Embed Size (px)
Citation preview

Special Article
Immune Responses Against the Myelin/OligodendrocyteGlycoprotein in Experimental Autoimmune Demyelination
HANS-CHRISTIAN VON BUDINGEN,1 NAOYUKI TANUMA, 1 PABLO VILLOSLADA, 1,2
JEAN-CHRISTOPHE OUALLET,1,3 STEPHEN L. HAUSER,1 and CLAUDE P. GENAIN1,4
Accepted: January 16, 2001
Myelin/oligodendrocyte glycoprotein (MOG) is a surface-exposed antigen of myelin and an important target for autoim-mune responses which mediate inflammatory demyelination inthe central nervous system. Experimentally, MOG inducesstrong pathogenic T cell responses in many strains of labora-tory animals. Immunological studies in humans also identifyMOG as a surprisingly prevalent antigenic molecule among themyelin proteins. In addition, the encephalitogenic properties ofMOG are linked to the induction of antibody responses whichhave been demonstrated to directly promote central nervoussystem demyelination, a hallmark neuropathological feature indisorders such as human multiple sclerosis. Factors responsiblefor autoimmunity to MOG likely include genetic influences aswell as other mechanisms, which are the subject of intenseinvestigation. This article reviews experimental data currentlyavailable on specificity and pathogenic roles of T cell andantibody responses against MOG, which have implicationsrelevant to multiple sclerosis and related disorders.
KEY WORDS: Autoimmunity; myelin proteins; epitopes; rodents;nonhuman primates.
INTRODUCTION
Multiple sclerosis (MS) is a chronic, inflammatory de-myelinating disorder of central nervous system (CNS)
white matter of unknown etiology, characterized patho-logically by perivascular inflammation, destruction of themyelin sheaths, proliferation of astrocytes with gliosis,and loss of oligodendrocytes and axons (1–3). Studies ofexperimental allergic encephalomyelitis (EAE), an auto-immune CNS inflammatory disorder produced in labo-ratory animals after sensitization with myelin antigens,have provided many of the current concepts used tounderstand the pathophysiology of human MS. In EAE,CNS inflammation is initiated by activation of autoag-gressive, myelin-specific T cells, which disrupt theblood–brain barrier and locally trigger a cascade ofevents that together promote or entertain the tissuedestruction that translate into neurological dysfunction(reviewed in Ref. 4).
After it was discovered that immunization with CNSwhite matter could induce CNS inflammatory demyeli-nation, many investigators seeked to identify a singleantigen responsible for this destructive autoimmune re-sponse. Studies have initially focused on the majormyelin proteins, myelin basic protein (MBP) and prote-olipid apoprotein (PLP) (5, 6). Observations that EAEcould be adoptively transferred by lymphoid cells (7),and the subsequent discovery that encephalitogenic (e.g.,disease-inducing) T cell populations were targeting arestricted repertoire of myelin epitopes, provided furtherimpulse for neuroimmunologists to elucidate the patho-physiologic role of myelin-specific T cell responses inEAE and MS (8, 9). Effector mechanisms of cellularimmunity, for example, proinflammatory cytokines suchas IFN-g and TNF-a, are regarded as major factors thatpromote CNS myelin damage (10) and are targets for
1Department of Neurology, University of California, San Francisco,California 94143. USA.
2Department of Neurology, University of Navarra, Pamplona, Spain.3Department of Neurology, Hopital Tenon, Paris, France.4To whom correspondence should be addressed at NeuroimmunologyLaboratory, Room C-440, University of California, San Francisco.505 Parnassus Avenue, San Francisco, California 94143-0435. Fax:(415) 502 5899. E-mail: [email protected].
Journal of Clinical Immunology, Vol. 21, No. 3, 2001
1550271-9142/01/0500-0155$19.50/0 © 2001 Plenum Publishing Corporation

disease-modifying therapies in MS (11). One looking inperspective at preclinical trials, however, quickly real-izes that therapeutic success in EAE has not been a verygood predictor of efficacy in MS trials. Perhaps thisemphasizes the likelihood that other mechanisms areinvolved in the pathogenesis of autoimmune CNS disor-ders, separately or in combination with autoagressive Tcell responses. Indeed, as suggested by recent analyses oflarge series of biopsy and autopsy material, MS may infact represent a group of very heterogeneous disorderswith distinctive neuropathological features that suggestdifferent pathophysiological mechanisms (12, 13). Thediversity of mechanisms that may result in CNS inflam-matory demyelination is also illustrated by the existenceof many different models of EAE, each characterized byvarious degrees of inflammation and demyelination,depending on the species studied and the method usedfor induction of disease. Thus, while it is established thatcellular autoimmunity most often leads to blood–brainbarrier disruption and perivascular inflammation, patho-genic T cell responses may not necessarily be associatedwith permanent tissue destruction which may requireadditional mechanisms (14).
Long before the discovery of MHC restriction and theelucidation of the trimolecular complex, earlier studies hadsuggested humoral factors as effector mechanisms for im-mune-mediated demyelination (15, 16). It was also recog-nized that the target for these antibody responses wasunlikely to be MBP (17–19). Lebaret al. (20, 21) were thefirst to propose that the prominent demyelination observedin guinea pigs immunized with whole myelin was producedby antibodies to a protein expressed by oligodendrocytes,identified as M2 and different from MBP and the proteo-lipid proteins. The M2 protein was isolated by Liningtonand colleagues in the mid 1980s, with the use of amonoclonal antibody raised against rat cerebellar glycopro-teins and termed myelin/oligodendrocyte glycoprotein, orMOG (22). Although quantitatively minor compared toother myelin proteins, MOG has, within the last decade,emerged as a prime candidate target for pathogenic CNS-specific autoimmune responses.
In murine EAE, the pathogenicity of immune re-sponses to MBP and PLP (which are abundant CNSantigens, together representing.75% of total myelinproteins) is restricted to susceptibility conferred byappropriate MHC class II elements. For example, theSJL/J mouse (H-2s) is responsive predominantly toimmunodominant epitopes of PLP, while the PL/J mouse(H-2u) will respond preferentially to the peptide 1–11 ofMBP. In contrast, MOG or MOG-derived peptides ap-pear to have a strong encephalitogenic potential in mostrodent strains and species studied to date. In addition to
the ability of MOG to produce vigorous CNS-specific Tcell responses (23), numerousin vitro andin vivo studieshave confirmed that anti-MOG antibodies have demyeli-nating properties (24–30). In humans, heightened T cellreactivity to MOG is observed in peripheral blood,compared to MBP and PLP (31–33). Furthermore,MOG-specific immunoglobulin deposition has recentlybeen described within actively demyelinating lesions ofMS and causally associated with irreversible vesiculartransformation of myelin sheaths (34, 35).
These observations highlight the unique antigenicproperties of MOG amongst myelin proteins, the reasonsfor which are not entirely understood. As methods for thepurification of native forms of MOG and the expressionof recombinant MOG have been developed, a substantialamount of information is now available on cellular andhumoral responses against this protein in several species,including mice and rat strains of different genetic back-grounds and outbred nonhuman primates. This articlebriefly outlines the biochemical characteristics of MOG,reviews our current knowledge of B cell and T cellresponses against MOG in EAE, and discusses how thisinformation can be used to better our understanding ofCNS autoimmune demyelination.
BIOCHEMICAL AND BIOLOGICAL PROPERTIES OFMOG (REFER ALSO TO TABLE I)
Cloning of cDNA encoding for MOG in human(36–38), rat (39), mouse, bovine (40, 41), and marsupialspecies (42) has shown that the protein contains 218amino acids with a high degree of conservation acrossspecies and is a member of the immunoglobulin (Ig)superfamily. The genes encoding for MOG have beenmapped at the distal end of the MHC class I region inhuman, mouse, and rat (43, 44). MOG is unique in the Igsuperfamily of proteins, since hydropathicity predictionsshow two hydrophobic domains (39, 45). The firsthydrophobic domain is located between amino acid (aa)residues 122 and 150 and spans the myelin membranelipid bilayer, while the second domain (aa residues174–200) most likely does not span the plasma mem-brane but may be associated with it or embedded withinit (46, 47). MOG contains a possible intracytoplasmicphosphorylation site (Thr 167) and anN-glycosylationsite (Asn 31) within the extracellular domain.In vivo,MOG is glycosylated and has been shown to contain theHNK-1 carbohydrate epitope, as do other members of theIg superfamily (48–50). The structure of MOG has notbeen experimentally obtained and its function(s) is cur-rently unknown, although its sequence and homologycharacteristics suggest that it may act as an adhesion
156 VON BUDINGEN ET AL.
Journal of Clinical Immunology, Vol. 21, No. 3, 2001

