Embed Size (px)
Citation preview

IDENTIFICATION OF A STARCH-BRANCHING ENZYME ANDCOEXISTING STARCH BIOSYNTHETIC ENZYMES FROMPARTIALLY PURIFIED MUNG BEAN (VIGNA RADIATA L.)
FRACTIONS
YUAN-TIH KO1,4, JIN-YI CHANG2, CHIEN-CHEN LAI3, MAO-RONG CHEN2
and JIA-WEI CHANG1
1Department of Food ScienceBiotechnology Division
National Taiwan Ocean University2 Pei-Ning Rd.
Keelung 20224, Taiwan
2Institute of NutritionChina Medical University
91 Hsueh-Shih Rd.Taichung 40421, Taiwan
3Institute of Molecular BiologyNational Chung-Hsing University
250 Kuokuang Rd.Taichung 40227, Taiwan
Accepted for Publication August 6, 2007
ABSTRACT
Three starch-branching enzyme (BE) bands (I, II and III) were detectedby zymogram in the soluble extract of developing mung bean (Vigna radiata L.cv KPS1), and radioactive method was used to trace BE during purification byliquid chromatography. Protein profiles of bands I and II were analyzed bysodium dodecyl sulfate-polyacrylamide gel electrophoresis and identified byproteomic analysis with liquid chromatography–mass spectroscopy/massspectrometry and bioinformatic searching. The 96 kDa BE possessing specificamino acid sequences of the A-type family of BEs was identified from band II.Mung bean 92 kDa sucrose synthase and the previously identified 105 kDastarch phosphorylase (SP) and its 55 kDa fragments were also found inboth bands I and II. Western blotting with SP antibodies showed that SPwas present in all of the purified BE fractions. In summary, with the aid of
4 Corresponding author. TEL: 886-2-2462-2192 ext. 5132; FAX: 886-2-2463-4203; EMAIL: [email protected]
Journal of Food Biochemistry 32 (2008) 122–141. All Rights Reserved.© 2008, The Author(s)Journal compilation © 2008, Blackwell Publishing
122

proteomic analysis, we have identified a major mung bean BE and coexistingstarch biosynthetic enzymes from partially purified fractions.
PRACTICAL APPLICATIONS
Mung bean starch possesses a unique structure of 45% dry weight ofbranched amylose content and is expected to be synthesized by unusual bio-logical machinery. Among the enzymes in the starch biosynthesis pathway ofhigher plants, branching enzyme (BE) is considered responsible for producingbranched structure from amylose and amylopectin chains. We utilized zymo-gram and radioactive methods to trace BE during purification by liquid chro-matography. Protein profiles of active bands were analyzed by sodium dodecylsulfate-polyacrylamide gel electrophoresis and identified by proteomic analy-sis with liquid chromatography–mass spectroscopy/mass spectrometry andbioinformatic searching. A mung bean 96 kDa A-type BE and coexistingstarch biosynthetic enzymes, starch phosphorylase and sucrose synthase, ableto synthesize starch in vitro, were identified from partially purified fractions.The internal sequences found in mung bean BE enabled the design of degen-erate primers for the cloning and expression of BE by molecular techniques tofurther investigate its physiological role and biotechnology applications in thefood industry.
INTRODUCTION
Starch is the principal reserve of carbohydrate biopolymers in plants thatare consumed as energy sources for human nutrition. There is also wideapplication of raw starch materials by nonfood industries such as textilesand plastics. Their biosynthesis occurs in plastids, i.e., chloroplasts in leavesand the amyloplasts of nonphotosynthetic tissues in higher plants (Martin andSmith 1995). The stroma of amyloplasts in sink tissues, e.g., seeds, tuber andendosperm, contains a group of enzymes which contribute to starch biosyn-thesis, mainly involving ADP glucose pyrophosphorylase (AGPase), starchsynthase (SS) and starch branching enzyme (BE) in the AGPase pathway.AGPase, which catalyzes the production of ADP–glucose from glucose-1-phosphate (G-1-P), is the substrate donor for SS. Granule-bound SS is for theproduction of linear amylose, and soluble SS in conjunction with BE is usedto produce amylopectin. Recently, increasing evidence has shown that starchphosphorylase (SP) and debranching enzyme, which used to be considered asstarch-degrading enzymes, showed that they are involved in starch synthesis(Hsu et al. 2004) and starch granule formation (Myers et al. 2000). They act
123IDENTIFICATION OF A STARCH-BRANCHING ENZYME

sequentially and cooperatively to synthesize helical amylose/amylopectinwhich is further deposited in semicrystalline starch granules.
Starch BE (EC 2.4.1.18) possesses both a-amylolytic hydrolase andglucosyl-transferase activities. It catalyzes the cleavage of a-1,4 linkageswithin a chain and the transfer of the released reducing end to a C6 hydroxylof the hydrolyzed chain or a new chain, creating an a-1,6 linkage. Multipleforms of BE have been found in maize (Boyer and Preiss 1978; Guan andPreiss 1993), peas (Matters and Boyer 1981; Smith 1988), rice (Nakamuraet al. 1992), potatoes (Khoshnoodi et al. 1993), sweet potatoes (Nakayamaand Nakamura 1994), barley (Sun et al. 1996), wheat (Morell et al. 1997), redalgae (Lluisma and Ragan 1998) and kidney beans (Nozaki et al. 2001).Although different isoform names and numbers have been designated amongspecies, they are generally classified into two classes in higher plants: familyA (e.g., potato BE A, maize BEII, pea BEI and rice RBE3) and family B (e.g.,potato BE B, maize BEI, pea BEII and rice RBE1) by phylogenic analysisaccording to amino acid sequence comparisons (Burton et al. 1995). Isoformsin the two classes are found in either starch-granule-associated or solubleforms, and the expression of family A is earlier than family B during the starchaccumulation period (Burton et al. 1995; Hamada et al. 2001). However, theprecise roles of individual BE isoforms and their regulation in the biosyntheticpathway are not fully understood (Tetlow et al. 2004). Nevertheless, it iscertain that the enzymatic properties of BE, such as the preferred length ofchain transfer and substrate specificity, determine the structure of starches,which implies its potential use in biotechnology applications and continuedresearch interest (Binderup et al. 2002).
The mung bean (Vigna radiata L.) is an essential ingredient used inmaking traditional oriental foods such as green bean cake and bean vermi-celli. It contains up to 45% dry weight of branched amylose content, whichmakes the retrogradation characteristics of mung bean starch severer amonglegume starches. In addition, its long amylose chain and amylopectin struc-ture make mung bean vermicelli appear translucent and thus more persistentto heating without breaking or deformation during cooking (Hoover et al.1997). Mung bean starch is not replaceable by other starchy raw materials innoodle manufacture (AVRDC 1975). Such a unique starch structure isexpected to be synthesized by an unusual biological machinery; however, upto date little research work has been performed on the mung bean (Tsay et al.1983) until our laboratory recently pursued this attempt to investigate SS(Ko et al. 2005a), SP (Ko et al. 2005b) and BE (Ko and Huang 2004; Chiang-Hsieh and Ko 2005). Among the enzymes in the starch biosynthesis pathwayof higher plants, the enzyme primarily responsible for producing such highlybranched structure within amylose chains in the mung bean has been thoughtto be BE. Determining whether mung bean BE possesses distinctive
124 Y-T. KO ET AL.

