Embed Size (px)
Citation preview

REVIEW 1157
Lesser-Known Enabling Technologies for Organic SynthesisLesser-Known Enabling Technologies for Organic SynthesisMatthew O’Brien,a Ross Denton,b Steven V. Ley*a
a Whiffen Laboratory, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UKE-mail: [email protected]
b School of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UKReceived 24 September 2010; revised 1 November 2010
SYNTHESIS 2011, No. 8, pp 1157–1192xx.xx.2011Advanced online publication: 30.03.2011DOI: 10.1055/s-0030-1259979; Art ID: E28310SS© Georg Thieme Verlag Stuttgart · New York
Abstract: In this review we summarize both new emerging tech-nologies as well as some overlooked methods which are beneficialto organic synthesis chemists.
1 Introduction2 Separation Methods2.1 Low-Temperature Chromatography2.2 Reflux Chromatography2.3 Phase-Switching Methods2.3.1 Affinity-Based Phase-Switching2.3.1.1 Fluorous Tags2.3.1.2 Acid–Base and Polarity Tags2.3.1.3 Transition-Metal and Ligand Affinity Tags2.3.1.4 Hydrogen-Bonding Interactions2.3.1.5 Polyaromatic Affinity Tags2.3.1.6 Crown Ether Tags2.3.1.7 Redox-Switchable Tags2.3.2 Reactive and Irreversible Tags2.3.2.1 ‘Click’ Tags2.3.2.2 Diels–Alder Tags2.3.3 Phase-Switching Involving Precipitation2.3.3.1 Precipitation by Polymerisation2.3.3.2 Solubility-Switching2.3.3.3 Temperature-Dependent Solubility2.3.4 Size-Exclusion Tags3 Synthesis Methods3.1 Cooled Microwave Heating3.2 Ball-Milling3.3 Design of Experiments and Related Techniques4 Conclusion
Key words: separation methods, synthesis methods, new enablingtechnologies
1 Introduction
The art and craft of organic synthesis has reached a veryhigh level of sophistication. However, in order to meet thedemands and respond to the massive number of potentialreaction variants and the workup protocols that are neces-sary to obtain pure products, highly elaborate equipmentfor analysis and separation have evolved over the years.The skills of the modern synthesis chemist encompass anenormous array of techniques and knowledge-based pro-tocols. Moreover, the need for greater speed in the discov-ery process has driven change at a phenomenal pace; somuch so that it is difficult not to be overwhelmed by, let
alone evaluate, the sheer volume of literature or to fullyassess the new tools available. In light of these problems,for this review we have brought together a selection oftopics that we believe can aid the synthesis process. Thesemethods, which have not been covered extensively in oth-er articles, are newly emerging, or in some cases older,overlooked technologies. The list of enabling processes isby no means exhaustive, but is intended to address someof the more challenging aspects of synthesis that relate toseparation, reactivity and the optimisation of a process.
2 Separation Methods
Since its invention over a century ago,1 liquid columnchromatography has become one of the most widely em-ployed techniques in the arsenal of the chemist, both foranalytical and preparative purposes. Although other tech-niques, such as distillation or recrystallisation, can oftenbe more efficient in specific preparative cases, they placecertain requirements on the compound to be isolated. Ifthe chemist wishes to distil a product to separate it fromthe other components of a mixture then it has not only tobe volatile at practical temperatures and pressures, but italso has to be stable to those conditions. Crystallisationobviously requires that the product is crystalline but, evenif this is so, the compound has first to be dissolved in asuitable solvent, perhaps at an elevated temperature, andis again required to be stable to these conditions. Liquidcolumn chromatography, on the other hand, has a muchbroader range of applicability and can often be relied uponto effect a separation with a minimum of developmentaleffort. However, there are certain limitations to the tech-nique which have to be addressed. Firstly, as with distilla-tion and recrystallisation, the compound(s) to be separatedmust be stable to the conditions. This is not always thecase. The stationary phase must have some affinity for thecompounds to be separated, otherwise they will simplypass straight through the column. Where delicate func-tionality is present, this affinity can often be so strong thatthe stationary phase acts as a catalyst for a reaction or pro-motes decomposition of the substrate. This is often seenwith silica gel, perhaps the most frequently used station-ary phase for preparative chromatography. The solventcan also be responsible for the decomposition of the sub-strate. Secondly, the amount of solvent used in a chro-matographic separation can often be significant,particularly if the compounds to be separated elute at avery similar rate. In large-scale preparative work this can
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

1158 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
be prohibitive in terms of cost, safety and the environmen-tal impact of the process. These are perhaps the principlereasons that preparative chromatographic separations arenot favoured on an industrial scale.
The first section of this review will focus on the use oflow-temperature chromatography as a means of purifyingreactive species that would otherwise decompose or react,and also as a way of increasing the chromatographic reso-lution, both for preparative and analytical purposes. Thefollowing section highlights the use of reflux chromatog-raphy as a way of dramatically reducing the amount ofboth solvent and labour used in chromatographic separa-tions.
2.1 Low-Temperature Chromatography
The technique of cooling the chromatography columnduring separation/purification has been known for sometime. In a seminal early example, Clements was investi-gating the chromatographic separation of a mixture of thecitrus-oil terpene hydrocarbons p-menthane, a-pinene,b-pinene and d-limonene (stereochemistry unspecified)on calcined silicic acid with various solvents (Figure 1).2
Figure 1
It was found initially that there was very little differencein the elution rate of all four components on standard col-umns. However, when the column was submerged in avessel containing dry ice and acetone (–78 °C) (‘…on theassumption that migration would be retarded at lowertemperatures’), not only were the elution rates generallyslower, but the difference between the elution rates wasthen sufficient to obtain complete separation of the com-ponents using petroleum ether as eluant. This separation isremarkable, particularly for the pinenes which differ onlyin the position of the double bond. Clements also re-marked that ‘in addition, the low temperature should in-hibit the chemical changes often observed in adsorptionchromatography.’
Dr Matthew O’Brien ob-tained his PhD under the su-pervision of Professor E.James Thomas (Universityof Manchester) working onthe total synthesis of thebryostatins. Following apostdoctoral position in thegroup of Professor IanPaterson FRS (University of
Cambridge) he was appoint-ed as the Kinerton Lecturerof Organic Chemistry atTrinity College Dublin.Matthew returned to Cam-bridge in 2006 to join the re-search group of ProfessorSteven V. Ley CBE FRSFMedSci and is currently afellow and director of stud-
ies for Natural Sciences atPembroke College. His cur-rent research interests in-clude the total synthesis ofnatural products and the de-velopment of new technolo-gies and techniques fororganic synthesis.
Dr Ross Denton began hisresearch career at the Uni-versity of Nottingham com-pleting his PhD in 2004under the supervision ofProfessor J. C. Anderson.Postdoctoral research at TheScripps Research Institute
with Professor K. C. Nico-laou (2005–2007) and TheUniversity of Cambridgewith Professor S. V. LeyCBE, FRS (2007–2008)then followed. He startedhis independent research ca-reer in 2008 as a Lecturer in
Organic Chemistry at TheUniversity of Nottingham.At present his research is fo-cussed on the developmentof catalytic phosphorus-mediated reactions and thesynthesis of polyphenolicnatural products.
Professor Steve Ley CBEFRS FMedSci has been theBP (1702) Professor ofChemistry at the Universityof Cambridge (where he is afellow of Trinity College)since 1992. Steve obtainedhis PhD from Loughbor-ough University with Pro-fessor Harry Heaney andafterwards carried out post-doctoral research with Pro-
fessor Leo Paquette (OhioState University) then Pro-fessor Sir Derek Barton(Imperial College). He wasappointed as a lecturer atImperial College in 1975,promoted to Professor in1983, and then to Head ofDepartment in 1989. In1990 he was elected to theRoyal Society (London) andwas President of The Royal
Society of Chemistry from2000–2002. Steve’s re-search interests are variedand span many disciplinesincluding new syntheticmethodologies, the totalsynthesis of natural prod-ucts, and the developmentof enabling technologies forchemical synthesis.
Biographical Sketches
1 2 3 4
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1159
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
The effect of temperature on the chromatographic resolu-tion of compounds has since been investigated by severalworkers and has even been given mathematical treatment.In many cases it appears that the resolution is enhanced atlower temperatures.3 This phenomenon has particularlyfound application in the field of biomolecule separation.For instance, it was found that phosphoglycerides contain-ing only long-chain saturated fatty acids could be com-pletely separated from those containing any degree ofunsaturation by performing thin-layer chromatography atlow temperature (–70 to –25 °C).4 In this case, it appearsthat the enhanced separation is related to the pronounceddifference in solubility of the compounds in the eluant atlower temperature. In the extreme case, one of the compo-nents precipitates out of solution and the technique ismore of a filtration than a chromatographic separation.Others have investigated the effect of temperature on theresolution of steroids in reverse-phase analytical HPLCand a significant improvement in resolution was foundwhen the chromatography was carried out at low temper-ature (–50 °C) enabling complete separation of the com-pounds.5 Remarkably, however, the application of low-temperature chromatography has seen little general appli-cation by the synthetic chemistry community despite itspotential. This is presumably due to the perceived compli-cation of the need for more advanced equipment.
Chromatographic separation, both preparative and analyt-ical, becomes more involved when one or more of thecompounds react under the separating conditions. Thesereactions can either be irreversible or reversible and ex-amples of both cases are known.
An early example of the use of low-temperature chroma-tography for the purification and isolation of unstable spe-cies was reported in 1975. In this case, it was found thatmicrosomal cytochrome P-450 could be isolated by per-forming column chromatography on DEAE cellulose gelat –20 °C. Both the low temperature and the solvent used(ethylene glycol) were thought to be equally important incontributing to the stabilisation of the enzyme, which isusually considered to be unstable in the purified state.6
During investigations into the mechanism of the thermalisomerisation of cis-bicyclo[6.1.0]nona-2,4,6-trienes, aseries of aza-analogues 7 and 8 were prepared by thedichloromethane-mediated cyclopropanation of the azoci-nyl dianion 6 (Scheme 1). These thermally labile homo-azocines were found to be very sensitive to columnchromatography under normal conditions. However, byperforming the chromatography on basic alumina at –60 °C,the compounds could be separated, which then enabled in-vestigation of the regioselectivity of their thermal isomer-isations.7
Ley and co-workers also reported in 1981 that iron-carbo-nyl insertion into allylic epoxides such as 11, followed byoxidative removal of the tricarbonyliron group in the com-plex 12 led to the formation of highly reactive b-propio-lactones, in which the carbonyl group originated from thepentacarbonyliron. As these lactones tended to be unsta-
ble, they were generally characterised as the diols after re-duction with lithium aluminium hydride. However, it wasfound that compound 13 could be isolated in practicallyquantitative yield by careful chromatography at 0 °C onsilica gel (Scheme 2).8
Scheme 2
A typical apparatus used for such low-temperature sepa-rations is shown in Figure 2. It consists of a chromatogra-phy column that has been constructed with a surroundingjacket through which a refrigerant circulates at the appro-priate temperature. Thermostatic refrigerant pumps arecommercially available and allow controlled refrigeranttemperatures down to –120 °C.9 The additional stopcocks(B and G) allow for the entire chromatographic process totake place under an inert atmosphere for air-sensitivecompounds.
2,3-Naphthoquinones (15a–c, Scheme 3) have generatedinterest as high-potential quinones which can act as dehy-drogenation reagents. However, they are highly reactiveand unstable. For example, the parent molecule 15a(R = H), is too unstable to be observed but can be trappedwith cyclopentadiene as its Diels–Alder adduct after po-tassium iodate oxidation of the precursor naphthalene-2,3-diol 14a.10 The diphenyl derivative 15b is more stableand can survive for 45 minutes at –20 °C, as observed bythe disappearance of the associated green colour.11 In anattempt to prepare stable derivatives of 15a, the oxidationof 14c with lead tetraacetate was carried out in anticipa-
Scheme 1
N
OMe
N
OMe
N
OMe
H
N
OMe
+
CH2Cl2
H
N
OMe
H
N
H
MeO
100 °C
87
9
2 K
100 °C
2-
10
65
O
Fe
OO
O
OFe(CO)5
(CO)3
Ce(IV)
11 12 13
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

1160 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
tion that the o-tolyl groups (presumably lying orthogonalto the o-quinonoid system) would present a significantsteric barrier to further reaction. This proved to be thecase, the deep green colour of the solution of 15c persist-ing unchanged for over two hours at –20 °C. However, inorder to isolate the compound for full characterisation (byIR spectroscopy), low-temperature chromatography hadto be carried out on a short path of silica gel eluting withdichloromethane–diethyl ether (95:5) at –30 °C.12
Scheme 3
During work on synthesis of fulvalene 19, an intermediatewas found to be highly unstable with respect to thermaldouble-bond isomerisation. 9,10-Dihydrofulvalene (17)was formed by the oxidative dimerisation of cyclopenta-dienyl sodium with copper(II) chloride at –30 °C. Al-though this intermediate was stable at or below –20 °C, itrapidly equilibrated with the isomeric dihydrofulvalenes
20, 21 and 22 at or above 0 °C (Scheme 4). Attemptedchromatography of 17 on alumina at 0 °C led to the for-mation of the 7,8-dihydrofulvalene 23. However, 17 couldbe purified without isomerisation using silica gel chroma-tography at –20 °C eluting with pentane–diethyl ether(1:1). 17 was then doubly deprotonated with n-butyllithi-um and the resulting dianion oxidised with copper(II)chloride to afford fulvalene 19 in good yield.13
Scheme 4
Perhaps one of the most notorious functional groups interms of sensitivity to hydrolysis is the silyl enol ether.Under normal circumstances it would be foolhardy to at-tempt purification on silica gel. However, Stoodley andco-workers wished to synthesise the glycosyl silyl enolether 25 (Scheme 5) for use as a chiral diene in Diels–Alder cyclisations. 25 could be formed from the methylketone 24 using standard silylating conditions but thecrude material was not sufficiently pure for further stud-ies. Fortunately, despite the delicate functionality present,25 could be purified by chromatography on silica gel at–78 °C (column jacketed with dry ice in acetone) elutingwith light petroleum–diethyl ether (1:1).14
Scheme 5
Another noteworthy example of the chromatography of asilyl enol ether is shown in Scheme 6. Here the requiredsilyl enol ether 28 was needed during investigation of thetransition state of the Mukaiyama aldol reaction. Thishighly labile compound was synthesised in two steps fromthe ketone 26. Treatment of 26 with potassium hexameth-yldisilazide and tert-butyldimethylsilyl trifluoromethane-
Figure 2 Apparatus for low-temperature chromatography; A:ground glass joint, B: straight bore PTFE stopcock, C: refrigerant out-let, D: jacketed column, E: refrigerant inlet, F: screwthread-typePTFE stopcock, G: straight bore PTFE stopcock, H: ground glassjoint
OH
OH
R
R
O
O
R
R
[O]
14a–c 15a–c
a R = Hb R = Phc R = o-tolyl
2 M+-
-
-
Li+
Li+
CuCl2
84%
BuLi
100%
CuCl2
73%
0 °C
+ +
Al2O3 column75%
16
17 18 19
20 21 22 23
O
O
OAc
OAc
OAc
OAc
O
O
O
OAc
OAc
OAc
OAc
OTMS
Et3N, TMSCl
24 25
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1161
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
sulfonate afforded the intermediate 27 which was purifiedby chromatography at –78 °C on activity III neutral alu-mina in very good yield. The dioxolanone protectinggroup was then carefully hydrolysed using sodium hy-droxide in methanol–tetrahydrofuran to afford the targetcompound 28 in good yield. Again, low-temperature col-umn chromatography at –78 °C was used to purify thisdelicate compound, in this case using activity V basic alu-mina.15
Scheme 6
1,2-Siloxetanes are another example of unstable siliconcompounds whose successful isolation required low-tem-perature chromatography. Leigh reported a photochemi-cal synthesis of the siloxetane 31 by irradiating thedisilane 29 in the presence of acetone (Scheme 7). It isthought that the reaction proceeds via the highly reactivesilene 30. However, all attempts to isolate 31 by the usualchromatographic means resulted in decomposition. But,by the careful use of radial chromatography at 5 °C (car-ried out in a coldroom using a chromatotron), 31 was suc-cessfully isolated.16
Scheme 7
An interesting sequence of transformations was used tosynthesise an advanced intermediate towards the synthe-sis of the clinical anticancer natural product mitomycin C(Scheme 8). Mercury-promoted cyclisation of the second-ary amine onto the alkyne in 33 led to the formation of theintermediate enamine 34. This was then coupled to thequinone 35 to afford a deep blue solution of 36. This un-stable product proved difficult to purify but it was foundthat by performing careful chromatography on alumina at0 °C (using the blue colour as a guide), 36 could be isolat-ed in 25% yield for the two-step sequence. The half-life of36 was only 1.5 days in toluene-d8 at –15 °C.17
Scheme 8
Carreira and co-workers synthesised the bisnaphtho-pyrans 37 to investigate their potential in optical materialsapplications. These compounds were found to display in-teresting temperature-dependent photochromism. Irradia-tion of these molecules at room temperature induces oneof the pyran rings to open, affording the transient isomericspecies 38 and 39 (Scheme 9). Irradiation at lower tem-peratures leads to the opening of both pyrans giving 40,which has different UV-VIS absorption characteristics.For 37 (R = F), the half-life of the ring-opened products38 and 39 is only 93 minutes at room temperature. Re-markably, despite their ephemeral nature, 38 and 39(R = F) were successfully separated by low-temperaturecolumn chromatography on silica gel at –78 °C, elutingwith dichloromethane, thus enabling their UV-VIS spec-tra to be recorded (at –78 °C).18
Organometallic chemistry is an area that abounds withtransient and reactive complexes, many of which are air-and moisture-sensitive, so it is perhaps not surprising thatlow-temperature chromatography has found useful appli-cation in this field. A very early example was that ofSellmann and co-workers who used the technique to sep-arate C5H5Mn(CO)2N2 from C5H5Mn(CO)3 during theirmechanistic investigation of nitrogen fixation.19 Investi-gating the activation of carbon–hydrogen bonds, Bergmanand co-workers prepared the cyclopropyl-hydrido rheni-um complexes 43a,b via irradiation of 41 or 42, respec-tively (Scheme 10). Despite its thermal lability and air-sensitivity, compound 43a was isolated in pure form bychromatography on activity III neutral alumina usingdeoxygenated eluents at –100 °C.20
The same research group successfully utilised low-temperature chromatography during an investigation intothe insertion of iridium into several hydrocarbons. For in-stance, when the dihydrogen species 44 was irradiated inthe presence of 1,1-dimethylcyclopropane (45), a mixtureof compounds was formed, consisting of a pair of diaste-reoisomers 46a,b and the regioisomer 47 which could beconverted by heating into 46a,b (Scheme 11). These com-
O O
O
O
O O
O
TBSOTBSO
O
TBSOTfKHMDS–95 °C
92%
NaOHMeOHTHF
83%
26 27
28
SiSiMe3
PhPhSi
Ph
Ph
H
SiMe3
Si
O
HPh
PhSiMe3
SiO
PhPh
SiMe3
hυ
C6H12
O
+29 30 31
32
N
HN
Ph
N
Ph
N
DPM
DPM
O
O
Br
OMeMeO O
O
Br
MeO N
Ph
N
DPM
HgCl2
Et3N
35
33 34
chromatographyon alumina at 0 °C
25%2 steps
t1/2 = 1.5 d in toluene-d8 at –15 °C
36
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

