Embed Size (px)
Citation preview

Introduction to R / sma / Bioconductor
Statistics for Microarray Data Analysis
The Fields Institute for Research in
Mathematical Sciences
May 25, 2002

Web sites + References
• http://www.R-project.org/
An introduction to RW.N.Venables, D.M.Smith and the R Development
Core Team
• http://lib.stat.cmu.edu/R/CRAN/• http://www.bioconductor.org/

Need to read files such as “swirl1.spot” or “samples.swirl” into the R programs. Functions:
read.tablescan
Save your workspace in RUsing the function
save.imageYou will only see
name.RData or.RData
In your directory

Download ?
Download SetupR.exe from http://cran.r-project.org/,

A few basics
• Working Directory- getwd()- setwd() or click on File and then click on Change Dir,
use Browse to determine your working directory.
• Workspace- save(a, b, file=“my.RData”) : save objects a and b into
the workpace “my.RData”- save.image(“my.RData”) : click on File and then click
on Save Workspace- load(“my.RData”) : click on File and then click on
Load Workspace
• Help- help.start()- help(): e.g. help(plot)

Search paths + packagessearch()> search()[1] ".GlobalEnv" "package:ctest" "Autoloads" "package:base"
library(cluster)search()> library(cluster)Loading required package: mva > search()[1] ".GlobalEnv" "package:mva" "package:cluster" "package:ctest" [5] "Autoloads" "package:base"
ls() : list objects in the GlovalEnvls(3) : list objects in search position number 3, in the above example, it is
package:cluster
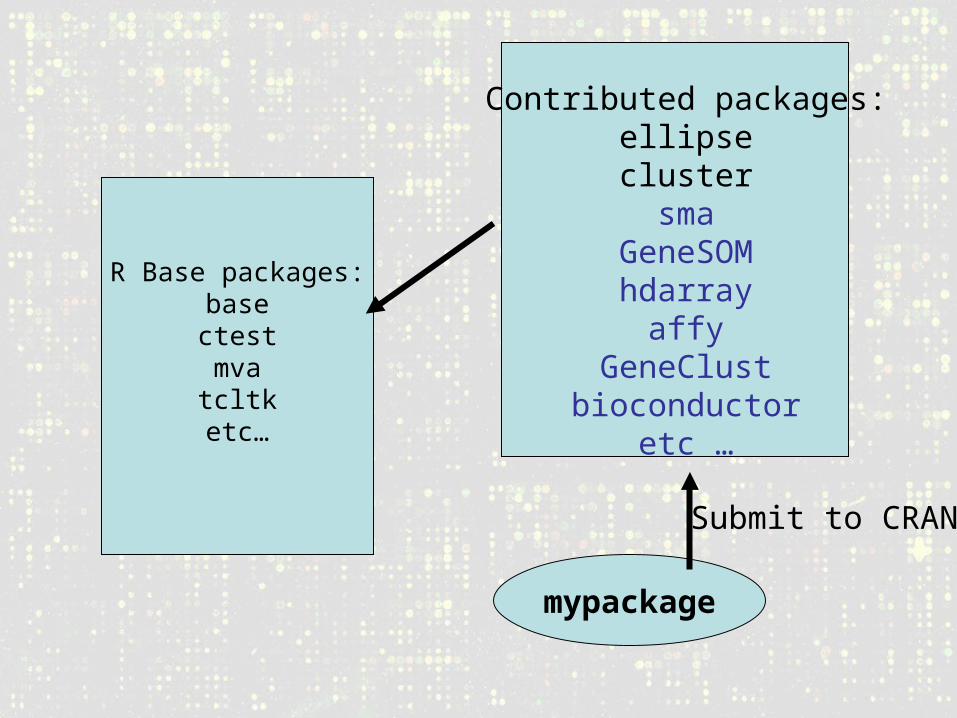
R Base packages:basectestmvatcltk
etc…
Contributed packages:ellipseclustersma
GeneSOMhdarray
affyGeneClust
bioconductoretc …
mypackage
Submit to CRAN

