Embed Size (px)
Citation preview

Mol Divers (2014) 18:829–840DOI 10.1007/s11030-014-9545-3
FULL-LENGTH PAPER
LBVS: an online platform for ligand-based virtual screeningusing publicly accessible databases
Minghao Zheng · Zhihong Liu · Xin Yan ·Qianzhi Ding · Qiong Gu · Jun Xu
Received: 24 April 2014 / Accepted: 12 August 2014 / Published online: 3 September 2014© Springer International Publishing Switzerland 2014
Abstract Abundant data on compound bioactivity andpublicly accessible chemical databases increase opportuni-ties for ligand-based drug discovery. In order to make fulluse of the data, an online platform for ligand-based virtualscreening (LBVS) using publicly accessible databases hasbeen developed. LBVS adopts Bayesian learning approachto create virtual screening models because of its noise tol-erance, speed, and efficiency in extracting knowledge fromdata. LBVS currently includes data derived from Bind-ingDB and ChEMBL. Three validation approaches have beenemployed to evaluate the virtual screening models createdfrom LBVS. The tenfold cross validation results of twentydifferent LBVS models demonstrate that LBVS achieves anaverage AUC value of 0.86. Our internal and external testingresults indicate that LBVS is predictive for lead identifica-tions. LBVS can be publicly accessed at http://rcdd.sysu.edu.cn/lbvs.
Keywords Ligand-based virtual screening ·Virtual screening · Public database · Bayesian learning
Minghao Zheng and Zhihong Liu have Equally contribution to thiswork.
M. Zheng · Z. Liu · X. Yan · Q. Ding · Q. Gu (B) · J. Xu (B)Research Center for Drug Discovery, School of PharmaceuticalSciences, Sun Yat-sen University, 132 East Circle at University City,Guangzhou 510006, Chinae-mail: [email protected]
J. Xue-mail: [email protected]
Introduction
Virtual high-throughput screening (vHTS) is a commonprocess in drug discovery [1–3]. Numerous successful vir-tual screening campaigns have been reported, and con-firmed with bioassays [3]. Various computational methods,including ligand- and structure-based strategies, were uti-lized to identify leads for a specific target. Usually, ligand-based approaches, such as QSAR, machine learning, areused for lead optimization. Recently, however, ligand-basedapproaches (such as naïve Bayesian classifiers, support vec-tor machines, decision trees, random forests, clustering meth-ods, and neural networks) are increasingly employed for druglead identification [4–7].
Notably, naïve Bayesian learning is widely used inchemoinformatics, since the Laplacian-corrected modifiednaïve Bayes approach was first applied to kinase inhibitorclassification in 2004 [8,9]. The widely used Bayesian learn-ing yields respectable biological target predictions [10,11],virtual screenings [8,12–16], and ADMET properties estima-tions [17,18]. Bayesian learning is widely employed in drugdiscovery primarily because it has the following advantages.First, Bayesian learning shows the tolerance of noise. Errorsin the bioactivity databases [19], the imbalance betweenactive and inactive compounds and the arbitrary definition ofbioactivity for activity and inactivity make HTS data noisyand sparse. The characteristic of Bayesian learning enablesit to handle HTS data analyses [9,15,20]. Second, Bayesianlearning is free of over fitting. Bayesian models are not con-structed via a fitting approach, which frees them from the“curse of dimensionality” when using numerous descriptors[8]. Third, Bayesian learning is rapid and efficient in process-ing large amounts of data [8].
Abundant data on compound bioactivity and publiclyaccessible chemical databases have the characteristics of big
123

830 Mol Divers (2014) 18:829–840
Table 1 Some popular bioactivity database in drug discovery
Name Compounds Targets Activities Website
BindingDB 427,325 6,589 1,009,290 http://www.bindingdb.org
ChEMBL 1,324,941 9,356 12,077,491 https://www.ebi.ac.uk/chembldb
PubChem 48,781,965 No data 739,658a http://pubchem.ncbi.nlm.nih.gov
Drugbank 7,677 4,092 14,785 http://beta.drugbank.ca
TTD 20,667 2,360 No data http://bidd.nus.edu.sg/group/ttd
The statistic data were from the website of databases by December of 2013a The number denoted here is the number of assays
data, which have drawn industrial, academic, and govern-mental attention [21–23] in recent years. The major chal-lenge is to extract knowledge from the data, and solve drugdiscovery problems. A significant amount of bioactivity dataat the molecular, cellular, and animal levels has been gener-ated due to the advent of high-throughput screening. ZINC,a free database of commercially available compounds forvirtual screening, contains over 35 million purchasable com-pounds [24]. ChEMBL is a publicly accessible database con-taining over 12 million compounds with biological activities[25]. Table 1 summarizes some well-known databases widelyaccessed for drug discovery. There is an increasing need fora platform for ligand-based virtual screening (LBVS) usingthese publicly accessible databases. The platform should takeover the routine tasks, such as importing data from differentsources, cleaning data, and creating predictive model [26].
The flowchart of LBVS is depicted in Fig. 1. Currently,LBVS collects BindingDB and ChEMBL databases for vir-tual screening modeling, employs Bayesian learning to buildvirtual screening models, which are validated by a numberof model validation methods. Atom center fragments (ACFs)
BindingDBChEMBL……
Targetrelateddata
Target-basedData selection
Virtualscreeningmodels
BayesianLearning
Validatedvirtualscreeningmodel
Validating models
Virtuallibrary
Virtualscreening
Virtualhits
Hit selection criteria
Privilegedfragments
Fig. 1 Schematic flow diagram of the LBVS platform. User can selecta target through web browser and the target-related ligands bioactivitydata can be retrieved from the public database BindingDB or ChEMBLand divided into active and inactive sets. Subsequently, naïve Bayesianmodel was constructed online and validated by various metrics. With thevalidated model, a virtual library (a commercial available or in-houselibrary) can be screened to generate virtual hits
are used as structural features for the Bayesian modeling. TheBayesian models are validated with a number of validationmethods, such as tenfold cross-validations.