1162 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
pounds were air-sensitive and required careful handling.However, diastereomers 46a,b could be separated usinglow-temperature chromatography on activity III neutralalumina at –80 °C. In this example, a double-jacketed col-umn was used with a stream of nitrogen gas (cooled bybubbling through liquid nitrogen) passed through the in-ner jacket while being insulated by the outer vacuum jack-et. The apparatus was flushed with argon prior tochromatography and a positive pressure of argon wasmaintained to prevent the oxidation of these delicate orga-nometallic complexes.21
In addition to the obvious benefits of using low-tempera-ture chromatography for the isolation of sensitive com-pounds, the technique has also found success in variousanalytical applications.
Whilst investigating a synthetic sequence that involvedmesylation of a secondary alcohol followed by cyclisationto a seven-membered ether en route to a pharmaceuticalcompound (Scheme 12), chemists at Merck had difficultyin monitoring the progress of the reaction by HPLC.22
Scheme 12
Using reverse-phase HPLC, compound 49 was found todecompose completely, mainly to the cyclised product 50with some of the eliminated compound 51 also beingformed. Changing to normal-phase HPLC (Chromega-bond Diol column) improved the situation, but significantcyclisation was still apparent. An investigation of the ef-fect of temperature found that only by running the columnat –30 °C could the on-column reaction be suppressedcompletely. Interestingly, analysis of the chromatogramsat several temperatures enabled a kinetic analysis and al-
Scheme 9
O
R
O
R
O
R
R
O
O
R
R
O
R
O
R
O
+
37
38
39
40
hυ
hυ
Scheme 10
CpRe(PMe3)3 Cp*Re(CO)(PMe3)2
Cp'L1L2ReH
hνcyclopropane cyclopropane
41 42hν
43a Cp' = C5H5, L1 = L2 = PMe343b Cp' = C5Me5, L1 = CO, L2 = PMe3
Scheme 11
(Cp*)(PMe3)Ir(H)2
hυ(Cp*)(PMe3)Ir
H
44
45 46
(Cp*)(PMe3)Ir
H
47
+
R
OHOH
R
OOHS
Me
O
O
MeSO2Cl
O
R
R
OH
NClR =
48 49
51
50 Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1163
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
lowed pseudo-first-order rate constants for the reaction tobe obtained.
The same research group also ran into difficulty monitor-ing the propargylic aldehyde 52 by reverse-phase HPLC(C18 hypersil). In 0.1% trifluoroacetic acid in tetrahydro-furan–acetonitrile at room temperature, severe fronting ofthe peak associated with 52 was observed. This wasthought to be due to the equilibrium shown between thealdehyde 52 and its hydrate form 53 (Scheme 13). At45 °C this equilibrium was rapid and no peak correspond-ing to 52 could be observed. However, it was found that awell-resolved peak for 52 could be observed when thechromatography was carried out at –5 °C.23
Scheme 13
An interesting example of a series of compounds that in-terconvert rapidly (compared with the timescale of the an-alytical chromatography) are cis/trans peptide rotamers,particularly those containing proline residues. Normally,the trans rotamers are far more stable thermodynamicallythan their cis counterparts which are usually only presentin trace amounts. However, as proline is a secondaryamine, peptides containing this residue have rotamers ofcomparable energy and the interconversion between themis relatively slow (Scheme 14).24 In an early example itwas found that chromatographic purification of alanyl-proline was complicated by the on-column interconver-sion of the cis and trans isomers. Only by performing thechromatography at elevated temperatures (which in-creased the rate of equilibration) could the compound beeluted as a single well-resolved peak.25 Later, Hendersonand Horvath found that the rotamers of valyl-prolinecould be separated as single compounds using reverse-phase chromatography at 0 °C at varying pH.26 The iso-mers could be kept at low temperature without any notice-able interconversion. With a reliable analytical protocol inhand, accurate measurements of the equilibrium constantof the cis–trans isomerism could be made.27 This low-temperature separation approach has since been applied toseveral other proline-containing peptides and other mole-cules.28
Several thorough mathematical treatments of low-temper-ature chromatograpy have also been proposed.29 Indeed,the modeling and analysis of chromatogram peak shapesat various temperatures has developed as ‘dynamic chro-
matography’ and is considered to be a valuable method ofobtaining kinetic data for a variety of equilibrium pro-cesses.30
In summary, while low-temperature chromatography hasfound some useful applications, particularly in analyticallaboratories, the technique has not attracted the attentionwhich it surely merits. We hope that the examples shownwill serve to encourage further consideration of the tech-nique, especially as a method of preparative separation ofrelatively labile compounds.
2.2 Reflux Chromatography
Assuming that compounds to be purified/separated do notreact or decompose significantly on the column supportmaterials, the chemist can sometimes encounter other re-stricting problems during chromatography. In cases wherethe elution rates of the compounds to be separated are verysimilar, a commonly employed tactic is to use a moreweakly eluting solvent and a longer elution time. Asidefrom the increased duration of the operation, this inevita-bly leads to larger amounts of solvent being used. On asmall scale this is not usually a significant problem but itcan be prohibitive for economic, safety and environmentalreasons on a larger scale. In an effort to minimise both thelabour (i.e., refilling solvent reservoirs and fraction col-lecting) and solvent involved in such separations/purifica-tions, the technique of ‘reflux chromatography’ has beendeveloped, wherein the solvent is continuously recycledfrom the collected fraction(s) to the solvent reservoir bymeans of distillation. Although the apparatuses used differsomewhat in their configuration, the method is a logicalextension of the Soxhlet extraction process. Whilst we usethe term ‘reflux chromatography’ here, other terms suchas ‘Soxhlet chromatography’, ‘continuous chromatogra-phy’ and ‘heisschromatographie (hot chromatography)’have been used to describe processes which operate by es-sentially the same principles.
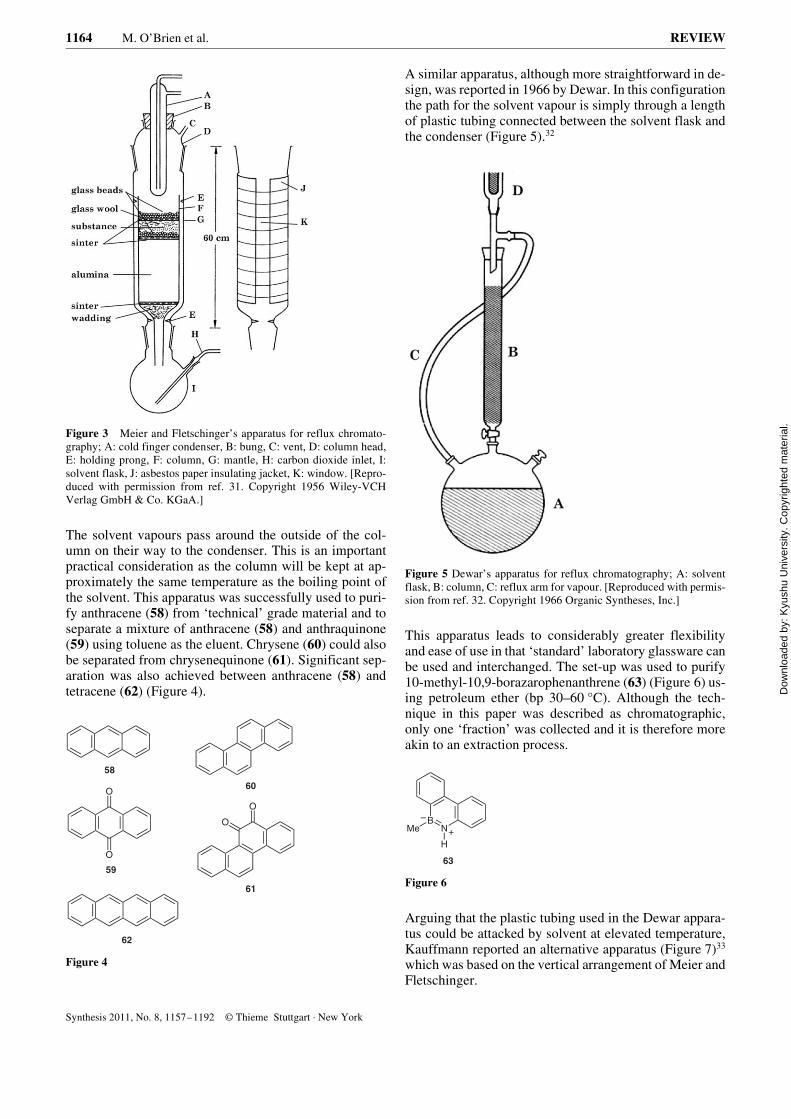
A very early example of such an apparatus is that reportedby Meier and Fletschinger in 1956.31 Very similar in de-sign to the classical Soxhlet apparatus, the column is sup-ported vertically above the collecting flask and thecondenser is supported vertically above the column(Figure 3).
N
O O
CF3
CF3
F
O
N
O O
CF3
CF3
F
OH
HO
H2O
52 53
Scheme 14
N
O
R
H2N
HN
O
R1
H2N
N
O OHO
R
H2N
R2
CO2H NH
O
R1
H2N
R2 CO2H
O
OH
54 55
5756
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.
1164 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
Figure 3 Meier and Fletschinger’s apparatus for reflux chromato-graphy; A: cold finger condenser, B: bung, C: vent, D: column head,E: holding prong, F: column, G: mantle, H: carbon dioxide inlet, I:solvent flask, J: asbestos paper insulating jacket, K: window. [Repro-duced with permission from ref. 31. Copyright 1956 Wiley-VCHVerlag GmbH & Co. KGaA.]
The solvent vapours pass around the outside of the col-umn on their way to the condenser. This is an importantpractical consideration as the column will be kept at ap-proximately the same temperature as the boiling point ofthe solvent. This apparatus was successfully used to puri-fy anthracene (58) from ‘technical’ grade material and toseparate a mixture of anthracene (58) and anthraquinone(59) using toluene as the eluent. Chrysene (60) could alsobe separated from chrysenequinone (61). Significant sep-aration was also achieved between anthracene (58) andtetracene (62) (Figure 4).
Figure 4
A similar apparatus, although more straightforward in de-sign, was reported in 1966 by Dewar. In this configurationthe path for the solvent vapour is simply through a lengthof plastic tubing connected between the solvent flask andthe condenser (Figure 5).32
Figure 5 Dewar’s apparatus for reflux chromatography; A: solventflask, B: column, C: reflux arm for vapour. [Reproduced with permis-sion from ref. 32. Copyright 1966 Organic Syntheses, Inc.]
This apparatus leads to considerably greater flexibilityand ease of use in that ‘standard’ laboratory glassware canbe used and interchanged. The set-up was used to purify10-methyl-10,9-borazarophenanthrene (63) (Figure 6) us-ing petroleum ether (bp 30–60 °C). Although the tech-nique in this paper was described as chromatographic,only one ‘fraction’ was collected and it is therefore moreakin to an extraction process.
Figure 6
Arguing that the plastic tubing used in the Dewar appara-tus could be attacked by solvent at elevated temperature,Kauffmann reported an alternative apparatus (Figure 7)33
which was based on the vertical arrangement of Meier andFletschinger.
O
O
O
O
58
60
61
59
62
BNMe
H+
–
63
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1165
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
Figure 7 Kauffmann’s apparatus for reflux chromatography; A:ground glass joint (for connection of reflux condenser), B: inner co-lumn, C: ground glass joint, D: 2 holes of 17–22 mm diameter, E:outer jacket, F: extra course glass frit of 30 mm diameter, G: groundglass ball joint, H: working length of 40 cm. [Reprinted (adapted)with permission from ref. 33. Copyright 1976 American ChemicalSociety.]
Certain practical improvements were made to increase themechanical strength and reliability of the apparatus. Thechromatography column no longer rests on glass fingerswithin the outer jacket but is attached to the joint connect-ing the condenser. These arrangements were sold com-mercially as ‘Kauffmann’ columns.34 These columnsseem to have found greatest use in the purification of or-ganic molecules with materials properties, where very lowlevels of impurity can significantly affect these properties.Examples include the scintillation fluors dimethyl-p-sexi-phenyl (64)35 and 9,10-bis(3-methylphenyl)anthracene(65),36 the ‘organic metal’ tetrathiafulvalene derivative6637 and ‘hole-conducting’ triarylamines 6738 (Figure 8).A medicinal chemistry application of the Kauffmann col-umn is the purification of pyridyl aryl ketone 68 whichwas an intermediate in the synthesis of dopamine reuptakeinhibitors by Froimowitz and co-workers.39
A very nice further application of reflux chromatographyis the separation and purification of the fullerenes C60 andC70. After the initial excitement of the discovery of buck-minsterfullerene, it was soon realised that further researchinto the properties and applications of the material would
require an economic and practical access to significantquantities. This prompted research into the efficient pro-duction of the material. Generally, the material is made byheating graphite or soot with an electric arc. Although theyields are low, the starting material is very cheap. A moresignificant problem, however, is the separation of thefullerenes from the rest of the material. Simple extractionwith hydrocarbon solvents removes most of the soot andwhilst chromatography on neutral alumina achieves ex-cellent separation of C60 and C70, the process is exceeding-ly time-consuming and requires excessive volumes ofsolvent. This was clearly a bottleneck in the developmentof fullerene research and several investigators sought amore efficient alternative. The research groups of Wudland Chatterjee independently reported in 1992 (back-to-back publications) very similar approaches to this prob-lem, both using ‘reflux chromatography’ on neutral alu-mina.40 Chatterjee40a used a commercially availableKaufmann column whilst Wudl40b used a custom-built ap-paratus similar in design to that reported previously byDewar. Using this technique, both groups were able to ob-tain analytically pure samples of C60 and C70 with signifi-cant reductions in the amount of time and solventrequired. Others later found that using a mixture of Norit-A alkaline decolourising carbon and silica gel led to an in-crease in the yield of the fullerenes.41 This successful sep-aration process has been pivotal to the development offullerene chemistry.42
A novel development of this type of reflux chromatogra-phy separation recently reported by our laboratory in-volves the use of a mixture of solvents as the eluant,combined in a ratio by which they form a low-boiling
Figure 8
2
OMe
MeO
S
SS
S
N
R1
R2
R3
R4
R5
R6
O
N
Cl
64
65
66
6867
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

