Embed Size (px)
Citation preview

02. Mai 2018 Management im Gesundheitswesen - Industrie 1
Management im Gesundheitswesen IIIIndustrie
Prof. Dr. med Reinhard Busse
FG Management im Gesundheitswesen, Technische Universität Berlin (WHO Collaborating Centre for Health Systems Research and Management)
&European Observatory on Health Systems and Policies
Medizintechnik: Regulatorische Rahmenbedingungen I

202. Mai 2018 Management im Gesundheitswesen - Industrie
Datum Inhalt der Lehrveranstaltung18.04.2018 Einführungsveranstaltung
25.04.2018Medizintechnik-Industrie
Marktentwicklung
02.05.2018 Regulatorische Rahmenbedingungen I
09.05.2018 Regulatorische Rahmenbedingungen II
16.05.2018 Kundenmanagement
23.05.2018 Telemedizin und e-Health
30.05.2018Pharmazeutische Industrie
Marktentwicklung
06.06.2018 Regulatorische Rahmenbedingungen I
13.06.2018 Regulatorische Rahmenbedingungen II
20.06.2018 Preisbildung
27.06.2018 Evaluation und Pharmakoökonomie
04.07.2018 Kundenmanagement
11.07.2018 Vorbereitung schriftlicher Test
18.07.2018 Schriftlicher Test

3
• Voraussetzung für das Inverkehrbringen (Marktzulassung):CE-Kennzeichnung
• Anbringung der CE-Kennzeichnung erfordert:- Erfüllung der grundlegenden Anforderungen durch Anwendung von Normen- Durchführung vorgeschriebener Konformitätsbewertungsverfahren
• ALT: verbindliche Richtlinien für MP:- 90/385/EWG (aktive implantierbare Medizinprodukte) (AIMD)- 98/79/EWG (In-vitro-Diagnostika) (IVD)- 93/42/EWG (sonstige Medizinprodukte) (MDD)
Management im Gesundheitswesen - Industrie
Inverkehrbringung von Medizinprodukten
02. Mai 2018
In-vitro-Diagnostikum (IVD) ist jedes Medizinprodukt, das als Reagenz, Reagenzprodukt, Kalibriermaterial, Kontrollmaterial, Kit, Instrument, Apparat, Gerät oder System — einzeln oder in Verbindung miteinander — nach der vom Hersteller festgelegten Zweckbestimmung zur In-vitro-Untersuchung von aus dem Körper stammenden Proben,
einschließlich Blut- und Gewebespenden, verwendet wird und ausschließlich oder hauptsächlich dazu dient, Informationen zu liefern:
- über physiologische oder pathologische Zustände oder- über angeborene Anomalien oder
- zur Prüfung auf Unbedenklichkeit und Verträglichkeit bei den potentiellen Empfängern oder-zur Überwachung therapeutischer Maßnahmen.
CAVE: In-vivo-Diagnostika (z.B. Kontrastmittel) fallen in Deutschland unter das Arzneimittelgesetz!
Ein „Aktives Medizinprodukt“ ist ein Medizinprodukt, dessen Betrieb von
einer Stromquelle oder einer anderen Energiequelle (mit Ausnahme der direkt vom
menschlichen Körper oder durch die Schwerkraft erzeugten Energie)
abhängig ist.

4
• Voraussetzung für das Inverkehrbringen (Marktzulassung):CE-Kennzeichnung
• Anbringung der CE-Kennzeichnung erfordert:- Erfüllung der grundlegenden Anforderungen durch Anwendung von Normen- Durchführung vorgeschriebener Konformitätsbewertungsverfahren
• ALT: verbindliche Richtlinien für MP:- 90/385/EWG (aktive implantierbare Medizinprodukte) (AIMD)- 98/79/EWG (In-vitro-Diagnostika) (IVD)- 93/42/EWG (sonstige Medizinprodukte) (MDD)
• NEU (Inkrafttreten 25. Mai 2017): Richtlinien ersetzt durch– Verordnung über Medizinprodukte 2017/745
(Medical Device Regulation, MDR)– Verordnung über In-vitro-Diagnostika 2017/746
(In-Vitro Diagnostic Medical Devices Regulation, IVDR)� haben als Verordnungen direkten Gesetzescharakter, d.h. deutsche MPG ist für viele Bereich dann nicht mehr nötig!
Management im Gesundheitswesen - Industrie
Inverkehrbringung von Medizinprodukten
02. Mai 2018
MDR: gilt ab 26. Mai 2020IVDR: gilt ab 26. Mai 2022� d.h. es gibt eine 3-jährige bzw. 5-jährige Übergangsfrist

