Embed Size (px)
Citation preview

MOLECULAR GENETICS AND METABOLISM 64, 108–118 (1998)ARTICLE NO. GM982683
Molecular Pathophysiology of Cystic Fibrosis Basedon the Rescued Knockout Mouse Model
J. Craig Cohen,* Susan L. Morrow,† R. John Cork,‡ Joseph B. Delcarpio,‡ and Janet E. Larson†
Departments of *Medicine, *Biochemistry and Molecular Biology, and ‡Cell Biology and Anatomy, Louisiana State University,School of Medicine, New Orleans, Louisiana 70112; and †Laboratory of Molecular Biology, Alton Ochsner Medical Foundation,
1516 Jefferson Highway, New Orleans, Louisiana
Received December 19, 1997, and in revised form January 7, 1998
The cystic fibrosis transmembrane conductanceCystic fibrosis transmembrane conductance regu-regulator (cftr) gene was the first gene cloned with-lator (cftr) gene mutations are thought to result inout prior knowledge of the structure and function ofcystic fibrosis due to an absence of the protein’sthe protein and without a chromosomal rearrange-chloride channel. Recently, the lethal intestinalment involving the gene (1). Structural analysis sug-blockage in the cftr knockout mouse was reversed
by a single in utero dose of a recombinant adenovi- gested that it was a cAMP-regulated chloride chan-rus containing the human cftr gene. The rescue of nel. Reports examining CFTR’s chloride channelthese animals did not require continuous expres- function and its relation to disease, however, havesion of the gene and the cAMP-dependent chloride overshadowed observations of the protein’s numer-channel was not permanently restored. These data ous regulatory abilities. CFTR regulates other secre-suggested that cftr was required for normal devel- tory channels (2), mediates vesicular trafficking (3),opment of the intestine but not for normal function
and affects glycosylation (4,5). The two nucleotideof the adult organ. Phenotypic changes in the intes-binding domains (NBD1 and 2) of CFTR share struc-tines and lungs of in utero cftr-treated knockout andtural homology with conserved sequences of G-pro-heterozygous mice revealed that altered develop-teins (6). CFTR is degraded by the ubiquitin–protea-ment was induced. The intestines of the untreatedsome pathway, usually reserved for proteins with aknockout mice were shown to be deficient in both
intracellular calcium and UTP receptors. Both of regulatory role that require rapid degradation (7).these deficiencies were partially corrected in the CFTR expression is greater in the fetus than inrescued knockout mice, whereas treatment of het- the adult. CFTR mRNA and protein show temporalerozygous animals disrupted the normal pattern of and tissue-specific distribution during developmentthese markers. Examination of the lungs of knock- (8–12). The protein is present as early as 7 weeksout cftr (0/0) mice with lectins showed an increase
in the yolk sac, and is expressed in the developingin secreted glycoconjugates containing a(2,6)-sialicrespiratory epithelium starting at 7 weeks of gesta-acid and fucose as compared with control heterozy-tion. In situ hybridization shows CFTR mRNAgotes. The in utero-treated knockouts showed an in-throughout the lung epithelium in the first and sec-crease in this material as well, but it was containedond trimesters (11).in intracellular vesicles. Electron microscopy of
these tissues confirmed the developmental alter- In the developing airways, CFTR distribution fol-ation of secretory cell differentiation in the lungs. lows the cephalocaudal pattern of maturation andThese data show that cftr is required in both the differentiation of epithelial cells (10). The distribu-lung and intestines for normal differentiation of a tion of both CFTR protein and mRNA is nonpolar-secretory cell population and that in its absence ized to one domain of epithelial cells in more imma-these cells fail to develop properly. q 1998 Academic Press
ture lungs. During the terminal saccular phase of
1081096-7192/98 $25.00Copyright q 1998 by Academic PressAll rights of reproduction in any form reserved.
