Embed Size (px)
Citation preview

498 www.thelancet.com/neurology Vol 12 May 2013
Review
Human rabies: neuropathogenesis, diagnosis, and managementThiravat Hemachudha, Gabriella Ugolini, Supaporn Wacharapluesadee, Witaya Sungkarat, Shanop Shuangshoti, Jiraporn Laothamatas
Rabies is an almost invariably fatal disease that can present as classic furious rabies or paralytic rabies. Recovery has been reported in only a few patients, most of whom were infected with bat rabies virus variants, and has been associated with promptness of host immune response and spontaneous (immune) virus clearance. Viral mechanisms that have evolved to minimise damage to the CNS but enable the virus to spread might explain why survivors have overall good functional recovery. The shorter survival of patients with furious rabies compared with those with paralytic rabies closely corresponds to the greater amount of virus and lower immune response in the CNS of patients with the furious form. Rabies virus is present in the CNS long before symptom onset: subclinical anterior horn cell dysfunction and abnormal brain MRI in patients with furious rabies are evident days before brain symptoms develop. How the virus produces its devastating eff ects and how it selectively impairs behaviour in patients with furious rabies and the peripheral nerves of patients with paralytic rabies is beginning to be understood. However, to develop a pragmatic treatment strategy, a thorough understanding of the neuropathogenetic mechanisms is needed.
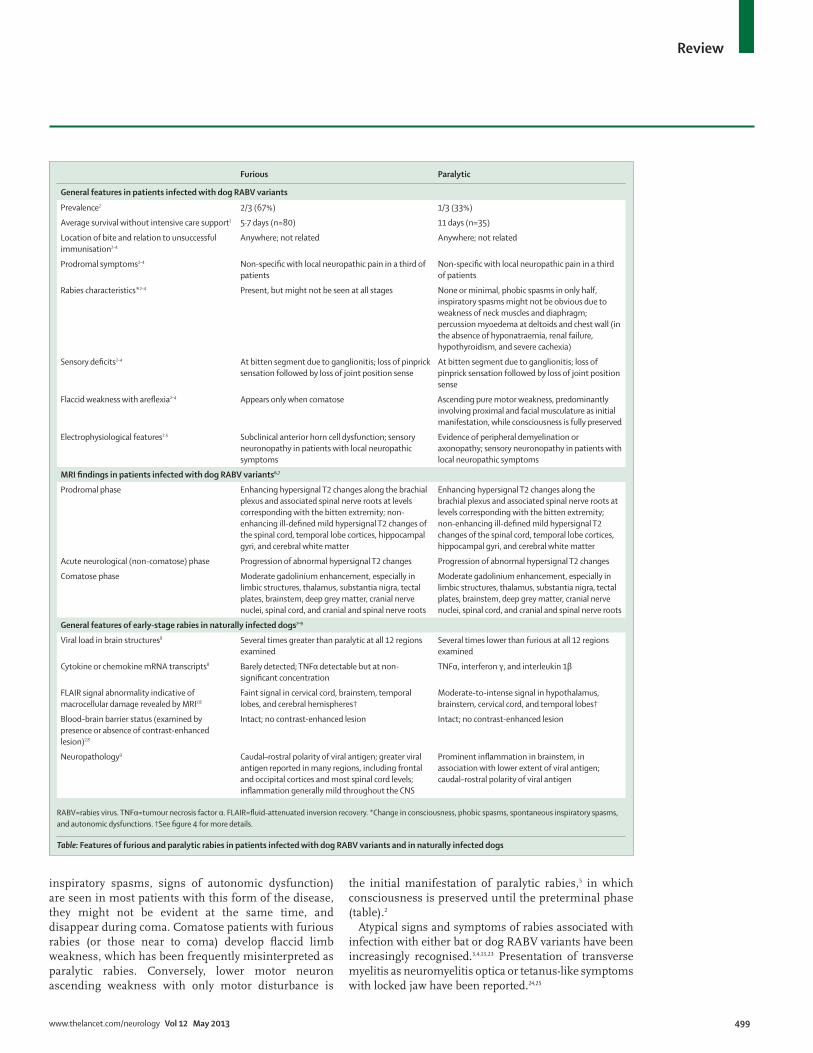
IntroductionRabies caused by rabies virus (RABV) genotype 1 is one of the most common fatal infections worldwide. It is mainly associated with dog bites in Europe, Asia, and Africa and with bats in the Americas.1 Two-thirds of patients infected with dog RABV variants present with classic furious rabies (characterised by fl uctuating consciousness and changed mental status, phobic or inspiratory spasms, and autonomic stimulation signs). The remaining third develop paralytic rabies, which resembles Guillain-Barré syndrome, although progression to coma, myoedema, and bladder incontinence clearly diff erentiate these two disorders (table).2,3 Rabies associated with bat RABV variants has atypical features, such as focal brainstem signs, myoclonus, hemichorea, and signs and symptoms of Horner’s syndrome.3,4
In other infectious encephalitides, damage can be shown by gross pathology, whereas in rabies the CNS seems healthy post mortem, with only minimal neuronal loss and varying degrees of evidence of infl ammatory reactions. This fi nding might be explained by the unique multilevel strategy that RABV has evolved to prevent viral clearance; RABV uses replication mechanisms that delay severe compromise of host cell metabolism and has the capacity to prevent apoptosis and evade innate and adaptive immune responses, which otherwise would promote changes in blood–brain barrier permeability and combat viral progression.10 MRI of the brain of patients with rabies reveals only subtle changes and the absence of gadolinium-enhanced lesions.6,7 With contrast imaging, mild to intense infl ammation is noted mainly in patients with rabies contracted after organ transplantation and in comatose patients infected with dog RABV variants.6,7,11,12
Attempts to treat symptomatic patients infected with dog RABV variants with therapeutics and intensive care support are usually unsuccessful. A few patients who survived infection with bat RABV variants with good functional recovery had evidence of an early immune
response, measured by neutralising or non-neutralising antibody to RABV in blood and CSF, with no RNA or virus detected in samples of biological fl uids or hair follicles.13–18 Therefore, a good outcome might depend on the promptness of the host response in the eradication of virus in both the CNS and periphery.14
In this Review, we summarise relevant data acquired from our studies of patients infected with dog RABV variants and from animal models associated with RABV genotype 1 infection. We extrapolate these data to explain the various clinical phases in furious and paralytic rabies caused by infection with dog RABV variants. We use a dog model with naturally acquired rabies to explain clinical diversities; this model exhibits clinical patterns similar to those seen in man, with an incubation period of weeks to months, and it allows in-vivo studies of CNS abnormalities and blood–brain barrier status by MRI. Results suggest that the immune system is unlikely to have a role in the acceleration of death, especially in furious rabies.3,19
Clinical features The clinical stages of rabies are: incubation, prodrome, acute neurological signs, coma, and death.3 Average survival (from clinical onset to death, with partial or no intensive care support) of 80 patients with furious rabies and 35 with paralytic rabies after infection with a dog RABV variant in Thailand between 1988 and 2004 was 5·7 and 11 days, respectively (data compiled from previously published reports; table).2 These survival times did not substantially diff er from those reported in India between 1980 and 2007.20,21 Survival can be extended to 1 month or longer in patients receiving intensive care support.12
Weakness at the bitten extremities might be evident at initial presentation, although subsequent progression can be in the form of either furious or paralytic rabies.5,22 Although all cardinal features of furious rabies (eg, fl uctuating consciousness, hydrophobia or aerophobia,
Lancet Neurol 2013; 12: 498–513
WHO Collaborating Centre for Research and Training on Viral
Zoonoses, Faculty of Medicine, Chulalongkorn University and King Chulalongkorn Memorial
Hospital, Bangkok, Thailand (Prof T Hemachudha MD, S Wacharapluesadee PhD, Prof S Shuangshoti MD);
Neurobiology and Development (UPR3294), Institute of Neurobiology
Alfred Fessard, Centre National de la Recherche Scientifi que
(CNRS), Gif-sur-Yvette, France (G Ugolini PhD); Advanced
Diagnostic Imaging (AIMC) and Department of Radiology, Ramathibodi Hospital, Faculty
of Medicine, Mahidol University, Bangkok, Thailand
(W Sungkarat PhD, J Laothamatas MD)
Correspondence to:Prof Thiravat Hemachudha,
WHO Collaborating Centre for Research and Training on Viral
Zoonoses, Faculty of Medicine, Chulalongkorn University and King Chulalongkorn Memorial
Hospital, Bangkok 10330, Thailand
www.thelancet.com/neurology Vol 12 May 2013 499
Review
