Embed Size (px)
Citation preview

Impaired Cell Adhesion and Apoptosis in aNovel CLN9 Batten Disease Variant
Angela Schulz, MD,1 Sumeer Dhar, PhD,1,2 Svetlana Rylova, PhD,1,3 Ghassan Dbaibo, MD,4
Joseph Alroy, DVM,5 Christian Hagel, MD,6 Isabelo Artacho, MD,7 Alfried Kohlschutter, MD,8
Simon Lin, MD,9 and Rose-Mary Boustany, MD1
We describe the ninth variant of neuronal ceroid lipofuscinosis (NCL) or Batten disease, due to defects in a putative newgene, CLN9. We therefore refer to the new variant as CLN9-deficient. Two Serbian sisters and two German brothers aredescribed. Their clinical history is characteristic for juvenile NCL. They show similar gene expression patterns. Theexistence of this variant is supported by the presence of curvilinear inclusions, fingerprint profiles, and granular osmi-ophilic deposits in neurons, lymphocytes, and conjunctival cells. Enzyme screening and sequencing of the coding regionsof other NCL genes was negative. CLN9-deficient cells have a distinctive phenotype. They have rounded cell bodies, haveprominent nucleoli, attach poorly to the culture dish, and are sensitive to apoptosis but have increased growth rates.Gene expression of proteins involved in cell adhesion and apoptosis is altered in these cells. Sphingolipid metabolism isalso perturbed. They have decreased levels of ceramide, sphingomyelin, lactosylceramide, ceramide trihexoside, and glo-boside and increased activity of serine palmitoyl transferase.
Ann Neurol 2004;56:342–350
A novel neuronal storage disease is described in twoSerbian sisters and two German brothers. The clinicalpresentation is similar to juvenile neuronal ceroid lipo-fuscinosis (JNCL). The NCLs are a group of inheritedneurodegenerative disorders. Clinical features includevisual loss, mental and motor deterioration, seizures,and early death.1,2 The diagnosis of NCL was based onclinical course and appearance of inclusions in cells andnow is confirmed by enzyme and/or genetic testing.
Classic variants with known gene defects include in-fantile NCL (INCL), late infantile NCL (LINCL),JNCL, and a rare adult variant.3–6 Atypical variantsinclude the Finnish variant, the Costa Rican or Portu-guese variant, Northern epilepsy with mental retarda-tion (EPMR), and the Turkish variant, with some casesallelic to EPMR.7–11 Another atypical variant is juve-nile variant with GRODS (granular osmiophilic depos-its).12,13 Tissues from NCL patients contain autofluo-rescent membrane bound inclusions with variableultrastructural characteristics. These inclusions aregranular, curvilinear, or fingerprint-like. The genes
CLN1 (INCL and juvenile variant with GRODS) andCLN2 (LINCL) both code for lysosomal enzymes. TheCLN1 gene product is lysosomal palmitoyl-proteinthioesterase 1 (PPT1).14 The CLN2 gene product istripeptidyl-peptidase 1 (TPP1).15 The CLN3 (JNCL),CLN8 (EPMR), and CLN6 (Portuguese/Costa Ricanvariant) genes code for novel transmembrane pro-teins.16–19 The CLN5 protein is a soluble glycopro-tein.20,21 The CLN8 protein possesses a Lag1 motiflike other TLC proteins (TRAM-Lag1p-CLN8).22 TheLag1 motif imparts ceramide synthase activity toyeast.23,24
The patients we describe had typical autofluorescentinclusions in brain, lymphocytes, and conjunctiva. En-zyme assays as well as molecular tests for known vari-ants of NCL were normal. Other storage diseases withneuronal involvement were ruled out. Fibroblasts frompatients have a distinctive phenotype. This study de-scribes clinical characteristics of the disease and pro-vides biological, biochemical, and gene expression cluesto its pathogenesis.
From the 1Departments of Pediatrics and Neurobiology, Duke Uni-versity Medical Center, Durham, NC; 2Division of Clinical Phar-macology, Department of Medical Sciences, Uppsala UniversityHospital; 3Institute of Molecular Biosciences, Section of VeterinaryMedical Biochemistry, Biomedical Center, Uppsala, Sweden; 4De-partments of Pediatrics and Biochemistry, American University ofBeirut, Beirut, Lebanon; 5Department of Pathology, Tufts Univer-sity School of Medicine and Veterinary Medicine, New EnglandMedical Center, Boston, MA; 6Department of Neuropathology,University of Hamburg, Germany; 7University Medical Center,University of California San Francisco, Fresno, CA; 8Department ofPediatrics, University of Hamburg, Hamburg, Germany; and 9Duke
Comprehensive Cancer Center and Duke Center for Bioinformaticsand Computational Biology, Duke University Medical Center,Durham, NC.
Received Sep 10, 2003, and in revised form Feb 5 and Apr 27,2004. Accepted for publication Mar 8, 2004.