properties motivated us to pursue a basic understanding of its purification andidentification.
We adopted three common methods to assay the presence of BE activitiesin our previous studies. By using an activity staining assay on a renaturedsodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gelin conjunction with a radioactive phosphorylase stimulation assay on theeluent of gel slices, two granule-bound 84 and 64 kDa proteins extracted frommature bean starch were identified to be associated with starch branchingactivities. They are considered to be family B-type BE isoforms that areexpressed in the late growth period and trapped within the starch granule (Koand Huang 2004). As to BE isoforms expressed during seedling growth, wedetected three apparent activity peaks in the crude soluble extract by screeningBE activities in sucrose gradient fractions with an amylose-branching assay(Chiang-Hsieh and Ko 2005). In the present report, the objective was toidentify mung bean BE. We utilized our previously established zymogram andradioactive methods to trace BE during purification by liquid chromatography(LC). Furthermore, using a proteomics approach, we have identified a familyA BE and its coexisting proteins from partially purified fractions of the devel-oping mung bean.
MATERIALS AND METHODS
Materials
Immature mung bean pods (cultivar KPS1) were collected at day 14 afterflowering (DAF 14) in a field at the Asian Vegetable Research and Develop-ment Center (Shanhua, Taiwan) and stored at -80C before use. Most reagent-grade chemicals were purchased from Sigma (St. Louis, MO) or MerckChemicals (Darmstadt, Germany). Diethylamino ethanol (DEAE) Fast Flowcolumn, ECL detection kit, hyperfilm, radioactive 14C-G-1-P, Sephacryl S-200HR column, Sephadex G-25 superfine beads and scintillation cocktail werefrom Amersham Biosciences (Taipei, Taiwan). Polyvinylidene difluoride(PVDF) membrane (Immobilon-P) was from Millipore (Bedford, MA). Bio-Safe Coomassie, glycine, broad-range molecular weight standard, the proteinassay standard II dye reagent concentrate, Q2-Sepharose column, SDS, SilverStain Plus and TEMED were purchased from Bio-Rad (Hercules, CA).Acrylamide/bis 37.5:1 was from Ameresco (Solon, OH). Tris base was fromUSB (Cleveland, OH). Trypsin was from Promega (Madison, WI).
Soluble Extract Preparation
Mung beans of 82 g from bean ovaries were carefully collected on ice.The crude extract was prepared in 150 mL of ice-cold homogenizing buffer
125IDENTIFICATION OF A STARCH-BRANCHING ENZYME

(50 mM Tris–HCl, 1.25 mM DTT, 10 mM ethylenediamine tetraacetic acid(EDTA), 10% glycerol and 5 mM PMSF; pH 7.5) in a -20C precooled mortarand minced until consistent. The homogenate was centrifuged at 10,000 ¥ gfor 10 min at 4C. The resulting supernatant was the crude soluble extract.
In Situ BE and SP Activity Staining
Protein samples were separated by native polyacrylamide gel electro-phoresis (native-PAGE), and either strips or whole gel were equilibrated with0.1 M sodium citrate buffer in pH 6.0 or 7.0 for 1 h before incubation. Fourassay conditions were used: condition 1, 20 mM G-1-P and 0.1 M sodiumcitrate at pH 6.0 (SP staining) (Yu et al. 2001); condition 2, 20 mM G-1-P and0.1 M sodium citrate at pH 7.0; condition 3, 50 mM G-1-P, 0.1 M sodiumcitrate, 1 mM AMP and 6 units/mL phosphorylase a at pH 7.0 (BE staining)(Nakamura et al. 1992; Sun et al. 1996); and condition 4 was similar to that ofBE but no phosphorylase a was added (endogenous phosphorylase was used toassay BE). All reactions were conducted for 24 h at 30C with mild agitationexcept those specified in Fig. 1. After incubation, the gel was stained with20 mL Lugol’s solution (10 mM I2 and 14 mM KI). The corresponding posi-tions of certain visualized gel bands were excised for SDS-PAGE and pro-teomic analysis.
BE Assays
Two methods were used. The first was activity staining as describedearlier. The second was a radioactive phosphorylase-stimulation assay (Ko andHuang 2004). Briefly, the 200 mL reaction mixtures contained the same com-ponents as in the BE activity staining buffer except that 50 mM hot/cold[14C]-G-1-P (14 mCi/mmol) was used to react at 30C with 200 rpm shaking for30 min. One unit of activity was defined as the incorporation of 1 nmol glucoseinto the ethanol-insoluble glucan polymer per minute.
Purification of BE by LC
All chromatographic procedures were conducted at 0~4C. TE buffer(10 mM Tris–HCl and 1 mM EDTA; pH 7.5) was used for ammonium sulfate(AS) fractionation. Buffer A (50 mM Tris–HCl, 1 mM DTT, 1 mM EDTA and5% glycerol; pH 7.5) and buffer B (buffer A containing 1 M NaCl) were,respectively, used in the DEAE Fast Flow and Q2 chromatography steps.Buffer C (buffer A containing 0.15 M NaCl) was used for the Sephacryl S-200gel filtration. All buffers were degassed by a Millipore filtration apparatusthrough 0.22 mm membranes (47 mm in diameter, Advantec MFS, Pleasanton,CA), and all samples were passed through 0.22 mm filter disks. All of the
126 Y-T. KO ET AL.

