Embed Size (px)
Citation preview

MARTIN-LUTHER-UNIVERSITÄT
HALLE-WITTENBERG
INSTITUT FÜR PHYSIK
GRUNDPRAKTIKUM
PRAKTIKUM
PHYSIK
FÜR
PHARMAZIESTUDENTEN
10. AUFLAGE (2011)

VorwortDas Praktikum „Physik für Pharmazeuten“ ist im Sinne der Approbationsordnung für Apothekereine scheingebende Veranstaltung, deren regelmäßiger und erfolgreicher Besuch für dieTeilnahme am Ersten Abschnitt der Pharmazeutischen Prüfung nachgewiesen werden muss.Nach Einschreibung zum Praktikum sind innerhalb des laufenden Semesters 7 Versuche zuerbringen und eine Abschlussklausur zu schreiben. Das Praktikum kann an der Martin-Luther-Universität nur einmal wiederholt werden.In den einführenden Kapiteln des Praktikumsheftes finden Sie Hinweise zu Praktikumsablaufund Versuchsführung sowie zur Protokollierung und Auswertung der Messergebnisse.Kontrollfragen und Literaturangaben am Ende jeder Versuchsanleitung sind als Hilfestellungzur Vorbereitung gedacht. Weitere Hinweise finden Sie auch auf der Webseite des Praktikums.
Martin-Luther-Universität Halle-WittenbergInstitut für PhysikPhysikalisches Praktikum für Pharmazeuten
http://www.physik.uni-halle.de/Lehre/Grundpraktikum
Herausgeber: Autoren:Martin-Luther-Universität Halle-Wittenberg A. Christ, K.-H. Felgner, W. Frän-
zel, K.-V. Jenderka, A. Klemenz,J. Leschhorn,
Institut für Physik, Grundpraktikum M. StölzerTel.: 0345 55-25551, -25550Fax: 0345 55-27300Email: [email protected] Praktikumsleiter: W. Fränzel [email protected]
10. Auflage Halle, April 2011

Einführung Laborordnung für das Praktikum
Inhaltsverzeichnis
ALLGEMEINE EINFÜHRUNG
Laborordnung für das Praktikum . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1Hinweise zum Ablauf des Praktikums . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2Richtlinien für die Protokollführung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4Fehlerrechnung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
MECHANIK
M2 Dichtebestimmung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10M4 Oberflächenspannung von Flüssigkeiten . . . . . . . . . . . . . . . . . . . . . . 13M13 Dehnung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16M14 Viskosität . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19M19 Ultraschall-Abbildungsverfahren . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
WÄRMELEHRE
W1 Lineare Ausdehnung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27W6 Spezifische Wärme von Metallen . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28W12 Luftfeuchtigkeit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31W17 Spezifische Wärme von Flüssigkeiten . . . . . . . . . . . . . . . . . . . . . . . . 35W25 Diffusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
ELEKTRIZITÄTSLEHRE
E7 Innenwiderstand von Spannungsquellen . . . . . . . . . . . . . . . . . . . . . . 41E8 Leitfähigkeit von Elektrolyten . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44E10 Thermospannung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48E34 Elektrolyse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51E39 Messwerterfassung mit dem Computer: EKG . . . . . . . . . . . . . . . . . . 53
OPTIK UND STRAHLUNG
O4 Mikroskop . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57O10 Polarimeter und Refraktometer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65O16 Radioaktivität . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70O20 Spektralphotometer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73O22 Röntgenverfahren . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
ANHANG
Hinweise zu Bedienung von Cassy-S . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83Kurzanleitung ORIGIN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86

Einführung Laborordnung für das Praktikum
1
Laborordnung für das Praktikum
Allgemeines Verhalten
1 Die Praktikanten haben sich in denPraktikumsräumen so zu verhalten, dassPersonen nicht gefährdet sowie Einrichtun-gen, Geräte und Versuchsaufbauten nichtbeschädigt werden.
2 Die von den betreuenden Assistenten,vom Praktikumspersonal sowie die in denVersuchsanleitungen gegebenen Hinweisezur Handhabung der Geräte und Versuchs-anordnungen sind unbedingt zu beachten.
3 Auftretende Störungen und Unregel-mäßigkeiten bei der Durchführung der Versu-che, Beschädigungen und Funktionsstörungenan Geräten und Einrichtungen sowie Unfällesind sofort zu melden. Es ist nicht zulässig,Geräte selbst zu reparieren!
4 Für grob fahrlässig verursachte Schädenan Geräten und Arbeitsmaterialien könnendie Praktikanten zur Verantwortung gezogenwerden.
5 Den Praktikanten steht jeweils nur dieam Arbeitsplatz befindliche Ausrüstung zurVerfügung. Es ist nicht gestattet, Geräte vonfremden Arbeitsplätzen zu benutzen.
6 Zur Auswertung von Messergebnissenkann jeder freie Computer genutzt werden.Dazu haben sich die Praktikanten mit derVersuchsbezeichnung anzumelden und beiBeendigung der Arbeit wieder abzumelden.
7 Nach Beendigung des Versuches ist derArbeitsplatz aufgeräumt und sauber zuverlassen.
8 Essen und Trinken ist in den Prakti-kumsräumen nicht erlaubt. Rauchen ist imgesamten Gebäude untersagt.
9 Die Benutzung von Handys ist in denPraktikumsräumen untersagt!
10 Das Praktikum beginnt pünktlich zu derim Stundenplan angegebenen Zeit. Mehr als
15 Minuten nach Praktikumsbeginn könnenkeine Versuche mehr begonnen werden.
11 Für einen erfolgreichen Abschlussmüssen Sie alle Praktikumstermine wahr-nehmen. In sehr dringenden Fällen sowie beiKrankheit können mit dem Praktikumsperso-nal Ersatztermine innerhalb der regulärenZeiten des Physikpraktikums für Pharmazeu-ten vereinbart werden.
Arbeiten mit elektrischen Schaltungen
12 Der Auf- und Abbau elektrischer Schal-tungen hat stets im spannungslosen Zustandzu erfolgen (Stromversorgungsgeräte aus,Batterien und Steckernetzteile nicht an-geschlossen). Die Schaltungen sind übersicht-lich aufzubauen.
13 Bei elektrischen Messgeräten ist auf dierichtige Polung, auf die Einstellung desrichtigen Messbereiches und die Verwendungder richtigen Messeingänge zu achten. (Über-lastungsgefahr!)
14 Elektrische Schaltungen müssen vor derInbetriebnahme vom zuständigen Assistentenüberprüft werden!
15 Unter Spannung stehende Anlagenmüssen ständig überwacht werden.
16 Spannungsführende Teile dürfen nichtberührt werden. Gefährliche Spannungen(> 42 V) sind in jedem Fall durch Schutzvor-richtungen vor Berührung gesichert. Es istuntersagt, solche Schutzvorrichtungen außerBetrieb zu setzen!
17 Bei Unfällen ist die Spannung sofortabzuschalten (Notausschalter: gelb-roteTastschalter in jedem Raum). Der Unfallmuss unverzüglich gemeldet werden.

Einführung Hinweise zum Ablauf des Praktikums
2
Arbeiten mit Chemikalien
18 Bei der Arbeit mit Chemikalien ist aufSauberkeit zu achten. Verwenden Sie Trichterzum Umfüllen und Fließpapierunterlagenbeim Abwiegen von Chemikalien!
19 Mit dem Versuchszubehör ausgegebeneArbeitsschutzmittel (z. B. Schutzbrille)müssen getragen werden!
20 Bei Unfällen oder bei Verschüttengefährlicher Substanzen (z. B. Quecksilber)muss sofort ein Assistent verständigt werden!Es sind keine eigenständigen Beseitigungs-versuche zu unternehmen!
21 Alle Chemikalien befinden sich inGefäßen mit eindeutiger Kennzeichnung desInhaltes. Dies ist besonders zu beachten,wenn Chemikalien nach der Verwendung indie Aufbewahrungsgefäße zurückgegossenwerden müssen.
22 Nach Beendigung des Versuches sindalle verwendeten Gefäße (außer Vorrats-gefäßen) sorgfältig auszuspülen.
Arbeiten mit radioaktiven Präparaten
23 Im Praktikum wird ausschließlich mitumschlossenen Präparaten unterhalb derFreigrenze laut Strahlenschutzverordnunggearbeitet. Die Strahlenbelastung währendeines Versuches ist 100...1000 mal geringerals bei einer Röntgenaufnahme.
24 Vermeiden Sie trotzdem jede unnötigeBestrahlung. Abstand ist der beste Strahlen-schutz! Halten Sie radioaktive Präparate nichtunnötig in der Hand. Halten Sie während derMessung einen Abstand von 0,5 m zumPräparat ein.
25 Es ist untersagt, die Präparate aus ihrenAcrylglashüllen zu entfernen.
Brandschutz
26 Bunsenbrenner und elektrische Heizge-räte sind so aufzustellen, dass sich keinebenachbarten Gegenstände entzünden kön-nen. Offene Flammen und eingeschalteteHeizgeräte müssen ständig beaufsichtigtwerden!
27 Abgebrannte Streichhölzer gehörennicht in Papierkörbe! Noch glimmendeStreichhölzer dürfen nicht weggeworfenwerden.
28 Vorsicht beim Umgang mit brennbarenFlüssigkeiten (z. B. Ethanol)! Sie sind vonoffenen Flammen fernzuhalten.
29 Wird ein Brand bemerkt, so ist diessofort zu melden und es sind nach Möglich-keit Löschmaßnahmen einzuleiten.
30 Jeder Praktikant hat sich über die Lageund Funktionsweise der Handfeuerlöschersowie über die vorhandenen Fluchtwege zuinformieren.
Hinweise zum Ablauf des Praktikums
1 Vorbereitung
Das Versuchsthema ist der Praktikums-Homepage im Internet oder dem Aushang imPraktikum (nur am vorherigen Praktikum-stag) zu entnehmen.
Zu Hause werden die physikalischen Grund-lagen zum Versuch studiert (Literaturangabenam Ende jeder Versuchsanleitung) und das
Protokoll vorbereitet (siehe auch „Richtlinienfür die Protokollführung“).
2 Versuchsausgabe
Das benötigte Zubehör wird an der Ausgabegegen Hinterlegung eines Studentenaus-weises pro Versuchsgruppe abgeholt.

Einführung Hinweise zum Ablauf des Praktikums
3
3 Kontrolle der Versuchsvorbereitung
Vor Versuchsbeginn erfolgt die Kontrolle derProtokollvorbereitung und ein kurzes mc-Antestat beim zuständigen Assistenten. Beiungenügender Vorbereitung darf der Versuchnicht durchgeführt werden und muss zueinem späteren Termin innerhalb der Vorle-sungszeit des laufenden Semesters nachgeholtwerden. Hierzu ist ein Termin zu verein-baren!
4 Versuchsdurchführung
Die Versuche werden in der Regel vonZweiergruppen durchgeführt. Dreiergruppensind nicht zulässig. Jeder Student führt eineigenes Protokoll.
Aufbau des Versuches.
Elektrische Schaltungen müssen vorInbetriebnahme vom zuständigen Assi-stenten überprüft werden!
Durchführung der Messungen und Protokoll-führung (siehe „Richtlinien zur Protokoll-führung“).
Kontrolle der Messwerte und Kurzunter-schrift des Assistenten am Tage der Ver-suchsdurchführung (falls die Auswertungnicht sofort fertiggestellt wird).
5 Versuchsauswertung
Die Versuchsauswertung wird, wenn mög-lich, während der Praktikumszeit durch-geführt bzw. begonnen. Sie ist bis zumnächsten Praktikumstermin fertigzustellen.
Hinweise zur Auswertung geben die „Richt-linien zur Protokollführung“.
Es werden Taschenrechner, Lineal, evtl.Kurvenlineal und Millimeterpapier benötigt.Computer können eingesetzt werden undstehen im Praktikum zur Verfügung. Milli-meterpapier und zu manchen Versuchenerforderliche Spezialpapiere können imPraktikum erworben werden.
6 Kontrolle der Versuchsauswertung
Die Bestätigung der erfolgreichen Versuchs-durchführung incl. Auswertung erfolgt durchdie Eintragung von Datum und Langunter-schrift des betreuenden Assistenten in dasProtokoll (in der Regel zum nächsten Praktik-umstermin).
Liegt die Auswertung nicht spätestens amübernächsten Praktikumstag vor, so gilt derVersuch als nicht bestanden.
7 Versuchsabgabe
Abgabe des Versuchszubehörs und Vorlageder Lang- oder Kurzunterschrift an derAusgabe.
8 Fehlversuche
Zum Nachholen versäumter oder nicht be-standener Versuche werden jedem Studieren-den zwei zusätzliche Praktikumsterminegarantiert. (ein “Nachhol-Termin” für alle amSemesterende, weitere Termine nach Mög-lichkeit im laufenden Semester) In jedem Fallmüssen Sie den Termin an der Versuchsaus-gabe vereinbaren, damit sichergestellt ist,dass der Arbeitsplatz frei ist! Zu einemPraktikumstermin kann jeweils nur einVersuch durchgeführt werden.
Studenten, die mit mehr als zwei Versuchenin Verzug sind, können das Praktikum indiesem Semester nicht beenden und erhaltenkeinen Schein.
9 Leistungskontrollen
Das Bestehen eines kurzen Antestates zuBeginn des Praktikums (siehe oben) ist Vor-aussetzung für die Zulassung zum Versuch.
Nach Abschluss aller Versuche wird eine mc-Klausur geschrieben. Voraussetzung zurZulassung zur Klausur ist, dass alle Versuchedurchgeführt wurden. Die Klausur kanngemäß Studienordnung wiederholt werden

Einführung Richtlinien für die Protokollführung
4
10 Abschluss des Praktikums
Das Praktikum gilt als erfolgreich absolviert,wenn die Abschlussklausur bestanden wurde.
Der erfolgreiche Abschluss des Praktikums
wird bescheinigt.
Gemäß Studienordnung kann das Praktikumeinmal als Ganzes wiederholt werden.
Richtlinien für die Protokollführung
Allgemeines
1 Jeder Student führt während des Versu-ches und unmittelbar ein Protokoll. DasProtokoll enthält - die Versuchsvorbereitung,- das Messprotokoll (alle Messwerte und
Beobachtungen in chronologischer Reihen-folge),
- die Auswertung.
2 Das Protokoll wird handschriftlich mitnicht löschbarem Stift geführt. Bleistift istnur für Diagramme und Skizzen zulässig.Fehlmessungen werden mit Angabe desGrundes durchgestrichen und dürfen nichtunlesbar gemacht werden. Fehler sind Teilder Arbeit, das Ausradieren oder Löschenvon Messdaten ist schlechter wissenschaftli-cher Stil!
3 Alle Protokolle des Praktikums sind ineinem gebundenen Heft der Größe A4 oder ineinem Schnellhefter zu führen und zu jederVeranstaltung mitzubringen.
4 Lose Blätter (auch Computerausdrucke)sind mit Name und Datum zu beschriften undin das Protokoll einzufügen.
Vorbereitung zu Hause
5 Jedes Protokoll muss einleitend enthal-ten: - Datum, - Versuchsbezeichnung und Aufgaben-
stellung (wörtlich), - kurze Beschreibung des Versuches mit
geplanter Durchführung (z. B. Schalt-skizze) und vorgesehener Auswertung ein-
schließlich der dafür benötigten Formeln, - vorbereitete Urlisten (Tabellen) für die
Aufnahme der Rohmessdaten.Dieser Teil des Protokolls ist Bestandteil derVersuchsvorbereitung und wird vor Ver-suchsbeginn vom Assistenten kontrolliert.
Protokollführung während des Versuches
6 Es werden alle Versuchsgeräte regi-striert (Versuchsaufbau).
7 Das Protokoll soll übersichtlich und gutlesbar sein, z.B durch eine klare Gliederungmit Zwischenüberschriften (“Messwerte zuAufgabe 1" oder ähnlich).
8 Alle physikalischen Größen sind voll-ständig mit Zahlenwert und Einheit anzuge-ben. Tabellen müssen eine Überschrift oderLegende besitzen, die Spalten sind mit physi-kalischer Größe und Einheit zu beschriften.
9 Es werden grundsätzlich alle Messdatenvor jeglicher rechnerischer Aufbereitungprotokolliert (Urlisten).
10 Das Messprotokoll ist dann vollständig,wenn nur mit seiner Hilfe auch eine Person,die den Versuch nicht selbst durchgeführt hat,die vollständige Auswertung des Versuchesvornehmen kann!
11 Wenn die Auswertung nicht währendder Praktikumszeit erfolgt, muss das Mess-protokoll (jedes einzelne Blatt!) vom Assi-stenten mit Datum und Kurzunterschriftabgezeichnet werden.

Einführung Fehlerrechnung und Statistik
5
Zur Auswertung
12 Alle Rechnungen müssen anhand derFormeln und der Messdaten im Protokollnachvollziehbar sein.
13 Diagramme werden auf Millimeter-papier mit Lineal bzw. Kurvenlineal gezeich-net oder mit dem Computer erstellt. Es sindgeeignete übersichtliche Maßstäbe zu wählenund die Achsen sind mit Größe und Einheitzu beschriften. In der Regel ist eine Legendeerforderlich.
14 Zu jedem Versuch gehört eine Fehler-diskussion. Fehler (geschätzt oder mit statisti-schen Mitteln berechnet) sind immer quanti-tativ anzugeben; bei manchen Versuchen
wird eine Fehlerrechnung gefordert. (Detailsim Kapitel “Fehlerrechnung“)
15 Die Versuchsergebnisse werden ineinem verbalen Ergebnissatz zusammenge-fasst, eingeschätzt (z. B. bezüglich ihrerMessgenauigkeit) und, wenn möglich, mitLiteraturwerten verglichen.
16 Das komplette Protokoll wird in derRegel (siehe „Hinweise zum Ablauf desPraktikums”) am nächsten Praktikumstagdem verantwortlichen Assistenten vorgelegt,von diesem durchgesehen und bewertet.Durch das Datum und die Langunterschriftwird der erfolgreiche Versuchsabschlussbescheinigt.
Fehlerrechnung und Statistik
Jede Messung einer physikalischen Größe istmit mehr oder weniger großen Messfehlernbehaftet. Misst man eine Größe mehrmals, soweichen die Ergebnisse im Allgemeinensowohl voneinander als auch vom zu be-stimmenden „wahren Wert“ ab. Ziel derFehlerrechnung ist das Ermitteln des bestenSchätzwertes für den wahren Wert (Mess-ergebnis) und für die Größe der Abweichung(Messunsicherheit).
1 Begriffsbestimmungen
Messgröße:Die zu messende physikalische Größe, z.B. Spannung U, Strom I, Masse m
Messwert:Der gemessene Wert einschließlich Ein-heit, z. B. U = 220 V, I = 2 A, m = 2 kg
Messergebnis:Das aus mehreren Messwerten berechneteErgebnis, z. B. P = U A I = 220 V A 2 A =440W
Messabweichung (früher Fehler genannt):
Differenz zwischen Messwert (odereinem aus mehreren Messungen gewon-nenen Wert) und wahrem Wert. Manunterscheidet zufällige und systematischeMessabweichungen. Die Messabwei-chung ist im Allgemeinen nicht genaubekannt, weil der wahre Wert nicht genaubekannt ist.
Zufällige oder statistische Messabweichun-gen (Fehler):
Sie treten unregelmäßig auf; sie schwan-ken in der Größe und im Vorzeichen.Hervorgerufen werden sie z. B. durchnicht beeinflussbare unsystematischeÄnderungen der Versuchs- und Umge-bungsbedingungen sowie durch Unvoll-kommenheiten beim subjektiven Erfassenvon Messwerten durch den Praktikanten.Durch mehrfaches Messen und Bildungdes arithmetischem Mittelwertes kann derEinfluss zufälliger Messabweichungenminimiert werden.
Systematische Messabweichungen (Fehler):Sie beeinflussen bei gleichen Versuchs-bedingungen die Messung in der gleichen

Einführung Fehlerrechnung und Statistik
6
s
x x
n
ii
n
=
−
−=
∑ ( )
.
2
1
1
(2)
′ =ss
n. (3)
∆ x
x x
n n
ii
n
=
−
⋅ −=
∑ ( )
( ).
2
1
1
(4)
xn
xii
n
==
∑1
1
. (1)
Weise. Hervorgerufen werden sie z. B.durch Unvollkommenheiten der Mess-geräte, der Maßverkörperungen und derMessverfahren sowie durch systema-tische Änderungen der Versuchsbedin-gungen. Sie setzen sich aus einem be-kannten und einem unbekannten Anteilzusammen. Das Messergebnis ist umbekannte systematische Messabwei-chungen zu korrigieren.
Messunsicherheit:Schätzung der Messabweichung. Gibteinen Bereich (Intervall) an, in dem der„wahre“ Wert einer Messgröße oder einesMessergebnisses mit hoher Wahrschein-lichkeit liegt. Sie wird auf der Grundlagevon Messwerten (m. H. statistischerMethoden) und vorliegender Kenntnissezu systematischen Messabweichungengeschätzt. Beispiel (für den Messwert U = 220 V):∆U = 2,4 V (absolute Messunsicher-heit), ∆U/U = 1,1% (relative Messunsi-cherheit)Der wahre Wert liegt mit großer Wahr-scheinlichkeit im Intervall (U!∆U,U+∆U).
Vollständiges MessergebnisMessergebnis mit Messunsicherheit; z.B.U = 220,0 V ± 2,4 VU = (220,0 ± 2,4) VU = 220,0 V und ∆U/U = 1,1 %
2 Ermittlung von Messunsicherheiten
2.1 Berechnung von Messunsicherheitenbei zufälligen Fehlern
Eine Messgröße x werde n mal gemessen; dieeinzelnen Messwerte xi (i = 1 ... n) streuenum einen Mittelwert
Wenn bei dieser Messreihe nur zufällige(statistische) Fehler auftreten, so ist die Ver-teilung der Messwerte eine Normalverteilung
(Gaußverteilung). Die graphische Darstellungeiner solchen Verteilung ergibt die soge-nannte „Glockenkurve“.Als Maß für die Streuung der Messwerte wirddie Standardabweichung s eingeführt:
Für die Gaußverteilung ergibt sich, dass68,3 % der Messwerte im Intervall ± sxliegen, d. h. die Wahrscheinlichkeit, einenMesswert in diesem Intervall anzutreffen,beträgt 68,3 %.Im Intervall ± 2s liegen 95,5 % und imxIntervall ± 3s 99,7 % aller Messwerte.xWerden von der Messgröße weitere Mess-reihen vom Umfang n aufgestellt, so sind diedazugehörigen Mittelwerte ebenfalls normal-verteilt; die Standardabweichung s' für dieStreuung der Mittelwerte ist dann:
Ist das Messergebnis ein Mittelwert einerxMessreihe mit n $ 10 Messwerten xi undkönnen dabei die systematischen Fehlergegenüber den zufälligen Fehlern vernachläs-sigt werden, so wird als Messunsicherheit ∆xdie Standardabweichung s' für den Mittel-wert gewählt:x
Ist die Messung eine Zählung zufälliger Er-eignisse (z. B. radioaktiver Zerfallsereig-nisse), x = N, so beträgt die Messunsicher-heit (bei Vernachlässigung systematischer
Fehler) ∆x = (siehe Versuch O16).N
2.2 Die Garantiefehlergrenze als Messun-sicherheit
Die Hersteller von Messgeräten geben in der

Einführung Fehlerrechnung und Statistik
7
y a b x= + ⋅ (5)
[ ]∆ y y a bxi
n
i ii
n2
1
2
1= =
∑ ∑= − + →( ) min. (6)
b = ∆y/∆x
∆y
∆xa
0 x
y
Abbildung 1 Lineare Regression mit y = a +b@x
Regel Garantiefehlergrenzen an (Beispiele:1,5 % vom Messbereich; 0,1 % vom Mess-wert + 2 Digit). Auf manchen Geräten ist die“Genauigkeitsklasse” angegeben. Das ist diemaximale Messabweichung in % vom End-wert des Messbereichs bzw. vom Wert derMaßverkörperung. Bei einer Genauigkeits-klasse von 1,5 und einem Messbereich von30V beträgt die Garantiefehlergrenze∆U = (1,5 % von 30 V) = 0,45 V.
2.3 Angabe einer geschätzten oberenFehlergrenze als Messunsicherheit
Liegen keine Angaben vor, so ist die Mess-unsicherheit zu schätzen:- Faustregel beim Ablesen von Skalen: ∆x =
(0,5 ... 1) Skalenteil- Längenmessungen mit einem Messschieber
(Noniusablesung): ∆l = 0,1 mm- Messung einer Schwingungsdauer T an-
hand von 20 Schwingungen: ∆(20T) = 0,2s. Für ∆T ergibt sich dann ∆T = 0,01 s.
- Die Messunsicherheit bei digital anzei-genden Messgeräten beträgt mindestens 1Digit (Digitalisierungsfehler), ist abermeist größer.
3 Anpassung einer Funktion an eineMessreihe (Regression)
3.1 Lineare Regression
Häufig besteht zwischen verschiedenenMessgrößen x und y ein linearer Zusamm-enhang
oder es wird ein solcher Zusammenhang ver-mutet.
Beispiel:Bei der thermischen Ausdehnung von Metal-len gilt für die Länge l = l0 + α@l0@∆T, α istder lineare thermische Ausdehnungskoeffi-zient, l0 die Länge bei der Temperaturdiffe-renz ∆T=0 (siehe Versuch W1).
Die eigentliche Messaufgabe besteht in derBestimmung der (konstanten) Parameter aund b. Grundsätzlich können a und b durchMessung von zwei Wertepaaren (x, y) be-stimmt werden. Meist wird jedoch eine ganzeMessreihe mit n Wertepaaren (xi, yi) (i = 1 ...n) aufgenommen, um zunächst den linearenZusammenhang nachzuweisen, ehe a und bermittelt werden.
Werden die Messwerte graphisch dargestellt,so streuen die Messpunkte wegen der unver-meidlichen statistischen Messabweichungenum eine ausgleichende Gerade, siehe Abb. 1.
Die Aufgabe besteht nun darin, die Gerade zufinden, die „am besten“ an die Messpunkteangepasst ist. Hierfür gibt es ein mathemati-sches Verfahren, das man als lineare Regres-sion, Ausgleichsrechnung, Geraden-An-passung oder auch (englisch) linear curve fitbezeichnet. Das Verfahren nach GAUSS
beruht auf der Minimierung der Summe derAbweichungsquadrate
und heißt deshalb auch Methode der kleinstenQuadrate.
Mit geeigneter Software (z.B. Excel, Origin,CassyLab) und auch mit manchen Taschen-rechnern kann man diese Methode benutzen,ohne dass man den mathematischen Forma-

Einführung Fehlerrechnung und Statistik
8
∆ ∆ ∆ ∆
∆ ∆
yy
xx
y
xx
y
xx
yy
xx
nn
ii
i
n
= + + +
==
∑
∂
∂
∂
∂
∂
∂
∂
∂
11
22
1
...
.
(7)
lismus kennen und verstehen muss. Dabeisind die n Messwertpaare (xi, yi) einzugeben,danach werden die Parameter a und b derangepassten (“besten”) Geraden und je nachSoftware auch die Standardabweichungen derParameter sa und sb berechnet.
Wenn keine geeigneten Rechenhilfsmittel zurVerfügung stehen, nimmt man die Anpassunggraphisch vor: Die Messpunkte werden in einmöglichst großes Diagramm auf Millimeter-papier gezeichnet und die Regressionsgerademit einem durchsichtigen Lineal „nachAugenmaß“ eingezeichnet. Die Parameter aund b werden als y-Achsenabschnitt bzw.Anstieg wie in Abb. 1 gezeigt abgelesen undihre Unsicherheiten werden geschätzt.
3.2 Regression mit anderen Funktionen
Das Regressionsverfahren mit der Methodeder kleinsten Quadrate kann nicht nur aufeine lineare Funktion (5), sondern auf beliebi-ge Funktionen mit mehreren Parameternangewendet werden. Im Allgemeinen istdieses Problem jedoch nicht mehr analytischlösbar, sondern muss mit Hilfe numerischerMethoden iterativ gelöst werden. Die imPraktikum eingesetzten ComputerprogrammeOrigin und CassyLab bieten diese Möglich-keit. (Stichworte: non-linear curve fit bzw.Freie Anpassung)Einige Funktionen können durch Transfor-mation bequem in eine lineare Funktionüberführt werden. In solchen Fällen kann dielineare Regression mit der transformiertenFunktion durchgeführt werden. Alternativkann die Funktion in einem Koordinatensys-tem mit geeignet nichtlinear (z.B. logarith-misch) geteilten Achsen dargestellt werden,so dass der Graph eine Gerade ergibt.
Beispiel:Beim Durchgang radioaktiver Strahlungdurch Materie der Dicke d gilt für die Intensi-tät I = I0@e
-µd, der “Schwächungskoeffizient µsoll aus mehreren Messwerten für I und dbestimmt werden. Logarithmiert man die
Gleichung, ergibt sich ln I = ln I0 - µ@d . Setztman ln I = y und d = x und vergleicht mit(5), sieht man dass a = ln I0 und b = !µ. DerSchwächungskoeffizient µ kann also durchlineare Regression mit den Wertepaaren (lnI,d) bestimmt werden. Alternativ wählt man fürdie graphische Darstellung I = f(d) Milli-meterpapier mit logarithmischer Teilung fürI und normaler (linearer) Einteilung für d,dann ergibt sich ebenfalls eine Gerade.
4 Messunsicherheiten für Messergeb-nisse (Fehlerfortpflanzung)
Es sei y = f(x1, x2, ..., xn) ein Messergebnis,das aus den Messwerten x1, x2, ..., xn mit denMessunsicherheiten ∆x1, ∆x2, ..., ∆xn zuberechnen ist. Wie groß ist dann die Mess-unsicherheit ∆y des Messergebnisses?
Für kleine Messunsicherheiten ∆xi kannman die Unsicherheit des Messergebnissesals “totales Differential” von y berechnen:
Dabei ist My/Mxi die partielle Ableitung von ynach der Messgröße xi. Bei dieser Art der Berechnung wird ange-nommen, dass sich die Einflüsse aller Mess-unsicherheiten auf die Unsicherheit desErgebnisses addieren - es ergibt sich diegrößtmögliche Messunsicherheit ∆y.
Für das Physikpraktikum und für die al-lermeisten Anwendungen in der Medizin istes ausreichend, folgende Spezialfälle zukennen:
Spezialfall 1:
Das Messergebnis ist eine Summe (oderDifferenz) aus den gewichteten Einzelmes-sungen:

Einführung Fehlerrechnung und Statistik
9
∆ ∆ ∆y
yn
x
xm
x
x= +
1
1
2
2
. (11)
y y y y± ∆ ∆und , (12)
y c x c x= +1 1 2 2 (8)
∆ ∆ ∆y c x c x= +1 1 2 2 . (9)
y c x xn m= ⋅ ⋅1 2(10)
(c1, c2 sind Konstanten). Durch Einsetzen in(7) ergibt sich die Unsicherheit des Ergeb-nisses zu
Die absolute Unsicherheit des Ergebnisses istgleich der Summe der absoluten Unsicherhei-ten der Messwerte gewichtet mit den Fakto-ren c1 und c2.
Spezialfall 2:
Das Messergebnis ist ein Produkt (oderQuotient) aus den Messwerten mit ganzzah-ligen Exponenten:
(c eine reelle und n, m ganzzahlige Kon-stanten). Durch Einsetzen in (7) ergibt sichdie Unsicherheit des Ergebnisses zu
Die relative Unsicherheit des Ergebnisses istgleich der Summe der relativen Unsicherhei-ten der Messwerte, gewichtet mit den Expo-
nenten mit denen sie in der Gleichung (10)für das Messergebnis auftreten.
Beispiel: Gleichmäßig beschleunigte Bewe-gung s = a/2 @ t2 ; Weg s und Zeit t werdengemessen und die Beschleunigung a ist zuberechnen:
as
t
aa
ss
tt
= ⋅ = +2 22 ,∆ ∆ ∆
5 Angabe der Messergebnisse mit ihrenMessunsicherheiten
Es ist immer das vollständige Messergebnisanzugeben:
wobei die Messunsicherheit ∆y nur ein oderzwei zählende (signifikante) Ziffern habendarf. Entsprechend ist die Zahl der Ziffern fürdas Messergebnis y zu wählen.
Beispiele:
y = (531,4 ± 2,3) mm; ∆y/y = 0,43 %
U = (20,00 ± 0,15) V; ∆U/U = 0,12 %
R = 2,145 kΩ ± 0,043 kΩ; ∆R/R = 2,0 %

Mechanik M 2 Dichtebestimmung
10
ρ =m
V. (1)
F m g V gA M M= ⋅ = ⋅ ⋅ρ . (2)
m g V g m g V gL N N L⋅ − ⋅ ⋅ = ⋅ − ⋅ ⋅ρ ρ (3)
m m L
L N
* //
.= ⋅−
−
11
ρ ρρ ρ
(5)
m m** W
L N
= ⋅−
−
1
1
ρ ρ
ρ ρ
/
/. (6)
m mNL N
L
= ⋅−
−
11
ρ ρρ ρ
//
. (4)
1 Aufgabenstellung
1.1 Die Dichte von drei Probekörpern istnach der Auftriebsmethode zu bestimmen.
1.2 Die Dichte von Ethanol ist mit Hilfeeines Pyknometers zu bestimmen.
1.3 Die Dichten von Ethanol und NaCl-Lösung sind mit der Dichtewaage nach Mohr-Westphal und mit dem Aräometer zu be-stimmen.
2 Grundlagen
Die Dichte ρ eines homogenen Stoffes ist dasVerhältnis seiner Masse m zu seinem Volu-men V :
In der Pharmazie spielen unterschiedlicheDichten z.B. eine große Rolle bei der Sedi-mentation. Je größer der Unterschied derDichten der Flüssigkeit und der Teilchen,desto schneller werden die Teilchen absin-ken. Ähnlich sind die Verhältnisse bei Zen-trifugationsprozessen, allerdings gestatten diedabei erreichbaren großen Zentrifugalkräftehöhere Sedimentationsgeschwindigkeiten alsallein unter Wirkung der Gravitation.Anwendung findet dies in der Arzneiformen-lehre (z.B. Suspensionen) und bei der analyti-schen Zentrifugation
2.0 AnalysenwaageDie Bestimmung der Masse zählt zu dengenauesten Messverfahren der Physik. Mitden Analysenwaagen im Praktikum lassensich relative Genauigkeiten bis zu 10-6 erzie-len. Bei solchen Präzisionsmessungen mussder Auftrieb in Luft berücksichtigt werden.Jeder Körper, der sich in einem Medium(Flüssigkeit oder Gas) befindet, erfährt einenAuftrieb. Die Auftriebskraft FA ist gleich der
Gewichtskraft des vom Körper verdrängtenMediums (ARCHIMEDESsches Prinzip):
Dabei sind mM und ρM Masse bzw. Dichte desvom Körper verdrängte Mediums, V seinVolumen und g = 9,81 ms-2 die Fallbeschleu-nigung. Bei einer Analysenwaage (Balkenwaage)wirkt der Auftrieb in Luft der Dichte ρL so-wohl auf den zu wiegenden Körper (Masse m,Dichte ρ) als auch auf die Wägestücke (Mas-se mN, Dichte ρN). Im Kräftegleichgewicht ist
bzw. mit V = m/ρ und VN = mN /ρN
Diese Formel für die Korrektur des Luftauf-triebs gilt auch für moderne elektronischeAnalysenwaagen. Hier werden zwar bei derWägung keine Gewichtstücke mehr benutzt,jedoch wird die Anzeige der Waage (ent-spricht mN) mit Hilfe von Normalgewicht-stücken mit der standardisierten Dichte vonρN = 8000 kg/m3 geeicht bzw. justiert.
2.1 Auftriebsmethode Mit Hilfe des Auftriebes lässt sich relativeinfach die Dichte eines Körpers mit unbe-kanntem Volumen V bestimmen. Dazu wirdder Körper mit Hilfe einer Analysenwaage anLuft und in Wasser eingetaucht gewogen.m* bezeichne den Anzeigewert der Waage beiWägung in Luft. Nach (4) ist
Danach wird der Körper völlig unter Wassergetaucht und erneut gewogen, Anzeige m**:
Dichtebestimmung M 2

