Embed Size (px)
Citation preview

Kinase regulation of MHC-I in tumors Brea et. al
Kinase regulation of Human MHC Class I Molecule Expression on Cancer Cells
Elliott J. Brea1,6, Claire Y. Oh1,6, Eusebio Manchado3, Sadna Budhu2, Ron S.Gejman1,6, George Mo1, Patrizia Mondello1, James E. Han1,6, Casey A. Jarvis1, David Ulmert1, Qing Xiang4, Aaron Y. Chang1,6, Ralph J. Garippa4, Taha Merghoub2, Jedd D. Wolchok2,6,Neal Rosen1,6, Scott W. Lowe3,5,6, David A. Scheinberg1,6* Affiliations: 1Molecular Pharmacology Program, 2Immunology Program, 3Cancer Biology and Genetics Program,4RNAi Core Facility, Memorial Sloan-Kettering Cancer Center New York, NY USA, 10065. 5Howard Hughes Medical Institute, New York, NY, 10065, USA. 6Weill Cornell Medicine, New York, New York, USA, 10021 *To whom correspondence should be addressed: [email protected] Running title: Kinase regulation of MHC-I in tumors Keywords: Immunotherapy, Kinases, Antigen presentation, TCR mimic, T cell therapy, MAPK Funding: The study was supported by US National Institutes of Health grant R01 CA 55349 (D.A.S.), P01 CA23766 (D.A.S.), Diversity Research Supplement for the P01CA023766 (E.J.B., D.A.S), MARF (D.A.S.), P30 CA008748, NCI Grant NIH T32CA062948 (C.Y.O.), NIGMS T32GM07739 (E.J.B), Memorial Sloan Kettering Cancer Center’s (MSKCC’s) Experimental Therapeutics Center and the Lymphoma Foundation and Tudor and Glades funds. Competing interests: D.A.S. is an inventor of the ESKM technology described in this paper and licensed by Memorial Sloan Kettering Cancer Center to Novartis. Corresponding author: David A. Scheinberg, MD, PhD; Molecular Pharmacology Program; Experimental Therapeutics Center; Leukemia Service; Memorial Sloan Kettering Cancer Center; 1275 York Avenue, NY, NY, 10065; 646-888-2190 phone 646-888-2195 office 646-422-0296 fax [email protected]
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
2
Word count: 5586 Figures: 5
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
3
Abstract: The major histocompatibility complex I (MHC-I) presents antigenic peptides to
tumor-specific CD8+ T cells. The regulation of MHC-I by kinases is largely unstudied,
even though many patients with cancer are receiving therapeutic kinase inhibitors.
Regulators of cell surface HLA amounts were discovered using a pooled human kinome
shRNA interference–based approach. Hits scoring highly were subsequently validated
by additional RNAi and pharmacologic inhibitors. MAP2K1 (MEK), EGFR, and RET
were validated as negative regulators of MHC-I expression and antigen presentation
machinery in multiple cancer types, acting through an ERK output–dependent
mechanism; the pathways responsible for increased MHC-I upon kinase inhibition were
mapped. Activated MAPK signaling in mouse tumors in vivo suppressed components of
MHC-I and the antigen presentation machinery. Pharmacologic inhibition of MAPK
signaling also led to improved peptide/MHC target recognition and killing by T cells and
TCR-mimic antibodies. Druggable kinases may thus serve as immediately applicable
targets for modulating immunotherapy for many diseases.
Introduction Major histocompatibility complex class I molecules (MHC-I) generally present
short peptides from either foreign or native intracellular proteins on the cell surface in an
HLA-restricted manner for recognition by CD8+ T cells via their T cell receptor (TCR)
(1). MHC-I is an essential protein for CD8+ cytotoxic T cell responses, effective
vaccination, adoptive T cell therapies, hematopoietic stem cell transplantation, and
organ rejection, among many important physiologic processes and therapeutic
manipulations. In addition, recently developed therapeutic TCR-based constructs and
TCR-mimic antibodies are directed to MHC/peptide complexes (2–5).
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
4
Although immunotherapies for cancer, infectious disease, and autoimmune
disease continue to gain use as effective therapeutic strategies, the mechanisms
underlying the control of presentation of foreign antigens or self-tumor antigens are only
partially understood and currently not exploited clinically (6). Reduced cell surface
presentation of tumor antigens on MHC-I is an important obstacle to effective
immunotherapy with adoptively transferred T cells, TCR constructs, tumor vaccines, and
TCR-mimic antibodies (7–12).
We hypothesized that signaling pathways driven by kinases also may regulate
surface MHC-I expression and that these could identified in loss- or gain-of-function
genetic screens using specific antibodies to detect MHC-I cell surface expression.
Previously, a genome-wide screen provided evidence that regulators of MHC-II could be
identified by RNAi knockdown (13). We decided to target a mesothelioma cell line for
our proof of concept, due to its robust expression of HLA and the need for more
effective therapies for this disease. Moreover, immunotherapies, such as the CTLA-4
blocking antibody tremelimumab, that rely on antigen presentation on MHC-I, are
currently being tested in mesothelioma (14). To identify signaling pathways that regulate
HLA expression in this model, we conducted an shRNA screen of currently annotated
human kinases, as it affords the immediate possibility of targeting identified kinases for
which inhibitors already exist. Among ‘hits’ identified in the screen were kinases that
negatively regulate HLA, including MAP2K1 (MEK1) and EGFR. In addition, we
discovered that DDR2 and MINK1 increase surface MHC-I. These pathways, the effects
of their inhibition, and the positive consequences of inhibition on MHC antigen
presentation, TCR-based recognition of the MHC/peptide complexes and subsequent
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
5
killing were explored. The use of loss- and gain-of-function screens to uncover
regulators of MHC-I could have broad implications for understanding and treating
multiple diseases with pathophysiology related to antigen presentation.
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
6
Materials and Methods Cell lines and culture conditions: After informed consent on Memorial Sloan-Kettering
Cancer Center (MSK) Institutional Review Board–approved protocols, peripheral blood
mononuclear cells (PBMCs) from HLA-typed healthy donors and patients were obtained
by Ficoll density centrifugation. The sources for obtaining human mesothelioma cell
lines JMN and Meso34 are described previously and were verified as unique cell lines
by IMPACT sequencing (Supplementary Table 1) (3). HEK293T, PC9, SKMEL5,
UACC257, SW480, CFPAC1, H827, H1975, H1299, and A549 were obtained from
ATCC between the years 2012 and 2016 and were not further validated. The TPC1 cell
was obtained from Dr. James Fagin lab, where the cell line was validated by IMPACT
sequencing, and used from 2014-2016. (Memorial Sloan-Kettering Cancer Center). The
B16-F10 melanoma line was originally obtained from I. Fidler, and used from 2015-
2016, and was not further validated (MD Anderson Cancer Center). Cell lines were
maintained 2-3 months in RPMI supplemented with 10% FBS and 2 mM L-glutamine
unless otherwise mentioned. HEK293T were grown in Dulbecco’s modified media with
10% FBS and 2 mM L-glutamine. Cells were checked regularly for mycoplasma.
ADCC: The HLA-A*02:01 positive mesothelioma cell lines JMN and Meso34, along with
the melanoma cell line SK-MEL5 were used in the ADCC assay as a target. Antibodies
(3 μg/ml) ESKM (15), PRAME, or its isotype control hIgG1 were incubated with target
cells and fresh healthy donor PBMCs at different effector/target ratios for 6 hours, along
with indicated doses of vehicle or trametinib in RPMI supplemented with 10% FBS. The
supernatant was harvested, and the cytotoxicity was measured by a 51Cr release assay
(Perkin Elmer).
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
7
Clonogenic killing assay: B16F10 cells were treated with either 0.1% DMSO or 1 uM
trametinib for 72 h. B16F10 cells (1 x 104) were then used as targets and in vitro–
activated Pmel T cells (5 x 104) as effectors isolated from the spleen of pmel
(GP100) B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J mice (Jackson Labs).