column chromatography steps (DEAE Fast Flow, Sephacryl S-200 and Q2)were performed in the BioLogic FPLC system (Bio-Rad). A 0.5 mL volumefrom each chromatographic fraction was desalted by a spin column (Bio-Rad)before measuring the protein concentration and BE activity by a radioactiveassay. The protein concentration was determined by protein assay standard IIdye reagent concentrate using bovine serum albumin for the standard curve inthe 1–10 mg/mL microquantification range.
AS Fractionation
The crude soluble extract was supplemented with 20% (w/v) solid ASwith stirring in an ice bath until it had completely dissolved. The mixture was
FIG. 1. EFFECTS OF REACTION CONDITIONS AND TIME ON THE APPEARANCE OFBRANCHING ENZYME (BE) BANDS DURING IN SITU ACTIVITY STAINING
(A) Effects of reaction conditions. (B) Effects of reaction time. The crude soluble extract, loadedwith the same 160 mg of protein in the 10 lanes, was run on a 7.5% native-polyacrylamide gel
electrophoresis. One lane was sliced and stained with Coomassie blue (labeled as crude). Four laneswere individually sliced and incubated in four different conditions separately for 24 h at 30C as
shown in panel A. The other five lanes were again individually sliced but incubated together usingthe BE condition (condition 3), in which the gels were removed periodically at 2, 4, 8, 16 and 24 hfor I2/KI staining as shown in panel B. Condition 1, 20 mM glucose-1-phosphate (G-1-P) and 0.1 M
sodium citrate, pH 6.0 (starch phosphorylase condition); condition 2, 20 mM G-1-P and 0.1 Msodium citrate, pH 7.0; condition 3, 50 mM G-1-P, 0.1 M sodium citrate, 1 mM AMP and
6 units/mL phosphorylase a, pH 7.0 (BE condition); and condition 4 was similar to that of BE, butno phosphorylase a was added (endogenous phosphorylase was used to assay BE).
127IDENTIFICATION OF A STARCH-BRANCHING ENZYME

allowed to set for 20 min with intermittent stirring every 5 min, and then wascentrifuged at 10,000 ¥ g for 10 min. The supernatant was supplemented with40% solid AS and then treated in the same manner as described earlier; similarprocedures were used for the 60 and 80% fractionations. The 20, 40, 60 and80% precipitates dissolved in TE were dialyzed overnight at 4C and concen-trated by Centricon (10,000 MWCO) for assaying BE activity.
DEAE Fast Flow Anion Exchange Chromatography
The active dialyzed 40~60% AS fraction precipitate, dissolved in bufferA, was applied to an equilibrated HiPrep 16/10 DEAE Fast Flow anionexchange column. The column was washed to release unbound proteins, fol-lowed by a linear 0~0.5 M NaCl gradient of buffer A/B at a flow rate of5 mL/min. Every 5 mL of the eluted fraction was separately collected.
Sephacryl S-200 Gel Filtration
Active fractions 58~61 from the DEAE Fast Flow column were pooledand concentrated by Amicon (model 8550, Millipore) using membrane PM 10(10,000 MWCO) to a volume of 4.5 mL. After centrifugation at 10,000 ¥ g for10 min and passing through a 0.22 mm filter disk, the concentrated sample wasapplied to an equilibrated Sephacryl S-200 (HiPrep 16/60) column in a bufferC system. The elution was run at a steady flow rate of 0.4 mL/min, and every2.5 mL of eluent was separately collected.
Q2-Sepharose Anion Exchange Chromatography
The active PI peak (fractions 13 and 14) and PII peak (fractions 19 and20) from the Sephacryl S-200 column were separately pooled. Each peakfraction was concentrated and desalted by Amicon Ultra-4 centrifugal filterdevices to 0.5 mL. After centrifugation at 10,000 ¥ g for 10 min and passingthrough a 0.22 mm filter, each concentrated sample was applied to an equili-brated Q2-Sepharose column with buffer A, and unbound proteins werewashed away. Then, the elution was run in a linear 0~0.5 M NaCl gradient ofbuffer A/B at a flow rate of 5 mL/min. Every 5 mL eluted fraction was sepa-rately collected.
PAGE
PAGE was conducted in a Mini PROTEAN II apparatus (Bio-Rad).Native-PAGE and SDS-PAGE were prepared as described previously (Koet al. 2005a). Native-PAGE gels were used for BE activity staining, andSDS-PAGE gels were subjected to Coomassie blue or silver staining, followedby Western blotting. Gel photo documentation and protein molecular size
128 Y-T. KO ET AL.

calculations were carried out with an Alpha Innotech ChemiImager IS4400system (San Leandro, CA) loaded with AlphaEase v. 2.3 software.
Western Blotting
Proteins were first separated by 0.75 mm thick SDS-PAGE using a Mini-gel system, and then electroblotted onto a PVDF membrane by Mini-transblotin transfer buffer. The membrane was successively hybridized with a 1:10 ratioof the first anti-55 kDa SP antibodies (Ko et al. 2005b) and a 1:1,000 ratio ofthe second antibodies, followed by ECL detection.
In-Gel Trypsin Digestion
The protein gel band (2 mm diameter) was cut with a pipette tip andtransferred into a 0.6 mL microcentrifuge tube. The gel pieces were washedtwice with 50 mL of a 1:1 ratio of acetonitrile (ACN) and 200 mM ammoniumbicarbonate for 5 min, shrunk with 100% ACN until the gels turned white, thendried for 5 min in a speed vacuum at room temperature. The gel pieces wererehydrated in 15 mL of 50 mM ammonium bicarbonate at 37C for 4 min, andthen supplemented with an equal volume of trypsin solution (20 ng/mL in50 mM ammonium bicarbonate), followed by incubation at 37C for 4 h or 30Cfor at least 16 h. After digestion, the mixture was sonicated, and the gelpieces were spun down to recover the peptide supernatant. The supernatantswere stored at -20C until the LC–mass spectroscopy/mass spectrometry(LC–MS/MS) analysis.
Nanoelectrospray Mass Spectrometry
Nanoscale capillary LC–MS/MS analysis was performed using anUltimate capillary LC system (LC Packings, Amsterdam, The Netherlands)coupled to a QSTARXL quadrupole-time of flight (Q-TOF) mass spectrometer(Applied Biosystem/MDS Sciex, Foster City, CA). The nanoscale capillary LCseparation was performed on an RP C18 column (15 cm, 75 mm i.d.) with aflow rate of 200 nL/min and a 70 min linear gradient of 5–50% buffer B.Buffer A contained 0.1% formic acid in 2% aqueous ACN; buffer B contained0.1% formic acid in 98% aqueous ACN.
A nanoelectrospray interface was used for LC–MS/MS analysis. Ioniza-tion (2.0 kV ionization potential) was performed with a coated nanoLC tip.The nanoLC tip for on-line LC–MS used was a PicoTip (FS360-20-10-D-20,New Objective, Cambridge, MA). The optimum sprayer position was typicallyflushed with or slightly inserted (approximately 1 mm) into the curtainchamber. The temperature of the heated laminar flow chamber was set at100 C. The potential of the curtain plate was set at 250 V, and the curtain gaswas set at 1.3 L/min.
129IDENTIFICATION OF A STARCH-BRANCHING ENZYME

