Embed Size (px)
Citation preview

E L S E V I E R Journal of Molecular Structure 444 (1998) 13-20
Journa l of
MOLECULAR STRUCTURE
Molecular structures of recognition system host + guest and DNA complex
Linjing Yang, Xizeng Feng, Imshik Lee, Chunli Bai* Institute of Chemistry, The Chinese Academy of Sciences, Beijing, 100080, China
Received 23 April 1997; accepted 26 June 1997
Abstract
Investigations were carried out to look into the system of host + guest as a ligand targeted to the duplex DNA. The possible structures of host RhB-~-CDen were studied by minimization and molecular dynamics simulation. In the association state of host + guest ( 1 : 1 ) system, the guest molecule of 1 -borneol could insert into the cyclodextrin cavity of the host molecule RhB-~- CDen. The interactive sites of the RhB-/3-CDen + borneol system as a ligand targeting to the duplex DNA d(C)8.d(G)8 were obtained by docking interaction. It was found that the ligand was probably bound to the duplex DNA in the minor groove. The conformations of DNA varies noticeably to accommodate the binding groups of the ligand. Water molecules bridge between the bases of duplex DNA and/or ligand by hydrogen bonds to make the entire system more stable. © 1998 Elsevier Science B.V.
Keywords: Host + guest system; Duplex DNA; RhB-~-CDen; l-Borneol; Molecular dynamics
1. Introduct ion
Deoxyribonucleic acid (DNA) is increasingly being seen as a target endpoint in the development of drug- based therapy [1]. Studies on ligands binding to DNA are important in the design of new and efficient drugs targeted to DNA [2]. Ligands can selectively bind to the major or minor groove of the helix at particular locations with respect to base-pair sequence. It was found that intercalating agents usually bind via the minor groove, while there are reported cases in which intercalation occurs in the major groove [3].
Molecules which possess large planar ring systems can be ligands to DNA by inserting the planar ring systems between two adjacent base-pair of the DNA.
*Corresponding author. Fax: +86 010 62557908; e-mail: clbai @infoc3.icas.ac.cn
0022-2860/98/$19.00 © 1998 Elsevier Science B.V. All rights PH S0022-2860(97)00276-7
For example, minor groove intercalation is charac- terized by the anthraquinone ring of doxorubicin inserting between base-pairs with its long axis nearly perpendicular to the long axes of the base pairs [4]. Both therapeutic (anticancer, antiviral and anti- bacterial drugs) and toxic (mutagenic, probably carcinogenic) behavior has been observed for compounds which intercalate to DNA.
Dervan et al. [5] have developed simple rules to rationally control the sequence specificity of minor- groove binding polyamides containing N-methylimi- dazole and N-methylpyrrole amino acids. Two eight-ring pyr ro le - imidazo le polyamides differing in sequence by a single amino acid bind specifically to respective six-base-pair target sites which differ in sequence by a single base pair. For side-by-side complexes of pyr ro le - imidazo le polyamides in the minor groove of DNA, the DNA-binding sequence
reserved

14 L. Yang et al./Journal of Molecular Structure 444 (1998) 13-20
specificity depends on the sequence of side-by-side amino-acid pairing [6-8].
The complexity of the ligands makes their mode of interaction with DNA a matter of considerable interest. In this paper, we studied a host + guest system which could target the double-helix DNA. The host molecule is rhodamine B-ethylenediamine- i3-cyclodextrins (RhB-13-CDen), the guest molecule is 1-borneol. Cyclodextrins (CDs) can accommodate a variety of organic compounds in their central cavities in aqueous solution [9]. According to the experimen- tal results, it appears that RhB-13-CDen forms associa- tion and exists as the modes of dimmer or monomer in the absence of guests. In the presence of guests, the association state would convert into a host + guest (1:1) complex [10]. The entire host + guest system was seen as a ligand targeting duplex DNA. Experi- mental evidence suggested that the base-pair sequence does not play a major role in the specific nature of most intercalation complexes [11-14]. For example, the interaction of doxorubicin with dinucleotide dimer sequences was modelled by molecular mechanics energy calculation. The results showed that the inter- calating geometry is relatively insensitive to base-pair sequence. However, the binding energy is moderately dependent upon base-pair sequence with CpG/GpC sites being energetically preferred [1]. Thus we
simply selected d(C)8-d(G)8 base pairs as the target DNA. In this paper, the most possible conformations of RhB-t3-CDen were determined and the interactive site of host and guest was investigated. Furthermore, we studied the interactive ways of host + guest system as a ligand targeting to the duplex DNA in detail.
