Embed Size (px)
Citation preview

Implications of Local Puumala Hantavirus Genetics and Epidemiology for Diagnostics and
Vaccine Development


UMEÅ UNIVERSITY MEDICAL DISSERTATIONS New Series No 964 – ISSN 0346-6612 - ISBN 91-7305-878-5
From the Department of Clinical Microbiology, Division of Virology Umeå University, Umeå, Sweden
Implications of Local Puumala Hantavirus Genetics and Epidemiology for Diagnostics and
Vaccine Development
Patrik Johansson
Umeå 2005

Copyright © 2005 Patrik Johansson ISBN 91-7305-878-5
Printed in Sweden by
Solfjädern Offset AB, Umeå, 2005

To my family

Abbreviations used in this thesis
a.a. Amino acid bp Base pair cRNA Complementary RNA CTL Cytotoxic T-lymphocyte DNA Deoxyribonucleic acid ELISA Enzyme-linked immunosorbent assay ER Endoplasmic reticulum FRNT Focal reduction neutralization test GC Glycoprotein from the C-terminal half of the polyprotein. (also known as G2) GN Glycoprotein from the N-terminal half of the polyprotein. (also known as G1) GPC Glycoprotein precursor HFRS Hemorrhagic fever with renal syndrome HLA Human leukocyte antigen, see MHC HPS Hantavirus Pulmonary Syndrome IFA Indirect immunofluorescence assay IFN Interferon IgA Immunoglobulin A IgE Immunoglobulin E IgG Immunoglobulin G IgM Immunoglobulin M Kb Kilo base KDa Kilo dalton L The large sized viral genome segment M The medium sized viral genome segment mab Monoclonal antibody MHC Major histocompatibility complex mRNA Messenger RNA N Nucleocapsid protein NE Nephropathia epidemica nt Nucleotide ORF Open reading frame PCR Polymerase chain reaction PTO Phosphorothioate nucleotide RdRp RNA dependent RNA polymerase, the viral polymerase RNA Ribonucleic acid rRNA Ribosomal RNA RT-PCR Reverse transcriptase PCR S The small sized viral genome segment TNF Tumor necrosis factor VLP Virus-like particle vRNA Viral RNA Abbreviation of virus names can be found in Table 1
vi

Table of contents Abbreviations used in this thesis vi Abstract ix Papers in this thesis xiii Sammanfattning på svenska xv Short introduction to hantaviruses and their diseases 1 The nucleocapsid protein 6 The glycoproteins 12 The RNA dependent RNA polymerase 16 The replication cycle 17 Hantaviruses and their hosts 22 Hantavirus caused diseases 31 Diagnostics of NE 34 The immune system 39 Vaccines 43 Aims of this thesis 54 Results and discussions 55 Paper I 55 Paper II 62 Paper III 65 Paper IV 71 Conclusions 77 Acknowledgements 78 References 79
vii

Patrik Johansson 2005
ignoramus et ignorabimus
viii

Abstract
Abstract
Puumala virus is a member of the hantavirus genus in the Bunyaviridae family.
Hantaviruses are enveloped by a lipid bilayer and possesses a tripartite single
stranded RNA genome with negative polarity. The hantaviruses encode four
proteins: a nucleocapsid protein (N), two membrane spanning glycoproteins (GN
and GC) and a RNA dependent RNA polymerase (RdRp). Most hantaviruses are
borne by rodent hosts, and found in most parts of the world. Hantaviruses cause
two forms of human disease, haemorrhagic fever with renal syndrome (HFRS) on
the Eurasian continents, and hantavirus pulmonary syndrome (HPS) in the
Americas. The majority of human hantavirus infections occur in Europe and
Eastern Asia, and the viruses are mainly transmitted to humans by inhalation of
aerosolized excreta from infected rodents.
Human Puumala virus infection results in nephropathia epidemica (NE), a mild
haemorrhagic disease with a mortality of about 0.1%. NE is the the second most
prevailing serious febrile viral infection in Northern Sweden, after influenza. The
yearly incidence is approximately 20 cases per 100 000 inhabitants in Northern
Sweden, and the seroprevalence, in this region reaches 18% for risk groups such as
forestry workers and farmers. It is thus of importance to have a good
understanding of the epidemiology and genetics of these viruses for the
development of new diagnostic methods and for future vaccine development.
The objectives of this thesis are: to characterize a local Puumala virus isolated from
a human patient; to characterize the genetic variability among local Puumala viruses
and their host, the bank vole, Clethrionomys glareolus; to use this information to
develop and evaluate genetic vaccines and to develop diagnostic and
epidemiological tools.
In this thesis, the complete genetic sequence of Puumala Umeå/hu was established,
and genetic analysis revealed that this human isolate clusteres together with other,
bank vole derived, Puumala viruses from that region. Two nucleotides that
ix

Patrik Johansson 2005
interfere with the double stranded RNA, panhandle structure were found in the S-
segment. These mismatches resulted in structural changes in this region, but did
not affect the biological activity of the virus. The N, GN and GC proteins were
expressed as full-length proteins and as overlapping peptides in mammalian cells.
Mutational mapping of the potential glycosylation sites identified four functional
N- and one O-linked glycosylation sites, in the GN and GC proteins.
Genomic analysis of local Puumala viruses and their individual rodent host,
Clethrionomys glareolus, from six different locations was performed. There were no
changes in the viral sequence over the five years that spanned this study, or over
short distances within one capture location. Interestingly, there was no correlation
between viral sequences and proximity of locations. While there could be
significant variation in viral genome sequences over relatively short distances, it
could also be relatively stable over longer distances. In earlier studies the degree of
such changes in the virus genome has been correlated with a similar degree of
changes in the rodent host. However, the phylogenetic analysis of the bank voles in
this study did not show evidence of this or any clear genetic distinction among
bank voles from different capture locations.
Based on the viral sequence information, PUUV-specific, PCR-generated linear
DNA vaccines were developed and tested in mammalian cells and on Balb/c mice.
We demonstrated that appropriate modifications in the flanking regions of these
linear PUUV-specific DNA constructs could protect these fragments against
degradation and is important for development of rapid and long lasting immune
responses. This is probably effected by prolonged biological half life of the
constructs within the cells. A nuclear localization signal peptide further improved
the immune response.
We also designed and fabricated a cDNA microarray for detection and
identification of hantaviruses. The design consisted of more than 2000 genetic
probes of the S and M-segments from twelve hantavirus isolates of six different
hantaviruses. This cDNA microarray was shown to be able to discriminate among
x

Abstract
different hantaviruses as well as individual strains of Puumala virus. Furthermore, it
was able to handle complex samples that were not possible to analyze using
conventional methods.
xi

Patrik Johansson 2005
xii

Papers in this thesis
Papers in this thesis
The thesis is based on the following papers. I. Johansson P, Olsson M, Lindgren L, Ahlm C, Elgh F, Holmström A, Bucht G. Complete gene sequence of a human Puumala hantavirus isolate, Puumala Umeå/hu: sequence comparison and characterisation of encoded gene products. Virus Res. 2004. 105(2):147-55. II. Johansson P, Olsson G, Ahlm C, Juto P, Bucht G, Elgh F. Genetic variability among Puumala viruses and bank voles in an endemic area (Northern Sweden). Manuscript. III. Johansson P, Lindgren T, Lundström M, Holmström A, Elgh F, Bucht G. PCR-generated linear DNA fragments utilized as a hantavirus DNA vaccine. Vaccine. 2002. 20(27-28):3379-88. IV. Nordström H, Johansson P, Li QG, Lundkvist Å, Nilsson P, Elgh F. Microarray technology for identification and distinction of hantaviruses. J. Med. Virol. 2004. 72(4):646-55.
xiii

Patrik Johansson 2005
xiv

Sammanfattning på svenska
Sammanfattning på svenska
Puumalavirus tillhör en grupp av zoonotiska virus i familjen Bunyaviridae som kallas
hantavirus. Alla virus tillhörande denna familj har en arvsmassa som består av tre
olika RNA segment med negativ polaritet. Denna arvsmassa är bunden till ett
nucleokapsidprotein och är tillsammans med ett viralt RNA-beroende RNA-
polymeras, inneslutna i ett lipidmembran, i vilket det sitter två virala transmembran-
proteiner. De senare binder till en cell som därefter blir infekterad. Hantavirus kan
ge upphov till två olika sjukdomar hos människa. Blödarfeber med njurengagemang
(hemmorhagic fever with renal syndrome, HFRS) i Europa och Asien och
hantavirus lungsyndrom (hantavirus pulmonary syndrome, HPS) i Amerika. De
flesta hantavirus har gnagare som värddjur och människan blir sjuk av att inandas
damm som innehåller virus i intorkat sekret från infekterade gnagare. I Europa och
Asien insjuknar årligen 60 000-150 000 personer i hantavirusinfektioner. Dessa
sjukdomar har en dödlighet som varierar mellan 0.1 till 20%, beroende på vilket
hantavirus man är infekterad av. I Amerika smittas betydligt färre personer, och
HPS ger en hög dödlighet (över 40%).
Värddjuret för Puumalavirus är skogssorken, Cletrionomys glareolus, som återfinns i
större delen Europa. Puumalavirus ger en mild variant av HFRS, kallad
nephropathia epidemica (NE) eller sorkfeber. Denna sjukdom har en dödlighet på
cirka 0.1%. Trots att skogssorken finns i hela landet rapporteras fall av sorkfeber
nästan bara norr om Limes Norrlandicus, den kulturhistoriska norrlandsgränsen, en
S-formad linje som sträcker sig från Gävle längsmed Dalälven, genom Västmanland
och ner till Dalsland. Det finns två kända populationer av skogssork i Sverige som
möter varandra i en kontaktzon norr om Sundsvall. Man tror att detta beror på att
sorkarna återinvandrade från två olika håll när glaciärerna smälte efter den sista
istiden, för 6-10 000 år sedan. De två typerna av skogssork kan särskiljas genom
studier av deras mitokondrie-DNA. De Puumalavirus som de olika
sorkpopulationerna bär skiljer sig också och tros ha följt sorkarna under deras
xv

Patrik Johansson 2005
vandring. I norra Sverige varierar antalet sorkar cykliskt med toppar var 3-4 år.
Insjuknandefrekvensen av sorkfeber följer också dessa cykler. I Sverige rapporteras
årligen cirka 150-500 fall med en årlig incidens av ca 20 fall per 100 000 invånare
för norrlandslänen (1998 var ett toppår med 562 rapporterade fall). Västerbottens
och Norrbottens län, som har flest fall av sorkfeber, har en årlig incidens på ca
35/100 000 invånare (67/100 000 för 1998). Antalet infekterade personer är
mycket högre än det registrerade antalet sjuka eftersom man kan se att bara ett fall
av åtta diagnostiseras och rapporteras. Det är också så att vissa yrkergrupper, som
skogsarbetare och bönder, vilka har en seroprevalens upptill 18%, är extra utsatta
för smitta.
Målet med denna avhandling har varit att i detalj karakterisera ett unikt
Puumalavirus, Puumala Umeå/hu, som isolerats från en patient i Umeå. Andra mål
har varit att undersöka den genetiska variabiliteten bland lokala Puumalavirus och
dess värddjur skogsorken och att använda resulteranade informationen för att
förbättra genetisk diagnostik och öka den epidemiologiska kunskapen om sorkfeber
samt att utveckla och utvärdera nya typer av genetiska vacciner mot sorkfeber.
Den fullständiga genetiska sekvensen för Puumala Umeå/hu har bestämts och en
genetisk analys visade att viruset är mycket nära besläktat med andra Puumalavirus
från näraliggande områden. Virusets tre stukturella proteiner producerades i
cellkultur och två typer av sockerstrukturer på membranproteinerna kartlades.
Analys av lokala Puumalavirus och deras värdjur, skogsorken, från sex områden i
Västerbottens kustland, visar att virusets arvsmassa kan skilja sig markant mellan
två närliggande fångstplatser, medan det kan vara relativt oförändrat över längre
avstånd. Tidigare studier har visat att skillnaderna i virusets arvsmassa har
samvarierat med liknande skillnader i värdjuret. Den genetiska analysen av
skogssorkarna visade dock väldigt få genetiska skillnader mellan djuren. Det var
heller ingen genetisk skillnad hos virusen över de fem år som studien varade eller
över korta avstånd inom en fångslokal.
xvi

Sammanfattning på svenska
Linjära DNA-vacciner ämnade att skydda mot Puumalavirusinfektion, framtagna
med s.k. PCR-teknik, utvecklades och undersöktes, dels i cellkultur och därefter i
möss. Vi visade att ett snabbt och långvarigt immunsvar erhölls när de linjära
DNA-fragmenten hade skyddats i flankerna mot DNA-nedbrytande enzymer. När
en kärnlokaliseringssignal kopplades till dessa DNA-fragment förbättrades DNA-
vaccinets egenskaper ytterligare.
Slutligen utvecklade och skapade vi ett hantavirusspecifikt cDNA-microarraychip
och visade att man med hjälp av cDNA-microarrayteknik kan skilja mellan olika
hantavirus och även mellan olika virus i Puumalavirusgruppen. Microarray-tekniken
kan också hantera komplexa prov som inte kan analyseras med konventionella
metoder.
xvii

Patrik Johansson 2005
xviii

Short introduction to hantaviruses and their diseases
Short introduction to hantaviruses and their diseases
Hantaviruses belong to the family of Bunyaviridae. In nature, hantaviruses are
maintained in persistently infected rodents (the only known exception is the
Thottapalayam virus that is carried by an insectivore). The virus is transmitted to
humans through inhalation of infectious material containing the virus, e.g. rodent
urine, excreta or by direct physical contact with infected animals [1]. Hantaviruses
cause hemorrhagic fever with renal syndrome (HFRS) on the Eurasian continents,
and hantavirus pulmonary syndrome (HPS) on the American continents. Currently
four hantavirus serotypes are associated with HFRS, with varying morbidity and
mortality. HFRS cases infected with Hantaan (HTNV) or Seoul (SEOV) viruses are
mainly found in Asia, whereas Dobrava (DOBV) and Puumala (PUUV) viruses
circulate primarily in Europe. A large variation in the mortality has been observed
(PUUV < 0.1%, SEOV<1%, HTNV=5-10% and DOBV<20% [2-4]) among the
60 000-150 000 yearly HFRS cases, a great majority of which are from China [1, 5,
6]. HPS, which can be divided into two variants [Sin Nombre (SNV) and New
York (NYV) virus (prototype) and Bayou (BAYV), Black Creek Canal (BCCV) and
Andes virus (ANDV) (renal variants)], often have a more severe outcome (both
variants show mortality rates of > 40%) [7, 8], but does not infect as many people,
with only a total of 384 cases reported, in the United States, through 1993-2004 [9].
ANDV, on the other hand causes around 200-300 cases annually in areas of South
America [10]. In Europe three hantavirus serotypes are represented. PUUV which
is endemic in many parts of Northern, Central and Eastern Europe [11], DOBV,
found in many parts of Central and Eastern Europe [12], and SEOV that has
recently been found in European rats [13]. In Scandinavia, the only hantavirus
described so far is the PUUV, which is of great concern in Northern Sweden where
it is second to the influenza virus, the most prevailing serious viral infection. There
are about 20 cases per 100.000 people and year and a seroprevalence of up to 18%
for risk groups such as forestry workers and farmers has been reported [14, 15].
The PUUV is carried by the bank vole, Clethrionomys glareolus, which is distributed in
1

Patrik Johansson 2005
most part of Europe, from Britain to the Urals, and from the far north of
Scandinavia down to the northern parts of the Mediterranean regions [16].
History of the diseases
Medical records from a hospital in Vladivostok, indicate that HFRS could be traced
back to 1913 in Russia [17]. In Sweden, in the early 1930´s a disease that was later
named Nephropathia Epidemica (NE) was described independently by two
Swedish physicians, G. Myhrman in Östersund and S.G. Zetterholm in Skellefteå
[18, 19]. Later, records from the Second World War clearly indicated that Finnish,
Russian, German and Japanese soldiers suffered from HFRS [1, 20-22]. In the hunt
for the mysterious pathogen, some gruesome events took place. Japanese soldiers
conducted experiments on Chinese prisoner “volunteers” while trying to develop
the disease-causing agent into a biological weapon [5, 23], while the Soviets infected
“psychiatric patients” in need of “pyrogenic treatment” with the disease [24, 25].
Although the viruses, and the subsequent diseases that they are responsible for, had
been around for a long time, not much was published until UN troops, during the
Korean War 1950-1953, fell ill from a, then unknown and deadly disease called
Korean Hemorrhagic fever. Both UN, South Korean and North Korean soldiers
suffered from the disease that was later confirmed to be HFRS [22, 24]. The disease
causing agent remained unknown until 1976, when Lee HW and co-workers
managed to immunostain lungs from black-striped field mouse (Apodemus agrarius)
but not lungs from other rodents, with sera from patients that had suffered from
Korean Hemorrhagic fever [26]. In 1978, Lee and co-workers isolated the virus
from an immunofluroescent positive Apodemus agrarius rodent by injecting a
suspension of the lung tissue to uninfected Apodemus agrarius rodents. The virus
they isolated was named Hantaan virus (HTNV) after the Hantaan River that flows
close to the demilitarized zone between South and North Korea. [27]. Later, Lee
and others successfully isolated another hantavirus from rats (Rattus norvegicus and
2

Short introduction to hantaviruses and their diseases
Rattus rattus), and this virus was named Seoul virus (SEOV), after the capital city of
South Korea [28]. In 1981, the HTNV was adapted for growth in cell culture.
Human alveolar epithelial cells were incubated with lung tissues of infected
Apodemus agrarius [29]. By using similar techniques, the causative agent for NE was
identified in the lungs of bank voles captured in or near the Finnish village
Puumala, which also gave its name to the NE causing virus, now called Puumala
virus (PUUV) [30]. In 1984, two groups independently succeeded in adapting
PUUV to cell culture, but this time in kidney epithelial cells of African green
monkey kidney (Vero E6 cells), which until today is the most commonly used cell
line for culturing hantaviruses [31, 32].
In 1992, the Dobrava virus (previously named Belgrade virus) was isolated from
yellow-necked mouse, Apodemus flavicollis, as well as from patients suffering from
HFRS in Yugoslavia [2, 33]. Soon after, the first American hantavirus, the Prospect
Hill virus (PHV), was isolated from the meadow vole, Microtus pennsylvanicus, in
Frederick, Maryland, USA [34]. Yet another virus, the Thailand virus (THAIV) was
isolated in Thailand from the greater bandicoot rat, Bandicota indica [35].
Although hantaviruses had been found in rodents in the USA, human illness caused
by hantaviruses had only been observed on the Eurasian continents in 1993.
During that year, young, fit people from the Four Corners region in the south west
of USA, became seriously ill and died soon after, in what was quickly recognized as
a new hantavirus caused disease [36]. This “new” virus, named Sin Nombre virus
(SNV), affected the victim’s heart and lungs in a much more severe way than
hantaviruses of the old world. This virus has been isolated from the deer mouse,
Peromyscus maniculatus. The illness caused by SNV is now called Hantavirus
Pulmonary Syndrome (HPS) or Hantavirus Cardio Pulmonary Syndrome (HCPS).
The discovery of this new deadly disease became a starting point for a new era of
hantavirus research, especially in the Americas where several new hantaviruses were
found and characterized soon after. However, the most notably viruses of the new
world are the SNV in North America and Andes in South America. ANDV,
3

Patrik Johansson 2005
isolated from the long-tailed pygmy rice rat, Oligoryzomys longicaudatus, is the only
hantavirus for which human to human transmission has been reported [37].
Interestingly, it was not realized until 1985 that the Thottapalayam virus (TPMV)
was the first hantavirus to be isolated. It was isolated from the spleen of an
apparently healthy shrew (Suncus murinus) in India already in 1964 [38]. However, at
that time, it was not possible to identify the virus. The virus was re-discovered in
1985, this time in China, by intra-muscular passaging in Apodemus agrarius rodents
[39-41].
The Bunyaviridae virus family
The Bunyaviridae family is one of the largest groups of animal and plant viruses and
comprises more than 300 recognized viruses. The family, based on antigenic
relationships, is divided into five genera (Bunyavirus, Phlebovirus, Uukuvirus,
Nairovirus and Hantavirus) with the first four genera being typical arthropod borne
viruses. The Hantaviruses are the only viruses in the Bunyaviridae family that are not
spread by arthropods [42].
The Bunyaviridae virion is spherical and the diameter is usually between 70-200 nm,
and with a mean diameter of 122 nm [43, 44]. The variation in size could be
explained by the variation of the number of genome segments enclosed in the
virion [45]. The hantavirus particle contain three genome segments, large (L),
medium (M) and small (S), of negative single stranded RNA. The three RNA
segments are surrounded by subunits of the N-protein, and form circular coils
complexed with the RNA dependent RNA polymerase (RdRp). These structures
are enveloped in a bilipid membrane, derived from the host cell ER-Golgi complex
from which the two heterodimeric viral glycoproteins are projecting out. In
electron microscopy (EM) this is visible as a well-defined, grid-like, arrangement of
6-7 nm long spikes (Fig. 1, 2) [43, 44, 46-48].
4

RdRp
N-proteintrimer
GN
GC
Glycosylations
Short introduction to hantaviruses and their diseases
Figure 1. EM picture of Hantaan virus. Amplification is 135 000 times [49].
Figure 2. Schematic picture of the composition of a hantavirus.
In hantaviruses, the small segment (S) is about 1.8 kb and encodes the nucleocapsid
protein (N), and in some bunyaviruses, also a non-structural protein (NSs). The
medium segment (M) (~3.6 kb) encodes a polyprotein that is processed into the
two glycosylated, membrane spanning proteins GN and GC (also known as G1 and
G2). The large segment (L) (~6.4 kb) encodes the viral RNA dependent RNA
polymerase (RdRp) [50]. The 5’ and 3’ ends of each of the three segments are
5

Patrik Johansson 2005
thought to hybridize with each other and form a panhandle structure, previously
shown also for other Bunyaviridae viruses (Fig 3) [51]. These conserved panhandle
structures vary in length among the three segments, being more elongated for the
longer segments. This could compensate for the lower possibility of the longer
fragments ends to find its complementary end in comparison to the shorter
segments, giving them a similar probability of circularization despite the difference
in length [52].
Figure 3. EM picture of Uukuniemi virus RNA. Notice the circular structures, indicating panhandle formation [51].
The nucleocapsid protein
The approximately 50 kDa hantavirus nucleocapsid protein (N), (429-433 a.a.), is
the most abundant protein, both in the virus infected cell and in the virus particle
where the copy number of the N protein is in the order of 3x107 molecules per cell
[53, 54].
The N protein has been reported to have many different functions. In the virus
particle, the primary role is to associate with the viral RNA (vRNA) and to form
the ribonucleoprotein complex, a stable structure necessary for the RNA to be
6

The nucleocapsid protein
transcribed by the virus encoded RNA dependent RNA polymerase (RdRp), once
internalized into the cell cytoplasm [55-60]. The N protein is also thought to be
involved in the interactions between the glycoproteins and the ribonucleoprotein,
most likely by binding to a YRTL-motif in the cytoplasmic tail of GN when forming
the virion [61, 62]. Furthermore, the N protein is assumed to facilitate the binding
between the RdRp to the ribonucleoprotein [54].
In the infected cell, the major role of the N protein is to bind to the vRNA and
cRNA, which is needed for the RdRp to be able to perform replication and
transcription. Another important function is to regulate the transcription of the
different forms of viral RNA’s (vRNA to mRNA’s, or to cRNA and back to
vRNA) [63]. Beside that, the N protein has been reported to have a role as a
chaperon during the folding of the genome segments into the typical panhandle
structure [64], and also in the binding of the newly synthesized viral
ribonucleoproteins to actin filaments that might facilitate transportation of the viral
components [65].
It is thought that the N protein is expressed in the cytosol where it can be observed
within two hours after infection. The protein is retained in the cytosol for 12 hours
post infection, possibly involved in viral RNA synthesis and interacting with actin
filaments [66]. Thereafter, the N protein is found to be transported, and localised at
the perinuclear structures surrounding the Golgi [53, 66], where the viral replication
is assumed to take place. The ribonucleoproteins are later assembled into virions in
the Golgi or at the plasma membrane [44, 65].
The main interaction between the N protein and the vRNA is thought to be
mediated by a homo-trimeric form of the molecule, formed through intermolecular
interactions between the amino- and carboxyl-terminal parts of three N monomers,
(Fig. 4) [67], and this trimeric form of the N-protein is more stable than the mono-
and dimeric forms. The trimers has a high specific affinity for the panhandle
structure of the genome segments, while the mono- and dimeric forms show more
unspecific affinities for viral RNA structures in general [68]. It has been shown that
7

Patrik Johansson 2005
the N-protein contains a conserved, central region that preferentially binds to the 5’
terminal HTNV vRNA [63].
Although it has not been shown exactly how, and which molecular form of N-
protein that binds to the vRNA first (Fig 5), there are indications that the trimer
binds initially to the vRNA panhandle structure in an entropy-driven process, and
that the vRNA’s primary sequence and secondary structures are of importance for
the binding. The monomeric form is then recruited by its affinity for the already
bound trimer and the RNA motifs itself [64].
One interesting observation is that in a mixed population, where different
molecular forms of N molecules exist simultaneously, the binding affinity between
the trimer and the RNA is lowered. This phenomena could be explained by a more
competitive binding of N monomer- and dimer-binding to the vRNA structures
compared to the trimer, or perhaps structural of property changes of the trimeric
form effected by the monomers or dimers [68]. These and other observations
suggest that the trimer binding to the panhandle structure of the RNA resulting in
encapsulation is complicated, and perhaps not the only biological function of the
N-protein/RNA interactions.
The N-protein has been reported to bind to the cRNA. The plus-stranded
complementary RNA (cRNA), which also can form similar panhandle structures as
those of the vRNA, is also encapsidated by the N protein [69], however with a
lower affinity, than that observed between the N trimer and the vRNA panhandle
[64]. In contrast to vRNA and cRNA, mRNA is not encapsidated [70]. This is
logical since the vRNA and cRNA is supposed to be handled by the viral
polymerase, while the mRNA is translated by cellular ribosomes.
Other interesting observations are that the N-protein binds to the YRTL-motif in
the cytoplasmic tail of GN [62], and that production of N-protein and the
glycoproteins in a cell is enough for the formation of virus-like particles [61].
Finally, since the N-protein is needed for transcription of both the vRNA and
8

The nucleocapsid protein
cRNA, it can be assumed that there is interaction between the N-protein and the
RdRp [62].
A number of publications have suggested that the N-proteins have interactions
with a number of non-viral components. One interesting observation is that, the N
protein is modified by the SUMO-1 (Small Ubiquitin Modifier 1 also called sentrin)
protein. The function of sumoylation is not clearly understood, but it has been
suggested to have a role in protein transportation and targeting to, and formation
of, certain subnuclear structures such as nuclear bodies (NBs). Other speculations
are that a variety of processes including the inflammatory response, including the
response to TNF-α, are regulated through sumoylation [62, 71-73]. Intriguingly the
N-protein also bind to another protein associated with the NBs, the DAXX
protein, a Fas mediated apoptosis enhancer [74]. Altogether, these different
specificities of the intensively studied multifunctional N protein suggest that the N-
protein could be interfering with the antiviral countermeasures of the host. Other
interesting features are the apparent targeting of the antiviral Myxovirus protein A
(MxA) to the N-protein, and that the N-protein has been shown to interact with
actin-filaments [75-77]. Finally, the N-protein has been suggested to interact with
cellular membranes localized in perinuclear regions of the cell [53].
9
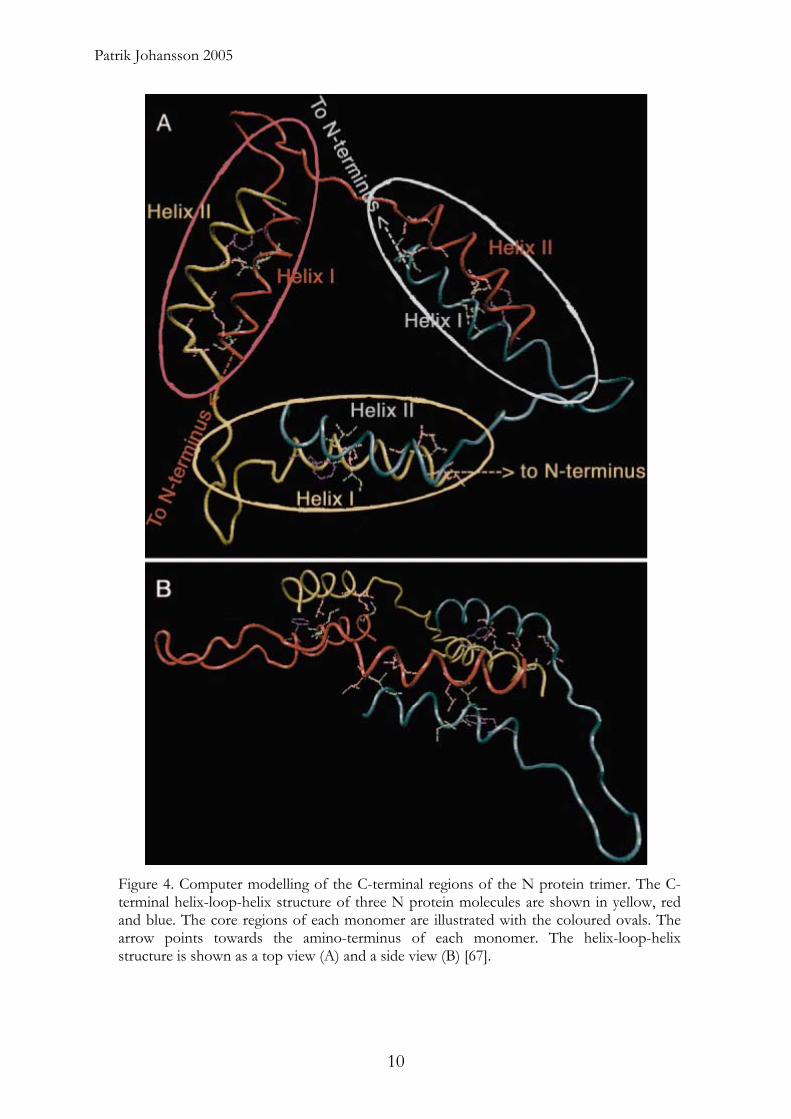
Patrik Johansson 2005
Figure 4. Computer modelling of the C-terminal regions of the N protein trimer. The C-terminal helix-loop-helix structure of three N protein molecules are shown in yellow, red and blue. The core regions of each monomer are illustrated with the coloured ovals. The arrow points towards the amino-terminus of each monomer. The helix-loop-helix structure is shown as a top view (A) and a side view (B) [67].
10

