Embed Size (px)
Citation preview

Hybridization of a Direct Numerical Simulation Software for Massively Parallel
Accelerator-based Architectures
Ramanan Sankaran Computational Scientist Oak Ridge National Laboratory Joint Research Faculty University of Tennessee, Knoxville
Jackie Chen (SNL), Ray Grout (NREL) and John Levesque (Cray)
2
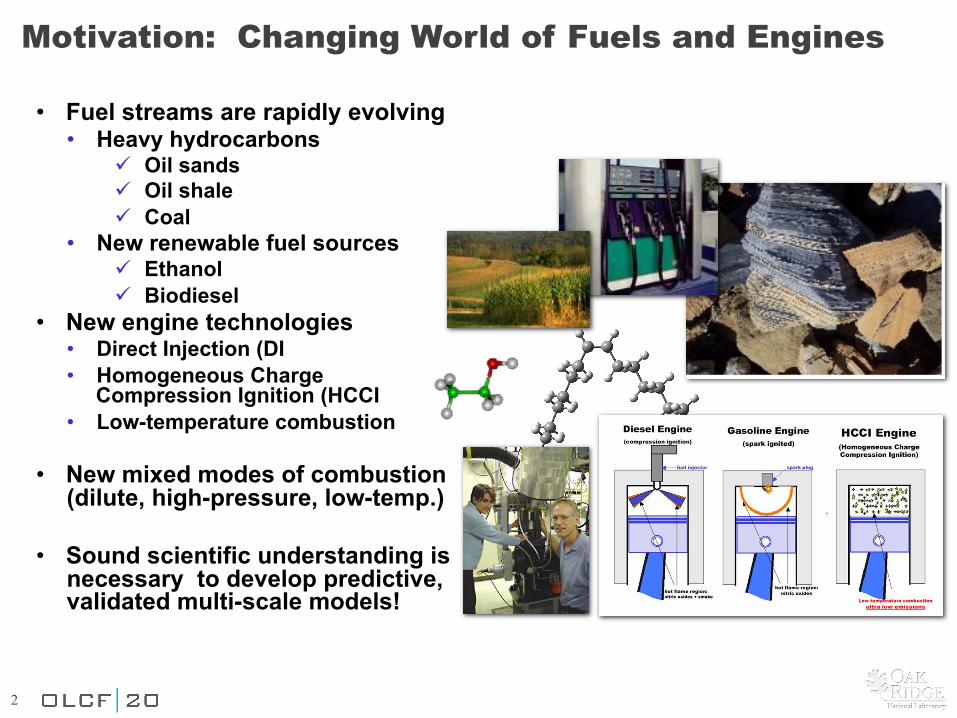
Motivation: Changing World of Fuels and Engines
• Fuel streams are rapidly evolving • Heavy hydrocarbons
! Oil sands ! Oil shale ! Coal
• New renewable fuel sources ! Ethanol ! Biodiesel
• New engine technologies • Direct Injection (DI • Homogeneous Charge
Compression Ignition (HCCI • Low-temperature combustion
• New mixed modes of combustion (dilute, high-pressure, low-temp.)
• Sound scientific understanding is
necessary to develop predictive, validated multi-scale models!

3
Combustion is a Complex, Multi-physics, Multi-scale Problem
Diesel Engine Autoignition, Soot Incandescence!Chuck Mueller, Sandia National Laboratories!
• Stiffness : wide range of length and time scales • In-cylinder geometry (cm) • Turbulence-chemistry (mm) • Soot inception (nanometer)
• Chemical complexity • large number of species and
reactions (100 s of species, thousands of reactions) !
• Multi-Physics complexity • multiphase (liquid spray, gas
phase, soot, surface)! • thermal radiation • acoustics ...
All these are tightly coupled

4
Direct Numerical Simulations (DNS) • Turbulent combustion occurs over a wide range of scales
– Device sizes are O(1m) – Diffusive scales and flame thickness O(10-100 µm) – Non-linear coupling and interaction among the entire range of
scales
• Combustion CFD approaches
• Direct numerical simulation (DNS) – No sub-grid models, but limited on range of scales – Simulations limited to canonical research configurations
Small scales Large scales
DNS LES RANS !

