Embed Size (px)
Citation preview

⎯ Tudományos Diákköri Konferencia ⎯
ROVÓ PETRA
Ismeretlen ónorganikus molekulák intramolekuláris kölcsönhatásának
jellemzése ismert molekulák szerkezete alapján
Témavezető: Sztáray Bálint (adjunktus)
Készült az Eötvös Loránd Tudományegyetem Általános Kémia Tanszékén
⎯ Budapest, 2005 ⎯

Tartalomjegyzék
1. Bevezetés............................................................................................................................ 3
2. Irodalmi előzmények.......................................................................................................... 5
2.1. Ónvegyületek jellemzése............................................................................................ 5
2.2. Oligomer struktúrák ................................................................................................... 6
2.3. Geometriai kontrol ..................................................................................................... 7
2.4. Kémiai affinitás ........................................................................................................ 10
2.5. Germánium-, óntartalmú heterociklusok.................................................................. 11
3. Eredmények és értelmezésük ........................................................................................... 12
3.1. Célkitűzés: Szerkezet-előrejelzés............................................................................. 12
3.2. Ónorganikus vegyületek........................................................................................... 13
3.2.1. A fenil → metil szubsztitúció........................................................................... 13
3.2.2. Az alkil → halogenid szubsztitúció.................................................................. 16
3.3. Germánium-organikus vegyületek ........................................................................... 18
3.3.1. Kémiai affinitás ................................................................................................ 19
3.4. Konkrét példák a szerkezet előrejelzésére ............................................................... 20
4. Összefoglalás.................................................................................................................... 25
5. Köszönetnyilvánítás ......................................................................................................... 26
6. Irodalomjegyzék............................................................................................................... 27
2

1. Bevezetés
Az intra- és intermolekuláris kölcsönhatások vizsgálata, és a molekulaszerkezet kutatása
egyre nagyobb hangsúlyt kap a tudományban.[1-4] Ezen belül a molekulák közti, és a
molekulán belül kialakuló másodlagos kötések utalnak az elemek közti elektron-affinitására.
A negyedik-hatodik főcsoport elemei közül a szilícium-oxigén másodlagos kölcsönhatásáról
az irodalomban részletes leírás található [5-8]. Ellenben keveset tudunk a 14. csoport
fémjeinek oxigén, illetve kalkogén kölcsönhatásáról [9-13]. Jelen dolgozatban részletesebben
foglalkozom a karboxil, tiokarboxil, xantát, ón és germánium észterek, ill. a heterociklusos
vegyületek másodlagos kölcsönhatásainak kialakulási körülményeivel, a kölcsönhatás
jellegével, valamint az ezekből a molekulákból származtatható vegyületek szerkezetének
előrejelzésével.
Az ónorganikus vegyületek kutatása igen nagy érdeklődésre tart számot, részben
biológiai aktivitásuk [14-16], másrészt ipari felhasználásuk miatt: hatékony stabilizátorok,
katalizátorok, vulkanizálószerek, biocidok, sőt, fakonzerválószerként is használhatóak [17-
19], de újabban ónorganikus reagenseket növekvő mértékben kezdenek alkalmazni szerves
szintéziseknél is [20-22]. Ezen vegyületek különleges szerkezete és reaktivitása általában az
úgynevezett hiperkötésnek, más szóval a hiperkoordinációnak köszönhető [23-29]. Az ón
hiperkoordinációjával először részletesebben Williem et al. [28-29] foglalkoztak,
munkájukban az OH ···· Sn kölcsönhatást részletesen ők írták le először. A kötés természetét
behatóan vizsgálták az ónorganikus centrum Lewis-sav erősségének függvényében.
Az eddig ismeretlen – még nem szintetizált – molekulák szerkezetének előrejelzésére
manapság a legelterjedtebb módszer a kvantumkémiai számítások alkalmazása. Ez azonban
sok esetben nem nyújt kellően hű képet az ismeretlen molekula szerkezetéről, mivel a
számítás általában gáz fázisú, izolált molekulára vonatkozik, noha gyakran a kristályos
molekula szerkezetére vagyunk kíváncsiak. További hátránya a módszernek, hogy nagyobb
molekulák esetén meglehetősen költséges lehet, valamint gyakran nem nyújt szemléletes
képet a molekula szerkezetét meghatározó kölcsönhatásokról, ezért is célszerű néhány esetben
alternatív megoldásokhoz fordulni. Egy ilyen eljárást szeretnék dolgozatomban vázolni, és
gyakorlati alkalmazására néhány példát hozni.
Munkámban a Cambridge Krisztallográfiai Adatbázisban (CCDC) fellelhető O, S ····
Ge, Sn másodlagos kölcsönhatás kialakulására képes vegyületeinek szerkezeti sajátosságait
3

hasonlítom össze, és úgy csoportosítom, hogy egy-egy szerkezetelem megváltozásának hatása
nyomon követhető legyen.
4

2. Irodalmi előzmények
2.1. Ónvegyületek jellemzése
Az ónt valószínűleg egyetlen más fém sem múlja felül fémorganikus vegyületei
alkalmazásának sokrétűségében. Az első ónorganikus vegyületet 1849-ben állították elő, de
nagymértékű alkalmazására csak a közelmúltban került sor. Szemléletesen mutatják ezt a
világtermelésre vonatkozó adatok, miszerint 1950 és 1980 között több mint 700-szorosára
nőtt az ónorganikus vegyületek termelése.
Az ón-oxigén oligomerek nagyarányú ipari felhasználása miatt döntő jelentőségű, hogy
előre pontosan meg lehessen határozni a reakcióterméket. Mivel számos ónorganikus vegyület
reakciójának mechanizmusa máig nem ismert, ezért a termék keletkezése gyakran
kiszámíthatatlan. Ebből, valamint az ipari felhasználásból következően fontossá vált a
reakciómechanizmusok, és a reaktáns anyagok tulajdonságainak minél részletesebb ismerete,
többek között az ónra ható intramolekuláris kölcsönhatások befolyásának vizsgálata.
Az ónorganikus vegyületeket aszerint csoportosíthatjuk, hogy hány kovalens kötésben
vesznek részt. Mivel az ón a negyedik főcsoport eleme, így a legtöbbször négy kovalens
kötést hoz létre. Erre a legegyszerűbb példa a tetrametil-ón, vagy a tetrafenil-ón. Ezek
szerkezete tökéletesen tetraéderes, a benne levő kovalens kötéshosszak megfelelnek az Sn-C
kovalensrádiuszösszegnek.
A tetrakoordinált-ónorganikus vegyületek több érdekes sajátságot mutatnak, a bennük
fellépő X····Sn gyenge másodlagos kölcsönhatásnak köszönhetően – ilyen például a
regióspecifikusan lejátszódó hidrolízis [11, 31], vagy a nagymértékű láncképző hajlam [9].
Általános jellemzője ezeknek a vegyületeknek, hogy az intermolekuláris donoratom – és
ezáltal a koordináló csoport – a központi fématom geometriai kontrolja alatt áll, hozzá egy
koordinatív kötéssel kapcsolódik, így az ónatom koordinációs száma ötre, hatra, sőt hétre
növekszik, ennek megfelelően ezeket a vegyületeket penta-, hexa-, vagy heptakoordinált
molekuláknak nevezzük. A hiperkoordináció következtében a tertaéderes ónorganikus
vegyület tetraéderes szerkezete trigonális bipiramisossá torzul, és az Sn-C kovalens
kötéshosszak valamelyest növekednek. Ennek a hiperkoordinációnak a következményeként
értelmezhető az is, ha a vegyület nem alkot oligomer láncot, hanem különálló molekulákként
van jelen a kristályban, ekkor molekulán belüli, nem pedig molekulák közötti
hiperkoordinációs kötés jön létre [9].
5

