Embed Size (px)
Citation preview

[CANCER RESEARCH 54, 17(17-1714. April 1, W4|
Induction of Apoptotic DNA Fragmentation and Cell Death in HL-60 Human
Promyelocytic Leukemia Cells by Pharmacological Inhibitors ofProtein Kinase C1
W. David Jarvis, Amy J. Turner, Lawrence F. Povirk, Rebecca S. Traylor, and Steven Grant2
Department of Medicine, Dinsitm of Hematologv ami Oncoloitv /1V. D. J., A. J. T.. R. S. T., S. (!.], and Department of Pharmacology/Toxicology ¡L.F. P., S. G.¡,MedicalCollege of Virginia, Richmond, Virginia 23298-0230
ABSTRACT
The present .studies were undertaken to characterize further the potential role of protein kinase C i I'M i in the regulation of apoptosis in
HL-60 promyelocytic leukemia cells. The capacity of acute exposure tospecific and nonspecific pharmacological inhibitors of I'M to promote
apoptotic DNA fragmentation was examined both quantitatively and qualitatively and correlated with effects on cellular differentiation and proliferation. Incubation of HL-60 cells for 6 h with chelerythrine and calphos-
tin C (highly specific inhibitors that act at the regulatory domain) or H7and gossypol (nonspecific inhibitors that act at the I'M catalytic domain)
produced concentration-dependent increases in DNA fragmentation. In
duction of DNA fragmentation by chelerythrine, calphostin C, and gossypol was biphasic, resulting in a sharp decline in effect at concentrationsabove 5 /IM, 0.1 ju\r. and 100 /IM. respectively, whereas maximal and morestable effects were observed in response to H7 11(HI/IMI.A 6-h exposure to
staurosporine, a nonspecific but potent PKC inhibitor, failed to induceDNA fragmentation at concentrations generally used to achieve maximalinhibition of enzyme activity (e.g., 50 n.vt) but promoted fragmentation atconsiderably higher concentrations (e.g., 200 IIMI. In contrast, 6-h ex
posures to the nonspecific protein kinase inhibitor hypericin (0.1 to 100/JMI or to the nonspecific inhibitor of protein kinase A, HA1004 (50 /KM).
were without effect on DNA fragmentation. DNA obtained from cellsexposed to chelerythrine (5 //MI, calphostin C (100 MM). H7 (50 /IM),gossypol (50 /IMI, and staurosporine (200 IIMI—but not hypericin (25/IM)—exhibited clear evidence of internucleosomal DNA cleavage on aga-
rose gel electrophoresis; moreover, these cells exhibited the classical morphological features of apoptosis (cell shrinkage, nuclear condensation, andthe formation of apoptotic bodies). All of the PKC inhibitors that inducedapoptosis, and one of the inhibitors that did not (hypericin), substantiallyinhibited HL-60 cell clonogenicity at the concentrations evaluated. None of
the agents tested induced cellular maturation as assessed by nonspecificesterase and nitro-blue tetrazolium positivity. DNA fragments obtained
from cells exposed to specific and nonspecific PKC inhibitors possessedpredominantly 5'-phosphate termini, consistent with the action of a Ca2+-
/Mg2*-dependent endonuclease. Finally, Northern blot analysis revealed
that exposure to calphostin C at a concentration that induced apoptosis(100 nM) failed to alter expression of bcl-2, an oncogene known to block
apoptosis in both lymphoid and myeloid leukemia cells. Together, theseobservations suggest that certain inhibitors of PKC, administered aloneand on a transient basis, are capable of inducing apoptotic DNA fragmentation and cell death in HL-60 promyelocytic leukemia cells in a highlyconcentration-dependent manner. These findings also suggest a protective
involvement of basal PKC activity in the regulation of programmed celldeath in myeloid leukemia cells.
Received 9/13/93; accepted 2/3/94.The cosi of publication of the article were defrayed in part by the payment of page
charges. This article must therefore he hereby marked advertisement in accordance with18 U.S.C. Section 1734 solely to indicate this fact.
1This work was supported by Grant CH-523 from the American Cancer Society
(S. G.), Grant CA-40615 (L. F. P.) and Cancer Center Support Core Grant CA-16059 fromthe National Cancer Institute, and the Bone Marrow Transplantation Core ResearchLaboratory of the Medical College of Virginia. W. D. J. is recipient of a Cancer BiologyFellowship supported by USPHS Training Grant CA-09564-05 and of National ResearchService Award CA-093NO. Portions of this material were presented in preliminary form atthe 82nd Meeting of the American Association for Cancer Research. Inc.
2 To whom requests for reprints should be addressed, at Department of Medicine.
Division of Hematology/Oncology, Box 230 MCV Station. Richmond. VA 23298-0230.
INTRODUCTION
Participation of the Ca2 +-/phospholipid-dcpendent protein kinase(protein kinase C; PKC)' in intracellular signaling processes has been
demonstrated in many cell types, including those of hematopoieticorigin (1). PKC-mediated phosphorylation of numerous protein sub
strates is associated with a wide range of biological effects, includingstimulus-secretion coupling, induction of cellular proliferation anddifferentiation, activation of nuclear transcription factors and cell-surface receptors, and tumor promotion (2-5). PKC is expressed inmammalian systems as a family of diverse serine-threonine kinases,
consisting of at least nine isoforms differing in both substrate specificity and dependence upon Ca +2 availability. Differential activation
of PKC isoforms has been postulated to account for the divergentactions of different enzyme activators (6). In hematopoietic cell systems, pharmacological agents that inhibit PKC activity, such as theisoquinolines staurosporine and H7, inhibit the growth of both normal(7) and leukemic (8) progenitors, suggesting the involvement of basalPKC activity in cell proliferation. In addition, PKC inhibitors blockleukemic cell differentiation in response to tumor-promoting phor-
boids (9, 10). Since cell differentiation is associated with a loss ofproliferativc potential, these observations indicate that PKC inhibitorsmay exert pleiotropic effects on hematopoietic cell behavior.
Recently, considerable attention has focused on the sequence ofevents referred to as programmed cell death, or apoptosis, and the rolethat this process may play in mediating the lethal effects of diverseantineoplastic agents in leukemic cells (11). Apoptosis represents anactive, energy-dependent process in which the cell participates in its
own destruction and is characterized by stereotypical morphologicalchanges, such as cell shrinkage, nuclear condensation, and the formation of membrane-bound apoptotic bodies (12). A salient character
istic of apoptotic cell death is the degradation of genomic DNA tooligonucleosomal fragments, presumably a consequence of activationof one or more of a family of Ca+2-/Mg ' 2-dependent endonucleases
(13). Apoptosis is distinct from necrosis, a passive process that resultsfrom altered osmoregulation and/or breaches of membrane integrityand is characterized by random degradation of DNA by lysosomalenzymes (14). Efforts to define a role for PKC in the induction ofapoptosis have been complicated by conflicting reports. For example,the observations that activation of PKC by exposure to PMA, eitheralone or in conjunction with Ca*2 ¡onophore, induces apoptosis in
cells of lymphoid origin (15) and that inhibition of PKC by exposureto H7 prevents glucocorticoid-induced apoptosis in murine thymo-
cytes (16) suggest that PKC activation promotes this process. On theother hand, the ability of PMA to oppose steroid-induced apoptosis inthymic lymphocytes (17) and to prevent growth factor-deprived he
matopoietic cells from undergoing apoptotic cell death (18) is, instead, consistent with an antagonistic influence. It is conceivable that
'The abbreviations used are: PKC, Ca-*-/phospholipid-dcpendcnt protein kinase.
protein kinase C; PMA. phorbol 12-myristate 13-acetatc; ara-C, 1-ß-D-arabinofuranosyl-cytosine; cGMP, guanosine-3'.5'-monophosphate; EC,,,, effective concentration (at 50%
maximal response): H7, [(±)-l-(5-isoquinolinesulfonyl)-2-mcthylpipcra/inc; HA-KKI4.W-(2'-guanidinoethyl)-5-isoquinolinesulfonamidc; NBT, nilro-blue letra/olium; PKA,
cAMP-dependent protein kinase. protein kinase A.