molecule; act as a receptor for complement binding, orparticipate in signaling processes within oligodendro-cytes (42, 51). MOG is developmentally regulated andappears to be a marker of oligodendrocyte maturation(52–55). Expression of MOG outside the CNS has notbeen firmly documented to date, unlike other proteins ofmyelin such as MBP and PLP (56–58).
The extracellular portion of MOG contains the Ig-likedomain, between cysteine residues at positions 24 and98, which suffices for induction of EAE in all specieswhen expressed in nonglycosylated recombinant form inbacteria. Factors that could explain the high encephali-togenicity of MOG include the extracellular location ofthe Ig-like domain and the fact that MOG is expressedlargely on the outermost lamellae of the myelin sheath,where it is accessible to autoimmune attack, especially toautoantibodies (59, 60). In addition, MOG shares a highamino acid sequence homology with other proteins suchas butyrophilin, a protein expressed in the lactatingmammary gland that is encoded by a gene which alsomaps in the MHC region. Autoimmunity triggered bymolecular mimicry is indeed possible in the case ofMOG, as cross-reactivity has been found between MOGpeptides and viral and butyrophilin peptides (61, 62).
MOG-INDUCED EAE IN VARIOUS STRAINSAND SPECIES
MOG was first characterized as an immunodominantautoantigen responsible for the demyelinating antibodyresponse in guinea pig EAE induced by immunizationwith spinal cord tissue in adjuvant (21, 26). Subse-quently, it was also demonstrated, in several species andmultiple antigen systems, that prominent demyelinationcould be induced by exogenous administration of MOG-specific antibodies (25, 27–30).
Several studies have now shown that MOG by itself isable to generate encephalitogenic T cell responses and to
induce EAE in inbred mice, rats, and nonhuman primates(Table II). All investigators used recombinant forms oftruncated MOG encompassing the Ig-like extracellulardomain (rMOG), with the exception of Johnset al. (63),who employed native MOG (aa 1–218) purified tohomogeneity to immunize Lewis rats in the absence ofBordetella pertussis (B. pertussis). In this particularstudy, human MOG appeared to be more efficient than ratMOG in terms of the incidence of EAE. It is possible thatthe greater immunogenicity of human MOG in Lewisrats was linked to different epitopes outside the Ig-likeextracellular domain. Amino acid sequence comparisonindicates that there are more substitutions between hu-man and rat in the transmembrane domains of MOG(Fig. 1). However, the C-terminal moiety of MOG (aa121–218) has not been separately shown to have enceph-alitogenic potential. All other studies indicate that rMOG(the Ig-like domain of MOG) suffices in most cases forreproducible induction of EAE in mice, rats, and nonhu-man primates and that human and rat rMOG are equiv-alent in potency (Table II). One exception concerns theSJL mouse, which is reportedly highly susceptible torecombinant human rMOG (aa 1–120, produced in insectcells) (64), and not to recombinant rat rMOG (aa 1–125,produced inE. coli) (65). The latter studies both reportedthat this mouse strain develops severe EAE when immu-nized with the immunodominant peptide MOG peptide92–106, which contains only one substitution betweenhuman (F96) and rat (Y96). Therefore, it is unlikely thatthe differences in susceptibility to EAE are explained bythe different amino acid sequences. On the other hand,these findings possibly suggest that glycosylation of theIg-like domain of MOG, and/or solubility of the antigen,may be important for encephalitogenicity in the SJLmouse (64).
To summarize these observations, it appears that mostspecies and strains studied develop moderate to severe
Table I. Properties of the MOG Antigen
Ref. Nos.
Integral membrane protein specific to CNS myelin 20, 22, 39, 102Located on the outermost lamellae of the myelin sheath and on the surface of oligodendrocytes 46, 47, 59, 60Surface marker of oligodendrocyte maturation 54, 55Expression parallels myelination 52, 53Quantitatively minor component of myelinMolecular size of 26–28 kD on SDS-PAGE and tendency to form dimers 21, 22, 60, 102, 103Glycosylated in native form. N-linked glycosylation at asp 31 39, 48, 60, 102Highly conserved among mouse, rat, guinea pig, bovine, marsupial, and human, but absent in
avian and reptiles brains 37–40, 42, 104Maps to the MHC region in the mouse, rat, and human 36, 43, 44Alternative mRNA splicing variants in humans, not mice 104, 105Nonglycosylated extracellular domain produces CNS demyelinating disease in guinea pigs,
mice, rats, and primates See this review
AUTOIMMUNITY TO MOG 157
Journal of Clinical Immunology, Vol. 21, No. 3, 2001

Tab
leII.
Enc
epha
litog
enic
Pro
pert
ies
ofN
ativ
eor
Rec
ombi
nant
MO
G(r
MO
G)
inV
ario
usS
trai
nsan
dS
peci
es,
and
Clin
ical
and
Pat
holo
gica
lPhe
noty
pes
a
Spe
cies
MH
Ccl
ass
IIre
stric
tion
elem
ent
Str
ain
Ant
igen
No.
ofim
mun
izat
ions
PT
Inci
denc
eof
EA
EC
linic
alco
urse
Rel
apse
sa
-MO
GA
bsD
emye
linat
ion
Ref
.N
o.
Mou
seH
-2gS
JL/J
Hum
anrM
OG
(1–1
20)
b1
115
/15
Chr
onic
,se
vere
No
1Y
es64
SJL
Rat
rMO
G(1
–125
)c2
11/
5A
cute
,m
ildN
DN
o65
H-2
dq
1B
iozz
iAB
/HR
atrM
OG
(1–1
25)c
21
5/6
Acu
te,
mod
erat
eN
oN
DN
o65
H-2
bC
57/B
L/6
Hum
anrM
OG
(1–1
21)
c1
16/
7C
hron
ic,
seve
reN
o1
Yes
106
H-2
bC
57/B
L/6
Rat
rMO
G(1
–122
)c1
14/
4C
hron
ic,
seve
reN
o1
Yes
79H
-2b
,p,q
C57
/BL/
10R
atrM
OG
(1–1
25)
c1
110
0%C
hron
icN
oN
DN
D10
7H
-2q
DB
A/l
Rat
rMO
G(1
–125
)c1
1/2
5/5
Chr
onic
,se
vere
No
1Y
es10
7H
-2g
7N
OD
/Lt
Rat
rMO
G(1
–122
)c1
14/
4C
hron
ic,
seve
reY
es1
Yes
79H
-2g
7N
OD
/Lt
Hum
anrM
OG
(1–1
21)c
11
9/9
Chr
onic
,se
vere
Yes
1Y
es10
6
Rat
RT
-1l
Lew
isR
atM
OG
(1–2
18)d
12
3/8
Chr
onic
,se
vere
Yes
1Y
es63
Hum
anM
OG
(1–2
18)d
12
10/1
0C
hron
ic,
seve
reY
es1
Yes
63R
atrM
OG
(1–1
25)c
12
9/11
Acu
te,
mod
erat
eY
es1
Yes
82R
T-1
nB
row
n-N
orw
ayR
atrM
OG
(1–1
25)
c1
27/
7A
cute
,le
thal
No
1Y
es67
Mar
mos
etO
utbr
edC
.ja
cch
us
Rat
rMO
G(1
–125
)c1
110
0%A
cute
,m
oder
ate
tose
vere
and
chro
nic/
rela
psin
g
1Y
es30
,66
,70
,71
,10
8
Rat
rMO
G(1
–125
)c1
23/
3A
cute
,se
vere
1Y
esun
publ
ishe
dH
uman
rMO
G(1
–125
)c1
23/
3C
hron
ic,
seve
reN
o1
Yes
72
Mac
aque
rhes
usO
utbr
edM
.m
ula
tta
Hum
anrM
OG
(1–1
25)c
11
8/8
Hyp
erac
ute,
fulm
inan
tN
o1
Yes
74
aW
here
avai
labl
eth
epr
esen
ceof
ahu
mor
alre
spon
seis
indi
cate
d.P
T,
B.
pert
ussi
s;N
D,
not
done
.bB
acul
oviru
sex
pres
sion
syst
emin
inse
ctce
lls;
glyc
osyl
ated
reco
mbi
nant
prot
ein.
c Exp
ress
edinE
.co
li;no
ngly
cosy
late
dpr
otei
n.dN
ativ
epr
otei
npu
rified
from
brai
ntis
sue.
158 VON BUDINGEN ET AL.
Journal of Clinical Immunology, Vol. 21, No. 3, 2001

EAE after immunization with the whole MOG moleculeor its extracellular domain. Where reported, EAE waspathologically characterized by significant demyelina-tion associated with perivascular infiltrates, with theexception of mice baring the H-2s (SJL) and H-2dq1
(Biozzi AB/H) MHC class II restriction haplotypes.Demyelination appeared to be associated with the pres-ence of autoantibodies to rMOG, especially in rats andprimates, where amplification of these antibody re-sponses resulted in hyperacute, lethal forms of EAE (66,67). The different origins of the MOG antigen did notresult in major alterations of the clinical or neuropatho-logical characteristics of EAE. Rather, the differences inthe incidence of disease and severity reported in thesestudies could be due largely to additional factors, such asvariations in housing environment, dose of antigen,adjuvants (presence or absence ofB. pertussis), and routeof immunization. In support of this hypothesis, a system-atic study of EAE conducted with nonglycosylated ratrMOG (aa 1–125) demonstrated that significant variationin EAE phenotype and lesion distribution could beobserved in a single rat strain, depending on the adjuvantregimen and physicochemical state of the rMOG em-ployed for immunization (68).
SPECIFICITIES OF PRIMARY T CELL AND B CELLRESPONSES IN rMOG-INDUCED EAE
Several studies have analyzed the fine specificity of Tcell and antibody responses using linear peptides over-lapping the sequence of the Ig-like domain of MOG in
animals actively immunized with rMOG. These data aresummarized in Fig. 2. Immune responses to the C-terminus portion of MOG, if any, have not been reportedto date.
Rodents
In inbred rodents, three major regions were identifiedas immunodominant for T cell responses. These includean N-terminus peptide (SJL mouse and Lewis rat), apeptide located within MOG aa 35–55 (CH3.SW,C57BL/6, NOD/Lt, Lewis rat), and one or more epitopeswithin the C-terminus portion of rMOG (SJL, C57BL/6,and NOD/Lt). It is clear that in most strains, the primaryT cell response is restricted to only one of these epitopes.This is also apparent when examining the encephalito-genic potential of individual peptides (as assessed byability to induce EAE by active immunization with shortpeptides or adoptive transfer of T cell lines), whichshows that a given strain generally responds only to asingle MOG peptide (see below). The specificity ofantibody responses has also been explored using linearpeptides of MOG and seems to correspond to the mainepitopes targeted by T cells.
Nonhuman Primates
In contrast to inbred rodents, immune responses toMOG appear to be more diverse in both New World andOld World outbred primates (C. jacchusmarmosets and
Fig. 1. Comparative deduced amino acid sequences of rat and human MOG. MOG shows a high degree of conservation between species, with mostsubstitutions located within the transmembrane domain. However, several substitutions located within the Ig-like, extracellular domain involveimmunodominant epitopes (see text). Shaded in gray are substitutions and boxed are transmembrane domains.
AUTOIMMUNITY TO MOG 159
Journal of Clinical Immunology, Vol. 21, No. 3, 2001
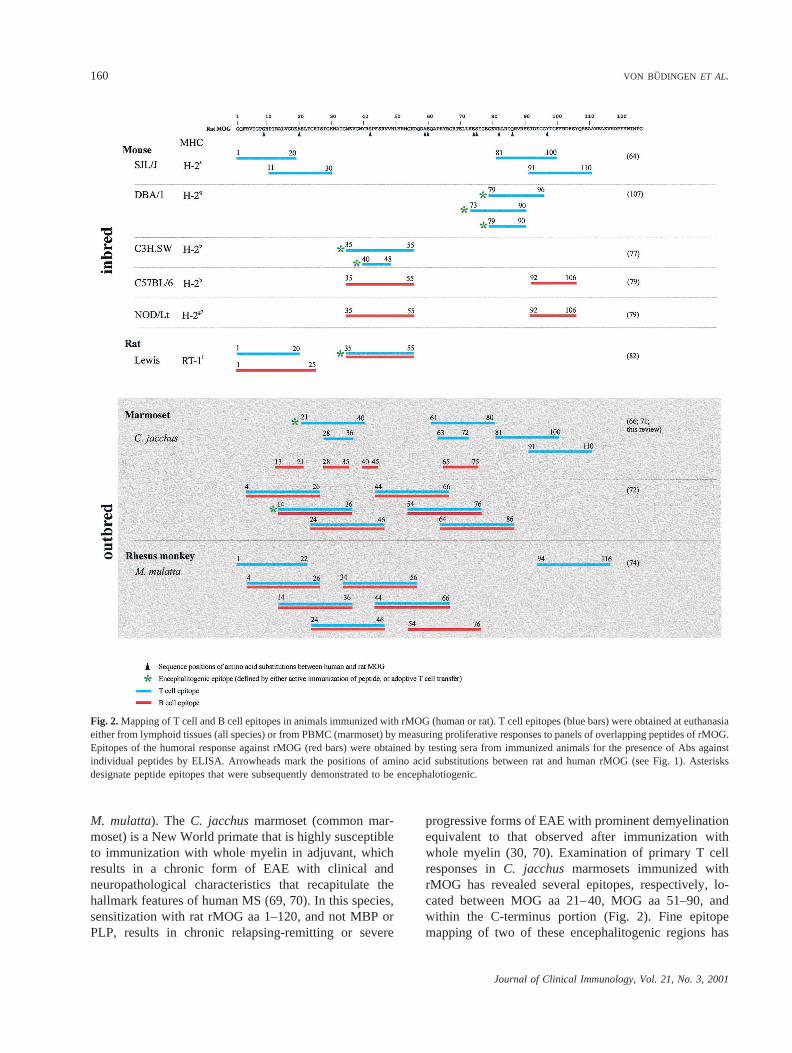
M. mulatta). The C. jacchusmarmoset (common mar-moset) is a New World primate that is highly susceptibleto immunization with whole myelin in adjuvant, whichresults in a chronic form of EAE with clinical andneuropathological characteristics that recapitulate thehallmark features of human MS (69, 70). In this species,sensitization with rat rMOG aa 1–120, and not MBP orPLP, results in chronic relapsing-remitting or severe
progressive forms of EAE with prominent demyelinationequivalent to that observed after immunization withwhole myelin (30, 70). Examination of primary T cellresponses inC. jacchus marmosets immunized withrMOG has revealed several epitopes, respectively, lo-cated between MOG aa 21–40, MOG aa 51–90, andwithin the C-terminus portion (Fig. 2). Fine epitopemapping of two of these encephalitogenic regions has
Fig. 2.Mapping of T cell and B cell epitopes in animals immunized with rMOG (human or rat). T cell epitopes (blue bars) were obtained at euthanasiaeither from lymphoid tissues (all species) or from PBMC (marmoset) by measuring proliferative responses to panels of overlapping peptides of rMOG.Epitopes of the humoral response against rMOG (red bars) were obtained by testing sera from immunized animals for the presence of Abs againstindividual peptides by ELISA. Arrowheads mark the positions of amino acid substitutions between rat and human rMOG (see Fig. 1). Asterisksdesignate peptide epitopes that were subsequently demonstrated to be encephalotiogenic.
160 VON BUDINGEN ET AL.
Journal of Clinical Immunology, Vol. 21, No. 3, 2001