Data acquisition was performed by automatic Information DependentAcquisition (IDA) (Applied Biosystem/MDS Sciex). The IDA automaticallyfinds the most intense ions in a TOF MS spectrum, and then performs anoptimized MS/MS analysis on the selected ions. The product ion spectragenerated by nanoLC–MS/MS were searched against NCBI databases forexact matches using the ProID program (Applied Biosystem/MDS Sciex)and the MASCOT search program. A Viridiplantae (green plants) taxonomyrestriction was used, and the mass tolerance of both precursor ion and frag-ment ions was set to 0.3 Da.
RESULTS AND DISCUSSION
Effects of Reaction Conditions and Time on BE Zymograms
To respond to the presence of BE in the soluble extract, its activity wasassayed in situ on native-PAGE gels by phosphorylase-stimulation method. Itvisualized the formation of newly synthesized amylose, catalyzed by a coupledaction of an exogenous phosphorylase from G-1-P, into amylopectin by BEactivity staining. The amylopectin product–enzyme complex was visualizedby the purplish color derived from the a-glucan binding with iodine on the gelzymogram (Sun et al. 1996; Ko and Huang 2004).
The phosphorylase-stimulation method for BE, however, was similar tothe assay for SP. In order to distinguish signals exclusively from the action ofBE rather than from SP, four different reaction conditions were examinedduring the activity staining (Fig. 1A). Condition 1 was a typical SP assay,condition 2 measured SP but used the optimal pH for BE, condition 3 was atypical BE assay and condition 4 used endogenous SP in the crude extract toproduce amylose upon which BE could act (no extra phosphorylase wasadded). The SP assay visualized a major starch–SP–I2 blue complex as shownin condition 2 except that a couple of extra-high-molecular-weight polymerswere detected (Fig. 1A, labels 1 and 2). The BE assay visualized three popu-lations designated I (the first blue band), II (the second heavy bluish purpleband) and III (the third light bluish purple band) (Fig. 1A, label 3). It is clearthat band I was detected at the same position as the major band shown incondition 1 and as previously reported for SP (Ko et al. 2005b). A weak bandI was apparently shown again in condition 4 (Fig. 1A, label 4); however, itscomplex color was of a typical amylopectin–I2 purple. Because condition 4responded to products catalyzed by endogenous SP to form amylose for BE toproduce amylopectin, this purplish band I comprised proteins of the samemobility, implying the combined action of endogenous SP and BE in the bandI complex. The effect of the reaction time on the appearance of BE bands
130 Y-T. KO ET AL.
showed that the detection of bands II and III required longer incubations of upto 24 h (Fig. 1B). This time length was adapted for the in situ assays through-out the study.
Purification of BE by LC
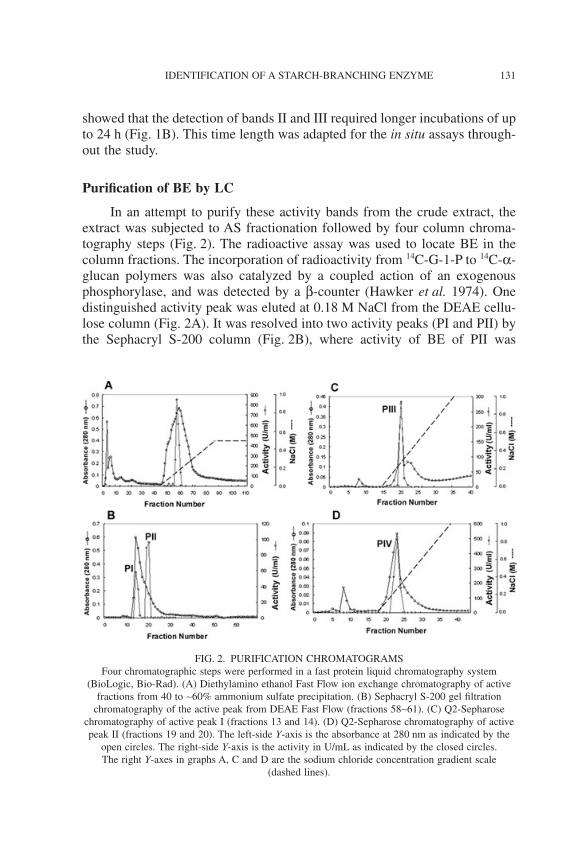
In an attempt to purify these activity bands from the crude extract, theextract was subjected to AS fractionation followed by four column chroma-tography steps (Fig. 2). The radioactive assay was used to locate BE in thecolumn fractions. The incorporation of radioactivity from 14C-G-1-P to 14C-a-glucan polymers was also catalyzed by a coupled action of an exogenousphosphorylase, and was detected by a b-counter (Hawker et al. 1974). Onedistinguished activity peak was eluted at 0.18 M NaCl from the DEAE cellu-lose column (Fig. 2A). It was resolved into two activity peaks (PI and PII) bythe Sephacryl S-200 column (Fig. 2B), where activity of BE of PII was
FIG. 2. PURIFICATION CHROMATOGRAMSFour chromatographic steps were performed in a fast protein liquid chromatography system
(BioLogic, Bio-Rad). (A) Diethylamino ethanol Fast Flow ion exchange chromatography of activefractions from 40 to ~60% ammonium sulfate precipitation. (B) Sephacryl S-200 gel filtration
chromatography of the active peak from DEAE Fast Flow (fractions 58~61). (C) Q2-Sepharosechromatography of active peak I (fractions 13 and 14). (D) Q2-Sepharose chromatography of activepeak II (fractions 19 and 20). The left-side Y-axis is the absorbance at 280 nm as indicated by the
open circles. The right-side Y-axis is the activity in U/mL as indicated by the closed circles.The right Y-axes in graphs A, C and D are the sodium chloride concentration gradient scale
(dashed lines).
131IDENTIFICATION OF A STARCH-BRANCHING ENZYME

