Embed Size (px)
Citation preview

INFORMAnON TO USERS
This manuscript has been reproduced from the microfilm master. UMI films
the text directly trom the original or c:opy submitted. Thus, sorne thesis and
dissertation copies are in typewriter face, white 0Chers may be from any type of
computer printer.
The quality of this ntproduetion is dependent upon the quality of the
copy submitted. Broken or indistinct print, coIor8d or po« quality illustrations
and photographs, print bleedlhrough, substandard margins, and improper
alignment can adverselyaffect Atproduction.
ln the unlikely event that the author did not send UMI a complete m..uscript
and there are missing pages, these will be notect. AJso, if unauthorized
copyright material had to be removed, a note will indicate the deletion.
Oversize materials (e.g., maps. drawings, charts) are reproduced by
sectioning the original, begiming al the upper Ieft-hand camer and continuing
from left to right in equal sections with small overtaps.
Photographs inducted in the original manuscript have been reproduced
xerographically in this copy. Higher quality 6- x 9" black and white
photographie prints are available for any photographs or illustrations appearing
in this copy for an additional charge. Contad UMI directly to order.
Bell & Howell Information and Leaming300 North Zeeb Raad. Ann Arbor, MI 48106-1346 USA
800-521-0600


•
•
.'
The M3 Muscarinic Acetylcholine Receptor Mediates p42mapk
Activation and c-fos mRNA Expression in Oligodendrocyte
Progenitors
By
FADIRAGHEB
Department of Pharmacology and TherapeuticsMcGill UniversityMontreal, Canada
March,1999
A thesis submitted to the Faculty of Graduate Studies and Research in partial fulfillmentof the requirements for the degree of
Master of Science
Copyright © Fadi Ragheb) 1999
1

1+1 National Libraryof Canada
Acquisitions andBibliographie services
395 Wellington StreetOttawa ON K1A OPUCanada
Bibliothèque nationaledu Canada
Acquisitions etservices bibliographiques
395. rue wellingtonOttawa ON K1 A. 0N4canada
The author bas granted a nonexclusive licence allowing theNational Library ofCanada toreproduce, loan, distribute or sencopies of this thesis in microfo~paper or electronic formats.
The author retains ownership of thecopyright in this thesis. Neither thethesis nor substantial extracts from itmay be printed or otherwisereproduced without the author' spermission.
L'auteur a accordé une licence nonexclusive permettant à laBibliothèque nationale du Canada dereproduire, prêter, distribuer ouvendre des copies de cette thèse sousla forme de microfiche/film, dereproduction sur papier ou sur fonnatélectronique.
L'auteur conserve la propriété dudroit d'auteur qui protège cette thèse.Ni la thèse ni des extraits substantielsde celle-ci ne doivent être imprimésou autrement reproduits sans sonautorisation.
0-612-50862-5
Canad~

•
•
•
TABLE OF CONTENTS
ABSTRACT•..•••...••.•.•.••.•••••.•••..••••.•••.•••.•••..••.•••••.•.•.••.•..••••••..••..•••.••...•.•..•••••••••••2ABSTRAIT•••••••••••••.•..•.•••...•••••••••.••.••••••••••••••••••••.•••.•••.••••••••.••.•••••••.•••.••..•..••••.••4ACKNOWLEDGMENTS.....•••••....•••..•.••.•••••••••••.•..•••.•••.•••••••••••••••.••••••...••....•••.••••••7ABBREVIATIONS••.••.••.•••.•••••..•.•••...•......•...••.•.••....••••••••••••••••••••••.•••.•••••.••••.••••••••9
CHAPTER 1: INTRODUCTION 12
BRJEF OVERVIEW OF HISTORICAL DEFINITIONS AND FUNCTION Of OL•..•...•.•..••.•13Introduction , _ 13
STAGES OF OLiGODENDROCYTE DEVELOPMENT 15Origin of Oligodendrocytes , , , ." ,. '" '" , 15Overview of O/igodendrocyte Developmental Lineage _ , 16
OLIGODENDROCYTE TYPE 2A A5TROCYTE PROGENITOR , , 19Developmenta/ Regulation , , 20Ofigodendrocyte Progenitor Differentiation and Maturation , , , 22
THE MUSCARINIC ACETYLCHOLINE RECEPTOR••.•.••....•.••••.........•••••..••.•.......•....••24Introduction , '" ., , , '" 24Molecular Bi%gy of the mAChR. , '" " _ 26Pharmacological Definitions of Subtypes , -28
MUSCARINIC COUPLING Ta G-PROTEINS AND EFFECTOR MOLECULES•.•••..•.•.•.••••31Introduction , ., , , '" ,_ , '" 31Composition and Structure ofHeterotrimeric G-proteins , , 31Adenylate Cyc/ase , 34Phospholipase C '" , , '" ., 36Phospho/ipase A2 _ , 37
MITOGEN-ACTIVATED PROTEIN KINASE•...••.••..•.........1••••••••••••••••••••••••••••••••••••••••••39Brief Overview: The MAPK Classes , , , , 39G-protein Coupled Receptors Signaling to MAPK. _ , , 40Novel Signaling Pathways Linking G-protein Coup/ed Receptor to MAPK Pathway.. .42Mapk Regulation of AP-1 Transcription Factor and the Induction of c-fos geneExpression , , , '" 45
CHAPTER TWO: STATEMENT OF PURPOSE..•.•...•••.••..•..••••..............•.49
CHAPTER THREE: MATERIALS AND METHODS••...•••••••.•••..................53Materia/s '" '" _.. , '" '" '" , , , '" , .54Cell Culture , '" .. , " , , , ,.. ,.54Immunocytochemistry , , , '" , , 56Western /mmunoblot Analysis _ '" .. ' , .. ' '" , ,.. ,..57Data Ana/ysis '" '" _ '" 58
CHAPTER FOUR: RESULTS....•.•.••••••••••••••...••.•••...•.••••.•••••...•.........•.•.60Resu/ts 61Figures 70
CHAPTER FIVE: DiSCUSSiON..•.•••.••••••••••••••••.••••••••••••••••••.••...•......••••82
CtiAf»1rE:Ft 51)(: FtE:f=E:FtE:..c:E:!) St6

•
•
•
ABSTRACT
Oligodendrocytes, the ceUs that produce and maintain myelin in the central
nervous system CCNS), express several muscarinic acetylcholine (ACh) receptor
(mAChR) subtypes. Carbachol (CCh), a stable analogue of ACh, stimulates various
intracellular signais through mAChRs, including p42-mitogen-activated protein kinase
(p42mapk) activation and c-fos gene expression. The present study examined the mAChR
subtype(s) and the signaling pathway(s) involved in both responses using purified cultures
of oligodendrocyte progenitors from newbom rats.
The M3 mAChR selective antagonist 4-DAMP and its irreversible analogue 4
DAMP-mustard, but not pirenzepine (M1) or methoctramine (M21M4), prevented p42mapk
activation and c-fos mRNA expression. Pretreatrnent with a phospholipase C (PLC)
inhibitor U73122 but not the inactive analogue U73343 blocked both responses, while the
phosphoinositide 3-kinase (PI3K) inhibitor wortmannin had no effect, suggesting the
involvement ofPLC but not PI3K.
In view of the fact that the M3 mAChR mediates p42mapk activation as weB as c
fos mRNA expression, the MAPK Kinase (MEK) inhibitor PD 098059 was used to
establish a link between both responses. PD 098059 pretreatment caused no significant
attenuation in c-fos mRNA levels, demonstrating that this response may not be dependent
on p42mapk activation. However, the phospholipase Al (PLA2) inhibitors quinacrine and
AACOCf3 abolished c-fos mRNA expression, implicating arachidonic acid and/or its
bioactive eicosanoid metabolites.
2

• ln summary, these results demonstrate that in oligodendrocyte progenitors, the M3
mAChR is mediating both p42D1apk activation and c-fos gene expression via PLC but not
PI3K, whereby c-fos gene expression is not regulated by p42mapk but by PLA2.
•
•3

•
•
•
RÉSUMÉ
Les oligodendrocytes produisent et maintiennent la myéline dans le système
nerveux central (SNC). Ces cellules possèdent plusieurs sous-types des récepteurs
acétylcholine (Ach) muscariniques (mAChR). Un analogue stable de l'acétylcholine,
carbachol (CCh) stimule de nombreux signaux intracellulaires à partir des mAChRs, dont
l'activation de la kinase p42mapk et l'expression du gène c-fos. La présente étude a
exanliné le(s) sous-type(s) des mAchRs et ces deux réponses intracellulaires, en utilisant
des cultures purifiées des prédécesseurs d'oligodendrocytes provenant de rats nouveaux
nés.
4-0AMP et son analogue irréversible 4-0A~IP-mustard sont des antagonistes
sélectifs du sous-type M3 des mAChRs et ont obvié l'activation de p42mapk et l'expression
de c-fos. à l'opposé de pirenzine (Ml) et methoctramine (M2/M4). Pré-traitement avec
U73122, qui inhibe la phospholipase C (PLC), a supprimé les deux réponses.
Wortmannin, un inhibiteur de la phosphoinositide 3-kinase (PI3K), et U73343, un
analogue inactif d'U73 122, n'ont eu aucuns effets sur ces deux événements. Ces résultats
suggèrent que PLC, mais pas PB-K, est impliqué dans la signalisation intracellulaire à
partir du récepteur M3 avec l'utilisation du CCh.
Étant donné que le récepteur M3 de cette famille cause à la fois l'activation de
p42mapk et l'expression de c-fos, PD 098059, un inhibiteur de la MAPK Kinase (MEK),
fut utilisé pour déterminer si un lien existe entre ces deux réponses. Pré-traitement avec
4

•
•
•
PD 098059 n'a pas causé d'atténuation significative sur l'expression d'ARN messagères,
démontrant que cette réponse ne dépend pas de l'activation de p42mapk• D'autre p~ les
inhibiteurs de la phospholipase A2 (PLA2) quinacrine et AACOCf3 ont aboli
l'expression d'ARN messagères du gène c-fos. Ces résultats impliquent l'acide
arachidonique et/ou ses métabolites eicosanoïdes qui sont bioactifs.
En sommaire, ces résultats démontrent que dans les prédécesseurs
d'oligodendrocytes, le M3 mAChR est responsable pour l'activation de p42mapk et
l'expression du gène c-fos. PLC, mais pas PI3-K, est impliqué dans ces deux réponses.
Par contre, l'expression de c-fos dépend de PLA2 mais pas de p42mapk•
5

•
•
•
Dedicated to my parents, for their love, confidence, and support that
they have given me during these years
6

•
•
•
ACKNOWLEDGMENTS
1 would like to thank tirst and foremost my supervisor Dr. Guillermina Almazan
for developing a scientist out of me, committed, dedicated, and inspired. Her constant
support, help, and contribution in my work are of priceless value, and her leading
example as both a scientist and a compassionate individual is of lasting influence and
guidance.
1 would also like to thank Arnani Khorchid and H. N. (Shirley) Liu for their
valuable contribution and assistance in my project , and for the lasting friendship that has
been formed during these years. 1 would like to also acknowledge Ana-Katherine
Martinez for her appreciated input and advice.
My special thanks go to my adviser, Dr. Barbara Esplin for her continuous
support and interest throughout my years al the department, and to Dr. L.f. Congote and
his Ph.O. student Marco of the Endocrinology Unit of the Royal Victoria Hospital for
providing me with the opportunity to believe in research and to allow me to observe and
experience it at first hand.
My gratitude extends to my parents that have always provided me with the
comfort and support, especially during our hard times at our early years in Canada. Their
love and guidance solidified my determination and perseverance. 1 would like to express
my deep gratitude for offering me the opportunity to have a solid education that wi 11
guide me for the rest of my life.
My expressed gratitude is also extended to my sister Shourouk, for her love,
confidence in me, and her patience, for it couldn't have been the same without her. 1aJso
express my admiration to my brother Loay for his role model personality, determination,
7

•
•
•
perseverance, and for bis brotherly guidance and support. 1 wish to express my love to
my aIder sister Loma, for her well appreciated support and guidance throughout these
years.
1 wouid like to also extend my deep gratitude to Barbara Milosevic, for her
greatly appreciated encouragement and for her continuous support. 1 also extend my
appreciation to her valuable contribution in the construction of this thesis and my ASN
poster. 1 am also very grateful to Anne Morinville for her assistance in translating my
abstract, to Martin Gagnon for his technical support for constructing my figures, and to
Alan Forster for the valuable work on the photographs.
Last but not least, 1 would like to thank the Department of Pharmacology and
Therapeutics for offering me the opportunity to complete my project in a stimulating
scientific environment.
8

• ABBREVIATIONS
AA Arachidonic acid
ACh Acetylcholine
ATP Adenosine triphosphate
Atr Atropine
bFGF Basic fibroblast growth factor
CaMK Calciurn/calmodulin-dependent protein kinases
cAMP 3',5' -cyclic adenosine monophosphate
CBP CREB-binding protein
CCh Carbachol
CNP 2'-3'-Cyclic nucleotide 3'phosphohydroIase
CNS Central nervous system
cPLA2 Cytosolic phospholipase A2
• CRE cA.MP response element
CREB cAMP response element binding protein
DAO Diacy19lycerol
DMEM Dulbecco's modified eagle medium
EDTA Ethylenediamine tetraacetic acid
EGF Epidermal growth factor
E-NCAM Embryonic neural cell adhesion molecule
ERK Extracellular-signal regulated kinase
FAK Focal adhesion kinase
FCS Fetai calf serum
GC Galactocerebroside
GDP Guanosine diphosphate
GFAP Glial fibrillary acidic protein
G-protein Guanosine triphosphate-binding protein
GTP Guanosine triphosphate• HBSS Hanks balanced salt solution
9

• HETE Hydroxyeicosatetraeinoic acid
HHSiD Hexahydro-sila-difenidol
ICso 500/0 inhibitory concentration
IGF I~II Insulin-like growth factor l, II
IP3 Inositol trisphosphate
JNK NHrterminal Jun kinase
mAb Monoclonal antibody
mAChR Muscarinic acetylcholine receptor
MAG Myelin-associated glycoprotein
MAPK Mitogen-activated protein kinase
MBP Myelin basic protein
MEK MAP kinase kinase
Meth Methoctramine
min minutes
MOG Myelin/oligodendrocyte glycoprtoein• nAChR nicotinic acetylcholine receptor
NT-3 Neurotrophin.3
0-2A Oligodendrocyte-type 2A astrocyte progenitors
aL a ligodendrocyte
aLP Oligodendrocyte progenitors
p42ffiapk p42-mitogen-activated protein kinase
PDGF Platelet-derived growth factor
PI Phosphatidylinosito1
PI3K PhosphatidyIinosito1-3-kinase
PIK Phosphatidylinositol kinase
PIP2 Phosphatidylinositol-4,5-bisphosphate
Pir Pirenzepine
PKA Protein kinase A
PKC Protein kinase C
PLA2 Phospholipase A2• PLC Phospholipase C
10

• PLD Phospholipase D
PLP Proteolipid protein
PMA Phorbol-12,13 myristate acetate
PNS Peripheral nervous system
POA Proligodendroblast antigen
Pre-Gale Pre-Galactocerebroside
Pro-OL Pro-oligodendroblast
PTX Pertussis toxin
Quin Quinacrine
Raf MAP Kinase kinase kinase
SAPK Stress-activated protein kinase
SClP Suppressed cAMP-inducible POU
SFM Serum-free medium
SIE Sis-inducible element
SRf Serum response factor• TCf Temary complex factor
Thr Threonine
TM Transmembrane
TPA Phorbol-12, 13 myristate acetate
TRE TPA response clement
Tyr Tyrosine
Wort Wortmannin
•11

•
•
•
CHAPTERONEINTRODUCTION
12

•
•
•
INTRODUCTION
(1) HISTORICAL DEFINITION AN}) FUNCTION OF
OLIGODENDROCYTES
(A) INTRODUCTION
Oligodendrocytes, which with astrocytes fonn the subclass of macroglia and
together with the microglia subclass fonn the family of neuroglial cells, are derived from
stem cell precursors arising from the subventricular zones of the developing central
nervous system (eNS). Astrocytes contribute to the structural support of the central
nervous system and function as a source of growth factors and cytokines, while microglia
are bone·marrow derived macrophages which serve as immunocompetent cells in the
central nervous system. Oligodendrocytes serve the specialist role of synthesizing and
maintaining the myelin sheath of neuronal axons. Myelin, a compact and well-organized
membranous structure. is an insulating sheath that wraps around electrically conductive
axons to maintain the electrical propagation down the axon and to prevent its dissipation
in both the CNS and the peripheral nervous system (PNS). The process of myelination
depends on glial-neuronal interaction, whereby a stable relationship between each
differentiated oligodendrocyte and short segments of several adjacent axons regulate this
process.
Rudolph Virchow (Virchow, 1858) was among the pioneers to describe neuroglia
and propose two functions for these cells: mechanical support of nerve cells and tissue
repaîr. He had also introduced the term myelin and described sheaths around nerve
13

•
•
•
fibers. Discoveries achieved in the twentietb century have confirmed the assigned
functions by Virchow, and have separated the functional roles played by astrocytes,
oligodendrocytes, and microglia in the central nervous system. Oligodendrocytes were
not identified as a distinct neuroglial subpopulation until many years after by Robertson
(Robertson, 1899), Cajal (Cajal, 1913), and Hortega (Hortega, 1921; 1928). Through
silver nitrate impregnation techniques, Hortega described the existence ofthese ceUs and
explained their morphology as oligo-dendro-glia, which translates from the Greek
language as literally "few-tree-glue". Hortega's contribution ificluded the recognition of
oligodendrocytes as the ceUs responsible for myelin formation that enwrap axons in the
white matter of the CNS.
CNS myelination is crucially dependent on the interaction of oligodendrocytes
and axons. Myelination occurs when the membranous processes of oligodendrocytes
contact and ensheath axons, and compact to fonn the myelin lamellae needed for
saltatory axonal conduction. This process also relies on cell surface and extracellular
matrix molecules that promote interactions between myelin and axons (Bartsch et al.,
1993). ln the CNS, one oligodendrocyte can myelinate more than one axonal segment,
while Schwann ceUs, the myelinating ceUs of the PNS, cao myelinate only one axon
(Morrell et al., 1993).
Due to their critical role embodied in myelin production and thus nerve impulse
maintenance, oligodendrocytes' death, degeneration, or myelin integrity aberration
results in debilitating sensory or motor conditions or diseases in humans, namely multiple
sclerosis (Quarels et al., 1993). Therefore, understanding the cellular homeostasis of
oligodendrocytes and myelin production, in addition to the environmental cues that
14

•
•
•
contribute to their development is of crucial importance. The development of severaJ in
vitro culture techniques for oligodendrocytes (McCarthy and de Vel1is, 1980; Szuchet
and Steffanson, 1980; Szuchet et al., 1980) simplified the study of their development,
metabolism, and myelin production, and facilitated the isolation and purification of
progenitor and mature oligodendrocytes from mammalian tissue.
(B) STAGES OF OLIGODENDROCYTE DEVELOPMENT
• Origin of Oligodendrocytes
Oligodendrocytes are generated over a short period of brain development during the
late gestational and early postnatal periods in mammals. In the forebrain and cerebellum,
oligodendrocyte progenitors (OLPs) commence their life as the immature
neuroectodermaI ceUs of the subventricular zone in the subependymal layer on the
surface of the lateral ventricles (Paterson et al., 1973; for review see Goldman, 1992;
Goldman, 1995). The subventricular zone is the germinal matrix area adjacent to
cerebral ventricles. Subsequently, these primordial glial ceUs migrate into gray and white
matter, whereby they differentiate and proliferate during migration. Their migration
occurs in a posto-anterior gradient (relative from the brainstem to the forebrain) to the
various areas of the brain, and also move radialy from the subventricular zone into thcir
terminal destination, the white matter nerve tracts. OLPs migrate along these nenTe
tracts, whereby they are responsive to environmental cues, leading them to proliferate,
differentiate, and synthesize myelin sheaths to enwrap adjacent axons.
15

•
•
•
• Overview of the OUlodendrocyte Developmental Lineale
Oligodendrocytes undergo controlled migration, proliferation and differentiation.
They both secrete and respond to severai growth factors, largely influencing their cellular
architecture and morphology. From the subventricular zones~ oligodendrocytes originate
as neuroectodermal ceUs that migrate and mature into postmitotic myelin-producing cells.
Oligodendrocyte progenitors expand from approximately 500~OOO at birth to a terminal
population of 60 million oligodendrocytes in the adult rat brain (Pfeiffer et al.~ 1993).
Confocal microscopy and image reconstruction studies of oligodendrocytes both in
culture and in situ contributed to oligodendrocyte developmental lineage investigation
(Warrington and Pfeiffer~ 1992; Warrington et al., 1993; Morgan et aL, 1992). During
their development, oligodendrocytes express developmental markers that are identified
by cell-specific antibodies. The expression of developmental markers provides a tool to
divide the lineage into distinct phenotypic stages characterized by the capabilities of the
celis to divide, migrate, differentiate, and produce myelin.
The youngest precursor ceUs are the Pre-GD3 ceUs (see fig. 1). The Pre-GD3
precursor cells are proliferative, monopolar ceUs expressing the embryonic neural cell
adhesion molecule (E-NCAM) (Hardy and Reynolds, 1991). These precursor cells
differentiate into proliferative and migratory bipolar GD3+ precursors that express
gangliosides recognized by the monoclonal antibodies (mAb) anti-GD3 and A2B5. They
are referred to as the o/igodendrocyte-type 2A astrocytes (O-2A) progenitors, due to their
bipotential to differentiate into either mature oligodendrocytes or type 2A astrocytes
(identified by the expression of glial fibrillary acidic protein, an intermediate filament
protein) (Raff, 1989) via different culturing factors. Eventhough the identity and
16

•
•
•
existence of the OLs in vivo is confirmed, there is no evideoce of the existence of the type
2A astrocytes in vivo (Skoff and Knapp, 1991; Norton and Farooq, 1993). A
developmental progression proceeds from the 0-2A stage to the Pro-o/igodendroblast
(Pro-DL) stage. Pro-OL cells possess multipolar, postmigratory, proliferative potentials,
and the phenotypic stage is characterized with the mAbs 04 and AOO? that recognize the
sulphated surface antigeo POA (Bansal et al., 1992). A transient developmental stage
follows the Pro-aL stage, named the Pre-Galactocerebroside (Pre-Gale). The Pre-GalC
ceUs stain positive to the mAb R but oot to the anti-GalC mAb 01. The GD3, Pro-DL,
and Pre-GaiC stages have often been labeled collectively as the "O-2A" progenitor;
however, they are functionally distinct in terms of electrical activities and responses to
neural-cell-derived mitogens and phorbol esters (Warrington et al., 1993; Gard and
Pfeiffer, 1993).
Yet to commence myelin synthesis, the onset of differentiation is necessary.
Terminal differentiation of oligodendrocyte progenitors is identified by the synthesis and
transport to the surface of GalC and sulphatides, and the synthesis of 2'-3' -Cyclic
nucleotide 3'phosphohydrolase (CNP), and later myelin basic protein (MBP), proteolipid
protein (PLP), myelin-associated glycoprotein (MAG), and myelin/oligodendrocyte
glycoprotein (MaG), hence the synthesis ofmyelin membranes (Pfeiffer et al., 1993).
17

•THE OLIGODENDROCYTE LINEAGE
MATURE Dla.cSULCNPPLPM8PYcG
IUM.lTUSE CL
Gale (A.mAt>. 01)
SUL (04/A007. R~)CNP
Pfe·~aIC
R-Ag (~·mAb)
FOAG03"-G03Yim+- '{imSCIP+-- SCIP-
Pro-OL
POA ((j.4,'AOC7)A2SSGD3VimSCI?
POGF+oFG~ bF3F A·mA!)
IlJ<l~l~_~GQ3 rQ-~~
Ci03A2BSVlmSCIP
-,?re-GD3
E·NC':'MVIM•
•

•
•
•
FIGURE 1: The Oligodendrocyte Lineage. The figure shows the stages of
development that an oligodendrocyte progenitor undergoes before becoming myelin
producing mature oligodendrocyte. Commonly used stage-specifie markers (antibodies
identifying sorne markers are shown in parenthesis) help identify the different stages
during oligodendrocyte development. Figure is adapted from Pfeiffer et al. (1993>.
18

•
•
•
(C) OLIGODENDROCYTE-TYPE 2A ASTROCYTE PROGENITOR
• Phenotype and Characteristics
Extensive in vi/ro studies have been completed on the OL lineage, including
studies on their migration, differentiation, and proliferation. Rat brain oligodendrocyte
precursor ceUs, also named 0-2A progenitors, were first isolated and characlerized in
vitro from the optic nerve by Raff and colleagues (Raff et al., 1983). Consequently, most
attention has been focused on this precursor cell. In culture~ the 0-2A progenitor features
a simple bipolar morphology, and contains a small soma (lO - 15 J,LM). ft is highly
proliferative. It binds the mAb A2B5, a mAb that reacts with a tetrasialoganglioside
(Eisenbarth et al., 1979; Raff et al., 1983). The 0-2A progenitor expresses the
ganglioside GD3 (Goldman et al., 1984) and the NG-2 chondroitin sulfate, in addition to
the intermediate filaments vimentin (Raff et al., 1984; Dubois-Dalcq~ 1987)~ nestin
(Almazan et al., 1993; Gallo and Annstrong, 1995), but does not express antigens of
mature astrocytes or mature oligodendrocytes. It also transiently expresses a seminolipid
and an unknown antigen called "proligodendroblast antigen" (POA) recognized by both
the A007 or 04 monoclonal antibodies (Bansal et al, 1992). 0-2A progenitors express
amply specific transcription factors characteristic of the phenotype: the transcription
factors SCIP/tst-l (Collarini et al., 1992), and myelin transcription factor 1 (MyTl)
(Annstrong et al., 1995). These transcription factors control myelin gene expression~ and
are downregulated as the ceUs develop into post-mitotic oligodendrocytes.
19

