Embed Size (px)
Citation preview

A Quick Guide to the
MutationR553X
C F T R S C I E N C E
© 2016 Vertex Pharmaceuticals Incorporated | VXR-HQ-02-00045a(1) | 03/2016

Loss of CFTR activity is the underlying cause of cystic fibrosis (CF)1
• People with 2 CFTR mutations resulting in loss of CFTR activity generally have a CF phenotype, which may include1-3,6
– Elevated sweat chloride (>60 mmol/L)
– Pancreatic insufficiency
– CBAVD a
– Lung function decline over time
– Pseudomonas aeruginosa colonization
aCBAVD, congenital bilateral absence of the vas deferens.
References: 1. Davis PB et al. Am J Respir Crit Care Med. 1996;154(5):1229-1256. 2. Rowe SM et al. Proc Am Thorac Soc. 2007;4(4):387-398. 3. Zielenski J. Respiration. 2000;67(2):117-133. 4. Sheppard DN et al. Nature. 1993;362(6416):160-164. 5. Welsh MJ, Smith AE. Cell. 1993;73(7): 1251-1254. 6. Castellani C et al. J Cyst Fibros. 2008;7(3):179-196.
Some CFTR mutations result in little to no CFTR activity3-5
0% 100%
Total CFTR Activity
0% 100%
Total CFTR Activity
Some CFTR mutations result in residual or partial CFTR activity3-5
Spectrum of Phenotypes Associated With Total CFTR Activity1,2
Total CFTR Activity
% of Normal
No CF Disease
CFTR-relatedDisorders
Cystic Fibrosis
Individuals with 2 normal alleles and CFTR mutation carriers
Clinical entities associated with CFTR dysfunctionthat do not fulfill diagnostic criteria for CF, e.g., CBAVD, acute recurrent or chronic pancreatitis and bronchiectasis
Typical phenotype: sinopulmonary disease, sweatchloride >60 mmol/L, pancreatic insufficiency, appearing early in life. In some patients, progressionof disease is delayed or not all features are present
100%
0%
Clinical Phenotype
2
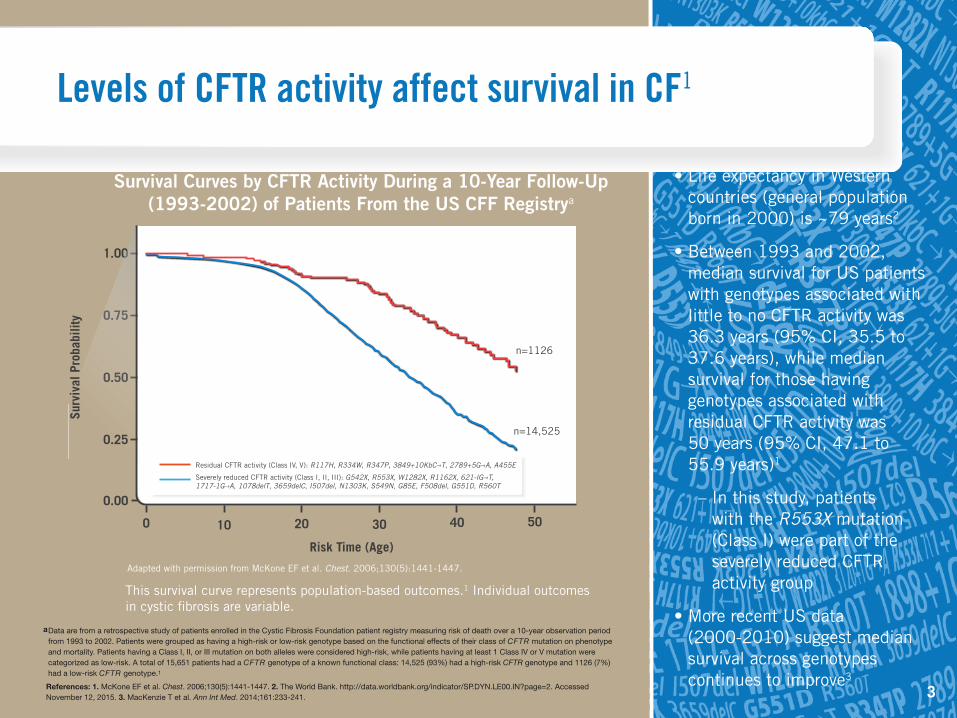
Levels of CFTR activity affect survival in CF1
References: 1. McKone EF et al. Chest. 2006;130(5):1441-1447. 2. The World Bank. http://data.worldbank.org/indicator/SP.DYN.LE00.IN?page=2. Accessed November 12, 2015. 3. MacKenzie T et al. Ann Int Med. 2014;161:233-241.
1.00
0.75
0.50
0.25
0.00
0 10 20
Risk Time (Age)
Surv
ival
Pro
babi
lity
30 40 50
Residual CFTR activity (Class IV, V): R117H, R334W, R347P, 3849+10KbC–›T, 2789+5G–›A, A455E
Severely reduced CFTR activity (Class I, II, III): G542X, R553X, W1282X, R1162X, 621-IG–›T, 1717-1G–›A, 1078delT, 3659delC, I507del, N1303K, S549N, G85E, F508del, G551D, R560T
n=1126
n=14,525
• Life expectancy in Western countries (general population born in 2000) is ~79 years2
• Between 1993 and 2002, median survival for US patients with genotypes associated with little to no CFTR activity was 36.3 years (95% CI, 35.5 to 37.6 years), while median survival for those having genotypes associated with residual CFTR activity was 50 years (95% CI, 47.1 to 55.9 years)1
– In this study, patients with the R553X mutation (Class I) were part of the severely reduced CFTR activity group
• More recent US data (2000-2010) suggest median survival across genotypes continues to improve3
Survival Curves by CFTR Activity During a 10-Year Follow-Up (1993-2002) of Patients From the US CFF Registrya
a Data are from a retrospective study of patients enrolled in the Cystic Fibrosis Foundation patient registry measuring risk of death over a 10-year observation period from 1993 to 2002. Patients were grouped as having a high-risk or low-risk genotype based on the functional effects of their class of CFTR mutation on phenotype and mortality. Patients having a Class I, II, or III mutation on both alleles were considered high-risk, while patients having at least 1 Class IV or V mutation were categorized as low-risk. A total of 15,651 patients had a CFTR genotype of a known functional class: 14,525 (93%) had a high-risk CFTR genotype and 1126 (7%) had a low-risk CFTR genotype.1
This survival curve represents population-based outcomes.1 Individual outcomes in cystic fibrosis are variable.
Adapted with permission from McKone EF et al. Chest. 2006;130(5):1441-1447.
3

