Embed Size (px)
Citation preview

doi:10.1006/jmbi.2001.4670 available online at http://www.idealibrary.com on J. Mol. Biol. (2001) 309, 737±749
Molecular Basis for Immune Complex Recognition: AComparison of Fc-Receptor Structures
Peter Sondermann*, Jens Kaiser and Uwe Jacob
Max-Planck-Institut fuÈ rBiochemie, AbteilungStrukturforschung, AmKlopferspitz 18aD-82152 MartinsriedGermany
E-mail address of the [email protected]
Abbreviations used: FcR, Fc-recepimmunoglobulin.
0022-2836/01/030737±13 $35.00/0
Once antigen is opsonised by IgG it is removed from the circulation byFcg-receptor expressing cells. Fcg-receptors are type I transmembranemolecules that carry extracellular parts consisting of two or three immu-noglobulin domains. Previously solved structures of Fc-receptors revealthat the N-terminal two Ig-like domains are arranged in a steep angleforming a heart-shaped structure. The crystal structure of the FcgRIII/hIgG1-Fc-fragment demonstrated that the Fc-fragment is recognisedthrough loops of the C-terminal receptor domain of the FcgRIII. As theoverall structure of the FcRs and their Ig ligands are very similar wemodelled the Ig complexes with FcgRI, FcgRII and FceRIa based on theFcgRIII/hIgG1-Fc-fragment structure. The obtained models are consistentwith the observed biochemical data and may explain the observed speci-®city and af®nities.
# 2001 Academic Press
Keywords: Fc-receptor; IgG; Fc-fragment; refolding; crystal structure
*Corresponding authorIntroduction
Immune complexes are rapidly removed fromthe blood circulation by Fc-receptor (FcR) expres-sing cells. This process guarantees that germsrecognised by antibodies do not spread in thebody. Besides endocytosis of these particles alsoother ef®cient defence mechanisms are triggeredby the Fc-part of the antibody, like the regulationof antibody production by B cells, antibody depen-dent cell-mediated cytotoxicity (ADCC) or therelease of in¯ammatory mediators that may acti-vate other immune competent cells.1 These effectsensure an ef®cient and broad immune responsemediated by the membrane-bound FcRs, which areexpressed on all immune competent cells in a dis-tinct pattern. It depends on the respective cell type,the IgG subclass and the type of FcR as to whicheffector function is ®nally elicited.
At the same time FcRs are in clinical focus sincethey are also important mediators of the disregu-lated immune system. FceRIa is the high af®nityIgE receptor expressed on mast cells. It is the pri-mary effector in allergic reactions and asthma.FcgRs play a role in infections, autoimmune reac-tions and cancer. Finally, it is discussed that thesemolecules may modulate antibody-mediated
ing author:
tor; Ig,
enhancement of viral infections.2 Thus, the positionof FcRs as a gateway to both cellular and humoralaspects of the immune cascade makes them anattractive target for immunotherapies.
The FcRs for the main serum immunoglobulinIgG can be subdivided into three classes and rep-resent type I transmembrane proteins. Their ecto-domains consist of either two (FcgRII, CD32;FcgRIII, CD16; KD � 10ÿ5-10ÿ7 M) or three (FcgRI,CD64; KD � 10ÿ8 M) related immunoglobulindomains, whereby the higher af®nity of the FcgRIis attributed to the third extra domain. FcgRII hasa wide distribution on immunocompetent cells andoccurs in two forms, FcgRIIa and FcgRIIb, theirextracellular regions sharing 93 % sequenceidentity. These two forms of the receptor can bedistinguished by their binding characteristics toIgG subclasses. In addition, two allotypes of theFcgRIIa isoform are known. The FcgRIIa highresponder (HR)3 carries in comparison to theFcgRIIa low responder (LR) the single amino acidexchange Arg131His in the proposed bindingregion, which alters its af®nity to mouse IgG1 andhuman IgG2. Besides the transmembrane form ofFcgRIII (FcgRIIIa) present on T cells and NK cells,a glycosyl phosphatidyl inositol (GPI) anchoredform is expressed in high numbers on neutrophils(FcgRIIIb). Two speci®c receptors for IgE exist: thelow af®nity receptor FceRII, CD23 (KD � 10ÿ6 M)and the two Ig-domain containing high af®nityreceptor FceRIa (KD � 10ÿ10 M). Only the lattershares homology with the FcgRs. Additionally to
# 2001 Academic Press
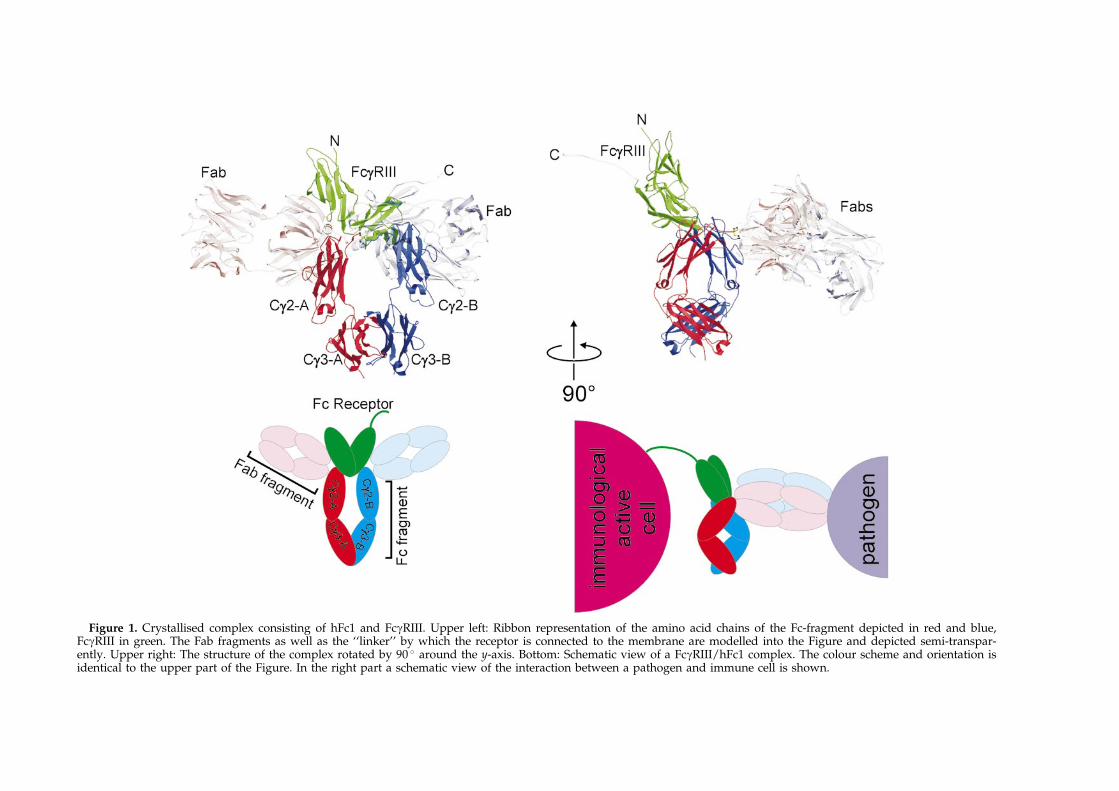
Figure 1. Crystallised complex consisting of hFc1 and FcgRIII. Upper left: Ribbon representation of the amino acid chains of the Fc-fragment depicted in red and blue,FcgRIII in green. The Fab fragments as well as the ``linker'' by which the receptor is connected to the membrane are modelled into the Figure and depicted semi-transpar-ently. Upper right: The structure of the complex rotated by 90 � around the y-axis. Bottom: Schematic view of a FcgRIII/hFc1 complex. The colour scheme and orientation isidentical to the upper part of the Figure. In the right part a schematic view of the interaction between a pathogen and immune cell is shown.