Materials and methods
Web implementation
The web platform was constructed from three components:the interface, the application server, and the database. Theweb interface was implemented using HTML, CSS andJavaScript, ChemDoodle, and D3.js for visualization. Theapplication server was implemented in the Go language(a new programming language that was initially devel-oped at Google). Bioactivity data from the public data-bases BindingDB (2013/6/13) and ChEMBLdb (release 17)were imported into the MongoDB and MySQL databases.Data for the ligand structure, target name, source organ-ism, and ligand–target affinity parameters (IC50, Ki, andKd) were extracted to create the active and inactive sub-sets. The nanomolar affinity values were used as the stan-dard unit. The user can select the target of interest in bind-ingDB or ChEMBL or alternatively upload a private datasetof bioactivity data in the SDF file format. Three thresh-olds for activity (100, 1,000, and 10,000 nM) were chosenbased on user demand. The common 5- and 10-fold crossvalidation methods can be used, and the atom center frag-ment layer can be set from 1 to 4. The optional test set canbe uploaded for online modeling. The active and inactivesets were generated based on the selected target and thresh-old. The naïve Bayesian classifier and atom center frag-ment generation methods were implemented in-house, anda binary executable program was called using the Go lan-guage. The model validation results, including ROC curves,privileged structures and virtual screening hits, are avail-able for download. Snapshots of the major pages are shownin Fig. 2.
Feature calculations
The structural features for Bayesian learning are the ACFs[27,28], which are computed with an in-house program. Vari-ous ACFs can be generated based on the ACF-layer (the num-
123

Mol Divers (2014) 18:829–840 831
Fig. 2 Snapshots of model building, virtual screening, and results pages
ber of bonds from an edge atom to the central atom). ACF-nwas derived from a heavy atom and n-bonds away neighboratoms. The generation of ACF is illustrated in Fig. 3. Detailedinformation on ACFs can be found in the references [27,28].
Naïve Bayesian classifier
The naïve Bayesian classifier is a method based on the Bayestheorem for conditional probability.
Fig. 3 An example of ACFs generation
123
832 Mol Divers (2014) 18:829–840
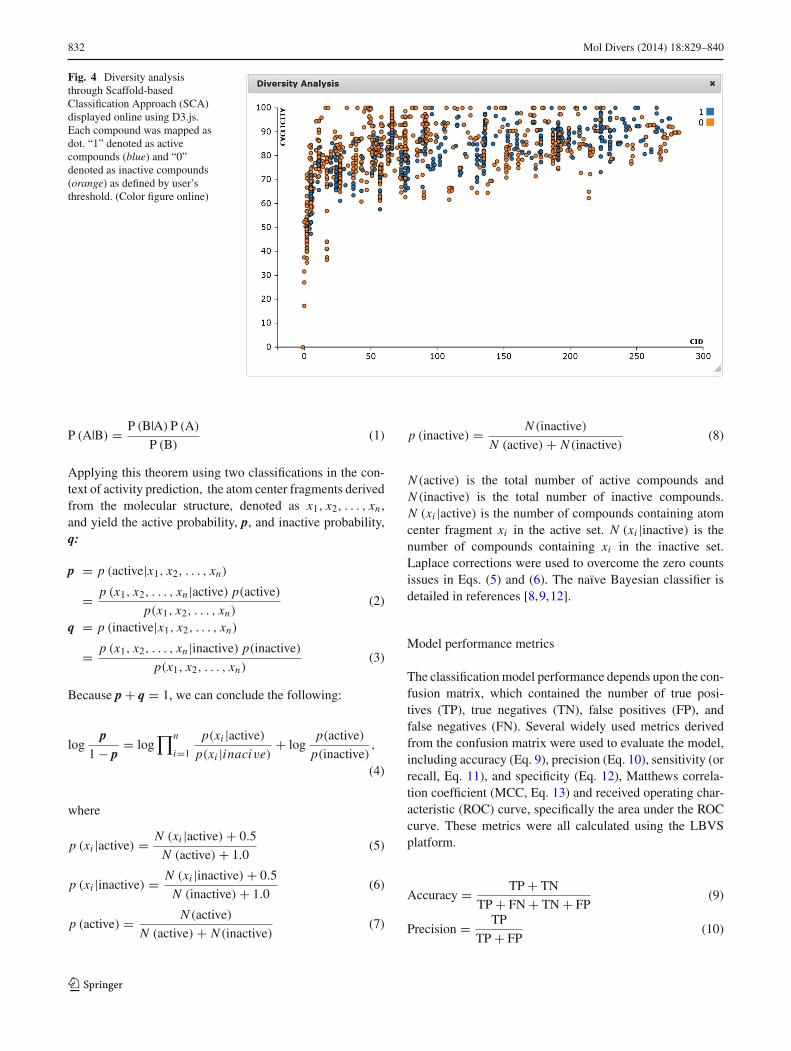
Fig. 4 Diversity analysisthrough Scaffold-basedClassification Approach (SCA)displayed online using D3.js.Each compound was mapped asdot. “1” denoted as activecompounds (blue) and “0”denoted as inactive compounds(orange) as defined by user’sthreshold. (Color figure online)
P (A|B) = P (B|A) P (A)
P (B)(1)
Applying this theorem using two classifications in the con-text of activity prediction, the atom center fragments derivedfrom the molecular structure, denoted as x1, x2, . . . , xn ,and yield the active probability, p, and inactive probability,q:
p = p (active|x1, x2, . . . , xn)
= p (x1, x2, . . . , xn|active) p(active)
p(x1, x2, . . . , xn)(2)
q = p (inactive|x1, x2, . . . , xn)
= p (x1, x2, . . . , xn|inactive) p(inactive)
p(x1, x2, . . . , xn)(3)
Because p + q = 1, we can conclude the following:
logp
1 − p= log
∏n
i=1
p(xi |active)
p(xi |inacive)+ log
p(active)
p(inactive),
(4)
where
p (xi |active) = N (xi |active) + 0.5
N (active) + 1.0(5)
p (xi |inactive) = N (xi |inactive) + 0.5
N (inactive) + 1.0(6)
p (active) = N (active)
N (active) + N (inactive)(7)
p (inactive) = N (inactive)
N (active) + N (inactive)(8)
N (active) is the total number of active compounds andN (inactive) is the total number of inactive compounds.N (xi |active) is the number of compounds containing atomcenter fragment xi in the active set. N (xi |inactive) is thenumber of compounds containing xi in the inactive set.Laplace corrections were used to overcome the zero countsissues in Eqs. (5) and (6). The naïve Bayesian classifier isdetailed in references [8,9,12].