2.2. Oligomer struktúrák
Az ónorganikus észterek láncképző hajlamával Smith és munkatársai [32-34]
foglalkoztak. A korábbi megfigyeléseik szerint az ón atomra vonatkozólag trigononális
bipiramisos transz-R3SnX2 (A) és cisz-R3SnX2 (B) geometria alakul ki; az előbbiben a
karbonil oxigén intermolekuláris kötéssel kapcsolódik a következő ónatomhoz, míg az
utóbbiban intramolekuláris kötés jön létre ugyanezen két atom között. Ezt szemlélteti az
alábbi ábra.
1. ábra Oligomer és diszkrét geometria
Megállapításaik szerint az oligomer lánc kialakulásában trimetilón-észterek vesznek
részt, úgymint trimetilón-acetát, vagy trimetil-ónglicinát. Ez utóbbi molekulában azonban
nem az acil-oxigén, hanem a nitrogén atom viselkedik intermolekuláris hídként.
Feltételezésük szerint azért, mert az NH2 csoport és a karboxil csoport oxigénje között
intermolekuláris H-híd jön létre, minek következtében a nitrogénnek megnő az elektron
donori jellege, így az intermolekuláris kötésben könnyebben vesz részt, mint a H-híd
donorjaként meggyengült donori tulajdonságú oxigén.
2. ábra Intermolekuláris N-kötés
6

A H-hidak vizsgálata érdekében 119mSn Mössbauer felvételt készítettek a trimetil-ón-
szalicilsavról 1 (DOZDEB) és ennek metoxi-származékáról 2, valamint a trimetilón-o-
aminobenzoesavról 3 és ennek dimetilamino-származékáról 4, illetve hasonló molekulákról
(majd a későbbiekben a trifenil analógokról is (lásd: CUNJOK, CUNJUQ CUNJAW,
CUNJEA [33]). Mind a négy lánc-polimer struktúrájú, de míg a 2 és 4 molekula a karboxil-
oxigénjén keresztül képez láncot, a szalicilát 1 molekulában a hidroxil csoport a karboxil
csoporttal hidrogénhíd-kötésben van, ezáltal a hidroxil-oxigén vesz részt az intermolekuláris
kötésben. Hasonló a struktúra az aminoszármazéknál 3, itt a nitrogénen keresztül létesül a
molekulák közötti kölcsönhatás. Az említett H-híd-kötést tartalmazó molekulákban az Sn····O
másodlagos kötés hosszabb, mint azokban, amelyekben nincs lehetőség a hidrogénkötésre –
ez a megállapítás összefügg az eddigi ismeretekkel. Ez utóbbi molekulánál kimutatható
azonban egy gyenge inramolekuláris kölcsönhatás is a központi fématom és az acil-oxigén
között, a kötéshossz értéke 2,98 Å – 3,11 Å közötti (van der Waals rádiuszösszeg 3,58 Å
[35]).
Összefoglalva tehát munkáikban külön-külön megvizsgálták a trimetil, és trifenilón
észterekben a H-híd kötés szerepét mind a donor, mind az akceptor jelleg szempontjából,
azonban nem tértek ki arra, hogy milyen különbsége van az inter- (és intra-) molekuláris
Sn····X kölcsönhatás erősségében ugyanazon vegyület trimetil és trifenil-származékában. A
metil → fenil ligandumcserével ezért a későbbiekben részletesebben foglalkozom.
2.3. Geometriai kontrol
Az ónorganikus vegyületek sokszínű geometriája nemcsak a különböző láncformákban,
hanem a diszkrét molekuláknál is megfigyelhető. A kétfogú diszubsztituált ónorganikus
vegyületek családjának részletes geometriai analízisével Mohamed-Ibrahim és munkatársai
foglakoztak [37-42]. Összesen négy különböző geometriai mintázatot figyeltek meg
diorganikus-ónbisz(xantát) molekulákban.
7
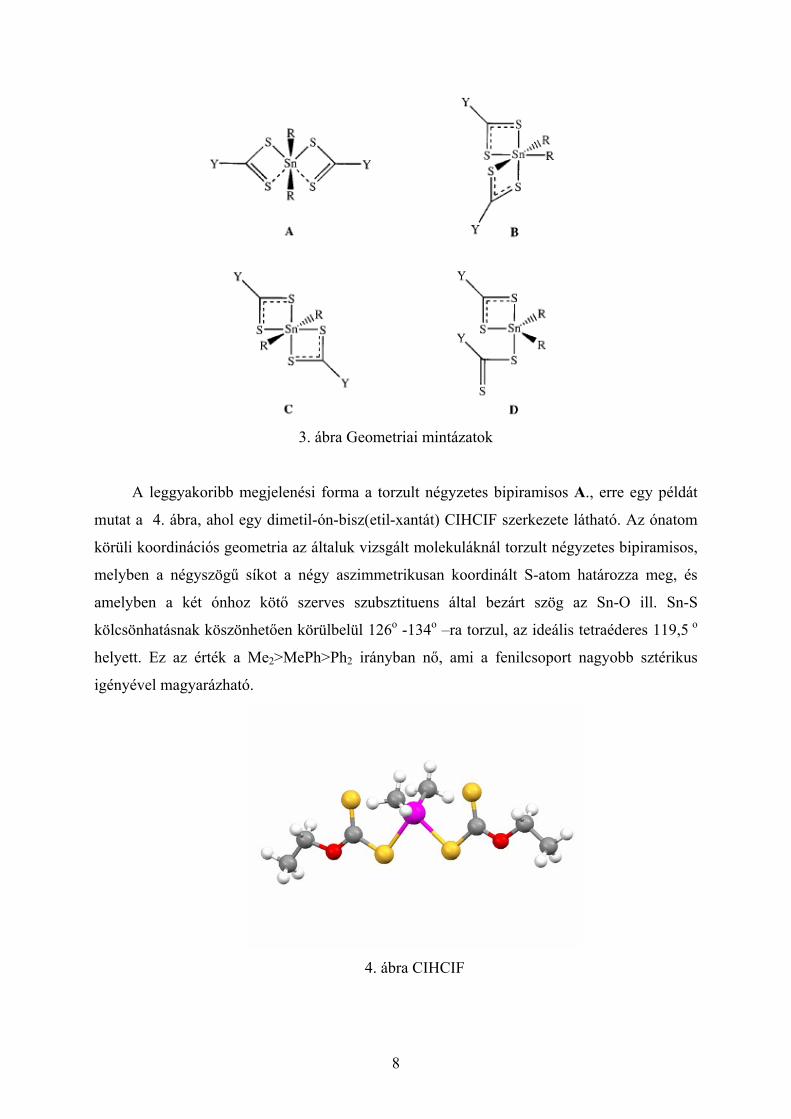
3. ábra Geometriai mintázatok
A leggyakoribb megjelenési forma a torzult négyzetes bipiramisos A., erre egy példát
mutat a 4. ábra, ahol egy dimetil-ón-bisz(etil-xantát) CIHCIF szerkezete látható. Az ónatom
körüli koordinációs geometria az általuk vizsgált molekuláknál torzult négyzetes bipiramisos,
melyben a négyszögű síkot a négy aszimmetrikusan koordinált S-atom határozza meg, és
amelyben a két ónhoz kötő szerves szubsztituens által bezárt szög az Sn-O ill. Sn-S
kölcsönhatásnak köszönhetően körülbelül 126o -134o –ra torzul, az ideális tetraéderes 119,5 o
helyett. Ez az érték a Me2>MePh>Ph2 irányban nő, ami a fenilcsoport nagyobb sztérikus
igényével magyarázható.
4. ábra CIHCIF
8