5
S3D –DNS solver
• Structured Cartesian mesh flow solver • Solves compressible reacting Navier-Stokes, energy and species
conservation equations. – 8th order explicit finite difference method – 4th order Runge-Kutta integrator with error estimator
• Detailed gas-phase thermodynamic, chemistry and molecular transport property evaluations
• Multi-physics: sprays, radiation and soot • Lagrangian particle tracking • MPI-1 based spatial decomposition and parallelism • Fortran code. Does not need linear algebra, FFT or
solver libraries.

6
Fundamental Insights on Turbulent Combustion
• DNS is a tool for fundamental studies of the micro-physics of turbulent reacting flows – Full access to time resolved 3D fields – turbulence-chemistry interactions
• Develop and validate reduced model descriptions used in macro-scale simulations of engineering-level systems
DNS Physical Models
Engineering CFD codes
(RANS, LES)

7
!"#"$%&%"'()*+,-$%)*"#.-%((/"&)0$&/1"&))2!**03)4&$/&%()
• Potential for high diesel-like efficiencies but low soot and NOx emissions
• Fuel-lean and at low temperatures – no flame, spontaneous autoignition
• Hard to control ignition timing, sensitive to fuel chemistry, need to moderate burn rate (high load)
• Better understand ignition chemistry of fuel blends and oxygenated hydrocarbon molecules in biomass derived fuels

8
International Journal of Engine Research, 2002. 3(4): p. 185-195.

9
Fuel chemistry and mixing control the rate of combustion in HCCI engines
• Inhomogeneities (thermal or composition) lead to sequential ignition front propagation down the gradient - combustion modes ranging from homogeneous explosion to propagating flames
• New modes operate far from equilibrium with highly transient intermittent ignition occurring at multiple sites
• Better understand and predict behavior of alternative fuels in HCCI engines
Optical engine experiments by Walton et al. show front-like propagation

10
DNS of DME HCCI Autoignition (G. Bansal et al. 2011)
• Turbulence and scalars initialized using an energy spectrum
• Initial turbulence integral time-scale and scalar RMS values – guided from practical engine experiments
• Reduced DME chemistry – 30 species • Initially homogeneous composition • (" = 0.3) with Gaussian temperature
distribution, T’ = 25K • Isentropic compression simulates HCCI
engine operation from 36 CAD to TDC
Vorticity
Temp
Initial Condition)
Temperature

Existence of highly wrinkled thin “cool flame” fronts – first ignition stage
!!"
Vorticity Temp
YCH2O YCH3OCH2O2
(Key intermediate)

Close proximity of IInd and IIIrd stage waves – inter-diffusion of heat and radicals IInd stage is chemistry driven spontaneous front; IIIrd stage is a deflagration wave
II III
III II No
diffusion
III
II
A twin-ring structure of heat release
Simultaneous Existence of Flames and Spontaneous Ignition

PDF modeling of molecular mixing in flames with differential diffusion
• The DNS data reveal individual species mixing at vastly different rates – due to species diffusivities and flame structure.
• Predictions of the state-of-the-art EMST model: Accounts for flame structure but unable to account for differential diffusion.
• New PDF modelling developed by Richardson and Chen (Combustion and Flame 2012) includes species diffusivities in a rigorous manner and correctly predicts the physics observed in the DNS.
Variation of normalised species mixing rates versus time:
Conventional EMST model
New EMST- model
DNS data
Richardson, Bansal and Chen in prep 2012
EMST model

Summary of DME HCCI DNS and Modeling
• DME autoignition occurs in three distinct chemical stages
• 2nd and 3rd stage can occur in close physical proximity
• Due to strong reaction generated gradients –scalar dissipation due to reaction
• Multi-scalar mixing models treating localness and differential diffusion (EMST-DD)
))
• 2nd stage is predominantly
spontaneous ignition front; 3rd stage is predominantly premixed deflagration
New EMST model
Diffusion-reaction Balance (OH)


16 8 Buddy Bland – CUG 2012
• Upgrade of Jaguar from Cray XT5 to XK6
• Cray Linux Environment operating system
• Gemini interconnect • 3-D Torus • Globally addressable memory • Advanced synchronization features
• AMD Opteron 6274 processors (Interlagos) • New accelerated node design using NVIDIA
multi-core accelerators • 2011: 960 NVIDIA x2090 “Fermi” GPUs • 2012: 14,592 NVIDIA “Kepler” GPUs
• 20+ PFlops peak system performance • 600 TB DDR3 mem. + 88 TB GDDR5 mem
ORNL’s “Titan” System
!"#$%&'()*+&
!"#$%&'()"*'+( ,-./--(
0"123(4(567()"*'+( 8,9(
:'#";<($';(3"*'( =9(>?(@(/(>?(
A("B(C';#2(DE2$+(F9G,9H( I/G(
A("B()J5K5L(MN'$O';P(F9G,=H(
,Q.8I9(
R"&SO(T<+&'#(:'#";<( /--(R?(
R"&SO(T<+&'#(U'SV(U';B";#S3D'(
9G@(U'&SBO"$+(
!;"++(T'D&2"3(?S3*W2*&E+(
XY,QZQ(R?6+([Y,,Z=(R?6+(\Y9QZG(R?6+(