Published online Jul 27, 2004 in Wiley InterScience(www.interscience.wiley.com). DOI: 10.1002/ana.20187
Address correspondence to Dr Boustany, Duke University MedicalCenter, MSRB Box 2604, Research Drive, Durham, NC, 27710.E-mail: [email protected]
342 © 2004 American Neurological AssociationPublished by Wiley-Liss, Inc., through Wiley Subscription Services

Materials and MethodsCell LinesCLN9-deficient fibroblast lines were derived from skin biop-sies of one Serbian sister and one German brother. TheCLN1-deficient fibroblast lines were provided by Dr S. Hoff-man (University of Texas, Southwestern Medical Center).CLN2-, CLN3-, and CLN6-deficient fibroblast lines werefrom patients at Duke University Medical Center. Use ofhuman cell lines in research is covered by an approved Dukeinstitutional review board protocol.
Tissue CultureFibroblasts were grown at 37°C with 5% CO2 in Dulbecco’smodified Eagle medium (DMEM) (Gibco-Invitrogen, GrandIsland, NY) containing 10% fetal bovine serum (FBS)(Gibco-Invitrogen) and 1% antibiotics/antimycotics.
Affymetrix GeneChip AnalysisPreparation of Total RNA/Probe LabelingRNA was isolated using RNeasy Mini Kit (Qiagen, Valencia,CA). Double-strand cDNA then was synthesized using Super-script II reverse transcriptase (Gibco-Invitrogen) (Affymetrix,Santa Clara, CA). Biotin-labeled cRNA was produced fromcDNA by an in vitro transcription reaction. Twenty microgramsof fragmented cRNA was hybridized at 45°C for 16 hours withthe hybridization target kit, and then probes were washed andstained in Streptavidin Phycoerythin solution (SAPE, final con-centration 10 �g/ml, Molecular Probes, Eugene, OR) solutionfor 30 minutes, with a final wash at 25°C at 10 cycles (fourmixes/cycle), in the Affymetrix Fluidics Station 400. Probe ar-rays were scanned at 570nm wavelength (pixel size, 3�m).
Data AcquisitionGeneChip analysis was run four times using the AffymetrixHuFL 6900 GeneChip and twice using the HG-U95Av2GeneChip. The Affymetrix Test 2 array was used to assessquality of target RNA. After scanning probe arrays, data werestored, converted to .txt files, imported into Microsoft Excel,and used for data analysis and interpretation.
Data AnalysisGenechip data were analyzed with the Affymetrix software,Microsoft Excel, and Access, Cluster, and S-plus (Seattle, WA)software. The raw measurements of expression of each genewere taken (AvgDiff), and a cubic root transformation was ap-plied to the raw data. This permits inclusion of negative valuesand reduces larger variation at higher expression levels. Thetransformed data were normalized by a weighted linear leastsquares fit. This procedure calibrates individual hybridizationswith a reference set, which was generated by averaging the ex-pression of each gene over all hybridizations. This weightedlinear fit method takes all genes into consideration but isweighted toward 47 housekeeping genes. After data normaliza-tion, fold changes were computed. Genes with negative inten-sity were reset to a minimum value of 20 to eliminate infinite-fold changes. Only genes without absent calls (AbsCall fromAffymetrix software) from all replicates, and with greater thantwofold changes in all possible pairs of comparison of CLN9-deficient versus normal samples, were included. Expression
levels were visualized by Cluster software with an establishedcolor code (red for upregulation, green for downregulation).
Neuronal Ceroid Lipofuscinosis GenesThe coding sequences for CLN3, CLN5, CLN6, and CLN8genes were amplified as previously described.25 Products weresequenced using an automated sequencer (377XL PrismDNA Sequencer; PE Biosystems, Foster City, CA).
Northern BlotA Northern blot was hybridized according to the manufac-turer’s protocol (Clontech, Palo Alto, CA). Probes were gen-erated by polymerase chain reaction (PCR) amplification ofthe CLN3 gene. PCR products were purified by QIAQuickPCR purification kit (Qiagen) and were [32P] labeled withthe RadPrime DNA labeling system (Gibco-Invitrogen). Hy-bridization was performed with ExpressHyb HybridizationSolution (Clontech) at 65°C.
Immunocytochemical Analysis of Subunit c ofMitochondrial ATP SynthaseParaffin-embedded slides of frontal cortex tissue from one ofthe CLN9-deficient and from one age-matched control pa-tient were used for staining. Immunocytochemical stainingwas performed as previously described.26 The primary anti-body used was raised by immunizing rabbits with a syntheticpeptide corresponding to N-terminal amino acid residues32-45 of subunit c of mitochondrial ATP synthase. Theslides were developed with 3,3�-diaminobenzidine (Sigma,St. Louis, MO) at 1mg/ml in phosphate-buffered saline(PBS) containing 0.003% H2O2 and then counterstainedwith hematoxylin (Sigma). The stained tissue sections wereexamined under �100 and �150 magnification.
Live and Dead Attached and Detached FibroblastsEqual numbers of normal and CLN9-deficient fibroblasts (1 �107 cells) were grown in 100 � 20mm dishes for 24 hours,after which either adherent live and dead cells or those in thesupernatant were harvested and counted by the trypan blue dyeexclusion method. The experiment was performed in triplicate.Counts of live and dead control fibroblasts were compared withthose of live and dead patient fibroblasts using Student’s t test.
Growth RateCells (7 � 104/well) were plated in 24-well plates and thenharvested and counted at 24, 48, 72, 96, and 124 hours intriplicate for each time point using the trypan blue dye ex-clusion method.