Ettől eltérő geometriai mintázata van a kisebb koordinációs számú, a központi atomhoz
egy-foggal kötő ditiokarbamát származékoknak D. Amennyiben nincs mód a központi
fématom körül további koordinációra, akkor ezeknek a molekuláknak a szerkezete torzult
trigonális bipiramisos.
Kevésbé gyakori az oktaéderes szerkezet, ilyenkor a szerves csoport cisz B, vagy transz
C állású. Általában cisz geometriát öltenek a dihalogén ón-bisxantát vegyületek is.
Feltételezésük szerint a különböző geometriai struktúrák kialakulásának oka az
elektronszerkezeti, a sztérikus ill. a kristálybeli illeszkedési (packing) hatással van
kapcsolatban, ennek érdekében ab initio és sűrűségfunkcionál (DFT) számításokat és
sűrűségfüggvény analízist végeztek 3-21G** bázison. A számolt gázfázisú és a mért
szilárdfázisú geometria közötti különbség az intermolekuláris kölcsönhatásokra vezethető
vissza. Megfigyelésük szerint a gyakran mellőzött szilárdfázis hatás igenis szignifikáns
szerepet játszik a koordinációs geometria, és a kötéshosszak kialakulásában. Például sok
aszimmetrikus geometriájúnak mért molekula szerkezete gáz fázisban mindig
szimmetrikusnak adódott.
(Többek között ezért is kell nagyon odafigyelnünk arra, hogy mikor mit mivel
hasonlítunk össze a munkánk során, mert már ugyanannak a molekulának a szerkezete
jelentős eltérést mutat gázfázisban a szilárdfázisú szerkezethez képest. Különösen igaz ez,
amikor másodlagos effektusokat vizsgálunk.)
Munkájukban a dimetil- (ACELID); fenil,metil- (ACELOJ) illetve a difenil–
ónbisz(metilxantát) (ACELUP) molekulák szerkezetét vizsgálták a geometria torzulása
szempontjából, valamint a mért adatokat hasonlították össze a számított paraméterekkel.
Emellett megemlítik – mint egy példát a geometriai diverzitásra, hogy az eddig publikált
dimetil, difenil-ónxantátok mindegyike két S,S-kelátgyűrűt tartalmaz, viszont az általuk
szintetizált dimetil-ónxantátban (ACELID) egy S,O-kelátgyűrű is van, az Sn-S(2) kötés körüli
rotáció következtében – ez a gyűrű az 5. ábrán is megfigyelhető.
9

5. ábra ACELID
Nem tértek ki azonban az általuk felrajzolt különböző geometriai formák (A, B, C, D)
közötti eltérések elemzésére, arra például, hogy milyen mértékű és jellegű változással jár egy
metil → halogenid szubsztitúció. A későbbiekben ezzel én részletesebben foglalkozom, és
összehasonlítom más alkil → halogenid ligandumcserével is.
2.4. Kémiai affinitás
A 14. csoport fématomjainak kémiai affinitásával többek között Kato és kollégái
foglalkoztak [43-47]. Munkájuk során trifenil-germánium, -ón, -ólom ditiokarboxilátokat
szintetizáltak, és az ezekben fellépő fém-kén másodlagos kötéserősség változásából
következtettek a fémek affinitására. Vizsgálataik szerint a van der Waals rádiuszösszeghez
képest a M····S másodlagos kötéstávolság csökkenésének aránya az ón esetén volt a
legnagyobb; Sn (19%) > Pb (12%), ≥ Ge (11%), vagyis relatíve az ón hoz létre legerősebb
másodlagos kötést, ahogy az elektron affinitás a 14. csoport fémjei közt Sn>Pb>Ge irányba
csökken. Ugyanez a kutatócsoport előállította a trifenil-ón, -germánium, -ólom-metilbenzol-
tiokarboxilátokat is, amely molekulákban észlelték a fém-oxigén másodlagos kötést, azonban
ekkor nem tértek ki a fématom elektronaffinitására, vagy ezek ditiokarboxilszármazékaival
történő összehasonlítására [44]. Részletesebben a számolt pályaenergiákat hasonlították össze,
és a számolt eredmények szerint a nO→σ*MS pályák átfedésének van nagy szerepe a kialakuló
rövid M-O kötésben.
Az ismert szerkezeti adatok alapján összehasonlítom a germánium és az ón, oxigén- és
kén-affinitását.
10

2.5. Germánium-, óntartalmú heterociklusok
Ilyen heterociklusos fémorganikus vegyületeket hosszű idővel ezelőtt szintetizáltak már.
A X2M-1,3,6-tritio-2-sztannaocének (X=R, Aril, Cl), és ezek germánium ill. oxo
származékainak vizsgálatával M. Dräger foglalkozott 1975-től kezdve [47-52]. Ezekben a
molekulákban a nyolctagú gyűrűben fellépő fém-oxigén, fém-kén másodlagos kötés miatt híd
alakul ki a gyűrűben, ami meghatározza a molekula geometriáját – egy ilyen szerkezetet
mutat be a 6. ábra.
6. ábra YIFLUU
A szerkezetvizsgálat során megállapították, hogy az erős intermolekuláris kölcsönhatás
nemcsak a rövid másodlagos kötéshosszban, hanem a geometria torzulásában is
megmutatkozik, a kötés erősödésével a szék-kád konformáció szék-székké ill. korona alakúvá
torzul. A konformáció változást tanulmányozta az akceptor és a donor atom függvényében,
valamint a fémhez kötő ligandumok szerint, azonban munkája során végig külön kezelte az
ónorgaikus és a germánium-organikus vegyületeket, azok szerkezeti adatait nem vetette össze.
Ezért jelen munkában az ón-germánium vegyületek szerkezetének összehasonlításával
részletesen foglakozom, különösen olyan szempontból, hogy milyen összefüggés van az
észterekben végbemenő ón → germánium csere, és a heterociklusos rendszerben lejátszódó
fém csere között. Ennek analógiájára összevethetőek ezen vegyületcsaládok a metil → fenil
szubsztitúció vagy a halogénszubsztitúció szempontjából is. Ezekre a továbbiakban hozok
egy-egy példát.
11

3. Eredmények és értelmezésük
3.1. Célkitűzés: Szerkezet-előrejelzés
A Cambridge-i Krisztallográfiai adatbázis hivatott az eddig szintetizált és röntgen
diffrakciós módszerrel megmért molekulák geometriai adatainak tárolására. Az adatbázist
minden évben frissítik, így mára már összesen sok tízezer (több százezer) molekula szerkezeti
paraméterei, és kristálytani besorolása található meg benne. Ennek az információáradatnak
köszönhetően lehetőségünk van arra, hogy a molekulák között párhuzamokat vonjunk, és
származtatási láncokat, hálókat építsünk fel közöttük. Ezek a hálóképek megmutatják, hogy
az adott molekula, mely más molekulákból származtathatót, illetve ebből még melyek
vezethetőek le egyszerűen, például egy ligandumcserével, emellett felderíthető az is, hogy
mennyire feltérképezett a vizsgált molekulacsalád – sok-e a „hiányzó láncszem”, van-e elég
alternatíva ahhoz, hogy az egyik szerkezetből a másikhoz jussunk stb.
Annak köszönhetően, hogy az ónorganikus vegyületek használata ennyire elterjedt az
iparban, sokan szintetizálták ezeket, így az adatbázisban mintegy 2900 db molekula található,
amely már kellő mennyiségű információ egy szerkezeti gráf felrajzolásához.
Megpróbálkoztam két egyszerűbb háló összeállításával: az egyikben – amelyen az
ónorganikus észterek vannak feltüntetve – összesen 30 ismert molekulaszerkezet van
felrajzolva, a másikban – amely nem észteri, de arra igen hasonló rendszert, egy
heterociklusos sztannaocán-germocán rendszert ábrázol – ennél ugyan kevesebb, mindössze
15 ismert molekulaszerkezet látható, de azok egy jóval összetettebb rendszerben vannak –
azaz jól feltérképezett a család. A második hálóban feltüntettem három ismeretlen molekulát,
amelyeket egy-egy kérdőjellel jelöltem. Ezek közül az Ismeretlen 1, és Ismeretlen 2
elnevezésű molekulákat a későbbiekben meghatározom. Az első szerkezet-gráf nem teljes –
nem tüntettem fel rajta minden általam vizsgált ónorganikus észtert, illetve a germánium
organikusokat sem –, ez csupán a váza a hálózatnak. A második ellenben teljesen
következetes, a hiányzó láncszemeket is feltüntettem.
A molekulaháló felhasználása többrétű lehet – egyik legfontosabb ezek közül talán a
szerkezet-előrejelzés. Amennyiben ismerjük több rokon vegyület szerkezeti paramétereit, az
atomi koordinátáikat – vagyis elég kompakt a hálónk –, akkor ezek összehasonlításával
konkrétan kiszámíthatjuk egyes szerkezetelemek változásának hatását, illetve
következtethetünk a kötésben résztvevő atomok elektronvonzó vagy elektronküldő
tulajdonságára. Ezek ismeretében gyakran előre jelezhető egy még nem ismert – de az
12

