Embed Size (px)
Citation preview

Interstitial Lung DiseaseApproach to Diagnosis
Sandra B.Weibel MD

General
• Broad category of diseases that cause inflammation and/or fibrosis of the lung
• May be up to 15% of pulmonary appts
• Need for make accurate diagnosis for therapeutic implications
• Many different disease entities

ILD
• Due to increasing numbers of cytotoxic drugs, increased detection of occupational lung disease and increasing life expectancy, as well as better imaging and diagnostic testing the incidence of these diseases is expected to rise

Definition
• “Interstitial lung disease”- a misnomer• Few cells in the interstitium of the normal lung• Injury to basement membrane shared by
epithelium and endothelium• Increased alveolar permeability and spillage of
serum contents into the alveolar space and recruitment of fibroblasts
• Collagen deposition• Also injury to small airways=respiratory
bronchioles, alveolar ducts and terminal bronchioles

• Interstitial compartment is the portion of the lung sandwiched between the epithelial and endothelial basement membrane
• Expansion of the interstitial compartment by inflammation with or without fibrosis– Necrosis– Hyperplasia– Collapse of basement
membrane– Inflammatory cells
What is the Pulmonary Interstitium?

Pathogenesis
• UNKNOWN!!!• Some understanding of the mechanism of
injury• Initiating injuries are likely multiple=
inhaled, sensitization to allergens, circulatory
• With continued injury, “repair” process continues with additional fibroproliferation that is unchecked

Incident Cases of ILD
Sarcoidosis8%
Occupation11% DILD
5% DAH4%
CTD9%
Other11%
Pulmonary Fibrosis52%
Coultas AJRCCM 1994; 150:967
(Incidence of IPF=26-31 per 100,000)

Clinical Presentation
• Dyspnea on exertion or a persistent non productive cough
• Abnormal CXR
• Pulmonary symptoms associated with another disease, such as CVD
• PFT abnormalities

Clinical Assessment
• History• Physical Exam• Chest Radiograph• Pulmonary Function Testing
– At Rest– Exercise
• Serologic Studies• Tissue examination

General
• Extensive history very important
• Symptoms often generic with dyspnea and cough
• Systemic symptoms often very important
• Timing and pace
• Demographics

History
• Review of systems very important for extrapulmonary complaints
• Smoking status
• Hobbies
• Medication list
• Occupation
• Exposure history
• Family history


History: Smoking• All of the following
DPLD are associated with smoking
a) IPF
b) RBILD
c) DIP
d) Histiocytosis X
• In Goodpasture’s syndrome– 100% of smokers vs. 20%
of nonsmokers experience pulmonary hemorrhage
• Individuals exposed to asbestos who smoke are more likely to develop asbestosis

3. Subacute Diseases (weeks to months)
• HSP, Sarcoid, Cellular NSIP, Drug,
“Chronic” EP, Bronchiolitis __________________________________________________________________________________________________________________
4. Chronic Diseases (months to years)
• UIP, Fibrotic NSIP, Pneumoconioses,
CVD-related, Chronic HSP
Smoking (RBILD and PLCH)
2. Acute Diseases (Days to weeks)
• DAD (AIP), EP, Vasculitis/DPH, Drug, CVD________________________________________________________________________________________________________________
History: Duration of Illness

History: Age and Gender
– LAM– Tuberous sclerosis– Pneumoconiosis
Age Gender

ORGANIC: Hypersensitivity Pneumonitis

Occupational ????