5
� Art des Konformitätsbewertungsverfahren sowie den Umfang derBeteiligung einer unabhängigen Prüf- und Zertifizierungsstelle (BenannteStelle) ist vom potenziellen (Gesundheits-)Risiko der Produkte – nicht vonihrer Wirksamkeit – abhängig (Logik des Binnenmarktes!)
• aktive implantierbare Medizinprodukte (90/385/EWG): keine weitere Unterscheidung nach Risikogesichtspunkten
• MP nach RL 93/42/EWG (und nach VO 2017/745): Produktdifferenzierungnach festgelegten Kriterien in Klassen I, IIa, IIb, III
• In-vitro-Diagnostika (98/79/EG): Einteilung in Gruppen (nach VO 2017/746 inKlassen A, B, C, D)
� Konformitätsbewertungsverfahren sowie deren Durchführung sind in derdt. Verordnung über Medizinprodukte (MPV)* geregelt. Diese verweist auf entsprechende Anhänge der europäischen Richtlinien
(* Achtung: diese darf nicht mit der neuen europ. Medizinprodukte-Verordnung verwechselt werden!)
Inverkehrbringung von Medizinprodukten
Management im Gesundheitswesen - Industrie02. Mai 2018

6
• einheitliches Zulassungsverfahren in Europa
• wird jeweils von „Benannten Stellen“ (Zertifizierungsstellen) durchgeführt• in Deutschland derzeit 10, z. B. TÜV, Materialprüfstellen, BerlinCert (an der TUB)
• in der EU insgesamt ca. 50 (in vielen Ländern nur 1 oder gar keine)
• Benennende Behörden der Benannten Stellen in Dtl: Benennung und Überwachung der Zertifizierungsstellen
• ZLG - Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten: nichtaktive Medizinprodukte und In-vitro-Diagnostika
• ZLS - Zentralstelle der Länder für Sicherheitstechnik: aktive Medizinprodukte
• Ziel: Risiko für den Verbraucher bzw. den Patienten zu minimieren• 3
• Hersteller muss Produktsicherheit und medizinisch-technische Leistungsfähigkeit nachweisen
• Zulassung basiert auf Überprüfung der Qualität und der Qualitätssicherungsmaßnahmen
Marktzulassung von Medizinprodukten
Management im Gesundheitswesen - Industrie02. Mai 2018

7
Benannten Stellen
Management im Gesundheitswesen - Industrie02. Mai 2018
http://www.dimdi.de/static/de/mpg/adress/benannte-stellen/

Management im Gesundheitswesen - Industrie 802. Mai 2018
https://www.zlg.de/medizinprodukte/dokumente/stellenlaboratorien/benannte-stellen.html
Benannten Stellen durch ZLG

02. Mai 2018 Management im Gesundheitswesen - Industrie 9
https://www.zlg.de/medizinprodukte/dokumente/stellenlaboratorien/benannte-stellen-9342ewg.html
Benannten Stellen nach § 15 MPG – RL 93/42/EWG

Management im Gesundheitswesen - Industrie 10
Ausschnitte des Geltungsbereiches – Berlin Cert
https://www.zlg.de/index.php?eID=tx_nawsecuredl&u=0&file=fileadmin/downloads/BS_MDD_0633.pdf&hash=a10b343b2ed87c48341084bb12d7ad285ecdf8bf
02. Mai 2018

11
• Wozu?
� zur Abschätzung des potentiellen Risikos
• Wie?
� durch die Einteilung in Risikoklassen (I, IIa, IIb, III)
• Wonach?
� Anwendung der 18 Regeln im Anhang IX der Richtlinie (93/42/EWG) unter Berücksichtigung
Klassifizierung von Medizinproduktennach Richtlinie 93/42/EWG
� aller Charakteristika des MP
� Zweckbestimmung
� Angewandte Technologie (aktiv, nicht aktiv ...)
� Anwendungsart, -dauer (<1h, <30d, >30d), -ort
Management im Gesundheitswesen - Industrie02. Mai 2018

