Embed Size (px)
Citation preview

Molecular Modeling:Molecular Modeling:Molecular MechanicsMolecular Mechanics
C372C372
Introduction to Introduction to Cheminformatics IICheminformatics II
Kelsey ForsytheKelsey Forsythe

Guidelines for UseGuidelines for Use
What systems were used to parameterizeWhat systems were used to parameterize How is energy calculatedHow is energy calculated What assumptions are used in the force What assumptions are used in the force
fieldfield How has it performed in the pastHow has it performed in the past

TransferabilityTransferability
AMBER (Assisted Model Building AMBER (Assisted Model Building Energy Refinement)Energy Refinement) Specific to proteins and nucleic acidsSpecific to proteins and nucleic acids
CHARMM (Chemistry at Harvard CHARMM (Chemistry at Harvard Macromolecular Mechanics)Macromolecular Mechanics) Specific to proteins and nucleic acids Specific to proteins and nucleic acids Widely used to model solvent effectsWidely used to model solvent effects Molecular dynamics integratorMolecular dynamics integrator

TransferabilityTransferability
MM? – (Allinger et. al.) MM? – (Allinger et. al.) Organic molecules Organic molecules
MMFF (Merck Molecular Force MMFF (Merck Molecular Force Field)Field) Organic moleculesOrganic molecules Molecular Dynamics Molecular Dynamics
Tripos/SYBYLTripos/SYBYL Organic and bio-organic moleculesOrganic and bio-organic molecules

TransferabilityTransferability
UFF (Universal Force Field)UFF (Universal Force Field) Parameters for all elements Parameters for all elements Inorganic systemsInorganic systems
YETI YETI Parameterized to model non-bonded Parameterized to model non-bonded
interactionsinteractions Docking (AmberDocking (AmberYETI)YETI)

How is Energy Calculated How is Energy Calculated
Valence TermsValence Terms Cross TermsCross Terms Non-bonding TermsNon-bonding Terms
Induced Dipole-Induced DipoleInduced Dipole-Induced Dipole Electrostatic/Ionic (Permanent Dipole) System not far Electrostatic/Ionic (Permanent Dipole) System not far
from equilibrium geometry (harmonic)from equilibrium geometry (harmonic) Energy is ? Energy is ?
Strain Energy (E=0 at equilibrium bond length/angle)Strain Energy (E=0 at equilibrium bond length/angle) Field Energy (Energy due to Non-bonding terms)Field Energy (Energy due to Non-bonding terms) Atomistic Heats of Formation (Parameterized so as to Atomistic Heats of Formation (Parameterized so as to
yield chemically meaningful values for yield chemically meaningful values for thermodynamics)thermodynamics)
K. Gilbert: This is only in the MM?-type force fieldsK. Gilbert: This is only in the MM?-type force fields

AssumptionsAssumptions
Hydrogens often not explicitly Hydrogens often not explicitly included (intrinsic hydrogen methods)included (intrinsic hydrogen methods) ““Methyl carbon” equated with 1 C and 3 Methyl carbon” equated with 1 C and 3
HsHs System not far from equilibrium System not far from equilibrium
geometry (harmonic)geometry (harmonic) Solvent is vacuum or simple dielectricSolvent is vacuum or simple dielectric

Modeling Potential energyModeling Potential energy
€
U(r) U(req ) −dU
dr r= req
(r − req ) +1
2
d2U
dr2
r= req
(r − req )2
€
−1
3
d3U
drr= req
(r − req )3 ....+1
n!
dnU
drn
r= req
(r − req )n€
=
€
≈