+ +
5’
3’
5’3’
Monomer Dimer Trimer
The nucleocapsid protein
Figure 5. Association of N protein subunits with the viral RNA. The monomers and dimers require the 5’ terminus of the vRNA for binding and are sensitive to increased ionic strength. The trimeric form of N bind specifically to the 3’/5’ panhandle structure and is resistant to high salt concentration [68].
Antigenic regions on the N-protein
When sera from PUUV IFA positive humans and polyclonal rabbit sera, raised
against the N-protein, were analysed by ELISA, the IgG and IgM antibody profiles
were almost exclusively directed against the amino-terminus of the N-protein [78].
However, when the B-cell epitopes were mapped by using short (10 a.a.) peptides
(PEPSCAN), the results were more inconsistent. Some studies indicate that the
epitopes are relatively evenly spread out over the N-protein, while others pointed
to the middle portion of the protein, and others the amino-terminal [79-81]. When
the PEPSCAN method were used to pin-point IgA binding sites, the carboxyl-
terminus of the protein was suggested to harbour the major binding epitopes [82].
However, the amino-terminal part has been successfully used for detection of
PUUV specific IgA antibodies [83].
T-helper cell epitopes have been identified in the amino-terminal part of the N
protein, as well as some additional epitopes were identified in the central and
11

Patrik Johansson 2005
carboxyl-terminal parts of this protein [81, 84]. Cytotoxic T-cell (CTL) epitopes
have been described throughout the primary sequence of the N protein [84, 85].
A nonstructural S (NSs) protein is encoded by the S-segment in several viruses of
the Bunyaviridae family, and it has been shown to mediate a variety of functions. In
Phleboviruses the NSs protein acts as an antagonist of interferon (IFN) induction,
possibly by shutting down the host cell transcription [86-88]. In Orthobunya
viruses it has been shown to suppress the translation in the host cell and viral RNA
synthesis [86, 89]. In Californian serogroup of Orthobunya viruses, the NSs also
suppresses host response against double stranded RNA, and induce apoptosis [90,
91]. The existence of a NSs protein has also been proposed for hantaviruses
harboured by Arvicolinae and Sigmodontinae, but not the Murinae, rodent. The
expression of the suggested 290 nucleotide NSs ORF, from both the Tula virus
(TULV) and the PUUV, inhibited promoter activities of IFN-β, NF-κB and IRF-3
in cell culture [92, 93].
The glycoproteins
The two glycosylated envelope proteins GN and GC, (N for amino terminal and C
for carboxy terminal) are encoded within a single ORF of the M-segment. The
mRNA encoded by the M-segment is translated into a polyprotein, cotranslationaly
imported into the ER were it is cleaved into the GN and GC proteins. This cleavage
is thought to occur directly after the conserved WAASA motif by a signal peptidase
present on the lumenal side of the ER [46, 94]. The processing procedure is very
rapid and efficient since no full length precursor form of the polyprotein has yet
been identified in hantavirus infected or cDNA transfected cells [95-98]. In the ER
the folding of GN and GC is most likely catalysed by chaperones, and they are
thought to form heterodimers here as well [48, 99, 100].
The molecular weight of the unglycosylated GN is about 64 kDa, and approximately
655 a.a. long (for PUUV), while the, GC is smaller, about 54 kDa and approximately
12

The glycoproteins
490 a.a. long (for PUUV). When glycosylated their weights are 68 and 57 kDa,
respectively [101]. The 1148 a.a. (for PUUV) polyprotein contain four major
hydrophobic regions, one within the first twenty a.a. residues and one region near
the carboxy terminus of GN, which are thought to act as signal peptides for the GN
and GC proteins respectively. The other two hydrophobic regions are thought to be
the transmembrane regions for GN and GC, respectively [46, 94, 102].
Both proteins are type I integral transmembrane proteins with carboxy-terminal
hydrophobic anchor domains facing the cytoplasm, and the amino-terminus
pointing towards the ER lumen. GC has a short cytoplasmatic region (< 9 a.a.)
while GN has an unusually long tail (~150 a.a.). This tail has been suggested to serve
many different functions such as acting as a substitute for a matrix protein by
interacting with the N-protein via its conserved cytoplasmic YRTL motif [46, 61,
62]. The YRTL motif has also been suggested to interact with a cellular AP-2
clathrin-associated adapter protein complex, which is thought to be involved in
regulating its budding from the Golgi [103-105]. The hantavirus glycoproteins, as
well as those from the other Bunyaviridae viruses, show a high number of conserved
cysteine residues, which indicate that cystine bridge formations in the ER may be
important for keeping the correct 3-dimensional structure and conformation [94].
Whether GN and/or GC are retained in the ER, when expressed individually, has
been debated for some time. Some studies claim that both proteins are trapped in
the ER when expressed individually [106-108], while others report that only GC is
retained and GN is transported to the Golgi [107, 109]. However, once in the Golgi
both proteins are retained on site, although it is unclear whether one or both
proteins have Golgi retention signals [46, 109].
Because GN and GC colocalize with a cis-Golgi matrix protein (GM130), and
undergo a high-mannose type of glycosylation, they are not believed to be
transported any further than to the cis-Golgi compartment [99]. Within this
location they are found mainly in lipid rafts [54]. One interesting observation is that
the Old World hantavirus glycoproteins are proposed to be accumulated in the
13

Patrik Johansson 2005
Golgi, where the complete virion assembles [98, 107], while the glycoproteins of
the New World hantaviruses, are proposed to be transported to the plasma
membrane and where their virions are eventually assembled [109, 110]. There is
however no complete consensus on the topic of plasma membrane assembly, and
the discussion is ongoing [46].
Glycosylation
Protein glycosylation is associated with several functions. For the envelope proteins
of hantaviruses, glycosylation has been shown to be essential for folding, targeting
and intracellular sorting and transport of the hantavirus glycoproteins [98, 99]. It
has also been shown that glycosylation is important and involved in recognition
phenomena, signal transduction after receptor binding, and mediation of
membrane fusion and penetration of cell membranes. Glycosylated proteins are
also involved in stabilizing certain structures, recruiting chaperones and to confer
resistance to proteolysis of stem regions in membrane proteins. Furthermore, they
are thought to be directly involved in virus morphogenesis at the budding site, and
are able to elicit a protective immune response [111-114].
The glycoproteins of hantaviruses have four potential N-glycosylation sites that are
conserved among all hantaviruses, three in GN (142, 357 and 409 in PUUV) and
one in GC (937 in PUUV) [115]. When these and other residues were further
investigated it is observed that GN has three or four functional N-glycosylations but
no O-glycosylation while GC has one N-glycosylation as well as one O-
glycosylation [99], [Paper I].
All HPS causing hantaviruses contain an immunoreceptor tyrosine-based activation
motif (ITAM) signalling element in the cytoplasmatic tail of the GN protein. For
the NY-1 virus, it has been shown to be involved in the binding and activation of
cellular kinases from both the src and syk families. Furthermore, it has been
suggested to dysregulate normal immune and endothelial cell responses. This
14

The glycoproteins
ITAM element is not present in HFRS-causing or non-disease-causing viruses of
the hantavirus genus (with TULV as an exception) [116]. In other viruses, including
HIV and SIV, the ITAM elements have been shown to play an immunoregulatory
role [117-119]. It is also thought that pathways that are regulated by ITAM
elements play a crucial role in endothelial cell function [120]. This ITAM element is
ubiquitinated, making the GN very difficult to express in vitro [46, 121-124], [Paper
I]. Destruction of this site greatly stabilizes the protein [121].
During the initial phase of the infection the virus particles attach to the cell surface
through the glycoproteins’ affinity for the receptor β-3 integrin receptor molecules
and possibly to an unidentified 30 kDa receptor [125-127].
The glycoproteins are suggested to undergo conformational changes at low pH
[128]. It has also been shown that hantavirus infected cells fuse under acidic
conditions [129, 130]. As the hantaviruses are internalized by clathrin coated
vesicles and transported into the acidic environment in the lysosomes [131], these
findings indicate that the glycoproteins could facilitate virus release from the
lysosome into the cytoplasm.
The glycoproteins are of high importance for protection and prophylaxis against
hantaviral infection since they contain the only known structure on the virus
particle that, when targeted against, results in a neutralizing immune response and
protection against hantaviral infection. It has also been shown that passive
immunization with immune sera and monoclonal antibodies with neutralizing
capacity, protects against SEOV, HTNV and PUUV infections [96, 132, 133].
Several neutralizing domains have been identified both in GN and GC. Many
different immunoglobulin classes (IgM, IgG and IgA) have been suggested to be
involved in neutralization of the virus [82, 134-136]. However, it seems like IgM is
the most important isotype for neutralization during the acute phase of PUUV
infection, and most likely for the other hantavirus infections as well [82].
15

Patrik Johansson 2005
Most of the epitopes in the glycoproteins seem to be conformational, since few or
none of the currently available monoclonal antibodies (MAbs) are reactive in
immunoblots carried out on denatured proteins [136].
The GC protein contains MAb epitopes that are shared by several hantaviruses,
whereas the epitopes of GN seem to be more virus type specific [137].
The RNA dependent RNA polymerase
Since the RNA genomes of hantaviruses are of negative polarity, they cannot be
transcribed and/or translated by the machinery of the host cell. To overcome this
problem, the virus encodes its own RNA dependent RNA polymerase (RdRp), a
multifunctional enzyme participating in both transcription and replication. To
execute all these activities, it must possess several different enzymatic activities
such as endonuclease, transcriptase, replicase and possibly also RNA helicase
activity [138]. This 2150 a.a. long protein, with a molecular mass of about 250 kDa,
is encoded by the L-segment, and its role as a RdRp has been known for at least
thirty years [139]. The functions of the RdRp has been described by several groups
[140-143]. The RdRp is the least abundant protein that is encoded by the virus,
with about 104 copies per cell, and it has been shown to be associated to
perinuclear membranes [54].
The N protein and the RdRp are the only proteins that are required for replication
of the hantavirus genomes [58].
The error rate during transcription has been estimated to be 10-3 miss-
incorporations per nucleotide position for the RdRp proteins of both PUUV and
TULV [144].
The flanking regions of both vRNA and cRNA are conserved and the 3’ and 5’ end
have been shown to hybridize with each other to form intra-strand panhandle
structures. It has been suggested that the RNA synthesis and RNA replication are
16

U A G U A G U A G/U Pu C U C C ---
A U C A U C A U C A G A Pu G ---
A U
C
AU
G
U A G U
A U C A
G/U
U
APu
C
C C
Pu G
A
U C
U
G
---
---
The RNA dependent RNA polymerase
most efficient when the panhandle forms cork-screw-like structures (Fig 6) [145,
146], indicating that these structures could be recognition and binding sites for the
RdRp. Since both vRNA and cRNA need to be in complex with the N-protein for
transcription and replication by the RdRp [55-60], it can be assumed that there is
some kind of direct interaction between the RdRp and the N-protein.
Figure 6. Suggested folding of hantavirus S-segment panhandle structure. Modified from [145].
The replication cycle
Virus attachment to the cell
The first step in virus entry into the cell involves an interaction between cell-
surface receptors and the viral attachment proteins, GN and/or GC. For pathogenic
hantaviruses it has been shown that virus entry is mediated through αv β3-integrins
[125, 126], while non-pathogenic hantaviruses (PH, TUL and TPM) probably bind
to αv β1-integrins [147, 148]. However, the virus does not bind to the RGD motif as
observed for many other integrin ligands such as vitronectin and fibronectin.
However, the αv β3-integrin binding vitronectin, and the αv β1-integrin binding
fibronectin block the binding of pathogenic and non-pathogenic hantaviruses,
respectively. The underlying mechanism is probably through steric obstruction
rather than direct blocking of the binding domains [126]. Another, still unidentified,
17

Patrik Johansson 2005
30 kDa cellular protein has been suggested as a putative receptor for HTNV entry
[127]. Finally, it has been shown that BCCV infects polarized epithelial cells more
efficiently from the apical side than through the basolateral membranes [110].
Virus entry into the cell
After viral attachment to the cell surface structures, the virus is taken up through
receptor-mediated endocytosis by clathrin-coated vesicles. These vesicles enter the
endocytotic pathway and the viral glycoproteins are thought to undergo a
conformational change induced by acidification of the endosome compartment,
promoting fusion of the viral and cell membranes [128, 131]. This event could
facilitate the escape of the viral ribonucleoprotein (RNP) from the
endosome/lysosome into the cytoplasm.
Figure 7. The infection cycle. Modified from [158].
18

The replication cycle
Transcription
After the entry of viral ribonucleoproteins into the cytoplasm, an initial “burst” of
transcription into positive stranded cRNA and mRNA transcripts starts. For
SEOV, that has been observed as soon as one hour after infection, and the number
of positive-stranded S RNA molecules increases logarithmically up to twelve hour
after infection. On the other hand, the concentration of negative-stranded
transcripts decreases during the first four hours, after which it starts to increase in
number. This initial period corresponds to the production of N-protein [66] which
is essential for further transcription since the RdRp can only transcribe and
replicate RNA in association with N-protein [55-59].
There are two hypothesizes about where transcription takes place. The most
accepted idea is based on the fact that the two proteins needed for transcription
and replication (the RdRp and the N-protein) are located at or near the perinuclear
membranes, and that the replication probably takes place there. [53, 54].
Furthermore, it has been shown for positive stranded RNA viruses that RNA
replication is associated with intracellular membranes [149]. One appealing idea is
that maintaining a high local concentration of the viral components in a specific
compartment or cellular structure would enhance the rate of viral replication and
assembly. This idea is consistent with the first hypothesis described above [150].
The other hypothesis is based the fact that the SEOV nucleoprotein is found in the
cytoplasm early after infection, and retains there for up to 12 hours before being
transported to the Golgi region where the ribonucleoproteins then gradually
accumulates [65, 66]. This argument suggests that transcription and replication
takes place in the cytoplasm, possibly in association with actin filaments.
19

3’ 5’(-) strand vRNA
Replication Transcription
Replication
5’ 3’ 3’5’(+) strand cRNA (+) strand mRNA
(-) strand vRNA
Translation
-NH2 -COOH
Packagening and assembly
} NTR
Cap
Patrik Johansson 2005
Figure 8. Hantavirus transcription and replication strategy. Modified from [138].
Transcription of vRNA into mRNA
The viral mRNA transcription is believed to start with an RdRp associated
endonuclease cleavage of 8-17 bases from the 5’-end of cellular mRNA’s “cap-
snatching”. These short fragments, containing a cap structure at the 5’ end, are then
used as primers for the transcription of the viral RNA (vRNA) into viral mRNA
according to the “prime and realign” principle [151]. The transcription of the three
different mRNA species differs in quantity, with most mRNA produced from the
S-segment and the least for the long L-segment. However the mechanism behind
this regulated transcription is not clearly understood [152].
It has also been suggested that there are different ways to terminate the
transcription of each of the three different segments. The N and L encoding
mRNA transcripts are probably not polyadenylated. Instead a 3’ flanking stem-loop
structure has been predicted to be formed. Interestingly, the mRNA transcribed
from the M-segments have been suggested to have a polyadenylation sequence of
about 70 bases, very similar to those found in other negative stranded RNA viruses,
and this poly-A tail has been shown to be functional [152, 153].
20

The replication cycle
Transcription of vRNA into cRNA and then back to vRNA
After the primary transcriptions (by the RdRp), when mRNA is the main transcript
to be produced, the transcription switches into producing cRNA, the template for
making new viral RNA (Fig. 7). It is speculated that the accumulation of N protein
might trigger this switch in direction of transcription [138]. This phenomenon has
also been noticed for other negative stranded RNA viruses [154, 155].
The initiation of transcription for other viral members of the Bunyaviridae family is
very similar to the transcription of mRNA, except that instead of using capped
RNA primers, derived from cellular mRNA, a single GTP is used. Both genomic
RNA (vRNA) and antigenomic cRNAs are thereafter encapsidated, but only vRNA
is packaged into virions [142].
Translation
As soon as the mRNA molecules appear after transcription, the translation of viral
proteins starts. The N-protein and the RdRp remains in the cytoplasm after
translation, while the glycoprotein precursor, which has a localization signal, is
cotranslationaly cleaved into GN and GC during the passage and entry into the ER
compartment [46]. The N-protein can be seen within two hours after infection,
while GN and GC are observed in Golgi only after about eight hours post infection
[66].
Virus assembly and release
Since hantaviruses do not have matrix proteins that facilitate binding between the
N-protein and the glycoproteins, it is speculated that the N-protein binds directly
to the one of the glycoproteins. The most likely candidate is the cytoplasmic tail of
GN (~150 a.a.), since the cytoplasmic tail of GC is deemed too short (9 a.a. long) for
21

Patrik Johansson 2005
this interaction. The idea that the N protein binds to one highly conserved YRTL-
motif in the cytoplasmic tail of GN has also been suggested by Kaukinen [62].
Until recently it was generally believed that all Bunyaviridae viruses bud into Golgi
compartment and subsequently get transported out to the plasma membrane in
vesicles. While this is still true for many viruses, several studies indicate that
Sigmodontinae- borne viruses bud at the plasma membrane [44, 110]. It has also
been suggested that the completed BCCV viral ribonucleoproteins are transported
along actin filaments from the place of synthesis to the plasma membrane before
virus assembly [65].
In cell culture released virus particles have been observed 24 hours after infection
with SEOV [66], but not much is known about the mechanisms involved in the
trafficking of viral components, or the final release of new infectious hantaviruses.
For BCCV, electron microscopy and titration of viral particles have demonstrated
their release from the apical side of polarized Vero kidney epithelial cells [110].
Hantaviruses and their hosts
The actual number of different hantavirus serotypes are being discussed, but the 23
officially recognized serotypes can be divided into three groups defined by both
their rodent host and their phylogenetic relationships: those carried by Murinae (Old
World mice and rats), Arvicolinae (voles and lemming, which are spread all over the
northern hemisphere) and those carried by Sigmodontinae (New World mice and rats)
rodents, all of the order Rodentia and family Muridae [156, 157]. Beside these, there
is also one hantavirus, Thottapalayam, which is carried by a shrew, Suncus murinus,
of the order Insectivora.
Of the four hantaviruses carried by the Murinae rodents, three cause disease in
humans; the HTNV that has the striped field mouse, Apodemus agrarius, as its host;
the SEOV that is carried by the black and the brown rats (Rattus rattus and Rattus
22

Hantaviruses and their hosts
norvegicus) and the DOBV that has the yellow-necked mouse, Apodemus flavicollis, or
the striped field mouse, Apodemus agrarius, as its host. It has been suggested that the
DOBV carried by Apodemus agrarius should be classified as a separate virus named
Saaremaa virus (SAAV) [158].
Among the six Arvicolinae borne viruses, only the PUUV causes disease in man. It is
carried by the bank vole, Clethrionomys glareolus.
For the eleven Sigmodontinae borne viruses, at least six cause human disease. The
most prominent are the SNV that is carried by the deer mouse, Peromyscus
maniculatus, and the ANDV by the long-tailed pygmy rice rat, Oligoryzomys
longicaudatus.
All known hantaviruses, with the exception of Thottapalayam, are maintained by
one (or a few closely related) rodent species that are believed to be persistently
infected [159]. Other animals can be infected, but they are all accidental “dead end”
hosts for the hantaviruses [160-164]. Human to human transmission usually does
not occur, although an exception might be the ANDV, for which this has been
described [37].
Like other persistent viruses, hantavirus infections appear to consist of a short
acute phase followed by a prolonged persistent phase [165]. However, very little is
known about the mechanism of this persistence.
It has been observed that during the persistent phase of the infection the virus
disappears from some organs, and comes back in a cyclic fashion [166]. It has also
been suggested that the virus attenuates and becomes less infectious during the
persistent phase [167]. One interesting observation is that deletions in the 3’ ends
of the vRNA occur just before a decline in the vRNA and cRNA concentrations in
persistently SEOV infected cells. One speculation is that these truncated segments
compete with the full size RNA segments and down regulate viral replication.
Other similar observations were noticed in the 3’ L cRNA and 5’ L vRNA, where
deletions also occurred in a cyclic fashion [165]. Furthermore, it has been observed
23
Patrik Johansson 2005
that in SEOV infected rats, only the viral cRNA was found during the persistent
phase of the infection [66].
Virus Abbreviated name
Animal host Source location of reference strain
Disease Ref
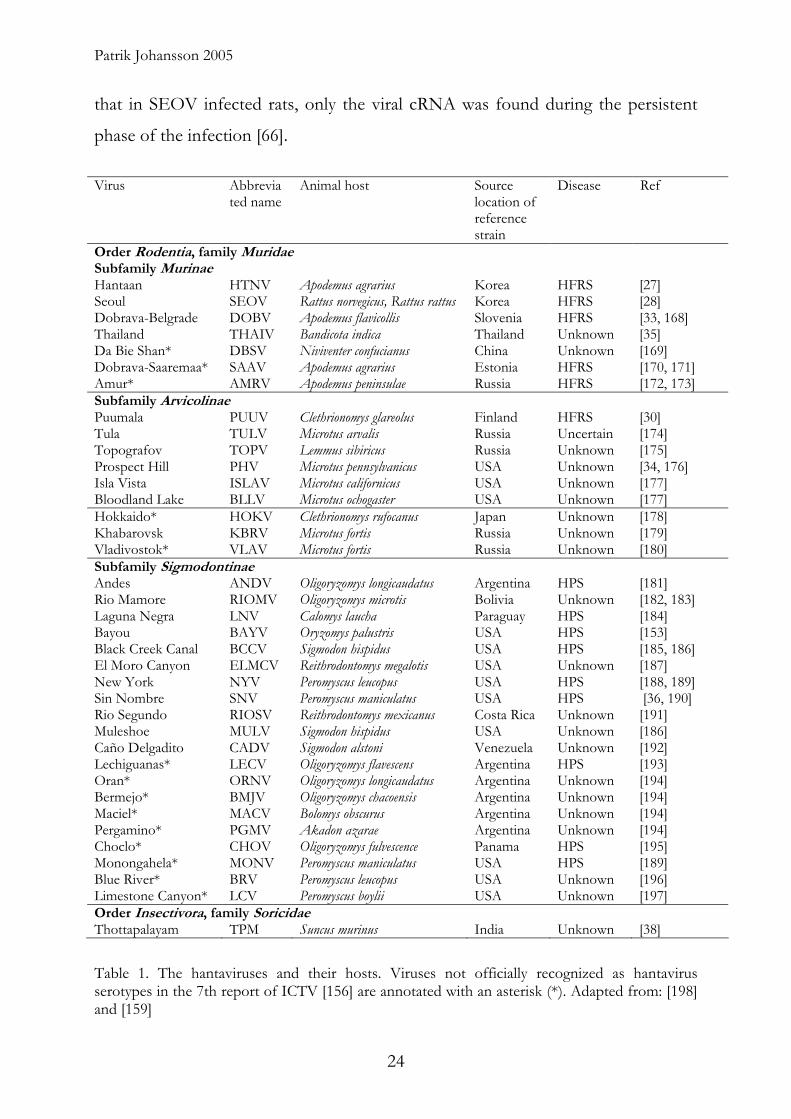
Order Rodentia, family Muridae Subfamily Murinae Hantaan HTNV Apodemus agrarius Korea HFRS [27] Seoul SEOV Rattus norvegicus, Rattus rattus Korea HFRS [28] Dobrava-Belgrade DOBV Apodemus flavicollis Slovenia HFRS [33, 168] Thailand THAIV Bandicota indica Thailand Unknown [35] Da Bie Shan* DBSV Niviventer confucianus China Unknown [169] Dobrava-Saaremaa* SAAV Apodemus agrarius Estonia HFRS [170, 171] Amur* AMRV Apodemus peninsulae Russia HFRS [172, 173] Subfamily Arvicolinae Puumala PUUV Clethrionomys glareolus Finland HFRS [30] Tula TULV Microtus arvalis Russia Uncertain [174] Topografov TOPV Lemmus sibiricus Russia Unknown [175] Prospect Hill PHV Microtus pennsylvanicus USA Unknown [34, 176] Isla Vista ISLAV Microtus californicus USA Unknown [177] Bloodland Lake BLLV Microtus ochogaster USA Unknown [177] Hokkaido* HOKV Clethrionomys rufocanus Japan Unknown [178] Khabarovsk KBRV Microtus fortis Russia Unknown [179] Vladivostok* VLAV Microtus fortis Russia Unknown [180] Subfamily Sigmodontinae Andes ANDV Oligoryzomys longicaudatus Argentina HPS [181] Rio Mamore RIOMV Oligoryzomys microtis Bolivia Unknown [182, 183] Laguna Negra LNV Calomys laucha Paraguay HPS [184] Bayou BAYV Oryzomys palustris USA HPS [153] Black Creek Canal BCCV Sigmodon hispidus USA HPS [185, 186] El Moro Canyon ELMCV Reithrodontomys megalotis USA Unknown [187] New York NYV Peromyscus leucopus USA HPS [188, 189] Sin Nombre SNV Peromyscus maniculatus USA HPS [36, 190] Rio Segundo RIOSV Reithrodontomys mexicanus Costa Rica Unknown [191] Muleshoe MULV Sigmodon hispidus USA Unknown [186] Caño Delgadito CADV Sigmodon alstoni Venezuela Unknown [192] Lechiguanas* LECV Oligoryzomys flavescens Argentina HPS [193] Oran* ORNV Oligoryzomys longicaudatus Argentina Unknown [194] Bermejo* BMJV Oligoryzomys chacoensis Argentina Unknown [194] Maciel* MACV Bolomys obscurus Argentina Unknown [194] Pergamino* PGMV Akadon azarae Argentina Unknown [194] Choclo* CHOV Oligoryzomys fulvescence Panama HPS [195] Monongahela* MONV Peromyscus maniculatus USA HPS [189] Blue River* BRV Peromyscus leucopus USA Unknown [196] Limestone Canyon* LCV Peromyscus boylii USA Unknown [197] Order Insectivora, family Soricidae Thottapalayam TPM Suncus murinus India Unknown [38]
Table 1. The hantaviruses and their hosts. Viruses not officially recognized as hantavirus serotypes in the 7th report of ICTV [156] are annotated with an asterisk (*). Adapted from: [198] and [159]
24

?
Presence of hantavirus
ANDV
SNV
BCCVDOBV
PUUV
HTNVTULV
SEOV
Hantaviruses and their hosts
Geographic spread
Hantaviruses are found in most parts of the world, with a few exceptions like
Antarctica, Greenland and possibly parts of Oceania and Africa. Since hantaviruses
are limited to the geographical region habited by their specific rodent they cannot
extend further than their host. SEOV is spread to most parts of the world since its
host, the rat, has spread with the help of the shipping industry [199].
Figure 9. Map illustrating the regions affected by hantaviruses. SEOV is probably present in most of the major ports around the world. For a more complete list of hantaviruses see Table 1.
Hantaviruses in North and South America
Hantaviruses currently identified in the Americas are either carried by Arvicolinae or
Sigmodontinae rodents. In North America there are, apart from PHV which is carried
by Microtus pennsylvanicus, several closely related viruses that are carried by other
Microtus rodents.
The Sigmodontinae borne viruses of Northern America can be divided into three
subgroups, reflected by the evolutionary relations between their rodent hosts. The
25