Pooled RNAi screening: A custom shRNA library targeting the full complement of 526
human kinases was designed using miR30-adapted DSIR predictions refined with
“sensor” rules (six shRNAs per gene) and constructed by PCR-cloning a pool of
oligonucleotides synthesized on 12k customized arrays (Agilent Technologies and
CustomArray) as previously described (16). The list of genes was obtained from
KinBase Database (http://kinase.com/human/kinome/) and was manually curated. After
sequence verification, 3156 shRNAs (5-6 per gene) were combined with positive control
HLA-A– and negative-control Renilla–targeting shRNAs at equal concentrations in one
pool. JMN mesothelioma cells stably expressing the Tet-On rt-TA3 gene were used.
This pool was subcloned into the TRMPV-Neo vector and transduced in triplicates into
Tet-on JMN mesothelioma cancer cells using conditions that predominantly lead to a
single retroviral integration and represent each shRNA in a calculated number of at
least 1,000 cells (Fig. 1A). Transduced cells were selected for 6 days using G418
(1 mg ml−1, Invitrogen); at each passage more than 3 x 107 cells were maintained to
preserve library representation throughout the experiment. After induction, T0 samples
were obtained (~3 x 107 cells per replicate, n = 3) and cells were subsequently cultured
in the presence of doxycycline (2 μg ml−1) to induce shRNA expression. After four days
(Tf), about 3 x 106 shRNA-expressing (dsRed+/Venus+) cells were sorted for each
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
8
replicate using a FACSAriaII (BD Biosciences). DAPI negative, dsRed+/Venus+ cells
were sorted by FACS into three populations of BB7 low, BB7 middle, and BB7 high
binding (Fig. 1). Genomic DNA from Tf samples was isolated by two rounds of phenol
extraction using PhaseLock tubes (5prime) followed by isopropanol precipitation. Deep-
sequencing template libraries were generated by PCR amplification of shRNA guide
strands as previously described (10). Libraries were analyzed on an Illumina Genome
Analyzer at a final concentration of 8 pM; 50 nucleotides of the guide strand were
sequenced using a custom primer (miR30EcoRISeq,
TAGCCCCTTGAATTCCGAGGCAGTAGGCA). Hits with lower than 100 reads from the
Illumina HiSeq were eliminated because they were not above background.
Relative representations of each individual shRNA were determined and compared in
each given sorted population. We separated hits phenotypically into negative regulators
(the population one standard deviation below the mean fluorescence intensity) or
positive regulators (the population one standard deviation above the mean fluorescence
intensity) of HLA-A*02:01. The ratio of the shRNA ranking between the high and low
population was compared, with a high ratio indicating a putative negative regulator of
surface HLA-A*02:01. The scoring criteria for a gene being a negative regulator of HLA-
A*02:01 was based on having two or more shRNA constructs score in the top 5% for
fold difference in relative representation between BB7 high population and BB7 low
population, with other constructs scoring within 1 SD of the mean fold change. The gene
products with at least two shRNA sequences in the top 5% ratio were selected for
further validation by other methods. The same discovery pipeline was used for
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
9
identifying positive regulators of HLA-A*02:01. For validation, the LT3GEPIR shRNA
vector was used (17) ( Supplementary Table S2). Cells were transduced and selected
with puromycin, then induced with doxycycline (2 μg/ml) for 96 h before evaluating BB7,
W6/32, ESK, or PRAME expression by flow cytometry.
Antibodies: Antibodies used for flow cytometry and western blot analysis are described
in Supplementary Table S3. Monoclonal antibodies (mAbs) used for flow cytometry
were specific for HLA-A02 (BB7.2), pan–HLA-ABC (W6/32), WT1 peptide RMF bound
to HLA-A02 (ESK1), PRAME peptide ALY bound to HLA-A02 (Pr20), H2-Kb (AF6-
88.5.5.3), and H2-Kq (KH114). Other antibodies used in this report are also listed in
Supplementary Table S3.
Real-Time PCR: Total RNA was extracted using Qiagen RNA Easy Plus(Qiagen;
#74134) after cells were treated for 48 h with indicated inhibitor. RNA
was converted into cDNA using qScript™ cDNA SuperMix (Quanta Biosciences
Gaithersburg, MD USA). Real-time assays were conducted using TaqMan realtime
probes (Life Technlogies) for human HLA-A (Hs01058806_g1), B2M
(Hs00187842_m1), TAP1 (Hs00388677_m1), TAP2 (Hs00241060_m1), and TBP
(Hs00427620_m1) with 50 ng cDNA. For assessment of gene expression using
RT-PCR PerfeCTa. FastMix. II (Quanta), reactions were carried out in
triplicates using standard thermocycling conditions (2 min at 50 °C, 10
min at 95 °C, 40 cycles of 15 sec at 95.C, and 1 min at 60 °C). TBP
was used as internal control and the ΔΔCT method was used for relative mRNA
calculations.
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
10
Promoter based studies: GLuc luciferase promoter was obtained from Genecoepia
(GeneCoepia Rockville, MD USA) with the B2M promoter cloned upstream of the GLuc
enzyme. Normalization was done to SEAP (under the constitutively active SV40
promoter). Cells were seeded at 5E3 cells/well and treated with indicated drugs for 72
hours. Luminescence quantitation was assayed using the Secrete-Pair Dual
Luminescence Assay Kit (GeneCoepia Rockville, MD USA).
Flow cytometric studies: Cell lines were seeded in triplicate in a 6-well tissue culture
plate at a density of 1E5 cells/well, and allowed to adhere overnight. The next day, cells
were treated with either vehicle control (0.1% DMSO), drugs or inhibitors at indicated
concentrations. Cells were then isolated at 72 hours after inhibitor treatment, and
washed with PBS. Cells were subsequently stained with BB7.2 (HLA-A02–specific
mAb), W6/32 (HLA-ABC–specific mAb), or AF6-88.5.5.3 (H2-Kb–specific mAb,
Ebiosciences). Cells were stained with propidium iodide for viability. Cells were
analyzed on BD Accuri C6 flow cytometer.
Overexpression of β2M: Human β2M cDNA was cloned into the MSCV Puromycin vector
Overexpression of mutant EGFR and NRAS: The pBABE retroviral vector encoding
either EGFR harboring the L858R mutation was used to stably transduce H1299 cell
line using HEK293T/Amphoteric cells and were selected in puromycin (2.5 μg/ml) for 5
days. EGFR L858R was a gift from Matthew Meyerson (Addgene plasmid # 11012).
For overexpression of NRAS the pBABE NRAS Q61K plasmid was used to transduce
H827 cells similar to described above, and selected in puromycin (2 μg/ml). pBabe N-
Ras 61K was a gift from Channing Der (Addgene plasmid # 12543).
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
11
Small molecule inhibitor studies: Compounds were obtained from SelleckChem
(Houston, TX USA). Drugs were used at sub-cytostatic doses by titration using the Cell
Titer Glo assay (Promega). All drugs were used in vitro at indicated doses in 1% DMSO.
Experiments were performed at least twice with similar results, and data shown are
representative.
siRNA knockdown: The JMN cell line was treated with a control scrambled siRNA, or
siRNA against STAT1, STAT3, and RelA. Cells were treated with the indicated drug 24
h after siRNA knockdown for 72 h before assaying for surface HLA-A by flow cytometry.
shRNA construct details are in Supplementary Table S2.
Transgenic EGFR L858R mouse model: FVB CC10-rtTA/EGFR-L858R mice were
obtained as a kind gift from the Harold Varmus lab. Mice were bred in accordance with
MSKCC institutional review board under protocol 96-11-044. Mice used for the
experiment were heterozygous for CC10-rtTA and EGFR-L858R as detected by
quantitative PCR genotyping. At 4-6 weeks of age, mice were put on doxycycline via
food pellets (625 mg/kg) (Harlan-Teklad) for > 6 weeks. Mice were imaged by
anesthetizing under 2% isoflurane and lung field images were acquired on a Bruker
4.7T Biospec scanner (Bruker Biospin Inc.) magnetic resonance imager (MRI) in the
small animal imaging core at MSKCC. Images were analyzed with Osirix Imaging
Software (Geneva, Switzerland). Once confirmed to have reticulonodular appearances
and consolidations by axial and coronal MR images, consistent with previous data
published on the transgenic mice (18).
