Embed Size (px)
Citation preview

David A. Lynch, MDWilliam D. Travis, MDNestor L. Muller, MD, PhDJeffrey R. Galvin, MDDavid M. Hansell, MDPhilippe A. Grenier, MDTalmadge E. King, Jr, MD
Published online10.1148/radiol.2361031674
Radiology 2005; 236:10–21
Abbreviations:AIP � acute interstitial pneumoniaCOP � cryptogenic organizing
pneumoniaDIP � desquamative interstitial
pneumoniaIIP � idiopathic interstitial
pneumoniaLIP � lymphoid interstitial
pneumoniaNSIP � nonspecific interstitial
pneumoniaRB-ILD � respiratory bronchiolitis–
associated interstitial lung diseaseUIP � usual interstitial pneumonia
1 From the Department of Radiology,University of Colorado Health SciencesCenter, CB A-030, 4200 E Ninth Ave,Denver, CO 80262 (D.A.L.); Depart-ments of Pulmonary and MediastinalPathology (W.D.T.) and Radiology(J.R.G.), Armed Forces Institute of Pa-thology, Washington, DC; Depart-ment of Radiology, Vancouver GeneralHospital, University of British Colum-bia, Vancouver, British Columbia,Canada (N.L.M.); Department of Radi-ology, Royal Brompton Hospital, Lon-don, England (D.M.H.); Departmentof Radiology, Hopital Pitie-Salpetriere,Paris, France (P.A.G.); and Depart-ment of Medicine, San Francisco Gen-eral Hospital, San Francisco, Calif(T.E.K.). Received October 15, 2003;revision requested January 12, 2004;revision received April 29; acceptedJuly 29. Address correspondence toD.A.L. (e-mail: [email protected]).
Authors stated no financial relation-ship to disclose.© RSNA, 2005
Idiopathic InterstitialPneumonias: CT Features1
Idiopathic interstitial pneumonias comprise usual interstitial pneumonia (UIP), non-specific interstitial pneumonia (NSIP), desquamative interstitial pneumonia (DIP),respiratory bronchiolitis–associated interstitial lung disease (RB-ILD), cryptogenicorganizing pneumonia (COP), acute interstitial pneumonia (AIP), and lymphoidinterstitial pneumonia (LIP). Each of these entities has a typical imaging and histo-logic pattern, although in practice the imaging patterns may be variable. Each entitymay be idiopathic or may be secondary to a recognizable cause such as collagenvascular disease or inhalational exposure. The diagnosis of idiopathic interstitialpneumonia is made by means of correlation of clinical, imaging, and pathologicfeatures. The characteristic computed tomographic (CT) features of UIP are pre-dominantly basal and peripheral reticular pattern with honeycombing and tractionbronchiectasis. NSIP is characterized by predominantly basal ground-glass opacityand/or reticular pattern, often with traction bronchiectasis. DIP and RB-ILD aresmoking-related lung diseases characterized by ground-glass opacity and centri-lobular nodules. COP is characterized by patchy peripheral or peribronchovascularconsolidation. AIP manifests as diffuse lung consolidation and ground-glass opacity.LIP is associated with a CT pattern of ground-glass opacity sometimes associatedwith perivascular cysts.© RSNA, 2005
The idiopathic interstitial pneumonias (IIPs) are a group of diffuse parenchymal lungdiseases that share many features but are sufficiently different from one another to bedesignated as separate disease entities (1). The general term idiopathic interstitial pneumoniaincludes usual interstitial pneumonia (UIP), nonspecific interstitial pneumonia (NSIP),desquamative interstitial pneumonia (DIP), respiratory bronchiolitis-associated interstitiallung disease (RB-ILD), cryptogenic organizing pneumonia (COP), acute interstitial pneu-monia (AIP), and lymphoid interstitial pneumonia (LIP). These entities can be easilydistinguished from other forms of diffuse parenchymal lung disease by clinical methods,including history, physical examination, laboratory studies, imaging, and pathologicanalysis. However, patterns of lung injury similar or identical to those seen in the IIPs arefound in many other conditions, including collagen vascular disease, drug reactions,asbestosis, and chronic hypersensitivity pneumonitis. The term idiopathic is reserved forthose conditions in which the cause of the lung injury pattern is unknown. The classifi-cation does not include other morphologically distinct idiopathic lung diseases such assarcoidosis and the eosinophilic pneumonias.
There have been several previous classifications of the IIPs (2–4), but none of these hasclearly delineated the complementary roles of the pathologist, radiologist, and clinician indiagnosing these conditions. Because of substantial variation in the definition and termi-nology of the IIPs, the American Thoracic Society and the European Respiratory Societyconvened an international committee of pulmonologists, thoracic radiologists, and pul-monary pathologists to clarify the nomenclature and typical patterns of these conditions.The classification was published in full in the American Journal of Respiratory and CriticalCare Medicine in 2002 (1). The purpose of the present review is to illustrate the aspects ofthis classification that are of importance to the radiologist. In particular, we will delineatethe typical radiologic features of these entities, with radiologic-pathologic correlation, andreview the radiologic differential diagnoses.
Although the new classification is based on histologic criteria, there is a clear recogni-tion that the pattern at thin-section computed tomography (CT) is important in delineat-
Review
10
Ra
dio
logy

ing the macroscopic morphology of theIIPs (Table 1). The prototypic CT featuresof each IIP are distinct, though withsome overlap. Each IIP pattern seen athistologic examination or CT is linked toa specific idiopathic clinical syndrome(Table 2). However, the differential diag-nosis of IIPs usually includes underlyingcollagen vascular disease or inhalationexposures, and the clinician has a criticalrole in identifying these causes of lunginjury.
Because morphologic patterns identi-fied by pathologists and imagers can bedue to a variety of causes, clinical evalu-ation is essential to prove that the mor-phologic pattern is truly idiopathic. Theterminology used in reporting pathologicand radiologic images should clearly in-dicate the differential diagnosis of themorphologic pattern. Terms such as DIPpattern and NSIP pattern can be helpful inindicating that one is discussing the his-tologic or radiologic pattern rather thanthe clinical syndrome. This conventionwill be used throughout this review.While each pattern is pathologically dis-tinct, two or more patterns may bepresent in a single biopsy specimen,which can sometimes lead to diagnosticdifficulty (eg, NSIP and UIP). Nonspecific
terms such as alveolitis or fibrosing alveoli-tis should not be used.
UIP is the most common of the IIPs (5).NSIP is the next most frequent, followedby COP. DIP, RB-ILD, and AIP are lesscommon, while LIP is rare.
Distinction among the IIPs is impor-tant largely because of the differences inprognosis associated with these condi-tions (5). Because UIP is associated with asharply decreased survival, comparedwith that associated with the other con-ditions, the most important task for theradiologist and pathologist is to distin-guish individuals with this morphologicpattern from those with the other enti-ties.
USUAL INTERSTITIALPNEUMONIA AND IDIOPATHICPULMONARY FIBROSIS
The terms usual interstitial pneumonia andidiopathic pulmonary fibrosis have becomemuch more narrowly defined since theywere originally proposed several decadesago. The term idiopathic pulmonary fibrosisis now applied solely to the clinical syn-drome associated with the morphologicpattern of UIP and specifically excludesentities such as NSIP and DIP (6). At his-tologic examination, the fibroblastic fo-cus—a cluster of fibroblasts and imma-ture connective tissue within the pulmo-nary interstitium (Fig 1)—has beenrecognized as a key early lesion of UIP (7).Because UIP is primarily a fibrotic condi-tion, the concept of alveolitis as an in-flammatory phase of UIP is no longervalid. The histologic diagnosis of UIP isbased on temporal heterogeneity: theidentification of fibrotic lesions of differ-ent stages (fibroblastic foci, mature fibro-sis, and honeycombing) within the samebiopsy specimen (Fig 1) (3). In additionto the temporal heterogeneity, the histo-logic abnormality is spatially heteroge-neous, with patchy lung involvementand normal lung adjacent to severely fi-brotic lung.
Patients with idiopathic pulmonary fi-brosis are usually over 50 years of age atthe time of presentation, with men beingaffected slightly more often than women(6). In most patients, symptoms havebeen present for more than 6 monthsbefore presentation. Patients usuallypresent with progressive shortness ofbreath and nonproductive cough. Finecrackles may be found during clinical ex-amination, and physiologic evaluationusually shows lung restriction. The clini-cal course of idiopathic pulmonary fibro-
sis is invariably one of gradual deteriora-tion, sometimes interspersed with peri-ods of more rapid decline. The mediansurvival from time of diagnosis varies be-tween 2.5 and 3.5 years (8). Idiopathicpulmonary fibrosis, as currently defined,does not usually respond to steroid treat-ment, in contrast to the other IIPs.
