Embed Size (px)
Citation preview

HIGHLIGHT
Angew. Chem. 2002, 114, Nr. 8 ¹ WILEY-VCH Verlag GmbH, 69451 Weinheim, Germany, 2002 0044-8249/02/11408-1391 $ 20.00+.50/0 1391
Neue Methoden f¸r das Hochdurchsatz-Screening von enantioselektivenKatalysatoren und Biokatalysatoren
Manfred T. Reetz*
Die Bedeutung der asymmetrischen Katalyse ist unum-stritten, nicht zuletzt belegt durch die Verleihung des Nobel-preises f¸r Chemie 2001 an K. Barry Sharpless, Ryoji Noyoriund William S. Knowles. Grunds‰tzlich stehen den Chemi-kern dabei zwei Mˆglichkeiten zur Verf¸gung: die Nutzungvon ‹bergangsmetallkatalysatoren in homogener Lˆsung[1]
und Biokatalysatoren.[2] Im ersteren Fall ist der Schl¸ssel zumErfolg das auf Intuition basierende πLiganden-Tuning™,Molecular Modeling und bis zu einem gewissen Grad Versuchund Irrtum.
In den 1990er Jahren wurde eine neue Technik entwickelt,die, etwas salopp, kombinatorische asymmetrische Katalysegenannt wurde.[3] Diese umfasst die zeitsparende paralleleSynthese und das Testen einer hohen Anzahl von chiralenKatalysatoren. Die Herausforderungen in diesem interessan-ten neuen Feld der asymmetrischen Katalyse erstrecken sichauf zwei Gebiete: zum einen auf Strategien zur modularenSynthese von chiralen Liganden, zum anderen auf dieEntwicklung von Hochdurchsatz-Assays zur Bestimmungdes Enantiomeren¸berschusses (ee).[4] Bislang war die Grˆ˚eder Katalysatorbibliotheken normalerweise auf weniger als100 Katalysatoren beschr‰nkt, die fast immer auf konventio-nelle Weise analysiert wurden.
Das Potential der kombinatorischen asymmetrischen Ka-talyse wurde also nur unvollst‰ndig genutzt. Eine Erhˆhungdes Erfolges bei der Identifizierung von Treffern (Hits) istdurch Vergrˆ˚erung der Bibliotheken wahrscheinlich. ImFalle der Enzymkatalyse wurde die Idee der Anwendungmolekularbiologischer Methoden der gerichteten Evolutionzur Erzeugung enantioselektiver Enzymmutanten (Varian-ten) k¸rzlich in die Praxis umgesetzt.[5, 6] Das zugrundeliegende Konzept geht weit ¸ber das der kombinatorischenKatalyse hinaus, da es auf wiederholten Zyklen von Muta-genese und Screening beruht, durch die ein evolution‰rerDruck erzeugt wird. Die Kenntnis der Struktur oder desMechanismus des Enzyms ist dabei nicht erforderlich. DieGrˆ˚e jeder durch Anwendung von fehlerhafter PCR oderDNA-Shuffling erzeugten Bibliothek von Enzymmutanten
bel‰uft sich auf mehrere tausend Komponenten. Dies wirftein schwieriges analytisches Problem auf, wenn von jederKomponente der Bibliotheken die Enantioselektivit‰t ineiner bestimmten Reaktion ermittelt werden soll.