Trypan Blue Dye ExclusionHarvested cells were centrifuged at 1,200 rpm for 5 minutesand pellets resuspended in 1 to 1 mixture of 4% trypan bluedye (Gibco-Invitrogen) and 1 � PBS, loaded onto a hemo-cytometer, and counted. Viable cells are white, and dead cellsare blue.
[3H]Thymidine IncorporationCells were plated in six-well plates at a density of 1 � 105
cells per well. After 0, 6, 12, 24, and 48 hours, cells from
Schulz et al: Adhesion and Apoptosis in CLN9 343

triplicate wells were incubated with 2�Ci/ml [3H]thymidine(Perkin Elmer, Boston, MA) in medium for 2 hours, washedtwice with ice-cold PBS, and DNA-precipitated with 5% tri-chloroacetic acid. The DNA precipitate was dissolved in0.2ml of 0.25M NaOH. Incorporated [3H]thymidine wasdetermined by liquid scintillation counting.
Etoposide TreatmentCells were treated with 10�g/ml etoposide (Sigma) for 18hours. Apoptosis was determined by propidium iodide (PI)staining and expressed as a ratio of red apoptotic cells tototal cells.
Propidium Iodide StainingEqual numbers of cells treated/not treated with etoposide weregrown on cover slips and stained with PI (5�g/ml) for 5 min-utes. Three fields of vision were chosen randomly at �100magnification. The total number of cells/field of vision(20–60 cells/field) was counted. The number of PI-positivered apoptotic cells was determined under fluorescence (excita-tion wavelength, 525nm; emission wavelength, 600nm). Thepercentage of PI-positive cells/total cells/field of vision was cal-culated, as well as average and standard deviation. Statisticalsignificance was determined using the Student’s t test.
Filipin StainingNiemann-Pick type C and normal and CLN9-deficient fibro-blasts were maintained in McCoy’s medium (Gibco-Invitrogen)supplemented with 10% LPDS (Intracel Corporation, Freder-ick, MD) for 5 days, harvested, seeded on cover slips, and in-cubated at 37°C for 24 hours in Dulbecco’s modified Eagle me-dium (DMEM) (Gibco-Invitrogen) containing 10% FBS and1% antibiotics/antimycotics. Cover slips were rinsed three timeswith Dulbecco’s PBS and fixed with 10% formalin (pH 7.4;Sigma) at room temperature for 1 hour. Monolayers were rinsedthree times with PBS and stained with 800 �l filipin solutionfor 60 minutes. The filipin solution was prepared by dissolving2.5mg of filipin complex (Sigma) in 1ml of dimethylformamide(Sigma). This was added to 50ml of PBS. Cells were rinsedthree times with 2ml PBS, and the cover slips were mounted onmicroscope slides with fluoromount (Southern Biotechnology,Birmingham, AL). Slides were examined under fluorescence (ex-citation wavelength, 372nm; emission wavelength, 446nm) at�100 and �400 magnification.
Sphingolipid LevelsCeramide and sphingomyelin levels in CLN9-deficient andnormal fibroblasts were measured as previously described andalso were quantified by mass spectrometry in the LipidomicsCore at the Medical University of South Carolina.27 Glyco-sphingolipid levels were measured after [14C]-galactose label-ing according to published methods.28 Results were normal-ized to lipid phosphate.
Serine Palmitoyl Transferase ActivityTwo hundred micrograms of protein was dissolved in 0.1MHepes, pH 8.0, 5mM DTT, 5mM EDTA, pH 7.4, and50�M pyridoxal-5-phosphate and reaction-initiated by add-ing 0.2mM Palmitoyl-CoA, 1mM Serine (Sigma), and16.75�l [14C]-serine (179.2mCi/mmol; Sigma) per 100�l of
sample. Reactions were incubated for 15 minutes at 37°Cand then terminated with 1.5ml chloroform-methanol (1:2).Organic soluble counts were extracted and quantified by liq-uid scintillation counting.27,29
ResultsIdentification of the CLN9-Deficient GenotypeAffymetrix GeneChip analysis of known NCL and un-known cases led to the discovery of this novel CLN9-deficient variant. RNA from the four cases had identi-cal gene expression profiles, which were distinctlydifferent from normal and from CLN1-, CLN2-,CLN3- and CLN6-deficient RNA. This pattern was re-produced six times. Figure 1 shows a partial dendro-gram of gene expression analyses performed with theHuFL 6900 GeneChip. Upregulated genes are labeledred; downregulated genes are green. The color blackdenotes no significant change in gene expression com-pared with control. Expression of genes involved in celladhesion and apoptosis was dysregulated in CLN9-deficient cells. These are shown in the table posted atwww.dbsr.duke.edu/pub/cln9/ and referred to below inthe discussion.
Clinical CourseThe clinical course of the CLN9-deficient patients isquite similar to that in JNCL patients. The two broth-ers presented at age 4 years with declining vision and
Fig 1. The CLN9-deficient genotype. Partial dendrogram de-picting gene expression patterns of CLN1-, CLN2-, CLN3-,CLN6-, and CLN9-deficient and normal fibroblast RNA.Upregulated genes are red, and downregulated genes are green.No change from control is black. Note similarity of gene ex-pression patterns in both CLN9-deficient cell lines [CLN9(1)Serbian patient, CLN9(2) German patient] in two experi-ments. Lanes 1, 2, and 9 and lanes 3, 4, and 10, respec-tively, represent experiments performed on two separate days.
