Embed Size (px)
Citation preview

Introduction to Newbler
NextGen BUG Assembly Workshop
Dec 2009Stephen Bridgett

Aims
• What is Newbler• How to Install• Input files• Flowgrams• Using sff tools• Output files• Command line options• GUI interface• Experiments with sample data

What is “Newbler” ? Roche's “GS De Novo Assembler” (where “GS” = “Genome Sequencer”)
Designed to assemble reads from the Roche 454 sequencer. Accepts: 454 Flx Standard reads, and the longer 454 Titanium reads. both single and paired-end reads.Optionally can include Sanger reads.
Initial versions focused on assembling Genomic reads. Latest version (2.3) includes optimisations for transcriptome data.
Runs on Linux, and has 32 bit and 64 bit versions. Has Command-line and Java-based GUI interface. Rarely called “Newbler” in Roche's documentation, rather “runAssembler”,
or “gsAssembler”.
Installing Newbler & .sff utilities• Previous versions had separate installation files for 32 bit or 64 bit operating
systems.• Latest 2.3 version has one installation file (installs 32 or 64 bit as
appropriate):“DataAnalysis_2.3.tgz”
• You can download it from: http://xyala.bio.ed.ac.uk/Gene_Pool/454_software/Version_2.3
• We got permission from Roche for NextGen BUG members to use Newbler.• To install:
tar zxvf DataAnalysis_2.3.tgzbash ./INSTALL
• Then choose to install:• (1) Locally, just for current user, • (2) System-wide for all users, use root password, default directory:
/opt/454/• Newbler 2.0.1 and 2.3 are already installed on your computer here.


Installing Newbler (cont. 2)• May request several extra libraries to be installed: zlib.i386
libXi.i386libXtst.i386libXaw.i386
• To install these: sudo yum install zlib.i386 libXi.i386 libXtst.i386 libXaw.i386Note: These are 32-bit libraries, even if installing on 64-bit operating system.
• To confirm installation, type: cat /opt/454/config/releaseTags.parse
• You should see: version = “2.3”

Installing Newbler (cont. 3)• The installation instructions are also on the website as a .pdf file:
TCB-09015_Software_Installation_2.3.pdf • See Page 15, ('Part B step 2' only, Skip steps 1 and 3, as the “DataProcessing” software mentioned
in step 1 and Cluster Support Utilities in step 3 are only needed for signal-processing of the raw images from the sequencer.)
• Also on website are the manual for the Newbler assembler, Reference mapper, Sff-tools, Amplicon variant analyzer, and File format info:
SW-Manual_PartC_Assembler-Mapper-SFFTools_Oct2009.pdf
SW-Manual_PartD_AmpliconVariantAnalyzer_Oct2009.pdf
SW-Manual_Overview-FileFormats_Oct2009.pdf
• Manual Parts A and B are for acquiring the data on the 454 sequencer and for signal-processing, and aren't on the website at present.

How does Newbler work?During the assembly process, the software:
• Identify pairwise overlaps between reads
• Constructs multiple alignments of overlapping reads, and divides or introduce breaks into the multiple alignments in regions where consistent differences are found between different sets of reads. (This step results in a preliminary set of “contigs” that represent the assembled reads.)
• Attempts to resolve branching structures between contigs
• Generates consensus basecalls of the contigs by using quality and flow signal information for each nucleotide flow included in the contigs’ multiple alignments
• Outputs the contig consensus sequences and corresponding quality scores, along with an ACE file of the multiple alignments and assembly metrics files.
• You will see message about these steps as assembly progesses.
Paired End data is available, the assembler performs these extra steps:
• Organizes the contigs into scaffolds using Paired End information to order and contigs and to approximate the distance between contigs.

Challenges• Contaminants in samples.
• Primers and adapters still present.
• Sequencing errors.
• “Homopolymer” errors – when eg. 5+ run of same base,
• Repeats in genome, - make assembly more difficult.
• Large genomes.a
• Transcriptomes (cDNA).