identified MOG aa 28–36 and MOG aa 63–72 as the coreT cell epitopes, the sequence of which is totally con-served among rat, human, and marmoset (71) (unpub-lished data). A separate study has identified MOG aa24–36 as a major T cell epitope of MOG in marmosetsimmunized with human rMOG. Interestingly, thisepitope of MOG could be presented to T cells by amonomorphic MHC class II molecule that is peculiar tothis species (72, 73). In rhesus monkeys (M. mulatta)immunized with human rMOG, T cell responses werefound to be directed against several epitopes encom-passed within MOG aa 4–20, MOG aa 34–56, and MOGaa 94–116 (74). Although two of these epitopes (MOGaa 4–20 and MOG aa 34–56) have been proposed asimmunodominant for T cell responses in patients withMS (75), this has not been confirmed by additionalstudies (33). Collectively, these data indicate that T cellautoimmunity to MOG encompasses a broadly heteroge-neous repertoire in outbred populations.
Similar to rodents, mapping studies with linear pep-tides of rMOG in primates indicate that humoral re-sponses generally recognize amino acid sequences thatcorrespond to those of T cell epitopes (Fig. 2). Inmarmosets, however, analysis of fine specificities for theantibody response has identified two additional regionsof rMOG which so far have not been recognized as T cellreactive, e.g., aa 13–21 and aa 40–45. As is discussedlater, although these observations indicate that multipleregions of the protein may be targeted by autoantibodyresponses, they provide little information on the exis-tence of conformational antibody epitopes of MOG.
ENCEPHALITOGENIC POTENTIAL OFMOG-DERIVED PEPTIDES
The ability of synthetic peptides derived from theamino acid sequence of MOG to induce EAE by activeimmunization with adjuvant has been tested in manyspecies. Results are summarized in Table III.
Rodents
In most inbred strains, the immunodominant enceph-alitogenic epitope identified in rMOG-immunized ani-mals was necessary and sufficient in order to induce EAE(see also Fig. 2). For example, MOG aa 35–55 or its coreepitope was found encephalitogenic in H-2b (C57BL/6,C3H.SW, and C57L), H-2u (PL/J), and H-2g7 (NOD/Lt)mice (76–79). This peptide was also found to be themajor encephalitogenic region of MOG in the Lewis rat,in association with the RT11 class II restriction element(63, 80). In contrast, the encephalitogenic peptides in
H-2s (SJL/J and A.SW) and H-2q (DBA/1) mice arelocated within the C-terminus moiety, corresponding,respectively, to aa 92–106 and aa 79–96 (65, 81). Inaddition to immunodominant epitopes, additional pep-tides of the N terminus of MOG were found to inducesubclinical EAE in the Lewis rat (82). The repertoire ofencephalitogenic MOG peptides appears somewhat morediverse in the Biozzi AB/H mouse, where severalepitopes including MOG aa 1–15, aa 8–22, aa 43–57,and aa 134–148 (outside the Ig-like domain) have beenshown to produce EAE, although disease incidence wasnot predictable (65). These results confirm a strongassociation between MHC haplotypes and selective en-cephalitogenic regions of MOG in mice, with MOG aa35–55 clearly being immunodominant in several differ-ent strains.
In contrast to extensive investigations of T cell recep-tor (TCR) usage in MBP- or PLP-induced EAE, little isknown of the TCR Vb gene usage of MOG peptide-specific T cells. Mendelet al. (76, 77) have reporteddiverse TCR Vb gene usage by MOG aa 35–55-specificT cells derived from H-2b mice. The encephalitogenicMOG aa 35–55-reactive T cell lines expressed Vb1,Vb6, Vb8, Vb14, and Vb15 gene segments in C57BL/6and Vb1, Vb2, Vb6, Vb8, Vb10, Vb14, and Vb15 genesegments in C3H.SW. A predominant expression of TCRVb8.2 (40–48%) is observed in both strains. However,the C57L mouse bares the H-2b haplotype and has adeletion of Vb8 genes, is equally susceptible to MOG aa35–55, and, compared to the C3H.SW mouse, does notshow major alterations in TCR Vb gene usage (Vb1,Vb2, Vb6, Vb14, and Vb16) (77). These studies suggestthat the encephalitogenic response to MOG in H-2b mice,although restricted in epitope recognition, is diverse atthe level of TCR utilization.
The clinical course of EAE induced with peptideepitopes of MOG was variable, depending on the strainstudied. MOG aa 35–55-induced chronic progressive,nonremitting EAE in C57BL/6, C3H.SW, and C57Lmice, and clinical signs were highly reproducible fromanimal to animal (76, 77). In PL/J mice, clinical mani-festations of EAE appeared unpredictable, of delayedonset, and often started abruptly with severe impairment(78). Although the factors underlying such differencesare unclear, one explanation may be that the coreencephalitogenic epitope in PL/J (H-2u) mice is differentfrom that in H-2b mice. MOG aa 40–55 is stronglyencephalitogenic in mice, and MOG aa 35–55-reactive Tcell lines restricted by H-2b react with MOG aa 40–55but not with MOG aa 36–45 (76). The core encephali-togenic T cell epitope in H-2b mice appears to be MOGaa 40–48 (77). In PL/J mice, MOG aa 35–55-reactive T
AUTOIMMUNITY TO MOG 161
Journal of Clinical Immunology, Vol. 21, No. 3, 2001