History: Occupational and Environmental
INORGANIC

www.pneumotox.com
History: Medications
Schwartz, ILD text book, 4th edition


Physical Findings
• Resting Tachypnea• Shallow breathing• Dry crackles• Digital clubbing• Pulmonary HTN• Non-pulmonary
findings



Laboratory

ILD: Evaluation
• Radiographic– CXR – HRCT
• Physiologic testing– PFT– Exercise test
• Lung Sampling– BAL– Lung biopsy: (TBBx, Surgical)

CXR: LlMITATIONS
• CXR is normal:– in 10 to 15 % of symptomatic patients with
proven infiltrative lung disease– 30% of those with bronchiectasis– ~ 60 % of patients with emphysema
• CXR has a sensitivity of 80% and a specificity of 82% percent for detection of DPLD
• CXR can provide a confident diagnosis in ~ 23 % of cases

A normal CXR does not rule out the presence of DPLD

CXR CLUES
Alveolar Filling
• Air-bronchograms
• Acinar rosettes
• Diffuse consolidation
• Nodule like, poor boarder definition
• Silhouetting: obliteration of normal structures

Interstitial Infiltrates
• Nodular
• Linear or reticular
• Mixed
• Honeycomb
• Cysts and traction bronchiectasis
• GGO
CXR CLUES

Radiographic Patterns in ILD
Pleural Involvement
Lymphangitic Carcinomatosis LAMDrug InducedRadiation PneumonitisAsbestosis
EffusionThickeningPlaquesMesothelioma
Collagen vascular disease
Kerley B lines
Chronic LV failureLymphangitic CALymphomaLAMVeno-occlusive diseaseAcute Eosinophilic Pneumonia
Adenopathy
SarcoidosisLymphomaLymphangitic CALIPAmyloidosisBerylliosisSilicosis

HRCT• 2 essential technical factors:
– Narrow collimation– Use of a high spatial frequency reconstruction
algorithm
• Does not use contrast
• Prone and supine
• Inspiratory and expiratory

The terminal bronchiole in the center divides into respiratory bronchioles with acini that contain alveoli. Lymphatics and veins run within the interlobular septa
Centrilobular area in blue (left) and perilymphatic area in yellow (right)

HRCT Clues
• What is the dominant HR-pattern: – Reticular – Nodular – High attenuation (ground-glass, consolidation) – Low attenuation (emphysema, cystic)
• Where is it located within the secondary lobule (centrilobular, perilymphatic or random)
• Is there an upper versus lower zone?• Central versus peripheral predominance • Are there additional findings (pleural involvement,
lymphadenopathy, traction bronchiectasis)

HRCT: Radiographic Pattern

Ground Glass Pattern• HP• PCP pneumonia• DIP• NSIP• PAP• DAH• Fluid

Pulmonary Function in IPF
• “Restrictive” impairment• Decreased FVC due to decreased
compliance of the lung• Normal to increased FEV1 /FVC ratio due
to traction on airways which maintains patency even at low lung volumes

Cysts or Cyst Like
LAMBronchiectasis
E
EG

Probability of Histologic Diagnosis of Diffuse Diseases
Surgical Biopsy
1. Granulomatous diseases
2. Malignant tumors/lymphangitic
3. DAD (any cause)
4. Certain infections
5. Alveolar proteinosis
6. Eosinophilic pneumonia
7. Vasculitis
8. Amyloidosis
9. EG/HX/PLCH
10. LAM
11. RB/RBILD/DIP
12. UIP/NSIP/LIP COP
13. Small airways disease
14. PHT and PVOD
Often
Sometimes
Never
Transbronchial Biopsy
Courtesy of Kevin O. Leslie, MD.

Approach to the ILD Patient
Martinez F, Flaherty K. Available at: http://www.chestnet.org/education/online/pccu/vol18/lessons03_04/lesson03.php.
Patient with Suspected ILD
Hx, PE, CXR, PFT, Labs
STOPHRCT
Hx and HRCT consistentwith IPF
Hx and HRCT Dx of other
ILD
Suspected other ILD
Atypical clinical or CT features of IPF
STOP STOP
STOPVATS
UIP Non IIPLIPOPDADDIPNSIP RBILD
Yes
No
Yes
No
Dx likely by bronch?
Is bronch diagnostic?
Dx likely by bronch?
Is bronch diagnostic?
Yes
Yes
No

