Embed Size (px)
Citation preview

Manifestations dermatologiquesdes maladies du système
hématopoïétique etoncologie dermatologique
Dermatologie et médecine, vol. 3

SpringerParisBerlinHeidelbergNew YorkHong KongLondresMilanTokyo

Didier Bessis
Manifestations dermatologiquesdes maladies du systèmehématopoïétique etoncologie dermatologiqueDermatologie et médecine, vol. 3
avec la collaboration deCamille Francès, Bernard Guillot et Jean-Jacques Guilhou

Didier Bessis
DermatologuePraticien hospitalierCentre hospitalier et universitaireHôpital Saint-Éloi80, avenue Augustin-Fliche34295 Montpellier cedex 5
Camille Francès
Professeur de dermatologie-vénérologieHôpital Tenon4, rue de la Chine75020 Paris
Bernard Guillot
Professeur de dermatologie-vénérologieChef du service de dermatologieCentre hospitalier et universitaireHôpital Saint-Éloi80, avenue Augustin-Fliche34295 Montpellier cedex 5
Jean-Jacques Guilhou
Professeur de dermatologie-vénérologieCentre hospitalier et universitaireHôpital Saint-Éloi80, avenue Augustin-Fliche34295 Montpellier cedex 5
ISBN-13 : 978-2-287-72091-8 Springer Paris Berlin Heidelberg New York
© Springer-Verlag France, Paris, 2009Springer-Verlag France est membre du groupe Springer Science + Business Media
Cet ouvrage est soumis au copyright. Tous droits réservés, notamment la reproduction et la représentation, la traduction, la réimpression, l’exposé, la reproduc-tion des illustrations et des tableaux, la transmission par voie d’enregistrement sonore ou visuel, la reproduction par microfilm ou tout autre moyen ainsi que laconservation des banques de données. La loi française sur le copyright du 9 septembre 1965 dans la version en vigueur n’autorise une reproduction intégrale oupartielle que dans certains cas, et en principe moyennant les paiements des droits. Toute représentation, reproduction, contrefaçon ou conservation dans une banquede données par quelque procédé que ce soit est sanctionnée par la loi pénale sur le copyright.
L’utilisation dans cet ouvrage de désignations, dénominations commerciales, marques de fabrique, etc. même sans spécification ne signifie pas que ces termessoient libres de la législation sur les marques de fabrique et la protection des marques et qu’ils puissent être utilisés par chacun.
La maison d’édition décline toute responsabilité quant à l’exactitude des indications de dosage et des modes d’emplois. Dans chaque cas il incombe à l’usager devérifier les informations données par comparaison à la littérature existante.
Couverture : Jean-François Montmarché

Auteurs
Sélim AractingiProfesseur des UniversitésPraticien hospitalierService de DermatologieHôpital Tenon4 rue de la Chine75020 Paris
Nicole BassetProfesseur des UniversitésPraticien hospitalierService de DermatologieHôpital Saint-Louis1 avenue Claude-Vellefaux75010 Paris
Frédéric BernardPraticien hospitalierService d’Hématologie pédiatriqueHôpital Arnaud-de-Villeneuve371 avenue du Doyen-Gaston-Giraud34295 Montpellier CEDEX 5
Didier BessisPraticien hospitalierService de DermatologieHôpital Saint-Éloi80 avenue Augustin-Fliche34295 Montpellier CEDEX 5
Marie Beylot-BarryProfesseur des UniversitésPraticien hospitalierService de DermatologieHôpital Haut-Lévêque33600 Pessac
Annie Bonnafoux-ClavèrePraticien hospitalierService de RadiothérapieHôpital Dupuytren2 avenue Martin-Luther-King87042 Limoges CEDEX
Jean-Marie BonnetblancProfesseur des UniversitésPraticien hospitalierService de DermatologieHôpital Dupuytren2 avenue Martin-Luther-King87042 Limoges CEDEX
Isabelle Bourgault-VilladaProfesseur des UniversitésPraticien hospitalierService de DermatologieHôpital Ambroise-Paré9 avenue Charles-de-Gaulle92100 Boulogne-Billancourt
Frédéric CambazardProfesseur des UniversitésPraticien hospitalierService de DermatologieHôpital NordAvenue Albert-Raimond42270 Saint-Priest-en-Jarez
Alain ClaudyProfesseur des UniversitésPraticien hospitalierService de DermatologieHôpital Édouard-Herriot5 place d’Arsonval69437 Lyon CEDEX 03
Pierre ClavèreProfesseur des UniversitésPraticien hospitalierService de RadiothérapieHôpital Dupuytren2 avenue Martin-Luther-King87042 Limoges CEDEX

VI Auteurs
Isabelle CoupierPraticien hospitalierService de Génétique médicaleHôpital Arnaud-de-Villeneuve80 avenue Augustin-Fliche34295 Montpellier CEDEX 5
Bernard CribierProfesseur des UniversitésPraticien hospitalierService de DermatologieHôpitaux universitaires de Strasbourg1 place de l’Hôpital67091 Strasbourg CEDEX
Michel DandurandPraticien hospitalierService de DermatologieHôpital CaremeauPlace du Professeur-Robert-Debré30029 Nîmes CEDEX
Olivier DereureProfesseur des UniversitésPraticien hospitalierService de DermatologieHôpital Saint-Éloi80 avenue Augustin-Fliche34295 Montpellier CEDEX 5
Brigitte DrenoProfesseur des UniversitésPraticien hospitalierService de DermatologieHôtel-DieuPlace Alexis-Ricordeau44093 Nantes CEDEX 1
Nicolas DupinProfesseur des UniversitésPraticien hospitalierService de DermatologieHôpital Cochin-Tarnier27 rue du Faubourg-Saint-Jacques75979 Paris CEDEX 14
Sylvie EuvrardPraticien attachéService de DermatologieHôpital Édouard-Herriot5 place d’ArsonvalLyon 69437 CEDEX 03
Florent GrangeProfesseur des UniversitésPraticien hospitalierService de DermatologieHôpital Robert-DebréAvenue du Général-Koenig51092 Reims CEDEX
Bernard GuillotProfesseur des UniversitésPraticien hospitalierService de DermatologieHôpital Saint-Éloi80 avenue Augustin-Fliche34295 Montpellier CEDEX 5
Jean KanitakisPraticien hospitalierService de DermatologieHôpital Édouard-Herriot5 place d’Arsonval69437 Lyon CEDEX 03
Nicolas KlugerChef de clinique des UniversitésAssistant des hôpitauxService de DermatologieHôpital Saint-Éloi80 avenue Augustin-Fliche34295 Montpellier CEDEX 5
Céleste LebbéProfesseur des UniversitésPraticien hospitalierService de DermatologieHôpital Saint-Louis1 avenue Claude-Vellefaux75010 Paris
MyriamMarqueChef de clinique des UniversitésAssistant des hôpitauxService de DermatologieHôpital CaremeauPlace du Professeur-Debré30029 Nîmes
Anne-Marie MohtyInterne des hôpitauxService de DermatologieHôpital Sainte-Marguerite270 boulevard Sainte-Marguerite13274 Marseille CEDEX 09

Auteurs VII
Gaëlle QuereuxPraticien hospitalierService de DermatologieHôtel-DieuPlace Alexis-Ricordeau44093 Nantes CEDEX 1
Catherine Renaud-VilmerPraticien hospitalierService de DermatologieHôpital Saint-Louis1 avenue Claude-Vellefaux75010 Paris
Marie-Aleth RichardProfesseur des UniversitésPraticien hospitalierService de DermatologieHôpital Sainte-Marguerite270 boulevard Sainte-Marguerite13274 Marseille CEDEX 09
Jean SibiliaProfesseur des UniversitésPraticien hospitalierService de RhumatologieHôpital de Hautepierre1 avenue de Molière67098 Strasbourg CEDEX
Agnès SparsaPraticien hospitalierService de DermatologieHôpital Dupuytren87042 Limoges CEDEX
Olivier VérolaMaître de conférences des UniversitésPraticien hospitalierService d’Anatomie pathologiqueHôpital Saint-Louis1 avenue Claude-Vellefaux75010 Paris
Dominique Vignon-PennamenPraticien attachéService d’Anatomie pathologiqueHôpital Saint-Louis1 avenue Claude-Vellefaux75010 Paris

Préface
J e suis très heureux de préfacer cette série de 5 volumes intitulée Dermatologieet Médecine. Le titre m’a d’abord un peu surpris. En effet, un lecteur profane
ou superficiel pourrait à première vue croire que la «Dermatologie » n’est pas de la«Médecine » et que, dans cette série publiée aux éditions Springer sous la directiondu docteur Bessis, les auteurs vont néanmoins s’évertuer à démontrer le contraire.Que c’est comme si l’on voulait démontrer que l’astrologie est vraiment une scienceen intitulant un ouvrage ou une série de publications « Astrologie et Sciences » !Fort heureusement, il n’en est rien. La «Dermatologie » est une science médi-
cale, celle de la pathologie du plus vaste et du plus lourd des organes humains,enveloppant le corps charnel, englobant les zones cutanéo-muqueuses transition-nelles oculaires, bucco-labiales et ano-génitales. Elle fut certes autrefois, et ellel’est encore des fois de nos jours, considérée par des confrères d’autres disciplinescomme une spécialité médicale à part, pas vraiment indispensable, pas vraimentsérieuse, où il n’y a pas d’urgence, où les soins locaux salissants inspiraient unecertaine répugnance, où la bénignité relative des affections traitées n’engageaitpas la santé publique, malgré l’appropriation par les dermatologues des maladiesdites vénériennes, où les pratiques médicales faisaient volontiers traiter les der-matologues de tanneurs ou de mégissiers.On a même failli craindre que la dermatologie ne soit entièrement « soluble »
dans les autres disciplines médicales, surtout après la création, notamment enFrance, de spécialités interdisciplinaires basées non sur la pathologie d’organe,mais sur le substrat étiologique ou pathogénique présumé des affections censéesêtre prises en charge par ces nouveaux spécialistes « transversaux », les infectio-logues, les immuno-allergologues, les généticiens, les cancérologues... Des pro-phètes inquiets voyaient déjà les eczémas et le psoriasis en immunologie clinique,les pyodermites et les mycoses en infectiologie, les acnés et les alopécies en endo-crinologie, les nævus et les carcinomes cutanés dans les centres anticancéreux... Ily eut de toute évidence quelques redistributions de rôles, notamment en matièrede MST, devenues des IST, davantage d’actes opératoires pris en charge par deschirurgiens plasticiens non dermatologues,mais aussi des réorientations internesdans notre spécialitémême, avec davantage de dermatologues se tournant versla médecine esthétique et se familiarisant plus avec les lasers, les fillings et lesminigrafts qu’avec les médicaments immunomodulateurs et les biothérapies. Aveccet argument imparable pour justifier cette orientation : « Il faut bien vivre de sonmétier ! » L’augmentation des servitudes administratives et déontologiques estsouvent invoquée comme une des causes déterminantes de ce choix.