inspiratory spasms, signs of autonomic dysfunction) are seen in most patients with this form of the disease, they might not be evident at the same time, and disappear during coma. Comatose patients with furious rabies (or those near to coma) develop fl accid limb weakness, which has been frequently misinterpreted as paralytic rabies. Conversely, lower motor neuron ascending weakness with only motor disturbance is
the initial manifestation of paralytic rabies,5 in which consciousness is preserved until the preterminal phase (table).2
Atypical signs and symptoms of rabies associated with infection with either bat or dog RABV variants have been increasingly recognised.3,4,13,23 Presentation of transverse myelitis as neuromyelitis optica or tetanus-like symptoms with locked jaw have been reported.24,25
Furious Paralytic
General features in patients infected with dog RABV variants
Prevalence2 2/3 (67%) 1/3 (33%)
Average survival without intensive care support2 5·7 days (n=80) 11 days (n=35)
Location of bite and relation to unsuccessful immunisation2–4
Anywhere; not related Anywhere; not related
Prodromal symptoms2–4 Non-specifi c with local neuropathic pain in a third of patients
Non-specifi c with local neuropathic pain in a third of patients
Rabies characteristics*2–4 Present, but might not be seen at all stages None or minimal, phobic spasms in only half, inspiratory spasms might not be obvious due to weakness of neck muscles and diaphragm; percussion myoedema at deltoids and chest wall (in the absence of hyponatraemia, renal failure, hypothyroidism, and severe cachexia)
Sensory defi cits2–4 At bitten segment due to ganglionitis; loss of pinprick sensation followed by loss of joint position sense
At bitten segment due to ganglionitis; loss of pinprick sensation followed by loss of joint position sense
Flaccid weakness with arefl exia2–4 Appears only when comatose Ascending pure motor weakness, predominantly involving proximal and facial musculature as initial manifestation, while consciousness is fully preserved
Electrophysiological features2,5 Subclinical anterior horn cell dysfunction; sensory neuronopathy in patients with local neuropathic symptoms
Evidence of peripheral demyelination or axonopathy; sensory neuronopathy in patients with local neuropathic symptoms
MRI fi ndings in patients infected with dog RABV variants6,7
Prodromal phase Enhancing hypersignal T2 changes along the brachial plexus and associated spinal nerve roots at levels corresponding with the bitten extremity; non-enhancing ill-defi ned mild hypersignal T2 changes of the spinal cord, temporal lobe cortices, hippocampal gyri, and cerebral white matter
Enhancing hypersignal T2 changes along the brachial plexus and associated spinal nerve roots at levels corresponding with the bitten extremity; non-enhancing ill-defi ned mild hypersignal T2 changes of the spinal cord, temporal lobe cortices, hippocampal gyri, and cerebral white matter
Acute neurological (non-comatose) phase Progression of abnormal hypersignal T2 changes Progression of abnormal hypersignal T2 changes
Comatose phase Moderate gadolinium enhancement, especially in limbic structures, thalamus, substantia nigra, tectal plates, brainstem, deep grey matter, cranial nerve nuclei, spinal cord, and cranial and spinal nerve roots
Moderate gadolinium enhancement, especially in limbic structures, thalamus, substantia nigra, tectal plates, brainstem, deep grey matter, cranial nerve nuclei, spinal cord, and cranial and spinal nerve roots
General features of early-stage rabies in naturally infected dogs7–9
Viral load in brain structures8 Several times greater than paralytic at all 12 regions examined
Several times lower than furious at all 12 regions examined
Cytokine or chemokine mRNA transcripts8 Barely detected; TNFα detectable but at non-signifi cant concentration
TNFα, interferon γ, and interleukin 1β
FLAIR signal abnormality indicative of macrocellular damage revealed by MRI7,8
Faint signal in cervical cord, brainstem, temporal lobes, and cerebral hemispheres†
Moderate-to-intense signal in hypothalamus, brainstem, cervical cord, and temporal lobes†
Blood–brain barrier status (examined by presence or absence of contrast-enhanced lesion)7,8
Intact; no contrast-enhanced lesion Intact; no contrast-enhanced lesion
Neuropathology9 Caudal–rostral polarity of viral antigen; greater viral antigen reported in many regions, including frontal and occipital cortices and most spinal cord levels; infl ammation generally mild throughout the CNS
Prominent infl ammation in brainstem, in association with lower extent of viral antigen; caudal–rostral polarity of viral antigen
RABV=rabies virus. TNFα=tumour necrosis factor α. FLAIR=fl uid-attenuated inversion recovery. *Change in consciousness, phobic spasms, spontaneous inspiratory spasms, and autonomic dysfunctions. †See fi gure 4 for more details.
Table: Features of furious and paralytic rabies in patients infected with dog RABV variants and in naturally infected dog s

500 www.thelancet.com/neurology Vol 12 May 2013
Review
Pathogenesis and pathophysiology The transfer of RABV-containing saliva from a bite from an infected animal is the most effi cient route of transmission. Other routes of transmission include: inhalation of aerosolised RABV; tissue and organ transplants; handling and skinning of infected carcasses; and contamination of an open wound, scratch, abrasion, or mucous membrane by infected saliva or neural tissue.3 The effi ciency of bite transmission depends on virus inocula and viral tissue tropism. The likelihood of infection with dog RABV variants is highest after deep bites that reach the muscle, because the virus can only infect motor endplates in the muscle, and uptake by sensory and sympathetic nerve endings in the muscle does not occur.26–34 Entry via the motor route is determined by the presence at the neuromuscular junction (but not at sensory or autonomic endings) of the nicotinic acetylcholine receptor (on the postsynaptic site), which binds RABV, promoting neural cell adhesion molecule-mediated uptake by motor endplates.28,35 Viraemia does not occur.36 Whether a sensory pathway occurs after infection with dog RABV variants through a cutaneous lesion (ie, no access to motor endplates) is still unclear, because confi rmatory experiments have not yet been done. Postexposure prophylaxis (PEP) with vaccine alone is needed in patients with minor skin lesions infl icted by a dog infected with RABV.