1166 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
azeotrope.43 The potential benefits of this enhancementare twofold. Firstly, it expands greatly the number ofavailable eluants, thus increasing the likelihood of obtain-ing an eluant of the required strength for the mixture to beseparated; the numbers of known azeotropes are vast andthere are possibly many more yet to be reported. Second-ly, lower boiling points can be obtained – an importantconsideration for mixtures of compounds which containdelicate functionality, especially if each fraction collectedwill be maintained at reflux for prolonged periods. In the
wake of our recently reported total synthesis of the anti-cancer marine macrolide spongistatin 1,44 work in our lab-oratory has been directed towards the scale-up of the syn-thesis with the aim of producing multi-gram quantities ofthe material for further biological and clinical evaluation.A key factor in our synthetic route was the recognition ofthe pseudo-symmetry about the C15 carbon in the ABCDfragment 69 (Scheme 15).
This allowed the synthesis of both the AB and CD frag-ments to proceed via the common intermediate 74, signif-
Scheme 15
C
D
O
O
O
H
H
TBSO
OMe
OPMB
OO
OO
AcO
H
H
OTES
OAll
A
B
OO
HO
O
O
O
O
AcO
H
H
OTES
H
H
TBSO
OMe
OAll
O
A
B
C
D
1
9
11
13
19
17
28
15
C
D
O
O
H
H
HO
OPMB
SS
OH
OO
HO
H
H
SS
OH
OPMB
A
B
carbonhomologation
carbonextrusion
PMBO
O O O SS O
OTESPMBO
O O O O
OTES
pseudoC2 axis
1:1 mixture of epimers
TESTES
11
19
55
aldol
11
19
69
70
71 73
72
747576
Scheme 16
C*
D*
O
O
H
H
HO
OPMB
SS
OH
OO
HO
H
H
SS
OH
OPMB
A
B
OPMB
O
O
O
SS
OTES
TESO
C
D
O
O
H
H
HO
OPMB
SS
OH
ABAA CDAE
74 72 77 73
CDAA
1:00 : 0.74 : 0.26
HClO4, MeCN H2O, CH2Cl2, 0 °C
+ +
HClO4, Ca(ClO4)2MeCN, H2O, CH2Cl2
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1167
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
icantly reducing the total number of synthetic steps. Thisapproach, however, relies heavily on the ability to sepa-rate the AB spiroketal 72 from the (interconvertible) CDspiroketal anomers 73 and 77 formed upon treatment of74 with aqueous perchloric acid in acetonitrile(Scheme 16).
Unfortunately, after an extensive search for suitable elu-ants, an efficient conventional chromatographic separa-tion could not be found. In the original synthesis(milligrams of spongistatin 1), separation was achieved bya laborious and painstaking chromatographic process in-volving repeated separations on HPLC columns. Thiswould clearly not be satisfactory for the scale-up synthesisand an azeotropic reflux chromatography separationmethod was developed for this purpose. It was found bymultiple elution analytical TLC studies that the approxi-mately 1:1 binary azeotrope of pentane and diethyl ethereluted the components of the mixture at a very slow ratewith complete resolution. After a successful preparativeTLC experiment confirmed this, reflux chromatographywas attempted. After small-scale prototypes proved suc-cessful, the apparatus shown in Figure 9 was constructedand the crude material from the reaction of 42 grams of 74was eluted over a period of 124 hours, collecting fractionsapproximately every 7 hours and monitoring by TLC.
The spiroketals were recovered in 93% yield and with al-most complete separation of 72 from 73/77. The calculat-ed total solvent flow over the time of the separation was590 L (80 mL/min), whilst the actual amount used was 17L, representing a significant reduction factor of 35. In fact,highlighting another advantage of using an azeotropicmixture, the distillate from the rotary evaporators hadconsistently the same 1:1 composition of pentane and di-ethyl ether and could therefore be recycled back throughthe system, reducing the amount of total solvent used evenfurther.
It can clearly be seen that, although largely unknown andseldom practiced, the relatively simple technique of refluxchromatography has huge potential as a means of separa-tion even though, compared with conventional chroma-tography, a little further groundwork is necessary todefine separation parameters.
2.3 Phase-Switching Methods
Following from the previous two topics, this section willdiscuss some aspects of an emerging area which is mainlyconcerned with separation and purification. However,whilst the former section dealt with practical techniques inisolation from the chemistry of the systems involved, thissection requires knowledge of the chemical properties andreactivity of the components of a system. In fact, the re-curring theme will be the strategy of incorporating theseparation into the chemical synthesis itself, by deliber-ately devising some chemical or physicochemical reactiv-ity or property into the component(s) of a system in orderto facilitate later separation. Many convenient nonchro-matographic methods of separation are based on partition-
ing of components into different phases (e.g., liquid–liquid, solid–liquid, liquid–gas) followed by mechanicalseparation of the phases. If the components are already indifferent phases, as in solid-supported synthesis (e.g.,Merrifield peptide synthesis)45 for instance, then separa-tion is greatly facilitated. However, there is often a signif-icant benefit, in terms of the kinetics of the chemicalreaction taking place, if the reacting components arepresent in the same phase (so that reaction does not takeplace only at the interface). In order to get ‘the best of bothworlds’, various strategies have evolved which allow thechemist to switch specific components between phases atwill. Although the various diverse elements of this rapidlyemerging field do not comfortably fit into a single classi-fication, they all share the common element of phase tran-sition. We have used the term ‘phase-switching’ in thisarticle;46 other terms such as ‘phase-trafficking’ have also
Figure 9 Ley’s apparatus for reflux chromatography; A: 20 cm jack-eted coil condenser, B: 80 mm jacket condenser, C: column, D: 150mm 14 G stainless steel needle, E: solvent flask, F: insulated side arm D
ownl
oade
d by
: Kyu
shu
Uni
vers
ity. C
opyr
ight
ed m
ater
ial.

1168 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
been used.47 We wish to highlight strategies where thefunctionality responsible for the phase transition (the‘phase-tag’) has been deliberately incorporated into acomponent in addition to the functionality required for thesynthetic reaction itself. This can be distinguished fromthose methods (which can be defined as ‘scavenging’) thatexploit the native reactivity of excess or unreacted reac-tants/reagents for their separation. To be generally appli-cable, such phase-tags need to be compatible with a widerange of functionalities yet have a specific, orthogonal re-activity/affinity which enables phase-switching. It is notour aim to review exhaustively any particular aspect ofthis area, but rather to cover a number of facets, highlight-ing the diversity and scope of this useful concept. Whilstspace will only allow a limited number of specific exam-ples to be discussed in detail, references to further exam-ples will be given. Additionally, the reader will be pointedin the direction of more focussed reviews where these areavailable. The organisation of the section is primarily ac-cording to the type of physicochemical process involvedin the phase transition. Each subsection is further dividedaccording to the type of chemical reactivity/affinity re-sponsible, whilst also recognising that there will be someoverlapping concepts.
2.3.1 Affinity-Based Phase-Switching
Every synthetic organic chemist is familiar with aqueous‘work-up’ in which the reaction mixture is partitioned be-tween immiscible aqueous and organic phases. The inor-ganic/ionic species present tend to go into the aqueousphase and less polar organic components tend to enter theorganic phase. In some cases, the organic component canalso be present in the aqueous phase, particularly if it ishighly polar and/or ionised. For instance, amines can of-
ten be extracted into aqueous acid in which they form asalt, and carboxylic acids can likewise be extracted intoaqueous alkali. By the deliberate incorporation of these(and other) functional groups (referred to as ‘tags’) intothe reactant/reagent, the chemist gains the ability to con-trol the phase location of the species and effect a phase-switch (e.g., from one liquid phase to another, or from aliquid phase to a solid phase).
2.3.1.1 Fluorous Tags
One of the main driving forces behind the advancement ofthe phase-switching concept over the last couple of de-cades has been the development of fluorous strategies, byCurran and others.48 The key recognition of the ability ofhighly fluorinated organic compounds to elicit strong in-termolecular interactions and to form a separate fluorousphase has led to several protocols for phase-switching inorganic synthesis. Variants include liquid–liquid andliquid–solid phase separations but they all share the keycomponent of the orthogonality of the fluorous interac-tion/separation, which makes this concept so useful for or-ganic synthesis. Excellent reviews have been written onthis subject and space does not permit us to discuss thearea in detail.49 In a recent example, a fluorous tag was in-corporated as the thiol component in a Pummerer-type ar-omatic substitution of glyoxylic amide 78 (Scheme 17).50
The product 79 was purified by fluorous solid-phase ex-traction (FSPE). In this technique, the fluorinated com-pound is phase-switched to a fluorinated solid support (inthis case perfluoroalkylated silica gel), by elution throughthe column with a highly fluorophobic solvent (e.g., 4:1MeOH–H2O) during which all non-fluorinated compo-nents are eluted whilst the fluorous-tagged compound re-mains firmly bound. The product can then be released
Scheme 17
Br
N
Pr
O
O Br
NO
SCH2CH2C8F171. HSCH2CH2C8F17 CH2Cl2, then TFAA, BF3⋅OEt2, r.t.
2. fluorous solid phase extraction (FSPE) Pr
Br
NO
SCH2CH2C8F17
Pr
O
O
1. MCPBA, CH2Cl22. FSPE
Br
NO
SCH2CH2C8F17
Pr
OO
NO
SCH2CH2C8F17
Pr
OO
Me3Si
1. MeI, K2CO3 DMF, 80 °C2. FSPE
1. Me3SiCCH, Et3N Pd(PPh3)4, CuI, 80 °C2. FSPE
NO
Pr
Me3Si
SmI2 THF–H2O
78 79
8081
82 83
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1169
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
from the solid support by elution with a less fluorophobicsolvent (e.g., MeOH). Oxidation, methylation andSonagashira coupling steps were each followed by FSPE,affording the respective products in high purity. Reduc-tive cleavage of the fluorous tag (which was extracted intothe aqueous layer on workup) was mediated by samari-um(II) iodide.
2.3.1.2 Acid–Base and Polarity Tags
A pioneering early and well-known example of this ideais the use of 3-ethyl-1-(N,N-dimethyl)aminopropylcarbo-diimide (EDCI, 84; Figure 10).51 This carbodiimide, in-troduced by Sheehan in 1956 for use in peptide synthesis,is soluble in dilute acid along with its urea byproduct andcan thus easily be removed from the peptide product bywashing with acid after reaction is complete, the peptideremaining in the organic solvent layer. A similar, more re-cent example is the use of diphenyl(2-pyridyl)phosphine(85).52 This phosphine, like triphenylphosphine, facili-tates Mitsunobu reactions. However, the incorporation ofthe basic pyridine nitrogen allows it and its phosphine ox-ide byproduct to be removed by selective extraction of theorganic phase with dilute aqueous acid, whereas the morecommonly used triphenylphosphine and triphenylphos-phine oxide generally requires extensive column chroma-tography to effect separation from products.53
Figure 10
Water-soluble reagents and by-products have been de-signed which lack ionisable groups but which incorporatepolar solubility tags instead. For instance, the di-2-meth-oxyethyl azodicarboxylate (DMEAD, 88) has been usedin place of the commonly employed diethyl azodicarbox-ylate (DEAD, 86) and diisopropyl azodicarboxylate (87)in Mitsunobu reactions (Figure 11).54 The presence of theextra polarity in the side chains imparts water solubilityon the reagent and its hydrazine dicarboxylate by-product89, thereby allowing removal by simple aqueous extrac-tion.
Acid- or base-affinity has often been used to effect aphase-switch from solution to a solid support which canthen be removed by simple filtration. For example, Moffatoxidation of a series of secondary alcohols 90 was carriedout using EDCI (84) and catalytic dichloroacetic acid indimethyl sulfoxide.55 After reaction was complete, a com-bination of solid-supported sulfonic acid 93 and solid-supported tertiary amine 94 were used to completely re-move the excess reagents and byproducts. The solid-supported amine acted to scavenge the dichloroacetic acid
and also deprotonated the hydrochloric acid salts of thecarbodiimide and its urea byproduct, which were subse-quently phase-switched to the sulfonic acid resin(Scheme 18). This example highlights an important ad-vantage of using solid-supported reagents/scavengers:due to the site isolation of the active species, combinationsof reagents that are normally mutually incompatible (e.g.,acid and base) can be used in the same vessel. Conversely,however, this can be seen as an important limitation of theuse of solid supports since two reagents cannot easily re-act if they are both on a solid support.
Organic acids have also been used as affinity tags to en-able sequestration to a basic phase such as aqueous alkalior a solid-supported base. However, the nature of the acid-ic functionality renders it incompatible with many func-tional groups. As a way around this problem, a maskedacidic group can be used. Recent work in our laboratory
NCN
N
Me
Me
HCl
EDCI (84)
P
N
85
Figure 11
N N
O
O
O
O
DEAD (86)
N N
O
O
O
O
DIAD (87)
N N
O
O
O
O
O Me
OMe
DMEAD (88)
HN NH
O
O
O
O
O Me
OMe
89
Scheme 18
NH
OH
NR2R1
NH
O
NR2R1
DMSO, CH2Cl2cat. dichloro-acetic acid
C NNNH
NH
O
Me2NHClHCl
90 91
84
92
SO3
NMe2
C NN
Me2N
SO3H
Me2HN+
–SO3
–
NH
NH
O
Me2HN+
NHMe2NHMe2
+Cl
–+
O
O
Cl
Cl–
93 94
95 96
97 98
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

1170 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
has made use of the phosphine 99 in a variety of reactions.The tert-butyl ester in the reagent and in the phosphine ox-ide by-product 105 is later cleaved by trifluoroacetic acidin dichloromethane to reveal the carboxylic acid which isthen readily sequestered by solid-supported sodium car-bonate.56 For example, using this reagent, the formation ofamides was carried out in a continuous flow reaction ap-paratus. Reaction of 99 with a series of azides 100 led tothe formation of intermediate iminophosphoranes 101which were coupled to various acid chlorides 102 to fur-nish, upon hydrolysis, the amides 107 (Scheme 19). Afterscavenging the excess acid chlorides with polymer-sup-ported benzylamine 103, the tert-butyl ester of 105 wascleaved to afford the carboxylic acid, which was removedby phase-switching to a column of polymer-supported so-dium carbonate followed by filtration through silica gel toafford the amides 107 in high purity.
Scheme 19
In a related process, the Kirschning group used the isobu-tyl-sulfonate group as a masked affinity tag in the hyper-valent iodide species 108.57 These were used in theruthenium(III)-catalysed oxidation of secondary alcohols110 to ketones 111 (Scheme 20). Following the reaction,the excess reagent 108 and the reduced form 109 wereconverted into the anionic sulfonate species 113 and 114by nucleophilic displacement with azide supported onion-exchange resin 112. The reagent and by-product werethus phase-switched to the solid support which was re-moved by simple filtration to reveal the product and thevolatile isobutyl azide (115) which could be removed byevaporation.
Boronic acids have also been used as affinity tags, bindingcovalently to a polymer-supported diethanolamine resinwhich can be washed to remove excess reagents and by-products. Release from the solid phase is effected byaqueous hydrolysis. This sequence has been used itera-tively to synthesise pentapeptides, the boronic acid tag be-
ing fully compatible with the usual peptide-couplingconditions.58
2.3.1.3 Transition-Metal and Ligand Affinity Tags
Our laboratory has reported several phase-switching pro-tocols based on the strong attraction between a solid-sup-ported copper(II) complex 116 and a complementary 2,2-bipyridyl affinity tag 118 (Scheme 21).59 The interactioncan be reversed by the addition of N,N,N¢,N¢-tetramethyl-ethylenediamine and the tags can be present in either thedesired product (‘catch and release’ strategy) or in the re-agent (‘scavenging’ strategy).
Scheme 21
For instance, the 2,2-bipyridyl-tagged phosphonate ester120 was used to effect Horner–Wadsworth–Emmons ole-finations with a series of aldehydes 121 (Scheme 22).60
After the reaction was complete, the products 122 werephase-switched to a solid support using the copper(II)-impregnated IRC-718 resin 116. Filtration and washing ofthe resin thus enabled removal of excess base and alde-hyde. The product was subsequently released by
Ph2P Ot-Bu
O
Ph N3
Ph2P Ot-Bu
O
N Ph
PPh2
Ot-Bu
O
O
NH
PhR
O
1. RCOCl 102, 80 °C mix on chip2.
NH2
1. CH2Cl2, TFA, Me3SiH
2. NaCO3
99
100
NH
PhR
O
104
105
107
101
DMAP (cat.)
MeCN
103
106
Scheme 20
R1 R2
OH
i-BuO3S I(OAc)2
RuCl3 (cat.),MeCN–H2O
R1 R2
O
i-BuO3S I
NMe3 N3+ –
O3S I(OAc)2–
NMe3+
O3S I–
NMe3+
i-BuN3+
110 111
108 109112
113
114 115
N
N
CO2–
N
CO2–
Cu2+
R
CO2–
N
CO2–
Cu2+
CO2–
N
CO2–
Cu2+
N
N
Me Me
Me Me
N
N
R
TMEDACu2+
116
117
118119
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1171
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
N,N,N¢,N¢-tetramethylethylenediamine in dichloro-methane. The solvent and residual N,N,N¢,N¢-tetramethyl-ethylenediamine were simply removed by evaporation toafford the olefinic products 122 in high yield and high pu-rity.
Scheme 22
These enoate products were then elaborated in a numberof ways, including a microwave-assisted tandem Michael–Claisen addition of butanone 123. This reaction effectedcleavage of the 2,2-bipyridyl tag which was selectivelysequestered from the reaction mixture by solid-supportedcopper(II) to afford the 1,3-diketone product 124. The bi-pyridyl tag could be recovered from the resin for re-use bysubsequent treatment with N,N,N¢,N¢-tetramethylethyl-enediamine.
Work by others has shown that monodentate pyridyl tagscan be used to phase-switch reagents from a chloroformsolution into a fluorous phase by coordination to a per-fluorocarboxylate–copper(II) species. The interactionhere is much weaker than that observed with the bidentateligands and can be reversed simply by adding excess tet-rahydrofuran, which is a competitive ligand for cop-per(II).61
2.3.1.4 Hydrogen-Bonding Interactions
Perhaps due to its importance in biological systems, hy-drogen bonding is one of the most widely studied intermo-lecular interactions62 and it is not surprising that it hasfound application in phase-switching protocols. In a re-cent example, two self-complementary hydrogen-bondingureidopyrimidinone units were used, one present as a tagon the molecule to be phase-switched, 125, and onepresent on the solid support, 126 (Scheme 23).63 Thedimerisation constants of 6 × 107 M–1 in chloroform and6 × 108 M–1 in toluene allow complete sequestration of thetagged species in these non-protic solvents. A series of nu-cleophilic aromatic substitutions were carried out usingthe affinity tag as a means to effect ‘catch and release’ pu-
rification of the tagged products and as a means to sepa-rate the tag after subsequent cleavage.
Another useful hydrogen-bonding phase-switch has alsobeen reported which involved six hydrogen-bonding in-teractions between the barbituric acid tag in 128 and apolymer-supported Hamilton-type bis(2,6-diaminopy-ridyl)isophthalamide receptor 129 with which it forms aparticularly strong complex (Figure 12).64
Figure 12
2.3.1.5 Polyaromatic Affinity Tags
A traditional purification technique in the organic synthet-ic chemistry laboratory is the use of ‘decolourising’ char-coal to nonspecifically remove unwanted colouredimpurities. Different forms of charcoal and activated car-bon are also used in large-scale industrial purifications.
N
2
Cu(II)
1. t-BuOK, THF, 0 °C2. RCHO 121, 0 → 25 °C
3.
4. filtration and wash5. TMEDA release
1. t-BuOK
0 °C then MW at 70 °CO
Cu(II)3.
4. filtration and wash5. TMEDA release
OO
R
124
123
92% yeild(R = Ph)
O O
P(O)(OEt)2
N
2
O O
R
N
2
O O
R
120
116
122
122
116
Scheme 23
NH
NH
O
N
HN
O
Et Bu
substrate
HN
HN
O
N
NH
O
+
N N
O
N
HN
O
Et Bu
substrate
NN
O
N
NH
O
H H HH
non-polar solvent polar solvent
125
126
127
O
NH
N
NH
O
O
NH
N
NH
O
HN
HN
O
O
O
O
substrate
129
128
O
NH
O
O
NHD
ownl
oade
d by
: Kyu
shu
Uni
vers
ity. C
opyr
ight
ed m
ater
ial.