1707
Research. on February 24, 2020. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from

INDUCTION OF APOPTOSIS BY PKC INHIBITORS
conflicting observations regarding the apparent role of PKC in theregulation of apoptosis reflect cell type-specific responses or, alter
natively, the influence of other factors, including concentration andscheduling considerations.
In a recent communication, we reported that bryostatin 1, a non-tumor-promoting PKC activator derived from the marine bryozoanBugula neritina, sensitizes human promyelocytic HL-60 leukemiacells to the lethal actions of ara-C by potentiating ara-C-induced
apoptosis (19); a logical extension of these studies would entaildetermining the influence of reduced PKC activity on the interactionbetween these two agents. Such an analysis would first require a morecomplete characterization of the effects of pharmacological PKCinhibitors administered alone, however. Presently, little information isavailable concerning the relationship between PKC inhibitors and theinduction of programmed cell death in human myeloid leukemia cells,and the results described in existing reports are inconsistent. Forexample, the potent (but nonspecific) PKC inhibitor staurosporine hasbeen reported both to antagonize (20) and to initiate apoptosis inHL-60 cells (21); similarly conflicting reports of H7 action have also
appeared (16, 22). In addition, detailed comparisons of the concentration-response relationships of different PKC inhibitors in the mod
ulation of apoptosis are generally lacking. The present investigationswere, therefore, undertaken to characterize the relative capacities ofdifferent pharmacological PKC inhibitors administered for a brief(i.e., 6-h) interval to promote apoptosis in human promyelocytic
leukemic cells. Our findings demonstrate that, while the effects ofthese agents are variable and highly dependent upon concentration,transient exposure of HL-60 cells to a subset of PKC inhibitors
unambiguously induces apoptotic DNA fragmentation and cell deathin HL-60 cells. In addition, these observations suggest indirectly that
basal activity of PKC prevents the occurrence of programmed celldeath in myeloid leukemic cells.
MATERIALS AND METHODS
Drugs and Reagents
Chelerythrine [EC50 (PKC) = 0.66 H.M;EC50 (PKA) = 170 ¡J.M;Ref. 23],H7 [EC50 (PKC) = 6 H.M;EC50 (PKA) = 3 H.M;EC50 (protein kinase G) = 5.8/¿M;Ref. 24], hypericin [ECsn (PKC) = 3.3 /¿M;Ref. 25], the relativelyinactive isomer iso-H7 [EC5() (PKC) = 60 ¡J.M;Ref. 26], and HA-1004 [EC50(PKC) = 40 /¿M,ECM) (PKA) = 2.3 /J.M;Ref. 24] (all from LC Laboratories,Woburn, MA) were diluted in sterile water; calphostin C [EC5I) (PKC) = 50
nM, EC50 (PKA) 2: 50 /J.M; Ref. 27] (LC Laboratories), staurosporine [EC50(PKC) = 0.7 nM, EC50 (PKA) = 7.0 nM; Ref. 28] (all from Sigma ChemicalCorp., St. Louis, MO) were dissolved in sterile DMSO; gossypol [EC50 = 50
to 100 /IM; Ref. 29] and the biologically inactive isomer apogossypol (Sigma)were dissolved in acetone. Stock preparations of all reagents were stored at-20°under light-free conditions. All reagents were diluted to appropriate final
concentrations in medium. Vehicle controls of water, DMSO, or acetone wereincluded in multiple studies (never exceeding a final concentration of 0.01%)and were consistently found to be ineffective in the induction of DNA fragmentation in HL-60 cells.
Cell Culture and Test Exposures
The human leukemic cell line HL-60 was derived from cells obtained from
a patient with acute promyelocytic leukemia as originally described byGallegher et al. (30); cells from passage numbers 74 and below were usedduring the course of these studies. HL-60 cells were grown in RPMI 1640(phenol red-free formulation) supplemented with 1.0% sodium pyruvate, non-
essential amino acids, L-glutamine, penicillin, and streptomycin (all fromGIBCO, Grand Island, NY) and 10% heat-inactivated fetal bovine serum
(Hyclone, Ogden, UT); all cultures were maintained under a fully humidifiedatmosphere of 95% room air-5% CO2 at 37°C.All cultures were passaged
twice weekly and exhibited a doubling time of approximately 24 h. HL-60 cell
cultures were routinely screened for Mycoplasma contamination by a rapid
hybridization assay selective for Mycoplasma ribosomal RNA (Gen-Probe,
San Diego, CA) and were consistently found to be free of contamination.Where appropriate, cell densities were determined by Coulter counter, and thenumber of viable cells were assessed by hematocytometer and trypan blueexclusion. For experimental incubations, HL-60 cells in log-phase growth weresuspended at a density of 4.5 x 10s cells/ml in 25 cm2 sterile polystyrene
T-flasks (Corning Industries, Corning, NY) and maintained as described
above. Cells were exposed to pharmacological agents in complete medium for
6 h; test exposures to calphostin C were conducted in the continuous presenceof light to promote photoactivation of this compound (31). Test incubationswere terminated by gentle pelleting of the cells at 400 X g for 10 min andwithdrawal of the medium. Following determination of cell density and viability, the cells were prepared variously for assessment of DNA fragmentationby spectrofluorophotometry or agarose gel electrophoresis, cloning efficiency,and examination of cell morphology and differentiation as described below.
Analysis of DNA Damage
Spectrofluorophotometric Quantitation of DNA Fragments. Followingexposure to test agents, cells in suspension (4.5 X 10s cells/ml; 5.0 x IO6 cells,total) were centrifuged at 2500 rpm for 15 min at 4°C.Aliquots of the
supernatant were withdrawn and adjusted to 5 mM Trizma-20 mM EDTA for
direct assay of released DNA fragments. The remainder of the medium wasaspirated, and the pellets were resuspended in lysis buffer (0.05% fully reducedTriton X-100, 5 mM Trizma, 20 mM EDTA, pH 8.0; 100 /xl/106 cells); cell
lysates and medium samples were then centrifuged at 30,000 X g for 40 minat 4°Cto separate fragmented DNA from intact chromatin and other cell
materials. The presence of small molecular weight DNA fragments in celllysate and medium samples was determined by a miniaturized adaptation ofquantitative ¿¿sbenzimidespectrofluorophotometry originally described byCesarone et al. (32); we have previously determined that the extent of DNAfragmentation detected in this assay correlates closely with induction ofapoptosis in HL-60 cells (33). Lysate and medium preparations were diluted inmodified TNE buffer (3 mM NaCl, 10 mM Tris-HCl, l mM EDTA, pH 7.4)
containing 1 fAg/ml feisbenzimide trihydrochloride (Sigma). DNA was thenquantified by spectrofluorophotometry with excitation at 365 nm (100-nmband width) and filtered emission at 460 nm (10-nm peak width). All DNAvalues in HL-60 cell lysate and medium preparations were standardized against
highly purified calf thymus DNA calibration standard; values for all suchresponses are expressed in the text as ng DNA per 1 x IO6 cells and reflect the
absolute amount of nonsedimenting, small (i.e., S3000 base pairs) fragmentsof DNA present in preparations of cell lysate and incubation medium.