17
T` = 3.75K T` = 7.50K T` = 15.0K T` = 30.0K
Increasing stratification H
omog
eneo
us!
Fron
t-lik
e!
Results from a 2D parametric study with hydrogen chemistry (9 chemical species), Chen et al. 2003.
" Objective: 3-dimensional DNS of HCCI combustion in a high-pressure stratified turbulent dimethyl ether (DME) blended iso octane/air mixture using detailed chemical kinetics (60 chemical species)
Grid: 2D O(106) 3D O(109). Chemical complexity: 9 60 species. " Goals: To investigate
# Interaction of 3D turbulence with important chemical kinetic pathways leading to ignition # Effects of charge stratification on heat release modes, pressure rise rates, and pollutant
formation # Generate a high-fidelity database for use as a benchmark to validate sub-grid combustion
models for mixed-mode combustion in LES and RANS
What do we want to simulate on Titan?

18
Acceleration strategy for Titan 1. Define target science problem 2. Profile legacy code 3. Identify key kernels for optimization 4. Requirements for host/accelerator work distribution 5. Prototype and explore performance bounds using cuda 6. “Hybridize” legacy code: MPI for inter-node, OpenMP
intra-node 7. OpenACC for GPU execution 8. Restructure to balance compute effort between accelerator
and host 5..6/7,1"&)8%,9/&%(():%,#
#$"%&'(&)*"+$",-))&*".$"%/&0&12&)&*"3$"+$"4/5' 678 9$":$"#;-<1 79=8 9$"6&'>&;&'*",$"6?&@-;A*":$":&'0 B978 7$"3<@&*"#$"9<51(C/*"4$":--))5D*"6$"E-(5D 7FGAG& 3$"85F5(H<5*"3$"6C/2&;IJ5G5; 4;&D

19
Performance Profile for Legacy S3D • A benchmark problem was defined to closely resemble the
target simulation – 52 species n-heptane chemistry and 483 grid points per node – 483 * 12,000 nodes = 1.5 billion
grid points
• Code was benchmarked and profiled on dual-hexcore XT5
• Several kernels identified and extracted into stand-alone driver programs
Chemistry
Core S3D

20
S3D readiness for Titan
Chemistry
Core S3D
• S3D refactoring started out with a CUDA approach for several key kernels
• Initial CUDA porting established the performance bounds and expectations
• Later we focused on refactoring S3D to a compiler directive approach – Portability to non-accelerator
platforms and non-CUDA architectures
• Currently, all of S3D has been ported to the GPU using OpenACC

21 13
Hierarchical Parallelism • MPI parallelism between nodes (or PGAS) • On-node, SMP-like parallelism via threads (or
subcommunicators, or…) • Vector parallelism
– SSE/AVX on CPUs – GPU threaded parallelism
• Exposure of unrealized parallelism is essential to exploit all near-future architectures.
• Uncovering unrealized parallelism and improving data locality improves the performance of even CPU-only code.
11010110101000 01010110100111 01110110111011
01010110101010
Disclaimer: No contract with vendor is in place

22
Hybridization of all MPI S3D
• Creation of an application that exhibits three levels of parallelism, MPI between nodes, OpenMP on the node and vectorizable loops
• OpenMP and OpenACC compiler directives are used to run the same application on CPU or accelerator
• Compiler directives do not imply “automatic”. Software refactoring was necessary. – to have high level OpenMP structures – remove loop dependencies that inhibit vectorization – Ensure data locality – Overlap computation with communication through host
• Currently achieving 1.2X speedup on Fermi-XK6 vs CPU

23
RHS Reorganization

24
Chemistry Kernels • Reaction rates, thermodynamic properties and transport
coefficients account for 55% of time in DNS – Complex chemical kinetic models needed to address multi-stage ignition
and flame dynamics
• Point-wise functions that are independent of DNS software’s mesh data structure and MPI-layer – Uses Chemkin API
• Porting of the chemistry kernels began a year before OLCF-3 was planned – Keiki software was developed for computing chemical kinetics on GPU
systems such as OLCF Titan
• How can this software impact other combustion codes that want to use accelerators?