12
Einteilung der Medizinprodukte nach Art in Klassen:
I Produkte mit niedrigem Risiko, die meisten nicht-invasivenProdukte und wiederverwendbare chirurgische Instrumente (z.B. Stethoskope, Spatel)
IIa nicht aktive Produkte mit mittlerem Risiko, invasive und nicht-invasive Produkte für kurzzeitige Benutzung (z.B. Kanülen)
IIb aktive Produkte mit mittlerem Risiko, die Substanzen oderEnergie mit potentiellem Risiko emittieren (z.B. Röntgengeräte),und Produkte für längere Nutzung
III Produkte mit hohem Risiko und solche, die mit demGefäßsystem oder dem zentralen Nervensystem in Kontakt kommen(z.B. Gefäßtransplantate)
Zu
neh
men
des G
efä
hrd
un
gsp
ote
nzia
l d
es
Med
izin
pro
du
kte
s
Klassen von Medizinprodukten nach Richtlinie 93/42/EWG
Management im Gesundheitswesen - Industrie02. Mai 2018

13
Klassifizierungsregeln
Nicht invasive Produkte
Invasive Produkte
Zusätzliche Regeln füraktive Produkte
Regel 1-4 Regel 5-8 Regel 9-12
Besondere Regeln
Regel 13-18
Klassifizierungsregeln – Anhang IX der RL 93/42/EWG
Management im Gesundheitswesen - Industrie02. Mai 2018

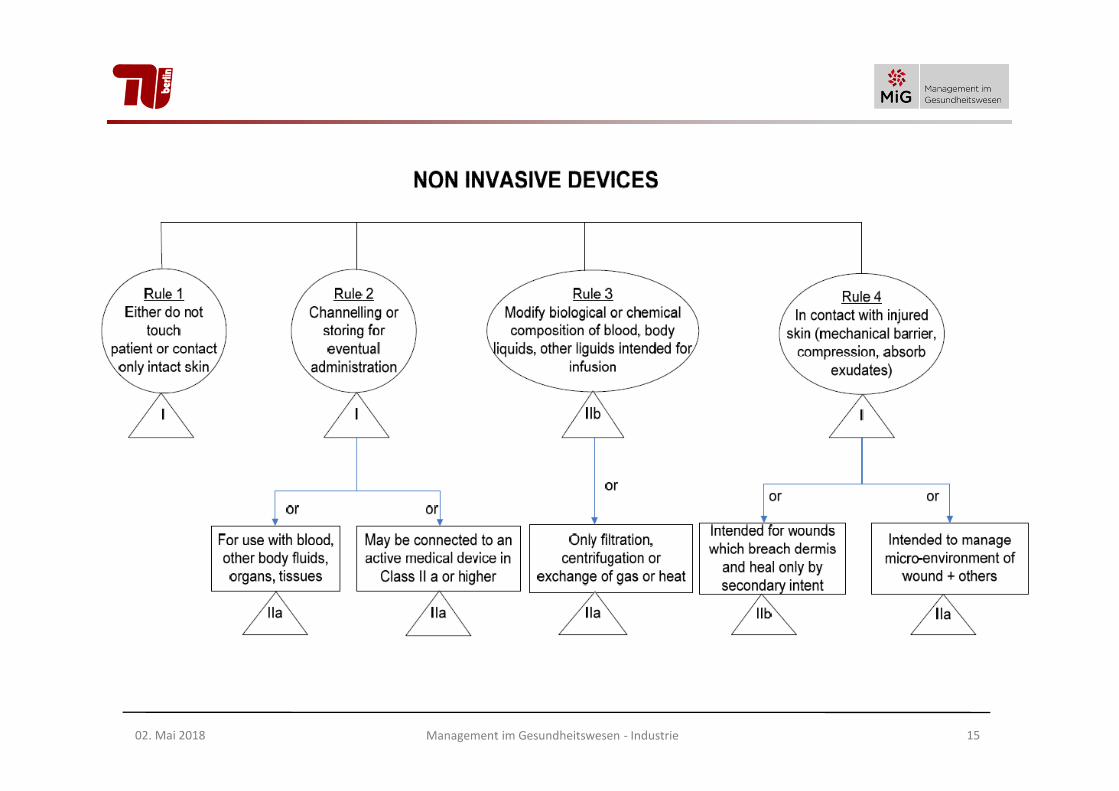
Wesentliche Klassifizierungsmerkmale vonnicht-invasiven Medizinprodukten
Regel 1:
alle nicht invasiven Produkte, für die keine andere Regel zutrifft: Klasse I
Regel 2:
Produkte zur Durchleitung von Blut oder zur Aufbewahrung von Blut etc., oder Einsatz mit Produkten der Klasse IIa oder höher: Klasse IIa
Regel 3:
Produkte zur Veränderung der biologischen und chemischen Zusammensetzung des Blutes, wenn in den Körper perfundiert: Klasse IIb
Regel 4:
Produkte, die in Berührung mit verletzter Haut kommen als mechanische Barriere: Klasse IIa
- mit Beeinflussung der Mikroumgebung einer Wunde: Klasse IIa
- bei Durchtrennung der Dermis: Klasse IIb
14Management im Gesundheitswesen - Industrie02. Mai 2018
Management im Gesundheitswesen - Industrie 1502. Mai 2018