Model performance metrics
The classification model performance depends upon the con-fusion matrix, which contained the number of true posi-tives (TP), true negatives (TN), false positives (FP), andfalse negatives (FN). Several widely used metrics derivedfrom the confusion matrix were used to evaluate the model,including accuracy (Eq. 9), precision (Eq. 10), sensitivity (orrecall, Eq. 11), and specificity (Eq. 12), Matthews correla-tion coefficient (MCC, Eq. 13) and received operating char-acteristic (ROC) curve, specifically the area under the ROCcurve. These metrics were all calculated using the LBVSplatform.
Accuracy = TP + TN
TP + FN + TN + FP(9)
Precision = TP
TP + FP(10)
123

Mol Divers (2014) 18:829–840 833
Fig. 5 ROC curves for selected targets retrieved from BindingDB. The tenfold cross validation method was employed with the level 2 ACFs asthe features for Bayesian learning
Sensitivity (Recall) = TP
TP + FN(11)
Specificity = TN
TN + FP(12)
MCC = TP × TN − FP × FN√(TP + FN) (TP + FP)(TN + FN)(TN + FP)
(13)
Results and discussions
Chemical space exploration
The chemical space of the training set directly influencedthe model performance and virtual screening application. Ascaffold-based classification approach [29] was used to ana-lyze the chemical space of the training set, including both
123
834 Mol Divers (2014) 18:829–840
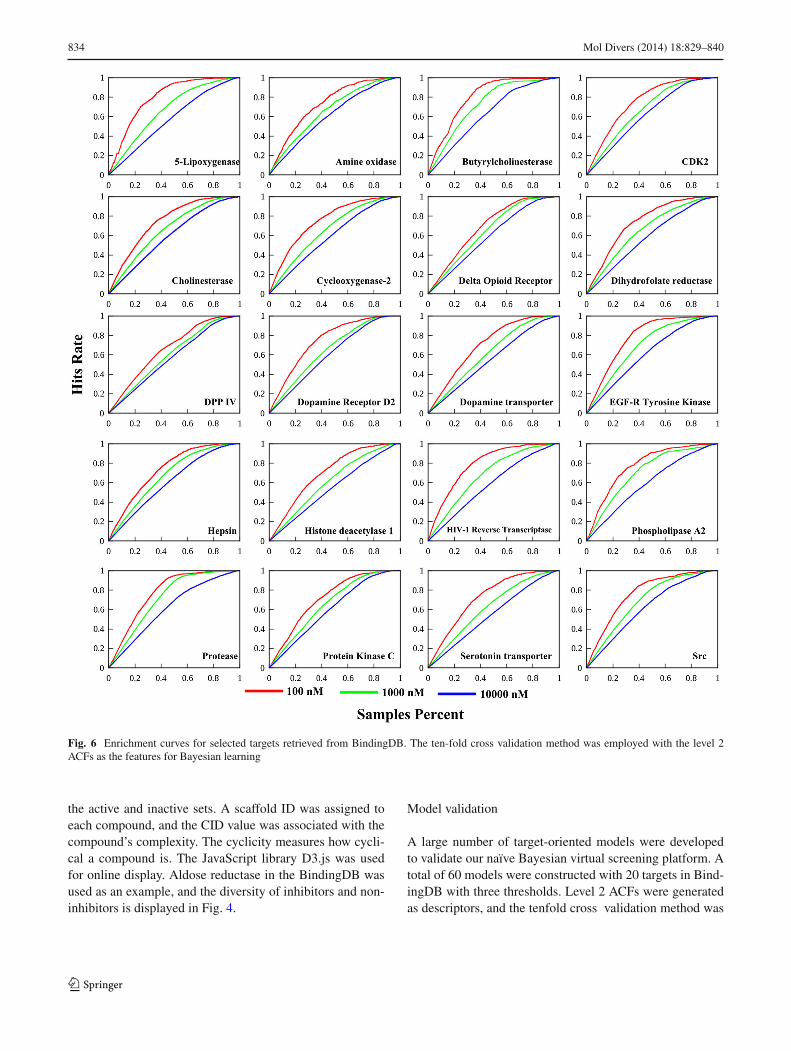
Fig. 6 Enrichment curves for selected targets retrieved from BindingDB. The ten-fold cross validation method was employed with the level 2ACFs as the features for Bayesian learning
the active and inactive sets. A scaffold ID was assigned toeach compound, and the CID value was associated with thecompound’s complexity. The cyclicity measures how cycli-cal a compound is. The JavaScript library D3.js was usedfor online display. Aldose reductase in the BindingDB wasused as an example, and the diversity of inhibitors and non-inhibitors is displayed in Fig. 4.