AID MGM 2683 / af13$$$261 06-17-98 05:49:07 bmmas AP: MGM

109MOLECULAR PHYSIOLOGY OF CYSTIC FIBROSIS
development, the localization of the mRNA and pro- ther heterozygous or previously rescued homozygousknockout mice. At 15–16 days of gestation the ani-tein shifts to the more apical distribution of ciliated
cells, similar to that seen in the adult lung (8,10). mals (14) were anesthetized and the fetuses surgi-cally exposed for injection. Virus (109 particles/ml ofThe significance of the presence of CFTR during
development was unclear until our laboratory tran- amniotic fluid) was injected into the amniotic fluid.Animals were allowed to deliver normally followingsiently expressed cftr in utero and permanently cor-
rected the lethal phenotype of the cftr0/0 mouse the surgery.(13). In utero gene therapy did not permanently re-
Virusesplace the cAMP-dependent chloride channel and con-tinuous functioning CFTR was not required for cor- Ad5.CMVlacZ, an adenovirus recombinant car-rection of the lethal intestinal obstruction of cftr rying the lacZ gene with a cytomegalovirus promo-0/0 mice. The first animals rescued by in utero tor, and Av1CF2 (generously provided by GeneticCFTR lived to be older than 1 year. Their untreated Therapy Inc., Gaithersburg, MD), an adenoviruslittermates did not survive into adulthood (ú45 days containing the human cftr gene with a Rous sarcomaof age). These results suggested that the rescue of virus promotor, were grown in S49 cells. Virus puri-the knockout mouse was due to a requirement of fication was accomplished by CsCl centrifugation.CFTR for normal epithelial development in the in-testines. Histologic Examination
Transfer to the developing epithelium was accom-All tissues were fixed in paraformaldehyde for 12–plished by direct injection of a first-generation, repli-
18 h followed by embedding in paraffin for thin sec-cation-defective adenovirus into individual amniotictioning. Periodic acid–Schiff base (PAS) and Alciansacs of rodent fetuses. At the time of infection, rodentblue stains were performed as described by Eversonlung and intestine development was comparable toPearse (16). SNA (Sambicus nigra, elderberry bark)that of a 10- to 20-week-gestation human (14). Thelectin conjugated to biotin (E-Y Laboratories) wasmajor airways had formed, allowing the virus to tar-used to stain lung sections and was developed by theget multipotential columnar and cuboidal stem cells.method of Castells et al. (17).Further differentiation and growth occurred after
the infection. During this period of development, en-Confocal Microscopic Examination of Tissuesdogenous CFTR is highly expressed in these cell
types. Columnar cells are the precursors of the secre- All tissues were sectioned and washed in phos-tory and ciliated epithelium of the conducting air- phate-buffered saline. The incubation with calciumways, and cuboidal cells are the precursors of the green (Molecular Probes) was done with 2 ml/ml 1respiratory epithelium (15). Differentiation of the al- mM Calcium Green-AM (Molecular Probes). Rhoda-veolar epithelium to surfactant-producing type II mine green-UTP (Molecular Probes) was also addedcells started 3–4 days following infection. The alve- at 2 ml/ml 1 mM stock solution. Incubations wereoli in these lungs do not develop until 10 days after for 1–2 h. All samples were examined on a Noraninfection (5 days after birth). Odyssey CLSM with a Nikon Diaphot inverted mi-
In this study, the characterization of lungs and croscope. The software used for the imaging wasintestines from cftr0/0 mice is used to identify Noran’s Intervision running on a SGI Indy worksta-changes in cell structure and function resulting from tion.transient expression of the cftr gene in utero. Thesein utero cftr-dependent alterations demonstrate this Electron Microscopic Examination of Tissuesgene’s essential role in the differentiation of secre-tory cells. Evidence is presented to show that gene Isolated lungs were dissected and immersion fixed
with gentle agitation in 100 ml of 0.1 M sodium caco-dosage of cftr is directly related to normal productionof mucus and lung function. dylate (pH 7.2)-buffered 2.5% glutaraldehyde at am-
bient temperature. Tissue slices were further fixedMATERIALS AND METHODSovernight in fresh fixative, then rinsed three times
In Utero Gene Transfer for 30 min each in chilled 0.1 M cacodylate buffer.Washed tissue blocks were cut into 1-mm cubesFemale heterozygous UNC knockout 1 C57bl/6
fifth-generation backcross mice were bred with ei- and postfixed in 1.0% osmium tetroxide/0.1 M caco-
AID MGM 2683 / af13$$$262 06-17-98 05:49:07 bmmas AP: MGM

110 COHEN ET AL.
dylate buffer (pH 7.2), en bloc stained using 0.5% Intestines from the in utero cftr-treated cftr0/0mice did not exhibit the pathology previously de-aqueous uranyl acetate, dehydrated in acetone, and
infiltrated and embedded in Polybed 812 (Poly- scribed for untreated cftr0/0 mice (18). A compari-son of intestines from the cftr//0 mice (Figs. 1A,sciences). Thins sections were obtained on a Reichert–
Jung Ultracut E ultramicrotome equipped with a 1C) with those of in utero cftr-treated cftr0/0 mice(Figs. 1B, 1D) showed similar intestine histology.diamond knife (Diatome). Thin sections were col-
lected on 150 slotted mesh copper grids and No goblet cell hypertrophy, no crypt dilation, anda normal amount of PAS-positive secretions werepoststained with lead citrate. Sections were exam-
ined in a JEOL 1210 transmission electron micro- observed in the in utero cftr-treated cftr0/0 mice.Thus, rescue from the lethal CF phenotype by inscope (Japanese Electron Optics Laboratory) at 60
kV. Images were recorded on Kodak 4489 EM film. utero cftr treatment is characterized by correction ofthe dysfunctional secretory cell pathology withoutpermanent replacement of the cAMP-dependentRESULTSchloride channel (14).