2. Method and results
In this section, the results are presented in three parts. In the first part, we studied the possible struc- tures of RhB-/3-CDen. Then, using the structure of RhB-t3-CDen as the host molecule and 1-borneol as the guest molecule, the interactive sites between host and guest were determined in the second part. The host + guest system is considered as the preliminary introduction of the ligand structure. In-depth discus- sions on the ligand will be presented in another paper [15]. Based on the results of the first two parts, we investigated the interactive characteristics of the above mentioned host + guest system as a ligand targeting the duplex DNA d(C)8.d(G)8 in the third part.
2.1. Structures of RhB-13-CDen
Considering all possible conformations of the host
Fig. 1. Three possible structure models of RhB-B-CDen. (a) One side of the three-aromatic-ring of the rhodamine group inserting into the cavity of the CD. (b) Benzene ring of rhodamine group and ethylene diamine being positioned in the cavity of CD, with the three-aromatic-ring being outside of CD cavity. (c) The entire rhodamine group being outside of CD cavity.

L. Yang et al./Journal of Molecular Structure 444 (1998) 13-20 15
Table 1 Conformational energies of the three structures of RhB-~-CDen (kcal mol -I)
Etotal Ebond EO E¢ Ex-term Evdw Eel
Fig. l(a) - 2 8 . 5 18.1 78.5 -229 .7 - 5 1 . 5 19.4 136.7 Fig. l ( b ) - 2 2 . 7 18.5 77.0 -232 .1 - 5 0 . 3 27.3 136.9 Fig. l(c) - 6 . 4 17.9 72.7 - 2 3 6 . 4 - 4 8 . 9 44.1 144.2
RhB-/3-CDen, three molecular models were con- structed using Biosym software. The three models were minimized over 1000 cycles by molecular mechanics and dynamics separately. Based on the conformational energies, the most stable model was selected as the host structure interacting with the guest. The rms values were all less than 0.01 kcal- mol-1 ,~-i at the end of calculation. The parameters .used in the force field were those of the cff91 force field.
The three possible structure models are all shown in Fig. 1: (a) one side of the three-aromatic-ring of RhB inserts into the cavity of CD; (b) the benzene ring of RhB and ethylene diamine are positioned in the cavity of CD, and the three-aromatic-ring is outside of CD cavity, (c) the three-aromatic-ring and the benzene ring of RhB are apparently outside the hydrophobic cavity of CD. From the three conformations of RhB- /3-CDen, we found that the benzene ring placing vertical to the plane of three-aromatic-ring of rhoda- mine B is most favorable in the RhB structure. According to the conformational energy summary (Table 1), the order of the conformational stability is Fig. l(a) > l(b) > l(c). The greatest differences between the conformational energies are contributed by van der Waals energies; the other energies are very similar. The stronger the van der Waals attraction, the more stable is the entire molecule. From the result it can be concluded that the conformations of RhB groups in the CD's cavity is crucial to the stability improvement of host structure RhB-/3-CDen.
than 0.01 kcal mol -~ ,~-1. The conformational energy of borneol in the free state is - 9.6 kcal mol -~.