Agarose Gel Electrophoresis. DNA fragmentation was analyzed by conventional agarose gel electrophoresis as reported previously (19), with minormodifications. Suspension cultures of HL-60 cells (2 X IO7 cells/group) were
treated with various agents as described above; following test incubations, thecells were washed several times in serum-free medium, and the final pellet wasresuspended in phosphate-buffered saline. Cells were lysed by incubation in10 mM Tris-HCl (pH 7.4) containing 20 mM EDTA and 0.1% Triton X-100
(fully hydrogenated) with brief mechanical agitation; the resulting lysate wasthen treated with 100 fig/ml proteinase-K (Sigma) for 16 h at 55°C.Thepartially deproteinated extract was centrifuged at 30,000 X g for 45 min at 4°C
to separate unfragmented bulk DNA from small, nucleosomal DNA fragments;the resulting pellet was discarded, and the supernatant was treated withribonuclease-A (Sigma) for 4 h at 37°C.Small molecular weight DNA residing
in the final extract was resolved by electrophoresis at 85 to 115 V/cm for 90to 180 min on 2.0% agarose gels impregnated with ethidium bromide; laneswere loaded with a fixed volume of extract, and electrophoretic gel profilesthus reflect DNA from a constant number of cells rather than a fixed quantityof DNA. Routinely, multiple DNA molecular weight reference preparations(100-, 123-, and 1000-base pair ladders; GIBCO-BRL) were run in parallel.
DNA fragments were visualized under UV light.
Other Studies
Clonogenicity. To assess cloning efficiency, HL-60 cells were plated in
soft agar as described previously (34). Briefly, cells were pelleted, washedtwice, and resuspended in cold RPMI 1640. After determining cell densities byCoulter counter, 400 cells were seeded in 35-mm tissue culture plates containing 1 ml of RPMI, 20% fetal calf serum, 10% 5637-CM, and 0.3% Bactoagar (Difco, Detroit, MI). The plates were placed in a 37°C,5% CO2, fully
1708
Research. on February 24, 2020. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from

INDUCTION OF APOITOS1S BY I'KC INHIBITORS
humidified incubator for 10 to 11 days, after which colonies, consisting of
groups of 50 or more cells, were scored on an inverted microscope.Cell Morphology. For examination of altered morphology associated with
contrasting modes of cell deaths (i.e., apoptosis versus necrosis), cells werewashed twice with cold serum-free RPMI 1640. and cytocentrifuge slides wereprepared and stained with 20% Wright-Giemsa stain; morphological assess
ments were performed. The mode of cell death in drug-treated populations was
determined based on the expression of morphological features characteristiceither of apoptosis (e.g., cell shrinkage, nuclear condensation, and extensiveformation of membrane blebs and apoplotic bodies) or of necrosis (e.g., cellswelling, nuclear expansion, and gross cytolysis).
Cell Differentiation. Cells were washed twice with coki RPMI 1640 andcytocentrifuge slides prepared and stained either with (a) n-naphthyl acetateesterase (Sigma kit No. °1-A)for determination of monocytic differentiation;
or with (/)) NBT for determination of neutrophilic differentiation, and thepercentage of positive cells were enumerated (at least 200 cells per experimental condition) as previously described (I1)).
Expression of bcl-2 Oncogene Expression. Other studies evaluated theeffect of PKC inhibitors on expression of the bcl-2 oncogene, which encodesa protein that putatively antagonizes the induction of apoplosis in nonhcma-
topoietic cells as well as in hemalopoietic cells of both myeloid (35) andlymphoid (36) origin. Northern analyses were performed by the method ofChirgwin el al. (37) and adapted as previously described (3S). Total cellularRNA was extracted by lysis of cell pellets in 4.0 M guanidine isothiocyanate.0.5% sodium luuryl sulfate, and ultracentrifugation through a 5.7 M CsCIcushion at 41,000 X g for 20 h at 20°C;RNA pellets were washed sequentially
in 95% elhanol and 70% ethanol before resuspension in water. RNA was thendenatured in 20 mM morpholino-propane sulfonic acid, 5 mM sodium acetate.
1 mM EDTA. 2.2. M formaldehyde, 50% formamide, pH 7.4. and resolved byelectrophoresis on 6.6% formaldehyde-1.0% agarose gels impregnated with
ethidium bromide; RNA bands were then transferred to nylon membranes(Nylran; Schleicher and Schuell. Keene. MD) by transphoresis. Radiolaheledbcl-2 complementary DNA probes (obtained from the pFL-1 plasmid contain
ing a 1.5-kilobase Hinti\\\-Eci>R\ insert: provided by Dr. Michael Cleary.Stanford University, Stanford, C'A) were prepared by standard nick translation
using [-y-3::P|ATP (NEN) and materials supplied by GIBCO-BRL. Hybridiza
tion to RNA blots was performed by incubation in 50 mM sodium phosphate.5X Denhardt's buffer, 5X saline sodium citrate buffer, 0.1% sodium dodecyl
sulfate, 250 /¿g/mlyeast RNA, 50% formamide. and 5.5% dextran sulfate for16 h at 42°C;the membranes were then washed extensively, and radioactivity
in each band was determined by conventional radioautography.Identification of 5'-Phosphate Oligonucleosomal DNA Fragments. To
characterize DNA cleavage products, pelleted cells were lysed and centrifugedas described above. DNA in the high-speed supernatant was extracted with
phenohchloroform (1:1), precipitated in ethanol, and dissolved in 250 fil 10mM Tris-HCI, 0.1 mM EDTA. pH 8; 3-/il aliquots of each sample (or 0.15 figof blunt-ended fragments generated by cleavage of pUC19 with Dpn as apositive control) were treated with either 0 or 3 units calf intestinal phos-phatase (U.S. Biochemicals) for 30 min at 37°Cand then for 30 min at 55°C
in a reaction of 25 ji.1. Samples were diluted to 50 ;xl in 25 mM EDTA,re-extracted in phenol, ethanol-precipitated, and then phosphorylated by incubation for 30 min at 37°Cin the presence of 20 /xCi [-y-'2P]ATP (3,000
Ci/mmol; NEN) and 5 units T4 polynuclcotide kinase (New England Biolabs)in a reaction volume of 20 /¿I.The samples were again diluted in EDTA,extracted in phenol, and then diluted to 0.5 ml and concentrated to approximately 0.1 ml four times using a Microcon-30 ultrafiltration device (Amicon)centrifuged at 5,(XK) X g to remove unreacted [•y-'-PJATP.Aliquots of each
sample were electrophoresed at 10 V/cm for 2 h on a 2% agarose-1% NuSicve(FMC) agarose gel. The gel was dried under vacuum at 60°Cand subjected to
autoradiography.
RESULTS
Quantitative analysis of DNA fragmentation was performed using6/sbenzimidc spcctrotluorophotomctry of nonscdimenting, low-molecular-weight fragments of double-stranded DNA. Under basal conditions, such fragments were present in HL-60 cells at a level ofapproximately 250 ng/10" cells. A 6-h exposure of HL-60 cells to two
highly specific inhibitors known to act at the diglyceride binding site
n
£ 500
V B 8 7 0 5 4-log M chelerythrine
V 9 6 7 8 5 4-log U calphoBtin C
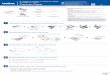
Fig. 1. Induction of DNA fragmentation by calphostin C and chdcrythrinc. HL-60 cellswere exposed to Ihc selective inhibitors of PKC' chelcrythrine (0.01 to 100 /AM;A) andealphostin C"(11.11(11to 10 /¿M:ß)in complete medium for 6 h; and the formation (T) and
release (A) of nonsedimenting DNA fragments were determined by />/.vben/imide spec-Irofluoropholometry. Points, mean of quadruplicate determinations from a representativeexperiment performed four limes with comparable resulls. Kurs, SEM.