előzőekből egyszerűen származtatható – molekula szerkezete, így az abban levő
intramolekuláris kölcsönhatás erőssége is. Az általam vizsgált molekulák mindegyikében van
egy vagy több intramolekuláris fém-oxigén vagy fém-kén kötés. Részletesebben ennek a
másodlagos kölcsönhatásnak a változásával foglalkozom, és ezen a példán keresztül
bemutatom, hogyan és mire lehet egyszerűen felhasználni ezt az elsőnek bonyolultnak tűnő
hálót – természetesen ennek analógiájára más paraméter változása is nyomon követhető.
3.2. Ónorganikus vegyületek
3.2.1. A fenil → metil szubsztitúció
A szerkezeti hálóban kék vonallal kötöttem össze azokat a molekulapárokat, amelyek
egymás fenil → metil szubsztitúciójával nyerhetőek. Ezen cserék mindegyike a központi
fématomon játszódik le, ezek közül három párnál monoszubsztitúció (ACELUP→ACELOJ;
ACELOJ→ACELID; DELXAT→YIFLEE), másik négynél diszubsztitúció
(PEJFIT→CIHCIF; ETCPSN→ETCMSN; XAVYUO→COLQEZ; PHTHSN→YIFLUU) és
egy párnál triszubsztitúció játszódik le (CUNJOK→DOZDEB). A fentebb említett
egyedülálló S,O kelátgyűrű miatt az ACELUP→ACELOJ összehasonlítás csak akkor lenne
elvégezhető, ha ismernénk az előbbi molekulának azt a rotációs izomerjét, amelyben már két
db S,S-kelátgyűrű található. Ez sajnos nem ismert.
A fenil→metil ligandumcsere másodlagos kötéshossz módosító hatását
tanulmányozhatjuk a 7. ábra két molekulájában (CUNJOK→DOZDEB). Az ábrázolt
példában az Sn····O kötéshossz változás -0,041 Å-nek adódott, vagyis a trifenil-szubsztituált
ónorganikus vegyületben a másodlagos kötéshossz hosszabb, mint a trimetil-származékban.
A kötéshossz-módosulásról összességében a 1. táblázat tájékoztat.
13
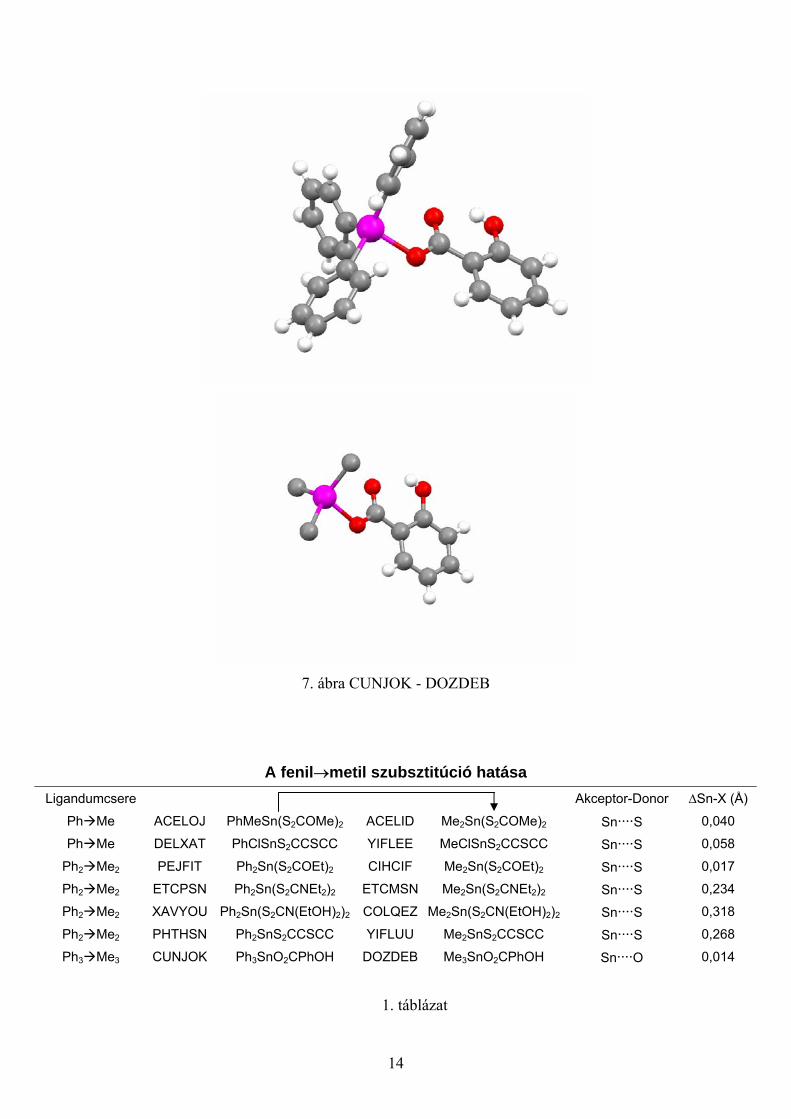
7. ábra CUNJOK - DOZDEB
A fenil→metil szubsztitúció hatása Ligandumcsere Akceptor-Donor ∆Sn-X (Å)
Ph Me ACELOJ PhMeSn(S2COMe)2 ACELID Me2Sn(S2COMe)2 Sn····S 0,040
Ph Me DELXAT PhClSnS2CCSCC YIFLEE MeClSnS2CCSCC Sn····S 0,058
Ph2 Me2 PEJFIT Ph2Sn(S2COEt)2 CIHCIF Me2Sn(S2COEt)2 Sn····S 0,017
Ph2 Me2 ETCPSN Ph2Sn(S2CNEt2)2 ETCMSN Me2Sn(S2CNEt2)2 Sn····S 0,234
Ph2 Me2 XAVYOU Ph2Sn(S2CN(EtOH)2)2 COLQEZ Me2Sn(S2CN(EtOH)2)2 Sn····S 0,318
Ph2 Me2 PHTHSN Ph2SnS2CCSCC YIFLUU Me2SnS2CCSCC Sn····S 0,268
Ph3 Me3 CUNJOK Ph3SnO2CPhOH DOZDEB Me3SnO2CPhOH Sn····O 0,014
1. táblázat
14

