Embed Size (px)
Citation preview

Eur. J. Biochem. 262, 117±126 (1999) q FEBS 1999
Manipulating the coordination mumber of the ferric iron within thecambialistic superoxide dismutase of Propionibacterium shermanii bychanging the pH-valueA crystallographic analysis
Marius Schmidt
Physikdepartment E17, Technische UniversitaÈt MuÈnchen, James Franck Straûe, Garching, Germany
The structure of the Propionibacterium freudenreichii subspecies shermanii superoxide dismutase (SOD) was
determined at various pH values. As a comparison, the structure of the fluoride coordinated SOD was solved. The
SOD crystallizes at pH 6.1 in the space group C2221 with two subunits, A and B, in the asymmetric unit. An
increase of the pH value changes the cell parameters slightly but not the symmetry of the crystals. The overall
structure of the SOD remains a compact tetrameter and is comparable to that at pH 6.1 no matter whether the pH
increases or fluoride is added. At values above pH 7.4, an additional hydroxide ion can bind to the active center. Its
position is similar to the binding site of the fluoride. The coordination number changes from five to six if the pH
increases or fluoride is added. The binding behavior of the hydroxide ion is different for subunit A and B. Structures
at different pH-values are comparable with models derived by spectroscopic methods. The influence of temperature
on the binding properties of the hydroxide ion was investigated using analysis of an X-ray structure solved at pH 8.1
and 140 K. Compared to the structure at room temperature, the structural changes are observable but remain small.
The consequences of hydroxide binding to the iron are discussed.
Keywords: cambialistic; superoxide dismutase; Propionibacterium; X-ray structure analysis; coordination number.
Superoxide dismutases (SODs) catalyze the degradation of thetoxic superoxide radical O2
´± in a two step cycle that involves areduction and a reoxidation of a central metal atom:
Meox � O2´2 ! Mered � O2
Mered � O2´2 � 2H� ! Meox � H2O2
At present, two structurally distinct families are known; theCu/Zn SODs which contain copper and zinc and the iron ormanganese SODs with either Fe or Mn in their active center.The activity of the Fe-Mn SODs is pH-dependent. Full activityis usually achieved at acidic and neutral pH values whereas adecrease of the activity occurs at pH values larger than pH 9[1,2]. Thus, these SODs will be referred to as low-pH SODs(lpH-SODs). In contrast there are SODs that change their rateconstant within the entire pH range [3,4]. In this group,increasing the pH above 6 causes the rate constant to decreasemonotonically by several orders of magnitude. At pH 10 nearlyfull inhibition is achieved. These SODs will be referred to as full
pH SODs (fpH-SODs). The mechanism of the reduction ofactivity is not fully understood. Kinetic measurements [2] favorthe existence of a protonized group near the active center thatchanges its charge if the pH value is changed. Therefore, thereduction of the activity might be due to changes of theelectrostatic interaction with the substrate. X-ray absorptionspectroscopy (XAS), MoÈssbauer and EPR measurementsperformed on both lpH-SODs and fpH-SODs [5±7] giveevidence for a change of the central metal's coordinationnumber from five to six due to an increase in the pH value. Thiscould favor the hypothesis that at high-pH values, inhibitionmay occur via a molecule that enhances the coordinationnumber of iron. X-ray structures are mainly available onlpH-SODs at low-pH values. Hence, the central metal of theseSODs is fivefold coordinated ([8] and references therein). AnX-ray crystallographic study that proves the sixfold coordinationstate can only succeed if the additional ligand accumulates at the6th coordination site in a sufficient amount that a significantelectron density can be observed. Using lpH-SODs this mightonly be possible if one increases the pH far above pH 9.0. Athigh-pH values the crystals normally decompose. Using anfpH-SOD, the change of the coordination number is expected tooccur at a more neutral pH value. The SOD of P. shermanii is anfpH-SOD. XAS measurements have shown [7] that the iron ofthis SOD alters its coordination state from 5 to 6 atapproximately pH 7.8. At this moderate pH value a largechange of the interacting forces between the molecules in thecrystal, and therefore a cracking of the crystal, is less probable.The X-ray structure of the P. shermanii Fe-SOD was solvedrecently [9] at pH 6.1. The iron is in the ferric Fe(III) statewithin the entire pH range [6] and is pentacoordinated atlow-pH. To show the complete structure of the hexacoordinated
Correspondence to M. Schmidt, The University of Chicago, Department
of Biochemistry and Molecular Biology, Cummings Life Science Center,
920 East 58th Street, Chicago, IL 60637, USA. Fax: +1 773 702 0439,
Tel: +1 773 702 1801, E-mail: [email protected]
Abbreviations: SOD, superoxide dismutase; lpH-SODs, low-pH SODs;
fpH-SODs, full pH SODs; XAS, X-ray absorption spectroscopy.
Enzymes: Superoxide dismutase (EC1.15.1.1)
Note: The coordinates of the SOD at pH 10 and at pH 8.1 (140 K) and those
of the fluoride coordinated SOD are available from the Protein DataBank,
with code numbers 1BT8, 1BSM and 1BS3, respectively.
(Received 4 January 1999, revised 18 February 1999, accepted
22 February 1999)

118 M. Schmidt (Eur. J. Biochem. 262) q FEBS 1999
state predicted by XAS, the X-ray structures of the Fe-SOD ofP. shermanii at different pH values and at different temperaturesare presented. For comparison, the structure of the fluoridecoordinated Fe-SOD was solved at low-pH. Fluoride is acompetitive inhibitor [3] that coordinatively binds to a free sixthcoordination site of the iron atom.
MATERIALS AND METHODS
X-ray structure determination
The Fe-SOD of P. shermanii was purified and crystals weregrown as in [9]. The crystals belong to space group C2221.Crystals were soaked at pH 7.4, 8.2, 9.3 and 10.0 in (NH4)2SO4
solutions. In order to maintain the stability of the crystals, the(NH4)2SO4 concentration has to be increased with increasingpH. At pH 7.4 and pH 8.1, 3.2 mol´L21 (NH4)2SO4 in50 mmol´L21 potassium phosphate (KPi) buffer or25 mmol´L21 Tris buffer, respectively, was used. At pH 9.3and 10.0, a 3.5-mol´L21 (NH4)2SO4 solution buffered by50 mmol´L21 sodium phosphate was prepared. Due to theevaporation of NH3, the pH is very unstable above pH 9.0.Therefore, care has to be taken not allow the NH3 to evaporate inthe soaking experiments. In all experiments, a crystal grown atpH 6.1 was transferred into a glass capillary. For experiments atpH 7.4 and 8.1, the capillary was washed several times andfinally filled with 80 mL of the soaking buffer. The capillary wasstored for 14 h in a closed box at 4 8C. No significant pHchanges occurred. The experiments at pH 9.3 and pH 10 wereperformed at room temperature. The capillaries containing thecrystals were filled initially by 100 mL of 3.2 mol´L21
(NH4)2SO4, pH 8.1 for 5 h. Subsequently, the buffer in thecapillaries was exchanged for 80 mL soaking liquor of thedesired pH value. The capillaries were transferred into a sealedtube containing 18 mL of the equivalent soaking buffer for 6 h.