Tab
leIII
.E
ncep
halit
ogen
icE
pito
pes
ofM
OG
inV
ario
usS
trai
nsan
dS
peci
esa
Spe
cies
MH
Ccl
ass
IIre
stric
tion
elem
ent
Str
ain
Ant
igen
No.
ofim
mun
izat
ions
PT
Inci
denc
eof
EA
EC
linic
alco
urse
Rel
apse
sa
-MO
GA
bsa
-pep
tide
Abs
Dem
yelin
atio
nR
ef.
Mou
seH
-2sS
JLaa
92–1
06b2
114
/16
Acu
te,
seve
reY
esN
DN
DY
es65
H-2
sS
JL/J
aa92
–106
12
15/1
7C
hron
ic,
seve
reY
esN
D92
–106
(1
)M
ild81
00
aa92
–106
11
12/1
3C
hron
ic,
seve
reY
esN
D92
–106
(1
)M
ild0
H-2
sA
.SW
aa92
–106
12
14/1
4C
hron
ic,
seve
reN
oN
D92
–106
Yes
00
0aa
92–1
061
18/
8C
hron
ic,
seve
reY
es,
with
prog
essi
onN
D92
–106
(1)
Yes
0
H-2
dq
lB
iozz
iAB
/Haa
1–15
c2
12/
11A
cute
,m
oder
ate
No
ND
ND
No
650
0aa
8–2
22
14/
6A
cute
,m
oder
ate
No
ND
ND
No
00
0aa
43–5
72
11/
5S
ever
eN
oN
DN
DN
o0
00
aa13
4–1
482
11/
5A
cute
,m
ildN
oN
DN
DN
o0
H-2
bC
57/B
L/6
aa35
–55
11
11/1
3C
hron
ic,
seve
reN
o1
35–5
5Y
es79
,10
90
0aa
35–5
51
14/
5-5/
5C
hron
ic,
seve
reN
oN
DN
DY
es76
00
aa40
–55
21
4/4
Chr
onic
,m
oder
ate
No
ND
ND
ND
00
0aa
1–21
,10
4–1
172
10/
?N
oE
AE
NA
ND
ND
ND
0H
-2b
C3H
.SW
aa35
–55
21
34/5
5C
hron
ic,
seve
reN
oN
DN
D?
00
0aa
40–5
52
14/
4C
hron
ic,
seve
reN
oN
DN
DN
D0
00
aa40
–48
21
4/5
Chr
onic
,se
vere
No
ND
ND
ND
77H
-2b
C57
Laa
40–
482
12/
6C
hron
ic,
mod
erat
eN
oN
DN
DN
D0
H-2
qD
BA
/1aa
73–9
0d1
12/
8C
hron
ic,
mild
No
ND
1(n
otsh
own)
ND
107
00
aa79
–90
11
3/5
Chr
onic
,m
ildN
oN
D1
(not
show
n)N
D0
00
aa79
–96
11
/212
/13
Chr
onic
,m
oder
ate
No
ND
1(n
otsh
own)
Mild
0aa
85–1
021
11/
6M
ildN
oN
D1
(not
show
n)N
D0
H-2
g7
NO
D/L
taa
35–5
51
111
/11
Chr
onic
,se
vere
Yes
135
–55,
92–1
06Y
es79
H-2
uP
L/J
aa35
–55e
32
3/13
Chr
onic
Yes
ND
ND
Mild
78aa
35–5
53
19/
14C
hron
icY
esN
DN
DM
ild0
Rat
RT
-1l
Lew
isaa
1–20
1–2
20/
4S
ubcl
inic
alN
/A1
1–20
,9
–25
No
820
0aa
9–2
51–
22
0/4
Sub
clin
ical
N/A
11–
20,
9–2
5N
o0
00
aa35
–55
12
14/1
8C
hron
ic,
seve
reY
es1
35–5
5Y
es63
00
aa35
–55
12
21/2
5M
ild–s
ever
eY
es1
37–
46,
43–5
0Y
es;
not
show
n80
00
aa35
–55
1–2
20/
4S
ubcl
inic
alN
/A1
35–5
5N
o82
Mar
mos
etou
tbre
dC
.ja
cch
us
aa21
–40
11
2/2
Chr
onic
,m
ildN
o1
21–
40,
31–5
0M
ildT
his
revi
ew0
0aa
1–20
,11
–30,
21–
401
12/
2C
hron
ic,
mild
No
11–
20,
11–3
0,21
–40
,31
–50
00
00
aa51
–70,
61–
80,
71–9
01
11/
1C
hron
ic,
mild
No
(2)
—0
00
0aa
81–1
00,
91–1
10,
101–
120
11
1/1
Chr
onic
,m
ildN
o1
91–1
10,
101–
120
00
00
Ove
rlapp
ing
20m
erpe
ptid
es1–
120
11
2/2
Chr
onic
,m
ildN
o1
1–20
,(1
1–30
),21
–40
,31
–50,
101–
120
00
aE
ncep
halit
ogen
icity
was
asse
ssed
byon
eor
mor
eim
mun
izat
ions
inth
epr
esen
ceor
abse
nce
ofB
.p
ert
uss
is(P
T).
Whe
reav
aila
ble,
info
rmat
ion
onan
tibod
ies
agai
nst
rMO
G(
a-r
MO
GA
bs),
MO
Gpe
ptid
es(a-p
eptid
eA
bs),
and
dem
yelin
atio
nis
incl
uded
.N
D,
not
done
;N
/A,
not
appl
icab
le.
bP
eptid
esal
sote
sted
but
not
foun
dto
indu
ceE
AE
incl
ude
over
lapp
ing
15m
erpe
ptid
esra
ngin
gfr
omaa
1to
92an
daa
106
to21
8.c P
eptid
esal
sote
sted
but
not
foun
dto
indu
ceE
AE
incl
ude
over
lapp
ing
15m
erpe
ptid
esra
ngin
gfr
omaa
15to
50,
aa50
to14
1,an
daa
141
to21
8.dP
eptid
esal
sote
sted
but
not
foun
dto
indu
ceE
AE
incl
ude
pept
ides
rang
ing
from
aa1
to84
and
aa91
to10
8.e P
eptid
esal
sote
sted
but
not
foun
dto
indu
ceE
AE
incl
ude
pept
ides
aa1–
21,
aa67
–87
,an
daa
202–
218.
162 VON BUDINGEN ET AL.
Journal of Clinical Immunology, Vol. 21, No. 3, 2001

cell lines react with MOG aa 36–45 but not with MOGaa 40–55 (78). This suggests that different epitopesencrypted within MOG aa 35–55 may influence diseasephenotype in these mouse strains. The influence of MHCgenes on EAE phenotype extends beyond the control ofT cell responses and may also determine the develop-ment and the magnitude of antibody responses as dis-cussed further below.
Nonhuman Primates
Data from our laboratory indicate that multiple peptideepitopes of MOG are encephalitogenic inC. jacchusmarmosets (Table III). Active immunizations with 100mg of MOG aa 21–40 (which contains a major T cellepitope), alone or in combination with equal amounts ofaa 1–20 and 11–30, reproducibly induced mild, chronicEAE. Neuropathologically, rare (1–3) inflammatory in-filtrates were observed, accompanied by moderate, albeitsignificant demyelination (Genainet al., in preparation).This syndrome could also be produced by immunizationwith a combination of peptides spanning residues aa51–90 and aa 81–120. Immunization with 100mg each of11 overlapping peptides corresponding to the entiresequence of human rMOG (aa 1–120) tended to producelesions that were more destructive than with immuniza-tion with individual groups of peptides but remainedscarce and mostly confined to the cervical spinal cord.No peptide or combination of peptides was capable ofreproducing the protracted, multifocal lesions associatedwith prominent demyelination that result from immuni-zation with whole rMOG in this species.
ANTIBODY RESPONSES IN MOGPEPTIDE-INDUCED EAE
Rodents
Immunization with rMOG and its peptide MOG35-55leads to demyelinating forms of EAE in C57BL/6 (H-2b)and NOD/Lt (H-2g7) mice. Antibody responses in thesemodels were found to be directed against whole rMOGas well an epitope located within MOG aa 35–55. Inaddition, epitope spreading can be observed in NOD/Ltmice which develop an antibody response against MOGaa 92–106 after immunization with aa 35–55 (79). Inboth A.SW and SJL mice (H-2s), immunization againstMOG aa 92–106 leads to a demyelinating form of EAEwith a humoral response directed against MOG aa92–106, however, the presence of antibodies against theentire extracellular domain of MOG has not been as-
sessed (81). Other studies of mouse susceptibility toimmunization against MOG peptides (H-2dq1, H-2b,p,q,H-2q, and H-2u) have not focused on analytical descrip-tion of antibody responses against either rMOG orMOG-derived peptides.
In Lewis rats, s.c. immunization with rMOG in thehind foot pad induced an acute, moderate form ofdemyelinating EAE with humoral responses directedagainst rMOG, MOG aa 1–20, MOG aa 9–25, and MOGaa 35–55. Animals immunized with MOG aa 1–20 orMOG aa 35–55 developed antibody responses directedagainst rMOG and the sensitizing antigen, without evi-dence of epitope spreading (82).
Nonhuman Primates
Immunization ofC. jacchusmarmosets with rMOGinvariably leads to a demyelinating form of EAE, with ahumoral immune response directed against severalepitopes contained within MOG1-50 and 51–80 (see Fig.2). Analysis of serum reactivity in animals immunizedwith single or groups of individual peptides revealed thatantibodies developed in these animals with specificity forthe sensitizing antigen, without evidence of epitopespreading. One exception was observed with the group ofpeptides corresponding to MOG aa 51–90, which did notgive rise to significant antibody responses. Immunizationof marmosets with the 11 overlapping 20mer peptidesspanning the entire sequence of the extracellular domainof MOG, induced antibody responses that were directedat epitopes within MOG1-50 and MOG101–120 (TableIII). It is noteworthy that humoral reactivity to peptidescontaining MOG aa 63–72 could be detected only inanimals immunized with rMOG, and not in animalsimmunized with any combination of the overlappinglinear peptides. The latter observation may indicate thatMOG aa 63–72-specific T cells are not able to providesufficient B cell helper function or that development ofan antibody response against this domain of MOG isdependent upon the recognition of conformationalepitopes present only in rMOG.
RESPECTIVE PATHOGENIC ROLES OF T CELL ANDANTIBODY RESPONSES TO MOG
The respective roles of T cell and antibody responsesin EAE pathogenesis can be studied using adoptive T celltransfer and/or passive antibody transfer systems withsingle or multiple antigens. Adoptive transfer experi-ments involving MOG-specific T cells have been per-formed in several species (Table IV). The results of theseexperiments are discussed in context of observations in
AUTOIMMUNITY TO MOG 163
Journal of Clinical Immunology, Vol. 21, No. 3, 2001
actively immunized animals and differ between strainsand species.
Rodents
Adoptive transfer of MOG aa 35–55-specific T cellsresulted in clinical EAE in H-2b (C57BL/6 and C3H.SW)mice and H-2g7 (NOD/Lt) mice, as did active immuni-zation with this peptide (76, 77, 79). Histologically,inflammatory foci could be seen throughout the CNS inC57BL/6 mice, but the presence of demyelination wasnot assessed (76). In PL/J mice, transfer of lymph nodeor spleen cells derived from animals sensitized withMOG aa 35–55 failed to induce clinical disease but wasable to produce CNS inflammation, and not demyelina-tion (78). Adoptive transfer experiments have not beenreported for other MOG-susceptible mouse strains, in-cluding Biozzi AB/H (H-2dql), C57/BL/1 (H-2b,p,q)DBA/1 (H-2q), and SJL (H-2s). Although adoptivelytransferred mice of the PL/J (H-2u) strain were observedfor only a short period of time (2 weeks), the discrepancybetween clinical EAE and neuropathology is reminiscentof adoptively transferred EAE in rats. In both Lewis andBrown Norway rats, adoptive transfer of MOG-specificor aa 35–55-reactive spleen cells or T cell lines failed toinduce significant neurological impairment (23, 63, 67,82, 83) but resulted in severe inflammation with promi-nent perivascular cuffing (23, 82, 83). These experi-ments, together with the occurence of EAE after activeimmunization with MOG peptides, unequivocally dem-onstrate a pathogenic role for MOG-specific T cells inCNS inflammation. However, the severity of EAE and
neuropathological phenotypes appear dependent on thestrain and the sensitizing antigen, especially with regardto the presence of primary demyelination.
What variables determine the severity of the inflam-matory response against MOG in rodents and the inci-dence of demyelination or irreversible CNS tissue de-struction? As outlined earlier, MHC class II genes playan important role in restricting encephalitogenic epitopespresented to T cells, which may have different disease-inducing potential (Table III). However, the variableEAE phenotypes observed in the different strains mayalso be linked to additional factors. In two mouse strainsbaring H-2s (SJL/J and A.SW), MOG aa 92–106 caninduce either chronic, relapsing–remitting EAE (SJL/Jmice) or nonremitting EAE (A.SW mice) (81). Thedifferent responses of SJL/J mice and A.SW mice toimmunization with the same MOG peptide suggest thatgenes outside of the MHC region contribute to thephenotype of EAE. Non-MHC genes can influenceclinical disease, T cell functions such as the secretion ofIFN-g (84), or other mediators contributing to CNSinflammation and/or demyelination. Of particular impor-tance in determining the course of EAE are the timing ofcytokine release in relationship to inflammatory events inthe CNS (85) and the presence and pathogenicity ofMOG-specific antibody responses.
We have examined correlations among clinical signsof EAE, the presence of MOG- and MOG peptide-specific antibodies, and demyelination reported in studiesof rodent EAE. In EAE actively induced by immuniza-tion with MOG or rMOG, it is clear that severe clinicalEAE and demyelination is consistently associated with
Table IV. Adoptive Transfer of MOG-Specific T Cells in Various Strains and Speciesa
SpeciesMHC class II
restriction element StrainImmunzing
antigenMethod ofinduction Clinical disease Inflammation Demyelination
Ref.No.(s.)
Mouse H-2u PL/J aa35–55 Lymph node orspleen cells
No Yes ND 78
H-2b C57/BL/6 aa35–55 T cell lines Yes (chronic) Yes ND 76H-2b C3H.SW aa35–55 T cell lines Yes (chronic) ND ND 77H-2g7 NOD/Lt aa35–55 Spleen cells Yes (chronic) ND ND 79
Rat RT-11 Lewis Rat MOG(1–218)
Spleen cells No ND ND 63
RT-11 Lewis a35–55 T cell lines No Yes No 23RT-11 Lewis aa1–20 T cell blasts No (weight loss) Minimal No
aa35–55 T cell blasts Yes (mild) Yes No 82,83aa60–67 T cell blasts No No No
RT-1n Brown-Norway Rat rMOG,(1–125,E. coli)
T cell blasts No Yes ND 67
Marmoset Outbred C. jacchus None (naive) T cell lines(aa21–40)
Yes (acute,monophasic)
Yes Minimal 95
aSummarized are adoptive transfer experiments reported in mice, rats, and marmosets, with the resulting clinical and pathological phenotypes.Prominent demyelination is not typically present in MOG adoptive transfer EAE. ND, not done.
164 VON BUDINGEN ET AL.
Journal of Clinical Immunology, Vol. 21, No. 3, 2001