Three possible interactive positions between host and guest were obtained using the docking method, calculating over 3500 cycles. The objective of a dock- ing calculation is to evaluate the interaction energies of many orientations of one molecule relative to the other, while searching for the orientations that results in low interaction energies. In docking, the interaction energy is computed by summing the energy contribu- tions between all atoms of the two molecules. The contribution between atoms interacting with other atoms in the same molecule is ignored. For example, for cff91 :
I/ Aij Bij qiqj~ Einteracti°n= ~i ~j ~ i j - ~ i j ' t - ~rij/i
The position with the strongest interaction between RhB-~-CDen and 1-borneol was selected, and the entire system was minimized by molecular mechanics and dynamics over 1000 cycles separately. Fig. 2 shows the initial structure determined by the docking method and the final structure after minimization. In the initial structure shown in Fig. 2(a), the interactive orientation between host and guest can be seen; bor- neol is at the edge of CD opposite to RhB groups. In the final structure shown in Fig. 2(b), borneol inserts into the cavity of CD and the three-aromatic-ring is completely expelled from the cavity. The intermole- cular energy of host and guest is - 25.1 kcal tool -~. The van der Waals energy between RhB-/3-CDen and
2.2. Structures of host + guest recognition system
Since the structure of RhB-/3-CDen in Fig. l(a) is the most stable of the three models, it is selected as the host molecule. The guest molecule is 1-borneol. The model of 1-borneol was built in Biosym software and minimized over 300 cycles with the rms value less
Fig. 2. The structures of host + guest system. (a) The initial structure determined by docking interaction. (b) The final structure after energy minimization.
16 L. Yang et al./Journal of Molecular Structure 444 (1998) 13-20
side view side view
Top view
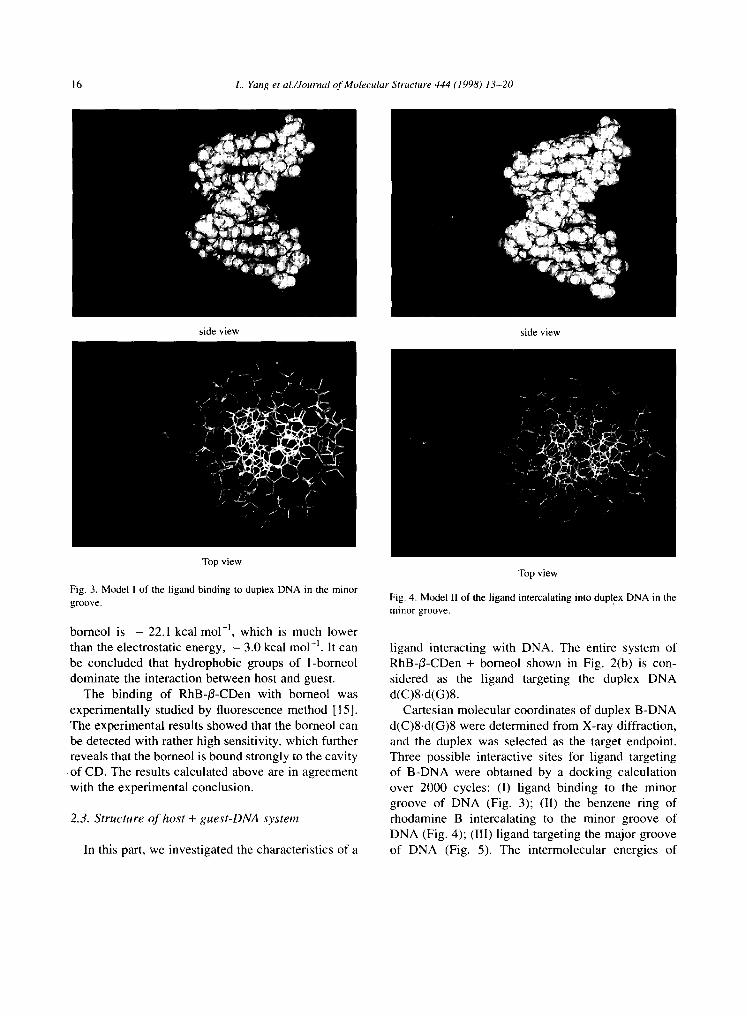
Fig. 3. Model I of the ligand binding to duplex DNA in the minor groove.
borneol is - 22.1 kcal mol-~, which is much lower than the electrostatic energy, - 3.0 kcal mol -l. It can be concluded that hydrophobic groups of 1-borneol dominate the interaction between host and guest.
The binding of RhB-/3-CDen with borneol was experimental ly studied by fluorescence method [ 15]. The experimental results showed that the borneol can be detected with rather high sensitivity, which further reveals that the borneol is bound strongly to the cavity of CD. The results calculated above are in agreement with the experimental conclusion.