Wesentliche Klassifizierungsmerkmalevon invasiven Medizinprodukten I
Regel 5:
Produkte (außer chirurgisch-invasiven) für
- vorübergehende Anwendung: Klasse I
- kurzzeitige Anwendung und bei Verwendung zusammen mit aktiven Produkten der Klasse IIa oder höher: Klasse IIb
Regel 6:
- Wiederverwendbare chirurgische Instrumente: Klasse I
- chirurgisch-invasive Produkte für die vorübergehende Anwendung: Klasse IIa
- Produkte zur Energieabgabe (ionisierende Strahlung): Klasse IIb
- Produkte mit biologischer Wirkung: Klasse IIb
- Produkte zur Verabreichung von Arzneimitteln mit potentieller Gefährdung: Klasse IIb
- Produkte, die resorbiert werden: Klasse IIb
- Produkte für den Einsatz am Herzen oder am zentralen Kreislaufsystem: Klasse III
16Management im Gesundheitswesen - Industrie02. Mai 2018

Wesentliche Klassifizierungsmerkmalevon invasiven Medizinprodukten II
Regel 7:
- Chirurgisch-invasive Produkte für kurzzeitige Anwendung: Klasse IIa
- Produkte zur Energieabgabe (ionisierende Strahlung) und zur Abgabe von Arzneimitteln: Klasse IIb
- Produkte für den Einsatz am Herzen oder am zentralen Kreislaufsystem: Klasse III
- Produkte in Kontakt mit dem zentralen Nervensystem: Klasse III
- Produkte mit biologischer Wirkung oder Resorption: Klasse III
Regel 8:
- Produkte zur Implantierung in Zähne: Klasse IIa
- alle implantierbaren Produkte und chirurgisch-invasiven Produkte zur langzeitigen Anwendung, wenn keine andere Regel zutrifft: Klasse IIb
- Produkte für den Einsatz am Herzen oder am zentralen Kreislaufsystem: Klasse III
- Produkte in Kontakt mit dem zentralen Nervensystem: Klasse III
- Produkte mit biologischer Wirkung oder Resorption: Klasse III
- Produkte zur Abgabe von Arzneimitteln: Klasse III
17Management im Gesundheitswesen - Industrie02. Mai 2018

Zusatzregeln für aktive (energiebetriebene) Produkte –Hierzu gehört auch eigenständige Software!
Regel 9 (aktive therapeutische Produkte, um „biologische Funktionen oder Strukturen im Zusammen-hang mit der Behandlung oder Linderung einer Krankheit, Verwundung oder Behinderung zu erhalten, zu verändern, zu ersetzen oder wiederherzustellen“):
- grundsätzlich :Klasse IIa
- bei potentieller Gefährdung: Klasse IIb
- zur Leistungssteuerung oder Leistungskontrolle von aktiven therapeutischen Produkten: Klasse IIb
Regel 10 (aktive diagnostische Produkte, um „eine direkte Diagnose oder Kontrolle von vitalenKörperfunktionen zu ermöglichen“):
- grundsätzlich: Klasse IIa
- mit potentieller Gefährdung (z. B. Änderung der Herzfunktion, der Atmung oder der Aktivität des zentralen Nervensystems): Klasse IIb
- zur Aussendung ionisierender Strahlung und Produkte zur Steuerung oder Kontrolle solcher Produkte: Klasse IIb
Regel 11 (aktiven Produkte zur Abgabe von Arzneimitteln, Körperflüssigkeiten und anderen Stoffen an den Körper oder/und für deren Entfernung):
- grundsätzlich: Klasse IIa
- bei potentieller Gefährdung: Klasse IIb
Regel 12:
alle anderen aktiven Produkte: Klasse I
18Management im Gesundheitswesen - Industrie02. Mai 2018