Rescue from the Lethal CF Phenotype by in UteroGene Therapy Altered Physiology of the Intestines following
in Utero cftr Gene TherapyHeterozygous females of the partially inbredstrain of cftrmlUnc129 1 C57Bl/6 knockout mouse The reversal of the knockout mouse intestinal pa-(fifth-generation backcross) were mated with either thology to that of a normal, heterozygous animal andheterozygous or previously rescued homozygous their survival for more than 1 year without specialknockout males for these experiments. Animals were dietary and environment intervention suggestedtreated in utero with either Av1CF2 (containing hu- that in utero cftr altered secretory cells and/or theirman cftr, provided by Gene Therapy Inc., Gaithers- function. Because calcium plays an integral role inburg, MD) or Ad5.CMVlacZ (expressing b-galactosi- secretion, intracellular calcium stores were evalu-dase) as control. The fetuses were treated at 15– ated. Calcium green-AM, a fluorescent stain for in-16 days of gestation, which is comparable to 10–20 tracellular calcium, was applied to serial crossweeks in human fetal development. This gestation sections from multiple animals of both controlwas shown to result in optimal uptake of the virus (Ad5CMV.lacZ-treated) and in utero cftr-treatedand transgene expression in both the rat and mouse cftr0/0 and cftr//0 mice. All samples were exam-fetus (14). To date, 28 animals in 10 independent ined by confocal microscopy and the results are pre-experiments over the past 12 months were rescued sented in Fig. 2.by in utero gene therapy with cftr. Treatment of knockout mouse intestines with cal-
Repeated characterization of chloride channels in cium green revealed little if any stores of intracellu-the intestines of in utero cftr-treated cftr0/0 mice lar Ca2/ (Fig. 2A). Examination of sections from bothconfirmed our previous finding (13) that no function- the jejunum and ileal regions failed to show any buting cAMP-regulated chloride channel was present. random background staining. In contrast, the intes-Continuous function of CFTR was not required for tines from heterozygous animals showed extensivereversal of the obstructive intestine phenotype of the Ca2/ stores (Fig. 2C). The cells in the intestinalknockout mice. crypts strained intensely, whereas those of the villi
did not stain in control cftr//0 mice. Thus, theReversal of Abnormal Intestine Morphology in thecftr0/0 mice exhibited a marked decrease in intra-cftr0/0 Mousecellular Ca2/ consistent with abnormal secretoryfunction and with improper differentiation of stemThe cftr0/0 mouse dies of intestinal obstruction
before reaching adulthood. Other investigators have cells in the intestinal crypts.Examination of the intestines from multiple inshown that increased mucus secretion and goblet
cell hypertrophy are associated with the obstruction utero cftr-treated cftr0/0 mice rescued from the CFphenotype demonstrated restored intracellular Ca2/(18). Intestines from rescued cftr0/0 and control
cftr//0 mice were fixed, embedded in paraffin, and in cells of the crypt (Fig. 2B). Importantly, the Ca2/-positive cells were located in foci. Because in uteroexamined following Alcian blue–PAS staining. All
animals examined were greater than 75 days of age. gene therapy with a nonintegrating adenovirus can
AID MGM 2683 / af13$$$262 06-17-98 05:49:07 bmmas AP: MGM

111MOLECULAR PHYSIOLOGY OF CYSTIC FIBROSIS
affect only individual stem cells and their progeny, genic receptors were clonally distributed on the villiin sections from both jejunum and ileal regions ofthe focal distribution of crypts positive for intracellu-
lar Ca2/ was consistent with the clonal rescue of a the intestine from rescued knockout mice ranging inage from 75 to 270 days. Thus, in utero cftr genesecretory cell population.
In utero cftr treatment of cftr//0 mice (Fig. 2D) therapy restored normal differentiation of the Ca2/-positive stem cells in the crypt to the P2u receptor-confirms the altered developmental programming of
Ca2/-storing crypt cells. CFTR overexpression dur- positive epithelial cells of the villi.This pattern of intestinal stem cell differentiationing intestinal development disrupted the normal
pattern of differentiation. In contrast to the control was disrupted in the in utero cftr-treated heterozy-gous mice (Fig. 3D). The altered pattern of intracel-heterozygous mice, the in utero cftr-treated cftr//0
mice exhibited a loss of the normal specificity for lular Ca2/-positive cells resulted in UTP receptorshomogeneously distributed throughout the intes-Ca2/-positive cells to the crypt cells. Ca2/-positive
cells were detected throughout the intestines. tines and not localized to the villi as seen in thenormal heterozygous mouse intestines.
Effect of in Utero cftr on Purinogenic Receptorsin the Intestines Mucous Secretions in the Lung following
in Utero RescuePurinogenic receptors in the airways and intestine
are potent regulators of intracellular trafficking and The lungs of control and in utero cftr-treated micewere examined for changes in secretory function.Ca2/ homeostasis (19). Given the results on altered
intracellular Ca2/ in the intestines it was important Histological evaluation with lectins specific for car-bohydrate components of mucus was used to evalu-to evaluate changes in these receptors in in utero
cftr-treated and control intestines, Fluorescent rho- ate changes in knockout, treated knockout, heterozy-gous, and treated heterozygous lungs (Table 1).damine green-labeled UTP, a potent inducer of P2u
receptors, was used to stain sections of intestines Changes in the secretory cells of the airway epi-thelium were obtained as exemplified by stainingfor confocal microscopic examination. Serial sections
from multiple animals were examined and the con- with the lectin SNA (Fig. 4). SNA is specific fora(2,6)-sialic acid, which is known to mediate adher-sistent results are presented in Fig. 3.