Model validation
A large number of target-oriented models were developedto validate our naïve Bayesian virtual screening platform. Atotal of 60 models were constructed with 20 targets in Bind-ingDB with three thresholds. Level 2 ACFs were generatedas descriptors, and the tenfold cross validation method was
123
Mol Divers (2014) 18:829–840 835
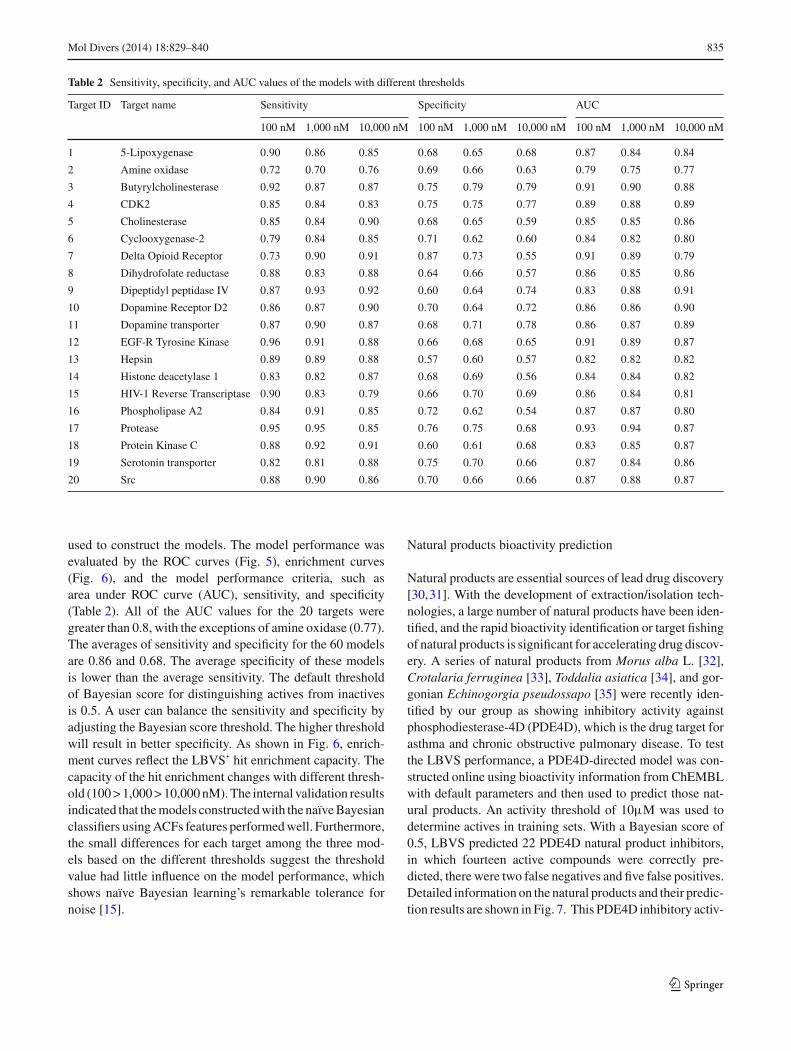
Table 2 Sensitivity, specificity, and AUC values of the models with different thresholds
Target ID Target name Sensitivity Specificity AUC
100 nM 1,000 nM 10,000 nM 100 nM 1,000 nM 10,000 nM 100 nM 1,000 nM 10,000 nM
1 5-Lipoxygenase 0.90 0.86 0.85 0.68 0.65 0.68 0.87 0.84 0.84
2 Amine oxidase 0.72 0.70 0.76 0.69 0.66 0.63 0.79 0.75 0.77
3 Butyrylcholinesterase 0.92 0.87 0.87 0.75 0.79 0.79 0.91 0.90 0.88
4 CDK2 0.85 0.84 0.83 0.75 0.75 0.77 0.89 0.88 0.89
5 Cholinesterase 0.85 0.84 0.90 0.68 0.65 0.59 0.85 0.85 0.86
6 Cyclooxygenase-2 0.79 0.84 0.85 0.71 0.62 0.60 0.84 0.82 0.80
7 Delta Opioid Receptor 0.73 0.90 0.91 0.87 0.73 0.55 0.91 0.89 0.79
8 Dihydrofolate reductase 0.88 0.83 0.88 0.64 0.66 0.57 0.86 0.85 0.86
9 Dipeptidyl peptidase IV 0.87 0.93 0.92 0.60 0.64 0.74 0.83 0.88 0.91
10 Dopamine Receptor D2 0.86 0.87 0.90 0.70 0.64 0.72 0.86 0.86 0.90
11 Dopamine transporter 0.87 0.90 0.87 0.68 0.71 0.78 0.86 0.87 0.89
12 EGF-R Tyrosine Kinase 0.96 0.91 0.88 0.66 0.68 0.65 0.91 0.89 0.87
13 Hepsin 0.89 0.89 0.88 0.57 0.60 0.57 0.82 0.82 0.82
14 Histone deacetylase 1 0.83 0.82 0.87 0.68 0.69 0.56 0.84 0.84 0.82
15 HIV-1 Reverse Transcriptase 0.90 0.83 0.79 0.66 0.70 0.69 0.86 0.84 0.81
16 Phospholipase A2 0.84 0.91 0.85 0.72 0.62 0.54 0.87 0.87 0.80
17 Protease 0.95 0.95 0.85 0.76 0.75 0.68 0.93 0.94 0.87
18 Protein Kinase C 0.88 0.92 0.91 0.60 0.61 0.68 0.83 0.85 0.87
19 Serotonin transporter 0.82 0.81 0.88 0.75 0.70 0.66 0.87 0.84 0.86
20 Src 0.88 0.90 0.86 0.70 0.66 0.66 0.87 0.88 0.87
used to construct the models. The model performance wasevaluated by the ROC curves (Fig. 5), enrichment curves(Fig. 6), and the model performance criteria, such asarea under ROC curve (AUC), sensitivity, and specificity(Table 2). All of the AUC values for the 20 targets weregreater than 0.8, with the exceptions of amine oxidase (0.77).The averages of sensitivity and specificity for the 60 modelsare 0.86 and 0.68. The average specificity of these modelsis lower than the average sensitivity. The default thresholdof Bayesian score for distinguishing actives from inactivesis 0.5. A user can balance the sensitivity and specificity byadjusting the Bayesian score threshold. The higher thresholdwill result in better specificity. As shown in Fig. 6, enrich-ment curves reflect the LBVS’ hit enrichment capacity. Thecapacity of the hit enrichment changes with different thresh-old (100 > 1,000 > 10,000 nM). The internal validation resultsindicated that the models constructed with the naïve Bayesianclassifiers using ACFs features performed well. Furthermore,the small differences for each target among the three mod-els based on the different thresholds suggest the thresholdvalue had little influence on the model performance, whichshows naïve Bayesian learning’s remarkable tolerance fornoise [15].
