Embed Size (px)
Citation preview

Multiple Sequence Alignment (MSA)
1. Uses of MSA
2. Technical difficulties
1. Select sequences
2. Select objective function
3. Optimize the objective function
1. Exact algorithms
2. Progressive algorithms
3. Iterative algorithms
1. Stochastic
2. Non-stochastic
4. Consistency-based algorithms
3. Tools to view alignments
1. MEGA
2. JALVIEW
(PSI-BLAST)
Function prediction
Fig. from Boris Steipe
U. of TorontoSequence
relationships
If the MSA is incorrect, the
above inferences are incorrect!
Chapter 12

Multiple sequence alignment: definition
• a collection of three or more protein (or nucleic acid)
sequences that are partially or completely aligned
• homologous residues are aligned in columns
across the length of the sequences
• residues are homologous in an evolutionary sense
• residues are homologous in a structural sense

ClustalW
Note how the region of a conserved histidine (▼) varies
depending on which algorithm is used

Praline

MUSCLE

Probcons

TCoffee

Multiple sequence alignment: properties
• not necessarily one “correct” alignment of a protein family
• protein sequences evolve...
• ...the corresponding three-dimensional structures
of proteins also evolve
• may be impossible to identify amino acid residues that align properly (structurally) throughout a multiple
sequence alignment
• for two proteins sharing 30% amino acid identity,
about 50% of the individual amino acids are superposable in the two structures

Multiple sequence alignment: features
• some aligned residues, such as cysteines that formdisulfide bridges, may be highly conserved
• there may be conserved motifs such as a transmembrane domain
• there may be conserved secondary structure features
• there may be regions with consistent patterns ofinsertions or deletions (indels)

Multiple sequence alignment: uses
• MSA is more sensitive than pairwise alignmentto detect homologs
• BLAST output can take the form of a MSA,and can reveal conserved residues or motifs
• Population data can be analyzed in a MSA (PopSet)
• A single query can be searched against
a database of MSAs (e.g. PFAM)
• Regulatory regions of genes may have consensussequences identifiable by MSA

I HvCO4
I OsC
I HvCO7
I OsF
I HvCO3
I OsB
I AtCO
I AtCOL3
I OsHd1
I HvCO1
I HvCO8
I OsG
II AtCOL6
II OsJ
IV HvCO9
IV OsH
IV OsI
ZCCT2 Ha
ZCCT2 Hb
ZCCT2 Tm
ZCCT2 Td
ZCCT1 TmG3116
ZCCT1 TmDV92
ZCCT1 Td
III AtCOL9
III OsN
83
82
91
99
60
66
73
94
56
53
99
I HvCO4
IV OsI
I OsC
III OsN
I HvCO7
I OsF
I HvCO3
I OsB
I AtCO
I AtCOL3
I OsHD1
I HvCO1
I HvCO8
I OsG
II AtCOL6
II OsJ
IV HvCO9
IV OsH
III AtCOL9
HvZCCT-Ha
HvZCCT-Hb
TmZCCT2
TdZCCT2
TmZCCT1-G3116
TmZCCT1-DV92
TdZCCT1
Photoperiod
response
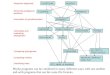
MSA of ZCCT genes
Cluster analysis of the CCT domains
Putative Zinc fingers
Exon 1 Exon 2CCTZF
IV
II
III
CCT domains
EMS
co-7
Yan et al. 2004. Science 303:1640

Fig. from Boris Steipe Univ. of Toronto
MSA. Technical difficulties

MSA. Technical difficulties

Multiple Sequence Alignment (MSA)
1. Uses of MSA
2. Technical difficulties
1. Select sequences
2. Select objective function
3. Optimize the objective function
1. Exact algorithms
2. Progressive algorithms
3. Iterative algorithms
1. Stochastic
2. Non-stochastic
4. Consistency-based algorithms
3. Tools to view alignments
1. MEGA
2. JALVIEW