The intestines from Ad5CMV.lacZ-treated, cftr ence of Pseudomonas aeruginosa to the respiratorymucosa (20). In utero treatment with cftr resulted in0/0 mice showed little if any specific staining with
the fluorescent labeled UTP (Fig. 3A). Similarly a change in the cellular location of the SNA-positiveglycoconjugates. As seen in Fig. 4A, the SNA-labeledtreated intestines from cftr//0 mice, however,
showed extensive staining (Fig. 3C). Thus, this assay glycoconjugates were localized to the surface epithe-lium in the cftr0/0 animals and were consistentlydemonstrates decreased P2u receptors in the intes-
tine of the CF knockout mouse. In contrast to the increased as compared with heterozygous mice (Fig.4B). In utero treatment with cftr of the cftr0/0 micedistribution in the control heterozygote intestines
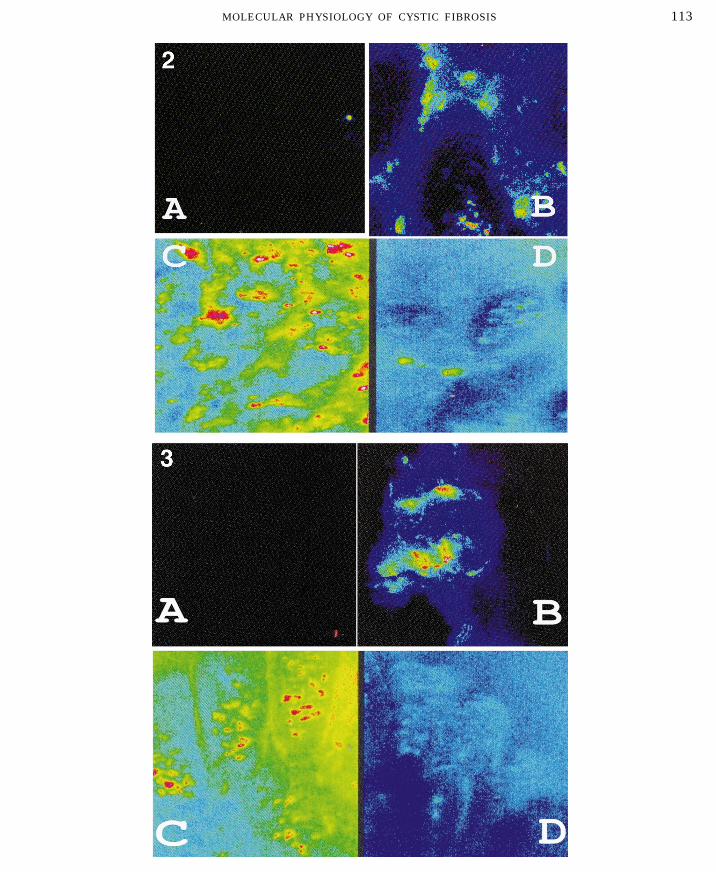
of Ca2/-positive cells localized to the crypt (Fig. 2), resulted in production of cells that stored SNA-posi-tive glycoconjugates as intracellular granules (Fig.fluorescent labeled UTP specifically stained the villi.
P2u receptors were a marker for differentiation of 4C). Cells containing these granules were presentthroughout the airways of the rescued cftr0/0 lungs.stem cells in crypt to mature epithelial cells lining
the villi. In utero cftr treatment of the heterozygotes (Fig. 4D)also resulted in an increase in these glycoconjugates;The in utero cftr-rescued cftr0/0mice showed res-
toration of the P2u receptors (Fig. 3B). Like the intra- however, in these mice the glycoconjugates were se-creted onto the cell surface.cellular Ca2/ restored to the crypt cells, the purino-
FIG. 1. Alcian blue–PAS stain of intestines from normal heterozygous and in utero cftr-treated cftr0/0 mice. The intestines fromanimals greater than 75 days of age were fixed and paraffin blocks sectioned for staining with Alcian blue–PAS and microscopic analysisat 401 (A, B) and 2501 (C, D). (A, C) Normal cftr//0; (B, D) in utero cftr-treated cftr0/0.
FIG. 4. SNA-positive glycoconjugates in the lungs from untreated and in utero cftr-treated heterozygous and knockout mice. Lungsfrom fetuses treated at 15–16 days of gestation with either Ad5.CMVlacZ (A, B) or Av1CF2 (C, D) were fixed and embedded in paraffinblocks, then stained by immunohistochemistry with the lectin SNA. (A, C) cftr0/0; (B, D), cftr//0.