344 Annals of Neurology Vol 56 No 3 September 2004

Fig 2. (A–C) Electron micrographs of brain sections from CLN9-deficient patients. (A) Electron micrograph of brain tissue fromone of the German CLN9-deficient brothers demonstrating the presence of secondary lysosomes containing curvilinear bodies. Magni-fication �20,000. (B, C) Electron micrograph of right frontal lobe brain biopsy of one of the Serbian CLN9-deficient patients il-lustrating granular osmiophilic deposits (GRODS) (arrows) and curvilinear bodies (arrow in frame). (B) Magnification �54,000.(C) (inset) Magnification �100,000. (D, E) Immunohistochemical analysis for subunit c of mitochondrial ATP synthase of normal(D) and CLN9-deficient (E) frontal cortex tissue sections. Note gray staining of neuronal cytoplasm in CLN9-deficient brain tissueindicating increased amounts of subunit c, as opposed to the normal blue stain of control brain tissue (arrows). Magnification�150; scale bar � 50�m
Schulz et al: Adhesion and Apoptosis in CLN9 345

seizures. Cognitive decline was apparent at age 6 years,with ataxia and rigidity at age 9 years. They developeddysarthria and scanning speech and were mute by age12 years. The younger brother died at age 15 yearsfollowing a bout of pneumonia. The older brother de-veloped hallucinations, intractable seizures, and diffi-culty swallowing and died at age 19 years. We are un-aware of any history of consanguinity in the Germanbrothers. Although there is no history of close consan-guinity in the Serbian sisters, the great-grandmotherscame from adjacent villages. The clinical course of theSerbian sisters is very similar to that of the Germanbrothers. They developed declining vision, progressiveataxia, and seizures with onset at age 4 years. By 9years, they could not ambulate independently. Theybecame mute at the age of 10 years and suffered fromfrequent generalized and myoclonic seizures. Electroen-cephalograms in all cases showed slowing, with fre-
quent polyspike wave discharges. The older sister wascompletely bedridden by age 14 years, requiring a feed-ing tube. She is now 19 years old. The younger sister,now 10 years old, is following an identical course.
Diagnostic WorkupFunduscopy in both brothers showed thinned vesselsand optic nerve atrophy. The older brother did not havesignificant pigmentary changes at age six. The youngerbrother had significant pigmentary changes in the retinaat the same age. Electroretinograms showed diminishedwave amplitudes. Electron micrographs of lymphocytesin both brothers showed numerous membrane-boundlysosomal vacuoles, most empty with some containingelectron-dense storage material with a fingerprint patterntypical for JNCL (H. H. Goebel, University of Mainz,and Dr Schwendemann, University of Hamburg). Atautopsy of the older brother, brain weight was 1,140gm,and neurons were ballooned with fine granular material.Dilatation of large neurons was seen in the cerebral cor-tex, basal ganglia, thalamus, and cerebellar cortex. Theprocess was less marked in the red nucleus, locus cer-uleus, and the lower olive. The substantia nigra wasatrophic with moderate astrogliosis and slight vascularproliferation. Atrophic changes also were seen in the nu-clei of the thalamus with moderate to high-grade astro-gliosis. Lipopigment material was seen in neurons in thepyramidal band of Ammon’s horn. Cerebellar Purkinjecells were dilated by storage material. Moderate sub-ependymal astrogliosis was seen in brain and spinal cord.The storage material stained gray with Sudan black andhad a yellow autofluorescence (H. J. Colmant, Univer-sity of Hamburg).
Diagnostic workup of the sisters showed progressivecerebral and cerebellar atrophy, predominantly involv-ing gray matter, by cranial computed tomography andmagnetic resonance imaging. Abnormal signal intensitywas seen in the periventricular white matter. Thesefindings were consistent with a diagnosis of NCL. Aright frontal brain biopsy from the older sister was sub-jected to electron micrograph examination. The neu-rons contained inclusions characteristic for NCL.There were a combination of membrane-bound gran-ular and curvilinear bodies (Fig 2A–C). Neurons
Fig 3. Morphology. (A) CLN9-deficient fibroblasts have arounded cell body and are small. (B) Normal fibroblasts areelongated. Magnification �400, scale bar � 50�m.
Table. Live and Dead Attached and Detached Fibroblasts(patients and controls)
TypeLive Cells
(%)Dead Cells
(%)
DetachedNormal fibroblasts 0 � 0 100 � 0CLN9-deficient fibroblasts 36.2 � 0.4 63.8 � 0.4
AttachedNormal fibroblasts 95.8 � 0.5 4.2 � 0.3CLN9-deficient fibroblasts 85.9 � 1.1 14.1 � 1.0
346 Annals of Neurology Vol 56 No 3 September 2004

stained positively with an antibody to subunit c of mi-tochondrial ATP synthase (see Fig 2D, E). Apoptoticneurons with nuclei containing aggregates of chroma-tin were present (not shown). The presence of apopto-tic neurons was verified by terminal deoxynucleotidyl-transferase–mediated dUTP nick end labeling orTUNEL staining. Enzyme screening for the CLN1-and CLN2-deficient variants was negative. The se-
quences of coding regions of CLN3, CLN5, CLN6,and CLN8 genes were normal. A normal Northern blotrules out the possibility of defects in the intronic orpromoter region of the CLN3 gene. The blot had aCLN3 mRNA band of normal size and intensity com-pared with the CLN3 band from control RNA.