Es ist offensichtlich, dass beide Zug‰nge zu asymmetri-schen Katalysatoren Hochdurchsatz-ee-Screeningsysteme er-fordern. Der erste f¸r die Handhabung von akzeptabel gro˚enEnzymbibliotheken entwickelte ee-Assay war ein relativgrobes, auf UV/Vis-Spektroskopie basierendes Screening-system zur Erfassung einer Lipase-katalysierten kinetischenRacematspaltung eines chiralen p-Nitrophenolesters. Die(R)- und (S)-Ester werden getrennt voneinander paarweisein einer 96er-Mikrotiterplatte unter Verwendung eines ein-fachen UV/Vis-Leseger‰ts getestet.[5a]
Das Konzept, die (R)- und (S)-Enantiomere bei einerkinetischen Racematspaltung getrennt zu vermessen, hateinige Nachteile.[4] Dennoch basieren andere Tests ebenfallsauf diesem Konzept,[4] darunter eine erst vor kurzem entwi-ckelte Methode, die gekoppelte enzymatische Umsetzungennutzt.[7] Mehrere allgemeinere Screeningsysteme sind erson-nen worden. Dazu z‰hlen Methoden, die auf der massen-spektrometrischen Analyse von Deuterium-markierten Sub-straten basieren und 1000 ee-Bestimmungen pro Tag erlauben(dies ist mittlerweile durch den Gebrauch eines Acht-Kanal-Multiplex-Einspritzsystems auf 10000 Bestimmungen erwei-tert worden),[8] sowie die Verwendung von Kapillarelektro-phorese (bis zu 30000 ee-Bestimmungen pro Tag)[9] undHPLC/Circulardichroismus(CD)-Spektroskopie (typischer-weise 1000 Proben pro Tag).[10]
Diese und andere Methoden wurden im vergangenen Jahrin mehreren ‹bersichtsartikeln behandelt.[4] Da aber keineinziger dieser Assays universell anwendbar ist, sind weitereBem¸hungen in diesem faszinierenden Forschungsbereichnotwendig. Feringa et al. berichteten k¸rzlich ¸ber einenraffinierten Farbtest f¸r Enantioselektivit‰t, der auf derchiralit‰tsabh‰ngigen Farberzeugung in dotierten Fl¸ssigkris-tallfilmen beruht.[11] Es bleibt abzuwarten, ob dieses inte-ressante System zu einem Hochdurchsatz-ee-Assay modifi-ziert werden kann.
K¸rzlich wurden mehrere neue Mˆglichkeiten zum Hoch-durchsatz-Screening von enantioselektiven Katalysatorenbeschrieben. So beschrieben Shair et al. eine neue Technik,die DNA-Mikroarrays nutzt.[12][19] Diese neue Technik wurdefr¸her zur relativen Bestimmung von genomweiten Genex-
[*] Prof. Dr. M. T. ReetzMax-Planck-Institut f¸r KohlenforschungKaiser-Wilhelm-Platz 1, 45470 M¸lheim an der Ruhr (Deutschland)Fax: (�49)208-306-2985E-mail : [email protected]

HIGHLIGHT
1392 ¹ WILEY-VCH Verlag GmbH, 69451 Weinheim, Germany, 2002 0044-8249/02/11408-1392 $ 20.00+.50/0 Angew. Chem. 2002, 114, Nr. 8
pressionsraten genutzt, wobei das Verh‰ltnis von Fluoreszenz-reportern betrachtet wurde.[13] Das Ziel in dem neuentwi-ckelten ee-Assay war die Bestimmung der Enantiomeren-reinheit von chiralen Aminos‰uren.[12] Solche (R)/(S)-Mi-schungen kˆnnen z.B. durch Rh-katalysierte Hydrierung derkorrespondierenden N-Acylaminoacrylate erhalten werden,wobei eine Bibliothek von kombinatorisch hergestelltenKatalysatoren verwendet wird. In einer Modellstudie wurdenMischungen von (R)- und (S)-Aminos‰uren zun‰chst an derAminofunktion zu den Boc-gesch¸tzten Derivaten acyliert(Boc� tert-Butoxycarbonyl). Die Proben wurden anschlie-˚end durch kovalente Verkn¸pfung in r‰umlich definierterAnordnung auf aminfunktionalisierte Glastr‰ger aufgebracht(Abbildung 1). In einem zweiten Schritt wurden die freienAminofunktionen der Oberfl‰che erschˆpfend acyliert. Derdritte Schritt umfasste die Entsch¸tzung der Aminos‰uren,
um die freie Aminofunktion der Aminos‰ure zu erhalten.Schlie˚lich wurden in einem vierten Schritt zwei pseudoe-nantiomere Fluoreszenzsonden an die freien Aminofunk-tionen auf der Arrayoberfl‰che gebunden.