Patrik Johansson 2005
first group, which consists of SNV and SNV-like viruses (MONV, NYV and BRV),
are carried by Peromyscus maniculatus or P. leucopus. The second group consists of
ELMCV and RIOSV that are carried by Reithrodontomys species. The third group
consists of BCCV and MULV that are carried by Sigmodon hispidus and BAYV,
carried by Oryzomys palustris [157].
The Sigmodontine rodents, that carry all hantaviruses of South America, are
believed to have immigrated to South America about 5-10 million years ago, which
is relatively recent on the evolutionary time scale. This has resulted in a fast
diversification among both the rodents and the hantaviruses they brought along. At
least three different groups of Sigmodontine rodents carry hantavirus; the
Phyllontini, Akodontini and Oryzomyini. However, all hantaviruses of South
America are closely related and have a 75-90% sequence identity [157].
Hantaviruses on the Eurasian continent
Hantaviruses in Eurasia are hosted by Arvicolinae, which are found in the northern
and eastern regions of Eurasia as well as in North America, and Murinae rodents
that are found through out both Asia and Europe [200].
Hantaviruses in Asia
The hantaviruses carried by Arvicolinae rodents, can be further divided into PUUV-
like viruses hosted by Clethrionomys rodents (Hokkaido and others, carried by C.
rufocanus and possibly other voles), those carried by microtine rodents (TULV and
KBRV, hosted by Microtus arvalis and M. fortis), and TOPV that is carried by Lemmus
sibiricus.
The Hantaviruses carried by Murinae rodents in Asia are SEOV (carried by Rattus
norvegicus and R. rattus), HTNV (carried by Apodemus agrarius), DBSV (carried by
Niviventer confucianus) and THAIV that is carried by Bandicota indica.
26

Hantaviruses and their hosts
The only hantavirus not harboured by a rodent, Thottapalayam virus (TPMV)
which is carried by a shrew, Suncus murinus, is also found in Asia [38].
Of these viruses only HTNV and SEOV are thought to cause human disease.
Hantaviruses in Europe
In Europe the Arvicolinae borne hantaviruses are PUUV and TULV carried by
Clethrionomys glareolus and Microtus arvalis, respectively. The Murinae borne
hantaviruses are DOBV and SAAV, carried by Apodemus flavicollis and A. agrarius,
respectively [157]. SEOV that also is Murinae borne (Rattus Norvegicus and R. rattus)
has been found here, but it is doubtful if it should be considered to be a naturally
European virus [13].
In Europe two major types of disease causing hantaviruses dominate, PUUV,
endemic in many parts of Central, Eastern and Northern Europe, and DOBV, that
can be found in many parts of Central and Eastern Europe [12].
TULV are generally not believed to cause human disease, but at least one case of
HFRS caused by TULV has been described [201]. TULV has been identified at
several locations in Central and Eastern Europe [202-207].
Hantaviruses in Fennoscandia
PUUV is endemic among the bank voles in all of Finland and Norway [7, 167], but
only in the northern two thirds of Sweden. Surprisingly, bank voles in the southern
part of Sweden do not seem to be infected with the virus [208]. The border
between infected and non-infected animals roughly coincides with Limes
Norrlandicus (Fig 9). Further to the south, the virus has been found in some
locations in Denmark. [209, 210]. Why southern Sweden present this gap in the
distribution of PUUV is not known. From investigations of mitochondrial DNA of
bank voles from Fennoscandia it can be seen that there are two different bank vole
27

Sundsvall
Sotkamo
Puumala
Helsinki
Stockholm
Oslo
100 km
Contact zone
”Southern” bank vole population
”Northern” bank vole population
Umeå
Patrik Johansson 2005
populations, one southern and one northern that in turn carry a southern and
northern variant of the PUUV (Fig 10) [211]. The reason for this is unclear, but is
probably related to the repopulation following the deglaciation after the last ice age
that happened 6-10 000 years ago [209]. Today the 50 km contact zone between the
southern and northern population can be found just north of the city of Sundsvall
in Northern Sweden [211]. Similar zones can be found in Finland and in Russia
[157].
Figure 10. Illustration of the distribution of the northern and southern bank vole populations. Black rodent figures represent the southern bank vole population and the white represents the northern. Modified from [157].
Coevolution of the viruses with their host animal
The hantaviruses are thought to have coevolved with their rodent hosts over a long
time, possibly for more than 50 million years [212, 213]. This can be seen in the
congruence of the hantavirus and rodent phylogenetic investigations [214].
Exceptions are seen and are thought to be caused by host switching. An example is
28

Hantaviruses and their hosts
the KBRV and TOPV that are closely related while their hosts the reed vole
(Microtus fortis) and the Siberian lemmings (Lemmus sibiricus), respectively, are only
distantly related [215]. An other example of possible host switching is found for the
DOBV that can be found in both Apodemus agrarius and Apodemus flavicollis [216].
Classification of hantaviruses
The genus hantavirus can be divided into at least 23 serotypes (Table 1). A serotype
is defined as fourfold or greater two-way difference in a virus neutralization assay,
between the virus in question and previously recognised related virus serotypes
[157]. This is not always enough, since a single amino acid change could lead to loss
of neutralization [136]. Therefore other criteria have been suggested. These include:
1) a unique ecological niche for each virus species (a clear association of a new
hantavirus with a different primary rodent host species or subspecies); 2) at least
7% difference in amino acid sequence from previously identified hantaviruses (on
comparison of complete glycoprotein precursor and N-protein sequences); 3) at
least fourfold difference in cross neutralization tests; and 4) absence of genetic
reassortment with other hantaviruses in nature [157].
Evolution of Hantaviruses
The major part of hantavirus evolution is caused by genetic drift, where nucleotide
changes, insertions and deletions are caused by errors made by the RdRp. The error
rate for RNA viruses usually varies between 10-3 and 10-6 mutations per site, and
for hantaviruses has been shown to be 10-3 [144, 217]. This high mutational rate
results in the formation of complex quasispecies populations within a single host
animal, that can evolve rapidly when a mutant becomes more fit than its “parent”
virus [217]. However, when the virus has adapted to its host over a long time, as
for the hantaviruses, it becomes increasingly difficult to outmatch the adapted
master genotype. The evolution for hantaviruses is now relatively slow, with only
29

Patrik Johansson 2005
0.7x10-7 to 2.2x10-6 nucleotide substitutions per site and year for the PUUV. This
rate is comparable with the estimated rate for the host rodents themselves (1.71-
7.31x10-7) and for other stable RNA viruses. This indicates that it might be the
evolution of the hosts that drives the evolution of the hantaviruses [213].
In addition to genetic drift, there are two other possible ways of evolution, which
are in more dramatic: recombination and segments reassortment.
Recombination, where parts of gene segments from two different viruses
recombine and for a chimeric segment, has been show for several RNA virus [218],
among them TULV [205]. Natural recombination events have been suggested for
PUUV also, and the possibility has been demonstrated in vitro [213, 219].
Segment reassortment, which involves a switch of complete virus genome
segments between viruses, has also been evident in nature [220-222], [Paper I].
Both recombination and reassortment events occur in host that are infected by two
different viruses simultaneously. Since the rodents can be persistently infected by
hantaviruses, the situation of co-infection can be readily achieved by a second
infection with a different virus than the primary infection.
Phenotypical changes in the virus induces by changes in the host
When PUUV was forced to change host, e.g. from bank vole to Vero 6 cells, there
were no changes in the amino acid sequence of the virus proteins, but changes in
the untranslated regions of the S-segment was observed. The cell culture adapted
viruses were also less infectious in their natural host, the bank vole [223]. In the
case of TULV, forced adaptation to cell culture led to the loss of the viral putative
NSs-ORF [93].
30

Hantavirus caused diseases
Hantavirus caused diseases
The severe symptoms of a hantavirus infection is assumed to be the result of
immunopathogenesis due to the host immune response induced by the virus
infection and probably not caused directly by the virus [84, 224]. One indication of
this is that T-cell deficient mice and severe combined immunodeficiency (SCID)
mice survive HTNV or SEOV infection better than immunocompetent control
mice [225, 226]. These experiments, and others, indicate that there might be a
connection between T-cell response and the obtained clinical manifestations.
Furthermore, human leukocyte antigen (HLA)-B8-DR3 and DQ2 alleles have been
associated with a more severe form of NE and HIV in humans [227, 228], while
HLA-B27 has been associated with more benign variants of NE and HIV [228,
229].
Both HFRS and HPS are associated with acute thrombocytopenia and changes in
vascular permeability [230].
The main effects of HFRS causing viruses are seen in the kidneys and the
hemostatic balance, while the symptoms of HPS are mainly correlated to the lungs
and heart. These general features are not absolute, as patients with HPS may also
have symptoms associated to the renal function [10, 231, 232], and about half of
NE patients have pulmonary involvement [233, 234]. However, HFRS patients do
not show any cardiac involvement [235].
Hemorrhagic Fever with renal Syndrome (HFRS)
HTNV, SEOV and PUUV cause 60 000-150 000 cases of HFRS each year, and the
great majority of these cases are found in China [5, 6]. The mortality of HFRS
varies from about 0.1% for PUUV to 20% for DOBV and about 1% for SEOV
and 5-10% for HTNV [2-4]. In Northern Sweden infections caused by PUUV
(NE) is the second most prevailing serious viral infection after influenza virus, with
31

Patrik Johansson 2005
about 20 cases per 100.000 people and year [14]. However, the number of sero-
converted people is higher. In a randomized and stratified study performed in
Northern Sweden, the prevalence of people having PUUV-specific IgGs was found
to be 5.4% among adult humans. When compared with the number of cases
reported, it indicates that only one of eight infected people have been diagnosed
and reported [236].
The incubation period of HFRS is usually 2-4 weeks (1-8 weeks), after which the
clinical progress of HFRS can be divided into five different phases based on the
clinical characteristics (Fig. 11). The febrile phase usually lasts 5-6 days, and is
characterized by high fever, chills with headache, myalgia and nausea. Flushing of
the face, erythematosus rashes, conjunctival injections and abdominal and/or back
pain represents other characteristic findings during this phase. The hypotensive
phase is characterized by a sudden drop in blood pressure and thrombocytopenia
that might contribute to a gradual appearance of bleeding manifestations. In the
most severe cases, shock could also occur during this phase. The oliguric phase has
a duration of 3–7 days associated with nausea, vomiting, and acute renal failure,
often in combination with hypertension due to simultaneous hypervolemia.
Approximately 50% of the lethal HFRS cases appear during this phase. The
polyuric phase is the first stage of recovery, and it is characterized by diuresis and
electrolytic imbalance. The convalescence phase can last for weeks to months while
the renal function gradually recovers [7, 237-239].
Since the virus is believed to show high virus titer only for a couple of days after
the symptoms appear, the treatment of HFRS is mostly based on relieving the
symptoms of the disease, and supportive care. However, trials have been
performed with the antiviral drug Ribavirin which seemed to be relatively
successful in the treatment of HFRS in China [240]. Treatment with NE specific
immune sera has also been tested, however with little effect [241]. In severe cases
dialysis is performed [242]. Plasmapheresis, where the part of the patient’s plasma is
32

Hantavirus caused diseases
exchanged for isotonic solution or albumin, managed to shorten the duration of the
illness in a study consisting of 54 patients [243].
Figure 11. Clinical characteristics of HFRS. The duration of the subsequent phases are shown as they would commonly occur in patients suffering from NE. The percentage shows the proportion of patients showing each symptom or finding in NE/KHF. Modified from [244].
Hantavirus Pulmonary Syndrome (HPS)
In North America about 30 cases of HPS are reported per year [245]. According to
the Pan American Health Organization (PAHO) ANDV causes 200-300 cases of
HPS annually in South America [246]. HPS, both in North and South America,
shows mortality rates of more than 40% [7].
HPS usually has an incubation period of 2-3 weeks (1-5 weeks) [247, 248], after
which the clinical features of HPS can be divided into four phases. A febrile phase
(usually 3-5 days, but varies from 1-12 days), characterized by fever, myalgia and
malaise. Other symptoms include headache, dizziness, anorexia, nausea, vomiting
and diarrhea, and in some patients abdominal pain. The cardiopulmonary phase is
characterized by shock and pulmonary edema. Bleeding manifestations are not the
33

Patrik Johansson 2005
major concern for the North American variant of HPS, however that has been
described in South America types of HPS [10]. The diuretic phase is characterized
by clearance of pulmonary edema and recovery from fever and shock. This phase is
followed by a convalescent phase. The recovery after HPS can take up to two
months and a slight pulmonary dysfunction can be observed for some time [248,
249].
For HPS, supporting treatments with oxygen treatment and in more severe cases,
mechanical ventilation has been conducted [10]. Antiviral treatment with Ribavirin
has been investigated but, in contrast to HFRS, it had limited effect [250].
Diagnostics of NE
Clinical diagnostics
In endemic areas, the most severe cases are diagnosed by physicians by
interpretation of the clinical findings, as those described above. However, for
milder or suspected cases, PUUV-specific laboratory tests are needed for
confirmation or verification before the diagnosis is given. Laboratory findings are
usually low platelet count, hematuria and proteinuria. Renal function is impaired
and the serum creatinine level is elevated.
Specific tests for suspected NE
The incubation period for a hantavirus infection is normally between 2-4 weeks,
and the appearance of symptoms is often accompanied by the development of a
significant immune response, with high levels of hantavirus specific antibodies
[224]. In most cases a single IgM test (IFA or ELISA) is sufficient to confirm a
hantavirus infection. However, during the early stages of the disease, the level of
IgM response might be below the detection limit. Therefore, a negative IgM result
can only exclude hantavirus infection six days or later after the onset of symptoms
34

Diagnostics of NE
[251]. During these early time-points, direct detection of viral RNA, usually RT-
PCR, could be undertaken. Nevertheless, most NE patients would have developed
high enough titers of IgG and IgM antibodies in the first serum sample taken [224].
Serological methods
Indirect immunofluorescence assay (IFA) is a common serological method for the
detection of virus specific antibodies. The patient serum is allowed to react with
virus-infected cells prepared on glass slides. These cells are from either an infected
tissue sample (most often rodent lung), or more commonly virus infected cell
cultures (Vero E6 cells are the most commonly used cell line). Fluorescent labelled
anti-human antibodies are then applied to detect anti-hantavirus antibodies (if
present) that has bound to the virus infected cells. A fluorescence microscope is
used for visualizing the result [26, 29]. The major advantage of IFA is that it utilizes
virus infected cells containing all the viral antigens which are in their natural
conformation, in contrast to purified antigens. However, the IFA analysis requires
an experienced operator to interpret the results which could sometimes be difficult
to evaluate. Furthermore, the production of virus infected cells is complicated since
hantaviruses are risk group 3 pathogens, that require the use of a biosafety level 3
laboratory (BSL3). On top of this, hantaviruses propagate slowly and give low virus
titers in cell culture [252]. However, despite these obstacles, the sensitivity offered
by IFA has led to its popularity in hospitals world-wide.
Enzyme-linked immunosorbent assay (ELISA) is another commonly used
serological method. This method is based on the immobilization of viral protein
antigens (most commonly the N-protein) to ELISA-plate wells. Patient sera are
then applied and if the serum samples contain antigen specific antibodies, they are
detected by the addition of enzyme labelled antibodies specifically directed against
the patient’s antibodies. The enzyme that is conjugated to the secondary anti-
human antibody in-turn breaks down an added substrate reagent, leading to a
35

Patrik Johansson 2005
change in colour. The change in colour can be quantitated by a specialised
spectrophotometer (ELISA reader). There are several variants of the ELISA-
method, and the ELISA as a method has the great advantage of having a high
throughput possibility. It is also sensitive, quantitative, relatively fast and does not
rely on any subjective analysis from the operator [253, 254].
Plaque reduction neutralization test (PRNT) and focal reduction neutralization test
(FRNT) are widely used methods for a more precise identification of the hantaviral
strains, viruses and serotype. These neutralization tests are performed by incubating
hantavirus containing samples with patient sera at various serial dilutions. After a
specified period of incubation, this mixture is seeded on susceptible cells (most
often Vero E6). The neutralizing capacity of the patient’s serum is then analyzed as
the end-point dilution needed to neutralize the virus in the tube, thus inhibiting
viral infection of the tissue culture cells. Since hantaviruses show cytopathic effect
(CPE) only under certain circumstances, and the commonly recognized sign of
cytopathocity, plaques or foci are difficult to be distinguished, techniques have
been developed to improve the visibility of such effect. These often involves
specific staining by fluorescent or enzymatically labelled specific antibodies [255-
257]. To facilitate high throughput assays, these methods have also been adapted to
the 96-well cell culture plate format [179, 258].
In lateral flow immunodiffusion immunochromatic assay, the sample, which could
be a drop of whole blood, is allowed to flow along a filter paper. When the sample
crosses a line pre-loaded with immobilized antigen (the N-protein is currently used)
the specific IgM antibodies, if present in the sample, binds to this antigen to give a
coloured line appearing on the test kit [259-261]. This method is very convenient,
rapid and does not require trained operator to handle. However, this method is less
sensitive in comparison with the local in-house methods, such as IFA and ELISA,
used in Northern Sweden [262].
Examples of other serological methods that have been used for hantavirus
diagnostics are: Hemagglutination-inhibition assay (HI) [263, 264]; Immune
36

Diagnostics of NE
adherence Hemagglutination assay (IAHA) [265, 266], and Complement fixation
test (CFT) [267, 268].
Virus isolation
The best way, and the classical way, of proving that a virus infection has taken
place is by isolating the responsible virus in an appropriate cell line. However, due
to the immunopathogenic characteristics of the disease, it has proven to be very
difficult to isolate viable hantavirus particles from infected patients, since the onset
of symptoms is an indication of an immune response active in neutralizing the
viruses and eliminating virus infected cells. Even when successful, it takes weeks of
blind passaging of the viruses before the virus can be detected [269]. Hantaviruses,
particularly PUUV, are usually very difficult to isolate in cell culture [167].
Electron microscopy (EM)
Electron microscopy has been used successfully for diagnosis of a number of viral
diseases. However, this is not the case for the hantaviruses, mainly because of the
low concentration of virus in the patients, and secondly because hantavirus particle
morphology is not distinctive enough to provide a straightforward morphological
diagnosis [270].
37

Patrik Johansson 2005
Detection of viral antigen
Methods that detect the actual viral antigens have been described as diagnostic
methods for other viral diseases [271], but since the amount of virus particles is
usually very low it is not used for diagnosis of hantaviruses.
Genetic assay
Reverse transcription polymerase chain reaction (RT-PCR) is one genetic method
developed to target the existence of viral RNA (vRNA) in a variety of samples
where the presence of virus particles is suspected. Viral RNA is converted to
complementary DNA by the use of a reverse transcriptase enzyme. This cDNA is
thereafter amplified by PCR and analyzed by agarose gel-electrophoresis. Real time
PCR that relies on fluorescence generated by the presence of PCR product has also
become available. Even though this method is extremely sensitive it is only useful
during early infection, since the virus particles are often eliminated within days after
the appearance of the first symptoms. One disadvantage of this method is the
reliance on specific primer sequences that could lead to false negative due to the
large sequence variability among the different hantavirus species or even among
isolates of the same serotype. Therefore it is of great importance to know the
genetic variability in the local hantavirus population, information of such will
enable good design of specific primers that are useful for the PCR-reaction in
question [272, 273].
Genetic microarray
An interesting method for further analysis of material amplified with PCR is the
microarray technique, where specific DNA probes are spotted onto a glass chip.
The number of individual probes, that can be spotted onto a single chip, range
from 10 000 to more than 100 000. The PCR amplified DNA is labelled with a
fluorescent dye, and hybridized to the DNA probes on the chip. Spots where
38

Diagnostics of NE
labelled sample DNA has hybridized will fluoresce when analyzed in a microscopic
microarray reader. The multiple probes render high specificity by eliminating false
positives due to non-specific PCR amplification. Good design of probes can also
provide detailed information with regards to the genotype of the virus sample [274,
275], [Paper IV].
DNA sequencing
Nucleotide sequencing of amplified DNA provides exact nucleotide sequence, thus
allowing identification of the precise genotype of the virus [Paper I, II]. Several new
methods for sequencing, such as oligonucleotide microarray, mass spectrometry,
and pyrosequencing are now emerging [276-278].
The immune system
The immune response is the mechanism by which the body recognizes and defends
itself against microorganisms, viruses and substances recognized as foreign and
potentially harmful to the body. The response against an infection can be divided
into two major parts; innate and acquired immunity. The innate immunity forms
the first line of defence in the immune response. It is non-specific and recognizes
general patterns of different pathogens, such as double stranded RNA and
unmethylated DNA CpG-motifs. The innate part of the immune system acts soon
after an infection, and the main components of the innate immune system are
granulocytes, mast cells, monocytes, and natural killer cells. Other components are
parts of the complement system and cytokines.
The development of adaptive immunity is delayed when compared with innate
immunity. However, when developed, it becomes very efficient against specific
pathogens, and, due to its memory function, it could respond more rapidly when
39

Patrik Johansson 2005
encountered with the same pathogen subsequently. The adaptive immunity can be
divided into two parts, the humoral (B-lymphocytes) and the cellular (T-
lymphocytes).
B-lymphocytes produce antibodies that bind to a variety of structures on the
pathogens, either in solution or directly on the surface of a pathogen. Antibodies
can neutralize viruses through agglutination or by blocking receptor molecules. It
can also mediate killing of cellular pathogens and infected cells.
The cellular immune response is dependent on T-lymphocytes that circulate in the
body, scanning major histocompability complex (MHC) molecules at the cell
surface. There are two types of MHC molecules, MHC I and MHC II. MHC I is
mostly involved in presenting degraded intracellular proteins while MHC II present
degraded, external, proteins that the cell has phagocytized. The T-lymphocytes will
recognize MHC molecules in complex with foreign proteins. There are two main
types of T-lymphocytes, the CD4+ T-helper (Th) cells that have an
immunoregulatory role, and the CD8+ cytotoxic T-lymphocytes (CTLs) whose role
is to kill infected cells.
Immunity to hantavirus infection
Infection with a hantavirus in believed to result in a life long acquired immunity,
and no reinfection has yet been reported [279, 280]. However, the immune
response is believed to play a significant role in the pathogenesis of hantavirus
diseases (see “Hantavirus caused diseases”). Neutralizing antibodies are thought to
be exclusively directed against the viral glycoproteins, and have been shown to be
able to protect against the disease by inactivating any circulating virus [96]. The N-
protein is thought to induce a cellular response, and it has been shown that
antibodies against the N-protein can protect against hantavirus infection as well
[96, 281, 282].
40

The immune system
Innate Immunity
TNF-α and interleukin (IL)-6 levels are both increased in all, and IL-10 in most
patients, during an infection with PUUV. While IL-6 and IL-10 levels return to
normal, the TNF-α values are still increased one week after the onset of disease.
This is of interest since TNF-α has been associated with symptoms like those
experienced for HFRS [224].
PUUV, as well as the other hantaviruses or other human pathogens of the
Bunyaviridae family are sensitive to interferon-α, and MXA, an interferon induced
dynamin-like GTPase, which is induced by interferon- α [75-77].
Acquired immunity
B-cell response
Hantavirus infections in humans initially induce high levels of virus IgM antibodies
directed against the viral N-protein and two envelope glycoproteins. This response
is soon followed by IgA, IgE and IgG antibodies [83, 280, 283]. The major
response of the IgM, A and G antibodies seems to be directed against the amino-
terminal part of the N-protein [78, 281].
The IgA antibody, which is a crucial part of the mucosal immunity, is detected
during the acute phase of the HFRS and HPS infections. However, the level of IgA
drop after the acute phase in SNV infected patients [284]. Interestingly, neutralizing
levels of IgA antibodies specific for PUUV are still found in sera from NE-patients
2 and 10 years after infection [81].
An IgE response is presumably initiated in HFRS before clinical symptoms are
found, and it has been speculated that they could be involved in the pathogenesis
of the disease. However, no correlation between IgE levels and clinical severity of
HFRS has been described [283].
41

Patrik Johansson 2005
IgG, which is the most abundant antibody type, appears after IgM, during the acute
phase of HFRS and HPS, and these antibodies are mainly directed against the N-
protein, while IgG antibodies directed against the GN and GC appear later, in the
beginning of the convalescence phase [230]. High levels of IgG are found in NE
patients more than 50 years after the recovery from the disease [279].
IgM antibodies are the most important antibody class during the early phase of
infection [280]. GN specific IgM antibodies have been observed before those
directed against the N-protein [285].
Neutralizing antibodies are found soon after the symptoms of NE appear, and
these, early, antibodies have been described as highly cross-reactive IgM and IgA1
antibodies reactive against many hantavirus serotypes [286].
Cross-reactivity
The serological cross-reactivity between different hantaviruses is high, especially
among viruses carried by related rodents (the Murinae, Arvicolinae and Sigmodontinae
rodent groups). This is expected since at the amino acid sequence level, the
nucleocapsid protein displayed more than 79% homology among viruses of the
same rodent group; and displayed between 60-77% homology among viruses from
the different groups [286]. The high degree of cross-reactivity has resulted in
difficulties in serotyping different viruses by ELISA, especially for the
discrimination of SEOV, HTNV and DOBV. This problem has more recently
been was solved by modification of the antigens used [287].
It has also been suggested that the complement system might be involved in the
pathogenesis of NE, and possibly in the damage of tubili and/or functional
changes in glomeruli of the kidneys [288]. The effect of the complement system
could possibly also explain the protective antibody mediated effect observed after
immunization with the N-proteins [289].
42

The immune system
T-cell response
T-cell activation occurs very early in HFRS and is associated with an increase in the
amount of neutrophils, monocytes, B- and T-cells [230, 290]. The T-cell response is
long-lived after a hantavirus infection, with a virus specific CTL responses in
peripheral blood mononuclear cells (PBMCs) of most patients, 6-15 years after
being infected with the virus [124, 291].
Immunogenetics
It has been observed that the severity of the NE disease can be coupled to the
MHC haplotype of the patients, where HLA-B8-DR3 gives a more severe outcome
and a higher viremia [227, 292], and HLA-B27 is associated with milder form of the
disease [229]. In SNV infection, HLA-B*3501 is associated with severe HPS [293].
Vaccines
History of vaccination and prevention
Long before vaccination was described and established as a method to enhance the
immune response and to prevent people from acquiring specific diseases, there
were variolation. People were given a mild infection of a disease, which would then
protect them from a more serious infection in the future. The first written account
of variolation tells about a Buddhist nun that practiced this around 1022 to 1063
AD. She blew powdered scabs of smallpox into the nose of non-immune people
[294]. By the 16th century variolation against smallpox was common practice in
China, India and Turkey, and at that time it was also introduced in Europe and
North America. Although variolation with smallpox is hazardous, the mortality
could be as low as one in two thousand [295].
43

Patrik Johansson 2005
Smallpox was not the only disease that people tried to prevent by variolation. Other
examples are: syphilis, measles, scarlet and yellow fever, muco-cutaneous
leishmaniasis, bovine pleuropneumonia, ruminant anthrax and bovine plague [295].
Vaccination
In the rural parts of the United Kingdom, and perhaps elsewhere too, it was widely
believed that cowpox gave protection against smallpox and vice versa. People that
had survived smallpox were sought for milking herds of cows, since they did not
get infected with, or spread cowpox to other animals. Inoculation of cowpox was
carried out at that time with varying results but no one had yet tried to challenge
any cowpox inoculated patients with smallpox, before Jenner. In 1796 he
inoculated an eight year old boy with lymph from cowpox lesions of an infected
milkmaid. He then challenged the boy with smallpox, and the boy did not develop
the disease. Jenner’s vaccination technique was to transfer lymph from a pustule of
a vaccinated person to a non-immunized person. This was called arm to arm
vaccination. However, sometimes this inoculum could get contaminated with other
diseases, e.g. syphilis. Later vaccines were produced directly from young cows
which resulted in a better and safer vaccine and with a greater availability.
The next step in vaccination did not occur until the end of the 19th century when
Pasteur and co-workers in 1880 managed to permanently attenuate Pasteurella
multocida, that causes fowl cholera, and in 1881 developed a vaccine against Bacillus
anthracis, the cause of anthrax. He later also developed a vaccine against rabies that
consisted of dried spinal cord of rabies infected rabbits. The advantage of the
rabies vaccine was its effectiveness even when given after being infected with
rabies.
Further advances in vaccine development was made in 1886 by Salmon and Smith,
who showed that heat killed salmonella bacteria could be used for immunization.
44

Vaccines
During this period of time vaccines against several infectious diseases were
developed: e.g. Vibrio cholerae, Salmonella typhi, Salmonella paratyphi and Yersina pestis.
Glenny and Hopkins discovered that formalin could be used to detoxify bacterial
toxins. However, the efficiency of the immunization was rather low. In 1926,
Glenny discovered that potassium and aluminium hydroxide, which is still used,
acts as an adjuvant to increase the potency of the immunization.
Other important vaccine discoveries are the BCG vaccine against tuberculosis,
developed in 1921 by Guérin and Calmette, the vaccine against influenza by Francis
and Salk in 1944, the dead Salk-vaccine (1954) and the live Sabin-vaccine (1959)
against polio [295].
The tremendous success of the vaccines in preventing diseases is demonstrated by
the eradication of smallpox and the great reduction in tuberculosis, polio and
measles cases. The latter two could be the next diseases to be eradicated [296].
Short introduction to the immune response to different vaccines
There are two main routes of antigen presentation to the immune system. One is
the MHC class I restricted antigen processing pathway when the antigens are
produced by intracellular parasites, such as viral antigens. Within the cell these
proteins are tagged by ubiquitin, which is a signal for degradation, and then
proteolytically digested by the 26S proteasome. The resulting 8-10 a.a. peptides are
then transported into ER by the TAP-transporter system, where they are bound to
MHC class I molecules. After this binding, the peptide/MHC I complex is
transported to the plasma membrane and presented to the immune systems CD8+
T-cells [297, 298]. This pathway of antigen degradation and presentation results in a
mostly cellular immune response with TNF-α as the main cytokine mediator.
The other route is the MHC class II restricted antigen processing pathway that
processes and presents antigens that are internalized from the exterior of the cell by
endocytosis. While the antigens are degraded to peptides, vesicles containing MHC
45