2.3. S t ruc ture o f hos t + g u e s t - D N A sys tem
In this part, we investigated the characteristics of a
Top view
Fig. 4. Model II of the ligand intercalating into duplex DNA in the minor groove.
ligand interacting with DNA. The entire system of RhB-i3-CDen + borneol shown in Fig. 2(b) is con- sidered as the ligand targeting the duplex DNA d(C)8.d(G)8.
Cartesian molecular coordinates of duplex B-DNA d(C)8-d(G)8 were determined from X-ray diffraction, and the duplex was selected as the target endpoint. Three possible interactive sites for ligand targeting of B-DNA were obtained by a docking calculation over 2000 cycles: (I) l igand binding to the minor groove of DNA (Fig. 3); (II) the benzene ring of rhodamine B intercalating to the minor groove of DNA (Fig. 4); (Ill) l igand targeting the major groove of DNA (Fig. 5). The intermolecular energies of

L. Yang et al./Journal o f Molecular Structure 444 (1998) 13-20 17
Table 2
In terac t ive energies o f l i g a n d - D N A after dock ing (kcal tool ~)
Mode l Etotal Evdw E~l~
I - 114.98 - 30.16 - 84.82
II - 28.62 - 19.49 - 9.13
III - 1.08 - 0 .49 - 0.58
side v i e w
Top v i ew
Fig. 5. Mode l I11 o f the l igand ta rge t ing to dup lex D N A in the ma jo r
groove .
l igand- recep tor in the three models are all shown in Table 2.
From Table 2, the interaction of ligand and DNA in model Ili, in which the ligand targets to DNA in the major groove, is apparently very weak, and the ligand is obviously observed far away from DNA (Fig. 5). Therefore we consider that it is unlikely that the ligand binds to DNA in the major groove, and we thus concentrated on studying the interactive sites of ligand targeting DNA in the minor groove.
We further studied the l i g a n d - D N A systems of models I and II surrounded by water molecules. The thickness of water molecules around ligand and DNA is chosen as 10 A [16]. The two systems of models 1
and II were minimized in the cff91 force field. Each residue of the duplex DNA has an net charge of - 1 assigned by the parameters of the cff91 force field. The comparat ive experiments were carried out in a phosphate buffer, and the concentration of Na + was too low to have an appreciable impact on the stability of duplex DNA. In the simulation we con- centrated on the ways of interaction between ligand and DNA; therefore we did not add the counterions during the calculation. However, they could be included if more experimental evidence were avail- able. Before calculation, the end residues of DNA were fixed and the entire system was minimized over 300 cycles by steepest descent and conjugate gradient methods, respectively. Then the end residues were released, and the system were subsequently calculated by steepest descent over 300 cycles and conjugate gradient over 1000 cycles without any restrictions. A linearly distance-dependent dielectric constant (e = 1 × r) was used in the calculations. The rms value was less than 0.1 kcal mol -~ ,~-i at the end of calculations.
The inter- and intramolecular energies in the two systems under water are all reported in Table 3. The intermolecular energy of l i g a n d - D N A in model I is - 137.05 kcal tool -], which is much lower than that in model II, - 41.59 kcal mol -~. In model I, the electro- static energy is apparently the main contributor to the
Table 3
The inter-
mol i) and in t ra -molecu la r energies o f the two sys tems (kcal -
Mode l Inter- or intra- Et,,tal Evd,~ Eele
molecu le
I l i g a n d - D N A - 137.05 - 25.41 - 111.64
in t r a -DNA - 207.72 - 140.66 - 67.07
in t ra - l igand 14.94 - 4.81 19.75
II l i g a n d - D N A - 41 .59 - 30.08 - 11.51
in t r a -DNA - 211.28 - 142.39 - 68.89
in t ra - l igand - 15.22 - 0 . 0 3 - 15.18

18 L. Yang et al./Journal of Molecular Structure 444 (1998) 13-20
Fig. 6. Model I of the ligand binding to duplex DNA in the minor groove with some water molecules bridging between bases of DNA and ligand (diagram shows: water molecules; I-borneol; RhB-~3- CDen; duplex DNA).