A különféle eseteket szemlélve megállapíthatjuk, hogy szubsztitúció hatására
mindegyik esetben megnőtt a vizsgált intramolekuláris kötés hossza, ami a központi fématom
Lewis-aciditásának csökkenésével magyarázható. A fenil → metil ligandumcserének azonban
eltérő nagyságú befolyása van az intramolekuláris kölcsönhatásra. Míg a már említett
szalicilsav-észter származékban (CUNJOK) és a xantát-vegyületekben (ACELOJ, PEJFIT)
nem mutatott nagymértékű Sn····X kötésmódosulást a ligandumcsere, addig a karbamát
vegyületeknél 0,23 – 0,32 Å (!) változás figyelhető meg. Hasonlóan nagy az intramolekuláris
kötéshossz-különbség a heterociklusos tritio-sztannaocánoknál is (PHTHSN). A feltűnően
nagy kötéshossz módoulás az első két esetben a geometriai változásnak a következménye. A
dimetilón-xantát (PEJFIT) és dimetilón-karbamát (ETCMSN) molekulák geometriája,
szerkezete is igen hasonló – négyzetes bipiramisos, a difenil származékok geometriája
azonban eltérő, a xantátoké négyzetes bipiramisos, tehát nem vátozik, míg a karbamátoké
oktaéderes, cisz állású fenilcsoportokkal. A molekuláris felépítésükben az egyedüli különbség
a ditiokarboxil-csoporthoz kötő atom anyagiminőségében van: a xantát molekulában etoxid
(OEt) csoport köt, a karbamátoknál pedig dietilamin NEt2. Ha részletesebben megnézzük a
karbamátok szerkezetét, akkor feltűnik, hogy a nitrogén planáris, és egy síkban van az egyik
fenil csoporttal. Feltételezhetően a nitrogén π pályája és a fenil csoport π rendszere között
átfedés jön létre ilyenkor, amely stabilizálja a molekulát.
A heterociklusos tritio-sztannaocának kötéshossz módosulására két példát hoztam, az
egyik esetben fenil,klór → metil,klór (DELXAT→YIFLEE) rendszerben figyelhetjük meg a
kötésváltozást, a másik esetben difenil → dimetil (PHTHSN→YIFLUU) szubsztitúció megy
végbe. Az ónhoz kötő klór miatt az ón Lewis- aciditása jelentősen megnő, így a Sn····S
koordinációs kötés igen erős (hossza 2,805 Å), ez a kötéshossz valamelyest gyengül a metil-
szubsztitúció révén, de a kötéshosszra nem hat olyan nagymértékben (hossza 2,863 Å), azt
leginkább a klorid ligandum elektronszívó tulajdonsága határozza meg. A dialkil
ligandumcsere esetén a másodlagos kötés gyenge – a difenil-származéknál 3,246 Å, azaz több
mint 0,4 Å-mel hosszabb, mint a klorid vegyület kötése –, a metil szubsztituensnél ez a kötés
tovább gyengül a központi atom Lewis-aciditásának csökkenése miatt. Ebben az esetben nem
lépett fel szerkezetmódosulás.
15

3.2.2. Az alkil → halogenid szubsztitúció
A többi diorganikus, dihalogén ón vegyületet megvizsgálva feltűnt, hogy mindegyik
esetben az ón atom körüli koordináció halogén szubsztituens esetében oktaéderes, és a
halogén atomok cisz állásúak, metil szubsztituensnél pedig négyszögalalpú bipiramis
konformációt tölt be. Azaz az alkil → halogén ligandumcserét követő konformációs
átalakulás nemcsak a xantát származékok jellemzője [37], hanem minden diszubsztituált –
hexakoordinált – ónorganikus vegyületben megfigyelhető. A heterociklusos sztannaocánok
esetén is fellép egy kis mértékű konformáció változás, ilyenkor szék-kád formából korona
alakú lesz a molekula.
Az alkil → halogenid ligandumcserék a szerkezeti gráfon piros vonallal vannak
feltüntetve.
Az említett konformáció-változást szemlélteti a 8. ábrán bemutatott két molekula, ahol a
diklór-ón-ditioetilkarbamát WIBTEG és a dimetil-ón-ditioetilkarbamát ETCMSN
molekulapár szerkezetének különbsége jól tanulmányozható. Ez, illetve másik két különböző
vegyület metil → klór cseréjének szerkezetmódosító hatását követhetjük nyomon a 2.
táblázatban.
8. ábra WIBTEG és ETCMSN
(A hidrogén atomokat – a tisztább szerkezeti kép érdekében – mellőztem)
16

Kiválasztott interatomi paraméterek (Å, fok)
CIHCIF CIHDEC ETCMSN WIBTEG YORJUK RAXWOC Me2Sn(S2COEt)2 Cl2Sn(S2COEt)2 Me2Sn(S2CNEt2)2 Cl2Sn(S2CNEt2)2 Me2Sn(SOCPh)2 Cl2Sn(SOCPh)2
X=Me X=Cl X=Me X=Cl X=Me X=Cl Y=S Y=S Y=S Y=S Y=O Y=O
Sn-S(1) 2,486 2,500 2,517 2,503 2,488 2,468 Sn-Y(1) 3,088 2,623 2,961 2,572 2,689 2,268 Sn-S(2) 2,501 2,518 2,528 2,511 2,488 2,468 Sn-Y(2) 3,151 2,554 2,916 2,572 2,689 2,268
X(1)-Sn-X(2) 130,09 93,80 142,62 91,86 123,21 98,23 S(1)-Sn-S(2) 83,52 160,56 83,29 160,38 89,72 146,71 Y(1)-Sn-Y(2) 150,31 87,57 147,73 90,18 150,60 83,35 X(1)-Sn-S(1) 108,33 99,86 108,04 89,68 111,07 101,16 X(1)-Sn-S(2) 108,35 94,26 101,79 103,33 111,07 100,44
2. táblázat
Az első pár a már említett dimetil-ón-bisz(dietilxantát) CIHCIF és diklór-ón-
bisz(dietilxantát) CIHDEC, a második két molekula ón-bisz(dietil,ditiokarbamát) metil
ETCMSN illetve klorid származéka WIBTEG, a harmadik pár pedig dimetil- YORJUK ill.
diklór-ón-bisz(S-monotiobenzoát) RAXWOC. Azaz az első két párnál az Sn····S másodlagos
kötés, az utolsó párnál pedig az Sn····O kölcsönhatás gyengülése vizsgálható.
Mindhárom molekulapárnál azonos jellegű a szerkezetváltozás, és a mértéke is igen
hasonló, mind az adott kötésszögek változását, mind a hiperkoordinációban résztvevő oxigén,
kén másodlagos kötés távolságának növekedését tekintve. A konformáció-átalakulásban a
kovalens kötéshosszak számottevően nem változnak. Meg kell említeni azonban, hogy ezek a
kötéshosszak a normális Sn-O, Sn-S kötésnél a hiperkoordináció következtében átlagosan
0,05 Å-mel hosszabbak [36]. A másodlagos kötéshosszak viszont szignifikáns csökkenést
mutatnak a metil→klorid szubsztitúció hatásaként – részben a konformáció módosulás,
részben az ón növekvő Lewis-aciditásának növekedése következtében – átlagos változásuk az
alábbiak szerint alakul: ∆Sn-S CIHCIF-CIHDEC: -0,531 Å; ∆Sn-S ETCMSN-WIBTEG: -0,367 Å; ∆Sn-O
YORJUK-RAXWOC: -0,421 Å. Ezek az általam – az ón-észter családban – megfigyelt legnagyobb
kötéshossz-csökkenések. A halogén-szubsztituált vegyületekben fellépő Sn····S hiperkötés
hossza igen rövid, összemérhető – szinte azonos – a kovalens kötéssel kötő Sn-S távolságával,
annál átlagosan mindössze 0,07 Å – 0,08 Å-mel hosszabbak, míg metil ligandum esetén a
(Sn-S(1)) – (Sn-S(3)) különbség 0,42 Å – 0,63 Å körül alakul. Ugyanez elmondható a S-
17

monotiobenzoát hiperkoordináló oxigénjének kötéstávolságairól, csak ott ez a rövidülés nem
ennyire feltűnő.
A heterociklusos sztannaocánok esetén feltűnően nagy a dimetil → diklorid
szubsztitúciót követő Sn····S másodlagos kötéshossz-rövidülés. A dimetil-származékban
(YIFLUU) az Sn-S másodlagos kötéstávolság 3,514 Å, a diklorid vegyületben ez
mindösszesen 2,760 Å (CLTHSN), tehát a különbség 0,754 Å (!). Ez a változás egyedül a
Lewis-aciditás változásának köszönhető, a koordináló ként nem befolyásolja a környezete, a
molekula geometriáját legfőképp az Sn-S másodlagos kötés határozza meg. Az említett két
molekula szerkezetének változása az alábbi ábrán tanulmányozható.
9. ábra CLTHSN és YIFLUU
(A hidrogén atomokat – a tisztább szerkezeti kép érdekében – mellőztem)
3.3. Germánium-organikus vegyületek
Az eddigiek során megvizsgáltam az ónorganikus észterekben a másodlagos
kötéstávolság alakulását a koordináló csoport és az akceptor atomhoz kötődő ligandumok
ismeretében. A továbbiakban általánosítom az eddigi megállapításokat a germánium-
18

