Embed Size (px)
Citation preview

34°
Methods for the Study of the Cytology of Bacteria andPleuropneumonia-like Organisms
By EMMY KLIENEBERGER-NOBEL(From the Lister Institute, London)
With one Plate
SUMMARY
Methods for the demonstration of the various cytological elements of bacteria aredescribed. The specialized techniques applicable to pleuropneumonia-like organismsand L-forms of bacteria are also presented.
DURING the last decade new methods for the study of various morpho-logical features of bacteria and other minute organisms have been
developed including techniques for the demonstration of nuclear structures,cytoplasm, cell walls, flagella, and capsular and slimy envelopes of bacteria.It has been shown that bacteria may possess a life-cycle and may have definitestages which differ widely in appearance. For example, they may resemblepleuropneumonia-like organisms in their so-called L-form (Klieneberger-Nobel, 1949). As an aid to students unfamiliar with the methods of morpho-logical study of bacteria a number of fixing and staining procedures now inuse have been collected below. These are preceded by a brief description ofsome forms of the bacterial cell as well as of pleuropneumonia-like organisms.
According to Robinow (1946) the usual bacterial cell consists of a cell walllined with a cytoplasmic layer which may be slightly thicker at the two cellpoles. The smallest bacterial cell has no transverse septum and no transversecytoplasmic layer; it contains only one nuclear structure which, in a youngculture, may be on the point of dividing into two. With further developmentthe nuclear structure divides and the division-products move apart. The cellelongates at the same time, and a cytoplasmic septum is formed across thecentre. With further growth a cell-wall septum may be formed which cuts thecytoplasmic septum into two layers and the cell into two compartments, eachof which may now contain one big nuclear structure or two smaller ones. A celltreated with a cytoplasmic stain is characterized by darkly stained cell tips(PL I, fig. 2); in preparations stained for nuclear structures the extreme tipsare light (PI. I, fig. 1). In older cells nuclear structures as well as cytoplasmicparts are often irregularly arranged. The shape, dimension, and arrangementof the nuclear structures seem to bear a relationship to the changes whichbacterial cultures undergo. For example, in myxococci (Klieneberger-Nobel,19476) the young cells are slender with two to four small, transversely arrangednuclear structures. The swarming cells possess two fatter nuclear bodies,arranged centrally, leaving fairly large parts of the cell free of nuclear matter.[Quarterly Journal of Microscopical Science, Vol. 91, part 3, September 1950.]

Emmy Klieneberger-Nobel—Methods for Study of Cytology of Bacteria 341
The nuclear bodies of the swarming cells join together before the microcystsdevelop. Nuclear fusion seems to be frequent in bacterial cells and oftenprecedes spore formation (Klieneberger-Nobel, 1945). It can also occur as aphysiological reaction. When young cells are transferred to a lower tempera-ture a fusion may take place (Klieneberger-Nobel, 1947c). When anaerobes ina young stage are exposed to the air the single nuclear structures collect in themedian axis of the cell and combine to form a nuclear cylinder (Klieneberger-Nobel, 1945).
The location of the transverse septa may be of importance in the study ofmicrobial development. Thus, for example, the sporulating hyphae of soilActinomycetes (Klieneberger-Nobel, 1947a) possess very pronounced trans-verse septa while the primary or substratum mycelia have very delicate septaonly.
Furthermore, some species of bacteria possess mucoid envelopes which maybe regarded as capsules proper or as amorphous slime (Klieneberger-Nobel,1949). Flagella are generally difficult to stain; nevertheless, those of theso-called 'swarmers' in Proteus vulgaris are easy to stain.
When bacteria change from the A-form into the L-form (Klieneberger-Nobel, 1949), a marked transformation in their morphology takes place. Inthe cells of the L-form the nuclear material is often very finely dispersed andcell walls proper are absent (PI. I, fig. 4). In consequence the individual unitsof the culture, which vary greatly in size and shape, are of a very fragilenature. Therefore special methods for the preparation of microscopicalspecimens must be used.
The pleuropneumonia-like organisms are often composed of even morefragile elements than those of the L-growth of bacteria. Their colonies may bebuilt up of small granules, fine flexible filaments, globular forms and flat,amoeboid elements of various sizes (Klieneberger and Smiles, 1942). Theglobules as well as the flat forms produce fine, darkly staining granules insidetheir lumen which are regarded as the reproductive units and are able toinitiate new growth (PI. I, fig. 5). The morphology of these organisms isstudied to advantage in special preparations carried out as described later.