AID MGM 2683 / af13$$$262 06-17-98 05:49:07 bmmas AP: MGM

112 COHEN ET AL.
06-17-98 05:49:07 bmmas AP: MGM
113MOLECULAR PHYSIOLOGY OF CYSTIC FIBROSIS
06-17-98 05:49:07 bmmas AP: MGM

114 COHEN ET AL.
TABLE 1Lectin Staining of Lung Sections from Normal and in Utero cftr-Treated cftr0/0 and cftr//0 Mice
CarbohydrateLectin specificity Specificity Result
SNA (EBL) a(2,6)-sialic acid Affinity to Pseudomonas Increased surface staining in knockout and alteredin mucusa cellular location in rescued knockout
UEA Fucose Possibly increased in CFb Increased surface staining in knockout and alteredcellular location in rescued knockout
MAA a(2,3)-sialic acid Affinity to Pseudomonas No differences in cellular location; possiblein mucusa increase in surface staining in untreated
knockoutMPA N-Acetylgalactosamine Clara and type II cell No differences observed
differentiationRCA Lactose and galactose Type I cell differentiation No differences observedWGA N-Acetylglucosamine Goblet cellsc No differences observedHPA N-Acetylgalactosamine Mucous cellsc No differences observed
a References (20–22).b Reference (23).c Reference (24).SNA (EBL), Sambicus nigra agglutinin (elderberry bark lectin); UEA, MAA, MPA, RCA, Ricinus communis agglutinin; WGA, wheat
germ agglutinin; HPA,
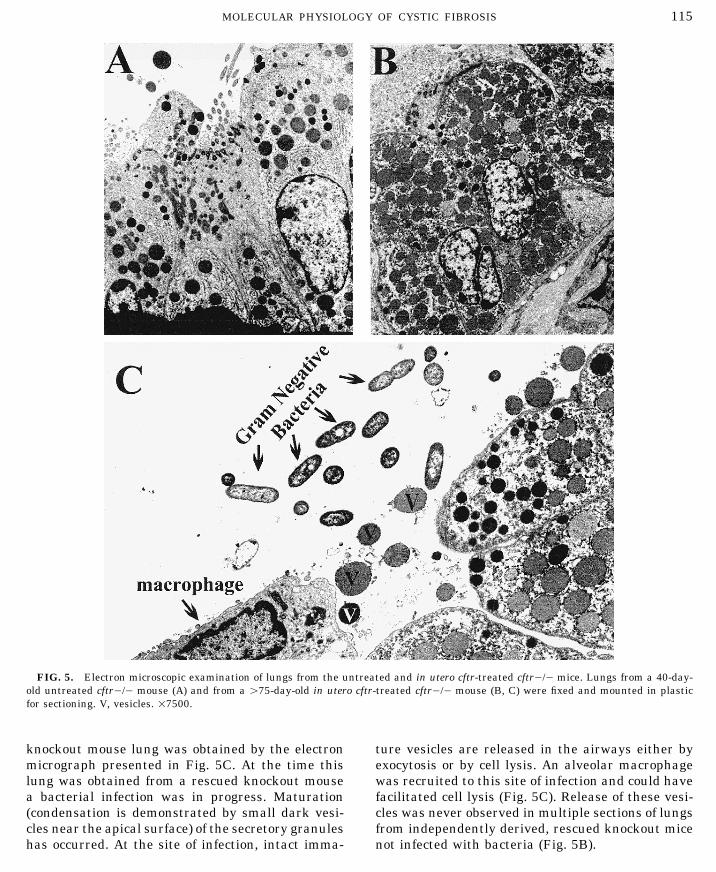
Identical changes were obtained after staining an electron microscope. As shown in Fig. 5, sig-with UEA, a lectin specific for fucose (Table 1). There nificant differences were found between treatedwere no qualitative or quantitative differences be- and untreated tissues. Secretory cells in the smalltween the experimental groups following staining airways of untreated cftr0/0 mice (40 days of age)with the lectins WGA, HPA, MPA, and RCA. WGA exhibited the ultrastructure features typical ofstained both the airway and lung parenchyma dif- Clara cells. Clara cells, the predominant secretoryfusely, whereas RCA stained only the parenchyma cell in the lower airways of mammals, have charac-and very little of the airways. There was a slight teristic secretory granules, a large amount ofincrease in MAA staining in the homozygous knock- smooth endoplasmic reticulum, and a smallout animals as compared with the other groups, but amount of rough endoplasmic reticulum in a basalchanges in cellular location did not occur. location near the nucleus (Fig. 5A). In contrast,
These data showed that in utero cftr treatment examination of airways from in utero cftr-treatedof cftr0/0 mice alters development in the lungs. A mice (ú75 days of age) revealed Clara-like cellspopulation of secretory cells were produced that with a significant increase in vesicles and dilatedstore glycoconjugates within intracellular vesicles. smooth endoplasmic reticulum (Fig. 5B). TheseThese glycoconjugates contain both fucose and cells had little rough endoplasmic reticulum.a(2,6)-sialic acid sugar moieties. These cells were These data are consistent with in utero cftr treat-not prominent in lungs from either cftr//0 or ment alteration of development of the secretorycftr0/0 mice. cells of the cftr0/0mouse lung and confirm resultsElectron Microscopic Examination of Lungs from light microscopy and lectin staining studies
(Fig. 4).Lungs from the rescued and control knockoutmice were fixed and sectioned for examination in Insight into the pathophysiology of the rescued
FIG. 2. Intracellular calcium stores in the intestines from rescued knockout mice. The intestines from cftr0/0 (A), in utero cftr-treated cftr0/0 (B), cftr//0 (C), and in utero cftr-treated cftr//0 (D) were cross sectioned and stained with calcium green for intracellularcalcium. All samples were examined by confocal microscopy. 1100.