Aus8: 0.7%
US1: 2%
Brazil2: 0.2%
Country registries listing the R553X mutation report 0.2% to 2% prevalence among patients with CF1-8
Prevalence of the R553X Mutation in Patients With Cystic Fibrosis (% of Patients With at Least 1 Allele)
References: 1. Cystic Fibrosis Foundation. Cystic Fibrosis Foundation Patient Registry 2013 Annual Data Report. Bethesda, MD. © 2014; Cystic Fibrosis Foundation. 2. The Brazilian Cystic Fibrosis Study Group. Brazilian Cystic Fibrosis Patient Registry. 2012 Annual Report. 3. Belgisch Mucoviscidose Register – Registre Belge de la Mucoviscidose. The Belgian Cystic Fibrosis Registry. Summary Report 2011. © 2014; Institute of Public Health (WIV-ISP), Brussels, Belgium. 4. Cystic Fibrosis Trust. UK Cystic Fibrosis Registry Annual Data Report 2014. © 2015; Cystic Fibrosis Trust; London, UK. 5. French Cystic Fibrosis Registry. Annual Report 2013. © Vaincre la Mu-coviscidose and Ined, 2015; Paris, France. 6. Mukoviszidose e.V. und Mukoviszidose Institut gemeinnützige Gesselschaft für Forschung und Therapienntwicklung mbH. Beriichtsband Qualitätssicherung Mukoviszidose 2012. © 2013, Bonn, Germany. 7. Nederlandse Cystic Fibrosis Stichting. Dutch Cystic Fibrosis Patient Registry. Report on the Year 2014. © 2015; NCFS: Baarn, Netherlands. 8. Cystic Fibrosis Australia. Australian Cystic Fibrosis Data Registry 2013–16th Annual Report. © 2015; Cystic Fibrosis Australia; Baulkham Hills NSW, Australia. 9. US CF Foundation, Johns Hopkins University, The Hospital for Sick Children. The Clinical and Functional TRanslation of CFTR (CFTR2). http://www.cftr2.org. Accessed November 12, 2015. 10. Bobadilla JL et al. Hum Mutat. 2002;19(2):575-606. 11. European Cystic Fibrosis Society. ECFS Patient Registry 2010 Annual Data Report. 2014. 12. World Health Organization. The molecular genetic epidemiology of cystic fibrosis: report of a joint meeting of WHO/ECFTN/ICF(M)A/ECFS; June 19, 2002; Genoa, Italy. © World Health Organization; 2004.
Europe:Belgium3: 2%UK4: 0.8%France5: 1%Germany6: 2%Netherlands7: 1%
• In the CFTR2 global database, ~1% of patients with CF have at least 1 copy of the R553X mutation9
4%
4%
4%
4%
3%
0.4%
Lithuania10
Russia10
Ukraine10
Slovakia10
Switzerland11
Canada12
Additional sources report frequency of the R553X mutation on CF alleles
Country % of Alleles
4