X Préface
Cette évolution n’a en fin de compte pas eu d’effets pervers sur le contenu etsur la pratique de la spécialité. Elle a en revanche nettement fait apparaître quel’abondance des lésions et des syndromes cutanés élémentaires et des entitésqu’elles expriment, leur reconnaissance facile par les spécialistes formés à cettediscipline, et leur accès direct à l’inspection et au prélèvement rendaient l’avis desdermatologues indispensable dans les disciplines transversales dans lesquelleson craignait de voir fondre la nôtre. Les dermatologues ont acquis avec cetteévolution, en quelques décennies, un état d’esprit de plus en plus « interniste »et ont pu se convaincre et convaincre autrui que la grande majorité des maladiescutanées, hormis quelques dermatoses exogènes ou mécanogènes, s’inscriventdans le contexte d’affections systémiques. Ils sont souvent aux avant-postes dansla suspicion puis la reconnaissance diagnostique de ces affections, par la démarcheséméiologique et nosologique propre à la spécialité, qui n’a pas vieilli,mais s’estau contraire enrichie par les contacts multidisciplinaires. N’était-il d’ailleurs paslogique de prévoir que la pathologie de l’enveloppe du corps entier ne pouvait querenforcer le concept et le besoin d’une pratique médicale dite de l’«homme global »,qui reviennent sans cesse dans les propos de l’éthiquemédicale et dans les objectifsd’enseignement et de formation professionnelle ?
L’ouvrage collectif coordonné par Didier Bessis avec la collaboration de BernardGuillot et de Jean-Jacques Guilhou, tous les trois de Montpellier, et de CamilleFrancès de Paris, avec de très nombreux auteurs, une centaine au total, presquetous français, est exemplaire de cette évolution de notre spécialité. Les nombreuxchapitres, plus de 120 répartis en 5 volumes,montrent qu’elle interfère sans arrêtavec les autres spécialités pour l’identification et la prise en charge d’innombrablesmaladies générales, depuis le lupus érythémateux jusqu’aux états psychotiques. La« Dermatologie », c’est vraiment de la «Médecine » de l’homme global. La lectureet la consultation fréquente de cette série d’ouvrages sauront vous en convaincre.
Professeur Édouard GrosshansStrasbourg, France

Avant-propos
C e troisième volume de Dermatologie etMédecine est consacré auxmanifesta-tions cutanées etmuqueuses des maladies du système hématopoïétique et à
l’oncologie dermatologique. Poursuivant l’esprit des deux premiers volumes, cetouvrage se veut avant tout original, à destination du médecin clinicien dermato-logue, interniste ou oncologue.
L’ensemble des affections du système hématopoïétique à expression dermatolo-gique (syndromes myéloprolifératifs etmyélodysplasiques, leucémies) ou à pointde départ cutané (lymphomes et histiocytoses cutanés) est largement détaillé àla lumière des dernières avancées cliniques (classification, nouvelles entités) etthérapeutiques. La rédaction des chapitres traitant des cancers cutanés, incluantles carcinomes basocellulaires et épidermoïdes, la maladie de Bowen, la maladie dePaget, le carcinome neuro-endocrine, les mélanomes, les sarcomes, les carcinomesannexiels, les cancers post-transplantation et les métastases, a été confiée à despraticiens français, tous référents dans leur domaine.
Plusieurs chapitres synthétiques et originaux en langue française constituentl’opportunité pour le praticien de se familiariser avec des affections rares, com-plexes et, de ce fait, souventméconnues. Les déficits immunitaires congénitauxsont abordés sous un angle dermatologique et replacés de façon simplifiée dans leurcontexte général immuno-hématologique. Les génodermatoses prédisposant auxcancers constituent également un domaine où le dermatologue doit pleinements’impliquer à l’heure de lamise en place de centres de références et de compétencesnationaux et multidisciplinaires. L’abondante iconographie illustrant ces patholo-gies, parfois exceptionnelles,mais reconnaissables « au coup d’œil », prend ici toutson intérêt dans le cadre de leur dépistage.
Enfin, les chapitres consacrés aux effets secondaires des traitements oncolo-giques intègrent les réactions cutanées induites par la radiothérapie, l’actualisationdes effets indésirables cutanéo-muqueux des chimiothérapies, ainsi qu’un sujetd’actualité et en constante évolution sur les effets secondaires dermatologiquesdes cytokines et des nouvellesmolécules anticancéreuses (inhibiteurs du récepteurà l’EGF, de tyrosine-kinase et des protéasomes).
Qu’ilme soit permis d’adressermes vifs remerciements à l’ensemble des auteurs,collaborateurs, ainsi qu’à l’ensemble des collègues quim’ont confié sans réserveleur iconographie.

XII Avant-propos
Je n’oublie pas une équipe enthousiaste resserrée autour de Gilles Pérez(www.octidi.fr)pourunemise enpages toujours particulièrement attractive,Mapiepour ses figures talentueuses et Nathalie Huilleret des éditions Springer pour sapatience bienveillante.
Didier Bessis
Toute référence à un chapitre issu de cet ouvrage devra porter la mention : Nom de l’auteur, titre du chapitre.In : Bessis D, Francès C, Guillot B, Guilhou JJ, éds, Dermatologie et Médecine, vol. 3 : Manifestationsdermatologiques des maladies du système hématopoïétique et oncologie dermatologique. Springer-VerlagFrance, 2009 suivi des numéros de pages.

Sommaire
49 Hémopathies myéloïdes,lymphoïdes et leucémiesNicolas Kluger, Myriam Marque,Sélim Aractingi
50 Classification des lymphomescutanésOlivier Dereure
51 Lymphomes cutanés TépidermotropesOlivier Dereure
52 Lymphomes T cutanés (horsmycosis fongoïde et syndrome deSézary)Marie Beylot-Barry
53 Lymphomes B cutanésFlorent Grange
54 HistiocytosesDidier Bessis, Frédéric Bernard,Frédéric Cambazard
55 Troubles de l’hémostaseAgnès Sparsa
56 Déficits immunitaires primitifsAgnès Sparsa, Jean Sibilia, DidierBessis
57 Maladie du greffon contre l’hôteAnne-Marie Mohty, Marie-AlethRichard
58 Carcinomes basocellulairesMichel Dandurand
59 Carcinomes épidermoïdesNicole Basset, Catherine Renaud-Vilmer
60 Maladie de BowenIsabelle Bourgault-Villada
61 Maladie de PagetJean Kanitakis
62 Carcinomes annexielsBernard Cribier
63 Carcinome neuro-endocrinecutanéAlain Claudy
64 MélanomesGaëlle Quereux, Brigitte Dreno
65 Maladie de KaposiBernard Guillot, Nicolas Dupin
66 Sarcomes cutanésCéleste Lebbé,Catherine Renaud-Vilmer,Marie-Dominique Vignon-Pennamen,Olivier Vérola

XIV Sommaire
67 Cancers cutanés aprèstransplantation d’organeSylvie Euvrard, Jean Kanitakis,Alain Claudy
68 Génodermatoses prédisposant auxcancersDidier Bessis, Myriam Marque,Nicolas Kluger, Isabelle Coupier
69 Syndromes paranéoplasiquesdermatologiquesDidier Bessis
70 Métastases cutanéesBernard Guillot
71 Effets cutanéo-muqueux indésirablesdes chimiothérapies antitumoralesDidier Bessis, Bernard Guillot,Olivier Dereure
72 Effets cutanéo-muqueux indésirablesdes cytokines et des nouvellesmolécules anticancéreusesDidier Bessis, Olivier Dereure,Bernard Guillot
73 Réactions cutanées induites parles rayonnements ionisantsPierre Clavere, Annie Bonnafoux-Clavere,Jean-Marie Bonnetblanc

49Hémopathies myéloïdes, lymphoïdeset leucémiesNicolas Kluger, Myriam Marque, Sélim Aractingi
Définition et classification 49-1Hémopathies myéloïdes 49-1Leucémies aiguës 49-2
Lésions cutanées spécifiques 49-3Lésions typiques 49-3Lésions atypiques 49-4Lésions muqueuses 49-4Leucémies cutanées aleucémiques 49-4Vasculite leucémique 49-4
Hémopathies lymphoïdes 49-5
Lymphome T de type lymphadénopathie angio-immunoblastique49-5Leucémie lymphoïde chronique B 49-6
Lésions cutanées « satellites » 49-6Dermatoses neutrophiliques 49-6Érythème noueux 49-8Manifestations vasculaires 49-8Vasculites 49-9Autres lésions satellites 49-9Lésions cutanées infectieuses 49-11
Références 49-11
L es hémopathies malignes peuvent s’accompagner delésions cutanées ou muqueuses dont la connaissance
est importante à double titre : elles peuvent être les pre-mières manifestations de l’hémopathie permettant sa re-connaissance et certaines d’entre elles marquent un tour-nant significatif du pronostic de l’affection imposant unemodification thérapeutique. La difficulté clinique reposeprincipalement sur le polymorphisme lésionnel des lésionsdermatologiques à l’origine d’une importante diversité desprésentations cliniques. La classification historique oppo-sant les lésions cutanées dites « spécifiques » des « non spé-cifiques » est abandonnée au profit d’une classification plusfonctionnelle distinguant :− les lésions cutanées spécifiques liées à l’infiltration des
cellules tumorales dans la peau ou les muqueuses ;− les lésions cutanées dites « satellites », paranéopla-
siques, même si la définition stricto sensu du caractèreparanéoplasique n’est pas constante ;
− les infections cutanées ;− les effets secondaires cutanéo-muqueux des traite-
ments des hémopathies, en priorité les chimiothérapies,abordés en détail dans le chap. 71, « Effets cutanéo-muqueuxindésirables des chimiothérapies antitumorales ».
Définition et classification
Hémopathies myéloïdesSyndromes myéloprolifératifs Ils sont caractérisés parune hyperactivité de la moelle osseuse liée à une réponse
anormale des précurseurs myéloïdes aux régulations phy-siologiques. La prolifération cellulaire, sans blocage de ma-turation, sanguine et tissulaire (rate, foie) porte sur aumoins une des trois lignées myéloïdes érythrocytaire, pla-quettaire ou granulocytaire. Les principaux syndromesmyé-loprolifératifs sont rappelés dans le tableau 49.1. Les cellulescirculantes dans le sang sont aux stades terminaux nor-maux de la lignée ou des précurseurs très différenciés (myé-locytes,métamyélocytes).Une association de deux ou troislignées cellulaires n’est pas rare à l’origine de syndromesmyéloprolifératifs inclassables. Les principaux risques évo-lutifs comprennent la thrombose vasculaire, et à terme,une transformation en leucémie aiguë myéloblastique.Syndromes myélodysplasiques Ils forment un groupehétérogène d’affections hématologiques caractérisées pardes troubles de la maturation médullaire, ou dysmyélo-poïèse, d’au moins deux lignées cellulaires hématopoïé-tiques, au sein d’une moelle osseuse de densité le plussouvent normale ou augmentée. Ces anomalies de la ma-turation des précurseurs des lignées sanguines se tra-duisent à la fois par des cytopénies de profondeur variableet des anomalies fonctionnelles des cellules circulantes.En 1982, la classification franco-américano-britannique(FAB) a individualisé chaque myélodysplasie au sein dece groupe. Cette classification a été remodelée par l’Orga-nisation mondiale de la santé (OMS) en 1999, avec l’ap-parition de nouveaux critères pour les syndromes myé-loprolifératifs et les leucémies aiguës (tableau 49.2). Lessyndromes myélodysplasiques touchent surtout des per-

49-2 Hémopathies myéloïdes, lymphoïdes et leucémies
� LAM leucémie aiguë myéloblastique
Tableau 49.1 Principaux syndromes myéloprolifératifs
Leucémie chroniquemyéloïde
Hyperleucocytose > 50 000/mm3 avecprédominance de polynucléairesneutrophiles, myélocytes etmétamyélocytesCaryotype médullaire : translocationt(9;22) (« chromosome Philadelphie »)
Polyglobulie primitive(maladie de Vaquez)
Hématocrite > 54 % (homme), > 47 %(femme)Volume globulaire total > 36 ml/kg(homme), > 32 ml/kg (femme)± thrombocytoseÉlimination des causes de polyglobuliessecondaires
Thrombocytémieessentielle
Thrombocytose > 600 000/mm3
Splénomégalie primitive(ou myélofibroseprimitive)
Fibrose médullaire importanteHyperleucocytose < 50 000/mm3
± polyglobulie ± thrombocytoseHématies en « larmes » sur le frottisÉvolution vers une insuffisance médullaire
Leucémiemyélomonocytairechronique
Sujet > 60 ans, anémie, thrombopénie,monocytose
sonnes âgées avec une prédominance masculine (sex-ratiode 1,5). Leur pronostic, défini par le score IPPS, est condi-tionné par trois critères : la blastose médullaire, les ano-malies caryotypiques et la profondeur des cytopénies.Leur évolution est dominée par le risque infectieux etla transformation en leucémie aiguë myéloïde (30% descas).
Leucémies aiguësElles sont caractérisées par la prolifération de précurseurshématopoïétiques peu différenciés incapables d’acheverleur maturation. Les cellules se divisent d’abord dans lamoelle qu’elles envahissent jusqu’à occuper l’ensemble du
Tableau 49.2 Classifications des syndromes myélodysplasiques
Classification franco-américano-britannique (FAB) Classification de l’Organisation mondiale de la santé (OMS)
Anémie réfractaireblastose médullaire < 5 %
Anémie réfractaireSyndrome 5q−Cytopénie réfractaire avec atteinte multi-lignées
Anémie réfractaire sidéroblastiqueblastose médullaire < 5 %, sidéroblastes en couronne > 15 %
Cytopénie réfractaire multi-lignées et anémie réfractaire avecsidéroblastes en couronne
Anémie réfractaire avec excès de blastesblastose médullaire 5 à 20 %
Anémie réfractaire avec excès de blastes I et II
Anémie réfractaire en transformationblastose médullaire entre 21 et 30 %
Leucémie aiguë myéloïde * : blastose médullaire > 20 % selon l’OMS(30 % selon la FAB)
Leucémie myélomonocytaire chroniqueblastose médullaire < 20 % et monocytose > 1000/mm3
Forme frontière de syndrome myéloprolifératif/myélodysplasique *
Forme inclassable
* Formes n’appartenant plus aux syndromes myélodysplasiques selon les nouveaux critères OMS
Coll.
DrL.
Énau
d,Pe
rpig
nan
Fig. 49.1 Multiples papules infiltrées d’une cuisse au cours d’uneleucémie aiguë lymphoblastique
volume médullaire avant de passer dans la circulation etd’essaimer dans d’autres tissus.Les leucémies se manifestent schématiquement de troisfaçons :− un syndrome tumoral clinique, reflet de la diffusion de
la maladie : adénopathies, splénomégalie ou d’autrestissus ;
− un syndrome d’insuffisance médullaire, reflet de la dys-fonction hématopoïétique, car les cellules leucémiques— qui remplacent les cellules normales — contraire-ment aux cellules des syndromes myéloprolifératifs,n’ont aucune fonctionnalité ;
− un syndrome thrombotique — moindre que dans lessyndromes myéloprolifératifs.
En fonction des aspects cytologiques, on distingue deuxgrands groupes de leucémies : les leucémies aiguës myélo-blastiques (LAM), caractérisées par une prolifération decellules blastiques granuleuses classées en LAM 0, 1, 2, 3...en fonction du type cellulaire identifié des leucémies aiguëslymphoblastiques, issues de la prolifération de précurseurslymphoïdes bloqués à un stade précoce. La distinction deces formes a des implications épidémiologiques, évolutiveset thérapeutiques.