Voraussetzung f¸r den Erfolg dieses ee-Assays ist einemerkliche paralle kinetische Racematspaltung beim Prozessder Amidkupplung,[12] ‰hnlich wie bei einem auf Massen-spektrometrie basierenden System, das k¸rzlich von Finnet al. entwickelt wurde.[14] In dem hier vorliegenden Fall sinddie ee-Werte durch Messung des Verh‰ltnisses der relevantenFluoreszenzintensit‰ten zug‰nglich. 8000 ee-Bestimmungen proTag sind mˆglich bei einer Genauigkeit von �10%. Es bleibtabzuwarten, ob die DNA-Mikroarraytechnik in einer analogenWeise f¸r andere Substrattypen modifiziert werden kann.
Seto et al. beschrieben eine enzymatische Methode zurBestimmung von Enantiomeren¸bersch¸ssen, die sie als
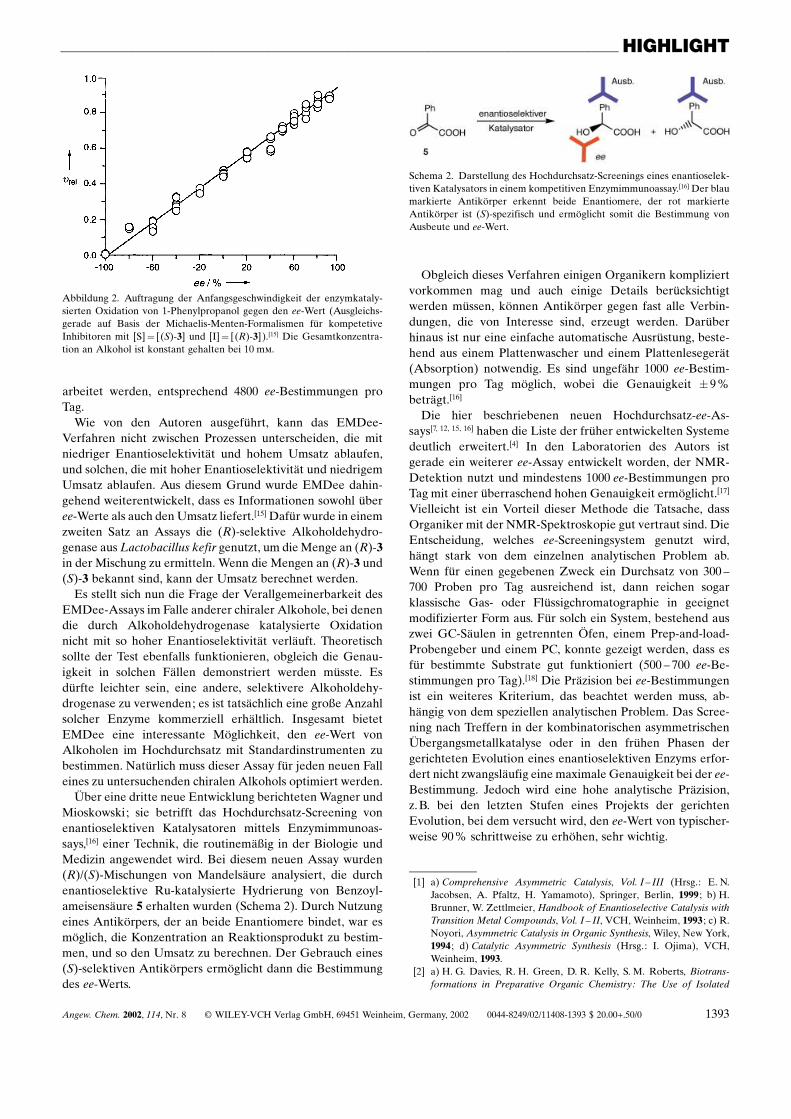
(EMDee) (πenzymatic method for determiningee™) bezeichneten.[15,20] Sie basiert darauf, dass eingeeignetes Enzym dazu genutzt werden kann, einEnantiomer des Produkts einer katalytischen Re-aktion selektiv umzusetzen. Die vielfach unter-suchte katalytische Addition von Diethylzink 2 anBenzaldehyd 1 wurde als Testsystem zur Demonst-ration der Leistungsf‰higkeit von EMDee ausge-w‰hlt. Das Reaktionsprodukt, 1-Phenylpropanol3, kann durch Verwendung der Alkoholde-hydrogenase aus Thermoanaerobium sp. zu Ethyl-phenylketon 4 oxidiert werden, wobei dieser Pro-zess vollst‰ndig (S)-selektiv ist (Schema 1). DieGeschwindigkeit dieser enzymatischen Oxidationkonnte durch UV-spektroskopische Verfolgungder Bildung von NADPH bei 340 nm gemessenwerden.