UIP is important for the radiologist be-cause it is one of the most common in-terstitial lung diseases and because a con-fident thin-section CT diagnosis of UIP isusually correct. The radiologist must befamiliar with the typical features of UIPand with the features that make UIP un-likely. UIP is characterized on thin-sec-tion CT images by the presence of retic-ular opacities, often associated withtraction bronchiectasis (Fig 2) (9,10). Hon-eycombing is common. Ground-glassopacity is common but is usually less ex-tensive than the reticular pattern. Archi-tectural distortion, which reflects lung fi-brosis, is often prominent. Lobar volumeloss is seen in cases of more advancedfibrosis. The distribution of UIP on CTimages is characteristically basal and pe-ripheral, though it is often patchy. Mi-cronodules, air trapping, nonhoneycombcysts, extensive ground-glass opacifica-tion, consolidation, or a predominantlyperibronchovascular distribution shouldlead to an alternative diagnosis.
The authors of several retrospectivestudies (11–15) have documented thatthe positive predictive value of a CT di-agnosis of UIP ranges from 70% to 100%,while the positive predictive value of aconfident CT diagnosis of UIP is 95%–100%. In a recent prospective study (16),the positive predictive value of a diagno-sis of UIP was about 90%, while the pos-itive predictive value of a confident diag-nosis of UIP was 96%. It should be notedthat, in general, these studies were per-formed by expert pulmonary radiolo-gists. Also, in these studies a confidentCT diagnosis of UIP was not made in25%–50% of cases of histologically dem-onstrated UIP. A confident CT diagnosisof UIP is difficult to make in patients whodo not show all of the typical features,particularly honeycombing.
Because of the high degree of accuracyof thin-section CT diagnosis in manycases of UIP, the diagnosis of UIP is com-monly based on clinical and imaging fea-tures, without the need for surgical bi-opsy. However, some cases of UIP have aCT appearance that overlaps with that ofNSIP. In such cases, the diagnosis of UIPcan only be made with the aid of lungbiopsy. The American Thoracic Societyhas published criteria for diagnosis of UIP
ESSENTIALS● The classification of IIPs is based on
histologic criteria; each histologic pat-tern is associated with a characteristicimaging pattern that correlates wellwith the histologic findings.
● Similar morphologic patterns of lunginjury may occur in other conditions,including collagen vascular disease,hypersensitivity pneumonitis, and drugtoxicity; these conditions must be ex-cluded clinically.
● In the correct clinical context, the CTfeatures of UIP and organizing pneu-monia are often diagnostic.
● Distinction of UIP from the other inter-stitial pneumonias is important be-cause UIP is associated with a substan-tially poorer prognosis than the otherentities.
● The role of the radiologist is to identifythe macroscopic morphologic patternand to work with the clinician and pa-thologist to generate an integratedclinical diagnosis.
Volume 236 � Number 1 Idiopathic Interstitial Pneumonias: CT Features � 11
Ra
dio
logy
in the absence of a surgical biopsy (Table3) (6). Flaherty et al (17) recently suggestedthat the patients with histologicallyproved UIP who had definite or probableUIP according to thin-section CT criteriahad a shorter survival than did those withindeterminate thin-section CT findings.This is most likely because the typical thin-section CT criteria for diagnosis of UIP in-clude the presence of honeycombing andmay thereby result in selection of patientswith later or more severe disease. Theirstudy reemphasizes the importance ofseeking lung biopsy in patients in whomCT findings are not diagnostic of UIP.
On serial CT scans in patients with id-iopathic pulmonary fibrosis, the areas of
ground-glass opacity may regress, butthese areas more commonly progress tofibrosis with honeycombing (Fig 2)(18,19). Honeycomb cysts usually en-large slowly over time.
Important complications of idiopathicpulmonary fibrosis include infection,lung cancer, and accelerated deteriora-tion (20). Since most treatments for idio-pathic pulmonary fibrosis cause immu-nocompromise, a variety of opportunis-tic infectious organisms may be presentin these patients, including Pneumocystiscarinii, Mycobacterium avium-intracellularecomplex (Fig 3), and mycetoma due toAspergillus species or other organisms.The reported frequency of lung cancer in
TABLE 1American Thoracic Society andEuropean Respiratory SocietyClassification of IIPs
Morphologic PatternClinical
Diagnosis
UIP Idiopathicpulmonaryfibrosis
NSIP NSIPDIP DIPRespiratory bronchiolitis RB-ILDOrganizing pneumonia COPDiffuse alveolar damage AIPLIP LIP
Note.—Adapted and reprinted, with permis-sion, from reference 1.
TABLE 2IIP Patterns
Morphologic Pattern Histologic Features Imaging Features Imaging Differential Diagnosis
UIP Spatial and temporal heterogeneity, dense fibrosis,fibroblastic foci, honeycombing
Basal, peripheral predominance,often patchy, reticularabnormality, honeycombing
Collagen vascular disease,asbestosis, chronichypersensitivity pneumonitis
NSIP Spatially and temporally homogeneous lungfibrosis or inflammation
Basal predominance, ground-glass abnormality, reticularabnormality
Collagen vascular disease, chronichypersensitivity pneumonitis,DIP
DIP Diffuse macrophage accumulation in alveoli Basal, peripheral predominance;ground-glass attenuation;sometimes cysts
Hypersensitivity pneumonitis,NSIP
Respiratorybronchiolitis
Peribronchiolar macrophage accumulation,bronchiolar fibrosis; macrophages have dustybrown cytoplasm
Centrilobular nodules, ground-glass attenuation
Hypersensitivity pneumonitis
Organizingpneumonia
Patchy distribution of intraluminal organizingfibrosis in distal airspaces; preservation of lungarchitecture; uniform temporal appearance; mildinterstitial chronic inflammation
Ground-glass attenuation;consolidation basal, peripheralpredominance
Collagen vascular disease,infection, vasculitis, sarcoidosis,lymphoma, alveolar carcinoma
Diffuse alveolardamage
Diffuse distribution, uniform temporal appearance,alveolar septal thickening due to organizingfibrosis, airspace organization, hyalinemembranes
Diffuse, ground-glass attenuation,consolidation
Acute respiratory distresssyndrome, infection,hydrostatic edema,hemorrhage
LIP Diffuse lymphoplasmacytic infiltration of alveolarsepta
Ground-glass attenuation, cysts DIP, NSIP, hypersensitivitypneumonitis
Source.—Reference 4.
TABLE 3American Thoracic Society Criteria for Diagnosis of IPF in Absence of Surgical Biopsy
Criterion Type Criterion Definition*
Major† Exclusion of other known causes of interstitial lung disease (eg, certain drug toxicities, environmental exposures, connectivetissue disease)
Abnormal pulmonary function studies that include evidence of restriction (reduced vital capacity often with increased FEV1/FVC)and impaired gas exchange (increased P(A � a)O2 with rest or exercise or decreased DLCO)
Bibasilar reticular abnormalities with minimal ground-glass opacities at thin-section CTTransbronchial lung biopsy or bronchoalveolar lavage specimens that show no features supporting alternate diagnosis
Minor‡ Age � 50 yInsidious onset of otherwise unexplained dyspnea on exertionIllness duration � 3 moBibasilar, inspiratory crackles (dry or “Velcro”-type in quality
Source.—Reference 15.* DLCO � diffusing capacity of carbon monoxide, FEV1 � forced expiratory volume in 1 second, FVC � forced vital capacity, P(A � a)O2 �
alveolar-arterial oxygen pressure difference.† All must be present.‡ Three of four must be present.
12 � Radiology � July 2005 Lynch et al
Ra
dio
logy
idiopathic pulmonary fibrosis varieswidely from series to series but is proba-bly about 10%–15% (21). When canceroccurs, it seems to predominantly affectthe lower lobes (Fig 4).
Accelerated deterioration (“acute exac-erbation”) of idiopathic pulmonary fibro-sis (22) manifests with a relatively shortonset of progressive dyspnea or cough,occasionally associated with systemicsymptoms, in a patient with underlyingidiopathic pulmonary fibrosis. There isusually a short prodrome of 4–8 weeksduration. On CT images, the accelerateddeterioration is characterized by diffuseor peripheral ground-glass opacification(Fig 5) (22), which must be distinguishedclinically from opportunistic viral orPneumocystis infection.