25
Detailed chemical kinetics are expensive
component in the simulation of chemically reacting flows. It isimportant because the fidelity of all subsequent steps of mecha-nism reduction depends on the fidelity of the detailed mechanism.In other words, the comprehensiveness of a reduced mechanismcannot exceed that of the detailed mechanism from which it isdeduced. This is a challenging task because, firstly, it is difficult tobe certain that all possible important species and reactions areidentified and included in the detailed mechanism. Furthermore,the number of reactions and species involved is large, and thedetermination of the rate constants of each of the identified reac-tions, either experimentally or computationally, is not a trivial task.
Lacking a systematic, first-principle procedure to identify allrelevant species and reactions that would render a mechanismcomprehensive, comprehensiveness can be considered based on theability of the mechanism to describe combustion phenomena asextensively as possible. There are two levels of considerations. First,since the nature of the collision dynamics is determined by theidentity of the colliding molecules as well as the frequency andenergetics of the collision, a comprehensive chemical description interms of themacroscopic thermodynamic properties would requireextensive coverage in the range of temperature, pressure, andcomposition of the reacting mixture. Second, in terms of combus-tion phenomena, comprehensiveness would require considerationsof homogeneous and diffusive ignition which cover low-, interme-diate- and high-temperature chemistry, steady burning andextinction which cover high-temperature chemistry, and premixedand nonpremixed flames which cover the relative concentrationsand mixedness of fuel and oxidizer. The global combustionresponses of interest would include the laminar flame speed, igni-tion and extinction strain rates, detonation induction length,detailed thermal and concentration structures of flames and deto-nations, oscillatory and pulsed unsteady effects to potentiallydiscriminate reactions of different time scales, and pollutantchemistry.
A final requirement for comprehensiveness is fuel hierarchy. Forexample, since hydrogen and CO oxidation constitute a part ofmethane oxidation, a methane mechanism must degenerate tothose for hydrogen and CO when all elementary reactions notrelated to them are stripped away. Thus amechanism developed fora fuel must contain descriptions of its intermediates and simplerfuels as its sub-mechanisms.
It is clear that since the size of a mechanism depends on theextent of comprehensiveness, some reduction can be achieved forrestricted comprehensiveness. Perhaps the most obvious restriction
is to fix the pressure to atmospheric because many fundamentaland practical combustion phenomena and processes take placeunder atmospheric pressure. Other restrictions can also beimposed, such as lean combustion, high-temperature flameswithout considering the possible presence of ignition described bylow-temperature chemistry, and homogeneous charge combustion.However, except for well-controlled laboratory-scale experiments,the combustion mode is frequently a mixed one in most complexand practical combustion situations, involving for example bothpremixed and nonpremixed reactants, or both ignition and flames.Consequently it is more conservative to apply unrestrictedcomprehensive mechanisms in simulations of complex flows.
4. Overview of mechanism reduction andfacilitated computation
The availability of a comprehensive detailed reaction mecha-nism does not mean that it can be readily adopted for computa-tional simulation. In fact, except for the smallest of fuels such ashydrogen andmethane, and for such simple combustion systems asthe 1-D laminar flame, detailed mechanisms of the larger fuels aresimply too large for simulation without substantial reduction.Fig. 10 shows the size of more than 20 detailed and moderatelyreduced skeletal mechanisms for hydrocarbon fuels of variousmolecular complexities compiled over the last two decades [15].Several interesting observations can be made here. First, thenumber of species, K, and reactions, I, increase with the size of themolecule, roughly in an exponential trend. Specifically, it is seenthat while typical mechanisms for C1 and C2 species consist of lessthan about a hundred species, those for realistic engine fuels consistof hundreds of species and thousands of reactions. Mechanisms ofsuch sizes are even difficult to apply in 1-D flame simulations. As anextreme example, the size of the compiled detailed mechanism formethyl decanoate [16], a biomass fuel surrogate, consists of 3036species and 8555 reactions. Computation using this mechanism istime consuming even for 0-D simulations.
The second observation from Fig. 10 is that the size of themechanisms tends to grow with time, as new discoveries inchemical kinetics are continuously being made. Furthermore, theemergence of computer-aided automatic mechanism generation[17–20] and computer software for rate parameter evaluation, such
10-5
10-4
10-3
10-2
10-1
0.5 0.6 0.7 0.8 0.9 1.0
Methane/Air, !=1.0, p=1 atm
GRI-Mech 1.212-Step10-Step4-Step
Auto
-Igni
tion
Del
ay (s
ec)
1000/T (1/K)
Fig. 9. Comparison of predicted ignition delay times of atmospheric, stoichiometricmethane–air mixtures using various reduced mechanisms and the detailed mecha-nism, showing the inadequacy of the four-step class of mechanisms.
101 102 103 104
102
103
104
before 20002000 to 2005after 2005
iso-octane (LLNL)
iso-octane (ENSIC-CNRS)
n-butane (LLNL)
CH4 (Konnov)
neo-pentane (LLNL)
C2H4 (San Diego)
CH4 (Leeds)
MethylDecanoate(LLNL)
C16 (LLNL)
C14 (LLNL)C12 (LLNL)
C10 (LLNL)
USC C1-C4USC C2H4
PRF
n-heptane (LLNL)
skeletal iso-octane (Lu & Law)skeletal n-heptane (Lu & Law)
1,3-ButadieneDME (Curran)C1-C3 (Qin et al)
GRI3.0
Num
ber o
f rea
ctio
ns, I
Number of species, K
GRI1.2
I = 5K
Fig. 10. Size of selected detailed and skeletal mechanisms for hydrocarbon fuels,together with the approximate years when the mechanisms were compiled.
T.F. Lu, C.K. Law / Progress in Energy and Combustion Science 35 (2009) 192–215196
From Lu and Law, PECS, 2009
• Chemical source term evaluation is computationally intensive
• Thousands of elementary reaction steps accumulated to global species reaction rates
• Often the target for model reductions or algorithmic improvements
• How fast can we compute detailed chemical kinetics on accelerators?