An introduction to R
based on the documents produced by
W.N.Venables, D.M.Smith and the R Development Core Team

Vectors and assignment
R operates on named data structures. The simplest such structure is the numeric vector, which is a single entity consisting of an ordered collection of numbers.
To set up a vector named x, say, consisting of five numbers, namely 10.4, 5.6, 3.1, 6.4 and 21.7, use the R command x <- c(10.4, 5.6, 3.1, 6.4, 21.7) orassign(“x”, c(10.4, 5.6, 3.1, 6.4, 21.7))
This is an assignment statement using the function c()
This is a numeric vector> is.numeric(x)[1] TRUE

Character
numeric
vector
logical
X <- c(1:5, 6, 9,3, 10)
X <- c(“a”, “b”, “c3”, “4”)
X <- c(1, 1, 0, TRUE, FALSE)

Other types of objects
matrices or more generally arrays are multi-dimensional generalizations of vectors.
lists provide a convenient way to return the results of a statistical computation.
data frames are matrix-like structures, in which the columns can be of different types. Think of data frames as `data matrices' with one row per observational unit but with (possibly) both numerical and categorical variables.
functions are themselves objects in R which can be stored in the project's workspace. This provides a simple and
convenient way to extend R.

Introduction to Bioconductor(taken from http://www.bioconductor.org)
The packages in the initial release include tools which facilitate:
- annotation (AnnBuilder, annotate)
- data management and organization through the use of the S4 class structure (Biobase, marrayClasses)
- identification of differentially expressed genes and clustering (edd, genefilter, geneplotter, multtest, ROC)
- analysis of Affymetrix expression array data (affy)
- diagnostic plots and normalization for cDNA array data (marrayInput, marrayNorm, marrayPlots)
- storage and retrieval of large datasets (rhdf5).

Character numericlogical
Slots
Most packages rely on the class/method mechanism provided by John Chambers’ R methods package, whichallows object-oriented programming in R
Class

marrayInfo
maLabelscharacter
maInfodata.frame
maNotescharacter
This class can be used to store either the gene names Information or samples information
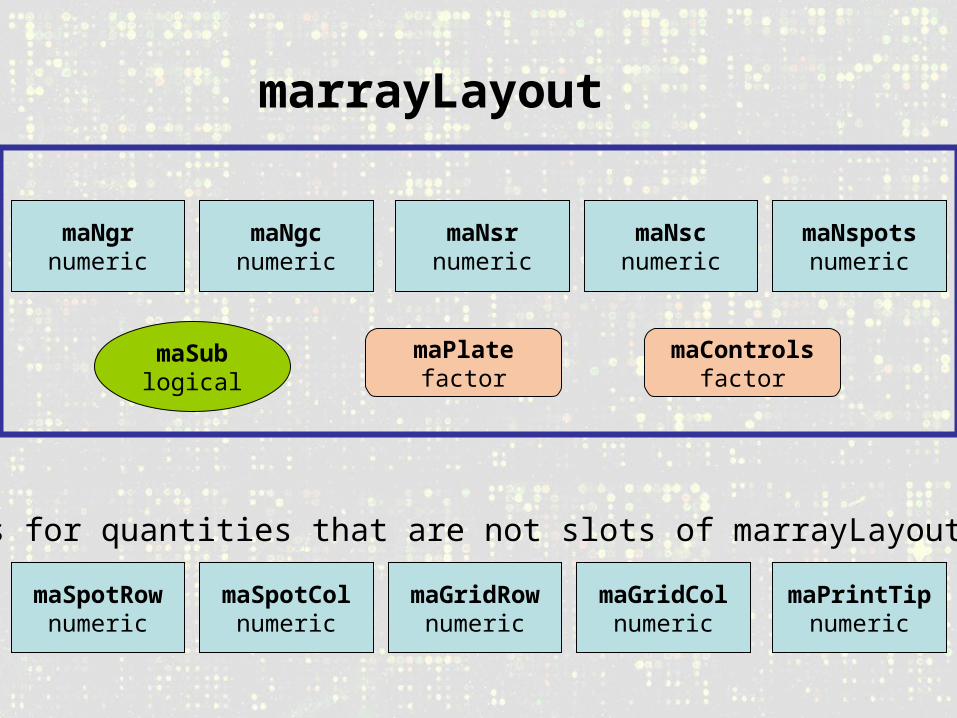
marrayLayout
maNscnumeric
maNsrnumeric
maNgcnumeric
maNgrnumeric
maNspotsnumeric
maSublogical
maPlatefactor
maControlsfactor
maSpotRownumeric
maSpotColnumeric
maGridRownumeric
maGridColnumeric
maPrintTipnumeric
Methods for quantities that are not slots of marrayLayout