of the PKC regulatory domain, chelerythrine (0.1 to 100 /XM;Fig. LA)and ealphostin C (0.001 to 100 JU.M;Fig. Iß),produced biphasic.concentration-dependent increases in the degradation of cellular intosmall double-stranded fragments, with maximal fragmentation occur
ring at 5 UM and 100 n.M. respectively. In neither case could theprogressive decline in DNA fragmentation noted at higher concentrations be attributed to loss of DNA during the physical dissolution ofapoptotic cells inasmuch as release of DNA fragments into the medium was minimal under these conditions. Parallel exposure of HL-60
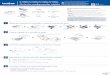
cells to H7, a nonspecific isoquinoline inhibitor of PKC acting at theATP-binding site of the catalytic domain (0.1 to 100 JJ.M;Fig. 2A) or
gossypol, a PKC inhibitor that acts at both the catalytic and regulatorydomains (0.01 to 200 /UM; Fig. 2B), also resulted in substantial,concentration-related inductions of DNA fragmentation, although at
considerably higher concentrations than required in the case of moreselective compounds (e.g., chelerythrine and ealphostin C").Whereas
gossypol-related DNA fragmentation declined as the concentration
was increased from 100 to 200 p,M, the response to H7 remainedstable over this concentration range. Exposure of HL-60 cells to theinactive analogues iso-H7 and apogossypol at equimolar concentra
tions failed to promote DNA fragmentation (not shown), as didHA-1004, an isoquinoline inhibitor of both adenosine-3'.5'-mono-phosphate-dependent protein kinase and guanosine-3',5'-monophos-
phate-dependent protein kinase (protein kinascs A and G; not shown).Six-h exposure of HL-60 cells to staurosporine (0.1 to 500 nM; Fig.2C), a potent but nonspecific inhibitor that acts at the catalytic-
domain, failed to induce DNA fragmentation at concentrations generally used to inhibit PKC activity [i.e., 50 to 100 nM). althoughextensive DNA fragmentation was observed at higher concentrations(>2()() nM)]. In contrast, a 6-hr exposure to hypericin, another non
specific inhibitor, failed to promote DNA fragmentation at all concentrations tested (0.01 to 200 /LLM).As noted in the studies usingspecific PKC inhibitors, none of these responses entailed the releaseof DNA fragments.
Qualitative analysis of DNA fragmentation was performed usingconventional agarose gel electrophoresis to resolve oligonucleosomalDNA fragments and yielded results consistent with the quantitativestudies described above (Fig. 3). The concentration of each PKC'
inhibitor used in these exposures was selected to correspond to: (a)levels associated with maximal or near-maximal degrees of DNA
1709
Research. on February 24, 2020. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from

INDUCTION OF APOPTOSIS BY PKC INHIBITORS
fragmentation as determined by spectrofluorophotometry; and/or (b)concentrations previously shown to be in excess of previously reported ECM, values for that agent. Unambiguous laddered electro-
phorctic patterns of oligonucleosomal DNA fragments were observedfollowing a 6-hr exposure of HL-60 cells to chelerythrine (5 JAM;Fig.
3, Lane 5) and calphostin C (100 nM; Fig. 3, Lane 6), as well as to the955-585-
123456 789 10
-
I
A3000
2•a
1000
B3000
-T'V
V 7 B S 4 3-lof " B7
V 8
-loi
7 S i 4 3
If goaiypol
D3000
-log II itauroiporinoV 7 8 5 4 3
-log II hypericin
Fig. 2. Effects of nonselective PKC inhibitors on the induction of DNA fragmentation.HL-60 cells were exposed lo H7 (0.1 to 200 /IM; A), gossypol (0.01 to 200 ¿IM;ß),staurosporine (0.1-100 nw; C), or hypericin (0.01-2IX) JIM;/)) in complete medium for 6h; and the formation (V) and release (A) of DNA fragments were determined spectroflu-
orophotomelrically as before. In each instance, points represent the mean ±SEM oftriplicate determinations from representative experiments performed four times withcomparable results.
bp
1200-600-400-
100-
Fig. 3. Induction of internucleosomal DNA fragmentation by pharmacological inhibitors of PKC'. HL-60 cells were exposed to the agents indicated for 6 h, and the formation
of oligonucleosomal fragments was determined by agarose gel clectrophoresis as described in the text. Lane /, control; Lane 2, H7 (50 JAM);Lane 3, staurosporine (50 nM);Lane 4. staurosporine (2(K) nM); Lane 5, chelerythrine (5 JAM);Lane 6, calphostin C (100nM); Lane 7, gossypol (1(X( JAM);Lane 8, hypericin (25 /U.M).The results are from arepresentative study performed four times with comparable outcomes. Intense fluorescence, in the absence of discernible DNA fragments, was noted in extracts from cellstreated with hypericin.
341-258-
141-105-
75-
IliFig. 4. Characterization of apoptotic DNA cleavage products. HL-60 cells were ex
posed to PKC inhibitors for 6 h. and the products of DNA fragmentation were characterized by phosphatase/kinase labeling as described. DNA in even-numbered lanes waspretreated with calf intestinal phosphatase; ail samples were then subjected to 5'-phos-phorylation by polynucleotide kinase in the presence of [y-32P]ATP and resolved on 3%agarose gels and subjected to autoradiography. Lanes 1 and 2, fragments of "55. 585, 341,258, 141. 105, and 75 base pairs; Lanes 3 and 4, low-molecular-weight DNA from
untreated cells; Lanes 5 and 6, H7 (50 ¿tw);Lanes 7 and 8, gossypol (50 fiw); Lanes 9 and70, chelerythrine (5 /¿M).
less selective compounds H7 (50 JAM;Fig. 3, Lane 2) and gossypol (50JU.M;Fig. 3, Lane 7); iso-H7 and apogossypol were both ineffective
(not shown). In addition, there was no evidence of oligonucleosomalfragmentation following parallel exposure to hypericin (25 JLLM;Fig. 3,Lane 8) or staurosporine (50 nM; Fig. 3, Lane 3), although ladderingwas observed at higher concentrations of the latter agent (e.g., 200 nM;Fig. 3, Lane 4).
DNA cleavage products were also characterized (Fig. 4). Internu-
cleosomal DNA cleavage associated with apoptosis has generallybeen attributed to a Ca"l"+/Mg+"^-dependent endonuclease, but re
cently this assignment has come under question (39), and DNase IIhas been proposed as an alternative endonuclease. Unlike most nucle-ases, DNase II leaves 3'-phosphate and 5'-hydroxyl termini (40); thus,
by examining the termini of fragments released by internucleosomalcleavage during apoptosis, it was possible to determine whetherDNase II (or a functionally similar enzyme) is involved. Low-molecular-weight DNA from cells exposed to H7, gossypol, or chelerythrinewas subjected to 5'-[12P] end-labeling by polynucleotide kinase, withor without prior removal of any 5'-phosphates with calf intestinalphosphatase. Virtually all of the [32P] incorporation into oligonucleo
somal fragments was phosphatase dependent, indicating that thesefragments possessed 5'-phosphate termini and thus were presumably
not generated by DNase II. Similar results were obtained using calphostin C (not shown).
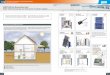
The appearance of cytoarchitectural features characteristic of apoptotic cell death was also monitored, and revealed that apoptoticmorphology was elicited by PKC inhibtors only at concentrationsassociated with induction of extensive DNA fragmentation (Fig. 5).HL-60 cells from untreated control groups are shown in Fig. 5A.
Exposure to the highly selective PKC inhibitor chelerythrine (5 /UM)for 6 h produced the characteristic features of apoptosis in the majority of cells, including cell shrinkage, nuclear condensation, membraneblebbing, and the appearance of membrane-bound apoptotic bodies; in
addition, some cytoplasmic granularity was noted. Apoptotic traitswere also observed in cells exposed either to calphostin C (100 nM;Fig. 5C) or to gossypol (50 /J.M;Fig. 5D). Exposure to H7 (50 nM; Fig.5£) induced even more prominent apoptotic changes than did chelerythrine, whereas parallel exposure to the inactive isomer iso-H7 at
1710
Research. on February 24, 2020. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from

INDUCTION OF APOPTOSIS BY PKC INHIBITORSI*»'.••'
Fig. 5. Expression of apoptolic morphology. HL-60 cells were exposed to PKCinhibitors in complete medium for 6 h. and cellular morphology was evaluated inpreparations stained with Wright-Giemsa. A, untreated control; B, chelerythrine (5 U.M);C, calphostin C (100 nM); D. gossypol (50 p.»);E, HI (50 ¿IM);F. iso-H7 (50 JIM);C. staurosporinc (50 nM); //, hypericin (25 U.M).
equimolar concentrations produced little effect on HL-60 cell mor
phology (Fig. 5F). In contrast, cells exposed to staurosporine (50 nM)exhibited some evidence of membrane blebbing but did not displayother changes typically associated with apoptosis (Fig. 5G); it shouldbe noted, however, that apoptotic morphology was elicited by exposure to staurosporine at much higher concentrations (e.g., 2:200-500nM; not shown). Similarly, 6-h exposure to hypericin (25 /XM;Fig. 5H)
produced some membrane blebbing but otherwise did not induceexpression of apoptotic morphology. A 6-h exposure to HA1004 alsofailed to produce alterations of HL-60 morphology (not shown).