Mechanik M 2 Dichtebestimmung
11
ρρ ρ
=−
−
m m
m m
W L**
**
*
*. (7)
m m mL Py
L N
L W
L N2 1 2
1
111
* /
///
=−
−+
−
−
ρ ρ
ρ ρρ ρρ ρ
(9)
m m mL Py
L N
L
L N3 1 3
1
11
1* /
//
/=
−
−+
−
−
ρ ρ
ρ ρ
ρ ρ
ρ ρ(10)
Abbildung 2 Mohr-Westphalsche Waage
m mL Py
L N1 1
1
1* /
/= ⋅
−
−
ρ ρ
ρ ρ(8)
mm
3
2
3
2
=ρρ
. (11)
( )ρ ρ ρ ρ=−
−⋅ − +
m m
m mW L L
3 1
2 1
* *
* *. (12)
(ρW - Dichte des Wassers).
Aus den Gleichungen (5) und (6) folgt:
2.2 Pyknometer
Ein Pyknometer ist ein Gefäß, mit dem einFlüssigkeitsvolumen sehr genau reproduzier-bar ist, da der durch die Krümmung derOberfläche verursachte Volumenfehlerwegen des geringen Kapillarquerschnittessehr klein ist (Abb.1). So kann man damitüber eine Messung der Masse sehr einfachund genau die Dichte von Flüssigkeitenbestimmen. Dabei erfolgt erst eine Messungmit Luft, um die Leermasse m1 des Pykno-meters zu erhalten:
Danach wird die Masse des Pyknometers mitdestilliertem Wasser der Masse m2 und dannseine Masse mit der Messflüssigkeit derMasse m3 bestimmt (m* ist jeweils Anzeigeder Waage):
Wegen des konstanten Pyknometervolumensist außerdem
Mit Hilfe der Gleichungen (8) bis (11) lässtsich ρ berechnen:
2.3 Mohr-Westphalsche WaageDie Mohr-Westphalsche Waage ist eineungleicharmige Hebelwaage. Der rechteHebelarm ist durch Kerben in 10 gleicheTeile geteilt. Am Ende des Hebelarmesbefindet sich ein Senkkörper mit sehr genaudefiniertem Volumen. Wird der Senkkörperin eine Flüssigkeit getaucht, erfährt er einenAuftrieb, der durch die Gewichtskraft ent-sprechend aufgelegter Reiter kompensiertwerden kann. Zur original Mohr-Westphal-schen Waage (Abb. 2) gehören große, mitt-lere und kleine Reiter, deren Gewichtskräftesich wie 100 : 10 : 1 verhalten. Die relativeDichte ergibt sich aus der Position der Reiterin den Kerben 1 bis 10.Die moderneren, im Praktikum eingesetztenDichtewaagen verwenden einen in 100 Teileeingeteilten Waagebalken, zwei Reiter imVerhältnis 100 : 1 und ein zusätzlichesAnhängegewicht. Die Dichte kann direkt ander Stellung der Reiter abgelesen werden.
2.4 Aräometer
Abbildung 1 Pyknometer

Mechanik M 2 Dichtebestimmung
12
ρ ~ ,1
VM
(13)
Abbildung 3Aräometer
Mit Hilfe eines Aräometers (Abb. 3) kannebenfalls die Dichte einer Flüssigkeit be-stimmt werden. Schwimmt das Aräometer inder Messflüssigkeit, ist die Gewichtskraft desAräometers FG gleich der Auftriebskraft FA.Nach (2) folgt für die Dichte der Messflüssig-keit
wobei VM das eingetauchte Volumen desAräometers ist. Die Skala, die sich am Aräo-meter befindet, zeigt unmittelbar die denEintauchtiefen entsprechende Dichte an.
3 Versuchsaufbau
3.1 Geräte zu Aufgabe 1:1 Analysenwaage mit Dichtebestimmungs-
einrichtung (Tauchkorb, Brücke, Becher-glas)
3 Probekörper1 Pinzette1 Spritzflasche mit H2O dest.
3.2 Geräte zu Aufgabe 2:1 Analysenwaage1 Pyknometer1 Pipette1 Spritzflasche mit H2O dest. 1 Flasche mit Ethanol
3.3 Geräte zu Aufgabe 3:1 Dichtewaage mit Senkkörper1 Senkglas (ca. 130 ml)2 Standzylinder (100 cm3)2 Aräometer
2 Flaschen mit Ethanol und NaCl-Lösung1 Thermometer
4 Versuchsdurchführung
Hinweise zur Handhabung der elektronischenWaage werden vom zuständigen Assistentengegeben bzw. der Gerätebeschreibung ent-nommen!
4.1 Zur Bestimmung der Dichte festerKörper nach der Auftriebsmethode werdendie beiliegenden Probekörper zunächst inLuft gewogen (Bestimmung von m*). Danachwird das Becherglas mit Wasser auf dieBrücke gestellt, der Tauchkorb vorsichtigeingehängt und die Waage auf Null abgegli-chen (mit Tariertaste T). Jetzt stellt man denProbekörper mit der Pinzette auf den Tauch-korb, so dass der Körper völlig im Wassereintaucht. Das Ablesen der Waage ergibt m**.Dies ist für alle Probekörper durchzuführen.Hinweis zur Fehlerbetrachtung: In Formel (6)ist nicht berücksichtigt, dass durch das Ein-tauchen des Probekörpers der Wasserspiegelim Becherglas steigt und somit ein geringerzusätzlicher Auftrieb an den Halterungen desTauchkorbes entsteht.
4.2 Zur Ermittlung der Flüssigkeitsdichte istdie Brücke mit dem Becherglas zu entfernen.Es werden die Leermasse des Pyknometers(m*
1), die Masse des Pyknometers mit destil-liertem Wasser (m*
2) und die Masse desPyknometers mit der Messflüssigkeit (m*
3)bestimmt.
4.3 Messung mit der Dichtewaage:Zunächst wird die Nullpunktseinstellung derDichtewaage kontrolliert und ggf. nach-justiert. Verfahren Sie hierzu entsprechendder ausliegenden Bedienungsanleitung.Für die Bestimmung der Dichte der beidenFlüssigkeiten muss der Senkkörper voll-ständig in diese eingetaucht sein. VermeidenSie Luftbläschen sowie die Berührung derGefäßwand! Die Waage wird durch Ver-schieben der Reiter abgeglichen. Dabei bleibt

Mechanik M 4 Oberflächenspannung von Flüssigkeiten
13
für Dichten < 1 g/cm3 das Anhängegewichteingehängt, für Dichten > 1 g/cm3 wird esausgehängt.Die Temperaturen der Messflüssigkeit sindzu messen.Bei der Dichtebestimmung mit dem Aräo-meter werden die Messflüssigkeiten in dieentsprechenden Standzylinder, in denen sichdie Aräometer befinden, gegossen und dieDichten abgelesen.
Die Messflüssigkeiten sind im Anschluss indie richtigen Behälter zurückzufüllen!
5 Auswertung
5.1 Die Dichte der Probekörper ist nach derGleichung (7) zu berechnen. Das Ergebnis istmit Tabellenwerten zu vergleichen. Umwelches Material könnte es sich bei denProbekörpern handeln?Die Dichte des Wassers kW für die Raum-temperatur wird einer Tabelle entnommen.Für die Dichte der Luft wird der bei Normal-druck geltende Wert kL = 0.0013 g/cm3
verwendet.
5.2 Die Dichte der Flüssigkeit ist nachGleichung (12) zu berechnen und mit demErgebnis der anderen Methoden sowie mitdem Tabellenwert zu vergleichen.
5.3 Die Dichten, die nach den zwei Metho-den ermittelt wurden, sind zu vergleichen.
Die Messunsicherheiten der verschiedenenMethoden zur Bestimmung der Dichte sindzu vergleichen.
6 Literatur
Geschke, D.(Hrsg.): Physikalisches Prakti-kum. B.G. Teubner Stuttgart Leipzig 2001
Kamke/Walcher: Physik für Mediziner. B.G.Teubner, Stuttgart 1994
Hellenthal, W.: Physik. Thieme, Stuttgart1998
7 Kontrollfragen
7.1 Welche Methoden zur Bestimmung derDichte von festen Körpern und Flüssigkeitenkennen Sie?
7.2 Welchen Einfluss hat der Auftrieb inLuft auf Wägungen?
7.3 Erläutern Sie die Messmethode mit demPyknometer! Worauf ist dabei besonders zuachten?
1 Aufgabenstellung
1.1 Die Oberflächenspannung von ver-schiedenen Flüssigkeiten ist mit Hilfe derAbreißmethode zu bestimmen.
1.2 Die Oberflächenspannung ist mit Hilfeder Steighöhe in Kapillaren zu bestimmen.
2 Grundlagen
2.1 Oberflächenspannung:Jedes einzelne Molekül einer Flüssigkeitwirkt innerhalb eines gewissen kugelförmi-gen Bereiches anziehend auf seine Nachbar-moleküle (Kohäsion). Ein Molekül im Inne-ren der Flüssigkeit übt nach allen Seitengleiche anziehende Kräfte aus und wird von
Oberflächenspannung von Flüssigkeiten M 4

Mechanik M 4 Oberflächenspannung von Flüssigkeiten
14
Abbildung 1 Kohäsionskräfte und Ober-flächenenergie von Flüssigkeitsmolekülen.
∆ ∆W A= ⋅σ . (1)
den umgebenden Molekülen ebenfalls gleich-mäßig angezogen, so dass die Resultierendedieser Kohäsionskräfte gleich Null ist. Befin-det sich das Molekül an der Oberfläche derFlüssigkeit, so werden die Kräfte nichtvollständig durch die Wechselwirkungskräftemit dem angrenzenden Medium (Adhäsion)kompensiert (Abb.1). Es ergibt sich eineresultierende Kraft in das Innere der Flüssig-keit. Möchte man jetzt ein Molekül aus demInneren an die Oberfläche bringen, muss mandie nach innen wirkenden Kräfte überwinden.Ein Molekül an der Oberfläche besitzt des-halb eine höhere potentielle Energie. DieEnergie der Gesamtheit aller in der Oberflä-che sitzenden Moleküle ist der Größe derOberfläche proportional und kann als Ober-flächenenergie bezeichnet werden. Will mandie Oberfläche um ∆A vergrößern, muss mander Flüssigkeit Energie zuführen, also Arbeit∆W verrichten:
Dabei ist σ die Oberflächenspannung, einevon der Temperatur abhängige Materialgröße.
Die SI-Einheit ist N/m. In der Natur ist jedesSystem bestrebt, den Zustand kleinster poten-tieller Energie zu erreichen. Die Oberflächeeines Flüssigkeitsvolumens wird daher immereinen möglichst kleinen Wert annehmen.Die Oberflächenspannung wirkt sich auf dieTropfenbildung an einer Pipette aus. EinTropfen reißt ab, wenn seine Gewichtskraft
die ihn haltende Kraft überschreitet. Letztereist proportional zur Oberflächenspannung derFlüssigkeit, so dass z.B. eine mittels Pipettedurch Tropfenzählung zu dosierende Sub-stanzmenge für verschiedene Flüssigkeitenzunächst mit anderen Verfahren kalibriertwerden muss. Die Messung der Oberflächenspannung miteinem Stalagmometer nutzt eben diesenEffekt: Es wird die Anzahl der Tropfen ineinem Volumen gezählt; sie ist proportionalzur Oberflächenspannung. Weitere Beispiele für den Einfluss der Ober-flächenspannung sind die Randkrümmungvon Flüssigkeiten in Gefäßen, die Kapillar-wirkung, die Wirkung von Waschmitteln unddas wasserabweisende Gefieder vonSchwimmvögeln.
2.2 Kapillarität:An einer Grenzfläche zwischen zwei Medienüben auch die verschiedenartigen MoleküleAnziehungskräfte aufeinander aus. Im Gegen-satz zur Kohäsion bezeichnet man die Anzie-hungskraft zwischen den Molekülen ver-schiedener Stoffe als Adhäsion.Die Oberfläche eines Festkörpers wird voneiner Flüssigkeit benetzt, wenn die Adhä-sionskräfte größer sind als die Kohäsions-kräfte innerhalb der Flüssigkeit. Die Flüssig-keit versucht dann, sich möglichst weit aufder Oberfläche auszubreiten. Taucht man eineKapillare mit dem Innendurchmesser 2r ineine benetzende Flüssigkeit (z.B. Glas inWasser), so steigt diese in der Kapillare nachoben (Kapillaraszension im Gegensatz zurKapillardepression bei nicht benetzendenFlüssigkeiten). Das ist folgendermaßen zuerklären:Die Benetzung der Kapillarinnenwand führtzu einer Vergrößerung der freien Flüssig-keitsoberfläche und damit der Oberflächen-energie. Durch das Aufsteigen der Flüssigkeitim Inneren der Kapillare wird die Größe derfreien Oberfläche wieder verringert, dafürerhöht sich aber die potentielle Energie. DerGleichgewichtszustand ist der Zustand mini-maler Energie. Die Höhe der Flüssigkeits-

Mechanik M 4 Oberflächenspannung von Flüssigkeiten
15
( )∆ ∆A r h= ⋅ ⋅ ⋅2 2 π . (5)
∆ ∆W F h= ⋅ . (6)
σπ
=⋅ ⋅
F
r4. (7)
∆ ∆ ∆W A r h1 2= ⋅ = ⋅σ σ π . (2)
∆ ∆ ∆W m g h r h g h22= ⋅ ⋅ = ⋅ ⋅π ρ . (3)
hr g
=2σ
ρ. (4)
säule im Gleichgewicht kann man deshalbmit folgender Überlegung berechnen:Steigt die Flüssigkeit um den kleinen Betrag∆h, so verringert sich die freie Oberfläche umden Betrag 2πr∆h und damit nach (1) dieOberflächenenergie um
Dabei vergrößert sich die potentielle Energiedurch das Anheben der Flüssigkeit in derKapillare um
(∆m ist der Massenzuwachs und ρ die Dichteder Flüssigkeit in der Kapillare, g = 9,81 m/s2
die Fallbeschleunigung.)Im Energieminimum ist ∆W = ∆W2 !∆W1 =0,daraus ergibt sich die Steighöhe h zu
Gl. (4) kann auch mit Hilfe des Gleichge-wichtes aus dem Druck durch die gekrümmteOberfläche und dem Schweredruck derFlüssigkeitssäule hergeleitet werden (sieheLiteraturangaben).
3 Versuchsaufbau
3.0 Geräte:1 Federkraftmesser 50 mN1 Glasgefäß1 Messring2 KapillarenGefäße mit Flüssigkeiten1 höhenverstellbarer Tisch
3.1 Zur Messung der Oberflächenspannungnach der Abreißmethode wird ein Messringmit dem Durchmesser 2@r verwendet, derzunächst völlig in die Flüssigkeit eingetauchtund damit vollständig benetzt wird. Beimlangsamen Herausziehen aus der Flüssigkeitbildet sich am Ring ein zylindrischer Flüssig-keitsfilm mit dem Durchmesser 2@r und derHöhe ∆h. Da sich der Film sowohl an der
Innen- als auch an der Außenfläche desRinges bildet, ergibt sich die Vergrößerungder Flüssigkeitsoberfläche zu:
Die dafür erforderliche Arbeit ist:
Setzt man Gleichung (5) und (6) in (1) ein,erhält man:
Die Kraft F wird mit einem Federkraftmessergemessen.
4 Versuchsdurchführung
4.1 Um Verunreinigungen auszuschließen,sollten Messring und Glasgefäß zu Beginnunter fließendem Wasser gründlich abgespültwerden.Der trockene Messring (evtl. Tropfen abtup-fen) wird an den Federkraftmesser angehängt.Der Federkraftmessers ist durch Verschiebendes Außenmantels auf Null (oder, falls nichtmöglich, auf einen ganzen Wert, z. B. 10 mN) zu stellen. Das mit der zu untersuchen-den Flüssigkeit gefüllte Glasgefäß wird aufden höhenverstellbaren Tisch gestellt. DurchAnheben der Tischfläche lässt man denMessring vollständig in die Flüssigkeiteintauchen.Zur Messung wird der Tisch langsam undohne Erschütterung abgesenkt und dabei dieAnzeige des Federkraftmessers beobachtet.Beim Abreißen der Verbindung zwischenFlüssigkeit und der Unterkante des Mess-ringes wird die Kraft F abgelesen. Die Mes-sung ist für jede Flüssigkeit 10 mal durch-zuführen. Beim Wechsel der Flüssigkeit sindGefäß und Bügel gründlich abzuspülen.
4.2 Die Kapillaren werden sorgfältig unterfließendem Wasser gespült und dann entleert(schräg halten und langsam auslaufen lassen,den Rest Wasser mit feuchtem Zellstoffaussaugen). Danach taucht man sie in die

Mechanik M 13 Dehnung
16
σ =FA
. (1)
Messflüssigkeit. Die Höhe der senkrechtenFlüssigkeitssäule über der äußeren Flüssig-keitsoberfläche wird mit einem Lineal gemes-sen. Die Messung wird mit beiden Kapillarenund mit jeder Flüssigkeit fünf mal durch-geführt. Der Innendurchmesser ist auf denKapillaren angegeben mit einer Genauigkeitvon 2∆r = 0,02 mm.
5 Auswertung
5.1 Aus den zehn Messwerten für die Kraftist jeweils der Mittelwert zu bilden und nachGleichung (7) die Oberflächenspannung σund die zugehörige Messunsicherheit zuberechnen. Der mittlere Radius des Mess-ringes beträgt r = ( 14,85 ± 0.05 ) mm.
5.2 Aus den Mittelwerten der gemessenenSteighöhen ist mit Hilfe von Gl. (4) jeweilsdie Oberflächenspannung einschließlich derstatistischen Messunsicherheit zu berechnen.(Die Dichte von Wasser und Seifenlösungbeträgt 1,0 g/cm3.)
Für beide Teilversuche ist eine Fehlerrech-nung durchzuführen. Vergleichen Sie alleErgebnisse miteinander!
6 Literatur
Haas, U.: Physik für Pharmazeuten undMediziner. WVG Stuttgart 2002
Trautwein, Kreibig, Oberhausen: Physik fürMediziner, Biologen, Pharmazeuten, W. deGruyter, Berlin 1987
Fercher, A.F.: Medizinische Physik, Springer,Wien 1992
7 Kontrollfragen
7.1 Welche Form nimmt ein Wassertropfenan, wenn keinerlei äußere Kräfte auf ihnwirken? Warum? Worauf beruht die „norma-le“ Tropfenform?
7.2 Wovon hängt die Steighöhe einerFlüssigkeit in einer Kapillare ab?
7.3 Was ist Benetzung, wovon hängt sie ab?
1 Aufgabenstellung
Es ist der Elastizitätsmodul E von zweiMetallen und von Polyamid (Perlon) durchDehnungsmessungen zu bestimmen.
2 Physikalische Grundlagen
Eine wichtige Eigenschaft von Festkörpernist die Elastizität. Ein Körper ist elastisch,wenn er nach einer durch äußere Kräftehervorgerufenen Gestaltsänderung seineursprüngliche Gestalt wieder annimmt,
sobald diese Kräfte wegfallen.Die elastischen Formänderungen könnendurch Dehnung, Stauchung, Biegung oderDrillung (Torsion) auftreten.Unter (mechanischer) Spannung versteht mandas Verhältnis aus Kraft und der Quer-schnittsfläche, an der die Kraft angreift:
Bei Normalspannungen σ steht die Kraft Fsenkrecht auf der Fläche A (Abb.1 links), bei
Dehnung M 13

Mechanik M 13 Dehnung
17
Abbildung 2 Spannungs-Dehnungs-Dia-gramm (schematisch)
1 Verhalten nach Hookeschem Gesetz2 Metall im Bereich (a) elastischer und (b)
plastischer Verformung3 Blutgefäß. (c) Verhalten nach Hooke-
schem Gesetz, (d) Verfestigung
ε σ=1
E. (3)
ε =∆ l
l0
. (2)
Tangentialspannungen τ verläuft sie parallelzur Fläche (Abb.1 rechts). Normalspannun-gen können als Zug- oder Druckspannungenwirksam werden, Tangentialspannungen alsScher- oder Torsionsspannungen.
Die relative Längenänderung ∆l/l0 bei Wir-kung einer Zugspannung (vergl. Abb.1) nenntman Dehnung:
Das elastische Verhalten bei Zug- und Druck-belastungen wird im Spannungs-Dehnungs-Diagramm (Abb.2) dargestellt.
Innerhalb des Proportionalitätsbereiches giltdas HOOKEsche Gesetz:
Die elastische Verformung ist der verformen-den mechanischen Spannung proportional.
Feste Körper haben eine Elastizitätsgrenze.Bei Überschreiten dieser Grenze kommt es jenach Material zu unterschiedlichen Abwei-chungen vom HOOKEschen Gesetz. BeiMetallen tritt meist eine irreversible Verfor-mung auf (Plastizität). Bei biologischenMaterialien und bei Polymeren ist bei hohenSpannungen aufgrund des makromolekularenAufbaus oft eine Verfestigung zu beobachten.Das Überschreiten der Zerreißgrenze führtzum Materialbruch.Die meisten Polymere und viele Biomateria-lien (z. B. Muskelfasern) zeigen kein reineselastisches sondern sogenanntes viskoelasti-sches Verhalten. Im Materialinneren tretengeschwindigkeitsabhängige Reibungskräfteauf (vergl. Versuch M14: Viskosität). DieDehnung hängt nicht nur von der Spannungsondern auch von der Zeit ab, wobei diemaximale Dehnung bei Wirkung einer kon-stanten Spannung asymptotisch erreicht wird.
Der Elastizitätsmodul E ist eine Material-größe. Im Bereich der Gültigkeit des HOO-KEschen Gesetzes stellt sein Kehrwert denProportionalitätsfaktor zwischen der relativenDehnung ∆l/l0 eines Stabes und der anlie-genden mechanischen Spannung F/A dar:Mit (1) und (2) ergibt sich eine allgemeineForm des HOOKEschen Gesetzes zu:
Mit der Längenausdehnung verbunden isteine Verringerung des Querschnitts, die mitHilfe der elastischen MaterialkonstantenPOISSONsche Querkontraktionszahl beschrie-ben werden kann.
3 Versuchsaufbau
3.0 Geräte:- Wandhalterung mit Messuhr- Inbusschlüssel für die Messuhr- 2 Metalldrähte und Perlonfaden mit Haken
F
Al0
∆l
F
A
Abbildung 1 Dehnung (Zug) und Scherung

Mechanik M 13 Dehnung
18
und Messmarke- Bandmaß- Mikrometerschraube- Massestücke- mit Schaumstoff gefüllter Eimer
Der zu vermessende Draht bzw. Faden kannin die obere Aufhängung der Apparatureingehängt werden. Auf ihm ist eine Mess-marke befestigt, diese wird unter den Fühlerder Messuhr geklemmt. Dazu ist der Fühlervorsichtig von Hand anzuheben. Sollte derMessbereich der Messuhr (0 bis 10 mm) nichtausreichen, so kann sie mittels einer Klemm-schraube in der Höhe verstellt werden. Am unteren Ende des Drahtes ist ein Hakenzum Einhängen der Massestücke angebracht.
4 Versuchsdurchführung
Der zu vermessende Metalldraht wird einge-hängt und mit einem Massestück von 500 gvorbelastet. Es ist darauf zu achten, dass derMessbereich ausreicht, gegebenenfalls mussdie Messuhr in ihrer Höhe verstellt werden.Diese Stellung der Messuhr entspricht ∆l = 0.Die Anfangslänge l0 wird mit dem Bandmaßbestimmt (freie Drahtlänge von der Klemm-schraube der oberer Aufhängung bis zurKlemmschraube der Messmarke). Der Durch-messer des Drahtes d wird an 5 verschiede-nen Stellen mit der Mikrometerschraubebestimmt.Nun wird der Draht mit verschiedenen Mas-sestücken (200g bis 2000g in 200g-Schritten)belastet und die zugehörigen Längenänderun-gen ∆l gemessen (∆l bezieht sich hierbeiimmer auf die Stellung der Messuhr imvorbelasteten Zustand l0).Der gesamte Messvorgang ist für beideMatalldrähte durchzuführen.
Die Längenänderung des Perlonfadens istwesentlich größer als die der Metalldrähte,sie wird nicht mit der Messuhr sondern miteinem Lineal, Zeichendreieck oder mit demBandmaß gemessen. Der Perlonfaden wirdeingehängt und mit 100 g vorbelastet. Sein
Durchmesser wird an 5 verschiedenen Stellenmit der Mikrometerschraube und die Länge l0
mit dem Bandmaß bestimmt.
Es kann vorkommen, dass der Perlonfadenreißt! Stellen Sie deshalb den Eimer mitSchaumstoff-Füllung unter die Apparatur,ehe Sie weitere Massestücke anhängen!
Der Abstand a zwischen der Messuhr-Halte-rung und der Messmarke ist mit einem Linealzu messen. Dieser Abstand entspricht ∆l=0.Danach wird der Faden schrittweise belastet(400 g bis 2000 g in 400 g-Schritten) und(nach 5 min) der Abstand a bestimmt. Unmittelbar nach dem Anhängen der Mas-sestücke kann man beobachten, dass dieLänge nicht sofort konstant ist, sondern,immer langsamer werdend, noch etwasanwächst (Viskoelastizität). Zwischen jederÄnderung der Gewichte und der Messungvon a ist deshalb eine Wartezeit von 5 mineinzuhalten.
5 Auswertung
Aus den Mittelwerten der Drahtdurchmessersind die Querschnittsflächen zu berechnen.Im Fall des Perlonfadens sind aus den gemes-senen Abständen a die Längenänderungen ∆lzu berechnen.Für jeden Messschritt werden die anliegendeZugspannung σ nach (1) und die Dehnung gnach (2) berechnet. Die Kraft in Gleichung(1) ist die Gewichtskraft der Massestücke(g = 9,81 m/s2).Für jedes Material wird die Zugspannung σals Funktion der Dehnung des g grafischdargestellt und der Elastizitätsmodul E alsKurvenanstieg aus dem Diagramm ermittelt.Mit Hilfe der ausliegenden Tabelle ist ausden ermittelten Werten auf das Material derMetalldrähte zu schließen.Die möglichen Messfehler sind zu benennen(z.B. Längenmessung; Kraftbestimmungusw.) und in ihrem Einfluss abzuschätzen.

Mechanik M 14 Viskosität
19
Abbildung 1 Laminare Strömung durch einRohr
F F FG R A= + . (1)
F r gA =4
33
2π ρ . (2)
6 Literatur
Geschke (Hrsg.): Physikalisches Praktikum.B. G. Teubner, Stuttgart Leipzig 2001
Haas, U.: Physik für Pharmazeuten undMediziner. WVG Stuttgart 2002
Kamke/Walcher: Physik für Mediziner. B.G.Teubner, Stuttgart 1994
7 Kontrollfragen
7.1 Was bedeutet elastisches Material-verhalten?
7.2 Was besagt das HOOKEsche Gesetz?Welche Abweichungen vom HOOKEschenGesetz können auftreten?
7.3 Wo spielen Dehnungen im mensch-lichen Organismus eine Rolle?
1 Aufgabenstellung
Es ist die Viskosität η von Rhizinusöl alsFunktion der Temperatur mit einem HÖPP-LER-Viskosimeter (Kugelfallmethode) zubestimmen.
2 Grundlagen
Reale Flüssigkeiten und Gase sind durchWechselwirkungskräfte zwischen den Mole-külen innerhalb des Stoffes (Kohäsion) undzu Molekülen anderer Stoffe an Grenzflächenwie z.B. festen Wandungen (Adhäsion)gekennzeichnet. Bei idealen Flüssigkeitenoder Gasen werden solche Kräfte vernachläs-sigt.Strömt eine benetzende reale Flüssigkeitdurch ein starres konzentrisches Rohr, sostellt sich im Falle einer laminaren stationä-ren Strömung ein parabolisches Strömungs-profil (die Verteilung der Strömungsge-schwindigkeiten entlang des Rohrradius) ein(Abb. 1). Durch Adhäsionskräfte haftet dieFlüssigkeit am Rand und strömt in der Mitteam schnellsten. Zur Modellierung stellt mansich die Strömung als ineinander gleitendeZylinder vor, die sich mit geringenGeschwindigkeitsunterschieden gegeneinan-der bewegen. Zwischen diesen Schichten tritt
durch Kohäsionskräfte Reibung auf. Ein Maßfür diese innere Reibung ist die Viskosität η.
Eine Flüssigkeit, bei der die Viskosität nichtvon der Strömung selbst sondern nur von derTemperatur abhängt, nennt man eineNEWTONsche Flüssigkeit.
Sinkt ein kugelförmiger Körper (Radius r,Dichte ρ1) in einer viskösen Flüssigkeit(Dichte ρ2), so wirken die Gewichtskraft FG,die Auftriebskraft FA und die ReibungskraftFR und es gilt im stationären Zustand:
Nach ARCHIMEDES ist der Auftrieb gleichdem Gewicht des von der Kugel verdrängtenFlüssigkeitsvolumens:
Viskosität M 14

Mechanik M 14 Viskosität
20
( )η ρ ρ= −2
9
2
1 2
r
sgt. (8)
( )η ρ ρ= −K t1 2 . (9)
F r gG =4
33
1π ρ . (3)
F rvR = 6πη (4)
4
36
4
33
13
2π ρ πη π ρr g rv r g= + (5)
( )64
33
1 2πη π ρ ρrv r g= − . (6)
( )η ρ ρ= −2
9
2
1 2
r
vg . (7)
Für die Gewichtskraft FG gilt:
Da die STOKESsche Reibungskraft FR nach
proportional zur Geschwindigkeit v der Kugelist, stellt sich nach kurzer beschleunigterBewegung ein stationärer Zustand mit kon-stanter Fallgeschwindigkeit ein (wenn FR =FG ! FA erreicht ist). Aus Gleichung (1) folgt:
und:
Aus der Fallgeschwindigkeit v einer Kugel ineiner unendlich ausgedehnten ruhendenNEWTONschen Flüssigkeit kann demnach dieViskosität η der Flüssigkeit bestimmt werden.Die Umstellung von Gleichung (6) nach ηliefert:
Ersetzt man die Geschwindigkeit v durch denFallweg s und die Fallzeit t [v = s/t], so erhältman:
Alle unveränderlichen Größen können ineiner Konstanten K vereinigt werden, so dassfolgt:
In einem HÖPPLER-Viskosimeter fällt dieKugel nicht in einer unendlich ausgedehntenFlüssigkeit, sondern in einer Röhre, derenDurchmesser wenig größer als der Kugel-durchmesser ist. Um eine definierte Abroll-bewegung zu erzielen, wird der Zylinder um10° gegen die Normale geneigt. Beidesbeeinflusst die Kugelkonstante K, so dass beiindustriell gefertigten Viskosimetern experi-
mentell bestimmte Kugelkonstanten angege-ben werden.
Die Viskosität von Flüssigkeiten nimmt mitzunehmender Temperatur sehr stark ab;näherungeweise gilt
(10)η = ⋅a ebT
(a, b Konstanten, T in K). Die Ursache hierfürist die thermische Bewegung der Teilchen.Wenn sich die Moleküle der Flüssigkeitstärker bewegen, können sie sehr viel leichteraneinander vorbeigleiten.Im Gegensatz dazu steigt in Gasen die Visko-sität mit zunehmender Temperatur sogar an,
es gilt .η ~ T
3 Versuchsaufbau
3.0 GeräteHÖPPLER-Viskosimeter2 StoppuhrenThermostat
3.1 Das HÖPPLER-Viskosimeter ist einPräzisionsmessinstrument. Es besteht auseinem drehbar gelagerten geneigten zylindri-schen Fallrohr, das mit der zu untersuchendenFlüssigkeit gefüllt ist. Das Fallrohr wird voneinem Wasserbad umgeben, dessen Tempera-tur durch einen Thermostaten geregelt wird.Am Fallrohr befinden sich ringförmigeMessmarken, der Abstand zwischen derobersten und der untersten beträgt 100 mm.Die Messanordnung kann um einen im Fußgelagerten Führungszapfen in die Messlage(mit Arretierung) oder in die Rücklauflagegeschwenkt werden.Im Praktikum kann mit ausreichender Genau-igkeit in beiden Richtungen gemessen wer-den; Präzisionsmessungen mit der in derPrüfbescheinigung angegebenen Kugelkon-stante dürfen nur in Messposition durch-geführt werden.

Mechanik M 14 Viskosität
21
4 Versuchsdurchführung
Studieren Sie die am Arbeitsplatz ausliegen-den Kurzanleitungen zum Thermostaten undzum Viskosimeter. Schalten Sie zu Beginnauf keinen Fall die Thermostatheizung ein,das Wasserbad benötigt viel Zeit um wiederabzukühlen!Die Viskosität soll im Temperaturbereich vonRaumtemperatur bis 50°C bestimmt werden(etwa ein Messpunkt aller 5 K).Das Viskosimeter ist m. H. der Libelle hori-zontal auszurichten. Vor der ersten Messungmuss die Kugel einmal die Messstreckedurchlaufen, um die Messflüssigkeit zudurchmischen.Die Messung der Fallzeiten zwischen deroberen und der unteren Ringmarke wird vonbeiden Studenten durchgeführt. Damit siesich gegenseitig nicht beeinflussen, startet(stoppt) der erste Student seine Uhr, wenn dieKugel mit ihrer unteren Fläche die obere(untere) Ringebene berührt. Der zweiteStudent beginnt die Messung, wenn die obereFläche der Kugel die obere Ringebene ver-lässt.Die Messungen sind bei jeder Temperaturfünf mal durchzuführen. Bei Messzeiten über2 min kann die halbe Messstrecke verwendetwerden. Falls das Fallen der Kugel durch einesehr große Luftblase behindert wird, ist derzuständige Assistent zu verständigen. Siedürfen das Viskosimeter nicht selbst öffnen!Man beginnt zweckmäßigerweise bei Raum-temperatur. Der Thermostat ist einzuschalten,die Solltemperatur wird auf einen Wert unterRaumtemperatur gestellt, damit der Thermo-stat nur umwälzt. Falls erforderlich, muss vorder Messung ein Temperaturausgleich zwi-schen Thermostat und Viskosimeter abgewar-tet werden (etwa 10 min). Danach wird die Temperatur schrittweise (inSchritten von etwa 5 K) bis auf 50°C erhöhtund die Fallzeiten werden bestimmt. Nachjedem Erreichen der Solltemperatur amThermostaten muss jeweils etwa 10 mingewartet werden, da die Wärmeübertragungauf die Flüssigkeit im Messzylinder eine
gewisse Zeit erfordert. Damit wird außerdemberücksichtigt, dass für die Regelung dieTemperatur im Thermostat selbst verwendetwird, während für das Experiment die Tem-peratur der Thermostatflüssigkeit im Viskosi-meter gemessen wird.
5 Auswertung
Berechnen Sie die Viskosität η nach Glei-chung (9) und stellen Sie η als Funktion derTemperatur graphisch dar.
Die Dichte der Kugel ρ1 und der Wert derKonstanten K sind der Prüfbescheinigung desjeweiligen Viskosimeters zu entnehmen.(Die Bescheinigung liegt am Messplatz aus,die verwendete Kugel ist unterstrichen.)Dichte von Rhizinusöl: ρ2 = 0,96 g cm -3
Weisen Sie nach, dass die Viskosität ent-sprechend Gl. (10) von der absoluten Tempe-ratur T abhängt. Stellen Sie hierzu ln(η) alsFunktion von 1/T graphisch dar und diskutie-ren Sie die so erhaltene Kurve.
6 Literatur
Haas, U.: Physik für Pharmazeuten undMediziner, WVG Stuttgart, 2002
Hellenthal, W.: Physik, Thieme, Stuttgart,1988
Trautwein, Kreibig, Oberhausen: Physik fürMediziner, Biologen, Pharmazeuten. W. deGruyter, Berlin 1987
7 Kontrollfragen
7.1 Wodurch unterscheiden sich reale undideale Flüssigkeiten?
7.2 Was ist innere Reibung, wie kann mansie messen?
7.3 Wie beeinflusst innere Reibung dieStrömung in einem Rohr?