MSA: Selection of sequences
• Use database searching to identify related proteins.
• Select homologous sequences (sequences sharing common ancestor).
• Global MSA programs are designed to align sequences that are similar over their entire
length. If sequences are different in length trim the parts that are not similar based on the
database search.
• PSI-Blast can help to define these region.
• Alternatively use local MSA programs (Gibbs sampler, Match-Box, or MACAW)
• Select a “representative” group of sequences. Alignments with large number of
sequences are slow to compute and hard to analyze.
• The inclusion of a large number of a particular group of closely related proteins
will dominate the alignment or profile (in spite of weighting schemes).
REMEMBER: MSA programs will produce alignments even
if you provide unrelated sequences!
Past problem: not enough related sequences.
Today’s problem: too many related sequences!

Multiple Sequence Alignment (MSA)
1. Uses of MSA
2. Technical difficulties
1. Select sequences
2. Select objective function
3. Optimize the objective function
1. Exact algorithms
2. Progressive algorithms
3. Iterative algorithms
1. Stochastic
2. Non-stochastic
4. Consistency-based algorithms
3. Tools to view alignments
1. MEGA
2. JALVIEW

Objective functions (OF) Define the mathematical objective of the search
A biologically ideal OF should
• Maximize similarity
• Minimize the number of gaps (over their length)
• Retain conserved motifs and patterns
• Retain functionally important alignments
• Recapitulate phylogeny
• Concentrate on alignable regions, not in gapped regions
• Consider the limitations imposed by the 3D structure
Most widely used MSA packages use a simple sum-of-pairs OF
• Define a mathematical optimum
• Use sum-of-pairs and affine gaps
• Use a context-independent Mutation Data Matrix (e.g. Blosum 62)
• Some add weighting proportional to the information in the seq.
It is a non-trivial task to test the biological correctness of an
objective function.

Seq.1 AT-AATG
Seq.2 CTGAG-G
Seq.3 ATGAA-G
Sum-of-pairs (SP) Objective Function
Induced pairwise alignment: After the best MSA is obtained, other sequences are
removed, spaces facing spaces are removed and a score is calculated using any
chosen scoring scheme (distance or similarity).
Seq.1 AT-AATG Induced Seq. 3-4 alignment
Seq.2 CTGAG-G Seq.3 CTG-GG Distance scheme
Seq.3 ATGAA-G Seq.4 ATGAAG # mismathes (including -)
43 2 Sum-of-pairs distance = 4 + 3 + 2 = 9
Sum-of-pairs score: The SP of a MSA is the sum of the scores of all the scores of
the induced pairwise global alignments
3
Weighted Sum-of-pairs score: each score can be multiplied by a weight.
Weights are often intended to reflect evolutionary distances to induce the
MSA to more accurately reflect known evolutionary history, or the
information carried by the sequences being aligned.

Sum-of-pairs (SP) Objective Function
Multiple MSA: Depending on the Mutation Data matrix selected (e.g. PAM or
BLOSUM) and on the selected gap penalties (opening and extension)
different MSA will be obtained. Which one is the correct one?
New Objective functions: less sensitive to gap penalty estimations thanks to the
incorporation of local information
• Segment-to-segment comparisons of the sequences (instead of character-to-
character) without gap penalties is the strategy used by DiAlign. This approach is
efficient where sequences are not globally related but share only local similarities,
(genomic DNA, many protein families) http://bibiserv.techfak.uni-bielefeld.de/dialign/.
• Consistency objective function: (e.g. T-Coffee)The optimal MSA is defined as the
one that agrees the most with all the optimal pair-wise alignments. Given a set of
independent observations the most consistent are often closer to “the truth”.
Seq.1 AT-AATG Seq.1 ATAATG Clustal
Seq.2 CTGAG-G Distance scheme Seq.2 CTGAGG Gap open= 11
Seq.3 ATGAA-G # mismathes (including -) Seq.3 ATGAAG Gap ext.= 1