Natural products bioactivity prediction
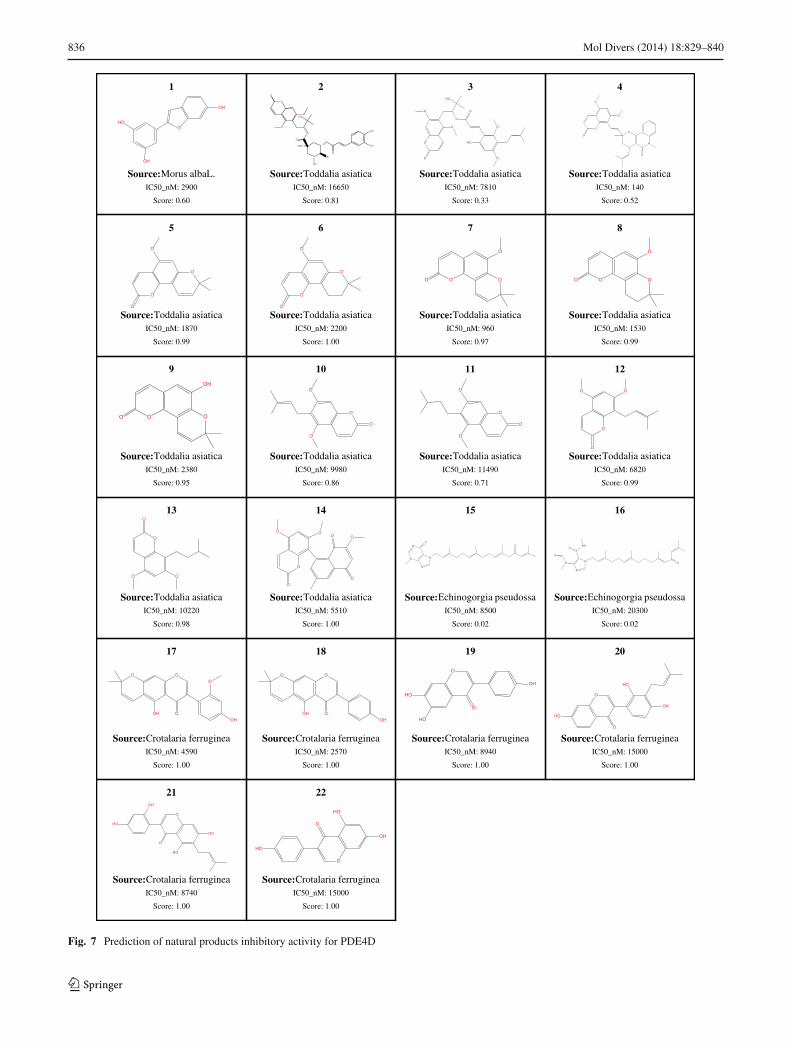
Natural products are essential sources of lead drug discovery[30,31]. With the development of extraction/isolation tech-nologies, a large number of natural products have been iden-tified, and the rapid bioactivity identification or target fishingof natural products is significant for accelerating drug discov-ery. A series of natural products from Morus alba L. [32],Crotalaria ferruginea [33], Toddalia asiatica [34], and gor-gonian Echinogorgia pseudossapo [35] were recently iden-tified by our group as showing inhibitory activity againstphosphodiesterase-4D (PDE4D), which is the drug target forasthma and chronic obstructive pulmonary disease. To testthe LBVS performance, a PDE4D-directed model was con-structed online using bioactivity information from ChEMBLwith default parameters and then used to predict those nat-ural products. An activity threshold of 10µM was used todetermine actives in training sets. With a Bayesian score of0.5, LBVS predicted 22 PDE4D natural product inhibitors,in which fourteen active compounds were correctly pre-dicted, there were two false negatives and five false positives.Detailed information on the natural products and their predic-tion results are shown in Fig. 7. This PDE4D inhibitory activ-
123
836 Mol Divers (2014) 18:829–840
O
O
O
O
H
H
H
1
Source:Morus albaL.IC50_nM: 2900
Score: 0.60
O
O
O
O
O
O
O
O
O
O
O
O
O
O
H
H
H
H
H
H
2
Source:Toddalia asiaticaIC50_nM: 16650
Score: 0.81
O
O
O
O
O
O
O
O
O
O
H
H
3
Source:Toddalia asiaticaIC50_nM: 7810
Score: 0.33
N
O
O
O
O
O
O
4
Source:Toddalia asiaticaIC50_nM: 140
Score: 0.52
O
O
O
O
5
Source:Toddalia asiaticaIC50_nM: 1870
Score: 0.99
O
O
O
O
6
Source:Toddalia asiaticaIC50_nM: 2200
Score: 1.00
O OO
O
7
Source:Toddalia asiaticaIC50_nM: 960
Score: 0.97
O OO
O
8
Source:Toddalia asiaticaIC50_nM: 1530
Score: 0.99
O OO
OH
9
Source:Toddalia asiaticaIC50_nM: 2380
Score: 0.95
O
O
O
O
10
Source:Toddalia asiaticaIC50_nM: 9980
Score: 0.86
O
O
O
O
11
Source:Toddalia asiaticaIC50_nM: 11490
Score: 0.71
O
O
OO
12
Source:Toddalia asiaticaIC50_nM: 6820
Score: 0.99
O
O
OO
13
Source:Toddalia asiaticaIC50_nM: 10220
Score: 0.98
O
O
OOOO
O
14
Source:Toddalia asiaticaIC50_nM: 5510
Score: 1.00
N
N
O
N
N
O
15
Source:Echinogorgia pseudossaIC50_nM: 8500
Score: 0.02
N
NN
ON
O
O
H
16
Source:Echinogorgia pseudossaIC50_nM: 20300
Score: 0.02
O O
O
OO
O
H
H
17
Source:Crotalaria ferrugineaIC50_nM: 4590
Score: 1.00
O O
O
OO
H
H
18
Source:Crotalaria ferrugineaIC50_nM: 2570
Score: 1.00
O
O
O
O
O
H
H
H
19
Source:Crotalaria ferrugineaIC50_nM: 8940
Score: 1.00
O
O
O
O
O
H
H
H
20
Source:Crotalaria ferrugineaIC50_nM: 15000
Score: 1.00
O
O
O
O
O
O
H
H
H
H
21
Source:Crotalaria ferrugineaIC50_nM: 8740
Score: 1.00
O
O
O
O
O
H
H
H
22
Source:Crotalaria ferrugineaIC50_nM: 15000
Score: 1.00
Fig. 7 Prediction of natural products inhibitory activity for PDE4D
123

Mol Divers (2014) 18:829–840 837
Table 3 Reported virtual screening hits validation results
No. Target name VS methods Hits Correctlypredicted
Hit rate (%) Reference
1 Kappa-opioid receptor Docking 4 3 75 [36]
2 Mycobacterium tuberculosisInhA
Pharmacophore, docking 3 2 67 [37]
3 Aldose reductase Docking 26 16 62 [38]
4 Sumoylation activating enzyme1
Docking, similarity 5 5 100 [39]
5 5-HT1A Pharmacophore 10 8 80 [40]
6 TTK Scaffold similarity 8 5 63 [41]
7 5-HT1A QSAR model 9 6 67 [42]
8 Liver X receptor Pharmacophore, shapesimilarity
3 2 67 [43]
9 Hsp90 Docking 4 2 50 [44]
10 5-HT transporter Similarity, pharmacophore,docking
46 30 65 [45]
11 Hepatitis C virus NS5Bpolymerase
Pharmacophore, 3D-QSAR,docking
2 2 100 [46]
12 PDE4D Pharmacophore, docking,molecular dynamics
15 13 87 [47]
ity prediction case study demonstrates the potential power ofour LBVS platform.