METHODS
Coverslips, Slides, and Mounting of Preparations
For practical reasons the use of a coverslip is necessary in order to obtainoptimal resolution. The use of coverslips allows further all the necessaryoperations to be carried out in small containers such as watch-glassesand small block dishes. This is very convenient and saves reagents andstains.
Stained coverslip films may be mounted in weak stain or in water. Thepreparation (avoid air bubbles) is gently pressed between layers of filter-paperand then sealed with a rim of paraffin wax. The thinner the liquid layerbetween coverslip and slide the better the resolution.

342 Emmy Klieneberger-Nobel—Methods for the Study of
When preparations are mounted in Canada balsam after staining in waterysolutions, they must be dehydrated. As a rule dehydration has to be carriedout rapidly. For dehydration the following solutions used in histology arerecommended:
1. Acetone, 19 ml.; xylene, 1 ml.2. Acetone, 14 ml.; xylene, 6 ml.3. Acetone, 6 ml.; xylene, 14 ml.4. Two or three changes of xylene.5. Canada balsam.
In some cases the staining qualities are improved by mounting in Canadabalsam, but often the cells are slightly reduced by shrinkage which may be adisadvantage.
Occasionally a neutral mounting medium which can be applied withoutdehydration and which, therefore, does not produce shrinkage may be wel-come. The following medium, devised by Romeis and Wiist (Romeis, 1932),has proved to be fairly satisfactory in some cases as, for example, in themounting of stained encapsulated organisms:
Components
Gelatine.Glycerine, twice distilled.MJ5 solution of borax (1-24 gm. borax crystals are dissolved in 10 ml.
M-NaOH and brought up to 100; o-i ml. of a concentrated aqueoussolution of HgCl2 is then added).
Bromthymolblue solution (o-i gm. bromthymolblue in 3-2 ml. M/20 NaOHbrought up to 250 ml. with distilled water).
M-NaOH solution.
Method
Seven gm. gelatine are added to 42 ml. of M/5 solution of borax. Place thegelatine in a hot-water bath until dissolved and add 42 ml. glycerine. Add10 drops of bromthymolblue solution to the warm glycerine-gelatine whichshould become yellow. Now add drop by drop M-NaOH until the colourchanges to blue. Filter the hot solution immediately (pH approximately 7*7).For use the glycerine-gelatine has to be liquefied by warming.
Preparation of the film
For the demonstration of details of bacterial structure it is of the greatestimportance to produce, whenever possible, a one-cell layer of organisms, wellspaced and with each cell spread flatly on the surface of the coverslip. Thebest way of achieving this is to grow the organisms between a layer of solidmedium and the coverslip. Clean, well-flamed coverslips (flame twenty timesquickly) are placed on sterile filter-paper disks in sterile Petri dishes. A platecontaining the solid medium is inoculated evenly from a fresh culture by

the Cytology of Bacteria and Pleuropneumonia-like Organisms 343
means of a spreader. Inocula taken from a broth culture can be more evenlyspread than those from cultures on solid media and will, therefore, yield moreconsistent results. Squares, smaller than the coverslips used, are cut from thefreshly inoculated plate by means of a flamed knife or a Hagedorn needle.The squares are placed, inoculated side downwards, upon the coverslips. ThePetri dishes containing the coverslip micro-cultures are incubated at thetemperature required in a moist chamber which ought to close fairly tightlyto allow for even humidity throughout. After growth has progressed to thestage desired, the micro-cultures are fixed and stained as described later. It isnot necessary to grow the bacteria between agaT and the coverslip for allpurposes. When dealing with ordinary bacteria, good staining results may beobtained when the squares are cut from cultures already developed to thepoint desired. However, the coverslip should not be pressed down on themedium and lifted up, a manipulation which involves gross displacement;a cut-out piece placed cautiously on the coverslip is always preferable. Yetit should be realized that when dealing with soft and fragile material and whenvery fine detail of structure is desired, the micro-culture is the best method.
Only sparsely grown cultures will yield the required one-cell layer. Yet theperiphery of colonies or the growth developing on the coverslip in the humidzone round the edge of the agar square often supply suitable material for thestudy of older growth. The development in this zone may be encouraged byrunning a drop of liquid round the edge of the freshly inoculated square orby using a medium of soft consistency. It must be realized that suitable growthon the coverslip of the organism under observation is of greater importancethan the fixing and staining methods applied later and that each new subjectstudied involves a little ingenuity to ensure the necessary flat, thin, and well-spaced growth on the coverslip.