Besondere Regeln
Regel 13: Produkte zu deren Bestandteil ein Stoff gehört, der bei gesonderter Verwendung als Arzneimittel angesehen werden kann: Klasse III
Regel 14:
- Produkte zur Empfängnisverhütung und zum Schutz vor der Übertragung sexuell übertragbaren Krankheiten: IIb
- Produkte zur Empfängnisverhütung und zum Schutz vor der Übertragung sexuell übertragbaren Krankheiten ,wenn diese implantiert werden oder wenn es sich um invasive Produkte zur langzeitigen Anwendung handelt: Klasse III
Regel 15:
- Produkte speziell zur Desinfektion von Produkten: Klasse IIa (Desinfektionsmittel für invasive MP: IIb)
- Produkte zur Desinfektion, Reinigung, Spülung und zum Hydratisieren von Kontaktlinsen: Klasse IIb
Regel 16: Nicht-aktive Produkte zur Aufzeichnung von Röntgendiagnosebildern: Klasse IIa (digitaler Röntgenbildempfänger: IIb)
Regel 17: Produkte, die unter Verwendung von tierischen Geweben oder Folgeerzeugnissen hergestellt werden: Klasse III
Regel 18:
Blutbeutel: Klasse IIb
19Management im Gesundheitswesen - Industrie02. Mai 2018

20
1. Welche Richtlinie gilt? � Ausschlussverfahren
- Eine Gehhilfe ist kein aktives implantierbares Medizinprodukt (keine
Energiequelle, keine Implantation) � 90/385/EWG ist nicht anwendbar
- Eine Gehhilfe ist kein in vitro Diagnostikum � 98/79/EG ist nicht anwendbar
- Nach dem Ausschlussverfahren ist die Richtlinie 93/42/EWG anzuwenden
2. Klassifizierung? � Überprüfen der Klassifizierungsregeln
- Gemäß den Klassifizierungsregeln des Anhangs IX der Richtlinie 93/42/EWG ist eine Gehhilfe ein Produkt der Klasse I, denn
- Regel 1: alle nicht invasiven Produkte gehören der Klasse I an, es sei denn, es findet eine der folgenden Regeln Anwendung
- Regel 2: nicht invasives MP dient der Durchleitung oder Aufbewahrung von
Körperflüssigkeiten ... IIa �trifft nicht zu
- Regel 3: nicht invasives MP zur Veränderung der biologischen oder chemischen
Zusammensetzung des Blutes ... IIa �trifft nicht zu
- Regel 4: Nicht invasive MP die mit verletzter Haut in Berührung kommen ... IIa, IIb
� trifft nicht zu
Klassifizierung eines Medizinproduktes – Beispiel Gehhilfe
Management im Gesundheitswesen - Industrie02. Mai 2018