Crystal Structures of Fc -Receptors 739
membrane-bound forms, soluble FcRs (sFcgRI,sFcgRII, sFcgRIII4 and sFceRIa5) are present inserum and other body ¯uids. They are producedeither by proteolysis, alternative splicing or by sep-arate genes and a physiological relevance inimmune regulation seems possible.4
FcRs contain a presumably helical transmem-brane region and a cytoplasmic tail which mediatesthe signal into the cell after the receptors are cross-linked through binding of immune complexes. Thecytoplasmic regions contain either an immunore-ceptor tyrosine-based activation motif (ITAM) as inFcgRIIa or the respective inhibitory motif (ITIM) asin FcgRIIb. FcgRIIIa, FcgRI and FceRIa are associ-ated with ITAM containing g-chains which per-form the signalling activity of these receptors. Bothof these motifs interact with SH2 domain contain-ing proteins to initiate signal transduction.6 It hasbeen discussed whether these accessory chainsalter the af®nity of the receptor to the antibody.7
The g-chain is a homodimeric, small type Itransmembrane proteins that carry a short extra-cellular part consisting of only ®ve amino acid resi-dues, including the cysteine that mediateshomodimer formation. The transmembrane part ofthis molecule interacts via an Asp/Arg pair8 withthe receptor while the intracellular part is capableof signal transduction.
FcgRIIIb lacks a transmembrane and a cyto-plasmic region and an immune response cannot betriggered directly by this receptor. It is still notclear how FcgRIIIb transduces signals into the cellas no direct contact to the cytoplasm exists. Differ-ent proteins are discussed like complement recep-tor 3,9 the formyl peptide receptor10 or FcgRIIa11
that may associate with FcgRIIIb for signal trans-duction.
The FcgRs represent together with the FceRIa agroup of homologue receptors with sequence iden-tities in the range of 50 %. The crystal structures ofthe FcgRIIa12 FcgRIIb13 and FceRIa14 have recentlybeen solved and show that these receptors foldinto two Ig-like domains which arrange into astrongly bent overall structure. It has been demon-strated by mutagenesis studies that the amino acidresidues involved in Ig binding are located on theinterstrand loops of the C-terminal domain and onthe connector to the N-terminal domain.15,16 On theother hand comparative binding studies with IgGfrom different species suggested an involvement ofthe lower hinge of IgG (residues Leu234-Ser239)and a nearby region including the interstrand loopAla327-Ile333 relevant for FcgRI-binding.17 How-ever, the molecular basis of Ig binding could onlyrecently be shown by the solution of the crystalstructure of sFcgRIIIb in complex with the Fc-frag-ment of human IgG118 (hFc1, Figure 1). The com-plex structure shows that both hFc1 and FcgRIIIrearrange upon complex formation and that inaddition to the mentioned loops, parts of thestrands are also involved in binding.
Here we describe the crystal structure of the sol-uble FcgRIII, analyse the structural rearrangements
upon complex formation and discuss a new crystalform of FcgRIIa. Furthermore, we modelled therelated FcgRIIa/IgG1, FcgRI/IgG1 and the FceRIa/IgE complexes on the basis of the sFcgRIIIb/hFc1complex crystal structure.
Results and Discussion
Production, characterisation andcrystallisation of soluble FcgggRs
Until recently soluble FcRs could only be pro-duced using expensive, time consuming and com-plex eukaryotic expression systems. We recentlydescribed the ®rst approach to produce sFcgRIIb inprokaryotic cells employing inclusion bodyexpression and subsequent refolding events.19 Thepuri®cation procedure employs besides theinclusion body preparation and refolding pro-cedure an af®nity column on hIgG Sepharose anda size exclusion chromatography step.
sFcgRIIa and sFcgRIII can easily be producedusing the same approach. However, the overallyield varies due to different expression levels andrefolding recovery. While sFcgRIIb shows both ahigh expression and refolding rate, sFcgRIIa wasonly weakly expressed but with a comparablerefolding rate under identical conditions assFcgRIIb. sFcgRIII was expressed in large amountsbut the refolding rate was signi®cantly lower. ForsFcgRIIb, the overall yield was 6 mg per litre cul-ture volume and around 10 % of the total inclusionbody protein. The yield of FcgRIIa and FcgRIII was3 mg per litre culture volume and both moleculescould be produced in suf®cient amounts to allowtheir crystallisation and subsequent structure deter-mination.
The crystal structure of FcgggRIII
In Figure 1 we show the solved structure of theFcgRIII in complex with hFc118 with the Fab frag-ments modelled to the Fc-fragment. The orientationof the hinge region implies that the Fab arms arequite sharply bent and may obtain a perpendicularorientation towards hFc1. In such an arrangementwhich was already proposed by Burton20 the Fabarms would have maximum sterical freedom torecognise bulky antigens when the Fc-fragment isoriented parallel to the membrane of the FcgRIIIexpressing cell. The nine amino acid linkerbetween the transmembrane domain and domain 2of FcgRIII is long enough to allow for such a bentconformation (lower part of Figure 1).