The effects of these agents on HL-60 cell viability and proliferative
capacity were determined by trypan blue exclusion and assays ofclonogenicity (Fig. 6). None of the agents tested significantly impaired the ability of HL-60 cells to exclude trypan blue, with the
single exception of calphostin C (100 nM), which produced a 27%decrease in dye exclusion. A close association was noted between theability of an agent to induce apoptosis and its capacity to suppress
HL-60 cell clonogenicity, however. Exposure of HL-60 cells to chel
erythrine (5 (J.M),calphostin C (100 nM), H7 (50 JAM),or gossypol(50 JU.M)for 6 h. all of which promoted extensive DNA fragmentationand apoptosis, resulted in dramatic inhibition of clonogenicity thatranged from 91 to 100%; conversely, iso-H7 (50 JUM)and staurospo
rine (50 nM), which failed to promote DNA damage and elicit morphological features characteristic of apoptosis, were without effect onHL-60 cell clonogenicity. The only exception to this pattern was
hypericin, which failed to induce DNA fragmentation and characteristic apoptotic morphology but abrogated clonogenicity, suggesting analternative mechanism of cytotoxicity for this agent. Finally, exposure
to chelerythrine, calphostin C, and gossypol at higher concentrationsthat were associated with a decline in DNA damage also suppressedclongenicity (not shown).
Because several agents that induce differentiation in HL-60 cells
(e.g., PMA and retinoic acid) also promote apoptosis (41, 42), furtherstudies were undertaken to determine whether the actions of PKCinhibitors could be related to leukemic cell maturation (Fig. 7).Continuous 72-h exposure of HL-60 cells to ara-C substantially in
creased the percentage of cells staining positively for nonspecificesterase, a marker for monocytoid differentiation as reported previously (43); similar results were obtained with PMA (not shown).Exposure of HL-60 cells to various PKC inhibitors failed to produce
significant increases in esterase positivity. Similarly, while the majority of cells incubated with DMSO stained positively for NBT, amarker for granulocytic maturation, none of the PKC inhibitors evaluated significantly increased NBT positivity.
Expression of the bcl-2 oncogene and oncoprotein was monitored
following exposure to calphostin C at a concentration that unambiguously induced apopotic DNA fragmentation and cell death (100 nM).Northern analysis revealed that levels of mRNA for both bcl-2
(Fig. SA) and the housekeeping gene GAPDH (Fig. 8fl) were unchanged throughout a 6-h exposure to calphostin C. In parallel studies.flow cytometry demonstrated that expression of the bcl-2 oncoprotein
also remained stable over this exposure interval. Similar results wereobtained in cells exposed to other inhibitors of PKC (not shown).
150
Fig. 6. Effects of PKC inhibitors on HL-60 cellcloning efficiency and viability. HL-60 cells wereexposed to chelerythrine (5 JAM),calphostin C (l(K)nM), H7 (50 JIM), gossypol (50 u,M), staurosporine(50 nM), or hypericin (25 JIM) in complete mediumfor (>h. and clonogenicity (aingte-hutchcil bars) andviability (doitbh'-haicht'il burs) were determined as
described in the text. Point*, mean ±SEM of triplicale determinations from a representative experiment performed three times with comparableresults.
iaoo
Cl<*4
tÃtéOao
100
50 -
150
100
50
o14-t->
aoo
1I
oo
A
Iv—<OAo
ua
oAPi
n oPi
OM
Oa
•r+
IH
OPimo
I
a•i-«O
1711
Research. on February 24, 2020. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from

INDUCTION OF APOPTOSIS HY 1'KC' INHIBITORS
75 -,
o
i25K
24 48 72
exposure (hr)
24 48 78
exposure (hr)
Fig. 7. Induction of cellular differentiation hy pharmacological inhibitors of PKC.HL-60 cells were exposed to PKC inhibitors for varied intervals (0 to 72 h) in completemedium and evaluated for expression of monocytic differentiation as assessed by nonspecific esterase (AT.V£)posilivily (A ) and for neutrophilic differentiation as assessed byAf/HTpositivily (fl): UK)cells were scored for each condition. D. control: A. chclerythrine(5 (i.M);T. calphostin C ( HXInM)i A. H7 (5(1 UM): ". staurosporinc (5(1 nM): •.gossypol
(50 M.M):O. hypericin (50 /¿M).Cells were also exposed to ara-C (100 nM; •)or DMSO(1.25%; •).respectively, as positive controls for NSE and NBT positivity. /-VÃÃ'/f/.v,mean
of triplicate determinations from a representative experiment performed four times withcomparable results. Bars, SEM.
15 60 120 180 360
b a bI I
b a11
b a
bcl228S
18S
B 15 60 120 180 360
OL
"
b al l
b a11
b a
GAPDH
Fig. 8. Northern blot analysis of bcl-2 oncogene expression. HL-60 cells were exposedto calphostin C (100 nM) for varied intervals (15 to 360 min), and steady-state mRNAlevels for the oncogene bcl-2 (A) and the house-keeping gene GAPDH (B) were monitored hy conventional Northern analysis as described in the text.
DISCUSSIONMultiple pathways exist for the regulation of apoptosis in mamma
lian cells. The present findings indicate that acute (i.e., 6-h) exposure
to PKC inhibitors is sufficient to induce apoptosis in the humanmyeloid leukemia cell line HL-60. This response was noted in re
sponse to both highly selective inhibitors (e.g., calphostin C andchelerythrine) that act at the diglyceride binding site within the PKCregulatory domain (23, 27), as well as by relatively nonspecificinhibitors (e.g., H7, staurosporine, and gossypol) that act at theATP-binding site within the catalytic domain of the enzyme (24, 28,
29). The cytotoxic efficacy of these agents varies considerably, however. The inability of both HA-1004, a moderately selective inhibitor
of protein kinases A and G, and of the biologically inactive isomeriso-H7 to induce apoptosis, strongly supports inhibition of PKC in the
action of H7. This consideration takes on greater significance in viewof evidence implicating other signaling systems (e.g., adenosine-3',5'-monophosphate) in the induction of programmed cell death in
lymphoid cells (44).Based upon our results, and those of several other laboratories, the
effect of PKC inhibitors on apoptotic events appears to vary with bothcell type and the cytotoxic insult. For example, H7 (50 to 100 /J.M)haspreviously been reported to induce apoptosis in HL-60 cells (45),MOLT-4 lymphoid leukemia cells and normal lymphocytes (46), andin concanavalin A-treated murine thymocytes (17). Conversely, H7
has also been reported to prevent apoptotic death in murine thymocytes exposed to corticosteroids (16). In an analogous fashion, staurosporine has been noted to trigger apoptosis in a variety of neoplasticcell lines (21) and to elicit the expression of apoptotic morphology inthe absence of characteristic DNA fragmentation in MOLT-4 cells
(47). On the other hand, exposure to staurosporine under conditionssimilar to those used in the present study (e.g., 86 nM for 4 h) has beenshown to suppress the morphological features but not the DNAdamage associated with apoptosis in UV radiation-treated HL-60cells, possibly by inhibiting (or retarding) PKC-dependent cytostruc-
tural alterations (20). In the current studies, staurosporine inducedapoptosis only at concentrations far in excess (e.g., 200 nM) of thereported K¡of this compound for PKC (0.7 nM) (28). Because staurosporine, like the antifungal agent cytochalasin B, interferes withother cellular functions (e.g., actin polymerization), such actions mayalso influence apoptotic processes, particularly at high inhibitor concentrations. Taken together, these observations suggest that drugconcentration, cell lineage, and the nature of a particular apoptoticstimulus all contribute to the extent to which programmed cell deathoccurs in response to PKC inhibitors.