•
•
•
• Developmental Regulation
The development of oligodendrocytes depends on intrinsic properties of each cell
and the availability of growth factors. The development of oligodendrocytes is Iargely
influenced by soluble factors released in their proximity by other components of the
CNS, such as neurons, astrocytes, and microglia. In addition, oligodendrocytes secrete
autocrine growth factors that can regulate their own development (Compston et al.,
1997). Studies performed in vitro have shown that platelet-derived growth factor (PDGF)
plays a major role in the control of 0-2A development. PDGF is a mitogenic factor for
0-2A progenitors, yet in the continued presence of PDGF, 0-2A progenitors eventually
stop proliferating and acquire mature differentiated oligodendrocyte characteristics
(Noble et al., 1988; Richardson et al., 1988). The source of PDGF has been linked to
neurons expressing PDGF in vivo (Yeh et al., 1991; Sasahara et al., 1991), while in vitro,
type 1 astrocytes have shown to secrete this growth factor (Richardson et al., 1988). In
addition, basic fibroblast growth factor (bFGF) is another growth factor that is involved
in regulating oligodendrocyte development. bFGF is also a mitogen for 0-2A
progenitors (Eccleston and Silberberg, 1985; Saneto and deVeL1is, 1985), whereby unlike
the effect of PDGF, progenitors' continuous exposure to bFGF prevents 0-2A
progenitor differentiation into mature oligodendrocytes (McKinnon et al., 1990), but
rnaintains ils proliferative state. The combined effect of bFGF and PDGF treatment of 0
2A progenitors result in long periods of proliferation without differentiation (Bogler et
al., 1990). bFGF is produced by astrocytes, microglia, and neurons (Pettmann et al.,
1986; Ferrara et al., 1988; Shimojo et al., 1991).
20

•
•
•
On the other hand~ neurotrophin-3 (NT-3) bas been shown to induce proliferative
effects on 0-2A progenitors (Barres et al.~ 1994). In addition, 0-2A progenitors are
influenced by epidermal growth factor (EGF), thyroxine and T3, retinoic acid, and
glucocorticoids, factors that promote progenitor differentiation and maturation in vitro
(Laeng et al., 1994). Other factors that drive progenitors into differentiation include
insulin-like growth factor 1 and Il (IGF-I~ IGF-II) (McMorris and Dubois-Dalcq~ 1988),
in addition ta IGF-I's role in promoting the survival ofprogenitors (Barres et al., 1992).
Moreover, studies performed in our laboratory have demonstrated a role for
glutamatergic, adrenergic, and muscarinic acetylcholine receptors (mAChRs) in
regulating proliferation of 0-2A progenitors. We have previously reported the block of
proliferation induced by stimulating the AMPA/Kainate receptors on 0-2A progenitors
(Liu and Almazan, 1995), while adrenergic stimulation caused no change in the
proliferative state of the progenitors (unpublished results). On the other hand,
pharmacological studies performed in our laboratory characterized the mAChR's
expressed on 0-2A progenitors and their developmental regulation, showing evidence for
the expression of several mAChR subtypes whereby the M3 subtype is the main subtype
expressed during development (Molina-Hoigado et al., unpublished). An interesting
finding was the extensive downregulation ofmAChRs during the differentiation of 0-2A
progenitors to mature differentiated oligodendrocytes, which May indicate a raie for these
receptors during their early development. Moreover, proliferation studies on 0-2A
progenitors showed that cholinergie stimulation of 0-2A progenitors increased the
proliferation of these cells, whereby the muscarinic antagonist atropine blocked this
21

•
•
•
response, demonstrating that muscarinic receptors are responsible for this cholinergie
response (Cohen et al., 1996).
The potentiaI of differentiating into type 2 astrocytes is a characteristic of 0-2A
progenitors. [t is an interesting developmentaI feature, whereby progenitors stop their
differentiation into oligodendrocytes and instead become astrocytes. ln vitro, the
maintenance of progenitors in serum can elicit this switch (Raff et al., 1983). In this
process, progenitors begin to express type 2 astrocyte characteristics~ such as stellate
shape morphology, binding of A2B5, expression of the astrocyte intermediate filament
protein, glial fibrillary acidic protein (GFAP), and failure to express gangliosides, lipids
and proteins characteristic of myelin (Raff et al., 1983). These ceUs have been labeled
type 2 astrocytes in order to he able to distinguish them from type 1 astrocytes, which are
A2B5 negative and display a flat shape.
• Oligodendrocyte Progenitor Differentiation and Maturation
Under certain culture conditions, oligodendrocyte progenitors enter the
maturation and differentiation stage. By withdra\\ing growth factors and maintaining
serum-free culture conditions, oligodendrocyte progenitors differentiate and lose their
proliferative state (Raff et al., 1983). ln vitro, this step is characterized by the loss of
nestin, vimentin, and the antigen A2B5, and the fonnation of multipolar processes, in
addition to the expression of a stage specifie modified lipid sulfatides recognized by the
04 (AOO?) antibody (Sommer and Schachner, 1981; Sarlieve et al., 1983; SansaI et al.,
1989; Knapp, 1991). The cells become reactive to the R-mAB, which reacts with not
only galactocerebrosides, but also to another epitope that is also recognized by the 01
22

•
•
•
mAb (Raff et al., 1978; Ranseht, 1982; Bansal and Pfeiffer, 1992; Sommer and
Schachner, 1981). Consequently, myelin specifie proteins are expressed to regulate and
commence the synthesis ofmyelin. Myelin specifie proteins inelude CNP, MAG, MOG~
MBP, and PLP (Pfeiffer et aI., 1981; Raff et al., 1983; Zurbriggen et al., 1984; Dubois
Dalcq et al., 1986; Dubois-Dalcq, 1987; Knapp et al., 1987, 1988; Amur-Umarjee et al.,
1990; Yim et al., 1995). These events signify the terminal differentiation of the
oligodendrocyte progenitor and preludes the post-mitotie mature oligodendrocyte. At
this stage, the morphology of the mature oligodendroeyte changes, whereby the cell body
darkens, and numerous processes form the stage-characteristic lace like lattice. This
··web" of processes supports the myelin membranous sheath. In the presence of neurons,
this membranous sheath forms a multilamellar~ spirally wrapped sheaths around the
axons, insulating their eiectrically conductivity, and ensuring fast electrical transmission.
23

• (II) THE MUSCARINIC ACETYLCHOLINE RECEPTOR
•
•
• Introduction
The classical neurotransmitter acetyIchoIine (ACh) transduces its signaIs to ceUs
via binding to either the ionotropic nicotinic receptor (nAChR) or the metabotropic
muscarinic receptor (mAChR). Nicotinic and muscarinic receptor subtypes are expressed
in both the PNS and the CNS. In the periphery, mAChRs regulate the synaptic
transmission in the ganglia of the autonomie nervous system, in addition to modulating
the functions of target organs of the parasympathetic nervous system (for review see
Cauifield and Birdsall, 1998). Such reguJation inciudes mediation of smooth muscle
contraction, glandular secretion, and modulation of cardiac rate and force. In the CNS,
mAChRs have been implicated in many important processes, including memory,
learning, temperature regulation, cardiovascular regulation, and control of movement (for
review see Nathanson, 1996). Therapeutically, the administration ofmuscarinic agonists
facilitate Iong-term potentiation and cause Parkinson's disease-like trernors, while
muscarinie antagonists interfere with leaming and memory and overcome the tremors in
Parkinson' s disease. Other therapeutic applications include treatments for asthma,
analgesia, and disorders of intestinal, cardiac, and urinary bladder function (for review
see Caulfield and Birdsall, 1998). In addition, several reports have indicated that
muscarinic agonists cao act as mitogens in cultured neural ceUs, whereby mAChRs may
play an important role in the development of the CNS (Ashkenazi et al., 1989; Gutkind et
al., 1991)
24

•
•
•
The mAChRs are members of the superfamily of hormones and neurotransmitter
receptors. The mAChR is a seven transmembrane domain receptor that is coupled to a
guanosine triphosphate (GTP) binding protein (G-protein). mAChRs regulate the
activities of intracellular second messenger pathways through ion channel and enzyme
activationldeactivation by the interaction with their coupied G-proteins. So far, there are
5 mAChR subtypes: the Ml, M2, M3, M4, and the MS subtypes. The MIIM31M5
mAChRs couple to pertussis toxin- (PTX) insensitive G-proteins (Gq/1t), while the
M2/M4 mAChRs bind PTX-sensitive G-proteins (GiJo)' The former group of subtypes
activate phospholipase C (PLC), phospholipase A2 (PLA2), phospholipase D (PLD),
tyrosine kinase, and calcium influx but do not inhibit adenylate cyclase, while the latter
group of receptors inhibits adenylate cyclase but do not stimulate PLC. However, this
coupling specificity of the mAChR subtypes is not absolute, for the M2 and the M4
subtypes can weakly activate PLC when expressed at high levels in certain cell types
(Ashkenazi et al. 1989a; Tietje et al., 1990; Tietje and Nathanson, 1991). The PTX
insensitive coupling to PLC is mediated by GCXq, Gall, Ga14, and Ga16, while the PTX
sensitive coupling to adenylate cyclase inhibition is mediated by Gai or Gao (Katz et al.,
1992; reviewed by Felder, 1995).
Historically, the initial studies indicating the existence of mAChR subtypes were
reported when gallamine showed cardioselective actions (Riker and Wescoe, 1951).
Eventually, Barlow et al. (1976) described significant distinctions in the pharmacological
properties of ileal and atrial muscarinic receptors. Pirenzepine was introduced as a
therapeutic agent for peptic ulcer disease, contributing to the appreciation of the existence
of mAChR subtypes. Pharmacological binding studies reported by Hammer et al. (1980)
25

•
•
•
and functional studies reported by Brown et al. (1980) and Hammer and Giachetti (1983)
provided an explanation for pirenzepine's in vivo selectivity, suggesting the existence of
more than one mAChR subtype
• Molecular Biology of the mAChR
Subsequent to the early pharmacologicaI findings, molecular cloning studies
identified 5 different that are products ofdistinct but homologous (Bonner, (989). Numa
and colleagues cloned the ml and the m2 genes (Kubo et al., 1986a,b), since the
pharmacologicaI profiles of the cloned receptors resembled the MI and the M2 subtypes
that have been pharmacologically characterized earlier. The m3, m4, and mS genes were
thereafter discovered (Bonner et al., 1987, 1988; Peralta et al., (987). The cloned
vertebrate receptor genes cIoned are intronless, and are similar across mammalian species
(Hall et al., 1993; Eglen et al., 1996). Each gene is approximately 460-532 amino acids
in length (m1=460; m2=466; m3=589-590; m4=478-490; m5=531-532). These five
genes encode mAChR glycoproteins that display the structural features of the seven
transmembrane helix G-protein-coupled receptor family. Based on the phannacological
binding studies ofeach receptor resembling the cloned genes, it is now recommended that
Ml, M2, M3, M4, and M5 be used to describe both the pharmacological subtypes and the
molecular subtypes encoded by the cloned genes.
Similar to most members of the G-protein-coupled receptor family whose ligand
recognition site binds small molecules, there are several major features of muscarinic
receptor structure. First, the ligand recognition site can he located within the outer half of
the membrane-embedded part of the protein. To bind the neurotransmitter ACh, aIl
26

•
•
•
mAChRs have an Asp residue in the distal N-tenninal part of the third transmembrane
domain which is thought to interact with the polar head group of the neurotransmitter and
other amine ligands (Curtis et al., 1989; Spalding et al., 1994). Second, the
transmembrane segments are a-helical, whereby three helices are oriented approximately
perpendicular to the membrane, and four helices are oriented at a more acute angle
(Baldwin et al., 1997). Furthermore, there are two conserved cysteine residues that forrn
a disulfide bond bet\veen the first and third extracellular loops (Kurtenbach et al, 1990;
Savarese et al., 1992). There exists also a conserved triplet of amino acids (Asp Arg Tyr)
at the cytoplasmic interface of TMIII with the second intracellular loop that is crucial for
both the expression and function of the receptor (Zhu et al., 1994; Jones et al .. 1995; Lu
et al., 1997). Moreover, the carboxy-terminus of the mAChR is located on the
intracellular side of the membrane, while the amino-terminus is located on the
extracellular side of the membrane containing one or more glycosylation sites. On the
other hand, the fourth intracellular region plus regions near the transmembrane area in the
second and third internaI loops seem to be targeted for phosphorylation by ligand
stinlulated negative feedback loop mediated by mAChR kinase (Haga et al., 1993). Yet
to distinguish between the different mAChR subtypes, a sequence divergence in the
postulated third internai (oops (i3) exists between the MIIM21M3 sequences compared
with the M21M4 sequences (Hulme et al., 1990; Wess, 1996; Wess et al., 1997). The
sequence divergence probably specifies the different coupling preferences of the two
categorized groups of subtypes (Wess, 1993). In addition, sequences \-vithin the i3 loop
differ sufficiently between each mAChR subtype that allows raising subtype specifie
antisera (see Levey, 1993 for explanation).
27

•
•
•
• Pharmacologieal Definitions of Subtypes
The task of pharmacologically characterizing the different mAChR subtypes has
been a difficult process, especially with the initial lack of agonists \vith any selectivity in
addition ta the lack of any antagonist with very high selectivity for any single receptor
subtype. Studies were focused on discovering natural agonists or antagonists plus
attempts ta synthesize selective agonists or antagonists that can bind selectively to each
subtype ta distinguish between each of the five mAChRs. Yet the binding sites for ACh
and other agonists were found to be similar among aIl of the mAChR subtypes.
However, the expression of more than one subtype in mammalian tissues emphasized the
concept of high selectivity for antagonists or agonists that are needed to be used in
clinical application for disorders resulting from muscarinic aberrations.
The definition of antagonist affinities for the five muscarinic receptors has been
aided amply by the use of radioligand binding techniques, with ligands such as
eH]pirenzepine and eH]N-methylscopolamine, in addition to the procedure of
membrane preparations from ceUs transfected with the gene for a specifie receptor,
achieving the expression of ooly one single subtype at a time. Binding studies using two
ne\V antagonists, hexahydro-sila-difenidol (HHSiD) and its para-fluoro analogue (p
HHSiD), enabled researchers to distinguish between the M2 muscarinic binding sites in
the heart and in the glandular tissues (for review see Caulfield and Birdsall, 1998). The
heart M2 receptor had a 70-fold lower affinity for p-HHSiD than for the "'M2" glandular
tissue receptor, which was then found to he distinct from the M2 subtype, and renamed
the M3 mAChR. Further studies developed two highly selective M2 antagonists that
display high selectivity between the Ml and the M4 subtypes (for review, see Grimm et
28

•
•
•
al., (994). On one hand, tripitramine displayed a 2000 fold higher binding affinity to the
Ml than to the MI, M3, and M4 subtypes, and (S)-dimethindene is an M2 selective
antagonist used in functional and binding studies.
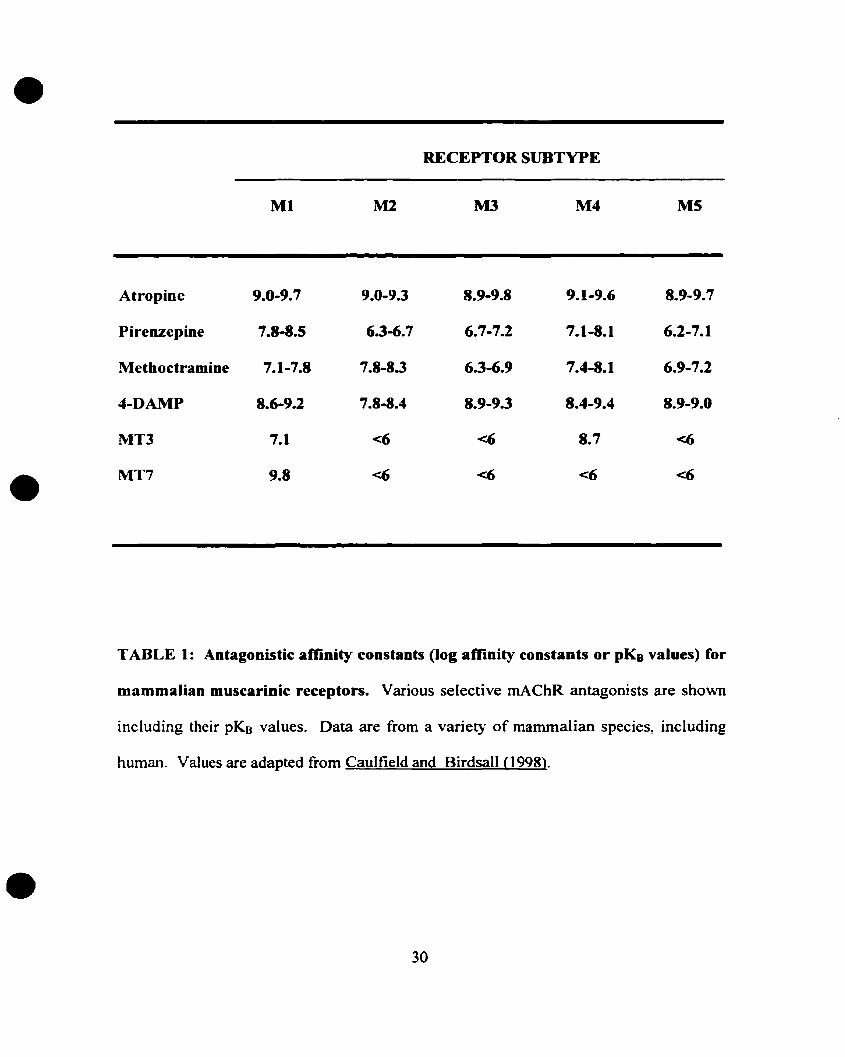
More selective antagonists have been recently synthesized (see Table 1),
including the M2 antagonist methoctramine, the M3 antagonist 4-DAMP and its
irreversible analogue 4-DAMP-mustard (Michel et al., 1989; Lazareno et al., 1990;
Caulfield 1993; Kondou et aL, 1994). In addition, n\'o muscarinic snake toxins have been
discovered, the MT3 and the MT7 toxins. They display very high affinity for muscarinic
receptors. MT3 and MT7 toxins belong to the many components of the venom of the
green (Dendroaspis angusticeps) and the black mamba (Dendroaspis polylepis) (Hulme
et al., 1990; Caulfield, 1993; Maggio et al., 1994; Eglen et al., 1996; Nunn et al., 1996;
Adem and Karlsson, 1997; Caulfield, 1997; Levine and Birdsall, 1997; Jolkkonen et al.,
1994). These two toxins show promising subtype selectivity (MT7 is highly selective to
the Ml subtype; MT3 is highly selective to the M4) and should be useful in muscarinic
receptor classification (for review see Caulfield and Birdsall, 1998). However, more
highly selective agonists are needed to activate preferentially each subtype in order to
implement the use of muscarinic agonists in clinical applications.
29
•RECEPTORSUBTVPE
MI Ml M3 M4 MS
Atropine 9.0-9.7 9.0-9.3 8.9-9.8 9.1-9.6 8.9-9.7
Pirenzepine 7.8-8.5 6.3-6.7 6.7-7.2 7.1-S.1 6.2-7.1
Methoctramine 7.1-7.8 7.8-S.3 6.3-6.9 7.4-S.1 6.9...7.2
4-DAMP 8.6...9.2 7.8...s.4 8.9-9.3 8.4-9.4 S.9-9.0
MT3 7.1 <6 <6 8.7 <6
• MT7 9.S <6 <6 <6 <6
TABLE 1: Antagonistic amnity constants (log affinity constants or pKB values) for
mammalian muscarinic receptors. Various selective mAChR antagonists are shown
including their pKB valueso Data are from a variet}° of mammalian species, including
human. Values are adapted from Caulfield and Birdsall (1998).
•30

•
•
•
111- MUSCARINIC COUPLING TO G-PROTEINS AND EFFECTOR
MOLECULES
• Introduction
Similar to most seven transmembrane receptors, the mAChRs are coupled to G
proteins that can transduce the exterior signal, in this case the binding of ACh to the
mAChR and the receptor's subsequent activation, into an intracellular signal govemed by
specifie second messenger cascades that cause many cellular responses. The link between
receptors and effectors is in many cases mediated by heterotrimeric G- proteins. G
protein transduced cellular responses include biochemical activities, metabolic changes,
enzyme activationldeactivation, downstream gene transcription, protein synthesis, cell
division, and celI motility (see Table 2).
• Composition and Structure of Heterotrimeric G-proteins
Heterotrimeric G-proteins consist of an a subunit (39-52kDa), a r3 subunit (35/36
kDa), and a y suhunit (6-8 kDa) (for review see Clapham and Neer, 1997; Muller and
Lohse, 1995). G-proteins can switch between an active foon and an inactive fonn
through binding to either a GTP or a GDP nucleotide. When bound ta the GTP
nucleotide. the G-protein is in its active stable state where it can activate directly diverse
effectors. G-proteins possess an intrinsic GTPase function in the a subunit which
hydrolyses GTP to GDP, and thus induce the GDP-binding inactive fonn. At this
inactive stage, the a. subunit associates with the f3y subunit. The ACh-bound fonn of the
mAChR induces the exchange of GDP to GTP al the a. subunit. The binding of GTP
31

•RECEPTOR SUBTYPE
Preferred G-proCein
Ml
q/ll
M2
iJo
M3
qlll
M4
i/o
MS
q/ll
Second messengers PLC IP3fDAG AC (-) PLC IP3IDAG AC(-) PLCCa2+/PKC Ca2+/PKC lP3/DAG
PLA2 PLA2 Ca2+IPKC
Locations Brain (conex.hippocampus) Heart.Hindbrain Smooth muscle Forebrain Substan.Glands, Sympathetic ganglia Smooth muscle Glands, Brain Striatum nigra
(f3y activates)Inhibits Ca2+ channelsDecrease hean rateDecrease heart forceDecrease neurotransm. release
• FunctionalResponses
M..current inhibition K+ channels Smooth musclecontraction
inhibitsCa2+ channels
•
TABLE 2: Muscarinic receptors: G-protein coupling, transduction pathways,
localization, and functional responses. rnAChRs couple selectively to different G-
proteins, triggering various intracellular responses. A variety of functional responses are
mediated by the different subtypes, and each subtype maintains a specifie anatomical
location. Refer to text for abbreviations. As adapted from Caulfield and Birdsall (1998).
32

•
•
•
results in the dissociation of the a subunit and the Py complex~ as a result both subunits
cao activate their own specific signaIs and events.
There bas been at least 21 different a subunits~ 6 13 subunits~ and 12 y subunits
c10ned (Hepler and Gilman, 1992; Rayet al., 1995; Simon et al., 1991; Watson et al.,
1996). A combination of ~y subunits reveal 72 potential f3y combinations that can bind
21 different a subunits. The large number of combinations raises questions about
specifie eoupling and function to different receptors. Since aU a subunits have
eharaeteristie functions, G-proteins have traditionally been defined by their cr subunits.
mAChR subtypes are eoupled to specifie G-proteins defined by their a subunits. The odd
numbered subtypes, Ml/M3/M5, are coupled to the PTX-insensitive GClq/ll protein, while
the even numbered subtypes~ M21M4, activated the PTX-sensitive Gailo protein.
Recently many (3y funetional attributes have been identified~ eliciting more
investigations in their structure and function. There are sorne restrictions to f3y
combinations. 132 does not fonn a stable complex with y}, and similarly for 133 and YI and
Y2 (Clapham and Neer, 1993; Pronin and Gautam, 1992; Schmidt et al.~ 1992), rendering
such combinations inactive (Iniguez-Lluhi et al., 1992). The region of Gy that defmes the
specificity of its interaction with GJ31 or G132, for example, is located in a 14 amino acid
segment close to the middle of the molecule (Spring and Neer, 1994). In addition, tissue
specifie loealization is another characteristic of the diversity of G-protein subunits,
whereby the YI isoform is restricted to retinal rods while 133 occurs preferentially in retinal
cones (Iniguez-Lluhi et al., 1993; Wilcox et al.~ 1994; Peng et al., 1992).
The J3y subunits are membrane bound with the exception of transducin-(3y (J31YI)~
whieh cao be found as a soluble complex (for review see Muller and Lohse, 1995;
33