1172 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
Although different types of adsorption are known, theytend to be dominated by Van der Waals and p–p interac-tions.65 This phenomenon has been utilised to good effectin phase-switching strategies. By incorporating a tetra-benzofluorene tag (130), Ramage and co-workers wereable to carry out a synthesis of the quinolone antibacterialciprofloxacin using a ‘catch and release’ purification pro-tocol after each synthetic step.66 In a polar solvent, thetagged intermediates were completely sequestered by thecharcoal solid support and later freed by washing the fil-tered solid with a nonpolar solvent (Scheme 24).
Another polyaromatic phase-switching tag was investi-gated in a series of amide-bond-forming reactions. Excessacid chloride was tagged with the pyrene triamine species133. After sequestering to charcoal and filtration of thesolid, the product amides 134 were recovered in high pu-rity (Scheme 25).67
2.3.1.6 Crown Ether Tags
The crown ether tag 135 was first used by Fukase and co-workers in the synthesis of a series of peptides(Scheme 26).68 At each stage, the intermediates were pu-rified by phase-switching to a polymer-supported ammo-nium ion 136. In dichloromethane (the reaction solvent) atightly bound complex was formed and all the impuritieswere easily removed simply by washing. Then, the puri-fied intermediates were readily recovered by elution fromthe solid support with a base (triethylamine) or a polar sol-vent (dichloromethane–methanol, 1:1).
The sequestration of an 18-crown-6 phase-switching tagby solid-supported potassium sulfonate has also beendemonstrated. Reaction of the tagged amine 140 with acidchloride 141 led to the formation of the amide 142(Scheme 27).69 Unreacted amine 140 was removed bypassing the reaction mixture through a column of solid-supported sulfonic acid 143. The tagged amide 142 wasthen purified using a ‘catch and release’ protocol by bind-ing to solid-supported potassium sulfate 144 after whichthe excess acid chloride 141 was removed by rinsing withmethanol. The pure amide 142 was subsequently dis-placed by washing with methanolic potassium carbonate.
2.3.1.7 Redox-Switchable Tags
An interesting redox-mediated phase-switching protocolinvolved the use of ferrocenyl-tagged phosphine 145 foruse in the Mitsunobu reaction (Scheme 28).70 After the re-
Scheme 24
substrate
charcoal
+
polar solvent non-polarsolvent
substrate
charcoal
130Scheme 25
N
NH2
NH2
R1NH2 Cl R2
O
1.
2. charcoal
NH
R1
R2
O
+
131 132
133
134
excess
Scheme 26
O O
O
O
O
O
O
O
O
O
O substrate
O O
O
O
O
O
O
O
O
O
O substrate
NH3+
NH3+
1.
2. wash off excess reagent and by-products
O O
O
O
O
O
O
O
O
O
O substrate
NH2
base orpolar solvent
135
136
137
138
139
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1173
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
action was complete, the mixture was treated withiron(III) chloride (readily soluble in tetrahydrofuran),which oxidised the ferrocene group in the phosphine ox-ide by-product (and any unused phosphine) to the ferroce-nyl cation, 146. This was then water-soluble by virtue ofits charge and, after the addition of diethyl ether, was eas-ily extracted by water. Treatment of the aqueous phasewith sodium thiosulfate reduced the cation back to theneutral ferrocene 147 which could then be extracted backinto an organic solvent. A trichlorosilane-mediated reduc-tion converted the phosphine oxide back into the phos-phine 145 ready for re-use.
Scheme 28
2.3.2 Reactive and Irreversable Tags
Many of the affinity-based phase-switching strategies dis-cussed previously involve reversible complexation. Al-though this is normally advantageous inasmuch as itpermits sequential phase-switches, ‘catch and release’ pu-rification strategies, and recovery of affinity tags for re-use, several successful phase-switching protocols makeuse of irreversible (or not easily reversed) covalent bind-ing through a specific type of reactivity. While there arenow innumerable examples of the selective removal of
one component of a mixture by covalent binding to anoth-er (usually solid) phase, most of these would be classifiedas scavenging – the sequestration of a reactive functionalgroup used to carry out the desired reaction. However,several examples of irreversible phase-switching proto-cols are known in which covalent binding to a specificallyincorporated phase-tag takes place.
2.3.2.1 ‘Click’ Tags
Given that the copper-catalysed Huisgen alkyne–azidecycloaddition has been shown to be an extremely mildprocess, it is no surprise that it has found its way into thearsenal of phase-switching techniques. Our laboratory re-cently reported the use of the alkyne-substituted phos-phine 148 in the synthesis of a series of guanidines(Scheme 29).56 The iminophosphorane 150, which wasgenerated in situ and used in slight excess, was mixed ona microfluidic chip with a series of isothiocyanates 151 toform the carbodiimides 153 and the phosphine sulfide 154(X = S). Excess 150 was removed by passing through acolumn loaded with polymer-supported isocyanate 152, toform the phosphine oxide 154 (X = O). After reactionwith excess volatile secondary amine 155 and evapora-tion, the guanidines 156 were formed in high yield.
Scheme 29
The phosphine byproducts 154 could be removed from thereaction mixture by a phase-switching cycloaddition ontothe polymer-supported azide 157 in the presence of cata-lytic amounts of copper salts. The copper salts were sub-sequently scavenged by solid-supported thiourea. Analternative phase-switch involved cyclisation of thealkyne onto the unsupported alkyl azide 160 (Scheme 30).The carboxylic acid in the product 161 then facilitatedphase-switching by immobilisation onto a polymer-supported sodium carbonate. It can be seen that, during
Scheme 27
O
OO
O
O
O
O
OO
O
O
O
O
Cl
Cl
Et3N, CH2Cl2
140
141
142+ unreacted 140 and 141
SO3H
SO3–K+
142 + 141
1.
2. wash with MeOH3. wash with K2CO3 in MeOH
pure 142
NH2
NH
O
Cl
144 143
Fe
PPh2
Fe
PPh21. Mitsunobu reaction
2. FeCl3+ + products
O
145 146
1. wash into aqueous2. aq Na2S2O3
Fe
PPh2
O
HSiCl3
147
PPh2
148
R1 N3
149
Ph2P NR1
150
1. R2-N=C=S, 151 mix on chip (< 1 equiv)2. 152 (scavenge excess 150)
R1N C NR2
PPh2X
PPh2X
NH
N
N
R2R1
R3 R4
NH
R4R3
evaporate
N C O152
++
153
154
156
154
X = O,S
155
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

1174 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
this two-step protocol, the phase-switching tag has beentransferred from the alkyne to the carboxylic acid.
The use of azide and alkyne scavenger resins for purifica-tion of excess reactants from cycloaddition reactions hasalso been reported.71
2.3.2.2 Diels–Alder Tags
Several examples of using the Diels–Alder reaction to ef-fect a phase-switch are known. Aryl bromide 162 wascoupled to a series of arylboronic acids to afford diaryl
compounds 163. Activating a ‘safety catch’ protocol, therobust aniline-amide, which had survived the Suzuki cou-pling conditions, was then converted into the more reac-tive indole-amide, 164, by acid-catalysed intramolecularcondensation with the acetal. Ladlow and co-workers thenused the anthracene-based tag to irreversibly phase-switch the compounds onto the polymer-supported male-imide 165 (Scheme 31).72 Reaction with a series ofamines 167 or alcohols 168 smoothly effected cleavagefrom the indole which remained bound to the resin.
The anthracene tag was also used in a related strategy dur-ing a series of Mitsunobu reactions. Anthracene-taggedtriarylphosphine 173 was used along with a polymer-supported azodicarboxylate 174 (Scheme 32).73 After re-action was complete, the hydrazine dicarboxylate and un-reacted azodicarboxylate were removed by filtration. Theunreacted phosphine 173 and phosphine oxide 175 werethen easily removed by a phase-switching Diels–Alder re-action, again onto a polymer-supported maleimide 177, toafford pure reaction products 176.
2.3.3 Phase-Switching Involving Precipitation
2.3.3.1 Precipitation by Polymerisation
An ingenious strategy for phase-switching to a solid phaseinvolves the actual creation of the solid phase itself fromthe reagent/component by polymerisation of reactivemonomer tags. Although a range of polymerisation meth-ods have been used for this purpose, ring-opening meta-thesis polymerisation (ROMP) has emerged as the mostprevalent.74 A pioneering example involved the use of
Scheme 30
PPh2X
N3 , CuI, DIPEA
then
NH
NH2
S
N NN
PPh2
X
CO2H
N3
Ph2P
X
NN
N
CO2H
154
157
158
159
160
161
CuI
Scheme 31
NMe
O
NH
O
OO
Br
TFA, H2Oacetone, 58 °C
activation of 'safety-catch'
N
O
O
N
Me O
N
O
N
O
O
N
Me O
NH O
Nu
Ar
+
RNH2 167 or ROH 168
100 °C
ArB(OH)2, Pd(Ph3P)4Cs2CO3, DME, 80 °C
N
O
O
162
164
163
166
169 170
NMe
O
NH
O
OO
Ar
NMe
O
N
O
165
ArAr
Nu = RNH, RO
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1175
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
norbornene-tagged azodicarboxylate 181 in combinationwith a polymer-supported triarylphosphine in theMitsunobu reaction (Scheme 33).75 After the reaction wascomplete, catalytic quantities of the Grubbs catalystCl2(Cy3P)2Ru=CHPh (185) were added, causing the rapidpolymerisation of the norbornene tags to afford a solidresidue 186. The reaction products 183 were removedfrom the solids in very high purity simply by washing.This mode of phase-switching has been dubbed ‘impurityannihilation’ by Barrett and co-workers.
In a slightly modified strategy, a reactive functionalitypresent in an excess reagent/reactant is covalently bound(scavenged) by an added compound which contains theappropriate complementary functionality as well as a nor-bornene tag after reaction is complete. The tagged impu-rity is then phase-switched into an insoluble resin byROMP and removed by filtration. An example is shown inScheme 34.76 A series of coupling reactions between var-ious alcohol nucleophiles 187 and electrophiles 188 (usedin excess) were carried out. The excess electrophiles werethen tagged by the addition of the norbornene-containingalcohol 190, marking them for the subsequent phase-switching polymerisation. After addition of the Grubbs II
catalyst (192), the products 189 were removed simply bywashing from the insoluble polymer 193.
A ‘catch and release’ variant of this strategy has also beendeveloped, an example of which is shown in Scheme 35.
Scheme 32
R1XH
R2OH
O
O NN
O
OEt
R1 X
R2
N
O
O
O
OR1 X
R2
N
O
O177
+
171
172
173
174
176
176
178
+
+
PPhPh
O O
175
PPhPh
O O
O
PPh
Ph
O
Scheme 33
R1XH R2OH R1 X
R2
R1 X
R2
179 180 183
N N
OO
OO
PPh2
HN
HN
OO
OO
HN
HN
OO
OO
RMMR
n n
Ru
PCy3
PCy3
Ph
Cl
Cl
183
186
184
couple
phase- switch
185
181
182
Scheme 34
NC
O
R OH HN
O
OR
HN
O
O
HN
O
OR
n
Ru
PCy3
Ph
Cl
Cl
N NMes Mes
HN
O
OR
NC
O
HO
HN
O
O
M R'
excess
unreacted
187
188
189
188
190
191
189
193
189
192
tag the unreacted isocyanate
phase-switch
reaction
192D
ownl
oade
d by
: Kyu
shu
Uni
vers
ity. C
opyr
ight
ed m
ater
ial.