No pH changes of the solution in the tubes could be observedeven after 1 week. Next, the soaking solution was removed fromthe capillary and one end sealed. The time elapsed during thisprocedure was <15 min. This might cause significant pH shiftsdue to the considerable evaporation of NH3, especially at pH 10.In order to allow the pH to equilibrate again, the capillary wasstored overnight in 15 mL of the soaking buffer. After that, thecapillary was quickly sealed within a few seconds. Toinvestigate a possible structural change due to the decrease ofthe temperature, a structure determination at 140 K and pH 8.1was performed. After the soaking procedure at pH 8.1equivalent to that described above, a crystal of dimensions0.5 £ 0.4 £ 0.4 mm3 was mounted in a silk loop and transferredto a cryoprotectant solution containing 250 mg´mL21 sucrose in3.2 mol´L21 (NH4)2SO4, 20 mmol´L21 Tris/KOH, pH 8.1 for10 s. The crystal was immediately frozen in a cold nitrogenstream at 140 K and exposed to X-rays. To solve the structure ofthe fluoride coordinated SOD, a crystals was soaked for 12 h atpH 6.1 and 4 8C in 3.0 mol´L21 (NH4)2SO4 and 200 mmol´L21
KF. Within a few seconds the color of the native crystalsvanishes completely, changing from weak yellow to transparent.All crystals were measured on an Enraf Nonius rotating anodeequipped by a Siemens Histar detector on a three circlediffractometer. One crystal was sufficient to record a completedata set. At pH 10.0, the crystals become sensitive towards X-ray radiation and typically decompose after 14 h in the X-raybeam. Nevertheless, data to 0.185 nm could be recorded duringthat time. Raw data were reduced and intensities were scaled bythe program saint. Mean values of the symmetrically equivalentreflections were calculated by agrovata/truncate [10]. Theresults are shown in Table 1. All data sets are of good statisticalquality, indicated by the Rsym and the completeness values. Theunit cell parameters differ slightly from those of the Fe-SOD atpH 6.1. No change in the space group was observed. To interpretthe data, subunits A and B of the Fe-SOD that are lying in the
Table 1. Data collection. Resol, maximum resolution of data collection; refs, number of unique reflections,
Rsym �
Phkl
Pi
j�Ihkl 2 Ii;hkljPhkl
Pi
Ii;hkl
where Ihkl is the mean value of symmetric equivalent Intensities Ii,hkl, comp: completeness; red: redundancy.
SOD a (nm), b (nm), c (nm) Resol. (nm) Refs Rsym Comp. Red.
Fe-SOD pH 6�.1 7�.983 8.569 10.859 0�.155
Fe-SOD 200 mm KF 7�.942 8.528 10.840 0�.155 45923 3�.6 78% 5�.1
Fe-SOD pH 7�.4 7�.982 8.580 10.851 0�.190 24891 3�.0 89% 3�.1
Fe-SOD pH 8�.1 7�.963 8.545 10.871 0�.180 27019 2�.8 78% 2�.7
Fe-SOD pH 9�.3 7�.994 8.580 10.900 0�.180 28568 2�.5 87% 3�.0
Fe-SOD pH 10�.0 7�.869 8.729 11.084 0�.185 22966 5�.9 71% 2�.8
Fe-SOD pH 8�.1 T = 140 K 7�.918 8.432 10.704 0�.135 72107 8�.3 92% 6�.1
Table 2. Refinement statistics. lc, linear compression from unit cell parameters at room temperature to those at 140K; rb, rigid body refinement; conv,
conventional refinement.
Model Fe-SOD fluoride Fe-SOD pH 7.4 Fe-SOD pH 8.1
Fe-SOD pH 8.1
T = 140 K Fe-SOD pH 9.3
First model Fe-SOD Fe-SOD Fe-SOD Fe-SOD Fe-SOD
pH 6�.1 pH 6�.1 pH 6�.1 pH 6�.1 pH 6�.1
Rcryst/Rfree 29�.6/29.8 24�.9/24.9 24�.5/24.9 48�.5/50.3 27�.1/27.4
Rigid body, Rcryst/Rfree 25�.8/25.8 23�.1/23.3 23�.9/24.1 lc: 33�.8/34.3
rb: 29.9/31.3
24�.3/24.5
Simulated annealing 1000K 1000K 1000K conv (0�.21 nm): 1000 K
Rcryst/Rfree 22�.6/26.1 21�.9/25.3 22�.1/25.0 27�.2/30.7 21�.3/24.9
Indiv. B-values Rcryst/Rfree 22�.0/26.4 21�.0/25.3 21�.6/25.8 26�.0/30.6 20�.7/24.3
NH2O, final model 542 349 376 659 495
Rcryst/Rfree 17�.0/20.8 15�.8/20.5 16�.3/20.9 20�.6/23.6 15�.2/20.4

q FEBS 1999 Coordination no. manipulation of SOD ferric iron (Eur. J. Biochem. 262) 119
asymmetric unit at pH 6.1 were used as a starting structure. Forthe refinement, similar protocols were followed for thestructures at pH 7.4, 8.1 and 9.3 and the fluoride coordinatedSOD (Table 2). In the first step, a rigid body refinement wasperformed on both subunits independently. Throughout, thisresulted in a decrease in the crystallographic Rcryst and Rfree [11]values. Then, a 1000 K simulated annealing protocol wasperformed followed by a conventional refinement. At the endthe individual B-values were refined. All refinements wereperformed with xplor [12]. At pH 10.0, the described approachfailed. Even though the cell constants differ only slightly and thespace group remains constant, the crystal at pH 10.0 does notappear to be isomorphous to crystals at lower pH values. Beforethe refinement was started, a molecular replacement wasperformed using AMoRe [13] in the resolution interval of1.50 nm to 0.32 nm (Table 3). The entire dimer that is lying inthe asymmetric unit at pH 6.1 was used as the search model.The correlation coefficient of the solution found was 80.3% andthe Rcryst was 27.1% at a resolution of 0.32 nm. In Table 3 therotational angles and the shifts necessary to place the pH 6.1
model in the unit cell at pH 10.0 are shown. After a rigid bodyrefinement performed on both subunits independently, theresulting dimer was refined using a 1500 K simulated annealingprotocol. At 140 K and pH 8.1, data to 0.135 nm were collected(see also Table 1). The refinement of the model towards the datawas initiated by a linear compression of the protein model atroom temperature (pH 6.1) to the unit cell parameters measuredat 140 K (see Table 2). At a resolution of 0.21 nm, the rigidbody refinement and 150 steps of conventional refinement couldlower the R-value to 27.2%. For the further refinement all datato 0.135 nm were used. All other refinement steps are similar forall structures reported here. From the resulting models,2FobsFcalc and FobsFcalc difference maps were calculated inorder to detect structural errors in the models or to find watermolecules. The corrected models were then conventionallyrefined. After three to four cycles of water searching, no furtherdifference electron peaks higher than about 3.5 s could bedetected outside the active center. In order to avoid model bias,special care was taken not