Modeling Potential energyModeling Potential energy
€
−dU
dr r= req
(r − req ) +
€
U(r) ≈1
2
d2U
dr2
r= req
(r − req )2 ≡1
2kAB (r − req )2
€
U(req )
€
U(r) 1
2
d2U
dr2
r= req
(r − req )2
€
≈
0 at minimum0
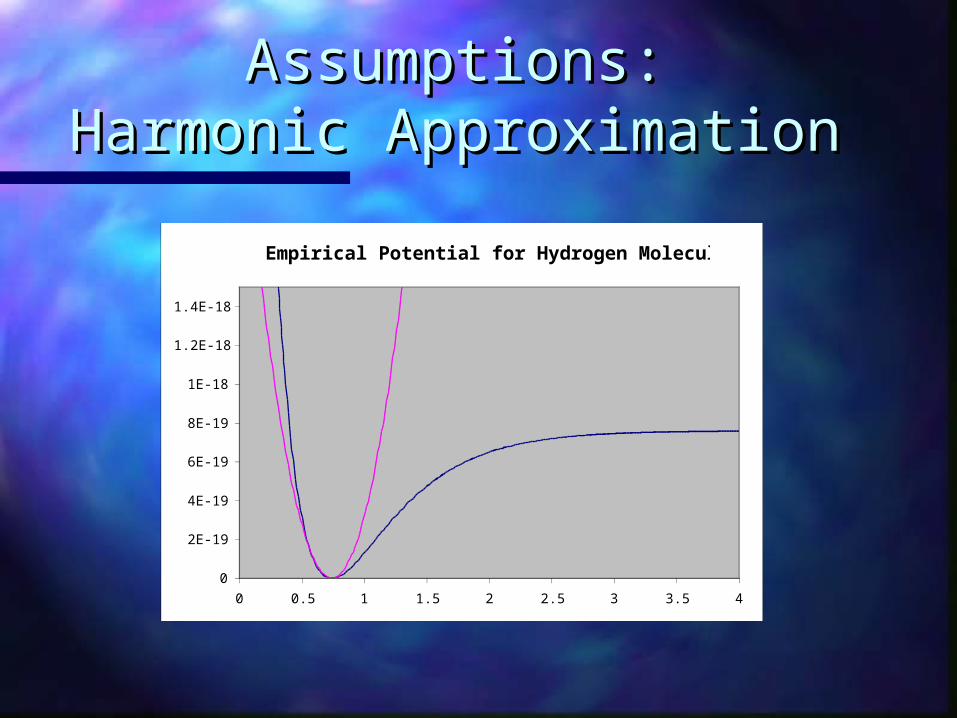
Assumptions:Assumptions:Harmonic ApproximationHarmonic Approximation
8.35E-28 8.77567E+14 20568787140 2.03098E-18 1.05374E-188.35E-28 8.77567E+14 20568787140 1.77569E-18 9.66155E-198.35E-28 8.77567E+14 20568787140 1.54682E-18 8.82365E-198.35E-28 8.77567E+14 20568787140 1.34201E-18 8.02375E-198.35E-28 8.77567E+14 20568787140 1.15913E-18 7.26185E-198.35E-28 8.77567E+14 20568787140 9.96207E-19 6.53795E-198.35E-28 8.77567E+14 20568787140 8.51451E-19 5.85205E-198.35E-28 8.77567E+14 20568787140 7.23209E-19 5.20415E-198.35E-28 8.77567E+14 20568787140 6.09973E-19 4.59425E-198.35E-28 8.77567E+14 20568787140 5.10362E-19 4.02235E-198.35E-28 8.77567E+14 20568787140 4.2311E-19 3.48845E-198.35E-28 8.77567E+14 20568787140 3.47061E-19 2.99255E-198.35E-28 8.77567E+14 20568787140 2.81155E-19 2.53465E-198.35E-28 8.77567E+14 20568787140 2.24426E-19 2.11475E-198.35E-28 8.77567E+14 20568787140 1.75987E-19 1.73285E-198.35E-28 8.77567E+14 20568787140 1.35031E-19 1.38895E-198.35E-28 8.77567E+14 20568787140 1.0082E-19 1.08305E-198.35E-28 8.77567E+14 20568787140 7.26787E-20 8.15147E-208.35E-28 8.77567E+14 20568787140 4.99924E-20 5.85247E-208.35E-28 8.77567E+14 20568787140 3.22001E-20 3.93347E-208.35E-28 8.77567E+14 20568787140 1.87901E-20 2.39447E-208.35E-28 8.77567E+14 20568787140 9.29638E-21 1.23547E-208.35E-28 8.77567E+14 20568787140 3.29443E-21 4.56475E-21
Empirical Potential for Hydrogen Molecule
0
2E-19
4E-19
6E-19
8E-19
1E-18
1.2E-18
1.4E-18
0 0.5 1 1.5 2 2.5 3 3.5 4