• Clapham and Neer, 1997). Ily complexes cannot he dissociated except with denaturants.
Isoprenylation of the y subunit at the C-tennÏnus is required for the complex' membrane
association. However, isoprenylation is not required for complex formation \Vith the ~
subWlit, but it is compulsory for ~y's interaction witll the a; subunit and the activation of
an effector molecule (Iniguez-Lluhi et al., 1992; Clarke et al., 1992; Higgins and Casey,
1994; Simonds et aL, 1991). On the other band, covalent modification of the a subunit
using fatty acids is required. \Vith the exception of ex l, all Cl subunits are palmitoylated
through a thioester bond to a cysteine residue in the third position of the N-terminus.
Palmitoylation appears to be necessary for membrane attachment of the a subunit, as
weil as for coupling to receptors (Linder et al., 1993; Veit et al., 1994; Wedegaertner et
al., 1993; Mumby et al., 1994; Parenti et al., 1993). Furthermore, a subunits are
• myristoylated at the N-terminal glycine via an amide linkage, and this modification
allows the a subunits to bind with an increasing affinity to the ~y complex (Buss et al.,
1987; Linder et al., 1991).
•
• Adenylate Cyclase
The decrease in adenylate cyclase activity induced by muscarinic activation has
been weil documented (for review see Felder, 1995). The expression of the M2 and the
rvl4 receptors in cell lines displayed coupling to adenylate cyclase inhibition (Buckley,
1990; Ashkenazi et al., 1989b; Hulme et al., 1991; Baumgold, 1992; Grimm et al., 1994;
Felder, 1995). In addition, G-protein reconstitution experiments showed that the G i
subtype coupled to the mAChRs is responsible for this response (Parker et al., 1991).
Adenylate cyclase catalyzes the breakdown of ATP into cAMP, which in tum cao
34

• activate cAMP-dependent protein kinases (pK.A). However, other studies have shown
that the expressed Ml subtype can weakly couple to adenylate cyclase inhibition through
a PTX·sensitive mechanism in RAT-l ceUs (Stein et al., 1988). On the other hand,
previous reports have shown that the endogenous M3 subtype can cause an accumulation
of cyclic-adenosine mono-phosphate (cAMP) in neuroblastoma ceUs (Baurngold and
Fishman, 1988), and also in other several ceUs such as the rat olfactory bulb (Olianas and
Onali, 1992)~ mouse parotid acinar ceUs (Watson et al., 1990), rat adrenal gland
(Regunathan et al. 1990), and rat sympathetic neurons (Suidan et al.~ 1991). Also, the f3y
subunits of the G-proteins coupled to the MI and the MS mAChRs can also weakly
stimulate adenylate cyclase types II and IV (Ashkenazi et al., 1989a; Pieroni et al., 1993).
The coupling of mAChRs to adenylate cyclase activation can be regulated through
• calcium and protein kinase mechanisms (Jansson et al., 1991; Baumgold et al., 1992).
cAMP production may occur through calciumlcalmodulin sensitive (types 1 and III) or
-insensitive (types II, IV, V, and VI) adenylate cyclases. The adenylate cyclase type
coupled to mAChRs is dependent on the cell line in which the mAChRs are expressed.
Nevertheless, the accumulation of cAMP may actually be the result of mAChR-mediated
phosphodiesterase inhibition in a variety of cell types including astrocytoma ceUs
(Meeker and Harden, 1982). In oligodendrocyte progenitors, previous studies from our
laboratory have reported the anenuation of J3-adrenergic-stimulated cAMP formation by
the M2 mAChR subtype, speculating a role for the Gai protein coupled to the M2
receptor in inhibiting adenylate cyclase (Cohen and Almazan, 1994).
• Phospholipase C (PLC)
•35

• The receptor-mediated activation of PLC leads to the hydrolysis of
phosphotidylinositol-4,5-bisphosphate (PIPz) to inositol trisphosphate (IP3) and
diacylglycerol (DAG). The family of PLC enzymes is grouped into three classes, J}, y,
and 8, with subtypes within each group (Rhee and Choi, 1992). Phosphoinositide
breakdown by ACh has been weil studied and linked to the M IIM31M5 mAChRs through
the coupling of Gqlll protein, while the M2 and M4 subtypes have been reported to
weakly activate PLC (Lambert et al., 1992). In the case of the Ml subtype,
phosphoinositide breakdown is mediated by PLC~1 through Gq/l l alpha subunits
(Berstein et al., 1992; Sawaki et al., 1993; Hildebrandt and Shuttleworth, 1993). On the
other hand, the M5 subtype have been linked to the activation of PLCf3 and
PLCy (44)PLCy activation is normally stimulated by tyrosine kinase-dependent
• phosphorylation, a mechanism induced by the M5-mediated calcium influx that activates
voltage-independent calcium channels and subsequent tyrosine kinase phosphorylation
(Gusovsky et al., 1993). However, the M2 subtype did not stimulate the phosphorylation
of PLCy or the mediation of calciwn influx. Later studies have shown, though, that the
M2 and M4 receptors cao also stimulate with lower efficiency phospholipase C through a
PTX-sensitive G-protein, namely through Gaz and Ga iJ (Ashkenazi et al., 1989;
Dell' Acqua et al., 1993). AIso, f3y subunits were shown to couple the M2 receptor and
phospholipase C-f32 (Katz et al., 1992). Studies in our laboratory have reported that in
oligodendrocyte progenitors, inositol trisphosphate accumulation was mediated by the
Ml and not by the M2 mAChR (Cohen and Almazan, 1994). Protein kinase C (PKC)
seemed to play a regulatory role in the mAChR-mediated accumulation of inositol
•36

•
•
•
trisphosphate, since an acute pretreatment with phorbol-12, 13 myristate acetate (PMA)
abolished the formation of inositol trisphosphates (Cohen and Almazan, 1994).
IP3 cao act 00 its respective receptor in the eodoplasmic reticulum. (an IP3
sensitive calcium channel), releasing calcium from its intracellular stores, while DAG cao
activate, with the cooperation of calcium, certain isozymes of PKC. PKC consists of
three subgroups: the classical, which include a, fil, 1311, and y, isozymes that are activated
by DAG and calcium; the novel, which inc1ude Ô, E, e, l'l, J.L, and are activated by DAG
alone; the atypica/, which include ç, À, 1., and are independent of both calcium and DAG
(for review see Mellor and Parker, 1998). The atypical isozymes are activated by
phosphatidylserine and phosphatidylinositol (3,4,5)-trisphosphate. Note that certain
isozymes are involved in stimulating proliferation (Stephens et al., 1993), while others
are involved in negatively regulating mAChR activity by phosphorylating sites on the i3
loop (Haga et aL, 1993).
• Phospholipase A2 (PLA2)
The enzymatic function of PLA2 is to catalyze the hydrolysis of membrane
phospholipids, mainly phosphatidylcholine, to generate free arachidonic acid (AA) and
the corresponding lysophospholipid. AA is a Mediator of many cell"Ular and
physiological mechanisms including inflammation, pain, and intracellular signaling
through itself and its bioactive eicosanoids. Lysophospholipids that are generated from
the hydrolysis of membrane phospholipids are recycled to the membrane. AA can he
converted to bioactive eicosanoids including prostaglandins, thromboxanes, leukotrienes,
37

•
•
•
epoxides~ and hydroxyeicosatetraeinoic acids (HETEs). Reports published have
described the release of AA througb mAChR stimulation, in addition to the release of
eicosanoid metabolites in a variety of tissues including heart, brain, and muscle.
Pharmacological studies linked the odd numbered mAChRs MIIM31M5 to the activation
of PLA2 (Conklin et al., 1988; Felder et al., 1990; Liao et al., 1990), whereby CCh
stimulated the release of AA in a PTX-insensitive manner, ruling out the involvement of
G i or Go- On the other band, M2 and M4 subtypes expressed at physiological levels
showed no coupling to PLA2 (Conklin et al., 1988). The activation of PLA2 has been
shown to require an IP3-independent calcium influx and protein kinase C (Felder et al.,
1990; Brooks et al., 1989). The high molecular weigbt cytosolic PLA2 (cPLA2) seemed
to he the isozyme that is involved in the mAChR-mediated AA release (Sharp et al.~
1991; Clark et al., 1991). It is activated with nanomolar levels of calcium, and has a
selectivity for phospholipids with arachidonic acid in the sn2 position.
38

• IV- MITOGEN-ACTIVATED PROTEIN KINASES
•
•
• Brief Overview: The MAPK Classes
The mitogen-activated protein kinase (MAPK) pathway is involved in cell
differentiation and proliferation. Il is activated by bath tyrosine kinase receptors as weil
as G-protein coupied receptors. Receptor tyrosine kinases activate MAPKs through a
rnultistep process. The binding of the ligand to the receptors leads to tyrosine
phosphorylation of a docking site on the receptor for the adapter protein GRB2/SEM-5
(for review see Schlessinger~ 1993). Subsequently~ the recruitment of a guanine
nucleotide exchange protein Sos occurs~ which recruits Ras to the plasma membrane~ and
results in the exchange of GDP for GTP by Ras. GTP-bound Ras then activates a
cascade of protein kinases listed sequentially as MAP kinase kinase kinase (A-Raf~ B
Raf, and Raf-I) and MAP kinase kinase (MEKI and MEK2). MEKs then phosphorylate
p44mapk and p42mapk MAPKs (ERK1 and ERK2) on both threonine and tyrosine residues.
As a result, MA.PKs phosphorylate and regulate the activity of key enzymes and nuclear
proteins, which can eventually regulate the expression of genes essential for proliferation.
However, the mechanism of G-protein coupled receptors· activation of MAPKs are less
understood, yet many groups have elucidated various signaling cascades.
In mammalian systems, II distinct MAPK and 7 MEK genes have been identified
to date. Members of the MAPK family include (i) extracellular signal-regulated kinases
(ERKI and ERK2) which are also known as p42mapk and p44mapk and are phosphorylated
by MEK 1 and MEK2; (ii) NH2-terminal Jun kinase/stress-activated protein kinases
(JNKlSAPK) a~ f} and~ y, which are phosphorylated by MEK4 and MEK7; and (iii) p38
39

•
•
•
MAPKs a~ 13, y, and Ô, which are phosphorylated by MEK3 and MEK6 (for review see
Lewis et al., 1998). MAPKs are activated by MEKs through phosphorylation at two
regulatory phosphorylation sites with sequence T(P)-X-Y(P) located in the 'activation
Hp' between subdomains 7 and 8 of the conserved kinase core sequence. In addition to
the conserved kinase core~ p42mapk and p44mapk contain C-terminal extensions and a 25
amino acid insert bet\veen subdomains 9 and 10. Both enzymes are activated by
phosphorylation within their activation Hp at Thr183-Glu-TyrI85 (as in rat sequence), a
reaction that has Tyr phosphorylation preceeding Thr phosphorylation (for review see
Lewis et al., (998). Both events of phosphorylation are needed to fully activate both
isozymes.
• G-protein Coupied Receptors Signaling to MAPK
Activation of MAPKs mediated by G-protein coupled receptors has been extensively
studied, including investigations on bombesin, endothelin-l, adrenaline, somatostatin,
LHRH, TRH, thromboxane A2, and muscarinic receptors. These receptors have been
shown to couple to PTX- sensitive and insensitive G-proteins, thus signaling to MAPK
using both G-proteins (Gilo and Gq/ll ). To study the mechanism of activation of MAPK
by G-protein coupled receptors such as the mAChRs, transfected cell lines that express
p42mapk (labeled as MAPK in this text) together with mAChR subtypes \-vere used (for
review see Gutkind et al., 1997). In transfected cell lines such as COS-7, CCh
stimulation increased MAPK activity in a concentration dependent manner through the
Ml and the M2 subtypes (Crespo et al., 1994), whereby the Ml-mediated activation was
PTX-insensitive, while the M2-mediated activation displayed PTX-sensitivity. To
40

•
•
•
elucidate the role of the a subunits of G-proteins, studies were performed in transfected
cells expressing wild type or activated G-protein a subunits, including Gaq and Gail
which couple MI and M2 subtypes respectively. However, these transfected ceUs did not
exhibit elevated MAPK phosphorylating activity (Crespo et al., 1994). As a result,
speculations of molecules in addition to a subunits of G-proteins play a role in the
activation of ~fl and M2-mediated MAPK activation.
Recently, available evidence has supported an active role for the GlJy complex in
signal transduction (Clapham and Neer, 1993). Since ex subunits did not stimulate the
activation of MAPK studies investigating the role of GlJy subunits were performed.
Indeed, transfecting ceUs with fll and y2 subunits and MAPK effectively elevated its
phosphorylating activity (Crespo et al., (994), demonstrating that the J3y-heterodimers
directly elicit biochemical pathways leading to MAPK activation. AIso, sequestration
studies performed on {3y complexes by overexpressing Gai proteins abolished the M2 and
~y-mediated activation of MAPK, and reduced this response when elicited by MI
stimulation. Therefore, these results indicate that Ml signaling to MAPK involves J3y
dependent and independent pathways, while the M2-mediated activation of MAPK
appears to be strictly dependent on GIJ1'
Mutant-ras studies were performed to investigate whether signaling from MI and
M2 receptors or {3y dimers involve p21 ras (Ras). These studies have sho\\'Il that mutant
ras expression abolished the elevated MAPK activity in response to Ml, M2, and J3y
dimer overexpression (for review see Gutkind, 1998; Lewis et al., 1998). Similar studies
were done to elucidate the role of PKC, and the results suggested that signaling from Ml
receptors to MAPK involves a PKC-dependent and PKC-independent pathway, and both
41

•
•
•
pathways converge at Ras. The PKC-independent pathway is mediated by the G13y
complex. On the other hand, the M2-mediated MAPK activation is PKC-independent,
and involves GitJy dimers, acting on a Ras-dependent pathway.
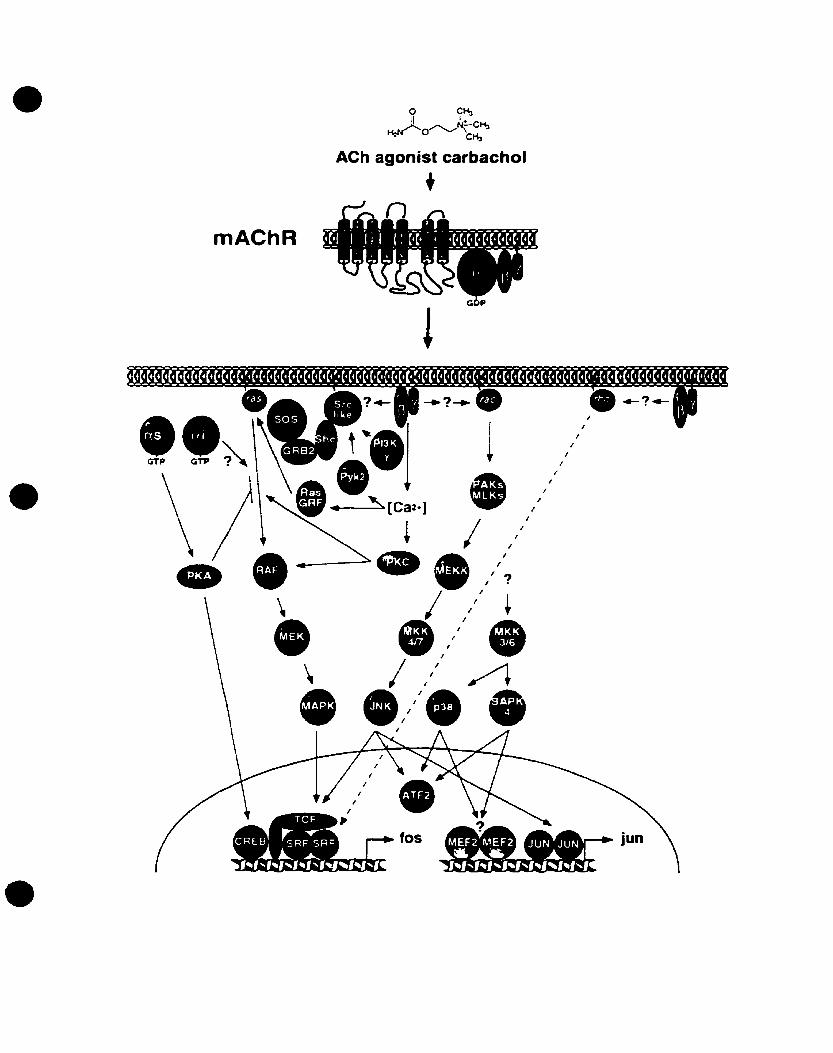
• Novel Signaling Pathways Linking G-protein Coupled Receptors to MAPK
Recent studies have shown that the activation of G-protein coupled receptors,
such as the mAChRs, induces the phosphorylation of the adapter protein Shc on tyrosine
residues and the formation of Shc-GRB2 complexes (see fig. 2) (Chen et al., 1996; van
Biesen et al., 1995). Other studies have demonstrated that Src, or a Src-like kinase,
mediates the ~y-induced phosphorylation of Shc, and eventually the recruitment of
GRB2 and Sos, consequently the activation of the Ras-MAPK pathway (Lutrell et al.,
1996). More studies have linked other non-receptor tyrosine kinases in signaling to the
MAPK pathway, including several Src-like kinases such as Fyn, Lyn. Yes, and Syk
(Ptasznik et al, 1995; Wan et al., 1996), in addition to the PKC-dependent Pyk2 (Della
Rocca et al., 1997; Dikic et al., 1996; Levet al., 1995). Pyk2 is related to the focal
adhesion kinase (FAK), a kinase involved in integrin signaling, where it contributes to
the formation of focal complexes in the celI.
Furthermore, reports have demonstrated that signaling from G-protein coupled receptors
to MAPK involves a wortmannin sensitive phosphatidylinositol-3-kinase (PI3K) (Hawes
et al., 1996; Lopez-Ilasaca et al., 1997). The PI3K family contains several species
including PI3Ka, PI3Kfi, and PI3Ky. The tyrosine kinase receptor-regulated
heterodimeric PI3Ka and PI3K(J consist of pllO catalytic subunits and different p85
42

•
•
•
adapter molecules. On the other band, the novel PI3Ky cao he activated by Py subunits of
heterotrimeric G proteins (Stoyanov et al., 1995). Lopez-Ilasaca et al. (1997)
demonstrated the activation of MAPK by the expression of PB Ky in Cos-7 ceUs, while
PI3Ka did not affect the activity of MAPK. The expression of a mutated PI3Ky that
lacks lipid kinase activity abolished MAPK activation mediated by the M2 receptor or by
Gllr expression (Lopez-Ilasaka et al., 1997).
43
•mAChR
o CH,
JL ~CtiJHzN" ~~ CH:J
ACh agonist carbachol
+
•
•

•
•
•
FIGURE 2: Divergent kinase cascades that Iink mAChRs to the MAPK pathways.
mAChRs couple to G-proteins that cao activate different signaling cascades upon
cholinergie stimulation of the receptor. Refer to text for discussion of pathways and for
abbreviations. Adapted and modified from Gutkind (1998>.
44

•
•
•
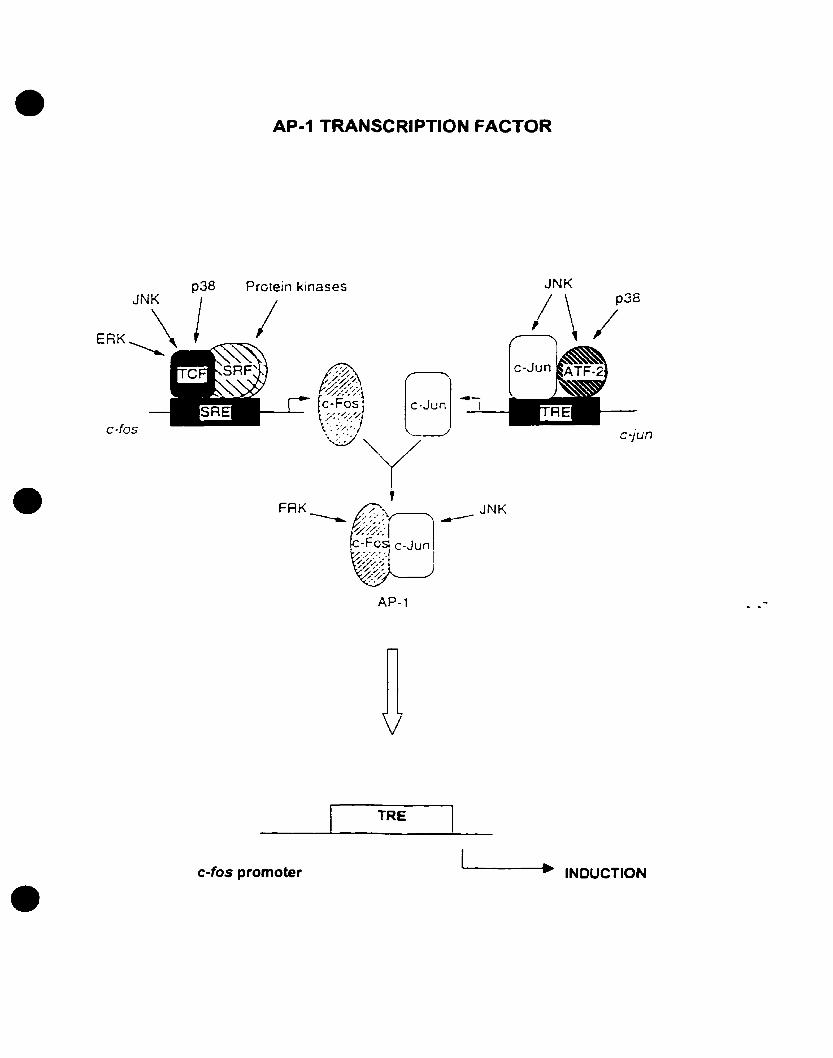
• MAPK Regulation of AP-l Transcription Factor and the Induction of c-fos Gene
Expression
One of the most investigated targets of MAPK signaling in mammals is the AP-l
transcription factor. AP-I consists of members of the Joo and Fos transcription factor
families which belong to the bZIP group of DNA binding proteins. Fos and Jun family
combine in various ways with other transcription factor families, including cAMP
response element binding protein (CREB)/ATF, NFAT, ETS, NFkB, and nuclear
hormone receptors. It has been shown that several signaIs can induce AP-l activity,
including growth factors, cytokines, neurotransmitters, and cellular stress (Davis, 1994;
Karin, 1995; Angel et al., 1991; for review see Whitmarsh and Davis, 1996; Gutkind,
1998). AP-l is able to bind the TPA-response element (TRE) with the consensus
sequence TGACTCA (Angel and Karin, 1991) (see fig. 3). The binding affinity for a
particular TRE sequence is dependent on the composition of the AP-l transcription
factor. Many immediate early genes, including c-fos, posses TRE sequences in their
promoter regions. On one hand, MAPK signaling pathways influence AP-I activity by
bath increasing the aboodance and activity of AP-l components (Karin, 1995).
On the promoter of the c·fos gene, there are three major transcriptional control
elements present that are targeted by inducible signaling pathways: the cAMP-response
element (CRE), the serum response element (SRE), and the sis-inducible element (SIE)
(Janknecht et al., 1995). The efficient regulation of these elements is required for the
expression of c-fas. The CRE is bound by the dimeric CREB and related transcription
45
• Ap·1 TRANSCRIPTION FACTOR
•
p38
JNK jERK~
C
Protein kinases
/
AP-1
JNK
/ \ p38, /
c-fos promoter
•___---L. T_R_E I_
10...----... INDUCTION

•
•
•
FIGURE 3: Regulation of AP-l transcription factor by MAP kinases. The AP-l
transcription factor is highly regulated by different MAPK's including ERKs, JNKs,
p38s, in addition to other protein kinases. The AP-l transcription is formed by the
dimerization of c-fos and c-Jun transcription factors. Both c-fos and c-jun gene
expression is highly regulated by protein kinases. Upon stimulation of their expression,
c-Fos and c-Jun proteins dimerize and regulate the expression ofother genes, including c
fos. Refer to text for abbreviations. Adapted and modified from Whitmarsh and Davis
(1996).
46

•
•
•
factors. CREB is activated by protein kinase A (PKA)7 a sereine/threonine kinase that is
activated by cAMP. Phosphorylated CREB binds CREB-binding protein (CBP) which
can facilitate the transcriptional machinery on the c-fos promoter. CaIciumlcalmodulin
dependent protein kinases (CalMK) have been impiicated in CRE-dependent c-fos
induction. by phosphoryiating CREB and therefore activating the transcription factor.
The c-fos SRE, on the other hand7 mediates serum induction of the c-fos
prornoter, hence the name. Yet SRE-mediated induction was shown to be also stimuIated
by growth factors, cytokines, and cellular stress (Cahill et ai., 1995; Cahill et al., 1996).
To start SRE-mediated transcription, the serum response factor (SRF) which recruits a
ternary complex factor (TCF) binds the SRE (Cahill et al., 1995). TCF proteins belong to
the ETS-domain family of DNA binding proteins that includes Elk-I, SAP l, and
SAP2INETIERP (Cahill et aI., 1995; Cahill et al., 1996; Price et al., 1995). In response
to several signais including growth factors and phorbol esters, the TCf Elk-I is
phosphorylated in its activation domain by members of ERK (Gille et aI., 1992; Marais
and Treisman, 1993; Janknecht et aI., 1993; Gille et al., 1995), JNK (Cavigelli et al.,
1995; Whitmarsh et al., 1995; Zinck et al., 1995; Gille et al., 1995), and p38 MAPKs
(Raingeaud et al., 1996), which increase its ability to form complexes with the SRf at the
SRE, therefore activating transcription (Gille et al., 1992; Gille et al., 1995). In
retrospect, the phosphorylation of the TCF Elk-l by ail three groups of MAPKs is crucial
for its transcriptional activity, whereby Elk-l acts to converge multiple MAPK signal
cascades at the SRE to increase c-fos transcription in response to a variety ofextracellular
signais, including the activation of G-protein coupled receptors such as mAChRs. Note
47