Lésions cutanées spécifiques 49-3
Coll.
D.Be
ssis
Fig. 49.2 Papules et nodules cutanés spécifiques de la face latérale ducou au cours d’une rechute de leucémie aiguë myéloblastique de type 4
Lésions cutanées spécifiques
Les lésions cutanées dites « spécifiques » sont définies par laprésence histologique d’une infiltration et d’une proliféra-tion de cellules hématopoïétiques malignes dans le derme,l’hypoderme et/ou l’épiderme. Dans le cadre des leucémies,lesAnglo-Américains utilisent la terminologie de « leukemiacutis ».
Coll.
DrV.
Riga
u,M
ontp
ellier
Fig. 49.3 Histologie d’une lésion cutanée spécifique de leucémie :infiltrat dermique de cellules hématopoïétiques tumorales
Coll.
PrO.
Dere
ure,
Mon
tpell
ier
Fig. 49.4 Purpura diffus et pigmenté d’une jambe au cours d’uneleucémie aiguë myéloblastique
Lésions typiquesClassiquement, elles sont constituées de papules,deplaquesinfiltrées, de nodules, voire de tumeurs indolores, de consis-tance ferme, voire dure, et de couleur rose à violacée (fig. 49.1et 49.2). La taille des lésions et leur distribution sont va-riables. La multiplication rapide du nombre de lésions, lecaractère purpurique et la fermeté des lésions sont évoca-teurs du diagnostic. Le caractère lupoïde à la vitropressionn’élimine pas le diagnostic. Des formes historiques ont étédécrites avec une infiltration du visage conférant un aspectléonin ou une atteinte élective des lèvres, des paupièreset de l’orbite. Une forme multinodulaire disséminée, ca-ractérisée par des petits nodules durs, indolores, roses oucyanotiques, enchassés dans le derme et l’hypoderme etd’évolution rapide est également décrit. Le diagnostic cli-nique est facilement évoqué quand l’hémopathie est déjàconnue. L’examen histologique met en évidence dans lederme un infiltrat de cellules hématopoïétiques de mêmeaspect cytologique et phénotypique que celui de l’hémopa-thie myéloïde (fig. 49.3). En revanche, si cette dernière n’estpas identifiée, l’examen anatomopathologique peut êtred’interprétation difficile et devra être complété systémati-quement par les immunomarquages de ces cellules ¹.
Coll.
PrO.
Dere
ure,
Mon
tpell
ier
Fig. 49.5 Multiples lésions papuleuses et purpuriques du tronc au coursd’une leucémie aiguë myéloblastique
49-4 Hémopathies myéloïdes, lymphoïdes et leucémies
� LAM leucémie aiguë myéloblastique
Lésions atypiquesIl existe une grande diversité de présentation clinique deslésions cutanées spécifiques des hémopathies myéloïdes :bulles ²,³, nécrose, prurigo, purpura ou ecchymoses (fig. 49.4et 49.5). Leur fréquence est plus élevée au cours des syn-dromes myélodysplasiques ³,⁴. Leur reconnaissance est es-sentielle car elles sont prédictives d’une acutisation du syn-dromemyélodysplasique en leucémie aiguë dans les 3moisqui suivent ⁴. Savoir répéter les biopsies et se méfier de lé-sions cutanées d’allure banale ou pseudo-infectieuse estune règle à garder à l’esprit chez tout patient porteur d’unemyélodysplasie. De nombreuses lésions atypiques à typed’érosion muqueuse, de vascularite ou de nécrose cutanéesont également décrites au cours des syndromes hyperéosi-nophiliques primitifs ⁵,⁶.
Lésions muqueusesUne hypertrophie gingivale est présente dans 40 à 50%des leucémies myélomonocytaires aiguës (LAM-4) etmo-noblastiques (LAM-5). Elle correspond histologiquementà un infiltrat tumoral dense. Des atteintes érosives superfi-cielles buccale (gingivite, stomatite), plus rarement analeou vulvaire ⁷,⁸, ont été décrites lors de leucémies aiguësou de transformation aiguës de leucémies chroniques. Desulcérations spécifiques du scrotum ont également été rap-portées ⁹. Les lésions muqueuses génitales peuvent parfoisfaire évoquer à tort unemaladie de Behçet ⁸,mais leur carac-tère hémorragique et la présence de pétéchies à proximitéorientent vers le diagnostic de lésions spécifiques d’hémo-pathies. Des tumeurs ulcérées des muqueuses labiales, lin-guales et palatines ainsi que des muqueuses ano-génitales,parfois compliquées de surinfection, de nécrose et d’hémor-ragies, ont également été décrites (fig. 49.6), le plus souventau cours des leucémies myélomonocytaires et monoblas-tiques ¹⁰. Exceptionnellement, un chlorome (sarcome myé-loïde) peut avoir une localisation muqueuse ¹¹.
Coll.
PrA.
-J.Ciu
rana
,Mon
tpell
ier
Fig. 49.6 Prolifération tumorale et nécrotique de la région anale aucours d’une leucémie aiguë lymphoblastique
Coll.
D.Be
ssis
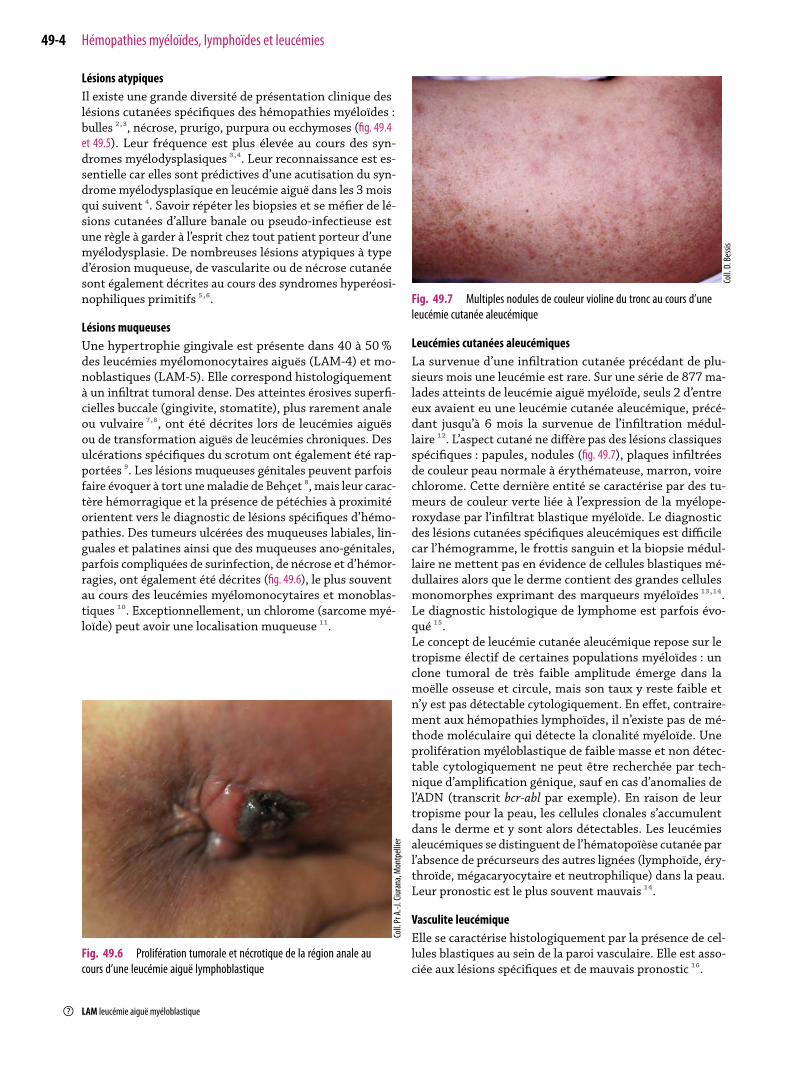
Fig. 49.7 Multiples nodules de couleur violine du tronc au cours d’uneleucémie cutanée aleucémique
Leucémies cutanées aleucémiquesLa survenue d’une infiltration cutanée précédant de plu-sieurs mois une leucémie est rare. Sur une série de 877ma-lades atteints de leucémie aiguëmyéloïde, seuls 2 d’entreeux avaient eu une leucémie cutanée aleucémique, précé-dant jusqu’à 6 mois la survenue de l’infiltration médul-laire ¹². L’aspect cutané ne diffère pas des lésions classiquesspécifiques : papules, nodules (fig. 49.7), plaques infiltréesde couleur peau normale à érythémateuse,marron, voirechlorome. Cette dernière entité se caractérise par des tu-meurs de couleur verte liée à l’expression de la myélope-roxydase par l’infiltrat blastique myéloïde. Le diagnosticdes lésions cutanées spécifiques aleucémiques est difficilecar l’hémogramme, le frottis sanguin et la biopsie médul-laire ne mettent pas en évidence de cellules blastiques mé-dullaires alors que le derme contient des grandes cellulesmonomorphes exprimant des marqueurs myéloïdes ¹³,¹⁴.Le diagnostic histologique de lymphome est parfois évo-qué ¹⁵.Le concept de leucémie cutanée aleucémique repose sur letropisme électif de certaines populations myéloïdes : unclone tumoral de très faible amplitude émerge dans lamoëlle osseuse et circule, mais son taux y reste faible etn’y est pas détectable cytologiquement. En effet, contraire-ment aux hémopathies lymphoïdes, il n’existe pas de mé-thode moléculaire qui détecte la clonalité myéloïde. Uneprolifération myéloblastique de faible masse et non détec-table cytologiquement ne peut être recherchée par tech-nique d’amplification génique, sauf en cas d’anomalies del’ADN (transcrit bcr-abl par exemple). En raison de leurtropisme pour la peau, les cellules clonales s’accumulentdans le derme et y sont alors détectables. Les leucémiesaleucémiques se distinguent de l’hématopoïèse cutanée parl’absence de précurseurs des autres lignées (lymphoïde, éry-throïde, mégacaryocytaire et neutrophilique) dans la peau.Leur pronostic est le plus souvent mauvais ¹⁴.
Vasculite leucémiqueElle se caractérise histologiquement par la présence de cel-lules blastiques au sein de la paroi vasculaire. Elle est asso-ciée aux lésions spécifiques et de mauvais pronostic ¹⁶.