Mice were sacrificed once confirmed to have lung tumors (non induced control
mice were also used, which genotypically were identical but did not receive dox diet).
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
12
The lungs were isolated and treated with collagenase IV in HBSS with Ca2+ and Mg2+
for 1 h 37ºC. Cells were then collected, blocked with mouse FcR block (Miltenyi),
counted, and stained with mouse CD45 (30-F11, Biolegend), human EGFR(AY13 clone,
Biolegend), and mouse H2-Kq (KH114, clone Abcam) antibodies. Flow cytometry
analysis was performed on Fortessa (BD Biosciences).
CC10/L858R microarray data: Expression data from tissue isolated from WT and EGFR
L858R transgenic mice were obtained from a previous study (GSE17373) and were
selected for statistically significant data (P < 0.05) for PDCD1 (PD-1), CD274 (PD-L1),
TAP1, TAP2, H2-Kd, and B2M gene expression between tumor bearing EGFR L858R
lung tissue and normal lung tissue (19,20).
Results
Pooled shRNA screen identified gene products regulating surface HLA-A*02:01 Loss or gain of function screens serve as starting points for identifying new
regulators of protein expression and function. We used an shRNA library against the
550 currently annotated human kinases to perform a custom pooled screen. For each
gene, six shRNA constructs were cloned into the TRMPV retroviral vector, a tetracycline
regulated vector that couples a mir30 based shRNA to a red fluorescent protein, which
allows easy tracking and sorting of cells productively expressing an shRNA (Fig. 1A)
(16). Knockdown of HLA-A*02:01 by use of an shRNA to this gene product in the same
vector was tested as a positive control and caused strong knockdown by both western
blot analysis and flow cytometry (Fig. 1B).
The amount of MHC-I and antigen presentation on surface HLA-A*02:01 is an
important determinant of efficacy for certain immunotherapies (21). We decided to use
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
13
the human mesothelioma cell line JMN as the target for these studies, which has stable
HLA-A*02:01 expression and which has been used as a target of MHC-I directed
therapies in vitro and in vivo (22). As a tool to show the impact of HLA modulation on
antigen recognition and potential for TCR-based killing, we used TCR mimic antibodies
that recognize peptide/MHC-I complexes. Knockdown of HLA-A substantially decreased
the killing efficacy of the TCR mimic antibody ESKM against the JMN mesothelioma cell
line (Supplementary Fig. S1). JMN was analyzed for presence of a predefined subset of
mutations using the MSK IMPACT platform (Supplementary Table S1). No mutations or
significant copy number alterations were observed in the HLA-A*02:01 or B2M genes.
The JMN cell line was screened with an shRNA library against the human
kinome, as described in the Methods, for genes acting as negative or positive regulators
of surface HLA-A, detected by flow cytometry with the HLA-A*02:01 specific mAb BB7.2
and fluorescence-activated cell sorting was used to sort populations based on HLA
expression (illustrated as in Fig. 1C). The top 5 hits are listed (Supplementary Table
S4).
Based on this analysis, MAP2K1 and EGFR were identified as important
negative regulators of surface HLA-A*02:01. We chose to further investigate EGFR and
MEK because of the availability of clinically approved drugs targeting these kinases
both in NSCLC and metastatic melanoma respectively (23), as well as extensive use of
immunotherapy.
EGFR is a receptor tyrosine kinase that binds epidermal growth factor and is
frequently found to be activated by mutation in NSCLC. Activated EGFR signals through
multiple downstream pathways, including the MAPK pathway. shRNA constructs
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
14
against MAP2K1 and EGFR showed a large increase in relative representation in the
BB7-high sorted population versus the BB7-low population, indicative of a negative
regulator of HLA-A*02:01 surface expression (Fig. 1D). We validated each of these
genes with independent shRNA knockdown to the gene products and saw significant
increases in HLA-A*02:01 by flow cytometry (Fig. 1E). These effects were seen not only
with HLA-A*02:01, but with total HLA-A, B, and C, suggesting coordinated control of all
HLA surface expression, as measured with the W6/32 mAb (Supplementary Fig. S4).
These findings were reproduced in multiple mesothelioma cell lines (Supplementary Fig.
S3A and B). The RET protooncogene was also identified as potential target, but was not
further studied at this time because no inhibitor of adequate specificity was available
(Supplementary Fig. S3C).
We identified examples of positive genetic regulators of HLA-A, including two
putative positive regulators DDR2 and MINK1 (Supplementary Table S4;
Supplementary Fig. S4A and S4B), and confirmed their activity as well using siRNA
knockdown (Supplementary Fig. S4C). Therefore, the kinase screen discovered multiple
positive and negative regulators of HLA expression, each of which in principle could be
explored further for mechanism and clinical utility. The top five negative regulators
evaluated were confirmed by additional study, whereas three of the five top positive
regulators were validated (SupplementaryTable S2).
The MAPK pathway regulates MHC-I Multiple potent small molecule inhibitors exist for EGFR and MEK, with several
already FDA approved, and others currently in clinical trials for various cancers (23). Of
note, the initial screen was performed in a cell line with EGFR activation and an
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
15
identified EGFR mutation (Supplementary Table S1) (24). We tested, in multiple cell
lines, the ability of inhibitors to phenocopy the loss of kinase expression leading to
increased HLA-A expression seen with shRNA. Cell surface HLA-A*02:01 expression
increased in response to MEK inhibition for 72 h with the selective MEK inhibitor
trametinib in mesothelioma cell lines with activated MAP kinase signaling (Fig. 2A). JMN
and PC9, a NSCLC cell line with an activating EGFR mutation (del E746-A750),
responded to the EGFR inhibitor afatinib, whereas the Meso34 cell line without an
EGFR mutation did not respond to afatinib at the same dose, demonstrating selectivity
for activation mutations in the MAPK pathway leading to a response to HLA-A up-
regulation (Supplementary Table 1). We detected an effect of MAP kinase pathway
inhibition on upregulation of HLA-A in the context of gain-of-function mutations or
activation of other targets in the MAP kinase pathway, such as the KRAS G12V
mutation in the SW480 and CFPAC-1 cell lines, the RET/PTC1 gene rearrangement in
the TPC1 thyroid cell line, and the BRAF V600E mutation seen in the UACC257 and
SK-MEL-5 melanoma cell lines (Fig. 2A). The MEKi trametinib did not affect surface
HLA-A expression on normal PBMC cells, showing that this effect is specifically seen in
cells with activated signaling.
To confirm that the increased HLA expression on the cell surface had important
functional significance for enhanced presentation of antigens, we quantified the cell
surface MHC/peptide epitope density by use of TCR mimic mAb selective for two well-
validated tumor-associated epitopes presented by HLA-A*02:01, a WT1 peptide and a
PRAME 300 peptide (25,26). Consistent with the increased surface HLA-A*02:01
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
16
expression, we also observed increased binding of the two TCR-mimic antibodies upon
inhibition of MEK and EGFR (Fig. 2B).
We confirmed the regulatory activity of the pathway in a gain-of-function
experiment by further stimulating the ERK pathway with EGF. The binding of EGF to the
EGFR suppressed surface HLA-A and HLA-A, B, C, providing additional confirmation of
the importance of the MAPK pathway in regulating surface MHC (Fig. 2C).
The mechanism by which the MAP kinase pathway suppresses HLA-A was
unknown. Given that many cancers have activating mutations in specific genes in the
MAP kinase pathway, we investigated inhibition of the identified hits in cell lines
harboring mutations in EGFR, or downstream in Ras. We used a panel of NSCLC cell
lines with activating mutations in EGFR, such as the delE746-A750 in H827, or
L858R/T790M mutation in H1975. The delE746-A750 confers sensitivity to erlotinib,
whereas the T790M confers resistance to erlotinib and to other first generation EGFR
inhibitors, but is sensitive to afatinib (27). We also used EGFR wild-type NSCLC lines
with downstream mutations, such as activating NRAS Q61K in H1299 or KRAS G12S in
A549.