1176 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
A mixed Linstead macrocyclisation between the maleoni-triles 194 (6 equiv) and 195 resulted in a mixture of thesymmetrical and unsymmetrical porphyrazines 196 and197.77 After filtration of solid impurities, polymerisationphase-switching was effected by the Grubbs II catalyst. Inthis case a cross-linking agent 198 was also used. Thesymmetrical porphyrazine 196 was then removed bywashing from the insoluble polymer. The desired productwas easily freed from the solid support by mercury(II)-mediated cleavage of the benzyl–sulfide bonds and wasthen further transformed into 200.
2.3.3.2 Solubility-Switching
A large number of precipitation-based phase-switchingstrategies that have emerged are based on differential sol-ubility of species in different solvents. Functionalisedpoly(ethylene glycol) (PEG) ethers, which are highly sol-uble in solvents such as dichloromethane and N,N-di-
methylformamide but insoluble in hexane and diethylether, for example, have received significant attention anduse as solubility-switchable ‘solid’ supports for organicsynthesis. These, and other polymer supports, have beenextensively reviewed and will not be discussed furtherhere.78
Certain tetraarylphosphonium salts have very differentsolubilities in different solvents. This phenomenon hasbeen exploited by incorporating them into solubility tags.For instance, the tetraarylphosphonium-tagged carbodi-imides 201 have been used recently in a series of reactionsincluding esterifications (Scheme 36), amidations and de-hydrations.79 The reactions were carried out in dichlo-romethane, in which the tetraarylphosphonium specieswere highly soluble, and after reaction was complete theaddition of diethyl ether caused their complete precipita-tion. The products 205 were then recovered in high yieldand high purity simply by filtration. These tags have also
Scheme 35
CN
SS
NC
OO
CN
PrPr
NC
N
N
N
N
N
N
N
N
Pr
Pr
Pr
Pr
Pr
PrPr
Pr
Mg
N
N
N
N
N
N
N
N
Pr
Pr
Pr
Pr
S
SPr
Pr
Mg
O
O
N
N
N
N
N
N
N
N
Pr
Pr
Pr
Pr
S
SPr
Pr
Mg
O
O
n
n
N
N
N
N
N
N
N
N
Pr
Pr
Pr
Pr
Pr
Pr
Zn
S Ni
S
Ph2P
PPh2
+
Mg(OBu)2
6 equiv
194
195
196 197
Ru
PhPCy3
Cl
Cl
MesN NMes
1. Hg(OAc)2, HOCH2CH2SH2. Ni(dppe)Cl23. Zn(OAc)2
192
199
198
200
i. phase-switchii. filter
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1177
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
been sucessfuly incorporated into other reagents includ-ing triarylphosphines, diazodicarboxylates, and tin hy-drides, and have been used as ‘solid’ supports for thesynthesis of small molecules.80
A solubility-switchable support has been developed basedon the precipitation of the bisulfate salts of quinoline tags.As shown in Scheme 37, the aryl bromide 206 was takenthrough a sequence of Suzuki coupling, nitro-group re-duction and amide formation.81 The purification at eachstage involved the addition of sulfuric acid, which proto-nated the quinoline causing complete precipitation, fol-
lowed by filtration. Subsequent treatment of thequinolinium salt with aqueous sodium bicarbonate re-vealed the free base. The quinolinic acid fragment 213was cleaved from the compound by alkali hydrolysis andcould be recovered from the aqueous phase by acidifica-tion and extraction.
Polymeric supports have also been developed that havewell-defined pH-dependent solubilities. For example, therhodium-based hydrogenation catalyst 214, which con-tains a polymeric ligand, was shown to be highly active inboth aqueous and acetonitrile solvents, in which it wassoluble (Figure 13).82 In aqueous systems, lowering thepH to 5 caused the protonation of sufficient carboxylategroups to effect complete precipitation of the polymerwhich could then be removed by filtration. Addition to afresh pH 7.5 solution caused the catalyst to redissolve andit could subsequently be re-used with retention of activity.In cases where acetonitrile was used as solvent, the com-
Scheme 36
N
C
NEt
PPh3
+–
PF6
R1
O
OH
R2 OH
R1
O
O
203
202
esterificationCH2Cl2
phase-switch:precipitate 204 byaddition of Et2O, filtration
R1
O
O R2
205
NH
PPh3
+–
PF6
204
NHEtO
R2
205
201
Scheme 37
N
O
OBr
N
O
O(HO)2BPd(PPh3)4, DME
1.
1. Fe, EtOH, H2O, NH4Cl2. H2SO4, filter3. NaHCO3, extract
N
O
O
N
O
O
1. PhCOCl (210), Et3N2. H2SO4, filter3. NaHCO3, extract
N
O
OLi
HO
LiOH, THF–H2O,then Et2O, extract
212 213
211
206
207
208
209
(in aqueous phase)isolated for re-useby acidification and extraction
NO2NO2
NH2
HN
O
NH
O
2. H2SO4, filter3. NaHCO3, extract
Figure 13
NaO2C
OMe
N
O
PPh2
Ph2P
OMe
NaO2C CO2Na
1n
Rh+
214
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.
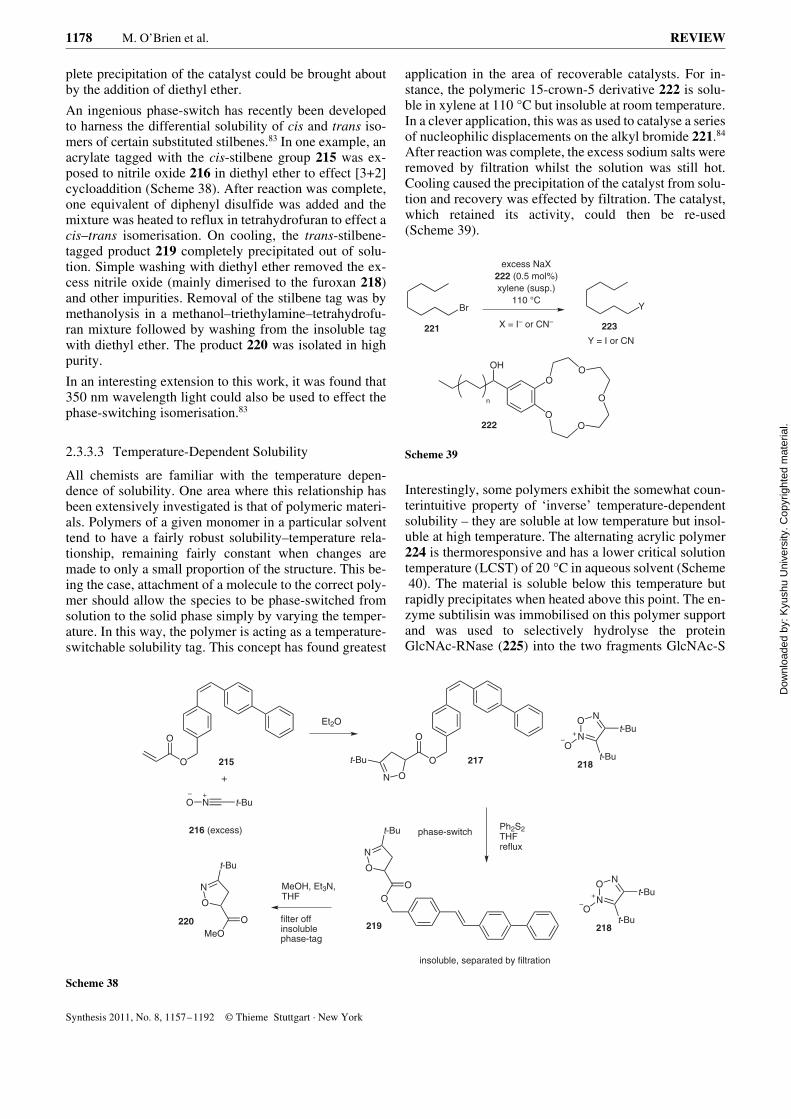
1178 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
plete precipitation of the catalyst could be brought aboutby the addition of diethyl ether.
An ingenious phase-switch has recently been developedto harness the differential solubility of cis and trans iso-mers of certain substituted stilbenes.83 In one example, anacrylate tagged with the cis-stilbene group 215 was ex-posed to nitrile oxide 216 in diethyl ether to effect [3+2]cycloaddition (Scheme 38). After reaction was complete,one equivalent of diphenyl disulfide was added and themixture was heated to reflux in tetrahydrofuran to effect acis–trans isomerisation. On cooling, the trans-stilbene-tagged product 219 completely precipitated out of solu-tion. Simple washing with diethyl ether removed the ex-cess nitrile oxide (mainly dimerised to the furoxan 218)and other impurities. Removal of the stilbene tag was bymethanolysis in a methanol–triethylamine–tetrahydrofu-ran mixture followed by washing from the insoluble tagwith diethyl ether. The product 220 was isolated in highpurity.
In an interesting extension to this work, it was found that350 nm wavelength light could also be used to effect thephase-switching isomerisation.83
2.3.3.3 Temperature-Dependent Solubility
All chemists are familiar with the temperature depen-dence of solubility. One area where this relationship hasbeen extensively investigated is that of polymeric materi-als. Polymers of a given monomer in a particular solventtend to have a fairly robust solubility–temperature rela-tionship, remaining fairly constant when changes aremade to only a small proportion of the structure. This be-ing the case, attachment of a molecule to the correct poly-mer should allow the species to be phase-switched fromsolution to the solid phase simply by varying the temper-ature. In this way, the polymer is acting as a temperature-switchable solubility tag. This concept has found greatest
application in the area of recoverable catalysts. For in-stance, the polymeric 15-crown-5 derivative 222 is solu-ble in xylene at 110 °C but insoluble at room temperature.In a clever application, this was as used to catalyse a seriesof nucleophilic displacements on the alkyl bromide 221.84
After reaction was complete, the excess sodium salts wereremoved by filtration whilst the solution was still hot.Cooling caused the precipitation of the catalyst from solu-tion and recovery was effected by filtration. The catalyst,which retained its activity, could then be re-used(Scheme 39).
Scheme 39
Interestingly, some polymers exhibit the somewhat coun-terintuitive property of ‘inverse’ temperature-dependentsolubility – they are soluble at low temperature but insol-uble at high temperature. The alternating acrylic polymer224 is thermoresponsive and has a lower critical solutiontemperature (LCST) of 20 °C in aqueous solvent (Scheme40). The material is soluble below this temperature but
rapidly precipitates when heated above this point. The en-zyme subtilisin was immobilised on this polymer supportand was used to selectively hydrolyse the proteinGlcNAc-RNase (225) into the two fragments GlcNAc-S
Scheme 38
O
O
NO t-Bu
215
216 (excess)
+–
O
O
217
N O
O
N
Nt-Bu
Ot-Bu
–+
218
Et2O
O
O
N
O
O
N
Nt-Bu
Ot-Bu
–+
218
Ph2S2THFreflux
MeO
O
N
O
insoluble, separated by filtration
219
MeOH, Et3N,THF
filter off insolublephase-tag
220
phase-switch
t-Bu
t-Bu
t-Bu
+
OH
n
excess NaX 222 (0.5 mol%)xylene (susp.)
110 °C
X = I– or CN–
Br Y
Y = I or CN221 223
222
O
O
OO
O
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1179
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
protein 226 and S peptide 227. Warming the solution to37 °C precipitated the immobilised protein which was iso-lated by centrifugation and could be re-used in further re-actions, retaining its activity.85
Scheme 40
2.3.4 Size-Exclusion Tags
A collection of related phase-transfer/separation tech-niques not commonly used in the synthetic organic chem-istry laboratory utilise size-dependent membranepermeation (e.g., dialysis or ultrafiltration). These meth-ods essentially rely on the inability of molecules to passthrough pores in a membrane which are smaller than themolecule itself. As such, a large group attached to a par-ticular molecule would act as a ‘size tag’, enabling aphase-switch to take place via a membrane with pores ofa size through which all other components would pass,leaving only the tagged component behind.
An example of the successful use of such a technique isshown in Scheme 41.86 Chloro ketone 229 was coupled tothe dentritic polyglycerol molecule 228. Purification fromall other components was then carried out by membraneseparation. A special parallel dialysis apparatus was de-signed that could enable 12 simultaneous separations to becarried out. Reaction with secondary amines 231 in thepresence of sodium iodide afforded the amino compounds232. Cleavage of the support was effected with trifluoro-acetic acid to furnish the keto amines 232. These wereseparated by dialysis or ultrafiltration from the dendrimersupport 228, which was recovered and could be re-used.Other separation techniques based on size (e.g., size-ex-clusion chromatography, centrifugation, gel-permeationchromatography) have also been used in a similar waywith dendrimers and polymers.87
In summary, it can be seen that a wide gamut of phase-switching strategies that offer great potential as enablingtechniques in organic synthesis have been developed. Wehope that this brief introduction to broadly related meth-odologies and concepts might serve as a reference andstarting point for further evolution and development of thefield at a time of rapid emergence.
3 Synthesis Methods
Up to this point in this review, we have chosen to focus onreaction clean-up technologies to solve downstream prob-lems of the synthesis process. In the final section, we com-ment briefly on some experimental technologies to aid thesynthesis component of the assembly process.
3.1 Cooled Microwave Heating
The field of focussed microwave-assisted organic synthe-sis has grown dramatically since the first reports on theuse of microwaves to accelerate organic reactions ap-peared in the mid-1980s.88 The improvements in reactionrates and product yields typically associated with micro-wave-assisted reactions, as compared to conventionalheating, coupled with the commercial availability of ded-icated instruments (Figure 14)89 has stimulated rapidgrowth in the field.
Several comprehensive reviews covering both theoreticaland practical aspects of microwaves have appeared re-cently.90 Here, however, we summarize, and highlight thepractical utility of, externally cooling while simultaneous-ly microwave heating samples as an emerging techniquewhich offers some advantages over conventional micro-wave heating for the synthetic chemist. Originally patent-ed as a concept by the CEM Corporation as PowerMAX,91
the technique allows a microwave reaction to be cooledwhilst simultaneously undergoing irradiation. This pro-cess results in the introduction of higher levels of micro-wave energy to a reaction mixture whilst maintaining aparticular temperature. At the present time the ‘cooled mi-crowave effect’ is debatable. Much of the debate is cen-
GlcNAc-RNaseGlcNAc-S protein
KETAAAKFERQHMDSSTSAA
+
(S peptide)
225226
227
Enz
Enz = subtilisin BPN' immobilised on 224
OO ONH NH O
NO O
20 5 2
224
Scheme 41
O
O OH
OH
1. PTSA, toluene reflux2. purify by dialysis or ultrafiltration
O
228
229
HNR1R2 231NaI, reflux
1. TFA–H2O (9:1)
2. separate (dialysis or ultrafiltration)
OH
OH
O
O
O
Cl
O
O
O
NR1R2
O
NR1R2
230
232
228
233
Cl
= dendrimer
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

1180 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
tered on how the temperature of the reaction is recordedand how rapidly changes in temperature can be detected.Furthermore, local heating effects arising in nonhomoge-neous reaction mixtures can also complicate matters.
Despite the recent growth in the use of cooled microwavemethods, a version of the technique was first described in1993.92 In this early work the use of microwave-transpar-ent solid carbon dioxide as a coolant allowed volatile liq-uids to be used in open-vessel reactions using domesticmicrowave ovens. This result represented a valuable andsafer alternative to the ‘microwave bombs’, as they werefirst described, which were in use at the time for reactionsinvolving volatile components. The reaction between fer-rocene 234 and benzene, or other arenes, to afford sand-wich complexes 235 was greatly facilitated by microwaveirradiation (Scheme 42). The yields obtained using cooledmicrowave irradiation compare very favourably to thoseobtained by conventional heating (in parentheses). More-over, under microwave conditions the number of equiva-lents of arene could be reduced.
Scheme 42
Another very early application of the method involved theoxidation of cis- and trans-4-tert-butylcyclohexanols 236with chromium trioxide (Scheme 43).93 At sub-ambienttemperatures the reaction in aqueous acetic acid is slow.However, the oxidation is rapid at temperatures above35 °C. Using the cooled microwave technique the reactionwas irradiated with varying microwave power to hold thereaction temperature at 25–28 °C. Without this degree ofcontrol, thermal runaway occurred. As the reaction ap-proached completion, the mixture was safely heated to110 °C and held for 15 minutes. Despite the above exam-ple being highlighted in a review along with the comment‘…concurrent heating and cooling were straightforwardin the microwave batch reactor, and should find other use-
ful applications,’ the technique remained overlooked untilthe advantages of the cooled microwave method usingmodern focussed microwave instrumentation was report-ed in 2003. The preparation of a-keto amides from acylchlorides and isonitriles was first described by Ugi duringthe 1960s.94
Scheme 43
However, this interesting protocol was limited by the slowhydrolysis of the intermediate a-keto imidoyl chlorides,such as 240 (Scheme 44). Such lengthy heating stepscompromise reaction throughput and often lead to valu-able reactions being underused in a drug discovery con-text. Both the addition (step 1) and hydrolysis (step 2)stages of the reaction were examined in a Discovery mi-crowave synthesiser in the absence of solvents, both withand without external cooling (Table 1).95 In an initial ex-periment an overall yield of 45% of a-keto amide 241 wasobtained.
Scheme 44
While this yield was comparable to that obtained usingconventional heating, the reaction time was decreasedfrom four hours to four minutes. In an attempt to improvethe yield the power was raised (step 1, entry b). However,this resulted in the formation of an intractable tar, presum-ably as a result of overheating. The overheating effect wasattenuated when the reaction vessel was cooled usingcompressed air during the course of irradiation (step 1, en-try c). By applying external cooling to both steps of the re-action the overall yield was improved and the reactiontime further reduced to a total of two minutes (entry d).This early work not only illustrates the advantages associ-ated with cooled microwave heating but also demon-strates how rapid reaction optimisation can be achieved
Figure 14
Fe
i) C6H6, AlCl3μ-wave, 3 min
Fe(ii) HPF6
PF6
88% (30–50%)
234 235
OHCrO3, AcOH (aq)
O
μ-wavesimultaneous
cooling
25–28 °C, 1 h;110 °C, 15 min
236 237
Cl
O
NC
238
239
N
O
Cl
HN
O
O
240
241
C6H6
μ-wave
CaCO3, H2O 50 °C
Δ or
orμ-wavesimultaneouscooling
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1181
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
using a microwave reactor due to the short reaction timesinvolved. The authors of this work, however, did not ex-haustively determine reaction temperatures within reac-tion vessels.
Although microwave methods are increasingly used topromote solution-phase reactions, there are relatively fewliterature examples of the use of polymer-supported re-agents acting as a heatsink in conjunction with microwaveheating. However, the combination of microwaves withexternal cooling can also be beneficial in this context. Forexample, the cyclohexane-1,3-dione resin 242 has beenused to prepare an amide library using a microwave-assisted catch and release protocol (Scheme 45).96 Boththe catch (O-acylation) and release (amide formation)steps were improved under microwave conditions. How-ever, higher release levels were observed when the resinmixture was subjected to continuous microwave irradia-tion with cooling as compared to runs in the absence ofcooling. A variety of amides including 245a–c were pre-pared in high yield and purity using this method.
Reactions using encapsulated reagents or catalysts canalso benefit from the additional cooling protocols. As a re-cent report on microwave-assisted Suzuki coupling reac-tions illustrates, the differences in product purity in thepresence and absence of external cooling can be dramatic.
A study concerning microwave-assisted Suzuki reactionsusing a polyurea microencapsulated palladium catalyst(PdEnCat)97 resulted in the synthesis of a biaryl library inmostly excellent purity.98 Under the original coupling pro-
tocol, biaryl 248 had been produced in low purity(Scheme 46, method a). However, when the Suzuki cou-pling was repeated with simultaneous cooling, the yieldwas significantly improved. The 1H NMR spectra of thecrude reaction mixtures for this Suzuki coupling, and fora further example from the library 249, with and withoutexternal cooling, are shown in Figure 15.
Scheme 46
Under method b, the reactions were heated at 50 W. How-ever, the internal temperature did not exceed 76 °C – sig-nificantly cooler that the 120 °C used in method a.Interestingly, repeating the reactions at 76 °C for the sameamount of time in the absence of external cooling resultedin incomplete conversion and the formation of by-products.
A further study involving three problematic aryl bromideswas then conducted with a range of boronic acids
Table 1 Microwave Synthesis of a-Keto Amides with and without Cooling
Entry Step 1 Step 2 Yield (%)
Time (min) Power (W) Cooling Tmax (°C) Time (min) Power (W) Cooling Tmax (°C)
a 2 60 off 149 2 60 off 195 45
b 1 100 off 150 2 60 off 195 0
c 1 100 on 100 2 60 off 180 69
d 1 100 on 100 1 100 on 125 74
Scheme 45
N
O
HO O
Me
(i) MeCO2H, CH2Cl2μ-wave, 100 °C
300 W, 2 x 10 min
(ii)N
R2
R3
HMeOH, μ-wave, 125 °C
200 W, 30 minsimultaneous cooling
R1 Cl
O
N
O
O OH
MeR1 N
O
R2
R3
242 242245
HN
O
245a, 98%, 100% puritya
N
O
245c, 63%, 85% puritya
OMe
HN
O
Cl
Cl
245b, 100%, 91% puritya
a Purity by LC-MS at 220 nm
+243
244
OCF3
B(OH)2
Br
F3CO
246
247
248
method a 48% puritymethod b >98% purity
PdEnCat (5 mol%)n-Bu4NOAc (200 mol%)
EtOH
method a: μ-wave, 120 °C, 50 W, 10 minmethod b: μ-wave, 50 W, 20 min, simultaneous cooling
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