The structure of sFcgRIII was solved by themethods of molecular replacement using the struc-ture of FcgRIIb as search model with one moleculein the asymmetric unit. The ®nal model containsthe amino acid residues Pro3 to Gly171 whichwere clearly de®ned in the electron density. Theterminal residues Met ÿ 4 to Leu2 could not betraced in the electron density and are disordered.The model is of good quality (see Table 1) and

Table 1. Data collection statistics
Type of receptor FcgRIIa (after one refinement round) FcgRIII
Space group C2 P22121
Cell constants a � 81.3 AÊ , b � 50.0 AÊ , c � 55.5 AÊ ,a � g � 90 �, b � 128.3 �
a � 36.7 AÊ , b � 60.3 AÊ , c � 85.6 AÊ ,a � b � g � 90 �
Completeness (Overall/last shell) (%) 90.5/69.0 94.7/72.3Rmerge overall/last shell (%) 11.1/24.2 11.4/50.0No. of unique reflections/multiplicity 3417/2.4 6725/3.0Resolution range used (AÊ ) 50-3.0 40-2.5Molecules per asymmetric unit/solventcontent (%)
1/46 1/48
Rfac/Rfreea (%) 24.6/32.7 19.5/26.1
RMSD bonds/angles n.d. 0.012/1.64RMSD bonded B-factors n.d. 5.857No. protein atoms/average B-factor (AÊ 2) 1365/20.8 1376/29.8No. solvent atoms/average B-factors (AÊ 2) 0/n.d. 67/41.0
a Rfree: 5 % of the re¯ections were used as a reference data set and were not included in the re®nement.
740 Crystal Structures of Fc -Receptors
none of the residues are located in the disallowedregion of the Ramachandran plot. FcgRIII obeysthe typical heart-shaped arrangement of the twoimmunoglobulin-like domains, as seen for otherFcRs (Figure 2). The two immunoglobulin domainsobey an identical overall fold with a ®ve-stranded(A0, C, C0, F, G) and a three-stranded (A, B, E)b-sheet facing each other to form a sandwich-likestructure. Strand A leaves the three-strandedb-sheet and crosses over to the ®ve-stranded b-sheet representing strand A0. This fold carries fea-tures of the Ig I-type and Ig C2-type but has so faronly been observed for the FcR family and the kill-er inhibitory receptors (KIR).21 The C0/E, F/G andB/C loops run next to each other to connect theb-strands from one b-sheet to the other. This loopregion and the C0 strand of domain 2 represent,together with the connector between the twoIg-like domains, the binding region for the Fc-fragment. These loops are clearly de®ned in the
Figure 2. Stereo ribbon representation of the sFcgRIII struacid residues of FcgRIII which contact IgG are shown in baldomain are coloured magenta and those contacting the Cg2-Bas cyan balls and the disulphide bridges in yellow. The termtively for the N-terminal domain in black and for the C-teFigure 1 (upper left), except that it is rotated by approximathe paper. The Figure was created using MOLSCRIPT37 and
electron density and a prominent patch of threehistidine residues (116, 131, 132) is found. Thereceptors are not glycosylated due to their Escheri-chia coli origin. Five potential glycosylation sitesexist but only Asn159 is found in the close vicinityof the binding region and the extent of glycosyla-tion added to this site might thus in¯uence theaf®nity to the Fc-fragment.22
The crystal structure of FcgggRIIa
The structure of FcgRIIa was already solved byMaxwell et al.12 and obeys the same overall fold asdescribed for FcgRIII. The reported structure orig-inates from crystals of space group P21212 and themolecules form a dimer about one of the crystallo-graphic dyads. This arrangement resulted in adominant crystal contact and it was speculatedthat a dimer of FcgRIIa recognises the IgG. In thisdimer the molecules contact each other via the
cture with residues relevant for IgG binding. The aminol-and-stick representation. Residues contacting the Cg2-A
domain green. Potential glycosylation sites are depictedini are labelled and the b-strands are numbered consecu-rminal domain in blue. The orientation is the same astely 180 � around the axis perpendicular to the plane ofRASTER3D.38

Crystal Structures of Fc -Receptors 741
CC0FG sheets and the C/C0 loop of the C-terminaldomain.
We started a crystallisation screen in an attemptto ®nd new crystallisation conditions and reportthe crystallisation of FcgRIIa in space group C2with one molecule in the asymmetric unit. Byapplying the symmetry operators of this spacegroup and inspection of the resulting dimers itbecomes evident that none of them resembles thedimer arrangement observed by Maxwell et al. Fur-thermore, the solved crystal structure of theFcgRIII/hFc1 complex shows involvement of theC0 strand of the receptor in complex formation.Due to the close homology of the FcgRs a commonbinding mode of Fc-fragment recognition can beassumed (see below). We showed that FcgRII ismonomeric in solution as proven by size exclusionchromatography of both baculo-derived,23 thusglycosylated, and E. coli material19 and concludethat the previously observed dimer originates fromthe requirements of crystal packing.
Our crystals of space group C2 diffracted to only2.8 AÊ in comparison to the crystals reported byMaxwell et al. that diffracted to 2.0 AÊ . Except forthe residues involved in crystal contacts essentiallythe same structure is found and due to the minorresolution the structure re®nement process was notcompleted.
FcRs obey similar structures
The overall fold of two Ig-like domains in a shar-ply bent structure is shared by the FcgRIII,FcgRIIa/b and FceRIa as shown by crystal struc-tures (Figure 3(a)). The binding region of the FcRto the Fc-fragments has been identi®ed by the sol-ution of the FcgRIII/hFc118 and the FceRIa/IgE-Fc-fragment24 complex structures. It consists mainlyof rather ¯exible loops that rearrange upon com-plex formation. The B/C, C0/E and F/G loops aswell as the C0 strand which carry the contact resi-dues in the FcgRIII/hFc1 structure contain con-served amino acid residues which realise commonbinding principles as well as sequence variationsfor the generation of speci®city (Figure 3(b)). Trp87and Trp110 are located on the B/C loop and theconnector to the ®rst domain and representtogether with Ile85 and Val155 a hydrophobicpocket. The tryptophan residues are conserved inthe FcR family including FceRIa and represent theprimary binding site for the Fc-fragment. Anexposed proline residue in the IgG subclasses(Pro329) and the IgE binds between these trypto-phan residues (``proline sandwich''). Sequence pos-ition 155 is a solvent-exposed hydrophobic residuein all FcRs and represents the binding site for theimportant IgG1 residue Leu235 (not in IgE).Another prominent region of FcgRIII consists ofthree histidine residues, His131 and His132 on theC0/E loop and His116 is the C-terminal residue ofstrand C. This region is the most variable amongthe FcRs and it is in contact with the lower hingeregion of the Fc-fragment (residues 234-238), which
is also not conserved among the IgGs and IgE. Thesite is used for the generation of speci®city in thecomplex structure and it is considerably morehydrophobic in other FcRs (see below for adetailed discussion). Finally the strand C0 ofFcgRIII contains the important solvent-exposedTyr129, which may vary to Phe in other FcRs andmakes multiple contacts to the Fc-fragment. Thisresidue is responsible for the tight binding of thereceptor to the hinge peptide and limits the spacefor the side-chains of Fc-fragment residues in somereceptor/Ig pairings (see below).