interaction of ligand and DNA; while the van der Waals interaction dominates in the model II. It is clear that in model II the benzene ring of rhodamine B group intercalates into the minor groove of DNA parallel to the base pairs. In both model 1 and II, the energies of intra-ligand are different. In model I, the intra-ligand energy is 14.94kcalmol i, which is higher than that in model II, - 15.22 kcal tool ~. The energy of the intra-ligand in the free state is - 22.7 kcal mol -~. Thus the intra-energies of the ligand in both models I and II are both higher than that in the free state. The main difference in
Fig. 7. Model II of the ligand intercalating into duplex DNA in the minor groove with some water molecules bridging between bases of DNA and/or ligand (diagram shows: water molecules; l-borneol: RhB-~-CDen; duplex DNA).
intra-ligand energy between the two models is that due to electrostatic interaction, from which it can be inferred that the conformation and charge distribution of the ligand in the two models are different. In order to obtain the strongest interaction between the ligand and DNA, the ligand should change the conformation and charge distribution. Therefore the intra-ligand energy would increase. The stronger the interaction between ligand and DNA, the higher is the intra- ligand energy in the ligand-DNA.
The stereodiagrams of the ligand-DNA system in models I and II with some water molecules are shown in Figs. 6 and 7 respectively. In Fig. 6, the CD cavity of the ligand with the guest borneol binds partially along the minor groove of duplex d(G)8.d(C)8. The CD cavity and rhodamine B group of the ligand form a ditch, which clips the backbone of d(G)8 strand. The minor groove structure is stabilized by complemen- tary electrostatic interactions between the adjacent base-pairs of DNA with the ligand. It can be seen that six hydrogen bonds form between hydroxyl H of CD and 04 of C7, hydroxyl O of CD and H21 of G5, and hydroxyl H and O1P of G7 backbone group. It is also observed that the base-pair planes of duplex DNA attacked by the ligand are distorted, and the end residues are also affected substantially. Most of the normal Watson-Crick hydrogen bonds are broken, which indicates that the duplex DNA is markedly modified to accommodate the ligand. There are some water molecules bridging the bases of the DNA and/or ligand by hydrogen bonds as shown in Fig. 6:
1. water molecules bridging the ligand and bases of DNA by two or three hydrogen bonds, for exam- ple, O(:/3-D-glucose)-.. H-O-H- . .N2:G4 and G6:O4-..H-O(...H:t3-D-glucose)-H---N3:G5;
2. water molecules bridging bases of duplex DNA, for instance, the hydrogen bonds of G3:O6.. .H- O(H)..-H41 :C5 and G3:H2 I. . .(H)O-H..-O4:G4 bridge two bases in different or same strands, water molecules even bridge three bases in the same d(G)8 strand by hydrogen bonds, such as, G6:O4.. .H-O(.. .H22:G4)-H.. .N3:G5;
3. water molecules along the backbone screening the repulsive electrostatic effect by bridging the oxygen atoms in same or different phosphate groups.

L. Yang et al./Journal o f Molecular Structure 444 (1998) 13-20 19
It seems that water molecules are more likely to form hydrogen bonds with bases in the d(G)8 strand than in the d(C)8 strand. From the analysis, we can conclude that water plays an important role in stabi- lizing the entire system of ligand and duplex DNA. In Fig. 7, there are no direct hydrogen bonds between DNA and ligand. However, some water molecules can bridge the ligand and DNA by hydrogen bonds the same as those discussed in Fig. 6.
3. D i s c u s s i o n
Usually, ligands which have a large planar ring insert between two adjacent base-pairs of the DNA. However, in the ligand of the RhB-CDen + borneol system, two end hydrogens of the three-aromatic-ring are replaced by -N(C2Hs)2 groups. The largest verti- cal distance between the end hydrogens of-N(C2Hs)2 group is about 4.50 ,~, while the normal distance between two adjacent base-pair planes is 3.38 ,~. The extending direction of ethyl groups is perpendi- cular to the three-aromatic-ring plane, which probably prevents the planar ring inserting into the adjacent base-pair of DNA. The depth of the CD cavity is
o
about 7.20 A (between two end hydrogens of the 3- glucose unit), while the minor groove width of duplex d(C)8.d(G)8 is about 9.98 A [17]. Thus the CD cavity of the ligand can partially bind in the minor groove of DNA and gives strong electrostatic/hydrogen bond interaction between the ligand and DNA. The benzene ring of rhodamine B is perpendicular to the three- aromatic-ring and has no replaced groups, thus the benzene ring can intercalate parallel into the minor groove. However, the three-aromatic ring could also prevent the benzene ring intercalating deeper.