Adapted from Ryu JH, et al. Mayo Clin Proc. 1998;73:1085-1101.Adapted from ATS/ERS. Am J Respir Crit Care Med. 2002;165:277-304.
1970Liebow and CaringtonLiebow and Carington
2002ATS/ERSATS/ERS
UIPNSIP
DIP-RBILDAIP
UIP/IPF
NSIP
DIPRB-ILD
AIP
Cellular
Fibrotic
COP
LIP
Historical Classification of IIP
UIPDIP
UIP-BOLIP
Giant cell IP
1997KatzensteinKatzenstein

Pathologic Classification

Modified Liebow classification of the idiopathic interstitial pneumonias (Katzenstein)
• Acute• Acute interstitial pneumonia (AIP)
• Chronic• Usual interstitial pneumonia (UIP)
• Subacute• Nonspecific interstitial pneumonia (NSIP)• Lymphocytic Interstitial Pneumonia (LIP)• Cryptogenic Organizing Pneumonia (COP)• Desquamative interstitial pneumonia/ (DIP) Respiratory bronchiolitis-associated interstitial lung disease (RBILD)


Idiopathic Pulmonary Fibrosis - Definition
• A distinct type of chronic fibrosing interstitial pneumonia of unknown cause, limited to the lungs, and associated with a surgical lung biopsy showing a histological pattern of usual interstitial pneumonia (UIP)

Clinical Features of IPF
• Fine inspiratory “Velcro” rales at lung bases are virtually universal at presentation
• Clubbing of fingers and toes 25-50%
• No skin or joint abnormalities

Criteria for Diagnosis of IPF
• Major criteria– Exclude known causes
of ILD– Abnormal PFT– Typical findings on
HRCT– No findings on
transbronchial lung biopsy or BAL supporting other dx
• Minor criteria– Age > 50 yr– Insidious onset of SOB– Durations of
symptoms in excess of 3 months
– “Velcro” rales at lung bases

Diagnosis of IPF
• In absence of surgical biopsy– All 4 major criteria AND three of four minor
criteria
• This emphasizes need for open lung biopsy but does not require it for diagnosis.

Risk Factors for IPF
• Age
• Smoking
• Male gender
• Exposure to metal dust and sawdust
• Genetics (2% of all cases are familial)

Epidemiology of IPF
Estimated 83,000 CurrentPatients in the United States
Prevalence
Weycker D, et al. Prevalence, Incidence, and Economic Costs of Idiopathic Pulmonary Fibrosis. Paper presented at: CHEST 2002, November 2-7, 2002; San Diego, California.
Estimated 31,000 NewPatients per Year in the United States
Incidence
0
50
100
150
200
250
300
45-54 55-64 65-74 75+
Male
Female
0
20
40
60
80
100
120
45-54 55-64 65-74 75+
Male
Female
Age Age
Per
100
,000

Radiographic Features of IPF
• PA Chest Radiograph– Reticulo-nodular
infiltrates with predilection for bases
– Decreased lung volumes
– Hazy cardiac and phrenic borders
– Normal in 2-10%
• HRCT of Chest– Subpleural and
paraseptal distribution of infiltrates
– Honeycomb fibrosis is frequent
– “Ground Glass” opacities are rare
– “Traction” bronchiectasis

Images courtesy of W. Richard Webb, MD.
IPF: CXR
Basal and peripheral reticulationReduced lung volume

IPF: Chest Radiograph vs. CT ScanIPF: Chest Radiograph vs. CT Scan

Classic IPF HRCT
Image courtesy of W. Richard Webb, MD.
Reticular opacities Tractionbronchiectasis
Honeycombing
Basal and subpleural predominance

Role of HRCT in Dx of IPF
• Useful when read by experienced clinician– Sensitivity 95%– Specificity 66%
• Less useful in inexperienced hands!• Extensive amount of ground glass opacities
strongly suggest another disease.• When a “confident diagnosis” cannot be made
by HRCT, a surgical biopsy should be strongly considered.