By contrast, patients with a negligible scratch to the skin infl icted by a bat infected with RABV are at very high risk of infection3 because bat RABV variants, unlike dog RABV variants, are able to multiply in epidermal cells in vitro.37 The higher incidence of local neuropathic pain in patients with bat RABV variant rabies (70%) than in dog-related cases (30%),3,4 and signs and symptoms of Horner’s syndrome and other atypical rabies features, might suggest additional (or alternative) transmission of bat RABV variants via sensory or sympathetic skin innervation, which should be investigated.28
Incubation periodThe incubation period or eclipse phase can vary from weeks to years, but lasts 1–2 months on average. The
incubation period is an intriguing feature in rabies and does not depend on bite location or clinical form.3 During most of the incubation, RABV lies in the muscle as a so-called smouldering, or low-replication-rate, infection38 that remains confi ned to the inoculated portion of the muscle.28,31 Immune recognition at this focal site might not be adequate. Dendritic cells—ie, antigen-presenting cells that have a key role in triggering both innate and adaptive immune responses39—are insuffi ciently activated by naturally acquired (street) RABV infection.40–42 The long incubation period is probably caused by low titre inocula and by the existence of endogenous RNA-silencing mechanisms or microRNAs (miRNAs) that slow down viral replication in the muscle.43 In fact, the muscle-specifi c miRNA mir-133, which is predicted to bind to both the nucleoprotein and glycoprotein transcripts of RABV,44 has been shown to substantially reduce expression of rabies viral protein in transfected Neuro-2a cells.45 With high titre inocula, however, RABV is able to infect motor endplates without previous replication in the muscle, as shown in rodent and primate models;28–31 moreover, with high titre inocula, uptake without previous replication can also occur by motor axons after inoculation directly into nerves.46,47 This fi nding might explain the exceptionally short incubation times in human rabies that usually occur in association with penetrating nerve injury.3
Mechanisms for clinical diversity in furious and paralytic rabies in manBy the time a patient develops the fi rst prodromal symptoms, such as fever, fl u-like symptoms, and gastrointestinal disturbances, the virus is already widely disseminated throughout the CNS.5,6 A more localised prodrome or neuropathic pain (eg, paraesthesia, allodynia, burning sensation) is a sign of dorsal root ganglia dysfunction as a result of immune attack (table).5 A case study of the clinical features of a patient with furious rabies is described in panel 1 and fi gure 1, and of a patient with the paralytic form in panel 2.
Pathways of propagation of RABV and relation with clinical prodromeThe spinal propagation sequence of RABV in man after a deep bite to the left wrist (as in the patient with furious rabies in panel 1 and fi gure 1) has been extrapolated from a combined analysis of studies of RABV propagation in animal models28,29,31,32,38,49–55 (ie, intramuscular inoculation of naturally acquired RABV in skunks38,53 and hamsters,50–52 and intramuscular inoculations of fi xed RABV challenge virus standard [CVS] strain or derived recombinants in rodents29,54,55 and primates28,31,32,49) and knowledge of spinal connectivity (fi gure 2).56–58 The relation between the spinal propagation sequence and prodromal symptoms and signs in the patient with furious rabies described in panel 1 and fi gure 1 is summarised (fi gure 3). Notably, when classic
Panel 1: Clinical features of a patient with furious rabies
A man aged 50 years who had been bitten by a dog on his left wrist 7 weeks earlier5 had prodromal symptoms of severe aching pain and paraesthesias on his left hand and arm (fi gure 1). MRI of the brain, 3 days after onset, showed an ill-defi ned mild hyperintensity change involving the deep and subcortical white matter, hippocampal gyri, brainstem, and cervical cord.6 Slight progression of MRI disturbances was noted on day 7. CSF examination showed two lymphocytes and 40 mg/dL protein. Rabies viral RNA was detected in his saliva 2 days after admission, but not in the CSF specimen.48 He survived for 8 days.

www.thelancet.com/neurology Vol 12 May 2013 501
Review
experimental studies of naturally acquired RABV propagation50–53 are revisited in the context of recent neuroanatomical investigations, taking into account current knowledge on CNS connectivity, the propagation properties of naturally acquired RABV of canine origin seem to be indistinguishable from those of fi xed RABV (CVS strain), which is consistent with retrograde transneuronal transfer via the motor route.
Centripetal propagation From the infected muscles, centripetal propagation of RABV occurs only via the motor route and is mediated exclusively by retrograde transneuronal transfer from motor neurons, which begins 2 days after uptake from motor endplates and proceeds at high speed (12-h intervals for each synaptic step, irrespective of distance).28,49 By day 4 after onset of infection in the motor neurons, connected spinal interneurons and ipsilateral (low cervical and upper thoracic) dorsal root ganglia innervating the bitten arm are heavily infected.53,54 Dorsal root ganglia infection fi rst involves large neurons in dorsal root ganglia (proprioceptive Ia and II aff erents and other large myelinated aff erents targeting infected motor neurons and interneurons; fi gure 2); proprioceptive dorsal root ganglia innervating motor neurons of antagonist muscles are also infected (via Ia inhibitory interneurons). Small dorsal root ganglia neurons (unmyelinated and small myelinated aff erents), which target higher-order interneurons in the dorsal horn, are infected later in the course of infection (fi gure 2). Via spinal interneuronal pathways, the infection rapidly involves bilateral cervical and thoracic dorsal root ganglia supplying the contralateral arm, the neck and back, and, later, lumbosacral dorsal root ganglia (fi gure 2).51,53 In parallel, retrograde transneuronal transfer leads to infection of brainstem and corticospinal pathways targeting the infected spinal motor neurons and interneurons, and higher-order CNS neurons.28,29,32,49,50,53,55
Centrifugal propagationA slow phase of centrifugal (anterograde) propagation can only begin 2 days after replication in each infected neuronal population, and leads to viral transport to the ventral and dorsal roots and centrifugal spread to extraneural organs via their sensory innervations38,50–52,59—ie, to muscle spindles (via large dorsal root ganglia neurons), skin (via cutaneous aff erents from large and small dorsal root ganglia neurons), and to immune and visceral organs, including the salivary glands, heart, and blood vessels (via small dorsal root ganglia neurons). Centrifugal viral propagation to visceral organs59 is probably mediated by dorsal root ganglia neurons that supply, via dichotomising axons, both somatic and visceral nerves and organs (eg, left ulnar nerve and the heart) and several visceral organs.60,61 These pathways form an anatomical substrate for referred pain and cross-organ sensitisation. Centrifugal propagation to extraneural
organs is related to the topography of viscerotopic sensory innervation60 and is distance-dependent; because anterograde axonal transport of RABV is ineffi cient,28,49 it might take weeks to reach remote organs. Progressive functional changes in infected sensory innervation of extraneural organs (including the heart and autonomic plexuses) might explain organ dysfunction and possibly
Panel 2: Clinical features of a patient with paralytic rabies
A man aged 34 years who had been bitten by a dog on his right ankle 2 months earlier had severe itching and piloerection on his right leg that progressed to his left leg within 2 days after presentation.5 On admission (day 3 after onset), he showed no detectable weakness. Refl exes were 1+ (diminished) in the right lower limb and 2+ (normal) in the other limbs. Hypoaesthesia to pinprick sensation was present on his right leg up to the groin. By day 4, paraparesis was noted, and refl exes were absent in both lower limbs and highly diminished in the upper limbs. On day 6, facial diaparesis and bulbar dysfunction were noted, and he was later intubated. He became agitated on day 7 and died on day 9. His CSF was acellular with 70 mg/dL protein. RNA was detected in the CSF on day 3 but not on day 7; saliva was negative for RNA on days 3 and 7.48
A B
Figure 1: Clinical diversity in a patient with furious rabies See panel 1 for background information. On day 3 after onset (A), the patient’s mental state was clear but he had pain in his left arm (arrows). The only abnormality on electrodiagnostic studies was the presence of abundant fi brillations and positive sharp waves in left C5–C8 limb and cervical paraspinal muscles (+). His motor and sensory functions were intact. Diminished-to-absent deep tendon refl exes were noted in the left arm. On day 5 (not shown), sensory nerve action potential (SNAP) amplitudes had reduced by about 50% in the left upper limb nerves compared with those on the right side. Diminished pinprick sensation up to his elbow was noted (shaded area in B) and the pain became intense. By day 6 (B), SNAP amplitudes had reduced further on the left upper extremity, and fi brillations and positive sharp waves involving bilateral C5–C7 limb and paraspinal muscles had progressed (+). Results of motor conduction studies, including F-waves, remained normal. Mild weakness of left hand and wrist muscles was detected. Pain was less severe than in previous days and tolerable. The diminished sensation in the area up to the left elbow (shaded area in B) had progressed slightly, along with absence of deep tendon refl exes and joint position sense of the left arm. He was confused and disoriented (circle in B), and he died on day 8. Reproduced from Hemachudha and colleagues,23 by permission of Springer.