the presence of serum MOG-specific antibodies (TableII). This is consistent with observations that passivetransfer of antibodies specific for whole MOG worsensthe course of MBP-induced EAE and induces demyeli-nation in SJL mice (86) and that EAE is also aggravatedin a mouse engineered to carry a transgene encoding fora monoclonal antibody against MOG (87). The associa-tion between demyelination and the humoral responses isless straightforward in the case of EAE induced withMOG peptides. Accounts of detailed neuropathologicalanalysis are given only for C57BL/6 (H-2b), NOD/Lt(H-2g7), SJL (H-2s), PL/J (H-2u) mice, and Lewis (RT-1l)rats (Table III). C57BL/6 and NOD/Lt mice developsevere chronic EAE after immunization with MOG aa35–55, in association with rMOG- and MOG aa 35–55-specific antibodies and neuropathologically, show demy-elination (79). SJL and A.SW mice, which respondexclusively to MOG aa 92–106, also develop chronicEAE with demyelination (65, 81), in association withanti-MOG aa 92–106 antibodies (81). One differencebetween these mouse strains is that the NOD, SJL, andA.SW develop relapses, in contrast to the B6 mouse.PL/J mice develop chronic relapsing EAE after immuni-zation with MOG aa 35–55 and display moderate demy-elination. Demyelination was not observed after adoptivetransfer of MOG aa 35–55-reactive T cells in this strain(78). This suggests that antibody responses to peptidescould play a role in the induction of demyelination,although antibody responses were not assessed in theactively immunized PL/J mice.
Thus, although these observations of mouse EAEindicate that development of antibody responses thattarget certain epitopes of MOG may influence clinicaland pathological phenotypes of MOG-induced EAE inmice, they fail to demonstrate a specific role of antibod-ies in promoting demyelination in all strains. A recentstudy has demonstrated that immunization of C57BL/6,B cell-deficient mice with MOG aa 35–55 is sufficient forthe development of severe clinical disease and CNSdemyelination that are comparable to those observed inwild-type animals (88). Unfortunately, the presence of
demyelination has not been assessed in this strain afteradoptive transfer of MOG aa 35–55-reactive T cells (76),thus it is not possible to understand to what extentantibodies specific for this peptide may contribute toEAE pathology. An additional important observation inthe C57BL/6 B-less mouse is that, while these animalsare fully susceptible to immunization with MOG aa35–55, they fail to develop clinical EAE and demyelina-tion if immunized with whole rMOG (Table V) (89).These findings have several possible explanations: thefirst is that, indeed, MOG-specific autoantibodies canproduce demyelination and worsen EAE in mice, butpathogenicity for these antibodies is linked to theirability to recognize conformational epitopes of MOG,and not linear determinants; second, this suggests that, inaddition to immunoglobulin production, B cells mayhave other important functions for the full developmentof EAE, including antigen processing and presentation.In the case of other strains such as Biozzi AB/H,insufficient information is available to understandwhether these mice fail to mount a pathogenic antibodyresponse to explain the absence of demyelination. Itwould be of interest to explore further the variouspathogenic mechanisms of MOG-induced EAE in mice,for example, by passive antibody transfer experimentsand/or cross-breeding of these strains with soluble im-munoglobulin-deficient mice, which retain expression ofthe B cell surface receptors (90).
In contrast to mice, a synergism between encephalito-genic T cells and pathogenic antibody is clearly involvedin the pathogenesis of demyelinating EAE in rats. InLewis rats, inflammation mediated by MOG-specific Tcells fails to induce gross neurological deficits anddemyelination (23, 63, 82, 83). Although Adelmanet al.were unable to induce severe and demyelinating EAEusing MOG aa 35–55 (82), in all other investigationsactive immunization with either whole MOG or MOG aa35–55 produce demyelinating EAE associated with an-tibody responses restricted to the immunizing antigen(Tables II and III). The demyelinating phenotype andsevere clinical EAE can be restored in recipients of
Table V. Immunogenic Properties of rMOG and MOG aa 35–55 in C57/BL/6 (H-2b) B Cell-Deficient Mice
Strain AntigenNo. of
immunizations PTIncidenceof EAE Inflammation Demyelination
T cell responseto antigen Antibody Ref. No.
B2/2 aa35–55 2 Yes 10/10 Yes Yes ND 2 88WT aa35–55 2 Yes 5/5 Yes Yes ND Yes
(aa35–55)B2/2 rMOG 2 Yes 0/20 Minimal Minimal 1 2 89WT rMOG 2 Yes 18/20 Yes Yes 1 NDB2/2 aa35–55 2 Yes 12/19 Yes Yes ND 2 0WT aa35–55 2 Yes 7/15 Yes Yes ND ND 0
aB2/2, targeted disruption of the IgM H-chain; WT, wild type; PT,B. pertussistoxin; ND, not done.
AUTOIMMUNITY TO MOG 165
Journal of Clinical Immunology, Vol. 21, No. 3, 2001

adoptive T cell transfer, by cotransfer of MOG-specificantibodies (23). This result is consistent with previousexperience that anti-MOG antibodies also aggravateMBP-induced EAE in Lewis rats (27, 29). Furthermore,in Brown Norway rats, MOG induces an acute and lethalform of EAE, which is linked to increased production ofautoantibodies (67). These findings, and other studies ofgenetic susceptibility to EAE, indicate that, in addition toMHC class II, autoimmunity to MOG is under thecontrol of multiple other genes in rats (91, 92).
Nonhuman Primates
Because of their close phylogeny to humans and thesimilarity of EAE in this species to human MS, themarmoset model has been instrumental in elucidatingimmunopathological mechanisms involved in the devel-opment of demyelinating lesions. Adoptive transfer of Tcells is possible inC. jacchusmarmosets, due to a naturalbone marrow chimerism. It has been possible to demon-strate an obligatory role for antibodies to MOG in thissystem, using a combination of adoptive transfer ofMBP-reactive T cells (or active immunization withMBP) and purified anti-MOG antibodies (30, 70). Theimportance of antibodies in marmoset EAE is alsoemphasized by observations that immune deviation ofMOG-specific responses toward a Th2 phenotype ofcytokines resulted in hyperacute, lethal forms of EAE(66) and that animals immunized with MP4, a chimericprotein of MBP and PLP, develop severe EAE anddemyelination only in association with determinantspreading of their antibody response to MOG (98).
The role of MOG-specific T cells and antibodies wasfurther explored by passive transfer of T cell clonesspecific for MOG aa 21–40 between syngeneic marmo-set siblings (Table IV). Interestingly, these T cell clonescould be readily derived from the PBMC of healthy,unimmunized animals, a finding that parallels our previ-ous observation that MBP-specific T cell clones arepresent in the natural circulating repertoire of theseprimates (94). Adoptive transfer of MOG-reactive T cellclones resulted in mild, acute monophasic clinical EAE,and minimal demyelination, albeit significantly moreCNS disease was present compared to recipients ofMBP-reactive T cell clones (94, 95). The extent andseverity of disease were similar to that induced by activeimmunization with MOG peptides, however, the clinicalcourse of EAE was monphasic and not chronic. Thissuggests that MOG-reactive T cells are significantlymore pathogenic in primates than are MBP-reactive Tcells but are not able to induce sustained CNS inflam-
matory responses and/or permanent demyelination withgliosis, which appear to be dependent on antibodies.
Because of the prime importance of humoral re-sponses to MOG in promoting or increasing demyelina-tion and tissue destruction, it is of interest to identify theepitopes that are targeted by pathogenic populations ofautoantibodies. Antibody responses in marmosets immu-nized with MOG-derived peptides are specific for theimmunizing antigen, and no determinant spreading hasbeen recorded to date. These antibodies most often alsorecognize rMOG (with the exception of those directedagainst peptides contained within MOG aa 51–90), butthe actual determinants may differ from those for anti-bodies present in rMOG-immunized marmosets (Fig. 2).The marked differences in clinical and neuropathologicalfeatures between MOG peptide- and rMOG-immunizedanimals suggest that pathogenic antibody populations areselectively produced in rMOG-immunized marmosets.Because of their cross-reactive specificities, these differ-ent antibody populations cannot be easily distinguishedone from another in standard ELISA assays.
In an attempt to clarify the epitopes targeted bydemyelinating autoantibodies inC. jacchus, we haveexamined the molecular diversity of the MOG-specificantibody repertoire in rMOG-immunized animals using acombinatorial library of phage-displayed antibody frag-ments. This analysis is in progress but, indeed, showsthat antibodies with a very high affinity to rMOG, whichare capable of competing with the native serum antibod-ies produced by rMOG-immunized marmosets, recog-nize exclusively structural epitopes of MOG, and not thelinear peptides (96) (von Bu¨dingenet al., in preparation).These data provide evidence that several populations ofantibodies with different pathogenic potential coexist inmarmoset EAE and that demyelinating antibody popula-tions could be directed against conformational epitopesof this protein. In support of this hypothesis, an obser-vation by Brehmet al. (97) has shown that a murinemonoclonal antibody which recognizes the linear epitopeMOG aa 1–26 fails to mediate demyelinationin vivo inrats, although this antibody is fully capable of recogniz-ing whole MOG. Further dissection of the specificities ofantibody responses to MOG and their pathogenic poten-tial will be of considerable interest, since it will helpdefine the targets for selective therapeutic interventionthat could prevent demyelination.
CONCLUDING REMARKS
The data summarized here highlight MOG as a uniqueCNS autoantigen. Although MOG is exposed at themyelin surface, expression has not been unequivocally
166 VON BUDINGEN ET AL.
Journal of Clinical Immunology, Vol. 21, No. 3, 2001