FIG. 3. UTP receptors in the intestines from rescued knockout mice. The intestines from cftr0/0 (A), in utero cftr-treated cftr0/0(B), cftr//0 (C), and in utero cftr-treated cftr//0 (D) were cross sectioned and stained with rhodamine green-UTP. All samples wereexamined by confocal microscopy. 1100.
AID MGM 2683 / af13$$$262 06-17-98 05:49:07 bmmas AP: MGM
115MOLECULAR PHYSIOLOGY OF CYSTIC FIBROSIS
FIG. 5. Electron microscopic examination of lungs from the untreated and in utero cftr-treated cftr0/0 mice. Lungs from a 40-day-old untreated cftr0/0 mouse (A) and from a ú75-day-old in utero cftr-treated cftr0/0 mouse (B, C) were fixed and mounted in plasticfor sectioning. V, vesicles. 17500.
knockout mouse lung was obtained by the electron ture vesicles are released in the airways either byexocytosis or by cell lysis. An alveolar macrophagemicrograph presented in Fig. 5C. At the time this
lung was obtained from a rescued knockout mouse was recruited to this site of infection and could havefacilitated cell lysis (Fig. 5C). Release of these vesi-a bacterial infection was in progress. Maturation
(condensation is demonstrated by small dark vesi- cles was never observed in multiple sections of lungsfrom independently derived, rescued knockout micecles near the apical surface) of the secretory granules
has occurred. At the site of infection, intact imma- not infected with bacteria (Fig. 5B).
AID MGM 2683 / af13$$$262 06-17-98 05:49:07 bmmas AP: MGM

116 COHEN ET AL.
DISCUSSION could be due to increased replication and/or survivalof stem cells necessary for population of the lungand intestines with sufficient numbers of secretoryThe cftr gene was the first gene cloned via a posi-
tional cloning strategy without a chromosomal rear- stem cells.A possible model for the role of cftr in cellularrangement involving the clone gene. Because of the
prominence of salty secretions in the clinical presen- differentiation is the glial cell missing gene (gcm)(28). This gene functions as a binary switch for glialtation of cystic fibrosis, the chloride channel function
was quickly identified in this protein. The dominant cell differentiation. In its absence, glial cells fail todevelop. Overproduction of gcm results in excess pro-paradigm for the molecular pathophysiology of this
disease then focused on this function and its replace- duction of glial cells.Data presented in this report document a secre-ment became the primary goal of gene therapy.
Perhaps a better perspective might be gained by tory cell missing (scm) function for the cftr gene. Inthe knockout animal and possibly the cystic fibrosisconsidering how one would view this cloned gene if
the association with cystic fibrosis and salt balance patient, the absence of functional CFTR in utero re-sults in a general dysfunction of secretory cells inwere not known. Genes known to be expressed at
high levels in the developing fetus include a long list multiple organs. In the mouse, the primary lethalphenotype is associated with development of the in-of growth factors and oncogenes. In fact, one of the
widely held criteria for developmental proteins is testines, but changes in the lung (Figs. 4, 5) andpancreas (unpublished observation of authors) alsotheir temporal- and spatial-specific pattern of ex-
pression in fetal tissues (25). Examination of the occur. In humans the lung, intestines, pancreas, andvas deferens all may be affected by failure of scm.published expression patterns of CFTR in fetal tis-
sues is consistent with a developmental function When scm activity is overproduced in heterozygousanimals, it results in altered secretory functions in(10,11).
Blau and Baltimore (26) suggested that an active both the lung and intestine (Figs. 2–4). In heterozy-gotes, overpopulation of these organs with scm-de-control mechanism governs differentiation, re-
sulting in the expression of specific genes through pendent secretory cells accounts for these changes.Several characteristics of the cell requiring scmthe dynamic interaction of regulator proteins at a
given time. The rescue of the cftr knockout mouse expression in utero were established by comparisonof the untreated and rescued knockout mice. Theseby transient in utero cftr expression reported here
and previously (13) suggests the participation of cells have a secretory function in both the lungs andintestines. In both of these tissues, the defect in theCFTR at 15–16 days of gestation in the develop-
mental regulation of the intestine, the organ most knockout mouse appears related to the regulationof glycoconjugate secretion. In the lung, both lectinaffected in the knockout mouse model.