•
•
•
that there are other TCF proteins that form temary complexes with the SRF at the SRE,
such as SAP-I. Its activity is also regulated by MAPKs including the ERK and the p38
MAPKs (Priee et al., 1995; Janknecht et al., 1995).
The formation of the AP-l transcription factor is limited by the induction of both
c-fos and c-jun genes, since AP-I dimers constitute the binding of c-fos and c-jun
transcriptional factors. The transcriptional response of the c-jun promoter is mediated by
two TREs, Jun 1 and Jun2 (Angel, 1995). These promoter elements are constitutively
bound where they preferentially bind c-Jun and ATF-2 (Angel, 1995). Both transcription
factors are phosphorylated on two sites by the JNK group of MAP kinases, therefore
enhancing the transcriptional activity and leading to the induction of the c-jun gene. As a
result, c-Jun protein synthesis is elevated. Subsequently, c-Jun transcription factors bind
to c-Fos transcription factors to fonn the AP-I complex, which in tum can regulate the
induction of c-fos and other immediate early genes at the AP-l response elements.
48

•
•
•
CHAPTERTWO
STATEMENT OF PURPOSE
49

•
•
•
STATEMENT OF PURPOSE
Earlier studies have demonstrated the ability of the oligodendrocyte lineage to
respond to cholinergie stimulation. Larocca et al. (1 987b) provided evidence for the
presence of the ~11 and the M2 mAChRs in pwified myelin isolated from adult rat brain~
eliciting the interest in investigating cholinergie responses and signaling in
oligodendrocytes. The purified myelin mAChRs couple to phosphoinositide hydrolysis
and inhibition of adenylate cyclase (Larocca et al.~ 1987a; Khan and Morell~ 1988).
Studies performed in our laboratory have pharmacologically characterized the mAChRs
present in 0-2A progenitors and in mature oligodendrocytes, displaying predominance of
the M3 subtype (see fig. 1) (Molina-Holgado et al., unpublished). However, the
pharmacological studies showed extensive downregulation of the mAChRs in mature
oligodendro'cytes during in vitro development, suggesting a role for mAChRs in their
progenitor stage (Molina-Holgado et al., submitted).
Cholinergie stimulation of 0-2A progenitors triggers various intracellular
responses that are mediated by mAChRs. Studies reported from our laboratory have
shawn that mAChR activation of progenitors causes an increase in their proliferation
(Cohen et al., 1996). Similarly, activation of mAChRs induees complex signal
transduction pathways in progenitors, including accumulation of 1P3 (Cohen and
Almazan~ 1994), increases in intracellular calcium levels (Cohen and Almazan~ 1994;
Kastritis et al., 1993; Takeda et al., 1995), production of calcium waves (Simpson and
Russell, 1996), inhibition of an inwardly rectifying potassium channel (Karschin et al.,
50

•
•
•
1994), and inhibition of f}-adrenergic-stimulated cAMP production (Cohen and Almazan,
1994).
We have also recently demonstrated the cholinergie activation of p42mapk in 0-2A
progenitors, an event that is mediated by mAChRs in the presence of extracellular
calcium (Larocca and Almazan, 1997). In addition, Pende et al. (1997) demonstrated the
phosphorylation of CREB by CCh stimulation, an event that is dependent on p42mapk
activation. Moreover, we have shown that cholinergie stimulation of 0-2A progenitors
increased c-fos mRNA expression, an event that is mediated by mAChRs as weIl (Cohen
et al., 1996). CCh stimulated p42IMPk activation and c-fos mRNA expression in a time
and concentration- dependent manner, involving DAG-independent PKC pathways
(Larocca and Almazan, 1997; Cohen et al., 1996). Yet the identity of the mAChR
subtype(s) that induce(s) the activation of p42IMPk and the expression of c-fos mRNA in
oligodendrocyte progenitors has not been determined. AIso, the signaling pathways that
connect mAChR activation to p42mapk activation and c-fos mRNA expression have not
been fully elucidated. Therefore, we decided to investigate the mAChR subtype(s)
involved in both responses, and to further define the intracellular effectors that link the
mAChR to the critical MAPK pathway and to the nucleus.
51
• AC) 120c:c.~ 100.cen~z-::r: 6
C")..-o~
"0(1)Coencft
i i-11 ..10
i·9
i-8
i..7
i·6
i-5
i-4
i-3
•• atropineo 4-DAMP• pirenzepine
o methoctramine
• tropicamide
B120
.,......cS 100Ü
:2 80E~
60~0'-
a. 40--J:C')
20-0
i i i i i i i , i• -11 -10 -9 -8 ..7 -6 -5 -4 ..3

•
•
•
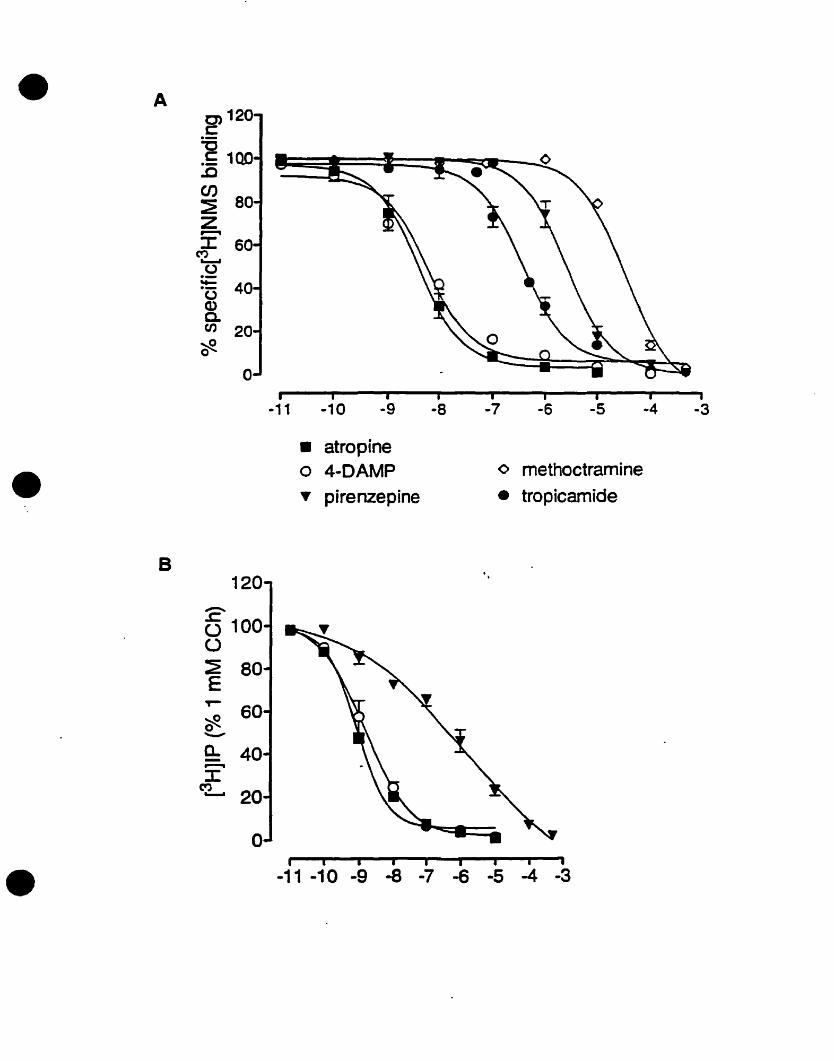
FIGURE 1: Competition and PI hydrolysis studies OD mAChRs in oligodendrocyte
progenitors. (A) Competition binding experiments were performed on whole cells.
Progenitors were exposed to increasing concentrations of muscarinic antagonists
[atropine (non-selective); pirenzepine (Ml selective); (methoctramine (M2 selective); 4
DAMP CM3 selective); tropicamide CM4 selective)] and 1 nM of the radiolabelled
acetylcholine analogue eH]NMS. (B) Inhibition of CCh-stimulated eHJIP
accumulation by increasing concentrations of muscarinic antagonists. Adapted and
modified from Molina-Holgado et al. {submittedl.
52

•
•
•
CHAPTER THREE
MATE~SANDMŒTHODS
53

•
•
•
MATERIALS AND METHOnS
Materials
The following reageots were obtained from the indicated supplier: carbachol~
atropine methyl bromide from Sigma-Aldrich Canada (Oakvil1e, Ontario); PD 098059,
methoctramine, 4-DAMP methiodide (4-DAMP), 4-DAMP-Mustard (4-DAMP-M),
pirenzepine, and wortmannin were purchased from RBI (Natick, MA); {1-[6-«(l7b-3
methoxyestra-I,3,5(lO)-trien-17-yl)amino)hexyl]-IH-pyrrole-2,5-dione} (U73122), {I
[6-«1 7b-3-methoxyestra-I,3,5(1O)-trien-17-yl)amino)hexyl]-2,5-pYrr0ldinedione}
(U73343) were from Calbiochem (La Jolla, CA). Phospho-specific p42mapk antibody
(Thrl83 and Tbr185) was obtained from Promega (Montreal, QC). The A2B5 mAb IgM
was from American Type Culture Collection CRL 1520. Second antibodies used for
immunofluorescence were purchased from Southern Biotechnology or Jackson
Immunoresearch Laboratories. The media Dulbecco's modified Eagle medium (DMEM),
Ham's FI2 medium and Hank's Balanced Salt Solution (HBSS) as weil as 7.5% bovine
serum albwnin (BSA) fraction V, fetal calf serum (FCS), penicillinlstreptomycin were
[rom Gibco BRL (Burlington, ON); PDGF AA, bFGF from UBI; poly-D-Iysine, poly-L
omithine, Triton-X-I 00. AlI other reagents were obtained from standard suppliers.
• CeU Culture
Oligodendrocyte progenitor primary cultures were prepared as described by
Almazan et al. (1993), modified from McCarthy and de Vellis (1980). The cultures were
prepared from the brains of newbom Sprague-Dawley rats. The meninges and blood
54

•
•
•
vessels were removed from the cerebral hemispheres in Ham's F12 medium. The tissue
suspension was passed through a 230 mm nylon mesh and collected by filtering through a
ISO mm and then a 100 mm nylon mesh. The resulting suspension was centrifuged for 10~
min at 1200 rpm and then resuspended in DMEM supplemented with 12 % FCS. Cells
were plated on poly-L-ornithine precoated 80 cm2 flasks and incubated at 37 oC with 5%
C02 in air. Culture medium was changed after 3 days and every two days thereafter. The
initial mixed glial cultures, gro\\-n for 9 to Il days, were placed on a rotary shaker at 225
rpm at 37°C for 3 br to remove loosely attached macrophages. Oligodendrocyte
progenitors were detached following shaking for 18 br at 260 rpm. Filtered through a 30
mm nylon mesh, the ceUs were plated on bacterial grade Petri dishes for 3 br. Under these
conditions, astrocytes and microglia become attached to the plastic surface and
oligodendrocyte progenitors remained in suspension. The final cell suspension was plated
on 6-well dishes (Falcon) pre-coated with poly-D-Iysine. Cultures were fed with serum
free medium (SFM) containing 2.5 ng/ml PDGF AA and 2.5 ng/ml bFGF to promote
self-renewal and prevent differentiation and changed every two days. SFM consisted ofa
DMEM-F 12 mixture (1: 1), 10 mM HEPES, 0.1 % bovine serum albumin (BSA), 25
mg/ml human transferrin, 30 nM triiodothyronine, 20 nM hydrocortisone, 20 nM
progesterone, 10 nM biotin, 5 mg/ml insulin, 16 mg/ml putrescine, 30 nM selenium, 50
units/ml penicillin and 50 mg/ml streptomycin as previously reported. The cultures were
immunocytochemically characterized by specifie markers for different ceU types. More
than 95% of the cells reacted positively with mouse monoclonal antibody A2B5, a
marker for oligodendrocyte progenitors in culture and less than 5% were
galactocerebroside positive oligodendrocytes, glial fibrillary acidic protein positive
55

•
•
•
astrocytes or complement type-3 positive microglia. These enriched oligodendrocyte
progenitor cell cultures were used in these studies.
• Immunocytochemistry
In order to determine the lineage composition of the cellular preparations,
immunocytochemistry was performed with A2B5 mAb that is specific to 0-2A
progenitors as shown by Cohen et al. (1996). Purified oligodendrocyte progenitors were
grO\\TI on poly-D-lysine-coated glass coverslips in 24-well tissue culture plates in SFM
plus growth factors for 3-5 days. For immunocytochemical detection "ith A2B5, cells
were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) for 20 min at
room temperature (RD. After fixation, the cells were washed severa! times with PBS
and blocked for 15 min in PBS containing 0.2% BSA, 5% goat serwn with 0.1% Triton
X-IOO for 45 min at RT. Monoclonal A2B5 mAb, applied as (1:500), was diluted in PBS
containing 10% FCS and 5% goat serwn with 0.1 % Triton X-IOO for 45 min at RT. The
second antibody SAM-IgM-FITC was applied (from Jackson Immunoresearch
Laboratories) and was diluted 1: 100. Coverslips were mounted in Immuno-mount and
were examined under a Leitz Diaplan epifluorescent microscope. On the day of the
experiment, 18 h after removal of the growth factors, more than 95% of the ceUs \vere
AlB5 positive oligodendrocyte progenitors (fig. 1)
• Western Immunoblot analysis
After preincubation with HBSS for 4 hr, cells were stimulated with
56

•
•
•
pharmaeologieal agents for the indieated time periode CeUs were lyzed in SDS sample
buffer (62.5 mM Tris-HCI, pH 6.8,2% w/v sodium dodeeyl sulfate (SOS), 10% glyeeroI,
50 mM dithiothreitol, 0.1 % w/v bromophenol blue) and boiled for 5 min. 30 J.lg of eaeh
protein extract were electrophoresed on 10% polyaerylamide gels and transferred to
Immobilon-P membranes (Millipore). Blots were blocked with 5% milk in Tris-buffered
saline containing 0.01% Tween 20 and then incubated overnight at 4 oC with anti
phospho-speeifie MAPK (1: 10000). Immunoreactivity was visualized by a
chemiluminescence deteetion system (Amersham) and quantified by densitometry using a
Master Scan Interpretative Densitometer.
• RNA extraction and Northem blot analysis
Total RNA was extracted from oligodendrocyte progenitors using a guanidium
thioeyanate method. The RJ."I'A pellets were resuspended in 50% formamide/2.2 M
formaldehyde/20 mM MOPS and denatured for 30 min at 65 oC. 10 J.lg of each extract
samples were eleetrophoresed on a 1.3% agarose-formaldehyde gel and transferred to
Hybond-N membranes. The c-fos probe was labeled with [a-32P]dCTP using a random
primer kit to a specifie aetivity of 108 epm/mg DNA. Following 2 hr of prehybridization,
membranes were ineubated at 42 oC for 48 hr with 106 epm/ml hybridization solution
(50% formamide, 25 mM sodium phosphate buffer (pH 6.5), 0.8 M NaCI, 0.5% SOS, 1
mM EDTA). Membranes were washed for 30 min at 50 oC in 0.1 X saline sodium citrate,
0.1 % SDS and exposed to X-ray film for 1-2 days. SignaIs were quantified using a
MaterSean Interpretive Oensitometer.
57

•
•
•
• Data analysis
One way analysis of variance followed by the Tukey-Kramer test was used to
detennine statistical significance: P values less than 0.05 were considered significant.
58

•
•
•

•
•
•
FIGURE 1: Purified oligodendrocyte progenitors (O-2A) were irnmunostained with the
A2B5 mAb, a marker for 0-2A progenitors in culture. Progenitor cultures showed 95%
purity.
59

1.
•
•
CHAPTER FOUR
RESULTS
60

•
•
•
RESULTS
• Activation of p42maPk by CCh is mediated through the M3 mAChR in
oligodendrocyte progenitors
The activation of p42maPk by cholinergie stimulation has been well documented in
other systems. Reports included studies on transfected cell lines and primary tissue
culture cells. We have shown that in cultured oligodendrocyte progenitors, CCh
stimulation activated p42nlapk via the rnAChRs (Larocca and Almazan, 1997). Larocca
and Almazan (1997) have demonstrated the expression of p42mapk by indirect
immunofluorescence using specifie anti_p42nlapk antibodies, whereby intense staining was
evident in the cytoplasm and the cellular processes but not in the nucleus. They have also
confirmed the expression ofp42mapk by Western blot analysis. In gel kinase activity using
MBP as a substrate demonstrated that CCh increased p42mapk, s kinase activity by two to
three above control levels. The activation of p42mapk by CCh was time- and
concentration-dependent, where maximallevels were reached at 2-5 min stimulation at an
optimal concentration of 10-100 J,lM. This response was blocked by atropine and EDTA
pretreatment, demonstrating that the activation of p42 mapL: by CCh is mediated by
mAChRs and is dependent on calcium.
Therefore, in view of these previous studies on p42mapk activation in addition to
the previous binding reports that pharmacologically characterized the mAChRs in
oligodendrocyte progenitors (Molina-Holgado et al., submitted), the present study
61

•
•
•
investigated the mAChR subtype that mediates the activation of p42mapk• Progenitor
cultures were pretreated with relatively selective mAChR antagonists at a concentration
of 1 JlM, including pirenzepine (Pir), an MI antagonist, methoctramine (Meth), an
M2/M4 antagonist, and 4-DAMP, an M3 antagonist, and the non-selective muscarinie
antagonist atropine (Atr) for 20 min. The 1 f.lM concentration of each muscarinic
antagonist used is sufficient to block its respective subtype(s) (Michel et al.~ 1989;
Lazareno et al., 1990; Caulfield 1993; Kondou et al., 1994; Molina-Holgado et al.,
submitted results). The cultures were then stimulated with 100 flM CCh for a period of 5
min, which corresponds to the optimal concentration and time period for CCh-induced
p42mapk activation (Larocca and Almazan, 1997). The levels of phosphorylated p42mapk
were detected using Western blot analysis and were quantified densitometrically (fig. 1).
Stimulating with 100 f.lM CCh increased p42mapk activation by 2-fold, while atropine
(non-selective) and 4-DAMP (M3-selective) blocked the response (fig. 2). On the other
hand, pirenzepine (Ml) and methoctramine (M21M4) had no effect on the CCh
stimulated activation of P42mapk•
To consolidate the results obtained with saturating concentrations of 4-DAMP,
cultures were pretreated with different concentrations of the antagonist, ranging from 1
nM to 1 JlM for 20 min. The cultures were then stimulated with 100 flM CCh for 5 min
(fig. 3). 4-DAMP reduced the activation of p42maPk dose dependently, with an
approximate ICso of 50 oM. Furthermore, an irreversible M3 selective antagonist, 4
DAMP-mustard (4-DAMP-M) (Barlow et al., 1991), blocked the activation of p42mapk,
thus confirrning that the M3 mAChR is the subtype responsible for the cholinergie
activation of P42mapk (Table 1).
62

•
•
•
• Carbaehol stimulates e-fos mRNA expression in oligodendro~yteprogenitors
through the M3 mACbR subtype
The activation of the M l/M3/M5 mAChRs activate PLC, leading to the activation
of PKC and the accumulation of intracellular calcium. Activated PKC cao potentially
stimulate a range ofcellular responses, including the induction of the proto-oncogenes c
fos and c-jun (Boyle et al., 1991; Distel and Spiegelman, 1990). These genes are a1so
referred to as immediate early genes, and their expression is regulated by transcription
regulatory factors that are activated by a variety of neurotransmitters, and are involved in
celI growth, proliferation, and differentiation.
Previous studies have shown that cholinergic stimulation of oligodendrocyte
progenitors increases c-fos mRNA expression in a time- and a concentration-dependent
manner (Cohen et al., 1996). This response was abolished by pretreatment with the non
selective muscarinic antagonist atropine. In addition, the expression of c-fos mRNA was
inhibited by the PKC inhibitor H7, but not by H-89, the PKA inhibitor. The induction of
c-fos mRNA was also dependent on the influx of extracellular calcium and the release of
calcium from intracellular stores, since both the extracellular calcium chelator EDTA and
the intracellular calcium chelator BAPTA blocked the response. However, Cohen et al.
(1996) did not address the identity of the mAChR mediating the induction of c-fos
rnRNA. Therefore, it was interesting to investigate whether the predominant mAChR
subtype, the M3, was also responsible for the induction ofc-fos.
Therefore, oIigodendrocyte progenitors were pretreated with selective mAChR
antagonists at 1 J.lM concentration for 20 min. The cultures were subsequently stimulated
with 100 J.lM CCh for 30 min, and the levels of c-fos mRNA levels expressed were
63

•
•
•
measured by Northern blot analysis (fig. 4). The autoradiograms were quantified
densitometrically (fig. 5). CCh alone increased the levels of c-fos mRNA by more than
six-fold above non-stimulated controls. Pretreatment of the cultures with either atropine
(non-selective) or 4-DAMP CM3 selective) abolished the CCh-stimulated c-fos mRNA
expression. On the other hand, 1 IJ.M of methoctramine (M21M4 selective) had no effect,
while pirenzepine reduced but did not entirely block c-fos expression. These resuIts
suggest that the M3 mAChR, which is also the predominant mAChR subtype found in
oligodendrocyte progenitors, is mediating the induction ofc-fos mRNA.
To define the ICso of 4-DAMP on c-fos mRNA expression, cultures were
pretreated with the antagonist (l DM to 1 J.l.M) for 20 min, followed by a 30 min
stimulation with 100 IJ.M CCh (fig. 6). 4-DAMP antagonized the effect of CCh on c-fos
rnRNA in a dose-dependent manner. A total block was obtained with 100 nM, and the
approximate ICso was calculated to be 10 nM. Furthermore, 1 J.LM 4-DAMP-mustard
(M3 irreversible) antagonized the CCh response (Table 1), confirming that the M3 is the
mAChR subtype responsible for the CCh-stimulated c-fos mRNA expression.
• The CCh-stimulated activation of p42mapk requires PLC
Previous studies have demonstrated the accumulation of inositol trisphosphates in
oligodendrocyte progenitors using [3H]-myo-inositol labeled young oligodendrocyte
cultures following stimulation with CCh (Cohen and Almazan, 1994). This response was
prevented by pretreatment with the non-selective mAChR antagonist atropine. The
accumulation of inositol trisphosphates was concentration- and time- dependent. In
64

•
•
•
addition, the same study reported a CCh-induced increase in the intracellular calcium
levels. The increase in intracellular calcium was dependent on bath the mobilization of
intracellular stores and the influx ofextracellular calcium.
Since mAChR stimulation increases the accumulation of calcium and inositol
trisphosphates, by the activation of PLC, and the mAChR-mediated activation of p42n13Pk
is calcium dependent and involves the M3 subtype which can couple to PLC via the Gqlll
G-protein (Larocca and Almazan, 1997), the present study investigated linking PLC
activation to the M3-mediated activation of p42mapk• To address this question, an
inhibitor of the enzyme PLC, U73122, and its close but inactive analogue U73343, were
used (Tatrai et al., 1994). Progenitor cultures were pretreated with 10 f.lM of either
U73122 or U73343 for 20 min prior to the 5 min treatment with 100 J.1M CCh (fig. 7).
Both inhibitors alone did not significantly reduce the levels of p42mapk activation.
However, pretreating the cultures with the active PLC inhibitor U73122 caused a
significant reduction in the CCh-stimulated leveIs of p42mapk activation, while the
inactive analogue U73343 had not effect. Therefore, these resuIts suggest that PLC
activation is implicated in the CCh-mediated activation of p42mapk
• The CCh-stimulated increase in c-fos mRNA expression requires PLC
The report of Cohen and Almazan (1994) demonstrated that cholinergie
stimulation of oligodendrocyte progenitors causes the accumulation of inositol
trisphosphates and intracellular calcium. Interestingly, the induction of the c-fos gene
was previously shown to be calcium dependent (Cohen et al., 1996). These response.r~
65

•
•
•
suggested a cholinergie activation of PLC via the odd numbered mAChRs, and the
activation of PLC may subsequently induce the expression of c-fos rnRNA through the
action of calcium.
To investigate the role of PLC in the cholinergie expression of c-fos m.RNA,
cultures were pretreated for 20 min with U73122 and its inactive analogue U73343,
followed by CCh stimulation for 30 min (fig. 8). The pretreatment with U73122 but not
U73343 caused a significant reduction in the levels of the CCh-stimuJated response,
displaying the involvement of PLC in the CCh-stimulated increase of c-fos mRNA
expression.
• Cholinergie p42..•pk activation and c-fos mRNA expression are Dot mediated by
PI3K
Phosphoinositide 3-kinase (PI3K) is a signaling Molecule that is regulated by
receptors with intrinsic or associated tyrosine kinase activities (Stoyanov et al., 1995; for
review, see Kapellar and Cantley, 1994). This enzyme phosphorylates the inositol ring of
phosphotidylinositol (PI) and phosphoinositides at the D3 position. There are several
species of PI3K that have been cloned and characterized including the heterodimeric
PI3K-a. and PI3K-f3, in addition to PI3K-y. PI3K-y can be activated in vitro by both Ct.
and f3y subunits of heterotrimeric G-proteins that are coupled to G-protein coupled
receptors (Stoyanov et al., 1995). In transfected cell lines, wortmannin, a PI3Ky
inhibitor, has been shown to inhibit p42maPk activation mediated by CCh. PI3Ky was
implicated in linking the f3y complex to the MAPK cascade (Lopez Ilasaca et al., 1997).
66