Patrik Johansson 2005
class II molecules are fused to the endosome/lysosome. The MHC II molecules
binds to the peptides and the complex is thereafter transported to cell surface
where the antigen fragments are presented to CD4+ T helper (Th) cells. The MHC
I pathway results in a essentially cell mediated immune response while the MHC II
pathway response is mainly a humoral response with IFN-γ being the main
cytokine mediator [299].
Vaccines against viral pathogens can be divided into either of the following
categories: live but attenuated pathogens, whole killed pathogens, chimeric viruses,
component vaccines that consist of one or several components of the pathogen, or
genetic vaccines where an antigen coding DNA or RNA molecules are used for
immunization.
The live but attenuated vaccines are the oldest and most tested. Examples from this
category are the vaccinia virus against smallpox, the Sabin polio vaccine, and the
new FluMist live influenza vaccine. These vaccines are supposed to give an
immune response that is similar to a natural infection, which is favourable if the
pathogens themselves induce a long immunity. However, that is not the case for all
diseases, e.g. human hepatitis C and rhinoviruses [300, 301]. A problem with these
vaccines are that although they are attenuated, they might still cause disease, (e.g.
the vaccinia virus that can cause severe infections in some patients) [302]. This is a
great concern for people with lowered immune capacity, such as transplanted
patients on immunosuppressive drugs or people suffering from HIV [303].
Another problem with the use of attenuated pathogens is the possibility of
mutations that might revert an attenuated organism back into a pathogenic strain
again [304].
Attenuated viral RNA genomes (infectious clones) have been suggested for use in
vaccination. This procedure results in an infection with a live virus where the
attenuation can be directly controlled. The attenuating factor is highly defined, in
contrast to most attenuated live vaccine pathogens [305].
46

Vaccines
Whole killed (inactivated) pathogen vaccines are used successfully against several
diseases, e.g. Japanese encephalitis, hepatitis A, rabies and polio. These vaccines are
proven and safe, however batches of incompletely killed vaccines have been
produced and distributed [306]. Another disadvantage of such an approach is that
the killed pathogens are not taken up by the host cells, or presented to the immune
system, by the same route as the corresponding live pathogens. This results in a less
protective immunity that usually requires compensation to some extent by the use
of an adjuvant to boost the immune response. The only adjuvant licensed for
human use is alum (aluminium hydroxide) which has a bias for MHC class II
antigen presentation, leading to a mainly humoral immune response and as a
consequence a lower cellular immune response [307].
Component vaccines, where one or a few selected components of a pathogen
antigens are used for inoculation, have been successfully used against several
diseases, such as tetanus, diphtheria, hemophilus influenzae and hepatitis B [308]. One
disadvantage with component vaccines is that many, especially neutralizing
antigenic epitopes of the pathogen are conformational, and a correct folding of the
antigen is of importance to obtain the required immune response. These
conformational structures are often dependent on the local environment and
modifications of the protein, such as protein-protein interactions, insertions into
lipid membranes or correctly glycosylated amino acid residues. Many of these
structures are difficult to mimic when using recombinantly produced antigens. A
second drawback is that these molecules are usually restricted to the MHC II
presentation pathway [308].
A variant of component vaccines are the recombinant fusion proteins where one
part of the protein self assembles to form virus like particles (VLPs) and the other
part of the protein contain the epitopes of interest. These chimeric VLPs, as
carriers of foreign epitopes and peptides, can help minimise or overcome the
conformational problem of the component vaccines, by giving them a basic
structure, and at the same time act as the antigenic partner (adjuvant). One example
47

Patrik Johansson 2005
is the hepatitis B core (HBc) proteins, that in itself forms VLPs, fused with the
antigenic protein epitopes [309]. One advantage of using HBc particles is that,
besides the humoral response, they also induce cellular immune responses [310].
Another interesting approach for component vaccines are the “edible” vaccines,
where the antigenic component is produced, often in a plant [311, 312]. For
hantavirus this technique is not yet functional [313-315].
Genetic vaccines
When performing genetic vaccination, DNA or RNA molecules are inoculated
directly into the cells of the vaccinees by different techniques, and the encoded
antigens are produced inside the vaccinees’s own cells. This is similar to how the
viral antigens are produced during an actual infection, and a MHC I type of antigen
presentation is favoured which would facilitate cell mediated killing of antigen
producing or virus infected cells. Another advantage of using genetic vaccines is
that they can be used on newborn children. Other types of vaccines are often
neutralized by maternal antibodies obtained before delivery or from breast milk.
Since genetic vaccine antigens are produced in the vaccinee’s own tissue, maternal
antibodies are not a problem [316].
RNA vaccines, where the mRNA coding for the antigen of choice is inoculated,
has the advantage, in comparison to DNA vaccines, of having little or no risk of
integration into the vaccinee’s genome. These mRNAs molecules are produced by
in vitro transcription and capping. However, the production of mRNA, in
comparison to plasmid DNA, is relatively cumbersome, and RNA is not as stable
as DNA.
48

Vaccines
DNA vaccines
The concept of DNA-vaccination has its root in the discovery that injected plasmid
DNA can induce long-term protein expression in muscle tissue [317]. It was soon
after shown that a plasmid that encoded an antigen could induce an immune
response when injected by the same way [318].
DNA vaccines have been shown to give protective immunity against several
infectious diseases, including hantavirus infections (see below) [319].
The main advantages with DNA-vaccines are, apart from the mechanism of their
function and level of protection, that they are cheap and easy to produce. DNA
preparations are done in large scale, and the purification is easy, giving a very high
yield and pure final preparations. This is in contrast to vaccines that are based on
whole pathogens or proteins. Furthermore, DNA is very stable and does not need
to be kept cool, which is a big advantage for a vaccine that needs to be stored and
transported.
The use of DNA vaccines has also been suggested, beside infectious, for the
treatment of cancer, allergies and autoimmune diseases [320-324].
Most DNA vaccines consist of plasmid DNA where the ORF of the antigen is
inserted after a strong mammalian promoter, usually the immediate-early promoter
of human cytomegalovirus (hCMV), and before a polyadenylation/termination
sequence, needed for efficient protein production in cells. Furthermore, these
plasmids usually contain an origin of replication and an antibiotic resistance gene,
which are needed for production of the plasmid it self.
A major concern with DNA vaccines is the risk of integration of the DNA
construct into the vaccinee’s genome, and thereby possibly disrupt or up/down
regulate a gene function that might lead to cancer. Though, this has not been seen
for the more common types of inoculation [325-327], it has been reported for
DNA inoculations followed by electroporation [328]. Another concern is the use of
an antibiotic resistance gene that is part of the plasmid back-bone.
49

Patrik Johansson 2005
Many different ways of inoculation and types of DNA constructs have been used,
and the types of DNA constructs can be divided into different groups, such as: if
the plasmid can replicate in the host cell or not, if there are any immunostimulatory
properties encoded in the DNA construct, if they encode one or more antigens or
ability induce apoptosis of the host cell.
Several different formulations of DNA-vaccines have been investigated. The most
important of these are pure DNA in solution, DNA bound to gold particles for use
with a gene gun, DNA in complex with cationic lipids or biodegradable polymers,
DNA contained in virus like particles or carried by virulence-attenuated infectious
bacteria such as Salmonella typhimurium or Listeria monocytogenes.
Many different routes of DNA administration have been used such as intradermal,
subcutaneous, intramuscular, mucosal (gut, vagina and oral) and intranasal [307].
However, the most frequently used techniques for DNA inoculation are
intramuscular injections of pure plasmid DNA and intradermal delivery with a gene
gun, where the DNA is coated on small gold particles and shot into the epidermis.
Part of the immune response elicited by DNA vaccines are attributed to
immunostimulatory non-methylated CpG motifs in the primary sequence of the
DNA that is being used for inoculation. CpG-motifs are abundant in bacterial
DNA, but not in mammalian DNA, and motifs stimulates the innate part of the
immune system in mammals [329].
Vaccines against hantaviruses
Currently only one vaccine against hantavirus infection is commercially available,
but several groups around the world are investigating both inactivated whole
viruses, recombinant and genetic vaccines.
The commercially available Hantavax™ consists of formalin inactivated HTNV
isolated from suckling mouse brain. It has been used for more than ten years
50

Vaccines
without any serious side effects, suggesting that it is safe to use. It has shown good
seroconversion with 100% seroconversion after 3 immunizations. However only
50% of the vaccinees possessed measurable neutralizing antibodies [230]. In a study
when South Korean soldier were vaccinated with Hantavax™, the vaccine showed
an approximately 75% protection after three immunizations [330]. A similar
vaccine was developed in North Korea where 1.2 million people were reported to
have been immunized in a field trial, with a protective rate of 88-100% [331].
Several vaccines that are based on inactivated HTNV and/or SEOV isolated from
cell culture have been developed in China. These vaccines have shown to have
fewer side effects and a more effective immune response with high levels of
neutralizing antibodies, when compared to rodent brain based vaccines [230].
HTNV N- , GN and GC-proteins, produced and purified from the baculovirus
system, protected hamsters against challenge with the live virus after inoculation
with the N-protein or a combined inoculation with both GN and GC. GN or GC
inoculations did not confer protection when used separately [96]. Baculovirus
derived PUUV N-protein has also been shown to give protection against challenge
in bank voles [281]. Similar results were seen when using E. coli or yeast derived N-
protein [281, 282, 332].
Recombinant vaccinia virus encoding the HTNV N, GN, GC or GN combined with
GC, where explored as a vaccine in hamsters and gerbils. It could be seen that only
the combined GN/GC variant resulted in neutralizing antibodies [96, 97]. Similar
results were obtained when using the SEOV N and GN/GC in a analogous fashion
[333]. When challenged with live virus it was seen that only the combination of
HTNV GN/GC gave full protection, while GN, GC and N-protein by themselves
only gave partial protection [96]. However, for SEOV both the GC/GN
combination and N-protein gave full protection [333]. When a vaccinia vaccine
expressing a combination of both the HTNV GC/GN and N-protein was tested on
humans in phases I and II clinical safety trials it was seen that an earlier immunity
to vaccinia greatly influenced the otherwise promising immunization. 100% of
51

Patrik Johansson 2005
vaccinia negative vaccinees seroconverted after two immunizations and 72%
showed neutralizing antibodies while only 50% of the vaccinia positive vaccinees
seroconverted after two immunizations and only 26% showed neutralizing
antibodies [334].
The use of packaged Sindbis virus replicons has also been evaluated. Here the
structural genes of the positive stranded RNA Sindbis virus was replaced by the
SEOV N-protein or GN/GC coding ORF. The Sindbis virus RNA could replicate
in the host cell, but since the structural genes were missing it could not produce
new viral particles. Hamsters immunized with the N-protein encoding Sindbis
replicon developed non-neutralizing antibodies, and obtained no protection against
the live virus. Most of the hamsters immunized with the GN/GC version developed
neutralizing antibodies and developed partial protection against the virus [335].
Genetic vaccines against hantaviruses
Several groups around the world have studied DNA vaccines against different
hantaviruses. Many vaccines encoding for the N-protein and the two glycoproteins,
independently or in combination, have been investigated. Genetic vaccination with
the gene encoding the N-protein have been shown to give a similar antibody
response as developed against live hantavirus infection [336]. In general, the N-
protein encoding vaccines does not produce neutralizing antibodies, but partial
protection against infection has been described [337, 338]. Constructs encoding
fragments of the SNV glycoproteins have also been investigated. Interestingly,
while none of the constructs resulted in significant levels of neutralizing antibodies,
several gave high level of protection against challenge with the virus [338]. The
most promising results have been obtained when constructs expressing the two
glycoproteins as a pair were used. Vaccines encoding HTN, SEO or AND virus
glycoproteins have all been shown to produce high titers of neutralizing antibodies,
and almost complete protection against challenge with the homologous virus. It
52

Vaccines
was further shown that the HTN based vaccine gave good cross protection against
challenge with both SEO and DOB virus, and that the sera obtained from rhesus
macaques immunized against ANDV or HTNV could be used for passive
immunization of hamsters, giving good protection against challenge against the
respective virus [123, 339, 340].
Animal models for hantavirus infection
For a long time, no adequate animal models for HFRS and HPS infection studies
have been available. One model based on bank voles, the host rodent for PUUV,
has successfully been used in challenge studies after vaccination [281]. However,
bank voles do not develop any illness after infection. To have a more human like
animal model, Groen et al. developed a Cynomolgus macaque model where the
symptoms of NE could be studied after infection with a cell culture adapted PUUV
strain [341]. This model was further improved by Klingström et al. who recorded
human like symptoms after infection with PUUV derived directly from infected
bank voles [342]. An excellent model for ANDV causing HPS is the Syrian
hamsters model developed by Hooper et al. in 2001 [343].
53

Patrik Johansson 2005
Aims of this thesis
The objectives of this thesis are: to characterize a local Puumala virus isolated from
a human patient; to characterize the genetic variability among local Puumala viruses
and their host, the bank vole, Clethrionomys glareolus; to use this information to
develop and evaluate genetic vaccines and to develop diagnostic and
epidemiological tools.
54

Paper I
Results and discussions
A unique PUUV strain, named PUUV Umeå/hu, was previously isolated from a
NE patient [269]. This thesis reports on the further investigations performed on
this isolate. The complete sequence of its genome has been determined and
analysed. Whole and parts of the encoded structural proteins have been expressed
in mammalian cells and their glycosylation patterns and expression profiles have
been determined. We have also studied the phylogenetic relationships of PUU
viruses and their host, the bank vole, from six different locations along the
coastland of the county of Västerbotten, Sweden. The viral sequences were used to
develop a novel microarray method for genetic identification and for future virus
diagnosis. A novel way of genetic immunization was also explored. Important
features of the genetic vaccine include: i) PUUV-specific PCR-produced linear
DNA fragments; ii) partial (5’-3’) exonuclease-protection; and iii) tagging with a
nuclear localization peptide. The approaches and results of the above investigations
are described in three publications and one manuscript summarised below.
Paper I
This paper describes the characterization of the PUUV Umeå/hu isolate. The virus
genome sequence was established and it represents the first complete sequence of a
Puumala virus isolated from a human. Sequence comparisons were made with
other hanta- and PUU-viruses; the N, GN and GC proteins and overlapping
peptides from these proteins were expressed, and the glycosylation patterns of the
GN and GC were investigated.
Comparison of PUUV Umeå/hu and the prototype strain PUUV Sotkamo
revealed that the overall sequence diversity over all three segments were
approximately 19%. However it was as high as 33% in the 3’ non-translated regions
55

Patrik Johansson 2005
of the S-and M-segments (Fig 12), and 5.5%, 8.4% and 6.6% for the N-protein, G-
proteins and polymerase protein amino acid sequences, respectively.
Figure 12. Similarity plot (Stuart Ray’s SimPlot 2.5) of full-length, antigenomic S-segment sequences of PUUV. The plot was calculated by using the PUUV Umeå/hu strain as query and the prototype strain PUUV Sotkamo, and two local PUUV strains, Vindeln and Hällnäs as references. The curves show nucleotide identity comparisons within a sliding window of 200 bases with a step size of 20 bases. The bases between the dotted lines indicate the open reading frame of the N-protein.
When comparing the 3’ and 5’ terminal sequences, which are believed to hybridize
with each other to form the panhandle structure, intrastrand base-pairing seems to
be impaired at distinct positions. There are two adjacent mismatches in position 9
from the 3’ end, and the 10th base from the 5’ end of the genomic sequence. When
sequences from different viral stocks were compared, the two nucleotides were
consistently an A or a C. Furthermore, the complementary nucleotides, in the
proposed panhandle structure, to the A or C were always uracils (U), suggesting
that the presence or absence of this bulge due to nucleotide mismatch, does not
interfere with the biological activity, as shown earlier for the Uukuniemi virus [145].
Another mismatch in position 15 of the S-segment was more consistent.
Phylogenetic trees were constructed from full-length sequences of 11 S and 4 M
full length PUUV sequences. As expected, PUUV Umeå/hu clusters together with
56

Paper I
the other PUUV of Northern Sweden (Fig 13 A). When comparing the
phylogenetic trees of three local PUUV S-segments and that of their M-segments, it
was observed that they where incongruent (Fig 13 B), suggesting that an S- and M
segment exchange might have occurred.
Figure 13. Phylogenetic trees based on complete genome segment sequences of PUUV strains from Northern Europe. (A) Radial tree constructed from PUUV S-segment sequences. (B) Phylograms of the S- and M-segments.
cDNA from the S ORF was expressed in mammalian cells, as the full-length N
protein and as four overlapping peptides (N1–N4). Western blot analysis with anti-
PUUV N serum revealed that all five gene products were present in samples from
transfected COS cells, albeit with different expression levels (Fig 14). The peptides
N1 and N3 and full-length N were detected in high levels both in the total cell
extract and in the tissue culture medium, in contrast to the peptides N2 and N4
that were exclusively detected in total cell extract (Fig 14 A), and in the tissue
culture medium, respectively (Fig 14 B).
57
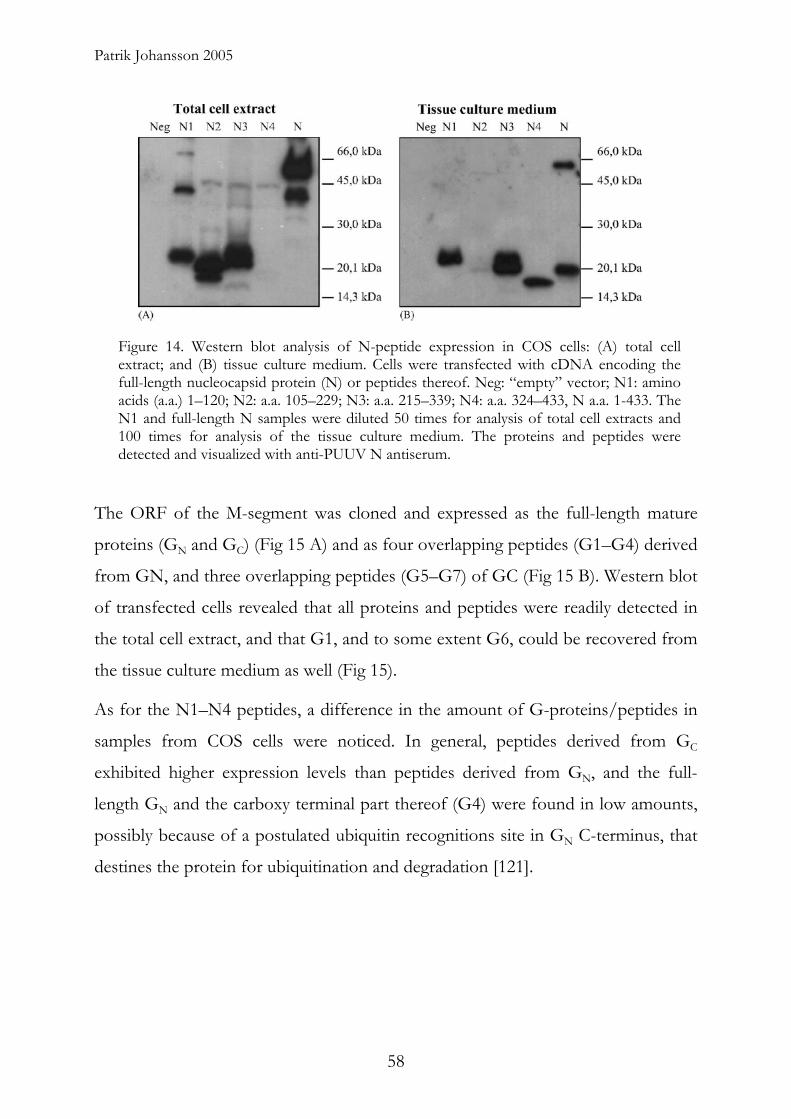
Patrik Johansson 2005
Figure 14. Western blot analysis of N-peptide expression in COS cells: (A) total cell extract; and (B) tissue culture medium. Cells were transfected with cDNA encoding the full-length nucleocapsid protein (N) or peptides thereof. Neg: “empty” vector; N1: amino acids (a.a.) 1–120; N2: a.a. 105–229; N3: a.a. 215–339; N4: a.a. 324–433, N a.a. 1-433. The N1 and full-length N samples were diluted 50 times for analysis of total cell extracts and 100 times for analysis of the tissue culture medium. The proteins and peptides were detected and visualized with anti-PUUV N antiserum.
The ORF of the M-segment was cloned and expressed as the full-length mature
proteins (GN and GC) (Fig 15 A) and as four overlapping peptides (G1–G4) derived
from GN, and three overlapping peptides (G5–G7) of GC (Fig 15 B). Western blot
of transfected cells revealed that all proteins and peptides were readily detected in
the total cell extract, and that G1, and to some extent G6, could be recovered from
the tissue culture medium as well (Fig 15).
As for the N1–N4 peptides, a difference in the amount of G-proteins/peptides in
samples from COS cells were noticed. In general, peptides derived from GC
exhibited higher expression levels than peptides derived from GN, and the full-
length GN and the carboxy terminal part thereof (G4) were found in low amounts,
possibly because of a postulated ubiquitin recognitions site in GN C-terminus, that
destines the protein for ubiquitination and degradation [121].
58
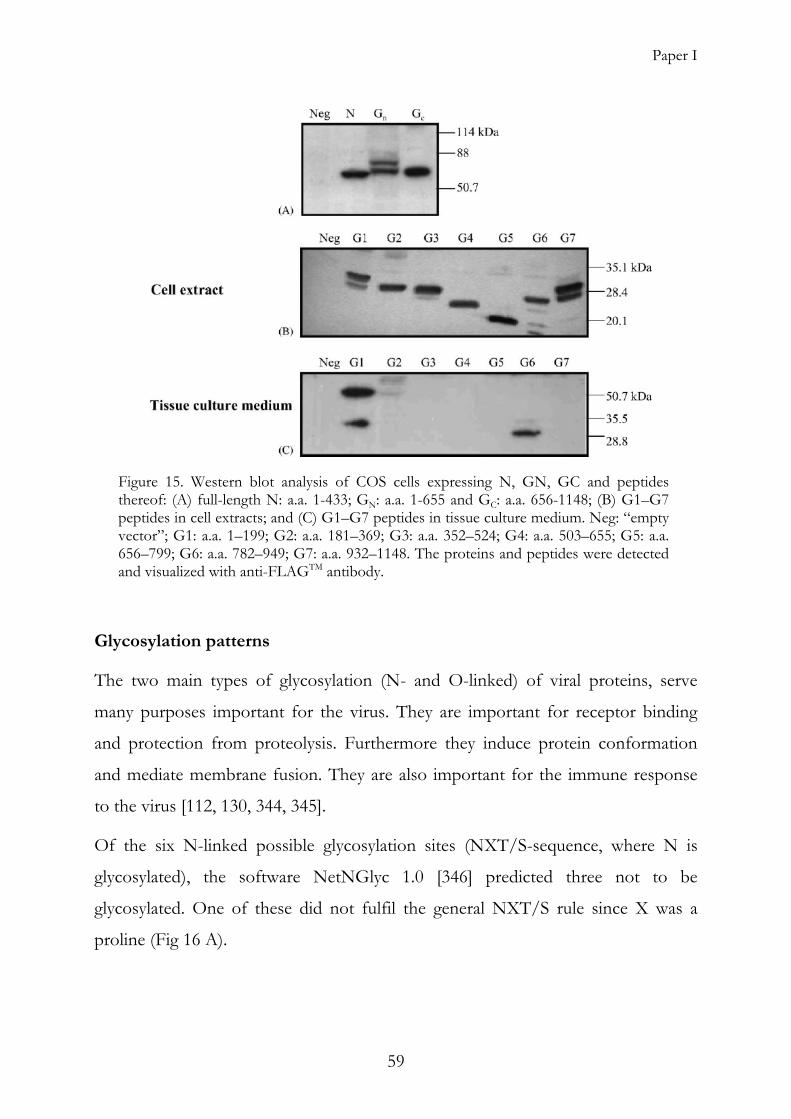
Paper I
Figure 15. Western blot analysis of COS cells expressing N, GN, GC and peptides thereof: (A) full-length N: a.a. 1-433; GN: a.a. 1-655 and GC: a.a. 656-1148; (B) G1–G7 peptides in cell extracts; and (C) G1–G7 peptides in tissue culture medium. Neg: “empty vector”; G1: a.a. 1–199; G2: a.a. 181–369; G3: a.a. 352–524; G4: a.a. 503–655; G5: a.a. 656–799; G6: a.a. 782–949; G7: a.a. 932–1148. The proteins and peptides were detected and visualized with anti-FLAGTM antibody.
Glycosylation patterns
The two main types of glycosylation (N- and O-linked) of viral proteins, serve
many purposes important for the virus. They are important for receptor binding
and protection from proteolysis. Furthermore they induce protein conformation
and mediate membrane fusion. They are also important for the immune response
to the virus [112, 130, 344, 345].
Of the six N-linked possible glycosylation sites (NXT/S-sequence, where N is
glycosylated), the software NetNGlyc 1.0 [346] predicted three not to be
glycosylated. One of these did not fulfil the general NXT/S rule since X was a
proline (Fig 16 A).
59

Patrik Johansson 2005
We investigated all the conserved N-glycosylation sites in peptides of PUUV
Umeå/hu GN and GC by substituting the specific asparagines (N) with serines (S).
In total, six single amino acid substitutions and one double mutant were generated
in the G1–G7 peptides (Fig 16 A).
Figure 16. Prediction and gel-shift analysis of possible N- and O-glycosylated residues. (A) Schematic representation of the poly-protein precursor of the glycoproteins GN and GC (indicated by bars of different colour). Positions where N- or O-linked glycosylations were predicted are indicated by the amino acids and the number of the residue. (B) Gel-shift analysis of glycoprotein (G) peptides, and (C) postulated O-glycosylation sites. Loss of N- or O-linked glycosylation was observed when mutated peptides migrated significantly faster than the corresponding wild-type peptide, indicated in panel A by underlining the modified amino acids. The proteins and peptides were detected and visualized with anti-FLAG™ antibody.
For O-linked glycosylation, there is no simple consensus sequence pattern. By
using the prediction software NetOGlyc 2.0 [112, 347, 348] three threonines (T):
were suggested to be O-glycosylated (Fig 16 A). Therefore, additional single amino
acid substitutions were made so that these three threonines were substituted with
alanines (A).
60

Paper I
When analyzing cell extracts from transfected cells, using Western blot, of (Fig 16
B), four of the six peptides, mutated for N-glycosylation, migrated significantly
faster than the corresponding wild-type peptides, indicating a loss of an N-linked
glycosylation site (underlined in Fig 16 A). Of the O-mutant peptides, only one
peptide displayed gel-shift in comparison to the corresponding wild-type peptide
(Fig 16 C). This was surprising since this site had been predicted with the lowest
probability of O-linked glycosylation among the postulated sites.
Here we have shown that PUUV Umeå/hu phylogenetically clusters with the other
PUUVs from Northern Sweden (Fig 13), and genetically differ significantly from
the PUU prototype virus Sotkamo. We have also shown that there is some
flexibility allowed in the panhandle structure, allowing both the presence and
absence of a small bulge while keeping it biological function. When peptides and
full length N- and G-proteins were expressed in mammalian cells, there are large
differences in expression levels and localization, for the different proteins and
peptides. (Fig. 14 and 15). Gel-shift analysis of the G1–G7 peptides revealed that
four of the six conserved N-glycosylation, and one of three O-glycosylation sites
were functional.
The presented characterization and use of endemic Puumala virus strains will
improve diagnostics and make it possible to perform the analyses with information
and reagents based on local, or closely related, strains of PUUV. This knowledge
combined with results from other investigations [337], [Paper III, IV], constitute
valuable information for future genetic vaccination studies.
61

Patrik Johansson 2005
Paper II
We wanted to investigate the genetic variability of Puumala viruses in local bank
vole populations to get a better understanding of it in general and particularly for
the requirements in the development of diagnostic methods and future vaccines.
Since the majority of NE patients in Västerbotten County, are believed to have
been infected in the coastal area, we wanted to see if there were any differences
among the local viruses in this area. PCR was run on cDNA from bank voles
collected from six different locations in the Västerbotten County, with primers
specific for the PUUV S-segment, as well as on DNA directly extracted from the
same bank voles using primers specific for the cytochrome B/D-loop region of the
bank vole mitochondrial genome. These PCR-products were then sequenced and
the phylogenetic trees for the two organisms were calculated. While investigating
the viral S-segment sequences it was seen that the phylogenetic relationships in the
northern part of Sweden consists of two branches, which from here on will be
called the southern branch and the northern branch, respectively. The southern
branch consists of PUUV sequences from the Bussjö location and two previously
described sequences from a location (Mellansel) to the south west of the study area.
The northern branch consists of the other five study locations as well as with all
previously described PUUV sequences from the region (with the exception of
Mellansel) (Fig 17). Interestingly the previously described human PUUV isolate
[Paper I], clustered tightly with other, bank vole derived, sequences from Bussjö,
where the patient lived, and probably acquired the disease.
62