Structural rearrangements uponFc-fragment binding
It has been shown that FcgRIII rearranges uponcomplex formation.18 The angle between the twoIg-like domains opens by 10 degrees and thesolvent-exposed Trp95 rotates into the opened cleftand forms a new hydrogen bond to Tyr14. Theinward rotation of that Trp liberates solvent mol-ecules but the opening of the interdomain cleftresults in an additional solvent-exposed surface.Taken together the solvent-exposed surface reducesby 137 AÊ 2 between the closed and the complexedform of the receptor when both effects are con-sidered. The contact area between the receptor andthe Fc-fragment is 895 AÊ 2. Table 2 compares thedistances of the atoms involved in hydrogenbridges across the domains. The observed contactsare modi®ed when the interdomain cleft opens.Furthermore, Table 2 shows that FcgRIII formsonly three hydrogen bonds close to the interdo-main hinge between the N and C-terminal domainin contrast to FcgRIIa/b (seven and eight hydrogenbonds, respectively), in which a tryptophan atposition 95 is not found. Therefore, it remainsquestionable whether the homologue receptorsundergo a comparable transformation upon com-plex formation (indeed the recently publishedFceRIa/IgE-Fc-fragment complex structure showsthat the receptor binds in a closed conformation, asdiscussed below).
The loops of FcgRIII involved in Fc-fragmentbinding are somewhat ¯exible and alter theirconformation upon complex formation. The B/Cloop of the C-terminal domain of FcgRIII is stabil-ised by a hydrogen bond between His108 andAsn112 in the complex which does not form in thefree structure. In the uncomplexed structures ofFcgRIIa and FcgRIIb this hydrogen bond is alreadyformed between the histidine and Asp112. How-ever, in the FcgRIII/hFc1 complex structure thecanonical interaction is found and the B/C loopfollows quite closely the path as in the freeFcgRIIa/b. Therefore we expect that the B/C loopsof the FcgRII isoforms adopt a similar confor-mation in respective Fc-fragment complexes. Theconformation of the C0/E loop is not signi®cantlychanged when FcgRIII binds to the Fc-fragment. Itis found in a slightly different conformation inFcgRIIa and FcgRIIb but it seems likely that this

Figure 3. (a) Superposition of FcR structures. The structures of the respective FcRs were superimposed with a leastsquare ®t algorithm using the Ca atoms of the molecules and depicted as coils. The FcRs are coloured as indicated inthe inset and the intradomain disulphide bridges are shown in yellow. The orientation is the same as in Figure 2. (b)Structure-based sequence alignment of FcgRIII and hIgG1 with their homologues. Amino acid residues of FcgRIII(upper part) that contact amino acid residues of hFc1 (lower part) in the FcgRIII/hFc1 structure are connected withred or blue and green lines for contacts of FcgRIII with Cg2-A or Cg2-B domains. Potential hydrogen bonds betweenFcgRIII and Cg2-B are shown in green. The homologue FcRs and immunoglobulins are aligned to both complex com-ponents. The degree of homology correlates with the depth of the colouring.
742 Crystal Structures of Fc -Receptors

Table 2. Interdomain contacts of FcRs
a Measured between the Ca atoms of residue 85 (centre), and residues 72 and 142.Non-productive hydrogen bonds above a threshold of 4.00 AÊ are shaded grey.
Crystal Structures of Fc -Receptors 743
loop will adopt the conformation found in thecomplexed FcgRIII as a reorientation would allowproductive interactions with the Fc-fragment(Figure 3(b), see below). In the free FceRIa thisloop with the amino acid residues 128 to 133adopts a very different conformation but in thecomplexed form the canonical structure is found.The backbone of the F/G loop varies only littlebetween the free and the complexed FcgRIII, butthe side-chains of the solvent-exposed residuesrearrange as in the other loops.
Models of FcR-Ig complexes
FcRs and Igs represent homologous proteinfamilies. Therefore, we modelled the possibleFcR-Ig complexes by replacing the differing aminoacids on the framework of the FcgRIII/hFc1complex structure. Figure 4 shows the modelledinteraction sites of the FcgRIIa-HR/IgG1, the
FcgRI/IgG1 and the FceRIa/IgE complex. In theFcgRIII/hFc1 complex structure18 FcgRIII binds theFc-fragment by inserting its F/G loop of the seconddomain deep into the cleft between the two CH2domains (Cg2-A and Cg2-B, Figure 1). This is theregion where the lower hinge residues Leu234 toPro238 approach the Fc-fragment. The horseshoe-shaped Fc-fragment opens asymmetrically by 7 AÊ
(distance measured between the Pro329 residues)and the FcgRIII rearranges (see above). The asym-metry introduced dictates that only one receptorcan be bound by the otherwise identical two chainsof the Fc-fragment. Three major sites of interactionwere mentioned: the proline sandwich whichhas been described above, the lower hinge of theCg2-A and the same region on the Cg2-B domain.The lower hinge region Leu234-Leu-Gly-Gly-Pro238 of the Cg2-A domain is bound with Leu235contacting a small hydrophobic pocket consistingof the methyl groups of Thr113 and Ala114 and

744 Crystal Structures of Fc -Receptors
the side-chain of Val155. On the Cg2-B domainLeu235 is in contact with the histidine patch ofFcgRIII and the signi®cance of this leucine for com-plex formation is not re¯ected. The two glycineresidues are bound in a narrow channel and areessential for steric and conformational reasons. InIgG2 the lower hinge residues are replaced byPro234-Val-Ala-Gly-Pro238 and consequently IgG2is bound with reduced af®nity to all FcgRs.
FcgRIIa shares 44 % sequence identity withFcgRIII in the extracellular region. In the recog-nition site for Leu235 of the Cg2-A hinge theFcgRIII-residues Thr113, Ala114 and Val155 arereplaced by Lys, Pro and Ile in all FcgRII isoforms(Figure 4(a)). The binding site on FcgRIIa forLeu235 of the Cg2-B hinge is due to the His116Valand His132Leu (Ser in FcgRIIb) more hydrophobicin comparison to FcgRIII. The replacement of theFcgRIII-histidines for smaller residues bringsLeu235 of Cg2-B closer towards FcgRII and intohydrophobic contact. This movement is hinderedby the His131Arg exchange in the FcgRIIa-HRallele. Arg131 does not ®nd suf®cient space in itsbinding pocket, thus explaining the generallylower af®nity of all hIgGs to the HR allele.25 AsFcgRIIb also carries an arginine at position 131 itslower af®nity towards hIgG23 can be explained ina similar way.