Metabolic labeling and two-dimensional gel electro-phoresis of mannose-6-phosphate glycoproteins isolatedfrom CLN9-deficient fibroblasts from one sister had apattern identical to the pattern seen in normal fibro-blasts. Testing was performed in the laboratory of P.Lobel. The following lysosomal storage diseases wereruled out by normal enzyme levels: Fabry disease (�-galactosidase), GM1-gangliosidosis (�-galactosidase),Tay–Sachs disease (�-hexosaminidase A), Niemann–Pickdisease types A and B (sphingomyelinase), Gaucher’s dis-ease (glucosylceramidase), mannosidosis, fucosidosis,mucopolysaccharidoses type I (�-iduronidase), type II(iduronate sulfatase), and type VII (�-glucuronidase).Niemann–Pick C disease was ruled out by negative fili-pin staining (see Fig 5). Assays for cathepsins A, B, C,D, H, and L were normal (laboratory of P. Lobel).
CLN9-Deficient Fibroblasts Have a DistinctMorphology and BiologyCells from the CLN9 patients have identical features. (1)CLN9-deficient fibroblasts have small and rounded cellbodies in contrast with the elongated, normal fibroblasts(Fig 3). Filipin staining, though not indicative of excesscholesterol storage, showed CLN9-deficient cells to haveprominent nucleoli (see Fig 5A–C). (2) CLN9-deficientfibroblasts attached poorly and piled up in mounds.This was seen by direct examination of the cultures withan inverted microscope. Numbers of detached liveCLN9-deficient cells were significantly higher than de-tached live normal fibroblasts (36.2% vs 0%, p � 0.05;Table). Numbers of attached dead CLN9-deficient fibro-blasts were higher than attached dead cells from controls(14.1% vs 4.2%, p � 0.05; see Table). (3) CLN9-deficient fibroblasts have a rapid growth rate comparedwith normal, and proliferation rates of CLN9-deficientfibroblasts compared with normal cells were increasedsuggesting increased DNA synthesis (Fig 4A, B). (4)CLN9-deficient fibroblasts have higher apoptotic ratescompared with normal cells. This was shown by PIstaining after treatment with proapoptotic etoposide. Anincreased number of apoptotic red-stained nuclei wasseen in CLN9-deficient cells (Fig 5D, E: 78.9% inCLN9-deficient fibroblasts vs 25.3% in control cells,p � 0.005).
Perturbation of Sphingolipid MetabolismMass measurements of ceramide and sphingomyelinand the glycosphingolipids lactosylceramide, ceramidetrihexoside, and globoside after metabolic labeling with[14C]-galactose were significantly decreased in CLN9-
Fig 4. (A) Growth curve of CLN9-deficient fibroblasts. Nor-mal fibroblasts (diamond) and CLN9-deficient fibroblasts(square: Serbian patient, triangle: German patient). Live cellswere counted at 24, 48, 72, 96, and 124 hours from tripli-cate experiments. A significant increase in growth rate inCLN9-deficient cells is seen after 48 hours, with continuedgrowth after 72 hours. Control fibroblasts became normallyconfluent because of contact inhibition. (B) [3H]Thymidineincorporation. Normal fibroblasts (diamond) and CLN9-deficient fibroblasts (square: Serbian patient, triangle: Ger-man patient). Proliferation was determined by [3H]thymidineincorporation at 0, 6, 12, 24, and 48 hours in triplicatemeasurements. An increase in [3H]thymidine incorporation inCLN9-deficient cells is seen at all time points and beyond 48hours. [3H]Thymidine incorporation in normal fibroblastsdecreases after 24 hours.
Schulz et al: Adhesion and Apoptosis in CLN9 347

Fig 5. (A–C) Filipin staining of (A) Niemann–Pick C fibroblasts, (B) normal fibroblasts, and (C) CLN9-deficient fibroblasts.Slides were examined under fluorescence (excitation wavelength, 372nm; emission wavelength, 446nm) at �400 magnification,scale bar � 50�m. (A) Niemann–Pick C fibroblasts exhibit accumulation of cholesterol within the cell body. (B) Normal fibro-blasts. (C) CLN9-deficient fibroblasts have prominent nucleoli (white arrow), but no excess cholesterol. (D, E) Increased apoptosisof CLN9-deficient fibroblasts. Propidium iodide staining of normal fibroblasts (D) and CLN9-deficient fibroblasts (E) after treat-ment with etoposide. Note increase in the number of red, apoptotic CLN9-deficient fibroblasts (25.27% represents number of apo-ptotic cells/total cells/vision field in normal fibroblasts vs 78.97% in CLN9-deficient cells, P � 0.005). Magnification �100; scalebar � 100�m.