21
Einige konkrete Beispiele für Medizinprodukte:
Klasse I Klasse IIa Klasse IIb Klasse III
• ärztliche Instrumente
• Gehhilfen• Rollstühle• Spitalbetten• Stützstrümpfe• Verbandmittel• wiederver-
wendbarechirurgische Instrumente
• Dentalmaterialien• diagnostische
Ultraschallgeräte• Einmalspritzen• Hörgeräte• Kontaktlinsen• Trachealtuben• Zahnkronen• Software zur The-
rapie/ Diagnostik
• Anästhesiegeräte• Beatmungsgeräte• Bestrahlungsgeräte• Blutbeutel• Defibrillatoren• Dialysegeräte• Kondome• Kontaktlinsenreiniger• Dentalimplantate• Software für
Intensivstationen
• Herzkatheter• künstliche
Gelenke• Stents• resorbierbares
chirurgisches Nahtmaterial
• Intrauterinpessar(Spirale)
• Brustimplantat
Management im Gesundheitswesen - Industrie02. Mai 2018
22
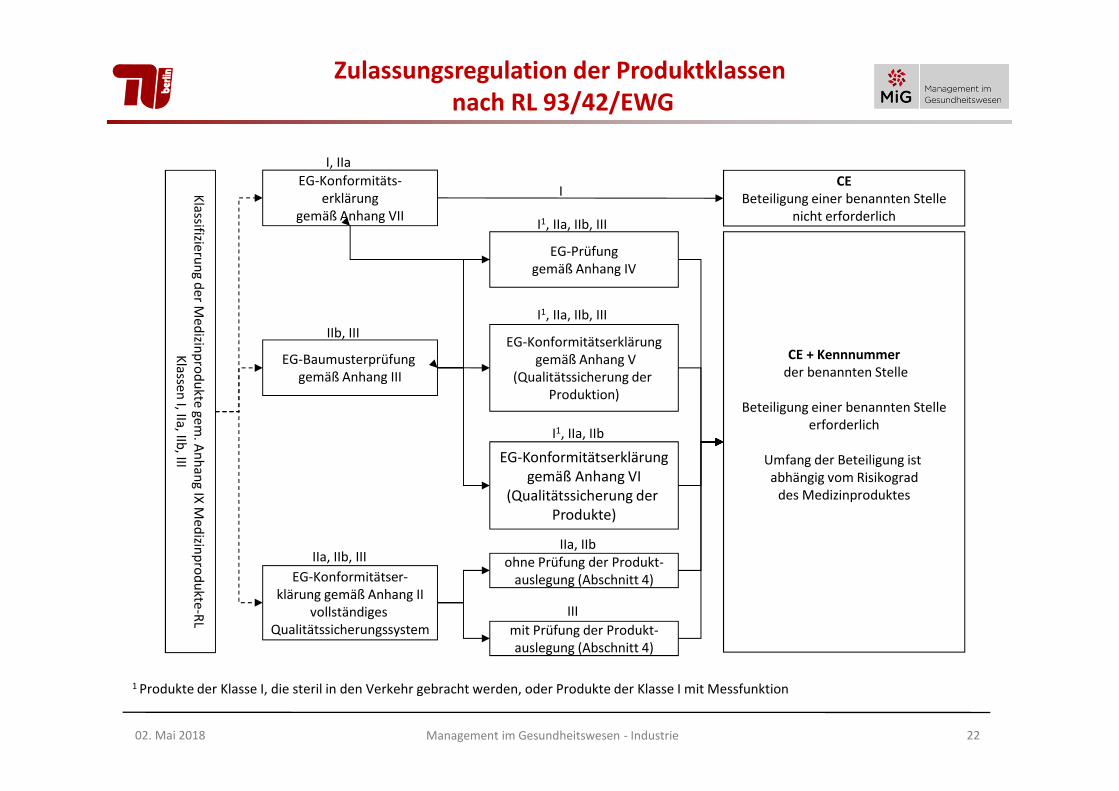
EG-Konformitäts-erklärung
gemäß Anhang VII
EG-Baumusterprüfung gemäß Anhang III
EG-Konformitätser-klärung gemäß Anhang II
vollständiges Qualitätssicherungssystem
EG-Prüfunggemäß Anhang IV
EG-Konformitätserklärunggemäß Anhang V
(Qualitätssicherung der Produktion)
EG-Konformitätserklärunggemäß Anhang VI
(Qualitätssicherung der Produkte)
CEBeteiligung einer benannten Stelle
nicht erforderlich
I, IIa
IIb, III
IIa, IIb, III
I
I1, IIa, IIb, III
I1, IIa, IIb, III
I1, IIa, IIb
CE + Kennnummerder benannten Stelle
Beteiligung einer benannten Stelleerforderlich
Umfang der Beteiligung ist abhängig vom Risikograd
des Medizinproduktes
ohne Prüfung der Produkt-auslegung (Abschnitt 4)
mit Prüfung der Produkt-auslegung (Abschnitt 4)
IIa, IIb
III
Klassifizie
run
g de
r Me
dizin
pro
du
kte ge
m. A
nh
ang IX
Me
dizin
pro
du
kte-R
LK
lassen
I, IIa, IIb, III
1 Produkte der Klasse I, die steril in den Verkehr gebracht werden, oder Produkte der Klasse I mit Messfunktion
Zulassungsregulation der Produktklassennach RL 93/42/EWG
Management im Gesundheitswesen - Industrie02. Mai 2018