The rigid Pro238 of the hinge motif of IgGs isreplaced by serine in mouse IgG1 (comparablewith the valine exchange in IgE, see below), whichliberates space for Arg131 of FcgRIIa-HR and adds¯exibility to the hinge peptide resulting in a higheraf®nity of the HR variant to mouse IgG1 comparedto the LR allele.3
FcgRI carries three Ig domains and the twoN-terminal domains are homologous to the tandemdomain receptors (45 % sequence identity withinits ®rst two domains to FcgRIII). Despite the con-siderably lower homology, the differing residuescould be modelled onto the FcgRIII/hFc1 frame-work with only minor adjustments. The prolinesandwich is conserved but the hydrophobic charac-ter of the FcgRI binding site for Leu 235 of theCg2-A hinge is pronounced (Figure 4(b)). Ala114,Val155 and Thr158 of FcgRIII are replaced by Leu,Met and His, respectively. The side-chain of His158may form additional polar interactions to Ser239and Gly237. The importance of the hinge residueLeu235 has been shown in mutagenesis experi-ments26 and with isolated oligopeptides.27 FcgRIdoes contribute hydrophobic residues to bindLeu235, on both the Cg2-A and the Cg2-B hinge.On the latter, Leu235 is bound to a considerablymore hydrophobic region in comparison to FcgRIII.The histidine residues 116 and 132 in FcgRIII arereplaced for tyrosine and tryptophan in FcgRI,which allows for a tighter binding of Leu235, andthe residues are larger as in FcgRII (valine and leu-cine). The hydrogen bond network between theCg2-B domain and the receptor is altered due tothe Lys117Asn exchange. Taken together, weassume that the hinge peptide is bound in a com-
parable conformation as in FcgRIII, but with ahigher af®nity due to the higher hydrophobicity inthis region.
The FceRIa/IgE-Fc-fragment complex structurewas solved by Garman et al.24 and a comparison ofthis structure with a model based on the FcgRIII/hFc1 structure is found in the next section.
A comparison of the FceeeRIaaa/IgE and theFcgggRIII/hFc1 structures
The ectodomains of FceRIa are related to FcgRIIIas are the other FcgRs (42 % identity). Besides arather unrelated N-terminal domain Ce2, the Fc-fragment of IgE consists of the domains Ce3 andCe4, which are homologous to the Ig domains Cg2and Cg3 of IgG. A related binding mode of FceRIato IgE as in the FcgRIII/IgG1 example was foundin the crystal structure with the receptor binding tothe connector between the Ce2 and Ce3 domains ofIgE.24 This is comparable to the recognition of thehinge peptide in the FcgRIII/hFc1 complex. Asuperposition of the two complex structures showsthat the IgE-Fc-fragment is found in a confor-mation related to the open form of hFc1 in thecomplex. The C-terminal domain of the complexedFcgRIII aligns with the C-terminal domain of com-plexed FceRIa but the angle between the N and C-terminal domains differs (Table 2) and FceRIa isfound in the same closed state as in the free formcomparable to the domain arrangement in the free,isolated FcgRIII structure.
Thus the overall binding mode of the two com-plex structures is related but a closer inspection ofthe conformation of the hinge residues in bothstructures reveals some differences. FceRIa is con-siderably more hydrophobic on its surface than theFcgRs and tends to aggregate in solution whendeglycosylated. A patch of four tryptophan resi-dues (Trp87, Trp110, Trp113 and Trp156) is foundin close vicinity. Trp110 and Trp87 belong to theproline sandwich. Trp113 replaces a lysine residuefound in FcgRIII and ®lls the space between thereceptor and the hinge of Ce3-A. The importantLeu235 of the IgG1 hinge is replaced by proline inIgE which liberates space between the two hingepeptides. This space is ®lled by Trp156 and Leu158of the FceRIa. The binding mode of the Ce3-Bhinge peptide is thought to be related to the bind-ing mode of mIgG1 to the FcgRIIa-HR. Pro238 isreplaced by valine which liberates space for Tyr131(corresponding to the His131Arg exchange in theFcgRIIa-HR/LR dualism) by increasing the confor-mational ¯exibility of the hinge peptide. The hingepeptide is moved away from the receptor due tothe rather bulky Tyr131 and can bend more shar-ply due to the Pro238Val replacement. Gly236 isno longer necessary and the replacing arginine isfound in an upward orientation. On the Ce3-A sidethe arginine residue orientates downwards andforms a polar interaction to Gln157. The hydrogennetwork around Gly237 is intact on the Ce3-B side.Lys117 of the receptor is conserved and forms

Figure 4. Models of FcR-Ig com-plexes. Close up of the modelledinteraction sites of IgG1 withFcgRIIa-HR (a) and FcgRI (b) andof IgE with FceRIa (c) depicted asstick models. The models were pro-duced by grafting mutations of theFcRs and IgE, respectively, onto theFcgRIII/hFc1 framework followedby minimisation of the resultingstructures. The colour scheme (FcR,green; Cg2-A/Ce3-A, red; Cg2-B/Ce3-B, blue) and the orientation ofthe structures is comparable to thatin the upper left part of Figure 1.Differing residues in FcgRIIa-HRand FcgRIII are labelled asmutations in (a) e.g. P114A. (a) Inaddition to the modelled inter-action site of FcgRIIa-HR with hFc1a superposition with the crystalstructure of the FcgRIII/hFc1 com-plex (grey) is shown. In (c) besidesthe coloured FceRIa/IgE-Fc-frag-ment complex structure only themost diverging Ce3-B hinge pep-tide of the model is shown in grey.The two CHAPS molecules foundin the structure of the FceRIa/IgE-Fc-fragment complex that wereused as crystallisation additive areshown in magenta. Possible hydro-gen bridges are depicted as brokenlines.
Crystal Structures of Fc -Receptors 745
polar interactions to Gly237 and Asp265, which arefound in IgE and IgG. The considerably bulkierand more hydrophobic residues of FceRIa can betolerated due to compensating replacements in theIgE molecule and are nicely buried from solvent inthe model.
However, as two CHAPS molecules used ascrystallisation additive are found within the bind-ing site which may alter the binding mode of thehinge peptides to FceRIa we started to model alsothis complex without the CHAPS molecules likethe FcgR/IgG complexes. Most of the predictionsare in agreement with the experimental structurewhich was not expected in view of the relativelylow degree of sequence similarity between IgG andIgE and the FcgRs and FceRIa, respectively. Onlythe proposed position of the Pro235 and succeed-
ing residues mainly in the Ce3-B hinge signi®cantlydiffer between the structure and our model butthis region is occupied by CHAPS molecules in thecrystal structure, which may distort the confor-mation of the contact interface (Figure 4(c)).