Multiple Sequence Alignment (MSA)
1. Uses of MSA
2. Technical difficulties
1. Select sequences
2. Select objective function
3. Optimize the objective function
1. Exact algorithms
2. Progressive algorithms
3. Iterative algorithms
1. Stochastic
2. Non-stochastic
4. Consistency-based algorithms
3. Tools to view alignments
1. MEGA
2. JALVIEW

MSA: Exact algorithmMSA program
• Multidimensional dynamic programming
• Optimizes sum-of-pairs
• More accurate than progressive methods
• BUT… Time proportional to Ln
• Practical to ~10 seq. of L<200-bp
DCA (Divide & Conquer Algorithm)
• Sits on top of MSA program
• Produces simultaneous MSA
• Cuts seq. in subsets, that are fed into MSA
• Practical to ~20-30 seq. of L<200-bp
• Easy WEB submission
OMA (Optimal Multiple Aligment)
• Iterative implementation of DCA
• Speeds up DCA
• Decreases memory requirements

Progressive algorithms (ClustalW, MultAlign, AMPS)
GARFIELDTHEFASTCAT---
GARFIELDTHELASTFATCAT
GARFIELD THE LAST FAST CAT GARFIELD THE FAST CAT GARFIELD THE VERY FAST CAT THE FAT CAT
GARFIELDTHEVERYFASTCAT
GARFIELDTHEFASTCAT----
GARFIELDTHELASTFAT-CAT
--------THEFAT-----CAT
GARFIELDTHEVERYFASTCAT
GARFIELDTHEFASTCAT---
GARFIELDTHELASTFATCAT
--------THEFA-----TCAT
GARFIELDTHEVERYFASTCAT
GARFIELDTHEFAS----TCAT
GARFIELDTHELASTFA-TCAT
DCA alignment
ClustalW Blosum62 Gap 11-1
Cheaper to open distal gap
than to align C and F
Example of Progressive algorithm
• Calculate distances/similarities
between sequences
• Construct a tree
• Add sequentially, following tree

Multiple sequence alignment: methods
Progressive methods: use a guide tree (a little like aphylogenetic tree but NOT a phylogenetic tree) to
determine how to combine pairwise alignments one by one
to create a multiple alignment.
Making multiple alignments using trees was a verypopular subject in the ‘80s. Fitch and Yasunobu (1974)
may have first proposed the idea, but Hogeweg andHesper (1984) and many others worked on the topic before
Feng and Doolittle (1987)—they made one
important contribution that got their names attached to thisalignment method.
Examples: CLUSTALW, MUSCLE

Multiple sequence alignment: methods
Example of MSA using ClustalW: two data sets
Five distantly related lipocalins (human to E. coli)
Five closely related RBPs
When you do this, obtain the sequences of interest in the FASTA format.

HomoloGene: an NCBI resource to obtain
multiple related sequences
[1] Enter a query at NCBI such as globin[2] Click on HomoloGene (left side)
[3] Choose a HomoloGene family, and view in the fasta format

Use ClustalW to do a progressive MSA
http://www2.ebi.ac.uk/clustalw/

Feng-Doolittle MSA occurs in 3 stages
[1] Do a set of global pairwise alignments
(Needleman and Wunsch’s dynamic programming
algorithm)
[2] Create a guide tree
[3] Progressively align the sequences

Progressive MSA stage 1 of 3:generate global pairwise alignments
five distantly related lipocalins
best score

Progressive MSA stage 1 of 3:generate global pairwise alignments
Start of Pairwise alignments
Aligning...
Sequences (1:2) Aligned. Score: 84
Sequences (1:3) Aligned. Score: 84
Sequences (1:4) Aligned. Score: 91
Sequences (1:5) Aligned. Score: 92
Sequences (2:3) Aligned. Score: 99
Sequences (2:4) Aligned. Score: 86
Sequences (2:5) Aligned. Score: 85
Sequences (3:4) Aligned. Score: 85
Sequences (3:5) Aligned. Score: 84
Sequences (4:5) Aligned. Score: 96
five closely related lipocalins
best score