P98-2
P98-3
Tavelsjö
Hällnäs
Vindeln
B95-3
B99-2B98-5
B98-4Umea/hu
B95-1
B98-3B98-1
B98-7B98-6B98-1
B99-1B98-2
Mellansel Cg49
Mellansel Cg47
G98-1
G98-4
G98-2
S98-3S98-1
S98-2 G98-3
G98-5
N98-2N98-1
P98-4
Hundberget
D98-1D98-2
D98-4
D98-3D98-5
P98-1N98-5
N98-4
N98-6N98-7
N98-3
B95-2
Bussjö
Gumboda/Skäran
Djäkneböle
Norum/Pålböle
Pålböle
Norum/Pålböle
Paper II
Figure 17. Phylogenetic tree of PUUVs of Västerbotten County. The tree is rooted to the other European PUUVs.
The sequences of the northern branch were very homogenous, even over a distance
of 80 km (Djäkneböle to Skäran). However, we could also see that large variation
(7%) could occur over a short distance (10 km). This was seen for Bussjö and
Djäkneböle, which are the closest locations between the northern and the southern
branch. This, for this short distance, rather dramatic difference in genetic sequence,
could indicate a contact zone. There is one previously described contact zone for
PUUV in Sweden. This contact zone is located approximately 250 km south of the
present study area, and correlates with the contact zone for the southern and
northern subspecies of bank voles.
To investigate if there was a similar difference between the bank voles in the study
area, as was seen for the PUU viruses they where carrying, a part of their
mitochondrial DNA was analyzed phylogenetically. Surprisingly, while there were
large and systematic differences in the viral genomes, there were no systematic
differences in the bank vole mitochondrial DNA sequences. Neither could we find
63

Patrik Johansson 2005
any differences in the virus sequence from viruses caught over a five year period,
nor over short distances (~300 meters).
The differences seen an the a.a. sequence of the N-protein for the previously
described PUUV and those presented in this study show very little variation, and
should not influence serological diagnostics or have any impact on a future vaccine.
However, the differences to the PUUV reference strain, PUUV Sotkamo, and to
the previously described PUUVs of Southern Sweden are more significant. The
impact of these differences in the N-protein, which is used for clinical diagnostics,
should be investigated further. Furthermore, the protein variation of the
glycoprotein encoding M-segment also needs further characterization to develop
better diagnostic methods, and for the development of vaccines.
For genetic diagnostics and epidemiology, the understanding of the local genetic
variation of PUUV is of great importance. As can be seen from this study, there
can be large variation over short distances, at the same time as there can be low
variation over longer distances. This variation has to be considered, when
developing new genetic diagnostic tools.
64

Paper III
Paper III
In this study, we investigated the use of linear DNA for genetic immunization
against the PUUV N-protein.
Earlier studies have shown that PCR-amplified DNA containing the promoter,
ORF and terminator sequence elicits an immune response when administered to
animals [349, 350].
Most genetic vaccinations, to date, involve immunization with circular plasmid
DNA containing genetic material from selected pathogens. In comparison to
plasmids, linear DNA fragments (LDF) containing only eukaryotic regulatory
elements, antigen encoding DNA and a poly-adenylation sequence could have
several advantages. Linear DNA fragments are smaller, more flexible and may,
therefore pass more easily through the nucleopores [351, 352]. In contrast to LDF,
plasmids usually contain other characteristics that are not desirable in DNA
vaccines (e.g. antibiotic resistance genes). Furthermore, plasmids are propagated in
bacteria and might therefore contain bacterial remnants such as lipopolysaccharides
or toxins that could cause unwanted side effects in the vaccinee. Such problems
would be avoided if PCR could be used to generate genetic vaccines.
However, in comparison to circular DNA, one major drawback of using linear
DNA is that they are more rapidly degraded, as their free ends are susceptible to
exonucleases. Earlier studies have shown that phosphorothioate (PTO)-
modifications in the backbone of synthetic oligonucleotides [353], or ligation of
closed loops of DNA to the free ends of linear DNA fragments [350] increases the
biological half-life by protecting the DNA against nuclease degradation.
Several studies have shown that nuclear localization signal peptides (NLS), when
conjugated to e.g. DNA, facilitate the transport of the macromolecules into the
nucleus, [350, 354, 355] which could be beneficial for the DNA half life and
expression.
65

Patrik Johansson 2005
We created PTO-protected DNA fragments with a single NLS-peptide from the
SV40 large T antigen, linked to its 3’-end, by the use of a single NLS-peptide linked
to the 3’ (PTO-modified) primer together with an unlinked PTO modified 5’
primer, during PCR-amplification.
As a template for the PCR an ORF encoding the gene of interest (GOI) was cloned
into a mammalian expression vector, under the control of a CMV promoter (Fig
18). We then designed primers specific for the region starting just upstream of the
CMV promoter and ending just downstream of the bovine growth hormone
(BGH) polyadenylation site.
Figure 18. Concept for generating linear DNA fragments. (A) A map of the eukaryotic expression vector pRc/CMV that was used as a template for generating linear DNA fragments. Control regions are indicated in grey and coding regions in black, and GOI refers to gene of interest. The arrows point out the directions of the genes. The two primer binding sites are denoted [5’] and [3’], and the resulting PCR product is also indicated. (B) The resulting linear DNA fragments obtained by PCR using the three different primer pairs. The non-modified LDF (top), the LDF containing the phosphorothioate-modified nucleotides at the 5’ ends, marked with the black box (middle), and finally (bottom), the PTO/NLS-modified LDF with the NLS peptide attached at the 3’ end (not in scale).
Three different sets of primer pairs for PCR amplification were used: ordinary
oligonucleotides without any modifications; oligonucleotides were the first five 3’
nucleotides had been replaced by a corresponding phosphorothioate (PTO)
nucleotide, and one pair that had the 3’ PTO modifications but where one of the
primers also had a nuclear localization signal (NLS) peptide attached.
66

Paper III
We first showed that the PTO-modified primer generated fragments were more
stable against 3’-5’ exonucleases, than non-modified (Fig 19). Following this, we
tested the different fragments for biological activity when being transfected into a
mammalian cell line, by measuring the activity of a control gene (acetylcholine
esterase) (Fig 20).
Figure 19. Exonuclease degradation test. The relative resistance of the LDF towards exonuclease degradation in vitro was examined using non-modified and phosphorothioate-modified LDF. The DNA fragments were incubated at 37 ◦C in a reaction buffer containing lambda exonuclease and exonuclease I.
Figure 19. Enzyme activity in LDF transfected HEK293 cells. The cells were transfected with LDF amplified with non-modified primers (white bar), PTO-modified primers (light grey bar) and PTO/NLS-modified primers (dark grey bar), and plasmid DNA encoding AChE (black bar). An empty plasmid was used as negative control. The enzyme activity in the cell extract from cells transfected the non-modified fragment was set to one. The diagram represents the average of four individual experiments using the same DNA construct. The error bars indicate the variation in enzymatic activity between the different transfections as standard deviation.
67

Patrik Johansson 2005
We then made a construct encoding the first 120 amino acids of the PUUV N-
protein, which has been shown to be the dominant region for the humoral immune
response towards the N-protein [78, 281]. Balb/c mice were immunized with the
use of a gene gun and received 3 booster shots with 2 week intervals. When
studying the response over the first 12 days, it was seen that circular plasmid DNA
gave the fastest response but that the NLS-modified fragments also were fast in
giving a significant antibody response. This occurred already on day 10, with the
PTO-only fragments also giving a fast response but not as strong as the NLS-
modified or the plasmid responses (Fig 21). Over a longer time period it could be
seen that the humoral immune response elicited by the PTO-modified fragments
caught up with the response of the NLS-fragments and the plasmid DNA at week
4, while it took another 2 weeks for the response of the unprotected fragments to
catch up (Fig 22). Also, the titers caused by the NLS-PTO fragments were more
long lasting than those from the PTO-only or non-modified fragments (Fig 23).
Figure 20. Kinetics of the early immune response. The figures represented by the bars indicate the ELISA reactivity against the antigen after the primary immunization. Serum samples were taken from two individuals of each group of immunized mice every second day. Different mice (two) were chosen at each occasion. From left—non-modified LDF (white bars), PTO-modified (light grey bars), PTO/NLS-modified (dark grey bars) and plasmid DNA (black bars). The lines indicate the suggested increase in antigen reactivity during the first 12 days after primary immunization.
68

0
0,5
1
1,5
2 A. ELISA
2 Weeks after primary immunization
Neg Neg
Abs
orba
nce
at 4
05 n
m
Non-modified LDFPTO-modified LDFPTO/NLS-modified LDFPlasmid DNA
B Western blot4 Weeks after primary immunization
0 2 4 6 8 Weeks after primary immunization
Paper III
Figure 21. Serological analysis of LDF immunized mice. (A) Mean ELISA values from LDF immunized mice. The ELISA was performed with mouse sera from individual animals of different vaccination groups. Blood samples were taken and prepared at the indicated time-points before each DNA vaccination, and analyzed for reactivity against the N peptide (the antigen). The bars at each time-point are: from left—non-modified LDF (white bars), PTO-modified (light grey bars), PTO/NLS-modified (dark grey bars) and plasmid DNA (black bars). (B) The Western blot analysis was performed with mouse sera from 20 mice (four mice per group) at two time-points (2 and 4 weeks after the primary immunization).
In our hands antigen stimulation of T-cells from N-protein immunized mice was
unsuccessful. However low levels of soluble cytokines (γ-INF and IL-4) were
found in tissue culture media from antigen-stimulated cells. Our findings suggest
that linear DNA fragment encoding the amino terminus of PUUV induces a strong
humoral immune response together with a low T-cell-mediated immune response.
These data were also supported by an isotype ELISA. Virtually, all antigen specific
antibodies were found to belong to IgG1 subspecies, correlating to a Th2 type of
immune response (data not shown).
69
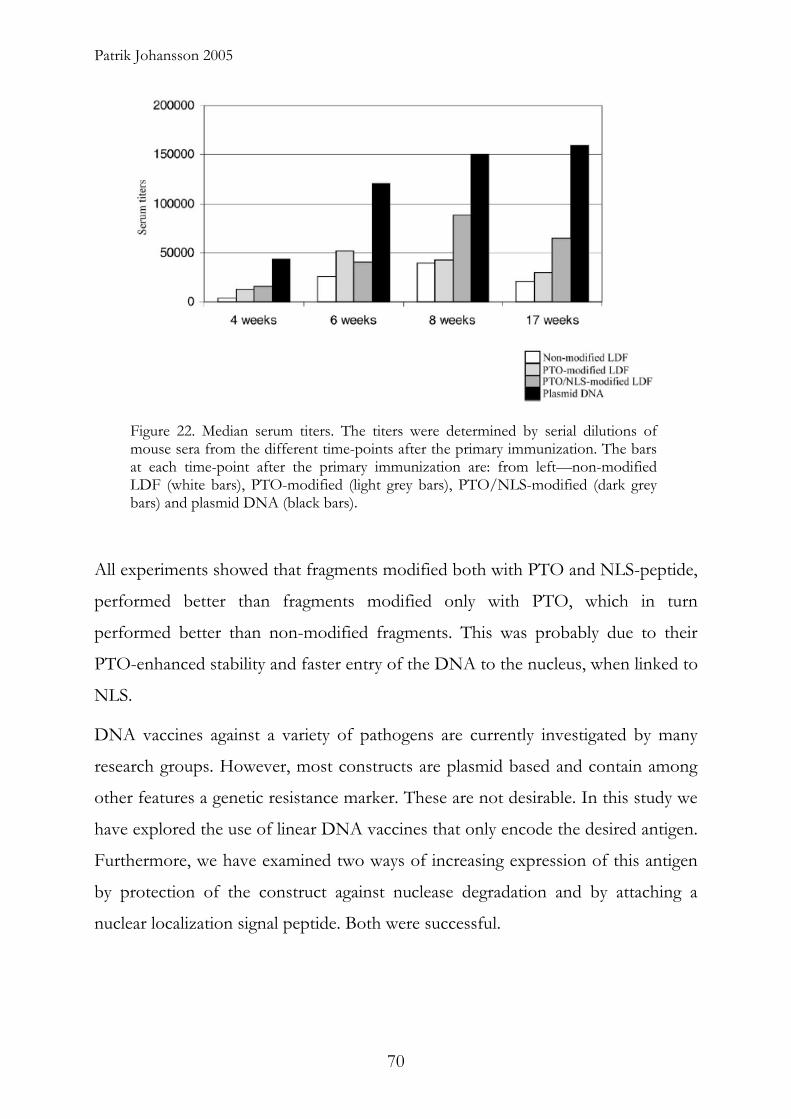
Patrik Johansson 2005
Figure 22. Median serum titers. The titers were determined by serial dilutions of mouse sera from the different time-points after the primary immunization. The bars at each time-point after the primary immunization are: from left—non-modified LDF (white bars), PTO-modified (light grey bars), PTO/NLS-modified (dark grey bars) and plasmid DNA (black bars).
All experiments showed that fragments modified both with PTO and NLS-peptide,
performed better than fragments modified only with PTO, which in turn
performed better than non-modified fragments. This was probably due to their
PTO-enhanced stability and faster entry of the DNA to the nucleus, when linked to
NLS.
DNA vaccines against a variety of pathogens are currently investigated by many
research groups. However, most constructs are plasmid based and contain among
other features a genetic resistance marker. These are not desirable. In this study we
have explored the use of linear DNA vaccines that only encode the desired antigen.
Furthermore, we have examined two ways of increasing expression of this antigen
by protection of the construct against nuclease degradation and by attaching a
nuclear localization signal peptide. Both were successful.
70

Paper IV
Paper IV
The objective of this study was to evaluate the potential of the cDNA microarray
technique to identify and discriminate PCR derived amplicons from genetically
highly similar hantaviruses.
Microarray is a hybridisation technique that permits simultaneous analysis of one
genetic sample against, up to tens of thousands of known probes [356, 357]. By
immobilizing various viral gene sequences on a glass slide, a sample can be assayed
for many different viruses simultaneously. These properties have been utilised for
identification of viral pathogens by two basically different approaches: the
oligonucleotide microarray [274, 358, 359], that provides a highly strain specific
virus identification, and the PCR fragment microarray [275], that gives a better
tolerance for inherent sequence variations.
We designed and printed a hantavirus specific microarray, consisting of DNA
fragments reverse transcribed and amplified from 12 virus isolates of six different
hantaviruses. The S and M-segment were represented by 500 bp overlapping and
250 bp non-overlapping DNA fragments. Each DNA fragment was printed as
duplicates in four subgrids, giving a supergrid. This supergrid was also replicated in
triplicate on the chip, giving 6 redundant spots for each individual DNA fragment
on the chip, giving a total of 2,304 spots per chip.
The overall pair-wise sequence identity among the 12 isolates varied between
54.3% (PHV vs. SEOV SR11) and 85.7% (PUUV Kazan vs. PUUV Cg1820) for
the M-segment, and 51.1% (PUUV Sotkamo vs. SEOV SR11) and 92.5% (PUUV
Kazan vs. PUUV Cg1820) for the S-segment.
After labelling, PCR fragments were then hybridized, either alone or in pairs, to the
microarray chip. It was observed that there were very little ambiguities in the results
for the different viruses (Fig 24, Table 2). However, for virus isolates that were
highly similar (PUUV Cg1820 vs. P360 and K27, and SEOV SR11 vs. SEOV 80-
39, which had nucleotide identities of 99% and 96% respectively), this method was
71

Patrik Johansson 2005
not elucidative enough (Table 2). Since these highly similar viruses gave a diluting
effect on the signals, one from each group was chosen, and the others excluded
from further analysis.
Figure 23. Hybridisation patterns of PUUV Umeå/hu represented by green spots, and HTNV 76-118 represented by red spots, to one supergrid of the hantavirus microarray. Yellow spots signify orientation spots.
Results and discriminative power obtained from analysis of M-segments spots were
similar to that derived from the analysis of both segments. However, when using
only spots corresponding to the S-segment, PUUV Cg1820 could not be
distinguished from PUUV Kazan, probably due to a combination of their genetic
similarity and inconsistencies in the amount of probe-DNA spotted on the chip.
To improve the resolution, we further investigated the use of only one relatively
conserved fragment (from the S-segment) of each of three closely related PUUV
(Umeå/hu, Sotkamo and Kazan). Two different fragment lengths from each strain
72

Paper IV
were used for hybridisation, to determine if fragment length would have an effect
on the outcome of analysis. Hybridization (%) a
Printed probe fragments
PUU
V U
meå
PUU
V S
otka
mo
PUU
V K
azan
PUU
V C
G18
20
HTN
V 7
6-11
8
SEO
V S
R11
DO
BV 3
970-
87
PHV
SNV
Con
vict
Cre
ek
Vira
l Con
trol b
Repl
icate
s c
PUUV Umeå 87 0 0 0 0 0 0 0 0 0 5 PUUV Sotkamo 1 82 2 0 0 0 0 0 0 0 2 PUUV Kazan 6 0 88 5 0 0 0 0 0 0 3 PUUV CG1820 6 3 31 52 0 0 0 0 0 0 2 PUUV P360 0 0 6 88 0 0 0 0 0 0 2 PUUV K27 6 6 16 68 0 0 0 0 0 0 2 HTNV 76-118 1 6 0 1 82 2 0 0 0 0 5 SEOV SR11 0 0 0 0 0 80 0 0 0 0 3 SEOV 80-39 0 0 0 0 0 88 0 0 0 0 4 DOBV 3970-87 0 0 0 0 0 0 89 0 0 0 2 PHV 0 0 0 0 0 0 0 100 0 0 3
Hyb
ridiz
ed ta
rget
s
SNV Convict Creek 1 1 4 1 8 1 0 0 72 0 4
Table 2. Specific Hybridisation Values for M and S Segments. a Calculated percentage values for the signals of the viral target sequence in the sample hybridised to the printed probe fragments. b Viral Control: haemorrhagic fever virus control DNA corresponding to the Ebola Zaire, Ebola Reston, Crimean Congo, and Marburg viruses. c Replicates: number of hybridisations of each target.
For a 456 bp fragment (position 43-499 of the S-segment, where the sequence
identities ranged from 88.4%, for Umeå/hu vs. Kazan, to 91.4% for Sotkamo vs.
Kazan) the different isolates could still be differentiated (Fig 25 A). However when
the shorter 250 bp fragment (position 249-499 of the S-segment, with sequence
identities ranging from 84.4%, for Umeå/hu vs. Kazan, to 89.6% for Sotkamo vs.
Kazan) was used, it was not longer possible to differentiate Sotkamo from Kazan
(Fig 25 B). Even though the percentage of sequence identity for the longer
fragment was lower, it performed better than the shorter fragment.
73
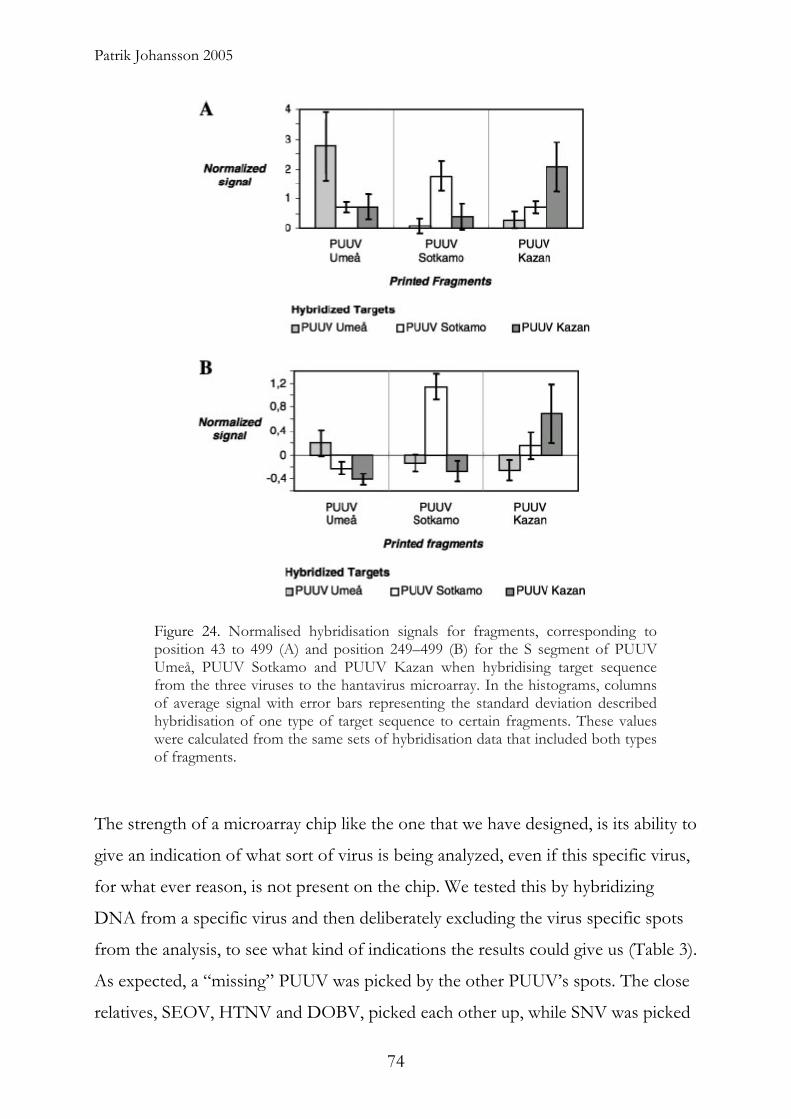
Patrik Johansson 2005
Figure 24. Normalised hybridisation signals for fragments, corresponding to position 43 to 499 (A) and position 249–499 (B) for the S segment of PUUV Umeå, PUUV Sotkamo and PUUV Kazan when hybridising target sequence from the three viruses to the hantavirus microarray. In the histograms, columns of average signal with error bars representing the standard deviation described hybridisation of one type of target sequence to certain fragments. These values were calculated from the same sets of hybridisation data that included both types of fragments.
The strength of a microarray chip like the one that we have designed, is its ability to
give an indication of what sort of virus is being analyzed, even if this specific virus,
for what ever reason, is not present on the chip. We tested this by hybridizing
DNA from a specific virus and then deliberately excluding the virus specific spots
from the analysis, to see what kind of indications the results could give us (Table 3).
As expected, a “missing” PUUV was picked by the other PUUV’s spots. The close
relatives, SEOV, HTNV and DOBV, picked each other up, while SNV was picked
74

Paper IV
up by HTNV and the PUUV. For unknown reasons, Prospect Hill virus did not
cross hybridise with any of the viruses tested.
Hybridization (%) a
Printed probe fragments
PUU
V U
meå
PUU
V S
otka
mo
PUU
V K
azan
PUU
V C
G18
20
HTN
V 7
6-11
8
SEO
V S
R11
DO
BV 3
970-
87
PHV
SNV
Con
vict
Cre
ek
Vira
l con
trol b
Repl
icate
s c
PUUV Umeå + 17 44 22 3 0 0 0 3 0 5 PUUV Sotkamo 17 + 41 25 0 10 0 0 9 0 2 PUUV Kazan 29 14 + 36 0 0 3 0 3 0 3 PUUV CG1820 18 15 56 + 0 0 3 0 0 0 2 HTNV 76-118 13 11 10 11 + 28 4 3 16 0 5 SEOV SR11 8 10 0 0 57 + 0 0 0 0 3 DOBV 3970-87 11 0 4 16 21 35 + 7 0 0 2 PHV 0 0 0 0 0 0 0 + 0 0 3 H
ybrid
ized
targ
ets
SNV Convict Creek 6 4 21 7 40 4 2 3 + 0 4
Table 3. Hybridisation values for removed viruses. a Calculated percentage values for the signals of the viral target sequence in the sample hybridised to the printed probe fragments. b,c Similar to Table 2. + Removed virus specific fragment
The ability to analyze complex samples, such as infected tissue from bank voles, is
also important. To test this we performed PCR on cDNA derived directly from six
bank voles caught in the wild just south of Umeå. When these PCR-reactions were
analyzed on agarose gel, only a smear could be seen, but when labelled and
hybridized to the chip they were all recognized as PUUV, and all but one was
recognized, unambiguously, as PUUV Umeå/hu (Table 4).
75

Patrik Johansson 2005
Hybridization (%) a
Printed probe fragments
Sam
ple
PUU
V U
meå
PUU
V S
otka
mo
PUU
V K
azan
PUU
V C
G18
20
HTN
V 7
6-11
8
SEO
V S
R11
DO
BV 3
970-
87
PHV
SNV
Con
vict
Cre
ek
Vira
l Con
trol b
Repl
icate
s c
1 82 0 10 0 0 0 0 0 0 0 1 2 20 17 10 17 10 0 0 7 9 0 2 3 76 7 0 2 0 0 0 0 0 0 2 4 83 1 0 3 0 0 0 0 0 0 2 5 84 0 0 0 0 0 0 0 0 0 2 6 71 6 5 6 0 0 0 0 0 0 2
Table 4. Hybridisation of six samples from lungs of bank voles wild caught outside of Umeå, Sweden. a, b, c similar to table 2.
Our approach of combining a well designed RT-PCR with a microarray of 500
nucleotide fragments covering large parts of the viral genomes, was able to render
surprisingly high discriminative power for differentiating viruses from complex
samples
The ability to discriminate and identify was accompanied by a tolerance for smaller
sequence differences. This makes the microarray suitable for testing samples
containing unknown viruses.
In spite of problems getting equal amounts of DNA in different spots, we have
demonstrated a good discriminative ability, partly by including many replicate spots
and partly by having an adaptable analysis method.
76

Conclusions
Conclusions
In this thesis, a local PUUV isolated from a human patient and PUUV RNA in
wild bank voles in the coastal region of Västerbotten County in Northern Sweden,
were characterised. We found that the genetic differences among hantaviruses
found in different location is not always gradual, but can show large variation even
when there is no obvious difference in their host genetics. We believe that the
resulting information will be of importance for development of both genetic and
serological diagnostic and epidemiological tools, as well as for future vaccine
development. We have also shown that the use of nuclease resistant, nucleotide
analogues and nuclear localization signal peptides greatly improve DNA vaccines
based on linear DNA, and that they could be an alternative to plasmid DNA for
vaccination purposes. Furthermore, the microarray technique was demonstrated to
be very suitable for differentiation among hantaviruses as well as among different
isolates of a specific hantavirus.
77

Patrik Johansson 2005
Acknowledgements
There are so many people that have helped me during the course of my Ph.D. It is impossible to mention everyone here.
So I would start by saying a big THANK YOU to you who are not mentioned on this page. Next, I would like to thank my two supervisors Fredrik and Göran, for making all this possible and for all the help and support that they have given me on and off work. I am also greatly indebted to: Marlene, for working long hard hours with me all the way from the Katab project. And to Bertil, for bringing food, so we wouldn’t starve; Lena, for helping me at work and at Kaptensgatan; Bosse, Anna, Marie, Henrik, Therese, Margaretha and everyone else, that still is or used to be part of the FOI virology group for helping and supporting me at FOI; Gert, for helping me with the bank voles and all the wilderness stuff; Clas for support and help with any clinical questions; Per, Anci, Göran, Ingrid, Katrine and Lena in the department of virology at Umeå University, for their invaluable help and support in all sort of ways; Oleg, for his deep knowledge and help during the write up; Linda Bladh, Henrik Nordström, Anna Wallin, Li Quan Gen and Åke Lundkvist, SMI, for generous help and support; Gunnar Sandström and Åke Sellström, FOI, for help and support; Department for Biomedical Sciences, for giving me support and shelter during my write up; I am also grateful to the CCD personnel at DSO National Laboratories, Singapore, for putting me up and allowing me to do research and writing in their facilities and helping me with all sorts of things. Thank you Mui Tiang, Michelle, Hwee Teng, , Yian Kim, Gek Kee, Jeffrey, Susie, Susan, Soo Sim, Yvonne, Chee Hwee and last but not least Dr. Lee. Christofer, Pär and Fabian, for making sure I had a life outside that lab as well. Pappa, syster yster and Gunborg, for all the help at home in Bankeryd. And last but not least my family, Lee Ching, Lei and Eigil, who have supported, cheered me up and put up with me during the whole journey. I love You!
78