•
•
•
To determine this link, cultures were pretreated for 20 min with 100 nM wortmannin
(Wort) (Arcaro and Wymann, (993) followed by 5 min stimulation with 100 J,lM CCh
(fig. 9A). Pretreatment with wortmannin had no effect on the levels of CCh-stimulated
p42mapk activation, demonstrating no dependence on PI3Ky action.
In addition, PI3K has been shown to have various different downstream targets
including PKC isozymes from both the conventional and novel families. in addition to
other kinases including Alet, Blet, and Rac (for review see Fukui et al., 1998). Many of
these targets are involved in cell growth, development. and motility. responses that
involve signaling to the nucleus. Interestingly, the c-fos proto-oncogene is also involved
in protein synthesis and cell proliferation, whereby its product, the transcription factor
Fos, is important in regulating the expression of other proteins via the AP-I transcription
factor. In view ofthese findings, the present study investigated whether PI3K May play a
role in regulating the cholinergie expression of c-fos mRNA (fig. 9B). However,
pretreating progenitor cultures with wortmannin did not significantly reduce the CCh
stimulated increase in c-fos mRNA expression, demonstrating that PI3K May also not he
implicated in this event.
• PD 098059, the MAPK Kinase (MEK) Inbibitor, reduces but does not abolish
tbe CCb-stimulated expression of c-fos mRNA in oligodendrocyte progenitors
Having established that both p42mapk activation and c-fos gene expression are
downstream from the M3 mAChR in oligodendrocyte progenitors, we attempted to
demonstrate a positive link between them. To address this issue, the MEK inhibitor PD
098059 (Alessi et al., 1995; Dudley et al., 1995) was added at different concentrations
67

•
•
•
ranging from 1 J..IM to 100 IJ.M for 20 min followed by a 30 min stimulation with 100 J,.lM
CCh (fig. 10). Pretreating the cultures with 10 J,.lM PD 098059, a concentration that is
sufficient to inhibit p42mapk activation (Larocca et Almazan, 1998), did not alter the CCh
stimulated levels of c-fos mRNA. Furthermore, a significant reduction was ooly attained
at 50 J.LM and 100 J,.lM concentrations of PD 098059. The inability of PD 098059 to
completely block the stimulation of c-fos mRNA expression by CCh indicates that the
MAPK pathway May not be the only pathway linking muscarinic activation of
oligodendrocyte progenitors to c-fos gene expression. Altematively, PD 098059 at SO
100 J.LM concentrations may be inhibiting other kinases since 10 J.lM concentration was
sufficient to completely block p42mapk activation (Larocca and Almazan, 1997).
• The CCh-mediated c-fos mRNA expression is abolished by PLA2 inhibitors
PLA2 catalyzes the hydrolysis of phosphatidylcholine to generate free
arachidonic acid (AA). An important domain of lipid research had focused on the
biochemistry of AA, and its cellular and physiological responses which include, but are
not limited to, inflammation, pain, and intracellular signaling. AA itself and its bioactive
eicosanoids are involved in many intracellular signaling pathways. The release of AA is
namely achieved in response to neurotransmitters, growth factors, or lipid stimulation of
cell systems. One such neurotransmitter is ACh.
Previous reports provided evidence for the release of AA via mAChR stimulation
in many systems, including the brain (for review see Felder, 1995), and phannacological
studies have demonstrated the coupling of the odd numbered mAChRs Ml /M3/M5 to the
activation of PLA2, namely to the cPLA2 (Conklin et al., 1988; Felder et al., 1990; Liao
68

•
•
•
et al., 1990; Sharp et al., 1991; Clark et al., 1989). Cholinergie stimulation of AA release
involved, as predicted, a PTX-insensitive G-protein, confirming the involvement of the
odd nwnbered mAChRs which couple to such G-proteins.
Many investigations have focused on the cell signaling function played by AA
and its metabolites in ceUs. Studies have reported the activation of members of the
MAPK family by AA and its Metabolites in several systems and have demonstrated the
co-compartmentalization of MAPKs with cPLA2 (Madamanchi et al., 1998; Hernandez
et al., 1998; Yu et al., 1998). In addition, recent reports provided evidence for PLA2 and
AA Metabolites in signaling to the c-fos serum response element (SRE) via Rac and Rho
dependent mechanisms (Kim et al., 1998; Kim and Kim, 1998; Kim et al., 1997).
Interestingly, it has been recently reported that the activation of JNK by G-protein
coupled receptors involved Rac and Rho-dependent signaling cascades (Coso et al.,
1995b; Coso et al., 1996; Minden et al., 1995; for review see Gutkind, 1998). These
results suggested a raie for PLA2 in signaling downstream of the mAChRs, and linking
its activation to nuclear responses, including the expression of c-fos mRNA.
Therefore, to investigate the role of AA in mediating the cholinergie stimulation
of c-fos mRNA expression, two cPLA2 inhibitors were used, quinacrine and AACOCf3
(Sakuta and Yoneda, 1994; Wissing et al., 1997). Progenitor cultures were pretreated
with 50 JlM quinacrine or 100 J..l.M AACOCf3 for 20 min followed by 100 J.lM CCh
stimulation for 30 min (fig. Il). The pretreatment of progenitors by either inhibitor
abolished the expression of c-fos mRNA induced by Ceh. These results suggest a role
for the activation of cPLA2 in mediating the increased expression of c-fos mRNA by
cholinergie stimulation in oligodendrocyte progenitors.
69
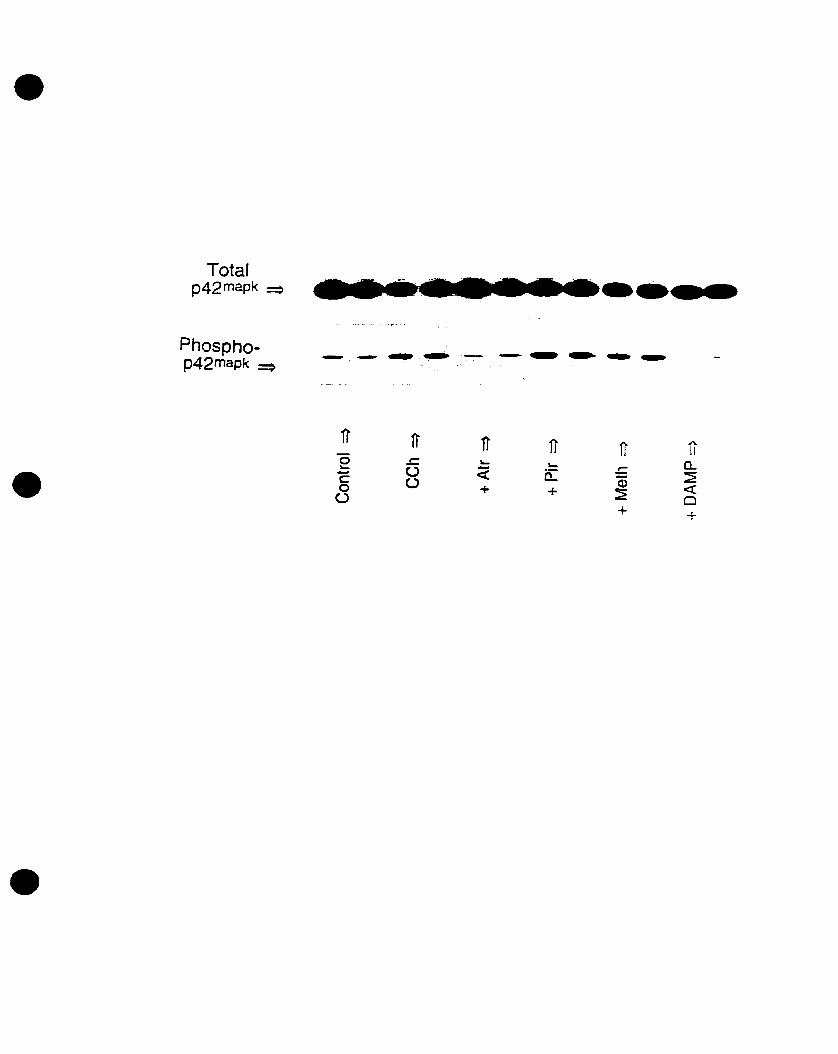
•
Totalp42mapk ~
Phosphop42mapk ~
·ta.......
11' 11' 11 ft Ir h
0II
~ ~
• ~ u < ~ a..ë a::: .r;
u êD ~0 + + «
ü ~ Q+
•

•
•
•
FIGURE 1: Western immunoblot autoradiogram demonstrating that p42mapk
activation is mediated by the M3 mACbR. An autoradiogram showing a Western
immunoblot analysis using phosphorylated p42mapk.recognizing antibody. The
membranes were also immunoblotted with non-phosphospecific p42mapk antibody that
recognizes total p42mapk, showing equal loading for each condition. AIl antagonists were
added for 10-20 min before CCh stimulation at a concentration of 1 J.1M (except for 4
DAMP, 100 nM). p42mapk activation is abolished by atropine (+ Atr) and 4-DAMP (+ 4
DAMP), but not by pirenzepine (+ Pir) or methoctramine (+ Meth). Note that the plus
sign (+) preceding the antagonists refers to pretreatment of progenitors with the
respective antagonists followed by the stimulation with 100 J.1M CCh.
70
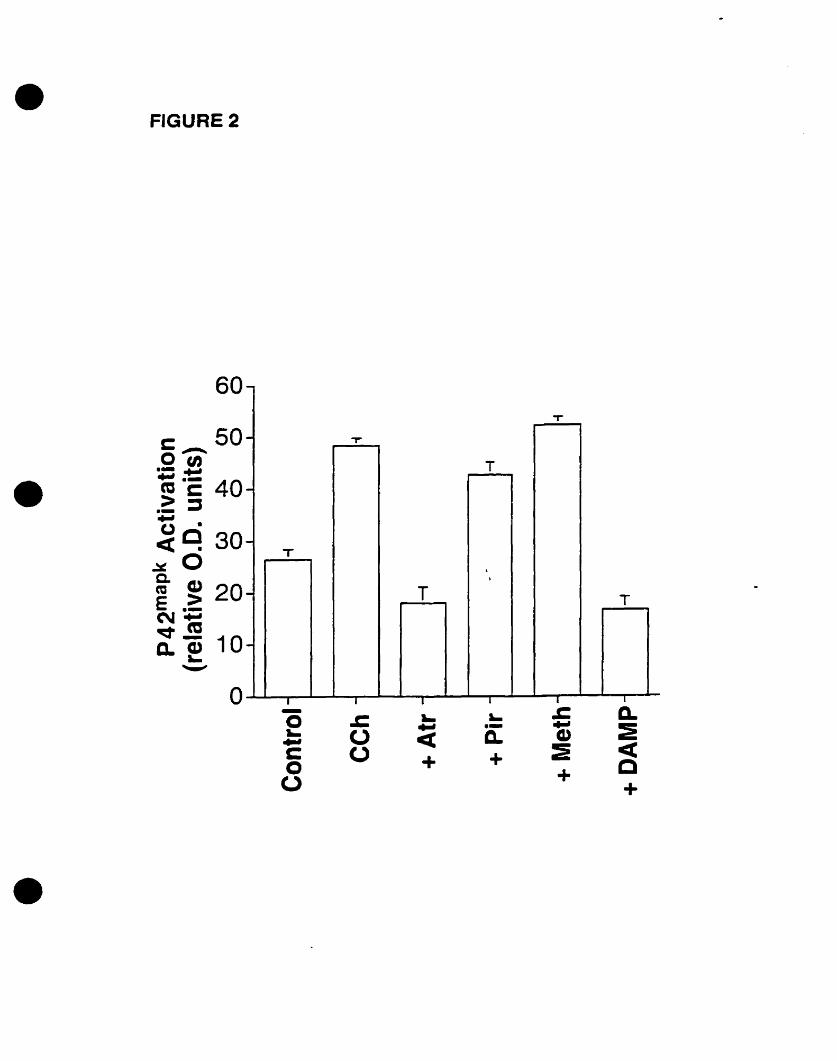
• FIGURE 2
D.:icec+
....-c.+
.cooo~..Coo
-
T
- T'
T
-
-T
-.
T T
-
1 1- 1 1 T la
60
•
•

• FIGURE 2: p4Z··pk activation is mediated by the M3 mAChR. Immunoblot
autoradiograms were quantified densitometrically and expressed in arbitrary optical
density (0.0.) units as mean ± SEM of two independent experiments perfonned in
triplicates. Muscarinic antagonists were preadministered for 10-20 min [Atropine (+ Atr,
1 flM); Pirenzepine (+ Pir, 1 J,lM); Methoctramine (+ Meth, 1 flM); 4-0AMP (+ DAMP,
100 nM)] followed by 100 J,lM CCh stimulation for S min. Atropine and 4-DAMP
blocked the CCh-stimulated levels. Statistical differences are indicated: control versus
(vs) CCh (p<O.OOI); control vs Atr + CCh (p>0.05, Dot significant); control vs Pif + CCh
(p<O.OI); control vs Meth + CCh (p<O.OOl); control vs 4-0AMP + CCh (p>O.OS); CCh
vs Atr + CCh (p<O.OOO1); CCh vs 4-DAMP + CCh (p<O.OOO 1).
•
•71
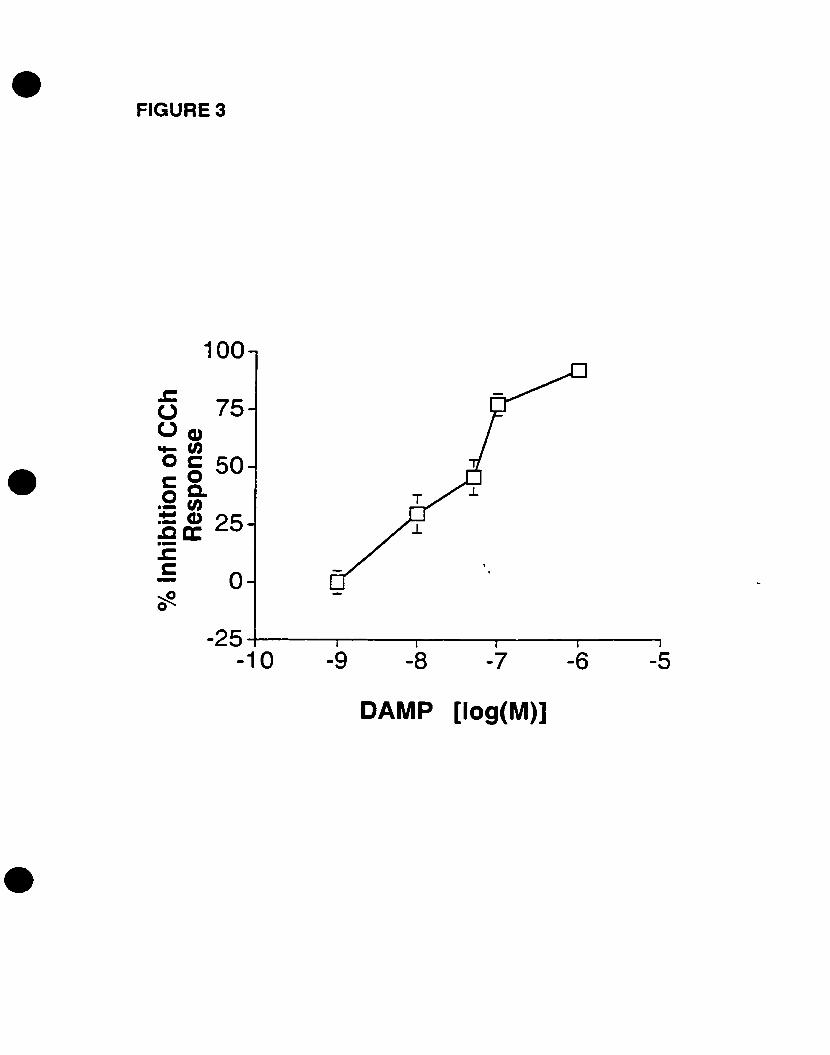
• FIGURE 3
-5-6-7-8-9
o
-25 4--------r----r--------r-----r----,
-10
100
cS 750(1)~'"o c: 50cOoc..- '":S~ 25.-oCc:
•
CAMP [log(M)]
•

•
•
•
FIGURE 3: 4-DAMP blo~ks the activation of p42 IDapk in a concentration-dependent
manDer. Progenitors were pretreated with increasing concentrations of the M3 more
selective antagonist 4-DAMP (1 nM - 1 J,lM), followed by 100 J.lM CCh stimulation for S
min. p42mapk activation was determined by Western immunoblot analysis, and
autoradiograms were quantified densitometrically. Values are expressed in arbitrary
O.D. units as percentage of inhibition of the CCh-stimulated activation of p42mapk
(Response) as mean ± SEM of two independent experiments performed in triplicates.
Statistical differences are indicated: Stimulated levels (Response, 100 J.lM CCh) vs 1 fJ.M
4-DAMP (p<O.OOI); Stimulated levels vs 100 nM + 4-DAMP (p<O.OOI); Stirnulated
levels vs 50 nM + 4-DAMP (p>O.OS); Stimulated levels vs 10 nM 4-0AMP + CCh
(p>O.OS); Stimulated levels vs 1 nM 4-DAMP + CCh (p>O.OS).
72

•28S rRNA
c-fos mRNA
• 11 11 11' 1J 11 110 .c ~ a. ~ ~~ u - ~ - CL- <: CDc (J <: ~0 + +(J C)
++
•

•
•
•
FIGURE 4: Northern blot autoradiogram demonstrating that e-fos mRNA
expression is mediated by the M3 subtype. Progenitors were pretreated with
muscarinic antagonists at 1 JlM (except for 4-DAMP, 100 nM) for 10 - 20 min, followed
by 100 ~M CCh stimulation for 30 min. Levels ofc-fos mRNA expressed were detected
by a standard Northem blot procedure. Ooly atropine and 4-DAMP pretreatment blocked
the CCh-stimulated c-fos mRNA expression levels. The membranes were also stained
with methylene blue to detect rRJ.'IA bands, demonstrating equal RNA loading in each
different condition.
73
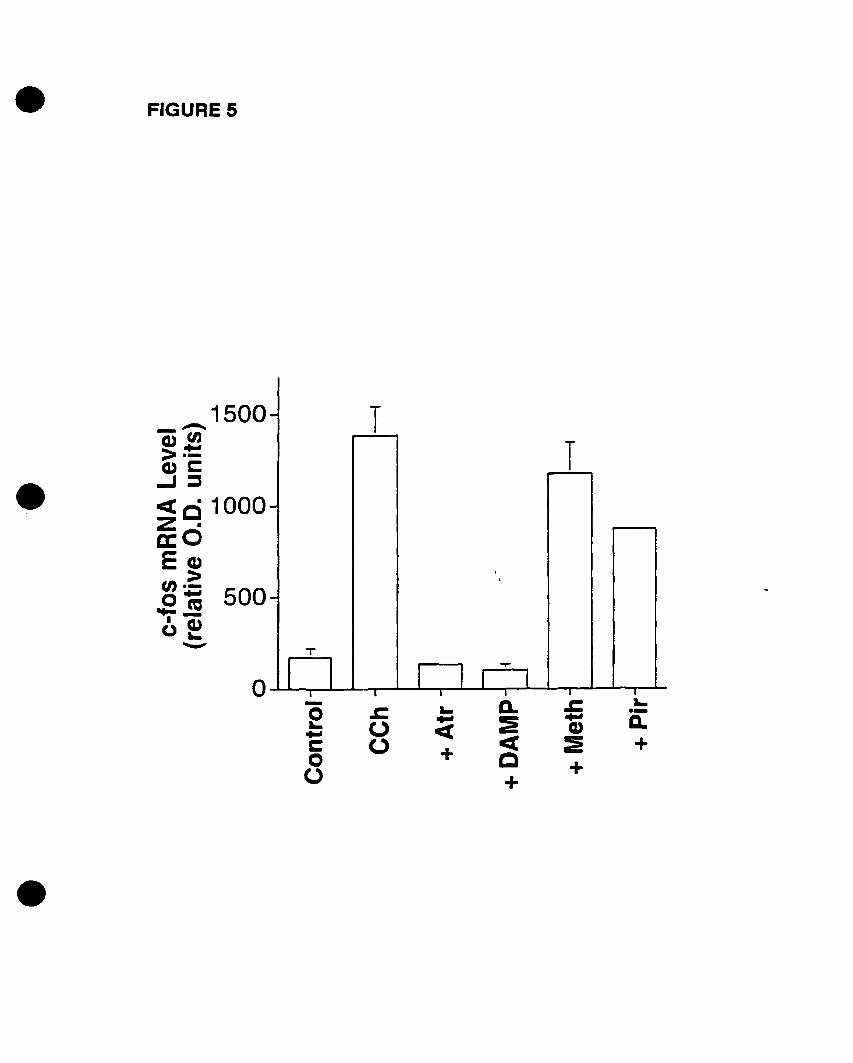
• FIGURE 5
.-D.+
.c...Cl)~
+
Q.:E<1:C+
~...<t+
.coo
-~...&:oo
- TT
-
,
-
T
r 1 r 1 r~
1T1 1T , 1~
o
1500.........-0Cl)..,>.-Cl)~
...J::I
<ci 1000~dECl)
>~; 500.... ~1 Cl)u~
"'-"
•
•

• FIGURE S: CCh..stimulated c..fos expression is mediated by the M3 mAChR. The
Northem blot autoradiogram was quantified densitometrically and ail values are
expressed in arbitrary 0.0. units as mean % SEM of two independent experiments
performed in triplicates. Only atropine and 4-DAMP blocked the CCh-stimulated c-fos
mR.~.A. levels. StatisticaJ differences are indicated: control vs CCh (p<O.OO1); control vs
Atr + CCh (p>O.OS); control vs 4-DAMP + CCh (P>O.OS); control vs Meth + CCh
(p<O.OOl); control vs Pir + CCh (p<O.OOI); CCh vs Atr + CCh (p<O.OOI); CCh vs 4
DAMP + CCh (p<O.OO 1); CCh vs Melh + CCh (p>O.OS); CCh vs Pir + CCh (p<O.O 1).
•
•74
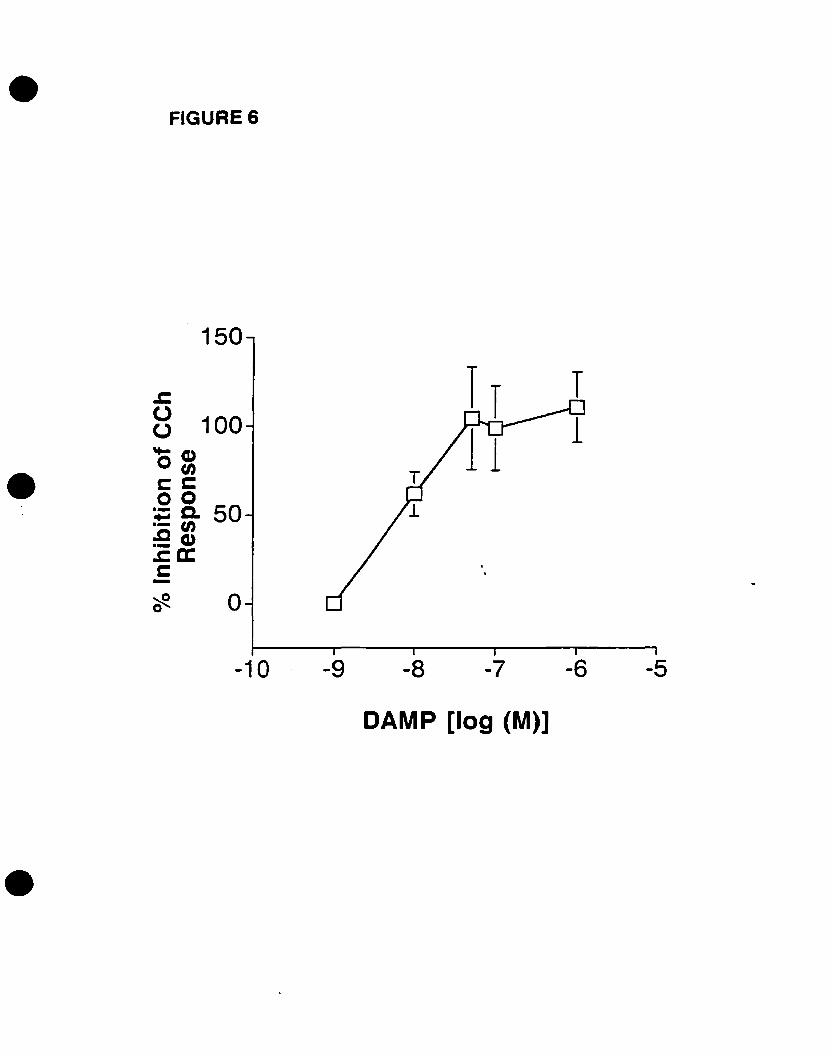
• FIGURE 6
•
150
.c:8 100
o~cC00; Cl. 50.- 0:eœ.ca:c-
cft. 0
-10 -9 -8 -7 -6 -5
•
DAMP [log (M)]