The differential diagnosis for the CTpattern of UIP includes collagen vasculardisease, chronic hypersensitivity pneu-monitis, and asbestosis (Fig 6). Featuresthat help to distinguish chronic hyper-
sensitivity pneumonitis from idiopathicpulmonary fibrosis include upper or mid-dle zone predominance, presence of mi-cronodules, absence of honeycombing(23), and presence of mosaic attenuationor air trapping (24). However, there are aminority of cases of chronic hypersensi-tivity pneumonitis with predominantlybasal reticular pattern and honeycomb-ing, which are radiologically indistin-guishable from UIP.
NONSPECIFIC INTERSTITIALPNEUMONIA
NSIP is a histologic entity characterizedby spatially homogenous alveolar wallthickening caused by inflammation and/
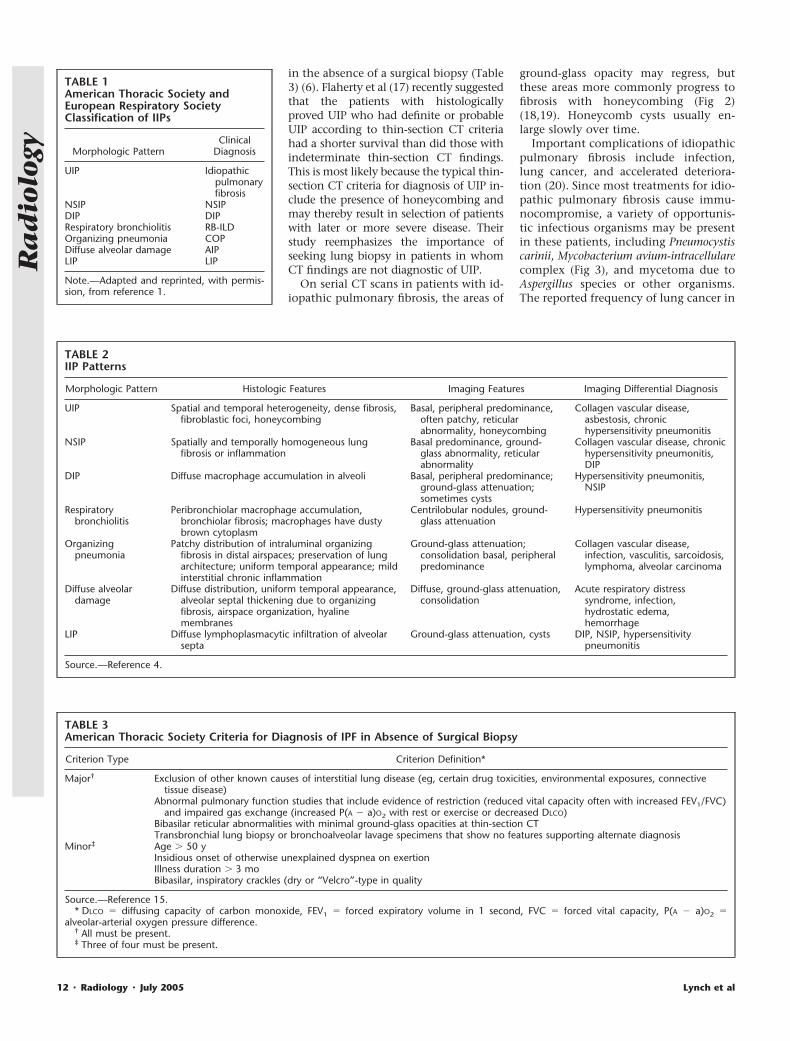
Figure 1. Photomicrographs show UIP pat-tern. (a) Patchy fibrosis with lung remodelingarchitecture and striking subpleural distribu-tion. Interstitial chronic inflammation is mild,with a few lymphoid aggregates (arrow). Areasof normal lung are present. There are no fea-tures of other interstitial lung disorders. Arrow-head � pleura. (Hematoxylin-eosin stain; orig-inal magnification, �4.) (b) Fibroblastic focusof loose organizing connective tissue (arrow) isseen adjacent to a dense collagenous scar. (Mo-vat stain; original magnification, �10.)
Figure 2. Transverse CT images in a 65-year-old man with progressive shortness of breath dueto UIP. (a) Left lower lobe shows peripheral ground-glass opacity and reticular patterns withtraction bronchiectasis (arrows). (b) Two years later, ground-glass opacification has progressed toreticular pattern and honeycombing, with progression of traction bronchiectasis.
Figure 3. Transverse CT images in a 78-year-old man with UIP complicated by infectionwith M avium-intracellulare complex. (a, b) Pre-dominantly basal subpleural reticular patternis present, with large left upper lobe cavity (�).
Figure 4. Transverse thin-section CT imagesin a 72-year-old man with UIP complicated bylarge cell neuroendocrine lung cancer, whichwas detected incidentally on chest radiograph(not shown). (a, b) Subpleural mass in rightupper lobe (arrow) is evident at two levels,with extensive, predominantly basal honey-combing.
Volume 236 � Number 1 Idiopathic Interstitial Pneumonias: CT Features � 13
Ra
dio
logy

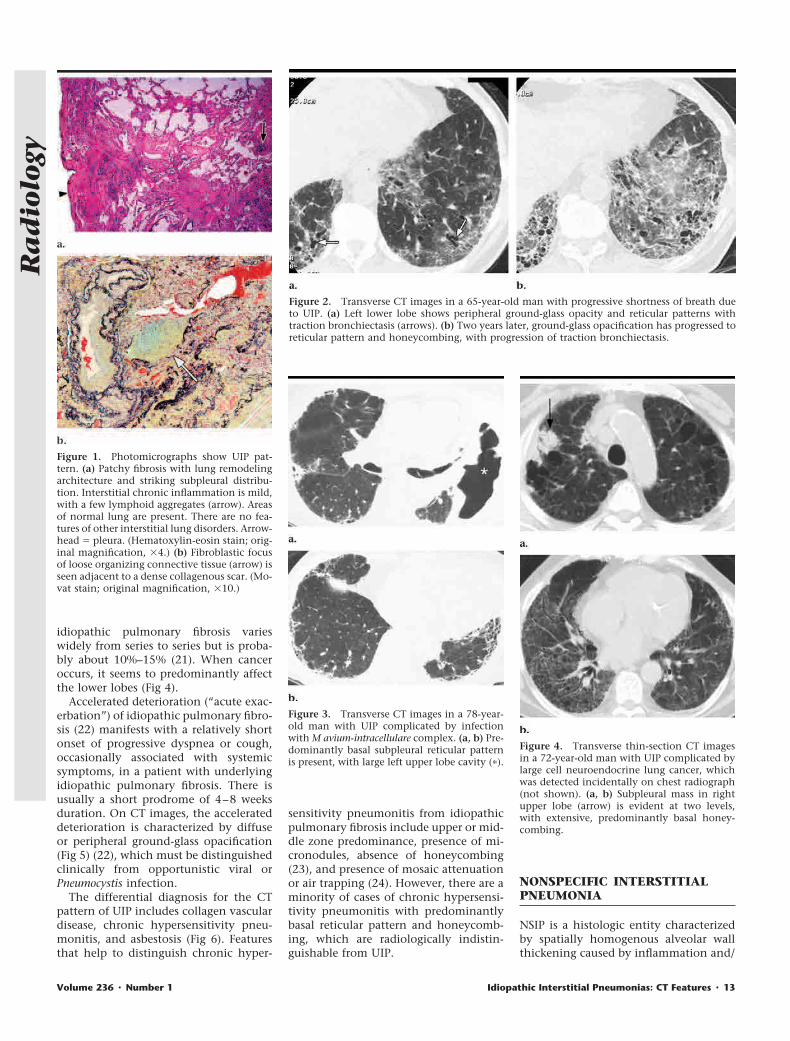
or fibrosis (25). The spatial and temporalhomogeneity of this pattern are impor-tant in distinguishing NSIP from UIP(Figs 7, 8). The most important clinicalfact about NSIP is that the prognosis issubstantially better than that of UIP(5,17,26) (Fig 9). NSIP may be classifiedon the basis of the relative amounts oflung fibrosis and inflammation. Patientswith predominant fibrosis (fibrotic NSIP)(Fig 7) have a poorer prognosis than dothose with inflammatory histologic find-ings (cellular NSIP) (Fig 8) (26). The clin-ical features of NSIP are similar to thoseof UIP, except that patients with NSIP aremore commonly female and generallyhave a younger mean age than do thosewith UIP.