Hémopathies lymphoïdes 49-5
� EBV Epstein-Barr virus · HHV human herpes virus
Coll.
DrC.
Gira
rd,M
ontp
ellier
Fig. 49.8 Éruption polymorphe maculeuse et papuleuse du tronc, par endroit nécrotique, au cours d’une lymphadénopathie angio-immunoblastique
Hémopathies lymphoïdes
Les localisations cutanées des hémopathies lymphoïdes seprésentent pour l’immense majorité d’entre elles commedes tumeurs, parfois à centre nécrosé ¹⁷. Deux types cli-niques méritent d’être soulignés.
Lymphome T de type lymphadénopathie angio-immunoblastique(LAI)
Il se caractérise par l’association de signes généraux (fièvre,sueurs nocturnes, amaigrissement), d’une hépatospléno-mégalie et d’adénopathies diffuses. Histologiquement, ilest défini par lamise en évidence sur biopsie ganglionnaire :1o d’une disparition de l’architecture ganglionnaire clas-sique, 2o d’un infiltrat polymorphe constitué d’immu-noblastes, de lymphocytes, de plasmocytes, d’éosinophileset d’histiocytes épithélioïdes, 3o et d’une hyperplasievasculaire arborescente post-capillaire. Les anomalies bio-logiques associées comprennent une anémie, une leucocy-tose et une hypergammaglobulinémie polyclonale. Cetteaffection peut toucher l’adulte à tout âge,mais concerne plu-tôt les sujets âgés (médiane de survenue de 64 ans environ)avec une prédominance masculine (sex-ratio de 1,4H/1 F).L’atteinte des enfants est rare mais possible. Son pronosticpéjoratif est principalement lié au risque d’infection et detransformation en lymphome de haut grade, avec un tauxde mortalité de 50 à 72% et une médiane de survie de 11 à30 mois.Des manifestations cutanées sont présentes dans la moi-
tié des cas et s’intègrent le plus souvent dans la présenta-tion clinique initiale. Il s’agit le plus souvent d’une érup-tion maculo-papuleuse, non spécifique, simulant un exan-thème viral ou une toxidermie ¹⁸,¹⁹.On peut aussi observerun purpura, des plaques infiltrées du tronc, des nodules,des papules de prurigo, des papules vésiculeuses ou urti-cariennes ou une vascularite (fig. 49.8). Des études clinico-histopathologiques ont mis en évidence l’existence de pos-sibles infiltrats spécifiques de lymphocytes atypiques aucours d’éruptions en plaques infiltrées d’allure spécifique,mais également au cours d’éruptions peu spécifiques d’al-lure virale.Des nodules granulomateux cutanés superficielset profonds ¹⁹, une nécrolyse épidermique toxique ²⁰, desdermatoses neutrophiliques polymorphes ²¹, un syndromede Wells ²², un eczéma craquelé ²³, une érythrodermie oudes lésions scléromyxœdème-like ²⁴ sont ponctuellementdécrits. Les lésions histologiques cutanées peuvent varierlors de biopsies répétées chez un même patient. L’étude deréarrangement TCR (TCell receptor) réalisée à partir des pré-lèvements cutanés peutmettre en évidence un clone ²⁵.Uneatteinte cutanée initiale est considérée comme un facteurde mauvais pronostic pour l’évolution de la LAI. Commedans les autres syndromes lymphoprolifératifs, le prurit estun symptôme fréquent, présent chez un tiers des patients.La pathogénie des LAI est encore débattue. L’implicationdemécanismes immunitaires en réaction à une stimulationtoxique (médicamenteuse : antibiotiques et anticonvulsi-vants) ou virale (principalement EBV, mais aussi HHV6et HHV8) avec prolifération lymphocytaire B secondaireet développement d’un clone lymphocytaire B a été initia-

49-6 Hémopathies myéloïdes, lymphoïdes et leucémies
� DN dermatose neutrophilique
Coll.
PrB.
Guill
ot,M
ontp
ellier
Fig. 49.9 Lésions papuleuses et nodulaires, nécrotiques du visage :lésions cutanées spécifiques d’une leucémie lymphoïde chronique
Coll.
D.Be
ssis
Fig. 49.10 Larges macules cutanées érythémateuses et squameuses :lésions cutanées spécifiques d’une leucémie lymphoïde chronique
lement postulée. Cependant la progression de la maladievers des lymphomes T de haut grade et l’évolution des tech-niques d’immunophénoypage, de biologie moléculaire etde cytogénétique font évoquer une prolifération clonalelymphocytaire T primitive. Les LAI sont ainsi actuellementclassées par l’OMS parmi les lymphomes T périphériques.
Leucémie lymphoïde chronique BLes manifestations cutanées sont présentes dans un quartdes cas et le plus souvent non spécifiques et rarement inau-gurales (15% des cas). Les lésions spécifiques sont consti-tuées par des papules érythémateuses localisées ou générali-sées (fig. 49.9), des plaques (fig. 49.10), des nodules et de largestumeurs (fig. 49.11). Une infiltration violine des oreilles etdu nez secondaire à l’envahissement tumoral du derme estévocatrice. Les lésions cutanées peuvent également se lo-caliser sur des cicatrices cutanées d’herpès ou de zona ²⁶.Du point de vue histologique, l’infiltrat lymphoïde est leplus souvent périvasculaire et périannexiel ou nodulairedermique et hypodermique, associé parfois à un infiltrat in-
Coll.
D.Be
ssis
Fig. 49.11 Large ulcération tumorale de la face postérieure du membresupérieur au cours de la transformation d’une leucémie lymphoïdechronique en syndrome de Richter
flammatoire non spécifique. Il est composé de lymphocytesmonomorphes, de petite taille et à chromatine condensée.Un contingent de cellules blastiques est présent lors detransformation en syndrome de Richter ²⁷. L’étude immu-nohistochimique met en évidence un marquage cellulairepar CD5, CD19, CD20 et CD43. Contrairement aux leu-cémies myéloïdes, l’atteinte cutanée ne constitue pas unmarqueur pronostic péjoratif.De nombreuses autres atteintes cutanées non spécifiquesont été décrites : pemphigus paranéoplasique (deuxièmeétiologie après les lymphomes malins non hodgkiniens),hypersensibilité aux piqûres d’insecte (fig. 49.12), érythèmeannulaire centrifuge ²⁸, augmentation de fréquence des car-cinomes cutanés, vasculite cutanée, mucinose folliculaire.
Lésions cutanées « satellites »
Dermatoses neutrophiliquesLes dermatoses neutrophiliques (DN) peuvent s’observeren dehors de toute hémopathie,mais leur prévalence estsignificativement accrue au cours des hémopathies myé-loïdes. Elles comprennent le syndrome de Sweet, le pyo-derma gangrenosum, l’hidradénite eccrine neutrophilique,l’erythema elevatum diutinum, le syndrome de Sneddon-

Lésions cutanées « satellites » 49-7
� G-CSF granulocyte-colony stimulating factor · GM-CSF granulocyte macrophage colony stimulating factor · HNE hidradénite neutrophilique eccrine · PCR polymerase chain reaction ·PG pyoderma gangrenosum · SyS syndrome de Sweet
Coll.
D.Be
ssis
Fig. 49.12 Lésions papuleuses et bulleuses de la face postérieure dubras après piqûre d’insecte (réaction d’hypersensibilité) au cours d’uneleucémie lymphoïde chronique
Wilkinson et les abcès aseptiques neutrophiliques. Ellessont caractérisées par une infiltration stérile de polynu-cléaires neutrophiles matures dans le derme.Le syndrome de Sweet (SyS) est associé à une néopla-sie dans 20% des cas, majoritairement des hémopathies(85%) ²⁹. Parmi celles-ci, la leucémie myéloïde aiguë estle plus fréquemment en cause. Les SyS associés aux hé-mopathies sont cliniquement caractérisés par une sur-représentation de lésions vésiculeuses, bulleuses (fig. 49.13),ulcérées ²⁹ ou parune association à des lésions de pyodermagangrenosum ³⁰. Une atteinte plus fréquente des membreset/ou de la muqueuse orale est également classique. La
Coll.
D.Be
ssis
Fig. 49.13 Syndrome de Sweet bulleux au cours d’un lymphomesplénique
présence d’une anémie (82%) ou d’un taux anormal deplaquettes (68%) est également évocateur du caractère pa-ranéoplasique du SyS et l’absence de fièvre ou d’hyperleu-cocytose ne doit pas faire remettre en cause le diagnos-tic. Le SyS survient le plus souvent de manière concomi-tante ou postérieure au diagnostic de l’hémopathie. LesSyS précédant l’hémopathie sont rares et surviennent dansun délai inférieur à 8 mois ³¹. Ils affectent aussi bien leshommes que les femmes. Près de 70% des patients onteu au moins une récurrence du syndrome, le plus souventen raison d’une décroissance trop rapide de la corticothé-rapie générale. Dans plus de 50% des cas, une atteinte ex-tracutanée est décrite, essentiellement des myalgies, desarthralgies, et plus rarement des atteintes osseuse, pulmo-naire et hépatique ²⁹. Le SyS peut être secondaire à un trai-tement de l’hémopathie : G-CSF ³² essentiellement, acidetout-transrétinoïque ³³, GM-CSF ³⁴ ou imatinib ³⁵. Sur leplan histologique, la présence de cellules myéloïdes imma-tures associées à des neutrophiles matures et le caractèreclonal des neutrophiles de l’infiltrat (hybridation in situ,PCR d’une mutation) ont pu être mis en évidence. Le SySassocié aux hémopathies pourrait donc être lié à la migra-tion dans le derme de neutrophiles clonaux issus de la dif-férenciation du clone myéloblastique. Enfin, au cours desSyS associés auxmyélodysplasies ou induits par le G-CSF,des infiltrats lympho-histiocytaires dermiques associés auxneutrophiles ont pu être mis en évidence et font l’objet dediscussions nosologiques ³⁶-³⁸.Les syndromes myéloprolifératifs se placent au premierrang des causes hématologiques de pyoderma gangreno-sum (PG) et, parmi eux, la polyglobulie primitive (maladiede Vaquez) ³⁹. La maladie hématologique est souvent an-cienne, bien que le diagnostic simultané des 2 pathologiessoit possible. Une atteinte de la muqueuse buccale et ocu-laire a été rapportée exceptionnellement ⁴⁰. Aucun cas dePG précédant le diagnostic hématologique, ni aucune loca-lisation viscérale de PG n’ont été rapportés au cours desPG associés aux hémopathies myéloïdes. L’apparition d’unPG au cours un syndrome myéloprolifératif connu est pré-dictif d’une myélofibrose (50%) ou d’une acutisation ⁴⁰,⁴¹.La corticothérapie générale est le traitement de premièreintention avec une réponse excellente et rapide. Des cas dePG ont été rapportés au décours de leucémies aiguës et chro-niques myéloïdes, plus rarement au cours de syndromesmyélodysplasiques et de leucémie à tricholeucocytes.L’hidradénite neutrophilique eccrine (HNE) est associéedans plus de 85% des cas à des hémopathies myéloïdes,essentiellement la leucémie aiguë myéloblastique ⁴²,⁴³. Ils’agit d’un tableau caractérisé par des plaques érythéma-teuses et infiltrées d’aspect proche du SyS. Une localisa-tion périorbitaire est classique. La différence avec le SySest histologique puisque l’infiltrat neutrophilique est élec-tivement localisé autour des glandes et des canaux sudo-raux eccrines. La biopsie est nécessaire et doit s’accompa-gner d’une mise en culture en cas de fièvre ou de cytopénieafin d’éliminer une infection. Deux particularités sont no-tables pour l’HNE : (a) un début fréquent en phase d’aplasiepost-chimiothérapie soulignant à nouveau le paradoxe de
49-8 Hémopathies myéloïdes, lymphoïdes et leucémies
� DN dermatose neutrophilique · EED erythema elevatum diutinum · HNE hidradénite neutrophilique eccrine · LAM leucémie aiguë myéloblastique · PG pyoderma gangrenosum ·SMD syndrome myélodysplasique · SyS syndrome de Sweet
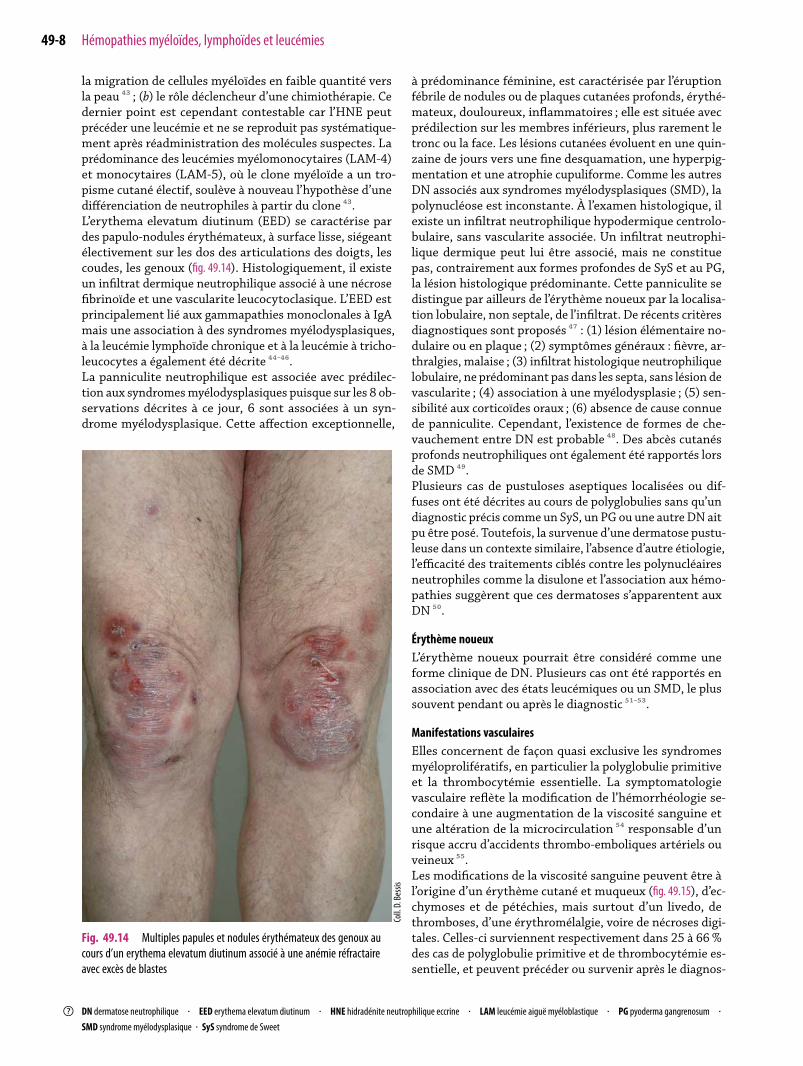
la migration de cellules myéloïdes en faible quantité versla peau ⁴³ ; (b) le rôle déclencheur d’une chimiothérapie. Cedernier point est cependant contestable car l’HNE peutprécéder une leucémie et ne se reproduit pas systématique-ment après réadministration des molécules suspectes. Laprédominance des leucémies myélomonocytaires (LAM-4)et monocytaires (LAM-5), où le clone myéloïde a un tro-pisme cutané électif, soulève à nouveau l’hypothèse d’unedifférenciation de neutrophiles à partir du clone ⁴³.L’erythema elevatum diutinum (EED) se caractérise pardes papulo-nodules érythémateux, à surface lisse, siégeantélectivement sur les dos des articulations des doigts, lescoudes, les genoux (fig. 49.14). Histologiquement, il existeun infiltrat dermique neutrophilique associé à une nécrosefibrinoïde et une vascularite leucocytoclasique. L’EED estprincipalement lié aux gammapathies monoclonales à IgAmais une association à des syndromes myélodysplasiques,à la leucémie lymphoïde chronique et à la leucémie à tricho-leucocytes a également été décrite ⁴⁴-⁴⁶.La panniculite neutrophilique est associée avec prédilec-tion aux syndromesmyélodysplasiques puisque sur les 8 ob-servations décrites à ce jour, 6 sont associées à un syn-drome myélodysplasique. Cette affection exceptionnelle,
Coll.
D.Be
ssis
Fig. 49.14 Multiples papules et nodules érythémateux des genoux aucours d’un erythema elevatum diutinum associé à une anémie réfractaireavec excès de blastes
à prédominance féminine, est caractérisée par l’éruptionfébrile de nodules ou de plaques cutanées profonds, érythé-mateux, douloureux, inflammatoires ; elle est située avecprédilection sur les membres inférieurs, plus rarement letronc ou la face. Les lésions cutanées évoluent en une quin-zaine de jours vers une fine desquamation, une hyperpig-mentation et une atrophie cupuliforme. Comme les autresDN associés aux syndromes myélodysplasiques (SMD), lapolynucléose est inconstante. À l’examen histologique, ilexiste un infiltrat neutrophilique hypodermique centrolo-bulaire, sans vascularite associée. Un infiltrat neutrophi-lique dermique peut lui être associé, mais ne constituepas, contrairement aux formes profondes de SyS et au PG,la lésion histologique prédominante. Cette panniculite sedistingue par ailleurs de l’érythème noueux par la localisa-tion lobulaire, non septale, de l’infiltrat.De récents critèresdiagnostiques sont proposés ⁴⁷ : (1) lésion élémentaire no-dulaire ou en plaque ; (2) symptômes généraux : fièvre, ar-thralgies,malaise ; (3) infiltrat histologique neutrophiliquelobulaire,ne prédominant pas dans les septa, sans lésion devascularite ; (4) association à une myélodysplasie ; (5) sen-sibilité aux corticoïdes oraux ; (6) absence de cause connuede panniculite. Cependant, l’existence de formes de che-vauchement entre DN est probable ⁴⁸. Des abcès cutanésprofonds neutrophiliques ont également été rapportés lorsde SMD ⁴⁹.Plusieurs cas de pustuloses aseptiques localisées ou dif-fuses ont été décrites au cours de polyglobulies sans qu’undiagnostic précis comme un SyS, un PG ou une autreDNaitpu être posé. Toutefois, la survenue d’une dermatose pustu-leuse dans un contexte similaire, l’absence d’autre étiologie,l’efficacité des traitements ciblés contre les polynucléairesneutrophiles comme la disulone et l’association aux hémo-pathies suggèrent que ces dermatoses s’apparentent auxDN ⁵⁰.
Érythème noueuxL’érythème noueux pourrait être considéré comme uneforme clinique de DN. Plusieurs cas ont été rapportés enassociation avec des états leucémiques ou un SMD, le plussouvent pendant ou après le diagnostic ⁵¹-⁵³.
Manifestations vasculairesElles concernent de façon quasi exclusive les syndromesmyéloprolifératifs, en particulier la polyglobulie primitiveet la thrombocytémie essentielle. La symptomatologievasculaire reflète la modification de l’hémorrhéologie se-condaire à une augmentation de la viscosité sanguine etune altération de la microcirculation ⁵⁴ responsable d’unrisque accru d’accidents thrombo-emboliques artériels ouveineux ⁵⁵.Les modifications de la viscosité sanguine peuvent être àl’origine d’un érythème cutané etmuqueux (fig. 49.15), d’ec-chymoses et de pétéchies, mais surtout d’un livedo, dethromboses, d’une érythromélalgie, voire de nécroses digi-tales. Celles-ci surviennent respectivement dans 25 à 66%des cas de polyglobulie primitive et de thrombocytémie es-sentielle, et peuvent précéder ou survenir après le diagnos-