HRCT in IPFHRCT in IPF
Paraseptal FibrosisSubpleural “honeycombing” fibrosis

““Traction” BronchiectasisTraction” Bronchiectasis

Geographic Heterogeneity of UIP

Bronchoscopy in IPF
• Transbronchial Lung Biopsy
• BAL findings


Histology of NSIP

UIP vs NSIP Pathology

Nonspecific Interstitial Pneumonia
• Younger age group than IPF (40-50 years)• Subtle onset is usual but may be subacute• No male predominance• Smoking is not a risk factor• Clubbing is less common than in IPF

Nonspecific Interstitial Pneumonia
• DLco is low and exercise induced desaturation is common
• Radiographic diagnosis is more challenging• Surgical biopsy is necessary to confirm the
diagnosis• NSIP may be seen in conjunction with UIP in
patients with IPF!

HRCT NSIP vs. UIP
NSIPNSIP UIPUIP

NSIP: Non Specific Interstitial PneumoniaCT features
Ground-glass opacity is the predominant finding: 75 to 100%.
Ground-glass opacity is the sole abnormality in 1/3 of cases.
Bilateral and symmetrical with subpleural predominance.
Cottin V et al. Am J Respir Crit Care Med 1998; 158: 1286-1293Kim TS et al. AJR 1998; 171: 949-953
Johkoh T et al. Radiology 1999; 211: 555-560Hartman TE et al. Radiology 2000; 217: 701-705

NSIP: Non Specific Interstitial PneumoniaCT features
• Ground glass attenuation 76%• Irregular linear opacities 46%• Honeycombing 30%• Consolidation 16%• Nodular opacities 14%• Interlobular septal thickening 6%• Traction bronchiectasis 36%
Hartman TE et al. Radiology 2000; 217: 701-705
50 patients with biopsy-proven NSIP
Wide variety of CT findings

NSIP vs IPF: Comparative Appearances at
and Diagnostic Accuracy of HRCT
MacDonald SLS et al. Radiology 2001; 221: 600-605
Accuracy of HRCT for the discrimination between NSIP and UIP: 66%
Subpleural
Basal no difference
Bronchocentric distributions
Greater proportion of GGO
Finer fibrosis in NSIP than UIP
Considerable overlap in thin-section CT patterns

Prognosis of NSIP
• Much better than in IPF
• Response to anti-inflammatory drugs is common
• Relapse is uncommon
• Outcome probably determined by the severity of fibrosis on biopsy and restrictive disease on PFT

Am J Respir Crit Care Med. 1999;160:899-905. Am J Respir Crit Care Med.1998;157:199-203.
Years
Survival for UIP Vs. NSIP
Years
76543210
0
20
40
60
80
100
UIP
NSIP
0 2 4 6 8 10
12
14
16
18
0
20
40
60
80
100
UIP
NSIPAli
ve (
%)
Ali
ve (
%)

NSIPNon Specific Interstitial Pneumonia
Katzenstein AL. Am J Surg Pathol 1994; 18: 136-147
Cellular pattern Fibrosing pattern