Use of the EGFRi erlotinib and afatinib upregulated surface MHC-I if the cell line
had the sensitizing mutation, whereas all responded to trametinib MEKi (Fig. 2D and
2E). The sensitivity to EGFRi erlotinib and afatinib upregulating surface MHC-I was not
observed with downstream activating RAS mutations. Expression of the activating
EGFR mutation L858R suppressed MHC-I in H1299 NRAS Q61K mutant cell lines (Fig.
2F).
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
17
H827 responded more strongly to EGFRi by erlotinib than MEKi by trametinib,
despite their similar suppression of pERK, a downstream marker of MEK activity. The
combination MEKi and EGFRi was equivalent to EGFRi alone. (Fig. 2G). We introduced
the NRAS Q61K mutation, shown to cause resistance to EGFRi and persistent
activation of the MAPK pathway in H827. Use of the EGFRi still had an effect on surface
MHC-I despite no change in pERK output on the H827 NRAS Q61K cell line (Fig. 2H).
This could be due to activation of parallel signaling pathways in EGFR mutant cancers
or differential stimulation of ERK. Thus, the MAPK pathway is not the only determinant
of EGFR-mediated regulation of surface MHC-I. Given that both EGFR and MEK are
involved in signaling via the MAP kinase pathway, these data validate the importance of
this pathway in regulating surface HLA-A and MHC-I.
Interferon-γ (IFNγ) is a well-known regulator of MHC-I via the JAK/STAT pathway
(28,29). We asked whether a combination of IFNγ with the kinase inhibitors would have
additive effects on HLA expression (Supplementary Fig. S5A, B, C). Both IFNγ and
afatinib (in EGFR mutant H1975 lung cancer cells), and IFNγ and trametinib (in Braf
mutant SK-MEL5 and UACC257 melanoma cells), alone, each increased expression of
cell surface HLA molecules, as measured by antibodies to HLA-A*02:01 or pan HLA-A,
B, C. The combination of the IFNγ and the drug had greater effects than either alone,
consistent with the involvement of two different pathways. PCR analysis of TAP1
showed that this internal component of the antigen presentation machinery was also
upregulated by IFNγ by 10 to 25 fold in all three cell lines. β2-Microglobulin was also
upregulated 4 to 7 fold with IFNγ treatment in all three lines. There were minimal
increases in these two proteins in response to the two kinase inhibitors in H1975 and
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
18
UACC257. However in SK-MEL5, trametinib increased both proteins alone and was
additive with interferon.
HLA-E is another component of the antigen presentation pathway that presents
MHC molecule–bound peptides and may be involved in downregulating NK cell immune
responses to cancers (30). IFNγ and afatinib (in EGFR mutant lung cancer cells) did not
affect HLA-E levels (Supplementary Fig. S5D). Trametinib, in Braf mutant SK-MEL5
melanoma cells, increased cell surface HLA-E molecules, but did not do so in UACC257
cells. IFNγ also variably upregulated HLA-E in the two melanoma lines, and the
combination of drugs was more effective in increasing HLA-E in UACC257 cells
(Supplementary Fig. S5E and F). Although an upregulation of HLA-E might be
expected to partially counter the effects of upregulation of classic MHC seen in these
cells, the net effect was to improve cytolytic activity.
Improving immunotherapy by inhibiting MAPK pathway We next tested the effects of modulating HLA-A*02:01 expression on the efficacy of
immunotherapies that depend on HLA-A*02:01 upregulation and antigen presentation,
by use of pmel-1 T cells expressing a TCR that reacts with gp100 and use of two
different TCR mimic antibodies whose function also depend on peptide/MHC-I
expression. The TCR mimic antibody ESKM, which targets a peptide from WT1 in the
context of HLA-A*02:01, and Pr20m, which targets a peptide from PRAME, were used
as easily quantifiable surrogate tools for measuring the potential therapeutic
consequences of upregulation of HLA-A*02:01-based antigen targets as a consequence
of MEK inhibition. The cytotoxicity of ESKM against the JMN and Meso34 human
mesothelioma cell lines were increased by MEK inhibition with trametinib (Fig. 3A and
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
19
B), which was used at a non-cytotoxic dose (Supplementary Fig. S6). Increased
cytotoxicity of the Pr20m mAb was also observed with use of the MEKi trametinib in the
SK-MEL-5 human melanoma cell line, validating this observation with multiple targets in
multiple cell lines (Fig. 3C).
Finally, specific killing by T cells increased after upregulatingMHC-I with MEKi.
The pmel-1 gp100-specific mouse T cells were more effective at killing of the gp100
positive target B16F10 melanoma cells following trametinib treatment, which correlated
with pERK inhibition and MHC-I upregulation (Fig. 3D, E, and F) (31). Therefore,
improved recognition as a result of the increased expression of MHC-I and its presented
peptides using three different target antigens by TCR or TCR mimics had significant
consequences for cytotoxic activity.
Mechanism of MAPK regulation of MHC-I We hypothesized that the inhibition of the MAP kinase pathway might act on
other components of the antigen presentation machinery in addition to MHC-I
molecules, thus allowing increased epitope expression in the more abundant cell
surface HLA molecules. Indeed, EGFR and MEK inhibition produced an increase in
mRNA gene expression of HLA-A along with other key components of the antigen
presentation pathway and MHC-I structure, such as TAP1, TAP2, and β2M, (Fig. 4A).
The JMN and Meso34 cells at t = 1 h were sensitive to trametinib at doses less than 10
nM as previously reported, but required higher doses to sustain inhibition of pERK at t =
72 h due to strong feedback (Supplementary Fig. S7A vs B). Doses of trametinib were
chosen over the IC50 of MEK by using pERK as a readout of MEK inhibition at 72 h
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
20
(Supplementary Fig. S7B and C). These data correlated with previous findings that
BRAF mutant cell lines are the most sensitive to MEK inhibition, when compared to
BRAF wild-type cell lines harboring further upstream mutations (32). A time course
showed maximal inhibition of MEK at 3 h, with maximal increases of HLA-A and β2M at
72 h (Supplementary Fig. S8). Surface HLA-A increased in a dose dependent manner
with increasing MEK inhibition in both melanoma and mesothelioma (Fig. 4B). The
phenotypes observed are unlikely from off-target effects of the drug, given the dose
response on pERK expression and the plateau of the dose response of surface HLA-A.
Antibodies against pERK, along with total ERK1/2, were used to show dose-
responsive increases in response to trametinib inversely correlated with HLA-A protein
expression. The increase of β2M much greater than that of HLA complexes in multiple
cell lines, consistent with the gene expression data (Fig. 4C). EGFRi with erlotinib also
caused a dose-dependent increase in HLA-A and β2M (Fig. 4D). Because β2M is
required for surface presentation of HLA, A,B, C and stability of the MHC-I molecules on
the cell surface, we investigated the potential role of β2M in controlling cell surface HLA-
A expression. Overexpression of β2M increased cell surface HLA-A and pan HLA-ABC,
phenocopying the effect of MEK inhibition (Fig. 4E), which was regulated by multiple
regulatory domains in the promoter region, including the ISRE site, E box, and NF-κB
sites. Using a luciferase-based promoter assay, we demonstrated that upon addition of
MEKi, a dose-dependent increase in activity on the HLA-A and B2M promoters was
observed (Fig. 4F). Knockdowns of STAT1, STAT3, and RelA (a component of NF-κB
complex) were performed on JMN cells, along with treatment with MEKi. STAT1
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
21
knockdown had the largest inhibition of upregulation of surface HLA-A after MEKi,
suggesting a role for STAT1 in responses to MEKi (Fig. 4G).
MAPK activation causes in vivo suppression of MHC-I and increased PD-1/L1 We confirmed that these observations on MHC regulation and antigen
presentation machinery were not limited to in vitro models. Microarray profiling of the
lung bearing tumors from transgenic EGFR L858R, which activates the MAPK pathway,
compared to normal lungs, demonstrated suppression of mouse MHC-I and antigen
presentation components H2-K/D, and β2M, thereby confirming the effects of this
pathway in vivo (Fig. 5A) (19). Upregulation of PD-1 and PD-L1 markers in the tumors
was also observed as previously published (20).