The highly selective PKC inhibitors calphostin C and chelerythrine,as well as the relatively nonspecific agent gossypol, exhibited biphasicconcentration-response relationships such that DNA fragmentation
declined and eventually subsided as drug levels were increased beyond maximally effective concentrations. Reductions in DNA damagewere not associated with restoration of cellular viability, however,indicating that other lethal events proceeded unimpaired. Similarly,administration of hypericin at high concentrations failed to promoteDNA fragmentation and the expression of apoptotic morphology,suggesting an independent mechanism of cytotoxicity for this agent,possibly related to inhibition of tyrosine kinase activity (48). Ofpotential relevance is the finding that rat brain PKC is inhibited bygossypol at high concentrations but stimulated at lower concentrationswhen assayed in a cell-free system (29). It would therefore be ofinterest to determine whether HL-60 cell PKC exhibits similar prop
erties and, if so, to define the potential relationship(s) to the inductionof apoptotic DNA fragmentation and cell death.
The present results suggest that triggering of apoptosis by pharmacological inhibitors of PKC is unrelated to cellular differentiation,although features common to both processes have been observed inhuman leukemia cells exposed to other classes of agents. For example,evidence of endonucleolytic DNA cleavage and apoptosis has beendescribed in HL-60 cells during both PMA-induced monocytic differentiation (41) and retinoic acid-induced neutrophilic differentiation
(42). Conversely, inhibition of PKC by various agents, including H7,staurosporine, and free sphingoid bases (e.g., sphingosine) has beenshown to antagonize phorboid-induced leukemic cell maturation (49,
50), suggesting that some aspect of PKC activity is essential forcellular differentiation. That induction of apoptosis in the HL-60 cell
line by H7, chelerythrine, calphostin C, and gossypol was unaccompanied by cellular differentiation provides indirect evidence that sep-
1712
Research. on February 24, 2020. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from

INDUCTION OF APOPTOSIS BY PKC INHIBITORS
arate differentiation-dependent and -independent pathways of apop-
tosis may exist; additional support for this position is provided by arecent report indicating that topoisomerase II-mediated apoptosis maybe antagonized by agents that induce HL-60 cell maturation (51). A
further possibility is that the apoptotic capacity of PKC inhibitorsinvolves cell cycle-related factors, as recently proposed (45).
Considerable attention has focused on the protooncogene bcl-2,
which encodes an inner mitochondria! protein that reportedly antagonizes apoptosis in nonhematopoietic cells and in cells of lymphoidand myeloid origin (35, 52, 53). Expression of bcl-2 correlates in
versely with the susceptibility of cells to apoptosis, and transfectedcell lines overexpressing bcl-2 exhibit greater resistance to drug-induced apoptosis than their wild-type counterparts (54). Based uponthese findings, increased expression of bcl-2 has been proposed as a
novel mechanism of drug resistance in neoplastic cells (55). Recently,induction of apoptosis in HL-60 cells by taxol (56) and ara-C (57) hasbeen temporally associated with down-regulation of bcl-2, raising the
possibility that decreased expression of this oncogene might contribute to drug-mediated lethality. In the present studies, however,
calphostin C clearly induced apoptosis but failed to modify levelsof bcl-2 message or protein. PKC-mediated phosphorylation ofbcl-2 oncoprotein in vitro on serine residues was described in a
recent preliminary communication (58); if phosphorylation is necessary for the cytoprotective properties of bcl-2, interference with
this process could account for the ability of calphostin C (or otherPKC inhibitors) to induce apoptosis in the absence of reducedoncogene expression.
The nature of the endonuclease(s) responsible for catalyzing theapoptotic degradation of genomic DNA remains the subject of debate.In addition to the Ca2+-/Mg2+-dependent activity classically associ
ated with apoptotic responses (13), participation of other enzymes hasbeen implicated; these include the action of DNase II in etoposide-induced apoptosis in human leukemia cells (39) and of a Zn2 +-
resistant endonuclease that catalyzes degradation of DNA to highmolecular weight fragments in steroid-treated thymocytes (59). Because PKC has been shown to activate the Na2+-/H+-antiporter,
thereby increasing intracellular pH (60), pharmacolgical inhibition ofPKC activity may antagonize this process, resulting in increasedDNase II activity. Nonetheless, the formation of oligonucleosomalDNA fragments bearing 5'-phosphate termini demonstrates that
DNase II is not primarily responsible for the induction of DNAdamage by inhibitors of PKC (61), although the participation of thisenzyme at earlier stages of DNA fragmentation cannot be ruled out.
In summary, the present studies provide evidence that acute exposure of myeloid leukemia cells to PKC inhibitors of varying specificities is sufficient to trigger internucleosomal DNA fragmentation andcell death. In addition, these results indirectly suggest that the basalactivity of one or more isoforms of PKC protects these cells fromapoptosis. For example, we have recently demonstrated that experimental increases in the availability of intracellular free diglyceridepotently antagonize ara-C-mediated apoptosis in HL-60 cells (33) andceramide-induced apoptosis in both HL-60 and U937 cells (62).Similarly, suppression of apoptosis by \-abl in an IL-3-dependent
hematopoietic cell line has recently been associated with increasedintracellular diglyceride levels (63). Taken together, these observations suggest that endogenous diracylglycerols oppose the pharmacological and physiological induction of apoptosis, at least in continuously cultured cells. The apparently conflicting observation that thePKC activator bryostatin 1 enhances ara-C-related apoptosis in HL-60cells (19) apparently derives from extensive down-regulation of PKC
as a consequence of chronic treatment with this agent (33). Thus,augmentation of drug-induced apoptosis in leukemia cells may poten
tially be accomplished in at least two ways: (a) partial or complete
enzyme down-modulation; or (6) direct enzyme inhibition, as implied
by the present studies. Given recent interest in the possible usefulnessof PKC inhibitors as modulators of antineoplastic drug action (64) andas antileukemic agents in their own right (65), studies designed to testthis model are currently underway.
REFERENCES
i.
10.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
Nishizuka, Y. The family of protein kinase C for signal Iransduction. JAMA. 262;1826-1833, 1989.
Craven, P. A., and DeRuhertis, D. Role of activation of protein kinase C in thestimulation of colonie epithelial proliferation by unsaturatcd fatly acids. Gastroenter-ology, 95.' 676-685, 1988.Brach. M. A.. Herrmann. F.. and Kufe, D. W. Activation of the AP-1 transcriptionfactor by arabinofuranosylcytosine in myeloid leukemia cells. Blood, 79: 728-734.
1992.Rahmsdorf, H. Ì..and Herrlich. P. Regulation of gene expression by tumor promoters.Pharmacol. Ther., 48: 157-188, 1990.Bcrridge, M. J. Inositol triphosphate and diacylglyccrol: two interacting secondmessengers. Annu. Rev. Biochem., 56.- 159-193. 1987.
Nishizuka, Y. The molecular heterogeneity of protein kinase C and its implicationsfor cellular regulation. Nature (Lond.), 334: 661-665, 1988.Katayama. N., Minami. N., and Shirakawa, S. Putative involvement of proteinkinase C in proliferation of human myeloid progenitor cells. Blood. 73: 123-129,
1989.Tohda, S., Nara, N., Inai, Y., and Aoki, N. Effect of protein kinase inhibitors on theproliferation of leukemic cells stimulated by granulocyte colony-stimulating factor,granulocyte-macrophage colony-stimulating factor, or interleukin-3. Leukemia (Baltimore), 5: 813-814, 1991.Barendsen, N., Mueller, M.. and Chen, B. Inhibition of TPA-induced monocyticdifferentiation of THP-1 human monocytic leukemia cells by staurosporine, a potentprotein kinase inhibitor. Leuk. Res., 14: 467-474, 1990.