348 Annals of Neurology Vol 56 No 3 September 2004
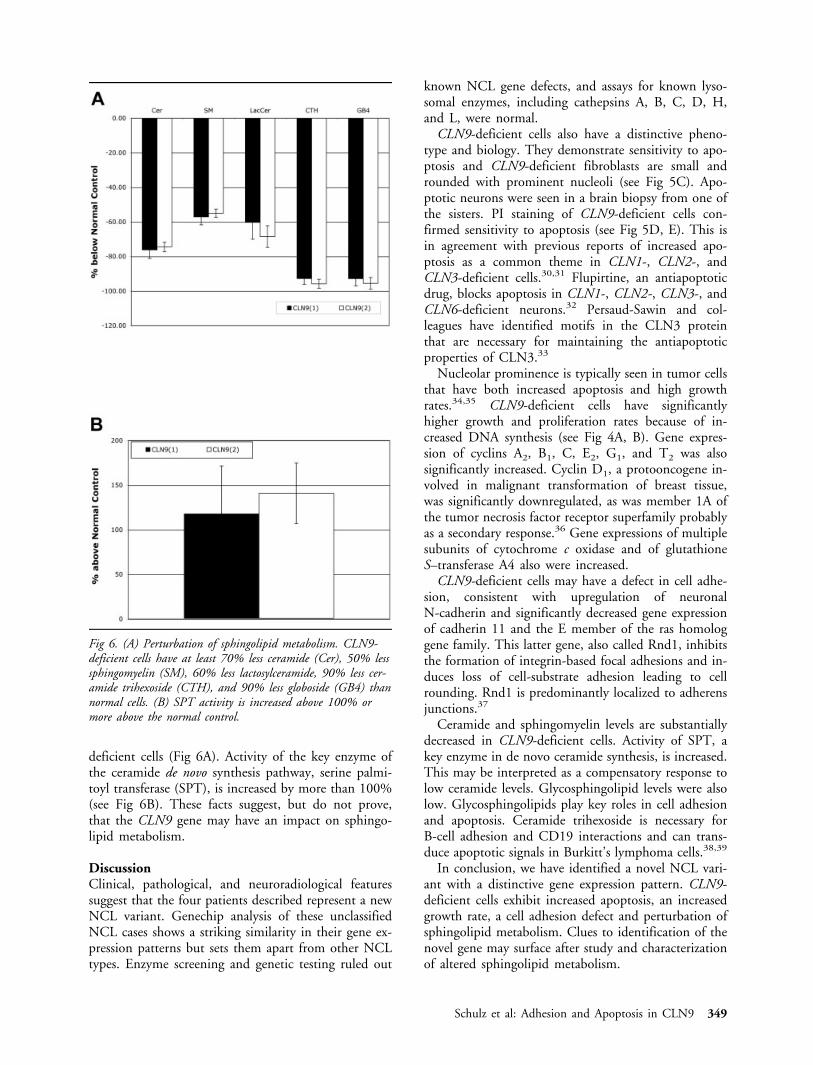
deficient cells (Fig 6A). Activity of the key enzyme ofthe ceramide de novo synthesis pathway, serine palmi-toyl transferase (SPT), is increased by more than 100%(see Fig 6B). These facts suggest, but do not prove,that the CLN9 gene may have an impact on sphingo-lipid metabolism.
DiscussionClinical, pathological, and neuroradiological featuressuggest that the four patients described represent a newNCL variant. Genechip analysis of these unclassifiedNCL cases shows a striking similarity in their gene ex-pression patterns but sets them apart from other NCLtypes. Enzyme screening and genetic testing ruled out
known NCL gene defects, and assays for known lyso-somal enzymes, including cathepsins A, B, C, D, H,and L, were normal.
CLN9-deficient cells also have a distinctive pheno-type and biology. They demonstrate sensitivity to apo-ptosis and CLN9-deficient fibroblasts are small androunded with prominent nucleoli (see Fig 5C). Apo-ptotic neurons were seen in a brain biopsy from one ofthe sisters. PI staining of CLN9-deficient cells con-firmed sensitivity to apoptosis (see Fig 5D, E). This isin agreement with previous reports of increased apo-ptosis as a common theme in CLN1-, CLN2-, andCLN3-deficient cells.30,31 Flupirtine, an antiapoptoticdrug, blocks apoptosis in CLN1-, CLN2-, CLN3-, andCLN6-deficient neurons.32 Persaud-Sawin and col-leagues have identified motifs in the CLN3 proteinthat are necessary for maintaining the antiapoptoticproperties of CLN3.33
Nucleolar prominence is typically seen in tumor cellsthat have both increased apoptosis and high growthrates.34,35 CLN9-deficient cells have significantlyhigher growth and proliferation rates because of in-creased DNA synthesis (see Fig 4A, B). Gene expres-sion of cyclins A2, B1, C, E2, G1, and T2 was alsosignificantly increased. Cyclin D1, a protooncogene in-volved in malignant transformation of breast tissue,was significantly downregulated, as was member 1A ofthe tumor necrosis factor receptor superfamily probablyas a secondary response.36 Gene expressions of multiplesubunits of cytochrome c oxidase and of glutathioneS–transferase A4 also were increased.