Because of the histologic spatial homo-geneity of NSIP, ground-glass opacity isits salient CT feature and is often associ-ated with evidence of fibrosis (lobar vol-ume loss, reticular pattern, and/or trac-tion bronchiectasis) (Fig 10) (10,27–31).
As with UIP, DIP, and COP, the abnor-mality usually shows a basal predomi-nance. The transverse distribution maybe subpleural, peribronchovascular, orboth. Consolidation is uncommon, andhoneycombing is rare. Variation amongCT features of NSIP reported in existingseries may be related to differences inhistologic diagnostic criteria for NSIP atdifferent centers. The CT features of cel-lular and fibrotic NSIP overlap consider-ably (32) (Fig 11).
The parenchymal abnormalities ofNSIP, including reticular pattern, trac-tion bronchiectasis, and ground-glassopacity, may all be reversible at follow-upexamination (Fig 12) (31). Indeed, itseems likely that many of the patientsincluded in previous series of fibrosingalveolitis who had a predominant pat-tern of ground-glass opacity had NSIPrather than UIP, which would therebyexplain the fact that these patients weremore likely to respond to steroid treat-ment (33,34).
Histologic and radiologic evidence ofthe NSIP pattern is commonly found inpatients with collagen vascular diseases
(Fig 13), hypersensitivity pneumonitis(Fig 14), and drug-induced lung disease.Therefore, the recognition of this patternshould prompt a search for the underly-ing cause. The CT pattern of NSIP mayoverlap with those of organizing pneu-monia and DIP. Because the thin-sectionCT features of NSIP may overlap withthose of organizing pneumonia, DIP, andUIP, a surgical lung biopsy should beconsidered when the thin-section CTpattern suggests NSIP.
DESQUAMATIVE INTERSTITIALPNEUMONIA
DIP is an uncommon condition that pri-marily affects cigarette smokers in their4th or 5th decades of life (35). It is char-acterized histologically by spatially ho-mogeneous thickening of alveolar septa,associated with intraalveolar accumula-tion of macrophages (Fig 15). The termdesquamative was applied to this entitybecause the intraalveolar macrophageswere initially thought to represent des-quamated alveolar cells.
DIP is more common in men than inwomen (male-to-female ratio, 2:1). A pro-gressive onset of dyspnea and dry coughis usual, and patients may progress torespiratory failure. Digital clubbing de-velops in about 40% of cases. Most pa-tients improve with smoking cessationand oral corticosteroids. The overall sur-vival is about 70% after 10 years.
Ground-glass opacification, present onCT images in all cases of DIP (Fig 16)(10,36), is due to the spatially homoge-neous accumulation of intraalveolarmacrophages and alveolar septal thicken-ing. The abnormality has a lower-zone
Figure 5. Transverse CT images of acceleratedUIP in a 78-year-old man with rapidly progres-sive shortness of breath. (a) Baseline imageshows subpleural reticular pattern and honey-combing (arrows), with ground-glass opacityin remainder of the lung. (b) Five monthslater, after 4 weeks of progressive breathless-ness, progression of honeycombing and exten-sive new ground-glass opacification are shown.Biopsy specimen showed organizing pneumo-nia pattern superimposed on background UIP.Patient responded to aggressive immunosup-pressive treatment.
Figure 6. UIP pattern in a 59-year-old manwith asbestosis. (a) Transverse thin-section CTimage shows predominantly peripheral honey-combing associated with reticular pattern anda lesser proportion of ground-glass opacifica-tion. (b) Transverse CT image (mediastinalwindow) shows bilateral noncalcified pleuralplaques (arrows).
Figure 7. Photomicrograph shows NSIP withfibrosing pattern. Alveolar walls (arrows) showdiffuse thickening caused by fibrosis and mildinterstitial inflammation. No fibroblastic fociare present. (Hematoxylin-eosin stain; originalmagnification, �10.)
14 � Radiology � July 2005 Lynch et al
Ra
dio
logy
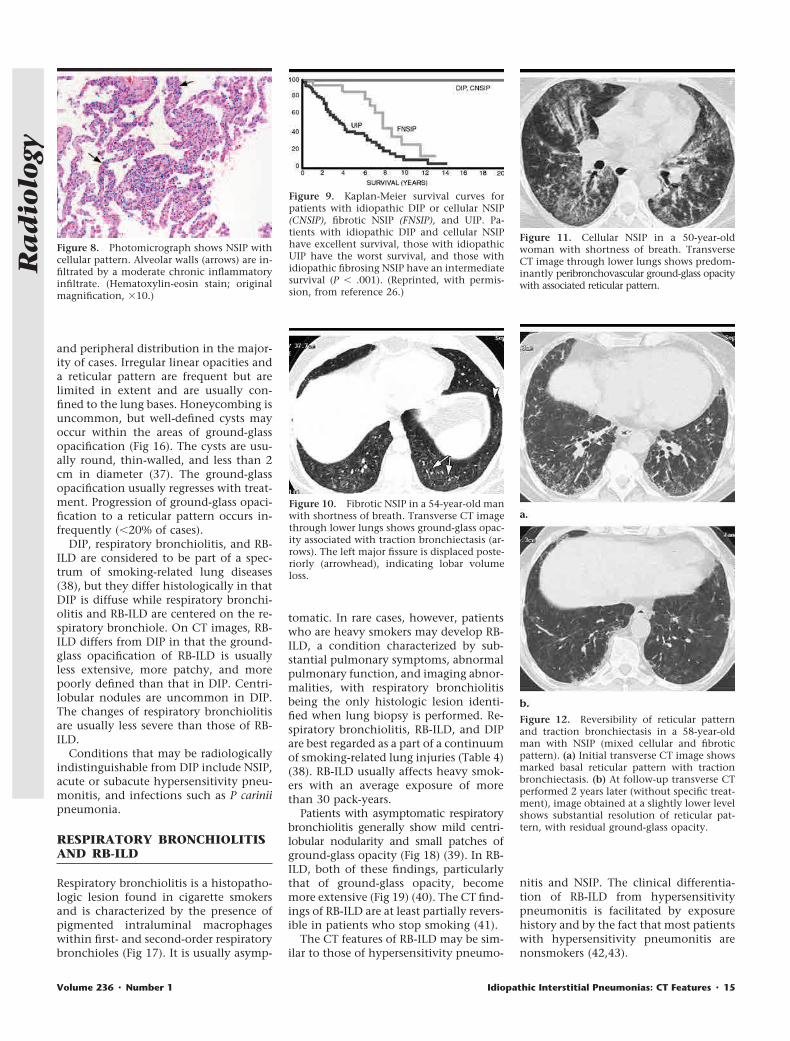
and peripheral distribution in the major-ity of cases. Irregular linear opacities anda reticular pattern are frequent but arelimited in extent and are usually con-fined to the lung bases. Honeycombing isuncommon, but well-defined cysts mayoccur within the areas of ground-glassopacification (Fig 16). The cysts are usu-ally round, thin-walled, and less than 2cm in diameter (37). The ground-glassopacification usually regresses with treat-ment. Progression of ground-glass opaci-fication to a reticular pattern occurs in-frequently (�20% of cases).
DIP, respiratory bronchiolitis, and RB-ILD are considered to be part of a spec-trum of smoking-related lung diseases(38), but they differ histologically in thatDIP is diffuse while respiratory bronchi-olitis and RB-ILD are centered on the re-spiratory bronchiole. On CT images, RB-ILD differs from DIP in that the ground-glass opacification of RB-ILD is usuallyless extensive, more patchy, and morepoorly defined than that in DIP. Centri-lobular nodules are uncommon in DIP.The changes of respiratory bronchiolitisare usually less severe than those of RB-ILD.
Conditions that may be radiologicallyindistinguishable from DIP include NSIP,acute or subacute hypersensitivity pneu-monitis, and infections such as P cariniipneumonia.
RESPIRATORY BRONCHIOLITISAND RB-ILD
Respiratory bronchiolitis is a histopatho-logic lesion found in cigarette smokersand is characterized by the presence ofpigmented intraluminal macrophageswithin first- and second-order respiratorybronchioles (Fig 17). It is usually asymp-
tomatic. In rare cases, however, patientswho are heavy smokers may develop RB-ILD, a condition characterized by sub-stantial pulmonary symptoms, abnormalpulmonary function, and imaging abnor-malities, with respiratory bronchiolitisbeing the only histologic lesion identi-fied when lung biopsy is performed. Re-spiratory bronchiolitis, RB-ILD, and DIPare best regarded as a part of a continuumof smoking-related lung injuries (Table 4)(38). RB-ILD usually affects heavy smok-ers with an average exposure of morethan 30 pack-years.