Expression of EGFR L858R in the transgenic mice, given doxycycline for > 6 wk,
was demonstrated by increased binding of a human EGFR-specific fluorescently
labeled mAb (Fig. 5B). Mice were confirmed to have development of lung
adenocarcinoma by MR, with development of a reticulonodular infiltrate in the lung,
consist with previous publications (18). The CD45–hEGFR+ population in the lung in the
EGFR L858R–expressing mice demonstrated decreased binding of a MHC-I–specific
mAb by flow cytometry, when compared to a wild-type mouse which did not express the
EGFR L858R mutation (Fig. 5C).
Discussion Immunotherapy of cancer is emerging as a successful and important component
of treatment. MHC molecules presenting antigens are the target of multiple therapeutic
strategies that involve vaccines, T cells or TCRs, TCR mimic antibodies, or T cell
checkpoint blockade. The latter, a highly effective recent example in cancer therapy,
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
22
appears to require presentation of neoantigens on MHC-I on the surface of cancer cells
(33–35). Most immunotherapies have focused on enhancing intrinsic effector cell
mechanisms for modulating the immune response, either by directly activating the
effector T cells or by relieving their suppression. In distinct contrast, here we propose an
alternative approach, whereby the antigenic targets on the cancer cells themselves are
modulated to improve TCR-based killing. The ability to regulate such responses by
selectively affecting target cells could have an important impact on both disease and
therapy. We propose that kinases are a readily druggable pathway that might be used in
conjunction with immunotherapy to enhance efficacy. The beneficial effect of the
combination of immunotherapy with kinase inhibition was shown in mouse models of
combined PD-1/PD-L1 blockade with MEKi (36). A second model, of adoptive T cell
therapy in combination with MEKi in BRAF mutant murine melanoma, has demonstrated
superiority to single agents alone (37). Our work has provided a new understanding of
another mechanism why these combination therapies may be more effective, wherein
upregulated MHC-I and antigen presentation on the target cells, essential for the
adaptive immune response, improves TCR-based recognition and killing. Indeed, many
of the patients treated currently with immunotherapies also receive kinase inhibitor
therapies as distinct treatments.
The loss- and gain-of-function screen described here allowed unbiased
interrogation of the currently annotated human kinases for their regulation of cell surface
MHC-I. We then explored mechanistically how such kinase regulators could be inhibited
for altering surface expression of MHC-I, as a way of validating the screen,
understanding the process, and also for extending the findings to functional modulation
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
23
of a model immunotherapy proof of concept that directly depends on MHC-I
presentation. In this case, we were able to specifically isolate the MHC as the sole
target of the therapy by use of therapeutic TCR mimic antibodies directed to antigens
presented by MHC.
This study also provides support for the use of a flow cytometry–based loss-of-
function pooled shRNA screen in the study of the regulation of other cell surface
molecules, and potentially intracellular antigens as well. This technique will allow many
laboratories without robotics and high-throughput flow cytometry equipment to
investigate pathways that can be easily perturbed with loss-of-function RNAi screens or
other techniques, such as CRISPR loss-of-function.
Here we have demonstrated the effect of the MAPK pathway on MHC-I, in vitro
and in vivo. However, therapeutic applications in humans of the combination of
available immunotherapies and pharmacologic kinase inhibition, to increase MHC-I
surface expression and antigen presentation, will be complicated and difficult to predict,
because T cells and NK cells also rely on similar kinase signaling pathways for
activation. More work needs to be done to determine optimal pathways or schedules or
doses to target MHC in tumor cells specifically, while sparing signaling pathways of the
effector cells (38). Some studies suggest conflicting effects of MEKi on T cell effector
function, which may be dependent on the tumor model evaluated (39,40). These effects
cannot be simply modeled in mice. Empirically derived optimal dosing and schedules
will be needed in in vivo models and in humans to show that use of kinase inhibition to
regulate immunotherapy has therapeutic benefits while sparing immune effector cells of
the detrimental effects (40–42). These investigations will be complicated by the effects
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
24
of some of the drugs on the cellular effectors themselves, the variable effects on the
cancer cells depending on their specific mutations, the time frames required to
upregulate the responses (about 3 days in the experiments here) and the time required
for the effects to wash out of the cancer cells and the effectors.
The data provide a mechanistic explanation of how MHC-I is regulated by the
MAPK pathway. MHC-I mRNA expression is regulated through upstream enhancer
elements, with involvement of the NF-κB transcription factor (43,44). MHC-I is also
induced by TNF, IL1, IFNβ, and IFNγ, which upregulates HLA-A via the JAK/STAT
pathway (28,29). The CIITA transcription factor can also act on MHC-I gene expression
(45). IFNγ can increase MHC-I and antigen presentation, but thus far its use has had
limited applications (46). We show here that combining the kinase inhibitors with IFNγ
in vitro can have additive effects on HLA expression, TAP1, and β2M. This may be of
benefit in vivo as IFNγ may be elaborated locally at tumor sites from tumor infiltrating
lymphocytes at steady state or in response to other immunotherapies, such as check
point blockade.
MEK has been proposed by others to be a regulator of MHC-I expression. EGFR
inhibition can augment MHC-I and MHC-II expression in keratinocytes (47). MEK was
previously identified as a negative regulator of HLA-A*02:01 in esophageal and gastric
cancer by Mimura et al. (48). We validated these targets in the screen as important
negative regulators of MHC-I and discovered a mechanistic role of the MAP kinase
pathway in regulating surface levels of MHC-I. Our data directly link to immune-
oncologic applications in humans, by demonstrating potent upregulation of MHC-I in a
wide variety of cancers, including melanoma and NSCLC, which are currently the
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
25
subject of FDA-approved therapies which depend upon on antigen presentation, such
as checkpoint blockade with mAbs ipilibumab, pembrolizumab, and nivolibumab. In
addition, by demonstrating that an FDA-approved MEKi upregulated MHC-I, the results
support the clinical testing of combination therapies, which could advance this concept
into human therapy. We characterized these effects on MHC-I in cells with activating
mutations in the MAP kinase pathway with various genotypic lesions, such as activating
EGFR mutations, BRAF mutations, and RAS mutations. Finally we also showed the
mechanism was active in RET-translocated thyroid cancer. The sum of these data
support the importance of MAPK pathway in regulating MHC-I quite broadly, while
providing new mechanistic insights.
Finally, our findings are immunologically significant. Upregulation induced by
MEK inhibition resulted in superior cytotoxic activity of TCR mimic antibodies (directed
to specific MHC presented antigens) and TCR-based therapy with a pmel-specific
murine T cell model. We further show, in a transgenically engineered mouse model in
vivo, that activating this pathway reduces expression of the components of the antigen
presentation machinery, along with MHC-I. This gain-of-function experiment is crucial to
proving that activation of MAPK can cause decreased MHC-I in vivo.
Activating EGFR mutations may contribute to immune escape, due to PD-L1
expression. Downregulation of MHC-I, which was observed from our study, may also
contribute to this finding (20). While demonstrating combination therapy with EGFRi and
checkpoint blockade would be rational, the transgenic EGFR mice have shown dramatic
tumor reduction and cures with EGFRi as monotherapy, leaving little window to show
synergism in currently existing mouse models with checkpoint blockade (18). Our data
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
26
also suggest that using combination therapy with MAP kinase inhibition can be
powerful, not only as a direct cancer therapy to prevent growth, but also indirectly to
promote immunotherapy.
HLA genes are a risk factor for autoimmune diseases such as ankolysing
spondylitis and multiple sclerosis (49–51). In addition to upregulation by certain kinases,
we showed downregulation of MHC-I through new kinase targets. These targets are not
currently addressed by immunosuppressive therapies, which inhibit the effector arm of
the immune response with concomitant toxicity. These new targets warrant additional
investigation into altering the course of autoimmune diseases by investigating the
efficacy of specific kinase inhibitors and developing appropriate mouse models.
A requirement of many immunotherapies therapies, particularly checkpoint
blockade, is the availability of recognizable antigens that are presented on MHC-I.