demonstrated outside the CNS, which implies that im-mune responses to this antigen may escape the control ofcentral thymic tolerance. It is clear that heightenedmemory responses to MOG exist in healthy individualsof outbred species (33, 95, 98, 99) (and Koehleret al., inpreparation), which have full capacity to induce CNSinflammatory disease as demonstrated by adoptive T celltransfer in the marmoset (95). These potentially autoag-gressive responses may arise early in life via mecha-nisms of mimicry to yet unknown self molecules, assuggested by the recent observation that receptor editingof MOG-specific immunoglobulins can take place in atransgenic mouse model that lacks the MOG gene (100).Mimicry to viral proteins has also been demonstrated forMOG, although this is not a feature unique to this antigen(101). The lack of spontaneous CNS-specific autoimmu-nity in most normal individuals implies the presence ofspecific regulatory mechanisms to maintain immunehomeostasis, which would be of interest to explore astargets for future therapeutic intervention.
Regardless of the mechanisms that give rise to andcontrol autoimmunity to MOG, the encephalitogenicproperties of this antigen probably lie within its abilityto induce immune responses that are highly patho-genic. Although some laboratory strains of mice ap-pear capable of developing CNS demyelination that ismediated by MOG-specific T cells, the role of auto-antibodies against MOG in tissue destruction has beenunequivocally demonstrated, especially in nonhumanprimates. Importantly, pathogenic antibody responsesagainst MOG have also been demonstratedin situ inMS brain (34). The identification of MOG and subse-quent studies of MOG-induced EAE have afforded thedemonstration that synergism between T cell andantibody responses to a single antigen of myelin canresult in CNS inflammatory lesions that recapitulatetypical lesions of MS. In addition, many experimentsreviewed here show that phenotypic expression ofautoimmunity to MOG may depend on specificitiesand molecular targets of the antibody response toMOG, rather than on factors controlled by cellularimmunity. This should stimulate further studies ofimmune responses to MOG, including the completecharacterization of structural epitopes targeted bypathogenic antibodies.
ACKNOWLEDGMENTS
This work was supported by grants from the NationalInstitutes of Health (AI 43073 to S.L.H.) and the Na-tional Multiple Sclerosis Society (JF2087-A-2 toC.P.G.). P.V. was a postdoctoral fellow of the Spanish
Ministry of Health (FIS 96/5122 and 97/5459). H.-C.v.B.is a postdoctoral fellow from the National Institutes ofHealth. N.T. is a postdoctoral fellow from the NationalMultiple Sclerosis Society. J.C.O. was a postdoctoralfellow from The Association pour la Recherche Contre laSclerose en Plaque and The Programme Lavoisier fromthe French Foreign Office.
REFERENCES
1. Lassmann H, Suchanek G, Ozawa K: Histopathology and theblood-cerebrospinal fluid barrier in multiple sclerosis. Ann Neu-rol 36 (Suppl):S42–S46, 1994
2. Raine C: The neuropathology of multiple sclerosis.In MultipleSclerosis: Clinical and Pathogenetic Basis, C Raine, H McFar-landand, W Tourtellotte (eds). London, Chapman & Hall, 1997,pp 151–171
3. Trapp B, Peterson J, Ransohoff R, Rudick R, Mork S, Bo L:Axonal transection in the lesions of multiple sclerosis. N EnglJ Med 338:278–285, 1998
4. Hohlfeld R: Biotechnological agents for the immunotherapy ofmultiple sclerosis. Principles, problems and perspectives. Brain120:865–916, 1997
5. Tabira T, Kira J-I: Strain and species differences of encephalito-genic determinants of myelin basic protein and proteolipidapoprotein.In Myelin: Biology and Chemistry, RE Martenson(ed). Boca Raton, FL, CRC Press, 1992, pp 783–799
6. Sobel RA, Greer JM, Kuchroo VK: Minireview: Autoimmuneresponses to myelin proteolipid protein. Neurochem Res 19:915–921, 1994
7. Paterson PY: Transfer of allergic encephalomyelitis in rats bymeans of lymph node cells. J Exp Med 111:119–135, 1960
8. Zamvil SS, Steinman L: The T-lymphocyte in experimentalallergic encephalomyelitis. Annu Rev Immunol 8:579–621, 1990
9. Martin R, McFarland, HF, McFarlin DE: Immunological aspectsof demyelinating diseases. Annu Rev Immunol 10:153–187, 1992
10. Brosnan CF, Raine CS: Mechanisms of immune injury in multi-ple sclerosis. Brain Pathol 6:243–257, 1996
11. Weinstock-Guttman B, Ransohoff R, Kinkel R, Rudick R: Theinterferons: Biological effects, mechanisms of action, and use inmultiple sclerosis. Ann Neurol 37:7–15 1995
12. Lucchinetti C, Bruck W, Rodriguez M, Lassmann H: Distinctpatterns of multiple sclerosis pathology indicates heterogeneity inpathogenesis. Brain Pathol 6:259–274, 1996
13. Lassmann H: Neuropathology in multiple sclerosis: New con-cepts. Multiple Sclerosis 4:93–98, 1998
14. Wekerle H, Kojima K, Lannes-Vierra J, Lassmann H, LiningtonC: Animal models. Ann Neurol 36:S47–S53, 1994
15. Bornstein M, Appel S: The application of tissue culture to thestudy of experimental “allergic” encephalomyelitis. I. Patterns ofdemyelination. J Neuropathol Exp Neurol 20:141–147, 1964
16. Brosnan CF, Stoner GL, Bloom BR, Wisniewski HM: Studies ondemyelination by activated lymphocytes in the rabbit eye. II.Antibody-dependent cell-mediated demyelination. J Immunol118:2103–2109, 1977
17. Seil FJ, Falk GA, Kies MW, Alvord EC: The in vitro demyeli-nating activity of sera from guinea pigs sensitized with wholeCNS and with purified encephalitogen. Exp Neurol 22:545–555,1968
AUTOIMMUNITY TO MOG 167
Journal of Clinical Immunology, Vol. 21, No. 3, 2001