Better prognosis than IPF
Responds to steroids
Overlap in CT appearance UIP – NSIP
Surgical biopsy, in atypical presentation of UIP
NSIP: Non Specific Interstitial Pneumonia In Summary
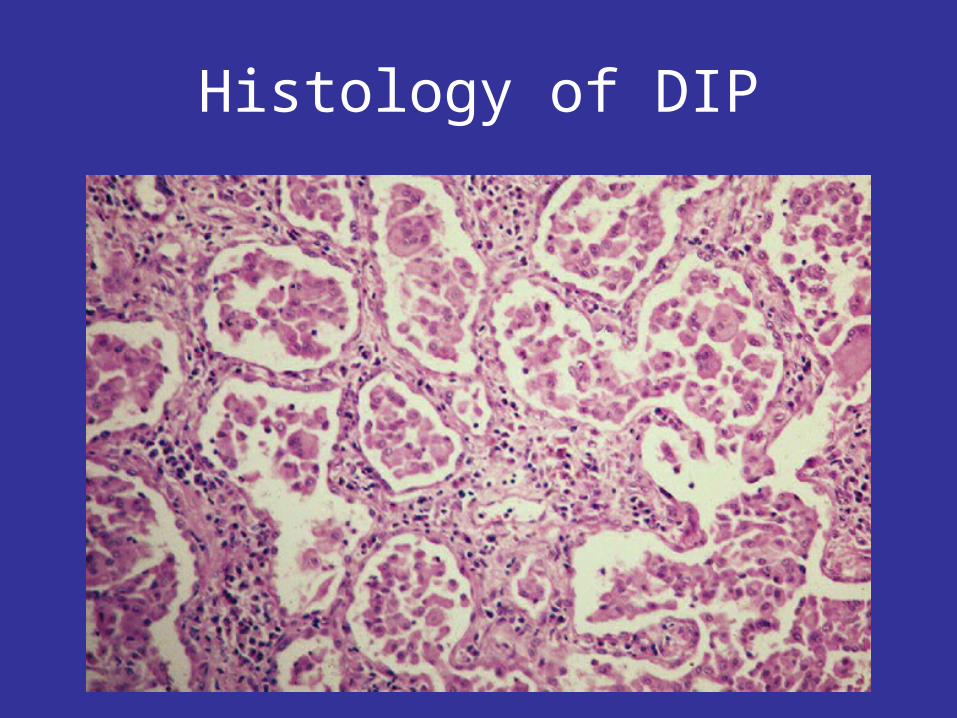
Histology of DIP

Desquamative Interstitial Pneumonia or DIP
• Male predominance• 90% of cases smoked• Less common than IPF• “Restrictive” PFT’s• Ground glass opacities• 20% respond to smoking
cessation• 75% respond to CS

Respiratory Bronchiolitis- ILD
• Smoking universal!• 3rd - 4th decade• Normal CXR• Reticular markings on
HRCT• Restriction + obstruction
on PFT• Smoking cessation!• Prognosis is variable

Acute Interstitial Pneumonia
• Abrupt onset often following viral illness
• No age predilection• Air space disease on
HRCT• Rapidly progresses to
death in 50%• Mimics ARDS but has no
apparent cause• Often a terminal event in
patients with IPF!

Summary of the IIP’s
• Causes remain unclear• Pathogenesis depends on conditions in the
alveolar microenvironment • Diagnosis requires surgical lung biopsy in most
cases• Natural history and response to anti-
inflammatory therapy is variable • IPF is clinically the most important type of IIP• UIP, NSIP, COP and AIP may occur in the
setting of connective tissue disease and drug induced disease


NSIP: Non Specific Interstitial Pneumonia In Summary
Basal-predominant ground glass attenuation
Reticular pattern
Traction bronchiectasis
Honeycombing ?

IPF: 47-64%
NSIP: 14 to 36%
RBILD/DIP: 10-17%
COP: 4-12%
AIP: 2%
LIP: 2%

Secondary pulmonary lobular anatomy

Supine Prone
Conventional HRCT


Pulmonary Function in IPF
• Abnormal oxygen exchange• Decreased DLco
– Diffusion block – increased distance between blood and oxygen
– Diffusion limitation – high velocity of blood flow limits how much oxygen can be transferred
• Widened (A-a)pO2 gradient– Almost always found with exertion
– May be present at rest in more advanced disease leading to hypoxemia
• Exchange of CO2 is normal until very late in the course of disease (hypercapnea is a terminal event)

PFT: Lung VolumesRestrictive Disease
TLC
RV
VC
TLC
RV
VCTLC
RV
VC
Normal ILD NM Disease

Peripheral Location
COP IPF

Genetics and IPF
• Familial disease accounts for up to 2% of cases– Autosomal dominant with variable
penetrance– M = F– Recent report of a mutation in surfactant C
protein in familial pulmonary fibrosis with UIP and in a kindred with NSIP*
*Am J Respir Crit Care Med 2002; 165:1322

NSIP: Non Specific Interstitial PneumoniaRadiographic features
Bilateral pulmonary infiltrates
Patchy parenchymal opacity
The lower zones are more frequently involved.
… No large detailed analysis of radiographic appearance!
ATS/ERS Multidisciplinary Consensus Classification of IIPs. Am J Respir Crit Care Med 2002; 165: 277-304