Lésions cutanées « satellites » 49-9
� LAM leucémie aiguë myéloblastique · SMD syndrome myélodysplasique
tic. Plusieurs cas de livedos ramifiés ont été rapportés, af-fectant les membres inférieurs, notamment les pieds et lesorteils etparfois les fesses ⁵⁶.Unenécrose etdes ulcérationscutanées ainsi qu’une aggravation liée auxmodifications dela température peuvent compliquer le livedo ⁵⁷. L’histologiepeutmettre en évidence des thrombi fibrineux, une proli-fération endothéliale et un infiltrat inflammatoire de lym-phocytes, d’histiocytes et de polynucléaires neutrophiles.Les saignées itératives peuvent permettre une régressiondu livedo ⁵⁶. La thrombocytémie et la polyglobulie de Va-quez sont les deux principales causes d’érythromélalgie,acrosyndrome rare où les extrémités sont rouges, chaudeset douloureuses lors de l’exposition au chaud ⁵⁸. Les argu-ments orientant vers une étiologie secondaire d’érythromé-lalgie sont un début tardif, l’absence d’antécédent familial,le caractère asymétrique de l’atteinte, la localisation auxmembres inférieurs et l’existence de troubles trophiques.Ainsi, une numération formule sanguine doit être réaliséesystématiquement devant toute manifestation de type éry-thromélalgique. De plus, en l’absence de cause secondaire,un suivi doit être mis en place, surtout en l’absence d’an-técédent familial. La prise en charge de l’érythromélalgierepose sur le traitement étiologique du syndrome myélo-prolifératif et l’acide acétylsalicylique per os. Plusieurs ob-servations d’engelures précédant la survenue de leucémiemyélomonocytaire chronique ont été rapportées ⁵⁹. La livi-diose acrale, ou ischémie acrale avec lividité, est une entitéconsécutive à des thrombi de cellules myéloblastiques. Elles’observe exceptionnellement au cours des LAM très hyper-leucocytaires (> 100 000 blastes/mm3). Cette leucostaseestmultisystémique et une atteinte cardiorespiratoire etneurologique accompagne l’atteinte cutanée ⁶⁰.Les phlébites superficielles compliquent 6% des polyglo-bulies et peuvent s’observer au cours d’autres syndromesmyéloprolifératifs.La survenue d’ulcérations de jambe est possible lors des syn-
Coll.
D.Be
ssis
Fig. 49.15 Érythrose palmaire au cours d’une polyglobulie primitive
dromesmyéloprolifératifs, soit comme conséquence de l’hy-perviscosité, soit comme conséquence d’éventuelles throm-boses profondes artérielles ou veineuses. Il convient de nepas oublier d’évoquer systématiquement, en cas d’ulcèredes membres inférieurs chez un patient avec un syndromemyéloprolifératif, la possibilité d’une ulcération infectieuse,d’un pyoderma gangrenosum et du rôle éventuel de théra-peutiques comme l’hydroxyurée. Enfin, des ulcères « idio-pathiques » avec hyalinisation du derme ont été rapportés.
VasculitesLe lien entre vasculite et hémopathies malignes est ap-porté par une revue ancienne de la littérature, où 41 vas-culites furent mises en évidence au sein d’un groupe de75 000 hémopathies contre 11 vasculites rapportées pour889 000 tumeurs solides ⁶¹. Les hémopathies les plus sou-vent inductrices de vascularites sont la leucémie à tricho-leucocytes et les syndromes myéloprolifératifs. L’aspectclinique est polymorphe : purpura infiltré, lésions maculo-papuleuses, urticariennes, vésiculo-bulleuses, pustuleusesou nodulaires, à type d’érythème polymorphe ou d’ulcé-rations cutanées ⁶². Histologiquement, la vascularite estleucocytoclasique dans la majorité des cas, plus rarementgranulomateuse. Le plus souvent, les vasculites précèdent(26% des cas) ou sont concomitantes (40%) du diagnos-tic de l’hémopathie ⁶³. Dans une série, une nette prédomi-nance des hémopathies lymphoïdes B (62,5% de 16 hé-mopathies avec vascularite) et de l’image de vasculariteleucocytoclasique (13/16) était observée, ainsi que la pré-sence de lésions de vascularite extracutanée dans un tiersdes cas ⁶³. Au cours des SMD, les vasculites cutanées consti-tuent lesmanifestations cutanées les plus fréquemment ob-servées et sont révélatrices de l’affection dans près de 40%des cas ⁶⁴. La recherche d’une cause associée médicamen-teuse, auto-immune ou infectieuse doit être systématique.À titre d’exemple, la présence d’une infection à mycobac-téries était mise en évidence dans 88% des leucémies àtricholeucocytes associées à une vasculite ⁶⁵. Le traitementétiologique de l’hémopathie est habituellement inefficacepour contrôler la vasculite, même si elle peut apparaîtremoins sévère ⁶¹. La sévérité de la vasculite pourrait dimi-nuer avec la progression de l’hémopathie ⁶⁶. Les vasculitesn’évoluent habituellement pas de façon parallèle à l’hémo-pathie, notamment celles associées aux leucémies à tricho-leucocytes. Certains auteurs ont suggéré que la présencede ces vasculites était associée à un mauvais pronostic ⁶⁷.
Autres lésions satellitesLe prurit peut concerner près de 50% des patients atteintsde polyglobulie primitive ⁶⁸. Les symptômes sont majoréslors d’un bain ou de la douche et, dans certains cas, lesimple contact avec l’eau déclenche un prurit irrépressibledéfinissant un prurit aquagénique secondaire ⁶⁹. Le pruritpeut durer de 15 à 60 minutes ⁶⁸ et peut être handicapantau point que le patient finit par se priver de bain ⁶⁹. Il peutprécéder de nombreuses années le diagnostic de polyglo-bulie primitive ⁷⁰. Sa sévérité n’est pas corrélée à la gravitéde cette dernière. Le mécanisme physiopathologique pré-