1182 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
(Scheme 47, Table 2). These substrates had been shownto produce multiple by-products in previous experiments.Again, significant improvements in product purity wereobserved for a range of cross-coupling reactions usingboth electron-rich and electron-poor arylboronic acids250 when method b (simultaneous cooling) was em-ployed.
Scheme 47
The cooled microwave method has also been shown to bebeneficial in Suzuki cross-coupling reactions of aryl chlo-rides. For example, 4-chlorotoluene and phenylboronicacid undergo an efficient Suzuki reaction in the presenceof Pd/C in an aqueous reaction medium (Scheme 48,Table 3).99 Initial results (entries 1–3) showed that theyield of biaryl 255 could be improved by increasing thetemperature; however, full conversion was not achieveddue to decomposition of the aryl halide 254. However, ap-plication of simultaneous cooling improved the yield of255 from 40% to 75% at the same temperature (entry 5 vs.entry 1).
Figure 15
Ar B(OH)2 Ar
R
method a or method b
PdEnCat (5 mol%)
EtOH
Br R
n-Bu4NOAc (200 mol%)
251
250 252
method a: μ-wave, 120 °C, 50 W, 10 minmethod b: μ-wave, 50 W, 20 min, simultaneous cooling
Table 2 Microwave-Assisted Suzuki Coupling of Aryl Bromides
Entry R Ar Purity (%)method a
Purity (%)method b
1 CH2OH Ph 46 >98
2 CH2OH 3,4,5-(MeO)3C6H2 <10 90
3 CH2OH 3-NO2C6H4 31 96
4 CH2OH 2-benzofuran 41 97
5 NHAc 3,4,5-(MeO)3C6H2 27 90
6 NHAc 3-NO2C6H4 32 96
7 NHAc 2-benzofuran <10 97
8 Ph 3,4,5-(MeO)3C6H2 <10 89
9 Ph 3-NO2C6H4 <10 93
10 Ph 2-benzofuran <10 86
Scheme 48
B(OH)2
Cl
254
Pd/C (1 mol%)Na2CO3 (3.7 mmol)n-Bu4NBr (1 mmol)
253 255μ-wave
300 W, 10 min
H2O
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1183
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
Further investigation showed that the effect of externalcooling is more significant for reactions involving elec-tron-rich aryl chlorides than electron-poor aryl chlorides.
In summary, the data for Suzuki cross-coupling reactionsis consistent with the hypothesis that the slower-reactingelectron-rich aryl chlorides undergo competitive thermaldecomposition in the absence of external cooling. The im-provements introduced by cooling are unlikely to be lim-ited to Suzuki reactions.
Indeed, the effect of cooling on microwave-promotedcopper-mediated Chan–Lam N-arylation reactions ofarylboronic acids 250 and pyrazinone 256 was recentlydisclosed (Scheme 49).100 In this instance, cooling to 0 °Cduring irradiation provided improved product yields for arange of arylboronic acids (Table 4).
Finally, the cooling method has also been of use in 1,3-di-polar cycloadditions. Microwave-promoted reactions be-tween bis(azidomethyl)benzene 258 and carbamoyl-
propiolates 259a,b (Scheme 50) resulted in the exclusiveformation of regioisomeric mono-cycloaddition products.Thus, 260a and 260b were isolated in 60% and 12%yields respectively, whilst 261a and 261b were obtainedin 54% and 18% yields.101
In an attempt to obtain the corresponding bis-triazoles thetemperature and microwave power were increased. How-ever, these increases resulted in significant decompositionof starting materials and charring. When simultaneouscooling was applied to the reactions, the desired double
Table 3 Microwave Conditions for the Suzuki Coupling of 4-Chlo-rotoluene
Entry Temp(°C)
Cooling Yield (%) of 255
Recovered 254 (%)
1 120 no 40 23
2 100 no 19 19
3 135 no 56 8
4 150 no 61 10
5 120 yes 75 7
6 100 yes 40 12
7 135 yes 71 10
8 150 yes 56 19
Scheme 50
N3
N3
O
N OEt
O
H
R
259a R = Ph259b R = p-tolyl
258
259
μ-wave, 55 °C
120 W, 30 min
μ-wave, 75 °C
120 W, 60 minsimultaneous cooling N
NN
N
N
N
CONHR
CO2Et
EtO2C
RHNOC
N
NN
N
N
N
CO2Et
CONHR
EtO2C
RHNOC
N
N3
260a 60%
N
N
EtO2C
RHNOC
N
N3
N
N
RHNOC
EtO2C
261a 12%
O
N OEt
O
H
R
259
260b 54% 261b 18%
262 R = Ph 54% 263 R = p-tolyl 65%
Scheme 49
R
B(OH)2
N
N O
Cl SPh
250
Cu(OAc)2 (200 mol%)Et3N (200 mol%)
method a or method b
257
N
HN O
SPhCl R
method a: r.t., 12 hmethod b: μ-wave, 0 °C, 300 W, 12 h, simultaneous cooling
256
CH2Cl2
Table 4 Microwave-Promoted Chan–Lam N-Arylation of Pyrazi-none 256
Entry R Yield (%)method a
Yield (%)method b
1 3-F3C 33 69
2 3-Cl 75 90
3 3-Br 36 49
4 3-EtO 28 64
5 4-MeO 44 69
6 H 64 87
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

1184 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
cycloaddition products 262 and 263 were isolated in 54%and 65% yields respectively, from regioisomeric mix-tures. In contrast to the examples above, where the cooledmicrowave method resulted in improved yields or puri-ties, Scheme 50 demonstrates how the technique can beused to obtain products which are inaccessible via con-ventional microwave heating.
Recently, reports on the use of the cooled microwavemethod in asymmetric organocatalysis have begun to ap-pear.102 For example, Bolm described the influence of mi-crowave irradiation on the proline-catalysed direct, three-component asymmetric Mannich reaction originally dis-closed by Córdova (Scheme 51).103 The authors were ableto demonstrate that the originally reported conditions forthis transformation could be significantly improved bymicrowave heating in conjunction with simultaneouscooling. Catalyst loadings as low as 0.5 mol% were usedto obtain products with up to 90% ee after three hours ofmicrowave heating. While the reductions in catalyst load-ing could be reproduced using conventional heating in anoil bath, the drop in reaction time could only be achievedvia microwave heating with simultaneous cooling(Scheme 51). It should be noted that fiber optic probeswere not used for temperature measurements in this study.
Scheme 51
This work was recently re-examined by Kappe who wasable to demonstrate that, in contrast to the original report,the results obtained either by microwave irradiation andmicrowave irradiation coupled with simultaneous coolingcould be reproduced by conventional heating for the sameamount of time and at the same temperature using an oilbath (Table 5).104
This study highlights the critical importance of accuratetemperature measurement using internal fiber opticprobes rather than the more commonly employed externalinfrared sensors. It remains to be seen, therefore, whetherthis heating and cooling method can effectively be con-trolled in synthesis devices or whether alternative tech-niques such as flow chemical methods will eventuallyprove to be superior.
3.2 Ball-Milling
In this section we highlight the synthesis applications ofball-milling. Ball-milling is a scalable mechanical tech-nique that has been used extensively for the preparationand modification of metals, hydrides, and other inorganicsolids. There are three main types of ball mills available:
the vibrating ball mill, the planetary ball mill and thestirred ball mill. All ball mills contain a grinding materialand many different materials can be used as media, in-cluding ceramic balls, flint pebbles, and stainless steelballs. In vibrational milling, agitation along the axis of themill causes the grinding material (e.g., ceramic ball bear-ings) to move and reduces the material inside to a finepowder.105 Planetary ball mills rely on the rotation of themill itself along its axis in combination with rotation ofthe platform to which the mill is attached. Finally, stirredball mills employ a mechanical stirrer to agitate the grind-ing medium. All three techniques have been employed ona laboratory scale using commercially available instru-ments such as that shown in Figure 16. At present, little isknown about ‘mechanochemistry’, that is, chemical reac-tions taking place during the mechanical milling of organ-ic solids.106 But, the grinding of particles results in largesurface areas for solids to mix and high temperatures andpressures can be obtained.
Reactions carried out in this manner are attractive from anenvironmental point of view since reactions between solidreactants are run in the absence of organic solvents. Someof the earliest examples documenting mechanical activa-
X
NH
O NH2
O
H H
264
266
265
267NH
CO2H
268 X = O
269 X = H, OHNaBH4
μ-wave orΔ, 65 °C
150–300 min
Table 5 Heating Methods in Asymmetric Organocatalysis of a Three-Component Mannich Reaction
Entry Heat methoda
267(mol%)
Power (W)
Time (h)
Yield (%)
ee (%)
1 oil bath 10 2.5 63 98
2 MW 10 6 2.5 66 97
3 MWair 10 15 2.5 61 98
4 MWliq 10 291 2.5 64 98
5 oil bath 1 5 79 97
6 MW 1 7 5 83 97
7 MWair 1 57 5 81 98
8 MWliq 1 232 5 81 97
a MW = microwave, MWair = microwave with simultaneous air cooling, MWliq = microwave with simultaneous liquid cooling.
Figure 16
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1185
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
tion of organic reactants can be found in the fullerenefield. For example, the dumbbell-shaped fullerene dimerC120 was prepared for the first time by the reaction of C60
with potassium cyanide in the solid state using a ‘vibrat-ing mill’.107 Rather than provide an exhaustive list of ap-plications, our intention here is to highlight key examplesrelevant to bond construction.108
Perhaps the most striking examples that have helped tobring the ball-milling method to the attention of the syn-thesis community are from the recent work of Bolm andco-workers.109,110
For example, the asymmetric opening of meso-anhydridesmediated by quinine or quinidine was effective under ball-milling conditions with a range of benzylic alcohols. Arepresentative example is depicted in Scheme 52. In mostcases reported yields were excellent and enantioselectivitiesup to 64% were obtained. No solvent was used in the re-actions except during workup and the use of equimolaramounts of the starting materials represented a significantpractical advantage over typical solution-phase condi-tions.109
Scheme 52
Asymmetric organocatalytic aldol reactions catalyzed byproline were also described (Scheme 53).110 Although sol-vent-free organocatalytic aldol reactions had been previ-ously reported, a comparative study showed that ball-milling reduced the reaction times dramatically. In the ex-ample shown the reaction time was reduced by 18.5 hourscompared to a solvent-free reaction conducted with con-ventional stirring.
Scheme 53
An interesting extension of the Knoevenagel–dehydrationreaction appeared recently. In this study mechanical ball-milling was used in a Michael addition–elimination–reduction sequence with solid dihydropyridine 278 as thereductant (Scheme 54).111,112
Scheme 54
Excellent yields were obtained for electron-deficient alde-hydes after chromatographic purification. Reduced, butnevertheless good, yields were obtained via an acidifica-tion and filtration protocol rendering the process free fromorganic solvents. This one-pot procedure represents animprovement over previous solution-phase syntheses inwhich a separate step was required for reduction of thebenzylidene dinitriles due to competing reduction of thealdehyde component.
Reductive amination of aromatic aldehydes and anilines isalso possible in the solid-state using dihydropyridine 278as the reductant in the presence of a Lewis acid catalyst(Scheme 55).112
Scheme 55
Again, excellent yields were obtained after chromato-graphic purification for electron-deficient aldehydes. Theuse of 1.02 equivalents of the dihydopyridine is notewor-thy. Solution-phase reactions of this type usually requireeither an inert atmosphere, or excess of the reductant inorder to prevent aerial oxidation of the dihydropyridine.
The mechanically induced Wittig reaction has also beendescribed recently. In a pioneering study, stabilised, semi-stabilised and non-stabilised phosphoranes were generat-ed in the solid-state by ball-milling the appropriate phos-
O
O
O
OHCO2H
O
O270
271
272 91%, 61% ee
quinidine (1.1 equiv)
ball-millingno solvent24–36 h
O CHO
NO2273
274
(S)-proline (10 mol%)
ball-milling, 5.5 h
O OH
NO2275
O
R
CN
CNNH
EtO2C CO2Et
ball-milling 30 Hz, 90 min
276 278277
N
EtO2C CO2Et
280
R
279
CN
CN
O
R
NH
EtO2C CO2Et
ball-milling 30 Hz, 90 min
281 278282
N
EtO2C CO2Et
280
R
283
NH
ZnCl2 (25 mol%)
NH2
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

1186 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
phonium salt with potassium carbonate.113
Characterisation of the derived phosphonium salts wasthen carried out using solid-state 31P NMR with magic an-gle spinning. Based on these initial results the mecha-nochemical Wittig reaction was achieved in the absenceof solvent (Scheme 56). It is noteworthy that an alkenesuch as 287 would typically be prepared in solution via amultistep process. Furthermore, strong bases are not re-quired for solid-phase ylide formation.
Scheme 56
Using the ball-milling technique, 287 was prepared ineight hours using a single reaction vessel. The stilbenewas isolated in 93% yield as an inseparable mixture ofgeometric isomers using column chromatography. Inter-estingly, the solid-state reaction afforded the thermody-namic E-configured stilbene selectively (E/Z = 3.5:1).Solution-phase reactions of this type typically afford theZ products or E/Z mixtures. Based on further investiga-tions, hidden liquid-state reactions, melting caused bylocal heating, as well as the formation of a low-meltingeutectic were ruled out.
Transition-metal-catalysed processes are also amenable toball-milling. For example, solid-state Heck reactions be-tween aryl halides and enamino esters have been report-ed.114 This work builds upon results obtained for Heckcouplings carried out in molten tetra-n-butylammoniumchloride.115 It is believed that in the molten state, alkylam-monium salts act as ionic liquids that have been claimedto have a stabilising effect on the catalytically active pal-ladium(0) species. Indeed, the tetra-n-butylammoniumion was essential for successful reactions (Scheme 57). Arange of aryl halides were examined with two differentalkenes in a planetary ball-mill. Variable yields of cou-pled products were obtained after chromatographic purifi-cation. Interestingly, electron-deficient aryl or heteroarylhalides, such as 4-iodonitrobenzene, did not undergo cou-pling whilst electron-rich substrates coupled efficiently.Prior protection of an amine functionality was not re-quired. This reactivity is in agreement with a study of sol-id-state Suzuki couplings where similar failure incoupling of electron-poor aryl halides was observed. Re-cently, the successful use of ball-milling in Sonagashiracouplings has also been reported.116
In an interesting recent study, the solvent-free reductionof aldehydes and ketones with sodium borohydride wasinvestigated using ball-milling conditions.117
Ketones and aldehydes 291 were ball-milled with stoichi-ometric (4:1) amounts of sodium borohydride (typically atambient temperature for 10 minutes to 1 hour) to form in-termediate tetralkoxyborates 292, where each hydrogenatom of the reducing agent has been transferred(Scheme 58). Typically, sodium borohydride reacts withmany of the protic solvents in which it dissolves with evo-lution of hydrogen gas, a disadvantage from both econom-ic and safety viewpoints. These intermediates were thenhydrolysed with water and the products 293 were isolatedin essentially quantitative yields. Interestingly, a,b-unsat-urated aldehydes were reduced with complete regioselec-tivity (293a), no saturated alcohols were observedalthough these are typically formed in significant amountswhen the reactions are performed in solution. Also, phen-acyl bromide was quantitatively reduced to form the bro-mohydrin with no loss of the bromine atom or epoxideformation.
Additionally, the quantitative mono-reduction of benzil294 could be achieved by using exactly 0.25 equivalents
BrPh3P
O
Br
Br
K2CO3 ball-milling8 h
284 285 286
287 (E/Z = 3.5:1)
93%
Scheme 57
X
R1
HN
O
O
O
t-Bu
R2
289a R2 = Bn
Pd(OAc)2, NaHCO3, HCO2Na n-Bu4NCl, NaCl
R1
290
HN O
O
O
O
R2
t-Bu
288
289b R2 = Me
ball-milling1 h
O
Scheme 58
R1 R2
O
BO
O
OO
R2
R1
R2
R1
R2
R1
R2
R1
H
H
H
H
–Na +
NaBH4(1.0 equiv)
ball-milling
4 x
291
292
R1 R2
OH
H2O
R1, R2 = alkyl, arylyeilds >99%
293
OH
100% 1,2 reduction
293a
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1187
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
of sodium borohydride (1 H per mole of substrate) to formbenzoin 295 (Scheme 59). This highly selective reactivityhas never been observed in solution. Using 0.5 equiva-lents of borohydride resulted in complete reduction af-fording an 80:20 ratio of the meso and rac stereoisomers296 and 297. Interestingly, when carried out in methanolas solvent, this reduction afforded the meso compound296 exclusively,118 illustrating the profound influence ofsolvent on the course of borohydride reductions.
Scheme 59
As the examples cited above illustrate, the ball-millingtechnique can be of value to synthetic chemists. At presentthe availability of dedicated apparatus for small-scale re-actions could be seen as limiting its use. Hopefully, in thefuture, dedicated ball-milling apparatus for organic reac-tions will be made available.
3.3 Design of Experiments and Related Tech-niques
We have discussed enabling technologies for both the pu-rification and the preparation of organic molecules. In thefinal section of this review we wish to discuss a techniquethat facilitates rapid optimisation of organic reactions, andthat, sadly, has rarely been applied outside the process-scale environment.
The optimisation of chemical reactions requires an under-standing of which variables are important to a particularoutcome. A traditional approach still used by most chem-ists, sometimes referred to as ‘the rule of one’, involvessystematic variation of one variable at a time during opti-misation (Figure 17, a). The axes of the reaction space de-fine variations in the nature of different constituents of thereaction system, such as reagent, solvent and catalystloading. In the traditional approach a single variable suchas ‘a’ might be changed whilst ‘b’ and ‘c’ remain con-stant. Then with ‘a’ fixed, optimisation of the other twovariables would be carried out in the same way in order todiscover an optimum combination of conditions. Whilsteffective, this is not the most efficient method by which toexplore experimental space. A more efficient approach in-volves the use of ‘design of experiments’ (DoE) tech-niques in which variables are first screened to determinewhich are important to the outcome. This is followed by‘optimisation’ where the best settings for the importantvariables are determined. In particular, response surfacemethods and factorial designs have been used.
For example, the two-level three-variable factorial exper-iment design (Figure 17, b) allows high and low settingsfor each variable to be selected. Experiments are then runin all possible combinations. In this way, the effect of eachvariable and the interaction between variables can be esti-mated. For a three-variable study a total of eight experi-ments will be required. The data can then be used togenerate a response surface graph (Figure 18). By usingsuch a graphical representation the interactions of vari-ables can be seen. Moreover, optimum conditions areclearly identified.
Figure 18 Response surface graph. [Reprinted from ref. 119h. Co-pyright (2010), with permission from Elsevier.]
Recent advances in supporting software, automated syn-thesis instrumentation and high-througput analytical tech-niques have led to the broader adoption in pharmaceuticaldiscovery and development laboratories. Indeed, by thelate 1990s the method was being employed by many pro-cess development groups and automated workstationswith on-line analysis were developed.119 However, de-spite the advantages associated with this method its use inacademic laboratories is very limited at present. Here wewish to showcase some of the recent applications of themethod for the optimisation of organic chemistry process-es using both conventional solution- and solid-phase re-agents.
A factorial experiment design was used by the processgroup at AstraZeneca for the optimization of an asymmet-ric sulfoxidation (Scheme 60).120 The use of the DoEmethod resulted in a 12% increase in enantiomeric excess.
Ph
O
Ph
O
Ph
OH
Ph
O
i. NaBH4 (0.25 equiv) ball-millingii. H2O
Ph
OH
Ph
OH
Ph
OH
Ph
OHi. NaBH4 (0.5 equiv) ball-millingii. H2O +
294
296 297
295
Figure 17
a
b
c
(a)
a
b
c
(b)
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