Number of pairwise alignments needed
For n sequences, (n-1)(n) / 2
For 5 sequences, (4)(5) / 2 = 10

Feng-Doolittle stage 2: guide tree
• Convert similarity scores to distance scores
• A tree shows the distance between objects
• Use UPGMA (defined in the phylogeny lecture)
• ClustalW provides a syntax to describe the tree
• A guide tree is not a phylogenetic tree

(gi|5803139|ref|NP_006735.1|:0.04284,
(gi|6174963|sp|Q00724|RETB_MOUS:0.00075,
gi|132407|sp|P04916|RETB_RAT:0.00423)
:0.10542)
:0.01900,
gi|89271|pir||A39486:0.01924,
gi|132403|sp|P18902|RETB_BOVIN:0.01902);
five closely related lipocalins
Progressive MSA stage 2 of 3:generate a guide tree calculated from
the distance matrix

Feng-Doolittle stage 3: progressive alignment
• Make a MSA based on the order in the guide tree
• Start with the two most closely related sequences
• Then add the next closest sequence
• Continue until all sequences are added to the MSA
• Rule: “once a gap, always a gap.”• To change the initial gap choices later on would be
to give more weight to distantly related sequences
• To maintain the initial gap choices is to trustthat those gaps are most believable

Progressive MSA stage 3 of 3:progressively align the sequences
following the branch order of the tree

Progressive MSA stage 3 of 3:CLUSTALX output

Clustal W alignment of 5 closely related lipocalins
CLUSTAL W (1.82) multiple sequence alignment
gi|89271|pir||A39486 MEWVWALVLLAALGSAQAERDCRVSSFRVKENFDKARFSGTWYAMAKKDP 50
gi|132403|sp|P18902|RETB_BOVIN ------------------ERDCRVSSFRVKENFDKARFAGTWYAMAKKDP 32
gi|5803139|ref|NP_006735.1| MKWVWALLLLAAW--AAAERDCRVSSFRVKENFDKARFSGTWYAMAKKDP 48
gi|6174963|sp|Q00724|RETB_MOUS MEWVWALVLLAALGGGSAERDCRVSSFRVKENFDKARFSGLWYAIAKKDP 50
gi|132407|sp|P04916|RETB_RAT MEWVWALVLLAALGGGSAERDCRVSSFRVKENFDKARFSGLWYAIAKKDP 50
********************:* ***:*****
gi|89271|pir||A39486 EGLFLQDNIVAEFSVDENGHMSATAKGRVRLLNNWDVCADMVGTFTDTED 100
gi|132403|sp|P18902|RETB_BOVIN EGLFLQDNIVAEFSVDENGHMSATAKGRVRLLNNWDVCADMVGTFTDTED 82
gi|5803139|ref|NP_006735.1| EGLFLQDNIVAEFSVDETGQMSATAKGRVRLLNNWDVCADMVGTFTDTED 98
gi|6174963|sp|Q00724|RETB_MOUS EGLFLQDNIIAEFSVDEKGHMSATAKGRVRLLSNWEVCADMVGTFTDTED 100
gi|132407|sp|P04916|RETB_RAT EGLFLQDNIIAEFSVDEKGHMSATAKGRVRLLSNWEVCADMVGTFTDTED 100
*********:*******.*:************.**:**************
gi|89271|pir||A39486 PAKFKMKYWGVASFLQKGNDDHWIIDTDYDTYAAQYSCRLQNLDGTCADS 150
gi|132403|sp|P18902|RETB_BOVIN PAKFKMKYWGVASFLQKGNDDHWIIDTDYETFAVQYSCRLLNLDGTCADS 132
gi|5803139|ref|NP_006735.1| PAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAVQYSCRLLNLDGTCADS 148
gi|6174963|sp|Q00724|RETB_MOUS PAKFKMKYWGVASFLQRGNDDHWIIDTDYDTFALQYSCRLQNLDGTCADS 150
gi|132407|sp|P04916|RETB_RAT PAKFKMKYWGVASFLQRGNDDHWIIDTDYDTFALQYSCRLQNLDGTCADS 150
****************:*******:****:*:* ****** *********
* asterisks indicate identity in a column