26
Partitioning at species/reaction level
• Similar to partitioning the grid for distributed memory parallelism (MPI)
• Why partition the computation at species/reaction level? – Asynchronous execution to hide latencies and data transfers
(memcpy across PCI) – Distribute work to multiple accelerators assigned to a single host – Allow finer grained parallelism at the chemistry level to multiply the
scalability of the flow solver
• Keiki treats the chemical kinetics as a graph and partitions it to minimize edgecut and maximize parallel performance

27
Reaction network as a graph
• Chemical reaction network is a bi-partite graph between two sets of vertices – The species form one set – The reactions form the second set – Stoichiometry of the reaction network defines the graph
• The adjacency matrix of the graph is
• Where B is the M x N stoichiometry matrix
A = 0 BBT 0
!
"##
$
%&&

28
Partitioning the graph
• Graph partitioning software Metis and PaToH were used to partition the bi-partite graph – A good quality partition minimizes edge-cut with maximum load balance – Reorders the network, without changing the answers
• Edge-cut induces redundant computation or synchronization points
• Partitions should be sized to meet the vector length and memory requirement – Large enough to have enough number of threads per thread block – Control shared memory requirement to obtain high occupancy
• Need a sufficient number of partitions that can execute concurrently

29
Partitioning iso-octane chemistry (contd)
• The quality of partitioning gets better as the chemistry model gets bigger

30
Keiki Performance
• Performance on dual 6-core Opteron CPU and Fermi GPU were compared – CPU peak = 2*62.4 = 125 GF – GPU peak = 515 GF
• The execution times on GPU were 2 ~ 3x faster than the CPU
• Much larger speedup expected with the Kepler GPU to be installed on Titan XK6 system
4/5JG(1;D"K-A5)"
L 4/5J>G'"61&'A&;A"J5C/&'G(J"&'A"1/5;J-AD'&JGC("A&1&"
E&;(5;M.'&)DI5;"
L E5;)"(-N2&;5"O-;"?&;(G'0"G'?<1"P)5("
L Q'15;O&C5"1-"0;&?/"&'&)D(G(M?&;RR-'G'0"
4ST."4-A5"#5'5;&1-;"
L K5C/&'G(JMO<5)"(?5CGPC"05'5;&15A"C-A5"
L E)<("#ESUC&?&V)5"(-)F5;"&'A"C-JV<(R-'"J-A5)"





