Entscheidend f¸r den Erfolg dieses Assays ist,dass die Geschwindigkeit der Oxidation ein di-rektes Ma˚ f¸r den ee-Wert ist (Abbildung 2).[15]
Der Hochdurchsatz wurde durch die Analyse von100 Proben im 384er-Format mittels eines UV/Fluoreszenz-Plattenleseger‰ts gezeigt. Jede Probeenthielt 1 �mol 1-Phenylpropanol 3 in einemGesamtvolumen von 100 �L. Die Genauigkeitdes ee-Werts bel‰uft sich auf �10%, wie inunabh‰ngigen GC-Untersuchungen ermittelt wur-de. Ungef‰hr 100 Proben kˆnnen in 30 min ver-
Abbildung 1. Reaktionsmikroarrays bei der Hochdurchsatz-ee-Bestimmung. Reagen-tien und Bedingungen:[12] Schritt 1: BocHNCHRCO2H, PyAOP, iPr2NEt, DMF;Schritt 2: Ac2O, Pyridin; Schritt 3: 10% CF3CO2H und 10% Et3SiH in CH2Cl2, dann3% Et3N in CH2Cl2; Schritt 4: Pentafluorphenyldiphenylphosphonit, iPr2NEt, 1:1-Mischung der beiden fluoreszierenden Prolinderivate, DMF, �20 �C. PyAOP� 7-Azabenzotriazol-1-yloxytris(pyrrolidino)phosphoniumhexafluorophosphat.
chiraler
Ph H
OEt2Zn
Ph Et
OH
Ph Et
OH
NADP+ NADPH
Ph Et
O
Ph Et
OH
Katalysator
(S)-3
+ +
(R)-321
Alkohol-dehydrogenase
4
+
(R)-3
Schema 1. Die Reaktionen, auf denen EMDee beruht, imFalle von 1-Phenylpropanol als Substrat f¸r die Alkohol-dehydrogenase.[15]
HIGHLIGHT
Angew. Chem. 2002, 114, Nr. 8 ¹ WILEY-VCH Verlag GmbH, 69451 Weinheim, Germany, 2002 0044-8249/02/11408-1393 $ 20.00+.50/0 1393
Abbildung 2. Auftragung der Anfangsgeschwindigkeit der enzymkataly-sierten Oxidation von 1-Phenylpropanol gegen den ee-Wert (Ausgleichs-gerade auf Basis der Michaelis-Menten-Formalismen f¸r kompetetiveInhibitoren mit [S]� [(S)-3] und [I]� [(R)-3]).[15] Die Gesamtkonzentra-tion an Alkohol ist konstant gehalten bei 10 m�.
arbeitet werden, entsprechend 4800 ee-Bestimmungen proTag.
Wie von den Autoren ausgef¸hrt, kann das EMDee-Verfahren nicht zwischen Prozessen unterscheiden, die mitniedriger Enantioselektivit‰t und hohem Umsatz ablaufen,und solchen, die mit hoher Enantioselektivit‰t und niedrigemUmsatz ablaufen. Aus diesem Grund wurde EMDee dahin-gehend weiterentwickelt, dass es Informationen sowohl ¸beree-Werte als auch den Umsatz liefert.[15] Daf¸r wurde in einemzweiten Satz an Assays die (R)-selektive Alkoholdehydro-genase ausLactobacillus kefir genutzt, um dieMenge an (R)-3in der Mischung zu ermitteln. Wenn die Mengen an (R)-3 und(S)-3 bekannt sind, kann der Umsatz berechnet werden.
Es stellt sich nun die Frage der Verallgemeinerbarkeit desEMDee-Assays im Falle anderer chiraler Alkohole, bei denendie durch Alkoholdehydrogenase katalysierte Oxidationnicht mit so hoher Enantioselektivit‰t verl‰uft. Theoretischsollte der Test ebenfalls funktionieren, obgleich die Genau-igkeit in solchen F‰llen demonstriert werden m¸sste. Esd¸rfte leichter sein, eine andere, selektivere Alkoholdehy-drogenase zu verwenden; es ist tats‰chlich eine gro˚e Anzahlsolcher Enzyme kommerziell erh‰ltlich. Insgesamt bietetEMDee eine interessante Mˆglichkeit, den ee-Wert vonAlkoholen im Hochdurchsatz mit Standardinstrumenten zubestimmen. Nat¸rlich muss dieser Assay f¸r jeden neuen Falleines zu untersuchenden chiralen Alkohols optimiert werden.