Tumors can decrease MHC-I expression, to avoid immune system detection of the rare
neoantigens created in tumors by mutations, and increase inhibitory receptor
expression. By modulating expression of these limited antigens, improved clinical
efficacy may be seen with certain immunotherapies in conjunction with current FDA-
approved small molecules targeting EGFR and MEK. The inhibition of kinase pathways
also caused a more general upregulation of the antigen presentation machinery,
including Tap (responsible for transporting peptides) and β2M (responsible for stabilizing
MHC-I). Many of the recently approved immunotherapies, such as blockade of CTLA-4
or PD-1, release the T cell inhibition promoted by target tumor cells. These
immunotherapies provide a promising approach to addressing multiple malignancies. By
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
27
rationally combining them with targeted small molecule inhibitors, novel synergistic
treatment strategies may be developed.
Acknowledgments: We thank T. Dao, L. Dubrovsky, D. Pankov, P Lito, D. Solit, E. Pamer, R. Brentjens, M. Will, A. Lujambia and A. Scott for their helpful discussions. We thank Y. Li and A. Younes for use of their equipment.
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
28
References: 1. Agrawal S, Kishore MC. MHC Class I Gene Expression and Regulation. J
Hematother Stem Cell Res. 2000;9:795–812.
2. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al.
Cancer regression in patients after transfer of genetically engineered
lymphocytes. Science. 2006;314:126–9.
3. Dao T, Yan S, Veomett N, Pankov D, Zhou L, Korontsvit T, et al. Targeting the
intracellular WT1 oncogene product with a therapeutic human antibody. Sci Transl
Med. 2013;5:176ra33.
4. Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell
therapy using antigen-specific CD8+ T cell clones for the treatment of patients
with metastatic melanoma: In vivo persistence, migration, and antitumor effect of
transferred T cells. Proc Natl Acad Sci. 2002;99:16168–73.
5. Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM,
et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity
and persist in post-transplant patients. Sci Transl Med. 2013;5:174ra27.
6. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat
Rev Cancer. 2012;12:252–64.
7. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature.
Nature Publishing Group; 2011;480:480–9.
8. Adams JL, Smothers J, Srinivasan R, Hoos A. Big opportunities for small
molecules in immuno-oncology. Nat Rev Drug Discov. Nature Publishing Group;
2015;14:603–22.
9. Chiappinelli KB, Zahnow CA, Ahuja N, Baylin SB. Combining Epigenetic and
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
29
Immunotherapy to Combat Cancer. Cancer Res. 2016;1683–90.
10. Campoli M, Ferrone S. HLA antigen changes in malignant cells: epigenetic
mechanisms and biologic significance. Oncogene. 2008;27:5869–85.
11. Leone P, Shin E-C, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC Class I
Antigen Processing and Presenting Machinery: Organization, Function, and
Defects in Tumor Cells. JNCI J Natl Cancer Inst. 2013;105:1172–87.
12. Kageshita T, Hirai S, Ono T, Hicklin DJ, Ferrone S. Down-Regulation of HLA
Class I Antigen-Processing Molecules in Malignant Melanoma. Am J Pathol.
1999;154:745–54.
13. Paul P, Van Den Hoorn T, Jongsma MLM, Bakker MJ, Hengeveld R, Janssen L,
et al. A Genome-wide Multidimensional RNAi Screen Reveals Pathways
Controlling MHC Class II Antigen Presentation. Cell. Elsevier Inc.; 2011;145:268–
83.
14. Calabrò L, Morra A, Fonsatti E, Cutaia O, Amato G, Giannarelli D, et al.
Tremelimumab for patients with chemotherapy-resistant advanced malignant
mesothelioma: an open-label, single-arm, phase 2 trial. Lancet Oncol. Elsevier;
2013;14:1104–11.
15. Veomett N, Dao T, Liu H, Xiang J, Pankov D, Dubrovsky L, et al. Therapeutic
Efficacy of an Fc-Enhanced TCR-like Antibody to the Intracellular WT1
Oncoprotein. Clin Cancer Res. 2014;20:4036–46.
16. Zuber J, McJunkin K, Fellmann C, Dow LE, Taylor MJ, Hannon GJ, et al. Toolkit
for evaluating genes required for proliferation and survival using tetracycline-
regulated RNAi. Nat Biotechnol. Nature Publishing Group, a division of Macmillan
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
30
Publishers Limited. All Rights Reserved.; 2011;29:79–83.
17. Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, et al. An
Optimized microRNA Backbone for Effective Single-Copy RNAi. Cell Rep.
2013;5:1704–13.
18. Politi K, Zakowski MF, Fan P-D, Schonfeld EA, Pao W, Varmus HE. Lung
adenocarcinomas induced in mice by mutant EGF receptors found in human lung
cancers respond to a tyrosine kinase inhibitor or to down-regulation of the
receptors. Genes Dev. 2006;20:1496–510.
19. Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A, et al. Dual
targeting of EGFR can overcome a major drug resistance mutation in mouse
models of EGFR mutant lung cancer. J Clin Invest. American Society for Clinical
Investigation; 2009;119:3000–10.
20. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al.
Activation of the PD-1 Pathway Contributes to Immune Escape in EGFR-Driven
Lung Tumors. Cancer Discov. 2013;3:1355–63.
21. Rivoltini L, Barracchini KC, Viggiano V, Kawakami Y, Smith A, Mixon A, et al.
Quantitative correlation between HLA class I allele expression and recognition of
melanoma cells by antigen-specific cytotoxic T lymphocytes. Cancer Res.
1995;55:3149–57.
22. Dao T, Yan S, Veomett N, Pankov D, Zhou L, Korontsvit T, et al. Targeting the
Intracellular WT1 Oncogene Product with a Therapeutic Human Antibody. Sci
Transl Med. 2013;5:176ra33-176ra33.
23. Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
31
Survival with MEK Inhibition in BRAF-Mutated Melanoma. N Engl J Med.
2012;367:107–14.
24. Brevet M, Shimizu S, Bott MJ, Shukla N, Zhou Q, Olshen AB, et al. Coactivation
of Receptor Tyrosine Kinases in Malignant Mesothelioma as a Rationale for
Combination Targeted Therapy: J Thorac Oncol. 2011;6:864–74.
25. Chang, Aaron, Dao, Tao, Scott, Andrew, Dubrovsky, Leonid, Liu, Cheng,
Scheinberg D. A Therapeutic TCR Mimic Monoclonal Antibody for Intracellular
PRAME Protein in Leukemias. Am Soc Hematol. 2015.
26. Krug LM, Dao T, Brown AB, Maslak P, Travis W, Bekele S, et al. WT1 peptide
vaccinations induce CD4 and CD8 T cell immune responses in patients with
mesothelioma and non-small cell lung cancer. Cancer Immunol Immunother.
2010;59:1467–79.
27. Cross DAE, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al.
AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to
EGFR inhibitors in lung cancer. Cancer Discov. American Association for Cancer
Research; 2014;4:1046–61.
28. Girdlestone J, Isamat M, Gewert D, Milstein C. Transcriptional regulation of HLA-
A and -B: differential binding of members of the Rel and IRF families of
transcription factors. Proc Natl Acad Sci U S A. 1993;90:11568–72.
29. Wolchok JD, Vilček J. Induction of HLA class I mRNA by cytokines in human
fibroblasts: comparison of TNF, IL-1 and IFN-β. Cytokine. 1992;4:520–7.
30. Kochan G, Escors D, Breckpot K, Guerrero-Setas D. Role of non-classical MHC
class I molecules in cancer immunosuppression. Oncoimmunology.
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
32
2013;2:e26491.
31. Budhu S, Loike JD, Pandolfi A, Han S, Catalano G, Constantinescu A, et al. CD8+
T cell concentration determines their efficiency in killing cognate antigen-
expressing syngeneic mammalian cells in vitro and in mouse tissues. J Exp Med.
2010;207:223–35.
32. Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, et al. BRAF
mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–62.
33. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al.
Mutational landscape determines sensitivity to PD-1 blockade in non-small cell
lung cancer. Science (80- ). American Association for the Advancement of
Science; 2015;348:124–8.
34. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al.
Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N Engl J
Med. 2014;371:2189–99.
35. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic
correlates of response to CTLA4 blockade in metastatic melanoma. Science (80-
). American Association for the Advancement of Science; 2015;350:207–11.
36. Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, et al. The BRAF and
MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in
Combination with Immunomodulatory Antibodies Targeting PD1, PD-L1 and
CTLA-4. Clin Cancer Res. 2015;
37. Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, et al.
Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
33
BRAF(V600E) melanoma. Sci Transl Med. 2015;7:279ra41.
38. Kortum RL, Rouquette-Jazdanian AK, Samelson LE. Ras and extracellular signal-
regulated kinase signaling in thymocytes and T cells. Trends Immunol.
2013;34:259–68.
39. Ebert PJR, Cheung J, Yang Y, Kim JM, Belvin M, Mellman I, et al. Article MAP
Kinase Inhibition Promotes T Cell and Anti- tumor Activity in Combination with
PD-L1 MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in
Combination with PD-L1 Checkpoint Blockade. Immunity. Elsevier Inc.; 2016;1–
13.
40. Vella LJ, Pasam A, Dimopoulos N, Andrews M, Knights A, Puaux A-L, et al. MEK
inhibition, alone or in combination with BRAF inhibition, affects multiple functions
of isolated normal human lymphocytes and dendritic cells. Cancer Immunol Res.
2014;canimm.0181.2013.
41. Callahan MK, Masters G, Pratilas CA, Ariyan C, Katz J, Kitano S, et al.
Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF
inhibitor. Cancer Immunol Res. 2014;2:70–9.
42. Vella LJ, Andrews MC, Pasam A, Woods K, Behren A, Cebon JS. The kinase
inhibitors dabrafenib and trametinib affect isolated immune cell populations.
Oncoimmunology. 3:e946367.
43. Gobin SJP, Biesta P, Elsen PJ Van den. Regulation of human β2-microglobulin
transactivation in hematopoietic cells. Blood. 2003;101:3058–64.
44. Wolchok D, Goodman A. induction cells of NF-xB may be necessary but is not
sufficient for of H-2 antigens by TNF in J558L murine myeloma. Cytokines.
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
34
1994;55:7–12.
45. Gobin SJP, Peijnenburg A, Van Eggermond M, Van Zutphen M, Van Den Berg R,
Van Den Elsen PJ. The RFX complex is crucial for the constitutive and CIITA-
mediated transactivation of MHC class I and B2-microglobulin genes. Immunity.
1998;9:531–41.
46. Zaidi MR, Merlino G. The Two Faces of Interferon-γ in Cancer. Clin Cancer Res.
2011;17:6118–24.
47. Pollack BP, Sapkota B, Cartee T V. Epidermal Growth Factor Receptor Inhibition
Augments the Expression of MHC Class I and II Genes. Clin Cancer Res.
2011;17:4400–13.
48. Mimura K, Shiraishi K, Mueller A, Izawa S, Kua L-F, So J, et al. The MAPK
pathway is a predominant regulator of HLA-A expression in esophageal and
gastric cancer. J Immunol. 2013;191:6261–72.
49. Fogdell-Hahn A, Ligers A, Gronning M, Hillert J, Olerup O. Multiple sclerosis: a
modifying influence of HLA class I genes in an HLA class II associated
autoimmune disease. Tissue Antigens. 2000;55:140–8.
50. Brown MA, Kenna T, Wordsworth BP. Genetics of ankylosing spondylitis-insights
into pathogenesis. Nat Rev Rheumatol. 2016;12:81–91.
51. Caroline Robert, M.D and Thomas S. Kupper M. Inflammatory Skin Diseases, T
Cells, and Immune Surveillance — NEJM. N Engl J Med. 1999;341:1817–28.
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
35
Figure 1. Screen for kinase regulators of surface HLA. A. A TRMPV inducible shRNA
retroviral vector was used for transducing JMN (HLA-A*02:01 positive human
mesothelioma line). TRE is the Tet responsive element, which drives expression of the
fluorophore dsRed and the shRNA hairpin. The constitutive PGK promoter drives the
Venus fluorophore along with Neomycin resistance cassette. B. Western blot and flow
cytometry data showing knockdown of HLA-A using TRMPV retroviral system with a
positive control shRNA to HLA-A02. The shRen is a negative control shRNA designed
against the Renilla gene C. Schema depicting the workflow pipeline for the screen of
regulators of surface HLA-A. D. Waterfall plot showing distribution of shRNA constructs
against MAP2K1 and EGFR as log fold difference between BB7 high sorted population
and BB7 low sorted population. E. shRNA knockdown of MAP2K1 and EGFR in JMN
cells validates them as a negative regulator of surface HLA-A. BB7.2 is a mAb specific
for HLA-A02. shRNA against Renilla was used as a negative control, whereas an
shRNA against HLA-A was used as a positive control. The screen was done in triplicate.
Inhibition experiments were performed at least twice with similar results, and data
shown are representative. Student’s t-test was done to compare each shRNA gene
knockdown MFI to the shRen control. (*≤0.05, **≤0.01, ***≤0.001, ****≤0.0001)
Figure 2. Use of selective EGFRi and MEKi increased cell surface HLA-A expression
and tumor antigen presentation, whereas activation of EGFR caused downregulation of
MHC-I. A. MEK inhibition and EGFR inhibition for 72 h with indicated inhibitors
increased HLA-A (BB7 binding) by flow cytometry in JMN, Meso34, PC-9, UACC257,
SK-MEL-5, SW480, and TPC1 cell lines. 1% DMSO was used as a vehicle control. B.
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
36
Binding of TCRm antibodies to peptide /MHC epitopes. In blue, use of ESK antibody to
a peptide derived from the oncoprotein WT1 that is presented on HLA-A0201. Binding
increased after inhibition of EGFR and MEK for 72 h in JMN, Meso34, and TPC1. In
red, the PRAME TCRm antibody to an epitope of PRAME tumor antigen presented on
HLA-A0201 on SKMEL5 cells. Experimental setup was similar to A. C. Treatment of
JMN with 10 nM EGF for 72 h, causing activation of the downstream MAPK pathway,
led to decreased surface HLA-A and total HLA-ABC. D. Use of EGFRi erlotinib, along
with MEKi trametinib, on H827 (EGFR E746del-A750 mutation), H1975 (L858R/T790M),
H1299 (EGFR wt, NRAS Q61K), and A549 (EGFR wt/KRAS G12S) to alter surface
HLA-ABC expression. Student t-test was done to compare each treatment to vehicle
control. *P values annotated as in figure 1. E. Western blot analysis showing degree of
inhibition of the MAP kinase pathway on a panel of NSCLC cell lines using 1% DMSO
(D), 100 nM erlotinib (E), 100 nM afatinib (A), or 500 nM trametinib (T). F. H1299 cells
were transduced with retroviral vectors expressing EGFR L858R and were analyzed for
surface pan HLA-ABC using W6/32. Activation of EGFR is demonstrated by western
blot G. EGFR inhibition upregulated surface HLA-ABC more than MEKi, despite
equivalent inhibition of pERK output. H. EGFRi upregulated MHC-I despite downstream
mutations causing constitutive MAPK activation. The NRAS Q61K mutation was
introduced into H827 and cells were treated with EGFRi or MEKi as done in 2G.
Experiments were performed 2-4 times with similar results, and data shown are
representative.
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
37
Figure 3. Improving cytolysis efficacy by up-regulating cell surface MHC-I.
A. Antibody dependent cellular cytotoxicity assay was performed on JMN human
mesothelioma cell line. Cells were incubated for 72 h with either vehicle control or
trametinib and subsequently exposed to either isotype antibody or ESKM in ADCC
assay B. ADCC assay on Meso34 (human mesothelioma). Experimental setup was
similar to 3A. C. ADCC assay on SKMEL5 (human melanoma) using TCRm mAb
PRAME against the PRAME epitope, experimental setup similar to 3A. D. B16F10 cells
were exposed to pmel-1 (gp100)–specific TCR T cells for 24 h, then killing was
assessed using a clonogenic assay described previously. E. B16F10 pERK protein, as
measured by pERK intracellular staining, in cells treated with vehicle or 1 μM trametinib.