In duplex DNA d(G)8.d(C)8, we simply define d(G)8 as the G strand and d(C)8 as the C strand.
Table 4 Interaction of strand-ligand in two positions (kcal mol -~)
Model Strand-ligand Etotal E~dw Eele
I C-ligand - 89.19 - 7.71 - 81.48 G-ligand - 47.86 - !7.7 - 30.16
II C-ligand - 20.03 - 14.72 - 5.32 G-ligand - 21.55 - 15.37 - 6.19
From Table 4, it can be seen that the C strand and G strand have different interaction with the ligand in model I, and the interaction between ligand and DNA mainly consists of electrostatic energies of the strand-ligand. The C strand-l igand energy is - 89.19 kcal mo1-1, which is about two times lower than the G strand-l igand energy, - 47.86 kcal mo1-1. The results are in agreement with the fact that the C strand has more direct hydrogen bonds bridging with the ligand than the G strand. It is obvious that in model I the ligand binds to the duplex DNA owing to the electrostatic interaction. In model II, the C strand-l igand energy is almost the same as the G strand-l igand energy. There are no direct hydrogen bonds between ligand and DNA strand, the interac- tions of the l igand-receptor system in model li are dominated by van der Waals energy.
The conformations of the DNA backbone is interesting because it does not fall into the classical category B-DNA, since the structure varies to accom- modate the ligand. Most of the sugar puckering mode of duplex d(C)8.d(G)8 are C2'-endo. The sugar puckering of three end residues of CI, C8 and G2 are all C3'-endo, G1, O4'-endo and G5, Cl ' -exo . The X values, which measure the rotation of the base around the sugar, vary somewhat. In B-DNA, for C2'-endo, - 144 ° -< X ----- - 115° [18]. In duplex d(C)8.d(G)8, the X angles are:
c1 c2 c3 c4 c5 c6 c7 c8 _ 7 7 ° _ 125 ° -80 ° _80 ° _ 131 ° _ 135 ° - 109 ° _68 °
G 1 G2 G3 G4 G5 G6 G7 G8 -162 ° - 109 ° -130 ° - 112 ° - 121 ° - 107 ° -96 ° -114 °
It appears that X for C3 and C4 in the interactive sites changes markedly, while X for C1, C8 and G1 also varies owing to their end position. The DNA molecule has thus changed in many ways to accom- modate the ligand with a bulky group binding in the minor groove.
According to the theoretical results, the system of RhB-3-CDen + borneol targets the duplex DNA d(C)8.d(G)8 in at least two different ways. The more likely way is that the CD of the ligand binds to DNA in the minor groove (model I). This interac- tive site is somewhat similar to the position of polyamides binding to the DNA proposed by Dervan

20 L. Yang et al./Journal of Molecular Structure 444 (1998) 13-20
et al. [6]. In both of them the ligand binds to double- helix DNA in the minor groove and there are some hydrogen bonds between the ligand and DNA. However, for a side-by-side complex of pyrrole- imidazole polyamides in the minor groove, the binding sequence has special recognition of the DNA; while for model I, there are no hydrogen bonds specific recognition between the ligand and DNA.
conformation and charge distribution of the ligand in the ligand-receptor system also change relative to those in free state.
Acknowledgements
This work was supported by the Foundation of Chinese Academy of Sciences and National Natural Science Foundation of China.