Blau (27) suggested that proteins involved in the staining (Fig. 4) and electron micrographs showedan increase in intracellular glycoconjugates follow-regulation of differentiation could be identified by
their ability to alter phenotype and produce a gain ing rescue by in utero cftr gene therapy. In the intes-tines, defects in both intracellular calcium and puri-in function rather than a loss of function. A distin-
guishing characteristic of in utero treatment with nogenic receptors of the knockout mouse are cor-rected following rescue (Figs. 2, 3) Intracellular Ca2/cftr was the correction of regulated secretion in the
epithelium. This function was rescued in both the is involved in maturation and packaging of glycocon-jugates (29), and purinogenic receptors are involvedlung and intestine regardless of the phenotypic clas-
sification of the secretory cell type. In the intestines, in the regulation of mucus release (30). Thus, find-ings in the intestines and lungs of in utero cftr-the normal intestinal histology was restored (Fig. 1).
Intracellular calcium stores and UTP receptors are treated cftr0/0mice are consistent with altered reg-ulation of glycoconjugate secretions.altered in the untreated knockout mice, and these
functions were clonally rescued in the in utero cftr- The developmental changes observed here follow-ing intrauterine cftr gene therapy in both the lungstreated cftr0/0 mice (Figs. 2, 3). In the lung, an
enhanced population of cells lining the airway store and intestines and those presented elsewhere in therat suggest a mechanism for the molecular physiol-glycoconjugate materials intracellularly (Figs. 4, 5).
The mechanism by which cftr influences differentia- ogy of cystic fibrosis that is independent of the chlo-ride channel deficiency. We propose that CFTR,tion was not revealed by these data. However, it
AID MGM 2683 / af13$$$262 06-17-98 05:49:07 bmmas AP: MGM

117MOLECULAR PHYSIOLOGY OF CYSTIC FIBROSIS
quence homologies between nucleotide binding regions ofthrough its scm function, plays an essential role inCFTR and G-proteins suggest structural and functional sim-the differentiation of mature secretory cells, e.g., theilarities. FEBS Lett 366:87–91, 1995.Clara cell in the mouse. In its absence, the gene(s)
7. Ward CL, Omura S, Kopito RR. Degradation of CFTR byrequired for mucus production is activated; however, the ubiquitin–proteasome pathway. Cell 83:121–127, 1995.the pathway(s) required for the regulation of mucous
8. McGrath SA, Basu A, Zeitlin PL. Cystic fibrosis gene andsecretion fails to develop. Evidence presented in the protein expression during fetal lung development. Am J Re-intestines suggests that purinogenic receptors and spir Cell Mol Biol 8:201–208, 1993.intracellular calcium regulatory pathways are es- 9. Tizzano EF, O’Brodovich H, Chitayat D, Benichou J-C,
Buchwald M. Regional expression of CFTR in developingsential components. The outcome of this improperhuman respiratory tissues. Am J Respir Cell Mol Bioldevelopment is a cell that continuously produces mu-10:355–362, 1994.cus, resulting in intestinal blockage in the knockout
10. Gaillard D, Ruocco S, Lallemand A, Dalemans W, Hinnraskymouse and perhaps enhanced lung infections in hu-J, Pucelle E. Immunohistochemical localization of cystic fi-mans. brosis transmembrane conductance regulator in human fe-
The electron micrograph in Fig. 5C illustrates our tal airway and digestive mucosa. Pediatr Res 36:137–143,proposed mechanism of regulated mucus production 1994.in the lung. The mature Clara-like cell from an in 11. McCray PB, Reenstra WW, Louie E, Johnson J, Bettencourt
JD, Bastacky J. Expression of CFTR and presence of cAMP-utero cftr-treated cftr0/0 mouse in this micrographmediated fluid secretion in human fetal lung. Am J Physiolappears to be induced by exposure to Gram-negative262:L472–L481, 1992.rods. Mature glycoconjugate-containing vesicles are
12. Harris A, Chalkely G, Goodman S, Coleman L. Expression ofreleased either by exocytosis or by cell lysis andthe cystic fibrosis gene in human development. Development
cause a local buildup of these products in the airway 113:305–310, 1991.where the bacteria are bound. The macrophage in 13. Larson JE, Morrow SL, Happel L, Sharp L, Cohen JC. Rever-this micrograph was either recruited by the released sal of the cystic fibrosis phenotype by somatic stem cell geneglycoconjugates or facilitated their release as an aid therapy in utero. Lancet 349:619–620, 1997.to phagocytosis. According to data presented in Figs. 14. Sekhon HS, Larson JE. In utero gene transfer into the pul-
monary epithelium. Nature Med 1:1201–1203, 1995.4 and 5 (A and B), in the cftr0/0 lung, this pathway15. Ten Have-Opbroek AAW. The development of the lung inis absent, leading to impaired innate immunity by
mammals: An analysis of concepts and findings. Am J Anatcontinuous overproduction that depletes reserves162:201–219, 1981.and impedes phagocytosis. Currently our laboratory
16. Everson Pearce AC. Histochemistry: Theoretical and Ap-is directed at further elucidating this pathway.plied. Vol. 2: Analytical Technology, New York: ChurchillLivingstone, 1985.