References
References
1. Lee, H.W. and G. van der Groen, Hemorrhagic fever with renal syndrome. Prog Med Virol,
1989. 36: p. 62-102. 2. Gligic, A., N. Dimkovic, S.Y. Xiao, G.J. Buckle, D. Jovanovic, D. Velimirovic, R.
Stojanovic, et al., Belgrade virus: a new hantavirus causing severe hemorrhagic fever with renal syndrome in Yugoslavia. J Infect Dis, 1992. 166(1): p. 113-20.
3. Clement, J., P. Heyman, P. McKenna, P. Colson, and T. Avsic-Zupanc, The hantaviruses of Europe: from the bedside to the bench. Emerg Infect Dis, 1997. 3(2): p. 205-11.
4. Lee, H.W., Hemorrhagic fever with renal syndrome in Korea. Rev Infect Dis, 1989. 11 Suppl 4: p. S864-76.
5. Lee, H.W., Epidemiology and Pathogenesis of Hemorrhagic Fever with Renal Syndrome, in The Bunyaviridae. 1996, Plenum Press: New York. p. 253-64.
6. Song, G., Epidemiological progresses of hemorrhagic fever with renal syndrome in China. Chin Med J (Engl), 1999. 112(5): p. 472-7.
7. Schmaljohn, C. and B. Hjelle, Hantaviruses: a global disease problem. Emerg Infect Dis, 1997. 3(2): p. 95-104.
8. Papadimitriou, M., Hantavirus nephropathy. Kidney Int, 1995. 48(3): p. 887-902. 9. CDC, http://www.cdc.gov/ncidod/diseases/hanta/hps/noframes/caseinfo.htm. Centers for disease
control and prevention, 2005. 10. Castillo, C., J. Naranjo, A. Sepulveda, G. Ossa, and H. Levy, Hantavirus pulmonary syndrome
due to Andes virus in Temuco, Chile: clinical experience with 16 adults. Chest, 2001. 120(2): p. 548-54.
11. Toivanen, A.L., L. Valanne, and T. Tatlisumak, Acute disseminated encephalomyelitis following nephropathia epidemica. Acta Neurol Scand, 2002. 105(4): p. 333-6.
12. Vapalahti, O., J. Mustonen, Å. Lundkvist, H. Henttonen, A. Plyusnin, and A. Vaheri, Hantavirus infections in Europe. Lancet Infect Dis, 2003. 3(10): p. 653-61.
13. Heyman, P., A. Plyusnina, P. Berny, C. Cochez, M. Artois, M. Zizi, J.P. Pirnay, et al., Seoul hantavirus in Europe: first demonstration of the virus genome in wild Rattus norvegicus captured in France. Eur J Clin Microbiol Infect Dis, 2004. 23(9): p. 711-7.
14. Olsson, G.E., F. Dalerum, B. Hornfeldt, F. Elgh, T.R. Palo, P. Juto, and C. Ahlm, Human hantavirus infections, Sweden. Emerg Infect Dis, 2003. 9(11): p. 1395-401.
15. Ahlm, C., A. Thelin, F. Elgh, P. Juto, E.L. Stiernström, S. Holmberg, and A. Tärnvik, Prevalence of antibodies specific to Puumala virus among farmers in Sweden. Scand J Work Environ Health, 1998. 24(2): p. 104-8.
16. Plyusnin, A., O. Vapalahti, H. Lehvaslaiho, N. Apekina, T. Mikhailova, I. Gavrilovskaya, J. Laakkonen, et al., Genetic variation of wild Puumala viruses within the serotype, local rodent populations and individual animal. Virus Res, 1995. 38(1): p. 25-41.
17. Casals, J., B.E. Henderson, H. Hoogstraal, K.M. Johnson, and A. Shelokov, A review of Soviet viral hemorrhagic fevers, 1969. J Infect Dis, 1970. 122(5): p. 437-53.
18. Zetterholm, S.G., Akuta nefriter simulerande akuta bukfall. Läkartidningen, 1934. 31: p. 425-9.
19. Myrhman, En njursjukdom med egenartad sjukdomsbild. Nordisk medicinsk tidskrift, 1934. 7: p. 793-4.
20. Stuhlfauth, K., Bericht uber ein neues schlammfieberähnliches Krankheitbild bei Deutschen Truppen in Lappland. Deutsche medizinische Wochenschrift, 1943. 69: p. 439.
21. Hortling, En epidemi av fältfeber (?) i finska Lappland. Nordisk medicinsk tidskrift, 1946. 30: p. 1001.
22. Smadel, J.E., Epidemic hemorrhagic fever. Am J Public Health, 1953. 43(10): p. 1327-30.
79

Patrik Johansson 2005
23. Traub, R. and C.L. Wisseman, Jr., Korean hemorrhagic fever. J Infect Dis, 1978. 138(2): p. 267-72.
24. Gajdusek, D.C., Virus hemorrhagic fevers. Special reference to hemorrhagic fever with renal syndrome (epidemic hemorrhagic fever). J Pediatr, 1962. 60: p. 841-57.
25. Johnson, K.M., Hantaviruses: history and overview. Curr Top Microbiol Immunol, 2001. 256: p. 1-14.
26. Lee, H.W. and P.W. Lee, Korean hemorrahagic fever. I. Demonstration of causative antigen and antibodies. Korean Journal of Internal medicine, 1976. 19: p. 371-83.
27. Lee, H.W., P.W. Lee, and K.M. Johnson, Isolation of the etiologic agent of Korean Hemorrhagic fever. J Infect Dis, 1978. 137(3): p. 298-308.
28. Lee, H.W., L.J. Baek, and K.M. Johnson, Isolation of Hantaan virus, the etiologic agent of Korean hemorrhagic fever, from wild urban rats. J Infect Dis, 1982. 146(5): p. 638-44.
29. French, G.R., R.S. Foulke, O.A. Brand, G.A. Eddy, H.W. Lee, and P.W. Lee, Korean hemorrhagic fever: propagation of the etiologic agent in a cell line of human origin. Science, 1981. 211(4486): p. 1046-8.
30. Brummer-Korvenkontio, M., A. Vaheri, T. Hovi, C.H. von Bonsdorff, J. Vuorimies, T. Manni, K. Penttinen, et al., Nephropathia epidemica: detection of antigen in bank voles and serologic diagnosis of human infection. J Infect Dis, 1980. 141(2): p. 131-4.
31. Niklasson, B. and J. Le Duc, Isolation of the nephropathia epidemica agent in Sweden. Lancet, 1984. 1(8384): p. 1012-3.
32. Yanagihara, R., D. Goldgaber, P.W. Lee, H.L. Amyx, D.C. Gajdusek, C.J. Gibbs, Jr., and A. Svedmyr, Propagation of nephropathia epidemica virus in cell culture. Lancet, 1984. 1(8384): p. 1013.
33. Avsic-Zupanc, T., S.Y. Xiao, R. Stojanovic, A. Gligic, G. van der Groen, and J.W. LeDuc, Characterization of Dobrava virus: a Hantavirus from Slovenia, Yugoslavia. J Med Virol, 1992. 38(2): p. 132-7.
34. Lee, P.W., H.L. Amyx, R. Yanagihara, D.C. Gajdusek, D. Goldgaber, and C.J. Gibbs, Jr., Partial characterization of Prospect Hill virus isolated from meadow voles in the United States. J Infect Dis, 1985. 152(4): p. 826-9.
35. Xiao, S.Y., J.W. Leduc, Y.K. Chu, and C.S. Schmaljohn, Phylogenetic analyses of virus isolates in the genus Hantavirus, family Bunyaviridae. Virology, 1994. 198(1): p. 205-17.
36. Nichol, S.T., C.F. Spiropoulou, S. Morzunov, P.E. Rollin, T.G. Ksiazek, H. Feldmann, A. Sanchez, et al., Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science, 1993. 262(5135): p. 914-7.
37. Wells, R.M., S. Sosa Estani, Z.E. Yadon, D. Enria, P. Padula, N. Pini, J.N. Mills, et al., An unusual hantavirus outbreak in southern Argentina: person-to-person transmission? Hantavirus Pulmonary Syndrome Study Group for Patagonia. Emerg Infect Dis, 1997. 3(2): p. 171-4.
38. Carey, D.E., R. Reuben, K.N. Panicker, R.E. Shope, and R.M. Myers, Thottapalayam virus: a presumptive arbovirus isolated from a shrew in India. Indian J Med Res, 1971. 59(11): p. 1758-60.
39. Tang, Y.W., Z.Y. Xu, Z.Y. Zhu, and T.F. Tsai, Isolation of haemorrhagic fever with renal syndrome virus from Suncus murinus, an insectivore. Lancet, 1985. 1(8427): p. 513-4.
40. Zeller, H.G., N. Karabatsos, C.H. Calisher, J.P. Digoutte, C.B. Cropp, F.A. Murphy, and R.E. Shope, Electron microscopic and antigenic studies of uncharacterized viruses. II. Evidence suggesting the placement of viruses in the family Bunyaviridae. Arch Virol, 1989. 108(3-4): p. 211-27.
41. Arthur, R.R., R.S. Lofts, J. Gomez, G.E. Glass, J.W. Leduc, and J.E. Childs, Grouping of hantaviruses by small (S) genome segment polymerase chain reaction and amplification of viral RNA from wild-caught rats. Am J Trop Med Hyg, 1992. 47(2): p. 210-24.
42. Schmaljohn, C.S., Molecular Biology of Hantaviruses, in The Bunyaviridae, R.M. Elliot, Editor. 1996, Plenum Press: New York and London. p. 63-90.
80

References
43. Hung, T., Z.Y. Chou, T.X. Zhao, S.M. Xia, and C.S. Hang, Morphology and morphogenesis of viruses of hemorrhagic fever with renal syndrome (HFRS). I. Some peculiar aspects of the morphogenesis of various strains of HFRS virus. Intervirology, 1985. 23(2): p. 97-108.
44. Goldsmith, C.S., L.H. Elliott, C.J. Peters, and S.R. Zaki, Ultrastructural characteristics of Sin Nombre virus, causative agent of hantavirus pulmonary syndrome. Arch Virol, 1995. 140(12): p. 2107-22.
45. Rodriguez, L.L., J.H. Owens, C.J. Peters, and S.T. Nichol, Genetic reassortment among viruses causing hantavirus pulmonary syndrome. Virology, 1998. 242(1): p. 99-106.
46. Spiropoulou, C.F., Hantavirus maturation. Curr Top Microbiol Immunol, 2001. 256: p. 33-46.
47. Murphy, F.A., A.K. Harrison, and S.G. Whitfield, Bunyaviridae: morphologic and morphogenetic similarities of Bunyamwera serologic supergroup viruses and several other arthropod-borne viruses. Intervirology, 1973. 1(4): p. 297-316.
48. Pettersson, R.F. and L. Melin, Synthesis, assembly and intracellular transport of Bunyaviridae membrane proteins, in The Bunyaviridae, R.M. Elliot, Editor. 1996, Plenum Press: New York. p. 159-88.
49. Hung, T., J. Zhou, S. Xia, and Z. He, Atlas of hemorrhagic fever with renal syndrome. 1988, Beijing: Science Press.
50. Schmaljohn, C.S., S.E. Hasty, S.A. Harrison, and J.M. Dalrymple, Characterization of Hantaan virions, the prototype virus of hemorrhagic fever with renal syndrome. J Infect Dis, 1983. 148(6): p. 1005-12.
51. Hewlett, M.J., R.F. Pettersson, and D. Baltimore, Circular forms of Uukuniemi virion RNA: an electron microscopic study. J Virol, 1977. 21(3): p. 1085-93.
52. Hacker, D., S. Rochat, and D. Kolakofsky, Anti-mRNAs in La Crosse bunyavirus-infected cells. J Virol, 1990. 64(10): p. 5051-7.
53. Ravkov, E.V. and R.W. Compans, Hantavirus nucleocapsid protein is expressed as a membrane-associated protein in the perinuclear region. J Virol, 2001. 75(4): p. 1808-15.
54. Kukkonen, S.K., A. Vaheri, and A. Plyusnin, Tula hantavirus L protein is a 250 kDa perinuclear membrane-associated protein. J Gen Virol, 2004. 85(Pt 5): p. 1181-9.
55. Bridgen, A. and R.M. Elliott, Rescue of a segmented negative-strand RNA virus entirely from cloned complementary DNAs. Proc Natl Acad Sci U S A, 1996. 93(26): p. 15400-4.
56. Dunn, E.F., D.C. Pritlove, H. Jin, and R.M. Elliott, Transcription of a recombinant bunyavirus RNA template by transiently expressed bunyavirus proteins. Virology, 1995. 211(1): p. 133-43.
57. Flick, R. and R.F. Pettersson, Reverse genetics system for Uukuniemi virus (Bunyaviridae): RNA polymerase I-catalyzed expression of chimeric viral RNAs. J Virol, 2001. 75(4): p. 1643-55.
58. Flick, K., J.W. Hooper, C.S. Schmaljohn, R.F. Pettersson, H. Feldmann, and R. Flick, Rescue of Hantaan virus minigenomes. Virology, 2003. 306(2): p. 219-24.
59. Blakqori, G., G. Kochs, O. Haller, and F. Weber, Functional L polymerase of La Crosse virus allows in vivo reconstitution of recombinant nucleocapsids. J Gen Virol, 2003. 84(Pt 5): p. 1207-14.
60. Accardi, L., C. Prehaud, P. Di Bonito, S. Mochi, M. Bouloy, and C. Giorgi, Activity of Toscana and Rift Valley fever virus transcription complexes on heterologous templates. J Gen Virol, 2001. 82(Pt 4): p. 781-5.
61. Betenbaugh, M., M. Yu, K. Kuehl, J. White, D. Pennock, K. Spik, and C. Schmaljohn, Nucleocapsid- and virus-like particles assemble in cells infected with recombinant baculoviruses or vaccinia viruses expressing the M and the S segments of Hantaan virus. Virus Res, 1995. 38(2-3): p. 111-24.
62. Kaukinen, P., Tula hantavirus nucleocapsid protein. Helsinki University Biomedical Dissertations. Vol. No. 53. 2004, Helsinki: Helsinki University.
63. Xu, X., W. Severson, N. Villegas, C.S. Schmaljohn, and C.B. Jonsson, The RNA binding domain of the hantaan virus N protein maps to a central, conserved region. J Virol, 2002. 76(7): p. 3301-8.
81

Patrik Johansson 2005
64. Mir, M.A. and A.T. Panganiban, The hantavirus nucleocapsid protein recognizes specific features of the viral RNA panhandle and is altered in conformation upon RNA binding. J Virol, 2005. 79(3): p. 1824-35.
65. Ravkov, E.V., S.T. Nichol, C.J. Peters, and R.W. Compans, Role of actin microfilaments in Black Creek Canal virus morphogenesis. J Virol, 1998. 72(4): p. 2865-70.
66. Kariwa, H., H. Tanabe, T. Mizutani, Y. Kon, K. Lokugamage, N. Lokugamage, M.A. Iwasa, et al., Synthesis of Seoul virus RNA and structural proteins in cultured cells. Arch Virol, 2003. 148(9): p. 1671-85.
67. Kaukinen, P., V. Kumar, K. Tulimaki, P. Engelhardt, A. Vaheri, and A. Plyusnin, Oligomerization of Hantavirus N protein: C-terminal alpha-helices interact to form a shared hydrophobic space. J Virol, 2004. 78(24): p. 13669-77.
68. Mir, M.A. and A.T. Panganiban, Trimeric hantavirus nucleocapsid protein binds specifically to the viral RNA panhandle. J Virol, 2004. 78(15): p. 8281-8.
69. Hacker, D., R. Raju, and D. Kolakofsky, La Crosse virus nucleocapsid protein controls its own synthesis in mosquito cells by encapsidating its mRNA. J Virol, 1989. 63(12): p. 5166-74.
70. Severson, W.E., X. Xu, and C.B. Jonsson, cis-Acting signals in encapsidation of Hantaan virus S-segment viral genomic RNA by its N protein. J Virol, 2001. 75(6): p. 2646-52.
71. Wilson, V.G. and D. Rangasamy, Viral interaction with the host cell sumoylation system. Virus Res, 2001. 81(1-2): p. 17-27.
72. Kaukinen, P., A. Vaheri, and A. Plyusnin, Non-covalent interaction between nucleocapsid protein of Tula hantavirus and small ubiquitin-related modifier-1, SUMO-1. Virus Res, 2003. 92(1): p. 37-45.
73. Maeda, A., B.H. Lee, K. Yoshimatsu, M. Saijo, I. Kurane, J. Arikawa, and S. Morikawa, The intracellular association of the nucleocapsid protein (NP) of hantaan virus (HTNV) with small ubiquitin-like modifier-1 (SUMO-1) conjugating enzyme 9 (Ubc9). Virology, 2003. 305(2): p. 288-97.
74. Li, X.D., T.P. Makela, D. Guo, R. Soliymani, V. Koistinen, O. Vapalahti, A. Vaheri, et al., Hantavirus nucleocapsid protein interacts with the Fas-mediated apoptosis enhancer Daxx. J Gen Virol, 2002. 83(Pt 4): p. 759-66.
75. Kochs, G., C. Janzen, H. Hohenberg, and O. Haller, Antivirally active MxA protein sequesters La Crosse virus nucleocapsid protein into perinuclear complexes. Proc Natl Acad Sci U S A, 2002. 99(5): p. 3153-8.
76. Kanerva, M., K. Melen, A. Vaheri, and I. Julkunen, Inhibition of puumala and tula hantaviruses in Vero cells by MxA protein. Virology, 1996. 224(1): p. 55-62.
77. Frese, M., G. Kochs, H. Feldmann, C. Hertkorn, and O. Haller, Inhibition of bunyaviruses, phleboviruses, and hantaviruses by human MxA protein. J Virol, 1996. 70(2): p. 915-23.
78. Elgh, F., Å. Lundkvist, O.A. Alexeyev, G. Wadell, and P. Juto, A major antigenic domain for the human humoral response to Puumala virus nucleocapsid protein is located at the amino-terminus. J Virol Methods, 1996. 59(1-2): p. 161-72.
79. Lundkvist, Å., S. Björsten, B. Niklasson, and N. Ahlborg, Mapping of B-cell determinants in the nucleocapsid protein of Puumala virus: definition of epitopes specific for acute immunoglobulin G recognition in humans. Clin Diagn Lab Immunol, 1995. 2(1): p. 82-6.
80. Tischler, N.D., H. Galeno, M. Rosemblatt, and P.D. Valenzuela, Human and rodent humoral immune responses to Andes virus structural proteins. Virology, 2005. 334(2): p. 319-26.
81. de Carvalho Nicacio, C., M. Sallberg, C. Hultgren, and Å. Lundkvist, T-helper and humoral responses to Puumala hantavirus nucleocapsid protein: identification of T-helper epitopes in a mouse model. J Gen Virol, 2001. 82(Pt 1): p. 129-38.
82. de Carvalho Nicacio, C., E. Björling, and Å. Lundkvist, Immunoglobulin A responses to Puumala hantavirus. J Gen Virol, 2000. 81(Pt 6): p. 1453-61.
82

References
83. Elgh, F., M. Linderholm, G. Wadell, A. Tärnvik, and P. Juto, Development of humoral cross-reactivity to the nucleocapsid protein of heterologous hantaviruses in nephropathia epidemica. FEMS Immunol Med Microbiol, 1998. 22(4): p. 309-15.
84. Ennis, F.A., J. Cruz, C.F. Spiropoulou, D. Waite, C.J. Peters, S.T. Nichol, H. Kariwa, et al., Hantavirus pulmonary syndrome: CD8+ and CD4+ cytotoxic T lymphocytes to epitopes on Sin Nombre virus nucleocapsid protein isolated during acute illness. Virology, 1997. 238(2): p. 380-90.
85. Van Epps, H.L., C.S. Schmaljohn, and F.A. Ennis, Human memory cytotoxic T-lymphocyte (CTL) responses to Hantaan virus infection: identification of virus-specific and cross-reactive CD8(+) CTL epitopes on nucleocapsid protein. J Virol, 1999. 73(7): p. 5301-8.
86. Bridgen, A., F. Weber, J.K. Fazakerley, and R.M. Elliott, Bunyamwera bunyavirus nonstructural protein NSs is a nonessential gene product that contributes to viral pathogenesis. Proc Natl Acad Sci U S A, 2001. 98(2): p. 664-9.
87. Bouloy, M., C. Janzen, P. Vialat, H. Khun, J. Pavlovic, M. Huerre, and O. Haller, Genetic evidence for an interferon-antagonistic function of rift valley fever virus nonstructural protein NSs. J Virol, 2001. 75(3): p. 1371-7.
88. Weber, F., A. Bridgen, J.K. Fazakerley, H. Streitenfeld, N. Kessler, R.E. Randall, and R.M. Elliott, Bunyamwera bunyavirus nonstructural protein NSs counteracts the induction of alpha/beta interferon. J Virol, 2002. 76(16): p. 7949-55.
89. Weber, F., E.F. Dunn, A. Bridgen, and R.M. Elliott, The Bunyamwera virus nonstructural protein NSs inhibits viral RNA synthesis in a minireplicon system. Virology, 2001. 281(1): p. 67-74.
90. Colon-Ramos, D.A., P.M. Irusta, E.C. Gan, M.R. Olson, J. Song, R.I. Morimoto, R.M. Elliott, et al., Inhibition of translation and induction of apoptosis by Bunyaviral nonstructural proteins bearing sequence similarity to reaper. Mol Biol Cell, 2003. 14(10): p. 4162-72.
91. Soldan, S.S., M.L. Plassmeyer, M.K. Matukonis, and F. Gonzalez-Scarano, La Crosse virus nonstructural protein NSs counteracts the effects of short interfering RNA. J Virol, 2005. 79(1): p. 234-44.
92. Tulimäki, K., P. Kaukinen, F. Weber, A. Vaheri, and A. Plyusnin. Tula and Puumala Hantavirus NSs ORFs are functional. in The 6th International Conference on Hemorrhagic Fever with Renal Syndrome (HFRS), Hantavirus Pulmonary Syndrome (HPS) and Hantaviruses. 2004. Seoul, Korea.
93. Plyusnin, A., Genetics of hantaviruses: implications to taxonomy. Arch Virol, 2002. 147(4): p. 665-82.
94. Lober, C., B. Anheier, S. Lindow, H.D. Klenk, and H. Feldmann, The Hantaan virus glycoprotein precursor is cleaved at the conserved pentapeptide WAASA. Virology, 2001. 289(2): p. 224-9.
95. Schmaljohn, C.S., S.E. Hasty, L. Rasmussen, and J.M. Dalrymple, Hantaan virus replication: effects of monensin, tunicamycin and endoglycosidases on the structural glycoproteins. J Gen Virol, 1986. 67 ( Pt 4): p. 707-17.
96. Schmaljohn, C.S., Y.K. Chu, A.L. Schmaljohn, and J.M. Dalrymple, Antigenic subunits of Hantaan virus expressed by baculovirus and vaccinia virus recombinants. J Virol, 1990. 64(7): p. 3162-70.
97. Pensiero, M.N., G.B. Jennings, C.S. Schmaljohn, and J. Hay, Expression of the Hantaan virus M genome segment by using a vaccinia virus recombinant. J Virol, 1988. 62(3): p. 696-702.
98. Ruusala, A., R. Persson, C.S. Schmaljohn, and R.F. Pettersson, Coexpression of the membrane glycoproteins G1 and G2 of Hantaan virus is required for targeting to the Golgi complex. Virology, 1992. 186(1): p. 53-64.
99. Shi, X. and R.M. Elliott, Analysis of N-linked glycosylation of hantaan virus glycoproteins and the role of oligosaccharide side chains in protein folding and intracellular trafficking. J Virol, 2004. 78(10): p. 5414-22.
83

Patrik Johansson 2005
100. Arikawa, J., A.L. Schmaljohn, J.M. Dalrymple, and C.S. Schmaljohn, Characterization of Hantaan virus envelope glycoprotein antigenic determinants defined by monoclonal antibodies. J Gen Virol, 1989. 70 ( Pt 3): p. 615-24.
101. Sheshberadaran, H., B. Niklasson, and E.A. Tkachenko, Antigenic relationship between hantaviruses analysed by immunoprecipitation. J Gen Virol, 1988. 69 ( Pt 10): p. 2645-51.
102. Schmaljohn, C.S., A.L. Schmaljohn, and J.M. Dalrymple, Hantaan virus M RNA: coding strategy, nucleotide sequence, and gene order. Virology, 1987. 157(1): p. 31-9.
103. Puffer, B.A., S.C. Watkins, and R.C. Montelaro, Equine infectious anemia virus Gag polyprotein late domain specifically recruits cellular AP-2 adapter protein complexes during virion assembly. J Virol, 1998. 72(12): p. 10218-21.
104. Ohno, H., J. Stewart, M.C. Fournier, H. Bosshart, I. Rhee, S. Miyatake, T. Saito, et al., Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science, 1995. 269(5232): p. 1872-5.
105. Scales, S.J., M. Gomez, and T.E. Kreis, Coat proteins regulating membrane traffic. Int Rev Cytol, 2000. 195: p. 67-144.
106. Deyde, V.M., A.A. Rizvanov, J. Chase, E.W. Otteson, and S.C. St Jeor, Interactions and trafficking of Andes and Sin Nombre Hantavirus glycoproteins G1 and G2. Virology, 2005. 331(2): p. 307-15.
107. Pensiero, M.N. and J. Hay, The Hantaan virus M-segment glycoproteins G1 and G2 can be expressed independently. J Virol, 1992. 66(4): p. 1907-14.
108. Shi, X. and R.M. Elliott, Golgi localization of Hantaan virus glycoproteins requires coexpression of G1 and G2. Virology, 2002. 300(1): p. 31-8.
109. Spiropoulou, C.F., C.S. Goldsmith, T.R. Shoemaker, C.J. Peters, and R.W. Compans, Sin Nombre virus glycoprotein trafficking. Virology, 2003. 308(1): p. 48-63.
110. Ravkov, E.V., S.T. Nichol, and R.W. Compans, Polarized entry and release in epithelial cells of Black Creek Canal virus, a New World hantavirus. J Virol, 1997. 71(2): p. 1147-54.
111. Helenius, A. and M. Aebi, Intracellular functions of N-linked glycans. Science, 2001. 291(5512): p. 2364-9.
112. Hansen, J.E., O. Lund, K. Rapacki, and S. Brunak, O-GLYCBASE version 2.0: a revised database of O-glycosylated proteins. Nucleic Acids Res, 1997. 25(1): p. 278-82.
113. Gabius, H.J., S. Andre, H. Kaltner, and H.C. Siebert, The sugar code: functional lectinomics. Biochim Biophys Acta, 2002. 1572(2-3): p. 165-77.
114. Opdenakker, G., P.M. Rudd, C.P. Ponting, and R.A. Dwek, Concepts and principles of glycobiology. Faseb J, 1993. 7(14): p. 1330-7.
115. Spiropoulou, C.F., S. Morzunov, H. Feldmann, A. Sanchez, C.J. Peters, and S.T. Nichol, Genome structure and variability of a virus causing hantavirus pulmonary syndrome. Virology, 1994. 200(2): p. 715-23.
116. Geimonen, E., R. LaMonica, K. Springer, Y. Farooqui, I.N. Gavrilovskaya, and E.R. Mackow, Hantavirus pulmonary syndrome-associated hantaviruses contain conserved and functional ITAM signaling elements. J Virol, 2003. 77(2): p. 1638-43.
117. Xu, X.N., B. Laffert, G.R. Screaton, M. Kraft, D. Wolf, W. Kolanus, J. Mongkolsapay, et al., Induction of Fas ligand expression by HIV involves the interaction of Nef with the T cell receptor zeta chain. J Exp Med, 1999. 189(9): p. 1489-96.
118. Dehghani, H., C.R. Brown, R. Plishka, A. Buckler-White, and V.M. Hirsch, The ITAM in Nef influences acute pathogenesis of AIDS-inducing simian immunodeficiency viruses SIVsm and SIVagm without altering kinetics or extent of viremia. J Virol, 2002. 76(9): p. 4379-89.
119. Luo, W. and B.M. Peterlin, Activation of the T-cell receptor signaling pathway by Nef from an aggressive strain of simian immunodeficiency virus. J Virol, 1997. 71(12): p. 9531-7.
120. Inatome, R., S. Yanagi, T. Takano, and H. Yamamura, A critical role for Syk in endothelial cell proliferation and migration. Biochem Biophys Res Commun, 2001. 286(1): p. 195-9.
84