Additional features of ClustalW improve
its ability to generate accurate MSAs
• Individual weights are assigned to sequences; very closely related sequences are given less weight,while distantly related sequences are given more weight
• Scoring matrices are varied dependent on the presenceof conserved or divergent sequences, e.g.:
PAM20 80-100% idPAM60 60-80% idPAM120 40-60% idPAM350 0-40% id
• Residue-specific gap penalties are applied

Iterative algorithmsRecurrent modifications of suboptimal solutions
Stochastic Iterative Algorithms
SAGA
• Uses a ‘Genetic Algorithm’
• Can use different objective functions (e.g. Coffee)
• Mutations randomly insertion or shift gaps
• Sequences can recombine
• Sequences evolve, higher OF scores survive
GAs and HMMs have probed rather
disappointing in ab initio alignments.
Better: Pre-compute MSA with other
program and then use this ones for
optimization
Evolution of a seq. alignment by recombination
Compatible ends

Consistency-based Algorithms
T-Coffee (Consistency Objective Function For alignmEnt Evaluation)
Version 2.00 and higher can mix
sequences and structures
Local and global pair-wise alignments
can come from different programs and
can be redundantThe EL is a position-specific substitution matrix
where the score associated with each pair of
residues depends on its compatibility with the
rest of the library. This library replaces the
Mutation data Matrix used in ClustalW.
Pair-wise distances are computed
A Neighbor joining tree is estimated
Sequences are aligned progressively following
the topology of the tree

Benchmark tests (from Notredame 2002)
ClustalW: performed well on Ref. sets 1-3, but poorly on 4-5 when long
internal or terminal gaps are required.
When large gaps required T-Coffee and DiAlign perform better

Summary strategies

Alignment Editors
Jalview
• Written in Java
• Input MSF, aligned FASTA
• ClustalW alignment
• Interactive alignment editor
• Multiple color schemes
• Can divide in sub-families
• Produces UPGMA, Neighbor-
joining trees and Principal
Component Analysis
• Incorporates information from
feature Table
• Incorporates structural inormation

Alignment Editors

Alignment Visualization

Multiple Sequence Alignment

Multiple Sequence Alignment

At the end of the day ...
• Use more than one alignment method,
• Make sure you have the right sequences.
• Don't align parts of sequences that can't be aligned
(because they are not homologuous).
• Realize problems from multi-domain proteins.
• Above all, use your common sense.
Multiple Sequence Alignment