502 www.thelancet.com/neurology Vol 12 May 2013
Review
even dysautonomia in patients with rabies. Although extraneural organs from which RABV can be detected also receive autonomic innervation, the autonomic nerve supply is unlikely to contribute to centrifugal spread. Experimental evidence after RABV inoculation into skeletal muscles shows that autonomic involvement (of spinal preganglionic neurons in the central autonomic area) is a rare and indirect event that is not indicative of peripheral uptake and might only begin quite late, via intraspinal pathways.28,29 Similarly, post-mortem ultrastructural fi ndings in a case report of a patient with furious rabies showed that cytoplasmic virus inclusions were abundant in sensory ganglia but rarely detected in sympathetic ganglia.62
During centrifugal spread, RABV antigen can also be carried to lymph nodes,63 both directly via their sensory innervation64 and indirectly via virus budding from
axons51 and draining of antigen to the lymph nodes. We postulate that high virus load in extraneural organs due to centrifugal spread38,50–52,59 helps dendritic cell activation and migration, triggering T-cell stimulation in lymph nodes and the adaptive immune response. In both cases (ie, direct or indirect transport of virus to lymph nodes), virus and activated dendritic cells will fi rst reach regional lymph nodes, which explains why T-lymphocyte activation occurs earlier in regional lymph nodes than in lymph nodes at other locations.65 Although the clinical stages of rabies are too advanced to enable any defi nite conclusions to be drawn, it is possible in a patient such as the one with furious rabies presented in fi gure 1 and 2 and panel 1, that once RABV has reached spinal motor neurons, it would take 3–4 days to infect the cervical dorsal root ganglia innervating the arm and skin of the neck,51,52 a further 2 days before centrifugal transport in sensory axons begins, another 3 days for transport to neck hair follicles and skin,51,52 and maybe 4 more days before reaching the regional (neck or axillary) lymph nodes. A further 4 days might then pass before T-cell activation in lymph nodes occurs66 and maybe another
Figure 2: Pathways of propagation of RABV to and from the spinal cord The diagram depicts propagation after a deep bite by a rabid animal on the left wrist (as in the patient described in panel 1 and fi gure 1). Spinal cord line drawing (top left) shows location of spinal segments. Fine dashed black lines in main image show borders of spinal laminae (I–X). Rabies virus (RABV) uptake from the muscle occurs exclusively via the motor route (fi rst-order neurons [1°]), with no infection of sensory (proprioceptive dorsal root ganglia) and autonomic neurons innervating the same muscle.28,29,49 Centripetal propagation to the spinal cord is represented by solid blue lines (solid green for antagonists at C8) and coloured circular markers (colour-coded according to synaptic order, see key on left side of fi gure); black arrows show transport direction. RABV propagates exclusively by retrograde transneuronal transfer, from infected motor neurons (1°, grey-coloured circular marker, C8 left) to monosynaptically connected spinal interneurons and dorsal root ganglia populations (second-order neurons [2°], black-coloured circular markers).29,54,55 Dorsal root ganglia populations include: large proprioceptive dorsal root ganglia neurons (group Ia and II aff erents) of low cervical or fi rst thoracic segments ipsilaterally;54,56,57 short propriospinal neurons ipsilaterally in laminae V–VII (lateral part) of cervical or upper thoracic segments, which receive both dorsal root ganglia aff erents and corticospinal, rubrospinal, and reticulospinal inputs and innervate arm muscles ipsilaterally;56,57 and long propriospinal neurons bilaterally in laminae VII, VIII, and X (including cholinergic populations in lamina X29 and partition cells54), which connect nearly the entire length of the spinal cord and innervate motor neurons of axial, proximal, and distal muscles bilaterally. The long propriospinal neurons receive both dorsal root ganglia aff erents and reticulospinal, vestibulospinal, and corticospinal pathways, are involved in a variety of refl ex pathways, and are part of spinal locomotor networks.56–58 Among 2° in lamina VII are Renshaw cells, Ia inhibitory interneurons, and Ib and II interneurons (in VII–VI). Further steps of retrograde transneuronal transfer from 2° result in infection of higher-order interneurons and dorsal root ganglia populations. Proprioceptive dorsal root ganglia innervating motor neurons of antagonist muscles (3°, solid green pathway at C8) are also infected (via Ia interneurons, 2°, which are pathways for reciprocal inhibition). Centrifugal (anterograde) propagation of RABV (open coloured circular markers and dashed red lines, or dashed green lines for antagonists at C8) is a slow (distance-dependent) process (see key on right side of fi gure), causing infection of other interneurons and motor neurons. Centrifugal transport of RABV from the spinal cord occurs in motor and sensory axons50–52 (large red arrows), leading to infection of extraneural organs via their sensory innervation.38,50–52,59 DRG=dorsal root ganglia. Ia IN=Ia inhibitory interneurons. IML=intermediolateral cell group. LPN=long propriospinal neurons. MN=motor neuron. pc=partition cell. rc=Renshaw cells. SPN=short propriospinal neurons.