to change any coordination number ofthe iron during the refinement. After completing the refine-ments, the resulting FobsFcalc difference maps were inspected atand near the active center. Two pronounced peaks, one in eachsubunit, appeared in the map that belonged to the fluoridesoaked Fe-SOD. Similar peaks were found in the map calculatedfrom the high-pH data. They were interpreted as fluoride anionsor as oxygen atoms of water or hydroxide ions. The coordinationnumber of the iron was increased from 5 to 6. At pH 7.4 and 8.1,the 6th coordinated molecule was manually oriented in thedifference electron peak. At pH 9.3, pH 10 and in the fluoridecoordinated SOD, the position of the sixth coordinated moleculewas refined conventionally. As the next step, an attempt to refinethe occupancy and the B-value of these molecules wasperformed. Due to the resolution limit of the data sets it wasnot possible to refine both. For example the s-value at the 6thcoordination site in subunit A at pH 9.3 is 5.9. If the B-valueand the occupancy are refined simultaneously the parametersincrease to values, 0.43 nm2 and 1.2, respectively, that areobviously too large. In order to extract a reasonable occupancyvalue, the B-value was fixed to the average value found in thefirst coordination shell of the iron (Table 4) and 30 cycles ofoccupancy refinement were performed.
RESULTS AND DISCUSSION
Overall structure
The overall structure of the P. shermanii SOD at pH 6.1 isshown schematically in Fig. 1. The enzyme is a compact
Table 3. Refinement against pH 10 data. MR, Molecular replacement;
eulerian angles a,b,g and shifts x,y,z. Transformation of the search model
from the unit cell at pH 6.1 to the unit cell at pH 10 preserving the same
origin.
Search model
Fe-SOD pH 10.0
Fe-SOD pH 6.1, subunit A and B
MR
Peak heights in rotation solution: 9�.9 s
Function next peak: , 5 s
Peak heights in translation solution: 16�.4 s
Function next peak: 9�.8 s
MR-solution to 0�.32 nm
Rcryst 27�.1
a, b, g [8] ±0�.24, 26.72, 0.046
x, y, z [nm] 0�.315, 20.067, 20.394
MR-solution to 0�.19 nm
Rcryst/Rfree 37�.0/37.8
Rigid body, Rcryst/Rfree 29�.5/29.5
Simulated annealing 1500 K
Rcryst/Rfree 22�.6/26.4
Indiv�. B-values
Rcryst/Rfree 22�.0/26.1
NH2O final model 292
Rcryst/Rfree 17�.4/22.7
Table 4. Peak heights at the sixth coordination site in s found in the FobsFcalc difference map and occupancy values of the corresponding sixth
coordinated molecule in subunit A and B at various pH values and with fluoride. The amount of active centers containing metal ions other than Fe was
estimated to be 0.2 per molecule using two different approaches. The occupancy of the sixth coordinated ion was corrected by this number (see text) to derive the
ligand : iron ratio. ligand : iron in parenthesis: the difference of this value with the theoretical value of 98.9% can be considered as an estimate of the error of the
method to determine the occupancy. , B . : Averaged B-value (in nm £ 100) determined from B-values of His27Ne2, His75Ne2, Asp161Od1, His165Ne2 and
the iron.
Subunit A Subunit B
s Occupancy Ligand:iron , B . s Occupancy Ligand:iron , B .
pH 6�.1 ± ± ± 8�.7 ± ± 6�.7
Fluoride 12�.1 0�.70 (0�.88) 11�.9 10�.9 0�.69 (0�.87) 9�.6
pH 7�.4 3�.5 0�.23 0�.29 7�.9 ± ± ± 5�.9
pH 8�.1 4�.2 0�.36 0�.45 5�.9 3�.1 0�.10 0�.13 8�.1
pH 8�.1 T = 140K 5�.2 0�.46 0�.58 7�.8 4�.6 0�.38 0�.47 7�.5
pH 9�.3 5�.9 0�.40 0�.50 10�.3 3�.9 0�.16 0�.20 8�.4
pH 10 6�.6 0�.68 0�.85 8�.4 6�.8 0�.63 0�.79 11�.0

120 M. Schmidt (Eur. J. Biochem. 262) q FEBS 1999
homotetramer (m = 90.5 kDa) that lies in two asymmetric unitshousing two noncrystallographically related subunits each. InFig. 2, the structure of one subunit consisting of 201 amino acidresidues is shown at pH 6.1. In order to visualize differences thestructure at pH 10 and the structure of the fluoride coordinatedSOD were overlaid onto the pH 6.1 structure by means of a leastsquares fit of the position of corresponding Ca-atoms. This wasperformed independently for both subunits in the asymmetricunit. In Fig. 3 the rmsd averaged over main chain atoms areplotted as a function of the residue number. As one can see, thedifferences between the structure at pH 6.1 and the fluoridecoordinated structure are small, averaging 0.016 nm. This is inthe range of the mean coordinate error that was estimated to be0.015 nm from a sA-plot [14]. At pH 10 the situation is slightlydifferent. The average deviation is 0.028 nm and 0.021 nm forsubunits A and B, respectively. These values are too small toindicate a large structural rearrangement, but are consideredsignificant. Larger deviations in the order of 0.06 nm occur nearresidue 50 or residue 90. These residues are exposed to thesolvent space and are affected by the pH changes preferentially.The question arises whether these structural changes also affectthe relative position of the subunits in the SOD tetramer. Toanswer this, the following was undertaken. The shortestdistances from all residues in one subunit to all residues in theother three subunits were calculated using the structure atpH 6.1. By deleting distances longer than 0.5 nm, a list ofresidues and their nearest neighbors in different subunits wasobtained. At pH 10, the shortest distances between residuesquoted in this list were determined again. Then, the differenceswith the equivalent distances at pH 6.1 were calculated. In thisway, a D-distance matrix was obtained constrained to interactingresidues. From Fig. 1 it can be seen that each subunit interactswith all others in the tetramer. Therefore, only those differencedistances starting from subunit A were calculated. The compar-ison of the structures at pH 6.1 and 10.0 is shown in Fig. 4;
interacting residues accumulate at the sequential positionsshown by the boxes. The distance deviations are quite smalland distribute around 0 nm. The average D-distance betweensubunit A and B is 20.0081 nm. This means that subunit B isrepelled only by this quantity at pH 10. Average distancedeviations for the other subunits are of the same order ofmagnitude. Consequently, the overall structure of the tetramer isalmost identical to that at pH 6.1.