F. B16F10 MHC-I expression assessed by flow cytometry after treatment with 1 μM
trametinib for 72 h. Experiments were performed 2-4 times with similar results, and data
shown are representative.
Figure 4. MAPK signaling suppresses antigen presentation machinery and MAPK
inhibition broadly up-regulates antigen presentation machinery. A. MEK and EGFR
inhibition for 48 h led to increased HLA-A, along with TAP1, TAP2, and β2M in JMN,
Meso34, SK-MEL-5 and UACC257, H827, and PC9 B. Dose dependent increase in
surface HLA-A with increasing MEKi in JMN and SKMEL5. Cells were analyzed by flow
cytometry at 72 h C. MEK inhibition leads to increasing amounts of HLA-A and β2M
protein. Cells were treated with indicating amounts of trametinib (MEKi) for 72 h and
specific antibodies to the indicated proteins were blotted. D. EGFR inhibition led to
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
38
increasing HLA-A and β2M protein. Experimental setup similar to Fig 4C. E.
Overexpression of β2M led to increased surface HLA-A and HLA-ABC. F. Treatment of
JMN with trametinib for 72 h led to increased activity on the HLA-A and B2M promoters.
The HLA-A and B2M promoter was cloned upstream of the Gaussian Luciferase gene.
SEAP under the CMV promoter was used as a normalization factor. G. Knockdown of
STAT1, on JMN cells treated with MEKi demonstrates role in mediating surface HLA-A
up-regulation. JMN cells were transfected with siRNA against the genes shown and
treated with either DMSO or 1 μM trametinib 24 h after siRNA transfection, then
assayed by flow cytometry for surface HLA-A expression 72 h after treatment.
Experiments were performed 2-4 times with similar results, and data shown are
representative.
Figure 5. Activation of MAPK pathway via activating EGFR mutations causes in vivo
suppression of MHC-I in addition to upregulation of checkpoint blockade. A.
Unsupervised hierarchical clustering microarray expression profiling analysis of lung
tumors from CC10/L858R mice with EGFR L858R tumor bearing lungs (right side,
black) or normal lungs (left side, green) focusing on H2-KD, B2M, TAP1, TAP2, PD-
L1(PDCD1), and PD-1 (CD274) gene expression. B. Flow cytometry data of FVB CC10-
rtTA/TetO EGFR L858R expressing mice. Mice were induced with doxycycline for >6
weeks before sacrificed (mice E-G). Control mice were kept on normal diet, but
genotypically identical (A-D). Lungs were isolated and stained with markers for CD45
(pan leukocyte), hEGFR, and H2-Kq (MHC-I). C. The CD45- lung population was
stained with mouse H2-kq specific mAb. CD45–hEGR– population shows higher MHC-I
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Kinase regulation of MHC-I in tumors Brea et. al
39
expression than CD45–hEGFR+ population. Representative MRI images of mouse lungs
are shown for two samples.
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177
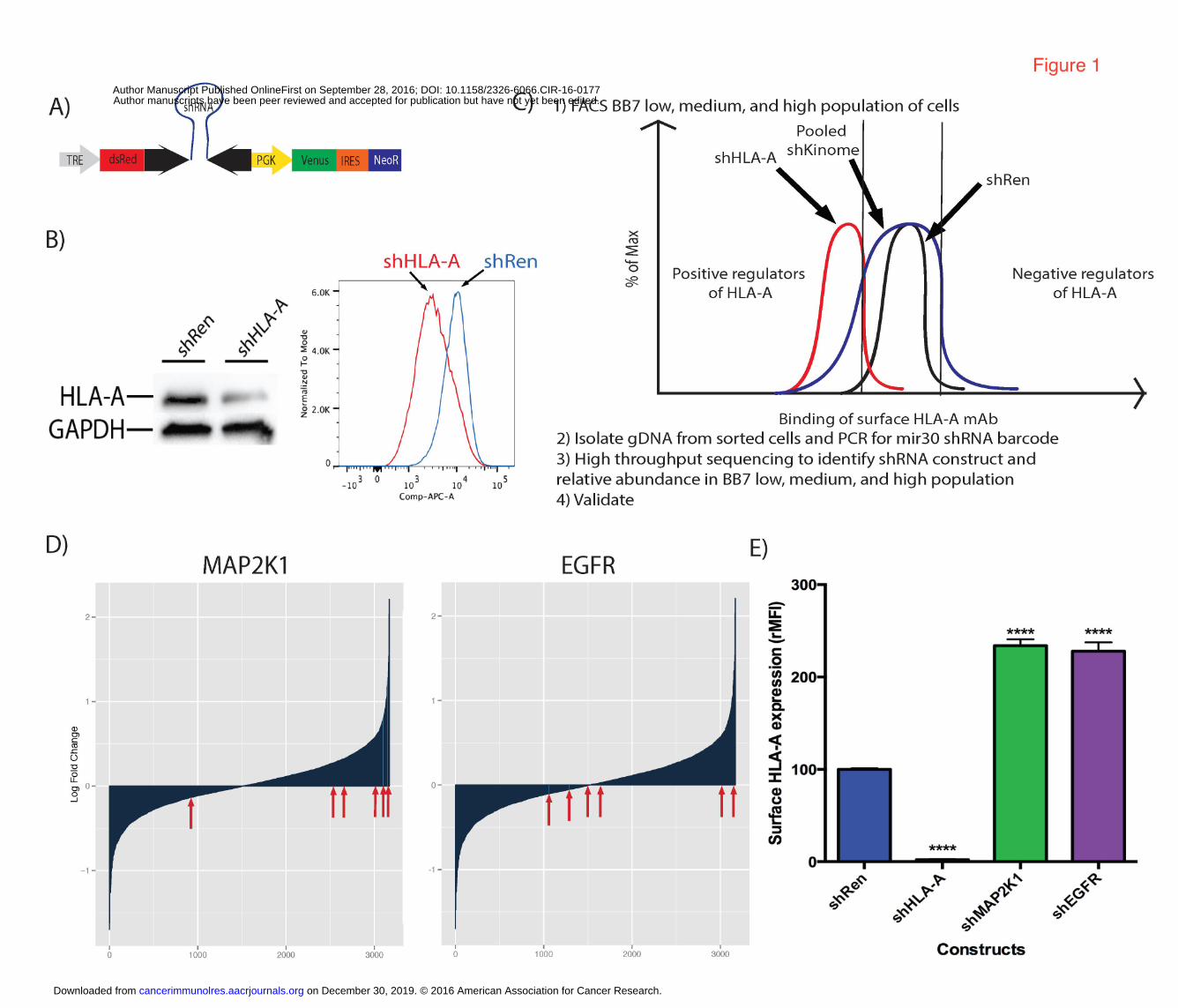
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177

Published OnlineFirst September 28, 2016.Cancer Immunol Res Elliott J Brea, Claire Y Oh, Eusebio Manchado, et al. on Cancer CellsKinase regulation of Human MHC Class I Molecule Expression
Updated version
10.1158/2326-6066.CIR-16-0177doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerimmunolres.aacrjournals.org/content/suppl/2016/09/28/2326-6066.CIR-16-0177.DC1
Access the most recent supplemental material at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerimmunolres.aacrjournals.org/content/early/2016/09/28/2326-6066.CIR-16-0177To request permission to re-use all or part of this article, use this link
on December 30, 2019. © 2016 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on September 28, 2016; DOI: 10.1158/2326-6066.CIR-16-0177









![MANUAL DE USUARIO MÁQUINAS DE HIELO...MANUAL DE USUARIO [AUTOCONTENIDAS Y REMOTAS ] MHC-230/506MA - MHC-235/517MA - MHC-280/625MA - MHC-320/706MA MHC-500/1109MAR - MHC-680/1466MAR](https://img.dokumen.tips/doc/110x75/5e93db5530a5a625c35ecff2/manual-de-usuario-mquinas-de-hielo-manual-de-usuario-autocontenidas-y-remotas.jpg)



