FIXING AND STAINING
A. Chromatinic Structures
Osmium tetroxide j Giemsa method. Remove the agar square with the tip ofa knife while holding the coverslip with a pair of forceps. Take care that thepiece does not slip along the coverslip. Put the coverslip with adherent filmquickly into osmium tetroxide vapour. Use for fixation a small glass jar withground-in, greased lid. Fill it with glass beads up to three-quarters of itsheight. Pour in 5 ml. of a 2 per cent, aqueous osmium tetroxide solution, bestprepared from crystals obtainable in sealed ampoules containing 1/10 gr. each.The jar must be refilled every fortnight or more often if much used. Fix the wetfilm in the vapour (film side upwards) for 3-5 minutes. Allow to dry in the air.
Cells of organisms poor in ribonucleic acid can be stained with Gicmsasolution alone (Gurr's R 66). Use two or three drops of Giemsa solution in3-5 ml. of a mixture of tap-water and freshly distilled water. Place the cover-slip film-side downwards into the Giemsa solution to prevent precipitationon the surface of the specimen. For example, certain stages of the vegetative

344 Emmy Klieneberger-Nobel—Methods for the Study of
forms of myxococci and some water organisms such as Sphaerotilus natansstain well by this simple method, showing up red chromatinic structures in abluish cytoplasm. Also very young cells of ordinary bacteria and L-forms canoften be stained in this manner quite satisfactorily. Mount in a drop of stainwithout washing or dehydrate and mount in Canada balsam.
Osmium tetroxide jHydrochloric acid/Giemsa method (Robinow). Mostbacterial organisms, however, possess a cytoplasm very rich in ribonucleicacid. Consequently they have a strong affinity for all basic dyes by which theyare stained so deeply that the nuclear structures are obscured. The ribonucleicacid is gradually decomposed when the fixed coverglass films are immersed innormal HC1 (film side up) at a temperature of approximately 550 C. The timeof this treatment varies with the species and the stage of the organism. Forexample, 4 minutes seem to be the correct time for 3-hour agar cultures ofB. coli or Proteus vulgaris. After treatment the films are rinsed in water andplaced in Giemsa solution (as above) and mounted as described. The periodsof staining must be determined by trial: 20 minutes should suffice for theabove-mentioned young cultures. Usually the nuclear structures appear deepred or black, and the cytoplasm takes a bluish colour. If applied too long, thehydrochloric acid produces an unstainable cytoplasm, yet the desoxypentose-nucleic acid contained in the chromatinic structures is very resistant to theaction of the acid. The staining method described above was first devised byPiekarski and introduced into bacteriology and widely used by Robinow.
Osmium tetroxide / Ribonuclease /Giemsa method (Boivin, modified). Afterfixing and drying as before, the coverslips are placed film side up in a watch-glass standing in a warm place (370 C. incubator or on top of water-bath).The film is covered with a 01 per cent, solution of ribonuclease which isallowed to remain for 4-20 minutes or longer according to the object. It isthen rinsed in water, stained with Giemsa and mounted as before. The effectof ribonuclease is similar to but slower than that of hydrochloric acid, but thestaining of the cells often shows a particularly beautiful contrast between thered nuclear structures and the blue cytoplasm. The ribonuclease has no effecton cells which are difficult to penetrate, such as spores, while the hydrochloricacid can be used for these as well as for vegetative cells. The ribonucleasetreatment first introduced by Boivin has been widely used by him and hiscollaborators. However, Boivin used it in combination with a particularfixation method outlined below.
Chabaud/Ribonuclease I Giemsa method (Boivin). Place the dried coverslipfilm into Chabaud's fixative (alcohol 80 per cent., 60 ml.; formalin, 5 ml.;glacial acetic acid, 2 ml.; phenol crystals, 15 gm.) for 10 minutes. Rinse inwater, apply ribonuclease, stain and mount as before. Preparations made inthis way show good staining contrast, but the fixative produces shrinkages ofthe cells, which may be of serious disadvantage when dealing with delicateelements of structure.