Reported virtual screening hits validation
The mission of a QSAR models is to predict the activity ofunknown compounds based on known actives and inactives.To further validate the hits enrichment power of our LBVSplatform in virtual screening, more restricted and larger-scalevalidation were performed using recently reported successfulvirtual screening cases. The target-directed virtual screeningprotocols reported in the Journal of Chemical Informationand Modeling over the last year were collected to evaluatethe hits enrichment power. Notably, non-drug targets anddrug targets with too small of a sample size in bindingDBand ChEMBL were excluded. Both structure-based virtualscreening methods such as docking and molecular dynamics,and LBVS methods such as pharmacophore, similarity, andQSAR modeling approaches were widely applied and suc-cessfully identified the hits. We collected 12 virtual screen-ing campaigns with 11 targets, and the hits for each targetwere extracted for the online LBVS predictions. The virtualscreening information is listed in Table 3. The true hit ratesranged from 50 to 100 % with an average value of 73 %,which suggested the strength of LBVS for virtual screening.The virtual screening validation set only contains the activecompounds. However, it is common for virtual screening thatonly active compounds were reported. The reported virtualhits were validated to demonstrate that the model can enrich
and predict bioactivities for unknown compounds (latest pub-lished active compounds not included in the training set),which is important for identifying new scaffolds for specifictarget. The user is advised to select the top score compoundsin the virtual screening rather than using the score thresholdof 0.5.
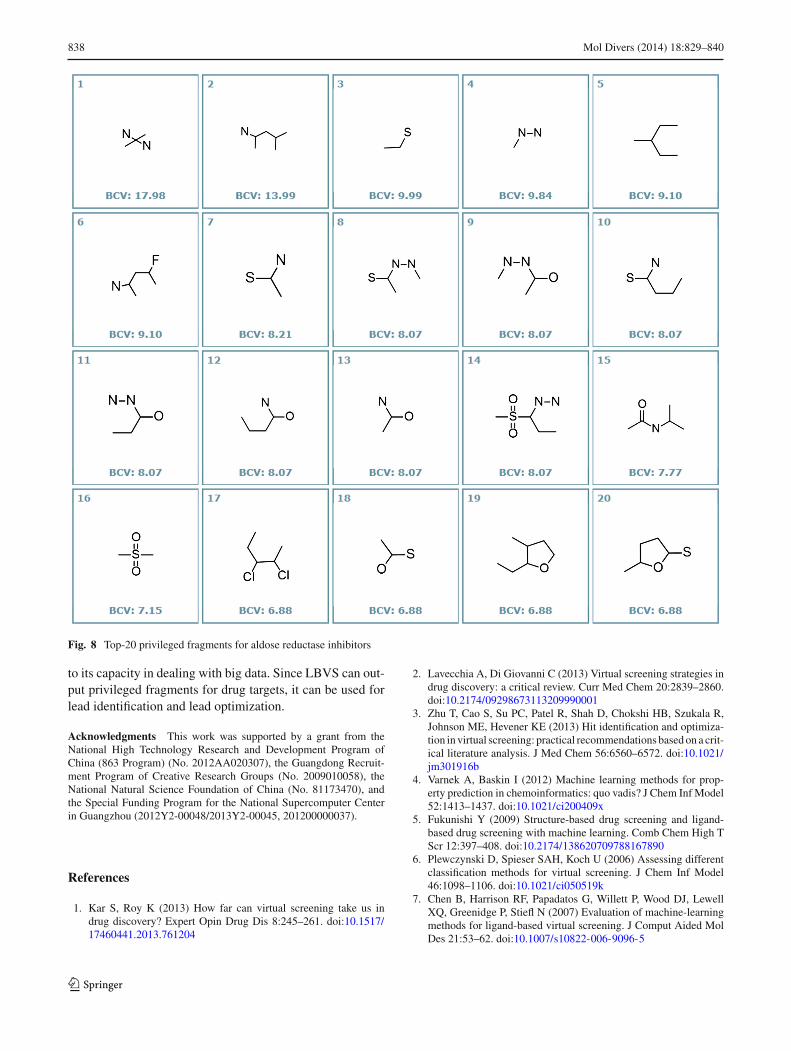
Privileged fragments for a target
It is worth to note that LBVS can output privileged frag-ments after the validated Bayesian model is generated for adrug target. These privileged fragments are associated withthe concept of privileged structures in medicinal chemistry[48–50]. A privileged fragment is an ACF with high bioac-tivity contribution value (BCV), which is calculated usingBCV = p (xi |active) /p (xi |inactive). The privileged frag-ments for aldose reductase inhibitors are listed in Fig. 8.These fragments can be used to guide lead optimization, orcompose new scaffolds for lead identification.
Conclusions
LBVS, an online platform for LBVS using publicly accessi-ble databases has been reported in this paper. This platformcan be a solution to manage and mine the increasingly avail-able data of compounds and their bioactivities for virtualscreening purposes. Bayesian learning is the main modelingtool to build target-oriented models for virtual screenings due
123
838 Mol Divers (2014) 18:829–840
Fig. 8 Top-20 privileged fragments for aldose reductase inhibitors
to its capacity in dealing with big data. Since LBVS can out-put privileged fragments for drug targets, it can be used forlead identification and lead optimization.
Acknowledgments This work was supported by a grant from theNational High Technology Research and Development Program ofChina (863 Program) (No. 2012AA020307), the Guangdong Recruit-ment Program of Creative Research Groups (No. 2009010058), theNational Natural Science Foundation of China (No. 81173470), andthe Special Funding Program for the National Supercomputer Centerin Guangzhou (2012Y2-00048/2013Y2-00045, 201200000037).