FcRs may contain multiple binding sites
Figure 5 shows a comparison of solid surfacerepresentations of the FcgRs emphasising the dif-fering surface hydrophobicity distribution. Thepart of the receptor surface involved in IgG1 com-plex formation is indicated and consists of anextended hydrophobic area which is consistentlyfound in all the solved receptor structures (Figure 5,top view) but also in the modelled structure ofFcgRI. The hydrophobicity in this area is especially

746 Crystal Structures of Fc -Receptors
pronounced in the FceRIa model. FceRIa shows thehighest af®nity to its binding partner and themajor part of the binding energy results from theliberation of the solvent molecules upon removalof that surface from solvent. FcgRIII carries aunique hydrophilic patch resulting from the threehistidine residues (Figure 5, front view) not presentin the other FcRs. Thus the binding region for theCg2-B hinge residue Leu235 is less hydrophobic(see also Figure 4). Consequently, therapeutic anti-bodies that are designed for low af®nity to theFcgRs by an exchange of this leucine to the chargedglutamate will affect FcgR af®nity but will notabolish FcgRIII binding. We therefore propose adifferent set of mutations that should completelyabrogate immune stimulation (see below). TheFceRIa-structure shows an overall higher surfacehydrophobicity in comparison to the other FcRs.This hydrophobicity is compensated by the exten-sive glycosylation of this molecule observed inmammalian expression systems. Indeed glycosyla-tion enhances solubility and stability of target mol-ecules and E. coli-expressed FceRIa can be refoldedlike the other FcRs but is not stable in solutionwhen the refolding buffer is removed.
Several interactions of FcRs with other proteinshave been described. Next to the common Fc-fragment recognition an association of FcRs withg-chains via the transmembrane region has beenshown.8 These g-chains cannot perform signaltransduction for the GPI anchored FcgRIIIb andsecondary contacts of FcRs with the complementreceptor 3,9 other FcRs11 or the formyl peptidereceptor10 are described. Possible binding sitesmight be extracted from Figure 5. For exampleFcgRIII shows an additional extended hydrophobicpatch on the N-terminal domain (indicated ascircles in the front and top view of Figure 5, con-sisting of Val6, Phe8, Trp13, Tyr30, Ala50, Tyr 53and Phe54). This continuous surface area is notinterrupted by glycosylation sites and is of a sizesuf®cient for a stable protein-protein interaction.
Considerations on clinical potential
With the solution of the FcgRIII crystal structurethe last of the homologous Fc-receptor structureshas been solved. Together with the structure sol-ution of the FcgRIII/hFc1 and the FceRIa/IgE-Fc-fragment complex which gave insights into thestructural rearrangements of the complex constitu-ents upon binding, a thorough understanding ofimmune complex recognition has been built up.Furthermore, FcRs are mediators in processes likeallergy, autoimmune diseases and cancer. Site-directed mutagenesis in the FcR binding region ofantibodies might result in molecules with higher ordiminished af®nity. Tumour cells could be moreef®ciently targeted to antigen presenting cells. Cur-now et al. summarised the use of bispeci®c anti-bodies that recognise a tumour antigen with oneFab and FcgRI with the other. Such bispeci®c anti-bodies are able to trigger an immune response
against the malignant cell.28 From our model wewould suggest Leu235, Pro329 and Leu328 as can-didates that should, when exchanged against morehydrophobic residues e.g. Trp or Phe, result inantibodies with higher af®nity to the receptor suchthat an engineered immunoglobulin would makethe production of bispeci®c antibodies obsoletedue to the increased af®nity of the Fc-fragmenttowards FcgRI.
On the other hand Duncan et al. could show thatLeu235 of the Fc-fragment affects the af®nity toFcgRI and an exchange of this residue for Gluresults in a 100-fold reduced af®nity.26 We wouldexpect residual af®nity to the FcgRIII which carriesa hydrophilic patch of three histidine residues inthe contact region for Leu235. Therefore, the IgGmutant Leu235Glu is supposed to be less effectivein suppressing the binding to this FcgR subtype.Antibodies with reduced af®nity to FcRs have beenraised against CD3 and CD4 by the groups ofWoodle29 and Reddy30 for the treatment of acuterenal allograft rejection or autoimmune diseases,respectively. Due to the Leu235Glu exchange theseantibodies cause a decreased expression of mousehistocompatibility complex (MHC) and T-cellreceptors and a reduced stimulation of the immunesystem in the patients. We would propose amutation of Gly237 to e.g. proline that shouldabrogate binding to all FcgRs for steric and confor-mational reasons.
Finally, the variety of FcR crystal forms availablewill certainly aid structure-based drug design pro-grams that might result in drugs that could glob-ally or speci®cally interfere with the immunestimulation via FcRs. Such FcR-based drugs wouldclearly represent an advantage over the immunesuppressive therapy commonly in use.
Material and Methods
Production, crystallisation and structuredetermination of sFcgggRIIa and sFcgggRIII
The expression of the soluble FcgRs was performed asdescribed previously.19 A new start methionine (close tothe cleavage site of the signal peptide) and a stop codon(between the putative extracellular part and the trans-membrane region) were introduced into the wild-typecDNA of FcgRIIa, FcgRIIb2 and FcgRIIIb. The mutantswere expressed under the control of a T7 promoter inE. coli BL21(DE3) inclusion bodies. After induction with1 mM IPTG (isopropyl-b-D-thiogalactopyranoside) atA600 nm � 1, and further growth at 37 �C for four hoursthe cells were harvested in soni®cation buffer (30 mMsodium phosphate, 300 mM sodium chloride and 0.02 %(w/v) sodium azide, pH 7.8), lysozyme treated andintensively soni®ed at 0 �C. The receptor fragmentsexpressed under these conditions were found exclusivelyin inclusion bodies which were puri®ed by repeatedresuspension with a Dounce homogeniser in soni®cationbuffer containing 0.5 % LDAO (N,N-dimethyldodecyla-mine N-oxide) and in two last steps in soni®cation bufferwithout LDAO. The puri®ed inclusion bodies were dis-solved to a protein concentration of 50 mg/ml in 6 Mguanidine chloride, 50 mM Tris-HCl (pH 8.0), 100 mM

Figure 5. Surface analysis of the FcR structures. A surface analysis was performed using the program GRASP39
with the crystal structures of FcgRIIa, FcgRIIb, FcgRIII and FceRIa as well as the modelled FcgRI. The colour codingspans from hydrophobic (green) to hydrophilic (red) with potential glycosylation sites indicated as blue balls. Theviewpoint is indicated with respect to the front view, which shows an identical orientation as in Figure 2. The IgGbinding site is surrounded by a black box in the FcgRIII molecule. The unique hydrophobic region on the N-terminaldomain of FcgRIII is circled.