threefold that of PI. The PI and PII peak fractions were further separatelypurified into peaks PIII (Fig. 2C) and PIV (Fig. 2D) by Q-Sepharose chroma-tography. Comparison with the crude extract showed that the BE purity wasenriched 30.4-fold in the PIII peak fractions, and 101.9-fold in the PIV peakfractions (Table 1).
Separation of Three BE Activity-stained Bands in the Purified Fractions
Separation of BE activities in the purified fractions was analyzed byactivity staining which showed differential product color and intensity (Fig. 3).The intensity was correlated with enzyme-specific activities (Fig. 3, lanes1~7). Apparently, most of band I was concentrated in the PI and PIII peakfractions, and bands II and III were concentrated in the PII and PIV peakfractions. The typical purple color of the amylopectin–I2 complex of bands IIand III, and the dark blue of band I were seen. However, a minor band II wasalso present in the PI and PIII peaks (Fig. 3, lanes 4 and 6). This zymogramalso showed that the signal from band III was low and undetected in the crude,AS and DEAE active fractions (lanes 1~3).
Protein Profiles in the BE Activity-stained Bands of Purified Fractions
The parallel positions of the activity-stained bands I and II from thepurified PI and PII peaks (Fig. 3, lanes 4 and 5) were individually excised from
TABLE 1.BRANCHING ENZYME (BE) PURIFICATION TABLE
Procedure Totalprotein(mg)
Totalactivity(U)
Specificactivity(U/mg)
Purification(fold)
Yield(%)
Crude extract 1,360 53,235 39 1 100.0Ammonium sulfate (AS) fractionation
(40–60% saluration)242 45,089 186 4.6 87.4
DEAE Fast Flow 27.7 13,386 483 12.3 25.1Sephacryl S–200
PI 3.49 2,598 744 19.0 4.9PII 0.96 2,288 2,383 60.7 4.3
Q2PIII 0.850 1,015 1,194 30.4 1.9PIV 0.226 905 4,001 101.9 1.7
Corresponding fractions with BE activities including fractions 58~61 in the DEAE Fast Flow column,fractions 13 and 14 (PI peak) and fractions 19 and 20 (PII peak) in the Sephacryl S-200 column,fractions 20 and 21 (PIII peak) and fractions 22~24 (PIV peak) in the Q2 chromatography columns(Fig. 2) were pooled in each step, then either desalted or Amicon-concentrated for assay by theradioactive method. Ninety percent of the pooled samples was used in the next purification procedure,and 10% was saved for the protein assay, in situ activity staining, and Western blotting.
132 Y-T. KO ET AL.

a Coomassie blue-stained native gel (Fig. 4A) to analyze their protein profilesby SDS-PAGE (Fig. 4B). Proteins were identified using a proteomicsapproach. Band III was of low quantity, sometimes undetected in the crude(Fig. 4A, lane 2), and could not be investigated further as described earlier(Fig. 3). Band I (Fig. 4B,C) contained not only our previously reported105 kDa SP and the 55 kDa N-terminal fragment of SP (Ko et al. 2005b), butalso a 62 kDa protein and minor bands. Band II clearly contained 96 and64 kDa proteins in addition to the 62 and 55 kDa proteins in band I, as well asminor proteins. As a result, it was obvious that these proteins were potentialBE candidates. A two-stage proteomics approach was used to identify mungbean BE and coexisting proteins. The first stage was an overall identification.The parallel bands of I and II were excised from a native gel (Fig. 4A,indicated by arrows) and subjected to trypsin digestion. Their tryptic frag-ments were analyzed by LC–MS/MS and matched using a bioinformaticsearch. The second stage was a confirmation step in which the targeted proteinspecies (Fig. 4B) were individually cut out for identification in a similarmanner.
Identification of Proteins in BE Activity-stained Bands
The first-stage identification result (Table 2) showed that trypticsequences in both bands I and II matched the 113 kDa fava bean SP with 12%coverage. This result indicated that the 105 kDa SP protein we previouslyidentified (Ko et al. 2005b) was present in these two activity-stained bands.
FIG. 3. ZYMOGRAM OF THE PURIFIED FRACTIONSSamples were from pooled fractions listed in Table 1. Lanes 1~5 were loaded with 20 mg, and lanes6 and 7 had 10 mg protein. They were run on 7.5% native-polyacrylamide gel electrophoresis gels,
and the activity was indicated by staining.
133IDENTIFICATION OF A STARCH-BRANCHING ENZYME

A novel finding was that the 92 kDa mung bean sucrose synthase (SucS;UDP–glucose: d-fructose 2-glucosyl-transferase, EC 2.4.1.13; 12% coverage)(Arai et al. 1992) was also found in both bands. The most important findingwas thus those tryptic fragments of band II exclusively matched the 96 kDa BEof kidney bean (16% coverage) in which the two fragments, DSIPAWIK andFDLGDADYLR, the specific sequences only found in family A-type BEs,were revealed by the NCBI blast search. In addition, no BE sequence wasmatched in band I, which indicated that the BE activity detected by theradioactive assay in PI peak from Sephacryl S-200 is not actually caused byBE, but because of the presence of endogenous phosphorylase in the homo-genate that also incorporated 14C-glucose in the linear polymer of glucose as itwas revealed by the staining assay in native-PAGE (Fig. 3).
When comparing its molecular size with BE species reported so far, theputative 96 kDa protein in the band II complex (Fig. 4B, band II lane) was
FIG. 4. PROTEIN PROFILES OF THE ACTIVITY BANDS I AND II(A) Corresponding positions of bands I and II in the active PII fraction on a native-polyacrylamide
gel electrophoresis (PAGE) gel. Lane 1 was stained with Coomassie blue, and lane 2 is anactivity-stained zymogram. (B) Protein profiles and molecular sizes in the activity bands I and IIfrom the corresponding PI and PII peak fractions in Fig. 3 (lanes 4 and 5). The gel was sodium
dodecyl sulfate (SDS)-PAGE and silver stained. (C) Band I was cut from the activity-stained sampleof the ammonium sulfate-precipitated fraction and analyzed by SDS-PAGE. The gel band was
well-minced with an equal volume of 2¥ SDS-PAGE sample buffer and sonicated for 1 min. Afterboiling the sample at 95C for 1 min and immediately placing it on ice, half of the sample was
loaded into the sample slot and run on 10% SDS-PAGE.
134 Y-T. KO ET AL.