References
1. Kar S, Roy K (2013) How far can virtual screening take us indrug discovery? Expert Opin Drug Dis 8:245–261. doi:10.1517/17460441.2013.761204
2. Lavecchia A, Di Giovanni C (2013) Virtual screening strategies indrug discovery: a critical review. Curr Med Chem 20:2839–2860.doi:10.2174/09298673113209990001
3. Zhu T, Cao S, Su PC, Patel R, Shah D, Chokshi HB, Szukala R,Johnson ME, Hevener KE (2013) Hit identification and optimiza-tion in virtual screening: practical recommendations based on a crit-ical literature analysis. J Med Chem 56:6560–6572. doi:10.1021/jm301916b
4. Varnek A, Baskin I (2012) Machine learning methods for prop-erty prediction in chemoinformatics: quo vadis? J Chem Inf Model52:1413–1437. doi:10.1021/ci200409x
5. Fukunishi Y (2009) Structure-based drug screening and ligand-based drug screening with machine learning. Comb Chem High TScr 12:397–408. doi:10.2174/138620709788167890
6. Plewczynski D, Spieser SAH, Koch U (2006) Assessing differentclassification methods for virtual screening. J Chem Inf Model46:1098–1106. doi:10.1021/ci050519k
7. Chen B, Harrison RF, Papadatos G, Willett P, Wood DJ, LewellXQ, Greenidge P, Stiefl N (2007) Evaluation of machine-learningmethods for ligand-based virtual screening. J Comput Aided MolDes 21:53–62. doi:10.1007/s10822-006-9096-5
123

Mol Divers (2014) 18:829–840 839
8. Xia X, Maliski EG, Gallant P, Rogers D (2004) Classification ofkinase inhibitors using a Bayesian model. J Med Chem 47:4463–4470. doi:10.1021/jm0303195
9. Mussa HY, Mitchell JB, Glen RC (2013) Full ”Laplacianised” pos-terior naive Bayesian algorithm. J Cheminform 5:37. doi:10.1186/1758-2946-5-37
10. Nidhi Glick M, Davies JW, Jenkins JL (2006) Prediction of bio-logical targets for compounds using multiple-category Bayesianmodels trained on chemogenomics databases. J Chem Inf Model46:1124–1133. doi:10.1021/ci060003g
11. Nigsch F, Bender A, Jenkins JL, Mitchell JB (2008) Ligand-targetprediction using Winnow and naive Bayesian algorithms and theimplications of overall performance statistics. J Chem Inf Model48:2313–2325. doi:10.1021/ci800079x
12. Sun HM (2006) An accurate and interpretable Bayesian classi-fication model for prediction of hERG liability. ChemMedChem1:315–322. doi:10.1002/cmdc.200500047
13. Prathipati P, Ma NL, Keller TH (2008) Global Bayesian modelsfor the prioritization of antitubercular agents. J Chem Inf Model48:2362–2370. doi:10.1021/ci800143n
14. Broccatelli F (2012) QSAR models for P-glycoprotein transportbased on a highly consistent data set. J Chem Inf Model 52:2462–2470. doi:10.1021/ci3002809
15. Klon AE (2009) Bayesian modeling in virtual high throughputscreening. Comb Chem High Throughput Screen 12:469–483.doi:10.2174/138620709788489046
16. Fang JS, Yang RY, Gao L, Zhou D, Yang SQ, Liu AL, Du GH (2013)Predictions of BuChE inhibitors using support vector machine andNaive Bayesian classification techniques in drug discovery. J ChemInf Model 53:3009–3020. doi:10.1021/ci400331p
17. Martins IF, Teixeira AL, Pinheiro L, Falcao AO (2012) A Bayesianapproach to in silico blood-brain barrier penetration modeling. JChem Inf Model 52:1686–1697. doi:10.1021/ci300124c
18. Xu C, Cheng F, Chen L, Du Z, Li W, Liu G, Lee PW, Tang Y (2012)In silico prediction of chemical Ames mutagenicity. J Chem InfModel 52:2840–2847. doi:10.1021/ci300400a
19. Tiikkainen P, Bellis L, Light Y, Franke L (2013) Estimating errorrates in bioactivity databases. J Chem Inf Model 53:2499–2505.doi:10.1021/ci400099q
20. Glick M, Jenkins JL, Nettles JH, Hitchings H, Davies JW (2006)Enrichment of high-throughput screening data with increasing lev-els of noise using support vector machines, recursive partition-ing, and Laplacian-modified naive Bayesian classifiers. J Chem InfModel 46:193–200. doi:10.1021/ci050374h
21. Marx V (2013) The big challenges of big data. Nature 498:255–260. doi:10.1038/498255a
22. Lynch C (2008) Big data: how do your data grow? Nature 455:28–29. doi:10.1038/455028a
23. Chute CG, Ullman-Cullere M, Wood GM, Lin SM, He M, PathakJ (2013) Some experiences and opportunities for big data in trans-lational research. Genet Med 15:802–809. doi:10.1038/gim.2013.121
24. Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG(2012) ZINC: a free tool to discover chemistry for biology. J ChemInf Model 52:1757–1768. doi:10.1021/ci3001277
25. Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A,Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, OveringtonJP (2012) ChEMBL: a large-scale bioactivity database for drugdiscovery. Nucleic Acids Res 40:D1100–D1107. doi:10.1093/nar/gkr777
26. Patiny L, Borel A (2013) ChemCalc: a building block for tomor-row’s chemical infrastructure. J Chem Inf Model 53:1223–1228.doi:10.1021/ci300563h
27. Kuhne R, Ebert RU, Schuurmann G (2009) Chemical domain ofQSAR models from atom-centered fragments. J Chem Inf Model49:2660–2669. doi:10.1021/ci900313u