Crystal Structures of Fc -Receptors 747
2-mercaptoethanol and separated from the insolublematter by centrifugation. Refolding was achieved byrapid dilution. Therefore 1 ml of the inclusion body sol-ution was added dropwise over 15 hours under stirringinto 400 ml of the refolding buffer (0.1 M Tris-HCl, 1.0-1.6 M arginine, 150 mM sodium chloride, 5 mM reducedglutathione, 0.5 mM oxidised glutathione, 0.1 mMphenylmethylsulphonyl ¯uoride, 0.02 % sodium azide,pH 8.0-8.5 at 4 �C). The mixture was stirred for severaldays until the concentration of free thiol groups wasreduced to 1 mM by air oxidation as measured accord-ing to Ellman.31 The solution was dialysed against PBSand sterile ®ltered before it was concentrated tenfold in astirring cell equipped with a 5 kDa molecular weight cutoff ultra®ltration membrane. The protein solution wasapplied to a hIgG Sepharose column (50 mg/ml hIgGper ml Sepharose 4B). Unbound protein was washed outwith 50 mM Tris (pH 8.0) before elution of the FcgR bylowering the pH (150 mM sodium chloride, 100 mM gly-cine, 0.02 % sodium azide, pH 3.0). The eluate wasimmediately neutralised with 1 M Tris-HCl (pH 8.0). TheFcgR-containing solution was concentrated and subjectedto gel ®ltration on a Superdex-75 column (Pharmacia/Germany) equilibrated with crystallisation buffer
(2 mM 3-[N-morpholino]propanesulphonic acid, 150 mMsodium chloride, 0.02 % sodium azide pH 7.2). The frac-tions containing the sFcgRs were pooled, concentrated to7 mg/ml and stored at ÿ20 �C. The identity with respectto size and folding was proven by N-terminal sequen-cing, ESI-MS and 1D-NMR.
Initial crystallisation experiments were performed insitting drops at 20 �C using the vapour diffusion methodby mixing 4 ml of the protein and 2 ml of the reservoirsolution. The ®nal optimised crystallisation conditionsfor FcgRIIa (0.2 M sodium acetate/acetic acid, pH 4.6,26 % PEG 8000) and for FcgRIII (0.1 M Mes/Tris, pH 7.8,22 % PEG 8000) yielded crystals which were used inX-ray diffraction experiments. The obtained datasetswere integrated using the program MOSFLM32 and sub-sequently scaled, reduced and truncated using routinesfrom the CCP4 program suite.33 Both structures weresolved by molecular replacement with the programAMoRe34 using the coordinates of sFcgRIIb (PDBentry 2fcb) yielding a clear solution. The model wascompleted in alternating cycles of model building andre®nement with the program O35 and the CNS programpackage.36

748 Crystal Structures of Fc -Receptors
Modelling
The FcgRIII/hFc1 structure was used as a base for themodelling of the other FcR-Ig complexes. Differing resi-dues in FcRs and Igs were grafted onto this framework.The resulting structures were minimised using the pro-gram SYBYL (Version 6.4, Tripos Inc. St. Louis/USA)with the TRIPOS force ®eld. Energy minimisation wasperformed with initial SIMPLEX minimisation with anenergy cut-off of 200 kcal/mol and alternating Powelland conjugate gradient methods. Van-der-Waals energiestypically decreased from 9 � 109 kcal/mol to 3 � 103
kcal/mol.
Atomic coordinates
The coordinates of the FcRs were taken from the Broo-khaven database with PDB entry codes: FcgRIIb, 2fcb;sFcgRIII, 1e4j; sFcgRIII/hFc1 complex, 1e4k; FceRIa,1f2q. Coordinates of FcgRIIa were deposited in the RCSBProtein Data Bank (PDB entry: 1h9v). The homologymodels are available as Supplementary Material.
Acknowledgement
We thank Vaughan Oosthuizen for helpful discus-sions.
References
1. Gessner, J. E., Heiken, H., Tamm, A. & Schmidt,R. E. (1998). The IgG Fc receptor family. Ann. Hema-tol. 76, 231-248.
2. Tortorella, D., Gewurz, B. E., Furman, M. H.,Schust, D. J. & Ploegh, H. L. (2000). Viral subversionof the immune system. Annu. Rev. Immunol. 18, 861-926.
3. Tax, W. J. M., Willems, H. W., Reekers, P. P. M.,Capel, P. J. A. & Koene, R. A. P. (1983). Polymorph-ism in mitogenic effect of IgG1 monoclonal anti-bodies against T3 antigen on human T cells. Nature,304, 445-447.
4. Fridman, W. H., Teillaud, J. L., Bouchard, C.,Teillaud, C., Astier, A., Tartour, E., Galon, J.,Mathiot, C. & Sautes, C. (1993). Soluble Fcg recep-tors. J. Leukoc. Biol. 54, 504-512.
5. Seminario, M. C., Saini, S. S., MacGlashan, D. W., Jr& Bochner, B. S. (1999). Intracellular expression andrelease of FceRla by human eosinophils. J. Immunol.162, 6893-6900.
6. Isakov, N. (1997). ITIMs and ITAMs. The Yin andYang of antigen and Fc receptor-linked signalingmachinery. Immunol. Res. 16, 85-100.
7. Suzuki, K., Hirose, T., Matsuda, H., Hasegawa, S.,Okumura, K. & Ra, C. (1998). The Fc receptor (FcR)gamma subunit is essential for IgE-binding activityof cell-surface expressed chimeric receptor moleculesconstructed from human high-af®nity IgE receptor(FceRl)a and FcRg subunits. Mol. Immunol. 35, 259-270.
8. Morton, H. C., van den Herik-Oudijk, I. E.,Vossebeld, P., Snijders, A., Verhoeven, A. J., Capel,P. J. & van de Winkel, J. G. (1995). Functionalassociation between the human myeloid immuno-globulin A Fc receptor (CD89) and FcR gamma
chain. Molecular basis for CD89/FcR gamma chainassociation. J. Biol. Chem. 270, 29781-29787.
9. Zhou, M., Todd, R. F., III, van de Winkel, J. G. &Petty, H. R. (1993). Cocapping of the leukoadhesinmolecules complement receptor type 3 and lympho-cyte functionassociated antigen-1 with Fcg receptorIII on human neutrophils. Possible role of lectin-likeinteractions. J. Immunol, 150, 3030-3041.
10. Kew, R. R., Grimaldi, C. M., Furie, M. B. & Fleit,H. B. (1992). Human neutrophil FcgRIIIB and formylpeptide receptors are functionally linked during for-myl-methionylleucyl-phenylalanine-induced chemo-taxis. J. Immunol. 149, 989-997.
11. Chuang, F. Y., Sassaroli, M. & Unkeless, J. C. (2000).Convergence of Fcg receptor IIA and Fcg receptorIIIB signaling pathways in human neutrophils.J. Immunol. 164, 350-360.