RL 2007/47/EGzur Änderung der RL 90/385/EWG u. 93/42/EWG
- Aufnahme von Software in die Definition MP
- Verschärfung der Konformitätsbewertung in den Anhängen der RL
- z.B. Benannte Stelle muss Stichprobe für jede generische Produktgruppe bei MP der Klasse IIa und IIb prüfen
- Längere Aufbewahrungsfristen für Herstellerunterlagen
- Änderung der Klassifizierungskriterien:
- Eigenständige Software � aktives MP
- Konzept der ununterbrochenen Anwendung
- Regel 15: Desinfektionsmittel für invasive MP � Klasse IIb
- Konsultationsverfahren für MP mit unterstützendem Arzneistoffanteil
- Anpassung der RL 90/385/EWG an die RL 93/42/EWG
23Management im Gesundheitswesen - Industrie02. Mai 2018

Gesetz zur Änderung medizinproduktrechtlicher Vorschriften
Ziel:
- Umsetzung der RL 2007/47/EG
Inhalte:
- Klinische Prüfung bzw. Leistungsbewertungsprüfungen:
- Zentrale Genehmigung durch BfArM /PEI (statt Anzeige bei LB) und
- positive Zustimmung einer nach Landesrecht gebildeten Ethik-Kommission
- Einführung von
- Meldepflicht bei allen schwerwiegenden unerwünschten Ereignissen in klinischen Studien (SAE – serious adverse event) von Prüfer und Sponsor (Doppelmeldung)
- entsprechenden Bewertungsverfahren im Rahmen von klinischen Prüfungen
24Management im Gesundheitswesen - Industrie02. Mai 2018

Gesetz zur Änderung medizinproduktrechtlicher Vorschriften
Klare Aufgabentrennung
- Zentrale Genehmigungsbehörden (BfArM und PEI) prüfen nach wissenschaftlichen und technischen Gesichtspunkten
- Technische Sicherheit
- Biokompatibilität
- Wissenschaftlichkeit des Prüfplans
- Generelle Nutzen-/ Risikobewertung
� Genehmigungs- statt Anzeigepflicht
- Landesrechtlich eingerichteten Ethik-Kommission prüft nach ethischen und rechtlichen Gesichtspunkten
- die Zulässigkeit der klinischen Prüfung
25Management im Gesundheitswesen - Industrie02. Mai 2018

Gesetz zur Änderung medizinproduktrechtlicher Vorschriften
Zweck einer klinische Prüfung:
- Erbringung des Nachweises, dass Leistungen des MP bei normalen Einsatzbedingungen den grundlegenden Anforderungen entsprechen(CAVE: Unterschied zu [klinischer] Wirksamkeit)
- Zweckbestimmung muss sehr genau beschrieben werden (Funktion, Indikation, Kontraindikation)
- Ermittlung unerwünschter Nebenwirkungen
- Sicherheitsregelungen zur Durchführung von klinischen Prüfungen verlangen, dass Produkte alle sicherheitstechnischen Eigenschaften aufweisen
26Management im Gesundheitswesen - Industrie02. Mai 2018

Management im Gesundheitswesen - Industrie 27
Der Hersteller hat für jedes MP u. a. den Nachweis zu erbringen, dass:
- die Anwendung des Medizinproduktes bei normalen Einsatzbedingungen
• den klinischen Zustand des Patienten
• die Sicherheit des Patienten
• die Sicherheit und Gesundheit des Anwenders und Dritter nicht gefährdet
- die angepriesene Leistungsfähigkeit des Produktes unter normalen
Bedingungen erreicht wird.
Dieser Nachweis kann durch eine Zusammenstellung der derzeit verfügbaren Literatur erbracht werden.
Für Hochrisikoprodukte der Klasse IIb und III sind klinische Prüfungen erforderlich.
Gesetz zur Änderung medizinproduktrechtlicher Vorschriften
02. Mai 2018

Management im Gesundheitswesen - Industrie 2802. Mai 2018
2017: Änderungen MDD + AIMD � MDR
• Einführung „Scrutiny-Verfahren“ (sog. Konsultationsverfahren) in den Zertifizierungsprozess:
– Geltungsbereich: implantierbare Produkte der Klasse III und aktive Produkte der Klasse IIb, die Arzneimittel an den Körper abgeben und/oder aus dem Körper entfernen
– Benannte Stelle erstellt Bericht über die Begutachtung der klinischen Bewertung („clinical evaluation assessment report“) und leitet diesen an die Kommission weiter
– Ggf. Weiterleitung durch die Kommission an ein Expertengremium
– Scientific assessment report: Expertengremium entscheidet über Notwendigkeit einer Erstellung eines wissenschaftlichen Gutachtens (innerhalb von 21 Tagen nach Auftragseingang durch Kommission)
– Ggf. Erstellung eines Gutachtens durch Expertengruppe (innerhalb von 60 Tagen nach Auftragseingang durch Kommission)