49-10 Hémopathies myéloïdes, lymphoïdes et leucémies
� PDGF platelet-derived growth factor · TGF transforming growth factor
cis est mal connu. La carence martiale pourrait favoriserce phénomène, une supplémentation en fer permettantl’amélioration des symptômes dans une petite série ⁷¹. Untraitement martial ne peut cependant être donné au longcours au risque d’une élevation du taux d’hématocrite. L’his-tamine, les prostaglandines et la sérotonine seraient égale-ment impliquées. Les traitements antihistaminiques sontinefficaces. L’aspirine est le plus constamment efficace enraison de son action antiprostaglandine. Les antiséroto-ninergiques, la cimétidine, la cholestyramine ou la photo-thérapie UVA ont également été proposés ⁷². Près de 20%des patients continuent de présenter un prurit malgré uncontrôle parfait de l’hématocrite.La polychondrite chronique atrophiante (PCA) est une ma-ladie systémique rare caractérisée par une inflammationrécidivante des cartilages du nez, des oreilles et des voiesrespiratoires, parfois associée à des manifestations articu-laires à type de polyarthrite, et dans 20 à 50% des cas, à dessignes cutanés extracartilagineux. Le développement d’unsyndrome myélodysplasique est noté dans environ 30%des PCA et la prévalence des PCA est d’environ 0,6% aucours des syndromes myélodysplasiques ⁷³. Ce tableau dePCA peut précéder le syndrome myélodysplasique de plu-sieurs mois ou années. Les manifestations cutanées sontbeaucoup plus fréquentes chez les patients atteints à la foisde PCA et de syndrome myélodysplasique qu’au cours desPCA isolées ⁷⁴. Elles comportent des nodules des membressimulant un érythème noueux avec, en histologie, un infil-trat neutrophilique septal le plus souvent, parfois lobulaire ;un purpura ; un livedo ; des papules urticariennes ; des ulcé-rations de jambe ; des phlébites superficielles ; des papulesviolines ; une aphtose orale plus ou moins associée à deslocalisations génitales ; des érythèmes annulaires centri-fuges.Les xanthomes plans diffus normolipémiques se caracté-risent par la présence de xanthomes plans du visage, ducou, du tronc, des plis, des fesses et des paupières avecdes taux plasmatiques de cholestérol et de triglycéridesnormaux ⁷⁵. L’apparition de plaques diffuses, extensives,jaune orangé à jaune marron, asymptomatiques, parfoissurélevées à la palpation, ainsi que l’aggravation brutale dexanthelasmas préexistants doit faire rechercher une hémo-pathie sous-jacente ⁷⁶. Néanmoins, il pourrait exister unbiais de publication et le nombre de patients avec un xan-thome plan diffus idiopathique serait sous-estimé ⁷⁷. Lespatients avec un xanthome plan diffus doivent être suivisrégulièrement car les lésions cutanées peuvent précéder lamaladie hématologique ⁷⁵.L’hématopoïèse extramédullaire cutanée est une manifes-tation rare survenant principalement lors de la myélofi-brose primitive ⁷⁸-⁸¹. Les aspects cutanés sont polymorphes.Dans lamajorité des cas, il s’agit de papules de couleur chair,érythémateuse ou cyanotique, de nodules ou de plaques.Les lésions peuvent être asymptomatiques ou sensibles,augmentant progressivement en taille et en nombre. Deslésions restreintes à des sites de traumatismes, des lésionsbulleuses, hémorragiques ou des ulcères de jambe bilaté-raux ont également été décrites ⁷⁸. Les manifestations cli-
Affections cutanées rares et syndromesmyélodysplasiques
49.A
Dermatoses inflammatoires et auto-immunesMaladie de Behçet (association à une trisomie 8)PanniculitesAcrodermatite continue de HallopeauPemphigoïde bulleuse
Dermatoses de surchargeMucinose folliculaire
Dermatoses par infiltrationFolliculite pustuleuse à éosinophilesPanniculite à éosinophilesSyndrome de Sézary (non secondaire aux chimiothérapies employéespour le traitement du lymphome)Leucémies/lymphomes cutanés à cellules blastiques NKSarcoïdose cutanée et pulmonaireGranulome annulaire disséminéGranulomatoses non spécifiques disséminées ou localisées
Troubles de la kératinisationPorokératose disséminée
AutresPruritÉrythrodermiePhotosensibilitéPorphyrie cutanéeAngio-endothéliomatose réactionnelle
Affections cutanées rares et polyglobulie primitive de Vaquez
49.B
Cellulite de WellsSpongiose à éosinophilesSarcoïdoseMastocytoseSyndrome POEMSXanthogranulome juvénileIchtyose acquiseDystrophie lamellaire
niques de la myélofibrose stricto sensu ne sont pas spéci-fiques. Elles reflètent l’anémie (pâleur), la thrombopénie(purpura pétéchial et ecchymotique), l’avortement intra-médullaire (ictère). La splénectomie pourrait être un fac-teur déclenchant le développement d’une hématopoïèsecutanée ⁷⁸,⁷⁹. L’histologie montre invariablement une in-filtration du derme par des cellules hématopoïétiques àdifférents stades de maturation et représentant diverse-ment les 3 lignées hématopoïétiques : érythroïdes, myé-loïdes etmégacaryocytaires. L’hématopoïèse extramédul-laire semble être la conséquence de la sécrétion inappro-priée de PDGF (platelet-derived growth factor), de FGF (fi-broblast growth factor) et de TGF-β (transforming growthfactor β) par des clones anormaux de mégacaryocytes et de

Références 49-11
monocytes médullaires. L’hématopoïèse extramédullaireest due à la croissance normale d’un clone hématopoïétiquemais en dehors de la moelle osseuse, liés au passage desprogéniteurs médullaires dans la circulation systémiqueet à leur survie dans certains sites extramédullaires. Letraitement est symptomatique : radiothérapie à faff ible dose,électronthérapie associée au traitement de la myélofibrose.De nombreuses autres affecffff tions sont rapportées, ponc-tuellement associées aux syndromes myélodysplasiques etmyéloprolifératifsff , dont la polyglobulie primitive, et sontmentionnés dans les encadrés 49.A et 49.B.
Lésions cutanées infectieusesElles sont fréquentes et potentiellement graves en raisonde l’immunodépression induite par la maladie (neutropé-nie) et par la chimiothérapie. Il existe en outre, chez ces ma-lades, une fréquente atrophie cutanée liée aux corticoïdeset aux chimiothérapies aplasiantes et des portes d’entrée in-fecff tieuses cutanées comme les cathéters. Les infecff tions cu-tanées sont primitives dans 68% des cas et possèdent deuxcaractères particuliers qui doivent être connus. Le premierest secondaire à la neutropénie qui est responsable de ta-bleaux cliniques très divers, parfoff is atténués et faussemenff trassurants sans inflammation,ni pus ou nécrose. Le secondest la très grande diversité des germes— notamment op-portunistes — chez ces patients. La conséquence de ces2 difficultés sémiologiques etmicrobiologiques est la réali-sation systématique de biopsies cutanées pour cultures bac-
tériologiques, virales etmycologiques et d’un examen histo-logique avec colorations spéciales (Gram, Giemsa-Gomori-Grocot, Ziehl) devant toute lésion suspecte chez unmaladeen aplasie ou leucémique-neutropénique.L’ecthyma gangrenosum se caractérise par une plaque éry-thémateuse à bordure annulaire et à évolution nécrotiqueunique ou multiple. Il est dû au pseudomonas aeruginosaou à d’autres bacilles à Gram négatif secrétant des toxinesnécrosantes. Un point particulier doit être mentionné surles infecff tions fongff iques cutanées ⁸². La principale caused’infecff tion fongff ique invasive chez les patients neutropé-niques en hématologie est le Candida, suivi d’Aspe’ rgr illus etde Fusarium. L’aspergillose invasive est la première causede décès d’origine infecff tieuse. D’autres germes comme Rhi-zopus et Mucor voient leur incidence augmenter. Le der-matologue peut être confronté à une infecff tion cutanéeprimitive ou une dissémination hématogène avec localisa-tion secondaire cutanée. Les arguments pour une originefongique sont : l’absence de réponse à une antibiothérapiebactérienne, la présence d’une lésion nécrotique, la neutro-pénie persistante, une onychomycose associée. Fusariumet Aspergillus peuvent donner des lésions escarotiques enl’absence de paronychie. Enfin, les points de ponction (ca-théters) peuvent être le site d’infection par Aspergillus ouRhizopus. Les autres manifesff tations cutanées des mycosessont les foff lliculites, les nodules sous-cutanés et les abcès.Le taux de mortalité en cas de dissémination systémiquedes mycoses est de l’ordre de 70 à 80% des cas.
1 SeppN,RadaszkiewiczT,MeijerCJ etal. Spe-cific skinmanifesff tations in acute leukemia withmonocytic differentiation. A morphologic andimmunohistochemical study of 11 cases. Cancer1993 ; 71:124-132.2 Ochonisky S, Aractingi S, Dombret H et al.Acute undiffeffff rentiated myeloblastic leukemiarevealed by specific hemorrhagic bullous le-sions. Arch Dermatol 1993 ; 129:512-513.3 Longacre TATT , Smoller BR. Leukemia cutis.Analysis of 50 biopsy-proven cases with an em-phasis on occurrence in myelodysplastic syn-dromes. Am J Clin Pathol 1993 ; 100:276-284.4 Aractingi S, Bachmeyer C, Miclea JM et al.Unusual specific cutaneous lesions in myelodys-plastic syndromes. J Am Acad Dermatol 1995 ;33:187-191.5 Aractingi S, Janin A, Zini JM et al. Spe-cific mucosal erosions in hypereosinophilic syn-drome. Evidence foff r eosinophilprotein deposi-tion. Arch Dermatol 1996 ; 132:535-541.6 Aractingi S, Bachmeyer C, Pautier P et al.Necrotic cutaneous lesions induced by hyper-eosinophilic syndrome secondary to a T-celllymphoma. J Am AcadDermatol 2002 ; 46:S133-136.7 Senti G, Schleiffenbaumb Bffff , Dummera R.VagVV inal ulcers as initial presentation of suba-
cute myelomonocytic leukemia. Dermatologygg1999 ; 199:346-348.8 Granel B, Serratrice J, Ene N et al. Genitalulcerations revealing an acute myelomonocyticleukaemia. Rev Med Interne 2004 ; 25:770-772.9 Zax RH, Kulp-Shorten CL, Callen JP. Leuke-mia cutis presenting as a scrotal ulcer. J AmAcad Dermatol 1989 ; 21:410-413.10 Wu J, Fantasia JE, Kaplan R. Oralmanifesff -tations of acute myelomonocytic leukemia : acase report and review of the classification ofleukemias. J Periodontol 2002 ; 73:664-668.11 Ersahin C, Omeroglu G, Potkul RK, Sal-hadar A. Myeloid sarcoma of the vulva as thepresenting symptom in a patient with acutemyeloid leukemia. GyG necol Oncol 2007 ; 106:259-261.12 BaerMR, Barcos M, Farrell H et al. Acutemyelogenous leukemia with leukemia cutis.Eighteen cases seen between 1969 and 1986.Cancer 1989 ; 63:2192-2200.13 Bachmeyer C, Turc Y, Fraitag S et al.Aleukemic monoblastic leukemia cutis. AnnDer-matol Venereol 2003 ; 130:773-775.14 Wilkins R, Janes S. Aleukaemic leukaemiacutis : case report and review of the literature.Clin Lab Haematol 2004 ; 26:73-75.15 Ohno S, Yokoo T, Ohta M et al. Aleukemic
leukemia cutis. J Am Acad Dermatol 1990 ; 22:374-377.16 Jones D, Dorfman DMff , Barnhill RL, Grant-er SR. Leukemic vasculitis : a feaff ture of leukemiacutis in some patients. Am J Clin Pathol 1997 ;107:637-642.17 Mansouri S, Aractingi S. Manifesff tationscutanées des leucémies. EncyclMéd Chir (Édi-tions scientifiques etmédicales, Elsevier SAS),Paris, Dermatologie, 98-710-A10, 2004, 7 p.18 Martel P, LPP aroche L, Courville P et al. Cu-taneous involvement in patients with angioim-munoblastic lymphadenopathy with dyspro-teinemia. Arch Dermatol 2000 ; 136:881-886.19 Suarez-Vilela D, Izquierdo-Garcia FM. An-gioimmunoblastic lymphadenopathy-like T-celllymphoma : cutaneous clinicalonsetwithpromi-nent granulomatous reaction. Am J Surgical Pa-thol 2003 ; 27:699-705.20 Jones B, Vun Y, Sabah M et al. ToTT xic epider-mal necrolysis secondary to angioimmunoblas-tic T-cell lymphoma. Austrarr las JDermatol 2005 ;46:187-191.21 Sellier S, Levesque H, Courville P et al.Cyclophosphamide-induced neutrophilic dis-ease in a patient with angioimmunoblastic lym-phadenopathy and myelodysplastic syndrome.Ann Dermatol Venereol 2006 ; 133:459-462.