I
L4
C3
C8
T4
2°3°4°5°6°
2.5 days3 days3.5 days4 days4.5 days
1° 2 days4.5 days5 days5.5 days6 days6.5 days
4 days
RABV
Muscle
DRG
2° 3°/5°
Sympatheticganglion
2°
2°
2°
2°
No uptake
No uptake
MN
MN
MN
MN
uptake
3°
4°
2°
3°
4°
3°
Ia IN
DRG
DRG
DRG
2°
MN1°
2°
IIIII
IVV
VI
VII
VIII
X
rc VIII
pc
rc
Ia IN
IIIIIIIV
V
VIIVIII
IMLX
IX
IIIIII
IVV
VII
VIII
X
IX
IX
VI
III
III
VVI
VII
VIII
X
IX
VII
IV
IX
IX
VIII
2°
DRG
DRG
DRG
DRG
5°
3°
4°
4°
LPN
SPN
3°
I
Temporal sequence of centripetal propagation
Onset of centrifugal propagation
L4
T4
C3
C8

www.thelancet.com/neurology Vol 12 May 2013 503
Review
3 days for ganglionopathy and neuropathic pain to begin (prodrome onset). The total estimated period (3 weeks) is well below the 7-week incubation in the patient described in fi gure 1 and panel 1, suggesting that he might have had low-level infection in his muscles for up to 4 weeks. The extensive centripetal and centrifugal viral propagation that had already occurred in this patient during incubation was shown by MRI changes (panel 1) and recovery of virus from his saliva at prodrome onset, which is mediated only by centrifugal spread to the salivary glands via their innervation.28,67
The sequence of prodromal symptoms and signs closely parallels the proposed viral propagation sequence. Motor neurons and connected proprioceptive dorsal root ganglia supplying the bitten arm are the fi rst nerve cells to be infected and the fi rst to show progressive functional changes (fi gures 2 and 3). Preferential entry via the motor route could explain why denervation potentials (detectable by day 3) precede sensory loss (day 5) and are initially localised to the bitten arm and ipsilateral neck paraspinal muscles (by day 3) but extend to the opposite arm and paraspinal muscles by day 6. Similarly, early and heavy infection of proprioceptive (Ia) dorsal root ganglia of agonist and antagonist muscles of the bitten arm (see above and fi gure 2), which mediate deep tendon refl exes, might explain the diminished deep tendon refl exes of the same arm by day 3 and their subsequent extinction (fi gures 1 and 3). Late onset of infection of cutaneous (vs proprioceptive) dorsal root ganglia might explain why sensory function in the bitten arm is still intact on day 3, but aff ected by day 5 (fi gure 3). Finally, focal defi cits (eg, weakness of left hand or wrist muscles, loss of deep
tendon refl exes and position sense, pain attenuation) by day 6 might signify progressive neuronal dysfunction and ganglionopathy. The same logic might also apply to prodromal symptoms and signs in patients with paralytic rabies. An association between the location of the bite and the topography of prodromal symptoms and signs has been noted, which could be explained by RABV propagation in spinal pathways and dorsal root ganglia that are synaptically connected with motor neurons innervating the bitten limb. For example, in the patient with paralytic rabies described in panel 2, RABV propagation from motor neurons supplying the bitten right ankle to connected spinal interneurons and (lumbosacral) dorsal root ganglia might explain why pain, diminished deep tendon refl exes, and hypoaesthesia are initially localised to the right leg. Preferential infection via the motor route might also explain why Wallerian-like degeneration and infl ammatory changes were much more severe in the ventral than in the dorsal spinal roots in a patient with paralytic rabies.68
Death in furious rabies: immune-mediated or virus-mediated mechanism?The immune response to RABV has been hypothesised to contribute to the disease process, on the basis of experimental evidence that survival can be prolonged in immunosuppressed mice, whereas paralysis and death ensue with the return of immune responsiveness after passive transfer of immune serum or cells.19 The fi nding that patients who had cellular immunity to RABV antigen and raised serum cytokine concentrations tended to have furious rather than paralytic rabies19,69 led to the belief
Figure 3: Association between prodromal symptoms and signs and spinal propagation of rabies virus in a patient with furious rabies1°=fi rst-order neuron. 2°=second-order neuron. 3°=third-order neuron. 4°=fourth-order neuron. 5°=fi fth-order neuron. DRG=dorsal root ganglia. SNAP=sensory nerve action potential.
Motor neurons(1° )
Proprioceptive DRG supplying the left arm (cervical-upper thoracic cord)(2°–3°)
Cutaneous and nociceptive DRG (4°–5°)
3 daysDenervation potentials at left C5–C8 arm and cervical paraspinal muscles
Diminished deep tendon reflexes in left arm
Pain in left arm
Pain in left armbecame intense
50% reduction of SNAP amplitudes in left arm
Diminished pinprick sensation up to the left elbow
Pain attenuationFurther reduction of SNAP amplitudes in left arm
6 daysDenervation potentials bilaterally in the arms and paraspinal musclesMild weakness of left hand and wrist muscles
Diminished sensation extending above the left elbow
Loss of deep tendon reflexes and joint position sense in left arm
5 days
2 days
2·5–3 days
3·5–4 days
Furious rabies in a man, aged 50 years, bitten by a dog on the left wrist 7 weeks earlier Sequence of symptoms and signs (number of days from prodrome)
Sequence of infection of motor neurons and sensory neurons (DRG)
innervating the bitten left arm(number of days after uptake of rabies
virus from motor endplates)

504 www.thelancet.com/neurology Vol 12 May 2013
Review
that furious manifestations are immune-mediated (and associated with production of proinfl ammatory cytokines and nitric oxide),3 and that vaccination should be withheld in patients with symptomatic rabies because it might accelerate death.70,71 However, accumulating evidence seems to prove otherwise. Attempts to prolong the clinical course of patients with rabies with high-dose steroids, antithymocyte globulin, or other immuno-suppressive drugs have failed.14,72 Moreover, studies in dogs with early-stage furious or paralytic rabies did not show an exaggerated immune response in infected brains.8 On the contrary, cytokine and chemokine mRNA transcripts were barely detectable in the brain, particularly in dogs with furious rabies.8 A greater amount of rabies viral RNA was reported in the brains of
furious dogs than in those of paralytic dogs, suggesting that furious manifestations are virus-mediated and associated with more extensive propagation of RABV to the brain than are paralytic manifestations.
Impaired neural tract integrity at brainstem and survival in paralytic rabiesIn-vivo MRI examination showed greater signal abnormalities in the brainstem, hypothalamus, and temporal lobe of dogs with paralytic rabies than in the same parts of the brain in dogs with furious rabies, despite paralytic dogs having a lower viral titre overall (table and fi gure 4).8 No evidence of blood–brain barrier leakage was reported in dogs with either furious or paralytic rabies. MRI studies showed that abnormalities in the brainstem were prominent in paralytic dogs with early-stage rabies,7,8 which might suggest that viral propagation is interrupted in paralytic dogs, resulting in less virus reaching the cerebral hemispheres. Post-mortem examination of brain (fi ve furious and fi ve paralytic cases) and spinal cord specimens (three of each clinical form) was done in ten naturally infected rabid dogs, including some that had MRI.9 To observe early pathological changes, all animals were killed shortly after developing signs of rabies. In both clinical forms, caudal–rostral distribution of RABV antigen, from greatest to least, was reported in the following order: spinal cord, brainstem, cerebellum, midline structures (caudate, thalamus), hippocampus, and cerebrum. By contrast, RABV RNA was most abundant in cerebral midline structures. More RABV antigen was detected in many more CNS regions in dogs with furious rabies than in those with paralytic rabies. Signifi cantly higher RNA concentrations were noted in the cerebral cortex, thalamus, midbrain, and medulla of dogs with the furious subtype than in those with the paralytic subtype, whereas RNA concentrations in the spinal cord were similar in both clinical forms. Infl ammation reported in the brainstem of dogs with paralytic rabies substantially correlated with increased MRI disturbances and increased extent of CNS immune response (table and fi gure 4).7–9 Thus, brainstem infl ammation might impede viral propagation towards the cerebrum, resulting in longer survival in patients with paralytic rabies.