Patients with asymptomatic respiratorybronchiolitis generally show mild centri-lobular nodularity and small patches ofground-glass opacity (Fig 18) (39). In RB-ILD, both of these findings, particularlythat of ground-glass opacity, becomemore extensive (Fig 19) (40). The CT find-ings of RB-ILD are at least partially revers-ible in patients who stop smoking (41).
The CT features of RB-ILD may be sim-ilar to those of hypersensitivity pneumo-
nitis and NSIP. The clinical differentia-tion of RB-ILD from hypersensitivitypneumonitis is facilitated by exposurehistory and by the fact that most patientswith hypersensitivity pneumonitis arenonsmokers (42,43).
Figure 8. Photomicrograph shows NSIP withcellular pattern. Alveolar walls (arrows) are in-filtrated by a moderate chronic inflammatoryinfiltrate. (Hematoxylin-eosin stain; originalmagnification, �10.)
Figure 9. Kaplan-Meier survival curves forpatients with idiopathic DIP or cellular NSIP(CNSIP), fibrotic NSIP (FNSIP), and UIP. Pa-tients with idiopathic DIP and cellular NSIPhave excellent survival, those with idiopathicUIP have the worst survival, and those withidiopathic fibrosing NSIP have an intermediatesurvival (P � .001). (Reprinted, with permis-sion, from reference 26.)
Figure 10. Fibrotic NSIP in a 54-year-old manwith shortness of breath. Transverse CT imagethrough lower lungs shows ground-glass opac-ity associated with traction bronchiectasis (ar-rows). The left major fissure is displaced poste-riorly (arrowhead), indicating lobar volumeloss.
Figure 11. Cellular NSIP in a 50-year-oldwoman with shortness of breath. TransverseCT image through lower lungs shows predom-inantly peribronchovascular ground-glass opacitywith associated reticular pattern.
Figure 12. Reversibility of reticular patternand traction bronchiectasis in a 58-year-oldman with NSIP (mixed cellular and fibroticpattern). (a) Initial transverse CT image showsmarked basal reticular pattern with tractionbronchiectasis. (b) At follow-up transverse CTperformed 2 years later (without specific treat-ment), image obtained at a slightly lower levelshows substantial resolution of reticular pat-tern, with residual ground-glass opacity.
Volume 236 � Number 1 Idiopathic Interstitial Pneumonias: CT Features � 15
Ra
dio
logy
CRYPTOGENIC ORGANIZINGPNEUMONIA
COP has also been called bronchiolitisobliterans organizing pneumonia (BOOP)or idiopathic BOOP. The term cryptogenicorganizing pneumonia is preferred because
its clinical, physiologic, and imaging fea-tures are unrelated to bronchiolar oblit-eration. For these reasons, COP is moreappropriately classified as an IIP than as asmall-airways disease. Although the orga-nizing pneumonia process is primarilyintraalveolar, it was included in the clas-sification of the interstitial pneumoniasbecause of its idiopathic nature and be-cause its appearance may overlap withthat of the other interstitial pneumonias.As with the other idiopathic pneumonias,the term organizing pneumonia is used to
refer to the morphologic pattern (whichmay occur in a wide variety of entities),while COP is used to indicate the associ-ated idiopathic clinical syndrome.
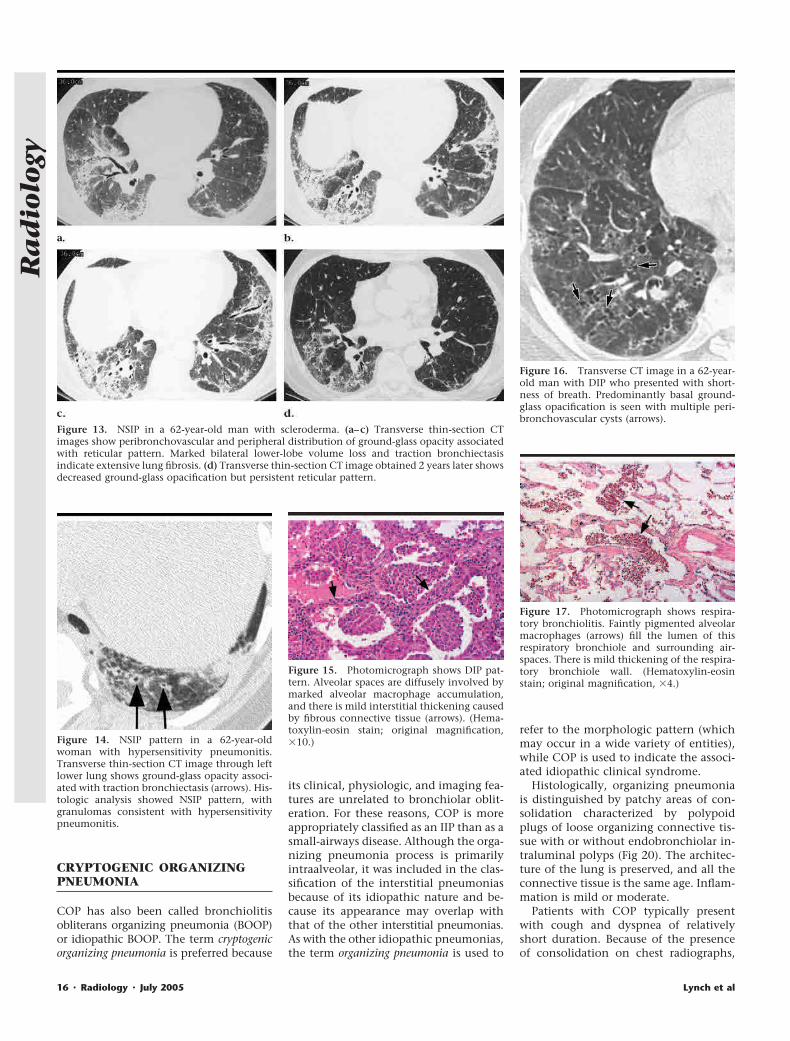
Histologically, organizing pneumoniais distinguished by patchy areas of con-solidation characterized by polypoidplugs of loose organizing connective tis-sue with or without endobronchiolar in-traluminal polyps (Fig 20). The architec-ture of the lung is preserved, and all theconnective tissue is the same age. Inflam-mation is mild or moderate.
Patients with COP typically presentwith cough and dyspnea of relativelyshort duration. Because of the presenceof consolidation on chest radiographs,
Figure 13. NSIP in a 62-year-old man with scleroderma. (a–c) Transverse thin-section CTimages show peribronchovascular and peripheral distribution of ground-glass opacity associatedwith reticular pattern. Marked bilateral lower-lobe volume loss and traction bronchiectasisindicate extensive lung fibrosis. (d) Transverse thin-section CT image obtained 2 years later showsdecreased ground-glass opacification but persistent reticular pattern.
Figure 14. NSIP pattern in a 62-year-oldwoman with hypersensitivity pneumonitis.Transverse thin-section CT image through leftlower lung shows ground-glass opacity associ-ated with traction bronchiectasis (arrows). His-tologic analysis showed NSIP pattern, withgranulomas consistent with hypersensitivitypneumonitis.
Figure 15. Photomicrograph shows DIP pat-tern. Alveolar spaces are diffusely involved bymarked alveolar macrophage accumulation,and there is mild interstitial thickening causedby fibrous connective tissue (arrows). (Hema-toxylin-eosin stain; original magnification,�10.)
Figure 16. Transverse CT image in a 62-year-old man with DIP who presented with short-ness of breath. Predominantly basal ground-glass opacification is seen with multiple peri-bronchovascular cysts (arrows).
Figure 17. Photomicrograph shows respira-tory bronchiolitis. Faintly pigmented alveolarmacrophages (arrows) fill the lumen of thisrespiratory bronchiole and surrounding air-spaces. There is mild thickening of the respira-tory bronchiole wall. (Hematoxylin-eosinstain; original magnification, �4.)
16 � Radiology � July 2005 Lynch et al
Ra
dio
logy
the initial diagnosis often is pneumonia,but the patients fail to respond to treat-ment with antibiotics.