‹ber eine dritte neue Entwicklung berichtetenWagner undMioskowski; sie betrifft das Hochdurchsatz-Screening vonenantioselektiven Katalysatoren mittels Enzymimmunoas-says,[16] einer Technik, die routinem‰˚ig in der Biologie undMedizin angewendet wird. Bei diesem neuen Assay wurden(R)/(S)-Mischungen von Mandels‰ure analysiert, die durchenantioselektive Ru-katalysierte Hydrierung von Benzoyl-ameisens‰ure 5 erhalten wurden (Schema 2). Durch Nutzungeines Antikˆrpers, der an beide Enantiomere bindet, war esmˆglich, die Konzentration an Reaktionsprodukt zu bestim-men, und so den Umsatz zu berechnen. Der Gebrauch eines(S)-selektiven Antikˆrpers ermˆglicht dann die Bestimmungdes ee-Werts.
Schema 2. Darstellung des Hochdurchsatz-Screenings eines enantioselek-tiven Katalysators in einem kompetitiven Enzymimmunoassay.[16] Der blaumarkierte Antikˆrper erkennt beide Enantiomere, der rot markierteAntikˆrper ist (S)-spezifisch und ermˆglicht somit die Bestimmung vonAusbeute und ee-Wert.
Obgleich dieses Verfahren einigen Organikern kompliziertvorkommen mag und auch einige Details ber¸cksichtigtwerden m¸ssen, kˆnnen Antikˆrper gegen fast alle Verbin-dungen, die von Interesse sind, erzeugt werden. Dar¸berhinaus ist nur eine einfache automatische Ausr¸stung, beste-hend aus einem Plattenwascher und einem Plattenleseger‰t(Absorption) notwendig. Es sind ungef‰hr 1000 ee-Bestim-mungen pro Tag mˆglich, wobei die Genauigkeit �9%betr‰gt.[16]
Die hier beschriebenen neuen Hochdurchsatz-ee-As-says[7, 12, 15, 16] haben die Liste der fr¸her entwickelten Systemedeutlich erweitert.[4] In den Laboratorien des Autors istgerade ein weiterer ee-Assay entwickelt worden, der NMR-Detektion nutzt und mindestens 1000 ee-Bestimmungen proTag mit einer ¸berraschend hohen Genauigkeit ermˆglicht.[17]
Vielleicht ist ein Vorteil dieser Methode die Tatsache, dassOrganiker mit der NMR-Spektroskopie gut vertraut sind. DieEntscheidung, welches ee-Screeningsystem genutzt wird,h‰ngt stark von dem einzelnen analytischen Problem ab.Wenn f¸r einen gegebenen Zweck ein Durchsatz von 300 ±700 Proben pro Tag ausreichend ist, dann reichen sogarklassische Gas- oder Fl¸ssigchromatographie in geeignetmodifizierter Form aus. F¸r solch ein System, bestehend auszwei GC-S‰ulen in getrennten ÷fen, einem Prep-and-load-Probengeber und einem PC, konnte gezeigt werden, dass esf¸r bestimmte Substrate gut funktioniert (500 ± 700 ee-Be-stimmungen pro Tag).[18] Die Pr‰zision bei ee-Bestimmungenist ein weiteres Kriterium, das beachtet werden muss, ab-h‰ngig von dem speziellen analytischen Problem. Das Scree-ning nach Treffern in der kombinatorischen asymmetrischen‹bergangsmetallkatalyse oder in den fr¸hen Phasen dergerichteten Evolution eines enantioselektiven Enzyms erfor-dert nicht zwangsl‰ufig eine maximale Genauigkeit bei der ee-Bestimmung. Jedoch wird eine hohe analytische Pr‰zision,z.B. bei den letzten Stufen eines Projekts der gerichtenEvolution, bei dem versucht wird, den ee-Wert von typischer-weise 90% schrittweise zu erhˆhen, sehr wichtig.