18. Raine CS, Hummelgard A, Swanson E, Bornstein MB: Multiplesclerosis: Serum-induced demyelination in vitro. A light andelectron microscope study. J Neurol Sci 20:127–148, 1973
19. Lebar R, Boutry JM, Vincent C, Robineaux R, Voisin G: Studies onautoimmune encephalomyelitis in the guinea pig. II. An in vitroinvestigation on the nature, properties, and specificities of theserum-demyelinating factor. J Immunol 116:1439–1446, 1976
20. Lebar R, Vincent C: Tentative identification of a second centralnervous system Myelin membrane autoantigen (M2) by a bio-chemical comparison with the basic protein (BP). J Neuroimmu-nol 1:367–389, 1981
21. Lebar R, Lubetzki C, Vincent C, Lombrail P, Boutry J-M: TheM2 autoantigen of central nervous system myelin, a glycoproteinpresent in oligodendrocyte membrane. Clin Exp Immunol 66:423–443, 1986
22. Linington C, Webb M, Woodhams PL: A novel myelin-associated glycoprotein defined by a mouse monoclonal antibody.J Neuroimmunol 6:387–396, 1984
23. Linington C, Berger T, Perry L, Weerth S, Hinze-Selch D, ZhangY, Lu H-C, Lassmann H, Wekerle H: T cells specific for themyelin oligodendrocyte glycoprotein, mediate an unusual auto-immune inflammatory response in the central nervous system.Eur J Immunol 23:1364–1373, 1993
24. Kerlero de Rosbo N, Honegger P, Lassmann H, Matthieu J-M:Demyelination induced in aggregating brain cell cultures by amonoclonal antibody against myelin/oligodendrocyte glycopro-tein. J Neurochem 55:583–587, 1990
25. Schluesener HJ, Sobel RA, Linington C, Weiner HL: A mono-clonal antibody against a myelin oligodendrocyte glycoproteininduces relapses and demyelination in central nervous systemautoimmune disease. J Immunol 139:4016–4021, 1987
26. Linington C, Lassmann H: Antibody responses in chronic relapsingexperimental allergic encephalomyelitis: Correlation of serum de-myelinating activity with antibody titre to the myelin/oligodendro-cyte glycoprotein (MOG). J Neuroimmunol 17:61–69, 1987
27. Linington C, Bradl M, Lassmann H, Brunner C, Vass K:Augmentation of demyelination in rat acute allergic encephalo-myelitis by circulating mouse monoclonal antibodies directedagainst a myelin/oligodendrocyte glycoprotein. Am J Pathol130:443–454, 1988
28. Linington C, Engelhardt B, Kapocs G, Lassman H: Induction ofpersistently demyelinated lesions in the rat following the repeatedadoptive transfer of encephalitogenic T cells and demyelinatingantibody. J Neuroimmunol 40:219–224, 1992
29. Lassmann H, Brunner C, Bradl M, Linington C: Experimentalallergic encephalomyelitis: The balance between encephalito-genic T lymphocytes and demyelinating antibodies determinessize and structure of demyelinated lesions. Acta Neuropathol(Berl) 75:566–576, 1988
30. Genain CP, Nguyen MH, Letvin NL, Pearl R, Davis RL,Adelman M, Lees MB, Linington C, Hauser SL: Antibodyfacilitation of multiple sclerosis-like lesions in a non humanprimate. J Clin Invest 96:2966–2974, 1995
31. Kerlero de Rosbo N, Milo R, Lees MB, Burger D, Bernard CCA,Ben-Nun A: Reactivity to myelin antigens in multiple sclerosis.Peripheral blood lymphocytes respond predominantly to myelinoligodendrocyte glycoprotein. J Clin Invest 92:2602–2608, 1993
32. Diaz-Villoslada P, Shih A, Shao L, Genain C, Hauser S: T-cellreactivity to myelin antigens: Myelin/oligodendrocyte glycopro-tein is a prevalent antigen. J Neuroimmunol 99:36–43, 1999
33. Lindert R, Haase C, Brehm U, Linington C, Wekerle H, HohlfeldR: Multiple sclerosis: B- and T-cell responses to the extracellular
domain of the myelin oligodendrocyte glycoprotein. Brain 122:2089–2100, 1999
34. Genain C, Cannella B, Hauser S, Raine C: Identification ofautoantibodies associated with myelin damage in multiple scle-rosis. Nature Med 5:170–175, 1999
35. Raine C, Cannella B, Hauser S, Genain C: Demyelination innon-human primate autoimmune encephalomyelitis and acutemultiple sclerosis lesions: A case for antigen-specific antibodymediation. Ann Neurol 46:144–160, 1999
36. Pham-Dinh D, Allinquant B, Ruberg M, Della-Gaspera B, Nuss-baum JL, Dautigny A: Characterization and expression of thecDNA coding for the human myelin/oligodendrocyte glycopro-tein. J Neurochem 63:2353–2356, 1994
37. Hilton A, Slavin A, Hilton D, Bernard C: Characterization ofcDNA and genomic clones encoding human myelin oligodendro-cyte glycoprotein. J Neurochem 65:309–318, 1995
38. Roth M, Malfroy L, Offer C, Sevin J, Enault G, Borot N,Pontarotti P, Coppin H: The human myelin oligodendrocyteglycoprotein (MOG) gene: Complete nucleotide sequence andstructural characterization. Genomics 28:241–250, 1995
39. Gardinier MV, Amiguet P, Linington C, Matthieu JM: Myelin/oligodendrocyte glycoprotein is a unique member of the immu-noglobulin superfamily. J Neurosci Res 33:177–187, 1992
40. Daubas P, Pham-Dinh D, Dautigny A: Structure and polymor-phism of the mouse oligodendrocyte glycoprotein gene. Genom-ics 23:36–41, 1994
41. Pham-Dinh D, Mattei M-G, Nussbaum J-L, Roussel G, PontarottiP, Roeckel N, Mather IH, Artzt K, Fisher Lindall K, Dautigny A:Myelin/oligodendrocyte glycoprotein is a member of a subset ofthe immunoglobulin superfamily encoded within the major his-tocompatibility complex. Proc Natl Acad Sci USA 90:7990–7994, 1993
42. Johns T, Bernard C: The structure and function of myelinoligodendrocyte glycoprotein. J Neurochem 72:1–9, 1999
43. Pham-Dinh D, Jones E, Pitiot G, Della Gaspera B, Daubas P,Mallet J, Le Paslier D, Fischer LK, Dautigny A: Physicalmapping of the human and mouse MOG gene at the distal end ofthe MHC class Ib region. Immunogenetics 42:386–391, 1995
44. Lambracht D, Prokop C, Hedrich H, Fischer LK, Wonigeit K:Mapping of H2-M homolog and MOG genes in the rat MHC.Immunogenetics 42:418–421, 1995
45. Hjelmstrom P, Penzotti J, Henne R, Lybrand T: A molecularmodel of myelin oligodendrocyte glycoprotein. J Neurochem71:1742–1749, 1998
46. Kroepfl J, Viise L, Charron A, Linington C, Gardinier M:Investigation of myelin/oligodendrocyte glycoprotein membranetopology. J Neurochem 67:2219–2222, 1996
47. Della Gaspera B, Pham-Dinh D, Roussel G, Nussbaum J-L,Dautigny A: Membrane topology of the myelin/oligodendrocyteglycoprotein. Eur J Biochem 258:478–484, 1998
48. Burger D, Steck AJ, Bernard CCA, Kerlero de Rosbo N: Humanmyelin/oligodendrocyte glycoprotein: A new member of theL2/HNK-1 family. J Neurochem 61:1822–1827, 1993
49. Schachner M, Martini R, Hall H, Orberger G: Functions of theL2/HNK-1 carbohydrate in the nervous system. Prog Brain Res105:183–188, 1995
50. Quarles R: Glycoproteins of myelin sheaths. J Mol Neurosci8:1–12, 1997
51. Dyer C, Matthieu J: Antibodies to myelin/oligodendrocyte-specific protein and myelin/oligodendrocyte glycoprotein signaldistinct changes in the organization of cultured oligodendroglialmembrane sheets. J Neurochem 62:777–787, 1994
168 VON BUDINGEN ET AL.
Journal of Clinical Immunology, Vol. 21, No. 3, 2001

52. Matthieu J, Amiguet P: Myelin/oligodendrocyte glycoproteinexpression during development in normal and myelin-deficientmice. Dev Neurosci 12:293–302, 1990
53. Slavin A, Johns T, Orian J, Bernard C: Regulation of myelinoligodendrocyte glycoprotein in different species throughoutdevelopment. Dev Neurosci 19:69–78, 1997
54. Scolding N, Frith S, Linington C, Morgan B, Campbell A,Compston D: Myelin-oligodendrocyte glycoprotein (MOG) is asurface marker for oligodendrocyte maturation. J Neuroimmunol22:169–176, 1989
55. Pfeiffer SE, Warrington AE, Bansal R: The oligodendrocyte andits many cellular processes. Trends Cell Biol 3:191–197, 1993
56. Pribyl T, Campagnoni C, Kampf K, Kashima T, Handley V,McMahon J, Campagnoni A: Expression of the myelin proteolipidprotein gene in the human fetal thymus. J Neuroimmunol 67:125–130, 1996
57. Campagnoni C, Garbay B, Micevych P, Pribyl T, Kampf K,Handley V, Campagnoni A: DM20 mRNA slice product of themyelin protein gene is expressed in the murine heart. J NeurosciRes 33:148–155, 1992
58. Voskuhl R: Myelin protein expression in lymphoid tissues:Implications for peripheral tolerance. Immunol Rev 164:81–82,1998
59. Brunner C, Lassman H, Waehneldt TV, Matthieu JM, LiningtonC: Differential ultrastructural localization of myelin basic protein,myelin/oligodendroglial glycoprotein, and 2939 cyclic nucleotidephosphodiesterase in the CNS of adult rats. J Neurochem 52:296–304, 1989
60. Birling M-C, Roussel G, Nussbaum F, Nussbaum J-L: Biochem-ical and immunohistochemical studies with specific polyclonalantibodies directed against bovine myelin/oligodendrocyte glyco-protein. Neurochem Res 18:937–945, 1993
61. Mokhtarian F, Zhang Z, Shi Y, Gonzales E, Sobel R: Molecularmimicry between a viral peptide and a myelin oligodendrocyteglycoprotein peptide induces autoimmune demyelinating diseasein mice. J Neuroimmunol 95:43–54, 1999
62. Stefferl A, Schubart A, Storch M, Amini A, Mather I, LassmannH, Linington C: Butyrophilin, a milk protein, modulates theencephalitogenic T cell response to myelin oligodendrocyteglycoprotein in experimental autoimmune encephalomyelitis.J Immunol 165:2859–2865, 2000
63. Johns TG, Kerlero de Rosbo N, Menon KK, Abo S, GonzalesMF, Bernard CCA: Myelin oligodendrocyte glycoprotein inducesa demyelinating encephalomyelitis resembling multiple sclerosis.J Immunol 154:5536–5541, 1995
64. Devaux B, Enderlin F, Wallner B, Smilek D: Induction of EAE inmice with recombinant human MOG, and treatment of EAE witha MOG peptide. J Neuroimmunol 75:169–173, 1997
65. Amor S, Groome N, Linington C, Morris MM, Dornmair K,Gardinier MV, Matthieu JM, Baker D: Identification of epitopesof myelin oligodendrocyte glycoprotein for the induction ofexperimental allergic encephalomyelitis in SJL and Biozzi AB/Hmice. J Immunol 153:4349–4356, 1994
66. Genain CP, Abel K, Belmar N, Villinger F, Rosenberg DP,Linington C, Raine CS, Hauser SL: Late complications ofimmune deviation therapy in a non human primate. Science274:2054–2057, 1996
67. Stefferl A, Brehm U, Storch M, Lambracht-Washington D,Bourquin CW, Lassmann H, Linington C: Myelin oligodendro-cyte glycoprotein induces experimental autoimmune encephalo-myelitis in the “resistant” Brown Norway rat: Disease suscepti-
bility is determined by MHC and MHC-linked effects on the Bcell response. J Immunol 163:40–49 1999
68. Storch MK, Stefferl A, Brehm U, Weissert R, Wallstrom E,Kerschensteiner M, Olsson T, Linington C, Lassmann H: Auto-immunity to myelin oligodendrocyte glycoprotein in rats mimicsthe spectrum of multiple sclerosis pathology. Brain Pathol 8:681–694, 1998
69. Massacesi L, Genain CP, Lee-Parritz D, Letvin NL, Canfield D,Hauser SL: Active and passively induced experimental autoim-mune encephalomyelitis in common marmosets: A new model formultiple sclerosis. Ann Neurol 37:519–530, 1995
70. Genain CP, Hauser SL: Allergic encephalomyelitis in commonmarmosets: Pathogenesis of a multiple sclerosis-like lesion.Methods Comp Methods Enzymol 10:420–434, 1996
71. Genain C, Belmar N, Diaz-Villoslada P, Hauser S: Fine specific-ities of T cell and B cell responses to myelin/oligodendrocyteglycoprotein in common marmosets. J Neuroimmunol 90:34,1998 (abstr)
72. Brok H, Uccelli A, Kerlero De Rosbo N, Bontrop R, Roccata-gliata L, de Groot N, Capello E, Laman J, Nicolay K, MancardiG, Ben-Nun A, ’t Hart B: Myelin/oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis in common marmosets:The encephalitogenic T cell epitope pMOG24–36 is presented bya monomorphic MHC class II molecule. J Immunol 165:1093–1101, 2000
73. Antunes S, de Groot N, Brok H, Doxiadis G, Menezes A, OttingN, Bontrop R: The common marmoset: a new world primatespecies with limited Mhc class II variability. Proc Natl Acad SciUSA 95:11745–11750, 1998
74. Kerlero de Rosbo N, Brok H, Bauer J, Kaye J, ’tHart B, Ben NunA: Rhesus monkeys are highly susceptible to experimentalautoimmune encephalomyelitis induced by myelin oligodendro-cyte glycoprotein: Characterization of immunodominant T and Bcell epitopes. J Neuroimmunol 110:83–96, 2000
75. Kerlero de Rosbo N, Hoffman M, Mendel I, Yust I, Kaye J,Bakimer R, Flechter S, Abramsky O, Milo R, Karni A, Ben-NunA: Predominance of the autoimmune response to myelin oligo-dendrocyte glycoprotein (MOG) in multiple sclerosis: Reactivityto the extracellular domain of MOG is directed against three mainregions. Eur J Immunol 27:3059–3069, 1997
76. Mendel I, Kerlero de Rosbo N, Ben-Nun A: A myelin oligoden-drocyte glycoprotein peptide induces typical chronic experimen-tal autoimmune encephalomyelitis in H-2b mice: fine specificityand T cell receptor Vb expression of encephalitogenic T cells.Eur J Immunol 25:1951–1959, 1995
77. Mendel I, Kerlero de Rosbo N, Ben-Nun A: Delineation of theminimal encephalitogenic epitope within the immunodominantregion of myelin oligodendrocyte glycoprotein: Diverse V betagene usage by T cells recognizing the core epitope encephalito-genic for T cell receptor V beta b and T cell receptor V beta aH-2b mice. Eur J Immunol 26:2470–2479, 1996
78. Kerlero de Rosbo N, Mendel I, Ben-Nun A: Chronic relapsingexperimental autoimmune encephalomyelitis with a delayed on-set and an atypical clinical course, induced in PL/J mice bymyelin oligodendrocyte glycoprotein (MOG)-derived peptide.Eur J Immunol 25:985–993, 1995
79. Bernard C, Johns T, Slavin A, Ichikawa M, Ewing C, Liu J,Bettadapura J: Myelin oligodendrocyte glycoprotein: A novel can-didate autoantigen in multiple sclerosis. J Mol Med 75:77–88, 1997
80. Ichikawa M, Johns TG, Liu J, Bernard CCA: Analysis of the fineB-cell specificity during the chronic/relapsing course of a multi-ple sclerosis-like disease in Lewis rats injected with the enceph-
AUTOIMMUNITY TO MOG 169
Journal of Clinical Immunology, Vol. 21, No. 3, 2001