Diff usion tensor imaging (DTI) provides quantitative values compared with normal controls and has greater sensitivity than basic MRI for the detection of microstructural and macrostructural damage (neural tract integrity), as shown by diminished fractional anisotropy and increased fl uid-attenuated inversion recovery (FLAIR) signals, respectively,7 and it is useful for the assessment of blood–brain barrier status. Our preliminary DTI data in rabid dogs suggest similar fi ndings to those seen on MRI, with evidence of compromised neural tract integrity in the brainstems of paralytic dogs.7 Mean diff usivity or blood–brain barrier leakage was not increased in either furious or paralytic
C
A
E
D
B
F
**
*
** * *
Figure 4: MRI in canine rabies Images are mid-sagittal fl uid-attenuated inversion recovery (FLAIR) MRI images of the hypothalamus, midbrain, brainstem, cerebellum, and upper cervical cord of a dog with early-stage paralytic rabies (A and C) and a dog with early-stage furious rabies (B and D). Also shown are coronal FLAIR images of the temporal lobe, frontal lobe, thalamus/hypothalamus, and basal ganglion of a dog with early-stage paralytic rabies (E) and a dog with early-stage furious rabies (F). The dog with paralytic rabies has moderate-to-substantial abnormal hypersignal T2 change at the hypothalamus (green arrow in A and E), dorsal midbrain (white arrow in A), brainstem (asterisk in A), and upper cervical cord (double white arrows in C). Also noted is moderate hypersignal T2 change at bilateral temporal lobes (asterisks in E) sparing the frontal and parietal lobes. The dog with furious rabies has less well defi ned mild-to-moderate diff use hypersignal T2 change involving the hypothalamus, midbrain, brainstem, and upper cervical cord (white arrows in B, D, and F) and cerebellum (asterisk in B). It has more diff use but less well defi ned mild hypersignal T2 change involving the temporal lobes, frontal lobes, and parietal lobes (asterisks in F) than the paralytic dog. Reproduced from Laothamatas and colleagues,8 by permission of Informa Healthcare.

www.thelancet.com/neurology Vol 12 May 2013 505
Review
dogs with early-stage rabies. However, cytotoxic brain oedema (decreased mean diff usivity) was noted almost exclusively in paralytic dogs.7
Specifi c virus variants in furious and paralytic rabiesOne hypothesis to explain clinical diversities in rabies would be the presence of furious and paralytic virus variants. However, a bite from the same dog was shown to have caused furious rabies in one patient and the paralytic form in another, which provides evidence against this hypothesis,19 although, theoretically, this fi nding would not rule out viral pathogenicity changes due to spontaneous point mutations after exposure. Mutations of the glycoprotein gene at amino acid position 333 (Arg) abolish virulence,73 whereas a substitution in the same gene at 194 (Asn) enhances pathogenicity.74 Other substitutions at positions 318 (Phe) and 352 (His), related to p75 neurotrophin receptor binding, might aff ect viral maturation and transport into the cell,35,75 whereas substitutions at positions 273 (Glu) and 394 (Gln) of the RABV nucleoprotein gene enhance immune evasion and increase pathogenicity.76
Analysis of RABV nucleoprotein, phosphoprotein, and glycoprotein genes from samples obtained from patients with either furious or paralytic rabies did not show specifi c patterns.77 Mutations in the RABV samples did not diff er between the two groups at positions 333 (Arg), 194 (Asn), 318 (Phe), or 352 (His) of the G gene, at positions 273 (Gln) and 394 (Glu) of the N protein gene, or at the carboxyl-terminal PDZ domain-binding motif, which is responsible for neuronal survival and apoptosis.78
However, this does not rule out the theory that mutations in the virus explain clinical diversity, because only the sequences of the glycoprotein, nucleoprotein, and phosphoprotein genes were analysed in brainstem samples, whereas other genes involved in pathogenicity (eg, the matrix protein gene) were not examined.79,80
Poor immune response in rabies-infected CNSThe CNS cannot mount an adaptive immune response to RABV because it does not contain any primary immune organs (ie, no antigen presenting cells and classic lymphatic drainage); the adaptive immune response has to be triggered in the periphery. Yet, infected neurons and glial cells are able to mount innate antiviral type I interferon (α or β) and infl ammatory cytokine responses after recognition of viral RNA by two classes of innate sensors: the endosomal transmembrane Toll-like receptors (TLRs) 3 and 7/8, and the cytoplasmic retinoic acid inducible gene 1-like helicases RIG-I (also known as DDX58) and MDA5 (also known as IFIH1).81–83 Interferon α and interferon β exert antiviral functions via JAK/STAT signal transduction pathways,84 whereas the major transcription factor for proinfl ammatory cytokines, such as tumour necrosis factor α (TNFα) and interleukins, is nuclear factor κB (NF-κB), which also supports early interferon transcription (interferon β, interferon α4).83,85
Despite being attacked by diff erent defence mechanisms, RABV successfully invades its host and reaches the brain (fi gure 5). Although neurons have the machinery to sense RABV infection and trigger innate immune responses,86 the virus has evolved several strategies to escape or lower activation of innate sensors and the antiviral eff ects of interferons. These strategies might allow preservation of neuronal integrity so that the virus can propagate between neurons. Human brain neurons express the innate sensor TLR3,87 which can trigger cell death in the presence of interferons.88 However, the virus uses TLR3 as an evasive strategy,10 by sequestering it in Negri bodies where it plays a part in RABV multiplication (fi gure 5).89 The presence of RABV also activates the innate sensors RIG-I and MDA590,91 and triggers type 1 interferon (via activation of interferon regulatory factor [IRF] 3 and 7 in association with activator protein 1 [AP-1] and NF-κB). However, the RABV phosphoprotein counteracts interferon antiviral eff ects, because it prevents transcription of interferon α/β genes by blocking phosphorylation of IRF3 and IRF7 by the kinases TBK1 and IKK-i (also known as IKBKE)92 and it also inhibits STAT signalling, the pathway by which interferon exerts antiviral activity,83,93–96 whereas activation of NF-κB is not disturbed (fi gure 5). The virus nucleoprotein (amino acid positions 273 and 394) is also important for evasion of host RIG-I-mediated antiviral response.76 Thus, interferon induction in RABV-infected CNS is reduced97 and neuroinfl ammation is moderate.