CLN9-deficient cells may have a defect in cell adhe-sion, consistent with upregulation of neuronalN-cadherin and significantly decreased gene expressionof cadherin 11 and the E member of the ras homologgene family. This latter gene, also called Rnd1, inhibitsthe formation of integrin-based focal adhesions and in-duces loss of cell-substrate adhesion leading to cellrounding. Rnd1 is predominantly localized to adherensjunctions.37
Ceramide and sphingomyelin levels are substantiallydecreased in CLN9-deficient cells. Activity of SPT, akey enzyme in de novo ceramide synthesis, is increased.This may be interpreted as a compensatory response tolow ceramide levels. Glycosphingolipid levels were alsolow. Glycosphingolipids play key roles in cell adhesionand apoptosis. Ceramide trihexoside is necessary forB-cell adhesion and CD19 interactions and can trans-duce apoptotic signals in Burkitt’s lymphoma cells.38,39
In conclusion, we have identified a novel NCL vari-ant with a distinctive gene expression pattern. CLN9-deficient cells exhibit increased apoptosis, an increasedgrowth rate, a cell adhesion defect and perturbation ofsphingolipid metabolism. Clues to identification of thenovel gene may surface after study and characterizationof altered sphingolipid metabolism.
Fig 6. (A) Perturbation of sphingolipid metabolism. CLN9-deficient cells have at least 70% less ceramide (Cer), 50% lesssphingomyelin (SM), 60% less lactosylceramide, 90% less cer-amide trihexoside (CTH), and 90% less globoside (GB4) thannormal cells. (B) SPT activity is increased above 100% ormore above the normal control.
Schulz et al: Adhesion and Apoptosis in CLN9 349

This work was supported by the Serbian Orthodox Church (R.-M.B.)and by the Deutsche Forschungsgemeinschaft (SCHU1597/1-1, A.S.).
We thank the affected families for participating in this study. We thankDrs P. Lobel and D. Sleat for performing metabolic labeling and two-dimensional gel electrophoresis of purified mannose-6-phosphate glyco-proteins and enzyme assays for cathepsins and other lysosomal enzymes.We thank A. Steed for performing the Northern blot for CLN3 onCLN9-deficient fibroblasts. We are grateful for measurements of sphin-gomyelin and ceramide levels performed by the Lipidomics Core of theMedical University of South Carolina, under the direction of Dr A.Bielawska. We are greatly indebted to Dr H. H. Goebel for kindlyreevaluating lymphocyte ultrastructure of the German CLN9-deficientpatients. We also thank D. Merz for help with the figures.
References1. Haltia M. The neuronal ceroid-lipofuscinoses. J Neuropathol
Exp Neurol 2003;62:1–13.2. Goebel HH, Mole SE, Lake BD (Eds.). The neuronal ceroid
lipofuscinoses (Batten disease). Amsterdam: IOS Press, 1999.3. Haltia M, Rapola J, Santavuori P, Keranen A. Infantile type of
so-called neuronal ceroid-lipofuscinosis-part 2. Morphologicaland biochemical studies. J Neurol Sci 1973;18:269–285.
4. Hassin G. Amaurotic family idiocy: late infantile type(Bielschowsky) with the clinical picture of decerebrate rigidity.Arch Neurol Psychiat 1926;16:708–727.
5. Harlem OK. Juvenile cerebroretinal degeneration (Spielmeyer-Vogt). Am J Dis Child 1960;100:918–923.
6. Goebel HH, Braak H. Adult neuronal ceroid-lipofuscinosis.Clin Neuropathol 1989;8:109–119.
7. Santavuori P, Rapola J, Sainio K, Raitta C. A variant of Jansky-Bielschowsky disease. Neuropediatrics 1982;13:135–141.
8. Sharp JD, Wheeler RB, Lake BD, et al. Genetic and physicalmapping of the CLN6 gene on chromosome 15q21–23. MolGenet Metab 1999;66:329–331.
9. Hirvasniemi A, Lang H, Lehesjoki AE, Leisti J. Northern epi-lepsy syndrome: an inherited childhood onset epilepsy with as-sociated mental deterioration. J Med Genet 1994;31:177–182.
10. Zhong N, Moroziewicz DN, Ju W, et al. Heterogeneity of late-infantile neuronal ceroid lipofuscinosis. Genet Med 2000;2:312–318.
11. Mitchell WA, Wheeler RB, Sharp JD, et al. Turkish variant lateinfantile neuronal ceroid lipofuscinosis (CLN7) may be allelicto CLN8. Eur J Paed Neurol 2001;5:21–27.
12. Lake BD, Bret EM, Boyd SC. A form of juvenile Batten diseasewith granular osmiophilic deposits. Neuropediatrics 1996;27:265–269.
13. Mazzei R, Conforti FL, Magariello A, et al. A novel mutationin the CLN1 gene in a patient with juvenile neuronal ceroidlipofuscinosis. J Neurol 2002;249:1398–1400.
14. Vesa J, Hellsten E, Verkruyse LA, et al. Mutations in thepalmitoyl protein thioesterase gene causing infantile neuronalceroid lipofuscinosis. Nature 1995;376:584–587.