Change of coordination number of the ferric iron
The structure of the Fe-SOD was initially solved at pH 6.1. Thestructure of the active center is also shown in Fig. 2. The iron isfivefold coordinated by three histidines, one aspartate and awater molecule or hydroxide ion at the 5th coordination site. AtpH 6.1, and presumably at all other pHs, a fluoride anion can bebound to the iron. Figure 5 shows the active center of thefluoride coordinated Fe-SOD. The iron is sixfold coordinated,with the fluoride occupying the free 6th coordination site. AtpH 7.4, 8.1, 9.3 and 10.0 the structure of the active center withinsubunit A is shown in Fig. 6a±6d, respectively. Above pH 8, asixfold coordination of the iron similar to that within thefluoride coordinated SOD is clearly visible in the electrondensity maps. The X-ray structure analysis can not distinguishbetween a water molecule or a hydroxide ion. Because the OH2
Fig. 2. Structure of the monomer at pH 6.1. Numbers denote the
beginning and the end of secondary structure elements within the sequence.
H1±H6, helices; B1±B3, b-sheet. Insert, pentacoordinated iron(III) and the
first coordination sphere.
Fig. 1. Structure of the homo-tetramer. Large spheres and arrows indicate
active centers within the subunits A, B, C and D. Each sphere contains one
iron(III) as the central metal ion.

q FEBS 1999 Coordination no. manipulation of SOD ferric iron (Eur. J. Biochem. 262) 121
concentration is the only parameter varied it is reasonable toassign a hydroxide ion to the electron density there. Onchanging to pH 6.1, all active centers lose their sixth coordinate,being fivefold coordinated below-pH 7.4. The disappearance ofthe electron density can also be monitored by the occupancy of
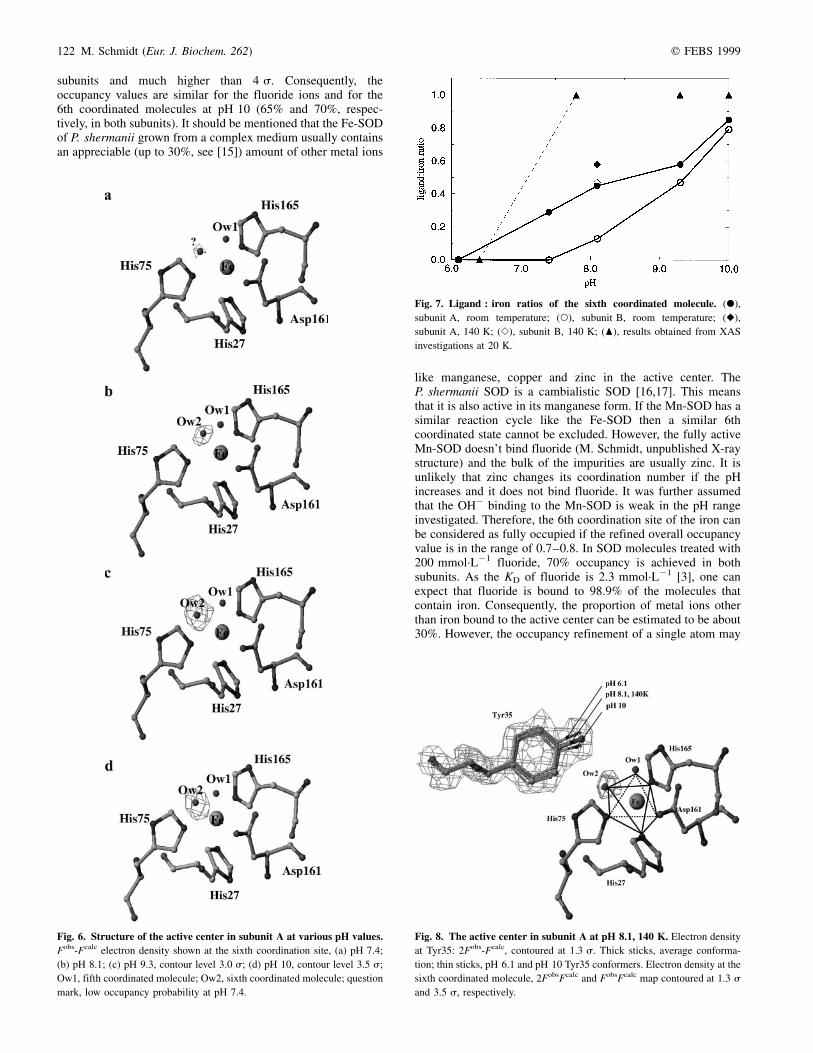
the corresponding solvent molecule (Table 4). When electrondensity peaks below 4 s were interpreted as water moleculestheir occupancy values refined to around 0.2. Differenceelectron density peaks having s-values higher than 4 wereinterpreted as significantly occupied 6th coordination sites. AtpH 7.4 no electron densities higher than this threshold werefound. Consequently, the occupancy refined to a vanishing smallvalue. At pH 8.1 a significant peak was only found in subunit A,whereas in subunit B the peak height was smaller than 3 s. AtpH 9.3 the height of the difference electron density was 5.9 and3.9 for subunit A and B, respectively. This means that at pH 8.1and 9.3 subunit A is partially sixfold coordinated, whereas forsubunit B insignificant occupancy values in the order of 0.2 arefound. For subunit A, the occupancy refinement yielded a valueof 0.36 and 0.4 for pH 8.1 and 9.3, respectively. At pH 10, aswell as near the fluoride-coordinated iron the difference electrondensity at the 6th coordination site was comparable in both
Fig. 3. (a) and (b) show rms-distances derived from averaging squared
distances between equal backbone atoms of corresponding amino acid
residues. (a) Solid line: subunit A pH 6.1/subunit A fluoride coordinated,
dashed line: subunit A pH 6.1/subunit A pH 10. (b) solid line: subunit A
pH 6.1/subunit B pH 6.1, dashed line: subunit A pH 9.3/subunit B pH 9.3.
(c) B-values. solid line: pH 6.1 subunit A, dashed line: pH 6.1 subunit B.
Fig. 4. Delta (D) distance plot calculated from the tetramer at pH 6.1
and the tetramer at pH 10 constrained to residues closer than 0.5 nm.