NSIP: Non Specific Interstitial PneumoniaCT features
Irregular linear and reticular opacities in ½ of cases,
may be associated with traction bronchiectasis.
Honeycombing and consolidation are relatively infrequent.
Cottin V et al. Am J Respir Crit Care Med 1998; 158: 1286-1293Kim TS et al. AJR 1998; 171: 949-953
Johkoh T et al. Radiology 1999; 211: 555-560Hartman TE et al. Radiology 2000; 217: 701-705

NSIP: Non Specific Interstitial PneumoniaCT – Pathologic Correlation
GGO = interstitial thickening• Varying amounts of interstitial inflammation and fibrosis
Irregular linear opacities and bronchial dilatation in GGO• Interstitial fibrosis and microscopic honeycombing
Consolidation• Area of organizing pneumonia, with or without microscopic honeycombing
Kim TS et al. AJR 1998; 171: 949-953

NSIP: Non Specific Interstitial PneumoniaSequential CT after steroids
Complete resolution of parenchymal abnormalities.
Bronchiectasis may resolve or may progress.
Bronchiectasis and bronchiolectasis may occurs after the
initial work-up.
Consolidation may decrease or evolves into honeycombing.
Akira M. Thorax 2000; 55: 854-859Nishiyama O et al. J Comput Assist Tomogr 2000; 24: 41-46
Complete recovery after steroids may occur.
50% have persistent abnormalities including bronchiectasis and honeycombing.

NSIP vs IPF: Different Survivals
Riha RL et al. Eur Respir J 2002; 19: 1114-1118
IPF NSIP P- value
Number of included patients
53 7
Median Survival (months)
78 178 0.03

NSIP: Non Specific Interstitial PneumoniaCT features
• Ground glass attenuation 76%• Irregular linear opacities 46%• Honeycombing 30%• Consolidation 16%• Nodular opacities 14%• Interlobular septal thickening 6%• Traction bronchiectasis 36%
Hartman TE et al. Radiology 2000; 217: 701-705
50 patients with biopsy-proven NSIP
Wide variety of CT findings

Hartman TE et al. Radiology 2000; 217: 701-705
Alternative Diagnosis n (39)IPF 16
EAA 10
BOOP (= COP) 7
DAD 1
LIP 1
Alveolar proteinosis 1
RB-AILD 1
Nondiagnostic 1
No disease 1
NSIP: Non Specific Interstitial PneumoniaAlternative CT diagnosis

Honeycomb lung

Chronic Alveaolar InfitratesW (Wegner’s) E (Eosinophilic pneumonia)B (BOOP, BAC)A (PAP, Aspiration)L (Lymphoma)L (Lipoid Pneumonia)S (Sacroidosis)
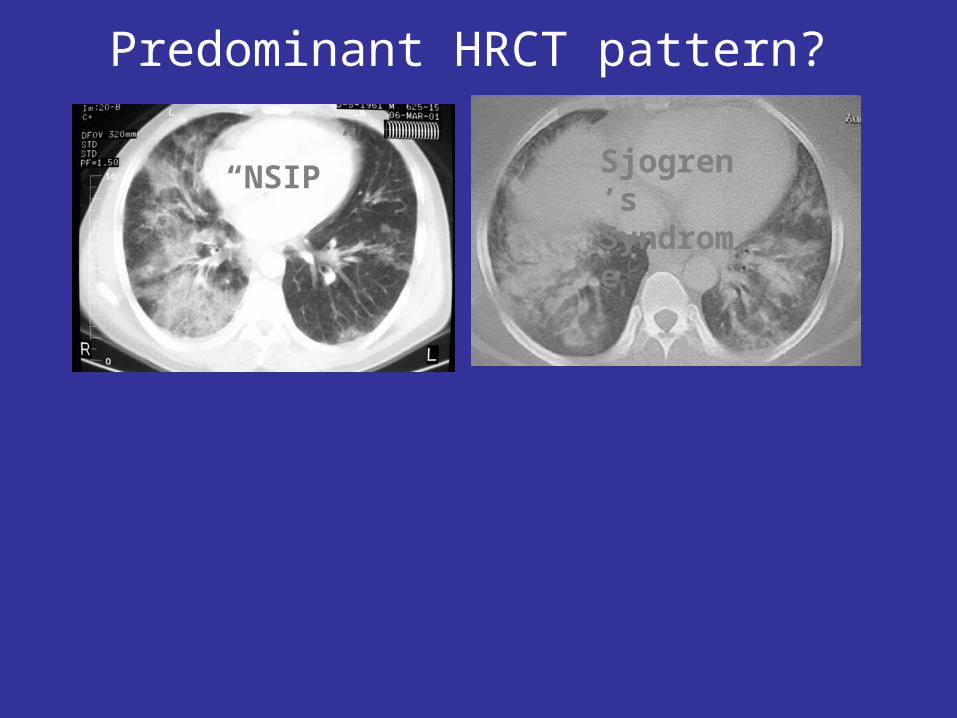
Predominant HRCT pattern?
“NSIP” Sjogren’s Syndrome