organikus vegyületekre is, illetve bemutatom a két fématom elektronvonzó tulajdonsága közti
különbséget.
3.3.1. Kémiai affinitás
10. ábra XICZUE
Kiválasztott interatomi paraméterek (Å)
XICZUE XATSIU XIDBAN XATSOA Ph3GeSOPhMe Ph3GeS2PhMe Ph3SnSOPhMe Ph3SnS2PhMe X = S X = S X = S X = S Y = O Y = S Y = O Y = S M-Y 3,003 3,371 2,907 3,207 M-X 2,255 2,253 2,445 2,446 M-C(1) 1,942 1,948 2,142 2,125 M-C(2) 1,944 1,943 2,131 2,137 M-C(3) 1,935 1,933 2,123 2,146
3. táblázat
Ebben a példasorban az intramolekuláris kölcsönhatás erősségét az adott molekulán
belüli donor és akceptor atomok anyagi minőségének függvényében vizsgáltam meg, ugyanis
ismert mind a trifenil-germánium-(4-metilbezotioát) (XICZUE) a trifenil-germánium-(4-
metilditiobenzoát) (XATSIU) és ezek ónszármazékainak (XIDBAN, XATSOA) térszerkezete.
Ezek a molekulák egymástól csak a kölcsönhatásban résztvevő donor és akceptoratomjaikban
különböznek, tehát tanulmányozható a Ge → Sn akceptoratom-csere, és a O → S donoratom-
csere hatása a molekulaszerkezetre. A kiválasztott atomi paramétereket a 3. táblázat
19

tartalmazza, és a vizsgált vegyületek egyikét (Ph3Ge(4-metilbezotioát)) az 10. ábra mutatja be
– a többi molekula is szerkezetileg nagyon hasonlóan néz ki. A molekulák felépítése igen
egyszerű, az intramolekuláris kölcsönhatás kialakulását nem befolyásolja egyéb szerkezeti
elem, csak közvetlenül a központi atomhoz kötő csoportok jellege. Szerkezetük a központi
fématom körül torzult tetraéderes. Mindegyik molekulában egy kén kovalens kötéssel köt a
központi fématomhoz, jelen esetben ennek a fém-kén kötésnek az erősségén még a
hiperkoordináció sem gyengít, hossza megegyezik az atomok kovelensrádiuszainak
összegével [36]. Nem változik a fém-szén kötés sem az egyes molekulákon belül, vagy a
szubsztitúció során. Hosszuk megegyezik a megfelelő tetrakoordinált-fém molekulában mért
kötéstávolságok hosszával.
Az adott négy molekulaszerkezet elegendő információt szolgáltat ahhoz, hogy a Ge és
az Sn oxigén- és kén-affinitását tanulmányozzuk. Az ismert másodlagos kötéstávolságok
alapján kiszámíthatjuk a van der Waals rádiuszra vonatkoztatott százalékos kötésrövidülést,
(vdWr-összeg – mért adat) / vdWr-összeg, pl. ∆SnS% = (3,96 Å – 3,207 Å)/ 3,96 Å = 19,0%.
Minél nagyobb ez az érték, annál nagyobb a fématom O- illetve S-affinitása. Kiszámolva a
többi lehetőségre a relatív kötésrövidülést a következőeket kaptam: ∆SnO%= 18,8%,
∆GeS%= 10,8%, ∆GeO%= 11,7%, vagyis ennek ismeretében a relatív affinitási sorrend
SnS≥SnO>>GeO≥GeS. Érdekes, hogy az ón kénaffinitása közel kétszerese a germániuménak,
míg az oxigén esetében nem ilyen nagy a különbség, de még így is igen jelentős.
3.4. Konkrét példák a szerkezet előrejelzésére
Kiválasztott interatomi paraméterek (Å)
CXTGER CXTHSN LIHRUP LIHSAW Cl2GeS2CCOCC Cl2SnS2CCOCC Br2SnS2CCOCC I2SnS2CCOCC M = Ge(1) M = Sn(1) M = Sn(1) M = Sn(1) X = O(1) X = O(1) X = O(1) X = O(1) M-X 2,355 - 2,393 2,359 2,410 2,431 M-S(1) 2,199 - 2,155 2,369 2,361 2,383 M-S(2) 2,168 - 2,216 2,369 2,361 2,38
4. táblázat
A megismert másodlagos kötéshossz-befolyásoló körülmények segítségével
megpróbálhatunk néhány molekulaszerkezet-adatra következtetni, azaz az ismereteink és a 4.
20

táblázat adatai segítségével kitölthetjük az 5. táblázat hiányzó adatait. Ebből a példasorból
megismerhetjük az ónatom elektronaffinitását a hozzá kötő különböző halogénatomok
függvényében. Az egyik felsorolt molekula szerkezeti képe a 11. ábrán látható. Ebben a
központi fém ón, amihez egy oxigén atom koordinál.
11. ábra CXTHSN
(A hidrogén atomokat – a tisztább szerkezeti kép érdekében – mellőztem)
A CXTGER molekulának kétféle röntgenfelvétele is megtalálható volt az adatbázisban,
ezért mindkettő szerkezeti paramétereit feltüntettem. Az ónvegyületekben az ónhoz kovalens
kötéssel kötő kénatomok távolsága kisebb a kovalensrádiusz-összegnél [36], de ezek a
hosszak nem változnak jelentősen más halogének alkalmazása esetén. A kialakuló
koordinációs kötés igen erős, mivel a két atom távolsága jóval a van der Waals rádiusz
összegen belül van [35]. Ez a kötés nagyobb rendszámú halogéneket alkalmazva egy kicsit
gyengül, ez egyértelműen a halogének csökkenő elektronegativitásával magyarázható.
Az elektronaffinitás számításánál felhasznált gondolatmenetet alkalmazva a felsorolt
molekulákban a relatív kötéserősödés a CXTGER molekulánál ∆GeO%= 29,6%–30,7%; a
CXTHSN molekulánál ∆SnO%=34,1%; a a LIHRUP molekulánál ∆SnO%= 32,7%; a
LIHSAW molekulánál pedig ∆SnO%= 32,1%. Tehát a relatív elektronaffinitás ezeknél a
molekuláknál a következőképpen alakul: Cl2-SnO>Br2-SnO>I2-SnO>Cl2-GeO; vagyis a
klorid tartalmú sztannaocán molekulában a legerősebb a másodlagos kötés, és minden vizsgált
ónorganikus halogenidben erősebb az Sn-S kötés, mint a germánium-organikusban. Ez az
elektronaffinitási sorrend megfelel a fentebb levezetettnek. Ha a központi atom cseréjét
21