Circles, difference distances between subunit A and subunit B; solid line
box, hot spot of interacting residues; squares, difference distances between
subunit A and subunit C; dashed line box, hot spot of interacting residues;
triangles, difference distances between subunit A and subunit D; dotted line
box, hot spot of interacting residues.
Fig. 5. The active center of the fluoride coordinated SOD in subunit A.
Electron density at Tyr35: 2Fobs-Fcalc contoured at 1.3 s; thick sticks show
main conformation, thin sticks show conformation observed at pH 6.1 (as
indicated); arrow, electron density not explained by the main conformation.
Electron density at the fluoride ion: Fobs-Fcalc map contoured at 3.5 s. Azi,
position of the azide ion if added.
122 M. Schmidt (Eur. J. Biochem. 262) q FEBS 1999
subunits and much higher than 4 s. Consequently, theoccupancy values are similar for the fluoride ions and for the6th coordinated molecules at pH 10 (65% and 70%, respec-tively, in both subunits). It should be mentioned that the Fe-SODof P. shermanii grown from a complex medium usually containsan appreciable (up to 30%, see [15]) amount of other metal ions
like manganese, copper and zinc in the active center. TheP. shermanii SOD is a cambialistic SOD [16,17]. This meansthat it is also active in its manganese form. If the Mn-SOD has asimilar reaction cycle like the Fe-SOD then a similar 6thcoordinated state cannot be excluded. However, the fully activeMn-SOD doesn't bind fluoride (M. Schmidt, unpublished X-raystructure) and the bulk of the impurities are usually zinc. It isunlikely that zinc changes its coordination number if the pHincreases and it does not bind fluoride. It was further assumedthat the OH2 binding to the Mn-SOD is weak in the pH rangeinvestigated. Therefore, the 6th coordination site of the iron canbe considered as fully occupied if the refined overall occupancyvalue is in the range of 0.7±0.8. In SOD molecules treated with200 mmol´L21 fluoride, 70% occupancy is achieved in bothsubunits. As the KD of fluoride is 2.3 mmol´L21 [3], one canexpect that fluoride is bound to 98.9% of the molecules thatcontain iron. Consequently, the proportion of metal ions otherthan iron bound to the active center can be estimated to be about30%. However, the occupancy refinement of a single atom may
Fig. 6. Structure of the active center in subunit A at various pH values.
Fobs-Fcalc electron density shown at the sixth coordination site, (a) pH 7.4;
(b) pH 8.1; (c) pH 9.3, contour level 3.0 s; (d) pH 10, contour level 3.5 s;
Ow1, fifth coordinated molecule; Ow2, sixth coordinated molecule; question
mark, low occupancy probability at pH 7.4.
Fig. 7. Ligand : iron ratios of the sixth coordinated molecule. (X),
subunit A, room temperature; (W), subunit B, room temperature; (V),
subunit A, 140 K; (S), subunit B, 140 K; (O), results obtained from XAS
investigations at 20 K.
Fig. 8. The active center in subunit A at pH 8.1, 140 K. Electron density
at Tyr35: 2Fobs-Fcalc, contoured at 1.3 s. Thick sticks, average conforma-
tion; thin sticks, pH 6.1 and pH 10 Tyr35 conformers. Electron density at the
sixth coordinated molecule, 2FobsFcalc and FobsFcalc map contoured at 1.3 s
and 3.5 s, respectively.

q FEBS 1999 Coordination no. manipulation of SOD ferric iron (Eur. J. Biochem. 262) 123
contain a large error. Therefore, another approach was used inaddition. The electron density at the Tyr35 site was used, withthe results listed in Table 5. As shown by Fig. 5, the electrondensity of the fluoride coordinated SOD is interpreted by onlyone main conformation of Tyr35. There is an additional electrondensity marked by the arrow that is not explained by thisconformation. A second Tyr35 conformation was also used. Thestructure of this conformer corresponds to that found in SODsubunits at pH 6.1 without fluoride. In order to extract thecoordinates of the Tyr35 in subunit A and B, these subunits wereoverlaid independently to corresponding subunits of the fluoridecoordinated SOD by means of the least square distancedeviation of the corresponding active center residues His27,His75, Asp161, His175 and the iron. This was performed withthe lsq_exp option in the program o [18]. The coordinates of theTyr35 found in the `uncoordinated' SOD were extracted andused as the second conformation to interpret the aforementionedadditional electron density. It will be shown later that theconformation of Tyr35 is also linked to the change of thecoordination state induced by changing the pH value. For theoccupancy refinement, the positions of both Tyr35 conforma-tions were fixed. The B-values of the atoms in both conforma-tions were derived from the refinement against their originaldata. Thirty cycles of grouped occupancy refinement wereperformed on both conformers. The fraction of the Tyr35conformation that corresponds to the five coordinated state wasdetermined to be between 15 and 20%. With these values, andtaking into account the values derived from the occupancyrefinement of the single fluoride ions described above, 80% of
Fig. 9. Structural changes in the first coordination shell at pH 10. Solid
line, His75 and His165 at pH 6.1; dashed line, His75 and His165 at pH 10.
Fobs-Fcalc electron density contoured at 3.5 s.
Fig. 10. Change of the symmetry from pH 6.1±
10.0, schematically. (a), Trigonal bipyramid, sixth
coordinated molecule absent; (b), octahedron,
sixth coordination site occupied. Important angles
are shown, see also text and Table 6.
Table 5. Occupancy of the Tyr35 conformation observed at pH 6.1 refined together with that found at pH 10 at various pH values. uncoordinated: Tyr35
conformation extracted from the pH 6.1 structure, coordinated: Tyr35 conformation extracted from the pH 10 structure. As a control the fraction of the Tyr35
conformation at pH 6.1 in the fluoride coordinated SOD was determined. All values are given separately for subunit A and B.