• FIGURE 6: 4-DAMP blocks the expression of c-fos mRNA in a concentration
dependent manDer. Progenitors were pretreated with increasing concentrations of 4
DA~1P (l nM - 1f.1M) for 10 - 20 min, followed by 100 f.1M CCh stimulation for 30 min.
e-fos mRNA levels were determined by Northem blot analysis, and were quantified
densitometrieally. Values are expressed in arbitrary 0.0. units of percentage inhibition
of CCh-stimulated c-fos mRNA expression levels (Response) as mean ± SEM of two
independent experiments performed in triplicates. Statistical differences are indicated:
CCh-stimulated levels (Response) vs 1 f.1M 4-DAMP + CCh (p<O.OO 1); ReSPOnse vs 100
nM 4-DAMP + CCh (p<O.OO1); Response vs 50 nM (p<O.OO1); Response vs 50 nM 4
DAMP + CCh (p<O.OOl); Response vs 10 nM 4-DAMP + CCh (p<O.OI); Response vs 1
nM 4-0AMP + CCh (p>O.OS).
•
•75

•Control
CCh
4-DAMP-M
4-DAMP-M + CCh
p42111apk Activation(relative O.D. units)
51.3 ± 4.2
122.S ± 0.2
41.3 ± 0.1
35.4 ± 3.1
c-fos mRNA Level(relative O.D. units)
45.4 ± 19.1
662 ± 107.7
49.2 ± 7.3
74.6 ± 7.4
•
•
TABLE 1: 4-DAMP-mustard (4-DAMP-M) blocks the CCh-stimulated p42maPk
phosphorylation and c-fos mRNA expression. Progenitors were preincubated with 1
JlM 4-DAMP-M for 20 min followed by 100 fJ.M Ceh. p42mapk activation levels are
detected using Western blot analysis, and c-fos rnRNA levels are detected using Northern
blot analysis. 4-DAMP-M blocked both CCh-stimulated responses. Statistical
differences indicated: control vs CCh (p<O.OS, for both responses); control vs 4-DAMP-
M + CCh (p>O.OS); CCh vs 4-DAMP-M + CCh (p<O.OS).
76

• FIGURE 7: CCh-stimulated p42-aPk activation requires PLC. Progenitors were
pretreated with 10)lM U73122 or 10 J..lM U73343 for 20 min prior to 5 min stimulation
with 100 J..lM CCh (+ Antagonist). Levels ofp42mapk activation were detected using the
Western blot analysis and were quantified densitometrically. Levels are expressed as
arbitrary optical density units (O.D.) as mean :i: SEM of triplicates. U73122 but not
U73343 blocked the stimulated levels. Statistical differences are indicated: control vs
CCh (p<O.OOl); control vs U73343 (p>O.OS); control vs U73122 (p<O.OS); control vs
U73343 + CCh (p<O.OOI); control vs U73122 + CCh (p>O.OS); CCh vs U73343 + CCh
(p>O.OS); CCh vs U73122 + CCh (p<O.OO 1).
•
•77
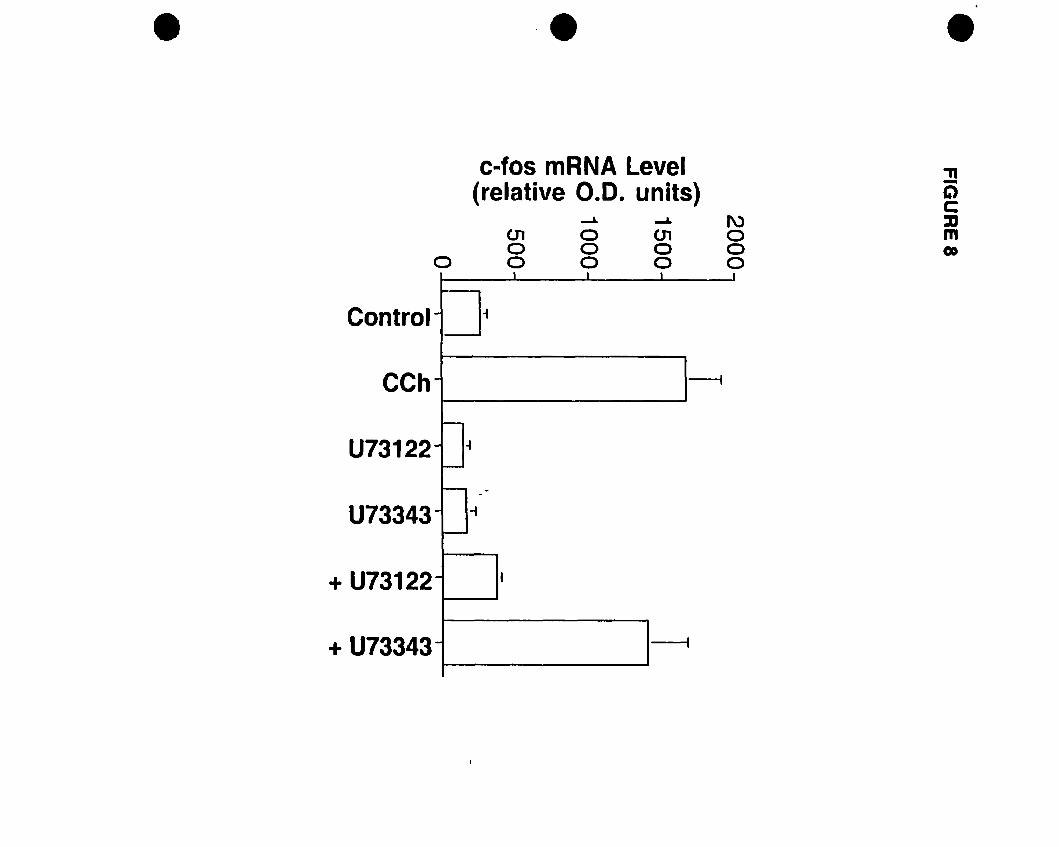
• '. •c-fos mRNA Level
(relative 0.0. units)
o01oo
--L
aoo
-L
01oo
1\)ooo
."-C)C::DmGO
Control
CCh
U73122
U73343
+ U73122
+ U73343
1 1 1 1
i
---i
1-
· il-
i'-- ~.· -i
-
· ,
----i
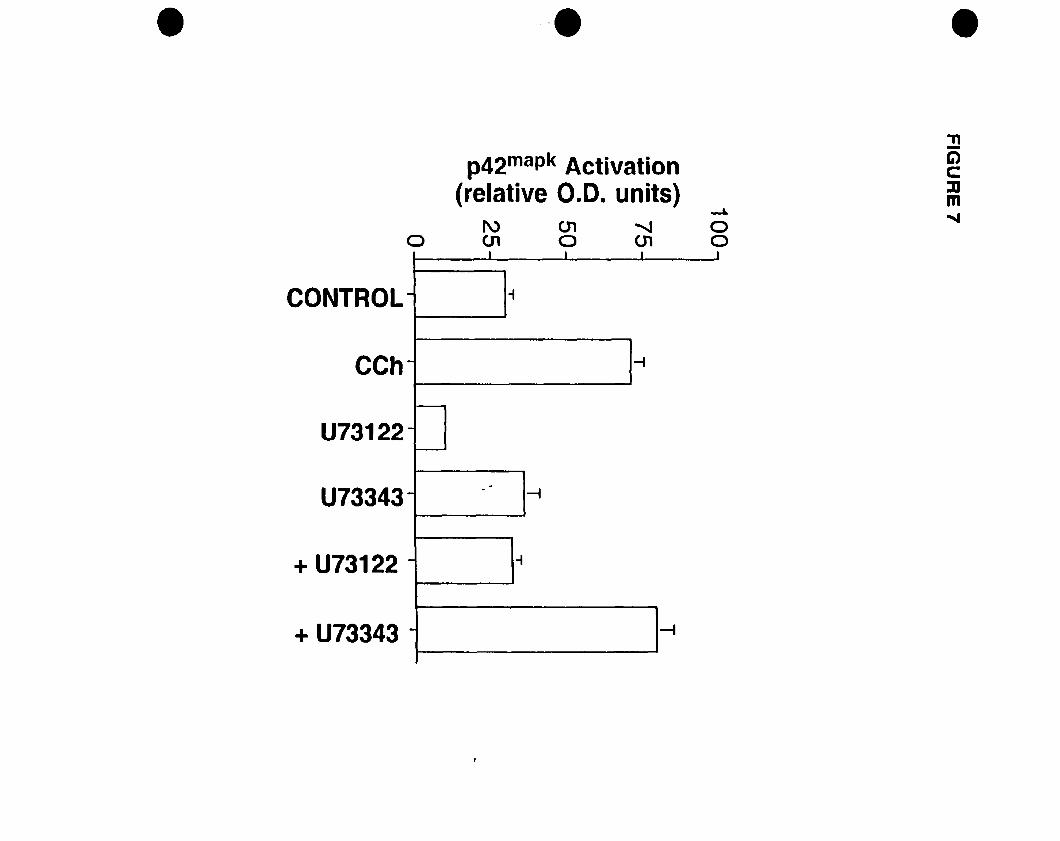
.'.p42mapk Activation
(relative 0.0. units)-L
1\) 01 ~ 00010010
•."-Clc:::am~
CONTROL
CCh
U73122
U73343
+ U73122
+ U73343
1 J1 11
-
-1
-r-
-fJ
t----1
f-J
. . -;
-l""-
r--1
- J-I
- 1
--1--

• FIGURE 8: CCb-stimulated c-fos expression requires PLC. Progenitors were
pretreated with 10 J.lM U73122 or U73343 for 20 min prior ta 100 J.1M CCh (+
Antagonist) stimulation for 30 min. c-fos mRNA levels were detected using a Northem
blot procedure and autoradiograms were quantified deositometrically. Levels are
expressed as arbitrary optical density (0.0.) units as Mean ~ SEM oftriplicates. U73122
but not U73343 blocked the CCh-stimulated levels. Statistical differences are indicated:
control vs CCh (p<O.OOI); cootrol vs U73122 (p>O.OS); control vs U73343 (p>O.OS);
control vs U73122 + CCh (p>O.OS); control vs U73343 + CCh (p<O.OI); CCh vs U73122
+ CCh (p<O.OO 1); CCh vs U73343 + CCh (p>O.OS).
•
•78
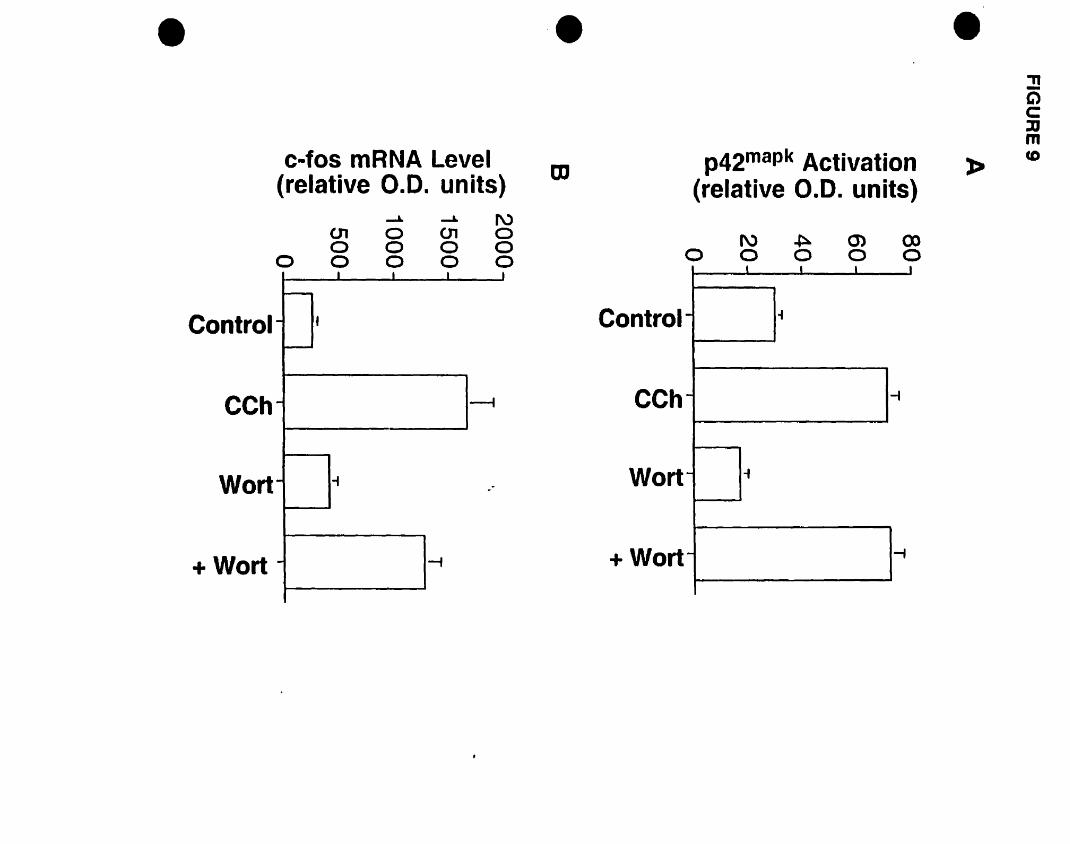
f\) ~ al 0000000
•
Control
CCh
Wort
+Wort
c-fos mRNA Level(relative 0.0. units)
-'" -'" f\)U1 0010o 0 0 0o 0 0 0 0
1 1 1 1
1--
f
~
. --1
- -i ..
- -f
'•
m
Control
CCh
Wort
+Wort
p42mapk Activation(relative 0.0. units)
1 1 1 1
. of
-i
of
-;
•»
:!!Clc::0mco

•
•
•
FIGURE 9: p42mapk activation and c-fos mRNA expression are Dot mediated by
PI3K. Progenitor cultures were pretreated with 100 nM wortmannin (Wort) for 20 min
followed by 100 J.lM CCh stimulation. A: p42maPk levels were detected using Western
blot analysis, and were quantified densitometrically and expressed as arbitrary optical
density (0.0.) units as mean ± SEM for two experiments in triplicates. Wortmannin
pretreatrnent did not significantly reduce stimulated levels. Statistical differences
indicated: control vs CCh (p<O.OOl); control vs Wort (p>O.OS); control vs Wort + CCh
(p<O.OOI); CCh vs Wort + CCh (P>O.OS). B: c-fos mRNA levels were detected using
Northem blot analysis, and were quantified densitometrically and expressed as arbitrary
0.0. units as Mean ± SEM of triplicates. Wortmannin (Wort) pretreatment did not
significantly reduce stimulated levels. Statistical differences indicated: control vs CCh
(p<O.OOI); control vs Wort (p>O.OS); control vs Wort + CCh (p<O.OOI); CCh vs Wort +
CCh (p>O.OS).
79
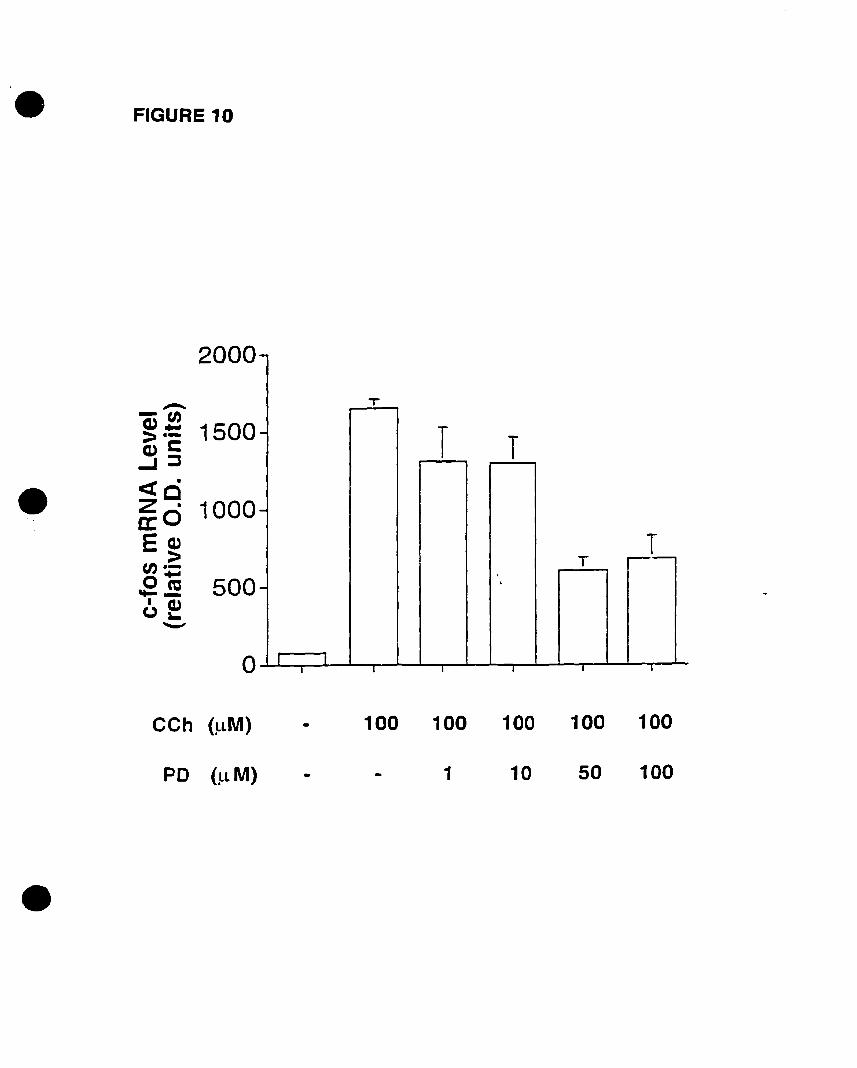
• FIGURE 10
2000
.........-U)aJ.., 1500:>.-aJl:-J::J
• <.zq 10000: 0Ecu::-C/).-0 ..... 500.... .!E1(1)u'-
'-'"
0
CCh (~lM)
-
T
-T T
-
TT
- ,
1 11 1 1 1 1 Î
100 100 100 100 100
•
PD (.lL M) 1 10 50 100

• FIGURE 10: PD 098059 does Dot block CCh..stimulated e..fos gene expressiOD.
Progenitors were pretreated with different concentrations of the MAPK Kinase inhibitor
PD 098059 (1 - 100 J-lM) for 20 min followed by 30 min 100 J.&.M CCh stimulation. c-fos
rnRNA levels \Vere detected using Northem blot analysis, and were quantified
densitornetrically. Values are expressed in arbitrary optical density (0.0.) units as mean
± SEM of triplicates. Only at high concentrations (SO - 100 J-lM) did PD 098059 reduce
the CCh-stimulated levels. Statistical differences are indicated: control vs CCh
(p<O.OOI); control vs 1 J.&.M PD 098059 (p<O.OOI); control vs 10 J.A.M PD 098059
(p<O.OOI); control vs 50 IJ.M PD 098059 (p<0.0l); control vs 100 J.A.M PD 098059
(p<O.Ol).
•
•80
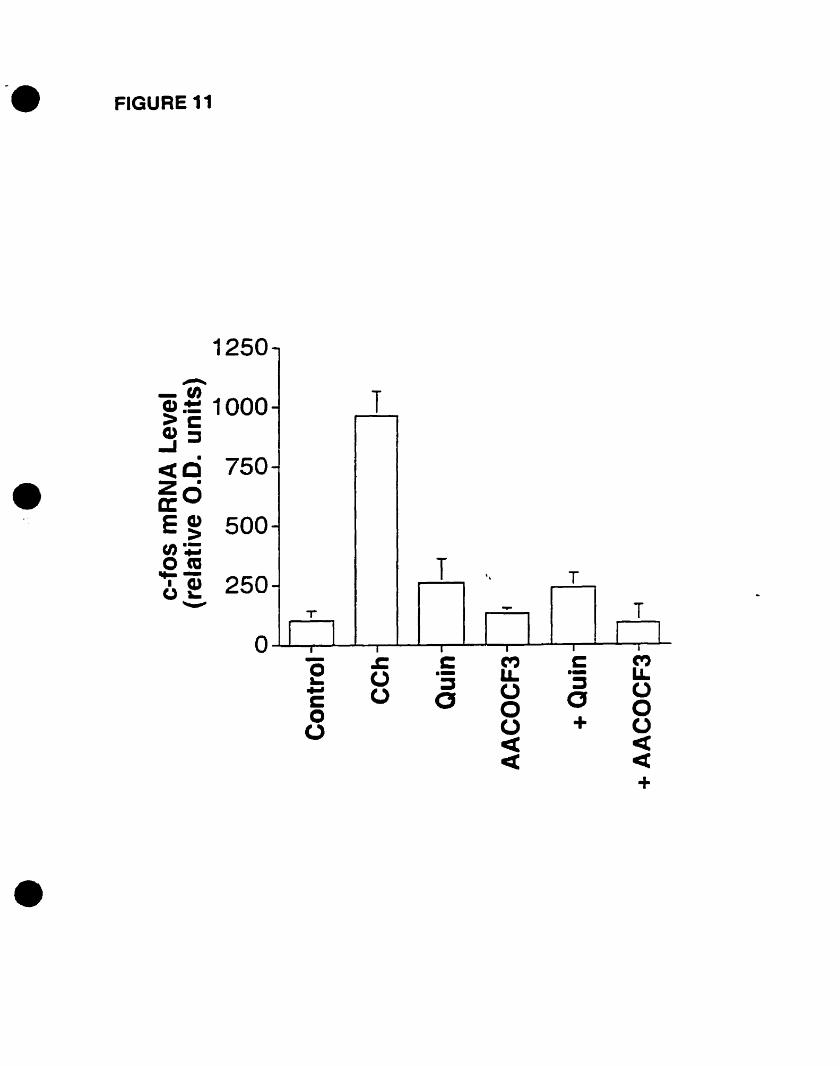
-. FIGURE 11
- ~ t:: M C M0 (.) .- LL -- LI.~ :::J :::J... (.) (J (Jc 0 a0 0 00 (J + (J
<t et<t et
+
-
- T
-
-
T " T-T -- T
1 11 1 r l-1 T 1 1 1 -1o
1250..--...
-0~~ 1000eue:..I::J
•<te 750
•Z·a: OE ~ 500(1);om.... -6 ~ 250
"'-'
•

•
•
•
FIGURE Il: PLA2 mediates the CCh-induced e-fos gene expression. Progenitors
were pretreated with 50 flM of quinacrine (Quin) or AACOCf3 for 20 min followed by
100 J.LM CCh stimulation for 30 min. mRNA levels were detected using Northem blot
analysis, and autoradiograms were quantified densitometrically and expressed as arbitrary
optical density (O.D.) units as mean % SEM of triplicates. Both antagonist pretreatments
bLocked the CCh-stimulated levels. Statistical differences are indicated: control vs CCh
(p<O.OOl); control vs Quin (p>0.05); control vs AACOCf3 (p>O.OS); control vs Quin +
CCh (p>O.OS); control vs AACOCfJ + CCh (p>O.OS); CCh vs Quin + CCh (p<O.OOI);
CCh vs AACOCfJ + CCh (p<O.OO 1).
81

•
•
•
CHAPTER FIVE
DISCUSSION
82

•
•
•
DISCUSSION
In summary~ the present study demonstrates that CCh~ a stable acetylcholine
analogue~ activates p42mapk via the M3 mAChR subtype in oligodendrocyte progenitors.
By pretreating oligodendrocyte progenitors with the M3 selective antagonist 4-DAMP
and its irreversible analogue 4-DAMP-mustard~ the CCh-stimulated p42mapk activation
was abolished. After establishing M3~s predominance in activating p42mapk~ the next step
was to use several pharmacological agents to unravel the pathway leading to its
activation. The present study demonstrated the involvement of PLC in the cholinergie
activation of p42mapk~ since a PLC inhibitor~ U73122, and not its close but inactive
analogue, U73343~ abolished this response. Moreover, pretreating with the PBK
inhibitor wortmannin did not reduce the levels of p42maPk activation~ indicating no role
for PBK in linking the M3 mAChR to p42mapk activation.
In addition, the present study has also focused on the pathways linking mAChR
stimulation to c-fos gene expression. We demonstrate that the cholinergie stimulation of
c-fos rnRNA expression is also mediated by the M3 mAChR subtype. This response was
blocked by 4-DAMP and 4-DAMP-mustard in a concentration-dependent manner.
Similar to p42mapkactivation~ PLC but not PI3K was implicated in the CCh-stimulated
response~ since the pretreatment with U73122 but not U73343 nor wortmannin blocked
the levels of c-fos mRNA expression. As both c-fos rnRNA expression and p42mapk
activation seem to be regulated by the same receptor and effector molecule~ it was
interesting to establish whether c-fos rnRNA expression was regulated by the activity of
p42mapk. Contrary to our predictions, pretreatment with PD 098059, a MAPK kinase
83

•
•
•
(MEK) inhibitor, did not decrease c-fos mRNA expression except at 50 and 100 flM
concentrations. At these high concentrations, the expression levels were reduced but
were not blocked. As a result, p42mapk activation may not be essential to the CCh
induced changes in c-fos mRNA expression. However, by pretreating progenitors with
the PLA2 inhibitors quinacrine and AACOCf3, c-fos mRNA levels were abolished,
suggesting a role for PLA2 in linking mAChR stimulation to c-fos gene expression.
mAChRs are G-protein coupled receptors, and the different subtypes couple to
different classes of G-proteins. The MIIM31M5 subtypes couple to the PTX-insensitive
class of G-proteins, namely the Gq/l t, while the t\·12/M4 subtypes couple to the PTX
sensitive class, mainly the Gilo (Offennanns, 1994). The stimulation of p42maPk is one of
the many signais mediated by G-protein coupled receptors such as the mAChRs. In
other systems, for example, the activation of p42mapk via the MI mAChR is mainly
mediated by the ex subunits of the Gq/11 protein (Hawes et al., 1995), whereby M IIM31M5
mediated p42mapk activation is PTX-insensitive (Wotta et al., 1998). On the other hand,
G iJo-coupled M2 mAChRs activate p42mapk primarily through the Gi/o rly subunit (Hawes
et al., 1995; Crespo et al., 1994).
Previous studies have shown that CCh activated p42mapk in oligodendrocyte
progenitors via the stimulation of mAChRs (Larocca and Almazan, 1997). Intense
immunostaining of oligodendrocyte progenitors demonstrated the presence of p42mapk in
the cytoplasm and in the cellular processes. However, Larocca and Almazan (1997) did
not account for the mAChR subtype that is involved in the activation of p42mapk. The
objective to identify the mAChR subtype stems from previous investigations that
characterized pharmacological1y and functional1y the mAChRs present on
84