RABV is also able to evade the adaptive immune response in the CNS. Activation and entry into the CNS are not limiting factors for T cells and monocytes, which can infi ltrate the CNS despite an intact blood–brain barrier (fi gure 5). After mice were infected with a virulent RABV strain, their brains were infi ltrated with T cells expressing a marker of activation (Cd69) and Crmp2 (also known as Dpysl2), a marker of T-cell polarisation and migration.98 Invading T cells and monocytes, however, undergo apoptosis shortly after entry into the infected brain parenchyma (fi gure 5). Post-mortem immuno-histochemical studies of the brains of human beings with rabies revealed that leucocytes were the only cells undergoing apoptosis.99 In post-mortem human brains infected with a vampire bat RABV variant, only infi ltrating adaptive immune T cells (CD4+ and CD8+) were apoptotic, whereas natural killer cells, macrophages, astrocytes, or neurons were not.100 Paradoxically, RABV uses the innate immune response to induce apoptosis of infi ltrating T cells. This strategy was demonstrated in a transgenic mouse model overexpressing Lgp2 (also known as Dhx58) to impair the RIG-I-mediated innate immune response; after RABV infection, lower morbidity and more viral clearance in the brain were noted in mice overexpressing Lgp2 than in a C57BL/6 strain of mice, with reduction of infi ltrating CD4+ T cells but less disappearance of infi ltrating CD8+ T cells,101 showing that host innate immune response favours the infi ltration of

506 www.thelancet.com/neurology Vol 12 May 2013
Review
T cells but simultaneously promotes CD8+ T-cell elimination. Elimination of infi ltrating T cells is mediated by upregulation of immunosubversive molecules on the neuronal surface, such as B7-H1 (also known as CD274)102 and HLA-G,103,104 which are interferon dependent, and Fas ligand, which is interferon independent.97,105 Surface expression of B7-H1, HLA-G, and Fas ligand by infected neurons triggers death of migrating T cells expressing the corresponding receptors (fi gure 5).97
Although an intact blood–brain barrier in rabies is not an obstacle for migration of activated lymphocytes into the CNS, effi cient entry, particularly of B cells, needs disruption of the blood–brain barrier.106–108 Failure to open
the blood–brain barrier and deliver immune eff ectors was a crucial factor in the lethality of the virus in mice infected with a silver-haired bat RABV variant.107 Intrathecal production of rabies antibody by infi ltrated B cells is needed for viral clearance from the CNS in mice infected with attenuated RABV.106
Neuroinfl ammation might inhibit the neuro-invasiveness (ie, ability to invade the CNS) of RABV. Comparative analysis of cytokine and chemokine mRNA transcripts in 12 brain regions of dogs with naturally acquired, early-stage rabies revealed greater infl ammatory cytokine transcription in the dogs with paralytic rabies than in those with the furious form (table).8 Interferon γ and interleukin 1β were detected exclusively in paralytic dogs. Granulocyte-macrophage colony-stimulating factor and interleukins 2, 4, 5, 8, and 10 were not detected in dogs with furious rabies. TNFα was detected in dogs with furious rabies and in those with the paralytic form, but was considered signifi cantly increased only in those with paralytic rabies, whereas only CCL2 and VEGF were signifi cantly increased in dogs with furious rabies. Interleukin 6, TNFα, interleukin 1α, interleukin 1β, and interferon γ cytokines have been reported to increase blood–brain barrier permeability in vitro and in vivo (by direct brain injection into piglets and rats) and to augment leucocyte CNS infi ltration.109 In the dogs depicted in fi gure 4, the blood–brain barrier remained intact despite the presence of cytokines. Infl ammatory cytokine trans-cription was associated with a lower viral load (in all CNS regions) in paralytic dogs than in furious dogs. Greater disturbances seen with MRI in dogs with paralytic rabies were associated with greater immune responses and less brain neuroinvasiveness (fi gure 4) than in furious dogs.8 During the late stage of disease, however, almost no cytokines were detected in the CNS in both rabies types, and similar viral quantities were detected in both forms.8
Proteomic profi ling studies of the brains of dogs with late-stage rabies showed upregulation of immuno-globulin heavy chain in the brainstem of paralytic dogs and interferon α4 and SARM1 protein in the hippocampus of dogs with furious rabies.110 CRMP-2, a marker of activated infi ltrating T cells, was down-regulated in the spinal cord in both rabies forms, but was upregulated in the brainstem of dogs with paralytic rabies. Examination of 25 brains of patients with furious and paralytic rabies revealed no correlation between infl ammation or viral antigen distribution and expression of interleukin 1β and TNFα (in microglia, macrophages, and lymphocytes).21 This might be explained by advanced stage of disease at examination.
A case study111 described a patient infected with a bat RABV variant who received treatment with a neuroprotective regimen of midazolam, ketamine, and propofol together with rabies immunoglobulin, ribavirin, and amantadine; the regimen was discontinued 48 days later because no neurological recovery occurred, and the patient died more than 2 months after disease onset.111
Figure 5: RABV immune-evasion mechanismsThe diagram depicts rabies virus (RABV) inhibition of innate immune response (A) and evasion of adaptive immune response (B) in the CNS. In infected neurons, RABV prevents activation of innate sensor Toll-like receptor 3 (TLR3) by sequestering it in Negri bodies (A). In the neuronal cytoplasm, viral RNA is recognised by immune sensors RIG-I and MDA5 (A, centre-left), which normally triggers transcription of innate antiviral type I interferon α/β via formation of a protein complex (not shown, including adaptor protein IPS-1 together with TRAF3, TBKBP1, NAP1, TANK, TRADD, RIP1, and FADD) that triggers phosphorylation of IRF3/7 by TBK1/IKK-i. Phosphorylated IRF3/7 is transported to the nucleus to induce transcription of interferon α/β in conjunction with activator protein (AP-1) and nuclear factor κB (NF-κB); AP-1 is activated via mitogen-activated protein kinases and NF-κB by NEMO (not shown). The RABV phosphoprotein (P, in red circle) alters transcription of interferon α/β genes by blocking phosphorylation of IRF3/7 and its nuclear import (red crosses), while activation of AP-1 and NF-κB is not disturbed. The RABV phosphoprotein also inhibits the STAT signalling pathway. Interferon α/β binds to cell surface receptors (grey, IFNAR1/2) and induces STAT phosphorylation (p) by kinases JAK1/TYK2 (not shown); phosphorylated STAT/IRF9 complex is imported to the nucleus, where it activates interferon-stimulated genes (ISGs) that have antiviral activities. The RABV phosphoprotein prevents transcription of ISGs by binding to phosphorylated STAT in the cytoplasm, blocking its nuclear import, and by binding to the STAT/IRF9 complex in the nucleus (mediated by a shorter version of phosphoprotein).83 Activated T cells and monocytes can cross the blood–brain barrier and invade the CNS (B), despite intactness of this barrier in patients with rabies. Surface expression of the immunosubversive molecules B7-H1, HLA-G, and Fas ligand (dark blue markers) by infected neurons leads to binding of T cells expressing the corresponding ligands, rapidly followed by death of migrating T cells (red cross).10
Cytoplasm
Activated T lymphocytes
Blood–brain barrier
Death of migratory T lymphocytes
TLR3 sequestration
in Negri bodies
Infected neuron
Surface expression of B7-H1, HLA-G,
Fas ligand
RIG-I MDA5NFκBAP-1
A Rabies virus inhibition of innate immune response B Evasion of adaptive immune response
STAT
Interferon α/β
ISGs
Infected neuron
NucleusNFκBAP-1
p
RABV RNA
TBK1/IKK-i
P
IRF3/7
p
IRF9
P
P
STAT
IRF9
IRF3/7
Interferon α/β
IFNAR1/2

www.thelancet.com/neurology Vol 12 May 2013 507
Review
CSF analysis before death (3 weeks after cessation of the regimen, which is potentially immunosuppressive) and post-mortem brain examin ation showed robust infl ammatory responses in the brain and CSF. The investigators propose that such a response at a very late stage might be related to immune reconstitution infl ammatory syndrome.111