Multiple Sequence Alignment
From Boris Steipe Univ. of Toronto

Hidden Markov Models
Bioinformatics, Sequence and Genome Analysis (pg205) , Mount.
The model accommodates the identities, mismatches, insertions, and deletions expected in a group of related proteins.
(A) MSA: Each column may include matches and mismatches (red positions), insertions (green positions), and deletions (purple positions).
(B) Each column in the model represents the possibility of a match, insert, or delete in each column of the alignment in A. The HMM is a probabilistic representation of the MSA. Sequences can be generated from the HMM by starting at the beginning state labeled BEG and then by following anyone of many pathways from one type of sequence variation to another (states) along the state transition arrows and terminating in the ending state labeled END. Any sequence can be generated by the model and each pathway has a probability associated with it. Each square match state stores an amino acid distribution such that the probability of finding an amino acid depends on the frequency of that amino acid within that match state. Each diamond-shaped insert state produces random amino acid letters for insertions between aligned columns and each circular delete state produces a deletion in the alignment with probability 1.
One of many ways of generating the sequence N K Y L T in the above profile is by the sequence BEG ->Ml ->11 ->M2 ->M3 :>M4 ->END. Each transition has an associated probability, and the sum of the probabilities of transitions leaving each state is 1. The average value of a transition would thus be 0.33, since there are three transitions from most states (there are only two from M4 and D4, hence the average from them is 0.5). For example, if a match state contains a uniform distribution across the 20 amino acids, the probability of any amino acid is 0.05. Using these average values of 0.33 or 0.5 for the transition values and 0.05 for the probability of each amino acid in each state, the probability of the above sequence N K Y L T is the product of all of the transition probabilities in the path and the probability that each state will produce the corresponding amino acid in the sequences, or 0.33 X 0.05 X 0.33 X
0.05 X 0.33 X 0.05 X 0.33 X 0.05 X 0.33 X 0.05 X 0.5 = 6.1 X 10-10. Since these probabilities are very small numbers, probabilities are converted to log odds scores, and the logarithms are added to give the overall probability score.
The secret of the HMM is to adjust the transition values and the distributions in each state by training the model with the sequences. The training involves finding every possible pathway through the model that can produce the sequences, counting the number of times each transition is used and which amino acids were required by each match and insert state to produce the sequences. This training procedure leaves a memory of the sequences in the model. As a consequence, the model will be able to give a better prediction of the sequences. Once the model has been adequately trained, of all the possible paths through the model that can generate the sequence N KY L T, the most probable should be the match-insert-3 match combination (as opposed to any other combination of matches, inserts, and deletions). Likewise, the other sequences in the alignment would also be predicted with highest probability as they appear in the alignment; i.e., the last sequence would be predicted with highest probability by the path match-match-delete-match. In this fashion, the trained HMM provides a multiple sequence alignment, such as shown in A. For each sequence, the objective is to infer the sequence of states in the model that generate the sequences. The generated sequence is a Markov chain because the next state is dependent on the current one. Because the actual sequence information is hidden with-in the model, the model is described as a hidden Markov model

Multiple sequence alignment to profile HMMs
► Hidden Markov models (HMMs) are “states”that describe the probability of having a
particular amino acid residue at arranged
in a column of a multiple sequence alignment
► HMMs are probabilistic models
► HMMs may give more sensitive alignmentsthan traditional techniques such as
progressive alignment

Simple Hidden Markov Model
Observation: YNNNYYNNNYN
(Y=goes out, N=doesn’t go out)
What is underlying reality (the hidden state chain)?
R
S
0.15
0.85
0.2
0.8
P(dog goes out in rain) = 0.1
P(dog goes out in sun) = 0.85

GTWYA (hs RBP)
GLWYA (mus RBP)
GRWYE (apoD)
GTWYE (E Coli)
GEWFS (MUP4)
An HMM is constructed from a MSA
Example: five lipocalins

GTWYA
GLWYA
GRWYE
GTWYE
GEWFS
PositionProb. 1 2 3 4 5p(G) 1.0p(T) 0.4p(L) 0.2p(R) 0.2p(E) 0.2 0.4p(W) 1.0p(Y) 0.8p(F) 0.2p(A) 0.4p(S) 0.2

GTWYA
GLWYA
GRWYE
GTWYE
GEWFS
Prob. 1 2 3 4 5p(G) 1.0p(T) 0.4p(L) 0.2p(R) 0.2p(E) 0.2 0.4p(W) 1.0p(Y) 0.8p(F) 0.2p(A) 0.4p(S) 0.2
P(GEWYE) = (1.0)(0.2)(1.0)(0.8)(0.4) = 0.064
log odds score = ln(1.0) + ln(0.2) + ln(1.0) + ln(0.8) + ln(0.4) = -2.75

GTWYA
GLWYA
GRWYE
GTWYE
GEWFS
P(GEWYE) = (1.0)(0.2)(1.0)(0.8)(0.5) = 0.08
log odds score = ln(1.0) + ln(0.2) + ln(1.0) + ln(0.8) + ln(0.5) = -2.53
G:1.0 W:1.0
T:0.4
L:0.2
R:0.2
E:0.2
Y:0.8
F:0.2
A:0.5
E:0.5
S:1.0