alitogenic myelin oligodendrocyte glycoprotein peptide 35–55.J Immunol 157:919–926, 1996
81. Tsunoda I, Kuang L, Theil D, Fujinami R: Antibody associationwith a novel model for primary progressive multiple sclerosis:Induction of relapsing-remitting and progressive forms of EAE inH2s mouse strains. Brain pathol 10:402–418, 2000
82. Adelmann M, Wood J, Benzel I, Fiori P, Lassmann H, MatthieuJ, Gardinier M, Dornmair K, Linington C: The N-terminaldomain of the myelin oligodendrocyte glycoprotein (MOG)induces acute demyelinating experimental autoimmune encepha-lomyelitis in the Lewis rat. J Neuroimmunol 63:17–27, 1995
83. Berger T, Weerth S, Kojima K, Linington C, Wekerle H,Lassmann H: Experimental autoimmune encephalomyelitis: Theantigen specificity of T lymphocytes determines the topographyof lesions in the central and peripheral nervous system. Lab Invest76:355–364, 1997
84. Weissert R, Wallstrom E, Storch MK, Stefferl A, Lorentzen J,Lassmann H, Linington C, Olsson T: MHC haplotype-dependentregulation of MOG-induced EAE in rats. J Clin Invest 102:1265–1273, 1998
85. Juedes AE, Hjelmstrom P, Bergman CM, Neild AL, Ruddle NH:Kinetics and cellular origin of cytokines in the central nervoussystem: Insight into mechanisms of myelin oligodendrocyteglycoprotein-induced experimental autoimmune encephalomyeli-tis. J Immunol 164:419–426, 2000
86. Schluesener HJ, Sobel RA, Linington C, Weiner HL: A mono-clonal antibody against a myelin oligodendrocyte glycoproteininduces relapses and demyelination in central nervous systemautoimmune disease. J Immunol 139:4016–4021, 1987
87. Litzenburger T, Fassler R, Bauer J, Lassmann H, Linington C,Wekerle H, Iglesias J: B lymphocytes producing demyelinatingautoantibodies: development and function in gene-targeted trans-genic mice. J Exp Med 188:169–180, 1998
88. Hjelmstrom P, Juedes A, Fjell J, Ruddle N: Cutting Edge: B celldeficient mice develop experimental allergic encephalomyelitiswith demyelination after myelin oligodendrocyte glycoproteinsensitization. J Immunol 161:4480–4483, 1998
89. Lyons J, San M, Happ M, Cross A: B cells are critical toinduction of experimental allergic encephalomyelitis by proteinbut not by a short encephalitogenic peptide. Eur J Immunol29:3432–3439, 1999
90. Chan OT, Hannum LG, Haberman AM, Madaio MP, ShlomchikMJ: A novel mouse with B cells but lacking serum antibodyreveals an antibody-independent role for B cells in murine lupus.J. Exp Med189:1639–48, 1999.
91. Dahlman I, Wallstrom E, Weissert R, Storch M, Kornek B,Jacobsson L, Linington C, Luthman H, Lassmann H, Olsson T:Linkage analysis of myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis in the ratidentifies a locus controlling demyelination on chromosome 18.Hum Mol Genet 8:2183–2190, 1999
92. Olsson T, Dahlman I, Wallstrom E, Weissert R, Piehl F: Geneticsof rat neuroinflammation. J Neuroimmunol 107:191–200, 2000
93. McFarland H, Lobito A, Johnson M, Nyswaner J, Frank J,Palardy G, Tresser N, Genain C, Mueller J, Matis L, Lenardo M:Determinant spreading associated with demyelination in a non-human primate model of multiple sclerosis. J Immunol 162:2384–2390, 1999
94. Genain CP, Lee-Parritz D, Nguyen MH, Massacesi L, Joshi N,Ferrante R, Hoffman K, Moseley M, Letvin NL, Hauser SL: Inhealthy primates, circulating autoreactive T cells mediate auto-immune disease. J Clin Invest 94:1339–1345, 1994
95. Villoslada P, Abel K, Heald N, Goertsches R, Hauser S, GenainC: Repertoire heterogeneity, encephalitogenicity and evidence forregulatory control of precursor T cells specific for myelinoligodendrocyte glycoprotein in naive populations of outbredprimates (submitted for publication)
96. von Budingen H-C, Hauser S, Genain C: Molecular analysis ofthe antibody response against myelin oligodendrocyte glycopro-tein in primate experimental autoimmune encephalomyelitis. AnnNeurol 48:A416, 2000
97. Brehm U, Piddlesden S, Gardinier M, Linington C: Epitopespecificity of demyelinating monoclonal autoantibodies directedagainst the human myelin oligodendrocyte glycoprotein (MOG).J Neuroimmunol 97:9–15, 1999.
98. Ouallet J-C, Heald N, Islar J, Hauser S, Genain C: Detection ofB-cells specific for the autoantigen myelin/oligodendrocyte gly-coprotein using a sensitive immunogold-labeling assay. Neurol-ogy 52(Suppl 2):A337, 1999
99. Reindl M, Linington C, Brehm U, Egg R, Dilitz E, Deisenham-mer F, Poewe W, Berger T: Antibodies against the myelinoligodendrocyte glycoprotein and the myelin basic protein inmultiple sclerosis and other neurological diseases: A comparativestudy. Brain 122:2047–2056, 1999
100. Litzenburger T, Blu¨thmann H, Morales P, Pham-Dinh D, Dau-tigny A, Wekerle H, Iglesias A: Development of myelin oligo-dendrocyte glycoprotein autoreactive transgenic B lymphocytes:Receptor editing in vivo after encounter of a self-antigen distinctfrom myelin oligodendrocyte glycoprotein. J Immunol 165:5360–5366, 2000
101. Wucherpfennig KW, Strominger JL: Molecular mimicry in T cellmediated autoimmunity: Viral peptides activate human T cellclones specific for myelin basic protein. Cell 80:695–705, 1995
102. Amiguet P, Gardinier MV, Zanetta JP, Matthieu JM: Purificationand partial structural and functional characterization of mousemyelin/oligodendrocyte glycoprotein. J Neurochem 58:1676–1682, 1992
103. Abo S, Bernard C, Webb M, Johns T, Alafaci A, Ward L,Simpson R, Kerlero de Rosbo N: Preparation of highly purifiedhuman myelin oligodendrocyte glycoprotein in quantities suffi-cient for encephalitogenicity and immunogenicity studies. Bio-chem Mol Biol Int 30:945–958, 1993
104. Pham-Dinh D, Della Gaspera B, Kerlero de Rosbo N, DautignyA: Sructure of the human myelin/oligodendrocyte glycoproteingene and multiple alternative spliced isoforms. Genomics 29:345–352, 1995
105. Ballenthin P, Gardinier M: Myelin/oligodendrocyte glycoproteinis alternatively spliced in humans but not mice. J Neurosci Res46:271–281, 1996
106. Bettadapura J, Menon K, Moritz S, Liu J, Bernard C: Expression,purification, and encephalitogenicity of recombinant human myelinoligodendrocyte glycoprotein. J Neurochem 70:1593–1599, 1998
107. Abdul-Majid K, Jirholt J, Stadelmann C, Stefferl A, Kjellen P,Wallstrom E, Holmdahl R, Lassmann H, Olsson T, Harris RA:Screening of several H-2 congenic mouse strains identifiedH-2(q) mice as highly susceptible to MOG-induced EAE withminimal adjuvant requirement. J Neuroimmunol 111:23–33, 2000
108. Genain CP, Hauser SL: Creation of a model for multiple sclerosisin Callithrix jacchusmarmosets. J Mol Med 75:187–197, 1997
109. Slavin A, Ewing C, Liu J, Ichikawa M, Slavin J, Bernard C:Induction of a multiple sclerosis-like disease in mice with animmunodominant epitope of myelin oligodendrocyte glycopro-tein. Autoimmunity 28:109–120, 1998
170 VON BUDINGEN ET AL.
Journal of Clinical Immunology, Vol. 21, No. 3, 2001











![Myelin oligodendrocyte glycoprotein-specific antibodies from ......protein (MBP)] used to induce experimental autoimmune encephalomyelitis (EAE) in rodent models through induction](https://img.dokumen.tips/doc/110x75/60ff0b7639f1f130b4007123/myelin-oligodendrocyte-glycoprotein-specific-antibodies-from-protein-mbp.jpg)

