Mass, R.. Pfannkuche. H.. Kharhanda. S., Gunji, H.. Meyer, G., Hartman. A.. Hidaka.A., Resch, K., Kufe, D., and Goppet-Strube. M. Protein kinase C activation andproto-oncogene expression in differcntiation/retrodiffcrcntiation of human U-937leukemia cells. Cell Growth & Differ., 2: 541-548. 1991.Kaufman. S. H. Induction by endonucleolytic DNA cleavage in human acute my-
elogenous leukemia cells by etoposide, camptothecin. and other cytotoxic anticancerdrugs: a cautionary note. Cancer Res., 49: 5870-5878, 1989.Wyllie, A. H. The significance of apoptosis. Int. Rev. Cytol., 6f<: 251-306, 1980.
Gaido, M. L.. and Cidlowski, J. A. Identification, purification, and characterization ofa calcium-dependent endonueleasc (NUC18) from apoptotic rat thymocytcs; NUC'IHis not histone H-B. J. Biol. Chem., 266.- 18580-18585, 1991.
Wyllie, A. H. Cell death: a new classification separating apoptosis from necrosis. In:I. D. Bowen and R. A. Lockshin (eds.). Cell Death in Biology and Pathology, pp.9-34. London: Chapman & Hall, 1981.Mercep, M., Noguchi, P. D.. Ashwell, J. D. The cell cycle block and lysis of anactivated T-cell hybridoma are distinct processes with different CA *2 requirements
and sensitivity to cyclosporine A. J. Immunol.. 142: 4085-4092, 1989.
Ojeda, F., Guarda, M. I., Maldonato, C., and Folch. H. Protein kinase C involvementin thymocyte apoptosis induced by hydrocortisone. Cell. Immunol., /25: 535-539,
1990.McConkey, D. J., Hartzell, P., Jondal. M., and Arrenius. S. Inhibition of DNAfragmentation in thymocytes and isolated thymocyte nuclei by agents that stimulateprotein kinase C. J. Biol. Chem., 264: 13399-13402. 1989.
Lotem, J., Cragoe, E. J.. and Sachs. L. Rescue from programmed cell death inleukemia and normal cells. Blood. 78: 953-960, 1991.
Grant. S., Jarvis, W. D., Swcrdlow, P. S., Turner, A. J., Traylor, R. S., Wallace.H. J., Lin. P-S., Petti!, G. R., and Gewirtz, D. A. Polentialion of the activity of1-0-D-arabinofuranosycytosine by the macrocyclic lactonc PK-C activator bryostatin
1 is associated with enhanced fragmentation of mature DNA. Cancer Res., 52:6270-6278, 1992.Cotter, T. G., Lennon, S. V., Clynn. J. M., and Green, D. R. Microfilament-disrupting
agents prevent the formation of apoptotic bodies in tumor cells undergoing apoptosis.Cancer Res., 52.' 997-1005, 1992.
Bertrand. R., Solary, E., Kohn. K. W., and Pommier. Y. Staurosporine mayactivate a common final pathway to apoptosis. Proc. Am. Assoc. Cancer Res., 34:1735, 1993.Forbes, I. J., Zalewski, P. D.. Grannakis, C., Cowled, P. A. Induction of apoptosis inchronic lymphocytic leukemia cells and its prevention by phorbol diester. Exp. CellRes., 198: 367-371, 1992.
Herbert. J. M.. Augereau, J. M., Gleye. J.. Maffrand, J. P. Chelerylhrine is a potentand specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun., 172:993_999, 1990
Hidaka, H., Inagaki, M.. Kawamoto, S., Sasski, Y. Isoquinolinc sulfonamidcs, novelpotent inhibitors of cyclic nucleotide protein kinase and protein kinase C. Biochemistry, 23: 5036-5041, 1984.
Takahashi. L, Nakanishi, S.. Hobayashi. E., Nakano, H., Suzuki, K., Tamaoki, T.Hypericin and pseudohypericin specifically inhibit protein kinase C: possible relationto their antirctroviral activity. Biochem. Biophys. Res. Commun., 765: 1207-1212,1989.Pelosin. J-M.. Keramidos, M.. Souvignet. C., and Chambaz, E. M. Differential
inhibition of protein kinase C subtypes. Biochem. Biophys. Res. Commun., 169:1040-1048, 1984.
1713
Research. on February 24, 2020. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from

INDUCTION OF APOPTOSIS BY PKC INHIBITORS
27. Kohayashi. E. et al. Calphoslin C (UCN-1028C). a novel microhial comptiund andhighly potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res.Commun.. 759: 548-553. 1989.
28. Schachtele, C., Seifen, R., and Osswald. H. Stimulus-dependent inhibition of platelet 48.aggregation by the protein kinase C inhibitors polymixin. H-7, and staurosporine.Biochem. Biophys. Res. Commun., I5I: 542-547, 1988.
29. Nakadatc, T.. Jeng, A. Y., and Blumherg. P. M. Comparison of protein kinase C 49.functional assays to clarify mechanisms til inhibitor action. Biochem. Pharmacol., 37:1541-1542, 1988.
3«.Gallcgher. R.. Collins, S.. Trujillo, J., McCredie. K., Ahearn, M., Tsai, S., Metzgar, 50.R.. Aulakh. G., Ting. R., Ruscelli. F.. and Gallo. R. Characterization of the continuousdifferentiating myeloid cell line (HL-60) from a patient with acute promyelocyticleukemia. Blood,'54: 713-733, 1979.
31. Bruns. R. F.. Miller. F. D.. Mcrriman, R. L. Howbert. J. J.. Heath. W. F.. Kobayashi. E.. 51.Takahashi. I.. Tamaoki. T., and Nakano. H. Inhibition of protein kinase C by calphostinC is light-dependent. Biochem. Biophys. Res. Commun.. 176: 288-293, 1991.
32. Cesaronc. C., Bolognesi, C., and Santi, L. Improved microfluorometric DNA deter- 52.minalion in biological material using Hoechst 33258. Anal. Biochem.. 100: 118-197,
1979.33. Jams. W. D.. Gcwirtz, D. A.. Povirk, L. R.. Turner, A. J., Traylor. R. S.. Pettit, G. R.. 53.
and Grant. S. Modulation of 1-ß-D-arabinofuranosylcytosine-induced apoptosis inhuman promyclocytic leukemia cells by bryostatin 1 and other activators of proteinkinase C. Biochem. Pharmacol.. in press. 1994.
34. Grant, S., Boise, L. L., Westin, E. L., Howe, C. S. W., Pettit. G. R.. Turner, A. J., and 54.McCrady. C. A. In vitro effects of bryostatin 1 on the metabolism and cytotoxicity ofl-/3-[>-arabinofuranosylcytosine in human leukemia cells. Biochem. Pharmacol., 42: 55.853-867, 1991.
35. totem. J.. Sachs, L. Regulation by be1-2. c-myc, and p53 of susceptibility to induction ofapoptosis by heat shock and cancer chemotherapy compounds in differentiation-compe- 56.tent and defective myeloid leukemia cells. Cell Growth & Differ.. 4: 41-47. 1993.
36. Miyashita. T.. and Reed, J. C. Bcl-2 oncoprotein blocks chemotherapy-inducedapoptosis in a human leukemia cell line. Blood, NI: 151-157, 1993.
37. Chirgwin. J.. Przybala. H.. McDonald. R.. and Rutter. W. Isolation of biologically 57.active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 18:5294-5299. 1979.
38. Boise. L. H.. Grant, S.. and Westin. E. H. Altered expression of c-rnyb in a subclone ofHL-60 that exhibits reversible differentiation. Cell Growth & Differ.,.?.- 53-61, 1992.
39. Barry. M. A.. Reynolds. J. E.. and Eastman. A. Etoposide-induced apoptosis in HL-60 58.
human leukemia cells is associated with intracellular acidification. Cancer Res., 5.ÃŽ:2349-2357, 1993.