HRCT Findings in Late IPF
Slide courtesy of G Raghu, MD.

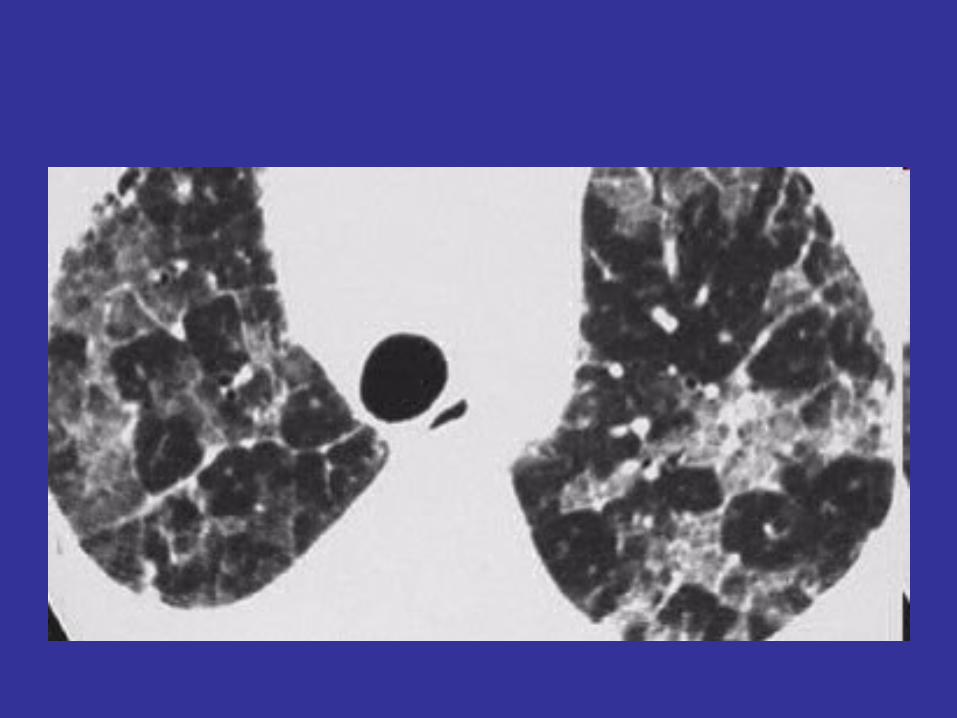


DIP: Radiographics
• CXR:– Normal: 3-22%– Patchy GGO– lower zone predilection
• HRCT:– GGO
• LL distribution (73 %)• peripheral distribution (59%)



Interstitial Lung Diseases
• Definition
• Clinical Presentation and evaluation
• Radiographic Features
• Classification