1188 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
Significantly, an optimal catalyst stoichiometry of 1.45equivalents (–)-diethyl tartrate and 0.95 equivalents titani-um(IV) isopropoxide was responsible for the increase inee. This may have been overlooked if more conventionaloptimization had been performed, since the literature sug-gests a 2:1 ratio of diethyl tartrate to titanium(IV) isopro-poxide is necessary for high enantioselectivities.
Scheme 60
The isolation of natural products can also benefit from theDoE method. For example, the extractive isolation of ole-oresin xanthophyll pigments from marigold mixed flourhas been studied (Figure 19).121 Extraction conditionswere identified that required 16% less solvent and resultedin a 2% increase in purity of the extracted pigment.
Figure 19
Although the improvements in the last two examples mayseem small, they are significant in the context of commer-cial-scale operations. More recently, the technique hasfound application for the optimization of polymer-supported reagents. Amide bond formation is widely em-ployed in the generation of combinatorial arrays. Howev-er, amide formation using the commercially availablepolymeric carbodiimide reagent 302 (Scheme 61) is noto-riously capricious. The variables considered were stoichi-ometry of resin, stoichiometry of amine, reaction time andconcentration. Initially, these variables were investigatedusing a single acid and amine. The statistical analysis re-
vealed optimal solvents, such as dichloromethane, whichcorrelated to their ability to swell polystyrene-based res-ins. Additionally, the detrimental effect of excess aminewas identified. It was also shown that the yield was notsensitive to concentration over the range studied. Underthe optimized conditions, a selection of amides 303 wasprepared (Scheme 61). Finally, an 80-member amide ar-ray was prepared using the optimized conditions.122
Scheme 61
A DoE optimisation of amide formation using polymer-supported hydroxybenzotriazole (PS-HOBT) 304 wasalso recently reported (Scheme 62).123 The optimisationwas carried out in two stages. Firstly, several reaction pa-rameters were screened to ascertain which were signifi-cant. These were found to be: order of addition, solventratio (DMF:CH2Cl2) and amount of diisopropylcarbodi-imide. A second set of experiments was then designed tooptimise within these variables. In comparison with theinitially reported conditions, it was found that the numberof equivalents of carboxylic acid could be reduced from 6to 1.5 and the reaction time was reduced from six hours totwo. The optimized conditions were used to synthesise a48-member library of structurally diverse amides 308 inyields of 54–93% with purities over 87%.
Scheme 62
Although the DoE approach reduces the total number ofexperiments required, optimisation over many variablesstill requires a significant number of data points. For thisreason, even higher efficiency can be obtained using auto-mation technologies. For example, the SNAr displacementof fluoride from 2,4-difluoro-1-nitrobenzene (309) byBoc-amino acids 310 was investigated using the ReactArray system124 to automate the sampling and monitoringof the reaction products for both yield and purity(Scheme 63).125 Variables considered were the stoichio-
Ntoluene
(–)-DET
N
OHN
N
OH
SEt
SEt
O298 299
variables: stoichiometry, addition temperature, addition rate, hold time, quench
ee improved from 60% to 92%
Ti(OiPr)4
t-BuOOH
R1 OH
O
R2R3NH
R1 N
O
R2
R3
variables: stoichiometry, concentration, reaction time, solvent character
85–100%5 examples
301
300303
C NN
c-Hex 302
+
R'HN
O
R
RCO2H305
coupling agent
wash
S
N N
N OH
OO
HN
S
N N
N O
OO
HN
O
R
RNH2
304 306
308
307
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1189
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
metry of the amino acid 310, stoichiometry of the polymer-supported base 311, reaction time and concentration.Analysis of the experimental data obtained was carriedout using Design Expert 5 software.126 The stoichiometryof the amino acid was found to be the most significantvariable. The number of equivalents of polymer-support-ed base also had an effect whilst reaction time and concen-tration were found to be relatively unimportant. Theoptimum conditions were found to be two equivalents ofamino acid and four equivalents of polymer-supportedbase. With these optimised conditions, yields were typi-cally greater than 90% and products were isolated ingreater than 95% purity without the need for chromato-graphic purification.
Scheme 63
Organic synthesis using microwave heating is a techniquethat is being used increasingly in medicinal chemistry lab-oratories. In 2003 the first report concerning the use ofDoE to optimize a microwave-promoted reaction ap-peared (Scheme 64).127
Scheme 64
Variables studied included reaction time, temperature andequivalents of amine base. The stoichiometry of the aminebase was found to be the most important variable. Onceoptimization was complete a 24-member library of 1,2,4-oxadiazoles 315 was prepared. The desired products wereisolated with an average purity of 68%.
A more recent example of DoE optimisation of micro-wave-promoted reactions is shown in Scheme 65. A Big-inelli three-component coupling of ethyl acetoacetate(316), thiourea (317) and 3-hydroxybenzaldehyde (318)was used in the synthesis of the mitotic kinesin Eg5 inhib-itor monastrol (319).128 Using the statistical DoE softwarepackage MODDE8,129 with an automated microwave re-actor, an initial screen of catalyst type and solvent wascarried out (16 reactions plus 3 centre points), which iden-tified the combination of ethanol and lanthanum(III) chlo-ride as optimal using HPLC as a rapid method of analysis(in terms of both conversion and purity). Ytterbium(III)trifluoromethanesulfonate performed comparably but wasruled out on the grounds of cost. A second set of 10 exper-
iments was then designed to optimise with respect to reac-tion time and temperature, which were both found to besignificant variables; conversions ranging from 67% to95% were obtained in the experiments carried out. Final-ly, optimisation over the variables of temperature and cat-alyst concentration was carried out, affording conditionswhich gave monastrol in 82% yield.
Scheme 65
From the foregoing discussion it is evident that applica-tions of the DoE technique in synthesis are expandingbeyond traditional process chemistry applications, partic-ularly in the areas drug discovery and parallel synthesis.We anticipate that the method will also be of considerableuse in more academic knowledge-based studies for reac-tion optimisation.
4 Conclusion
In this article we have brought together certain tools andtechnologies which on their own do not merit a distinct re-view. However, collectively these lesser-known and lessused methods enhance the armoury of the modern synthe-sis chemist. By bringing together these enabling conceptstogether in this way, we hope to promote their applicationin specific instances where the current more establishedprotocols are not always effective. Given the immense di-versity of structural types and phenomenal range of reac-tions and reagents needed to assemble these compounds,continued development of the tools of synthesis is essen-tial to meet these challenges.
References
(1) (a) Tswett, M. S. Ber. Dtsch. Bot. Ges. 1906, 316. (b) Tswett, M. S. Ber. Dtsch. Bot. Ges. 1906, 384. (c) Tswett, M. S. Proceedings of the Warsaw Society of Naturalists, Biology Section 1903, 14, 20.
(2) Clements, R. L. Science 1958, 128, 899.(3) (a) Gant, J. R.; Dolan, J. W.; Snyder, L. R. J. Chromatogr.
1979, 185, 153. (b) Norden, B.; Jonas, I. Inorg. Nucl. Chem. Lett. 1976, 12, 33. (c) Jonas, I.; Norden, B. Nature 1975, 258, 597. (d) Sheikh, S. U.; Touchstone, J. C. J. Chromatogr. 1988, 455, 327. (e) Henderson, D. E.; O’Connor, D. J. Adv. Chromatogr. 1984, 23, 65. (f) Teutenberg, T. Anal. Chim. Acta 2009, 643, 1. (g) Henderson, D. E.; O’Connor, D. J.; Kirby, J. F.; Sears, C. P. J. Chromatogr. Sci. 1985, 23, 477. (h) Hsu, H. W.; Davis, D. S.; Wei, C. H.; Yang, W. K. Anal. Biochem. 1976, 73, 513. (i) Jones, B. A. J. Liq. Chromatogr. Relat. Technol.
NO2
FF
NO2
NHFHCl⋅H2N CO2t-Bu
R
DIPEA
EtOH, reflux309
310
312
(311)
Rt-BuO2C
R1 NH2
NOH
R2 OH
O
313 314
μ-wave
HBTUN
N
OR2
R1
315
variables: stoichiometry, reaction time, temperature
i-Pr2NEt+
OH
O
O
O
EtO
H2N S
NH2
OH
NH
NH
EtO
O
S
+
catalystsolvent
timetemp
μ-wave
318
316317 319
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

1190 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
2004, 27, 1331. (j) Lerosen, A. L.; Rivet, C. A. Anal. Chem. 1948, 20, 1093. (k) Maggs, R. J. J. Chromatogr. Sci. 1969, 7, 145. (l) Yamasaki, T.; Sakai, M.; Kanamori, T.; Sogo, M. Chromatographia 1986, 21, 478. (m) Collins, F. D.; Shotlander, V. L. J. Lipid Res. 1960, 1, 352. (n) Liodakis, S.; Pappa, A.; Parissakis, G. J. Chromatogr. Sci. 1989, 27, 149. (o) Scott, R. P. W.; Lawrence, J. G. J. Chromatogr. Sci. 1969, 7, 65. (p) Stahl, E. Angew. Chem. Int. Ed. 1964, 3, 784. (q) Locke, D. C.; Martire, D. E. Anal. Chem. 1967, 39, 921. (r) Bolliet, D.; Poole, C. F. Analyst 1998, 123, 295. (s) Hesse, G.; Engelhar, H. J. Chromatogr. 1966, 21, 228. (t) Chang, L. T. Anal. Chem. 1953, 25, 1235. (u) Magalhaes, A. F.; Magalhaes, E. G.; Junior, V. N.; Filho, H. F. L. Phytochemistry 1989, 28, 2497.
(4) Henderson, R. F.; Clayton, M. H. Anal. Biochem. 1976, 70, 440.
(5) (a) Sheikh, S. U.; Touchstone, J. C. Anal. Chem. 1987, 59, 1472. (b) Touchstone, J. C.; Sheikh, S. U. J. High Resolut. Chromatogr. & Chromatogr. Commun. 1986, 9, 464. (c) Sheikh, S. U.; Touchstone, J. C. J. Liq. Chromatogr. 1987, 10, 2489.
(6) Balny, C.; Lepeuch, C.; Debey, P. Anal. Biochem. 1975, 63, 321.
(7) (a) Ewing, G. D.; Ley, S. V.; Paquette, L. A. J. Am. Chem. Soc. 1978, 100, 2909. (b) Paquette, L. A.; Ewing, G. D.; Ley, S. V.; Berk, H. C.; Traynor, S. G. J. Org. Chem. 1978, 43, 4712.
(8) Annis, G. D.; Ley, S. V.; Self, C. R.; Sivaramakrishnan, R. J. Chem. Soc., Perkin Trans. 1 1981, 270.
(9) Huber UK, Shire Hill, Saffron Walden, Essex CB11 3AZ, UK.
(10) Horak, V.; Foster, F. V.; de Levie, R.; Jones, J. W.; Svoronos, P. Tetrahedron Lett. 1981, 22, 3577.
(11) Jones, D. W.; Wife, R. L. J. Chem. Soc., Perkin Trans. 1 1974, 1.
(12) (a) Jones, D. W.; Pomfret, A. J. Chem. Soc., Chem. Commun. 1983, 703. (b) Jones, D. W.; Pomfret, A. J. Chem. Soc., Perkin Trans. 1 1991, 13.
(13) Escher, A.; Rutsch, W.; Neuenschwander, M. Helv. Chim. Acta 1986, 69, 1644.
(14) Gupta, R. C.; Larsen, D. S.; Stoodley, R. J.; Slawin, A. M. Z.; Williams, D. J. J. Chem. Soc., Perkin Trans. 1 1989, 739.
(15) (a) Denmark, S. E.; Lee, W. S. Chem. Asian J. 2008, 3, 327. (b) Denmark, S. E.; Lee, W. S. J. Org. Chem. 1994, 59, 707.
(16) Toltl, N. P.; Leigh, W. J. Organometallics 1996, 15, 2554.(17) Williams, A. L.; Srinivasan, J. M.; Johnston, J. N. Org. Lett.
2006, 8, 6047.(18) Zhao, W. L.; Carreira, E. M. Org. Lett. 2006, 8, 99.(19) Sellmann, D.; Jonk, E.; Reinecke, H. J.; Wurminghausen, T.
Fresenius Z. Anal. Chem. 1979, 294, 372.(20) Bergman, R. G.; Seidler, P. F.; Wenzel, T. T. J. Am. Chem.
Soc. 1985, 107, 4358.(21) (a) Mobley, T. A.; Schade, C.; Bergman, R. G. J. Am. Chem.
Soc. 1995, 117, 7822. (b) Mobley, T. A.; Schade, C.; Bergman, R. G. Organometallics 1998, 17, 3574.
(22) Egekeze, J. O.; Danielski, M. C.; Grinberg, N.; Smith, G. B.; Sidler, D. R.; Perpall, H. J.; Bicker, G. R.; Tway, P. C. Anal. Chem. 1995, 67, 2292.
(23) LoBrutto, R.; Bereznitski, Y.; Novak, T. J.; DiMichele, L.; Pan, L.; Journet, M.; Kowal, J.; Grinberg, N. J. Chromatogr., A 2003, 995, 67.
(24) Dugave, C.; Demange, L. Chem. Rev. 2003, 103, 2475.(25) Melander, W. R.; Jacobson, J.; Horváth, C. J. Chromatogr.,
A 1982, 234, 269.(26) Henderson, D. E.; Horváth, C. J. Chromatogr., A 1986, 368,
203.
(27) Jacobson, J.; Melander, W.; Vaisnys, G.; Horváth, C. J. Phys. Chem. 1984, 88, 4536.
(28) (a) Bouabdallah, S.; Trabelsi, H.; Bouzouita, K.; Sabbah, S. J. Biochem. Biophys. Meth. 2002, 54, 391. (b) Henderson, D. E.; Mello, J. A. J. Chromatogr., A 1990, 499, 79. (c) Lebl, M.; Fang, S.; Hruby, V. J. J. Chromatogr. A 1991, 586, 145. (d) Liberek, B.; Augustiniak, J.; Ciarkowski, J.; Plucinski, K.; Stachowiak, K. J. Chromatogr. 1974, 95, 223. (e) Henderson, D. E.; Saltzman, S. J.; Uden, P. C.; Cheng, Z. Polyhedron 1988, 7, 369. (f) Moriyasu, M.; Kato, A.; Hashimoto, Y. J. Chromatogr. 1987, 400, 143.
(29) Melander, W. R.; Lin, H. J.; Jacobson, J.; Horváth, C. J. Phys. Chem. 1984, 88, 4527.
(30) (a) Li, L.; Thompson, R.; Sowa, J. R.; Clausen, A.; Dowling, T. J. Chromatogr., A 2004, 1043, 171. (b) Trapp, O. Anal. Chem. 2006, 78, 189. (c) Trapp, O.; Schoetz, G.; Schurig, V. Determination of enantiomerization barriers by dynamic and stopped-flow chromatographic methods, 2nd International Symposium on Biological Chirality, Szeged, Hungary, Aug 27–31, 2000, 403 (d) Wolf, C. Chem. Soc. Rev. 2005, 34, 595. (e) Casarini, D.; Lunazzi, L.; Alcaro, S.; Gasparrini, F.; Villani, C. J. Org. Chem. 1995, 60, 5515.
(31) Meier, R.; Fletschinger, J. Angew. Chem. 1956, 68, 373.(32) Dewar, M. J. S.; Dewar, R. B. K.; Gaibel, Z. L. F. Org.
Synth. 1966, 46, 65.(33) Kauffman, J. M.; Bjorkman, C. O. J. Chem. Educ. 1976, 53,
33.(34) Ace Glass Inc., P.O. Box 688, 1430 North West Blvd.,
Vineland, NJ 08362-0688, USA (product no. 5879)(35) Kauffman, J. M. Synthesis 1999, 918.(36) Kauffman, J. M. Synthesis 2001, 197.(37) Faldt, A.; Pedersen, M. J.; Krogh, T. P.; Hviid, L.;
Christensen, J. B. Synth. Met. 1998, 94, 307.(38) Goodbrand, H. B.; Hu, N. X. J. Org. Chem. 1999, 64, 670.(39) Froimowitz, M.; Gu, Y. H.; Dakin, L. A.; Nagafuji, P. M.;
Kelley, C. J.; Parrish, D.; Deschamps, J. R.; Janowsky, A. J. Med. Chem. 2007, 50, 219.
(40) (a) Chatterjee, K.; Parker, D. H.; Wurz, P.; Lykke, K. R.; Gruen, D. M.; Stock, L. M. J. Org. Chem. 1992, 57, 3253. (b) Khemani, K. C.; Prato, M.; Wudl, F. J. Org. Chem. 1992, 57, 3254.
(41) Scrivens, W. A.; Bedworth, P. V.; Tour, J. M. J. Am. Chem. Soc. 1992, 114, 7917.
(42) (a) Darwish, A. D.; Kroto, H. W.; Taylor, R.; Walton, D. R. M. J. Chem. Soc., Chem. Commun. 1994, 15. (b) Grushko, Y. S.; Sedov, V. P.; Shilin, V. A. Russ. J. Appl. Chem. 2007, 80, 448.
(43) O’Brien, M.; Dieguez-Vazquez, A.; Hsu, D. S.; Kraus, H.; Sumino, Y.; Ley, S. V. Org. Biomol. Chem. 2008, 6, 1159.
(44) (a) Ball, M.; Gaunt, M. J.; Hook, D. F.; Jessiman, A. S.; Kawahara, S.; Orsini, P.; Scolaro, A.; Talbot, A. C.; Tanner, H. R.; Yamanoi, S.; Ley, S. V. Angew. Chem. Int. Ed. 2005, 44, 5433. (b) Gaunt, M. J.; Jessiman, A. S.; Orsini, P.; Tanner, H. R.; Hook, D. F.; Ley, S. V. Org. Lett. 2003, 5, 4819. (c) Gaunt, M. J.; Hook, D. F.; Tanner, H. R.; Ley, S. V. Org. Lett. 2003, 5, 4815.
(45) Merrifield, R. B. J. Am. Chem. Soc. 1963, 85, 2149.(46) Linclau, B.; Sing, A. K.; Curran, D. P. J. Org. Chem. 1999,
64, 2835.(47) Flynn, D. L. Med. Res. Rev. 1999, 19, 408.(48) (a) Zhu, D. W. Synthesis 1993, 953. (b) Curran, D. P.;
Hadida, S. J. Am. Chem. Soc. 1996, 118, 2531. (c) Studer, A.; Hadida, S.; Ferritto, R.; Kim, S. Y.; Jeger, P.; Wipf, P.; Curran, D. P. Science 1997, 275, 823. (d) Horvath, I. T.; Rabai, J. Science 1994, 266, 72.
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