TAB
LE
2.PR
OT
EIN
SID
EN
TIF
IED
INT
HE
BR
AN
CH
ING
EN
ZY
ME
(BE
)A
CT
IVIT
Y-S
TAIN
ED
BA
ND
SI
AN
DII
Iden
tity
Star
chph
osph
oryl
ase
Sucr
ose
synt
hase
Fam
ilyA
BE
Act
ivity
-sta
ined
band
I,II
I,II
IIO
bser
ved
size
(kD
a)~1
0592
~96
The
mos
tm
atch
edsp
ecie
sVi
cia
faba
L.(
fava
bean
)Vi
gna
radi
ata
L.(
mun
gbe
an)
Pha
seol
usvu
lgar
is(k
idne
ybe
an)
Reg
iste
red
ID/s
ize
P535
36(1
13kD
a)Q
0139
0(9
2kD
a)B
AA
8234
8(9
6kD
a)M
atch
edse
quen
ces
148Q
AY
YL
SME
FLQ
GR
160
62LT
DG
AFG
EV
LR
7216
7IY
EID
PSL
LA
HR
178
291A
VA
HD
VPI
PGY
K30
223
1FQ
EIG
LE
R23
819
1LH
DE
INK
YE
GG
LD
TFS
R20
733
1HT
EA
SEA
LA
NA
EK
343
441T
KY
PESD
IYW
K45
125
2NE
FGV
WE
IFL
PNN
VD
GSP
PIPH
GSR
276
397V
AV
QM
ND
TH
PTL
CI
PEL
MR
415
508V
VH
GID
VFD
PK51
828
9DSI
PAW
IK29
642
7DA
WN
ITQ
R43
458
4NIT
GLV
EW
YG
K59
439
9FG
TPE
EL
K40
647
7TII
AE
YG
TAD
SDL
LD
KK
493
600E
LVN
LVV
VA
GD
R61
148
0WW
LD
EY
K48
677
5AFA
TY
VQ
AK
783
718A
AD
LLV
EFF
EK
728
571L
QM
AIA
DK
578
911S
GV
FGSY
NY
DE
LIG
SLE
GN
EG
FGR
934
751Y
TW
QIY
SQR
759
608C
VA
YA
ESH
DQ
ALV
GD
K62
398
3TIH
EY
AR
989
784Y
LE
MFY
AL
K79
271
3FD
LG
DA
DY
LR
722
794I
VL
DSD
DA
LFG
GFN
R80
882
8SFL
VY
APS
R83
6
135IDENTIFICATION OF A STARCH-BRANCHING ENZYME

similar to the 97 kDa BE in potato tubers (Khoshnoodi et al. 1993), the99.7 kDa BEI in peas (Burton et al. 1995) and the 98 kDa BEII in kidneybeans (Hamada et al. 2001), all of which belong to family A-type BEs. This96 kDa band was excised from the gel and subjected to the second confirma-tion stage. In addition, the 64 kDa protein (Fig. 4B, band II lane), anotherpotential BE, was similar to the size of a reported minor 63 kDa protein thatwas recognized by potato BE antibodies and confirmed as being a proteolyticBE fragment during purification (Vos-Scheperkeuter et al. 1989). Such a trun-cated 64 kDa potato BE form was also reported as being predicted from theamino acid sequence after removing the transit peptide sequence from the BEcDNA of the potato (Khoshnoodi et al. 1993). Moreover, we observed thesimultaneous occurrence of 96 and 64 kDa proteins in the active fractions, theintensities in the PII or PIV peak fractions of which on SDS-PAGE werecorrelated with their corresponding BE activities (data not shown). Interest-ingly, a 64 kDa protein of similar size to BE activity-related proteins was alsofound in our previous study on mung bean starch granules (Ko and Huang2004). Therefore, based on the earlier observations, the 64 kDa protein in bandII (Fig. 4B, lane II) was also individually excised and identified in the secondstage. This 96 kDa protein was not merely confirmed to be the mung beanfamily A-type BE isoform, and the internal sequence of the 64 kDa protein wasderived from the 96 kDa BE as speculated. A similar speculation was madewhen examining the 62 kDa protein found in both bands I and II complexes,and the internal sequence was found to match with the SP. The quantity of theappearance of 64 and 62 kDa, however, was not stable from time to time,which indicated that they may be derived from eventual hydrolysis duringpurification as reported in potato (Vos-Scheperkeuter et al. 1989).
Western Blot Analysis of SP in Purified BE Fractions
Because SP was found in both bands I and II, the crude extract andpurified BE fractions (Table 1) were probed with an anti-55 kDa SP antibodyto detect their interactions with SP on a Western blot (Fig. 5). Results showedthat the 105 kDa SP was present in all fractions with strong signals in the PIand PIII peaks, where SP in the band I complex dominated the activity. Therewas more of the 55 kDa SP present in the crude extract and much less in theactive AS-precipitated fraction (Fig. 5, the AS lane). In addition to the identi-fied 62 and 55 kDa SP fragments, proteins with sizes of 58 and 52 kDa werealso detected in the PI and PIII fractions. These signals may also form thepartial degradation of the 105 kDa SP to various extents during purification bysome endogenous proteases, because SP was reported to be regulated byproteolytic control in the sweet potato (Chen et al. 2002). Nevertheless, in ourreports that 105 kDa bands were detected in the most BE-active PII and PIV
136 Y-T. KO ET AL.