•
•
•
oligodendrocyte progenitors. Competition binding studies demonstrated the presence of
several mAChRs~ with the M3 being the predominant subtype expressed in
oligodendrocyte progenitors (Molina-Holgado et al.~ submitted). CCh-mediated PI
hydrolysis inhibition using mAChR antagonists also confirmed the predominance of the
M3 subtype. Therefore, we inferred a role for the M3 subtype in mediating the activation
of p42mapk• Confrrming our predictions, we indeed demonstrate that the M3 selective
antagonist 4-DAMP and its irreversible analogue 4-DAMP-mustard blocked the CCh
stimulated activation of p42mapk• Both aotagonists were applied at low concentrations
(l00 nM for 4-DAMP, 1 J.lM for 4-DAMP-mustard) that yield more selectivity to the M3
subtype (Michel et al., 1989; Lazareno et al.~ 1990; Caulfield 1993; Kondou et al., 1994;
Molina-Holgado et al., submitted results). 4-DAMP inhibited P42milPk activation in a
concentration-dependent manner, showing that the ICso of 4-DAMP is at a relatively low
concentration of 50 oM. On the other hand, both 1 J.lM pirenzepine, an MI antagonist,
and 1 J.lM methoctramine, an M21M4 antagonist, did not affect the stimulated levels.
These results denote that the M3 is the mAChR responsible for the CCh-stimulated
p42ffiapk activation.
Subsequently~ we investigated the signaling pathway that links the M3 mAChR to
p42 mapk activation. The M3 mAChR has been shown to couple to PLe through the
interaction of the a subunit of the PTX-insensitive Gq/ 11 protein (for review see Exton,
1996; Lee and Rhee, 1995). The activation of PLC by CCh or other ACh analogues leads
to the generation of two second messengers, inositol 1,4,5-trisphosphate (IP3) and
diacylglycerol (DAG). It has been weil established that IP3 cao cause an increase in the
concentration of intracellular calcium as a result of intracellular storage mobilization and
85

•
•
•
DAG can activate the cIassical TPA-sensitive PKCs (Berridge, 1993). Previous reports
provide evidence for the activation of PLC by CCh stimulation of oligodendrocyte
progenitors (Cohen and Almazan, 1994). CCh induces the generation of IP3 through PI
hydrolysis as weil as the accumulation of intracellular calcium through the mobilization
of intracellular storage sites and the extracelluar influx through calcium ion channels
(Cohen and Almazan, 1994). Furthermore, another report demonstrated that chelation of
extracellular calcium by EDTA blocked the activation of p42mapk in oligodendrocyte
progenitors (Larocca and Almazan, 1997). However, the requirement of intracellular
calcium may not he ruled out, since the intracellular calcium chelator BAPTA was not
used in the study by Larocca and Almazan (1997). Therefore, we speculated a role for
PLC and its generated second messengers in linking the stimulation of mAChRs to
dO\-Vllstream activation ofp42mapk•
1t is interesting to note that other studies have shown that MEKs and MAPKs can
be stimulated by the elevation of intracellular calcium through calcium ionophores,
thapsigargin, elevated extracellular calcium, or membrane depolarization (for review see
Lewis et aL, 1998). On the other hand, the question of PKC stimulation of the MAPK
cascade has been addressed, yet in Many cases the mechanism is cell type dependent.
Phorbol esters activate MAPKs in nearly every cell (for review see Lewis et al., 1998).
Larocca and Almazan (1997) demonstrated that CCh-induced p42mapk activation in
oligodendrocyte progenitors involved TPA-insensitive (or DAG-independent) PKC
isozyme(s), since the PKC inhibitors H7 and bisindolylmaleimide GFlü9203X, but not
chronic pretreatment with TPA, abolished the mAChR-mediated response.
To investigate the role ofPLC, the present study used the PLC inhibitior U73122
86

•
•
•
and its inactive analogue U73343 (Tatrai et al.~ 1994). The active PLC inhibitor U73122,
but not the inactive analogue U73343, blocked the CCh-stimulated levels of p42mapk
activation. However, U73122 administration without CCh stimulation reduced the basal
levels significantly, in comparison to U73343 which had no effect on basal levels.
Previous studies have reported that in addition to PLC inhibition, U73122 may act on
other sites. Such mechanisms include the inhibiton of both intracellular calcium
channels, which are activated by fonnation of IP) by PLC, and plasma membrane bound
calcium channels (Pulcinelli et al., 1998). In the absence of CCh stimulation, muscarinic
receptor-coupled PLC is inactive, and thus Ïntracellular calcium channels activated by IP3
are closed. On the other hand, plasma membrane-bound calcium channels are
independent of PLC activation and may play a role in transient calcium currents.
Therefore, U73 122 may antagonize plasma membrane calcium channels and as a resuit
decrease the basal levels of p42maPk activation, while attenuating the CCh-stimulated
response by inhibiting both PLC activity and calcium channels.
Previously, Lopez-I1asaca et al. (1997) have demonstrated that p42mapk activation
by G-protein coupled receptors can be mediated by PI3K, a phosphatidylinositol kinase
(PIK), through a process that requires G-protein's f3y subunits. The wortmannin-sensitive
PI3K isozyme, PI3Ky, cao be activated by both the a and the f3y subunits of
heterotrimeric G-proteins (Stoyanov et al., 1995). Several studies have reported on the
regulation of differeot PIKs and PI pools by mAChRs. For example, wortmannin
sensitive PIKs, such as PI4K (Sorensen et al., 1998), play a role in the endocytosis of
mAChRs (Sorensen et al., 1998), in addition to the regulation of PI pools by mAChRs, a
mechanism that involves the activation of different PIKs (Willars et al., 1998). The
87

•
•
•
present study investigated whether a wortmannin sensitive PIK is required for stimulating
p42mapl.; by CCh. Pretreating progenitors with wortmannin showed that wortmannin was
not effective in reducing the levels of CCh-stimuIated p42mapk activation, and thus PI3K
or any other wortmannin-sensitive PIK May not be impIicated in the CCh-mediated
process. These results were similar to other recent studies that demonstrate wortmannin
insensitivity of the activation ofp42mapk by the M3 mAChR (Wotta et al., 1998).
Therefore~ in view of our present study, we conclude that the M3 subtype is
mediating the activation of p42mapk in oligodendrocyte progenitors via the action of PLC,
presumably by calcium mobilization, but not through PI3K. We specuIated that the
cascade linking the M3 mAChR to p42mapk activation does not require classical DAG
sensitive PKC isozymes. The requirement of calcium for the activation of p42mapk has
been previously reported in oligodendrocyte progenitors (Larocca and Almazan, 1997).
However, contrary to previous reports indicating the involvement of a classical TPA
sensitive PKC isozyme(s) (Schonwasser et al., 1998; Young et al., 1996), Larocca and
Almazan (1997) have shown that in oligodendrocyte progenitors, the activation of
p42mapk May require an atypical PKC isozyme that is TPA-insensitive. Ono et al. (1989)
demonstrated the expression of the atypical PKCÇ in oligodendrocyte progenitors.
Asotra and Macklin (1994) have aIso shown the expression of severaI phospholipid
dependent PKC isozymes in oIigodendrocyte progenitors, including PKCÔ and PKCE. In
other cell lines, studies have shown that atypical PKC isozymes can activate p42mapk oruy
at the level of MEK but not Raf-l, suggesting a different signaling pathway linking
atypical PKCs to MEK activation (Schonwasser et al., 1998). Therefore, it is tempting to
speculate that the CCh-induced activation of p42mapk in oligodendrocyte progenitors
88

•
•
•
involves an atypicaI PKC isozyme in addition to a PLC-dependent pathway that generates
calcium, and further studies are needed to elucidate the PKC isozyme(s) involved.
Moreover, the present study investigated nuclear responses downstream of the
mAChRs. Studies in cholinergie nuclear responses in oligodendrocyte progenitors have
been previously reported (Cohen et al., 1996). The investigation of nuclear responses
induced by cholinergie stimulation of oligodendrocyte progenitors can help unravel the
role of mAChRs in oligodendrocyte development. Previous pharmacological and
functional studies on progenitor and mature oligodendrocytes have displayed the
presence of several mAChRs subtypes in progenitors that undergo extensive
downregulation during the process of maturation (Molina-Holgado et al., submitted).
This interesting fmding poses the question of the developmental role pIayed by mAChRs
in the maturation of oligodendroeyte progenitors. Indeed, we have demonstrated that
cholinergie stimulation of oligodendroeyte progenitors induces several developmental
and signaling responses, including increased proliferation of progenitors, second
messengers accumulation, p42mapk activation, and c-fos gene expression (Cohen et al.,
1996; Cohen and Almazan, 1994; Larocca and Almazan, 1997; Cohen et al., 1996).
Expression of c-fos proto-oncogene has been implicated in cell growth, proliferation,
protein synthesis, and inhibition ofdifferentiation (for review see Janknecht et al., 1995).
The prevention of differentiation by c-fos gene expression is an interesting observation
because CCh can stimulate proliferation and not differentiation of progenitors in addition
to the expression of c-fos rnRJ.'lA (Cohen et al., 1996).
Therefore, we investigated the downstream signaling events linking mAChR
activation to nuclear responses such as c-fos gene expression. First, the mAChR subtype
89

•
•
•
that is mediating the expression of e-fos mRNA was pharmaeologieally detennined.
Sinee the predominant subtype expressed in oligodendrocyte progenitors is the M3
subtype, and this subtype was also shown to mediate the activation of p42mapk, we
postulated that the M3 is also the subtype responsible for the cholinergie stimulation ofc
fos mRNA expression. By using selective mAChR antagonists, we have provided
evidence in this study that the M3 mAChR does indeed mediate the CCh-stimulated
expression of c-fos mRNA. Both 4-DAMP and 4-DAMP-mustard bloeked this response.
Attenuation of c-fos gene expression by 4-DAMP demonstrated dose-dependency
whereby the ICso of 4-DAMP on c-fos mRNA expression is 10 nM. Both pirenzepine and
methoctramine had no effeet on the levels ofexpression.
Furthermore, since the M3 mAChR is coupled to a PTX-insensitive G-protein
which can activate PLC, we postulated a role for PLC in mediating the CCh-stimulated c
fos nlRNA expression. As we have demonstrated for the stimulation of p42mapk, U73122
but not U73343 was effective in blocking the CCh-stimulated response. Since Cohen and
Almazan (1994) have provided evidenee for the activation of PLC by CCh, our fmdings
support a role for PLC in mediating the CCh-induced c-fos gene expression. The
present study also confmns earlier reports by Cohen et al. (1996) which have shown that
CCh-induced c-fos gene expression is calcium dependent, since PLC can generate an
increase in intracellular calcium through the action of IP3 on the endoplasmic reticulum.
However, Cohen et al. (1996) have shown that only the atypical TPA-insensitive PKC
isozymes may be involved in this response. Our results therefore indicate a potential role
for PLC in mediating the CCh-induced response only through calcium and not through
the classical TPA-sensitive PKC isozymes. Further investigations are needed to elucidate
90

•
•
•
the different TPA-insensitive PKC isozyme(s) that may play a role in modulating the
CCh-stimulated expression ofc-fos mRNA.
Other signaling Molecules that cao potentially mediate c-fos mRNA expression
were investigated. Wortmannin, a PI3K inhibitor, was used to identify a possible role
for PI3K or any other wortmannin-sensitive PIK in mediating the CCh-stimulated c-fos
gene expression. Previous studies have shown that PI3K is a signaling molecule that is
regulated by receptors with intrinsic or associated tyrosine kinase activities, in addition to
its activation by both a and r3y subunits of heterotrimeric G-proteins (for review, see
Kapellar and Cantley, 1994). PI3K is involved in the regulation of protein synthesis and
cell proliferation by mitogens, whereby it can activate a variety of signaling Molecules
that include p70S6-kinase which activates the ribosomal 86 protein (Valius et al., 1993;
Seva et al., 1997), Many PKC isozymes from both conventional and novel families, plus
other kinases including Akt, Bkt, and Rac (for review see Fukui et al., 1998). The
different activation targets of PI3K lead us to speculate a role for PI3K in stimulating the
expression of proto-oncogenes such as c-fos. However, similar to the mode of p42mapk
activation by CCh, our present findings showed that wortmannin did not significantly
reduce the levels ofc-fos mRNA expression upon CCh stimulation.
The fact that both p42mapk activation and c-fos mRNA expression are mediated by
the same mAChR, the M3 subtype, suggests that this subtype cao play a major role in
cholinergie regulation of oligodendrocyte progenitors. It cao also he speculated that the
M3 mAChR cao also mediate the proliferative action of CCh on oligodendrocyte
progenitors (Cohen et al., 1996). Our findings denote that both responses are mediated
by similar pathways, involving PLC activation, calcium accumulation, and a TPA-
91

•
•
•
insensitive PKC isozyme, but not wortmannin-sensitive PIKs. As a resul~ we questioned
whether both CCh-induced responses are linked. Our results demonstrate that
pretreatment with PD 098059, a MEKI inhibitor, at concentrations 1 to 10 flM, did not
significantly reduce the CCh-stimulated levels of c-fos mRNA. ft is important to note
that PD 098059 cao inhibit MEK1, the kinase that activates p42Rl3Pk, at 1-10 IJ.M, since
the ICso of this inhibitor on MEKI is 2-7 flM (Alessi et al., 1995; Dudley et al., 1995).
At concentrations of 50 and 100 flM, however, PD 098059 caused a significant reduction
but not a total block of the response. Sïnce Larocca and Almazan (1997) have previously
demonstrated that 10 flM concentration of PD 098059 is sufficient to completely block
the levels of p42mapk activation in oligodendrocyte progenitors (Larocca and Almazan,
1997), our current results suggest that the MAPK pathway rnay not be the major pathway
linking mAChR stimulation to c-fos rnRNA expression. The attenuation of c-fos mRNA
levels at 50 and 100 J.lM PD 098059 concentration May result from non-selective action
of this inhibitor at other MEKs such as MEK2 (ICso = 50 flM). These findings are
intriguing, since Many studies have linked the stimulation of the MAPK pathway to
downstream regulation of the c-fos promoter via the activation of transcription factors by
severa! MAPK species (for review see Janknecht et al., 1995; Gutk.ind, 1998).
Recent studies in oligodendrocyte progenitors showed a CCh-induced activation
of the transcription factor cyclic-adenosine monophosphate (cAMP) response element
binding protein (CREB) and the activation of the calcium-calmodulin kinase II (CaMK
II) (Pende et al., 1997; Sato-Bigbee et al., 1999). Both effector Molecules have been
shown to regulate the promoter of immediate early genes, such as c-fos. Pende et al.
(1997) have displayed that both responses are calcium dependent (Pende et al., 1997).
92

•
•
•
Calcium cao trigger various intracellular events including both short term and long term
(Ghosh and Greenberg, 1995). It has also been postulated that calcium signaIs can
regulate the function of several transcription factors, including CREB (Sheng et al.,
1990), in addition to the modulation of several key regulatory enzymes such as PKC's
and CaMK's. Both of these families of kinases have been shown to be able to
phosphorylate CREB in vitro (Yamamoto et al., 1988; Sheng et al., 1991). Once
phosphorylated, CREB cao modulate the transcriptional activity of several genes by
binding to the cAMP response element (CRE) site at the promoter region. CREB can
modulate the expression of c-fos by binding to the CRE that is present on the c-fos
promoter (for review, see Karin.. 1995; Gutkind, 1998). Contrary to our speculations, the
study has aiso shown that pretreatment with the MEK inhibitor PD 098059 blocked the
activation of CREB by CCh, suggesting the involvement of the MAPK pathway in
stimulating CREB (Pende et al., 1997). Since our present study demonstrated no
significant decrease in the Ievels of c-fos mRNA expression upon PD 098059
pretreatment~ it can therefore be postuJated that while the CCh-stimulated activation of
both CaMKII and CREB are calcium dependent and may play a role in regulating
nuclear responses, the activation of CREB may not contribute to the CCh-stimulated
changes in c-fos gene expression.
The c-fos promoter contains several response elements such as SRE and SIE that
are regulated by transcription factors other than CREB, including AP-I, SRf, Elk-l, and
Sap-l (for review see Gutkind, 1998; Janknecht et al., 1995). Moreover, the c-Jun-NH2
terminal kinase (JNK) has been weB characterized and identified as a serine/threonine
kinase that cao phosphorylate transcription factors involved in c-fos gene regulation, such
93

•
•
•
as c-Jun, c-Fos, and Elk-l. Upon stimulation, c-Fos and c-Jun dimerize to fonn the AP-l
transcription factor. Furthermore, the activation of EIk-1 facilitates binding to the SRF.
The formation of the AP-l transcription factor and the binding of SRf to Elk-l are
crucial regulatory events for c-fos gene expression. Recently, the p38 species ofMAPKs
has aIso been implicated in regulating the activity of certain transcription factors such as
MEF2 and ATF2 that can modulate the c-jun promoter (Han et al., 1997; Gutkind, 1998).
This field of research has been poody understood and it is currently being investigated.
As a result, further work is required to identify a role for other pathways or other ~1APK
species in the transduction cascade that links modulations in c-fos gene expression to
receptor activation.
Our investigations of the pathways downstream of the mAChRs that cao lead to
c-fos rnRNA expression explored the role of PLA2 in linking receptor activation to gene
expression. The product of PLA2 enzymatic activity, arachidonic acid (AA), can be
converted to prostaglandins, thromboxanes, leukotrienes, and other bioactive eicosanoids.
This major eicosanoid pathway has been shown to be activated by cholinergie stimulation
via the odd numbered mAChRs in many systems (for review see Felder, 1995). Recent
reports have described a linkage between AA and its metabolites to the stimulation of the
c-fos SRE through Rac and Rho-dependent mechanisms (Kim et al., 1998; Kim et al.,
1997). Via the activation of Rac and Rho-related proteins, G-protein coupled receptors,
including the mAChRs, cao stimulate MAPK species such as JNK (Coso et al., 1995b;
Caso et al., 1996; Minden et al., 1995), and subsequently the modulation of gene
expression such as c-jun and c-fos (for review see Gutkind, 1998). These findings
provided ample evidence that c-fos gene expression may be PLA2-dependent. In
94

•
•
•
oligodendrocytes, studies have shown that AA cao Mediate severa! responses, including
MBP phosphorylation aod the inhibition of potassium conductances (Takeda and Soliven,
1997; Soliven and Wang, 1995). Our results confirmed our extrapolations, since the
pretreatment of progenitor cultures with either PLA2 inhibitors, quinacrine or AACOCF3,
abolished the stimulated levels of c-fos mRNA. However, further studies are needed in
order to determine whether AA itself or any of its Metabolites Mediate this response, and
therefore, more studies are needed to unravel the metabolite(s} leading to this nuclear
response.
In summary ~ our present study demonstrates that the M3 is the mAChR subtype
mediating the CCh-stimulated p42mapk activation and c-fos mRNA expression in
oligodendrocyte progenitors. The activation of p42mapk is dependent on PLC but not on a
wortmannin-sensitive PI3K. Similarly, the CCh-stimulated c-fos mRNA expression also
involves the activation of PLC and PLA2 but not a wortmannin-sensitive PI3K.
Although both p42rt141Pk activation and c-fos mRNA expression are mediated by the same
mAChR subtype, our study demonstrates that the activation of p42mapk is not necessarily
required for the CCh-mediated c-fos rnRNA expression. Further studies are needed to
identify the downstream signaling pathways that lead to the activation of p42mapk and the
increased expression ofc-fos mRNA, including the identification of the PKC isozyme(s)
that mediate(s) both responses, the investigation of other MAPK species that contribute
to the regulation of c-fos gene expression, in addition to the exploration of the role(s) of
AA and/or its Metabolites in mediating this nuclear response.
95

•
•
•
CHAPTERSIX
REFERENCES
96

•
•
REFERENCES
Adem A and Karlson E (1997). Life Sei 60, 1069-1076.
AgranoffBW, Albers RW, MolinoffPB (ed): "Basic Neurochemistry" New York: Raven
Press, pp 771-792.
Almazan G, Afar DEH, Bell JC. (1993). J Neurosei Res 36, 163-172.
Amur-Umarjee SG, Dasu RG, Ca..rnpagnoni AT. (1990). Dev Neurosci 12,251-262.
Angel P, Karin M. (1991). Bioehim. Biophys. Acta 1072, 129-157.
Angel P. (1995). The role and regulation of the Jun pro/eins in response to phorbol
ester and UV light. In: Baeuerle PA (ed). Inducible gene expression, vol. 1, Birkhauser,
Arcaro A, Wymann MP. (1993). Bioehemical J 296,297-301.
Armstrong Re, Kim JG, Hudson LD. (1995). GUa 14,303-321.
Ashkenazi A, Peralta EG, Winslow JW, Ramachandran J, and Capon DJ. (1989). Cell
• 56,487-493.
97

•
•
•
Ashkenazi A, Peralta EG, Winslow JW, Ramachandran J, Capon DJ. (1989a). Trend
Pharmacol Sci Supplement, 16-22.
Ashkenazi A, Ramachandran J, Capon DJ. (1989b). JVature 340,146-150.
Ashkenazi A, Winslow JW, Peralta EG, Peterson GL, Schimerlik MI~ Capon DJ, and
Ramachandran J. (1987). Science 238, 672-675.
Asotra K and rvlacklin WB. (1994). J Neurosci Res 39,273-289.
Baldwin Jrvl, Schertler GF and Unger VM. (1997). J .Voi Bio/272, 144 -164.
Bansal R, Stefansson K, Pfeiffer SE. (1992). J Neurochem 58, 2221-2229.
Bansal R, Warrington AE, Gard AL, Ranscht B, Pfeiffer SE. (1989). J Neurosci Res 24,
548-557.
Barlow RB, Berry lU, Glenton PAM, Nikolau NM and Soh KS. (1976). BrJ Pharmaco/
58, 613-620.
Barlow RB, McMillen LS, Veale MA. (1991). BritishJ Pharmaco/l02(3), 657-662.
98

•
•
•
Barres BA, et al. (1992). Cel/70, 31-46.
Barres BA, Raff MC, Gaese F, Bartke 1, Deschant G, Barde Y-A. (1994). Nature 367,
371-375.
Bartsch U, Peshava P, RaffM, Schachner M. (1993). GUa 9,57-69.
Baumgold J and Fishman PH. (1988). Biochem Biophys Res Commun 154, 1137-1143/
Baumgold J. (1992). Trends Pharm Sei 13, 339-340.
Berridge MJ. (1993). Nature 361,315-325.
Berstein G, Blank JL, Smrcka A, Higashijima, T. Stemweis PC, Exton JH, and Ross
BJ, de Jong P, Reed RR., Simon MI et al. (1992). Nat Genet 1,85-91.
Bogler 0, Wren D, Barnett SC, Land H, and Noble M. (1990). Proc Nat/ Acad Sci USA
87, 6368-6372.
Bonner TI, Buckley NJ, Young AC and Brann MR. (1988). Neuron 1,403-410.
Bonner TI, Buckley NJ, Young AC, Brann MR. (1987). Science 237, 527-532.
Boston, pp. 63-92.
99

•
•
•
Brooks RC, McCarthy K.O, Lapetina EG, Morell P. (1989). J Biol Chem 264, 20147
20153.
Brown DA, Forward A and Marsh S. (1980). BrJ Pharmacol71, 362 -364.
Buckley NJ. (1990). Molecular pharmacology of cloned muscarinic receptors.
lntracellular Messengers and Implications for Drug Development (Nahorski, S. ed) pp.
11-30, Wiley, New York.
Buss JE, Mumby SM, Casey Pl, Gilman AG, Sefton BM. (1987). Proc JVatl Acad Sci
USA 84, 7493-7497.
Cahill MA, lanknecht R, Nordheim A. (1995). Signal uptake by the c-fos serum
response element. In: Baeurle PA (ed). Inducible gene expression, vol. 2. Birkhauser,
Boston, pp39-72.
Cahill MA, Janknecht R, Nordheim A. (1996). Curr Biol 6, 16-19.
Cajal SR. (1913). Trab Lab invest Biol Univ Madrid 11,219-237.
Caulfield MP, and Birdsall JM. (1998). Pharmacological Reviews 50 (2) 279-290.
100