49-12 Hémopathies myéloïdes, lymphoïdes et leucémies
22 Renner R, Kaufer F, Treudler R et al.Eosinophilic cellulitis (Wells’ syndrome) in asso-ciation with angioimmunoblastic lymphadeno-pathy. Acta Derm Venerol 2007 ; 87:525-528.23 Van Voorst Vader PC, Folkers E, vanRhenen DJ. Craquelé-like eruption in angioim-munoblastic lymphadenopathy. Arch Dermatol1979 ; 115:370.24 Rahmani R, Brenner S, Krakowski A et al.Angioimmunoblastic lymphadenopathy withscleromyxedema-like lesions and serummono-clonal protein. Isr J Med Sci 1983 ; 19:235-239.25 Huang CT, Chuang SS. Angioimmunoblas-tic T-cell lymphoma with cutaneous involve-ment : a case report with subtle histologicchanges and clonal T-cell proliferation. Arch Pa-thol Lab Med 2004 ; 128:22-24.26 Cerroni L, Zenahlik P, Höfler G et al. Spe-cific cutaneous infiltrates of B-cell chronic lym-phocytic leukemia : a clinicopathologic andprognostic study of 42 patients. Am J Surg Pa-thol 1996 ; 20:1000-1010.27 Fraitag S, Bodemer C,Rousselot P et al. [Cu-taneous transformation of chronic lymphoidleukemia into immunoblastic lymphoma. Cuta-neousmanifestation ofRichter syndrome]. AnnDermatol Venereol 1995 ; 122:530-533.28 Stokkermans-Dubois J, Beylot-Barry M,Vergier B et al. Erythema annulare centrifugumrevealing chronic lymphocytic leukaemia. Br JDermatol 2007 ; 157:1045-1047.29 Cohen PR, Kurzrock R. Sweet’s syndromeand cancer. Clin Dermatol 1993 ; 11:149-157.30 Von den Driesch P. Sweet’s syndrome(acute febrile neutrophilic dermatosis). J AmAcad Dermatol 1994 ; 31:535-556.31 Cooper PH, Innes DJ Jr, Greer KE. Acutefebrile neutrophilic dermatosis (Sweet’s syn-drome) and myeloproliferative disorders. Can-cer 1983 ; 51:1518-1526.32 Hasegawa M, Sato S, Nakada M et al.Sweet’s syndrome associated with granulocytecolony-stimulating factor. Eur JDermatol 1998 ;8:503-505.33 Astudillo L, Loche F, Reynish W et al.Sweet’s syndrome associated with retinoic acidsyndrome in a patient with promyelocyticleukemia. Ann Hematol 2002 ; 81:111-114.34 Kumar G, Bernstein JM, Waibel JS, Bau-mann MA. Sweet’s syndrome associated withsargramostim (granulocyte-macrophage colonystimulating factor) treatment. Am J Hematol2004 ; 76:283-285.35 Ayirookuzhi SJ,Ma L, Ramshesh P,Mills G.Imatinib-induced sweet syndrome in a patientwith chronic myeloid leukemia. Arch Dermatol2005 ; 141:368-370.36 Wong GA, Guerin DM, Parslew R. Sweet’s
syndrome and polycythaemia rubra vera. ClinExp Dermatol 2000 ; 25:296-298.37 Vignon-Penamen MD, Juillard C, RybojadM et al. Chronic recurrent lymphocytic Sweetsyndrome as a predictive marker of myelodys-plasia. Arch Dermatol 2006 ; 142:1170-1176.38 Tomasini C, Aloi F, Osella-Abate S et al.Immature myeloid precursors in chronic neu-trophilic dermatosis associated with myelodys-plastic syndrome. Am J Dermatopathol 2000 ;22:429-433.39 Barriere H, Litoux P, Stalder JF, BarrierJ. [Pyoderma gangrenosum and haemopathy.Report of three cases]. Ann Dermatol Venereol1979 ; 106:427-435.40 Bertram-Callens A,Machet L, Vaillant L etal. [Buccal and ocular localizations of pyodermagangrenosum in Vaquez’s disease]. Ann Derma-tol Venereol 1991 ; 118:611-614.41 Ho KK, Otridge BW, Vandenberg E, Pow-ell FC. Pyoderma gangrenosum, polycythemiarubra vera, and the development of leukemia. JAm Acad Dermatol 1992 ; 27:804-808.42 Moisson YF, Aractingi S, Pinquier L et al.Neutrophilic eccrinehidradenitis.AnnDermatolVenereol 1992 ; 119:605-611.43 Aractingi S, Mallet V, Pinquier L et al. Neu-trophilic dermatoses during granulocytopenia.Arch Dermatol 1995 ; 131:1141-1145.44 Yiannias JA, el-Azhary RA, Gibson LE.Erythema elevatum diutinum : a clinical andhistopathologic study of 13 patients. JAmAcadDermatol 1992 ; 26:38-44.45 Aractingi S, Bachmeyer C, Dombret H et al.Simultaneous occurrence of two rare cutaneousmarkers of poor prognosis in myelodysplasticsyndrome : erythema elevatum diutinum andspecific lesions. Br J Dermatol 1994 ; 131:112-117.46 Morand JJ, Lightburn E, Richard MA et al.[Skin manifestations associated with myelodys-plastic syndromes]. RevMed Interne 2001 ; 22:845-853.47 Sutra-Loubet C, Carlotti A, Guillemette J,WallachD.Neutrophilic panniculitis. JAmAcadDermatol 2004 ; 50:280-285.48 Wallach D, Vignon-Pennamen MD. Fromacute febrile neutrophilic dermatosis to neu-trophilic disease : forty years of clinical research.J Am Acad Dermatol 2006 ; 55:1066-1071.49 Horiguchi Y, Lee SG, Matsumoto I et al.Abscess-forming neutrophilic dermatosis : re-port of three cases associatedwithhemopathies.Dermatology 1998 ; 197:174-177.50 Grob JJ, Mege JL, Prax AM, BonerandiJJ. Disseminated pustular dermatosis in poly-cythemia vera. Relationship with neutrophilicdermatosis of myeloproliferative disorders :
study of neutrophil function. J Am Acad Derma-tol 1988 ; 18:1212-1218.51 Anan T, Imamura T, Yokoyama S, Fuji-wara S. Erythema nodosum and granuloma-tous lesions preceding acute myelomonocyticleukemia. J Dermatol 2004 ; 31:741-747.52 Choi JH, Ahn MJ, Park YW et al. A caseof erythema nodosum and serositis associatedwithmyelodysplastic syndrome.Korean J InternMed 2005 ; 20:177-179.53 Sullivan R, Clowers-Webb H, Davis MD.Erythema nodosum : a presenting sign of acutemyelogenous leukemia. Cutis 2005 ; 76:114-116.54 Itin PH,Winkelmann RK. Cutaneous man-ifestations in patients with essential thrombo-cythemia. J Am Acad Dermatol 1991 ; 24:59-63.55 Hachulla E, Rose C, Trillot N et al. Whatvascular events suggest amyeloproliferative dis-order ? J Mal Vasc 2000 ; 25:382-387.56 Filo V, Brezova D, Hlavcak P, Filova A.Livedo reticularis as a presenting symptom ofpolycythaemia vera.ClinExpDermatol1999 ;24:428.57 Clarke J, Wells GC. Livedo reticularis withthrombocythaemia in association with poly-cythaemia rubra vera. Br JDermatol 1976 ; 95:63-64.58 Lazareth I, Fiessinger JN, Priollet P. Ery-thermalgia, rare acrosyndrome. 13 cases. PresseMed 1988 ; 17:2235-2239.59 Dreno B, Gandon P, Bureau B et al. Skinlesions from hypersensitivity to cold duringchronic myelomonocytic leukaemia. Br J Der-matol 1986 ; 115:607-609.60 Frankel DH, Larson RA, Lorincz AL. Acrallividosis— a sign ofmyeloproliferative diseases.Hyperleukocytosis syndrome in chronic myel-ogenous leukemia. Arch Dermatol 1987 ; 123:921-924.61 Greer JM, Longley S, Edwards NL et al. Vas-culitis associated with malignancy. Experiencewith 13 patients and literature review.Medicine(Baltimore) 1988 ; 67:220-230.62 Liozon E, Loustaud V, Fauchais AL et al.Concurrent temporal (giant cell) arteritis andmalignancy : report of 20 patients with reviewof the literature. J Rheumatol 2006 ; 33:1606-1614.63 Bachmeyer C, Wetterwald E, Aractingi S.Cutaneous vasculitis in the course of hemato-logic malignancies. Dermatology 2005 ; 210:8-14.64 Morand JJ, Lightburn E, Richard MA et al.[Skin manifestations associated with myelodys-plastic syndromes]. RevMed Interne 2001 ; 22:845-853.65 Farcet JP,Weschsler J,Wirquin V et al. Vas-

Références 49-13
Toute référence à ce chapitre devra porter la mention : Kluger N, Marque M, Aractingi S. Hémopathies myéloïdes, lymphoïdes et leucémies. In : Bessis D, Francès C, Guillot B, Guilhou JJ, éds, Dermatologie
et Médecine, vol. 3 : Manifestations dermatologiques des maladies du système hématopoïétique et oncologie dermatologique. Springer-Verlag France, 2007 : 49.1-49.13.
culitis in hairy-cell leukemia. Archrr Intern Med1987 ; 147:660-664.66 Longley S, Caldwell JR, Panush RS. Para-neoplastic vasculitis. Unique syndrome of cu-taneous angiitis and arthritis associated withmyeloprolifeff rative disorders. Am JMed 1986 ;80:1027-1030.67 Berthier S, Magy N, Gil H et al. [Myelodys-plasias and systemic diseases. A non-foff rtuitousassociation]. Rev Med Interne 2001 ; 22:428-432.68 Steinman HK, Kobza-Black A, Lotti TMet al. Polycythaemia rubra vera and water-induced pruritus : blood histamine levels andcutaneous fibrinolytic activity before and afterwater challenge. Br JDermatol 1987 ; 116:329-333.69 Bayrou O, Leynadier F. [FF Aquagenic pruri-tus]. Ann Dermatol Venereol 1999 ; 126:76-80.70 Archer CB, Camp RD, Greavaa es MW.WW Poly-cythaemia vera can present with aquagenic pru-ritus. Lancet 1988 ; 1:1451.71 Salem HH, Van deVV rWeyden MB, YoungYY IF,FF
Wiley JS. Pruritus and severe iron deficiencyin polycythaemia vera. BrMed J (Clin Res Ed)d1982 ; 285:91-92.72 Menage HD, Norris PG, Hawk JL, GravesMW. ThWW e efficacy of psoralen photochemother-apy in the treatment of aquagenic pruritus. BrJ Dermatol 1993 ; 129:163-165.73 Hebbar M, Brouillard M, Wattel E et al. As-sociation ofmyelodysplastic syndrome and re-lapsing polychondritis : fuff rther evidence. Leu-kemia 1995 ; 9:731-733.74 Frances C, el Rassi R, Laporte JL et al. Der-matologicmanifesff tations of relapsing polychon-dritis. A study of 200 cases at a single center.Medicine (Baltimore) 2001 ; 80:173-179.75 StockmanA,Delanghe J,GeertsML,Naeya-ert JM. Diffuse pffff lane normolipaemic xan-thomatosis in a patient with chronic lymphaticleukaemia and monoclonal gammopathy. Der-matology 2002 ; 204:351-354.76 KimKJ, LeeDP,PP Suh HS et al.Diffuse pffff lanexanthoma in a patient with chronic myeloidleukemia. J Dermatol 2004 ; 31:503-505.
77 Marcoval J,Moreno A, Bordas X et al. Dif-fuse pff lane xanthoma : clinicopathologic studyof 8 cases. J Am Acad Dermatol 1998 ; 39:439-342.78 MizoguchiM,Kawa Y,Minami T et al. Cuta-neous extramedullary hematopoiesis inmyelofi-brosis. J Am Acad Dermatol 1990 ; 22:351-355.79 Schofield JK, Shun JL, Cerio R,Grice K. Cu-taneous extramedullary hematopoiesis with apreponderance of atypical megakaryocytes inmyelofibrosis. J Am Acad Dermatol 1990 ; 22:334-337.80 Ohno S, Yokoo T, Ohta M et al. Aleukemicleukemia cutis. J Am Acad Dermatol 1990 ; 22:374-377.81 Haniffa MA, Wilkins BS, Blasdale C, Simp-sonNB.Cutaneous extramedullaryhemopoiesisin chronic myeloprolifeff rative and myelodysplas-tic disorders. J Am AcadDermatol 2006 ; 55:S28-31.82 Mays SR, Cohen PR. Emerging dermato-logic issues in the oncology patient. Semin Cu-tan Med Surg 2006 ; 25:179-189.