HMMER: build a hidden Markov model
Determining effective sequence number ... done. [4]
Weighting sequences heuristically ... done.
Constructing model architecture ... done.
Converting counts to probabilities ... done.
Setting model name, etc. ... done. [x]
Constructed a profile HMM (length 230)
Average score: 411.45 bits
Minimum score: 353.73 bits
Maximum score: 460.63 bits
Std. deviation: 52.58 bits

HMMER: calibrate a hidden Markov model
HMM file: lipocalins.hmm
Length distribution mean: 325
Length distribution s.d.: 200
Number of samples: 5000
random seed: 1034351005
histogram(s) saved to: [not saved]
POSIX threads: 2
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
HMM : x
mu : -123.894508
lambda : 0.179608
max : -79.334000

HMMER: search an HMM against GenBankScores for complete sequences (score includes all domains):
Sequence Description Score E-value N
-------- ----------- ----- ------- ---
gi|20888903|ref|XP_129259.1| [Mus] (XM_129259) ret 461.1 1.9e-133 1
gi|132407|sp|P04916|RETB_RAT Plasma retinol- 458.0 1.7e-132 1
gi|20548126|ref|XP_005907.5|[Homo] (XM_005907) sim 454.9 1.4e-131 1
gi|5803139|ref|NP_006735.1| (NM_006744) ret 454.6 1.7e-131 1
gi|20141667|sp|P02753|RETB_HUMAN Plasma retinol- 451.1 1.9e-130 1
.
.
gi|16767588|ref|NP_463203.1|[Salmonella] (NC_003197) out 318.2 1.9e-90 1
gi|5803139|ref|NP_006735.1|: domain 1 of 1, from 1 to 195: score 454.6, E = 1.7e-131
*->mkwVMkLLLLaALagvfgaAErdAfsvgkCrvpsPPRGfrVkeNFDv
mkwV++LLLLaA + +aAErd Crv+s frVkeNFD+
gi|5803139 1 MKWVWALLLLAA--W--AAAERD------CRVSS----FRVKENFDK 33
erylGtWYeIaKkDprFErGLllqdkItAeySleEhGsMsataeGrirVL
+r++GtWY++aKkDp E GL+lqd+I+Ae+S++E+G+Msata+Gr+r+L
gi|5803139 34 ARFSGTWYAMAKKDP--E-GLFLQDNIVAEFSVDETGQMSATAKGRVRLL 80
eNkelcADkvGTvtqiEGeasevfLtadPaklklKyaGvaSflqpGfddy
+N+++cAD+vGT+t++E dPak+k+Ky+GvaSflq+G+dd+
gi|5803139 81 NNWDVCADMVGTFTDTE----------DPAKFKMKYWGVASFLQKGNDDH 120

HMMER: search an HMM against GenBankmatch to a bacterial lipocalin
gi|16767588|ref|NP_463203.1|: domain 1 of 1, from 1 to 177: score 318.2, E = 1.9e-90
*->mkwVMkLLLLaALagvfgaAErdAfsvgkCrvpsPPRGfrVkeNFDv
M+LL+ +A a ++ Af+v++C++p+PP+G++V++NFD+
gi|1676758 1 ----MRLLPVVA------AVTA-AFLVVACSSPTPPKGVTVVNNFDA 36
erylGtWYeIaKkDprFErGLllqdkItAeySleEhGsMsataeGrirVL
+rylGtWYeIa+ D+rFErGL + +tA+ySl++ +G+i+V+
gi|1676758 37 KRYLGTWYEIARLDHRFERGL---EQVTATYSLRD--------DGGINVI 75
eNkelcADkvGTvtqiEGeasevfLtadPaklklKyaGvaSflqpGfddy
Nk++++D+ +++ +EG+a ++t+ P +++lK+ Sf++p++++y
gi|1676758 76 -NKGYNPDR-EMWQKTEGKA---YFTGSPNRAALKV----SFFGPFYGGY 116