A B
pH 6�.1 pH 10 pH 6.1 pH 10
Condition uncoordinated coordinated uncoordinated coordinated
Fluoride 0�.15 0�.85 0.20 0.80
pH 6�.1 1�.00 0 1.00 0
pH 7�.4 0�.70 0�.30 1.0 0
pH 8�.1 0�.58 0�.42 0.78 0.22
pH 8�.1 T = 140K 0�.48 0�.52 0.62 0.38
pH 9�.3 0�.48 0�.52 0.80 0.20
pH 10 0�.22 0�.78 0.21 0.79

124 M. Schmidt (Eur. J. Biochem. 262) q FEBS 1999
the active centers within the crystal were estimated to contain aniron. If the refined occupancy value is divided by this number,the ligand : iron ratio can be obtained (Table 4) in the presenceof an impurity. A comparison with results obtained by methodslike XAS, EPR and MoÈssbauer spectroscopy that specificallysense the iron even within an appreciable amount of other metalions becomes possible. In Fig. 7 the ligand : iron ratio is plottedas function of pH for both subunits. Both active centers arehexacoordinated at pH 10 and the ligand : iron ratio is nearly90%. At pH 9.3 the ligand : iron ratio is 50% in subunit Awhereas in subunit B only 20% remains. This is also visible bypeaks of different heights in the electron density. It seems that atthese pH values subunit B has a different binding constant forhydroxide ions compared to subunit A. As the overall structureof subunits A and B at pH 9.3 is similar (compare Fig. 3b), thereexists no structural reason for this behavior. Another explanationcould be that noncrystallographically related subunits sensedifferent chemical environments. Thus, the pH value within thecrystal might be different from that of the soaking solution. Thismakes comparison with other techniques that use frozensolutions difficult. Nevertheless, results obtained by XASinvestigations are reproduced at pH 10 and pH 6.1 (see [7]and Fig. 7, triangles). A discrepancy remains at about pH 8. Oneof the reasons for this might be that the present X-ray structuresare measured at room temperature whereas the XAS is measuredat 20 K. It was reported for another SOD that the cooling of theenzyme changes its visible absorption spectrum [19]. Thisthermodichroism was interpreted as a temperature dependentbinding of a water molecule to the 6th coordination site. As thevisible spectrum of the P. shermanii SOD is nearly featureless,an absorption change can hardly be monitored. In order tocompare the XAS results with the X-ray structure analysis, thedetermination of the structure at pH 8.1 was repeated at 140 K.The higher resolution was of great advantage with regard tointerpreting the result. The electron density at the 6thcoordination site was clearly observable in both subunits(Fig. 8). The ligand : iron ratio at the 6th coordination site is58% and 47% (Fig. 7) for subunit A and B, respectively. Insubunit A this ratio is only 10% higher compared to that at room
temperature, whereas in subunit B a significant increase isobservable. It is conspicuous that the ligand : iron ratio insubunit B at low temperatures match that observed in subunit Aat room temperature. This might be a further indication of a pHgradient between the subunits that may vanish during thecooling process. Therefore, it is likely that a temperaturedependent increase of the sixfold coordination is only small inthe SOD. Nevertheless, ligand : iron ratios of 60% found insubunit A at pH 8.1 and low temperatures are likely to besufficient to explain the results obtained by XAS.
The first coordination shell of the iron
The first coordination shell of the iron is determined by theamino acid residues directly bound to the iron (His27, His75,His165 and Asp161) in addition to one or more solventmolecules depending on the pH. If the ligand : iron ratio issmaller than 1, the electron density represents an admixture ofthe five coordinated impurity, the low-pH Fe(III) species and the6th coordinated high-pH Fe(III) species. At pH 10, the fractionof protein molecules in the high-pH state is so large thatsignificant structural changes within the first coordinationsphere are observable. These take place at the position ofHis75 and His165 (see Fig. 9). The iron, Asp161Od1, His75Ne2
and His165Ne2 are lying approximately in the same plane. Thesolid line shown in Fig. 9 connects the His75 and His165positions observable at pH 6.1. At pH 10, these histidines moveto new positions indicated by the dotted lines. In theaforementioned plane, all three angles change. This is shownschematically in Fig. 10a and 10b, respectively. A trigonalbipyramid is formed by the five atoms coordinated to the iron atpH 6.1. The sixfold coordination found at pH 10 is indicated bythe octahedron. The angles between the coordinated atoms andthe central iron are shown in Table 6. The angle enclosed byHis27, Fe and Ow1 is about 1808 at pH 6.1. It remains nearlyconstant over the pH range. If the iron is fivefold coordinated,the angles lying in the trigonal plane are expected to be close to1208. This is true only for the angle His165, Fe, Asp161. Theother ones deviate significantly. Nevertheless, in the transfer tohigh-pH the angles change towards a tetragonal symmetry.Asp161 hardly moves at all and His27 remains relatively stablewhereas His75 and His165 move relative to the iron. Angularand structural changes are equivalent to those induced by thebinding of fluoride and azide (see also Table 6, the structure ofthe azide coordinated SOD was taken from [15]). No matterwhat kind of molecule binds to the free 6th site, similarstructural changes occur. In Table 7 the distances of the residuesin the first coordination shell to the iron are shown. Distancesderived from the X-ray structure analysis are also comparedwith those found from the XAS spectra. The XAS indicate adistance increase of about 0.005 nm from pH 6.1 to pH 10. Thistendency is also found in the X-ray structures. Nevertheless,compared to the X-ray structures the XAS underestimates thedistances about 0.01 nm at pH 10. A possible explanation forthis discrepancy might be structural differences betweendifferent metal kinds present in the SOD. These cannot bedistinguished by X-ray structure analysis. In the X-ray structuresthe histidine iron distances are 0.215 nm at low-pH and0.223 nm at pH 10 in the average. The 5th coordinated watermolecule binds about 0.22 nm from the iron. At a similardistance, but at a different site, the 6th coordinated hydroxideion can be found at high-pH. This site can also be occupiedby an atom of the azide molecule. Fluoride occupies a place0.02±0.03 nm closer to the iron compared to the hydroxide ion.Thus, the position of the 6th coordination site is defined at a
Fig. 11. Structure of the active center at pH 10, subunit A. Fobs-Fcalc
electron density was calculated by omitting Ow1 and Ow2. Contour level,
3.5 s. Tyr35 conformations observed at pH 6.1, 8.1 and 9.3 were inserted for
comparison.

q FEBS 1999 Coordination no. manipulation of SOD ferric iron (Eur. J. Biochem. 262) 125
radial distance of 0.21 nm ^ 0.015 nm from the iron on astraight line from the iron to the fluoride ion. This location actsas the binding site for the substrate O2
´±.