40. Nakcmura, M.. Sakakaki, Y.. Watanabe, N., and Takagi, Y. Purification and charac- 59.tcri/ation of the Ca2 *-/Mg2 '-dependent endodeoxyribonucleasc from calf thymus
chromalin. J. Biochem., W: 143-152, 1981.41. Gunji. H.. Hass, R., and Kufe, D. Internucleosomal DNA fragmentation during 60.
phorbol ester-induced monocytic differentiation and G,,-G| arrest. J. Clin. Invest., 89:954-960, 1992.
42. Martin, S. J., Beardley, J. G.. and Cotter. T. G. HL-60 cells induced to differentiation 61.toward neutrophils subsequently die via apoptosis. Clin. Exp. Immunol.. 79: 448-
453, 1990.43. Griffin. J.. Munroe. D., Major. P.. and Kufe. D. Induction of differentiation of human 62.
myeloid leukemia cells by inhibitors of DNA synthesis. Exp. Hematol., 10: 774-781.1982.
44. McConkey. D. J.. Arrenuis, S.. and Jondal. M. Agents that elevate cAMP stimulate 63.DNA fragmentation in thymocytes. J. Immunol., 145: 1227-1230. 1990.
45. Gorczyca. W.. Gong, J., Ardelt, B.. Tráganos. F.. and Darzunkiewicz. Z. The cellcycle related differences in susceptibility of HL-60 cells to apoptosis induced by 64.various anlitumor agents. Cancer Res.. .5.Î:3186-3192. 1993.
46. Tráganos. E.. Knulti-Hotz. J.. Hotz, M., Gorczyca. W., Ardelt. B.. and Darzunkiewicz. Z. The protein kinase C inhibitor H7 blocks normal human lymphocyte 65.stimulation and induced apoptosis of both normal lymphocytes and MOLT-4 leukemia cells. Int. J. Oncol., 2: 47-59. 1993.
47. Falcicri. E.. Martelli. A. M., Bareggi. R.. Cataldi, A., and Cocco, L. The protein
kinase C inhibitor staurosporine induces morphological changes typical of apoplosisin MOLT-4 cells without concomitant DNA fragmentation. Biochem. Biophys. Res.Commun.. 193: 19-25, 1993.DeWitte. P.. Agostinis. P.. VanLint. J.. Merlevede, W., and Vandenheede, J. R.Inhibition of epidermal growth factor receptor tyrosine kinase activity by hypericin.Biochem. Pharmacol.. 46: 1929-1936, 1993.Matsui. T.. Nakao, Y., Koizumi, T., Katakami, Y., and Fujita, T. Inhibition of phorbolester-induced phenotypic differentiation of HL-60 cells by l-(isoquinolinylsulfonyl)-2-methylpiperazine, a protein kinase inhibitor. Cancer Res., 46: 583-587, 1986.
Merrill. A. H.. Sereni. A. M., Stevens, V. L., Hannun, Y., Bell, R. M., and Kinjade.J. M. Inhibition of phorbol diester-dcpendent differentiation of human leukemic(HL-60) cells by sphingosine and other long-chain sphingoid bases. J. Biol. Chem.,261: 12610-12615. 1986.
Solary. E., Bctrand, R.. Kohn. K. W.. and Pommier. Y. Differential induction ofapoptosis in undifferentiated and differentiated HL-60 cells by DNA topoisomerasesI and II inhibitors. Blood, 81: 1359-1368, 1993.
Nunez. G., London. L.. Hockenberg. D.. Alexander. M.. McKearn, J., and Korsmeyer,S. S. Deregulated bcl-2 gene expression selectively prolongs survival of growthfactor-deprived hemopoietic cell lines. J. Immunol., 144: 3607-3610, 1990.McDonnell. T. J.. Tronscosco. P., Brisbay. S. M., Logothetis, C.. Chung, L. K. W.,Hsieh, J-T.. Tu. S. M., and Campbell. M. L. Expression of the protooncogene bcl-2in the prostate and its association with the emergence of androgen-independentprostate cancer. Cancer Res., 52: 6940-6944, 1992.Miyashita. T.. and Reed, J. C. Bcl-2 oncoprotein blocks chemotherapy-inducedapoptosis in a human leukemia cell line. Blood, 81: 151-157, 1993.Walton. M. L. Whysong. D., O'Connor, P. M.. Hockcnbery, D.. Korsmeyer. S. J., and
Kohn. K. W. Constitutive expression of human hcl-2 modulates nitrogen mustard andcamptothccin induced apoptosis. Cancer Res., 53: 1853-1861, 1993.
Bhalla. K.. Ihrado. A. M.. Tourkina. E.. Tang, C.. Maloney. M. E.. and Huang. Y.Taxol induces internucleosomal DNA fragmentation associated with programmedcell death in human myeloid leukemia cells. Leukemia (Baltimore), 7: 563-568.
1993.Bhalla. K.. Tang, C., Ibrado. A. M., Grant, S., Tourkina, E., Holladay, C., Hughes, M.,Mahoney. M. E.. and Huang, Y. Granulocyte-macrophage colony stimulating factor/interleukin-3 fusion protein (PIXY 321) enhances high dose ara-C induced pro
grammed cell death or apoptosis in human myeloid leukemia cells. Blood, 80:2883-2890. 1992.May. W. S.. Tyler. P. G.. Armstrong. D. K.. and Davidson. N. E. Role for serinephosphorylation of Bcl-2 in an anti-apoptotic signaling pathway triggered by IL-3,erythropoictin, and bryostatin 1. Blood, 82 (Suppl.l): 1738, 1993.Brown. D. G.. Sun, X-M.. and Cohen, G. M. Dcxamethasone-induced apoptosis
involves cleavage of DNA to large fragments prior to internuceosomal fragmentation.J. Biol. Chem.. 26«:3037-3039, 1993.
Rosoff, P. M.. Stein. L. F., and Cantley. L. C. Phorbol dieslers induce differentiationin a pre-B-lymphocyte cell line by enhancing Na2+-/H +-exchange. J. Biol. Chem.,
259: 7057-7060, 1984.Alnemri. E. S.. and Litwack. G. Activation of internuclcosomal DNA cleavage inhuman CEM lymphocytes by glucocorticoids and novobiocin. J. Biol. Chem.. 265:17323-17333. 1990.
Jarvis. W. D.. Kolcsnick. R. N.. Fornari. F. A.. Traylor. R. S.. Gewirtz, D. A., andGrant. S. Induction of apoptotic cell death by sphingomyelinase and ceramidc in avariety of mammalian cells. Proc. Nati. Acad. Sci. USA, 91: 73-77, 1994.
Dive, C., Evans, C. A., Lynch, P. J., Musk, P., and Whetton, A. D. The role of proteinkinase C in the suppression of apoptosis by v-abl in hematopoietic cells. Proc. Am.Assoc. Cancer Res.. 34: 516. 1993.Sato. W.. Yusa, K., Naito. M.. and Tsurro. T. Staurosporine, a potent inhibitor ofc-kinase. enhances drug accumulation in multidrug-resistant cells. Biochem. Biophys.Res., 173: 1252-1257, 1990.Laredo. J.. Demur. C., Muller. C.. Saivin. S., Cassar, G.. Bousquet, C., Dastugue, N..Jeffrezou J-P.. Colomhres, O., and Laurent. G. Effects of H7 and staurosporine on theproliferation and self-renewal of acute myeloid leukemia progenitors. Leukemia(Baltimore), 7: 813-820. 1993.
1714
Research. on February 24, 2020. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from

1994;54:1707-1714. Cancer Res W. David Jarvis, Amy J. Turner, Lawrence F. Povirk, et al. Pharmacological Inhibitors of Protein Kinase CHL-60 Human Promyelocytic Leukemia Cells by Induction of Apoptotic DNA Fragmentation and Cell Death in
Updated version
http://cancerres.aacrjournals.org/content/54/7/1707
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/54/7/1707To request permission to re-use all or part of this article, use this link
Research. on February 24, 2020. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from