REFERENCES 17. Castells MT, Ballesta J, Madrid JF, Aviles M, Mavtinez-Menarguez JA. Characterization of glycoconjugates in devel-oping rat respiratory system by means of conventional and1. Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R,lectin biochemistry. Histochemistry 95:419–426, 1991.Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou J, Drumm
ML, Iannuzzi MC, Collins FS, Tsui LC. Identification of the 18. Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Bou-cystic fibrosis gene: Cloning and characterization of comple- cher RC, Smithies O, Koller BH. An animal model for cysticmentary DNA. Science 245: 1059–1065, 1989. fibrosis made by gene targeting. Science 257:1083–1088,
1992.2. Guggino WB, Egan M, Schwiebert E. Mechanisms for theinteraction of CFTR with other secretory Cl- channels. Ped- 19. Kim KC, Lee BC. P2 purinoceptor regulation of mucin re-atr Pulmonol S12:115, 1995. lease by airway goblet cells in primary culture. Br J Pharma-
col 103:1053–1056, 1991.3. Bradbury NA, Jilling T, Berta G, Sorscher EJ, Bridges RJ,Kirk KL. Regulation of plasma membrane recycling by 20. Plotkowski MC, Bajolet-Laudinat O, Puchelle E. CellularCFTR. Science 256:530–531, 1992. and molecular mechanisms of bacterial adhesion to respira-
tory mucosa. Eur Respir J 6:903–916, 1993.4. Kube D, Perez A, Davis PB. Quantitative fluorescent micros-copy reveals altered cell surface glycoconjugates on 9HTEo0 21. Ramphal R, Houdret N, Koo L, Lambin G, Roussel P. Differ-cells transfected with the regulatory domain of CFTR or ences in adhesion of Pseudomonas aeruginosa to mucin gly-DF508 CFTR. Pediatr Pulmonol S12:238, 1995. copeptides from sputa of patients with cystic fibrosis and
chronic bronchitis. Infect Immun 57:3066–3071, 1989.5. Weyer P, Barasch J, Al Awqati Q, Ausiello DA, Brown D.Immunolocalization of two sialyltransferases is altered in 22. Lambin G, Lhermitte M, Klein A, Houdret N, Scharfman A,polarized LLC-PK1 epithelial cells expressing DF508 CFTR. Ramplhal R, Roussel P. The carbohydrate diversity of hu-Pediatr Pulmonol S12:238, 1995. man respiratory mucins: A protection for the underlying mu-
cosa? Am Rev Respir Dis 144:S19–S24, 1991.6. Monavalin P, Dearborn DG, McPherson JM, Smith AE. Se-
AID MGM 2683 / af13$$$263 06-17-98 05:49:07 bmmas AP: MGM

118 COHEN ET AL.
23. Carnoy C, Ramphal R, Scharfman A, Lo-Guidice JM, Hou- 26. Blau HM, Baltimore D. Differentiation requires continuousregulation. J Cell Biol 112:781–783, 1991.dret N, Klein A, Galabert C, Lamblin G, Roussel P. Altered
carbohydrate composition of salivary mucins from patients 27. Blau HM. Hierarchies of regulatory genes may specify mam-with cystic fibrosis and the adhesion of Pseudomonas aerugi- malian development. Cell 53:673–674, 1988.nosa. Am J Respir Cell Mol Biol 9:323–334, 1993.
28. Hosoya T, Takizawa K, Nitta K, Hotta Y. Glial cell missing:24. Mazzuca M, Lhermitte M, Lafitte J-J, Roussel P. Use of
A binary switch between neuronal and glial cell determina-lectins for detection of glyconjugates in the glandular cells
tion in Drosophilia. Cell 82:1025–1026, 1995.of the human bronchial mucosa. J Histochem Cytochem
29. Thorn P. Spatial domains for Ca2/ signaling in secretory30:956–966, 1982.epithelial cells. Cell Calcium 20:203–214, 1996.25. Mercola M, Stiles CD. Growth factor superfamilies and
mammalian embryogenesis. Development 102:451–460, 30. Conigrave AD, Jiang L. Review: Ca2/-mobilizing receptorsfor ATP and UTP. Cell Calcium 17:111–119, 1995.1988.
AID MGM 2683 / af13$$$263 06-17-98 05:49:07 bmmas AP: MGM





