COP is characterized radiographicallyby unilateral or bilateral areas of consol-idation (44). Consolidation is present onCT images in 90% of patients with COP(Fig 20), with a subpleural or peribron-chial distribution in up to 50% of cases(45). The lower lungs are more frequentlyinvolved. Air bronchograms, with mildcylindric bronchial dilatation, are com-mon. Ground-glass opacities are presentin about 60% of cases. Reticular opacitiesare less common but, when present, areassociated with histologic evidence of fi-brosis (46). Pleural effusion may occur,although this is relatively uncommon(Fig 21).
Most patients with COP demonstrateradiologic improvement or resolutionwith steroid treatment. The parenchymalabnormalities may spontaneously resolveor migrate. If reticular opacities are presenton the chest radiograph or CT image of apatient with COP, the patient is less likelyto respond to steroids (47,48).
In addition to cases of COP, the orga-nizing pneumonia pattern may be foundin cases of collagen vascular diseases (par-ticularly rheumatoid arthritis and poly-myositis) (Fig 22). The differential diag-
nosis of COP includes bronchioloalveolarcarcinoma, lymphoma, vasculitis, sarcoid-osis, chronic eosinophilic pneumonia, andinfection. Most of these entities can be ex-cluded with the aid of clinical evaluation,bronchoalveolar lavage, and/or transbron-chial biopsy.
ACUTE INTERSTITIALPNEUMONIA
AIP is a rapidly progressive form of inter-stitial pneumonia. The histologic find-
ings are those of diffuse alveolar damage(Fig 23) indistinguishable from the histo-logic pattern found in acute respiratorydistress syndrome caused by sepsis andshock. Edema and hyaline membranesare prominent in the acute phase, andorganizing alveolar septal fibrosis andpneumocyte hyperplasia are conspicuousin the organizing phase. The term acuteinterstitial pneumonia is reserved for dif-fuse alveolar damage of unknown origin.
Patients with AIP often have a priorillness suggestive of a viral upper respira-tory infection with constitutional symp-
Figure 18. Respiratory bronchiolitis in a 34-year-old male cigarette smoker. Transversethin-section CT image obtained with patientin prone position shows small patch of focalground-glass opacity (arrowhead) in posteriorportion of right lung and some centrilobularnodules (arrows).
Figure 19. RB-ILD in a 41-year-old man with30 pack-year history of cigarette smoking.Transverse CT image shows widespreadground-glass opacification, with some poorlydefined centrilobular nodules (arrowheads).
Figure 20. Photomicrograph shows organiz-ing pneumonia pattern. Loose plugs of con-nective tissue (arrows) are present in an alveo-lar duct and adjacent alveolar spaces. Lungarchitecture is preserved, and connective tissueis all the same age. (Hematoxylin-eosin stain;original magnification, �10.)
Figure 21. COP in a 75-year-old man whopresented with recurrent “pneumonia.”(a, b) Transverse CT images show bilateral pul-monary consolidation with subpleural andperibronchovascular predominance and rightpleural effusion (arrows).
TABLE 4Continuum of Smoking-related Lung Diseases
Condition
Symptoms andPhysiologicImpairment Pathologic Feature
CT Feature
Ground-GlassOpacification
CentrilobularNodules
RB* Uncommon Bronchiolocentric Small patches MildRB-ILD Severe Macrophages extend into
peribronchioloar regionExtensive Extensive
DIP Severe Diffuse intraalveolarmacrophages
Extensive Uncommon
* RB � respiratory bronchiolitis.
Volume 236 � Number 1 Idiopathic Interstitial Pneumonias: CT Features � 17
Ra
dio
logy

toms. Hypoxemia progresses rapidly torespiratory failure. Mechanical ventila-tion is usually required. Most patientsfulfill the clinical criteria for acute respi-ratory distress syndrome: acute onset, ra-tio of arterial partial pressure of oxygento fraction of inspired oxygen of 200 mmHg or lower, diffuse bilateral opacities onchest radiographs, and pulmonary capil-lary wedge pressure of less than 18 mmHg. The mortality rate is 50% or higher.
The most common CT findings in pa-tients with AIP are areas of ground-glassopacity, bronchial dilatation, and archi-tectural distortion (Fig 24) (49). In theearly exudative phase, the lung showsareas of ground-glass opacity that aremost often bilateral and patchy, with ar-eas of focal sparing of lung lobules, thatproduce a geographic appearance (50).Consolidation is seen in most cases, par-ticularly in the dependent lung. The or-ganizing stage of diffuse alveolar damage
is associated with distortion of broncho-vascular bundles and traction bronchiec-tasis. The few patients who survive showprogressive clearing of the ground-glassopacity and consolidation. The mostcommon residual thin-section CT find-ings are areas of hypoattenuation, lungcysts, reticular pattern, and associated pa-renchymal distortion occurring mainlyin the nondependent lung (51).
Although the radiologic appearancesof AIP and acute respiratory distress syn-drome overlap, patients with AIP aremore likely to have a symmetric lower-lobe distribution of abnormalities and agreater prevalence of honeycombing(52). The reason for the increased preva-lence of honeycombing is unclear butmay be related to the presence of under-lying UIP in some cases. In addition toacute respiratory distress syndrome (Fig25), the radiologic differential diagnosisof AIP depends on the stage but can in-clude widespread infection, hydrostaticedema, acute eosinophilic pneumonia,and pulmonary hemorrhage.
LYMPHOID INTERSTITIALPNEUMONIA
Liebow and Carrington (53) introducedthe term lymphoid interstitial pneumonia
in 1973 to describe a diffuse lymphocyticinterstitial infiltrate that was distinctfrom other patterns of interstitial pneu-monia (Fig 26). The alveolar septal inter-stitium is infiltrated by lymphocytes andsmall to moderate numbers of plasmacells. Immunohistochemical analysis isimportant for distinguishing LIP fromlow-grade lymphoma. If LIP is proved tobe due to polyclonal lymphocyte prolif-eration, progression to lymphoma isquite uncommon. LIP is commonly asso-ciated with connective tissue disorders(particularly Sjogren syndrome), with im-munodeficiency (particularly acquiredimmunodeficiency syndrome), and withCastleman syndrome. Idiopathic LIP israre, but it was included in the AmericanThoracic Society and European Respira-tory Society classification because it mustbe considered in the clinical and radio-logic differential diagnosis of diffuse lungdisease, and its histologic pattern is un-equivocally that of an interstitial pneu-monia. The clinical manifestation of LIP
Figure 22. Organizing pneumonia pattern ina 55-year-old woman with rheumatoid arthri-tis. Transverse CT image shows focal consoli-dation (arrows) in lingula, with an air bron-chogram. The abnormality was not associatedwith symptoms of infection and resolvedspontaneously.
Figure 23. Photomicrograph shows diffusealveolar damage. Lung shows diffuse alveolarwall thickening caused by proliferating con-nective tissue and prominent hyaline mem-branes (arrows). (Hematoxylin-eosin stain;original magnification, �20.)
Figure 24. AIP in a 65-year-old woman whopresented with rapidly progressive shortness ofbreath. (a, b) Transverse CT images show ex-tensive ground-glass opacity, with consolida-tion in more dependent parts of the lung andlobular areas of sparing.
Figure 25. Acute respiratory distress syn-drome secondary to trauma in a 50-year-oldwoman. Transverse CT image shows patchygeographic lung consolidation on the rightwith more diffuse consolidation in the left up-per lung. A small left pneumothorax (arrow) ispresent.
Figure 26. Photomicrograph shows LIP. Al-veolar walls (arrows) are markedly infiltratedby lymphocytes and plasma cells. (Hematoxy-lin-eosin stain; original magnification, �20.)
18 � Radiology � July 2005 Lynch et al
Ra
dio
logy

is usually that of the underlying systemicdisease.
The dominant CT finding in LIP is usu-ally ground-glass opacity (Fig 27). Perivas-cular cysts or, less commonly, perivascularhoneycombing can also be seen (54,55).Reticular pattern is seen in about half ofpatients. Lung nodules and widespreadconsolidation may occur. Other findingsmay include thickening of the broncho-vascular bundles and interlobular septalthickening.
ACCURACY OF CT DIAGNOSISOF IIP
As discussed earlier in this review, theaccuracy of CT diagnosis of IIPs is great-est for UIP. The classic CT features of COP(subpleural or peribronchovascular con-solidation) can be diagnostic if infection,malignancy, and eosinophilic pneumo-nia are excluded.
Ground-glass opacity, with or withoutreticular pattern, is the salient feature ofNSIP, DIP, RB-ILD, and LIP. Apart fromthe presence of cysts in some cases of DIPand LIP, there are no firm criteria fordistinguishing among these entities. Al-though the prognosis of these non-UIPdiseases is similar, histologic evaluationis often important to help exclude othercauses of diffuse ground-glass opacitysuch as hypersensitivity pneumonitis.