HMMER: search an HMM against GenBank
Scores for complete sequences (score includes all domains):
Sequence Description Score E-value N
-------- ----------- ----- ------- ---
gi|3041715|sp|P27485|RETB_PIG Plasma retinol- 614.2 1.6e-179 1
gi|89271|pir||A39486 plasma retinol- 613.9 1.9e-179 1
gi|20888903|ref|XP_129259.1| (XM_129259) ret 608.8 6.8e-178 1
gi|132407|sp|P04916|RETB_RAT Plasma retinol- 608.0 1.1e-177 1
gi|20548126|ref|XP_005907.5| (XM_005907) sim 607.3 1.9e-177 1
gi|20141667|sp|P02753|RETB_HUMAN Plasma retinol- 605.3 7.2e-177 1
gi|5803139|ref|NP_006735.1| (NM_006744) ret 600.2 2.6e-175 1
gi|5803139|ref|NP_006735.1|: domain 1 of 1, from 1 to 199: score 600.2, E = 2.6e-175
*->meWvWaLvLLaalGgasaERDCRvssFRvKEnFDKARFsGtWYAiAK
m+WvWaL+LLaa+ a+aERDCRvssFRvKEnFDKARFsGtWYA+AK
gi|5803139 1 MKWVWALLLLAAW--AAAERDCRVSSFRVKENFDKARFSGTWYAMAK 45
KDPEGLFLqDnivAEFsvDEkGhmsAtAKGRvRLLnnWdvCADmvGtFtD
KDPEGLFLqDnivAEFsvDE+G+msAtAKGRvRLLnnWdvCADmvGtFtD
gi|5803139 46 KDPEGLFLQDNIVAEFSVDETGQMSATAKGRVRLLNNWDVCADMVGTFTD 95
tEDPAKFKmKYWGvAsFLqkGnDDHWiiDtDYdtfAvqYsCRLlnLDGtC
tEDPAKFKmKYWGvAsFLqkGnDDHWi+DtDYdt+AvqYsCRLlnLDGtC
gi|5803139 96 TEDPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAVQYSCRLLNLDGTC 145

Multiple Sequence Alignment

Multiple Sequence Alignment

Multiple Sequence Alignment

Multiple Sequence Alignment

Two kinds of multiple sequence alignment resources
Text-based or query-based searches:CDD, Pfam (profile HMMs), PROSITE
[2] Multiple sequence alignment by manual input
Muscle, ClustalW, ClustalX
[1] Databases of multiple sequence alignments

BLOCKS
CDD
Pfam
SMART
DOMO (Gapped MSA)
INTERPRO
iProClass
MetaFAM
PRINTS
PRODOM (PSI-BLAST)
PROSITE
Databases of multiple sequence alignments
These
Use
HMMs

PFAM (protein family) database:
http://pfam.sanger.ac.uk/

PFAM (protein
family) text search result

PFAM HMM for lipocalins
20 amino acids
position

PFAM HMM for lipocalins: GXW motif
G W
20 amino acids

PFAM GCG MSF format

Pfam (protein family) database

PFAM JalView viewer

PFAM JalView viewer


SMART: Simple Modular
Architecture Research Tool(emphasis on cell signaling)
Page 338

SMART: lipocalin result

BLOCKS
CDD
Pfam
SMART
DOMO (Gapped MSA)
INTERPRO
iProClass
MetaFAM
PRINTS
PRODOM (PSI-BLAST)
PROSITE
Databases of multiple sequence alignments
Conserved
Domain
Database
(CDD) at NCBI =
PFAM + SMART

[1] Go to NCBI � Structure[2] Click CDD
[3] Enter a text query, or a protein sequence
CDD: Conserved domain database

CDD: Conserved domain database

CDD=
PFAM+
SMART

Purpose: to find conserved domainsin the query sequence
Query = your favorite protein
Database = set of many position-specificscoring matrices (PSSMs), i.e. a set of MSAs
CDD is related to PSI-BLAST, but distinct
CDD searches against profiles generatedfrom pre-selected alignments
CDD uses RPS-BLAST: reverse position-specific