Osmium tetroxide/GiemsdjEosin (Badian). Fix with the vapour of osmiumtetroxide solution as above, stain with Giemsa solution (20 minutes are
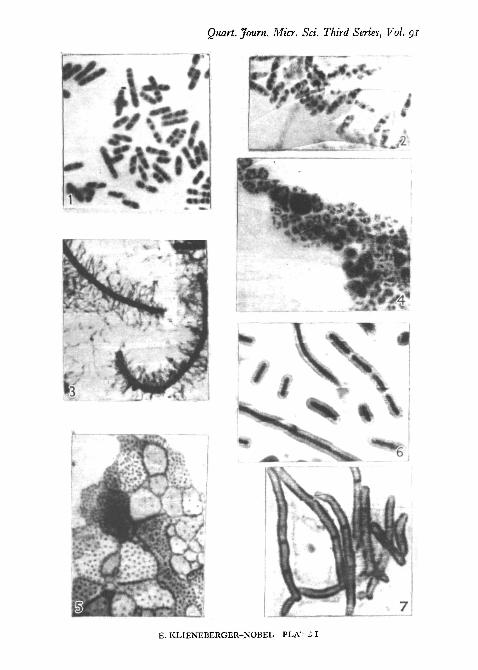
Quart. Journ. Micr. Set. Third Series, Vol. gi
E. KLIENEBERGER-NOBEL—PLA"? S I

the Cytology of Bacteria and Pleuropneumonia-like Organisms 345
sufficient for B. coli and Proteus vulgaris). Mix two drops of a 1 per cent,watery eosin solution with 5 ml. distilled water in a watch-glass. Put the stainedcoverslip, unrinsed, into the eosin solution and leave for 30 seconds, rinseand mount in water. This simple method often produces good contrasts, butlike the ribonuclease method has no effect on spores.
It should be mentioned that the Feulgen reaction, if applied in a suitableway, demonstrates clearly the chromatinic structures of bacteria, yet the cellas a whole remains unstained.
PL I, fig. 1, shows the regularly arranged chromatinic structures of aspore-bearing Gram-positive bacillus stained by the hydrochloric acid/Giemsamethod. PI. I, fig. 4, shows a water vibrio in the L-stage stained by thehydrochloric acid/Giemsa method.
B. Cytoplasm {Klieneberger-Nobel, Robinow)
Use either micro-cultures or squares cut out from plate cultures and placedon coverslips. Place the coverslip with adherent agar square upwards into awatch-glass and cover with Bouin's fixing solution (sat. solution of picric acid,20 ml.; formalin, 10 ml.; glacial acetic acid, 1 ml..). Leave for i | hours;remove the agar square. The bacterial film should adhere firmly to the surfaceof the coverslip. Wash well, stain in Giemsa solution for 30 minutes or asrequired. Mount in the dilute stain. The cytoplasmic areas, tips of the cells,cytoplasmic layers, and septa are stained bluish or purple. The areas of thecells occupied by the chromatinic structures remain unstained.
PI. I, fig. 2, shows a culture of B. coli with its cytoplasmic areas wellstained.
C. Cell Walls and Transverse Septa (Robinow)
Fix the wet film with the vapour of osmium tetroxide solution or useBouin's solution through the agar as described above. Place the preparation(film side downwards) into a 5 per cent, tannic acid solution for 20-30 minutes.Wash thoroughly under the tap and place the coverslip in a weak solutionof crystal violet for a few seconds or longer as required (two drops of a 05 percent, watery solution of crystal violet with 5 ml. of distilled water). Mountin a drop of stain. The tannic acid mordant renders the interior of the cellspractically unstainable while the walls and the transverse septa are distinctlystained. PI. I, fig. 7, shows cells of a Proactinomyces stained in the mannerdescribed.
D. Flagella of Proteus sp. (Klieneberger-Nobel)
Pour an agar plate and dry it in the incubator for several hours in an invertedposition with the lid ajar. Place a small inoculum of Proteus in the middle andleave the plate at warm room temperature for approximately 24 hours, bywhich time a swarming edge should have developed. Watch for motility underthe low power of the microscope. If motility is observed, cut out agar squares,half the surface of each of which is covered with growth. Place the squares on

346 Emmy Klieneberger-Nobel—Methods for the Study of
coverslips and fix with Bouin's solution as described before. Wash and stainwith Giemsa solution for approximately 18 hours. Mount in a drop of thestain or dehydrate and mount in Canada balsam. PL I, fig. 3, shows swarmingcells of Proteus sp. stained for flagella.
E. Capsules (Klieneberger-Nobet)
Use a nutrient agar containing 30 per cent, of horse serum. Plant theorganism to be examined on the surface of the plate and incubate in a CO2
atmosphere (about 10 per cent.) for a few hours or until a delicate layer ofgrowth is visible. In particular cases it may be of advantage to add carbo-hydrate to the medium. Cut out squares, put them on coverslips and fix withBouin's solution as above. Wash, place in Giemsa for several hours, and thenmount in the stain. The organisms should stain darkly, the capsules shouldstain a delicate blue or pink. A nice contrast may be obtained if staining withGiemsa is followed by counterstaining with a thin eosin solution (see above).Leave in the eosin for a few seconds only, wash and mount in water.
If tannic acid is applied for 30 minutes after the fixation and the film stainedin crystal violet for about 10 minutes the capsules stain a deep violet while theorganism remains unstained. Though giving occasionally very good contrastthis method is not consistently successful.