The second coordination shell of the iron
The second coordination shell consists of amino acids that shieldthe active center from the solvent and the protein matrix andmight contribute to the catalytic activity. As in other Fe or MnSODs, a small channel is left open that connects the activecenter with the solvent space. The 6th coordination site is lyingexactly in that channel. One of the residues that protrude intothis channel is Tyr35. Large structural changes in the secondcoordination sphere are only observable at Tyr35. Tyr35 isbelieved to be a crucial residue in the catalytic cycle [20,21].Located near the hydroxy group of the phenyl ring, this residuecan establish a hydrogen bond with the molecule bound to the6th coordination site. Typical H-bond lengths measured from thecenter of an oxygen (that of the Tyr35 hydroxyl group) to thecenter of the fluoride or hydroxide ion are in the order of0.28 nm to 0.35 nm [22]. To establish a hydrogen bond, theTyr35 conformer found at pH 6.1 has to swing into the activecenter (Fig. 11 and Table 7). It is assumed that the displacementof Tyr35 is correlated to the structural changes in the firstcoordination shell and to the binding of the 6th coordinatedmolecule. To prove this assumption, the Tyr35 conformations atpH 10 together with those found at pH 6.1 were used to interpretthe electron density at each particular pH value. The extractionof the Tyr35 coordinates together with their correspondingB-values was performed similar to the procedure describedabove at each pH value. The occupancy value for each of thetwo Tyr35 conformations was determined by performing30 cycles of grouped occupancy refinement in xplor(Table 5). If the swinging in of the Tyr35 is correlated to thebinding of a sixth coordinated molecule, then the fraction of
Tyr35 in the high-pH state must reproduce the occupancy valuecalculated in Table 4. Compared to the occupancy refinement ofa single atom this method tends to give slightly larger values forthe coordinated state (compare Table 4 and Table 5). However,occupancy values that differ in subunit A and B are also wellreproduced and distinguished at pH 8.1 and 9.3. At pH 10similar results are obtained for both subunits. It is of note thatidentical structural changes are observed in the fluoridecoordinated SOD (Fig. 5). This indicates that the Tyr35displacement is induced by the binding of the sixth coordinatedmolecule like fluoride or hydroxide at high-pH values. Theseions are likely to be bound by a coordinative bond to the ironand by a hydrogen bond to Tyr35 (Fig. 11). The hydrogen bondfurther stabilizes the hexacoordinated iron complex. It isnoteworthy that Tyr35 swings into the opposite direction ifazide is bound. Because the distance of Tyr35 to the azide islarger than 0.35 nm, a hydrogen bond is less probable. On theother hand, azide can be bound to His32 (Fig. 5) by one of its N-atoms and therefore can be stabilized further at its position. Inthe case of the hydroxide ion the strength of the hydrogen bondto Tyr35 might be the reason for the low dissociation constantthat is discussed in the next section. However, the exact structureof the hydrogen bonds within the active center remains unknowndue to the limited resolution.
Structural changes and activity
The steady state overall reaction rate decays from25 £ 108´mol s21 at pH 6.4 to about 1 £ 108´mol s21 atpH 10 [3]. The origin of this inhibition is controversial, and isdiscussed in [2,5,7]. This work and the XAS investigations giveclear evidence for a blocking of the substrate binding site byvarious anions. Azide and fluoride act as competitive inhibitors,with comparable dissociation constants to the Fe(III). For azidethe KD(azi) is 2.0 mmol´L21, for fluoride the KD(F2) is
Table 7. Distances (in nm) from atoms in the first and second coordination shell to the iron in subunit A. Last column: distance Tyr35Oh to sixth
coordination site defined by the hydroxide, fluoride or atom 1 of the azide ion. Number in brackets denote the site that is not significantly occupied. Comparison
with XAS at pH 6.1 and pH 10 (data from [7]): The assignment of HisNe2 and oxygen atomic distances to residue numbers is arbitrary.
His27
Ne2
His75
Ne2
Asp161
Od1
His165
Ne2
Ow1
fluoride
azide Ow2
Tyr35
Oh
Tyr35-Ow2
Tyr35-F2
Tyr35-azi1
pH 6�.1 0�.213 0�.215 0�.183 0�.217 0�.210 ± 0�.521 ±
pH 7�.4 0�.225 0�.222 0�.193 0�.215 0�.200 (0�.238) 0�.510 0�.353
pH 8�.1 0�.219 0�.219 0�.186 0�.220 0�.209 0�.183 0�.486 0�.315
pH 9�.3 0�.217 0�.214 0�.196 0�.221 0�.208 0�.214 0�.475 0�.315
pH 10 0�.223 0�.223 0�.185 0�.221 0�.222 0�.209 0�.459 0�.300
pH 8�.1140K 0�.223 0�.214 0�.192 0�.213 0�.213 0�.236 0�.468 0�.295
fluoride 0�.214 0�.214 0�.184 0�.210 0�.210 0�.189 0�.453 0�.309
azide 0�.218 0�.214 0�.190 0�.213 0�.215 0�.225 0�.525 0�.371
XAS pH 6�.4 0�.209 0�.211 0�.186 0�.211 0�.197 ±
XAS pH 10�.0 0�.213 0�.215 0�.193 0�.216 0�.198 0�.207
Table 6. Angles (in degrees) found within the first coordination sphere of the iron in subunit A. The histidines coordinate with their Ne2 atoms, the aspartate
with the Od1 atom. Ow1 denotes the fifth coordinated solvent molecule, also visible at low-pH. Ow2 defines the molecule, either hydroxide, azide (azi1: atom 1)
or fluoride (F-) at the sixth coordination site. Numbers in brackets denote angles derived from the insignificantly occupied 6th coordination site at pH 7.4.
pH 6.1 pH 7.4 pH 8.1 pH 9.3 pH 10 F- azi1 pH 8.1/140 K
His27-Fe-Ow1 179 176 174 176 174 177 175 172
His165-Fe-His75 134 135 138 143 147 144 149 147
His75-Fe-Asp161 107 108 104 105 103 102 98 99
Asp161-Fe-His165 120 117 117 111 109 113 112 114
His-165-Fe-Ow2 ± (68) 68 74 75 83 72 73
Ow2-Fe-His75 ± (67) 72 70 75 61 77 74

126 M. Schmidt (Eur. J. Biochem. 262) q FEBS 1999
2.3 mmol´L21 [3]. In agreement with the XAS data it is shownhere that OH2 binds to the iron of the SOD. From the kineticdata it seems that half maximal inhibition of the SOD occursapproximately at pH 7.5. Hence, the KD(OH2) of this reactionshould be in the order of 0.5 mmol´L21. Therefore, theKD(OH2) is about four orders of magnitude smaller comparedto the KD values of fluoride and azide. In this way, OH2 is amuch stronger ligand than these anions. For the Fe-SOD ofP. shermanii the Michaelis constant (Km) determined at low-pH(, pH 8) is not known. It can be assumed that it is comparableto the apparent Km values of lpH-SODs that are measured tobe about 100 mmol´L21 [2,23]. This value is two orders ofmagnitude larger than the KD of OH2 and 20 times smallercompared to the KD of fluoride and azide. Fluoride and azidecan easily be replaced competitively by small concentrations ofthe substrate. At high-pH the situation changes. The SOD formsa stable complex with OH2 that is catalytically inactive even athigh substrate concentrations. Consequently, if another inhibi-tory compound is added at high-pH the SOD can only slightly beinhibited further. This is consistent with the results obtained by[3]. If one wants to determine the KD(OH2) by crystallographyit has to be proven that the pH in the crystal exactly matches thatof the soaking solution. This is not achieved here. Nevertheless,the X-ray data clearly elucidates one, if not the only, possiblemechanism to inhibit the enzyme at high-pH values. A similarpH-dependence of the activity is found in the (weakly active)Fe(sub)-MnSOD of Serratia marcescens [4]. However, the sameiron(III)-hydroxo complex is found within the (inactive)Fe(sub)-MnSOD of Escherichia coli at pH 8.5 [24]. Seemingly,there are evidences that the cambialistic P. shermanii SODis a natural Fe(sub)-MnSOD but with similar activity withFe and Mn.