� MFmycosis fongoïde
50Classification des lymphomes cutanésOlivier Dereure
L a classification des lymphomes cutanés a beaucoupvarié au cours des vingt dernières années où elle est
passée d’une série très limitée de sous-catégories de lym-phomes extraganglionnaires à une classification beaucoupplus pragmatique et finalement nettement plus adaptéeà la pratique quotidienne, fruit d’une collaboration entrel’EORTC (European Organisation for Research and Treat-ment of Cancer) et l’OMS (Organisation mondiale de lasanté), mais également entre cliniciens et pathologistes ¹.Cette classification a deux buts principaux : faciliter la dé-marche diagnostique,mais également la prise en charge despatients en classant les différentes entités en fonction deleur pronostic. Il s’agit donc d’une classification à usage émi-nemment pratique,mais qui pourrait encore être remaniéedans l’avenir, notamment en devenant une classificationétiopathogénique si les mécanismes physiopathologiquesse précisent.Actuellement, elle distingue deux grandes catégories delymphomes cutanés : les lymphomes cutanés à cellules T età cellules NK (Natural Killer) d’une part, et les lymphomescutanés primitifs à cellules B d’autre part. Une troisièmecatégorie est représentée par une affection où les cellulestumorales dérivent de précurseurs hématologiques, l’hé-matodermie CD4+, CD56+ encore appelée lymphome à cel-lules NK « blastiques ». Cette classification est résuméedans l’encadré 50.A.Parmi les lymphomes cutanés T, deux grands types sontindividualisés : des lymphomes dits indolents, d’évolutionlente, et de bon pronostic : lymphomes cutanés T épidermo-tropes de type mycosis fongoïde (MF) et ses variants (my-cosis fungoïde pilotrope, réticulose pagétoïde, granuloma-tous slack skin), mais également les lympho-proliférationsCD30+ (papulose lymphomatoïde, lymphome anaplasiquecutané primitif à grandes cellules CD30+ et formes inter-médiaires). Sont également considérés comme de bon pro-nostic et d’évolution lente : les lymphomes T sous-cutanésalpha/bêta de type panniculite et les lymphomes T à pe-tites et moyennes cellules. Outre ce groupe de bon pro-nostic, d’autres entités, de classification ou de nosologiesouvent provisoire, sont nettement plus agressives : syn-drome de Sézary qui représente classiquement la forme leu-cémique des lymphomes T épidermotropes, lymphomesà cellules T/NK extranodaux de type nasal, souvent liés
au virus Epstein-Barr, lymphomes T épidermotropes agres-sifs CD8+, lymphomes T cutanés gamma/delta là aussi àtype de panniculite, mais nettement plus agressifs que lesformes alpha/bêta, lymphomes cutanés primitifs à grandescellules CD30− et lymphomes à grandes cellules CD30+ ounon compliquant un MF.Parmi les lymphomes cutanés primitifs B, deux groupessont également définis : des lymphomes d’évolution lenteou très lente, indolents, de bon pronostic : lymphomes àcellules de type centro-folliculaire incluant la classique réti-culose de Crosti et des lymphomes des zones marginales B(ex-immunocytome), ainsi que les plasmocytomes. En re-vanche, les lymphomes B cutanés à grandes cellules rondesBCL-2+ de type jambe et les autres lymphomes cutanés
Classification OMS-EORTC des lymphomes cutanés primitifs
50.A
Lymphomes cutanés primitifs T et NK− Mycosis fongoïde et ses variants (annexotropique, réticulose pa-
gétoïde, granulomatous slack skin)− Syndrome de Sézary− Lymphomes ni MF ni Sézary� Lymphoproliférations CD30+ incluant papulose lymphoma-
toïde et lymphome cutané primitif anaplasique à grandescellules CD30+
� Lymphome T sous-cutané à type de panniculite� Lymphome T/NK de type nasal� Lymphome cutané périphérique T sans autre précision� Lymphome T cutané primitif épidermotrope agressif (provi-
soire)� Lymphome T cutané gamma/delta (provisoire)� Lymphome cutané T pléomorphe à cellules petites et moyennes
(provisoire)Lymphomes cutanés primitifs B− Lymphomes des zones marginales− Lymphomes de type centro-folliculaires− Lymphomes cutanés diffus à grandes cellules de type jambe− Autres lymphomes cutanés diffus à grandes cellules− Lymphomes intravasculairesHématodermies développées aux dépens de précurseurs hé-matologiques− Hématodermie CD4+/CD56+ (lymphome « blastique »)

50-2 Classification des lymphomes cutanés
Toute référence à ce chapitre devra porter la mention : Dereure O. Classification des lymphomes cutanés. In : Bessis D, Francès C, Guillot B, Guilhou JJ, éds,Dermatologie etMédecine, vol. 3 : Manifestations
dermatologiques des maladies du système hématopoïétique et oncologie dermatologique. Springer-Verlag France, 2007 : 50.1-50.2.
primitifs à grandes cellules sont de pronostic intermédiaireou mauvais ainsi que les lymphomes endovasculaires àgrandes cellules B sauf dans leur variété cutanée pure quipeut être classée dans les formes indolentes.L’hématodermie CD4+ etCD56+ est également classée dansles formes agressives.En pratique, la classification des lymphomes cutanés primi-tifsff repose donc sur une analyse histologique et immuno-histochimique soigneuse avaa ec utilisation systématiqued’anticorps reconnaissant les molécules de surface Cff D(c(( luster diffeffff rencrr iation) 2, 3, 4, 8, 30, 19, 20, éventuelle-ment complétée en foncff tion de l’aspect anatomocliniquepar un marquage de CD56, BCL2, BCL6, CD25, CD7, etdes chaînes des récepteurs T alpha/bêta ou gamma/deltaou des chaînes légères des immunoglobulines kappa etlambda. Cette analyse doit être conduite sur des biopsies
fixées, mais également si possible sur des biopsies conge-lées puisque certains anticorps ne sont utilisables que dansces conditions. C’est dire que les biopsies doivent être debonne qualité, d’un volume suffisant et répétées si néces-saires en cas d’aspect non spécifique alors que la suspi-cion clinique est importante, notamment dans le cas deslymphomes cutanés T épidermotropes dont le diagnostichistologique initial peut se révéler particulièrement diffi-cile avaa ec un infiltrat non spécifique. Une étude en biolo-gie moléculaire à la recherche d’une population lympho-cytaire T ou B dominante grâce à l’étude du réarrange-ment des gènes codant pour les différentes chaînes durécepteur T ou du gène codant pour les chaînes lourdesdes immunoglobulines est souvent nécessaire, notammentpour appuyer un diagnostic anatomoclinique un peu hési-tant.
1 Willemze R, Jaffe ES, Burg G et al.WHO-EORTC classification for cutaneous lymphomas. Blood 2005 ; 105:3768-3785.

� MFmycosis fongoïde
51Lymphomes cutanés T épidermotropesOlivier Dereure
Mycosis fongoïde et ses variants 51-1Mycosis fongoïde classique 51-1Formes particulières 51-6
Syndrome de Sézary 51-7Références 51-8
L es lymphomes cutanés T épidermotropes font partiedes lymphomes cutanés primitifs de faible agressivité
clinique, même si certaines formes évoluées, nettementplus rares, ont un pronostic plus réservé. Ils représententla grande majorité des lymphomes T cutanés primitifs etpeuvent être séparés en deux grandes entités : le mycosisfongoïde (MF) « classique » et ses variantes, qui est de loinla forme clinique la plus fréquente, et le syndrome de Sé-zary, qui en est généralement considéré comme la contre-partie leucémique. D’autres formes, telles que la réticulosepagétoïde ou la granulomatous slack skin sont beaucoup plusrares. L’étiologie de ces lymphomes ainsi que leur méca-nisme physiopathologique ne sont pas connus avec préci-sion,même si certaines hypothèses commencent à se fairejour. Leur traitement n’est pas exactement codifié, maisdes algorithmes ont été élaborés récemment afin de guiderle clinicien dans ses choix thérapeutiques.
Mycosis fongoïde et ses variants
Mycosis fongoïde classiqueLe mycosis fongoïde est le lymphome cutané T primitif leplus fréquent, caractérisé par une prolifération de lympho-cytes T de taille petite à moyenne et dont le noyau peutparfois prendre un aspect cérébriforme,multi-encoché (cel-lules dites de Sézary). Décrit par Alibert en 1806, il repré-sente environ 50% de tous les lymphomes cutanés primi-tifs.L’épidémiologie de ce lymphome est mal connue, mais ilsemble que son incidence soit supérieure aux notions clas-siques. Cette incidence a été en augmentation,mais s’estapparemment stabilisée depuis 1984, du moins aux États-Unis ¹. Elle augmente clairement avec l’âge et atteint d’avan-tage les hommes (sex-ratio de 2,3/1 dans la populationblanche, 5,2/1 chez lesAsiatiques). Les facteurs épidémiolo-giques et de risque sontmal connus,mais il est possible quel’exposition à certains agents contaminants extérieurs tels
que les insecticides, les pesticides et les fungicides puissejouer un rôle même si ce point n’a jamais été démontréavec certitude. L’immunodépression congénitale ou induitene semble pas jouer de rôle particulier. Le MF atteint enpriorité les adultes au-delà de 50 ans, avec un âge médianau diagnostic de 55 à 60 ans. Toutefois, il peut atteindre,quoique rarement, les enfants et les adolescents.Les mécanismes moléculaires de cette affection ne sontpas connus avec certitude et plusieurs théories qui nesont d’ailleurs pas mutuellement exclusives ont été propo-sées ²-⁵ : stimulation antigénique chronique par un agent ex-térieur ;modification des voies d’apoptose post-activationlymphocytaire par l’antigène, notamment de la voie FAS-dépendante ; infection par un rétrovirus lymphotrope ; sti-mulation lymphocytaire par la sécrétion chronique de cyto-kines par d’autres cellules, etc. Il faut noter que la théoriede l’activation antigénique chronique et celle de l’anomalied’apoptose post-activation lymphocytaire sont tout à faitcomplémentaires et, par analogie aux lymphomes follicu-laires B avec translocation 14-18, pourraient parfaitementêtre cohérentes avec la faible évolutivité de cette affection,compatible avec un phénomène de lympho-accumulationet non de lympho-prolifération.Cliniquement, le mycosis fongoïde suit une progressiontrès lente s’étalant sur des années ou des décennies en pas-sant progressivement d’un stade de plaques érythémato-squameuses rouge bistre non infiltrées (fig. 51.1), parfoisstriées, à disposition arciforme, bien limitées, géomé-triques, parfois encochées (fig. 51.2), à des lésions qui s’épais-sissent progressivement pour aboutir à des plaques cui-vrées plus ou moins infiltrées (fig. 51.3), parfois érosives,souvent peu symptomatiques, à l’exception d’un pruritparfois féroce ⁶-⁸. Beaucoup plus rarement, des lésions au-thentiquement tumorales, souvent ulcérées, apparaissentsoit sur les plaques infiltrées (fig. 51.4), soit de novo (fig. 51.5).Chez un nombre très restreint de patients, une érythroder-mie, une atteinte ganglionnaire, voire viscérale, spécifique

51-2 Lymphomes cutanés T épidermotropes
Coll.
D.Be
ssis
Fig. 51.1 Macules érythémato-squameuses non infiltrées de la faceantérieure d’une cuisse : stage initial d’un mycosis fongoïde
Coll.
D.Be
ssis
Fig. 51.2 Large macule érythémato-squameuse à contour arciforme et àbord encoché : mycosis fongoïde
peut apparaître. À partir du stade tumoral, le pronostics’altère progressivement à la fois en qualité et en espé-rance de vie et le traitement devient souvent plus diffi-cile et nettement moins opérant tandis que l’évolutions’accélère de façon très sensible. L’étendue des lésionset le type lésionnel définissent la classification du myco-sis fongoïde en plusieurs stades (stade Ia à IV) commel’indique le tableau 51.1. Le pronostic dépend bien évidem-ment du stade de la maladie chez un patient donné.Toute la surface cutanée peut être atteinte,mais certaineszones sont particulièrement représentées telles que les
Coll.
D.Be
ssis
Fig. 51.3 Macules et papules érythémato-squameuses diffuses du dosau cours d’un mycosis fongoïde
Coll.
D.Be
ssis
Fig. 51.4 Lésion tumorale survenant sur une plaque infiltrée de mycosisfongoïde
fesses et les zones protégées du soleil. Les muqueusespeuvent également être touchées, en particulier buccaleset génitales sous forme de lésions souvent tumorales(fig. 51.6).Un certain nombre de formes cliniques particulières etsouvent trompeuses ont été rapportées : bulleuses, hypo-pigmentées (fig. 51.7) notamment chez les patients naturel-





