Mechanik M 19 Ultraschall-Abbildungsverfahren
22
λ =cf (1)
cE
L =−
+ −ρν
ν ν1
1 1 2( )( )(2)
y y x= ⋅ − ⋅0 e µ . (3)
1 Aufgabenstellung
1.1 Bestimmung der Schallgeschwindigkeitund der Wellenlänge von Longitudinalwellenin Polyethylen (PE), Berechnung des Elastizi-tätsmoduls von Polyethylen.
1.2 Bestimmung der Anzahl und Lage vonBohrlöchern in einem PE-Körper, Anferti-gung einer Lageskizze.
2 Grundlagen
Steht ein mechanischer Schwinger in Kontaktzu einem anderen Medium, so findet durchdie Kopplung zu diesem eine Energieüber-tragung statt, die sich als mechanische bzw.elastische Welle (Schallwelle) ausbreitet. Diein dem Medium entstehenden periodischenDruck- bzw. Dichteänderungen breiten sichmit einer Phasengeschwindigkeit (der Schall-geschwindigkeit) c aus. Die Wellenlänge λ imMedium wird nach
durch die Frequenz f der Schallquelle unddie von Stoffeigenschaften abhängige Aus-breitungsgeschwindigkeit c bestimmt. Diemechanischen Wellen treten in gasförmigenund flüssigen Stoffen infolge fehlenderScherelastizität stets als Longitudinalwellenauf, während in festen Körpern außer Longi-tudinalwellen auch Transversalwellen sowieVerkopplungen zwischen beiden (z.B. Ober-flächenwellen, Rayleighwellen) auftretenkönnen. In unendlich ausgedehnten, homogenen,isotropen Festkörpern ergibt sich die Schall-geschwindigkeit cL für Longitudinalwellenaus den mechanischen Eigenschaften desAusbreitungsmediums nach:
(E: Elastizitätsmodul, ν: POISSONscher Quer-kontraktionskoeffizient; ρ: Massendichte).
Durch inelastische Prozesse wird die Schall-welle im Medium gedämpft (absorbiert). Fürdie Abhängigkeit der Schwingungsamplitudey von der Ausbreitungsrichtung x gilt dasSchwächungsgesetz
Dabei ist y0 die Amplitude bei x = 0 und µ derSchwächungskoeffizient (auch Absorptions-koeffizient). Die Dämpfung kann in dermedizinischen Ultraschalldiagnostik zurUnterscheidung verschiedener Gewebeartendienen.
In der Akustik werden Frequenzen unterhalbdes menschlichen Hörbereichs (ca. 16 Hz -16 kHz) als Infraschall und oberhalb diesesBereiches als Ultraschall bezeichnet.Ultraschallwellen werden mit Hilfe desinversen piezoelektrischen Effektes erzeugt.Eine Scheibe aus piezoelektrischer Keramik -der Ultraschallwandler oder Transducer -wird elektrisch zu Schwingungen angeregt.Sie schwingt mit ihrer Resonanzfrequenz fr
und verursacht so eine sich im umgebendenMedium ausbreitende Schallwelle.Bei den Impuls-Echo-Verfahren A-Bild, B-Bild und TM (die Bezeichnungen kommenvon den engl. Begriffen Amplitude, Bright-ness und Time Motion) wird der Ultraschall-wandler durch einen elektrischen Spannungs-impuls zu einer kurzzeitigen mechanischenDickenschwingung und zum Aussenden einesUltraschallimpulses angeregt (reziprokerpiezoelektrischer Effekt). Aus dem angekop-pelten Medium auf denselben Wandlerauftreffende Ultraschallwellen bewirkengeringe Deformationen des Wandlers, die in
Ultraschall-Abbildungsverfahren M 19

Mechanik M 19 Ultraschall-Abbildungsverfahren
23
Z c= ⋅ρ . (4)
RII
Z ZZ Z
R= =−
+
0
1 2
1 2
2
(5)
I I ID R= −0 . (6)
Abbildung 2 Entstehung von A- und B-Bild
cl
t=
2(7)
dem piezoelektrischen Material in elektrischeSpannungen umgewandelt werden (direkterpiezoelektrischer Effekt).Ein und derselbe Wandler kann deshalbsowohl als Sender als auch als Empfängergenutzt werden.
Unter akustischer Impedanz (Schallkenn-impedanz, akustischer Widerstand) Z verstehtman das Produkt aus den MaterialkennzahlenMassendichte ρ und Schallgeschwindigkeit c:
Änderungen oder Sprünge der akustischenImpedanz (z.B. an Organgrenzflächen beimedizinischen Ultraschalluntersuchungen)längs der Ausbreitungsrichtung führen zueiner teilweisen Reflexion der Schallwelleund damit gleichzeitig zu einer Schwächungin Ausbreitungsrichtung (siehe Abb. 1). Fürden senkrechten Einfall einer Schallwelle aufeine Fläche gilt:
(R = Reflexionsgrad; I0, IR = einfallende undreflektierte Intensität; Z1, Z2 = akustischeImpedanzen).
Der durch die Fläche hindurchgehende AnteilID berechnet sich nach
Beim A-Bild-Verfahren wird die Amplitudeder vom Schallwandler gesendeten sowie der
empfangenen und verstärkten akustischenImpulse auf dem Monitor in Abhängigkeitvon der Zeit dargestellt. Die Echos vonStrukturgrenzen im Medium, an denen sichdie akustische Impedanz ändert, erscheinenim Bild als Zacken (Abb.2). Der zeitlicheAbstand zwischen Sendeimpuls und Emp-fangsecho entspricht der doppelten Laufzeit tdes akustischen Impulses zwischen Wandlerund reflektierender Struktur. Bei bekannterSchallgeschwindigkeit c kann damit nach
die Entfernung l zwischen beiden gemessenwerden.
Beim B-Bild- oder Schnittbildverfahren wirddie Amplitude des eindimensionalen A-Bild-Verfahrens in Grauwerte (Brightness) einerzweidimensionalen Hell-Dunkel-Darstellungumgesetzt. Durch Bewegung des Schallwand-lers (siehe Abb.2) erhält man ein Schnittbild.Moderne Schallwandler für B-Bild-Gerätesind sogenannte Multielementwandler. Siebestehen aus einer Zeile von vielen einzelnenWandlerelementen. Eine Bewegung desSchallkopfes ist nicht mehr erforderlich, siewird ersetzt durch die elektronische Ans-teuerung der einzelnen Wandlerelemente.
Z1 = D 1@ c1
Z 2 = D 2@ c2
I0
IR
ID
Abbildung 1 Reflexion von Ultraschall aneiner Grenzfläche zwischen zwei Stoffenunterschiedlicher Schallimpedanz

Mechanik M 19 Ultraschall-Abbildungsverfahren
24
Abbildung 3 Zum lateralen Auflösungsver-mögen [nach: Krestl, Bildgebende Systemefür die medizinische Diagnostik]
Die Qualität des Ultraschallbildes wird durchdas Auflösungsvermögen charakterisiert.Darunter versteht man den Kehrwert deskleinstmöglichen Abstandes zweier reflektie-render Strukturen, die bei der Wiedergabegerade noch als getrennte Punkte dargestelltwerden können. Man unterscheidet das axiale(in Ausbreitungsrichtung) und das lateraleAuflösungsvermögen (quer zur Ausbreitungs-richtung), siehe Abb.3.
Während das axiale Auflösungsvermögen vorallem durch die Dauer des Schallimpulsesbestimmt wird, hängt das laterale Auflö-sungsvermögen stark von der Schallfeld-geometrie ab. So sind die Querabmessungendes Schallfeldes in einer bestimmten Entfer-nung vom Wandler minimal (Fokussierung),danach wird der Schallstrahl mit zunehmen-dem Abstand breiter und die Auflösungschlechter. Die Impulsdauer, der Querschnittdes Schallfeldes und der Abstand des Fokus-bereiches vom Wandler werden mit wachsen-der Frequenz geringer. Daher werden mithöherer Ultraschallfrequenz sowohl dieaxiale als auch die laterale Auflösung besser.Jedoch wächst mit zunehmender Frequenzauch die Dämpfung der Ultraschallwellen,und damit wird der abbildbare Bereich(Eindringtiefe) kleiner.
Die Interpretation eines Ultraschall-B-Bildeswird durch verschiedene Effekte erschwert:- Schallschatten entstehen hinter stark
reflektierenden Strukturen. Objekte hinterder Struktur bleiben unsichtbar.
- Mehrfachbilder können durch Mehrfach-reflexion des Schalls zwischen einer starkreflektierenden Struktur und der Oberflä-che auftreten.
- Abbildungsfehler (Lagefehler) könnendurch Brechung der Schallwellen an Struk-turen mit unterschiedlicher Schallge-schwindigkeit entstehen.
3 Versuchsaufbau
3.0 Geräte:- Ultraschallgerät - Computer- 2 Schallköpfe (1 MHz; 2 MHz)- PE-Körper mit Fehlstellen- Messschieber
3.1 Das Ultraschallgerät ermöglicht ein A-Bild sowie (durch manuelle Bewegung desSchallkopfes) ein einfaches B-Bild. DieDarstellung erfolgt auf dem Computerbild-schirm.Zum Messen von Zeiten (bzw. Abständen) imA-Bild dienen zwei farbige Marker, die mitder Maus verschoben werden können. DieMessung der Amplitude erfolgt mit demMaus-Cursor.Regler an der Frontplatte des Ultraschall-gerätes dienen der Einstellung der Leistungder ausgesandten Ultraschallpulse (TRANS-
MITTER), der Verstärkung des empfangenenEchosignals (RECEIVER) sowie der laufzeit-abhängigen Verstärkung (TGC = Time GainControl). Weitere Hinweise zur Bedienung und Funk-tionsweise sind der am Platz ausliegendenBedienungsanleitung zu entnehmen.
4 Versuchsdurchführung
Der verwendete Schallkopf ist an der BuchsePROBE / REFLECTION anzuschließen, Kipp-schalter auf REFLEC. Die Ankopplung derSchallwandler an den PE-Körper erfolgt mit

Mechanik M 19 Ultraschall-Abbildungsverfahren
25
Wasser. (Es ist nur ein dünner Wasserfilmerforderlich!)
4.1 Für die Bestimmung der Schallge-schwindigkeit sind mit einem Messschieberdie Dicke l des PE-Körpers und aus der Zeit-skala bei aufgesetztem Schallkopf der zeitli-che Abstand t zwischen dem Beginn desSende- (oder Initial-) Echos und dem Beginndes Endecho (Rückwandecho) zu bestimmen.
4.3 Der PE-Körper ist mit beiden Schall-köpfen auf Fehlstellen zu untersuchen.Um die Messungen zu erleichtern, wirdzuerst die Schallgeschwindigkeit berechnet(siehe 5.1) und im Menüpunkt 'Einstellungen'eingegeben. Danach wird die x-Achse (durchKlick auf den Button 'Tiefe') von Laufzeit aufAbstand umgestellt. Kontrollieren Sie, ob dieTiefe des Rückwandechos gleich der gemes-senen Dicke des PE-Körpers ist!
Ein Schallkopf wird über die seitlichenFlächen des PE-Körpers geführt. Am Gerätsind dabei die Einstellungen für LAV, Lei-stung und Verstärkung nach folgendenGesichtspunkten zu variieren:
- Das gewünschte Echo darf nicht vomInitialecho überdeckt werden.
- Die mit zunehmender Eindringtiefe verbun-dene Schwächung muss ausgeglichenwerden.
- Das Echosignal darf nicht übersteuert sein,damit eine genaue Lokalisation auf demSchirm möglich ist.
Wenn Reflexe von Bohrlöchern gefundenund alle Einstellungen optimiert sind, schal-ten Sie um in den B-Bild-Modus. Mit dem B-Bild gewinnt man schnell einen Überblicküber die Lage der Löcher.
Stellen Sie die ungefähre Größe des Körperssowie Anfangs- und Endwert der Farbskalerichtig ein, drücken Sie den Start/Stop-Buttonund führen Sie den Schallkopf langsam undgleichmäßig über den PE-Körper. Die Mes-sung muss mit dem Start/Stop-Button been-det werden. Eventuell müssen Sie für ein
gutes Bild ein wenig üben und alle Einstel-lungen weiter verbessern.Das B-Bild kann ausgedruckt werden.
Bitte drucken Sie für jede Frequenz undpro Student nur einmal!
Die genaue Messung der Lage der Bohr-löcher muss im A-Bild erfolgen. Für alleLöcher sind die Abstände von der Oberflächezu bestimmen. Die zweite Koordinate jedesLoches wird ermittelt, indem man die Mes-sung nach Drehen des PE-Körpers um 90o
wiederholt. Mit Hilfe des höherfrequentenSchallkopfes (besseres Auflösungsvermögen)ist nachzuprüfen, ob die gefundenen Fehl-stellen möglicherweise zusätzlich strukturiertsind.
5 Auswertung
5.1 Die Schallgeschwindigkeit ist nachGleichung (7) zu berechnen. Die Berechnung der Wellenlänge λ erfolgt fürbeide Wandler über die Gleichung (1). DerElastizitätsmodul ist nach Gleichung (2) zuerrechnen.(ν = 0,45; ρ = 0,932 g cm-3)
5.3 Es ist auf Millimeterpapier im Maßstab1 : 1 ein Schnitt des PE-Körpers mit Ein-zeichnung der Fehlstellen (Bohrungen)darzustellen.Das B-Bild ist auf Artefakte (Schallschatten,Mehrfachbilder) zu untersuchen.
6 Literatur
Kamke/Walcher: Physik für Mediziner. B.G.Teubner, Stuttgart 1994
Fercher, A.F.: Medizinische Physik, Springer,1992
Millner, R.: Wissensspeicher Ultraschall-technik, Leipzig: Fachbuchverlag 1987

Mechanik M 19 Ultraschall-Abbildungsverfahren
26
7 Kontrollfragen
7.1 Welche physikalische Größe ist auf derSkale des Ultraschallgerätes dargestellt?
7.2 Warum ist eine Ankopplung von Ultra-schallwandlern mittels Wasser oder Gel
notwendig?
7.3 Welche Wellenlänge hat eine Ultra-schallwelle in Polyethylen bei einer Frequenzvona) 1 MHz b) 2 MHz ?

Wärmelehre W 1 Lineare Ausdehnung
27
( )[ ]( )[ ]
l l T T
l l T T
1 0 1 0
2 0 2 0
1
1
= + −
= + −
α
α(2)
l lT TT T2 1
2 0
1 0
11
=+ −
+ −
α
α
( )( )
. (3)
∆ l l T T0 0 0= −α ( ) (1)
( )[ ]l l T T2 1 2 11= + −α , (4)
∆ ∆l l T= ⋅ ⋅1 α . (5)
1 Aufgabenstellung
Der lineare Ausdehnungskoeffizient zweierunbekannter Materialien ist zu bestimmen.
2 Grundlagen
In Festkörpern und Flüssigkeiten führen dieTeilchen (Atome bzw. Moleküle) temperatur-abhängige Schwingungen aus. Wegen derunsymmetrischen Potentialkurve der zwi-schenatomaren Bindung werden die mittlerenAtomabstände mit zunehmender Schwin-gungsamplitude größer. Das äußert sich ineiner Volumenausdehnung, in festen Körpernauch in einer Längenausdehnung. Diese ist inhomogenen und isotropen Festkörpern (Me-tall, Polymere, Glas) in alle Richtungengleich groß.Bei der Erwärmung bzw. Abkühlung einesStabes ändert sich seine Länge mit der Tem-peratur T. Ist l0 die Länge des Stabes bei derTemperatur T0 = 0EC, so ist die Längen-änderung ∆l0 = l - l0
Dabei ist α der lineare Ausdehnungskoeffi-zient (SI-Einheit: 1/K), eine Materialgröße,die für das verwendete Metallrohr im Bereichvon 0EC bis 100EC nur geringfügig von derTemperatur abhängt. Ist l1 die Länge bei derTemperatur T1 und l2 die Länge bei derTemperatur T2, so ergibt sich:
und damit
Da α sehr klein ist, kann näherungsweise für
(3) gesetzt werden:
bzw. mit ∆l = l2 - l1
3 Versuchsaufbau
3.0 Geräte- Messuhr- Bandmaß- Halterung mit zwei eingespannten Rohren- Umwälzthermostat
3.1 Die Rohre sind einseitig eingespannt. ImAbstand l1 von der Einspannung drückt jedesRohr gegen den Stift einer Messuhr, so dassdie Längenänderung ∆l mit einer Genauigkeitvon 1/100 mm gemessen werden kann. DieTemperaturveränderungen werden mit Hilfeeines Thermostaten erzeugt, der temperiertesWasser durch die Rohre pumpt.
4 Versuchsdurchführung
Studieren Sie die am Arbeitsplatz ausliegendeKurzanleitung zum Thermostaten. SchaltenSie zu Beginn auf keinen Fall dieThermostatheizung ein, das Wasserbadbenötigt viel Zeit um wieder abzukühlen!
Der Thermostat wird ohne Heizung (vor-gewählte Temperatur unterhalb der Raum-temperatur) in Betrieb genommen, damit dieRohre die Temperatur annehmen, die dasThermometer am Thermostaten anzeigt.Diese Temperatur wird abgelesen und dieMessuhr auf Null gestellt.Die Längen l1 zwischen der Einspannung undder Stirnseite der Rohre ist mit dem Bandmaßzu messen.Danach ist am Thermostat eine Temperatur
Lineare Ausdehnung W 1

Wärmelehre W 6 Spezifische Wärme von Metallen
28
cCm
Qm T
= =∆
. (1)
einzustellen, die um ca. 10 K höher liegt alsdie Ausgangstemperatur. Etwa 5 min nachErreichen der Solltemperatur werden dieTemperatur T und die dazugehörigen Län-genänderungen ∆l abgelesen.Die Temperatur wird in Schritten von ca.10 K weiter erhöht; dabei sind jeweils dieTemperatur und die Längenänderungen ingleicher Weise zu messen, bis eine Tempera-tur von ca. 80EC erreicht ist.
Während der gesamten Messungen müssenErschütterungen vermieden werden!
5 Auswertung
Entsprechend der Gleichung (5) ist dieLängenänderung ∆l in Abhängigkeit von derTemperatur T graphisch darzustellen. DerAnstieg der Kurve ist durch lineare Regressi-on zu ermitteln. Aus dem Anstieg ist derlineare Ausdehnungskoeffizient α zu be-rechnen.
Es ist eine Fehlerrechnung durchzuführen.Anhand des Ausdehnungskoeffizienten istdas Material der Rohre zu bestimmen.
6 Literatur
Haas, U.; Physik für Pharmazeuten undMediziner, WVG Stuttgart, 2002
Harten, H.-U. Physik für Mediziner.Springer-Verlag, Berlin, 1993.
7 Kontrollfragen
7.1 Welche physikalischen Phänomenewerden zur Temperaturmessung genutzt?
7.2 Weshalb ist die Wärmekapazität einesThermometers für die Temperaturmessungvon Bedeutung?
7.3 Um wieviel wird ein 1 m langer Metall-stab bei Erwärmung um 10 K etwa länger?
1 Aufgabenstellung
Die spezifische Wärmekapazität von dreiverschiedenen Metallen ist zu bestimmen.
2 Grundlagen
Die Aufnahme oder Abgabe einer bestimmtenWärmemenge Q ist mit der Temperatur-änderungen eines Körpers verbunden, sofernnicht chemische Reaktionen oder Änderun-gen des Aggregatzustandes erfolgen. Dabeikann der Wärmetransport von einem Körperzum anderen über Wärmeleitung (Kondukti-on), Wärmeströmung (Konvektion) undWärmestrahlung erfolgen. Betrachtet man ein abgeschlossenes System
(kein Stoff- und Energieaustausch mit derUmgebung), dann gilt der Energieerhaltungs-satz, d. h. die Summe der Wärmeenergienaller am Wärmeaustausch beteiligten Körperist konstant. Ein Kalorimeter stellt die nä-herungsweise experimentelle Umsetzungeines abgeschlossenen Systems dar.
Die Wärmekapazität C ist definiert als dasVerhältnis zwischen der dem Körper zu-geführten Wärme Q und der dadurch hervor-gerufenen Temperaturerhöhung ∆T. Diespezifische Wärme c ist die Wärmekapazitätpro Masseneinheit:
Um eine Substanz der Temperatur T1 auf eine
Spezifische Wärme von Metallen W 6

Wärmelehre W 6 Spezifische Wärme von Metallen
29
( )( ) ( )
Q Q Q
m c T T
m c T T m c T T
M W K
M M H
W W K K
= +
− =
− + −
2
2 1 2 1
(3)
( )( )( )
cm c m c T T
m T TM
W W K K
M H
=+ −
−
2 1
2
. (4)
( )Q C T T m c T= ⋅ − = ⋅ ⋅2 1 ∆ (2)
Temperatur T2 = T1 + ∆T zu erwärmen, mussihr die Wärmemenge
zugeführt werden.
Die Messung der spezifischen Wärme ge-schieht in einem Mischungskalorimeter.Das Kalorimetergefäß hat die Masse mK unddie spezifische Wärmekapazität cK. Im Kalo-rimetergefäß befindet sich Wasser der MassemW und der spezifischen Wärmekapazität cW.Gefäß und Wasser haben zunächst die Tem-peratur T1. Dann wird ein heißes Metallstückder Masse mM mit der Temperatur TH in dasKalorimeter eingeführt. Durch Wärmeaus-tausch stellt sich nach einiger Zeit die Misch-temperatur T2 ein.Bei diesem Vorgang gibt der Metallkörperdie Wärmemenge QM ab, das Wasser nimmtdie Wärmemenge QW und das Kalorimeter dieWärmemenge QK auf. Nach dem Energie-erhaltungssatz gilt:
Für die spezifische Wärmekapazität cM desMetalls ergibt sich daraus:
Ein reales Kalorimeter ist kein vollständigabgeschlossenes System, sondern gibt imVerlaufe des Experiments Wärme an dieUmgebung ab bzw. nimmt Wärme auf. DerFehler, der durch den Wärmeaustausch mitder Umgebung entsteht, kann minimiertwerden, wenn man die Temperaturen T1 undT2 entsprechend Abb.1 graphisch aus einemTemperatur-Zeit-Diagramm bestimmt. Dazu wird der Temperaturverlauf im Kalori-meter während des gesamten Experiments inAbhängigkeit von der Zeit aufgetragen. Indas Diagramm wird dann eine Senkrechte so
eingetragen, dass die entstehenden FlächenABC und CDE etwa gleich groß sind. DieTemperaturen T1 und T2 ergeben sich dannaus den Schnittpunkten der an die gemesseneKurve angelegten Tangenten mit der Senk-rechten (siehe Abb.1). Die Senkrechte repräsentiert einen idealisier-ten adiabatischen Prozess, der in einem sehrkurzen Zeitraum (∆t60) abläuft, so dasswährend dieser Zeit kein Wärmeaustauschmit der Umgebung stattfinden kann.
3 Versuchsaufbau
3.0 Geräte- Kalorimeter- Magnetrührer, Rührstäbchen- Digitalthermometer- 3 Probekörper- Gefäß zur Erwärmung der Probekörper- Laborkocher- Stoppuhr- Waagen (0,1 g und 0,001 g Auflösung)
3.1 Das Kalorimeter besteht aus eineminneren und einem äußeren Gefäß mit Deckelzur Wärmeisolierung.Das Temperaturmessgerät hat eine Mess-genauigkeit von 0,2 K und eine Auflösungvon 0,1 K. Durch langes Drücken der TasteTn wird die Differenz zur augenblicklichenTemperatur mit einer Auflösung von 0,01 Kangezeigt.
Abb.1: Temperatur-Zeit-Diagramm

Wärmelehre W 6 Spezifische Wärme von Metallen
30
4 Versuchsdurchführung
Zunächst werden die Massen der Probekörper(mM) und die Masse des Kalorimeters mK
(inneres Gefäß) bestimmt. Das innere Gefäß des Kalorimeters wird mitetwa 600 ml Wasser gefüllt. Die Wassermas-se ist mit einer geeigneten Waage zu be-stimmen. Die Wassertemperatur sollte etwa2…3 K unter der Raumtemperatur liegen.Protokollieren Sie die Anfangstemperatur desWassers und schalten Sie dann das Digital-thermometer auf 1/100 K -Anzeige um.Üben Sie einmal, einen Probekörper mög-lichst schnell und ohne anzustoßen in dasKalorimeter einzuhängen.
Die Probekörper sind in siedendem Wasserzu erwärmen. Es ist darauf zu achten, dass dieKörper vollständig eintauchen und genügendlange (etwa 10 min) im Wasserbad verweilen,damit die Zuordnung der Siedetemperatur beigegebenem Luftdruck zur Körpertemperaturdes erwärmten Probekörpers gerechtfertigtist. Die Probekörper dürfen weder den Randnoch den Boden des Gefäßes berühren.Während des gesamten Experiments mussDer Rührer gleichmäßig laufen.
Bei der Bestimmung der Temperaturen T1
und T2 für alle drei Probekörper soll derWärmeaustausch zwischen Kalorimeter undUmgebung entsprechend Abb.1 berück-sichtigt werden. Deshalb ist der Temperatur-verlauf im Kalorimeter über einen längerenZeitraum zu registrieren, indem in Abständenvon 30 Sekunden die Anzeige des Thermo-meters protokolliert wird. Am besten gehtman nach folgendem Plan vor:
t / min 0 Beginn der Temperaturmessung
(Vorperiode) 5 Körper 1 in Kalorimeter tauchen10 Körper 1 herausnehmen
11 Körper 2 in Kalorimeter tauchen16 Körper 2 herausnehmen
17 Körper 3 in Kalorimeter tauchen23 Ende der Messung (Nachperiode)
Für die Bestimmung der Siedetemperatur TB
wird der Luftdruck am Barometer (an derWand an der Fensterseite) abgelesen undentsprechend der Umrechnungstabelle dieSiedetemperatur ermittelt.
5 Auswertung
Der Temperaturverlauf im Kalorimeter wirdin Abhängigkeit von der Zeit graphischdargestellt (vergl. Abb1). Die Bestimmungvon T1 und T2 kann für alle drei Probekörperin einem Diagramm vorgenommen werden. Die spezifische Wärmekapazität der Metalleist nach Gleichung (4) zu berechnen. Dabeisind folgende Werte zu verwenden:
spezifische Wärmekapazität des Wassers: cW = 4187 J kg-1 K-1
spezifische Wärmekapazität des Gefäßes: cK = 920 J kg-1 K-1
Für die Masse mK des Kalorimeters ist dieSumme aus der Masse des inneren Gefäßesund der Masse des Rührers einzusetzen. Die Siedetemperatur des Wassers ist anhanddes gemessenen Luftdrucks einer Tabelle zuentnehmen.
Mit Hilfe der Messergebnisse sind die unbe-kannten Metalle zu bestimmen.
6 Literatur
Bergmann, L., Schaefer, C., Lehrbuch derExperimentalphysik, 10 Aufl., de GruyterBerlin-New York 1990
Haas, U.; Physik für Pharmazeuten undMediziner, WVG Stuttgart, 2002
Trautwein, A., Kreibig, U., Oberhausen, E.;Physik für Mediziner, de Gruyter, 1987
7 Kontrollfragen
7.1 Erläutern Sie die Begriffe Wärmekapazi-

Wärmelehre W 12 Luftfeuchtigkeit
31
U TT = ⋅α ∆ . (1)
fp
prd
S
= . (3)
fm
Va
d= . (2)
tät, Temperatur, abgeschlossenes thermo-dynamisches System, thermodynamischesGleichgewicht!
7.2 Nennen Sie Temperaturmessverfahren!
7.3 Erläutern Sie den ersten und zweitenHauptsatz der Thermodynamik!
1 Aufgabenstellung
1.1 Ein Kupfer-Konstantan-Thermoelementist zu kalibrieren.
1.2 Die relative Luftfeuchtigkeit ist mitHilfe eines Taupunkt-Hygrometers zu be-stimmen.
1.3 Das RAOULTsche Gesetz (Dampfdruck-erniedrigung in Lösungen) ist qualitativ zubestätigen.
2 Physikalische Grundlagen
2.1 Thermoelement: In einem (isolierten)elektrischen Leiter wird durch einen Tempe-raturgradienten eine elektrische Potentialdif-ferenz erzeugt (absoluter SEEBECK-Effekt),die jedoch nicht unmittelbar messbar ist.
Verbindet man zwei verschiedene Leiter, z.B. Kupfer und Konstantan wie in Abb.1, zueinem Stromkreis und bringt die beidenKontaktstellen auf verschiedene Temperatu-
ren T0 und T1, so entstehen in den beidenLeitern unterschiedliche innere Potentiale.Zwischen den Punkten A und B tritt dieDifferenz der beiden Potentiale auf, diesogenannte Thermospannung. Dies wird alsSEEBECK-Effekt bezeichnet.Die Thermospannung UT ist näherungsweiseproportional zur Temperaturdifferenz ∆T =T1 - T0 :
Der Koeffizient α heißt Thermokraft oderSeebeckkoeffizient und ist von beiden Mate-rialien abhängig.Thermoelemente werden häufig zur Tempera-turmessung verwendet. Sie bieten den Vor-teil, dass sich die Thermospannungen un-mittelbar als Eingangssignale für Computer,Steuer- und Regelgeräte nutzen lassen.
2.2 Luftfeuchtigkeit nennt man den Gehaltder Luft an Wasserdampf. Die absolute Luftfeuchtigkeit fa ist dieWasserdampfmasse md pro Volumen V derLuft:
Die relative Luftfeuchtigkeit fr ist dasVerhältnis der vorhandenen Wasserdampf-menge zur Sättigungsmenge bzw. das Ver-hältnis des vorhandenen Dampfdruckes pd
zum Sättigungsdampfdruck pS bei dervorliegenden Temperatur T :
Abb.1: Kupfer-Konstantan-Thermoelement
Luftfeuchtigkeit W 12

Wärmelehre W 12 Luftfeuchtigkeit
32
fp T
p T
p
p Trd
S
S
S
= =( )
( )
( )
( ).
τ(5)
∆ p
p
p p
px
n
n nS
S S L
S
=−
= =+
,.2
1 2
(4)
Die relative Luftfeuchtigkeit wird meist inProzent angegeben.In einem geschlossenen Gefäß, in dem sichreines Wasser und darüber Luft befinden,verdampft ein Teil des Wassers, bis der Raumoberhalb der Flüssigkeit mit Wasserdampfgesättigt ist. Es bildet sich im thermodyna-mischen Gleichgewicht der Sättigungsdampf-druck pS aus, der nur von der Art der Flüs-sigkeit (hier Wasser) und von der Temperatur(etwa exponentiell) abhängt. Die relativeLuftfeuchtigkeit beträgt in diesem Fall100 %. Ist die Flüssigkeit im geschlossenen Gefäßeine wässrige Lösung, so ist der Sättigungs-dampfdruck über der Lösung entsprechenddem RAOULTschen Gesetz um ∆p verringert.Diese Dampfdruckerniedrigung ist unabhän-gig von der Art des gelösten Stoffes, siehängt nur von der Anzahl der gelösten Teil-chen ab:
Dabei ist x der Molenbruch des gelöstenStoffes (n2: Menge der gelösten Teilchen, n1:Teilchenmenge des Lösungsmittels), pS istder Sättigungsdampfdruck des reinen Lö-sungsmittels und pS,L der der Lösung. Bei derBestimmung von x muss die Dissoziation desgelösten Stoffes berücksichtigt werden. DasRaoultsche Gesetz gilt nur für n2 n n1, beihöheren Konzentrationen x ist die beobachte-te Dampfdruckerniedrigung geringer. Infolgedes Raoultschen Gesetzes ist die Luftfeuch-tigkeit über einer Lösung kleiner als 100 %.
Ist bei der Temperatur T die Luftfeuchtigkeitim Raum kleiner als 100 %, so kann man100 %ige Luftfeuchtigkeit erreichen, indemman die Temperatur erniedrigt. Ab einerbestimmten Temperatur τ, dem Taupunkt,kondensiert der Wasserdampf und schneidetsich z. B. auf einer Oberfläche ab. Dies dientzur Messung der Luftfeuchtigkeit mit einemTaupunkthygrometer: Anhand der Tempera-tur τ kann der zugehörige Sättigungsdampf-
druck pS(τ) ermittelt werden, der gleich demDampfdruck pd(T) ist. Die relative Luftfeuch-tigkeit fr ergibt sich dann aus
3 Versuchsaufbau
3.0 Geräte:- Taupunkthygrometer (Alu-Grundkörper
mit Peltierkühler, Metallspiegel, Thermo-element und Lichtschranke, Abdeckhaube)
- Steuergerät für Lichtschranke- Stromversorgungsgerät für Peltierkühler- Kupfer-Konstantan-Thermoelement, eine
Lötstelle in Röhrchen mit Gallium- Becherglas, Isoliergefäß, flache Schale- Flasche mit 3 molarer CaCl2-Lösung- Sensor-Cassy mit µV-BOX
- Computer, CassyLab-Software
3.1 Zur Kalibrierung des Kupfer-Konstantan-Thermoelementes dienen zweiFixpunkte: der Schmelzpunkt von Wasserund der von Gallium (TS = 29,76 °C). DieThermospannung wird mittels Sensor-Cassy/µV-Box und Computer gemessen.
Eine Kurzanleitung zur Benutzung der Cassy-Lab Software befindet sich im Anhang.
3.2 Das Taupunkthygrometer (Abb.2)besitzt eine abkühlbare, spiegelnde Metallflä-che (polierter Aluminiumblock), derenTemperatur gemessen und deren Bedeckungmit kondensiertem Wasserdampf beobachtetwerden kann. Zur Temperaturerniedrigungdient ein Halbleiterkühlelement (Nutzung desPeltier-Effektes), das mit Hilfe eines Strom-versorgungsgerätes (30V/1,5A; Betrieb alsKonstantstromquelle) betrieben wird. DieTemperaturmessung erfolgt mit einemKupfer-Konstantan-Thermoelement, dessenMessstelle sich im Aluminiumblock befindetund die Vergleichsstelle bei 0°C in einemEis-Wasser-Gemisch. Die Thermospannung

