
Lawrence M., Ho D., Sun G., Steinberg G. and Sapolsky R. (1996)
Overexpression of bcl-2 with herpes simplex virus vectors protects
CNS neurons against neurological insults in vitro and in vivo.
J. Neurosci. 16, 486–495.
Lawrence M. S., Sun G., Ho D., McIntosh L., Kunis D., McLaughlin J.,
Sapolsky R. and Steinberg G. (1997) Herpes simplex viral vectors
expressing Bcl-2 are neuroprotective when delivered following a
stroke. J. Cereb. Blood Flow Metab. 17, 740–747.
Lebel C., Ali S., McKee M. and Bondy S. (1990) Organometal-induced
incrase in oxygen reactive species: the potential of dichlorofluo-
rescein diacitate as an index of neurotoxic damage. Toxicol. Appl.
Pharm. 104, 17–25.
Lee M., Hun D., Marshall K., Ellerby L., Bredesen D., Jenner P. and
Halliwell B. (2001) Effect of overexpression of bcl-2 on cellular
oxidative damage, nitric oxide production, antioxidant defenses,
and the proteasome. Free Rad. Biol. Med. 31, 1550–1558.
Lezoualc’h F., Rupprecht R., Holsboer F. and Behl C. (1996) Bcl-2
prevents hippocampal cell death induced by the neuroleptic drug
haloperidol. Brain Res. 738, 176–181.
Lin Y. and Phillis J. (1991) Oxypurinol reduces focal ischemic brain
injury in the rat. Neurosci. Lett. 126, 187–190.
Linnik M., Zahos P., Geschwind M. and Federoff H. (1995) Expression
of bcl-2 from a defective herpes simplex virus-1 vector limits
neuronal death in focal cerebral ischemia. Stroke 26, 1670–1677/.
Liu Y., Rosenthal R., Haywood Y., Miljkovic-Lolic M., Vanderhoek J.
and Fiskum G. (1998) Normoxic ventilation ofollowing cardiac
922 S. Howard et al.
� 2002 International Society for Neurochemistry, Journal of Neurochemistry, 83, 914–923

arrest reduces oxidation of brain lipids and improves neurological
outcomes. Stroke 29, 1679–1686.
Luc Cadet J., Harrington B. and Ordonez S. (2000) Bcl-2 overexpression
attenuates dopamine-induced apoptosis in an immortalized neural
cell line by suppressing the production of reactive oxygen species.
Synapse 35, 228–235.
McLaughlin J., Roozendaal B., Gupta A., Ajilore O., Dumas T., Hsieh J.,
Ho D., Lawrence M., McGaugh J. and Sapolsky R. (2000) Sparing
neuronal function post-seizure with gene therapy. Proc. Natl Acad.
Sci. USA 97, 12804–12809.
Marin M., Fernandez A., Bick R., Brisbay S., Buja L., Snuggs M.,
McConkey D., Eschenbach A., Keating J. and McDonnell T.
(1996) Apoptosis suppression by bcl-2 is correlated with the
regulation of nuclear and cytosolic calcium. Oncogene 12, 2259–
2266.
Monyer H., Hartley D. and Choi D. (1990) 21-Aminosteroids attenuate
excitotoxic neuronal injury in cortical cell cultures. Neuron 5, 121–
128.
Murphy A., Bredesen D., Cortopassi G., Wang E. and Fiskum G. (1996)
Bcl-2 potentiates the maximal calcium uptake capacity of neural
cell mitochondria. Proc. Natl Acad. Sci. USA 93, 9893–9898.
Myers K., Fiskum G., Liu Y., Simmens S., Bredesen D. and Murphy A.
(1995) Bcl-2 protects cells from cyanide/aglycemia-induced lipid
oxidation, mitochondrial injury, and loss of viability. J. Neuro-
chem. 65, 2432–2439.
Oh J., Choi W., Kim J., Seo J., O’Malley K. and Oh Y. (1998)
Overexpression onf HA-Bax but not bcl-2 or bcl-XL attenuates
6-OHDA-induced neuronal apoptosisl. Exp. Neurol. 154, 193–
200.
Oh Y., Wong S., Moffat M. and O’Malley K. (1995) Overexpresion of
bcl-2 attenuates MPP+, but not 6-OHDA, induced cell death in a
dopaminergic neuronal cell line. Neurobiol. Dis. 2, 157–164.