15. Sleat DE, Donnelly RJ, Lackland H, et al. Association of mu-tations in a lysosomal protein with classical late-infantile neu-ronal ceroid lipofuscinosis. Science 1997;277:1802–1805.
16. International Batten Disease Consortium. Isolation of a novelgene underlying Batten disease, CLN3. Cell 1995;82:949–957.
17. Ranta S, Zhang Y, Ross B, et al. The neuronal ceroid lipofus-cinoses in human EPMR and mnd mutant mice are associatedwith mutations in CLN8. Nat Genet 1999;23:233–236.
18. Gao H, Boustany RM, Espinola JA, et al. Mutations in a novelCLN6-encoded transmembrane protein cause variant neuronalceroid lipofuscinosis in man and mouse. Am J Hum Genet2002;70:324–335.
19. Wheeler RB, Sharp JD, Schultz RA, et al. The gene mutated invariant late-infantile neuronal ceroid lipofuscinosis (CLN6) andin nclf mutant mice encodes a novel predicted transmembraneprotein. Am J Hum Genet 2002;70:537–542.
20. Savukoski M, Kestila M, Williams R, et al. Defined chromo-somal assignment of CLN5 demonstrates that at least four ge-netic loci are involved in the pathogenesis of human ceroid li-pofuscinosis. Am J Hum Genet 1994;55:695–701.
21. Isosomppi J, Vesa J, Jalanko A, Peltonen L. Lysosomal localiza-tion of the neuronal ceroid lipofuscinosis CLN5 protein. HumMol Genet 2002;11:885–891.
22. Winter E, Ponting CP. Tram, Lag1 and CLN8: members of anovel family of lipid sensing domains? Trends Biochem Sci2002;27:381–383.
23. Schorling S, Vallee B, Barz WP, et al. Lag1p and Lac1p are es-sential for the Acyl-CoA-dependent ceramide synthase reaction inSaccharomyces cerevisiae. Mol Biol Cell 2001;12:3417–3427.
24. Guillas I, Kirchman PA, Chuard R, et al. C26-CoA-dependentceramide synthesis of Saccharomyces cerevisiae is operated byLag1p and Lac1p. EMBO J 2001;11:2655–2665.
25. Teixeira CA, Espinola J, Liang H, et al. Novel mutations in theCLN6 gene cause a variant late infantile neuronal ceroid lipo-fuscinosis. Hum Mutat 2003;21:502–508.
26. Johnson DW, Speier S, Qian WH, et al. Role of subunit-9 ofmitochondrial ATP synthase in Batten disease. Am J MedGenet 1995;57:350–360.
27. Rylova SN, Amalfitano A, Persaud-Sawin DA, et al. The CLN3gene is a novel molecular target for cancer drug discovery. Can-cer Res 2002;62:801–808.
28. Uemura K, Sugiyama E, Tamai C, et al. Effect of an inhibitorof glucosylceramide synthesis on cultured rabbit skin fibro-blasts. J Biochem 1990;4:525–530.
29. Williams RD, Wang E, Merrill AH Jr. Enzymology of long-chain base synthesis by liver: characterization of serine palmi-toyltransferase in rat liver microsomes. Arch Biochem Biophys1984;228:282–291.
30. Lane S, Jolly R, Schmechel D, et al. Apoptosis as the mecha-nism of neurodegeneration in Batten’s disease. J Neurochem1996;67:677–683.
31. Cho S, Dawson PE, Dawson G. Role of palmitoyl-protein thio-esterase in cell death: implications for infantile neuronal ceroidlipofuscinosis. Eur J Paed Neurol 2001;5:53–55.
32. Dhar S, Bitting RL, Rylova S, et al. Flupirtine blocks apoptosisin Batten patient lymphoblasts and in human postmitotic CLN3-and CLN2-deficient neurons. Ann Neurol 2002;51:448–466.
33. Persaud-Sawin DA, Van Dongen A, Boustany RM. Motifswithin the CLN3 protein: modulation of cell growth rates andapoptosis. Hum Mol Genet 2002;11:2129–2142.
34. Smetana K. Are nucleoli participating in programmed celldeath? J Appl Biomed 2003;1:93–97.
35. Horky M, Kotala V, Anton M, Wesierska-Gadek J. Nucleolusand apoptosis. Ann NY Acad Sci 2002;973:258–264.
36. Yu Q, Geng Y, Sicinski P. Specific protection against breastcancer by cyclin D1 ablation. Nature 2001;411:1017–1021.
37. Nobes CD, Lauritzen I, Mattei MG, et al. A new member of therho family, Rnd1, promotes disassembly of actin filament struc-tures and loss of cell adhesion. J Cell Biol 1998;141:187–197.
38. Jackson T, Van Exel C, Reagans K, et al. Comparison of ad-hesion mechanisms and surface protein expression in CD77-positive and CD77-negative Burkitt’s lymphoma cells. Cell MolBiol 2001;47:1195–1200.
39. Mangeney M, Lingwood CA, Taga S, et al. Apoptosis inducedin Burkitt’s lymphoma cells via GB3/CD77, a glycolipid anti-gen. Cancer Res 1993;53:5314–5319.
350 Annals of Neurology Vol 56 No 3 September 2004





