Wärmelehre W 12 Luftfeuchtigkeit
33
Abb.2:
Versuchsaufbau zur Be-stimmung der Luftfeuchte
wird mit der µV-Box des Sensor-Cassygemessen und mit Hilfe des Computersregistriert.Zur reproduzierbaren Beobachtung derWasserdampf-Kondensation dient eineReflexlichtschranke mit Anzeige-LED undRelaisausgang. Das Relais der Lichtschrankeermöglicht eine einfache Temperaturregelungzum Erreichen des Taupunktes, indem es denKühlerstrom entsprechend der Betauung desSpiegels automatisch ein- und ausschaltet.
3.3 Das Taupunkthygrometer befindet sichzusammen mit einer Schale mit Wasser bzw.3 molarer CaCl2-Lösung unter einer Plastik-haube.
4 Versuchsdurchführung
4.1 Die Schaltung wird entsprechend Abb. 1aufgebaut. Als Spannungsmesser dient dieµV-Box des Sensor-Cassy. Starten Sie dasProgramm CASSYLab-W12 erst, nachdemdas Messgerät mit Strom versorgt ist. AlleEinstellungen im Programm (Messbereich,Messintervall etc.) sind bereits richtig vor-eingestellt. Mit der Taste F9 oder Mausklick
auf wird die Aufzeichnung einer Mess-reihe gestartet und auch wieder beendet.
Das Isoliergefäß wird mit gestoßenem Eisund (nicht zuviel) Wasser gefüllt, das Becher-glas mit heißem Wasser. Die Vergleichs-Lötstelle des verwendeten Thermoelementswird in das Eis-Wasser-Gemisch getaucht.
Zur Kalibrierung des Kupfer-Konstantan-Thermoelementes wird eine Messreihegestartet und das Röhrchen mit Gallium indas heiße Wasser getaucht. Bei einer Tempe-ratur von T1 = 29,76°C schmilzt das Gallium,so dass die Spannungs-Zeit-Kurve einenHaltepunkt aufweist, bis das gesamte Galliumgeschmolzen ist. Bringt man danach dasRöhrchen in das Becherglas mit dem Eis-Wasser-Gemisch, so zeigt der Spannungsver-lauf bei T1 = 29,76°C wieder einen Halte-punkt (evtl. erst nach einer Unterkühlung derGalliumschmelze). Aus dem Mittelwert derHaltepunkte beim Schmelzen und beimErstarren des Galliums wird mit (1) derSeebeck-Koeffizient α des Kupfer-Konstantan-Thermoelementes berechnet.
4.2 Die Versuchsanordnung ist entspre-chend Abb.2 aufzubauen. Die Vergleichs-Lötstelle des Thermoelements des Taupunkt-hygrometers muss sich im Eis-Wasser-Ge-misch befinden. Die Thermospannung wirdwie unter 4.1 mit Hilfe des Computers regi-striert.Der Schaltzustand der Lichtschranke wirddurch eine LED signalisiert. Die Komparator-schwelle wird so eingestellt, dass bei nichtbeschlagenem Spiegel gerade noch Reflexionangezeigt wird (LED leuchtet), bei beschla-genem Spiegel jedoch die LED erlischt. Nach dem Start einer Messreihe in CassyLabwird die Stromversorgung des Kühlerseingeschaltet und die Thermospannungregistriert (Kühlerstrom I .1 A mittels Strom-

Wärmelehre W 12 Luftfeuchtigkeit
34
n nV n M
M1 = =−
H2OCaCl2 CaCl2
H2O
ρ. (7)
ρ =+
=+
m m
V
n M n M
V
CaCl2 H2O
CaCl2 CaCl2 H2O H2O .
(6)
n n2 3= ⋅ CaCl2 . (8)
regler einstellen). Die LED der Lichtschrankeleuchtet. Wenn der Taupunkt unterschrittenwird, erlischt die LED und der Kühlerstromwird durch das Relais unterbrochen. DieTemperatur im Taupunkthygrometer steigtwieder; der Belag auf der Spiegelflächeverdampft, so dass die LED wieder aufleuch-tet und der Kühler automatisch wieder einge-schaltet wird. Auf diese Weise ergeben sich„Regelschwingungen“ um die Thermospan-nung, die dem Taupunkt entspricht. DieRegelung funktioniert am besten, wenn derKühlerstrom so eingestellt wird, dassAbkühl- und Aufheizgeschwindigkeit etwagleich sind. (Warum?) Es werden etwa 10Regelschwingungen aufgezeichnet. NachBeendigung der Messung ist der Kühlerauszuschalten.Die Thermospannung am Taupunkt ist durchMittelwertbildung im Programm CassyLab zubestimmen.
4.3 Zum Nachweis des Raoultschen Ge-setzes wird neben das Hygrometer eineSchale mit Wasser gestellt, dessen Tempera-tur gleich der Raumtemperatur ist. Hygro-meter und Wasserschale werden gemeinsammit der Plastikhaube abgedeckt. Die Luft-feuchtigkeit unter der Haube wird nun lang-sam bis auf nahezu 100 % ansteigen. Nach20…30 min wird die Messung gestartet undder Kühlerstrom eingeschaltet (günstig sindhier etwa 0,2…0,5 A). Die Thermospannungwird solange aufgezeichnet, bis sich derTaupunkt nicht mehr ändert (etwa 5 min). Der Versuch wird wiederholt, wobei dieSchale nun mit 3 molarer CaCl2-Lösunggefüllt ist. Die o.g. Wartezeit ist wiedereinzuhalten; die Messkurve wird in das selbeDiagramm geschrieben.Die Thermospannungen am Taupunkt werdendurch Mittelwertbildung im ProgrammCassyLab bestimmt.
Die CaCl2-Lösung wird nach Versuchs-ende wieder in die Flasche zurück gefüllt!
5 Auswertung
5.1 Das Messdiagramm UT(t) ist auszudru-cken. Der Seebeck-Koeffizient α des Kupfer-Konstantan-Thermoelementes ist nach Gl. (1)zu berechnen.
5.2 Das Messdiagramm UT(t) ist auszudru-cken. Die Raumtemperatur und der Taupunktsind aus UT zu ermitteln. Die relative Luft-feuchtigkeit fr wird nach (5) berechnet, wobeidie Sättigungsdampfdrücke der beiliegendenTabelle entnommen werden.
5.3 Die Taupunkte und die relativen Luft-feuchtigkeiten über Wasser und 3 molarerCaCl2-Lösung (Dichte: 1,25 g cm-3, Dissozia-tionsgrad 100%) werden wie unter 5.2 er-mittelt. Die durch Dampfdruckerniedrigungnach dem Raoultschen Gesetz zu erwartendeLuftfeuchtigkeit über der CaCl2-Lösung istmit Hilfe Gl. (4) zu berechnen. Die gemesse-nen Luftfeuchtigkeiten sind mit den theore-tisch erwarteten Werten zu vergleichen.
Hilfe zur Berechnung des Molenbruches:mCaCl2 und mH2O sind die Massen, nCaCl2 undnH2O die Stoffmengen und MCaCl2 und MH2O
die Molmassen der CaCl2- und der H2O-Moleküle in V = 1 l Salzlösung. Dann ist dieDichte der Lösung
Daraus ergibt sich die Teilchenmenge desLösungsmittels
Wegen der Dissoziation ist die Menge dergelösten Teilchen

Wärmelehre W 17 Spezifische Wärme von Flüssigkeiten
35
( )( )
cUIt C T T
m T T
K=
− −
−
2 1
2 1
. (6)
Q U I t= ⋅ ⋅ , (5)
6 Literatur
Bergmann - Schäfer: Lehrbuch der Experi-mentalphysik. de Gruyter Lehrbuch, 11.Auflage 1998
Kamke/Walcher: Physik für Mediziner. B.G.Teubner, Stuttgart 1994
Haas, U.: Physik für Pharmazeuten undMediziner. WVG Stuttgart 2002
7 Kontrollfragen
7.1 Erklären Sie die Entstehung von Wetter-erscheinungen wie Regen, Nebel, Tau!
7.2 Wie funktioniert ein Thermoelement?
7.3 Bei welcher Temperatur siedet eineSalzlösung (in Wasser)?
1 Aufgabenstellung
Die spezifische Wärmekapazität von Wasserist zu bestimmen.
2 Grundlagen
Siehe Grundlagen zum Versuch W 6 !
Im durchzuführenden Versuch befindet sichin einem Kalorimeter mit der Wärmekapazi-tät CK Wasser der Masse m und der spezi-fischen Wärmekapazität c. Durch eine elek-trische Heizung im Kalorimeter kann demWasser definiert Wärme zugeführt werden.Gefäß und Wasser haben zunächst die Tem-peratur T1. Wird die Heizung eingeschaltet,so werden Kalorimeter und Wasser bis zurTemperatur T2 erwärmt. Dabei ist die von derHeizung in der Zeit t abgegebene Wärme-menge Q
wobei U die Spannung und I die Stromstärkesind. Die vom Wasser aufgenommene Wär-memenge QW und die vom Kalorimeteraufgenommene Wärmemenge QK sind nachder Energieerhaltung gleich Q (siehe Gl.(3)).
Daraus ergibt sich für die spezifische Wärme-kapazität c des Wassers:
3 Versuchsaufbau
3.0 Geräte1 Kalorimeter mit elektrischer Heizung 1 elektronisches Thermometer1 Stoppuhr1 Magnetrührer1 Magnetrührstäbchen1 Stromversorgungsgerät2 Vielfachmessgeräte Verbindungskabel
3.1 Das Kalorimeter besteht aus eineminneren Gefäß, das gegenüber dem äußerenGefäß wärmeisoliert ist, sowie dem Deckel.Das Schaltbild für den Anschluss der Hei-zung ist in Abb. 2 gezeigt.
Spezifische Wärme von Flüssigkeiten W 17

Wärmelehre W 17 Spezifische Wärme von Flüssigkeiten
36
4 Versuchsdurchführung
Das Kalorimeter wird bis ca. 2 cm unter demRand mit Wasser gefüllt. Die Wassertempera-tur sollte etwa der Raumtemperatur entspre-chen. Die Wassermasse m ist mit einer ge-eigneten Waage zu bestimmen.Die elektrische Schaltung wird entsprechendAbb. 2 aufgebaut. Die Schaltung ist einemAssistenten vor Inbetriebnahme vorzuführen!Der Rührer muss während des gesamtenExperiments gleichmäßig laufen!Zur Bestimmung der Temperaturen T1 und T2
ist der Temperaturverlauf im Kalorimeter ineinem Zeitraum von 28 min fortlaufend zuregistrieren. In Abständen von 30 Sekundenist die Anzeige des elektronischen Thermo-meters abzulesen:
a) 10 min bevor die Heizung eingeschaltetwird,
b) während einer Heizperiode von 8 min und
c) 10 min nach Abschalten der Heizung.
Am Stromversorgungsgerät ist der Strom mitBeginn der Heizperiode auf 1,8 A einzustel-len. Es ist darauf zu achten, dass während derHeizperiode die Spannung U und der Strom Ikonstant bleiben. Sie werden mit den beidenVielfachmessgeräten in geeigneten Mess-bereichen bestimmt.
Nach Beendigung des Versuches bitte dasKalorimeter ausleeren und zum Trocknen
offen stehen lassen!
5 Versuchsauswertung
Zur Bestimmung von T1 und T2 wird einTemperatur-Zeit-Diagramm entsprechendAbb.1 (Versuch W 6) gezeichnet.Die spezifische Wärmekapazität der Flüssig-keit c ist nach Gleichung (6) zu berechnen.
Die Wärmekapazität des Kalorimeters CK
beträgt (66±10) J K -1..
Führen Sie eine Fehlerrechnung durch undvergleichen Sie Ihr Ergebnis mit dem Ta-bellenwert !
6 Literatur
Haas, U.; Physik für Pharmazeuten undMediziner. WVG Stuttgart, 2002
Trautwein, A., Kreibig, U., Oberhausen, E.:Physik für Mediziner. de Gruyter Berlin-NewYork 1987
7 Kontrollfragen
7.1 Erläutern Sie die Begriffe Wärme-kapazität, Wärmemenge, Temperatur, adiaba-tischer Prozess, thermodynamisches Gleich-gewicht!
7.2 Was besagen die Hauptsätze der Ther-modynamik?
7.3 Was muss bei der Temperaturmessungeines Objektes beachtet werden?
Abb.2: Schaltbild zum Versuchsaufbau

Wärmelehre W 25 Diffusion
37
J D Acx
= − ⋅ ⋅
d
d. (1)
1 Aufgabenstellung
Der Diffusionskoeffizient eines unbekanntenSalzes in Wasser soll bestimmt werden.Hierfür ist
1.1 die elektrische Leitfähigkeit zweierSalzlösungen in Abhängigkeit von der Kon-zentration zu messen (Erstellung von Kali-brierkurven),
1.2 die Apparatekonstante der Diffusions-zelle zu bestimmen durch Messung derDiffusion eines bekanntes Salzes (KCl),
1.3 der gesuchte Diffusionskoeffizient zubestimmen durch Wiederholung der Messungzu 1.2 mit dem unbekannten Salz.
2 Grundlagen
Diffusion ist eine Form des Massetransportsin Festkörpern, Flüssigkeiten und Gasen, derdurch die mikroskopische, ungeordneteBewegung der Teilchen (Brownsche Bewe-gung) hervorgerufen wird und so gerichtetist, dass sich ein lokales räumliches Konzen-trationsgefälle (Konzentrationsgradient)ausgleicht. Sie besitzt große Bedeutung in derzellulären und organismischen Physiologie.In einem einphasigen System bei konstanterTemperatur und dem Fehlen äußerer Kräftesorgt die Diffusion für eine gleichmäßigeKonzentration der Komponenten der Phaseim gesamten System.Diffusion durch eine semipermeable Wand(permeabel für das Lösungsmittel, nicht aberfür gelöste Stoffe) führt zur Osmose.Um den eindimensionalen Diffusionsvorgangquantitativ zu beschreiben, wird der Diffu-sionsfluss (auch Teilchenstrom) J definiertals die Stoffmenge, die netto pro Zeiteinheitin positive x-Richtung durch eine senkrechtzu dieser Richtung angeordnete Fläche Ahindurchtritt. Die Einheit des Diffusions-
flusses ist mol/s. Bezieht man den Diffusions-fluss auf die Fläche A, so spricht man vondem spezifischen Diffusionsfluss oder derDiffusionsstromdichte Φ = J/A.Der Diffusionsfluss ist im isothermen Fallproportional zur betrachteten Querschnittsflä-che A und zum Konzentrationsgradientendc/dx (1. FICKsches Gesetz):
Dabei ist D der Diffusionskoeffizient (Ein-heit: m2 s-1), eine Materialkennzahl für dieBeweglichkeit der Teilchen in der Phase. Dasnegative Vorzeichen in (1) gibt an, dass derTeilchenfluss in Richtung abnehmenderKonzentration verläuft. Der Diffusions-koeffizient ist abhängig von der Temperatur,der Konzentration der Komponente und vonder Art und Konzentration anderer Kompo-nenten. Er nimmt mit steigender Temperatur(häufig exponentiell) zu.
Als einfache Anwendung des 1. FICKschenGesetzes soll die stationäre Diffusion zwi-schen zwei Lösungsräumen untersuchtwerden, die durch eine feinporige Wand derDicke s getrennt sind (Abb.1). Wenn jederder beiden Lösungsräume gut durchmischtwird (z.B. durch einen Rührer), kann man injedem der beiden Räume eine ortsunabhängi-
Abb.1: Messkammer und Konzentrations-profil
Diffusion W 25

Wärmelehre W 25 Diffusion
38
( )dd
cx
c c
sI II
= −−
. (2)
dd
cx
c
sI= − (3)
cJ tVII =
⋅. (4)
β =⋅
A
V s (5)
c t c D tII I( ) = ⋅ ⋅ ⋅β (6)
d
dII
I
c
tc D= ⋅ ⋅β . (7)
ge Konzentration annehmen. Das gesamteKonzentrationsgefälle erstreckt sich dannüber die feinporige Trennwand, in dieser ist
Da bei der experimentellen Realisierung diebeiden Lösungsräume relativ groß sind unddie Bedingung cI o cII während der gesamtenMesszeit erfüllt ist, vereinfacht sich (2) zu
und der Diffusionsfluss J wird entsprechendGl. (1) zeitlich konstant.Die Konzentration cII ergibt sich bei kon-stantem Zustrom von Ionen in denLösungsraum II mit dem Volumen V zu
Fasst man die gerätespezifischen Größen zueiner Apparatekonstanten
zusammen, so ergibt sich aus (4) mit (1), (3)und (5) ein linearer Anstieg der Konzen-tration cII im Lösungsraum II in Abhängigkeitvon der Zeit
mit dem Anstieg
Misst man cII in Abhängigkeit von der Zeit t,so kann also aus dem Anstieg der Messkurvebei bekannter Apparatekonstante β undKonzentration cI der Diffusionskoeffizient Dbestimmt werden. Die Apparatekonstantekann durch die Messung mit einer Substanzmit bekanntem Diffusionskoeffizientenbestimmt werden.
Im 1. FICKschen Gesetz (Gleichung (1)) stehtder Gradient der Stoffmengenkonzentration(Molarität, Einheit mol/l). Da in (6) und (7)
die Konzentration auf beiden Seiten derGleichung steht, kann sie dort durch dieMassenkonzentration (Einheit g/l) ersetztwerden. Damit ist es für die Bestimmung desDiffusionskoeffizienten nicht notwendig, dieMolmasse des gelösten Salzes zu kennen. ImWeiteren werden deshalb für alle Konzen-trationsangaben Massenkonzentrationen in g/lverwendet.
3 Versuchsaufbau
3.0 Geräte- Einzelmesskammer- Doppelmesskammer- 2 Röhrchen für Stammlösung A und B- Leitfähigkeits-Messsonde mit Stativ- Generator 1 V, 130 Hz- Anschluss-Box mit Umschalter und Mess-
widerstand- Digitalmultimeter- Doppelmagnetrührer- 2 Magnetrührstäbchen- Pipette 100 µl, Pipettenspitzen- Mikrospatel, Pinzette- Stoppuhr- Verbindungskabel
3.1 Die Ionenkonzentration der beidenSalzlösungen (Substanz A: KCl, Substanz B:unbekanntes Salz) wird mit Hilfe Messungelektrischen Leitwertes bestimmt.Die Einzelmesskammer besitzt die gleichenMaße wie ein Teil der Doppelmesskammerund dient zur Kalibrierung der Konzen-trationsmessung, d. h., zur experimentellenBestimmung des Zusammenhangs zwischenLeitwert und Konzentration.Die poröse Wand der Doppelmesskammerwird durch ein Zellulosenitrat-Filter mit0,2 µm Porengröße gebildet. Abb.2 zeigt die Anordnung zur Messung derLeitwert-Konzentrationsabhängigkeit derLösung. In die Messkammer wird eineLeitfähigkeits-Messsonde, die aus zweiElektroden besteht, eingetaucht.Mit Hilfe eines Voltmeters kann über einen

Wärmelehre W 25 Diffusion
39
G cI
U UU
U U Rz i
i
z i
( ) .=−
=−
⋅1
(8)
Messwiderstand (R=100Ω; in die Anschluss-box eingebaut) der zwischen den Elektrodenfließende Strom I bestimmt werden. Dieserergibt sich aus der zu messenden SpannungUi nach dem Ohmschen Gesetz.
Mit Hilfe eines Umschalters kann mit demselben Voltmeter auch die Generatorspan-nung Uz bestimmt werden. Aus diesen beidenSpannungsmessungen und dem bekanntenWert des Messwiderstandes lässt sich der zurjeweiligen Konzentration gehörende elektri-sche Leitwert der Lösung G(c) berechnen:
4 Versuchsdurchführung
Bereiten Sie schon vor Beginn des Prakti-kums die Tabellen für die Aufnahme derMesswerte vor und berechnen Sie dieKonzentrationen der Messlösungen in g/lin den einzelnen Schritten!
Für das Versuchsergebnis sind Sauberkeit derArbeitsgeräte und Reinheit der Lösungen vongroßer Bedeutung. Nach jedem Benutzensind deshalb die Messkammern zu reinigen!Zum Einwiegen der Substanzen stehen 2Waagen (Genauigkeit 1 mg), Wägeschälchenund Wägepapier zur Verfügung. Am ein-fachsten ist es, die Substanzen direkt in die
(trockenen!) Glasröhrchen einzuwiegen.Achten Sie darauf, dass die Magnetrührerimmer funktionieren!
4.1 Messung der Kalibrierkurven:Sowohl für die KCl-Lösung als auch für dieLösung des unbekannten Salzes wird derLeitwert in Abhängigkeit von der Salzkon-zentration im Konzentrationsbereich von(0…0,5) g/l gemessen. Dazu sind von beiden Stoffen jeweils 3 mleiner Stammlösung der Konzentration 220 g/lherzustellen (Salz in ca. 2 ml aqua dest.auflösen, dann mit der Pipette auf 3 mlauffüllen). Die Einzelmesskammer ist mit 220 ml aquabidest. zu füllen. In 5 Schritten werdenjeweils 100 µl der KCl-Stammlösung zurMesslösung hinzugegeben. Nach dem Kon-zentrationsausgleich sind jeweils Ui und Uz
zu messen. Diese Prozedur ist mit demunbekannten Salz zu wiederholen.
Es ist empfehlenswert, die Auswertung zudiesem Teilversuch sofort durchzuführen(siehe 5.1), damit die Kalibrierkurven bereitswährend der Durchführung von 4.2 und 4.3zur Verfügung stehen.
4.2 Bestimmung der Apparatekonstante β:Beide Teile der Doppelmesskammer sind mitje 220 ml aqua bidest. zu füllen. Zum Zeit-punkt t = 0 wird durch Zugabe von KCl inKammer I eine Konzentration von 10 g/leingestellt. Der Leitwert in Kammer II ist15 min lang alle 3 min zu messen. Die Pausenzwischen den Messungen sollten Sie nutzenum den Leitwert sofort zu berechnen undanhand der Kalibrierkurve die Konzentrationzu bestimmen (siehe 5.2).
4.3 Messung des Diffusionskoeffizientender Substanz B:Die Messung erfolgt analog zu Punkt 4.2. Fürdas Salz mit dem unbekannten Diffusions-koeffizienten wird eine Ausgangskonzen-tration in Kammer I von 20 g/l eingestellt.
Abb.2: Messanordnung zur Bestimmung desLeitwertes der Lösung

Wärmelehre W 25 Diffusion
40
5 Auswertung
5.1 Aus den gemessenen Spannungen Ui
und Uz ist der Leitwert der Lösungen nachGl. (8) zu berechnen. Für beide Salze ist derLeitwert in Abhängigkeit von der Konzen-tration graphisch darzustellen.
5.2 Aus den gemessenen Spannungen Ui
und Uz ist nach Gl. (8) der Leitwert derLösung im Kammer II zu berechnen. MitHilfe der in 5.1 erstellten Kalibrierkurvensind aus den Leitwerten die Konzentrationenzu ermitteln. Die KCl-Konzentration inKammer II ist in Abhängigkeit von der Zeitgraphisch darzustellen. Mittels linearerRegression wird der Anstieg der Kurvebestimmt und aus diesem gemäß Gl. (7) dieApparatekonstante β berechnet.
Der Diffusionskoeffizient von KCl beträgtD0 = 1,996 @ 10-9 m² s-1
5.3 Die Konzentration des unbekanntenSalzes in Kammer II ist wie in 5.2 zu er-mitteln und in Abhängigkeit von der Zeitgraphisch darzustellen. Mittels linearerRegression wird der Anstieg der Kurve
bestimmt und aus diesem gemäß Gl. (7) derDiffusionskoeffizient D berechnet. Dabei istdie mit KCl ermittelte Apparatekonstante βaus 5.2 einzusetzen.
6 Literatur
Haas, U.; Physik für Pharmazeuten undMediziner. WVG Stuttgart, 2002
Adam, G., Läuger, P., Stark, G., Physika-lische Chemie und Biophysik, SpringerBerlin, 1995
7 Kontrollfragen
7.1 Welcher Zusammenhang besteht zwi-schen der Diffusion und der Wärmeleitung?
7.2 Warum erfolgt die Messung des Leit-wertes der Lösungen mit niederfrequentemWechselstrom?
7.3 Von welchen Größen hängt der Diffu-sionskoeffizient ab?

Elektrizitätslehre E 7 Innenwiderstand von Spannungsquellen
41
U I R I Ri L0 = ⋅ + ⋅ (1)
U I RK L= ⋅ . (2)
U U I RK i0 = + ⋅ . (3)
IU
R RL i
=+
0 . (4)
1 Aufgabenstellung
1.1 Die Strom-Spannungs-Kennlinienverschiedener Gleichspannungsquellen sindaufzunehmen.
1.2 Die Innenwiderstände dieser Span-nungsquellen sind zu bestimmen.
2 Grundlagen
Bei der Messung elektrischer Spannungen(besonders bei sehr kleinen Spannungen wieBiopotentialen) hängen die Messergebnisseentscheidend von der Wahl des Messverfah-rens ab. Genauso wie bei Spannungsmessun-gen an Akkumulatoren oder Batterien, kannauch bei Messungen an biologischen Objek-ten (z.B. EKG-Ableitung) nicht ohne Rück-wirkung auf die Spannung gemessen werden.Zwischen der zu messenden Spannungsquelle(U0 - Urspannung) und den Klemmen desMessgerätes (UK - Klemmenspannung)besteht ein zusätzlicher Innenwiderstand Ri,der vom “Innenleben” der Spannungsquelleabhängt. Der innere Widerstand des an-geschlossenen Messgerätes stellt den Last-widerstand RL dar, mit dem die Spannungs-quelle während der Messung belastet wird.
Dieser Zusammenhang wird im allgemeinenErsatzschaltbild für eine Spannungsquelledargestellt (Abb.1).Mit dem Maschensatz (ÿ KIRCHHOFFscheGesetze) folgt für diesen einfachen Strom-kreis:
mit I als fließendem Strom.Der Spannungsabfall am Lastwiderstand istgleich der Klemmenspannung:
Damit lässt sich (1) schreiben als:
Der Innenwiderstand kann aus der Abhängig-keit der Klemmenspannung UK vom Strombestimmt werden. Wenn die Größenordnung des Innenwider-standes bekannt ist, dann kann man meist eingeeignetes Spannungsmessverfahren (bzw.Messgerät) auswählen, bei dem RL o Ri ist.Dann gilt I@Ri n UK bzw. UK . U0, d. h. dieUrspannung U0 (auch Leerlaufspannunggenannt) wird nahezu unverfälscht gemessen.Mit Hilfe einer Kompensationsschaltung istes auch möglich, die Leerlaufspannungunmittelbar (bei I = 0) zu messen.
Entsprechend Gl. (3) liegt an den Polen einerSpannungsquelle im unbelasteten Zustand,das heißt wenn der Quelle kein Strom ent-nommen wird, die Urspannung U0 oderLeerlaufspannung (früher auch als elektro-motorische Kraft bezeichnet) an. Wird nun andie Spannungsquelle ein “Verbraucher”angeschlossen, so dass ein Strom fließt, sowird die Spannung an den Polen der Quelleauf einen Wert sinken, der als Klemmen-spannung UK bezeichnet wird.Für den Laststrom folgt aus (3):Abb.1: Ersatzschaltbild einer Spannungs-
quelle
Innenwiderstand von Spannungsquellen E 7

Elektrizitätslehre E 7 Innenwiderstand von Spannungsquellen
42
Abb.2: Strom-Spannungs-Kennlinie einerSpannungsquelle mit konstantem Innenwider-stand.
P P PU
R Rg i Li L
= + =+
02
. (8)
η = =+
=
+
P
P
R
R R R
R
L
G
L
i L i
L
1
1.
(9)
P U IL K= ⋅ (5)
( )P
U
R RRL
i L
L=+
⋅0
2
2 . (6)
( )P
U
R RRi
i L
i=+
⋅02
2 . (7)
Aus Gleichung (3) ist zu erkennen, dass dieKlemmenspannung linear mit der Strombela-stung sinkt. Die graphische Darstellung desZusammenhangs zwischen Klemmenspan-nung und Strom wird als Strom-Spannungs-Kennlinie einer Spannungsquelle bezeichnet.Abbildung 2 zeigt eine solche Kennlinie füreine Spannungsquelle mit konstantem Innen-widerstand. Aus der Darstellung geht hervor,dass bei I = 0 die Klemmenspannung UK
gleich der Urspannung U0 ist. Mit abnehmen-dem Lastwiderstand RL steigt der Strom unddie Klemmenspannung sinkt, bis der Span-nungsabfall über Ri den Wert der Urspannungerreicht hat. Jetzt ist der Lastwiderstand Nullund der Spannungsabfall am InnenwiderstandRi ist gleich der Urspannung U0. Diesen Fallnennt man Kurzschluss. Im Kurzschlussfallwird UK Null und es fließt der maximalmögliche Strom, der als Kurzschlussstrombezeichnet wird.Derartige Kennlinien weisen zum BeispielTrockenbatterien oder Akkumulatoren auf, dasie einen relativ konstanten Innenwiderstandbesitzen.
Die von einer Spannungsquelle gelieferteelektrische Energie soll der Verbraucher(Widerstand RL) nutzen. Da der Verbraucher-oder Laststrom I aber auch durch die Span-nungsquelle fließt, nimmt diese (über denInnenwiderstand Ri) einen Teil der am ge-samten Umsatz beteiligten Leistung auf. Diean den Verbraucher abgegebene Leistung
kann mit Hilfe der Gleichungen (2) und (4)umgeformt werden zu:
Analog gilt für die in der Spannungsquelleverbrauchte Leistung:
Die gesamte von der Spannungsquelle gelie-ferte Leistung ist damit:
Für den Wirkungsgrad η, das Verhältnis vonNutzleistung zur Gesamtleistung, folgt:
Nach Gleichung (9) steigt der Wirkungsgradmit wachsendem Verhältnis von RL zu Ri an.Um die innerhalb der Spannungsquelleverbrauchte Leistung gering zu halten, solltedie Bedingung RL >> Ri eingehalten werden,d.h. es muss in Leerlaufnähe gearbeitetwerden. Diese Forderung gilt für Primär- undSekundärelemente wegen deren endlicherKapazität (entnehmbare Ladung), die übli-cherweise in Ah (Amperestunden) angegebenwird und die nicht mit der Kapazität einesKondensators verwechselt werden sollte.Anders ist die Situation bei Solarzellen, dadie Sonne ja ständig Energie nachliefert. Hierbesteht das Ziel darin, dem Verbraucher einemöglichst hohe Leistung zuzuführen. DerLastwiderstand RL, für den die Leistung PL
maximal wird, kann bestimmt werden indemdie erste Ableitung von Gleichung (6)dPL/dRL = 0 gesetzt wird. Es ergibt sich dieeinfache Beziehung RL = Ri. In diesem Fall

Elektrizitätslehre E 7 Innenwiderstand von Spannungsquellen
43
spricht man von (Leistungs-) Anpassung.
3 Versuchsaufbau
3.0 Geräte:verschiedene Gleichspannungsquellen:
Trockenbatterie, Bleiakku, Solarzellen-modul
verschiedene Lastwiderstände:Schiebewiderstand 140 Ω, Drehwider-stand 5 Ω, 4 Dekadenwiderstände 1/10/100/1000 Ω
2 Digitalmultimeter1 SchalterVerbindungsleitungen
3.1 Schaltungsaufbau:Für die Aufnahme der Strom-Spannungs-Kennlinie wird die Schaltung nach Abb.3aufgebaut. Als Spannungsmesser wird einMessgerät mit hohem Eingangswiderstandverwendet, so dass die Spannungsquelle beigeöffnetem Schalter nur mit einem minima-len Strom belastet wird.
4 Versuchsdurchführung
Achtung!Bleiakkus liefern sehr hohe Ströme. Durcheinen Kurzschluss können Kabel ver-schmelzen! Falsch angeschlossene Mess-geräte können beschädigt werden! LassenSie die Schaltung vor dem Anschließen derSpannungsquellen kontrollieren!
Machen Sie sich zunächst mit der Funktions-weise der zu verwendenden Lastwiderständevertraut. Dazu stehen Ihnen die Multimeter
zur Verfügung, mit denen Widerstände direktgemessen werden können.Messen Sie die Leerlaufspannungen U0 fürden Bleiakku und für die Trockenbatterie.Damit diese beiden Spannungsquellen wäh-rend der Messung nicht zu stark entladenwerden, muss der Strom begrenzt werden auf3 A für den Bleiakku und 200 mA für dieTrockenbatterie. Berechnen Sie aus diesenWerten die erforderlichen Lastwiderstände RL
und entscheiden Sie dann, welche Wider-stände Sie für die Batterie und den Akkuverwenden wollen. Für das Solarzellenmodulwerden die vier Dekadenwiderstände (0 …10000 Ω) verwendet. Stellen Sie die ver-änderlichen Widerstände (den Schiebewider-stand bzw. die Dekadenwiderstände) vorBeginn der Messung so ein, dass die genann-ten Ströme in keinem Fall - auch nicht bei zuvernachlässigendem Innenwiderstand -überschritten werden.Zur Aufnahme der I-U-Kennlinie wird dieSchaltung nach Abbildung 3 aufgebaut. Für die Kennlinie des Akkus werden minde-stens 10 etwa äquidistante Messpunktezwischen 0 und 3 A gewählt. Die dazugehöri-gen Klemmenspannungen Uk sowie dieUrspannung U0 (bei I = 0) sind zu messen.Für die Kennlinie der Batterie werden min-destens 10 etwa äquidistante Messpunktezwischen 0 und 200 mA gewählt. Die da-zugehörigen Klemmenspannungen Uk sowiedie Urspannung U0 (bei I = 0) sind wieder zumessen.Für das Solarzellenmodul ist die gesamte I-U-Kennlinie, d.h. von der Leerlaufspannungbis zum Kurzschlussstrom, aufzunehmen. Eswerden mindestens 20, bezüglich der Stroms-tärke möglichst äquidistante Punkte gemes-sen. Hier sind neben der Stromstärke I undder Klemmenspannung Uk auch die Werte derzugehörigen Lastwiderstände RL zu erfassen.
5 Auswertung
Für alle Gleichspannungsquellen werden dieStrom-Spannungs-Kennlinien graphisch
Abb.3: Versuchsschaltung
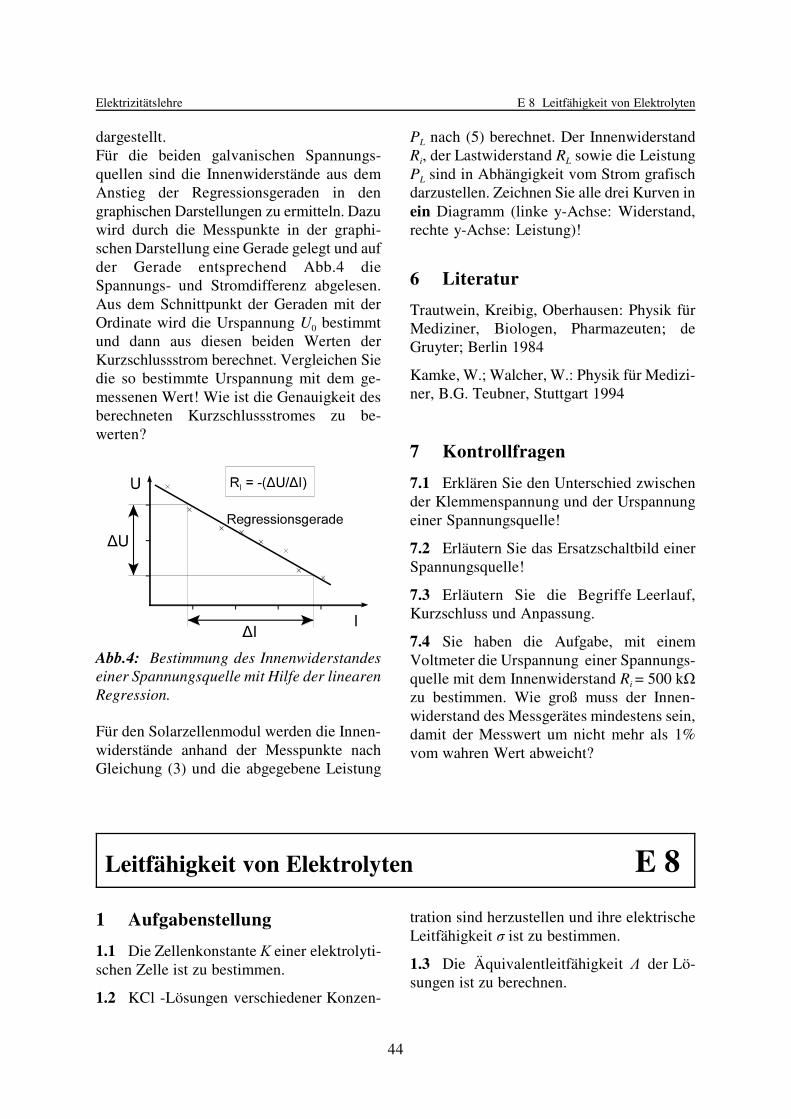
Elektrizitätslehre E 8 Leitfähigkeit von Elektrolyten
44
Abb.4: Bestimmung des Innenwiderstandeseiner Spannungsquelle mit Hilfe der linearenRegression.
dargestellt.Für die beiden galvanischen Spannungs-quellen sind die Innenwiderstände aus demAnstieg der Regressionsgeraden in dengraphischen Darstellungen zu ermitteln. Dazuwird durch die Messpunkte in der graphi-schen Darstellung eine Gerade gelegt und aufder Gerade entsprechend Abb.4 dieSpannungs- und Stromdifferenz abgelesen.Aus dem Schnittpunkt der Geraden mit derOrdinate wird die Urspannung U0 bestimmtund dann aus diesen beiden Werten derKurzschlussstrom berechnet. Vergleichen Siedie so bestimmte Urspannung mit dem ge-messenen Wert! Wie ist die Genauigkeit desberechneten Kurzschlussstromes zu be-werten?
Für den Solarzellenmodul werden die Innen-widerstände anhand der Messpunkte nachGleichung (3) und die abgegebene Leistung
PL nach (5) berechnet. Der InnenwiderstandRi, der Lastwiderstand RL sowie die LeistungPL sind in Abhängigkeit vom Strom grafischdarzustellen. Zeichnen Sie alle drei Kurven inein Diagramm (linke y-Achse: Widerstand,rechte y-Achse: Leistung)!
6 Literatur
Trautwein, Kreibig, Oberhausen: Physik fürMediziner, Biologen, Pharmazeuten; deGruyter; Berlin 1984
Kamke, W.; Walcher, W.: Physik für Medizi-ner, B.G. Teubner, Stuttgart 1994
7 Kontrollfragen
7.1 Erklären Sie den Unterschied zwischender Klemmenspannung und der Urspannungeiner Spannungsquelle!
7.2 Erläutern Sie das Ersatzschaltbild einerSpannungsquelle!
7.3 Erläutern Sie die Begriffe Leerlauf,Kurzschluss und Anpassung.
7.4 Sie haben die Aufgabe, mit einemVoltmeter die Urspannung einer Spannungs-quelle mit dem Innenwiderstand Ri = 500 kΩzu bestimmen. Wie groß muss der Innen-widerstand des Messgerätes mindestens sein,damit der Messwert um nicht mehr als 1%vom wahren Wert abweicht?
1 Aufgabenstellung
1.1 Die Zellenkonstante Κ einer elektrolyti-schen Zelle ist zu bestimmen.
1.2 KCl -Lösungen verschiedener Konzen-
tration sind herzustellen und ihre elektrischeLeitfähigkeit σ ist zu bestimmen.
1.3 Die Äquivalentleitfähigkeit Λ der Lö-sungen ist zu berechnen.
Leitfähigkeit von Elektrolyten E 8