12. Maxwell, K. F., Powell, M. S., Hulett, M. D., Barton,P. A., McKenzie, I. F., Garrett, T. P. & Hogarth, P. M.(1999). Crystal structure of the human leukocyte Fcreceptor, FcgRlla. Nature Struct. Biol. 6, 437-442.
13. Sondermann, P., Huber, R. & Jacob, U. (1999).Crystal structure of the soluble form of the humanFcg-receptor Ilb: a new member of the immunoglo-bulin superfamily at 1.7 AÊ resolution. EMBO J. 18,1095-1103.
14. Garman, S. C., Kinet, J. P. & Jardetzky, T. S. (1998).Crystal structure of the human high-af®nity IgEreceptor. Cell, 95, 951-961.
15. Tamm, A. & Schmidt, R. E. (1997). IgG binding siteson human Fcg receptors. Int. Rev. Immunol. 16, 57-85.
16. Hulett, M. D., Witort, E., Brinkworth, R. I.,McKenzie, I. F. C. & Hogarth, P. M. (1995). Multipleregions of human FcgRll (CD32) contribute to thebinding of IgG. J. Biol. Chem. 270, 21188-21194.
17. Burton, D. R., Jefferis, R., Partridge, L. J. & Woof,J. M. (1988). Molecular recognition of antibody (IgG)by cellular Fc receptor (FcRI). Mol. Immunol. 25,1175-1181.
18. Sondermann, P., Huber, R., Oosthuizen, V. & Jacob,U. (2000). The 3.2-AÊ crystal structure of the humanIgGl Fc fragment-FcgRlll complex. Nature, 406, 267-273.
19. Sondermann, P. & Jacob, U. (1999). Human FcgRllbexpressed in Escherichia coli reveals IgG bindingcapability. Biol. Chem. 380, 717-721.
20. Burton, D. R. (1990). Antibody: the ¯exible adaptormolecule. Trends Biochem. Sci. 15, 64-69.
21. Maenaka, K., Juji, T., Stuart, D. I. & Jones, E. V.(1999). Crystal structure of the human p58 killercell inhibitory receptor (KIR2DL3) speci®c forHLA-Cw3-related MHC class I. Structure Fold. Des.7, 391-398.
22. Edberg, J. C. & Kimberly, R. P. (1997). Cell type-speci®c glycoforms of FcgRllla (CD16): differentialligand binding. J. Immunol. 159, 3849-3857.
23. Sondermann, P., Jacob, U., Kutscher, C. & Frey, J.(1999). Characterization and crystallization of sol-uble human FcgRll (CD32) isoforms produced ininsect cells. Biochemistry, 38, 8469-8477.
24. Garman, S. C., Wurzburg, B. A., Tarchevskaya, S. S.,Kinet, J. P. & Jardetzky, T. S. (2000). Structure of theFc fragment of human IgE bound to its high-af®nityreceptor FceRla. Nature, 406, 259-266.
25. Warmerdam, P. A. M., van de Winkel, J. G. J., Vlug,A., Westerdaal, N. A. C. & Capel, P. J. A. (1991). Asingle amino acid in the second Ig-like domain of

Crystal Structures of Fc -Receptors 749
the human Fcg receptor II is critical for human IgG2binding. J. Immunol. 147, 1338-1343.
26. Duncan, A. R., Woof, J. M., Partridge, L. J., Burton,D. R. & Winter, G. (1988). Localization of the bind-ing site for the human high-af®nity Fc receptor onIgG. Nature, 332, 563-564.
27. Sheridan, J. M., Hayes, G. M. & Austen, B. M.(1999). Solid-phase synthesis and cyclization of alarge branched peptide from IgG Fc with af®nity forFcgRl. J. Pept. Sci. 5, 555-562.
28. Curnow, R. T. (1997). Clinical experience withCD64-directed immunotherapy. An overview.Cancer Immunol. Immunother. 45, 210-215.
29. Woodle, E. S., Xu, D., Zivin, R. A., Auger, J.,Charette, J., O'Laughlin, R., Peace, D., Jollife, L. K.,Haverty, T., Bluestone, J. A. & Thistlethwaite, J. R.,Jr (1999). Phase I trial of a humanized, Fc receptornonbinding OKT3 antibody, huOKT3g1(Ala-Ala) inthe treatment of acute renal allograft rejection.Transplantation, 68, 608-616.
30. Reddy, M. P., Kinney, C. A., Chaikin, M. A., Payne,A., Fishman-Lobell, J., Tsui, P., Monte Dal, P. R.,Doyle, M. L., Brigham-Burke, M. R., Anderson, D.,Reff, M., Newman, R., Hanna, N., Sweet, R. W. &Truneh, A. (2000). Elimination of Fc Receptor-depen-dent effector functions of a modi®ed IgG4 mono-clonal antibody to human CD4. J. Immunol. 164,1925-1933.
31. Ellman, G. L. (1959). Tissue sulfhydryl groups. Arch.Biochem. Biophys. 82, 70-77.
32. Leslie, A. G. W. (1997). Mos¯m User Guide, Mos¯mVersion 5.50, MRC, Laboratory of Molecular Biology,Cambridge, UK.
33. Collaborative Computational Project Number 4(1994). The CCP4 suite: programs for protein crystal-lography. Acta Crystallog. sect. D, 50, 760-763.
34. Navaza, J. (1994). AMoRe: an automated packagefor molecular replacement. Acta Crystallog. sect. A,50, 157-163.
35. Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard,M. (1991). Improved methods for building protein
models in electron density maps and the location oferrors in these models. Acta Crystallog. sect. A, 47,110-119.
36. Brunger, A. T., Adams, P. D., Clore, G. M., DeLano,W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S.,Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J.,Rice, L. M., Simonson, T. & Warren, G. L. (1998).Crystallography & NMR system: a new softwaresuite for macromolecular structure determination.Acta Crystallog. sect. D, 54, 905-921.
37. Kraulis, P. J. (1991). MOLSCRIPT: a program to pro-duce both detailed and schematic plots of proteinstructures. J. Appl. Crystallog. 24, 946-950.
38. Merritt, E. A. & Murphy, M. E. P. (1994). Raster3DVersion 2.0. A program for photorealistic moleculargraphics. Acta Crystallog. sect. D, 50, 869-873.
39. Nicholls, A., Sharp, K. A. & Honig, B. (1991). Proteinfolding and association: insights from the interfacialand thermodynamic properties of hydrocarbons.Proteins: Struct. Funct. Genet. 11, 281-296.
Edited by I. A. Wilson
(Received 31 October 2000; received in revised form 29March 2001; accepted 30 March 2001)
http://www.academicpress.com/jmb
Supplementary material is available on IDEAL





