References
121. Geimonen, E., I. Fernandez, I.N. Gavrilovskaya, and E.R. Mackow, Tyrosine residues direct the ubiquitination and degradation of the NY-1 hantavirus G1 cytoplasmic tail. J Virol, 2003. 77(20): p. 10760-868.
122. Bharadwaj, M., C.R. Lyons, I.A. Wortman, and B. Hjelle, Intramuscular inoculation of Sin Nombre hantavirus cDNAs induces cellular and humoral immune responses in BALB/c mice. Vaccine, 1999. 17(22): p. 2836-43.
123. Hooper, J.W., D.M. Custer, E. Thompson, and C.S. Schmaljohn, DNA vaccination with the Hantaan virus M gene protects Hamsters against three of four HFRS hantaviruses and elicits a high-titer neutralizing antibody response in Rhesus monkeys. J Virol, 2001. 75(18): p. 8469-77.
124. Terajima, M., H.L. Van Epps, D. Li, A.M. Leporati, S.E. Juhlin, J. Mustonen, A. Vaheri, et al., Generation of recombinant vaccinia viruses expressing Puumala virus proteins and use in isolating cytotoxic T cells specific for Puumala virus. Virus Res, 2002. 84(1-2): p. 67-77.
125. Gavrilovskaya, I.N., M. Shepley, R. Shaw, M.H. Ginsberg, and E.R. Mackow, beta3 Integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc Natl Acad Sci U S A, 1998. 95(12): p. 7074-9.
126. Gavrilovskaya, I.N., E.J. Brown, M.H. Ginsberg, and E.R. Mackow, Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by beta3 integrins. J Virol, 1999. 73(5): p. 3951-9.
127. Kim, T.Y., Y. Choi, H.S. Cheong, and J. Choe, Identification of a cell surface 30 kDa protein as a candidate receptor for Hantaan virus. J Gen Virol, 2002. 83(Pt 4): p. 767-73.
128. Pekosz, A. and F. Gonzalez-Scarano, The extracellular domain of La Crosse virus G1 forms oligomers and undergoes pH-dependent conformational changes. Virology, 1996. 225(1): p. 243-7.
129. Arikawa, J., I. Takashima, and N. Hashimoto, Cell fusion by haemorrhagic fever with renal syndrome (HFRS) viruses and its application for titration of virus infectivity and neutralizing antibody. Arch Virol, 1985. 86(3-4): p. 303-13.
130. Ogino, M., K. Yoshimatsu, H. Ebihara, K. Araki, B.H. Lee, M. Okumura, and J. Arikawa, Cell fusion activities of Hantaan virus envelope glycoproteins. J Virol, 2004. 78(19): p. 10776-82.
131. Jin, M., J. Park, S. Lee, B. Park, J. Shin, K.J. Song, T.I. Ahn, et al., Hantaan virus enters cells by clathrin-dependent receptor-mediated endocytosis. Virology, 2002. 294(1): p. 60-9.
132. Zhang, X.K., I. Takashima, and N. Hashimoto, Characteristics of passive immunity against hantavirus infection in rats. Arch Virol, 1989. 105(3-4): p. 235-46.
133. Arikawa, J., J.S. Yao, K. Yoshimatsu, I. Takashima, and N. Hashimoto, Protective role of antigenic sites on the envelope protein of Hantaan virus defined by monoclonal antibodies. Arch Virol, 1992. 126(1-4): p. 271-81.
134. Koch, J., M. Liang, I. Queitsch, A.A. Kraus, and E.K. Bautz, Human recombinant neutralizing antibodies against hantaan virus G2 protein. Virology, 2003. 308(1): p. 64-73.
135. Liang, M., M. Mahler, J. Koch, Y. Ji, D. Li, C. Schmaljohn, and E.K. Bautz, Generation of an HFRS patient-derived neutralizing recombinant antibody to Hantaan virus G1 protein and definition of the neutralizing domain. J Med Virol, 2003. 69(1): p. 99-107.
136. Hörling, J. and Å. Lundkvist, Single amino acid substitutions in Puumala virus envelope glycoproteins G1 and G2 eliminate important neutralization epitopes. Virus Res, 1997. 48(1): p. 89-100.
137. Chu, Y.K., C. Rossi, J.W. Leduc, H.W. Lee, C.S. Schmaljohn, and J.M. Dalrymple, Serological relationships among viruses in the Hantavirus genus, family Bunyaviridae. Virology, 1994. 198(1): p. 196-204.
138. Jonsson, C.B. and C.S. Schmaljohn, Replication of hantaviruses. Curr Top Microbiol Immunol, 2001. 256: p. 15-32.
139. Pettersson, R.F., The structure of Uukuniemi virus, a proposed member of the bunyaviruses. Med Biol, 1975. 53(5): p. 418-24.
140. Jin, H. and R.M. Elliott, Mutagenesis of the L protein encoded by Bunyamwera virus and production of monospecific antibodies. J Gen Virol, 1992. 73 ( Pt 9): p. 2235-44.
85

Patrik Johansson 2005
141. Jin, H. and R.M. Elliott, Expression of functional Bunyamwera virus L protein by recombinant vaccinia viruses. J Virol, 1991. 65(8): p. 4182-9.
142. Jin, H. and R.M. Elliott, Characterization of Bunyamwera virus S RNA that is transcribed and replicated by the L protein expressed from recombinant vaccinia virus. J Virol, 1993. 67(3): p. 1396-404.
143. Schmaljohn, C.S. and J.M. Dalrymple, Analysis of Hantaan virus RNA: evidence for a new genus of bunyaviridae. Virology, 1983. 131(2): p. 482-91.
144. Plyusnin, A., Y. Cheng, H. Lehvaslaiho, and A. Vaheri, Quasispecies in wild-type tula hantavirus populations. J Virol, 1996. 70(12): p. 9060-3.
145. Flick, R., F. Elgh, and R.F. Pettersson, Mutational analysis of the Uukuniemi virus (Bunyaviridae family) promoter reveals two elements of functional importance. J Virol, 2002. 76(21): p. 10849-60.
146. Flick, R., G. Neumann, E. Hoffmann, E. Neumeier, and G. Hobom, Promoter elements in the influenza vRNA terminal structure. Rna, 1996. 2(10): p. 1046-57.
147. Mackow, E.R. and I.N. Gavrilovskaya, Cellular receptors and hantavirus pathogenesis. Curr Top Microbiol Immunol, 2001. 256: p. 91-115.
148. Gavrilovskaya, I.N., T. Peresleni, E. Geimonen, and E.R. Mackow, Pathogenic hantaviruses selectively inhibit beta3 integrin directed endothelial cell migration. Arch Virol, 2002. 147(10): p. 1913-31.
149. den Boon, J.A., J. Chen, and P. Ahlquist, Identification of sequences in Brome mosaic virus replicase protein 1a that mediate association with endoplasmic reticulum membranes. J Virol, 2001. 75(24): p. 12370-81.
150. Kukkonen, S.K., A. Vaheri, and A. Plyusnin, L protein, the RNA-dependent RNA polymerase of hantaviruses. Arch Virol, 2005. 150(3): p. 533-56.
151. Garcin, D., M. Lezzi, M. Dobbs, R.M. Elliott, C. Schmaljohn, C.Y. Kang, and D. Kolakofsky, The 5' ends of Hantaan virus (Bunyaviridae) RNAs suggest a prime-and-realign mechanism for the initiation of RNA synthesis. J Virol, 1995. 69(9): p. 5754-62.
152. Hutchinson, K.L., C.J. Peters, and S.T. Nichol, Sin Nombre virus mRNA synthesis. Virology, 1996. 224(1): p. 139-49.
153. Morzunov, S.P., H. Feldmann, C.F. Spiropoulou, V.A. Semenova, P.E. Rollin, T.G. Ksiazek, C.J. Peters, et al., A newly recognized virus associated with a fatal case of hantavirus pulmonary syndrome in Louisiana. J Virol, 1995. 69(3): p. 1980-3.
154. Beaton, A.R. and R.M. Krug, Transcription antitermination during influenza viral template RNA synthesis requires the nucleocapsid protein and the absence of a 5' capped end. Proc Natl Acad Sci U S A, 1986. 83(17): p. 6282-6.
155. Patton, J.T., N.L. Davis, and G.W. Wertz, N protein alone satisfies the requirement for protein synthesis during RNA replication of vesicular stomatitis virus. J Virol, 1984. 49(2): p. 303-9.
156. Elliott, R.M., M. Bouloy, C.H. Calisher, R. Goldbach, J.T. Moyer, S.T. Nichol, R.F. Pettersson, et al., Bunyaviridae, in Virus Taxonomy: VIIth report of the International Committee on Taxonomy of Viruses, M.H.V. van Regenmortel, et al., Editors. 1999, Academic Press: San Diego, California. p. 599-631.
157. Plyusnin, A. and S.P. Morzunov, Virus evolution and genetic diversity of hantaviruses and their rodent hosts. Curr Top Microbiol Immunol, 2001. 256: p. 47-75.
158. Sjölander, K.B., I. Golovljova, V. Vasilenko, A. Plyusnin, and Å. Lundkvist, Serological divergence of Dobrava and Saaremaa hantaviruses: evidence for two distinct serotypes. Epidemiol Infect, 2002. 128(1): p. 99-103.
159. Meyer, B.J. and C.S. Schmaljohn, Persistent hantavirus infections: characteristics and mechanisms. Trends Microbiol, 2000. 8(2): p. 61-7.
160. Slonova, R.A., E.A. Tkachenko, E.L. Kushnarev, T.K. Dzagurova, and T.I. Astakova, Hantavirus isolation from birds. Acta Virol, 1992. 36(5): p. 493.
86

References
161. Malecki, T.M., G.P. Jillson, J.P. Thilsted, J. Elrod, N. Torrez-Martinez, and B. Hjelle, Serologic survey for hantavirus infection in domestic animals and coyotes from New Mexico and northeastern Arizona. J Am Vet Med Assoc, 1998. 212(7): p. 970-3.
162. Ahlm, C., K. Wallin, Å. Lundkvist, F. Elgh, P. Juto, M. Merza, and A. Tärnvik, Serologic evidence of Puumala virus infection in wild moose in northern Sweden. Am J Trop Med Hyg, 2000. 62(1): p. 106-11.
163. Danes, L., M. Pejcoch, K. Bukovjan, J. Veleba, and M. Halackova, [Antibodies against Hantaviruses in game and domestic oxen in the Czech Republic]. Cesk Epidemiol Mikrobiol Imunol, 1992. 41(1): p. 15-8.
164. Escutenaire, S. and P.P. Pastoret, [Hantavirus infection epidemiology in Belgium]. Bull Mem Acad R Med Belg, 2001. 156(1-2): p. 137-44; discussion 144-6.
165. Meyer, B.J. and C. Schmaljohn, Accumulation of terminally deleted RNAs may play a role in Seoul virus persistence. J Virol, 2000. 74(3): p. 1321-31.
166. Hutchinson, K.L., P.E. Rollin, and C.J. Peters, Pathogenesis of a North American hantavirus, Black Creek Canal virus, in experimentally infected Sigmodon hispidus. Am J Trop Med Hyg, 1998. 59(1): p. 58-65.
167. Lundkvist, Å., D. Wiger, J. Hörling, K.B. Sjölander, A. Plyusnina, R. Mehl, A. Vaheri, et al., Isolation and characterization of Puumala hantavirus from Norway: evidence for a distinct phylogenetic sublineage. J Gen Virol, 1998. 79 ( Pt 11): p. 2603-14.
168. Avsic-Zupanc, T., A. Toney, K. Anderson, Y.K. Chu, and C. Schmaljohn, Genetic and antigenic properties of Dobrava virus: a unique member of the Hantavirus genus, family Bunyaviridae. J Gen Virol, 1995. 76 ( Pt 11): p. 2801-8.
169. Wang, H., K. Yoshimatsu, H. Ebihara, M. Ogino, K. Araki, H. Kariwa, Z. Wang, et al., Genetic diversity of hantaviruses isolated in china and characterization of novel hantaviruses isolated from Niviventer confucianus and Rattus rattus. Virology, 2000. 278(2): p. 332-45.
170. Plyusnin, A., O. Vapalahti, V. Vasilenko, H. Henttonen, and A. Vaheri, Dobrava hantavirus in Estonia: does the virus exist throughout Europe? Lancet, 1997. 349(9062): p. 1369-70.
171. Nemirov, K., O. Vapalahti, Å. Lundkvist, V. Vasilenko, I. Golovljova, A. Plyusnina, J. Niemimaa, et al., Isolation and characterization of Dobrava hantavirus carried by the striped field mouse (Apodemus agrarius) in Estonia. J Gen Virol, 1999. 80 ( Pt 2): p. 371-9.
172. Yashina, L.N., N.A. Patrushev, L.I. Ivanov, R.A. Slonova, V.P. Mishin, G.G. Kompanez, N.I. Zdanovskaya, et al., Genetic diversity of hantaviruses associated with hemorrhagic fever with renal syndrome in the far east of Russia. Virus Res, 2000. 70(1-2): p. 31-44.
173. Lokugamage, K., H. Kariwa, D. Hayasaka, B.Z. Cui, T. Iwasaki, N. Lokugamage, L.I. Ivanov, et al., Genetic characterization of hantaviruses transmitted by the Korean field mouse (Apodemus peninsulae), Far East Russia. Emerg Infect Dis, 2002. 8(8): p. 768-76.
174. Plyusnin, A., O. Vapalahti, H. Lankinen, H. Lehvaslaiho, N. Apekina, Y. Myasnikov, H. Kallio-Kokko, et al., Tula virus: a newly detected hantavirus carried by European common voles. J Virol, 1994. 68(12): p. 7833-9.
175. Plyusnin, A., O. Vapalahti, Å. Lundkvist, H. Henttonen, and A. Vaheri, Newly recognised hantavirus in Siberian lemmings. Lancet, 1996. 347(9018): p. 1835.
176. Lee, P.W., H.L. Amyx, D.C. Gajdusek, R.T. Yanagihara, D. Goldgaber, and C.J. Gibbs, Jr., New hemorrhagic fever with renal syndrome-related virus in rodents in the United States. Lancet, 1982. 2(8312): p. 1405.
177. Song, W., N. Torrez-Martinez, W. Irwin, F.J. Harrison, R. Davis, M. Ascher, M. Jay, et al., Isla Vista virus: a genetically novel hantavirus of the California vole Microtus californicus. J Gen Virol, 1995. 76 ( Pt 12): p. 3195-9.
178. Kariwa, H., S. Yoshizumi, J. Arikawa, K. Yoshimatsu, K. Takahashi, I. Takashima, and N. Hashimoto, Evidence for the existence of Puumula-related virus among Clethrionomys rufocanus in Hokkaido, Japan. Am J Trop Med Hyg, 1995. 53(3): p. 222-7.
87

Patrik Johansson 2005
179. Hörling, J., V. Chizhikov, Å. Lundkvist, M. Jonsson, L. Ivanov, A. Dekonenko, B. Niklasson, et al., Khabarovsk virus: a phylogenetically and serologically distinct hantavirus isolated from Microtus fortis trapped in far-east Russia. J Gen Virol, 1996. 77 ( Pt 4): p. 687-94.
180. Kariwa, H., K. Yoshimatsu, J. Sawabe, E. Yokota, J. Arikawa, I. Takashima, H. Fukushima, et al., Genetic diversities of hantaviruses among rodents in Hokkaido, Japan and Far East Russia. Virus Res, 1999. 59(2): p. 219-28.
181. Lopez, N., P. Padula, C. Rossi, M.E. Lazaro, and M.T. Franze-Fernandez, Genetic identification of a new hantavirus causing severe pulmonary syndrome in Argentina. Virology, 1996. 220(1): p. 223-6.
182. Bharadwaj, M., J. Botten, N. Torrez-Martinez, and B. Hjelle, Rio Mamore virus: genetic characterization of a newly recognized hantavirus of the pygmy rice rat, Oligoryzomys microtis, from Bolivia. Am J Trop Med Hyg, 1997. 57(3): p. 368-74.
183. Hjelle, B., J. Krolikowski, N. Torrez-Martinez, F. Chavez-Giles, C. Vanner, and E. Laposata, Phylogenetically distinct hantavirus implicated in a case of hantavirus pulmonary syndrome in the northeastern United States. J Med Virol, 1995. 46(1): p. 21-7.
184. Johnson, A.M., M.D. Bowen, T.G. Ksiazek, R.J. Williams, R.T. Bryan, J.N. Mills, C.J. Peters, et al., Laguna Negra virus associated with HPS in western Paraguay and Bolivia. Virology, 1997. 238(1): p. 115-27.
185. Ravkov, E.V., P.E. Rollin, T.G. Ksiazek, C.J. Peters, and S.T. Nichol, Genetic and serologic analysis of Black Creek Canal virus and its association with human disease and Sigmodon hispidus infection. Virology, 1995. 210(2): p. 482-9.
186. Rawlings, J.A., N. Torrez-Martinez, S.U. Neill, G.M. Moore, B.N. Hicks, S. Pichuantes, A. Nguyen, et al., Cocirculation of multiple hantaviruses in Texas, with characterization of the small (S) genome of a previously undescribed virus of cotton rats (Sigmodon hispidus). Am J Trop Med Hyg, 1996. 55(6): p. 672-9.
187. Hjelle, B., F. Chavez-Giles, N. Torrez-Martinez, T. Yates, J. Sarisky, J. Webb, and M. Ascher, Genetic identification of a novel hantavirus of the harvest mouse Reithrodontomys megalotis. J Virol, 1994. 68(10): p. 6751-4.
188. Hjelle, B., F. Chavez-Giles, N. Torrez-Martinez, T. Yamada, J. Sarisky, M. Ascher, and S. Jenison, Dominant glycoprotein epitope of four corners hantavirus is conserved across a wide geographical area. J Gen Virol, 1994. 75 ( Pt 11): p. 2881-8.
189. Song, J.W., L.J. Baek, J.W. Nagle, D. Schlitter, and R. Yanagihara, Genetic and phylogenetic analyses of hantaviral sequences amplified from archival tissues of deer mice (Peromyscus maniculatus nubiterrae) captured in the eastern United States. Arch Virol, 1996. 141(5): p. 959-67.
190. Elliott, L.H., T.G. Ksiazek, P.E. Rollin, C.F. Spiropoulou, S. Morzunov, M. Monroe, C.S. Goldsmith, et al., Isolation of the causative agent of hantavirus pulmonary syndrome. Am J Trop Med Hyg, 1994. 51(1): p. 102-8.
191. Hjelle, B., B. Anderson, N. Torrez-Martinez, W. Song, W.L. Gannon, and T.L. Yates, Prevalence and geographic genetic variation of hantaviruses of New World harvest mice (Reithrodontomys): identification of a divergent genotype from a Costa Rican Reithrodontomys mexicanus. Virology, 1995. 207(2): p. 452-9.
192. Fulhorst, C.F., M.C. Monroe, R.A. Salas, G. Duno, A. Utrera, T.G. Ksiazek, S.T. Nichol, et al., Isolation, characterization and geographic distribution of Cano Delgadito virus, a newly discovered South American hantavirus (family Bunyaviridae). Virus Res, 1997. 51(2): p. 159-71.
193. Lopez, N., P. Padula, C. Rossi, S. Miguel, A. Edelstein, E. Ramirez, and M.T. Franze-Fernandez, Genetic characterization and phylogeny of Andes virus and variants from Argentina and Chile. Virus Res, 1997. 50(1): p. 77-84.
194. Levis, S., J.E. Rowe, S. Morzunov, D.A. Enria, and S. St Jeor, New hantaviruses causing hantavirus pulmonary syndrome in central Argentina. Lancet, 1997. 349(9057): p. 998-9.
88

References
195. Vincent, M.J., E. Quiroz, F. Gracia, A.J. Sanchez, T.G. Ksiazek, P.T. Kitsutani, L.A. Ruedas, et al., Hantavirus pulmonary syndrome in Panama: identification of novel hantaviruses and their likely reservoirs. Virology, 2000. 277(1): p. 14-9.
196. Morzunov, S.P., J.E. Rowe, T.G. Ksiazek, C.J. Peters, S.C. St Jeor, and S.T. Nichol, Genetic analysis of the diversity and origin of hantaviruses in Peromyscus leucopus mice in North America. J Virol, 1998. 72(1): p. 57-64.
197. Sanchez, A.J., K.D. Abbott, and S.T. Nichol, Genetic identification and characterization of limestone canyon virus, a unique Peromyscus-borne hantavirus. Virology, 2001. 286(2): p. 345-53.
198. Nemirov, K., Genetic Relationships of Saaremaa and Dobrava Hantaviruses, in Department of virology, Haartman Institute. 2003, Univeristy of Helsinki: Helsinki. p. 94.
199. Lednicky, J.A., Hantaviruses. a short review. Arch Pathol Lab Med, 2003. 127(1): p. 30-5. 200. Savela, M., Life. 2004. 201. Schultze, D., Å. Lundkvist, U. Blauenstein, and P. Heyman, Tula virus infection associated
with fever and exanthema after a wild rodent bite. Eur J Clin Microbiol Infect Dis, 2002. 21(4): p. 304-6.
202. Song, J.W., L.J. Baek, K.J. Song, A. Skrok, J. Markowski, J. Bratosiewicz-Wasik, R. Kordek, et al., Characterization of Tula virus from common voles (microtus arvalis) in Poland: evidence for geographic-specific phylogenetic clustering. Virus Genes, 2004. 29(2): p. 239-47.
203. Song, J.W., A. Gligic, and R. Yanagihara, Identification of Tula hantavirus in Pitymys subterraneus captured in the Cacak region of Serbia-Yugoslavia. Int J Infect Dis, 2002. 6(1): p. 31-6.
204. Plyusnin, A., Y. Cheng, O. Vapalahti, M. Pejcoch, J. Unar, Z. Jelinkova, H. Lehvaslaiho, et al., Genetic variation in Tula hantaviruses: sequence analysis of the S and M segments of strains from Central Europe. Virus Res, 1995. 39(2-3): p. 237-50.
205. Sibold, C., H. Meisel, D.H. Krüger, M. Labuda, J. Lysy, O. Kozuch, M. Pejcoch, et al., Recombination in Tula hantavirus evolution: analysis of genetic lineages from Slovakia. J Virol, 1999. 73(1): p. 667-75.
206. Bowen, M.D., W. Gelbmann, T.G. Ksiazek, S.T. Nichol, and N. Nowotny, Puumala virus and two genetic variants of Tula virus are present in Austrian rodents. J Med Virol, 1997. 53(2): p. 174-81.
207. Heyman, P., J. Klingström, F. de Jaegere, G. Leclercq, F. Rozenfeld, S. Escutenaire, C. Vandenvelde, et al., Tula hantavirus in Belgium. Epidemiol Infect, 2002. 128(2): p. 251-6.
208. Niklasson, B. and J.W. LeDuc, Epidemiology of nephropathia epidemica in Sweden. J Infect Dis, 1987. 155(2): p. 269-76.
209. Asikainen, K., T. Hanninen, H. Henttonen, J. Niemimaa, J. Laakkonen, H.K. Andersen, N. Bille, et al., Molecular evolution of puumala hantavirus in Fennoscandia: phylogenetic analysis of strains from two recolonization routes, Karelia and Denmark. J Gen Virol, 2000. 81(Pt 12): p. 2833-41.
210. Sironen, T., A. Plyusnina, H.K. Andersen, J. Lodal, H. Leirs, J. Niemimaa, H. Henttonen, et al., Distribution of Puumala hantavirus in Denmark: analysis of bank voles (Clethrionomys glareolus) from Fyn and Jutland. Vector Borne Zoonotic Dis, 2002. 2(1): p. 37-45.
211. Hörling, J., Å. Lundkvist, M. Jaarola, A. Plyusnin, H. Tegelstrom, K. Persson, H. Lehvaslaiho, et al., Distribution and genetic heterogeneity of Puumala virus in Sweden. J Gen Virol, 1996. 77 (10): p. 2555-62.
212. Hughes, A.L. and R. Friedman, Evolutionary diversification of protein-coding genes of hantaviruses. Mol Biol Evol, 2000. 17(10): p. 1558-68.
213. Sironen, T., A. Vaheri, and A. Plyusnin, Molecular evolution of Puumala hantavirus. J Virol, 2001. 75(23): p. 11803-10.
214. Jackson, A.P. and M.A. Charleston, A cophylogenetic perspective of RNA-virus evolution. Mol Biol Evol, 2004. 21(1): p. 45-57.
89

Patrik Johansson 2005
215. Vapalahti, O., Å. Lundkvist, V. Fedorov, C.J. Conroy, S. Hirvonen, A. Plyusnina, K. Nemirov, et al., Isolation and characterization of a hantavirus from Lemmus sibiricus: evidence for host switch during hantavirus evolution. J Virol, 1999. 73(7): p. 5586-92.
216. Nemirov, K., H. Henttonen, A. Vaheri, and A. Plyusnin, Phylogenetic evidence for host switching in the evolution of hantaviruses carried by Apodemus mice. Virus Res, 2002. 90(1-2): p. 207-15.
217. Holland, J.J., J.C. De La Torre, and D.A. Steinhauer, RNA Virus Populations as Quasispecies, in Genetic Diversity of RNA Viruses. 1992, Springer-Verlag: Berlin. p. 1-20.
218. Worobey, M. and E.C. Holmes, Evolutionary aspects of recombination in RNA viruses. J Gen Virol, 1999. 80 ( Pt 10): p. 2535-43.
219. Plyusnin, A., S.K. Kukkonen, A. Plyusnina, O. Vapalahti, and A. Vaheri, Transfection-mediated generation of functionally competent Tula hantavirus with recombinant S RNA segment. Embo J, 2002. 21(6): p. 1497-503.
220. Henderson, W.W., M.C. Monroe, S.C. St Jeor, W.P. Thayer, J.E. Rowe, C.J. Peters, and S.T. Nichol, Naturally occurring Sin Nombre virus genetic reassortants. Virology, 1995. 214(2): p. 602-10.
221. Klempa, B., H.A. Schmidt, R. Ulrich, S. Kaluz, M. Labuda, H. Meisel, B. Hjelle, et al., Genetic interaction between distinct Dobrava hantavirus subtypes in Apodemus agrarius and A. flavicollis in nature. J Virol, 2003. 77(1): p. 804-9.
222. Li, D., A.L. Schmaljohn, K. Anderson, and C.S. Schmaljohn, Complete nucleotide sequences of the M and S segments of two hantavirus isolates from California: evidence for reassortment in nature among viruses related to hantavirus pulmonary syndrome. Virology, 1995. 206(2): p. 973-83.
223. Lundkvist, Å., Y. Cheng, K.B. Sjölander, B. Niklasson, A. Vaheri, and A. Plyusnin, Cell culture adaptation of Puumala hantavirus changes the infectivity for its natural reservoir, Clethrionomys glareolus, and leads to accumulation of mutants with altered genomic RNA S segment. J Virol, 1997. 71(12): p. 9515-23.
224. Terajima, M., O. Vapalahti, H.L. Van Epps, A. Vaheri, and F.A. Ennis, Immune responses to Puumala virus infection and the pathogenesis of nephropathia epidemica. Microbes Infect, 2004. 6(2): p. 238-45.
225. Nakamura, T., R. Yanagihara, C.J. Gibbs, Jr., H.L. Amyx, and D.C. Gajdusek, Differential susceptibility and resistance of immunocompetent and immunodeficient mice to fatal Hantaan virus infection. Arch Virol, 1985. 86(1-2): p. 109-20.
226. Yoshimatsu, K., J. Arikawa, S. Ohbora, and C. Itakura, Hantavirus infection in SCID mice. J Vet Med Sci, 1997. 59(10): p. 863-8.
227. Mustonen, J., J. Partanen, M. Kanerva, K. Pietila, O. Vapalahti, A. Pasternack, and A. Vaheri, Genetic susceptibility to severe course of nephropathia epidemica caused by Puumala hantavirus. Kidney Int, 1996. 49(1): p. 217-21.
228. McNeil, A.J., P.L. Yap, S.M. Gore, R.P. Brettle, M. McColl, R. Wyld, S. Davidson, et al., Association of HLA types A1-B8-DR3 and B27 with rapid and slow progression of HIV disease. Qjm, 1996. 89(3): p. 177-85.
229. Mustonen, J., J. Partanen, M. Kanerva, K. Pietila, O. Vapalahti, A. Pasternack, and A. Vaheri, Association of HLA B27 with benign clinical course of nephropathia epidemica caused by Puumala hantavirus. Scand J Immunol, 1998. 47(3): p. 277-9.
230. Maes, P., J. Clement, I. Gavrilovskaya, and M. Van Ranst, Hantaviruses: immunology, treatment, and prevention. Viral Immunol, 2004. 17(4): p. 481-97.
231. Riquelme, R., M. Riquelme, A. Torres, M.L. Rioseco, J.A. Vergara, L. Scholz, and A. Carriel, Hantavirus pulmonary syndrome, southern Chile. Emerg Infect Dis, 2003. 9(11): p. 1438-43.
232. Passaro, D.J., W.J. Shieh, J.K. Hacker, C.L. Fritz, S.R. Hogan, M. Fischer, R.M. Hendry, et al., Predominant kidney involvement in a fatal case of hantavirus pulmonary syndrome caused by Sin Nombre virus. Clin Infect Dis, 2001. 33(2): p. 263-4.
90