Elektrizitätslehre E 8 Leitfähigkeit von Elektrolyten
45
Abb.1: WHEATSTONE-Brückenschaltung
RlA
= ⋅ρ , (3)
C
C
R
Rx
1
3
4
= . (2)
GAl
= ⋅σ , (4)
GR K
= = ⋅1 1
σ . (5)
R
R
R
RX
1 3
4
= . (1)
2 Grundlagen
2.1 Für eine genaue Widerstandsmessungund für die Bestimmung von kleinen Wider-ständen werden häufig Brückenschaltungenverwendet. Die Grundschaltung jeder Mess-brücke ist die Wheatstone-Brücke (Abb.1). Zwischen den Punkten A und B sowie zwi-schen C und D liegt die Spannung U an.Ohne Galvanometer zwischen den Mess-punkten E und F wird das Potential in diesenPunkten durch die Verhältnisse R1 : RX sowieR3 : R4 bestimmt (Spannungsteiler zwischenA und B sowie zwischen C und D).Besteht zwischen E und F kein Potential-unterschied, so sagt man, Brücke ist abgegli-chen. Über das Galvanometer (ein hoch-empfindliches Amperemeter) wird dann keinStrom fließen. In diesem Fall muss die fol-gende Bedingung erfüllt sein:
Wenn zwischen E und F ein Potentialunter-schied besteht, so fließt über das Galvano-meter ein Strom. Das Prinzip der Brücken-messung besteht darin, durch Änderung derbekannten Widerstände R1, R3 und R4 denabgeglichenen Zustand der Brücke zu finden.Die Größe des unbekannten Widerstandes RX
lässt sich dann nach (1) berechnen. Soll eine Messbrücke mit Wechselstrombetrieben werden, so ist eine mögliche Pha-senverschiebung zwischen Strom und Span-nung zu berücksichtigen. Für den Brücken-abgleich müssen in diesem Fall Betrag und
Phase der Spannung zwischen E und Fabgeglichen werden. Zu diesem Zweck wirddie einfache Wheatstone-Schaltung durchzusätzliche Kapazitäten ergänzt, siehe Ver-suchsschaltung Abb.2.Da die elektrolytische Zelle neben demWiderstand Rx auch eine Kapazität Cx besitzt,müssen für den Abgleich der Brücke zweiBedingungen erfüllt sein: der Amplituden-abgleich (1) und der Phasenabgleich
(1) und (2) gelten gleichzeitig, wenn dieBrücke vollständig abgeglichen ist.Die Brückenschaltung in Abb.2 wird alsWien-Brücke bezeichnet. Sie kann nach Gl.(2) auch zur Messung von Kapazitäteneingesetzt werden.
2.2 Der elektrische Widerstand eines Leitersmit der Länge l und dem Querschnitt A ist
er wird neben den geometrischen Größen lund A durch eine Materialkonstante, denspezifischen elektrischen Widerstand ρ,bestimmt. Der Kehrwert des elektrischenWiderstandes G = 1/R heißt elektrischerLeitwert, seine SI-Einheit ist das Siemens:[G] = 1/Ω = S. Analog zu (3) schreibt man
σ = 1/ρ heißt spezifische elektrische Leit-fähigkeit und hat die Einheit [σ] = S/m. DieAngabe eines Leitwertes ist insbesondere beielektrisch leitenden Flüssigkeiten üblich.
Elektrolyte sind elektrisch leitende Flüssig-keiten, bei denen der Ladungstransport durchIonen erfolgt.Eine elektrolytische Zelle, die mit einemElektrolyten gefüllt ist, hat den elektrischenWiderstand R. Für den elektrischen LeitwertG der Zelle schreibt man

Elektrizitätslehre E 8 Leitfähigkeit von Elektrolyten
46
Abb.2: Schaltung zur Leitfähigkeitsmessungin Elektrolyten (Wienbrücke).
Λ =σcq
(6)
cz nV
m zM Vq =
⋅=
⋅⋅
(7)
σ =KRx
. (8)In dieser Gleichung wurden gegenüber Gl.(4) die Größen A und l zur Zellkonstanten Kzusammengefasst. Ihre Einheit ist ”Κ› =1/m.Die Zellkonstante K wird nur vom Aufbau(Größe und Form) der elektrolytischen Zellebestimmt, während die elektrische Leitfähig-keit σ nur von der Art des gelösten Stoffes,seiner Konzentration und von der Temperaturabhängt. Die Leitfähigkeit von Elektrolyten steigt mitder Temperatur (wegen der thermischenBewegung der Teilchen) und mit der Ionen-konzentration an. In stark verdünnten Salzlö-sungen ist die elektrische Leitfähigkeit zurKonzentration proportional.
Die Äquivalentleitfähigkeit Λ ist
wobei cq die Äquivalentkonzentration (Nor-malität eines Elektrolyten) ist:
Dabei bedeuten: m = Masse, V = Volumen,M = Molmasse in g/mol, n = m/M = Stoff-menge (Anzahl der Mole), z = Wertigkeit(Ladungszahl). Die SI-Einheit von Λ istS m2/mol, die von cq ist mol/m3, meist wirdjedoch mol/l verwendet.Die Äquivalentleitfähigkeit hängt im All-gemeinen sowohl vom Dissoziationsgrad alsauch von der Konzentration ab. Durch elek-trostatische Wechselwirkung behindern sichdie wandernden Ionen gegenseitig. Dahernimmt die Äquivalentleitfähigkeit mit zuneh-mender Konzentration ab. Bei starker Ver-dünnung (÷ große Abstände zwischen denIonen) ist die Wechselwirkung vernachlässig-bar, die Äquivalentleitfähigkeit vollständigdissoziierter Elektrolyte wird dann von derKonzentration unabhängig.
Zur Bestimmung der elektrischen Leitfähig-keit σ wird der Widerstand Rx der elektrolyti-schen Zelle gemessen. Aus (5) ergibt sich
Die Zellkonstante Κ kann durch Messungeines Elektrolyten bekannter Leitfähigkeit(hier gesättigte NaCl-Lösung) ermitteltwerden.Die Leitfähigkeit von Elektrolyten mussgrundsätzlich mit Wechselstrom gemessenwerden, damit das Messergebnis nicht durchdie Elektrodenpolarisation verfälscht wird.Die Widerstände Rx der gefüllten elektrolyti-schen Zelle werden deshalb mit Hilfe einerWechselstrombrücke (Abb.2) ermittelt.
3 Versuchsaufbau
3.0 Geräte- elektrolytische Zelle mit Halterung- Generator 1 kHz- Vielfachmesser- Kurbelwiderstand (R 1)- Kapazitätsdekaden (C 1) - 2 Festwiderstände 100 Ω (R3, R4)- Thermometer- Flasche mit NaCl-Lösung- Flasche mit KCl- 3 Messzylinder 100 ml, 25 ml, 10 ml- Becherglas, Trichter, Spatel, Glasstab
3.1 Als Spannungsquelle dient ein Wechsel-spannungs-Generator mit der festen Frequenz1 kHz und einer Spannung von Ueff = 2 V.Für die Widerstände R3 und R4 werden zwei

Elektrizitätslehre E 8 Leitfähigkeit von Elektrolyten
47
gleiche Festwiderstände mit je 100 Ω und fürden Widerstand R1 ein Kurbelwiderstandverwendet. Die Kapazität C1 kann zwischen1 nF und 999 nF variiert werden. Zur An-zeige des Brückenstromes dient ein empfind-licher Vielfachmesser mit veränderlichenMessbereichen.
4 Versuchsdurchführung
Die Brückenschaltung ist nach Abb.2 auf-zubauen.
4.1 Bestimmung der Zellkonstante: Dieelektrolytische Zelle wird mit der gesättigtenNaCl-Lösung gefüllt, die Temperatur T derLösung ist zu messen. Am Vielfachmesserwird zunächst ein unempfindlicher Mess-bereich (z.B. 100 mA) eingestellt; Generatorund Vielfachmesser sind einzuschalten. Nunwerden der Widerstand R1 und die KapazitätC1 abwechselnd verändert, bis der Brücken-strom ein Minimum erreicht, wobei dieMessbereiche am Vielfachmesser immerempfindlicher geschaltet werden. Wenn dieBrücke abgeglichen ist, sollte der Brücken-strom kleiner als 3 µA sein. Der dazugehörigeWiderstand R1 = Rx (folgt aus (1), wennR3 = R4) ist abzulesen und zu protokollieren.
Achtung! Nach jeder Widerstandsmessungmuss am Vielfachmesser wieder ein un-empfindlicher Messbereich eingestelltwerden, damit beim Wechsel der Elek-trolyte keine Überlastung des Messgerätesauftritt.
4.2 Bestimmung der elektrischen Leitfähig-keit von KCl-Lösungen der Konzentration1N; 0,5N; 0,2N; 0,1N; 0,05N und 0,02N:Zunächst werden 100 ml KCl-Lösung mit derÄquivalentkonzentration cq = 1 mol/l (1Noder 1-normale Lösung) hergestellt. DieMolmasse von KCl beträgt 74,55 g/mol. Dieerforderliche Menge KCl ist abzuwiegen undim Messzylinder in etwa 90 ml destilliertemWasser aufzulösen (Rühren mit Glasstab).Danach wird die Lösung mit destilliertem
Wasser auf 100 ml aufgefüllt.Die elektrolytische Zelle ist mit dieser Lö-sung zu füllen, der dazugehörige Widerstandwird wie oben beschrieben gemessen. DieTemperatur der Lösung wird gemessen.Anschließend wird die 1N KCl-Lösung zurweiteren Verwendung in einem Becherglasaufbewahrt; die Reste können nach Abschlussdes Versuches entsorgt werden.Je 100 ml KCl-Lösung der Konzentration0,5N; 0,2N; 0,1N; 0,05N und 0,02N werdendurch Verdünnung eines Teils der 1N KCl-Lösung hergestellt. Der Widerstand und dieTemperatur dieser Lösungen in der elek-trolytischen Zelle werden gemessen. An-schließend sind die Lösungen zu verwerfen.Alle gemessenen Werte sind übersichtlichtabellarisch zu erfassen.
Hinweis:Es muss sauber gearbeitet werden. Dieelektrolytische Zelle und die Elektroden sindbei jedem Umfüllen sorgfältig zu spülen.Die gesättigte NaCl-Lösung wird nach derMessung in die Vorratsflasche zurückgegos-sen.
5 Auswertung
5.1 Die Zellenkonstante K ist mittels (8) zuberechnen. Die elektrische Leitfähigkeit σ dergesättigten NaCl-Lösung ist anhand dergemessenen Temperatur T einer Tabelle zuentnehmen.
5.2 Die elektrischen Leitfähigkeiten σ derKCl-Lösungen werden nach Gl. (8), dieÄquivalentleitfähigkeiten Λ nach (6) be-rechnet.Die Leitfähigkeit und die Äquivalentleit-fähigkeit sind in Abhängigkeit von derÄquivalenkonzentration (Normalität) derKCl-Lösung grafisch darzustellen und zudiskutieren.
6 Literatur
Gerthsen, Kneser, Vogel: Physik. Springer

Elektrizitätslehre E 10 Thermospannung
48
U TT = ⋅α ∆ . (1)
Verlag 1989
Beier, W.; Pliquett, F.: Physik für das Studi-um der Medizin, Biowissenschaften, Veteri-närmedizin, J.A.Barth, Leipzig 1987
7 Kontrollfragen
7.1 Erläutern Sie das Prinzip der Wider-
standsmessung mit einer Brückenschaltung!
7.2 Was versteht man unter Amplitude undPhase eines Wechselstromes?
7.3 Wie wird die Äquivalentleitfähigkeitdefiniert, und welche Eigenschaft des gelös-ten Salzes wird hierüber charakterisiert?
1 Aufgabenstellung
Der SEEBECK-Koeffizient eines Thermo-elementes ist mit Hilfe einer Kompensations-methode zu bestimmen.
2 Grundlagen
2.1 Thermoelement: In einem (isolierten)elektrischen Leiter wird durch einen Tempe-raturgradienten eine elektrische Potentialdif-ferenz erzeugt (absoluter SEEBECK-Effekt),die jedoch nicht unmittelbar messbar ist.Vereinfacht gesehen, verhalten sich dieLadungsträger im Leiter wie die Teilcheneines idealen Gases: Ladungsträger von derwärmeren Seite mit höherer kinetischerEnergie diffundieren schneller zur kaltenSeite als umgekehrt; auf diese Weise entstehtauf der kalten Seite ein Überschuss an La-dungsträgern, der durch die entstehendePotentialdifferenz begrenzt wird.Verbindet man zwei verschiedene Leiter zueinem Stromkreis (Abb.1) und bringt diebeiden Kontaktstellen auf verschiedeneTemperaturen T0 und T1, so entstehen in denbeiden Leitern unterschiedliche innere Poten-tiale. Zwischen den Punkten A und B tritt dieDifferenz der beiden Potentiale auf, diesogenannte Thermospannung. Dies wird als
SEEBECK-Effekt bezeichnet.
In vielen Lehrbüchern wird die Thermospan-nung falsch als „Kontaktspannung“ erklärt.Dies ist nicht richtig! (siehe Literaturhinweis)Die Thermospannung UT ist näherungsweiseproportional zur Temperaturdifferenz ∆T =T1 - T0 :
Der Koeffizient α heißt Thermokraft oderSeebeckkoeffizient und ist von beiden Mate-rialien abhängig.Thermoelemente werden häufig zur Tempera-turmessung verwendet. Sie bieten den Vor-teil, dass sich die Thermospannungen un-mittelbar als Eingangssignale für Computer,Steuer- und Regelgeräte nutzen lassen.
2.2 Kompensationsschaltung: Die Messungder Urspannung einer Spannungsquelle ist
Abb.1: Kupfer-Konstantan-Thermoelement
Thermospannung E10

Elektrizitätslehre E 10 Thermospannung
49
Abb.2: Kompensationsschaltung
1 2
10 1
1 2
.
h
E
U I R I R
RU I R U
R R
= ⋅ + ⋅
= ⋅ = ⋅+
(2)
Abb.3: Versuchsaufbau
IURh
h= . (3)
nur möglich, wenn der Quelle dabei keinStrom entnommen wird (siehe Versuch E 7).Stromlose Spannungsmessungen lassen sichmit Hilfe einer Kompensationsschaltungvornehmen (Abb.2).
Hierzu werden neben der zu messendenSpannungsquelle U0 eine Hilfsspannungs-quelle Uh (genau bekannte Spannung, gerin-ger Innenwiderstand), ein Spannungsteiler(bestehend aus den Teilwiderständen R1 undR2) und ein Galvanometer (hochempfindli-ches Amperemeter, Nullinstrument) benötigt.Wird jetzt durch Veränderung des Verhält-nisses zwischen R1 und R2 erreicht, dass derSpannungsabfall über R1 gleich der SpannungU0 ist, so wird der Spannungsquelle U0 keinStrom entnommen.Für die beiden Maschen in der Schaltung inAbb.2 gilt in diesem Fall nach der KIRCH-HOFFschen Regel:
Mit Hilfe des Galvanometers kann die Kom-pensationsschaltung genau abgeglichenwerden; U0 wird dann nach (2) berechnet.
3 Versuchsaufbau
3.0 Geräte- Halterung mit Thermoelement- Heizplatte mit Magnetrührer- Becherglas
- Alu-Gefäß- Rührstäbchen- Digitalthermometer- Stöpselwiderstand- elektronisches Galvanometer- Akkumulator- Vielfachmesser- Verbindungsleitungen
3.1 Zur Bestimmung der Thermospannun-gen UT wird eine Kompensationsschaltungnach Abb.3 verwendet. Die linke Lötstelledes Thermoelementes wird mit Hilfe einesEis-Wasser-Gemisches auf einer konstantenTemperatur von T0 = 0EC gehalten; dieTemperatur der rechten Lötstelle T1 kanndurch die Erwärmung des Wasserbades (Alu-Gefäß) verändert werden. Für T1-T0 = ∆T … 0entsteht eine Thermospannung UT .Die Widerstände R (Gesamtwiderstand) undR1 (Teilwiderstand von R) werden durch denPräzisions-Stöpselwiderstand gebildet. Dieserkann durch Einstecken von Metallstöpselnverändert werden (dabei werden jeweils dieaufgedruckten Teilwiderstände kurzgeschlos-sen). Mit Hilfe zweier Abgriffstöpsel kannein beliebiger, genau definierter Teilwider-stand abgegriffen werden.Der Stöpselwiderstand ist an einem Akkumu-lator mit der (Hilfs-) Spannung Uh ange-schlossen, es fließt ein Strom Ih:
Der Spannungsabfall U1 am Widerstand R1
zwischen den verstellbaren Abgriffspitzen(Abb.3) beträgt

Elektrizitätslehre E 10 Thermospannung
50
U R IR U
Rh
h
1 11= ⋅ =
⋅. (4)
Mit dieser (Gegen-) Spannung U1 kann dieThermospannung UT kompensiert werden; esist U1 = UT, wenn der Thermostrom IT, dervom Galvanometer G angezeigt wird, gleichNull ist.
4 Versuchsdurchführung
Achtung! Bevor der Akku an den Stöpsel-widerstand angeschlossen wird, muss dieSchaltung vom Assistenten überprüft werden!Beim Auf- und Abbau des Versuches sowiebeim Messen darf das Galvanometer nichtüberlastet werden!
Bauen Sie die Schaltung nach Abb.3 auf. DerGesamtwiderstand R des Stöpselwiderstandesist durch Einfügen von Messingstöpselngeeigneter Anzahl und Position so einzurich-ten, dass der Strom Ih = 0,05 mA beträgt.Dazu ist R zunächst mit Hilfe der aufgedruck-ten Akku-Spannung Uh zu berechnen. NachInbetriebnahme der Schaltung erfolgt einFeinabgleich m.H. des Amperemeters.
Das Becherglas ist mit einer Eis-Wasser-Mischung zu füllen, so dass die linke Löt-stelle eine Temperatur von T0 = 0EC an-nimmt. Dazu wird das Eis in der neben demWaschbecken bereitstehenden Keramik-schüssel fein zerstoßen. Das Alu-Gefäß istmit Wasser von ca. 5°C zu füllen (einigeEisstückchen hineingeben). Es ist ein Widerstand R1 so einzustellen, dassam Galvanometer G ein negativer Ausschlagentsteht, d.h. U1 > UT. Erwärmt man dann das Wasserbad (unterständigem Rühren), so steigt T1 und damit dieThermospannung UT. Wenn IT = 0 ist (Null-durchgang am Galvanometer), ist UT = U1. Indiesem Moment ist die Temperatur T1 ab-zulesen. Danach wird die Gegenspannung U1
mit Hilfe von R1 wieder erhöht (Linksaus-schlag am Galvanometer).
Die weitere Erwärmung führt erneut zu IT = 0(also UT = U1), wobei beim Nulldurchgangdes Zeigers am Galvanometer wieder dieTemperatur T1 gemessen wird. Dieses Ver-fahren wird bis zur Siedetemperatur fortge-setzt. Zur Einstellung der Gegenspannungen U1
empfiehlt es sich, folgende Widerstandswertefür R1 zu wählen: 5Ω, 10Ω, 15Ω, 20Ω, …
5 Auswertung
Für alle Messpunkte wird die Thermospan-nung UT berechnet und in Abhängigkeit vonder Temperatur T1 graphisch dargestellt. Aus der graphischen Darstellung ist dieThermokraft (der Seebeckkoeffizient) α durchlineare Regression zu ermitteln. VergleichenSie das Ergebnis mit Literaturwerten!
6 Literatur
Bergmann-Schäfer: Lehrbuch der Experimen-talphysik, Bd. II, de Gruyter, Berlin - NewYork 1987
http://www.uni-konstanz.de/FuF/Physik/Jaeckle/papers/thermospannung/
7 Kontrollfragen
7.1 Wie entsteht eine Thermospannung?
7.2 Welche Vorteile hat ein Thermoelementverglichen mit anderen Temperaturmess-verfahren?
7.3 Warum verwendet man zur Messung derThermospannung eine Kompensationsschal-tung?

Elektrizitätslehre E 34 Elektrolyse
51
m A Q= ⋅ . (1)
AM
z F=
⋅(4)
Q
nz N ea= ⋅ ⋅ . (3)
mM Qz F
=⋅
⋅. (5)
m
m
A
A
M z
M z1
2
1
2
1 1
2 2
= =/
/. (2)
H SO 2H SO2 4 4→ ++ −2 . (6)
2H O 2SO 2H SO O2 4 2
2 4 2+ → +− . (7)
p V n R T⋅ = ⋅ ⋅ . (8)
1 Aufgabenstellung
Die Faraday-Konstante ist durch Elektrolysein einem Hofmannschen Wasserzersetzungs-apparat zu bestimmen.
2 Physikalische Grundlagen
Elektrolyte sind elektrisch leitende Stoffe(meist Lösungen), bei denen der Ladungs-transport durch positiv geladene Ionen undnegativ geladene Ionen erfolgt. Unter Elek-trolyse versteht man die mit dem Stromdurch-gang verbundene Zersetzung des Elektrolytenund das Abschneiden der Reaktionsproduktean den Elektroden. Da die elektrolytischeLeitung eine Ionenleitung ist, ist der La-dungstransport mit einem Massetransportverbunden. Die an einer Elektrode abge-schiedene Masse m ist der transportiertenLadung Q proportional (1. FARADAYschesGesetz):
Dabei ist A ein stoffabhängiger Propor-tionalitätsfaktor (elektrochemisches Äquiva-
lent). Sind und zwei Massen ver-m1 m2
schiedener Stoffe, die durch eine gleicheLadung abgeschieden wurden, M1 und M2
ihre Molmassen sowie z1 bzw. z2 ihre Wertig-keit (Ladungszahlen der Ionen), so lautet das2. FARADAYsche Gesetz:
Betrachtet man den Fall, dass 1 mol einesStoffes abgeschieden wird, so ist die dabeitransportierte Ladung (Ladung pro Mol)
Es bedeuten n = m/M die Anzahl der Mole;Na = 6,022A1023 mol-1 die Avogadro-Kon-
stante und e = 1,602A10-19 As die Elementar-ladung.Das Produkt F = Na A e = 96484 As/molheißt Faraday-Konstante.
Aus den Gleichungen (1) und (3) folgen
und
Beim Hofmannschen Wasserzersetzungs-apparat (Abb.1) dient als Elektrolyt verdünn-te Schwefelsäure H2SO4 ; die Elektrodenbestehen aus chemisch inertem Platin. DieDissoziation der Schwefelsäure ergibt
Die H+-Ionen wandern zur Katode (negativeElektrode) und werden dort neutralisiert(Reduktion), so dass Wasserstoff H2 abge-schieden wird. SO4
2!-Ionen wandern zurAnode (positive Elektrode), geben dort ihreLadung ab (Oxydation) und verbinden sichmit Wasser zu Schwefelsäure; es entstehtSauerstoff:
Diese Elektrolyse stellt eine Wasserzerset-zung dar; das Volumen des abgeschiedenenWasserstoffs H2 ist doppelt so groß wie dasVolumen des abgeschiedenen SauerstoffesO2. Für die Bestimmung der Faraday-Konstante Fwird der abgeschiedene Wasserstoff verwen-det, da sich Sauerstoff relativ stark in Wasserlöst. Der Wasserstoff kann bei Zimmer-temperatur als ideales Gas angesehen werden,es gilt
Dabei bedeuten p: Druck; V: Volumen; T:
Elektrolyse E 34

Elektrizitätslehre E 34 Elektrolyse
52
FR T Q
p V=
⋅ ⋅
⋅ ⋅2. (9)
Q I dt= ∫ (10)
Q I t= ⋅ . (11)
Q I dt I tii
n
= ≈ ⋅∫ ∑=1
∆ . (12)
Temperatur; R: Gaskonstante.Berücksichtigt man, dass der Wasserstoff imElektrolyten atomar (H+), im Gasvolumenaber molekular vorliegt (H2), so dass
ist, dann folgt aus (5) und (8)M 2 MH H2=
3 Versuchsaufbau
3.0 Geräte:1 Hofmannscher Wasserzersetzungsapparat1 Stromversorgungsgerät1 Widerstand 10 Ω1 Schalter1 Sensor-CASSYVerbindungsleitungen1 Thermometer1 Computer mit CassyLab-Software
3.1 Für die Bestimmung der Faraday-Kon-stante nach (9) muss die Ladung Q gemessenwerden. Sind I der Strom und t die Zeit, so ist
bzw., wenn I = konstant,
Da aber der Strom durch den Elektrolyten
über eine längere Zeit nicht konstant ist,empfiehlt sich die Bestimmung der Ladungdurch Integration. Der Strom wird hierbei mitHilfe des Sensor-CASSY in kurzen Zeit-abständen (∆t = 0,2 s) gemessen und vomComputer aufgezeichnet. Dies ergibt eineMessreihe von n Einzelmessungen Ii. DieLadung kann dann durch numerische In-tegration bestimmt werden:
4 Versuchsdurchführung
Die Bedienungsanweisungen zur Benutzungder Geräte werden vom zuständigen Assi-stenten gegeben bzw. der ausliegendenGerätebeschreibung entnommen. Ausführ-liche Informationen zur Benutzung derCassyLab Software sind im Online-Handbuchund in der Hilfe zum Programm zu finden,eine Kurzanleitung befindet sich im Anhangdieses Heftes.
Die Schaltung nach Abb.1 ist aufzubauen. InCassyLab sind Messgröße, Messbereich undMessintervall (0,2 s) einzustellen. Es darfkeine Messzeit vorgegeben werden, da dieMessung manuell beendet werden soll.Zur Bestimmung der Faraday-Konstante F istzunächst durch vorsichtiges Öffnen derbeiden Hähne am Zersetzungsapparat dieFlüssigkeitshöhe mit Hilfe des Ausgleich-gefäßes auf Null (oder, wenn das nichtmöglich ist, auf 10 cm3) eingerichtet. DieHähne werden wieder geschlossen.
Vorsicht beim Umgang mit dem Elek-trolyten (verdünnte Schwefelsäure)!
Die Messung wird nun gestartet und danachder Schalter geschlossen. Es beginnt dieElektrolyse.Nach der Bildung von ca. 20 cm3 Wasserstoffist die Elektrolyse abzuschalten und danachdie Messung zu stoppen. Wenn die Gas-entwicklung aufgehört hat, werden die Flüs-sigkeitsspiegel des Ausgleichgefäßes und des
Abb.1: Versuchsaufbau mit HofmannschemWasserzersetzungsapparat

Elektrizitätslehre E 39 Messwerterfassung mit dem Computer (EKG)
53
H2-Rohres auf die gleiche Höhe gebracht(Abb.1), so dass der Druck im H2-Rohr gleichdem äußeren Luftdruck pb ist. Das durch dieElektrolyse abgeschiedene Volumen desWasserstoffes, die Temperatur T und der
Luftdruck (Barometer) werden abgelesen.pb
Die Messkurven I(t) der Stromstärke werdenabgespeichert.Die Elektrolyse und die Messungen zurBestimmung der Faraday-Konstante sind dreimal bei verschiedenen Stromstärken (etwa100 mA, 200 mA und 300 mA) durchzufüh-ren. Es sollen alle Messkurven in einer Grafikdargestellt werden!
5 Auswertung
Aus den Messkurven I(t) wird die Ladungs-menge durch Integration mittels CassyLabbestimmt.
Für jede Messreihe ist die Faraday-KonstanteF nach Gleichung (9) zu berechnen. Dabeimuss der Partialdruck des Wasserdampfes pW
im Wasserzersetzungsapparat berücksichtigt
werden: Für den H2-Partialdruck gilt p = pb -pW . Der Wasserdampfdruck ist der aus-liegenden Tabelle zu entnehmen.
Am Beispiel einer Messreihe ist eine Fehler-rechnung durchzuführen.
6 Literatur
Walcher, W.: Praktikum der Physik, B. G.Teubner, Stuttgart 1989
Hellenthal, W.: Physik und ihre Anwendungin der Praxis für Pharmazeuten, Medizinerund Biologen, G. Thieme, Stuttgart 1988
7 Kontrollfragen
7.1 Was sagen die beiden FARADAYschenGesetze aus?
7.2 Was ist ein ideales Gas?
7.3 Nennen Sie einige praktische Anwen-dungen der Elektrolyse.
1 Aufgabenstellung
1.1 Mit Hilfe des CASSY-Messwerterfas-sungssystems ist ein 3-Kanal-EKG auf-zunehmen.
1.2 Die Größe der R-Zacken-Potentiale unddie Pulsfrequenz sind zu bestimmen.
1.3 Die Lage der elektrischen Herzachse istzu bestimmen.
2 Grundlagen
Die rhythmische Kontraktion des Herz-muskels wird stimuliert durch eine elektri-sche Erregung der Herzzellen, die am Sinus-Knoten beginnt und sich in charakteristischerWeise über das gesamte Herz ausbreitet. DieAktionspotentiale (etwa 70 mV) aller Zellenergeben in Summe ein elektrisches Dipolfelddes Herzens. Dieses Feld breitet sich auch imgesamten Organismus aus, wobei es durchdie elektrische Leitfähigkeit geschwächtwird.
Messwerterfassung mit dem Computer (EKG) E 39

Elektrizitätslehre E 39 Messwerterfassung mit dem Computer (EKG)
54
Abb.1: EKG-Ableitungen nach Einthoven
Auf der Hautoberfläche können deshalb dieelektrischen Vorgänge bei der Reizaus-breitung im Herzen gemessen werden. Dabeiwerden eine Reihe Elektroden in normierterArt und Weise an den Armen und Beinensowie auf dem Brustkorb angebracht. Dieelektrischen Spannungen auf der Körperober-fläche liegen im Millivoltbereich und können,entsprechend verstärkt, gemessen und aufeinem Monitor oder mit Hilfe eines Schrei-bers oder Druckers dargestellt werden.
Bei den drei bipolaren Ableitungen nachEINTHOVEN (Abb.1) werden die folgendenPotentiale gemessen:
Abl. I zwischen linkem und rechtem Arm
Abl. II zw. linkem Bein und rechtem Arm
Abl. III zw. linkem Bein und linkem Arm.
Im so genannten EINTHOVEN-Dreieck(Abb.2) lässt sich aus den drei Potentialen U1,U2 und U3 der Dipolvektor den Herzens(genauer: die Projektion des Dipolvektors aufdie Frontalebene) konstruieren. Der Di-polvektor variiert im Rhythmus des Herz-schlages, seine Lage im Moment des größtenPotentials (R-Zacken-Potential) wird alselektrische Herzachse bezeichnet. Sie stimmtbei normaler Erregungsausbreitung etwa mit
der anatomischen Längsachse des Herzensüberein.
Neben der Kurvenform im EKG (siehe Abb.4in Versuch E23) ist die Lage der Herzachsebzw. der zeitliche Verlauf des Dipolvektorsvon diagnostischer Bedeutung (Vektor-EKG).
Messen mit dem Computer:
Moderne medizinische Diagnosegeräte (z. B.EKG, EEG, Ultraschall-A-Bild, Audiometer)werden heute oft auf der Basis handelsübli-cher Computer gebaut. Das hat sowohlpraktische Gründe (vielseitige Einsetzbarkeit,einfache Vernetzung mit anderen Geräten,mit der elektronischen Patientenkartei etc.)als auch ökonomische Gründe. Der Computer(bzw. die Software) übernimmt die Aus-wertung und die graphische Darstellung derMessergebnisse sowie ihre Archivierung.Lediglich die eigentliche Messwerterfassung,Signalverstärkung und die Umwandlung desanalogen Messsignals in digitale Daten (A/D-Wandlung) muss noch mit Hilfe speziellerHardware erfolgen.
Die Genauigkeit der Messung wird dabeidurch die sogenannte Auflösung oder Wand-lerbreite des A/D-Wandlers begrenzt. Bei-spielsweise bedeutet eine Auflösung von 12Bit, dass der Wandler 212 = 4096 verschiede-ne digitale Werte messen kann. Die kleinstenoch messbare Änderung des Messsignalsbeträgt also 1/4096 des Messbereichs.
Abb.2: Bestimmung der Lage der elektrischHerzachse im Einthofen-Diagramm

Elektrizitätslehre E 39 Messwerterfassung mit dem Computer (EKG)
55
f RG =12
. (1)
Ein zeitabhängiges Messsignal (z. B. einEKG-Signal) wird einfach in schneller Folgeimmer wieder gemessen („abgetastet“). DieAnzahl der Messungen pro Sekunde bezeich-net man als Messrate oder Abtastrate R, dieEinheit ist 1 Hz. (Beispiel: Bei einem Zeit-intervall ∆t von 10 ms zwischen zwei Mes-sungen beträgt die Messrate R = 1/∆t = 100Hz.) Technisch sind heute Messraten bis etwa1 GHz möglich.
Die Messrate bestimmt die Zeitauflösungbzw. die maximal messbare Frequenz fG
(Grenzfrequenz), es gilt
3 Versuchsaufbau
3.0 Geräte:
1 Sensor-CASSY
1 EKG/EMG-Box
1 Computer mit CASSYLab-Software
4 Klammerelektroden
Elektrodenspray
3.1 Die EKG/EMG-Box ist bereits auf dasSensor-CASSY aufgesteckt und darf nichtentfernt werden. Das Cassy ist mit demComputer über ein serielles Kabel verbunden.Beim Start des Programms CASSYLab mussdiese Konfiguration automatisch erkanntwerden.
4 Versuchsdurchführung
Versorgen Sie das CASSY mit Strom undstarten Sie danach am Computer das Pro-gramm CASSYLab. Aktivieren Sie dieEKG/EMG-Box, indem Sie mit der Maus aufdas Bild der Box klicken.
Machen Sie sich nun mit der Bedienung desProgramms vertraut. Benutzen Sie hierzu die
Kurzanleitung im Anhang und die Online-
Hilfe . Folgende Tätigkeiten müssen Siebeherrschen:
- Einstellung von Messbereich und -Intervall
- Start und Stopp einer Messung
- Vergrößern/Alles anzeigen (Zoom)
- Skalieren des Diagramms
- Einfügen von Text in das Diagramm
- Anzeigen der Messpunkte
- Messen von Differenzen (Spannung, Zeit)
- Speichern und Drucken der Messung
Zur Ableitung des EKG-Signals nach EINT-HOVEN (Abb.1) werden die Metallflächen derElektrodenklammern mit Elektrodenspraybesprüht und an den Innenseiten der Handge-lenke und oberhalb der Knöchel befestigt.(Das Spray dient zur Desinfektion und zurVerbesserung des elektrischen Kontaktes.)Die Elektrodenkabel werden wie folgt an-geschlossen: rot - rechter Arm, gelb - linkerArm, grün - linke Wade, schwarz - rechteWade.
Die Versuchsperson muss in ruhiger, ent-spannter Lage sitzen, die Unterarme auflie-gend, damit die EKG-Potentiale nicht durchandere Muskel-Aktionspotentiale verfälschtwerden.
Die Voreinstellungen der Messparameterwerden unverändert beibehalten. Das EKGwird etwa 10...20 s lang aufgezeichnet unddanach abgespeichert. Falls die Kurven sehrunregelmäßig sind, werden die Ursachenhierfür beseitigt und die Messung wirdwiederholt. Alle Versuchsbedingungen sindzu protokollieren!
Wenn möglich, wird das EKG an jedemStudenten gemessen; die Auswertung führtjeder an seinem EKG durch.
5 Auswertung
Die Messung des R-Zacken-Potentials der

Elektrizitätslehre E 39 Messwerterfassung mit dem Computer (EKG)
56
drei Ableitungen und der Pulsfrequenz sollmit CASSYLab durchgeführt werden. Benut-zen Sie hierfür die Zoom-Funktion; hilfreichsind außerdem die Funktionen „Koordinatenanzeigen“, „Differenz messen“ und „Werteanzeigen“. Falls die Zeit nicht reicht, wirddas EKG ausgedruckt und zuhause mittelsLineal ausgewertet.
Für die drei Ableitungen UI, UII und UIII wirdan jeweils 5 verschiedenen Stellen die Höheder R-Zacke abgelesen und daraus der Mittel-wert berechnet.
Die Pulsfrequenz f (in min-1) wird aus dermittleren Zeit zwischen zwei R-Zackenermittelt. Dafür sind aus 10 aufeinander
folgenden Pulsschlägen der Mittelwert undTdie Standardabweichung sT zu berechnen.Welche Bedeutung hat die Standardabwei-chung in diesem Zusammenhang?
Für die Bestimmung der Lage der elektri-schen Herzachse werden die Ableitungen Iund II verwendet. Erfassen Sie etwa fünfzeitgleiche Wertepaare (UI, UII) vom Beginnbis zum Ende einer R-Zacke. Die Wertepaarewerden im EINTHOVEN-Dreieck (Spezial-papier, im Praktikum erhältlich) auf denAbleitungslinien I und II abgetragen, derSchnittpunkt im Dreiecksgitter wird markiert.Die so entstandenen Punkte werden der Reihenach durch eine Linie verbunden. Diese Liniezeigt den Verlauf der Frontalprojektion desDipolvektors. Die Verbindung vom Null-punkt zum maximalen Ausschlag definiert
den elektrischen Herzvektor.
Zusätzliche Experimentiermöglichkeiten:
S Zeichnen Sie EKGs mit verschiedenenMessintervallen (5 ms, 10 ms, 20 ms,50 ms) auf. Welchen Einfluss hat dasMessintervall (bzw. die Messrate) auf dasMessergebnis?
S Messen Sie die Größe und des Muskel-Aktionspotentials am Unterarm. FolgenSie dazu der Anleitung in der Online-Hilfezu CASSYLab.
6 Literaturangaben
Kamke, W.; Walcher, W.: Physik für Medizi-ner, B.G. Teubner, Stuttgart 1994
www.neurop.ruhr-uni-bochum.de/lehre/Prak
tikum
7 Kontrollfragen
7.1 Was verstehen Sie unter dem elektri-schen Herzvektor und wie kann man ihnmessen?
7.2 Welche Messrate ist bei der Computer-gestützten Messung eines EKGs mindestenserforderlich?
7.3 Warum werden beim EKG besondershochohmige Spannungsmessgeräte eingesetzt?