nézzük, akkor elmondható, hogy a Ge→Sn csere hatására relatíve 4,5%-3,4%-ot erősödött a
másodlagos kötés. A bróm-szubsztitúciót követően 1,4%-ot, a további jód hatására 0,6%-ot.
Szinte már minden adat a rendelkezésünkre áll ahhoz, hogy megjósoljuk az 5. táblázat
„Ismeretlen 1” és „Ismeretlen 2” molekulájának térszerkezeti adatait. Ehhez még szükségünk
van a kén-analógok relatív kötéserősödési adataira. Ezt a CLTHSN molekulára kiszámítva
30,3%-ot kapunk, a LIHSEA-ban pedig 30,1%-os a kötéserősödés. Azaz itt az Sn-S affinitása
kisebb az Sn-O affinitásánál – a tioszalicilsav észternél ez fordítva volt.
A számolás menetét az alábbi diagramm szemlélteti:
CXTGER CXTHSN
Ge→Sn
Ge←Sn
O→S
? Ismeretlen 1 CLTHSN
A kérdőjellel jelölt „Ismeretlen 1” molekula a CLTHSN germánium analógja.
Valószínűleg a kovalens kötéshosszak itt sem változnak jelentősen, tehát a Ge-S(1) és Ge-
S(2) hossz 2,26 Å körülinek várható. Az intramolekuláris kötés hosszának számításához az
imént kiszámolt Ge→Sn csere eredményeit használjuk fel: akkor ez a kötéserősödés 3,4-
4,5%-os erősödéssel járt, ha itt is ezt körülbelül ennyinek tekintjük – amit megtehetünk,
hiszen más körülmény nem változott, akkor az intramolekuláris Ge-S(3) kötéshossz 2,763 Å.
22
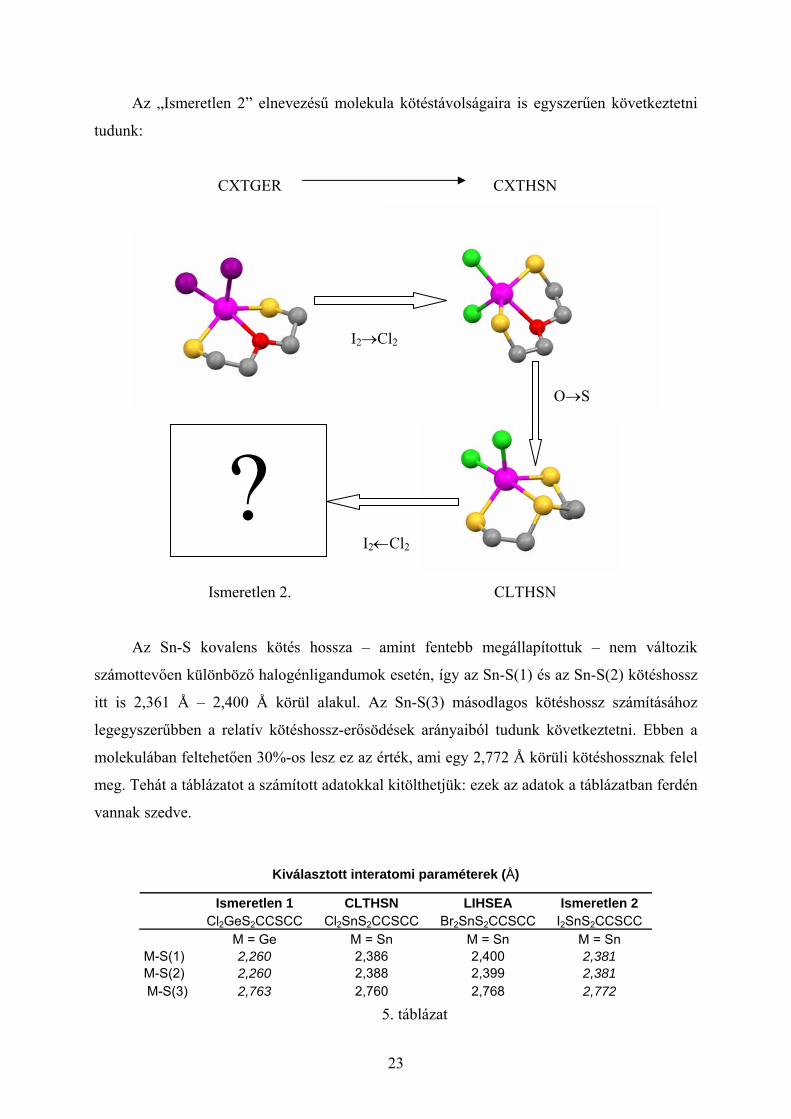
Az „Ismeretlen 2” elnevezésű molekula kötéstávolságaira is egyszerűen következtetni
tudunk:
CXTGER CXTHSN
I2→Cl2
O→S
? I2←Cl2
Ismeretlen 2. CLTHSN
Az Sn-S kovalens kötés hossza – amint fentebb megállapítottuk – nem változik
számottevően különböző halogénligandumok esetén, így az Sn-S(1) és az Sn-S(2) kötéshossz
itt is 2,361 Å – 2,400 Å körül alakul. Az Sn-S(3) másodlagos kötéshossz számításához
legegyszerűbben a relatív kötéshossz-erősödések arányaiból tudunk következtetni. Ebben a
molekulában feltehetően 30%-os lesz ez az érték, ami egy 2,772 Å körüli kötéshossznak felel
meg. Tehát a táblázatot a számított adatokkal kitölthetjük: ezek az adatok a táblázatban ferdén
vannak szedve.
Kiválasztott interatomi paraméterek (Å)
Ismeretlen 1 CLTHSN LIHSEA Ismeretlen 2 Cl2GeS2CCSCC Cl2SnS2CCSCC Br2SnS2CCSCC I2SnS2CCSCC M = Ge M = Sn M = Sn M = Sn M-S(1) 2,260 2,386 2,400 2,381 M-S(2) 2,260 2,388 2,399 2,381 M-S(3) 2,763 2,760 2,768 2,772
5. táblázat
23

Az itt kiszámított adatok természetesen csak közelítő értékek, de annál valósághűbb a
levezetett érték, minél több, hozzá igen hasonló molekula szerkezeti paramétereit használtuk
fel a számítás során.
Ennek analógiájára sok más számítást is végezhetnénk. Nagy mennyiségű adatból
kiindulva az első menetben következtetünk néhány molekulaparaméterre, majd többek között
ezek felhasználásával második menetben is számíthatunk más adatokat, és ezt így
folytathatjuk tovább – értelemszerűen egyre kisebb megbízhatósággal.
24

4. Összefoglalás
Munkám során ónorganikus és germánium-organikus vegyületekben kialakuló
intramolekuláris oxigén és kén kölcsönhatást vizsgáltam. Továbbá bemutattam egy egyszerű,
spekulatív számítási módszert, aminek segítségével ismeretlen molekula-szerkezet kötéshossz
adataira lehet következtetni.
A ón-dialkilészterek vizsgálatakor kiderült, hogy a kötéserősödés leginkább a
konformáció-változásban nyilvánul meg.
Úgy találtam, hogy mindegyik dialkil származék geometriája torzult négyzetes bipiramis,
a dihalogenideké, és a difenil ónkarbamátoké pedig oktaéderes, melyben a halogének
illetve a fenil csoportok cisz állásúak.
A Me2 → Cl2 ligandumcsere következtében az Sn····S koordinációs kötéshossz az észteri
reendszer két vizsgált esetében ∆Sn-SCIHCIF-CIHDEC= -0,531 Å, ∆Sn-S ETCMSN-WIBTEG= -
0,367 Å-mel változott, az Sn····O kölcsönhatás pedig ∆Sn-OYORJUK-RAXWOC= -0,421 Å-
mel. Heterocikulos sztannaocánok esetén ∆Sn-SYIFLUU-CLTHSN= -0,754 Å.
A klorid szubsztitúció során minden molekulapárnál kimutattam a hiperkoordniációt
követő kovalenskötés-hosszabbodást is, értéke 0,05 Å körül volt.
A következőkben az interakcióban résztvevő atomok anyagi minőségétől függően
vizsgáltam a másodlagos kötés erősségét. Úgy találtam, hogy az ónnak jelentősen
nagyobb a germániumnál az elektronaffinitása, a relatív affinitási sorrend
SnS≥SnO>>GeO≥GeS irányban csökkent a tioszalicilsav észtereknél.
Végül heterociklusos sztannaocán és germocán rendszerekben is tanulmányoztam a relatív
kötéserősödést. Felállítottam egy relatív affinitási sorrendet a fématomhoz kötő
halogenid függvényében; a sorrend Cl2-SnO>Br2-SnO>I2-SnO>Cl2-GeO.
Dolgozatom végén két példát hoztam még nem szintetizált molekulák szerkezeti
paramétereinek előrejelzésére.
25

5. Köszönetnyilvánítás
Szeretnék köszönetet mondani mindazoknak, akik hozzájárultak a dolgozatom elkészítéséhez.
Elsősorban Sztáray Bálint témavezetőmnek, aki munkám során hasznos tanácsokkal látott el,
Harmat Veronikának, akik megtanított a CCDC adatbázis kezelésére, valamint
kutatócsoportunk tagjainak – Bődi Andrásnak, Gengeliczki Zsoltnak, Pongor Csabának,
Hornung Balázsnak, Kiss Andrásnak, Révész Áginak, Rádi Krisztinának –, akik mindig
készek voltak nekem segíteni. Végül pedig Rácz Andrásnak mondok köszönetet, aki a
magyarról magyarabbra történő „fordításban” segített.
26