Because AIP usually manifests as acutehypoxemic respiratory failure, it does notenter into the clinical differential diagno-sis of the other IIPs.
Johkoh et al (10) reviewed the accuracyof CT diagnosis in 129 patients with UIP,NSIP, DIP, COP, or AIP. They found thatthe positive predictive value of CT fordiagnosis of each entity was 79% for
COP, 71% for UIP, 65% for AIP, 63% forDIP, and only 9% for NSIP. The low levelof accuracy for diagnosis of NSIP may bedue to the fact that the CT features ofNSIP were not well established at thetime the study was performed. Theirstudy may have included a relativelylarge number of cases of atypical UIP,since patients with typical UIP are usu-ally treated without surgical biopsy. Amore recent study (32) in patients withUIP and NSIP found that the positive pre-dictive value of a diagnosis of NSIP was67%. In about 25% of cases of UIP, how-ever, the CT appearances overlap withthose of NSIP. Since the prognosis ofNSIP is substantially different from thatof UIP, biopsy may be necessary to dis-tinguish these cases of “atypical UIP”from NSIP.
INTEGRATED DIAGNOSISOF IIP
Distinction among the IIPs is importantlargely because of the differences in prog-nosis associated with these conditions(5). Because UIP is associated withsharply decreased survival relative to thatof the other conditions, the most impor-tant task for the radiologist and patholo-gist is to distinguish individuals with this
morphologic pattern from those with theother entities.
The diagnosis of IIP requires integra-tion of the morphologic patterns identi-fied by the radiologist and pathologistwith the clinical features evaluated bythe clinician. A critical role for the clini-cian is to determine whether the intersti-tial abnormality is idiopathic or relatedto an inhalational exposure or to colla-gen vascular disease. The radiologistmust determine whether the CT featuresare typical for UIP or for organizingpneumonia or whether the features areless specific. The decision about biopsy inthe patient suspected of having an IIPshould be based on consultation betweenthe clinician and radiologist. Patientswith typical clinical and radiologic fea-tures of UIP will usually not need to un-dergo biopsy. Patients with typical clini-cal and radiologic features of organizingpneumonia may not require a biopsy ifinfection and neoplasm can be excludedafter bronchoscopy with lavage and bi-opsy. The other interstitial pneumoniasusually cannot be distinguished on thebasis of clinical and CT features, and tho-racoscopic biopsy will usually be neces-sary if a precise histologic diagnosis isrequired—particularly if hypersensitivitypneumonitis is included in the differ-ential diagnosis (unless the exposure his-tory provides compelling evidence for hy-
Figure 27. LIP caused by Sjogren syndromein a 62-year-old woman. Transverse thin-sec-tion CT image obtained with patient proneshows diffuse ground-glass opacification andmultiple lung cysts (arrows).
Figure 28. American Thoracic Society and European RespiratorySociety classification of IIPs: key points for the radiologist. ARDS �acute respiratory distress syndrome.
Volume 236 � Number 1 Idiopathic Interstitial Pneumonias: CT Features � 19
Ra
dio
logy

persensitivity pneumonitis). When surgi-cal biopsy is performed, the results shouldalways be interpreted in conjunction withthe CT findings, since CT shows the mac-roscopic morphology of the entire lungwhile biopsy reveals microscopic mor-phology in only one or two small periph-eral areas. CT may also be helpful in iden-tifying a suitable location for surgical bi-opsy.
SUMMARY
Figure 28 summarizes key points of theAmerican Thoracic Society and EuropeanRespiratory Society classification of IIPsthat are of relevance to radiologists. TheIIPs are each associated with typical his-tologic and imaging patterns, and accu-rate diagnosis of these disorders requiresa dynamic integrated approach correlat-ing clinical, radiologic, and pathologicfeatures. The CT appearances of UIP andCOP may be diagnostic in the correctclinical context, but there is substantialoverlap in the CT appearances of theother IIPs. The presence of cysts shouldsuggest the possibility of LIP or DIP.
References1. American Thoracic Society, European Re-
spiratory Society. American Thoracic Soci-ety/European Respiratory Society Interna-tional Multidisciplinary Consensus Classi-fication of the Idiopathic InterstitialPneumonias: this joint statement of theAmerican Thoracic Society (ATS), and theEuropean Respiratory Society (ERS) wasadopted by the ATS board of directors,June 2001 and by the ERS executive com-mittee, June 2001. Am J Respir Crit CareMed 2002; 165:277–304.
2. Liebow A. Definition and classification ofinterstitial pneumonias in human pathol-ogy. Prog Respir Res 1975; 8:1–33.
3. Katzenstein AL, Myers JL. Idiopathic pul-monary fibrosis: clinical relevance ofpathologic classification. Am J Respir CritCare Med 1998; 157:1301–1315.
4. Muller NL, Colby TV. Idiopathic intersti-tial pneumonias: high-resolution CT andhistologic findings. RadioGraphics 1997;17:1016–1022.
5. Collard HR, King TE Jr. Demystifying idio-pathic interstitial pneumonia. Arch InternMed 2003; 163:17–29.
6. American Thoracic Society. Idiopathic pul-monary fibrosis: diagnosis and treat-ment—international consensus statement:American Thoracic Society (ATS), and theEuropean Respiratory Society (ERS). Am JRespir Crit Care Med 2000; 161:646–664.
7. King TE Jr, Schwarz MI, Brown K, et al.Idiopathic pulmonary fibrosis: relation-ship between histopathologic features andmortality. Am J Respir Crit Care Med 2001;164:1025–1032.
8. Bjoraker J, Ryu J, Edwin M, et al. Prognos-tic significance of histopathologic subsetsin idiopathic pulmonary fibrosis. Am J Re-spir Crit Care Med 1998; 157:199–203.
9. Nishimura K, Kitaichi M, Izumi T, Nagai S,Kanaoka M, Itoh H. Usual interstitial pneu-monia: histologic correlation with high-resolution CT. Radiology 1992; 182:337–342.
10. Johkoh T, Muller NL, Cartier Y, et al. Idio-pathic interstitial pneumonias: diagnosticaccuracy of thin-section CT in 129 pa-tients. Radiology 1999; 211:555–560.
11. Grenier P, Valeyre D, Cluzel P, BraunerMW, Lenoir S, Chastand C. Chronic dif-fuse interstitial lung disease: diagnosticvalue of chest radiography and high-reso-lution CT. Radiology 1991; 179:123–132.
12. Mathieson JR, Mayo JR, Staples CA, MullerNL. Chronic diffuse infiltrative lung dis-ease: comparison of diagnostic accuracy ofCT and chest radiography. Radiology1989; 171:111–116.
13. Tung KT, Wells AU, Rubens MB, Kirk JM,du Bois RM, Hansell DM. Accuracy of thetypical computed tomographic appear-ances of fibrosing alveolitis. Thorax 1993;48:334–338.
14. Lee KS, Primack SL, Staples CA, Mayo JR,Aldrich JE, Muller NL. Chronic infiltrativelung disease: comparison of diagnostic ac-curacies of radiography and low- and con-ventional-dose thin-section CT. Radiology1994; 191:669–673.
15. Swensen S, Aughenbaugh G, Myers J. Dif-fuse lung disease: diagnostic accuracy ofCT in patients undergoing surgical biopsyof the lung. Radiology 1997; 205:229–234.
16. Hunninghake GW, Zimmerman MB,Schwartz DA, et al. Utility of a lung biopsyfor the diagnosis of idiopathic pulmonaryfibrosis. Am J Respir Crit Care Med 2001;164:193–196.
17. Flaherty KR, Thwaite EL, Kazerooni EA, etal. Radiological versus histological diagno-sis in UIP and NSIP: survival implications.Thorax 2003; 58:143–148.
18. Akira M, Sakatani M, Ueda E. Idiopathicpulmonary fibrosis: progression of honey-combing at thin-section CT. Radiology1993; 189:687–691.
19. Terriff B, Kwan S, Chan-Yeung M, MullerN. Fibrosing alveolitis: chest radiographyand CT as predictors of clinical and func-tional impairment at follow-up in 26 pa-tients. Radiology 1992; 184:445–449.
20. Panos RJ, Mortenson RL, Niccoli SA, KingTJ. Clinical deterioration in patients withidiopathic pulmonary fibrosis: causes andassessment. Am J Med 1990; 88:396–404.