Management im Gesundheitswesen - Industrie 2902. Mai 2018
2017: Änderungen MDD + AIMD � MDR
• Produktnummer (UDI - Unique Device Identification): leichtere Identifikation und Nachverfolgung fehlerhafter Produkte
• Europaweite Datenbank Eudamed (European Databank on Medical Devices): Daten des MP inkl. UDI, des Herstellers, der prüfenden Benannten Stelle, Daten zu klinischen Studien, Vigilance and post-market surveillance Daten
• Verschärfungen bei den klinischen Bewertungen und Prüfungen: Sammlung, Dokumentation und Auswertung klinischer Daten auch nach Markteinführung verpflichtend
• Benannte Stellen werden im Zuge der MDR erneut überprüft und ausgewählt

30
Ablauf: Entwicklung eines Medizinproduktes
Medizinprodukt Arzneimittel
Biokompatibilität
Technische Leistung
Anwendung an Patienten(partiell an Probanden)
Zertifizierung
Technische EntwicklungProduktion
Langzeitbeobachtung Post Market Clinical Follow-up
Toxikologie
PharmakokinetikPharmakodynamik
Anwendung an Probanden/ Patienten (Phase I-III)
Zulassung
Technische EntwicklungProduktion
MarktsüberwachungPhase IV
Schorn GH (2010): Klinische Prüfungen von Medizinprodukten: Fragen und Antworten zum geänderten MP-Recht. In: Medizinprodukte Journal 17 (3)190-196.
30Management im Gesundheitswesen - Industrie02. Mai 2018

31
Zertifizierung bedeutet
nicht automatisch
Kostenübernahme
durch die GKV
(� Reg. Rahmenbed. II)
Management im Gesundheitswesen - Industrie02. Mai 2018

32
Erweiterung des üblichen Dreiecks bei Medizinprodukten (I)
Zahler (GKV)
Patient
Hersteller
Beitrag/Steuern
Zuzahlungen
Hilfsmitteldistributeur(z.B. Sanitätshaus)
Vergütung für Produkt und/oder Dienstleistung
Regulierer
med. Leistungserbringer(z.B. Krankenhaus)
Management im Gesundheitswesen - Industrie02. Mai 2018

33
Strukturierung von Medizinproduktenunter Vergütungsgesichtspunkten
Management im Gesundheitswesen - Industrie
i.d.R. Standardprodukte, die einem individuellen Patienten verschrieben und gegeben werden (z.B. Inkontinenzunterlagen, Rollstühle) – Produkt, nicht Dienstleistung steht im Mittelpunkt
Kategorie IHilfsmittel
Kategorie III„Medizintechnik zurArzt-Unterstützung“
Technische Geräte, die Ärzte bei Diagnostik und Behandlung unterstützen (Röntgen, Endoskope); Finanzierung in zwei Stufen:
Kategorie IIImplantate & Endo-
Prothesen
Medizinprodukte, die einem individuellen Patienten implantiert oder angepasst werden (z.B. Gelenk-Endoprothesen, Stents, Herzschrittmacher) – zumeist steht die Dienstleistung im Vordergrund
Medizinprodukte
• IIIa: Investition• IIIb: Refinanzierung über Dienstleistungen
02. Mai 2018

34
Zahler (GKV)
Patient
Hersteller
Beitrag/Steuern
Zuzahlungen
Hilfsmitteldistributeur(z.B. Sanitätshaus)
Vergütung für Produkt und/oder Dienstleistung
(Investition)
med. Leistungserbringer(z.B. Krankenhaus)
NB: I, II, IIIa und IIIb beziehen sich auf die Kategorien in der vorherigen Abbildung
I
I
I
II
II
II
III b
III a
III b
Erweiterung des üblichen Dreiecksbei Medizinprodukten (II)
Management im Gesundheitswesen - Industrie02. Mai 2018






















![MR-Kontrastmittel-Untersuchungen bei eingeschränkter ... · Die alternativ empfohlene kontrastmittelverstärkte MR-Angiographie (ceMRA) kann […]das gesamte Gefäßsystem […]](https://img.dokumen.tips/doc/110x75/5ccefd3b88c993b6568b9cd1/mr-kontrastmittel-untersuchungen-bei-eingeschraenkter-die-alternativ-empfohlene.jpg)