ILD
• 81 in 100,000 prevalence in men• 67 in 100,000 in women
• 32.5 in 100,000 incidence in men• 26 in 100,000 in women
• 200 in 100,000 incidence in age >75• 30-40% of all ILD “IPF”

Evaluation of ILD
• EXTENSIVE HISTORY • AGE, GENDER, UNDERLYING
COMORBIDITIES, DRUGS, SMOKING, OCCUPATIONAL HISTORY, HOBBIES, PETS, FAMHX
• DURATION OF SYMPTOMS• PHYSICAL EXAM• LABORATORIES• IMAGING• SPIROMETRY, LUNG VOLUMES AND DLCO

Imaging
• PA/LAT CXR
• HRCT

Schwarz, ILD, 2003, RB
GROUND GLASS

Schwarz, ILD, 2003, HP
NODULAR GROUND GLASS

Schwarz, ILD, 2003, LC
INTRALOBULAR SEPTAL THICKENING

Schwarz, ILD, 2003, HP
RETICULAR INFILTRATES

Schwarz, ILD, 2003
HONEYCOMB LUNG

HONEYCOMB LUNG

Clinical Classification
• Connective Tissue Diseases
• Drug-induced
• Primary Unclassified
• Occupational
• Idiopathic Disorders

Clinical Classification-- CTD
• Scleroderma• Polymyositis-dermatomyositis• Systemic lupus erythematosus• Rheumatoid arthritis**• Mixed connective tissue disease• Ankylosing spondylitis• Behcet’s• Sjogren’s syndrome

Primary- Unclassified• Sarcoidosis• Eosinophilic granuloma• Amyloidosis• Lipoid pneumonia• Lymphangitic
carcinomatosis• Broncholaveolar carcinoma• Pulmonary lymphoma• Gaucher's disease• Niemann-Pick disease• Hermansky-Pudlak
syndrome• Neurofibromatosis
• LAM• Tuberous sclerosis• Acute respiratory distress
syndrome• AIDS• Bone marrow transplantation• Postinfectious• Eosinophilic pneumonia• Alveolar proteinosis• Diffuse alveolar hemorrhage
syndromes• Alveolar microlithiasis• Metastatic calcification

Occupational
• Silicosis- tile, glass • Asbestosis • Hard-metal pneumoconiosis • Coal worker's pneumoconiosis • Berylliosis-fluorescent bulbs, dental, electronics • Aluminum oxide fibrosis-abrasives • Talc pneumoconiosis • Siderosis (arc welder)-iron foundry, welders• Stannosis (tin)

Idiopathic
Acute interstitial pneumonitis (Hamman-Rich syndrome)
Familial idiopathic pulmonary fibrosis
Desquamative interstitial pneumonitis
Respiratory bronchiolitis
Cryptogenic organizing pneumonia
Nonspecific interstitial pneumonitis
Lymphocytic interstitial pneumonia (Sjögren's syndrome, connective tissue disease, AIDS, Hashimoto's thyroiditis)
Autoimmune pulmonary fibrosis (inflammatory bowel disease, primary biliary cirrhosis, idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia)

Idiopathic ILD
• Diagnosis is made based upon clinical history, radiographic findings and pathology
• Some pathologic findings are pathognomonic for a particular disease, while others are suggestive in the correct clinical setting

IPF• Major Criteria
– • Exclusion of other known causes of ILD, such as certain drug toxicities, environmental exposures, and connective tissue diseases
– • Abnormal pulmonary function studies that include evidence of restriction (reduced VC often with an increased FEV1 /FVC ratio) and impaired gas exchange [increased AaPo2 with rest or exercise or decreased DLCO ]
– • Bibasilar reticular abnormalities with minimal ground glass opacities on HRCT scans
– • Transbronchial lung biopsy or bronchoalveolar lavage (BAL) showing no features to support an alternative diagnosis
• Minor Criteria – • Age > 50 yr – • Insidious onset of otherwise unexplained dyspnea on exertion – • Duration of illness 3 mo – • Bibasilar, inspiratory crackles (dry or “Velcro” type in quality)
IPF consensus statement, 2000, AJRCCM