21. Bouros D, Hatzakis K, Labrakis H, Zeibeco-glou K. Association of malignancy withdiseases causing interstitial pulmonarychanges. Chest 2002; 121:1278–1289.
22. Akira M, Hamada H, Sakatani M, Koba-yashi C, Nishioka M, Yamamoto S. CTfindings during phase of accelerated dete-rioration in patients with idiopathic pul-monary fibrosis. AJR Am J Roentgenol1997; 168:79–83.
23. Lynch D, Newell J, Logan P, King T, MullerN. Can CT distinguish idiopathic pulmo-nary fibrosis from hypersensitivity pneu-monitis? AJR Am J Roentgenol 1995; 165:807–811.
24. Remy-Jardin M, Remy J, Wallaert B, MullerNL. Subacute and chronic bird breederhypersensitivity pneumonitis: sequentialevaluation with CT and correlation withlung function tests and bronchoalveolarlavage. Radiology 1993; 189:111–118.
25. Katzenstein AL, Fiorelli RF. Nonspecific in-terstitial pneumonia/fibrosis: histologicfeatures and clinical significance. Am JSurg Pathol 1994; 18:136–147.
26. Travis WD, Matsui K, Moss J, Ferrans VJ.Idiopathic nonspecific interstitial pneu-monia: prognostic significance of cellularand fibrosing patterns—survival compari-son with usual interstitial pneumoniaand desquamative interstitial pneumonia.Am J Surg Pathol 2000; 24:19–33.
27. Park JS, Lee KS, Kim JS, et al. Nonspecificinterstitial pneumonia with fibrosis: radio-graphic and CT findings in seven patients.Radiology 1995; 195:645–648.
28. Kim T, Lee K, Chung M, et al. Nonspecificinterstitial pneumonia with fibrosis: highresolution CT and pathologic findings. AJRAm J Roentgenol 1998; 171:1645–1650.
29. Nagai S, Kitaichi M, Itoh H, Nishimura K,Izumi T, Colby TV. Idiopathic nonspecificinterstitial pneumonia/fibrosis: compari-son with idiopathic pulmonary fibrosisand BOOP. Eur Respir J 1998; 12:1010–1019.
30. Hartman TE, Swensen SJ, Hansell DM, etal. Nonspecific interstitial pneumonia:variable appearance at high-resolutionchest CT. Radiology 2000; 217:701–705.
31. Nishiyama O, Kondoh Y, Taniguchi H, etal. Serial high resolution CT findings innonspecific interstitial pneumonia/fibro-sis. J Comput Assist Tomogr 2000; 24:41–46.
32. MacDonald SLS, Rubens MB, Hansell DM,et al. Nonspecific interstitial pneumoniaand usual interstitial pneumonia: compar-ative appearances and diagnostic accuracyof thin-section CT. Radiology 2001; 221:600–605.
33. Lee J, Im J-G, Ahn J, Kim Y, Han M. Fibro-sing alveolitis: prognostic implication ofground-glass attenuation at high-resolu-tion CT. Radiology 1992; 184:451–454.
34. Wells AU, Rubens MB, du Bois RM, HansellDM. Serial CT in fibrosing alveolitis: prog-nostic significance of the initial pattern.AJR Am J Roentgenol 1993; 161:1159–1165.
35. Ryu JH, Colby TV, Hartman TE, Vassallo R.Smoking-related interstitial lung diseases:a concise review. Eur Respir J 2001; 17:122–132.
36. Hartman TE, Primack SL, Swensen SJ, Han-sell D, McGuinness G, Muller NL. Desqua-mative interstitial pneumonia: thin-sec-tion CT findings in 22 patients. Radiology1993; 187:787–790.
37. Koyama M, Johkoh T, Honda O, et al.Chronic cystic lung disease: diagnostic ac-curacy of high-resolution CT in 92 pa-tients. AJR Am J Roentgenol 2003; 180:827–835.
38. Heyneman LE, Ward S, Lynch DA, Remy-Jardin M, Johkoh T, Muller NL. Respiratorybronchiolitis, respiratory bronchiolitis-as-sociated interstitial lung disease, and des-quamative interstitial pneumonia: differ-ent entities or part of the spectrum of thesame disease process? AJR Am J Roentge-nol 1999; 173:1617–1622.
39. Remy-Jardin M, Remy J, Gosselin B, Bec-ette V, Edme JL. Lung parenchymal changessecondary to cigarette smoking: patholog-ic-CT correlations. Radiology 1993; 186:643–651.
40. Holt R, Schmidt R, Godwin J, Raghu G.High resolution CT in respiratory bronchi-
20 � Radiology � July 2005 Lynch et al
Ra
dio
logy

olitis-associated interstitial lung disease.J Comput Assist Tomogr 1993; 17:46–50.
41. Park JS, Brown KK, Tuder RM, Hale VA,King TE Jr, Lynch DA. Respiratory bronchi-olitis-associated interstitial lung disease:radiologic features with clinical andpathologic correlation. J Comput AssistTomogr 2002; 26:13–20.
42. Dalphin JC, Debieuvre D, Pernet D, et al.Prevalence and risk factors for chronicbronchitis and farmer’s lung in Frenchdairy farmers. Br J Ind Med 1993; 50:941–944.
43. Warren C. Extrinsic allergic alveolitis: adisease commoner in nonsmokers. Thorax1977; 32:567–573.
44. Muller NL, Guerry-Force ML, Staples CA,et al. Differential diagnosis of bronchiolitisobliterans with organizing pneumoniaand usual interstitial pneumonia: clinical,functional, and radiologic findings. Radi-ology 1987; 162:151–156.
45. Lee KS, Kullnig P, Hartman TE, Muller NL.Cryptogenic organizing pneumonia: CTfindings in 43 patients. AJR Am J Roentge-nol 1994; 162:543–546.
46. Bouchardy LM, Kuhlman JE, Ball WC Jr,Hruban RH, Askin FB, Siegelman SS. CTfindings in bronchiolitis obliterans orga-nizing pneumonia (BOOP) with radio-graphic, clinical, and histologic correla-tion. J Comput Assist Tomogr 1993; 17:352–357.
47. Cordier JF, Loire R, Brune J. Idiopathicbronchiolitis obliterans organizing pneu-monia: definition of characteristic clinicalprofiles in a series of 16 patients. Chest1989; 96:999–1004.
48. Lee J, Lynch D, Sharma S, Brown K, MullerN. Organizing pneumonia: prognostic im-plication of high-resolution CT features.J Comput Assist Tomogr 2003; 27:260–265.
49. Johkoh T, Muller NL, Taniguchi H, et al.Acute interstitial pneumonia: thin-sectionCT findings in 36 patients. Radiology1999; 211:859–863.
50. Ichikado K, Johkoh T, Ikezoe J, et al. Acuteinterstitial pneumonia: high-resolutionCT findings correlated with pathology.AJR Am J Roentgenol 1997; 168:333–338.
51. Desai SR, Wells AU, Rubens MB, EvansTW, Hansell DM. Acute respiratory distresssyndrome: CT abnormalities at long-termfollow-up. Radiology 1999; 210:29–35.
52. Tomiyama N, Muller NL, Johkoh T, et al.Acute respiratory distress syndrome andacute interstitial pneumonia: comparisonof thin-section CT findings. J Comput As-sist Tomogr 2001; 25:28–33.
53. Liebow AA, Carrington CB. Diffuse pulmo-nary lymphoreticular infiltrations associ-ated with dysproteinemia. Med Clin NorthAm 1973; 57:809–843.
54. Ichikawa Y, Kinoshita M, Koga T, OizumiK, Fujimoto K, Hayabuchi N. Lung cystformation in lymphocytic interstitialpneumonitis: CT features. J Comput AssistTomogr 1994; 18:745–748.
55. Johkoh T, Muller NL, Pickford HA, et al.Lymphocytic interstitial pneumonia: thin-section CT findings in 22 patients. Radiol-ogy 1999; 212:567–572.
Volume 236 � Number 1 Idiopathic Interstitial Pneumonias: CT Features � 21
Ra
dio
logy







![Review Article Understanding Idiopathic Interstitial ...downloads.hindawi.com/journals/bmri/2015/304186.pdf · [, ]. ese numbers may even be an underestimation, because the studies](https://img.dokumen.tips/doc/110x75/5e30b3c58f86050a75604738/review-article-understanding-idiopathic-interstitial-ese-numbers-may-even.jpg)





