Optik und Strahlung O 4 Mikroskop
57
1 Aufgabenstellung
1.1 Einstellung des Mikroskops und derKÖHLERschen Beleuchtungseinrichtung.
1.2 Kalibrierung eines Okularmikrometersdurch Bestimmung des Abbildungsmaßstabesfür mehrere Objektive.
1.3 Justierung der Phasenkontrasteinrich-tung.
1.4 Beobachtung biologischer Präparate mitverschiedenen Verfahren und Ausmessungvon Strukturen.
2 Grundlagen
2.1 Aufbau des Mikroskops:Das optische System eines Lichtmikroskopsbesteht aus dem Objektiv und Okular. Umdas Prinzip der Bildentstehung besser erken-nen zu können, werden die Linsensysteme (eswerden zur Bildfehlerkorrektur jeweilsmehrere Linsen benötigt) zu je einer dünnenKonvexlinse zusammengefasst.Ein Gegenstand G zwischen einfacher unddoppelter Brennweite des Objektives wird alsumgekehrtes, vergrößertes und reelles Zwi-
schenbild B außerhalb der doppelten Brenn-weite des Objektives abgebildet, Abb.1. DasOkular wird als Lupe eingesetzt. Das reelleZwischenbild, das sich innerhalb der ein-fachen Brennweite des Okulars befindet, wirddadurch dem akkommodierten Auge alsdivergentes Strahlenbündel angeboten.Eine rückwärtige Verlängerung der Strahlenzeigt, in welcher Größe und Entfernung dasAuge etwa das virtuelle Bild erkennt. Eineeinfache Bildkonstruktion nach der geometri-schen Optik kann jeweils mit Hilfe zweierausgezeichneter Strahlen (Parallelstrahl,Brennpunktstrahl, Mittelpunktstrahl; sieheAbb.1) eines Bildpunktes gefunden werden.
Für ein Mikroskopieren mit entspanntem(d. h. nicht akkommodiertem) Auge soll dasvirtuelle Bild in großer Entfernung (“imUnendlichen”) entstehen. Abweichend vonAbb.1 fällt dann das reelle Zwischenbild indie Brennebene des Okulars; der Sehwinkelβ' bleibt gleich.In modernen Mikroskopen mit “Unendlichop-tik” (ICS, infinity corrected system) bildetdas Objektiv allein den Gegenstand nicht inder Zwischenbildebene sondern im Unendli-chen ab, d. h. der Gegenstand befindet sich in
Abb.1: Strahlengang eines Mikroskopes
Mikroskop O 4

Optik und Strahlung O 4 Mikroskop
58
V =′
≈′tan
tan.
β
β
β
β(1)
V V VOb Ok= ⋅ . (2)
BG
VOb= (3)
dn A
=⋅
=λ
αλ
sin. (4)
der Brennebene des Objektivs. Erst durcheine zusätzliche Tubuslinse (zwischen Objek-tiv und Okular) entsteht das reelle Zwischen-bild in der Brennebene des Okulars. Dasermöglicht u. A. den problemlosen Einbauvon Zubehör (z. B. für Fluoreszenz- undPolarisationsmikroskopie), da die Tubuslängevariabel ist.
Als Vergrößerung V bezeichnet man dasVerhältnis der scheinbaren Größe einesObjektes mit optischem Instrument zurscheinbaren Größe ohne Instrument in derdeutlichen Sehweite (auch Bezugssehweite,25 cm Entfernung vom Auge). Sie ergibt sichaus den Sehwinkeln β' und β mit und ohneoptisches Instrument zu:
Beim Mikroskop setzt sich die Gesamtver-größerung aus der Objektivvergrößerung VOb
und Okularvergrößerung VOk zusammen:
Will man die Größe eines Objektes im Mi-kroskop messen, dann bringt man an dieStelle des reellen Zwischenbildes einenMaßstab („Okularmessplatte” oder „Okular-mikrometer“). Dieser ist dann gemeinsam mitdem Bild scharf zu sehen und dient alsGrößenvergleich. Für exakte Messungenmuss also der Abbildungsmaßstab des Objek-tives, d.h. das Verhältnis von Bildgröße B zuGegenstandsgröße G
genau bekannt sein. Er steht in der Regel aufdem Objektiv, jedoch kann sich der tatsäch-liche Wert vom aufgedruckten aufgrund vonFertigungstoleranzen etwas unterscheiden.Der Abbildungsmaßstab wird bestimmt,indem man ein Objekt definierter Größe(„Objektmikrometer”) mit dem Okularmikro-meter vergleicht.
2.2 Auflösungsvermögen:Die Bildentstehung im Mikroskop kann
vollständig nur mit Hilfe der Wellennatur desLichtes verstanden werden. Das einfallendeLicht wird an den Strukturen des Objekts Ggebeugt (Abb.2). Das Objektiv O vereinigtgebeugte und ungebeugte Wellen in derBildebene B, wo durch Interferenz das reelleZwischenbild entsteht. Zur Verdeutlichungbetrachtet man als Modellobjekt G einBeugungsgitter, bei dem nur ein charakteristi-scher Abstand (die Gitterkonstante) auftritt.Ein heller (bzw. dunkler) Punkt entsteht imreellen Zwischenbild B nur, wenn durchInterferenz des ungebeugten Anteils mit dengebeugten Strahlen eine Verstärkung (bzw.Auslöschung) eintritt.Nach der Theorie des Auflösungsvermögensvon ABBE wird ein Bilddetail nur dann
aufgelöst wird, wenn neben dem ungebeugtenLicht wenigstens das Beugungsmaximumerster Ordnung in das Objektiv fällt und zurBildentstehung beiträgt. Daraus ergibt sichder kleinste Abstand d zweier Objektpunkte,die noch getrennt abgebildet werden können:
Dabei ist λ die Wellenlänge des Lichtes undA = n@ sinα die numerische Apertur desObjektives (mit dem halben Öffnungswinkelα des Objektives und dem Brechungsindex ndes Mediums zwischen Objekt und Objektiv).Ein Objektdetail wird um so objektähnlicherabgebildet, je mehr Beugungsmaxima im Bildinterferieren. Den Kehrwert von d bezeichnetman als Auflösungsvermögen. Die numeri-
Abb.2: Wellenoptische Erklärung derAbbildung am Mikroskop

Optik und Strahlung O 4 Mikroskop
59
dAmin .=
λ2
(5)
sche Apertur ist neben der Vergrößerung aufdem Objektiv angegeben.Die Wellenlänge λ ist durch den sichtbarenBereich des Spektrums bestimmt (Mittelwert550 nm). Eine Steigerung des Auflösungsver-mögens kann durch Verwendung von Öl-immersionssystemen erzielt werden. Dabeiwird das Medium Luft (n . 1) zwischenObjekt und Objektiv durch ein Immersionsöl(n .1,5) ersetzt; dafür sind spezielle Objekti-ve erforderlich. Außerdem ergibt sich eineSteigerung des Auflösungsvermögens, wenndas Objekt nicht mit parallelem Licht (wiebei der Herleitung von Gl. (4) vorausgesetzt),sondern aus verschiedenen Richtungenbeleuchtet wird. Wenn die Beleuchtungs-apertur (Sinus des halben Öffnungswinkelsdes Beleuchtungskegels) gleich der numeri-schen Apertur des Objektivs ist, ergibt sichein Grenzwert von
Dieser Wert gilt auch für die Mikroskopievon selbstleuchtenden Objekten (Fluores-zenzmikroskopie) und im Dunkelfeld.
2.3 Förderliche Vergrößerung: Ein Auge mitnormaler Sehschärfe kann zwei Punkte nochgetrennt wahrnehmen, wenn sie unter einemWinkel von zwei Bogenminuten erscheinen.In der deutlichen Sehweite von 25 cm ent-spricht das einem Abstand von 0,15 mm.Wird die Vergrößerung des Mikroskops sogewählt, dass die kleinsten trennbaren Ob-jektabstände d (durch das Auflösungsver-
mögen des Mikroskops gegeben) im virtuel-len Bild unter einem Winkel von zwei Bo-genminuten erscheinen, dann bezeichnet mandiese Vergrößerung als förderliche Ver-größerung VM . Als Faustregel gilt: VM =(500…1000)@A . Vergrößerungen über diesenBetrag hinaus bezeichnet man als „leereVergrößerung“, denn man erhält keine neuenInformationen von dem Objekt.
2.4 Köhlersche Beleuchtung:Beim Beleuchtungsverfahren nach KÖHLER
wird der Lichtkegel der Beleuchtung demÖffnungskegel des Objektivs angepasst.Dadurch wird das Auflösungsvermögens desObjektives vollständig ausgenutzt und über-flüssiges Licht, das als Streulicht den Kon-trast vermindert, wird vermieden.Abb. 3 zeigt den prinzipiellen Aufbau derBeleuchtungsanordnung. Sie besteht ausLichtquelle, Kollektorlinse und Leuchtfeld-blende, die sich in der Mikroskopierleuchtebefinden, sowie Aperturblende und Konden-sorlinse, die unterhalb des Mikroskoptischesangebracht sind.Der Kollektor bildet die Lichtquelle (Glüh-wendel) in die Ebene der Aperturblende ab.Der Kondensor bildet die Leuchtfeldblende,die sich unmittelbar neben dem Kollektorbefindet, in die Objektebene ab. Die Apertur-blende liegt in der vorderen Brennebene desKondensors. Dadurch werden alle von einemPunkt der Lichtquelle ausgehenden Strahlenzu Parallelstrahlen. Je kleiner die Apertur-blende eingestellt wird, um so kleiner ist derÖffnungswinkel des Strahlenbündels (die
Abb.3: KÖHLERsches Beleuchtungsverfahren

Optik und Strahlung O 4 Mikroskop
60
Beleuchtungsapertur).Durch diese Anordnung können sowohl dieGröße des ausgeleuchteten Feldes (mit derLeuchtfeldblende) als auch die Größe derBeleuchtungsapertur (mit der Aperturblende)unabhängig voneinander verändert werden.Die Aperturblende verändert außerdem dieBildhelligkeit.
2.5 Verfahren zur Kontraststeigerung:Viele biologische Präparate, vor allem Gewe-beschnitte, zeigen im einfachen Durchlicht-mikroskop wenig Kontrast, so dass man trotzausreichender Vergrößerung und Auflösungkaum etwas erkennen kann. Oft werdenPräparate deshalb mit verschiedenen Metho-den eingefärbt. Dies ist jedoch zeitaufwendig,außerdem lassen sich lebende Präparate kaumfärben und das Objekt wird durch die Fär-bung selbst verändert. Mit speziellen opti-schen Vorrichtungen am Mikroskop lässt sichder Kontrast ebenfalls steigern; es könnensogar Strukturen sichtbar gemacht werden,die im „normalen“ (Hellfeld-)Mikroskop un-sichtbar sind. Für Medizin und Biologie vonBedeutung sind die Dunkelfeldmikroskopie,die Phasen-kontrastmikroskopie, diePolarisations-mikroskopie und dieFluoreszenz-mikroskopie. Alle außer demletzten Ver-fahren können im Praktikumerprobt werden.
2.5.1 Dunkelfeld: Das bisher beschriebeneMikroskopierverfahren heißt Hellfeld, da dasGesichtsfeld ohne Präparat hell ausgeleuchtetist. Sorgt man durch eine Zentralblende in derMitte der Aperturblende (Abb.3) dafür, dasskein Licht auf direktem Wege in den Strah-lengang des Mikroskops gelangen kann, sobleibt das Gesichtsfeld ohne Präparat dunkel.Das Licht trifft nur aus solchen Winkeln aufdas Präparat, die größer sind als die Objekti-vapertur. Zur Abbildung trägt dann nur dasam Objekt gebeugte Licht bei.In diesem „Dunkelfeldkontrast“ erscheinenvöllig lichtdurchlässige und völlig licht-undurchlässige Bereiche des Objektes ingleicher Weise dunkel, die Kanten vonObjektstrukturen leuchten dagegen hell auf.
Besonders gut zu erkennen sind kleinstePartikel, die das Licht nach allen Seitenstreuen.
2.5.2 Phasenkontrast: Mikroskopische Präpa-rate unterscheidet man in Amplitudenobjekteund Phasenobjekte. Amplitudenobjektebesitzen unterschiedliche Lichtdurchlässig-keiten, was zu einem Hell-Dunkel-Kontrastführt. Phasenobjekte dagegen besitzen überall(etwa) gleiche Lichtdurchlässigkeit, aberverschiedene Brechzahlen. Im durchgehen-den Licht treten Gang- bzw. Phasenunter-schiede auf (siehe (6) und (7)), die das Augenicht wahrzunehmen kann. Typische Phasen-objekte sind ungefärbte Gewebeschnitte.Zur wellenoptischenen Beschreibung derBildentstehung im Mikroskop betrachtet manBeugungsgitter als einfache Modellobjekte.Bei einem Amplitudengitter (siehe Abb.4)haben die Gitteröffnungen und -stege unter-schiedliche Lichtdurchlässigkeiten, so dassdas im Zwischenbild Hellig-keitsunterschiedevorliegen, die das Auge wahrnimmt. BeimPhasengitter haben die Gitterelemente gleicheLichtdurch-lässigkeiten, aber verschiedeneBrechzahlen. Obwohl auch beim Phasengitterdurch Interferenz ein reelles Zwischenbildentsteht, treten dabei aber keine Helligkeits-unterschiede auf, d.h. es gibt keinen Bildkon-trast. Die mathematisch exakte wellenoptischeBeschreibung der Bildentstehung zeigt, dassbei einem Amplitudengitter das gebeugteLicht im reellen Zwischenbild gegenüberdem ungebeugten um 180° (λ/2) phasenver-schoben ist, bei einem Phasengitter dagegenum 90° (λ/4). Beim Phasenkontrastverfahrennach ZERNIKE (Nobelpreis 1953) wird durcheinen Trick die Phasendifferenz zwischengebeugtem und ungebeugtem Licht um 90°vergrößert - dadurch wird aus dem Phasen-kontrast ein Amplitudenkontrast, d.h. Phasen-objekte werden sichtbar.

Optik und Strahlung O 4 Mikroskop
61
An die Stelle der Aperturblende (siehe Abb.3) wird eine Ringblende in den Strahlenganggebracht, hierdurch wird das Präparat miteinem Strahlenbündel in Form eines Kegel-mantels beleuchtet. Die ungebeugten Strahlendurchlaufen in der hinteren Brennebene desObjektives eine Ringfläche. An dieser Stellebefindet sich im Objektiv ein λ/4-Phasenring,der die Phase der ungebeugten Strahlengegenüber dem überwiegendem Anteil dergebeugten Strahlen um -90° verschiebt.
Eine weitere Kontraststeigerung erzielt mandurch eine Schwächung des ungebeugtenLichtes durch den leicht grau getönten Pha-senring. (Nur deshalb kann man ihn sehen;siehe Abschnitt 4.3.)
2.5.3 Polarisationskontrast:Schickt man natürliches Licht durch einPolarisationsfilter (den Polarisator P), soerhält man linear polarisiertes Licht (sieheVersuch O 10).Lässt man dieses Licht auf ein zweites Polari-sationsfilter (den Analysator A) fallen, sowird es nur dann ungehindert hindurchgelassen, wenn die Durchlassrichtung desAnalysators parallel zu der des Polarisatorsist. Ist die Durchlassrichtung des Analysatorsdagegen um 90° gedreht („gekreuzte Stel-lung“ von P und A), so wird das Licht voll-ständig ausgelöscht.Für den qualitativen Polarisationskontrast istein Mikroskop mit einem Polarisator in derBeleuchtungseinrichtung und eine Analysatoroberhalb des Objektivs ausgerüstet. Beidebefinden sich in gekreuzter Stellung, so dassdas Gesichtsfeld ohne Präparat dunkel bleibt.Wenn aber Strukturen im Präparat die Polari-
sationsrichtung des Lichtes verändern, dannwird dieses Licht im Analysator nicht mehrausgelöscht, und die Strukturen erscheinenhell oder in charakteristischen Interferenz-farben.(Ein „Polarisationsmikroskop“ besitzt darü-ber hinaus Einrichtungen zum Messen vonWinkeln, einen drehbaren Probentisch, einespezielle Beleuchtung u. a.)
Die Polarisationsrichtung kann durch zweiphysikalische Effekte beeinflusst werden:(i) Optischer Aktivität ist die Eigenschaftbestimmter asymmetrisch aufgebaute Kristal-le oder organischer Stoffe mit asymmetri-schem Kohlenstoffatom (z.B. Zucker), beimDurchgang von linear polarisiertem Lichtdessen Schwingungs-richtung zu drehen.Siehe hierzu Versuch O10.(ii) Doppelbrechung ist eine Eigenschaftoptisch anisotroper Stoffe. Diese besitzen fürverschiedene Schwingungsrichtungen desLichtes unterschiedliche Brechzahlen. Da-durch werden unpolarisierte Lichtstrahlen inzwei zueinander senkrecht linear polarisierteTeilbündel aufgespalten. Ursachen für die Doppelbrechung sindasymmetrische Kristallgitter (z.B. bei Kalk-spat), mechanische Spannung („Spannungs-doppelbrechung“) oder ein submikroskopischanisotroper Aufbau („Formdoppelbrechung“).Letzteres tritt häufig bei biologischen Mate-rialien auf, die eine geschichtete- oder Faser-struktur besitzen(z.B. Muskelfasern, Nerven-fasern, Kollagenfasern).Abb.5 zeigt die Schwingungsrichtungen desLichtes, wenn eine dünne doppelbrechendePlatte der Dicke d zwischen gekreuzte Polari-satoren (P und A) gebracht wird. Hinter demPolarisator P hat das Licht die Schwingungs-richtung L. Beim Durchgang durch die Plattewird es in zwei Strahlen aufgespalten, diesenkrecht zueinander polarisiert sind(Schwingungsrichtungen L1 und L2). Auf-grund der für beide Strahlen unterschiedli-chen Brechzahlen n1 und n2 besteht zwischenihnen nach Austritt aus der Platte ein Gang-unterschied
Abb.4: Amplitudengitter (oben) und Phasengitter (unten)

Optik und Strahlung O 4 Mikroskop
62
( )δ = ⋅ −d n n1 2(6)
( )ϕδλ λ
= = ⋅ −2 2 1 2π πd
n n . (7)
bzw. eine Phasendifferenz von
Die Phasenverschiebung hängt also von derDicke der Platte und von der Wellenlänge λdes Lichtes ab.Die beiden Teilstrahlen können zunächstnicht miteinander interferieren, da sie senk-recht zueinander polarisiert sind. (BeideTeilstrahlen zusammen nennt man auchzirkular bzw. elliptisch polarisiert.)Der Analysator lässt von den beiden Strahlenjeweils die Anteile L1' und L2' hindurch. Dadiese jetzt in gleicher Richtung polarisiertsind, interferieren sie miteinander.Das Resultat hängt vom Gangunterschied δab. Für δ = λ, 2λ, 3λ, ... verstärken sich beideAnteile und für δ = 1/2 λ,
3/2 λ, 5/2 λ, ... löschen
sie sich aus. Im Spektrum des einfallendenweißen Lichtes werden sich folglich be-stimmte Farben auslöschen und andereverstärken. Gemäß der Physiologie desAuges entstehen charakteristische Misch-farben. Bei sehr kleinen Gangunterschiedenδ n λ entstehen keine Interferenzfarben.
Schwach doppelbrechende Strukturen wiez.B. Kollagenfasern führen also im Polarisa-tionskontrast zu einer Aufhellung und könnendadurch identifiziert werden. Stark doppel-brechende Strukturen wie manche Kristalleoder Knochen erscheinen farbig.
3 Versuchsaufbau
3.0 Geräte:- Mikroskop „Axiostar“ mit Phasenkontrast-
einrichtung, Okularmikrometer und Analy-sator eingebaut
- Hilfsmikroskop- Polarisationsfilter- Objektmikrometer- Objektträger- Deckgläser- Präparate: Blutausstrich, Schnitt einer
Maus oder Kaninchenzunge, Hautschnitt,Sehne, Knochenschnitt, Diatomeen
3.1 Beim Mikroskop Axiostar befindet sichdie Lampe hinter einer Streuscheibe im Mik-roskopfuß, sie muss nicht justiert werden. Am Kondensor befindet sich ein Blendenre-volver mit den Ringblenden Ph1, Ph2 undPh3 für den Phasenkontrast sowie den Stel-lungen H für Hellfeld und DF für Dunkelfeld.Die mit Ph gekennzeichneten Objektive sindfür Phasenkontrast geeignet.Im Tubus des Mikroskops ist der Analysatorfür den Polarisationskontrast fest eingebautDas Polarisator wird bei Bedarf in die Vertie-fung über der Leuchtfeldblende gelegt.
3.2 Das Objektmikrometer für die Be-stimmung des Abbildungsmaßstabes ist 1 mmgroß mit 0,01 mm Teilung, das Okularmikro-meter 10 mm mit 0,1 mm Teilung.
3.3 Diatomeen sind einzellige Kieselalgen,die in mehr als 12.000 Arten praktisch überallvorkommen. Sie weisen in ihrer Schalenkon-struktion winzige Feinststrukturen von hoherRegelmäßigkeit auf und dienen deshalb alsTestobjekte für das Auflösungsvermögen vonMikroskopobjektiven.
4 Versuchsdurchführung
Hinweis:Um Schäden an den Objekten und Objektivenzu vermeiden, führt man das Objektiv unterseitlicher Sicht dicht über das Präparat.Anschließend erfolgt die Scharfstellung
Abb.5: Durch P linear polarisiertes Licht Lwird an einer doppelbrechenden Platte in L1
und L2 zerlegt. Durch den gekreuzten Analy-sator A gelangen die Anteile L1' und L2'.

Optik und Strahlung O 4 Mikroskop
63
durch Vergrößerung des Abstandes.
4.1 Alle Einstellung werden zuerst mit demObjektiv 10× durchgeführt. Der Kondensorist zunächst ganz nach oben zu stellen, derRevolver muss sich in der Stellung H (Hell-feld) befinden und die Aperturblende (Hebelam Kondensorrevolver) soll etwa halb geöff-net sein.
1. Fokussieren des Okularmikrometers:Stellen Sie mit dem Beleuchtungsstärkereglerdie Helligkeit geeignet ein. Falls ein Präparatauf dem Objekttisch liegt, defokussieren Siees, so dass es nicht zu sehen ist. Stellen Siedurch Verdrehen der Augenlinse das Okular-mikrometer scharf; versuchen Sie dabeientspannt in die Ferne zu blicken. Das zweiteOkular ist etwa so einzustellen wie das erste.(Wenn keine Okularmessplatte verwendetwird entfällt dieser Schritt, statt dessen sinddie Augenlinsen etwa in Mittelstellung zubringen.)
2. Legen Sie ein kontrastreiches Präparat(am besten den Blutausstrich) auf den Ob-jekttisch. Blicken Sie mit einem Auge in dasOkular mit der Messplatte und fokussierenSie auf das Objekt. Blicken Sie nun mit demanderen Auge in das andere Okular undstellen Sie, falls erforderlich, die Bildschärfedurch Verdrehen der Augenlinse nach.
3. Schließen Sie die Leuchtfeldblende soweit, dass sie im Sehfeld (zunächst unscharf)erscheint. Stellen sie dann den Kondensor soein, dass die Leuchtfeldblende scharf in derBildebene abgebildet wird. Mit Hilfe derbeiden Stellschrauben am Kondensor wirddas Bild der Leuchtfeldblende zentriert.Danach öffnen Sie die Leuchtfeldblende soweit, dass sie gerade aus dem Bildfeld ver-schwindet.
4. Entfernen Sie ein Okular. Im Tubus siehtman nun das Bild der Aperturblende in derhinteren Brennebene des Objektivs. (Dasergibt sich aus Abb.3: Strahlen, die voneinem Punkt der Aperturblende ausgehen,sind im Objekt parallel und werden folglich
in der hinteren Brennebene des Objektivsvereinigt.) Schließen Sie nun die Apertur-blende so weit, bis ihr Rand gerade sichtbarwird. Jetzt ist die Beleuchtungsapertur gleichder Objektivapertur. Eine weitere Verringerung der Beleuchtungs-apertur kann - objektabhängig - erforderlichsein, um einen ausreichenden Kontrast zuerzielen. Ein zu großer Durchmesser derAperturblende führt zu überschüssigem Licht,das nicht zur Abbildung beiträgt, jedoch alsStreulicht den Kontrast vermindert. Ein zukleiner Durchmesser vermindert das Auflö-sungsvermögen. In den meisten Fällen erzieltman einen Kompromiss zwischen maxima-lem Auflösungsvermögen und maximalemKontrast durch folgende Faustregel:Die Beleuchtungsapertur soll etwa 2/3 der
Objektivapertur betragen.
Die Einstellung der Beleuchtung nach demKÖHLERschen Prinzip ist damit für diesesObjektiv gewährleistet. Die Zentrierung des Kondensors und dieEinstellung von Leuchtfeld- und Apertur-blende müssen nach jedem Objektivwechselwiederholt werden. Meist reicht es auch aus,den Kondensor nur für das stärkste Objektivzu zentrieren, für alle anderen Objektivestimmt die Zentrierung dann ungefähr.Die Bildhelligkeit sollte grundsätzlich nichtmit Hilfe der Aperturblende sondern mit demRegler (über dem Schalter) oder mit Hilfevon Graufiltern eingestellt werden.
4.2 Zur Bestimmung der Abbildungsmaßstä-be der Objektive wird das Objektmikrometerauf den Objekttisch gelegt und das Mikro-skop darauf scharf gestellt. Bei richtigerEinstellung ist keine Parallaxe (Verschiebungzwischen Okular- und Objektmikrometer beiÄnderung des Blickwinkels) mehr zu sehen.Beim Vergleich der beiden Skalen wird einemöglichst große Gegenstandsgröße G auf derObjektmikrometerskale gewählt; die da-zugehörige Bildgröße B ist auf der Okularmi-krometerskale abzulesen. Diese Messungensind für alle vier vorhandenen Objektivedurchzuführen.

Optik und Strahlung O 4 Mikroskop
64
4.3 Ein Phasenkontrastobjektiv wird in denStrahlengang eingeschwenkt; das Okularmi-krometer durch das Hilfsmikroskop ersetztund dieses auf das Phasenplättchen scharfeingestellt. Das Hilfsmikroskop verändert denStrahlengang im Mikroskop so, dass man indie hintere Brennebene des Objektivs sieht.Am Blendenrevolver ist die zum Objektivpassende Ringblende (Ph1 bzw. Ph2) auszu-wählen, die Aperturblende muss dabei vollgeöffnet sein.Es wird kontrolliert, ob beim Einrasten desBlendenrevolvers das Phasenplättchen dieRingblende vollständig überdeckt. Gegebe-nenfalls ist die Blende zu justieren. Die dazuerforderlichen Justierschlüssel hat der zu-ständige Assistent. Diese Kontrolle ist für alle drei Phasenkon-trastobjektive durchzuführen. Setzt mandanach wieder das Okular anstelle des Hilfs-mikroskops ein, ist das Phasenkontrastmikro-skop arbeitsfähig.
4.4 Bei der mikroskopischen Untersuchungvon Präparaten beginnt man grundsätzlichmit der schwächsten Vergrößerung. Ist einObjekt gefunden und fokussiert, werden biszur erforderlichen Vergrößerung schrittweisestärkere Objektive eingesetzt.
Um die verschiedenen Kontrastierungs-verfahren kennen zulernen, werden folgendeUntersuchungen vorgeschlagen (der Assistentlegt fest, welche durchzuführen sind):
Vergessen Sie dabei nicht, alle Beobach-tungen zu protokollieren!
1. Der Blutausstrich ist im Hellfeld zu be-trachten. In der stärksten Vergrößerung wer-den die Durchmesser von 10 Erythrozytenbestimmt. Dabei sollten einzelne, flachliegende Erythrozyten ausgewählt werden. Es werden die Bildgrößen in Skalenteilen(Skt.) gemessen. Später werden daraus mitHilfe des Abbildungsmaßstabes die Objekt-größen berechnet.
2. Im Diatomeen-Präparat sind an mindestenszwei Objekten die Größen der Feinstrukturen
unter Verwendung des Objektivs mit derstärksten Vergrößerung zu messen bzw. zuschätzen. Suchen Sie die kleinsten nocherkennbaren Strukturen und schätzen Sie ausderen Größe das Auflösungsvermögen desObjektivs. Beobachten (und protokollieren)Sie, wie sich die Änderung der Beleuchtungs-apertur auf Kontrast und Auflösungsver-mögen auswirkt. Betrachten Sie das Präparatauch im Dunkelfeld. (siehe Hinweis unter 5.)
3. Am Phasenkontrast-Präparat (junge Mausoder Kaninchen-Geschmacksknospen, ge-kennzeichnet mit PHAKO) ist mit und ohnePhasenkontrast zu untersuchen, welcheStrukturdetails erkennbar sind. (z.B. be-stimmte Organe, Zellen, Zellkerne)
4. Der gefärbte Schnitt einer Sehne ist imHellfeld und mit Polarisationskontrast zubetrachten. Dazu wird das Polarisationsfilterauf die Leuchtfeldblende gelegt und sogedreht, dass das Gesichtsfeld ohne Präparatbei maximaler Beleuchtungsstärke dunkel ist.Die Aperturblende muss dazu etwas ge-schlossen werden. Die Doppelbrechung imSehnengewebe wird durch Kollagen hervor-gerufen.
5. Der gefärbte Knochenschnitt ist im Hell-feld, Dunkelfeld und Polarisations-kontrastzu untersuchen. Für Dunkelfeldkontrast ist maximale Be-leuchtung erforderlich, die Aperturblendemuss vollständig geöffnet sein. Falls dasGesichtsfeld (mit Präparat) ungleichmäßigund/oder farbig ausgeleuchtet erscheint, mussder Kondensor etwas verstellt werden. (Dierichtige Zentrierung des Kondensors undEinstellung der Leuchtfeldblende entspr. 4.1wird immer vorausgesetzt.)
6. Ein Haar wird mit einem Tropfen Wasserauf einen Objektträger gelegt und mit einemDeckglas abgedeckt. (Ein Deckglas derStandarddicke 0,17 mm ist erforderlich beientsprechend korrigierten Objektiven, die mit/0.17 gekennzeichnet sind.)Der Durchmesser des Haares ist an 10 ver-schiedenen Stellen zu bestimmen.