Blast search to check for contaminants• Blastx search of 10,000 randomly picked reads against UniRef90 or
(Non-redundant dataset)
• Sorted by frequency of Description (or Tax) with evalue > e-8
Frequency Subject_description
1689 (16.9 %) Picea sitchensis
907 (9.1 %) Vitis vinifera
311 (3.1 %) Physcomitrella patens subsp. patens
282 (2.8 %) Arabidopsis thaliana
218 (2.2 %) Oryza sativa Japonica Group
153 (1.5 %) Zea mays
58 (0.6 %) Oryza sativa Indica
58 (0.6 %) Oryza sativa
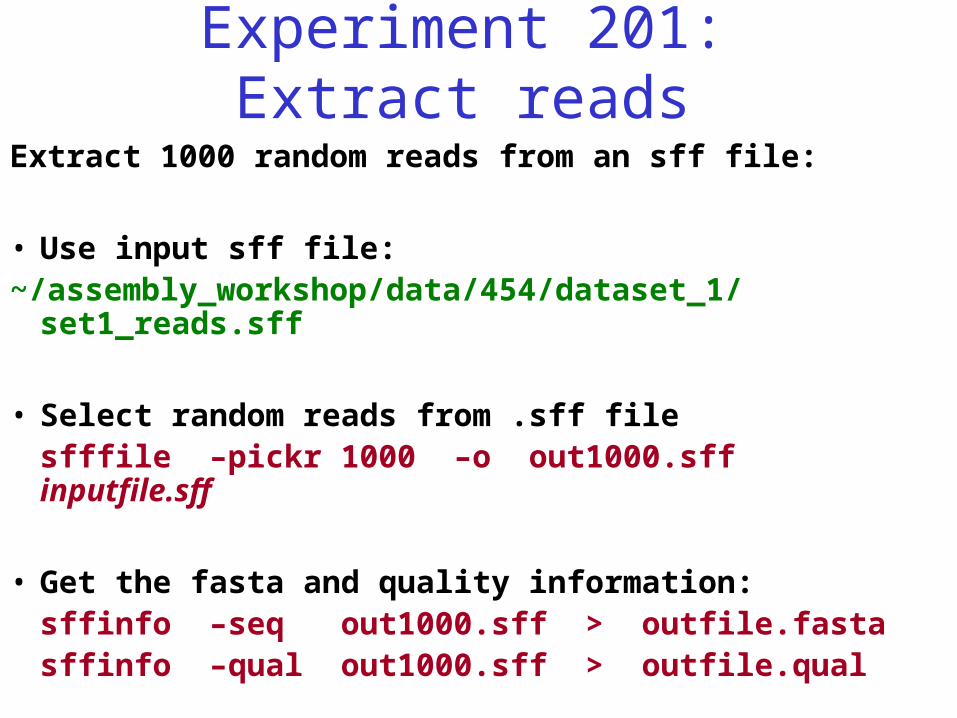
Experiment 201: Extract reads
Extract 1000 random reads from an sff file:
• Use input sff file: ~/assembly_workshop/data/454/dataset_1/set1_reads.sff
• Select random reads from .sff filesfffile –pickr 1000 –o out1000.sff inputfile.sff
• Get the fasta and quality information:sffinfo –seq out1000.sff > outfile.fastasffinfo –qual out1000.sff > outfile.qual

Flowgrams and Homopolymer error
• To help understand output from Newbler alignment.
• Animation of 454 sequencing to explain Flowgrams.

Homopolymer error
• Different between signal of 1 and signal of 2 = 100%.• Different between signal of 5 and 6 is 20% so errors more
likely after eg. AAAAA.
0
1
2
3
4
5
6
Cycle 1 Cycle 2 Cycle 3 Cycle 4 Cycle 5
ACTG
A ?c TT - AAAAA ?a