Papadopoulos M., Koumenis I. and Xu 1 Giffard R. (1998) Potentiation
of murine astrocyte antioxidant defence by bcl-2: protection in part
reflects elevated glutathione levels. Eur. J. Neurosci. 10, 1252–
1258.
Patel R., McIntosh L., McLaughlin J., Brooke S., Nimon V. and
Sapolsky R. (2002) Disruptive effects of glucocorticoids on
glutathione peroxidase biochemistry in hippocampal cultures.
J. Neurochem. 82, 118–125.
Phillips R., Lawrence M., Ho D. and Sapolsky R. (2000) Limitations in
the neuroprotective potential of gene therapy with Bcl-2. Brain
Res. 859, 202–209.
Rosse T., Olivier R., Monney L., Rager M., Conus S., Fellay I., Jansen
B. and Borner C. (1998) Bcl-2 prolongs cell survival after Bax-
induced release of cytochrome c. Nature 391, 496–499.
Saybasili H., Yuksel M., Haklar G. and Yalcin A. (2001) Effect of
mitochondrial electron transport chain inhibitors on superoxide
radical generation in rat hippocampal and striatal slices. Antioxi-
dants Redox Signaling 3, 1099–1104.
Sensi S., Yin H., Carriedo S., Rao S. and Weiss J. (1999) Preferential
Zn2+ influx through Ca2+-permeable AMPA/kainite channels trig-
gers prolonged mitochondrial superoxide production. Proc. Natl
Acad. Sci. USA 96, 2414–2419.
Shimazaki K., Uirabe M., Monahan J., Ozawa K. and Kawai N. (2000)
AAV vector-mediated bcl-2 gene tranfser into post-ischemic gerbil
brain in vivo: prospects for gene therapy of ischemia-induced
neuronal death. Gene Ther. 7, 1244–1251.
Shimizu S., Eguchi Y., Kosaka H., Kamiike W., Matsuda H. and Tsu-
jimoto Y. (1995) Prevention of hypoxia-induced cell death by
Bcl-2 and Bcl-Xl. Nature 374, 811–814.
Steinman H. (1995) The Bcl-2 oncoprotein functions as a pro-oxidant.
J. Biol. Chem. 270, 3487–3493.
Tamatani M., Mitsuda N., Matsuzaki H., Okado H., Miyake S., Vitek M.,
Yamaguchi A. and Tohyama M. (2000) A pathway of neuronal
apoptosis induced by hypoxia-reoxygenatrion: roles of NF-kB and
Bcl-2. J. Neurochem. 75, 683–690.
Traystman R., Kirsch J. and Koehler R. (1991) Oxygen radical mecha-
nisms of brain injury following ischemia and reperfusion. J. Appl.
Physiol. 71, 1185–1195.
Voehringer D., McConkey D., McDonnell T., Brisbay S. and Meyn R.
(1998) Bcl-2 expression causes redistribution of glutathione to the
nucleus. Proc. Natl Acad. Sci. USA 95, 2956–2961.
Wang H., Rapp U. and Reed J. (1996) Bcl-2 targets the protein kinase
Raf-1 to mitochondria. Cell 87, 629–636.
Xu L., Lee A. and Giffard R. (1999) Overexpression of bcl-2, bcl-x or
hsp70 in murine cortical astrocytes reduces injury of co-cultured
neurons. Neurosci. Lett. 277, 193–197.
Yamada M., Oligino T., Mata M., Goss J. R., Glorioso J. C. and Fink D.
J. (1999) HSV vector-mediated expression of Bcl-2 prevents
6-OHDA induced degeneration of neurons in the substantia nigra
in vivo. Proc. Natl Acad. Sci. USA 96, 4078–4083.
Yang L., Matthews R., Schulz J., Klockgether T., Liao A., Martinou J.,
Penney J., Hyman B. and Beal M. (1998) 1-Methyl-4-phenyl-
1,2,3,6-tetrahydropyride neurotoxicity is attenuated in mice over-
expressing Bcl-2. J. Neurosci. 18, 8145–8152.
Neuroprotective effects of bcl-2 overexpression 923
� 2002 International Society for Neurochemistry, Journal of Neurochemistry, 83, 914–923





