•
•
•
Caulfield MP. (1997). Muscarinic receptor classification, in Muscarinic Receptor
Subtypes in Smooth Muscle (Eglen RM ed) pp 1-37, CRC Press, New York.
Caulfield MP. (1993). Pharmacol Ther5S, 319-379.
Cavigelli rvl, Dolft F, Claret F-X, Karin M. (1995). EMBO J 14, 5957-5964.
Chen Y, Grall D, Saleini AE, Pelicci PG, Pouyssegur 1 and Van Obberghen-Schilling E.
(1996). EMBOJI5, 1037-1044.
Chou MM and Blems J. (1995). Curr Opin Cel/ Biol 7, 806-814.
Clapham DE and Neer El. (1997). Annu Rev Pharmacol Toxicol37, 167-203.
Clapham DE, and Neer El. (1993). Nature 365, 403-406.
Clark JD, Lin L-L, K.riz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, Knopf JL.
(1991). Ce1l65, 1043-1051.
Clarke S. (1992). Annu Rev Biochem 61, 355-386.
Cohen RI and Almazan G. (1994). Eur J Neurosci 6, 1213-1224.
101

•
•
•
Cohen RI, Molina-Hoigado E, Almazan G. (1996). Mol Brain Res 43, 193-201.
Collarini EJ, Kuhn R, Marshall CJ, Monuki ES, Lemke G, Richardson WD. (1992).
Development 116, 193-200.
Compston A, Zajicek J, Sussman J, Webb A, Hall G, Muir D, Shaw C, Wood A,
Conklin BR, Brann MR, Buckley, NJ MA AL, Bonner TI, and Axelrod J. (1988). Proc
Nat! Acad Sci USA 85, 8698-8702.
Coso DA, Chiariello M, Yu JC, Teramoto H, Crespo, Xu N, Miki T and Gutkind JS
(1995). Cel/ 81, 1137-1146.
Coso DA, Teramoto H, Simonds WF, Gutkind JS. (1996). J Biol Chem 271, 3963-3966.
Crespo P. Xu N, Simonds WF, and Gutking JS. (1994). Nature 369, 418-420.
Curtis CA, Wheatley M, Bansal S, Birdall NJM, eveleigh P, Pedder EK, Poyner D and
Hulme EC. (1989). J Biol Chem 264, 489-495.
Davis RJ. (1994). Trend Bioichem Sci 19,470-473.
Dell'Acqua ML, Carroll, Re and Peralta EG. (1993). J Biol Chem 258,5676-5685.
102

•
•
•
Della Rocca GJ, van Biesen T, Daaka Y, Luttrell DK, Luttrell LM, Lefkowitz RJ.
(1997). J Biol Chem 272, 19125-19132.
Dikic I. Tokiwa G, Lev S, Courtneidge SA and Schlessinger J. (1996). Nature 383, 547
50.
Dubois-Dalcq M, Behar T, Hudson L, Lazzarini RA. (1986). J Cell 8ioll02, 384-392.
Dubois-Dalcq M. (1987). EMBO 6, 2587-2595.
Eccleston PA, and Silberberg DH. (1985). Dev Brain Res 21, 2315-2318.
Eglen RM and Whiting RL. (1996). Pharmacol Toxico/ 78, 59-78.
Eisenbarth GS, Walsh fS, Nirenberg M. (1979). Proe Natl Acad Sei USA 76, 1286
1290.
EM..( 1992). J Biol Chem 267, 8081-8088.
Exton JH. (1996). Annu Reb Pharmaeo/ Toxicol36, 481-509.
FEBS Leu 352, 91-94.
103

•
•
•
Felder CC, Dieter P, Kinsella J, tamura K, Kanterman, RY, and Axelrod J. (1990). J
Pharmaco/ Exp Therap 255, 1140-1147.
Felder CC. (1995). FASEB 9,619-625.
Ferrara N, Ousley F, Gospodarawicz D. (1988). Brain Res 462, 223-232.
Fukui Y, Ihara S, Nagata S. (1998). J Biochem 124, 1-7.
Gallo V and Armstrong RC. (1995). J Neurosci 15, 394-406.
Gard AL and Pfeiffer. (1993). Deve/opmenta/ Bio/159(2), 618-630.
Gille H, Kortenjann M, Thomae 0, Moomaw C, Siaughter C, Cobb MH, Shaw PE.
e1995). EA1BO J 14, 951-962.
Gille H, Sharrocks AD, Shaw PE. (1992). Nature (London) 358, 414-417.
Gille H, StrahJ T, Shaw PE. (1995). Curr Bio/S, 1191-1200.
Goldrnan JE, Hirano M, Yu RK., Seyfried TN. (1984). J Neuroimmuno/7, 179-192.
Goldrnan JE. (1995). J Neuro-Onco/ 24, 61-64.
104

•
•
•
Goldman JE. (1992). TINS 15 (10), 359-362.
Gosh A, Greenberg ME. (1995). Science 268, 239-247.
Grimm U, Moser U, Mutsehler E, Larnbrecht G. (1994). Pharmazie 49, 711-726.
Gusovsky F, Leuden JE, Lohn EC, and Felder CC. (1993). J Biol Chem 268, 7768-7772.
Gutkind JS, Crespo P, Xu N, Teramoto H, and Coso OA. (1997). Lift Sciences 60, 999
1006.
Gutkind JS, Novotny EA, Brann MR and Robbins KC. (1991). Proc Nat! Acad Sei USA
88, 4703-4707.
Gutkind JS. (1998). Oncogene 17(11), 1331-1342.
Haesenbroeck B, Dhand R, Nurnberg B, Gierschik P, Seedorf K, Hsuan JJ, Waterfield
MD and Wetzker R. (1995). Science 269, 690-693.
Haga T, Haga K, Kameyama K, Nakata H. (1993). Lift SciS2, 421-428 .
105

•
•
•
Hall lM, Caulfield MP, Watson SP and Guard S. (1993). Trends Pharmacol Sei 14, 376-
383.
Hammer R, Berrie CP, BirdsaII NJM, Burgen ASV, Hulme EC. (1980). Nature 283, 90
92.
Han 1~ Jiang Y, Li Z, Kravchenko VV, Ulevitch RJ. (1997). Na/ure 386, 296-299.
Hardy R and Reynolds R. (1991). Developmentlll, 1061-1080.
Hawes BE, Luttrell LM, Van Biesen T, Luttrell DK, Lansing Tl and Lefkowitz RJ.
(1996). J Biol Chem 271, 19443-19450.
Hawes BE, van Biesen T, Koch Wl, Luttrell LM, Lefkowitz RJ. (1995). J Biol Chem
270, 17148-53.
Hepler lR and Gilman AG. (1992). Trens Biochem Sei 17, 383-387.
Hernandez M, Burillo SL, Crespo MS, Nieto ML. (1998). J Biol Chem 273(1), 606-612.
Higgins lB and Casey Pl. (1994). J Biol Chem 269, 8967-9073.
Hildebrandt ffi. (1994). J Biol Chem 269,12508-12513.
106

1.
•
•
Hildebrandt JP, and Shuttleworth TJ. (1993). J Membr Biol 133, 183-190.
Hortega P and Del R. (1921). Boletin de la Real Sociedad Espanola de la Historia
lvatural.
Hortega P and Del R. (1928). l\rfemoria de la Real Sociedad Espanola de la Historia
NaturaI14.5-122.
Hulme EC, Birdsali JNM and Buckley NJ. (1990). Ann Rev Pharmacol Toxicol 30,633
673 .
Hulme EC, Birdsall NMJ, Buckley NJ. (1991). Ann Rev Pharmacol Toxicol30, 633
673.
Ichiyama A, Kanagawa K, Matsuo H, Hirose T and Numa S. (1986b). FEBS Leif 209,
367-372.
Iniguez-Lluhi J, Kleuss C, Gilman AG. (1993). Trends Cel/Biol 3,230-236.
Iniguez-Lluhi J, Simon MI, Robishaw ID, Gilman AG. (1992(. J Biol Chem 267, 23409
23417.
107

•
•
•
Ho A~ Satoh T~ Kaziro y~ Itoh H. (1995). FEBS Letf 368, 183-187.
Janknecht R, Cahill MA, Nordheim A. (1995). Carcinogenesis 16,443-450.
Janknecht R., Ernst WH, Nordheim A. (1995). Oncogene 10.1209-1216.
Janknecht R., Ernst WH, Pingoud V, Nordheim A. (1993). ElvlBOJ 12,5097-5104.
Jansson CC, Kukkonen J, Akennan KE. (1991). Biochem Biophys Acta 1095,255-260.
Jolkkonen M, Van Giersbergen PLM, Hellman U, Wernstedt C, Karlsson E. (1994).
Jones PG, Curtis CAM and Hulme EC. (1995). EurJpharmaco/ 288,251-257.
Kapellar R and Cantley Le. (1994). Bioessays 16~ 565.
Karin M. (1995). J Biol Chem 270, 16483-16484.
Karschin A, Wischmeyer E, Davidson N, Lester HA. (1994). Eur J Neurosci 6~ 1756
1764.
Kastritsis CH, McCarthy KO. (1993). GUa 8, 106-113.
108

•
•
•
Katz A, Wu D, and Simon MI. (1992). Nature 360, 686-689.
Kim BC and Kim JH. (1998). BiochemJ330(2), 1009-1014.
Kim BC, Ha KS, Park lb, Kim JH. (1998). Biochem Biophys Res Comm 247(3),630
635.
Kim BC, Lim Cl, Kim lH. (1997). FEBS Leu 415(3), 325-328.
Knapp PE. Bartlett WP, SkoffRF. (1987). Dev Biol 120, 356-365.
Knapp PE, SkoffRP, Sprinkle Tl. (1988). J Neurosci Res 21,249-259.
Knapp PE. (1991). J Neurosei Res 30,336-345.
Koch WJ, Hawes B, Allen L, Lefkowitz RJ. (1994). Proc Natl Acad Sei USA 91,
12706-12710.
Koch Wl, Hawes BE, Allen LF, Lefkowitz RJ. (1994). Proc Natl Acad Sei USA 91,
12706-12710.
Kondou H, Inagaki N, Fukui H. (1994). Neurosci Letl180, 131-134.
109

•
•
•
Kubo T , Maeda A, Sugimoto K, Akiba 1, Mikami A, Takahashi H, Haga T, Haga K,
Kubo T, Fukuda k. Mikami A, Maeda A, Takahashi H, Mishina M, Haga T, Haga K,
Ichiyama A, Kanagawa K, Kojima M, Matsuo H, Hirose T and Numa S. (1986a). Nature
323,411-416.
Kurtenbach E~ Curtis CA, Pedder EK, Aitken A, Harris AC and Hulme EC. (1990). J
Biol Chem 265, 13702-13708.
Laeng P, Decimo D, Pettmann B, Janet T, Labourdette G. (1994). J Neurosci Res 39,
613-633.
Lambert DG, Burford NT, and Nahorski SR. (1992). Biochem Soc Trans 20, 130-135.
Larocca JN and Almazan G. (1997). J Neurosci Res 50, 743-754.
Lazareno S, Buckley NJ, Roberts FF. (1990). Mol Pharmacol38, 805-815.
Lee SB, Rhee SG. (1995). Curr Opinion Cell Biol 7, 183-189.
Leibowitz S, Kennedy MC. (1978). lValure 274, 813-816.
Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B,
110

•
•
•
Levey AI. (1993). Lift Sci 52~ 441-448.
Levine RR and Birdsall NJM (eds). (1997). Subtypes of muscarinic receptors.
Proceedings of the 71h international symposium on subtypes of muscarinic receptors. Lift
Sei 60, 963-1207.
Lewis TS, Shapiro PS, Ahn NG. (1998). Advances Cancer Res 74, 49-138.
Liao CF, Schilling WP, Bimbaumer M, and Bimbaumer L. (1990). J Biol Chem 265,
11273-11284.
Linder ME, Middleton P, Hepler JR~ Taussig ~ Gilman AG. (1993). Proc Natl Acad Sci
USA 90, 3675-3679.
Linder ME, Pang I-H, Duronio RJ, Gordon JI, Stemweis PC, Gilman AG. (1991). J Biol
Chem 266,4654-4659.
Liu H-N and Almazan G. (1995). Elir J Neurosci 7, 2355-2363.
Lopez-IIasaca M, Gutkind JS, Wetzker R. (1998). J Biol Chem 273(5), 2505*2508.
111

•
•
•
Lopez-Ilasaka M, Crespo P, Pellici PG, Gutkind JS and Wetzker R. (1997). Science,
275, 394-397.
Lu Z-L, Curtis CA, Jones PG, Pavia J and Hulme EC. (1997). Mol Pharmacol 51, 234
241.
Luttrell LM and Lefkowitz RJ. (1995). Nature 376, 781-784.
Luttrell LM, Hawes BE, van Biesen T, Luttrell OK, Lansing Tl, Lefkowitz RJ. (1996).
J Biol Chem 271, 19443-19450.
Maggio R, Barbier P, Bolognesi ML, Minarini A, Tedeschi 0, Melchiorre C. (1994).
Eur J Pharmacol 268, 459-462.
Marais ~ Wynne J, Treisman R. (1993). Cell73,381-393.
Matthews RP, Guthrie CR, Wailes LM, Zhao X, Means AR, McKnight as. (1994). Aloi
Cell 8io/14, 6107-6116.
McCarthy KD, de Vellis J. (1980). J Cel! Biol 85, 890-902.
McKinnon RD, Matsui T, Dubois-OaJcq M and Aaronson SA. (1990). Neuron 5, 603
614.
112

•
•
•
McMorris FA and Dubois-Dalcq M. (1988). J Neurosci Res 21, 199-209.
Meeker RB and Harden TK. (1982). Mol Pharmaco/22, 310-319.
Mellor H and Parker PI. (1998). BiochemJ332,281-292.
Michel AD, Stefanich E, Whiting RL. (1989). Eur J Pharmaco/166, 459-466.
Minden A, Lin A, Claret FX, Abo A and Karin M. (1995). Cel/SI, 1147-1157.
Molina-Holgado E, Ragheb F, Khourchid A, Hsueh-Ning L, Almazan G (submitted):
Characterization and regulation of rnuscarinic receptors coupled to phosphoinositide
hydrolysis in ü2A progenitors and differentiated oligodendrocytes.
~1organ F, Barbarese E, Carson I. (1992). Scanning Microsc 6,345-357.
Morrell P, Quarles RH, Norton WT. (1993). Myelin formation, structure and
biochemistry. In Siegel, GI, AgranoffBW, Albers RW, MolinoffPB (ed): "Basic Muller
Sand Lohse MI. (1995). Biochem Soc Trans 23, 141-148.
Mumby SM, Kleuss C, Gilman AG. (1994). Proc Nal/ Acad Sei 91, 2800-2804.
113

•
•
•
Nathanson NM. (1996). Progress in Brain Research 1099 165-168.
Noble M9 Murray ~ Stroobant P, Waterfield MD and Riddle P. (1988) Nature 333,560
562.
Norton WT and Farooq M. (1993). Dev Brain Res 72~ 193-202.
Nunn PA, Greengrass PM, Newgreen DT, Naylor AM, Wallis RM. (1996). BI J
Pharmacol117, 130P.
Offermanns S, and G. Schultz. (1994). Naunyn-Schmiedebers Arch. Pharmaco/289, 343-
351.
Offermanns S~ Wieland T, Homann D, Sandmann J, Bombien E, Spicher K, Schultz G,
and Jakobs KH. (1994). Mol Pharmacol4S, 890-898.
Olianas, MC and Onali P. (1992). J Neurochem 58, 1723-1729.
Ono Y, Fujii T, Ogita K, Kikkawa U, Igarashi K, Nishizuka Y. (1989). Proc. lVatl Acad
Sei USA 86,3099-3103.
Parenti M, Vigano MA9 Newman CMH, Milligan G 9 Magee AI. (1993). Bioehem J 291,
349-353.
114

•
•
•
Parker EM, Kameyama K, Higashijima T, and Ross EM. (1991). J Biol Chem 266,519
527.
Paterson lA, Privat A, Ling EA, Leblond CP. (1973). J Comp Neurol 149, 83-102.
Pende M, Fisher TL, Simpson PB, Russell lT, Blenis 1, Gallo V. (1997). J Neurosci
17(4), 1291-1301.
Peng YW, Robishaw ID, Levine MA, Vau K-W. (1992). Proc Natl Acad Sei USA 89,
10882-10886.
Peralta EG, Ahkenazi A, Winslow lW, Smith DH, Ramachandran 1 and Capon
Dl.(1987). EMBOJ6,3923-3929.
Pettmann B, Labourdette G, Weibel t\1, Sensenbrenner M. (1986). Neurosci Let/ers 68,
175-180.
Pfeiffer SE, Barbarese E, Bhat S. (1981). J Neurosci Res 6, 369-380.
Pfeiffer SE, Warrington AE, Bansal R. (1993). Trends Ce/l Biol 3, 191-197.
Pieroni lP, lacobowitz 0, Chen J, Iyengar R. (1993). Curr Opin Neurobio/3, 345-351 .
115

•
•
•
Priee MA, Rogers AE, Treisman R. (1995). EMBO J 14, 2589-2601.
Pronin AN and Gautam N. (1992). Proc Natl Acad Sci USA 89, 6220-6224.
Ptasznik A, Traynor-Kaplan A and Bokoeh GM. (1995). J Biol Chem 270, 19969
19973.
Pulcinelli FM, Gresele P, Bonuglia M, Gazzaniga PP. (1998). Biochem Pharmacol
56(11), 1481-1484.
Quarles RH, Morell P, McFarlin DE. (1993). Diseases involving myelin. In Siegel, GJ,
RaffMC, Miller RH, Noble M. (1983). Nature 303,390-396.
Raff MC, Mirsky R, Fields KL, Lisak RP, Dorfman SH, Silberberg DH, Gregson NA.
Raff MC, Williams BP, Miller RH. (1984). EMBO 3, 1857-1864.
RaffMC. (1989). Science 243, 1450-1455.
Ranseht B, Clapshaw PA, Noble M, Seifert W. (1982). Proc Natl Acad Sei USA 79,
2709-2713 .
116

•
•
•
Ray K~ Kunsch C, Bonner LM, Robishaw ID. (1995). J Biol Chem 270,21765-21771.
Regunathan S, Meeley MP~ and Reis DJ. (1990). Lift Sci 47, 2127 - 2133.
Rhee SG and Choi KD. (1992). J Biol Chem 267, 12393-12396.
Richardson WD, Pringle N, Mosley MJ, Wesermark B, Dubois-Dalcq M. (1988). Ce//
53, 309-319.
Riker WF and Wescoe WC. (1951). Ann NY Acad Sei 54, 373-394.
Robertson WF. (1899). Scottish Medical Surgical J 4,23-30.
Sakuta H and Yoneda 1. (1994). Eur J Pharmacol 252, 117-121.
Saneto RP, and deVellis J. (1985). Proc. Nall Acad Sei USA 82, 3509-3513.
Sarlieve LL, Fabre M, Sesz J, Matthieu lM. (1983). J Neurosci Res 10,191-216.
Sasahara M el al. (1991). Ce//64, 217-227.
Sato-Bigbee C, Pal S, Chu A K. (1999). J Neurochem 72(1), 139-147.
117

•
•
•
Savarese TM, Wang CD and Fraser CM. (1992). J Bio/ Chem 257, 11439-11448.
Sawaki K, Hiramatsu Y, Baum, BJ and Ambudkar IS. (1993). Arch Biochem Biophys
305, 546-550.
Schlessinger J. (1993). Trends Biochem Sei 18, 273-275.
Schlessinger J. (1995). Nature 376, 737-745.
Schmidt CJ, Thomas TC, Levine MA, Neer EJ. (1992). J Biol Chem 267, 13807-13810.
Schonwasser D~ Marais RM, Marshall CJ, Parker PJ. (1998). Aloi Cell Bio/18, 790-798.
Scolding N. (1997). JAna! 190, 161-200.
Seva C, Kowalski -Chauvel A, Daulhae L, Barthez C, Vaysse N~ Pradayrol L. (1997).
Biochem Biophys Res Comm 238, 202-206.
Sharp JD~ White DL, Chiou XG, Goodson T, Gamboa GC, McClure D, Burgett S,
Hoskins J, Skatrud PL et al. (1991). J Biol Chem 266, 14850-14853.
Sheng M, McFadden G, Greenberg ME. (1990). Neuron 4, 571-582.
118

•
•
•
Sheng M, Thompson M, Greenberg ME. (1991). Science 252, 1427-1430.
Shimojo M, Nakajima K, Takei N, Hamanoue M, Kohsaka S. (1991). Neurosci Letters
123, 229-231.
Simon MI, Strathmann MP and Gautam N. (1991). Science 252,802-808.
Simonds WF, Butrynski JE, Gautam N, Unson CG, Spiegel AM. (1991). J Biol Chem
266.5363-5366.
Simpson PB and Russell JT. (1996). J Biol Chem 271,33493-33501.
Skoff RP and Knapp PEe (1991). Glia 4, 165-1 74.
Soliven B and Wang N. (1995). Am J Physiol269, C341-C348.
Sommer r and Schachner M. (1981). Dev Biol 83, 311-327.
Sorensen SO, Linseman DA, McEwen EL, Heacock AM, Fisher SK. (1988). Mol
Pharm 53(5), 827-836.
119

•
•
•
Spalding TA, Birdsall NJM, Curtis CAM and Hulme EC. (1994). J Bio/ Chem 269,
4092-4097.
Spring DJ and Neer EJ. (1994). J Biol Chem 269, 22882-22886.
Stein R~ Pinkas-Kramarski ~ and Sokolovsky M. (1988). EMBOJ 7,3031-3035.
Stephens EV, Kalinec G, Brann MR Gutkind JS. (1993). Oncogene 8, 19-26.
Stoyanov B, Volinia S, Hanck T, Rubio l, Loubtchenkov M, Malek D, Stoyanova S, Van
Suidan HS, Murrell RD, Tolkovsky AM. (1991). Cell Regu/2, 13-25.
Szuchet S and Steffanson K. (1980). Adv Cel! JVeurobio/l, 313-346.
Szuchet S Stefanason K~ Wollman RL, Dawson G, Arnason BG. (1980). Brain Res 200,
151-164.
Takeda M and SoIiven B. (1997). GUa 21, 277-284.
Takeda M, Nelson DJ, SoIiven B. (1995). Glia 14,225-236.
Tatrai A, Lee SK, Stern PH. (1994). Biochim Biophysic Acta 1224(3), 575-582.
120

•
•
•
Tietje KM and Nathanson NM. (1991). J Biol Chem 266, 17382-17387.
Tietje KM, Goldman PS, and Nathanson NM. (1990). J Biol Chem 265, 2828-2834.
Valius M and Kazlauskas A. (1993). Ce// 73,321-334.
Van Biesen T, Hawes BE, Luttrell DK, Krueger KM, Touhara K, Porfiri E, Sakaue M,
Veit Ivl, Numberg B, Spicher K, Harteneck C, Ponimaskin E, Schultz G, Schmidt MFG.
(1994). FERS Leif 339, 160-164.
Virchow R. (1858). Cellularpathologie in ihre Begrundung aufphysiologische und
pathologische Gewebelehre. Berlin: A. Hirschwald, Berlin.
Wan Y, Kurosaki T and Huang XV. (1996). Nature 380,541-544.
Warrington AE and Pfeiffer SE. (1992). J Neurosci Res 33, 338-353.
Warrington AE, Barbarese E, Pfeiffer SE. (1993). J Neurosci Res 34, 1-13.
Watson Al, Aragay AM, Slepak VZ, Simon MI. (1996). J Biol Chem 271,28154
28169.
121

•
•
•
\Vatson EL, Singh Je, McPhee C, Beavo J~ and Jacobson KL. (1990). Mol Pharmacol
38, 547-553.
Wedegaertner PB, Chu DH~ Wilson P~ Levis Ml Boume HR. (1993). J Biol Chem 268,
25001-25008.
\Vess J (1993). Trends Pharmacol Sei 14, 308-313.
Wess J (1996). Crit Rev NeurobiollO, 69-99.
Wess J, Liu J, Blin N, Yun J, Lerche C and Lostenis E. (1997). Lift Sei 60, 1007-1014.
Whitmarsh Al, Shore P, Sharrocks AD, Davis RJ. (1995). Science 269, 403-407.
\Vilcox MD, Schey KL, Dingus J, Mehta ND, Tatum BS, Halushka M, Finch JW,
Willars GB, Nahorski SR, Challiss RA. (1998). J Bio/ Chem 273(9), 5037-5046.
Wissing D. Mouritzen H, Egeblad M, Poirier GG, Jaattela M. (1997). Proc Natl Acad
Sei USA 94, 5073-5077.
Wotta DR, Wattenberg EV, Langason RB, el-Fakahany EE. (1998). Pharmacology 56(4),
175-186.
122

•
•
•
Yamamoto KK., Gonzalez GA, Biggs III WH, Montminy MR. (1988). Nature 334, 494
498.
Yeh H-J. et al (1991). Ce1l64, 209-216.
Yim SH, Farrer RG, Quarles RH. (1995). Dev lveurosci 17, 171-180.
Young SW, Dickens M, Tavare JM. (1996). FEBS Lett 384, 181-184.
Yu W, Bozza PT, Tzizik DM, Gray JP, Cassara J, Dvorak AM, Weiler PF. (1998). Am J
PathoI152(3), 759-769.
Zhu 5Z, Wang SZ, Hu J and EI-Fakahany EE. (1994).1\10/ Pharmacol4S, 4170523.
Zinck R, Cahill MA, Kracht M, Sachsenmaier C, Hipskind RA, Nordheim A. (1995).
Afol Cell BiolIS, 4930-4938.
Zubriggen A, Vandevelde M, SteckA, ..c\ngst B. (1984). J Neuroimmunol6, 41-49.
123





