The R553X mutation results in defective biosynthesis of the CFTR protein1-5
References: 1. Zielenski J. Respiration. 2000;67(2):117-133. 2. Welsh MJ, Smith AE. Cell. 1993;73(7):1251-1254. 3. Rowntree RK, Harris A. Ann Hum Genet. 2003;67(Pt 5):471-485. 4. Hamosh A et al. J Clin Invest. 1991;88(6):1880-1885. 5. Stanke F et al. J Med Genet. 2008;45(1):47-54.
• R553X is a nonsense mutation, which produces a premature stop codon1-4
• The cell cannot synthesize a full-length CFTR protein, a Class I mutation1-4
• As a result, few to no CFTR proteins are present at the apical cell surface1,2,5
CFTR Processing
CFTR Synthesis
CFTR Trafficking
Endoplasmicreticulum
Proteosome
Golgicomplex
DNA
mRNA
Transcription
T
T
T
T
T
T
A
A
A
G
C
C
C
C
C
U
U
U
A
A
A
A
A
A
G
G
G
G
G
C
Startcodon
Prematurestop codon
mRNA strand
Rest ofmRNA strand
Rest ofCFTRgene
CFTR gene(DNA)
CFTR Turnover
CFTR Function
Illustrative Example of Class I Defect
5

The R553X allele results in little to no total CFTR activity1-6
Defective Synthesis (Class I)
R553X allele results in few to no CFTR channels at
apical surface
Channel-open Probability: N/A Conductance: N/A
Little to NoR553X-CFTR
Activity
A virtual absence of R553X-CFTR protein quantity…
…regardless of function since few to no CFTR proteins reach the surface…
…results in little to no total CFTR activity
References: 1. Zielenski J. Respiration. 2000;67(2):117-133. 2. Welsh MJ, Smith AE. Cell. 1993;73(7):1251-1254. 3. Rowntree RK, Harris A. Ann Hum Genet. 2003;67(Pt 5):471-485. 4. Hamosh A et al. J Clin Invest. 1991;88(6):1880-1885. 5. Stanke F et al. J Med Genet. 2008;45(1):47-54. 6. Sheppard DN et al. Nature. 1993;362(6416):160-164.
1
1
2
2
3
3
Total CFTR activity can be defined as total ion transport mediated by CFTR protein channels at the cell surface, depending on CFTR protein quantity and function.6
x x =
Channel-openProbability
Conductance
CFTR FunctionTotal
CFTR ActivityxCFTR Quantity x =
N/A, not applicable.
6
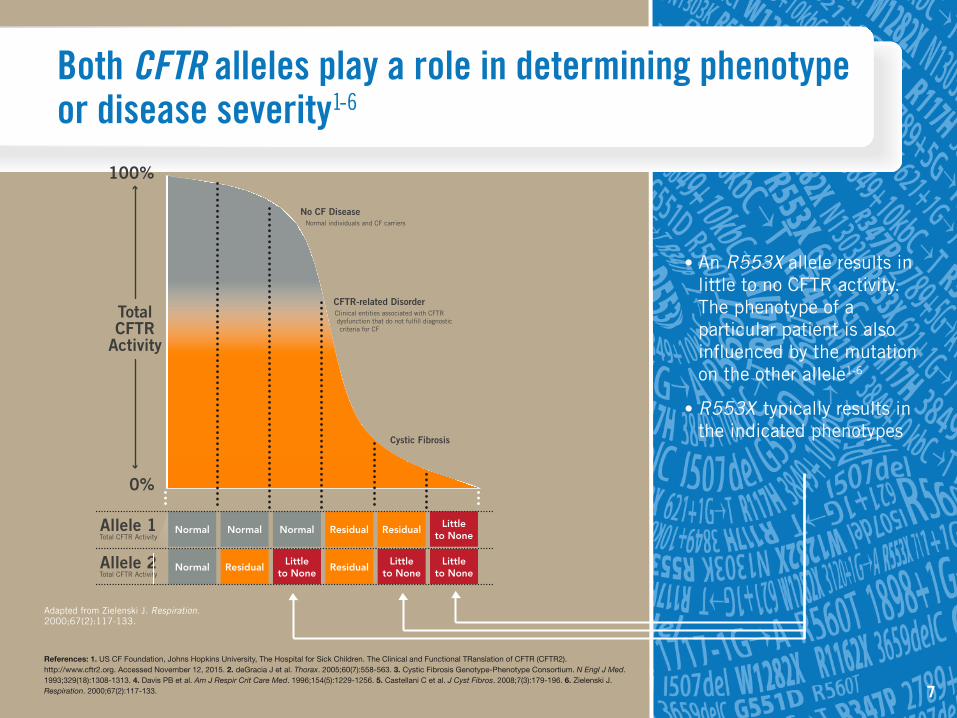
Both CFTR alleles play a role in determining phenotype or disease severity1-6
• An R553X allele results in little to no CFTR activity. The phenotype of a particular patient is also influenced by the mutation on the other allele1-6
• R553X typically results in the indicated phenotypes
Adapted from Zielenski J. Respiration. 2000;67(2):117-133.
References: 1. US CF Foundation, Johns Hopkins University, The Hospital for Sick Children. The Clinical and Functional TRanslation of CFTR (CFTR2). http://www.cftr2.org. Accessed November 12, 2015. 2. deGracia J et al. Thorax. 2005;60(7):558-563. 3. Cystic Fibrosis Genotype-Phenotype Consortium. N Engl J Med. 1993;329(18):1308-1313. 4. Davis PB et al. Am J Respir Crit Care Med. 1996;154(5):1229-1256. 5. Castellani C et al. J Cyst Fibros. 2008;7(3):179-196. 6. Zielenski J. Respiration. 2000;67(2):117-133.
No CF DiseaseNormal individuals and CF carriers
Cystic Fibrosis
CFTR-related DisorderClinical entities associated with CFTR dysfunction that do not fulfill diagnostic criteria for CF
Normal Normal Normal
Normal Residual
Residual Residual
Residual
Littleto None
Littleto None
Littleto None
Littleto None
Allele 1Total CFTR Activity
Allele 2Total CFTR Activity
TotalCFTR
Activity
0%
100%
7