Flowgrams
• Averaged flowgram signal for all reads in one lane.• This can be extracted from sfffile, using sffinfo then a script to average
and plot.• Should have reasonably flat along central region of flows.• Has MINT adapters, which contain 30T’s.
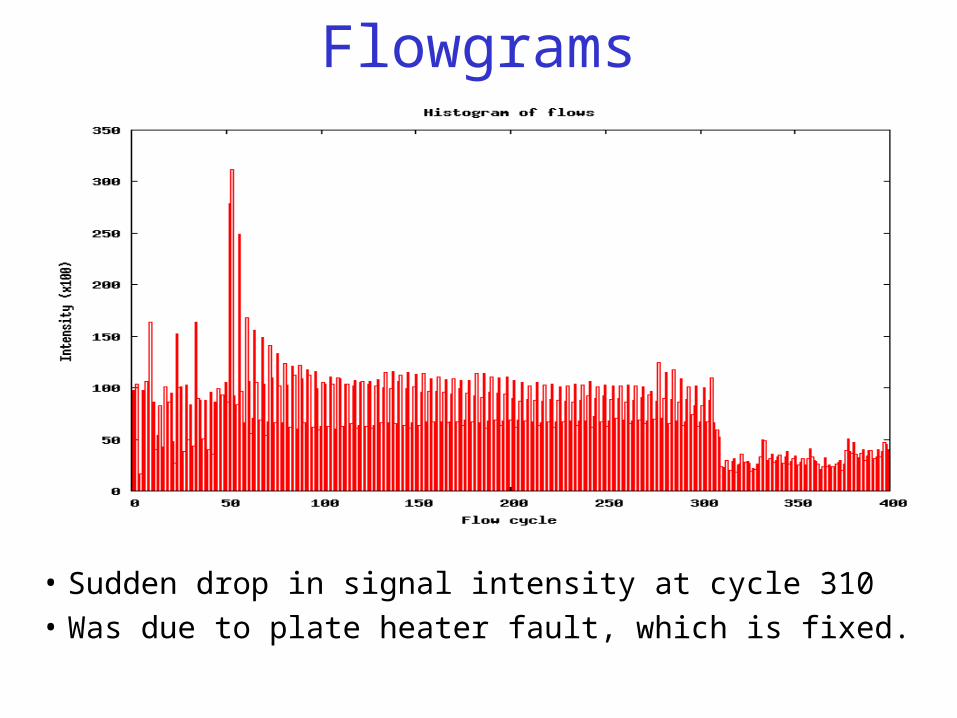
Flowgrams
• Sudden drop in signal intensity at cycle 310
• Was due to plate heater fault, which is fixed.

Experiment 202
• Get the flowgram information for the 1000 random reads selected earlier:
sffinfo –notrim –tab out1000.sff > out.flow
-notrim means don’t quality trim the reads
To view the flow information:more out.flow
(q to quit)

Extra challenges of transcriptome assembly
• Ribosomal RNA (small and large sub-units) and chimeras.
• Poly A’s, Poly T’s tails (added after gene transcription). …….ATGCTAAAAAAAAAAAAAAA-3'
• MINT or other adapter sequences:
At the 5' end: 5'-AAGCAGTGGTATCAACGCAGAGTACGGGGG-3'
At the 3' end: 5'-AAGCAGTGGTATCAACGCAGAGTAC(T)30VN-3'
Extra challenges with transcriptomes (2)
• Extra sample preparation steps, so more risk of cloning errors or contamination.
• Alternative splicing - differing reads from same part of genome.
• Wider range of read lengths.
• Roche’s Newbler assembler sometimes didn’t finish transcriptome assembly, seemed to get lost when “Detangling Alignments”

Inputs to Newbler assembler
Newbler accepts:
Roche's .sff files (standard flowgram format), which contain flowgrams, in addition to fasta and quality information.
Fasta files, with or without Quality files, such as Sanger reads, which can be used as a scaffolds.
Parameters specified by the user, to guide the assembly, (or parameters can all be left at their default values.)

Command-line interface
• Basic command-line in linux console:
runAssembler [options] reads.sff
• Which will create an the assembly in an output directory called:
P_yyyy_mm_dd_hh_min_sec_runAssemblywhere P_ = Project, followed by date and time
• You can use the default values for all options.
• But large number of optional parameters are available for controlling and refining the assembly:
Experiment 203: Genome assembly• dataset 1: 454 titanium genome reads for 6 Mb genome
~/assembly_workshop/data/454/dataset_1/set1_reads.sff
Get metrics for the raw reads:
sffinfo -seq > reads.fasta
process_contigs.pl –i reads.fasta –o process_reads (although these are reads, rather than contigs the same script can still be used.)
more process_reads/contig_stats.txt
Estimate the average read depth (genome 6Mb )

Experiment 204: AssemblyAssembly command:
runAssembly -o assembly1 reads.sff
• Where reads are: ~/assembly_workshop/data/454/dataset_1/set1_reads.sff
Look in the assembly1 subdirectory, and see what you think the files contain.

Common options
• -o output_directory to set name of output directory (overwrites existing directory without warning!)
• -vt trimmingFile.fasta to trim primers, adapters or polyA tails from start or end of reads
• -vs screeningFile.fasta to remove reads that closely matching a cloning vector such as E.Coli.
• (-vs and -vt will also match for the reverse-complements of the given sequences.)