Assumptions:Assumptions:Harmonic ApproximationHarmonic Approximation
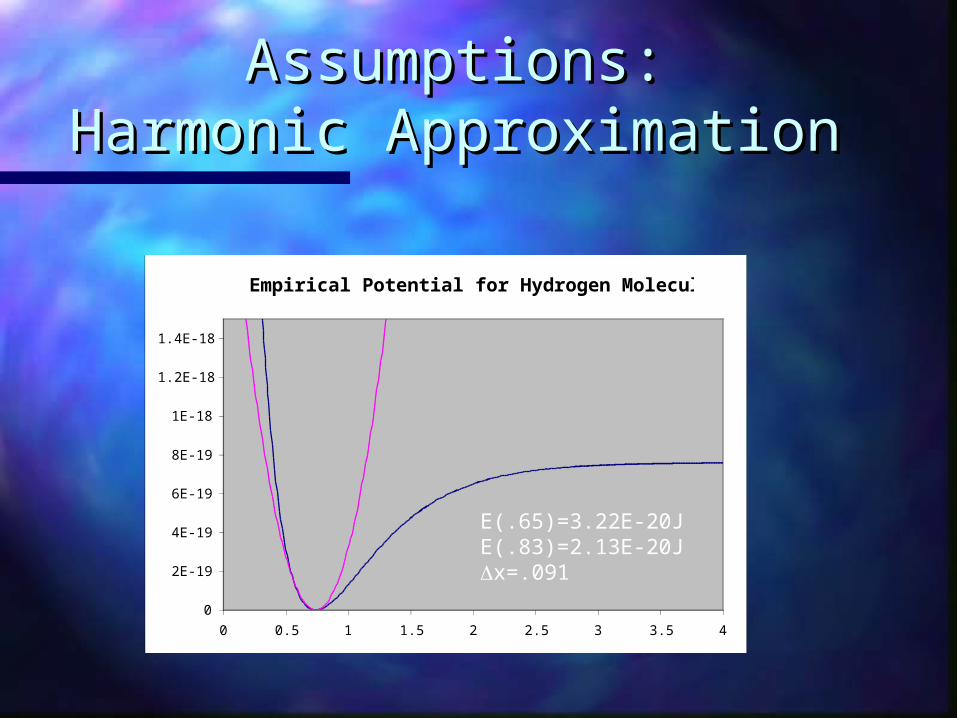
Determining k?
€
E(x0 − Δx) ≅ E(x0) +1
2
d2U
dx 2
x0
((x0 − Δx) − x0)2
E(x0 + Δx) ≅ E(x0) +1
2
d2U
dx 2
x0
((x0 + Δx) − x0)2
d2U
dx 2
x0
≅E(x0 − Δx) + E(x0 + Δx) − 2E(x0)
(Δx)2
8.35E-28 8.77567E+14 20568787140 2.03098E-18 1.05374E-188.35E-28 8.77567E+14 20568787140 1.77569E-18 9.66155E-198.35E-28 8.77567E+14 20568787140 1.54682E-18 8.82365E-198.35E-28 8.77567E+14 20568787140 1.34201E-18 8.02375E-198.35E-28 8.77567E+14 20568787140 1.15913E-18 7.26185E-198.35E-28 8.77567E+14 20568787140 9.96207E-19 6.53795E-198.35E-28 8.77567E+14 20568787140 8.51451E-19 5.85205E-198.35E-28 8.77567E+14 20568787140 7.23209E-19 5.20415E-198.35E-28 8.77567E+14 20568787140 6.09973E-19 4.59425E-198.35E-28 8.77567E+14 20568787140 5.10362E-19 4.02235E-198.35E-28 8.77567E+14 20568787140 4.2311E-19 3.48845E-198.35E-28 8.77567E+14 20568787140 3.47061E-19 2.99255E-198.35E-28 8.77567E+14 20568787140 2.81155E-19 2.53465E-198.35E-28 8.77567E+14 20568787140 2.24426E-19 2.11475E-198.35E-28 8.77567E+14 20568787140 1.75987E-19 1.73285E-198.35E-28 8.77567E+14 20568787140 1.35031E-19 1.38895E-198.35E-28 8.77567E+14 20568787140 1.0082E-19 1.08305E-198.35E-28 8.77567E+14 20568787140 7.26787E-20 8.15147E-208.35E-28 8.77567E+14 20568787140 4.99924E-20 5.85247E-208.35E-28 8.77567E+14 20568787140 3.22001E-20 3.93347E-208.35E-28 8.77567E+14 20568787140 1.87901E-20 2.39447E-208.35E-28 8.77567E+14 20568787140 9.29638E-21 1.23547E-208.35E-28 8.77567E+14 20568787140 3.29443E-21 4.56475E-21
Empirical Potential for Hydrogen Molecule
0
2E-19
4E-19
6E-19
8E-19
1E-18
1.2E-18
1.4E-18
0 0.5 1 1.5 2 2.5 3 3.5 4
Assumptions:Assumptions:Harmonic ApproximationHarmonic Approximation
E(.65)=3.22E-20JE(.83)=2.13E-20Jx=.091