4. Conclusion
The possible structures of RhB-13-CDen were investigated by molecular mechanics and dynamics simulation, and the most stable is that in which one side of the three-aromatic-ring exists in the cavity of CD. The guest molecule, 1-borneol, can insert into the CD cavity of the host RhB-~-CDen and the three- aromatic-ring group is completely expelled. The entire system of RhB-/3-CDen + borneol is seen as the ligand targeting the duplex DNA d(C)8.d(G)8. Three interactive sites were obtained using a docking calculation. By comparing the interaction energy, we can conclude that the most likely structure for the ligand targeting DNA is with the CD cavity binding along the minor groove of duplex DNA with hydrogen bonds bridging the ligand and DNA. The other possible interaction site is with the benzene ring of rhodamine B group intercalating into the minor groove parallel to the base pairs of the duplex, and no direct hydrogen bonds between the ligand and DNA. In model I, the interaction between the C strand and the ligand is much stronger than that between the G strand and the ligand, which is due to the electro- static/hydrogen bond interaction. In model II, the interaction between the C strand and the ligand is similar to that between the G strand and the ligand. In both models, there are some water molecules bridging bases of DNA and/or ligand. It is interesting that most water molecules bridge between the G strand of the duplex DNA and the ligand. Water plays an important role in stabilizing the entire system of ligand-receptor. From the analysis of sugar puck- ers and X dihedral angles of DNA, the conformation of DNA varies somewhat to accommodate the ligand with large groups binding in the minor groove. The
References
[ll A.J. Hopfinger, M.G. Cardozo, Y. Kawakami, J. Chem. Soc., Faraday Trans. 91 (1995) 2515.
[2] B. Lambert, J.B. Lepecg, in W. Guschlbauer, W. Saenger (eds.), DNA-Ligand Interactions, From Drugs to Proteins, Plenum, New York, 1986.
[3] A. Delbarre, M. Delepierre, C. Garbay, J. lgolen, J.B. Le Pecq, B.P. Roques, Proc. Natl. Acad. Sci. USA. 84 (1987) 2155.
[4] Y. Nakata, A.J. Hopfinger, Biochem. Biophys. Res. Commun. 95 (1980) 583.
[5] W.J. Trauger, E.E. Braid, B.P. Dervan, Nature (London) 382 (1996) 559,
[6] W.S. Wade, M. Mrksich, P.B. Dervan, J. Am. Chem. Soc. 114 (1992) 8783.
[7] M. Mrksich et al., Proc. Natl. Acad. Sci. USA. 89 (1992) 7586. [8] W.S. Wade, M. Mrkscich, P.B. Dervan, Biochemistry 32
(1993) 11385. [9] M.L. Bender, M. Komiyama, Cyclodextrin Chemistry,
Springer, New York, 1997. [10] Akihiko Veno, Iwao Suzukt, Tetsuo Osa, Chem. Lett., (1989)
1059. [11] A.J. Hopfinger, M.G. Cardozo, Y. Kawakami, in C.L. Propst,
T.L Perun (eds.), Nucleic Acid Targeted Drug Design, Dekker, New York, 1992, p. 151.
[121 W.A. Denny, Anti-Cancer Drug Design 4 (1989) 241. [13] J.W. Lown (ed.), Anthracycline and Anthracenedione-based
Anticancer Agents, Elsevier, Amsterdam, 1988. [14] L.P.G. Wakecin, Med. Res. Rev. 6 (1986) 275. [15] Linjing Yang, Xizeng Feng, Imshik Lee, Chunli Bai, Structure
studies on host + guest recognition sensory system, Journal of Inclusion Phenomena and Molecular Recognition in Chemis- try, in press.
[16] W. Saenger, Principles of Nucleic Acid Structure, Springer, New York, 1984.
[17] D.M. Soumpasis, T.M. Jovi, Computation of Biomolecular Structures, Springer, Berlin, 1993.
[18] G.J. Quigley, A.H.-J. Wang, G. Ughetto, G. van der Marel, J.H. van Boom, A. Rich, Proc. Natl. Acad. Sci. USA 77 (1980) 7204.



![Host–guest interactions between p-sulfonatocalix[4]arene and · Studies on the thermodynamic behavior and recognition pro- ... between the host and guest species. Knowledge (at](https://img.dokumen.tips/doc/110x75/5f0247987e708231d4037a04/hostaguest-interactions-between-p-sulfonatocalix4arene-and-studies-on-the-thermodynamic.jpg)

