More options• -a num minimum contig length for 454AllContigs (default 100)
• -l num mim contig length for 454LargeContigs (default=500)
• -large for large or complex genomes, speeds up assembly, but reduces accuracy. Not with -cdna option.
• -m keep sequence data in memory to speed up assembly, but needs sufficient RAM.
• -cpu num num CPU’s to use (default=all), to speed up the computing alignments and generating output steps.
• -minlen num minimum length of reads to use in assembly
• -rip output each read in only one contig.

Even more options
• -notrim disable default quality & primer trimming of input reads.
• -p filename specify input file contains paired-end reads.
• -ud treats each reads separately, not grouping duplicates.
• -ss set seed step parameter
• -sl set seed length parameter
• -sc set seed count parameter
• -ml set minimum overlap length
• -mi set minimum overlap identify
• -nobig skip output of large files (.ace, 454AlignmentInfo.tsv)
• -consed creates subdirectory, with .ace, and .phd files, and sff_dir for consed

Experiment 205: Using options• Use some of these options that you think may improve
assembly. runAssembly [your options] –o assembly2 reads.sff
• Change into subdir assembly2 • Look through some of the output files, eg:
less filename (or use a texteditor)process_contigs.pl -i 454AllContigs.fna –o stats
• Assembler Manual is available on the web links page so you can try different options
• Upload your results onto the webpage and see how compare.

Transcriptome optionsNewbler collects into “Isogroups”, then creates “Isotigs”
New options for transcriptomes:• -cdna = for transcriptome (cDNA assembly)
• -ig = max contigs in an isogroup (default 500 contigs)• -it = max number of isotigs in an isogroup (default 100)
• -icc = maximum number of contigs in one isotig (default 100 contigs)• -icl = isotig contig length threshold, below which traversal stops
(default 3 base pairs)
• Pages 142 to 146 of Part C of the Roche Assembly manual gives a good table of all the options.

Common options (again)• -o output_directory to set name of output directory
(overwrites existing directory without warning!)
• -vt trimmingFile.fasta to trim primers, adapters or polyA tails from start or end of reads
• -vs screeningFile.fasta to remove reads that closely matching a cloning vector such as E.Coli.
• (-vs and -vt will also match for the reverse-complements of the given sequences.)

Experiment 206: Transcriptome assemblyUsing: ~/assembly_workshop/data/454/dataset_2/
Enter the following on one line:runAssembly
-o assembly3 -vt MINTandPolyA.fna
–vs RNA.fna (groups at front half only) -cdna • -ig NUM (max contigs in an isogroup, default 500 contigs)• -it NUM (max number of isotigs in an isogroup, default 100)• -icc NUM (max contigs in one isotig, default 100 contigs)• -icl NUM (isotig contig length threshold, default 3 bp)
reads.sff

Incremental assembly
There are also alternative command-line commands (instead of ‘runAssembly’) that can perform incremental assembly, adding, or removing, runs to an existing project over time:
• newAssembly, • addRun, • removeRun, • runProject.

Default Output for Genome & Transcriptome projectsIn the Assembly subdirectory:
• 454AllContigs.fna (-a num) fasta file of all contigs of size >100 (or num)• 454AllContigs.qual quality scores (Phred-based) for each base in the '454AllContigs.fna'
contigs file. (eg: 20 = 1 in 100 probability of incorrect base call; 50 = 1 in 100,000)
• 454LargeContigs.fna (-l num) fasta file of contigs >500bp (or num)• 454LargeContigs.qual quality scores for '454LargeContigs.fna'
• 454NewblerMetrics.txt statistics of the assembly, eg: number of reads and bases aligned, overlaps found, mean contig sizes,
• 454NewblerProgress.txt log of assembly progress (same as console output)
• 454ReadStatus.txt status of each read in assembly, alignment 3' and 5' positions within contig.
• 454TrimStatus.txt = each read's original and revised trim-points used in the assembly.
• sff = subdirectory containing unix symbolic links to the .sff files used.

Default Output (cont. 2)
• 454AlignmentInfo.tsv (-infoall/-info/-noinfo) base consensus, quality, depth and flow-signal, at each position in each contig. A very useful file.
• eg:
Position Consensus Quality Unique Align Signal Signal Score Depth Depth StdDev (incl. duplicates)
>contig000081 G 64 26 32 0.98 0.052 A 64 27 33 0.94 0.133 T 64 27 33 1.97 0.144 T 64 27 33 1.97 0.145 G 64 27 33 0.97 0.06...etc...