Assumptions:Assumptions:Harmonic ApproximationHarmonic Approximation
€
d2U
dx 2
x0
≅E(x0 − Δx) + E(x0 + Δx) − 2E0
Δx 2
d2U
dx 2
x0
=(3.22 ×10−20 J) + (2.126 ×10−20 J)
(.091A ×1m
1×1010 A)2
d2U
dx 2
x0
= 6.45 ×102 kg
s2≡ k

Assumptions:Assumptions:Harmonic ApproximationHarmonic Approximation
€
d2U
dx 2
x0
= 6.45 ×102 kg
s2≡ k
HO − −− > k = mω2
∴ ω =k
μ=
6.45 ×102 kg
s2
1.67 ×10−27 kg= 6.215 ×1014 Hz
ν =ω
2π= 9.891×1013 Hz ≡ 3.30 ×103cm−1(Exp : 4.395 ×103cm−1)

AssumptionsAssumptions
Hydrogens often not explicitly Hydrogens often not explicitly included (intrinsic hydrogen methods)included (intrinsic hydrogen methods) ““Methyl carbon” equated with 1 C and 3 Methyl carbon” equated with 1 C and 3
HsHs System not far from equilibrium System not far from equilibrium
geometry (harmonic)geometry (harmonic) Solvent is vacuum or simple dielectricSolvent is vacuum or simple dielectric

Assumptions:Assumptions:solvent effectssolvent effects
Christensen, O. B. et. al, Phys. Rev. B. 40, 1993 (1989)
H2 in Pd
DFT
Empirical Potentials for Hydrogen Molecule
0
1
2
3
4
5
6
7
8
9
10
0 0.5 1 1.5 2 2.5

Intermolecular/atomic Intermolecular/atomic modelsmodels
General form:General form:
Lennard-Jones Lennard-Jones
€
V = V (r) + V (ri,rj ) + V (ri,rj ,rk ) + .....i< jj<k
N
∑i< j
N
∑
€
V (rij ) = 4εσ
r
⎛
⎝ ⎜
⎞
⎠ ⎟
12
1 2 3 −
σ
r
⎛
⎝ ⎜
⎞
⎠ ⎟6 ⎡
⎣ ⎢
⎤
⎦ ⎥
1 2 3
⎡
⎣
⎢ ⎢ ⎢ ⎢
⎤
⎦
⎥ ⎥ ⎥ ⎥
Van derWaals repulsionVan derWaals repulsion London AttractionLondon Attraction
0at which distance
depth well
=−−
ABVσε

MMFF EnergyMMFF Energy
Electrostatics (ionic compounds) Electrostatics (ionic compounds) D – Dielectric ConstantD – Dielectric Constant - electrostatic buffering constant- electrostatic buffering constant
( )nij
jiticelectrosta
RD
qqE
δ+=

MMFF EnergyMMFF Energy
Analogous to Lennard-Jones 6-12 Analogous to Lennard-Jones 6-12 potentialpotential London Dispersion ForcesLondon Dispersion Forces Van der Waals RepulsionsVan der Waals Repulsions
⎪⎭
⎪⎬⎫
⎪⎩
⎪⎨⎧
−+⎪⎭
⎪⎬⎫
⎪⎩
⎪⎨⎧
+= 2
07.0
07.1
07.0
07.17*7
7*7
*
*
ijij
ij
ijij
ijijVDW
RR
R
RR
RE ε
The form for the repulsive part has no physical basis and is for computational convenience when working with largemacromolecules. K. Gilbert: Force fields like MM2 which is used for smaller organic systems will use a Buckingham potential (or expontential) which accurately reflects the chemistry/physics.

Pros and ConsPros and Cons
N >> 1000 atomsN >> 1000 atoms Easily Easily
constructedconstructed
AccuracyAccuracy Not robust enough Not robust enough
to describe subtle to describe subtle chemical effectschemical effects HydrophobicityHydrophobicity Excited StatesExcited States RadicalsRadicals
Does not Does not reproduce quantal reproduce quantal naturenature

CaveatsCaveats
Compare energy differences NOT energiesCompare energy differences NOT energies Always compare results with higher order Always compare results with higher order
theory (theory (ab initioab initio) and/or experiments) and/or experiments





