ACKNOWLEDGEMENT
The author greatly acknowledges the support of Fritz Parak. The stimulating
discussions were very helpful.
REFERENCES
1. Terech, A., Pucheault, J. & Ferradini, C. (1983) Saturation behavior of
the manganese-containing superoxide dismutase from Paracoccus
denitrificans. Biochem. Biophys. Res. Commun. 113, 114±120.
2. Bull, C. & Fee, J.A. (1985) Steady states kinetic studies of superoxide
dismutases: properties of the iron containing protein from Escheri-
chia coli. J. Am. Chem. Soc. 107, 3295±3304.
3. Meier, B., Scherk, C., Schmidt, M. & Parak, F. (1998) pH dependent
inhibition by azide and fluoride of the iron superoxide dismutase from
Propionibacterium shermanii. Biochem. J. 331, 403±407.
4. Yamakura, F., Kobayashi, K., Harumi, U.E. & Konno, M. (1995) The
pH-dependent changes of the enzymic activity and spectroscopic
properties of iron-substituted manganese superoxide dismutase. Eur. J.
Biochem. 227, 700±706.
5. Tierney, D.L., Fee, J.A., Ludwig, M.L. & Penner-Hahn, J.E. (1995)
X-ray absorption spectroscopy on the iron site in E. coli Fe (III)
superoxide dismutase. Biochemistry 34, 1661±1668.
6. Iakovleva, O., Parak, F., Rimke, T., Meier, B., HuÈttermann, J. & Kappl,
R. (1996) The active center of superoxide dismutase from Propioni-
bacterium shermanii. Il Nuovo Cimento 18, 199±212.
7. Scherk, C., Schmidt, M., Nolting, H.F., Meier, B. & Parak, F. (1996)
EXAFS investigations of the active site of iron superoxide dismutase
of Escherichia coli and Propionibacterium shermanii. J. Eur. Biophys.
24, 243±250.
8. Lah, M.S., Dixon, M.M., Pattridge, K.A., Stallings, W.C., Fee, J.A. &
Ludwig, M.L. (1995) Structure-function in Escherichia coli iron
superoxide dismutase: comparisons with manganese enzyme from
Thermus thermophilus. Biochemistry 34, 1646±1660.
9. Schmidt, M., Meier, B. & Parak, F. (1996) X-ray structure of the
cambialistic superoxide dismutase from Propionibacterium shermanii
active with Fe or Mn. J. Biol. Inorg. Chem. 1, 532±541.
10. CCP4 (1994) CCP4, Collaborative computational project, number 4
Acta Crystallogr. D50, 760±763.
11. BruÈnger, A.T. (1992) The free R-value: a novel statistical quality for
assessing the accuracy of crystal structures. Nature 335, 472±474.
12. BruÈnger, A.T. (1996) X-plor (online), version 3.851, a system for X-ray
crystallography and NMR. Yale University.
13. Navaza, J. (1994) AMoRe: an automated package for molecular
replacement. Acta Crystallogr. A50, 157±163.
14. Read, R.J. (1986) Improved Fourier coefficients for maps using
phases from partial structures with errors. Acta Crystallogr. A42,
140±149.
15. Schmidt, M., Scherk, C., Iakovleva, O., Nolting, H.F., Meier, B. &
Parak, F. (1998) The structure of the azide coordinated superoxide
dismutase of Propionibacterium shermanii investigated by X-ray
structure analysis, extended X-ray absorption fine structure, MoÈss-
bauer and electron paramagnetic resonance spectroscopy. Inorganica
Chimica Acta 275±276, 65±72.
16. Meier, B., Barra, D., Bossa, F., Calabrese, L. & Rotilio, G. (1982)
Synthesis of either Fe- or Mn-superoxide dismutase with an apparently
identical protein moiety by an anaerobic bacterium dependent on the
metal supplied. J. Biol. Chem. 257, 13977±13980.
17. Martin, M.E., Byers, B.R., Olson, M.O.J., Salin, M.L., Arceneaux,
J.E.L. & Tolbert, C. (1986) A Streptococcus mutans superoxide
dismutase that is active with either manganese or ion as a
cofactor. J. Biol. Chem. 261, 9361±9367.
18. Jones, A., Zou, J.Y., Couran, S.W. & Kielgaard, M. (1991) Improved
methods for building protein models in electron density maps and
the location of errors in these models. Acta Crystallogr. A47,
110±119.
19. Whittaker, M.M. & Whittaker, J.M. (1996) Low-temperature thermo-
dicroismus marks a change in coordination for the metal ion in
manganese superoxide dismutase. Biochemistry 35, 6762±6770.
20. Hunter, T., Ikebukuro, K., Bannister, W.H., Bannister, J.V. & Hunter,
G.J. (1997) The conserved residue tyrosine 34 is essential for
maximal activity of iron-superoxide dismutase from Escherichia coli.
Biochemistry 36, 4925±4933.
21. Guan, Y., Hickey, M.J., Borgstahl, G.E.O., Hallewell, R.A., Lepock,
J.R., O'Connor, D., Hsieh, Y., Nick, H.S., Silverman, D.N. & Tainer,
J.A. (1998) Crystal structure of Y34F mutant human mitochondrial
manganese superoxide dismutase and the functional role of tyrosine
34. Biochemistry 37, 4722±4730.
22. Jeffrey, G.A. & Saenger, W. (1991) Hydrogen Bonding in
Biological Structures. Springer-Verlag, New York.
23. Bull, C., Niederhofer, E.C., Yoshida, T. & Fee, J.A. (1991) Kinetic
studies of superoxide dismutases: properties of the manganese-
containing protein from Thermus thermophilus. J. Am. Chem. Soc.
113, 4069±4076.
24. Edward, R.A., Whittaker, M.M., Jameson, G.B. & Baker, E.N. (1998)
Distinct metal environment in Fe-substituted manganese superoxide
dismutase provides a structural basis of metal specifity. J. Am.
Chem. Soc. 120, 9684±9685.





