fractions by Western blot analysis, and SP and its fragments were identified inthe band I and II complexes by the proteomic approach (Table 2), suggestingthat SP seemed to almost be accompanied by active BE bands even thoughsome of them might not be physically associated forms. This observationcorresponded to a recent report on wheat (Tetlow et al. 2004) which showedthat BEIIb and BEI were associated with SP in a complex whose integrity isdependent on protein phosphorylation. Thus, this study on mung bean rein-forces the scenario that BE and SP coexist together and could cooperate inamylopectin biosynthesis.
In the established starch biosynthetic pathway, BE and SS are believed toact together in forming amylopectin. This cooperative reaction was deducedfrom observations that the two enzymes co-migrated in partially purifiedfractions in early studies on the pea (Matters and Boyer 1981), sorghum andmaize (Boyer 1985). Because mung bean starch possesses a unique structureand properties, it is expected that it is synthesized by unusual biologicalmachinery. In this study, using the proteomics analysis, we observed thecoexistence of SP and SucS in the band I complex, and SP, SucS and BE in theband II complex, implying their collaborative roles in starch biosynthesis.The finding of mung bean SucS (Arai et al. 1992) in both the bands I and IIcomplexes is not surprising because its activity has been known to be relatedto sugar import, cell wall synthesis and starch biosynthesis (Heim et al. 1993).SucS activity is a reliable marker of sink activity (the relative rate of starch
FIG. 5. WESTERN BLOT ANALYSIS OF STARCH PHOSPHORYLASE (SP) IN THEPURIFIED BRANCHING ENZYME FRACTIONS
Samples were the same as those used in Fig. 4, but instead were run on 10% sodium dodecylsulfate-polyacrylamide gel electrophoresis gels, followed by blotting onto a polyvinylidene
difluoride membrane, hybridized with anti-55 kDa SP antibodies and ECL detected on X-ray film.
137IDENTIFICATION OF A STARCH-BRANCHING ENZYME

synthesis) in the developing pea seed (Dejardin et al. 1997). However, basedon their physical proximity and identification in the band complexes, starchsynthesis in mung beans may depend on a pathway which acts mainly by SP,SucS and BE, where SucS should still act as the first enzyme catalyzing thecleavage of sucrose to produce fructose and UDP–glucose. UDP–glucose isthen converted into G-1-P by UDP glucose pyrophosphorylase. However,G-1-P is favorably utilized by SP to directly synthesize amylose which is thenconverted into amylopectin by BE, rather than solely being used by AGPase toform ADP–glucose in the AGPase pathway. Therefore, either the coexisting ofSucS and SP found in band I or that of SucS, SP and BE found in band II maybe a typical event in mung bean physiology. Further investigations are requiredto address the questions of why UDP glucose pyrophosphorylase was notfound in the complex, how the three biosynthetic enzymes can meet togetherto synthesize starch if SucS was known to be a cytosolic enzyme rather thanamyloplastidic type, whether these bands were derived from different cellularcompartments, how they cooperate with other starch biosynthesis enzymes,how each of their isoforms participates in and is physically associated duringthe growth of seeds and what regulatory effectors are involved. In addition, theidentification of the internal sequence information of mung bean BE in thisstudy will be able to degenerate primers for the cloning and expression of BEby molecular techniques, and to further investigate its biotechnology applica-tions in the food industry.
In summary, this study showed that three BE activity-stained bandswere visualized in zymograms, and two bands I and II were further purifiedby chromatography in the developing mung bean. Using an innovative pro-teomics approach, we identified a major family A 96 kDa BE from thepurple band II of partially purified fractions, where SP and SucS were alsopresent. In addition, SP and SucS were identified in both bands I and II, andSP was detected in all of the active partial purified BE fractions. Moreover,we observed that multiple starch biosynthetic enzymes coexist together andsynthesize starch in vitro, which imply that they may cooperate to synthesizestarch in vivo.
ACKNOWLEDGMENTS
We thank the Plant Physiology Division of the Crop ImprovementProgram at the Asian Vegetable Research and Development Center for pro-viding the KPS1 cultivar and arranging the field growth of mung beans. Thiswork was supported by grants (NSC90-2313-B-039-001 and NSC94-2313-B-019-038) from the National Science Council of the R.O.C.
138 Y-T. KO ET AL.

REFERENCES
ARAI, M., MORI, H. and IMASEKI, H. 1992. Expression of the gene forsucrose synthase during growth of mung bean seedlings. Plant CellPhysiol. 33, 503–506.
AVRDC. 1975. Chemical Analysis of Mungbean Seeds, In Progress Report,Asian Vegetable Research and Development Center, Shanhua, Taiwan.
BINDERUP, K., MIKKELSEN, R. and PREISS, J. 2002. Truncation of theamino terminus of branching enzyme changes its chain transfer pattern.Arch. Biochem. Biophys. 397, 279–285.
BOYER, C.D. 1985. Soluble starch synthases and starch branching enzymesfrom developing seeds of sorghum-bicolor cultivar M-5186. Phytochem-istry 24, 15–18.
BOYER, C.D. and PREISS, J. 1978. Multiple forms of (1-4)-a-d-glucan,(1-4)-a-d-glucan-6-glycosyl transferase from developing Zea mays L.kernels. Carbohydr. Res. 61, 321–334.
BURTON, R.A., BEWLEY, J.D., SMITH, A.M., BHATTACHARYYA, M.K.,TATGE, H., RING, S., BULL, V., HAMILTON, W.D.O. and MARTIN,C. 1995. Starch branching enzymes belonging to distinct enzyme familiesare differentially expressed during pea embryo development. Plant J. 7,3–15.
CHEN, H.M., CHANG, S.C., WU, C.C., CUO, T.S., WU, J.S. and JUANG,R.H. 2002. Regulation of the catalytic behaviour of l-form starch phos-phorylase from sweet potato roots by proteolysis. Physiol. Plant. 114,506–515.
CHIANG-HSIEH, P.Y. and KO, Y.T. 2005. Incorporation of sucrose gradientcentrifugation with spectrophotometric assay for isolating mungbeanstarch branching enzyme. Taiwan J. Agric. Chem. Food Sci. 43, 419–427.
DEJARDIN, A., ROCHAT, C., WUILLEME, S. and BOUTIN, J.P. 1997.Contribution of sucrose synthase, ADP-glucose pyrophosphorylase andstarch synthase to starch synthesis in developing pea seeds. Plant CellEnviron. 20, 1421–1430.
GUAN, H.P. and PREISS, J. 1993. Differentiation of the properties of thebranching isozymes from maize (Zea mays). Plant Physiol. 102, 1269–1273.
HAMADA, S., NOZAKI, K., ITO, H., YOSHIMOTO, Y., YOSHIDA, H.,HIRAGA, S., ONODERA, S., HONMA, M., TAKEDA, Y. andMATSUI, H. 2001. Two starch-branching-enzyme isoforms occur in dif-ferent fractions of developing seeds of kidney bean. Biochem. J. 359,23–34.
HAWKER, J.S., OZBUN, J.L., OZAKI, H., GREENBERG, E. and PREISS, J.1974. Interaction of spinach leaf adenosine diphosphate glucose a-1,4-
139IDENTIFICATION OF A STARCH-BRANCHING ENZYME