References
233. Linderholm, M., A. Billström, B. Settergren, and A. Tärnvik, Pulmonary involvement in nephropathia epidemica as demonstrated by computed tomography. Infection, 1992. 20(5): p. 263-6.
234. Schutt, M., H. Meisel, D.H. Krüger, R. Ulrich, K. Dalhoff, and C. Dodt, Life-threatening Dobrava hantavirus infection with unusually extended pulmonary involvement. Clin Nephrol, 2004. 62(1): p. 54-7.
235. Clement, J., P. Colson, and P. McKenna, Hantavirus pulmonary syndrome in New England and Europe. N Engl J Med, 1994. 331(8): p. 545-6; author reply 547-8.
236. Ahlm, C., M. Linderholm, P. Juto, B. Stegmayr, and B. Settergren, Prevalence of serum IgG antibodies to Puumala virus (haemorrhagic fever with renal syndrome) in northern Sweden. Epidemiol Infect, 1994. 113(1): p. 129-36.
237. Settergren, B., Nephropathia epidemica (hemorrhagic fever with renal syndrome) in Scandinavia. Rev Infect Dis, 1991. 13(4): p. 736-44.
238. Settergren, B., C. Ahlm, O. Alexeyev, J. Billheden, and B. Stegmayr, Pathogenetic and clinical aspects of the renal involvement in hemorrhagic fever with renal syndrome. Ren Fail, 1997. 19(1): p. 1-14.
239. Mustonen, J., M. Brummer-Korvenkontio, K. Hedman, A. Pasternack, K. Pietila, and A. Vaheri, Nephropathia epidemica in Finland: a retrospective study of 126 cases. Scand J Infect Dis, 1994. 26(1): p. 7-13.
240. Huggins, J.W., C.M. Hsiang, T.M. Cosgriff, M.Y. Guang, J.I. Smith, Z.O. Wu, J.W. LeDuc, et al., Prospective, double-blind, concurrent, placebo-controlled clinical trial of intravenous ribavirin therapy of hemorrhagic fever with renal syndrome. J Infect Dis, 1991. 164(6): p. 1119-27.
241. Alexeyev, O., Personal communication. 2005. 242. Ahlm, C., Personal communication. 2005. 243. Sirotin, B.Z., I.M. Davidovich, and Y.L. Fedorchenko. Use of Plasmapheresis in Treatment of
Patients with HFRS. in The 6th International Conferance on Hemrrhagic Fever with Renal Syndrome (HFRS), Hantavirus Pulmonary Syndrome and Hantaviruses. 2004. Seoul: International Society for Hantaviruses.
244. Kanerva, M., J. Mustonen, and A. Vaheri, Pathogenesis of puumala and other hantavirus infections. Rev Med Virol, 1998. 8(2): p. 67-86.
245. Graziano, K.L. and B. Tempest, Hantavirus pulmonary syndrome: a zebra worth knowing. Am Fam Physician, 2002. 66(6): p. 1015-20.
246. PAHO, http://www.paho.org/English/AD/DPC/CD/hanta-americas-93-02.pdf. 1993, Pan American Health Organization.
247. Kitsutani, P.T., R.W. Denton, C.L. Fritz, R.A. Murray, R.L. Todd, W.J. Pape, J. Wyatt Frampton, et al., Acute Sin Nombre hantavirus infection without pulmonary syndrome, United States. Emerg Infect Dis, 1999. 5(5): p. 701-5.
248. Enria, D.A., A.M. Briggiler, N. Pini, and S. Levis, Clinical manifestations of New World hantaviruses. Curr Top Microbiol Immunol, 2001. 256: p. 117-34.
249. Nichol, S.T., T.G. Ksiazek, P.E. Rollin, and C.J. Peters, Hantavirus Pulmonary Syndrome and Newly Described Hantaviruses in the United States, in The Bunyaviridae, R.M. Elliott, Editor. 1996, Plenum Press: New York. p. 269-280.
250. Chapman, L.E., G.J. Mertz, C.J. Peters, H.M. Jolson, A.S. Khan, T.G. Ksiazek, F.T. Koster, et al., Intravenous ribavirin for hantavirus pulmonary syndrome: safety and tolerance during 1 year of open-label experience. Ribavirin Study Group. Antivir Ther, 1999. 4(4): p. 211-9.
251. Kallio-Kokko, H., O. Vapalahti, Å. Lundkvist, and A. Vaheri, Evaluation of Puumala virus IgG and IgM enzyme immunoassays based on recombinant baculovirus-expressed nucleocapsid protein for early nephropathia epidemica diagnosis. Clin Diagn Virol, 1998. 10(1): p. 83-90.
252. Sjölander, K.B., F. Elgh, H. Kallio-Kokko, O. Vapalahti, M. Hägglund, V. Palmcrantz, P. Juto, et al., Evaluation of serological methods for diagnosis of Puumala hantavirus infection (nephropathia epidemica). J Clin Microbiol, 1997. 35(12): p. 3264-8.
91

Patrik Johansson 2005
253. Elgh, F., M. Linderholm, G. Wadell, and P. Juto, The clinical usefulness of a Puumalavirus recombinant nucleocapsid protein based enzyme-linked immunosorbent assay in the diagnosis of nephropathia epidemica as compared with an immunofluorescence assay. Clinical and Diagnostic Virology, 1996. 6(1): p. 17-26.
254. Ivanov, A., O. Vapalahti, H. Lankinen, E. Tkachenko, A. Vaheri, B. Niklasson, and Å. Lundkvist, Biotin-labeled antigen: a novel approach for detection of Puumala virus-specific IgM. J Virol Methods, 1996. 62(1): p. 87-92.
255. Tanishita, O., Y. Takahashi, Y. Okuno, K. Yamanishi, and M. Takahashi, Evaluation of focus reduction neutralization test with peroxidase-antiperoxidase staining technique for hemorrhagic fever with renal syndrome virus. J Clin Microbiol, 1984. 20(6): p. 1213-5.
256. Niklasson, B., M. Jonsson, Å. Lundkvist, J. Hörling, and E. Tkachenko, Comparison of European isolates of viruses causing hemorrhagic fever with renal syndrome by a neutralization test. Am J Trop Med Hyg, 1991. 45(6): p. 660-5.
257. Heider, H., B. Ziaja, C. Priemer, Å. Lundkvist, J. Neyts, D.H. Krüger, and R. Ulrich, A chemiluminescence detection method of hantaviral antigens in neutralisation assays and inhibitor studies. J Virol Methods, 2001. 96(1): p. 17-23.
258. Hörling, J., Å. Lundkvist, J.W. Huggins, and B. Niklasson, Antibodies to Puumala virus in humans determined by neutralization test. J Virol Methods, 1992. 39(1-2): p. 139-47.
259. Hujakka, H., V. Koistinen, P. Eerikainen, I. Kuronen, A. Laatikainen, J. Kauppinen, A. Vaheri, et al., Comparison of a new immunochromatographic rapid test with a commercial EIA for the detection of Puumala virus specific IgM antibodies. J Clin Virol, 2001. 23(1-2): p. 79-85.
260. Hujakka, H., V. Koistinen, P. Eerikainen, I. Kuronen, I. Mononen, M. Parviainen, Å. Lundkvist, et al., New immunochromatographic rapid test for diagnosis of acute Puumala virus infection. J Clin Microbiol, 2001. 39(6): p. 2146-50.
261. Hujakka, H., V. Koistinen, I. Kuronen, P. Eerikainen, M. Parviainen, Å. Lundkvist, A. Vaheri, et al., Diagnostic rapid tests for acute hantavirus infections: specific tests for Hantaan, Dobrava and Puumala viruses versus a hantavirus combination test. J Virol Methods, 2003. 108(1): p. 117-22.
262. Juto, P., Personal communication. 263. Tsai, T.F., Y.W. Tang, S.L. Hu, K.L. Ye, G.L. Chen, and Z.Y. Xu, Hemagglutination-
inhibiting antibody in hemorrhagic fever with renal syndrome. J Infect Dis, 1984. 150(6): p. 895-8. 264. Lu, Q., Z. Zhu, and J. Weng, Immune responses to inactivated vaccine in people naturally infected
with hantaviruses. J Med Virol, 1996. 49(4): p. 333-5. 265. Inouye, S., S. Matsuno, and R. Kono, Difference in antibody reactivity between complement fixation
and immune adherence hemagglutination tests with virus antigens. J Clin Microbiol, 1981. 14(3): p. 241-6.
266. Tang, Y.W., Y.L. Li, K.L. Ye, Z.Y. Xu, S.L. Ruo, S.P. Fisher-Hoch, and J.B. McCormick, Distribution of hantavirus serotypes Hantaan and Seoul causing hemorrhagic fever with renal syndrome and identification by hemagglutination inhibition assay. J Clin Microbiol, 1991. 29(9): p. 1924-7.
267. Sugiyama, K., Y. Matsuura, C. Morita, S. Shiga, Y. Akao, T. Komatsu, and T. Kitamura, An immune adherence assay for discrimination between etiologic agents of hemorrhagic fever with renal syndrome. J Infect Dis, 1984. 149(1): p. 67-73.
268. Gresikova, M. and M. Sekeyova, A simple method of preparing a complement-fixing antigen from the virus of haemorrhagic fever with renal syndrome (Western type). Acta Virol, 1988. 32(3): p. 272-4.
269. Juto, P., F. Elgh, C. Ahlm, O.A. Alexeyev, K. Edlund, Å. Lundkvist, and G. Wadell, The first human isolate of Puumala virus in Scandinavia as cultured from phytohemagglutinin stimulated leucocytes. J Med Virol, 1997. 53(2): p. 150-6.
270. Biel, S.S. and H.R. Gelderblom, Diagnostic electron microscopy is still a timely and rewarding method. J Clin Virol, 1999. 13(1-2): p. 105-19.
92

References
271. Cao, C., C. Shi, P. Li, Y. Tong, and Q. Ma, Diagnosis of hepatitis C Virus (HCV) infection by antigen-capturing ELISA. Clin Diagn Virol, 1996. 6(2-3): p. 137-45.
272. Garin, D., C. Peyrefitte, J.M. Crance, A. Le Faou, A. Jouan, and M. Bouloy, Highly sensitive Taqman PCR detection of Puumala hantavirus. Microbes Infect, 2001. 3(9): p. 739-45.
273. Aitichou, M., S.S. Saleh, A.K. McElroy, C. Schmaljohn, and M.S. Ibrahim, Identification of Dobrava, Hantaan, Seoul, and Puumala viruses by one-step real-time RT-PCR. J Virol Methods, 2005. 124(1-2): p. 21-6.
274. Wilson, W.J., C.L. Strout, T.Z. DeSantis, J.L. Stilwell, A.V. Carrano, and G.L. Andersen, Sequence-specific identification of 18 pathogenic microorganisms using microarray technology. Mol Cell Probes, 2002. 16(2): p. 119-27.
275. Li, J., S. Chen, and D.H. Evans, Typing and subtyping influenza virus using DNA microarrays and multiplex reverse transcriptase PCR. J Clin Microbiol, 2001. 39(2): p. 696-704.
276. Karaman, M.W., S. Groshen, C.C. Lee, B.L. Pike, and J.G. Hacia, Comparisons of substitution, insertion and deletion probes for resequencing and mutational analysis using oligonucleotide microarrays. Nucleic Acids Res, 2005. 33(3): p. e33.
277. Amexis, G., P. Oeth, K. Abel, A. Ivshina, F. Pelloquin, C.R. Cantor, A. Braun, et al., Quantitative mutant analysis of viral quasispecies by chip-based matrix-assisted laser desorption/ ionization time-of-flight mass spectrometry. Proc Natl Acad Sci U S A, 2001. 98(21): p. 12097-102.
278. Ronaghi, M., M. Uhlen, and P. Nyren, A sequencing method based on real-time pyrophosphate. Science, 1998. 281(5375): p. 363, 365.
279. Settergren, B., C. Ahlm, P. Juto, and B. Niklasson, Specific Puumala IgG virus half a century after haemorrhagic fever with renal syndrome. Lancet, 1991. 338(8758): p. 66.
280. Lundkvist, Å., J. Hörling, and B. Niklasson, The humoral response to Puumala virus infection (nephropathia epidemica) investigated by viral protein specific immunoassays. Arch Virol, 1993. 130(1-2): p. 121-30.
281. Lundkvist, Å., H. Kallio-Kokko, K.B. Sjölander, H. Lankinen, B. Niklasson, A. Vaheri, and O. Vapalahti, Characterization of Puumala virus nucleocapsid protein: identification of B-cell epitopes and domains involved in protective immunity. Virology, 1996. 216(2): p. 397-406.
282. Dargeviciute, A., K. Brus Sjolander, K. Sasnauskas, D.H. Krüger, H. Meisel, R. Ulrich, and Å. Lundkvist, Yeast-expressed Puumala hantavirus nucleocapsid protein induces protection in a bank vole model. Vaccine, 2002. 20(29-30): p. 3523-31.
283. Alexeyev, O.A., C. Ahlm, J. Billheden, B. Settergren, G. Wadell, and P. Juto, Elevated levels of total and Puumala virus-specific immunoglobulin E in the Scandinavian type of hemorrhagic fever with renal syndrome. Clin Diagn Lab Immunol, 1994. 1(3): p. 269-72.
284. Bostik, P., J. Winter, T.G. Ksiazek, P.E. Rollin, F. Villinger, S.R. Zaki, C.J. Peters, et al., Sin nombre virus (SNV) Ig isotype antibody response during acute and convalescent phases of hantavirus pulmonary syndrome. Emerg Infect Dis, 2000. 6(2): p. 184-7.
285. Groen, J., J. Dalrymple, S. Fisher-Hoch, J.G. Jordans, J.P. Clement, and A.D. Osterhaus, Serum antibodies to structural proteins of Hantavirus arise at different times after infection. J Med Virol, 1992. 37(4): p. 283-7.
286. Vapalahti, O., Å. Lundkvist, and A. Vaheri, Human immune response, host genetics, and severity of disease. Curr Top Microbiol Immunol, 2001. 256: p. 153-69.
287. Araki, K., K. Yoshimatsu, M. Ogino, H. Ebihara, Å. Lundkvist, H. Kariwa, I. Takashima, et al., Truncated hantavirus nucleocapsid proteins for serotyping Hantaan, Seoul, and Dobrava hantavirus infections. J Clin Microbiol, 2001. 39(7): p. 2397-404.
288. Paakkala, A., J. Mustonen, M. Viander, H. Huhtala, and A. Pasternack, Complement activation in nephropathia epidemica caused by Puumala hantavirus. Clin Nephrol, 2000. 53(6): p. 424-31.
93

Patrik Johansson 2005
289. Yoshimatsu, K., J. Arikawa, M. Tamura, R. Yoshida, Å. Lundkvist, B. Niklasson, H. Kariwa, et al., Characterization of the nucleocapsid protein of Hantaan virus strain 76-118 using monoclonal antibodies. J Gen Virol, 1996. 77 ( Pt 4): p. 695-704.
290. Linderholm, M., L. Bjermer, P. Juto, G. Roos, T. Sandström, B. Settergren, and A. Tärnvik, Local host response in the lower respiratory tract in nephropathia epidemica. Scand J Infect Dis, 1993. 25(5): p. 639-46.
291. Van Epps, H.L., M. Terajima, J. Mustonen, T.P. Arstila, E.A. Corey, A. Vaheri, and F.A. Ennis, Long-lived memory T lymphocyte responses after hantavirus infection. J Exp Med, 2002. 196(5): p. 579-88.
292. Plyusnin, A., J. Hörling, M. Kanerva, J. Mustonen, Y. Cheng, J. Partanen, O. Vapalahti, et al., Puumala hantavirus genome in patients with nephropathia epidemica: correlation of PCR positivity with HLA haplotype and link to viral sequences in local rodents. J Clin Microbiol, 1997. 35(5): p. 1090-6.
293. Kilpatrick, E.D., M. Terajima, F.T. Koster, M.D. Catalina, J. Cruz, and F.A. Ennis, Role of specific CD8+ T cells in the severity of a fulminant zoonotic viral hemorrhagic fever, hantavirus pulmonary syndrome. J Immunol, 2004. 172(5): p. 3297-304.
294. Balasubramanian, A.V., Prevention of smallpox in ancient India. CURRENT SCIENCE, 2002. 83(9-10): p. 1055.
295. Bazin, H., A brief history of the prevention of infectious diseases by immunisations. Comp Immunol Microbiol Infect Dis, 2003. 26(5-6): p. 293-308.
296. Wright, P.F., Global immunization--a medical perspective. Soc Sci Med, 1995. 41(5): p. 609-16. 297. Kloetzel, P.M., The proteasome and MHC class I antigen processing. Biochim Biophys Acta,
2004. 1695(1-3): p. 225-33. 298. Lauvau, G. and N. Glaichenhaus, Mini-review: Presentation of pathogen-derived antigens in vivo.
Eur J Immunol, 2004. 34(4): p. 913-20. 299. Giese, M., DNA-antiviral vaccines: new developments and approaches--a review. Virus Genes, 1998.
17(3): p. 219-32. 300. Spengler, U., M. Lechmann, B. Irrgang, F.L. Dumoulin, and T. Sauerbruch, Immune
responses in hepatitis C virus infection. J Hepatol, 1996. 24(2 Suppl): p. 20-5. 301. Callow, K.A., H.F. Parry, M. Sergeant, and D.A. Tyrrell, The time course of the immune
response to experimental coronavirus infection of man. Epidemiol Infect, 1990. 105(2): p. 435-46. 302. Smallpox vaccine adverse events among civilians--United States, February 25-March 3, 2003. MMWR
Morb Mortal Wkly Rep, 2003. 52(9): p. 180-1, 191. 303. Castelli, F. and A. Patroni, The human immunodeficiency virus-infected traveler. Clin Infect Dis,
2000. 31(6): p. 1403-8. 304. Minor, P.D., G. Dunn, M.E. Ramsay, and D. Brown, Effect of different immunisation schedules
on the excretion and reversion of oral poliovaccine strains. J Med Virol, 2005. 75(1): p. 153-60. 305. Mandl, C.W., J.H. Aberle, S.W. Aberle, H. Holzmann, S.L. Allison, and F.X. Heinz, In
vitro-synthesized infectious RNA as an attenuated live vaccine in a flavivirus model. Nat Med, 1998. 4(12): p. 1438-40.
306. Offit, P.A., The Cutter incident, 50 years later. N Engl J Med, 2005. 352(14): p. 1411-2. 307. Davis, H.L. and M.J. McCluskie, DNA vaccines for viral diseases. Microbes Infect, 1999. 1(1):
p. 7-21. 308. Kaiser, B., Vaccinationer. 1997, Lund: Studentlitteratur. 309. Geldmacher, A., D. Skrastina, I. Petrovskis, G. Borisova, J.A. Berriman, A.M. Roseman,
R.A. Crowther, et al., An amino-terminal segment of hantavirus nucleocapsid protein presented on hepatitis B virus core particles induces a strong and highly cross-reactive antibody response in mice. Virology, 2004. 323(1): p. 108-19.
310. Milich, D.R., D.L. Peterson, J. Zheng, J.L. Hughes, R. Wirtz, and F. Schodel, The hepatitis nucleocapsid as a vaccine carrier moiety. Ann N Y Acad Sci, 1995. 754: p. 187-201.
94

References
311. Yu, J. and W.H. Langridge, A plant-based multicomponent vaccine protects mice from enteric diseases. Nat Biotechnol, 2001. 19(6): p. 548-52.
312. Webster, D.E., M.C. Thomas, Z. Huang, and S.L. Wesselingh, The development of a plant-based vaccine for measles. Vaccine, 2005. 23(15): p. 1859-65.
313. Kehm, R., N.J. Jakob, T.M. Welzel, E. Tobiasch, O. Viczian, S. Jock, K. Geider, et al., Expression of immunogenic Puumala virus nucleocapsid protein in transgenic tobacco and potato plants. Virus Genes, 2001. 22(1): p. 73-83.
314. Khattak, S., G. Darai, S. Sule, and A. Rosen-Wolff, Characterization of expression of Puumala virus nucleocapsid protein in transgenic plants. Intervirology, 2002. 45(4-6): p. 334-9.
315. Khattak, S., G. Darai, and A. Rosen-Wolff, Puumala virus nucleocapsid protein expressed in transgenic plants is not immunogenic after oral administration. Virus Genes, 2004. 29(1): p. 109-16.
316. Bot, A. and C. Bona, Genetic immunization of neonates. Microbes Infect, 2002. 4(4): p. 511-20. 317. Wolff, J.A., R.W. Malone, P. Williams, W. Chong, G. Acsadi, A. Jani, and P.L. Felgner,
Direct gene transfer into mouse muscle in vivo. Science, 1990. 247(4949 Pt 1): p. 1465-8. 318. Tang, D.C., M. DeVit, and S.A. Johnston, Genetic immunization is a simple method for eliciting
an immune response. Nature, 1992. 356(6365): p. 152-4. 319. Robinson, H.L. and C.A. Torres, DNA vaccines. Semin Immunol, 1997. 9(5): p. 271-83. 320. Coon, B., L.L. An, J.L. Whitton, and M.G. von Herrath, DNA immunization to prevent
autoimmune diabetes. J Clin Invest, 1999. 104(2): p. 189-94. 321. von Herrath, M.G. and J.L. Whitton, DNA vaccination to treat autoimmune diabetes. Ann Med,
2000. 32(5): p. 285-92. 322. Mimran, A., F. Mor, P. Carmi, F.J. Quintana, V. Rotter, and I.R. Cohen, DNA vaccination
with CD25 protects rats from adjuvant arthritis and induces an antiergotypic response. J Clin Invest, 2004. 113(6): p. 924-32.
323. Schoen, C., J. Stritzker, W. Goebel, and S. Pilgrim, Bacteria as DNA vaccine carriers for genetic immunization. Int J Med Microbiol, 2004. 294(5): p. 319-35.
324. Hartl, A., R. Weiss, R. Hochreiter, S. Scheiblhofer, and J. Thalhamer, DNA vaccines for allergy treatment. Methods, 2004. 32(3): p. 328-39.
325. Manam, S., B.J. Ledwith, A.B. Barnum, P.J. Troilo, C.J. Pauley, L.B. Harper, T.G. Griffiths, 2nd, et al., Plasmid DNA vaccines: tissue distribution and effects of DNA sequence, adjuvants and delivery method on integration into host DNA. Intervirology, 2000. 43(4-6): p. 273-81.
326. Kang, K.K., S.M. Choi, J.H. Choi, D.S. Lee, C.Y. Kim, B.O. Ahn, B.M. Kim, et al., Safety evaluation of GX-12, a new HIV therapeutic vaccine: investigation of integration into the host genome and expression in the reproductive organs. Intervirology, 2003. 46(5): p. 270-6.
327. Nichols, W.W., B.J. Ledwith, S.V. Manam, and P.J. Troilo, Potential DNA vaccine integration into host cell genome. Ann N Y Acad Sci, 1995. 772: p. 30-9.
328. Nishikawa, M. and L. Huang, Nonviral vectors in the new millennium: delivery barriers in gene transfer. Hum Gene Ther, 2001. 12(8): p. 861-70.
329. Krieg, A.M. and J.N. Kline, Immune effects and therapeutic applications of CpG motifs in bacterial DNA. Immunopharmacology, 2000. 48(3): p. 303-5.
330. Park, K., C.S. Kim, and K.T. Moon, Protective effectiveness of hantavirus vaccine. Emerg Infect Dis, 2004. 10(12): p. 2218-20.
331. Hooper, J.W. and D. Li, Vaccines against hantaviruses. Curr Top Microbiol Immunol, 2001. 256: p. 171-91.
332. de Carvalho Nicacio, C., M. Gonzalez Della Valle, P. Padula, E. Björling, A. Plyusnin, and Å. Lundkvist, Cross-protection against challenge with Puumala virus after immunization with nucleocapsid proteins from different hantaviruses. J Virol, 2002. 76(13): p. 6669-77.
333. Xu, X., S.L. Ruo, J.B. McCormick, and S.P. Fisher-Hoch, Immunity to Hantavirus challenge in Meriones unguiculatus induced by vaccinia-vectored viral proteins. Am J Trop Med Hyg, 1992. 47(4): p. 397-404.
95

Patrik Johansson 2005
334. McClain, D.J., P.L. Summers, S.A. Harrison, A.L. Schmaljohn, and C.S. Schmaljohn, Clinical evaluation of a vaccinia-vectored Hantaan virus vaccine. J Med Virol, 2000. 60(1): p. 77-85.
335. Kamrud, K.I., J.W. Hooper, F. Elgh, and C.S. Schmaljohn, Comparison of the protective efficacy of naked DNA, DNA-based Sindbis replicon, and packaged Sindbis replicon vectors expressing Hantavirus structural genes in hamsters. Virology, 1999. 263(1): p. 209-19.
336. Koletzki, D., R. Schirmbeck, Å. Lundkvist, H. Meisel, D.H. Krüger, and R. Ulrich, DNA vaccination of mice with a plasmid encoding Puumala hantavirus nucleocapsid protein mimics the B-cell response induced by virus infection. J Biotechnol, 2001. 84(1): p. 73-8.
337. Bucht, G., K.B. Sjolander, S. Eriksson, L. Lindgren, Å. Lundkvist, and F. Elgh, Modifying the cellular transport of DNA-based vaccines alters the immune response to hantavirus nucleocapsid protein. Vaccine, 2001. 19(28-29): p. 3820-9.
338. Bharadwaj, M., K. Mirowsky, C. Ye, J. Botten, B. Masten, J. Yee, C.R. Lyons, et al., Genetic vaccines protect against Sin Nombre hantavirus challenge in the deer mouse (Peromyscus maniculatus). J Gen Virol, 2002. 83(Pt 7): p. 1745-51.
339. Hooper, J.W., K.I. Kamrud, F. Elgh, D. Custer, and C.S. Schmaljohn, DNA vaccination with hantavirus M segment elicits neutralizing antibodies and protects against seoul virus infection. Virology, 1999. 255(2): p. 269-78.
340. Custer, D.M., E. Thompson, C.S. Schmaljohn, T.G. Ksiazek, and J.W. Hooper, Active and passive vaccination against hantavirus pulmonary syndrome with Andes virus M genome segment-based DNA vaccine. J Virol, 2003. 77(18): p. 9894-905.
341. Groen, J., M. Gerding, J.P. Koeman, P.J. Roholl, G. van Amerongen, H.G. Jordans, H.G. Niesters, et al., A macaque model for hantavirus infection. J Infect Dis, 1995. 172(1): p. 38-44.
342. Klingström, J., A. Plyusnin, A. Vaheri, and Å. Lundkvist, Wild-type Puumala hantavirus infection induces cytokines, C-reactive protein, creatinine, and nitric oxide in cynomolgus macaques. J Virol, 2002. 76(1): p. 444-9.
343. Hooper, J.W., T. Larsen, D.M. Custer, and C.S. Schmaljohn, A lethal disease model for hantavirus pulmonary syndrome. Virology, 2001. 289(1): p. 6-14.
344. Jentoft, N., Why are proteins O-glycosylated? Trends Biochem Sci, 1990. 15(8): p. 291-4. 345. Owens, R.J., J.W. Dubay, E. Hunter, and R.W. Compans, Human immunodeficiency virus
envelope protein determines the site of virus release in polarized epithelial cells. Proc Natl Acad Sci U S A, 1991. 88(9): p. 3987-91.
346. Gupta, R., E. Jung, and B. S., Prediction of N-glycosylation sites in human proteins. Manuscript in preparation, 2004. http://www.cbs.dtu.dk/services/NetNGlyc/.
347. Hansen, J.E., O. Lund, J. Engelbrecht, H. Bohr, and J.O. Nielsen, Prediction of O-glycosylation of mammalian proteins: specificity patterns of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase. Biochem J, 1995. 308 ( Pt 3): p. 801-13.
348. Hansen, J.E., O. Lund, J. Nilsson, K. Rapacki, and S. Brunak, O-GLYCBASE Version 3.0: a revised database of O-glycosylated proteins. Nucleic Acids Res, 1998. 26(1): p. 387-9.
349. Sykes, K.F. and S.A. Johnston, Linear expression elements: a rapid, in vivo, method to screen for gene functions. Nat Biotechnol, 1999. 17(4): p. 355-9.
350. Zanta, M.A., P. Belguise-Valladier, and J.P. Behr, Gene delivery: a single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc Natl Acad Sci U S A, 1999. 96(1): p. 91-6.
351. Allen, T.D., J.M. Cronshaw, S. Bagley, E. Kiseleva, and M.W. Goldberg, The nuclear pore complex: mediator of translocation between nucleus and cytoplasm. J Cell Sci, 2000. 113 ( Pt 10): p. 1651-9.
352. Salman, H., D. Zbaida, Y. Rabin, D. Chatenay, and M. Elbaum, Kinetics and mechanism of DNA uptake into the cell nucleus. Proc Natl Acad Sci U S A, 2001. 98(13): p. 7247-52.
353. Stein, C.A. and J.S. Cohen, Oligodeoxynucleotides as inhibitors of gene expression: a review. Cancer Res, 1988. 48(10): p. 2659-68.
96

References
354. Adam, S.A. and L. Gerace, Cytosolic proteins that specifically bind nuclear location signals are receptors for nuclear import. Cell, 1991. 66(5): p. 837-47.
355. Lewin, M., N. Carlesso, C.H. Tung, X.W. Tang, D. Cory, D.T. Scadden, and R. Weissleder, Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat Biotechnol, 2000. 18(4): p. 410-4.
356. Shalon, D., S.J. Smith, and P.O. Brown, A DNA microarray system for analyzing complex DNA samples using two-color fluorescent probe hybridization. Genome Res, 1996. 6(7): p. 639-45.
357. Kurian, K.M., C.J. Watson, and A.H. Wyllie, DNA chip technology. J Pathol, 1999. 187(3): p. 267-71.
358. Small, J., D.R. Call, F.J. Brockman, T.M. Straub, and D.P. Chandler, Direct detection of 16S rRNA in soil extracts by using oligonucleotide microarrays. Appl Environ Microbiol, 2001. 67(10): p. 4708-16.
359. Chizhikov, V., M. Wagner, A. Ivshina, Y. Hoshino, A.Z. Kapikian, and K. Chumakov, Detection and genotyping of human group A rotaviruses by oligonucleotide microarray hybridization. J Clin Microbiol, 2002. 40(7): p. 2398-407.
97





