[1] a) Comprehensive Asymmetric Catalysis, Vol. I ± III (Hrsg.: E. N.Jacobsen, A. Pfaltz, H. Yamamoto), Springer, Berlin, 1999 ; b) H.Brunner, W. Zettlmeier, Handbook of Enantioselective Catalysis withTransition Metal Compounds, Vol. I ± II, VCH, Weinheim, 1993 ; c) R.Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, New York,1994 ; d) Catalytic Asymmetric Synthesis (Hrsg.: I. Ojima), VCH,Weinheim, 1993.
[2] a) H. G. Davies, R. H. Green, D. R. Kelly, S. M. Roberts, Biotrans-formations in Preparative Organic Chemistry: The Use of Isolated

HIGHLIGHT
1394 ¹ WILEY-VCH Verlag GmbH, 69451 Weinheim, Germany, 2002 0044-8249/02/11408-1394 $ 20.00+.50/0 Angew. Chem. 2002, 114, Nr. 8
Enzymes and Whole Cell Systems in Synthesis, Academic Press,London, 1989 ; b) C. H. Wong, G. M. Whitesides,Enzymes in SyntheticOrganic Chemistry, Pergamon, Oxford, 1994 (Tetrahedron OrganicChemistry Series, Vol. 12); c) Enzyme Catalysis in Organic Synthesis:A Comprehensive Handbook, Vol. I ± II (Hrsg.: K. Drauz, H. Wald-mann), VCH, Weinheim, 1995 ; d) K. Faber, Biotransformations inOrganic Chemistry, 3. Aufl. , Springer, Berlin, 1997.
[3] Siehe z.B.: a) M. B. Francis, E. N. Jacobsen, Angew. Chem. 1999, 111,987 ± 991; Angew. Chem. Int. Ed. 1999, 38, 937 ± 941; b) S. R. Gil-bertson, C.-W. T. Chang, Chem. Commun. 1997, 975 ± 976; c) C.Gennari, S. Ceccarelli, U. Piarulli, C. A. G. N. Montalbetti, R. F. W.Jackson, J. Org. Chem. 1998, 63, 5312 ± 5313; d) K. Burgess, H.-J. Lim,A. M. Porte, G. A. Sulikowski, Angew. Chem. 1996, 108, 192 ± 194;Angew. Chem. Int. Ed. Engl. 1996, 35, 220 ± 222; e) C. A. Krueger,K. W. Kuntz, C. D. Dzierba, W. G. Wirschun, J. D. Gleason, M. L.Snapper, A. H. Hoveyda, J. Am. Chem. Soc. 1999, 121, 4284 ± 4285;f) J. Long, J. Hu, X. Shen, B. Ji, K. Ding, J. Am. Chem. Soc. 2002, 124,10 ± 11; g) A. Berkessel, R. Rield, J. Comb. Chem. 2000, 2, 215 ± 219;h) S. Dahmen, S. Br‰se, Synthesis 2001, 1431 ± 1449.
[4] a) ‹bersicht ¸ber kombinatorische und evolutionsgesteuerte Metho-den zur Bildung enantioselektiver Katalysatoren:M. T. Reetz,Angew.Chem. 2001, 113, 292 ± 320; Angew. Chem. Int. Ed. 2001, 40, 284 ± 310;‹bersicht ¸ber Screening von Biokatalysatoren: b) D. Wahler, J.-L.Reymond, Curr. Opin. Chem. Biol. 2001, 5, 152 ± 158; c) F. M.-V.Varas, L. Hartman, A.-Shah, D. C. Demirjian, ACS Symp. Ser. 2001,776, 41 ± 54; d) ‹bersicht ¸ber enantioselektive πDetektoren™ imMiniaturformat: T. J. Edkins, D. R. Bobbitt, Anal. Chem. 2001, 73,488A-496A.
[5] a) M. T. Reetz, A. Zonta, K. Schimossek, K. Liebeton, K.-E. Jaeger,Angew. Chem. 1997, 109, 2961 ± 2963; Angew. Chem. Int. Ed. Engl.1997, 36, 2830 ± 2832; b) M. T. Reetz, S. Wilensek, D. Zha, K.-E.Jaeger, Angew. Chem. 2001, 113, 3701 ± 3703; Angew. Chem. Int. Ed.2001, 40, 3589 ± 3591; c) D. Zha, S. Wilensek, M. Hermes, K.-E. Jaeger,M. T. Reetz, Chem. Commun. 2001, 2664 ± 2665.