It should be mentioned that the capsule staining methods described dependon the serum content of the culture medium. Probably any serum wouldserve the purpose though horse serum has been used here. PI. I, fig. 6, showsorganisms of the Klebsiella group prepared by the Bouin/Giemsa method.
F. Slime (Klieneberger-Nobel)
Mix a loop of the cultures to be examined and a loop of saline and spreadit on the coverslip. Dry in air and place into Chabaud's solution (see above)for 10 minutes. Rinse and place in 5 per cent, tannic acid or in Loffler'smordant for 30 minutes. Wash' thoroughly and stain in crystal violet forseveral hours. Mount in the stain or in water. The organisms should beunstained with dark contours; the amorphous slime should be stained pink.
While the capsules of all organisms examined could be well demonstratedby means of the methods described, the slime of some organisms staineddistinctly while in the case of others the staining was not so successful.
G. Pleuropneumonia-like organisms and L-forms of bacteria
For the study of pleuropneumonia-like organisms, micro-cultures are pre-pared from suitable medium. (From ox heart infusion-peptone agar, a boiledblood medium according to Levinthal (1921) is prepared. This is filteredunder sterile conditions. Sterile horse serum is added.) The micro-culturesare incubated in a moist chamber. They are fixed at appropriate times withBouin's solution, washed and stained in weak Giemsa solution for severalhours or, better, overnight. They can be dehydrated to advantage in theacetone-xylene mixtures mentioned above and mounted in Canada balsam.

the Cytology of Bacteria and Pleuropneumonia-Iike Organisms 347
Care should be taken that dehydration is carried out as rapidly as possible.It is advisable to blot the water off gently before the actual dehydration. Inpreparations made as described no appreciable dislocation of elements shouldhave occurred. They should show whole stained colonies, the delicate edgesof which can be examined under the high power of the microscope. Bodiescontaining granules are usually developed after about 48 hours of incubation.The vapour of osmium tetroxide also fixes pleuropneumonia-like organisms,but owing to their extreme delicacy they do not stand up well to the separationof the agar square from the coverslip, which has to precede fixation. L-growthis often more resistant. It usually withstands the separation process, particu-larly if it is not pure but interspersed with bacillary forms. Therefore osmiumtetroxide fixation can be used for the fixation of the L-growth. This fixationmay be followed by the hydrochloric acid treatment and by staining withGiemsa solution. PL I, fig. 5, shows the 2-day-old growth of the pleuro-pneumonia-like organism isolated from bronchopneumonic lungs of rats. Therelatively large bodies are filled with fine filterable granules. The preparationwas carried out by the Bouin/Giemsa method. The L-growth of the watervibrio of PL I, fig. 4, was demonstrated by the osmium tetroxide/hydrochloricacid/Giemsa method.
REFERENCESBADIAN, J., 1933. Arch. Mikr., 4, 409.BOIVIN, A., 1948. Revue medicale de Liege, 3, 237.KLIENEBERGER-NOBEL, E., 1945. J. Hyg. Camb.,' 44, 99.
i947<J. J- gen. Microbiol., r, 22.19476- Ibid., 1, 33.1949. Ibid., 3, 434.
KLIENEBERGER, E., and SMILES, J., 1942. J. Hyg., 43, no.LEVINTHAL, W., 1921. Ergebn. allg. Path. path. Anat. 19 Jahrg., II. Abt., 5, 878.ROBINOW, C. F., 1942. Proc. Roy. Soc. B, 130, 299.
1944. J. Hyg., 43, 413.1946. Addendum in Dubos, R. J., The Bacterial Cell. Harvard (University Press).
ROMEIS, B., 1932. Taschenbuch der mikroskopischen Technik, 13. Aufl., Miinchen und Berlin(Oldenburg).
EXPLANATION OF PLATE I(Magn. X 2,500-3,000)
FIG. 1. Gram-positive bacillus, young. Hydrochloric acid, Giemsa. (Chromatinic struc-tures.)
FIG. 2. Bad. colt, young. Bouin, Giemsa. (Cytoplasm.)FIG. 3. Proteus sp., swarming organisms. Bouin, Giemsa. (Flagella.)FIG. 4. Water vibrio growing completely in the L-phase. Hydrochloric acid, Giemsa.
(Chromatinic structures.)FIG. 5. Pleuropneumonia-like organism from bronchopneumonic lungs of rats. Bouin,
Giemsa prolonged. (Granules in large bodies.)FIG. 6. Organism of Klebsiella group. Capsule staining method t.FIG. 7. Proactinomyces. Bouin, tannic acid, crystal violet. (Transverse septa.)





