6. Irodalomjegyzék
1. Rosenfield, R. E.; Parthsarathy, R.; Dunitz, J. D. J. Am. Chem. Soc. 1977, 99, 486;
2. Row, T. N. G.; Parthsarathy, R. J. Am. Chem. Soc. 1981, 103, 477.
3. Wei, Y.; Hendicksen, W. A.; Crouch, R. J.; Satow, Y. Science 1990, 249, 1389.
4. Niyomura, O.; Kato, S.; Inagaki S., J. Am. Chem. Soc. 2000, 122, 2132.
5. Ishihara, H.; Kato, S. Tertahedron Lett. 1972, 3751.
6. Barrow, M. J.; Ebsworth, e. A. V.; Huntley, C. M.; Rankin, D. W. H. J. Chem. Soc.
Calton Trans, 1982, 1131.
7. Rohonczy, J.; Knausz, D.; Csákvári, B.; Sohár, P.; Pelczer, I.; Párkányi, L. J. Orgmet
Chem. 1988, 340, 293.
8. Kawahara, Y.; Kato, S.; Kanda, T.; Murai, T.; Ebihara, M.; Bull. Chem. Soc. Jpn.
1995, 68, 3507.
9. Holmes R. R.; Day, R. O.; Chandrasekhar, V.; Vollano, J. F.; Holmes, J. M.;, Inorg.
Chem. 1986, 25, 2490.
10. Drake, J. E.; Mislankar, A. G.; Wong, M. L. Y., Inorg. Chem. 1991, 30, 2174.
11. Dakternieks, D.; Zhu, H.; Masi, D.; Mealli, C. Inorg. Chem. 1992, 31, 3601.
12. Lockhart, T. P.; Manders W. F.; Schlemper E. O.; Zuckerman J. J., J. Am. Chem.
Soc. 1986, 108, 4074.
13. Haiduc, I., Rev. Inorg. Chem. 1981, 3, 353.
14. Barnes, J. M.; Magos, L. Organomet. Chem. Rev. 1968, 3, 137;
15. Swisher, R. G.; Vollano J. F.; Chandrasekhar V.; Day R. O.; Holmes R. R. Inorg.
Chem., 1984, 23, 3147.
16. Nath, M.; Yadav, R.; Eng, G.; Musingarimi, P.; Appl. Organomet. Chem. 1999, 13,
29.
17. Evans C. J.; Kaprel S., Organotin Compounds in Modern Technology ,Journal of
Organometallic Chemistrs Library, 16 Elsevier, Amsterdam, 1985, 280 pp.
18. Blunden, S. J.; Cusack, P. A.; Hill R., The Industrial Uses of Tin Chemicals, Royal
Society of Chemistry, London, 1985, 346 pp.
19. Das K.; Ng S. W.; Gielen M., Chemistry and Technology of Silicon and Tin, Oxford
University Press, Oxford, 1992, 608 pp.
27

20. M. Pereye; J.-P. Quintard; Rahm, Tin in Organic Synthesis, Butterworths, London,
1987, 342 pp.; J. K. Stille, Angew. Chem. Int. Edn. Engl., 1986, 25, 508-24.
21. Jousseaume, B.; Villeneuve, P. J. Chem. Soc., Chem. Commun. 1987, 513.
22. Pan, H.; Willem, R.; Meunier-Piret, J.; Gielen, M. Organometallics 1990, 9, 2199.
23. Akiba, K.; Ed. Chemistry of Hypervalent Compounds; Wiley-VCH; Weinheim,
Germany, 1999.
24. Jurkschat, K.; Pieper, N.; Seemeyer, S.; Schürmann, M.; Biesemans, M.;
Verbunggen, I.; Willem, R. Organometallics 2001, 20, 868.
25. Jastrzebski, J. T. B. H.; Boersma, J.; Esh, P. M.; van Koten, G. Organometallics
1991, 10, 930.
26. Mitzel, N. W.; Losehand, U.; Richardson, A.; Organometallics 1999, 18, 2610.
27. Buntine, M. A.; Kosovel, F. J.; Tieking, E. R. T.; Phosphorus, Sulfur Relat. Elem.
1999, 150, 261.
28. Willem, R.; Delmotte, A.; De Borger, I.; Biesemans, M.; Gielen, M.; Kayser, F.;
Tiekink, E. R. T. Organometallics, 1994, 480, 255.
29. Kayser, F.; Biesemans, M.; Delmotte, A.; Verbruggen, I.; De Borger, I.; Gielen, M.;
Willem, R.; Tieking, E. R.T. Organometallics, 1994, 13, 4026.
30. Smith P. J.; Day, R. O.; Chandrasekhar, V.; Holmes R. R; Inorg. Chem., 1986, 25,
2495.
31. Dakternieks, D.; Jurkschat K.; Tozer, R.; Hook J.; Tieking, E. R. T. Organometallics
1997, 16, 3696.
32. Smith, P. J; Day, R. O; Chandrasekhar, V.; Holmes, J. M., Holmes, R. R., Inorg.
Chem. 1986, 25, 2495.
33. Swisher, R. G.; Vollano, J. F.; Chandrasekhar, V.; Day, R. O.; Holmes, R. R., Inorg.
Chem. 1984, 23, 3147.
34. Vollano, J. F.; Day, R. O; Rau, D. N.; Chandrasekhar, V.; Holmes, R. H., Inorg.
Chem. 1984, 23, 3153
35. Bondi, A. J. Phys. Chem. 1964, 68, 441. Ge-O: 3,40 Å; Ge-S: 3,78 Å; Sn-O: 3,58 Å;
Sn-S: 3,96 Å
36. Pauling, L. The Chemical Bond; Corell University Press: Ithaca, NY, 1976; pp 146-
155 Csp2=O 1,20-1,23 Å, Csp2=S 1,61 Å, Sn-O 2,06 Å; Sn-S 2,44 Å; Ge-O 1,88 Å;
Ge-S 2,26 Å
37. Mohamed-Ibrahim M. I.; Chee S. S.; Buntine M. A.; Cox M. J.; Tieking E. R. T.
Organometallics, 2000, 19, 5410.
28

38. Tieking E. R. T., Trends in Organomet. Chem. 1994, 1, 71.
39. Hall, V. J.; Tieking, E. R. T.; Main Group Met. Chem. 1998, 21, 245.
40. Hook, J. M.; Linaham, B. M.; Taylor, R. L. Tieking, E. R. T.; van Gorkom, L.;
Webster, L. K., Main Group Met. Chem. 1994, 17, 293.
41. Buntine, M. A.; Hall, V. J.; Kosovel, F. J., Tieking, E. R. T., J. Phys. Chem. 1998,
102, 2472.
42. Tieking, E. R. T.; Winter, G. J. Organomet Chem. 1986, 314, 85
43. Kato, S.; Tani, K.; Kitaoka, N.; Yamada, K.; Mifune, H., J. Orgmet. Chem. 2000,
611, 190.
44. Tani, K.; Kato, S.; Kanda T.; Kanda T.; Inagaki S., Org. Lett. 2001, 3, 655.
45. Kato, S., Kato, T.; Shibahashi, Y.; Kakudo, E.; Mizuta, M.; Ishii, Y. J. Organomet.
Chem. 1974, 76, 215.
46. Kato, S.; Hori, A.; Shiotani, H.; Mizuta M.; Hayashi, N.; Takakuwa, T.; J.
Organomet. Chem. 1974, 82, 223.
47. Katada, T.; Kato, S.; Mizuta, M.; Chem. Lett. 1975, 1037.
48. Dräger M.; Engler R., Z. anorg. Allg. Chem. 1975, 413, 229.;
49. Dräger M., Z. anorg. Allg. Chem. 1976, 423, 53.
50. Dräger M., Z. anorg. Allg. Chem. 1977, 428, 243.
51. Dräger M., Z. Naturforsch. 1981, 36b, 437.
52. Dräger M., J. Organomet. Chem. 1983, 251, 209.
53. Dräger M., Z. anorg. Allg. Chem. 1985, 527, 169.
29





