[6] M. T. Reetz, K.-E. Jaeger, Chem. Eur. J. 2000, 6, 407 ± 412.[7] M. Baumann, R. St¸rmer, U. T. Bornscheuer, Angew. Chem. 2001,
113, 4329 ± 4333; Angew. Chem. Int. Ed. 2001, 40, 4201 ± 4204.
[8] M. T. Reetz, M. H. Becker, H.-W. Klein, D. Stˆckigt, Angew. Chem.1999, 111, 1872 ± 1875; Angew. Chem. Int. Ed. 1999, 38, 1758 ± 1761.
[9] M. T. Reetz, K. M. K¸hling, A. Deege, H. Hinrichs, D. Belder,Angew.Chem. 2000, 112, 4049 ± 4052; Angew. Chem. Int. Ed. 2000, 39, 3891 ±3893.
[10] a) K. Ding, A. Ishii, K. Mikami, Angew. Chem. 1999, 111, 519 ± 523;Angew. Chem. Int. Ed. 1999, 38, 497 ± 501; b) M. T. Reetz, K. M.K¸hling, H. Hinrichs, A. Deege, Chirality 2000, 12, 479 ± 482; c) K.Mikami, R. Angelaud, K. Ding, A. Ishii, A. Tanaka, N. Sawada, K.Kudo, M. Senda, Chem. Eur. J. 2001, 7, 730 ± 737; d) T. Hattori, Y.Minato, S. Yao, M. G. Finn, S. Miyano, Tetrahedron Lett. 2001, 42,8015 ± 8018.
[11] R. A. van Delden, B. L. Feringa,Angew. Chem. 2001, 113, 3298 ± 3300;Angew. Chem. Int. Ed. 2001, 40, 3198 ± 3200.
[12] G. A. Korbel, G. Lalic, M. D. Shair, J. Am. Chem. Soc. 2001, 123, 361 ±362.
[13] B. Phimister, Nat. Genet. 1999, 21, 1 (Suppl.).[14] J. Guo, J. Wu, G. Siuzdak, M. G. Finn, Angew. Chem. 1999, 111, 1868 ±
1871; Angew. Chem. Int. Ed. 1999, 38, 1755 ± 1758.[15] P. Abato, C. T. Seto, J. Am. Chem. Soc. 2001, 123, 9206 ± 9207.[16] F. Taran, C. Gauchet, B. Mohar, S. Meunier, A. Valleix, P. Y. Renard,
C. Cre¬minon, J. Grassi, A. Wagner, C. Miokowski, Angew. Chem.2002, 114, 132 ± 135; Angew. Chem. Int. Ed. 2002, 41, 124 ± 127.
[17] M. T. Reetz, A. Eipper, R. Mynott, P. Tielmann, unverˆffentlichteErgebnisse.
[18] M. T. Reetz, K. M. K¸hling, S. Wilensek, H. Husmann, U. W. H‰usig,M. Hermes, Catal. Today 2001, 67, 389 ± 396.
[19] Ein Aufsatz zum Thema πDNA-Mikroarrays™ findet sich in diesemHeft: M. C. Pirrung, Angew. Chem. 2002, 114, 1326 ± 1341; Angew.Chem. Int. Ed. 2002, 41, 1276 ± 1289.
[20] Anmerkung in der Druckfahne: Berkowitz et al. beschrieben in einerunabh‰ngigen Arbeit ein enzymatisches In-situ-Screeningsystem(ISES), das dem EMDee-Ansatz von Sato et al. ‰hnlich ist, alsHilfsmittel zur Katalysatorentdeckung. Enantioselektivit‰t wurdehierbei allerdings (noch) nicht ber¸cksichtigt: D. B. Berkowitz, M.Bose, S. Choi,Angew. Chem. 2002, 114, Heft 9;Angew. Chem. Int. Ed.2002, 41, issue 9.



