Optik und Strahlung O 10 Polarimeter und Refraktometer
65
5 Auswertung
5.2 Die Abbildungsmaßstäbe der Objektivewerden nach Gl. (3) berechnet und mit denAngaben auf den Objektiven verglichen.
5.4 Aus den 10 Einzelmessungen der Ery-throzyten und des Haares sind Mittelwert undStandardabweichung und daraus m.H. desAbbildungsmaßstabes nach Gl. (3) die Grö-ßen in µm zu berechnen. Welche Bedeutunghat hier die Standardabweichung?Aus der numerischen Apertur des verwende-ten Objektivs ist nach (4) und nach (5) dastheoretische Auflösungsvermögen zu be-rechnen und mit dem am Diatomeen-Präparatabgeschätzten Wert zu vergleichen.
6 Literaturangaben
Gerlach, D.: Das.Lichtmikroskop, Georg
Thieme Verlag, 1985
Trautwein, Kreibig, Oberhausen: Physik fürMediziner, Biologen, Pharmazeuten. deGruyter; Berlin 1984
7 Kontrollfragen
7.1 Wodurch werden die Vergrößerung unddas Auflösungsvermögen eines Mikroskopsbestimmt?
7.2 Was versteht man unter einer förderli-chen Vergrößerung, und wie groß ist sie beieinem Objektiv mit Ölimmersion (n@sin α =1,3 ; λ=500nm)?
7.3 Wozu dienen Leuchtfeld- und Apertur-blende?
7.4 Welche Kontrastierungsverfahren gibtes und wofür setzt man sie ein?
1 Aufgabenstellung
1.1 Die Konzentration einer wässrigenZuckerlösung (Saccharose) ist mit demPolarimeter zu bestimmen.
1.2 Die Brechzahl von Glycerol-Wasser-Gemischen ist in Abhängigkeit von derKonzentration mit dem Refraktometer zubestimmen.
1.3 Von einem vorgegebenen Glycerol-Wasser-Gemisch ist die Konzentration zuermitteln.
2 Grundlagen
Lichtwellen gehören zu den elektromagne-tischen Wellen. Jeder Lichtstrahl besteht
dabei aus einer Vielzahl einzelner Wellenzü-ge. Ein solcher Wellenzug besteht aus einemelektrischem und damit verkoppelten magne-tischem Wechselfeld, deren Lage jeweilstransversal (senkrecht) zur Ausbreitungs-richtung ist. Elektrische und magnetischeFeldstärke schließen einen Winkel von 90°ein (Abb.1).Besteht ein Lichtstrahl aus natürlichem(unpolarisiertem) Licht, können die elektri-schen und magnetischen Felder in beliebige,dabei aber immer zur Ausbreitungsrichtungtransversale Richtungen schwingen. Lichtheißt linear polarisiert, wenn alle elektrischenFelder nur noch in einer transversalen Rich-tung schwingen. Die zugehörigen magneti-schen Felder liegen dann senkrecht dazu auchnur noch in einer Richtung.
Polarimeter und Refraktometer O 10

Optik und Strahlung O 10 Polarimeter und Refraktometer
66
ncc
= 0 . (2)
ϕ = ⋅ ⋅k l c . (1)
n n1 2⋅ = ⋅sin sin .α β (3)
Die Richtung der elektrischen Feldstärke-vektoren heißt Schwingungsrichtung oderPolarisationsrichtung des Lichtes.
2.1 Aus normalem, unpolarisiertem Lichterzeugt man linear polarisiertes Licht durchReflexion an einem durchsichtigen Stoffunter dem BREWSTERschen Winkel, durchDoppelbrechung (NICOLsches Prisma, sieheLiteraturangaben) oder mit Hilfe von Polari-sationsfiltern auf der Basis dichroitischerFolien.Unter Dichroismus versteht man die Eigen-schaft mancher doppelbrechender Stoffe,einen der beiden senkrecht zueinander linearpolarisierten Teilstrahlen zusätzlich stark zuabsorbieren, während der andere fast un-geschwächt hindurchgeht. Dichroismus lässtsich künstlich erzeugen in Polymerfilmen,deren Makromoleküle parallel ausgerichtetsind (Formdoppel-brechung, siehe VersuchO4). Damit lassen sich preiswerte Polarisa-tionsfilter herstellen, die einen Polarisations-grad von bis zu 99% aufweisen.
Optisch aktive Substanzen sind Stoffe, diebeim Durchgang von linear polarisiertemLicht dessen Schwingungsrichtung drehen.Diese optische Aktivität kann hervorgerufenwerden durch asymmetrische Molekülstruk-turen (z.B. bei asymmetrischen Kohlenstoff-atomen) oder durch die schraubenförmigeAnordnung von Gitterbausteinen. MancheSubstanzen, von denen zueinander spiegel-bildliche Isomere existieren (chirale Verbin-dungen), gibt es in einer rechtsdrehenden (+)und einer linksdrehenden (!) Variante.Beispiele sind die verschiedenen Zucker und
Milchsäure.Bei Lösungen von optisch aktiven Sub-stanzen hängt der Drehwinkel von der Art desStoffes, von der Dicke der durchstrahltenSchicht (Länge l des Polarimeterrohrs), vonder Konzentration c und von der Wellenlängeλ ab. Diese Wellenlängenabhängigkeit nenntman Rotationsdispersion. Blaues Licht wirdstärker gedreht als rotes.Für den Drehwinkel φ gilt:
Die Materialgröße k heißt spezifische Dre-hung oder spezifisches Drehvermögen, siehängt von der Wellenlänge ab.
2.2 Die Brechzahl n eines Stoffes ist de-finiert als das Verhältnis der Lichtgeschwin-digkeit im Vakuum c0 zur Lichtgeschwindig-keit c im Stoff:
Sie ist abhängig vom Material und von derWellenlänge λ des Lichtes (Dispersion). Ineiner Lösung ist die Brechzahl von derKonzentration (d. h. vom Mischungsverhält-nis) abhängig. Die Messung der Brechzahleignet sich deshalb in manchen Fällen fürgenaue und einfach durchführbare Konzen-trationsmessungen. Anwendungen sind z. B.die Bestimmung des Gesamteiweißgehaltesim Blutserum in der Medizin oder die Be-stimmung des Zuckergehaltes im Traubensaftin der Winzerei.
Beim Übergang des Lichtes von einemoptisch dünneren Medium mit der Brechzahln1 zu einem optisch dichteren Medium mitder Brechzahl n2 (n2 > n1) werden die Licht-strahlen zum Einfallslot hin gebrochen(Abb.1). Bezeichnet man den Einfallswinkelmit α und den Brechungswinkel mit β, solautet das Brechungsgesetz:
Für den größtmöglichen Einfallswinkelα = 90° (streifender Lichteinfall) ergibt sichein maximaler Brechungswinkel βGr.
Abb.1: Lage des elektrischen und magneti-schen Feldstärkevektors für einen Wellenzug

Optik und Strahlung O 10 Polarimeter und Refraktometer
67
sin .βGr
n
n= 1
2
(4)
Den Strahlengang in Abb.2 kann man auchumkehren: vom optisch dichteren Medium(n2) zum optisch dünneren Medium (n1),Einfallswinkel β, Ausfallswinkel α. Fürβ > βGr wird kein Licht in das optisch dünnereMedium gebrochen, denn das Brechungs-gesetz (3) kann nicht erfüllt werden. Stattdessen wird das Licht an der Grenzflächevollständig reflektiert. βGr heißt deshalbGrenzwinkel der Totalreflexion. Aus dem Brechungsgesetz ergibt sich:
Bei bekannter Brechzahl n2 (Messprisma desRefraktometers) kann somit durch die Mes-sung des Grenzwinkels βGr die Brechzahl n1
des anderen Mediums (der Messflüssigkeit)bestimmt werden.Zur Messung des Grenzwinkels beleuchtetman die Grenzfläche durch eine Mattscheibemit rauer Oberfläche (siehe Abb.3). DieLichtstrahlen treffen dann unter allen mögli-chen Einfallswinkeln zwischen 0E und 90Eauf die Grenzfläche. Somit können alleBrechungswinkel zwischen 0E und βGr auf-treten. Wenn man durch ein Fernrohr unterdem Winkel βGr auf die Grenzfläche blickt,sieht man eine Hell-Dunkel-Grenze; dieselässt sich leicht ausmessen.
3 Versuchsaufbau
3.0 Geräte:- Polarimeter mit Natrium-Spektralleuchte- Polarimeterrohr- Flasche mit Zuckerlösung- Refraktometer nach ABBE
- 2 Büretten mit Glycerol und aqua dest.- diverse Glasgeräte- Fläschchen mit Glycerol-Wasser-Gemisch unbekannter Konzentration
3.1 Das Polarimeter besteht aus einer mono-chromatischen Lichtquelle (Na-D-Licht,λ = 589,3 nm), einem Polarimeterrohr, einemPolarisator und einem drehbaren Analysatormit Winkelmesseinrichtung. Stehen die Schwingungsrichtungen (Durch-lassrichtungen) von Polarisator und Analysa-tor senkrecht zueinander (gekreuzt), so ist dasGesichtsfeld im Polarimeter dunkel.Bringt man dann zwischen Polarisator undAnalysator das Polarimeterrohr mit derLösung des optisch aktiven Stoffes (Zucker-lösung), so wird das Gesichtsfeld aufgehellt,da die Schwingungsrichtung des linearpolarisierten Lichtes um den Winkel φ ge-dreht wurde. Dreht man den Analysator umdiesen Winkel φ nach, so ist das Gesichtsfeldwieder dunkel. Auf diese Weise lässt sich derDrehwinkel φ messen.Da die Einstellung des Gesichtsfeldes aufmaximale Dunkelheit oder Helligkeit ohneVergleich sehr ungenau ist, benutzt man imPolarimeter ein dreigeteiltes Gesichtsfeld(siehe Abb.2). Dazu besteht der Polarisatoraus zwei um 10E gegeneinander versetzten
Abb.2: Strahlengang der Brechung für n2>n-1. Links: allgemeiner Fall, rechts: streifenderLichteinfall.
Abb.3: Strahlengang am ABBE-Refraktometer

Optik und Strahlung O 10 Polarimeter und Refraktometer
68
Abb.4: Dreigeteiltes Gesichtsfeld im Polari-meter
ϕ ϕ ϕ= −m 0 . (5)
Polarisationsfolien (Halbschattenpolari-meter). Während des Messvorganges wird derDrehwinkel des Analysators so eingestellt,dass sich die drei Teile des Gesichtsfeldsnicht unterscheiden (gleiche Resthelligkeit)und die Trennlinien nahezu verschwinden.Bei einer geringen Drehung in eine Richtungmuss der mittlere Teil heller und bei geringerDrehung in die entgegengesetzte Richtungdunkler als die äußeren Teile werden.Zur genauen Ablesung des Winkels für denUmschlagpunkt ist die Winkelmessein-richtung am Analysator mit einem Noniusausgestattet, mit dessen Hilfe der Winkel auf0,05E genau abgelesen werden kann.
3.2 Das Refraktometer besteht im wesentli-chen aus
- einem Beleuchtungsprisma mit einer rauenOberfläche,
- einem Messprisma, dessen Brechzahl n2
größer sein muss als die Brechzahl n1 derMessflüssigkeit,
- einem schwenkbaren Fernrohr mit Winkel-messeinrichtung, wobei auf der gemäß (4)kalibrierten Skale Brechzahlen abgelesenwerden können,
- einer Einrichtung (AMICI-Prismenpaar) zurKompensation der Dispersion (Farbsäume).
Der streifend einfallende Lichtanteil (α.90E)wird unter dem Grenzwinkel βGr gebrochenund im Fernrohr als Grenzlinie zwischenHell- und Dunkelfeld beobachtet.
4 Versuchsdurchführung
4.1 Zu Beginn wird die Na-Spektralleuchteeingeschaltet; nach ca. 5 min erreicht dieLampe ihre maximale Helligkeit.Die Nullstellung des Polarimeters wirdbestimmt, indem man ohne Polarimeterrohrdie Einstellung des Gesichtsfeldes wie in 3.1beschrieben vornimmt (Umschlagpunkt ein-stellen) und den dazugehörigen Winkel φ0
abliest. Die Messung ist 5 mal zu wiederho-len.Das Polarimeterrohr soll möglichst blasenfreimit Zuckerlösung gefüllt werden. Dazu hältman das Rohr senkrecht und füllt es voll-ständig. Das Glasfenster (ohne Schraubkap-pe) wird seitlich über die Öffnung geschobenund überflüssige Lösung mit Zellstoff ent-fernt. Danach wird das Rohr (nicht zu fest)zugeschraubt und in das Polarimeter einge-legt. Eine verbleibende kleine Blase kann indie Verdickung des Polarimeterrohres ge-bracht werden. Nun wird der Analysator nachgedreht underneut der Umschlagpunkt eingestellt; derzugehörige Winkel φm ist abzulesen. Auchdiese Messungen werden 5 mal durchgeführt.Der Drehwinkel φ ergibt sich dann als Diffe-renz der Mittelwerte von φm und φ0:
Die Länge des Polarimeterrohres ist zumessen.Nach den Messungen wird die verwendeteZuckerlösung in die Flasche zurückgefülltund das Polarimeterrohr mit Wasser ausge-spült; das Rohr ist offen zu lassen.
4.2 Die beiden Prismen des Refraktometersmüssen sich auf der rechten Seite befinden,die Lichteintrittsöffnung der Skalenbeleuch-tung (links oben) muss aufgeklappt sein.Die Prismen werden auseinander geklappt,das Beleuchtungsprisma mit der rauen Ober-fläche (unten) wird etwa waagerecht einge-richtet. Nun bringt man 1-2 Tropfen derMessflüssigkeit auf das Beleuchtungsprisma,danach wird das Beleuchtungsprisma auf dasMessprisma geklappt und der Riegel ge-

Optik und Strahlung O 10 Polarimeter und Refraktometer
69
schlossen. Der Beleuchtungsspiegel ist so einzurichten,dass die rechteckige Lichteintrittsöffnungausgeleuchtet wird. Nun wird die Grenzliniezwischen Hell- und Dunkelfeld im Fernrohraufgesucht. Sie wird mit Hilfe des großenRändelknopfes auf die Mitte des Faden-kreuzes eingestellt und die dazugehörigeBrechzahl im Ablesemikroskop abgelesen.Die Scharfstellung auf das Fadenkreuz undauf die Skalen erfolgt durch Drehen derentsprechenden Okularlinsen am Fernrohrbzw. am Ablesemikroskop. Ein evtl. vorhan-dener Farbsaum lässt sich durch Verstellender AMICI-Prismen (kleiner Rändelknopf)weitgehend beseitigen.Es werden die Brechzahlen für folgendeFlüssigkeiten gemessen:
- aqua dest.,
- reines Glycerol,
- 5 Glycerol-Wasser-Gemische: 4:1, 4:2, 4:4, 4:8, 4:16 und
- ein Glycerol-Wasser-Gemisch unbekannterKonzentration.
Für das Gemisch 4:1 nimmt man 4 ml Glyce-rol und 1 ml aqua dest., die weiteren Ge-mische werden durch Verdünnung mit Was-ser hergestellt.Jede Brechzahl ist 5 mal zu messen (jeweilsNeueinstellung mit großem Rändelknopf).Bei Wechsel der Messflüssigkeit und amEnde sind die Prismen sorgfältig zu reinigen.
4.3 Die Brechzahl des Glycerol-WasserGemisches unbekannter Zusammensetzungist ebenfalls fünf mal zu messen.
5 Auswertung
5.1 Die Konzentration c (in g/l) der Zucker-lösung wird nach den Gleichungen (1) und(5) berechnet.Das spezifisches Drehvermögen von Sac-charose (C12H22O11) beträgt bei λ = 589,3 nm
k = 66,456 grad ml dm-1 g-1.Es ist eine Fehlerrechnung durchzuführen.Die Messunsicherheit des Drehwinkels ergibtsich aus der Summe der statistischen Unsi-cherheiten von Nullstellung und Drehwinkel.
5.2 Die Brechzahlen sind in Abhängigkeitvon der Volumenkonzentration grafischdarzustellen.
5.3 Mit Hilfe des Diagramms aus 5.2 wirddie Konzentration des unbekannten Glycerol-Wasser-Gemisches bestimmt. Die Konzen-tration ist in Vol.-% Glycerol anzugeben.
6 Literatur
Grimsehl, E.: Lehrbuch der Physik. Bd 3,B.G. Teubner, Leipzig 1978
Trautwein, A.; Kreibig, U.; Oberhausen, E.:Physik für Mediziner, de Gruyter, Berlin1987.
Kamke, D.; Walcher, W.: Physik für Medizi-ner, B.G. Teubner, Stuttgart, 1994.
7 Kontrollfragen
7.1 Was ist Licht?
7.2 Wie kann linear polarisiertes Lichterzeugt werden?
7.3 Was ist Brechung, wann tritt Totalrefle-xion auf?
7.4 Welche störenden Effekte kann Disper-sion im Refraktometer hervorrufen?

Atom- und Kernphysik O 16 Radioaktivität
70
d dN N t= − ⋅ ⋅λ (2)
N t N t( ) = ⋅− ⋅
0 e λ (3)
d .dNAt
= − (1)
1 Aufgabenstellung
1.1 Die Abhängigkeit der Strahlungsintensi-tät vom Abstand zur Strahlenquelle ist zuermitteln.
1.2 Der Schwächungskoeffizient und dieHalbwertsdicke von Blei für die Gamma-strahlung von Co-60 sind zu bestimmen.
2 Physikalische Grundlagen
Unter Radioaktivität versteht man die Eigen-schaft bestimmter Atomkerne, sich infolgeungünstiger Proton-Neutron-Verhältnissespontan in andere Atomkerne oder Atomker-ne anderen Energieinhalts unter Emissioncharakteristischer radioaktiver Strahlungumzuwandeln. Sie kommt natürlich vor, kannaber auch künstlich erzeugt werden (durchBeschuss stabiler Atomkerne). Abhängig vonder Art der Umwandlung entsteht dabei: α-Strahlung (Heliumkerne, bestehend aus 2Protonen und zwei Neutronen),β!-Strahlung (Elektronen),β+-Strahlung (Positronen),γ-Strahlung (elektromagnetische Wellen miteiner Quantenenergie über 100 keV),Neutronen und (selten) Protonen.γ-Strahlung entsteht als Folge von Kernre-aktionen, bei denen der Kern in einen ange-regten Zustand gelangt ist. Aus diesem kehrter durch Aussendung von γ-Strahlung wiederin den Grundzustand zurück. Auf der Messung radioaktiver Strahlungberuhen viele diagnostische Verfahren derNuklearmedizin. Z. B. können durch Aktivi-tätsmessungen nach Gabe von Radiopharma-ka Stoffwechselprozesse ohne Eingriff in denKörper untersucht werden (zeitlicher Verlauf,Anreicherung in Organen usw.). BildgebendeVerfahren, die auf der Messung von γ-Strah-lung basieren (Szintigraphie, Positronenemis-
sionstomographie), ergänzen die bekanntenRöntgen- und NMR-Methoden.Der therapeutische Einsatz radioaktiverStrahlung dient meist der Bestrahlung vonTumoren.Das im Praktikum untersuchte Co-60 ist einγ-Strahler, der früher in der Medizin für dieTelekobalttherapie verwendet wurde. Co-60entsteht durch Neutronen-Einfang aus Co-59und hat eine Halbwertszeit von 5,27 Jahren.
2.1 In einem radioaktiven Präparat ist dieZahl der sich pro Zeiteinheit umwandelndenAtomkerne proportional zur Zahl der vorhan-denen Kerne. Die (mittlere) Anzahl derKernumwandlungen pro Sekunde nennt manAktivität A:
Da alle Atomkerne mit der gleichen Wahr-scheinlichkeit zerfallen folgt, dass währenddes folgenden Zeitintervalls dt die Zahl derradioaktiven Kerne um
(λ - Zerfallskonstante) abnehmen wird. Fürdie Anzahl N gilt daher das Zerfallsgesetz
mit N0 der Zahl der radioaktiven Atomkernezur Zeit t = 0.
Wenn ein γ-Quant (oder auch ein α- oder β-Teilchen) von einem Strahlungsdetektor wiedem Geiger-Müller-Zählrohr registriert wird,löst es in diesem einen Stromimpuls aus; dieImpulse werden gezählt. Die pro Zeiteinheitregistrierte Impulsanzahl N heißt ImpulsrateI, sie ist proportional zur Strahlungsintensität.Außerdem hängt sie von den Eigenschaftendes Zählrohres und unter Umständen auchvon der Energie der Strahlung ab.Die von einem radioaktiven Präparat erzeugte
Radioaktivität O 16

Atom- und Kernphysik O 16 Radioaktivität
71
I Ix
= ⋅−µ⋅
0 e . (5)
I I IP L= − , (4)
µ µ µ µ µ= + + +S Ph C P . (6)
Impulsrate ergibt sich aus der Differenz derImpulsraten, die mit und ohne Präparatgemessen werden:
wobei IP die Messrate und IL den Leerwert(Nulleffektrate) bezeichnet. Der Nulleffektwird durch kosmische und Umgebungs-strahlung sowie durch Detektorstörimpulsebewirkt.
2.2 Beim Durchgang durch Materie wird dieIntensität der Gamma-Strahlung (gemessenals Impulsrate I) in Abhängigkeit von derDicke x des durchstrahlten Stoffes vermindert(Schwächungsgesetz):
I0 ist die Intensität der einfallenden und I dieIntensität der austretenden Strahlung. µ heißtSchwächungskoeffizient und hängt vom Stoffund von der Energie der Gamma-Quanten ab.Für die Schwächung sind neben der elasti-schen Streuung (µS) drei Absorptionseffektewesentlich: Der Photoeffekt (µPh), die une-lastische Streuung (Comptoneffekt, µC) undder Paarbildung (µP):
Der Einfluss dieser einzelnen Effekte auf denSchwächungskoeffizienten ist energieabhän-gig, wobei die elastische Streuung und derPhotoeffekt bei niedrigen und der Paar-bildungseffekt bei den höchsten Energiendominieren.Unter der Halbwertsdicke d1/2 eines Stoffesversteht man die Schichtdicke, nach der dieIntensität der Strahlung auf die Hälfte abge-sunken ist. Aus der Gleichung (5) folgt damit
3 Versuchsaufbau
3.0 Geräte:- Radioaktives Präparat Co-60 (γ-Strahler
1,17 MeV und 1,33 MeV; A = 74 kBq 2010;t1/2 = 5,27 a)
- Geiger-Müller-Zählrohr
- Digitalzähler- verschieden dicke Absorberplatten aus Blei
3.1 Das radioaktive Präparat Co-60 befindetsich in einer Bohrung in einem Acrylglas-block, der auf einem beweglichen Schlittenbefestigt ist. Zur Messung der Strahlungs-intensität (Impulsrate) wird ein Fensterzähl-rohr verwendet, das ein selbstlöschendesGeiger-Müller-Zählrohr zum Nachweis derGamma-Strahlung ist. Die Impulse werdenvon einem elektronischen Zähler registriert,der gleichzeitig die Betriebsspannung für dasZählrohr liefert. Präparat und Zählrohr sind so auf einerSchiene angeordnet, dass die zu messendeStrahlung vom Präparat durch eine Öffnungim Acrylglasblock auf das Fenster des Zähl-rohres trifft. In den Strahlengang könnenAbsorberplatten gestellt werden. Der Abstandzwischen Präparat und Zählrohr ergibt sichaus dem an der Schiene abgelesenem Abstandzwischen den grauen Schlitten + 10 mm.
4 Versuchsdurchführung
Die Co-60 - Quelle ist ein umschlossenesPräparat mit einer Aktivität unterhalb derFreigrenze laut Strahlenschutzverordnung.Die Strahlenbelastung (effektive Dosis) beider Durchführung des Versuches liegt in derGrößenordnung von 1 µSv, das entspricht0,1% der Dosis bei einer medizinischenRöntgenaufnahme.
Strahlenschutz
Entsprechend der Strahlenschutzver-ordnung ist jede Bestrahlung, auch un-terhalb der zulässigen Grenzwerte, zuminimieren. Abstand ist der beste Strah-lenschutz! Deshalb: Halten Sie das Prä-parat nicht unnötig in der Hand. HaltenSie beim Experimentieren einen Abstandvon etwa 0,5 m zum Präparat ein.

Atom- und Kernphysik O 16 Radioaktivität
72
ln ln
lg lg lg .
I I x
I I x
= − ⋅
= − ⋅ ⋅
0
0
µ
µ
oder
e
4.1 Für das Zählrohr ist eine Betriebs-spannung von 480 V einzustellen. Die Mess-zeit beträgt 60 s. Alle Messungen werdenjeweils fünf mal durchgeführt.Zu Beginn ist der Nulleffekt IL durch Mes-sung ohne radioaktives Präparat zu bestim-men.Zur Ermittlung der Abhängigkeit der Impuls-rate vom Abstand r wird das Präparat in denAbständen 40; 50; 70; 100; 140; 190 und 250mm zum Zählrohr positioniert und die zu-gehörigen Impulsraten werden bestimmt.
4.2 Zur Bestimmung des Schwächungs-koeffizienten µ für Blei wird das Präparat ineinem Abstand von 70 mm vom Zählrohrpositioniert und zwischen ihnen die Absor-berhalterung eingesetzt. Bleiplatten ver-schiedener Dicken x werden in den Strahlen-gang gebracht und die dazugehörigen Impuls-raten gemessen. Die Messungen sind für x =1; 2; 5; 10; 20; 30 mm je fünfmal durch-zuführen. Die Messung für x = 0 wurdebereits in 4.1 durchgeführt.
5 Auswertung
Aus den jeweils fünf Einzelmessungen in 4.1und 4.2 wird der Mittelwert gebildet. AlleWerte für die Impulsraten werden durchAbzug des Nulleffektes korrigiert.
5.1 Zur Bestimmung des AbstandsgesetzesI = I(r) werden die Impulsraten in Abhängig-keit vom Abstand r doppelt logarithmischdargestellt. Aus dem Anstieg ist der Exponentdes Abstandsgesetzes zu bestimmen. Verglei-chen Sie diesen mit dem theoretischen Wert!
5.2 Zur Bestimmung des Schwächungs-koeffizienten µ für Blei werden die Impuls-raten in Abhängigkeit von den Absorberdi-cken x graphisch dargestellt. Es ist einehalblogarithmische Darstellung zu wählen
(Zählrate logarithmisch als Ordinate, Dickelinear als Abszisse). Alternativ kann ln(I)berechnet und linear (d. h. auf normalemMillimeterpapier) dargestellt werden.Aus (5) erhält man durch Logarithmieren:
Der Schwächungskoeffizient µ ergibt sichfolglich aus dem Anstieg der Kurve. Die Halbwertsdicke von Blei ist nach Gl. (7)zu berechnen.
6 Literaturangaben
Haas, U.: Physik für Pharmazeuten undMediziner. WVG Stuttgart, 2002
Beier, W., Pliquett, F., Physik, Leipzig 1987
Fercher, A.F.: Medizinische Physik. SpringerVerlag 1992
7 Kontrollfragen
7.1 Was ist der Unterschied zwischenRöntgen- und γ-Strahlung?
7.2 Was versteht man unter den Begriffen„Halbwertsdicke“ und „Halbwertszeit“?
7.3 Nach welchem Gesetz nimmt die Strah-lungsintensität mit der Entfernung ab?
7.4 Welcher Zusammenhang besteht zwi-schen der Härte und der Durchdringungs-fähigkeit der γ-Strahlung?

Optik und Atomphysik O 20 Spektralphotometer
73
I I e d= −
0µ , (2)
I I e n c d= − ⋅ ⋅0
ε . (4)
TII
=0
. (1)
EI
Ic dn n= = ⋅ ⋅ln 0 ε . (5)
EI
Ic d= = ⋅ ⋅lg .0 ε (6)
ET
= lg .1
(7)
µ ε= ⋅n c . (3)
TI
IT Tges = = ⋅2
01 2 . (8)
1 Aufgabenstellung
Bringen Sie bitte zum Versuch zwei kleine,frische grüne Blätter mit!
1.1 Die Apparatefunktion eines Spektral-photometers ist aufzunehmen, das Gerät istzu kalibrieren.
1.2 Ein alkoholischer Chlorophyllextrakt istherzustellen und seine Extinktion bei ver-schiedenen Konzentrationen im Spektral-bereich von 400…750 nm zu bestimmen.
1.3 Die Konzentration von Chlorophyll aund Chlorophyll b sowie das Verhältnisbeider Konzentrationen sind zu ermitteln.
2 Grundlagen
2.1 Beim Durchgang durch Materie wirdLicht durch Absorption und durch Streuung(Brechung und Reflexion an kleinen Teil-chen) geschwächt. An Grenzflächen wird dasLicht partiell reflektiert. Alle drei Prozessesind abhängig von der Wellenlänge.Das Verhältnis der durch eine Probe hin-durchgehenden Lichtintensität I zu ein-fallender Intensität I0 wird als Transmissions-grad oder kurz Transmission T bezeichnet:
Die Abnahme der Intensität durch Absorptionkann in Abhängigkeit von der durchstrahltenMaterialdicke d mathematisch beschriebenwerden durch
wobei µ als Absorptionskoeffizient bezeich-net wird; siehe auch Versuch O16, Gl.(5).Wird in einer Lösung das Licht vom gelöstenStoff absorbiert, so ist µ proportional zudessen Konzentration c:
εn heißt (natürlicher) Extinktionskoeffizientund hängt von der Substanz und von derWellenlänge ab. Aus (2) und (3) ergibt sich das LAMBERT-BEERsche Absorptionsgesetz
Als (natürliche) Extinktion En bezeichnetman
Die Konzentration einer Lösung lässt sichdamit über die Extinktion bestimmen.
In der Praxis wird meist nicht mit dem natür-lichen, sondern mit dem dekadischen Loga-rithmus gerechnet, so auch bei dem hierverwendeten Spektralphotometer. Die Bezie-hung für die dekadische Extinktion E lautet:
Zwischen dem spezifischen dekadischen (ε)und dem spezifischen natürlichen Extink-tionskoeffizient (εn) besteht die Beziehung ε = εn / ln 10 = 0,4343 @εn
Durch Vergleich von (1) und (6) findet mandie wichtige Gleichung
Wir betrachten nun den Fall, dass das Lichterst durch eine Lösung des Stoffes 1 unddanach durch eine Lösung des Stoffes 2geschwächt werde. Die eingestrahlte Intensi-tät sei I0, die Intensität nach der 1. Lösung I1
und nach der 2. Lösung I2. Dann ist nach (1)die Transmission der 1. Lösung T1 = I1/I0, dieder 2. Lösung T2 = I2/I1 und die Trans-mission der gesamten Anordnung
Spektralphotometer O 20

Optik und Atomphysik O 20 Spektralphotometer
74
400 500 600 7000,0
0,2
0,4
0,6
0,8
1,0
Chlorophyll b
Extin
ktio
n
Wellenlänge λ / nm
Chlorophyll a
Abb.1: Extinktion von Chlorophyll a undChlorophyll b
lg lg lg
.
1 1 1
1 2 1 2
1 2
T T T T
E E Eges
⋅= +
= +
(9)
E c d c d
E c d c d
a a b b
a a b b
649 649 649
665 665 665
= ⋅ ⋅ + ⋅ ⋅
= ⋅ ⋅ + ⋅ ⋅
ε ε
ε ε
, ,
, ,
(10)
Für die Extinktionen gilt nach (7):
Diese Betrachtung gilt auch, wenn sich diebeiden Stoffe gemeinsam in einer Lösungbefinden (sofern sie sich nicht gegenseitigbeeinflussen). Zusammengefasst gilt deshalbfolgender Satz:
Wird Licht durch mehrere verschiedeneProzesse geschwächt, so multiplizieren sichdie Transmissionen und addieren sich dieExtinktionen der Einzelprozesse.
Im wissenschaftlichen Alltag wird die Ex-tinktion (englisch: absorbance) oft fälschlich“Absorption” genannt. Dies sollte im Inter-esse einer eindeutigen Sprache vermiedenwerden. Die Absorption bzw. der Absorp-tionsgrad A bezeichnet die Größe (1! T ).
2.2 Die photometrische Bestimmung desChlorophyllgehaltes in Biomasse ist eineStandardmethode in der Biologie. Dafür istunter anderem die vollständige und schonen-de Extraktion des Chlorophylls erforderlich,was im Rahmen des Physikpraktikums nichtmöglich ist. Hier liegt der Schwerpunkt aufder spektroskopischen Methode. Es soll dieKonzentration in einem selbst hergestellten
Chlorophyll-Rohextrakt gemessen werden.
Dieser enthält hauptsächlich Chlorophyll a(Chl.a) und Chlorophyll b (Chl.b), derenExtinktionsspektren bekannt sind (sieheAbb.1).In Ethanol liegt das Extinktionsmaximumvon Chl.a bei 665 nm und das von Chl.b bei649 nm. Die Extinktion der Rohextraktlösungist entsprechend Gl. (9) die Summe aus denAnteilen beider Chlorophylle. Misst man dieExtinktion in den beiden Maxima, so kannmit Hilfe der Gleichungen
und den vier Extinktionskoeffizienten fürChl.a und Chl.b bei 649 nm und 665 nm dieKonzentration von Chl.a und Chl.b berechnetwerden.
3 Versuchsaufbau
3.0 Geräte- Spektralphotometer NOVASPEC II- Computer mit Spektrometersoftware- 2 Küvetten (durchstrahlte Dicke: 10 mm)- Reibeschale, Pistill, Quarzsand- Ethanol 99% mit 1% MEK vergällt- 2 Bechergläser 50 ml und 100 ml- 1 Mischgefäß 4 ml- Filterpapier- Pasteurpipetten- Mikroliterpipette 750 µl
3.1 Die Abb.2 zeigt den optischen Aufbaudes Spektralphotometers NOVASPEC II. DieLichtquelle LQ sendet weißes Licht in denEintrittsspalt SP des Monochromators. Dieserbesteht aus dem Beugungsgitter G, demKondensorspiegel S2 und KollimatorspiegelS3, sowie dem Filter F zum Ausblendenhöherer Beugungsordnungen. Der Austritts-spalt wird durch die Lichteintrittsöffnung amKüvettenhalter Kü gebildet. Durch Drehendes Gitters wird Licht definierter Wellenlän-ge (zwischen 325 und 900 nm) durch dieK ü v e t

Optik und Atomphysik O 20 Spektralphotometer
75
Abb.2: Aufbau des Spektralphotometers
TTT
P
A
( )( )( )
.λλλ
= (11)
E E EP A( ) ( ) ( ) .λ λ λ= − (12)
te geschickt, dessen Intensität mit dem Photo-detektor PD gemessen wird.Das Gerät kann wahlweise Extinktion oderTransmission anzeigen (mit mode-Taste abs
bzw. trans auswählen oder im Programm dieBetriebsart umschalten).Nach dem Einschalten wird bei leeremProbenraum als “Transmission” lediglich einzur gemessenen Intensität proportionalerWert angezeigt, der von der Wellenlängeabhängt - die Apparatefunktion TA(λ). Sieenthält noch die Einflüsse aller Gerätekompo-nenten (Lampenspektrum, Reflexionsver-mögen des Gitters und der Spiegel, Filter-Absorption, Sensorkennlinie). Mit einer Probe misst man das SpektrumTP(λ). Daraus berechnet sich die (wellenlän-genabhängige) Transmission der Probeentsprechend (1) nach
Für die Extinktion gilt entsprechend:
Diese Rechnung kann das Gerät selbst durch-führen. Um die Transmission bzw. Extinktioneines gelösten Stoffes direkt zu messen, gehtman wie folgt vor:
- eine Küvette mit reinem Lösungsmittel inden Probenraum bringen,
- wenn nur bei einer Wellenlänge gemessenwerden soll: Wellenlänge einstellen undTaste “set ref” drücken,
- wenn ein Spektrum gemessen werden soll:Funktion “base line” starten bzw. im Pro-gramm Einstellungen/Basislinie auswählen.
Hierdurch wird die Apparatefunktion gemes-sen und anschließend entsprechend (11) bzw.(12) auf diese normiert; die Anzeige beträgtdanach T = 100,0 % bzw. E = 0,000.
4 Versuchsdurchführung
Zum Umgang mit den Küvetten:- Die Oberflächen im optischen Strahlengang
müssen klar sein; zerkratzte Küvetten sindauszusondern.
- Küvetten nicht an den optischen Flächenberühren!
- nasse Außenflächen vorsichtig trocknen!- Küvetten nur bis zur Hälfte füllen (1 ml)!
Vor Beginn der Messungen sollte das Gerätmindestens 5 Minuten eingeschaltet sein(Aufwärmzeit). Alle Messungen werden imSpektralbereich von 400 nm bis 750 nm miteiner Schrittweite von 2 nm durchgeführt.
4.1 Starten Sie das Programm “Spektro-meter” erst nach Einschalten des Mess-gerätes. Zeichnen Sie bei leerem Probenraumim Modus Transmission ein Spektrum auf.Bringen Sie eine Küvette mit dem Lösungs-mittel Ethanol in den Strahlengang undwiederholen Sie die Messung.Kalibrieren Sie nun das Messgerät, indem Sieeine Basislinie erstellen, und zeichnen Sieanschließend ein drittes Spektrum auf. DurchUmschalten der Betriebsart können dieTransmissionsspektren in Extinktionsspek-tren transformiert werden. Sichern Sie dieErgebnisse durch Speichern oder Ausdru-cken!
4.2 Zur Herstellung des Chlorophyll-Roh-extraktes werden etwa 0,2 g Blattmasse in derReibeschale mit etwas Quarzsand und einigenml Ethanol zerrieben. Dabei werden die

Optik und Atomphysik O 20 Spektralphotometer
76
( )
( )
c E E
c E E
a
b
= ⋅ − ⋅
= ⋅ − ⋅
13 53 5 20
22 43 7 07
665 649
649 665
, ,
, ,
mg / l
mg / l(13)
Zellen aufgebrochen und das Clorophyll gehtin Lösung. Fügen Sie anschließend weiteresEthanol hinzu, insgesamt etwa 10 ml. FaltenSie ein Rundfilter zu einer Tüte und filtrierenSie den Extrakt damit in das kleine Becher-glas. Füllen Sie 1 ml der Lösung in eine Küvetteund prüfen Sie die Transmission bei 665 nm.Falls diese kleiner als 2 % ist, verdünnen Siedie Lösung im Becherglas schrittweise mitjeweils 1 ml Ethanol und überprüfen Sieerneut. (Wenn die Transmission zu gering ist,wird die Messung sehr ungenau.)Messen Sie nun ein vollständiges Extink-tionsspektrum. Diese Messung ist mit den drei Verdünnun-gen 75 %, 50 % und 25 % zu wiederholen.Zur Herstellung der Verdünnungen sind dieauf 750 µl eingestellte Pipette und das Misch-gefäß zu verwenden. Küvette und Misch-gefäß sind nach jedem Schritt mit wenigEthanol zu spülen und zu trocknen. Dasgrößere Becherglas dient als Restbehälterzum Verwerfen gebrauchter Lösungen.
5 Auswertung
5.1 Die Spektren aus 4.1 sind zu diskutieren.
5.2 Stellen Sie die Wellenlängen der vierdeutlich erkennbaren Maxima in den Spek-tren fest und ordnen Sie diese den beidenFarbstoffen Chl.a und Chl.b zu.Die Extinktion in allen vier Maxima ist inAbhängigkeit von der Konzentration gra-phisch darzustellen und an Hand von Gl. (6)zu diskutieren.
5.3 Löst man das Gleichungssystem (10)
nach den Konzentrationen ca und cb auf, soerhält man
(Zahlenangaben nach R. J. Ritchie 2006).Bestimmen Sie für alle Verdünnungen ausden Spektren die Extinktion bei den Wellen-längen 649 nm und 665 nm. Berechnen Siemit Gl. (13) die Konzentrationen von Chloro-phyll a und Chlorophyll b sowie das Verhält-nis beider Konzentrationen.
6 Literatur
Hellenthal, W., Physik, Thieme 1988
Trautwein, Kreibig, Oberhausen: Physik fürMediziner, Biologen, Pharmazeuten. W. deGruyter, Berlin 1987
R. J. Ritchie, Photosynth. Res. (2006) 89 pp27-41
7 Kontrollfragen
7.1 Wie kann man mit einem Photometerdie Konzentration eines gelösten Stoffes ineiner Lösung unabhängig von weiterengelösten Stoffen bestimmen?
7.2 Warum erscheinen uns Gegenstände beiBeleuchtung mit weißem Licht farbig?
7.3 Wie groß ist die Transmission bei einerExtinktion von E = 0, E = 1 und E = 2?