marrayRaw
Methods for quantities that are not slots of marrayRaw
maLayoutmarrayLayout
maGnamesmarrayInfo
maTargetsmarrayInfo
maNotescharacter
maRfmatrix
maRbmatrix
maGf matrix
maGbmatrix
maWmatrix
maLRmatrix
maLG matrix
maMmatrix
maAmatrix
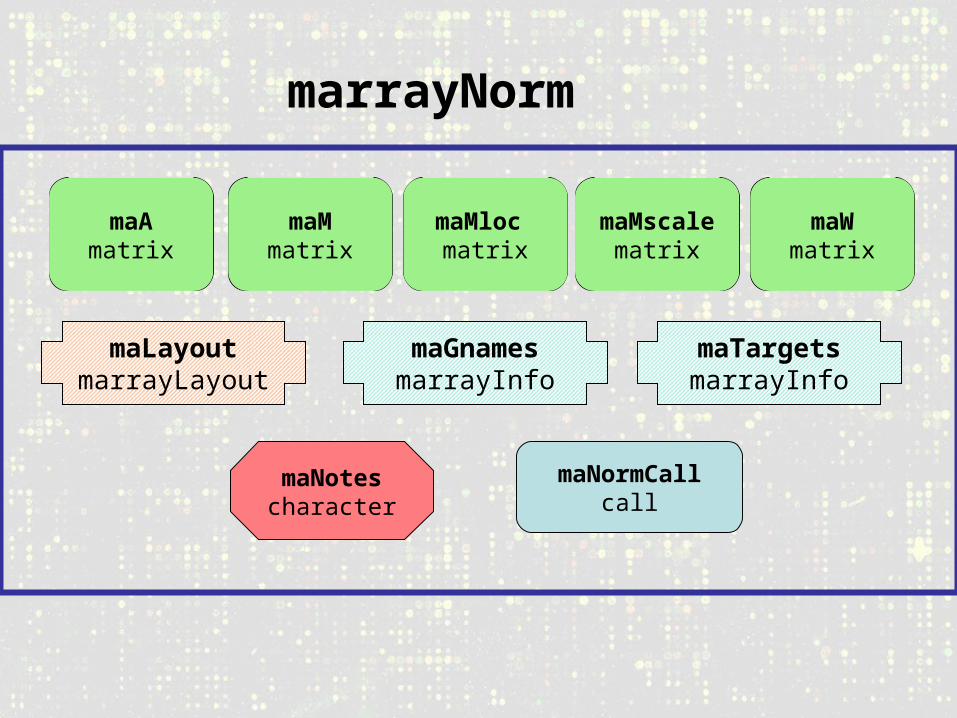
marrayNorm
maLayoutmarrayLayout
maGnamesmarrayInfo
maTargetsmarrayInfo
maNotescharacter
maNormCallcall
maAmatrix
maMmatrix
maMloc matrix
maMscalematrix
maWmatrix

Swirl data
Data (Spot Files)• swirl.1.spot• swirl.2.spot• swirl.3.spot• swirl.4.spot
Target information files• SwirlSample.txt
Gene List• fish.gal
Layout:Grid size: 4 by 4Spot matrix: 22 by 24





