R553X in combination with another allele that produces little to no CFTR activity usually results in a CF phenotype1-5
References: 1. US CF Foundation, Johns Hopkins University, The Hospital for Sick Children. The Clinical and Functional TRanslation of CFTR (CFTR2). http://www.cftr2.org. Accessed November 12, 2015. 2. deGracia J et al. Thorax. 2005;60(7):558-563. 3. Cystic Fibrosis Genotype-Phenotype Consortium. N Engl J Med. 1993;329(18):1308-1313. 4. Davis PB et al. Am J Respir Crit Care Med. 1996;154(5):1229-1256. 5. Castellani C et al. J Cyst Fibros. 2008;7(3):179-196. 6. Cleveland RH et al. Radiology. 2009;253(3):813-821.
CF Phenotype In patients registered in the CFTR2 database with an R553X mutation on 1 allele and a pancreatic insufficient mutation on the second allele1
• Elevated sweat chloride (average):103 mmol/L
• Lung function decline over time6
• Pseudomonas colonization: 59% of patients
• Pancreatic insufficiency: 97% of patients
Little to No Total CFTR Activity
EnvironmentalFactors
Modifier Genes
Little to NoCFTR Protein
Activity
Allele #1: R553X
Little to NoCFTR Protein
Activity
Allele #2
CFTR Genotype
8

Summary• Loss of CFTR activity is the underlying cause of CF
• Levels of CFTR activity affect survival in CF
• Country registries listing the R553X mutation report 0.2% to 2% prevalence among patients with CF
• The R553X mutation results in defective biosynthesis of the CFTR protein
• The R553X allele results in little to no total CFTR activity
• Both CFTR alleles play a role in determining phenotype or disease severity
• R553X in combination with another allele that produces little to no CFTR activity usually results in a CF phenotype
9















![Cystic Fibrosis Agents - Texas Health and Human Services · 3. Manual step – Does the client have at least one F508del mutation in the CFTR gene? [ ] Yes (Go to #4)](https://img.dokumen.tips/doc/110x75/5f105f3f7e708231d448c89f/cystic-fibrosis-agents-texas-health-and-human-services-3-manual-step-a-does.jpg)