REVIEW Lesser-Known Enabling Technologies for Organic Synthesis 1191
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
(49) (a) Curran, D. P. Angew. Chem. Int. Ed. 1998, 37, 1175. (b) Gladysz, J. A.; Curran, D. P.; Horvath, I. T. Handbook of Fluorous Chemistry; Wiley-VCH: Weinheim, 2004. (c) Zhang, W. Chem. Rev. 2004, 104, 2531. (d) Horvath, I. T. Acc. Chem. Res. 1998, 31, 641. (e) de Wolf, E.; van Koten, G.; Deelman, B. J. Chem. Soc. Rev. 1999, 28, 37.
(50) McAllister, L. A.; McCormick, R. A.; Brand, S.; Procter, D. J. Angew. Chem. Int. Ed. 2005, 44, 452.
(51) (a) Sheehan, J. C.; Hlavka, J. J. J. Org. Chem. 1956, 21, 439. (b) Sheehan, J. C.; Boshart, G. L.; Cruickshank, P. A. J. Org. Chem. 1961, 26, 2525.
(52) Camp, D.; Jenkins, I. D. Aust. J. Chem. 1988, 41, 1835.(53) (a) El Bakkari, M.; Vincent, J. M. QSAR Comb. Sci. 2006,
25, 689. (b) Yoshida, J.; Itami, K.; Mitsudo, K.; Suga, S. Tetrahedron Lett. 1999, 40, 3403.
(54) Sugimura, T.; Hagiya, K. Chem. Lett. 2007, 36, 566.(55) Flynn, D. L.; Crich, J. Z.; Devraj, R. V.; Hockerman, S. L.;
Parlow, J. J.; South, M. S.; Woodard, S. J. Am. Chem. Soc. 1997, 119, 4874.
(56) Smith, C. D.; Baxendale, I. R.; Tranmer, G. K.; Baumann, M.; Smith, S. C.; Lewthwaite, R. A.; Ley, S. V. Org. Biomol. Chem. 2007, 5, 1562.
(57) Kunst, E.; Gallier, F.; Dujardin, G.; Yusubov, M. S.; Kirschning, A. Org. Lett. 2007, 9, 5199.
(58) Mothana, S.; Chahal, N.; Vanneste, S.; Hall, D. G. J. Comb. Chem. 2007, 9, 193.
(59) Ley, S. V.; Massi, A.; Rodriguez, F.; Horwell, D. C.; Lewthwaite, R. A.; Pritchard, M. C.; Reid, A. M. Angew. Chem. Int. Ed. 2001, 40, 1053.
(60) Siu, J.; Baxendale, I. R.; Lewthwaite, R. A.; Ley, S. V. Org. Biomol. Chem. 2005, 3, 3140.
(61) El Bakkari, M.; Fronton, B.; Luguya, R.; Vincent, J.-M. J. Fluorine Chem. 2006, 127, 558.
(62) Fersht, A. R. Trends Biochem. Sci. 1987, 12, 301.(63) Gruijters, B. W. T.; Verkade, J. M. M.; van Delft, F. L.;
Sijbesma, R. P.; Hermkens, P. H. H.; Rutjes, F. Eur. J. Org. Chem. 2007, 4197.
(64) Zhang, S. Q.; Fukase, K.; Izumi, M.; Fukase, Y.; Kusumoto, S. Synlett 2001, 590.
(65) de Ridder, D. J.; Villacorte, L.; Verliefde, A. R. D.; Verberk, J. Q. J. C.; Heijman, S. G. J.; Amy, G. L.; van Dijk, J. C. Water Res. 2010, 44, 3077.
(66) Hay, A. M.; Hobbs-Dewitt, S.; MacDonald, A. A.; Ramage, R. Synthesis 1999, 1979.
(67) Warmus, J. S.; da Silva, M. I. Org. Lett. 2000, 2, 1807.(68) Zhang, S. Q.; Fukase, K.; Kusumoto, S. Tetrahedron Lett.
1999, 40, 7479.(69) Lepore, S. D. Tetrahedron Lett. 2001, 42, 6437.(70) Fleckenstein, C. A.; Plenio, H. Adv. Synth. Catal. 2006, 348,
1058.(71) Leeb, L.; Gmeiner, P.; Lober, S. QSAR Comb. Sci. 2007, 26,
1145.(72) Li, X.; Abell, C.; Congreve, M. S.; Warrington, B. H.;
Ladlow, M. Org. Biomol. Chem. 2004, 2, 989.(73) Lan, P.; Porco, J. A.; South, M. S.; Parlow, J. J. J. Comb.
Chem. 2003, 5, 660.(74) Harned, A. M.; Zhang, M. J.; Vedantham, P.; Mukherjee, S.;
Herpel, R. H.; Flynn, D. L.; Hanson, P. R. Aldrichimica Acta 2005, 38, 3.
(75) Barrett, A. G. M.; Roberts, R. S.; Schroder, J. Org. Lett. 2000, 2, 2999.
(76) Moore, J. D.; Harned, A. M.; Henle, J.; Flynn, D. L.; Hanson, P. R. Org. Lett. 2002, 4, 1847.
(77) Fuchter, M. J.; Hoffman, B. M.; Barrett, A. G. M. J. Org. Chem. 2005, 70, 5086.
(78) Dickerson, T. J.; Reed, N. N.; Janda, K. D. Chem. Rev. 2002, 102, 3325.
(79) Ginisty, M.; Roy, M. N.; Charette, A. B. J. Org. Chem. 2008, 73, 2542.
(80) (a) Stazi, F.; Marcoux, D.; Poupon, J. C.; Latassa, D.; Charette, A. B. Angew. Chem. Int. Ed. 2007, 46, 5011. (b) Poupon, J. C.; Boezio, A. A.; Charette, A. B. Angew. Chem. Int. Ed. 2006, 45, 1415. (c) Poupon, J. C.; Marcoux, D.; Cloarec, J. M.; Charette, A. B. Org. Lett. 2007, 9, 3591.
(81) Perrier, H.; Labelle, M. J. Org. Chem. 1999, 64, 2110.(82) Bergbreiter, D. E.; Liu, Y.-S. Tetrahedron Lett. 1997, 38,
3703.(83) (a) Bosanac, T.; Yang, J. M.; Wilcox, C. S. Angew. Chem.
Int. Ed. 2001, 40, 1875. (b) Bosanac, T.; Wilcox, C. S. Org. Lett. 2004, 6, 2321. (c) Bosanac, T.; Wilcox, C. S. J. Am. Chem. Soc. 2002, 124, 4194.
(84) (a) Bergbreiter, D. E.; Blanton, J. R. J. Org. Chem. 1985, 50, 5828. (b) Bergbreiter, D. E. Chem. Rev. 2002, 102, 3345.
(85) Huang, X.; Witte, K. L.; Bergbreiter, D. E.; Wong, C.-H. Adv. Synth. Catal. 2001, 343, 675.
(86) Haag, R.; Sunder, A.; Hebel, A.; Roller, S. J. Comb. Chem. 2002, 4, 112.
(87) (a) Malenfant, P. R. L.; Jayaraman, M.; Frechet, J. M. J. Chem. Mater. 1999, 11, 3420. (b) Haag, R. Chem. Eur. J. 2001, 7, 327. (c) van Heerbeek, R.; Kamer, P. C. J.; van Leeuwen, P.; Reek, J. N. H. Chem. Rev. 2002, 102, 3717. (d) Kim, R. M.; Manna, M.; Hutchins, S. M.; Griffin, P. R.; Yates, N. A.; Bernick, A. M.; Chapman, K. T. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 10012. (e) Lu, J.; Toy, P. H. Chem. Rev. 2009, 109, 815.
(88) (a) Giguere, R. J.; Bray, T. L.; Duncan, S. M.; Majetich, G. Tetrahedron Lett. 1986, 27, 4945. (b) Gedye, R.; Smith, F.; Westaway, K.; Ali, H.; Baldisera, L.; Laberge, L.; Rousell, J. Tetrahedron Lett. 1986, 27, 279.
(89) CEM Corporation, PO Box 200, 3100 Smith Farm Rd, Matthews, NC 28106, USA.
(90) (a) Kappe, C. O. Angew. Chem. Int. Ed. 2004, 43, 6250. (b) Lidstrom, P.; Tierney, J.; Wathey, B.; Westman, J. Tetrahedron 2001, 57, 9225. (c) Collins, J. M.; Leadbeater, N. E. Org. Biomol. Chem. 2007, 5, 1141. (d) Strauss, C. R.; Trainor, R. W. Aust. J. Chem. 1995, 48, 1665.
(91) Hayes, B. L.; Collins, M. US Patent 6,744,024, 2004.(92) Dabirmanesh, Q.; Roberts, R. M. G. J. Organomet. Chem.
1993, 460, C28.(93) Raner, K. D.; Strauss, C. R.; Trainor, R. W.; Thorn, J. S.
J. Org. Chem. 1995, 60, 2456.(94) (a) Ugi, I.; Steinbruckner, C. Angew. Chem. 1960, 72, 267.
(b) Ugi, I. Angew. Chem. 1962, 74, 9.(95) Chen, J. J.; Deshpande, S. V. Tetrahedron Lett. 2003, 44,
8873.(96) Humphrey, C. E.; Easson, M. A. M.; Tierney, J. P.; Turner,
N. J. Org. Lett. 2003, 5, 849.(97) (a) Lee, C. K. Y.; Holmes, A. B.; Ley, S. V.; McConvey, I.
F.; Al-Duri, B.; Leeke, G. A.; Santos, R. C. D.; Seville, J. P. K. Chem. Commun. 2005, 2175. (b) Yu, J. Q.; Wu, H. C.; Ramarao, C.; Spencer, J. B.; Ley, S. V. Chem. Commun. 2003, 678. (c) Ramarao, C.; Ley, S. V.; Smith, S. C.; Shirley, I. M.; DeAlmeida, N. Chem. Commun. 2002, 1132.
(98) Baxendale, I. R.; Griffiths-Jones, C. M.; Ley, S. V.; Tranmer, G. K. Chem. Eur. J. 2006, 12, 4407.
(99) Arvela, R. K.; Leadbeater, N. E. Org. Lett. 2005, 7, 2101.(100) Singh, B. K.; Appukkuttan, P.; Claerhout, S.; Parmar, V. S.;
Van der Eycken, E. Org. Lett. 2006, 8, 1863.(101) Katritzky, A. R.; Zhang, Y. M.; Singh, S. K.; Steel, P. J.
ARKIVOC 2003, (xv), 47.(102) Mosse, S.; Alexakis, A. Org. Lett. 2006, 8, 3577.
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.

1192 M. O’Brien et al. REVIEW
Synthesis 2011, No. 8, 1157–1192 © Thieme Stuttgart · New York
(103) (a) Rodriguez, B.; Bolm, C. J. Org. Chem. 2006, 71, 2888. (b) Ibrahem, I.; Zou, W. B.; Casas, J.; Sunden, H.; Cordova, A. Tetrahedron 2006, 62, 357. (c) Ibrahem, I.; Casas, J.; Cordova, A. Angew. Chem. Int. Ed. 2004, 43, 6528.
(104) Hosseini, M.; Stiasni, N.; Barbieri, V.; Kappe, C. O. J. Org. Chem. 2007, 72, 1417.
(105) (a) Suryanarayana, C. Prog. Mater. Sci. 2001, 46, 1. (b) Volkov, V. V.; Myakishev, K. G. Inorg. Chim. Acta 1999, 289, 51.
(106) Kaupp, G. CrystEngComm 2009, 11, 388.(107) (a) Wang, G.-W.; Komatsu, K.; Murata, Y.; Shiro, M.
Nature 1997, 387, 583. (b) Komatsu, K.; Wang, G.-W.; Murata, Y.; Tanaka, T.; Fujiwara, K.; Yamamoto, K.; Saunders, M. J. Org. Chem. 1998, 63, 9358.
(108) (a) Rodríguez, B.; Bruckmann, A.; Rantanen, T.; Bolm, C. Adv. Synth. Catal . 2007, 349, 2213. (b) Bruckmann, A.; Krebs, A.; Bolm, C. Green Chem. 2008, 10, 1131.
(109) (a) Rodriguez, B.; Rantanen, T.; Bolm, C. Angew. Chem. Int. Ed. 2006, 45, 6924. (b) Rantanen, T.; Schiffers, I.; Bolm, C. Org. Process Res. Dev. 2007, 11, 592.
(110) Rodriguez, B.; Bruckmann, A.; Bolm, C. Chem. Eur. J. 2007, 13, 4710.
(111) Kaupp, G.; Naimi-Jamal, M. R.; Schmeyers, J. Tetrahedron 2003, 59, 3753.
(112) Zhang, Z.; Gao, J.; Xia, J. J.; Wang, G. W. Org. Biomol. Chem. 2005, 3, 1617.
(113) Balema, V. P.; Wiench, J. W.; Pruski, M.; Pecharsky, V. K. J. Am. Chem. Soc. 2002, 124, 6244.
(114) (a) Tullberg, E.; Schacher, F.; Peters, D.; Frejd, T. Synthesis 2006, 1183. (b) Tullberg, E.; Peters, D.; Frejd, T. J. Organomet. Chem. 2004, 689, 3778.
(115) Calo, V.; Nacci, A.; Monopoli, A.; Lopez, L.; di Cosmo, A. Tetrahedron 2001, 57, 6071.
(116) (a) Fulmer, D. A.; Shearouse, W. C.; Medonza, S. T.; Mack, J. Green Chem. 2009, 11, 1821. (b) Thorwirth, R.; Stolle, A.; Ondruschka, B. Green Chem. 2010, 12, 985.
(117) Naimi-Jamal, M. R.; Mokhtari, J.; Dekamin, M. G.; Kaupp, G. Eur. J. Org. Chem. 2009, 3567.
(118) Prasad, K. R. K.; Joshi, N. N. J. Org. Chem. 1996, 61, 3888.(119) (a) Tye, H. Drug Discovery Today 2004, 9, 485.
(b) Montgomery, D. C. Design and Analysis of Experiments, 6th ed.; Wiley: New York, 2004. (c) Owen, M. R.; Luscombe, C.; Lai, L. W.; Godbert, S.; Crookes, D. L.; Emiabata-Smith, D. Org. Process Res. Dev. 2001, 5, 308. (d) Carlson, R. Design and Optimisation in Organic Synthesis; Elsevier: Amsterdam, 1992. (e) Rubin, A. E.; Tummala, S.; Both, D. A.; Wang, C. C.; Delaney, E. J. Chem. Rev. 2006, 106, 2794. (f) Carlson, R.; Carlson, J. E. Org. Process Res. Dev. 2005, 9, 680. (g) Stansbury, W. F. Chemometr.. Intell. Lab. Syst. 1997, 36, 199. (h) Gooding, O. W. Curr. Opin. Chem. Biol. 2004, 8, 297. (i) Guervenou, J.; Giamarchi, P.; Coulouarn, C.; Guerda, M.; Le Lez, C.; Oboyet, T. Chemometr. Intell. Lab. Syst. 2002, 63, 81. (j) Emiabata-Smith, D. F.; Crookes, D. L.; Owen, M. R. Org. Process Res. Dev. 1999, 3, 281.
(120) Hogan, P. J.; Hopes, P. A.; Moss, W. O.; Robinson, G. E.; Patel, I. Org. Process Res. Dev. 2002, 6, 225.
(121) Navarrete-Bolanos, J. L.; Jimenez-Islas, H.; Botello-Alvarez, E.; Rico-Martinez, R. Org. Process Res. Dev. 2002, 6, 841.
(122) Jamieson, C.; Congreve, M. S.; Emiabata-Smith, D. F.; Ley, S. V. Synlett 2000, 1603.
(123) Gooding, O. W.; Vo, L.; Battacharyya, S.; Labadie, J. W. J. Comb. Chem. 2002, 4, 578.
(124) Anachem Instruments Ltd., Anachem House, Charles Street, Luton, Bedfordshire LU2 0EB, UK.
(125) Jamieson, C.; Congreve, M. S.; Emiabata-Smith, D. F.; Ley, S. V.; Scicinski, J. J. Org. Process Res. Dev. 2002, 6, 823.
(126) Stat-Ease Inc., 2021 E. Hennepin Avenue, Suite 480, Minneapolis, MN 55413-2726, USA.
(127) Evans, M. D.; Ring, J.; Schoen, A.; Bell, A.; Edwards, P.; Berthelot, D.; Nicewonger, R.; Baldino, C. M. Tetrahedron Lett. 2003, 44, 9337.
(128) Glasnov, T. N.; Tye, H.; Kappe, C. O. Tetrahedron 2008, 64, 2035.
(129) Umetrics AB, Box 7960, 907 19 Umeå, Sweden.
Dow
nloa
ded
by: K
yush
u U
nive
rsity
. Cop
yrig
hted
mat
eria
l.





