Default output (cont. 3)• 54ContigGraph.txt = describes the branching structure between contigs.
• Has 3 sections:
•(1) Graph Node information (the contigs):ContigNum ContigName Length Average_depth1 contig00001 452 42.62 contig00002 603 253.9...etc...
•(2) Graph edges (C=contig edge; or S=scaffold edge for paired end reads, also S in -cdna graphs):Edge FromContigNum FromEnd ToContigNum ToEnd AlignmentReadDepth C 3 5’ 2639 5’ 24C 6 5’ 7 5’ 36...etc....S 1 1558 2560:+;2802:-;2872:-;2575:-;2783:-;2614:-S 2 671 2560:+;1327:-...etc....
•(3) More graph informationI 3 t 24:2639-5'..1284-3'I 9 atcgattgaaatcaatggagaaagatacTATAGAAAGTTAATAAAaGTATCTGTAGAGCCGACAGTTG....etc...F 2 2751/188/0.0;2931/8/0.0;2957/36/0.0;1242/226/6.0 -F 3 2639/24/0.0 1284/24/0.0

Output specific to Genome assembly
• 454Contigs.ace (-noace/-consed) = ACE format file, showing how reads were aligned to form contigs, viewable in eg. Tablet, EagleView, or Consed
• Unlike traditional ace files, here the same read can be in several contigs (but is given an extra suffix), if one contig is in a repeat region and the next is contig is a non-repeat region, and the read spans the junction.

Output from Transcriptome assemblyOnly in latest Newbler version 2.3, with new -cdna option:
• 454Isotigs.fna fasta file of Isotigs, from multiple-alignment graph (the isogroup)
• 454Isotigs.qual quality scores (Phred-based) for each base in 'Isotigs.fna‘.
• 454Isotigs.ace ACE format for cDNA Isotigs.
• 454IsotigLayout.txt how contigs are laid along each isotig in the isogroup,
• eg:
>isogroup00003 numIsotigs=8 numContigs=11 Length : 495 508 142 171 251 308 98 61 61 566 306 (bp) Contig : 02209 02600 02782 00425 02597 00426 02119 02340 02624 02132 02630 Total:isotig00004 >>>>> >>>>> >>>>> >>>>> >>>>> >>>>> 1484isotig00005 >>>>> >>>>> >>>>> >>>>> >>>>> >>>>> 1484isotig00006 >>>>> >>>>> >>>>> >>>>> >>>>> >>>>> 1497isotig00007 >>>>> >>>>> >>>>> >>>>> >>>>> >>>>> 1497isotig00008 >>>>> >>>>> >>>>> >>>>> >>>>> 1472isotig00009 >>>>> >>>>> >>>>> >>>>> >>>>> 1485

Experiment 207: Look at output files
• Look into the assembly3 subdirectory, especially at the Isotigs files

GUI interface to Newbler
• gsAssembler
• Which Urmi will explain now.

Experiment 208: Using the GUI
Run: gsAssemblerGraphical interface should appear
Can use: /dataset2 (or other dataset)
• Choose options and run the assembly
• Look at the resulting assembly in the viewing tab.• What do you think about the accuracy of the
assembly?

Roche's software also includes:• Gs Reference Mapper for mapping to reference (for
model organisms can specify file of known annotations and SNP's)
• Amplicon Variant analyser for analysing DNA variants (eg rare alleles) in ultra deep coverage of regions of interest. (see manual Part D on website for more information)
• File Tools: • sffinfo extract fasta, quality and flowgrams as text from .sff
files.• sfffile join sff files; extract part of sff file by MIDs, read names
or random reads; or trim reads in user-defined ways.• sff2scf converts one read from sff file into an SCF file (or
performs “call throughs” to access SCF data for Sanger reads)• fnafile Constructs a FASTA file (& quality file) from list of
FASTA, PHD and SCF files.

Experiments• Now have three assemblies.
• Also view your assemblies in the viewers which Ben will be discussing next.
• Final 4th assembly which is a more challenging transcriptome assembly so if time we can try it.

Viewing the assembly in GUI
• Finish looking at the assembly in the GUI.


Videos about 454 sequencing
• Pyrosequencing:
http://www.youtube.com/watch?v=kYAGFrbGl6E
• Genome Sequencer FLX System Workflow:
http://www.youtube.com/watch?v=bFNjxKHP8Jc





