Atom- und Kernphysik O 22 Röntgenverfahren
77
E e Um
v Em
vePh
e= ⋅ = = +2 21
222 (1)
E h f hc
Ph = ⋅ = ⋅λ
. (2)
1 Aufgabenstellung
1.1 Messung von Röntgenemissionsspektreneiner Molybdän-Anode mit Hilfe eines LiF-Kristalls und Bestimmung der maximalenQuantenenergie der Röntgenstrahlung inAbhängigkeit von der Anodenspannung.
1.2 Bestimmung der Ionendosisleistung derRöntgenröhre.
1.3 Durchleuchtung und Interpretationbiologischer Objekte.
2 Physikalische Grundlagen
Bei der diagnostischen Anwendung vonRöntgenstrahlen stehen photographischeVerfahren bei der Anfertigung von Röntgen-aufnahmen noch im Vordergrund. Abgebildetwird das unterschiedliche Absorptionsver-mögen verschiedener Gewebe. Diese Unter-schiede können durch die Applikation vonKontrastmitteln verstärkt werden. EineKontrasterhöhung bei geringen Differenzenin der Absorption erhält man mit computerto-mographischen Verfahren.In der medizinischen Forschung kommt derRöntgenstrukturanalyse hervorragendeBedeutung zu. Mit ihrer Hilfe konnte dieTertiärstruktur von Eiweißen und Nuklein-säuren mit einer hohen räumlichen Auflösung(< 0,3 nm) bestimmt werden.
2.1 Als Röntgenstrahlen werden Photoneneiner Wellenlänge zwischen 0,01 nm und10 nm bezeichnet. Sie entstehen beim Be-schuss einer Anode mit Elektronen, derenEnergie 10 keV überschreitet. Beim Aufprallentstehen neben ca. 98% Wärme zwei Artenvon Röntgenstrahlung:Bremsstrahlung: Die auftreffenden Elek-tronen werden im Kernfeld des Anodenmate-rials abgebremst. Die Differenz zwischen denkinetischen Energien des Elektrons vor und
nach der Wechselwirkung wird in Röntgen-strahlung der Frequenz f umgesetzt (Gl. 2).Mit E der kinetischen Energie der Elektronenbeim Aufprall auf die Anode nach der Be-schleunigung im elektrischen Feld ergibt sichfolgende Energiebilanz:
mit:e = 1,602 @10-19 C: ElementarladungU: Anodenspannungme: Elektronenmassev1: Geschwindigkeit des Elektrons vor dem
Aufprallv2: Geschwindigkeit des Elektrons nach dem
AufprallEph: Photonenenergie (Energie eines Rönt-
genstrahlungsquants).
Die Energie eines Strahlungsquants ist
h = 6,625 @10-34 Ws2: PLANCKsche Konstantec = 2,998 @108 ms-1: Vakuumlichtgeschwin-
digkeitf: Frequenzλ: Wellenlänge
Die Energie wird in diesem Zusammenhangmeist in eV (Elektronenvolt) angegeben. 1 eVist die kinetische Energie, die eine Elementar-ladung e bei der Beschleunigung durch eineSpannung von 1 V erhält. Die Energie inJoule erhält man folglich, indem man denWert in eV mit e multipliziert.
Die Bremsstrahlung hat ein kontinuierlichesSpektrum mit kurzwelliger Kante (sieheAbb.1). Letztere kommt dadurch zustande,dass die Elektronen beim Aufprall höchstensihre gesamte kinetische Energie in Röntgen-strahlung umsetzen können (vollständigeAbbremsung, v2 = 0). Die Röntgenstrahlung
Röntgenverfahren O 22

Atom- und Kernphysik O 22 Röntgenverfahren
78
E e U h f hc
Ph max maxmin
, .= ⋅ = ⋅ =λ
(3)
λ
λ
α
β
K
K
=−
=−
hcE E
hcE E
L K
M K
(4)
2d k⋅ = ⋅sin β λ (5)
hat dann eine maximale Energie, die Wellen-länge wird minimal:
Charakteristische Strahlung: Beim Aufprallkönnen Anodenatome ionisiert werden. Wenndadurch eine Leerstelle auf der dem Kern amnächsten liegenden K-Schale entsteht, sowird diese durch Elektronen der L- bzw. M-Schale sofort wieder besetzt und die Energie-differenz in Form von Röntgenstrahlungabgegeben. Die Photonen (Energiequanten),die während dieser Elektronensprünge freiwerden, bezeichnet man als Kα- bzw. Kβ-Photonen. Ihre Wellenlängen berechnen sichaus:
EL - EK : Differenz der Elektronenenergiender L- und K-Schale,
EM - EK : Differenz der Elektronenenergiender M- und K-Schale.
Da diese Energiedifferenz charakteristisch fürjedes Material ist, wird die Strahlung „cha-rakteristische Strahlung“ genannt. Sie hat einLinienspektrum, welches die Bremsstrahlungüberlagert (siehe Abb.1).
Röntgenbeugung:Die Wellenlänge von Röntgenstrahlen kannmit Hilfe der Beugung an einem Kristallgitterbei bekanntem Netzebenenabstand bestimmtwerden (Röntgen-Spektralanalyse). Um-gekehrt werden mit Röntgenstrahlung be-kannter Wellenlänge Atomabstände in Kris-tallgittern gemessen. (BRAGG-Verfahren).Gemäß dem Huygensschen Prinzip kannjedes Atom des von der Röntgenstrahlunggetroffenen Kristalls als Ausgangspunkt einerElementarwelle betrachtet werden. DieKristallatome lassen sich in einer Vielzahlvon hintereinander liegenden, zur Oberfläche(Spaltfläche) des Kristalls parallelen Ebenen,zusammenfassen. Man nennt diese EbenenNetzebenen. Im einfachsten Fall lässt sich dieBeugung (Diffraktion) von Röntgenstrahlenauf die Reflexion an Netzebenen eines Kris-tallgitters zurückführen. Jede Netzebenewirkt auf die einfallende Röntgenstrahlungwie ein partieller Spiegel, d. h. ein (sehrkleiner) Teil des auf die Ebene treffendenRöntgenstrahlenbündels wird reflektiert.
Abb.2 zeigt die grundlegenden Vorgänge beidiesem als BRAGG-Reflexion bezeichnetenVorgang: Die an den Netzebenen A und Breflektierten Strahlen 1 und 2 interferierenmiteinander. Konstruktive Interferenz (einsog. „Reflex“) tritt nur auf, wenn der Gang-unterschied der beiden Wellen gleich einemganzen Vielfachen der Wellenlänge ist:
mit k = 1, 2, … Dabei ist k die Beugungsordnung und d derNetzebenenabstand (d = 0,201 nm für denLiF-Kristall). Für die erste Beugungsordnung
Abb.1: Typisches Röntgenspektrum be-stehend aus Bremsstrahlung und charakte-ristischen Linien des Anodenmaterials
Abb.2: BRAGG-Reflexion

Atom- und Kernphysik O 22 Röntgenverfahren
79
EhcdPh =
21
sin.
β(6)
Abb.3: Messung der Ionendosisleistung miteiner Ionisationskammer
H w D= ⋅ (9)
jQ
m tIm
C= =∆
∆ ∆(10)
H J
h j
= ⋅
= ⋅
32 5
32 5
,
, .
SvAs kg
oder
SvAs kg
-1
-1
(11)
JQm
=∆
∆. (7)
DEm
=∆∆
, (8)
(k = 1) ergibt sich mit Gl. (2):
Durch Drehen des Kristalls wird der Einfalls-winkel der Röntgenstrahlung und damit auchder Phasenunterschied der interferierendenStrahlen verändert, so dass die Bedingung derkonstruktiven Interferenz (5) für jeweilsandere Wellenlängen des Primärstrahls erfülltwird (vergl. Abb.2). Gleichzeitig mit derRotation des Kristalls muss der Strahlungs-empfänger (Zählrohr) unter dem doppeltenBragg-Winkel β mitgeführt werden, so dassimmer die Reflexionsbedingung Zählrohr-winkel = 2×Kristallwinkel erfüllt ist. Damitkann das Spektrum einer Röntgenquellebestimmt werden.
2.2 Als Dosimetrie bezeichnet man dieMessung der Wirkung, die ionisierendeStrahlung (Röntgen und radioaktive Strah-lung) beim Durchgang durch Materie her-vorruft. Diese Wirkung kann entweder überdie Menge der in der Materie erzeugten Ionenoder über die von der Materie absorbierteEnergie gemessen werden.Die Ionendosis J ist definiert als Quotientaus der in einem Volumenelement erzeugtenLadung der Ionen eines Vorzeichens ∆Q undder Masse des durchstrahlten Volumen-elementes ∆m:
Die Einheit der Ionendosis ist As/kg oderC/kg (die alte Einheit 1 Röntgen = 2,58 @10!4
C/kg darf nicht mehr verwendet werden).
Die Energiedosis D ist der Quotient aus derim Volumenelement absorbierten Energieund der Masse des durchstrahlten Volumen-elementes ∆m:
ihre Einheit ist das Gray (1 Gy = 1 J/kg).Die biologische Wirkung ionisierenderStrahlung wird durch die Äquivalentdosis
angegeben (biologisch bewertete Energiedo-sis). Die Einheit ist das Sievert (1 Sv =1 J/kg). w heißt Strahlungswichtungsfaktor;w = 1 für Röntgen-, Gamma- und Beta-strahlung und w = 20 für Alphastrahlung.Die wirksame Intensität der Röntgenstrah-lung ist die Dosis pro Zeit, die als Ionendosis-leistung j (Einheit A/kg), Energiedosislei-stung d (Einheit Gy/s) bzw. Äquivalentdosis-leistung h (Einheit Sv/s) bezeichnet wird. Die Ionendosisleistung wird in einem mitLuft gefüllten Kondensator (“Ionisations-kammer”) entsprechend Abb.3 gemessen. Anden Kondensator wird eine Spannung ange-legt, die so groß ist (etwa 100...300 V), dassalle erzeugten Ionen zu den Kondensator-platten gelangen. Dann ergibt sich die Ionen-dosisleistung
aus der Stromstärke IC und der durchstrahltenLuftmasse m. Mit Hilfe der bekannten mitt-leren Ionisationsenergie der Luftmolekülekann die Ionendosis in die Äquivalentdosisumgerechnet werden; es gilt (für Luft ):

Atom- und Kernphysik O 22 Röntgenverfahren
80
Erklärung zur Strahlensicherheit:
Das Röntgengerät ist so aufgebaut, dassRöntgenstrahlung nur bei geschlossenenTüren von Röhren- und Experimentier-raum erzeugt wird. Dabei werden dieGrenzwerte der gemäß der Röntgenver-ordnung außerhalb des Gehäuses zulässi-gen Strahlung mit mehrfacher Sicherheitunterschritten.Das Röntgengerät ist gemäß der "Ver-ordnung über den Schutz vor Schädendurch Röntgenstrahlen" (Röntgenverord-nung-RöV) vom 8.1.1987 bauartlichzugelassen (Zulassungskennzeichen NW807/97 Rö).
Abb.4: Röntgengerät mit Goniometer.
a Netzanschlussfeld (Seite), b Bedienfeld, c Anschlussfeld, d Röhrenraum mit Mo-Röhre, e Experimentierraum mit Goniometer, f Leuchtschirm, g Leerkanal, h Verriegelungstaster
3 Versuchsaufbau
3.0 Geräte- Röntgengerät mit Goniometer incl. LiF-
Kristall, Zählrohr und Impulsratenmesser- PC mit Programm “Röntgengerät”- Plattenkondensator mit Röntgenblende für
Ionendosismessung- Spannungsquelle 0...450 V, Ri = 5 MΩ- Strom-Messverstärker- Vielfachmesser- Verbindungsleitungen- Biologische Objekte zur Durchleuchtung
3.1 Das Röntgengerät (siehe Abb.4) besitztein strahlenabschirmendes Gehäuse, das ausdrei getrennten Kammern besteht. Die größte(rechte) Kammer ist der Experimentierraum,der das Goniometer (für Röntgenbeugungs-untersuchungen) oder den Plattenkondensatoroder die zu durchleuchtenden Präparate ent-hält. In der mittleren Kammer befindet sichdie Röntgenröhre. Die linke Kammer enthältdie mikroprozessorgesteuerte Elektronik mitden Bedien- und Anzeigelementen.Die Schiebetüren und Sichtfenster des Gerä-
tes bestehen aus Bleiglas (Vorsicht, kratz-empfindlich!).
3.2 Die Hochspannungsquelle besitzt einensehr großen Innenwiderstand (Ri = 5 MΩ)und ist deshalb berührungsungefährlich. Zur

Atom- und Kernphysik O 22 Röntgenverfahren
81
Strommessung dient der Messverstärker miteinem Vielfachmesser als Anzeigegerät.
4 Versuchsdurchführung
Der Versuch darf erst nach Einweisung indie Bedienung des Gerätes und den Ver-suchsablauf durch den betreuenden Assi-stenten begonnen werden. Die in die Kristallhalter fest eingebautenKristalle sind sehr empfindlich. Bitteberühren Sie diese nicht!
4.1 Zur Aufnahme der Röntgenspektren inBRAGG-Anordnung sind folgende Betriebs-parameter einzustellen:Anodenstrom: I = 1,0 mAHochspannung: U = 20…35 kVMesszeit: ∆t = 5 sSchrittweite: ∆β = 0,1°Anfangswinkel: βmin = 4,0°Endwinkel: βmax = 12,0°Starten Sie das Computerprogramm „Rönt-gengerät“. Die Spektren werden im automatischen Scan-Modus mit 2:1-Kopplung von Zählrohr- undKristallbewegung (“COUPLED”) aufgenom-men und auf dem Computerbildschirmdargestellt. Man beginnt am besten mit der maximalenBeschleunigungsspannung von 35 kV. DurchDrücken des Knopfes “SCAN” wird dieAufzeichnung eines Spektrums gestartet.Weitere Spektren sind jeweils bei einerHochspannung von 30 kV, 25 kV und 20 kVaufzunehmen. Alle Spektren werden in dieselbe Grafik geschrieben.
4.2 Für die Messung der Ionendosisleistungwird das Röntgengerät mit eingebautemPlattenkondensator benutzt. VervollständigenSie die Schaltung gemäß Abb.3! Das Koaxi-alkabel von der unteren Kondensatorplatte istmit den Eingang I des Messverstärkers zuverbinden. Der Masseanschluss z des Ver-stärkers wird mit dem Minuspol und dieobere Kondensatorplatte mit dem Pluspol derSpannungsquelle verbunden. Es wird der
Messbereich 10!9A verwendet (1 V amVerstärkerausgang entspricht I = 1 nA) undeine Spannung U $ 200 V.Messen Sie den Ionenstrom IC bei der maxi-malen Beschleunigungsspannung von 35 kVund den Anodenströmen 1,0 mA; 0,8 mA;0,6 mA; 0,4 mA und 0,2 mA.Notieren Sie den Luftdruck p und die Tem-peratur T im Röntgengerät.
4.3 Zur Durchleuchtung von Objekten istdas dafür vorbereitete Röntgengerät zubenutzen. Die maximal mögliche Energiewird eingestellt (U = 35 kV, I = 1 mA), derRaum muss abgedunkelt werden.Interpretieren Sie den Schatten der Objekteauf dem Leuchtschirm. Untersuchen Sie dieVeränderung des Bildes bei Veränderung desPräparateortes. Sie können auch eigeneObjekte (z.B. Taschenrechner, Kugelschrei-ber) durchleuchten. Vergessen Sie nicht, alleBeobachtungen zu protokollieren!Nach Beendigung der Untersuchungen mussder Leuchtschirm mit dem zugehörigenDeckel wieder abgedeckt werden.
5 Auswertung
5.1 Die Wellenlängen und die Quanten-energien der charakteristischen Linien Kβ undKα der Molybdänanode sind nach Gleichung(5) bzw. (6) zu bestimmen. Die Quanten-energien sind in keV anzugeben. Für jede verwendete Anodenspannung Uwird die maximale Quantenenergie (in eV)nach (6) aus dem zur jeweiligen kurzwelligenKante gehörenden Winkel β berechnet. Ineiner Tabelle sind diese Energien mit der denElektronen im elektrischen Feld zugeführtenEnergie E = eAU zu vergleichen. Im Rahmen der Fehlerbetrachtung ist dasWellenlängen-Auflösungsvermögen desRöntgengerätes abzuschätzen.
5.2 Die Ionendosisleistung j ist nach (10)aus dem Ionenstrom IC und der Masse m desdurchstrahlten Luftvolumens V zu berechnenund in Abhängigkeit vom Anodenstromgraphisch darzustellen.

Atom- und Kernphysik O 22 Röntgenverfahren
82
m VT p
T p= ⋅ = ⋅ρ ρ ρ, 0
0
0
(12)
Die Luftmasse ergibt sich aus
mit V = 125 cm3, ρ0 = 1,293 kg/m3, T0 = 273 K und p0 = 1013 hPa.
Die maximale Äquivalentdosisleistung h imRöntgengerät (bei I = 1 mA) ist mit Hilfe vonGl. (11) zu berechnen und in der Einheit Sv/hanzugeben.
5.3 Die Beobachtungen bei der Durch-leuchtung von Objekten sind zu protokollie-ren.
6 Literatur
Fercher, A. F.: Medizinische Physik, Sprin-ger, 1992
Haas, U.: Physik für Pharmazeuten und
Mediziner, WVG Stuttgert, 2002
7 Kontrollfragen
7.1 Wie ist das Spektrum einer Röntgenröh-re zu erklären, welchen Einfluss haben dieBetriebsparameter U und I ?
7.2 Wie wird die biologische Wirkungionisierender Strahlung gemessen?
7.3 Auf welche Weise schädigen Röntgen-strahlen (wie auch Gammastrahlen) denOrganismus und wie kann man sich dagegenschützen?
7.4 Wie kann man Röntgenstrahlung nach-weisen?

Anhang Hinweise zur Bedienung von Cassy-S
83
Hinweise zur Bedienung des Computer-gesteuertenMesswerterfassungssystems Cassy-S
Allgemeines
Cassy-S, ein für die Lehre in Schulen und Universitäten konzipiertes universelles Messwert-Erfassungssystem, besteht aus verschiedenen Geräten mit serieller oder USB-Schnittstellesowie der Windows-Software CassyLab. Sensor-Cassy ist ein zweikanaliges Messgerät für Strom und Spannung, die mit einer Auflösungvon 12 bit (1:4096) und einer maximalen Messrate von 100 kHz (105 Messwerte pro Sekunde)erfasst werden. Mit Hilfe von Zubehör (aufzusteckende „Sensorboxen“ und verschiedeneSensoren) können damit fast alle denkbaren physikalischen Größen gemessen werden.Pocket- und Mobile-Cassy besitzen die gleiche Funktionalität, jedoch nur einen Messkanal,messen nur 104 bzw. 5 Werte pro Sekunde und benötigen keinen Stromanschluss. Power-Cassy ist ein Leistungs-Funktionsgenerator, also eine computersteuerbare Strom- oderSpannungsquelle, die bei einer Abtastrate von 100 kHz maximal 10V / 1A liefert.Cassy-Display ist ein großformatiges Anzeigegerät. Es zeigt in Verbindung mit Sensor-Cassy(ohne Computer) gleichzeitig zwei Messwerte an, kann Werte speichern und ermöglicht dieAuswahl von Messgröße und Messbereich.Die Software CassyLab steuert die Messung, stellt die Messergebnisse in einfacher Weisegrafisch dar und bietet viele Möglichkeiten zur mathematischen Auswertung. Die Bedienung istrelativ einfach und erfordert nur wenige Grundkenntnisse. CassyLab ist beim Hersteller unterhttp://www.ld-didactic.de frei verfügbar und kann daher auch zuhause zur Auswertung vomMessungen aus dem Praktikum verwendet werden.
Quick Start: Einschalten und Konfigurieren des Systems
Stellen Sie sicher, dass alle benötigten Cassy-Module zusammengesteckt, mit dem PCverbunden und mit Strom versorgt sind. Starten Sie nun das Programm CassyLab. Der DialogEinstellungen, Cassy wird präsentiert und zeigt die vorgefundene Gerätekonfiguration.
Um eine Messung durchzuführen, muss der entsprechende Eingang oder Ausgang ââââ angeklicktwerden. Es erscheint ein Fenster, in dem der Eingang bzw. Ausgang konfiguriert werden kann:
Messgröße (falls mehreremöglich sind), Stellgröße(bei einem Ausgang),Messbereich (dazu gehörtauch Nullpunkt rechts,links oder Mitte) und ggf.weitere Parameter müssenden Erfordernissen ent-sprechend eingestellt wer-den.
Nun müssen noch dieMessparameter eingestelltwerden (Button Messpara-meter anzeigen im FensterEinstellungen):

Anhang Hinweise zur Bedienung von Cassy-S
84
Automatische Aufnahme bedeutett, dass der Computer mehrere Messpunkte nacheinanderaufzeichnet. Neue Messreihe anhängen bewirkt, dass in einer Grafik mehrere Messreihen (d. h.Kurven) dargestellt werden können. Die wichtigste Größe, die hier auf einen sinnvollen Werteingestellt werden muss, ist das MessIntervall. Bei 10 µs entstehen pro Sekunde 100.000Messwerte! Aus Intervall und Messzeit ergibt sich die Anzahl der Einzelmessungen. Wenn beiMesszeit kein Wert eingetragen ist, wird die Messreihe fortgesetzt bis sie manuell gestopptwird.
Eine Messreihe kann mit F9 oder durch Klick auf gestartet und beendet werden.
Grundlegende Bedienelemente
Es gibt keine Menüzeile wie inden meisten Programmen üb-lich. Alle Funktionen lassensich entweder über die Button-
leiste ãããã bzw. die zugehörigenFunktionstasten oder überKontext-Menüs (Rechts-Klickauf Messwerte, Diagramm,Achsen, Messinstrumente etc.)erreichen.
ääää Anzeigeinstrument.Rechts-Klick: Messgrö-ße und Messbereich ein-stellen
åååå Umschalten zwischenmehreren Darstellungen(definieren in Einstel-lungen)
ææææ Messwerte-Tabelle kann editiert werden. Rechts-Klick: Messwerte und Messreihenlöschen
çççç Im Diagramm werden Punkte und Kurvenbereiche mit der Maus markiert. Rechts-Klick:Anzeige-Einstellungen, Anmerkungen und Markierungen einfügen, alle mathematischenAuswertungen
èèèè Achsen bzw. Skalen können verschoben und mit Rechts-Klick geändert werden
éééé Umschalten zwischen verschiedenen y-Achsen

Anhang Hinweise zur Bedienung von Cassy-S
85
êêêê Trennlinie verschieben
F4 Neue Messung (aktuelle Messung löschen)
F3 Gespeicherte Messung (mit allen Einstellungen und Auswertungen) laden
F2 Aktuelle Messung (mit allen Einstellungen und Auswertungen) speichern
Diagramm ausdrucken (Bitte drucken Sie keine Tabelle mit 10.000 Werten!!)
F9 Eine Messreihe starten oder beenden (Einzelmessung bei manueller Aufnahme)
F5 Das Fenster Einstellungen aufrufen; zweimal drücken für Messparameter
F6 Inhalt der Statuszeile (z. B. das Ergebnis einer Rechnung) groß darstellen
F1 Hilfe
F7 Alle Anzeigeinstrumente ein/aus schalten
Das Anzeigeinstrument IA1 ein/aus schalten
Tipps und Tricks zu CassyLab
• Klicken Sie mit der rechten Maustaste in das Diagramm um ein Menü mit allen Anzeige-und Auswertefunktionen zu erhalten.
• Mit Alt+T fügen Sie Text in das Diagramm ein, dabei ist immer das Ergebnis der letztenAuswertung voreingestellt. Machen Sie viel von dieser Möglichkeit Gebrauch!
• Skalierung der Achsen: Rechts-Klick auf eine Achse.
• Änderung des Messbereiches: Rechts-Klick auf das betreffende Anzeigeinstrument.
• Die Zoom-Funktion wird mit Alt+Z aufgerufen, Alt+A zeigt wieder alles an.
• In den Einstellungen auf der Seite Parameter/Formel/FFT kann eine neue physikalischeGröße erzeugt werden, die aus Messgrößen berechnet oder manuell in die Wertetabelleeingetragen werden kann. Auf diese Weise kann z. B. aus Strom I(t) und Spannung U(t) derWiderstand R(t) als Funktion der Zeit berechnet werden, oder aus dem gemessenen Weg s(t)durch Differentiation die Geschwindigkeit. Die Regeln für die Eingabe von Formeln findenSie in der Hilfe.
• Zusätzliche Diagramme können in den Einstellungen auf der Seite Darstellung als „neueDarstellung“ angelegt werden. Beispiel: Sie messen die Temperaturen T1 und T2 alsFunktion der Zeit; dann können Sie in einem weiteren Diagramm T2 als Funktion von T1
darstellen.
• Die Auswertefunktionen (z. B. Mittelwertbildung, Regressionsanalyse, Integration) sindausführlich in der Hilfe zum Programm dokumentiert. Jede mathematische Auswertungbezieht sich immer auf einen Kurvenbereich, der mit der Maus markiert werden muss.
• Das Programm ist frei verfügbar (s.o.), Sie können Ihre Messergebnisse aus dem Praktikum(*.lab Dateien) auch am eigenen PC zuhause oder im Computerpool auswerten. Messwertekönnen als *.txt Dateien exportiert werden (mit F2 speichern und Dateityp ASCII Exportauswählen) und mit anderen Programmen (Origin, Excel, ...) importiert werden.

Anhang Kurzanleitung zur Software ORIGIN
86
Kurzanleitung zur Software ORIGIN
Für die Auswertung von Messergebnissen und die Anfertigung grafischer Darstellungen stehtin allen Physikpraktika die professionelle Visualisierungs- und Datenanalysesoftware Origin 8.0zur Verfügung. Die Uni Halle besitzt eine Campuslizenz dieser Software, die sich auch vonzuhause aus nutzen lässt. Nähere Informationen hierzu gibt es in Stud.IP und auf den Webseitendes Universitäts-Rechenzentrums.Der Umgang mit Software ist nicht Gegenstand der Physikausbildung. Origin ist lediglich einAngebot an Sie. Sie können alle Auswertungen und Grafiken auch von Hand in Tabellen bzw.auf Millimeterpapier anfertigen oder beliebige andere Software (z.B. Microsoft Excel)verwenden.
1. Grundsätzliches
• Englisch ist die empfohlene und voreingestellte Sprache. Im Help Menü kann auf Deutschumgestellt werden, dann sind jedoch einige Begriffe ungünstig oder falsch übersetzt.
• Alle Daten, Rechnungen und Grafiken werden zusammen in einer Projektdatei gespeichert.Ein leeres Projekt (bei Programmstart) enthält nur die Arbeitsmappe Book1 mit einer x- undeiner y-Spalte zur Eingabe der Daten. Weitere Spalten erzeugt man mittels Rechtsklick in
den leeren Bereich oder mit ., weitere Arbeitsmappen mit File - New... oder .
• Eine Grafik erhält man am schnellsten, indem man eine oder mehrere y-Spalten markiert (in
den Spaltenkopf klicken) und Plot wählt oder auf einen der Buttons klickt.
• Alle Objekte (z.B. Spaltennamen, Beschriftungen von Achsen, Skalierung von Achsen,Aussehen einer Kurve, Legende) kann man bearbeiten, indem man darauf doppelklickt.
2. Arbeitsmappen (Workbook)
• Die Dateneingabe ist Windows-typisch. Weitere Spalten erhält man mit .
• In den Spaltenkopf sollte man immer Long Name und Units eingetragen - sie werdenautomatisch in die Achsen-Beschriftung und in die Legende einer Grafik übernommen.
• Denominierung von Spalten als x oder y: Rechtsklick in Spaltenkopf und Set As auswählen.
• Rechnen mit Spalten: Rechtsklick in Spaltenkopf und Set Column Values... wählen.
Syntax: Spalte A ! Spalte B col(A) !col(B)
a b / (c + d) a * b / (c + d)
x2 x^2
%&x sqrt(x)
ex exp(x)
π pi

Anhang Kurzanleitung zur Software ORIGIN
87
3. Grafiken (Graph)
• Verschönern einer Grafik: Doppelklick auf die zu ändernden Dinge (siehe oben).
• Hinzufügen einer weiteren Kurve zu einer bereits existierenden Grafik:
Weg1: in der Arbeitsmappe die zu zeichnenden Spalten auswählen (markieren); in dasgewünschte Koordinatensystem (“Layer”) klicken; im Menü Graph - Add Plot to Layer
wählen.
Weg 2: Doppelklick auf das “Layersymbol” links oben im Graph, unter Available Data
Spalten auswählen, den => Button klicken.
• Hinzufügen eines weiteren Koordinatensystems oder einer zweiten Achse zu einer bestehen-
den Grafik: Graph - New Layer (Axes) wählen oder einen der Buttons drücken.
• Eine Legende hinzufügen oder die bestehende Legende aktualisieren: Graph - New Legend
wählen oder drücken.
• Beliebigen Text in eine Grafik schreiben: drücken und in die Grafik klicken. Text
formatieren mit Format-Buttonleiste (z.B. hoch, tief, griechisch mit )
• Werte aus einer Grafik ablesen mit den Werkzeugen Screen Reader / Data Reader .
• Glatte (runde) Kurven durch die Messpunkte zeichnen: Kurve doppelklicken; denDiagrammtyp “Linie + Symbol” wählen; Linie - Connect - Spline oder B-Spline einstellen.
• Lineare Regression: Analysis - Fit Linear wählen. Bei mehreren Kurven vorher im MenüDaten die richtige Kurve auswählen. Soll nur ein Teil der Kurve angepasst werden, dann
vorher mit den Werkzeugen Data Selector und Mask Tool den Bereich einschränken.Mit Fit Linear: Grundpraktikum-Standard wird die Regression sofort ausgeführt, alleParameter sind bereits für die meisten Anwendungsfälle im Praktikum sinnvoll eingestellt.
4. Ausdruck von Grafiken und Arbeitsmappen
• Prüfen Sie Ihre Grafiken vor dem Ausdruck genau, vermeiden Sie mehrfache Korrektur-drucke! Im Praktikum werden mehrere 10.000 Seiten pro Jahr gedruckt - das kostet Geldund belastet die Umwelt. Arbeitsmappen mit sehr vielen Daten (mehrere Seiten) sollen imPraktikum nicht gedruckt werden. Solche mit nur drei Zahlen können Sie abschreiben!
• Mehrere Grafiken und/oder Arbeitsmappen und Text auf einem A4-Blatt anordnen:
File - New... Layout wählen oder klicken; Rechtsklick auf die Layoutseite um Grafikenoder Arbeitsmappen hinzuzufügen.
• druckt eine Grafik, Arbeitsmappe oder ein Layout unmittelbar auf ein A4-Blatt.

Anhang Naturkonstanten
88
Einige Naturkonstanten
Lichtgeschwindigkeit im Vakuum c = 2,997 924 58 @ 108 m/s. 300 000 km/s
Gravitationskonstante γ = 6,673 9 @ 1011 N m2 kg!2
Elementarladung e0 = 1,602 177 33 @ 10!19 C
Elektronenruhemasse me = 9,109 389 7 @ 10!31 kg
Atomare Masseneinheit u = 1,660 277 @ 10!27 kg
elektrische Feldkonstante ε0 = 8,854 187 817 @ 10!12 A s V!1 m!1
(Dielektrizitätskonstante des Vakuums)
magnetische Feldkonstante µ0 = 1,256 637 1 @ 10!6 V s A!1 m!1
(Permeabilität des Vakuums)
Planck-Konstante h = 6,626 075 5 @ 10!34 J s(Planckschen Wirkungsquantum) = 4,135 7 @ 10!15 eV s
Avogadro-Konstante NA = 6,022 136 7 @ 1023 mol!1
Boltzmann-Konstante k = 1,380 658 @ 10!23 J/K
Gaskonstante R = 8,314 510 J mol!1 K!1
Faraday-Konstante F = 9,648 4 @ 104 As/mol





