28. Yan X, Gu Q, Lu F, Li J, Xu J (2012) GSA: a GPU-acceleratedstructure similarity algorithm and its application in progres-sive virtual screening. Mol Divers 16:759–769. doi:10.1007/s11030-012-9403-0
29. Xu J (2002) A new approach to finding natural chemical structureclasses. J Med Chem 45:5311–5320. doi:10.1021/jm010520k
30. Newman DJ, Cragg GM (2012) Natural products as sources of newdrugs over the 30 years from 1981 to 2010. J Nat Prod 75:311–335.doi:10.1021/np200906s
31. Cragg GM, Newman DJ (2013) Natural products: a continuingsource of novel drug leads. Bba-Gen Subj 1830:3670–3695. doi:10.1016/j.bbagen.2013.02.008
32. Chen SK, Zhao P, Shao YX, Li Z, Zhang CX, Liu PQ, He XX,Luo HB, Hu XP (2012) Moracin M from Morus alba L. is anatural phosphodiesterase-4 inhibitor. Bioorganic Med Chem Lett22:3261–3264. doi:10.1016/j.bmcl.2012.03.026
33. Liu YN, Huang YY, Bao JM, Cai YH, Guo YQ, Liu SN, LuoHB, Yin S (2014) Natural phosphodiesterase-4 (PDE4) inhibitorsfrom Crotalaria ferruginea. Fitoterapia 94:177–182. doi:10.1016/j.fitote.2014.02.010
34. Lin TT, Huang YY, Tang GH, Cheng ZB, Liu X, Luo HB,Yin S (2014) Prenylated coumarins: natural phosphodiesterase-4inhibitors from toddalia asiatica. J Nat Prod 77:955–962. doi:10.1021/np401040d
35. Sun ZH, Cai YH, Fan CQ, Tang GH, Luo HB, Yin S (2014) Sixnew tetraprenylated alkaloids from the South China Sea gorgonianEchinogorgia pseudossapo. Mar Drugs 12:672–681. doi:10.3390/md12020672
36. Negri A, Rives ML, Caspers MJ, Prisinzano TE, Javitch JA, FilizolaM (2013) Discovery of a novel selective kappa-opioid receptoragonist using crystal structure-based virtual screening. J Chem InfModel 53:521–526. doi:10.1021/ci400019t
37. Pauli I, dos Santos RN, Rostirolla DC, Martinelli LK, DucatiRG, Timmers LF, Basso LA, Santos DS, Guido RV, Andricop-ulo AD, Norberto de Souza O (2013) Discovery of new inhibitorsof Mycobacterium tuberculosis InhA enzyme using virtual screen-ing and a 3D-pharmacophore-based approach. J Chem Inf Model53:2390–2401. doi:10.1021/ci400202t
38. Wang L, Gu Q, Zheng X, Ye J, Liu Z, Li J, Hu X, Hagler A,Xu J (2013) Discovery of new selective human aldose reduc-tase inhibitors through virtual screening multiple binding pocketconformations. J Chem Inf Model 53:2409–2422. doi:10.1021/ci400322j
39. Kumar A, Ito A, Hirohama M, Yoshida M, Zhang KY (2013) Identi-fication of sumoylation activating enzyme 1 inhibitors by structure-based virtual screening. J Chem Inf Model 53:809–820. doi:10.1021/ci300618e
40. Xu L, Zhou S, Yu K, Gao B, Jiang H, Zhen X, Fu W (2013)Molecular modeling of the 3D structure of 5-HT(1A)R: discoveryof novel 5-HT(1A)R agonists via dynamic pharmacophore-basedvirtual screening. J Chem Inf Model 53:3202–3211. doi:10.1021/ci400481p
41. Langdon SR, Westwood IM, van Montfort RL, Brown N, Blagg J(2013) Scaffold-focused virtual screening: prospective applicationto the discovery of TTK inhibitors. J Chem Inf Model 53:1100–1112. doi:10.1021/ci400100c
42. Luo M, Wang XS, Roth BL, Golbraikh A, Tropsha A (2014) Appli-cation of quantitative structure-activity relationship models of 5-HT1A receptor binding to virtual screening identifies novel andpotent 5-HT1A ligands. J Chem Inf Model 54:634–647. doi:10.1021/ci400460q
43. Temml V, Voss CV, Dirsch VM, Schuster D (2014) Discovery ofnew liver X receptor agonists by pharmacophore modeling andshape-based virtual screening. J Chem Inf Model 54:367–371.doi:10.1021/ci400682b
123

840 Mol Divers (2014) 18:829–840
44. Caroli A, Ballante F, Wickersham RB 3rd, Corelli F, RagnoR (2014) Hsp90 inhibitors, part 2: combining ligand-based andstructure-based approaches for virtual screening application. JChem Inf Model 54:970–977. doi:10.1021/ci400760a
45. Gabrielsen M, Kurczab R, Siwek A, Wolak M, Ravna AW, Kris-tiansen K, Kufareva I, Abagyan R, Nowak G, Chilmonczyk Z, SylteI, Bojarski AJ (2014) Identification of novel serotonin transportercompounds by virtual screening. J Chem Inf Model 54:933–943.doi:10.1021/ci400742s
46. Therese PJ, Manvar D, Kondepudi S, Battu MB, Sriram D, Basu A,Yogeeswari P, Kaushik-Basu N (2014) Multiple e-pharmacophoremodeling, 3D-QSAR, and high-throughput virtual screening ofhepatitis C virus NS5B polymerase inhibitors. J Chem Inf Model54:539–552. doi:10.1021/ci400644r
47. Li Z, Cai YH, Cheng YK, Lu X, Shao YX, Li X, Liu M, Liu P, LuoHB (2013) Identification of novel phosphodiesterase-4D inhibitorsprescreened by molecular dynamics-augmented modeling and val-idated by bioassay. J Chem Inf Model 53:972–981. doi:10.1021/ci400063s
48. Klekota J, Roth FP (2008) Chemical substructures that enrichfor biological activity. Bioinformatics 24:2518–2525. doi:10.1093/bioinformatics/btn479
49. Costantino L, Barlocco D (2006) Privileged structures as leadsin medicinal chemistry. Curr Med Chem 13:65–85. doi:10.2174/092986706775197999
50. Welsch ME, Snyder SA, Stockwell BR (2010) Privileged scaf-folds for library design and drug discovery. Curr Opin Chem Biol14:347–361. doi:10.1016/j.cbpa.2010.02.018
123





