glucan, a-1,4-glucosyl transferase and a-1,4-glucan, a-1,4-glucan-6-glucosyl transferase in synthesis of branched a-glucan. Arch. Biochem.Biophys. 160, 530–551.
HEIM, U., WEBER, H., BAUMLEIN, H. and WOBUS, U. 1993. A sucrose-synthase gene of Vicia faba L., expression pattern in developing seeds inrelation to starch synthesis and metabolic regulation. Planta 191, 394–401.
HOOVER, R., LI, Y.X. and SENANAYAKE, N. 1997. Physicochemical char-acterization of mung bean starch. Food Hydrocoll. 11, 401–408.
HSU, J.H., YANG, C.C., SU, J.C. and LEE, P.D. 2004. Purification andcharacterization of a cytosolic starch phosphorylase from etiolated riceseedlings. Bot. Bull. Acad. Sin. 45, 187–196.
KHOSHNOODI, J., EK, B., RASK, L. and LARSSON, H. 1993. Character-ization of the 97 and 103 kDa forms of starch branching enzyme frompotato tubers. FEBS Lett. 332, 132–138.
KO, Y.T. and HUANG, L.H. 2004. Mungbean (Vigna radiata L.) starchbranching enzyme activity-related proteins in SDS-PAGE gel after rena-turation. Taiwan J. Agric. Chem. Food Sci. 42, 215–223.
KO, Y.T., PAN, C.H., LEE, Y.T. and CHANG, J.Y. 2005a. Detection ofproteins related to starch synthase activity in the developing mungbean(Vigna radiata L.). J. Agric. Food Chem. 53, 4805–4812.
KO, Y.T., CHANG, J.Y., LEE, Y.T. and WU, Y.H. 2005b. The identification ofstarch phosphorylase in the developing mungbean (Vigna radiata L.).J. Agric. Food Chem. 53, 5708–5715.
LLUISMA, A.O. and RAGAN, M.A. 1998. Cloning and characterization of anuclear gene encoding a starch-branching enzyme from the marine redalga Gracilaria gracilis. Curr. Genet. 34, 105–111.
MARTIN, C. and SMITH, A. 1995. Starch biosynthesis. Plant Cell 7, 971–985.
MATTERS, G.L. and BOYER, C.D. 1981. Starch synthases and starchbranching enzymes from Pisum sativum. Phytochemistry 20, 1805–1809.
MORELL, M.K., BLENNOW, A., KOSAR-HASHEMI, B.K. and SAMUEL,M.S. 1997. Differential expression and properties of starch branchingenzyme isoforms in developing wheat endosperm. Plant Physiol. 113,201–208.
MYERS, A.M., MORELL, M.K., JAMES, M.G. and BALL, S.G. 2000.Recent progress toward understanding biosynthesis of the amylopectincrystal. Plant Physiol. 122, 989–997.
NAKAMURA, Y., TAKEICHI, T., KAWAGUCHI, K. and YAMANOUCHI,H. 1992. Purification of two forms of starch branching enzyme(Q-enzyme) from developing rice endosperm. Physiol. Plant. 84, 329–335.
140 Y-T. KO ET AL.

NAKAYAMA, S. and NAKAMURA, Y. 1994. Purification and some proper-ties of starch branching enzyme (Q-enzyme) from tuberous root of sweetpotato. Physiol. Plant. 91, 763–769.
NOZAKI, K., HAMADA, S., NAKAMORI, T., ITO, H., SAGISAKA, S.,YOSHIDA, H., TAKEDA, Y., HONMA, M. and MATSUI, H. 2001.Major isoforms of starch branching enzymes in premature seeds ofkidney bean (Phaseolus vulgaris L.). Biosci. Biotechnol. Biochem. 65,1141–1148.
SMITH, A.M. 1988. Major differences in isoforms of starch-branchingenzyme between developing embryos of round- and wrinkled-seededpeas (Pisum sativum L.). Planta 175, 270–279.
SUN, C.X., SATHISH, P., EK, B., DEIBER, A. and JANSSON, C. 1996.Demonstration of in vitro starch branching enzyme activity for a 51/50-kDa polypeptide isolated from developing barley (Hordeum vulgare)caryopses. Physiol. Plant. 96, 474–483.
TETLOW, I.J., MORELL, M.K. and EMES, M.J. 2004. Recent developmentsin understanding the regulation of starch metabolism in higher plants.J. Exp. Bot. 55, 2131–2145.
TSAY, J.S., KOU, W.L. and KUO, C.G. 1983. Enzymes involved in starchsynthesis in the developing mungbean seed. Phytochemistry 22, 1573–1576.
VOS-SCHEPERKEUTER, G.H., DE WIT, J.G., PONSTEIN, A.S.,FEENSTRA, W.J. and WITHOLT, B. 1989. Immunological comparisonof the starch branching enzymes from potato tubers and maize kernels.Plant Physiol. 90, 75–84.
YU, Y., MU, H.H., WASSERMAN, B.P. and CARMAN, G.M. 2001. Identi-fication of the maize amyloplast stromal 112-kDa protein as a plastidicstarch phosphorylase. Plant Physiol. 125, 351–359.
141IDENTIFICATION OF A STARCH-BRANCHING ENZYME





