Preservation of neuronal integrityRABV is transcribed and replicates in neuronal cell bodies and dendrites. Maintenance and preservation of these structures and the axon, where the complete enveloped particle is transported, might be as important for the virus as its ability to evade immune responses.10 RABV infection causes neuronal dysfunction rather than neuronal death.112,113 Survival of infected neurons is ensured by the capacity of virulent RABV strains to prevent apoptosis by maintaining viral gene expression below threshold levels and by interfering with proapoptotic factors.73,112,114 Prevention of apoptosis is the hallmark of naturally acquired RABV infection; it depends on restricted expression of the glycoprotein protein and the glycoprotein gene sequence, including four amino acids at the carboxyl-terminal PDZ domain-binding motif that can bind cellular PDZ proteins, which control cell polarity and apoptosis.73,78,115 The matrix protein amino acid residues Arg 77 and Glu 81 might be associated with delayed apoptosis and increased pathogenicity of naturally acquired RABV.80 Thus, neurons that have been infected with RABV for several days (asymptomatic period) do not exhibit cytopathic changes and remain metabolically viable in vivo, expressing their neurotransmitters and transporting other markers.29–31,33,49,116 Yet, when severe clinical disease has developed, wild-type RABV infection in mice results in changes to host protein expression, particularly expression of proteins involved in ion homoeostasis and docking and fusion of synaptic vesicles to presynaptic membranes,117 which might lead to the defective neurotransmission recognised in rabies.113 Neuronal dysfunction might also be related to oxidative damage.118,119 In moribund mice, mild structural damage involving exclusively neuronal processes (beading and fragmentation of axons and dendrites, with vacuoles that correspond with swollen mitochondria) was recognised;120 it was shown in vitro to be the result of oxidative stress, through virus-induced inhibition of NF-κB signalling, which plays a crucial role in axonal growth, neuronal survival, and antiviral responses.118,119
Studies of human brains infected with naturally acquired RABV show that the virus is capable of preserving neuronal integrity to support its propagation. Apoptosis was evident in infl ammatory cells but not in neurons.99,121 Cytochrome c leakage and evidence of mitochondrial membrane permeabilisation were absent in spinal cord and brainstem in patients with rabies, explaining the absence of anterior horn cell weakness and preservation of consciousness until the preterminal stage.122
Pathogenesis of paralytic rabies in man Peripheral nerve dysfunction, axonopathy, or myelinopathy causes weakness in patients with paralytic rabies.2,68,123 Axonopathy was reported in three patients with paralytic rabies (one Chinese, one Mexican, and one Thai, infected in their respective countries) who had an acute motor axonal Guillain-Barré syndrome variant.5,22,68 Wallerian-like degeneration and infl ammatory changes were more abundant in the ventral than in the dorsal nerve roots in the Chinese patient.68 Deposition of IgG and complement proteins on RABV-positive axons was also shown, suggesting an antibody-mediated complement attack. In support of this fi nding, one patient with furious rabies who received intravenous human rabies immunoglobulin developed weakness of the facial, limb, and neck fl exor muscles 36 h after administration.124 However, the mechanism is unclear, since CSF rabies neutralising antibodies were not detected in this patient, or in another 30 Thai patients infected with dog RABV variants, 14 of whom had paralytic rabies.2 Antiglycolipid antibodies were not detected in one patient with axonal and two with demyelinating paralytic rabies;5 this fi nding is important because the exact pathogenic mechanism in paralytic rabies is unknown, and one hypothesis is that the humoral immune response plays a part.
Segmental demyelination and remyelination, or myelinated nerve fi bre loss of the spinal roots and peripheral nerves, were characteristic fi ndings in 11 patients with paralytic rabies.123 None of these patients had Wallerian-like degeneration as the only pathological feature. Such demyelination was absent in patients with furious rabies.125 Another two patients with electrophysiological evidence of demyelinating Guillain-Barré syndrome variants had infl ammation of the dorsal and ventral spinal nerve roots that was more severe than in the spinal cord.5
Neuroimaging and molecular techniques in antemortem diagnosisMRI abnormalities provide clues for diff erential diagnosis with other encephalitides, in terms of preferential sites and extent of involvement (brain only or whole neuroaxis), presence of oedema (cytotoxic or vasogenic) or minute haemorrhage, and extent of signal intensity.7 MRI of patients with rabies can vary, since abnormalities can result from infection, host reaction, or complications (hypoxia, shock, bleeding, and metabolic derangements).3,5–7,126,127 Typically, MRI abnormalities are hypersignal T2 changes without contrast enhancement involving the spinal cord, brainstem, thalamus, limbic structures, and white matter during the non-comatose phase (fi gure 6). Both clinical forms of rabies in man have similar MRI features (table).6,7 Lesions in the brachial plexus, spinal cord, and nerve roots are already seen at the prodromal stage as signal intensity abnormalities or enhancement.6 During the comatose phase, widespread

508 www.thelancet.com/neurology Vol 12 May 2013
Review
T2 hyperintense lesions in the brainstem and forebrain can be seen; these are probably due to virus-induced neuronal injury and superimposed hypoxic insult.7 Neuronal injury might be related to oxidative stress, as shown in an animal model infected with fi xed RABV (CVS strain).118 Blood–brain barrier breakdown is evident only during the late or comatose phase of the disease, as gadolinium-enhanced lesions along the brainstem and other midline structures.6,7 MRI images are similar in patients with dog or bat RABV variants in terms of location and pattern of abnormal signal intensity.22,128,129 MRI might help with diagnosis when it is combined with clinical data and staging of disease severity and with knowledge of whether the patient is also aff ected with metabolic, electrolyte, or haematological disturbances or has a compromised cardiovascular status. Lesion characteristics at diff erent stages have been reviewed in detail elsewhere.7
Antibody assay might not be useful in the diagnosis of rabies associated with dog RABV variants. RABV neutralising antibody in serum (and not in CSF) was detected in only six of 31 Thai patients and in none of 43 unvaccinated patients with dog RABV variants from Cambodia, Madagascar, and Senegal.3,130 Immune
recognition of rabies nucleoprotein and neutralising antibody titre development is inadequate in these patients.131 21 of 43 patients in the USA (1960–2009) had neutralising serum antibody; 18 were infected with bat RABV variants, two with dog variants, and one with a bobcat variant.13 Only recently have antibody assays been considered in dog RABV variant endemic countries because of the potential to assess chance of recovery, particularly when viral RNA is not detected.15–18 However, patients with neutralising antibody in their serum or CSF but no detected viral RNA have died despite intensive care support.13
RABV RNA can be detected in saliva, extracted hair follicles or a biopsy sample of skin tissue from the nape of the neck that contains hair follicles, CSF, and urine.48,130,132–134 Descriptions of the biological samples and molecular methods for detection have been detailed elsewhere.135 Frequently used techniques, such as hemi-nested reverse transcription (RT) PCR (targeting the large polymerase [L] gene) and TaqMan real-time RT-PCR (nucleoprotein gene), had similar degrees of sensitivity when serial dilutions of purifi ed RNA from RABV-infected dog brain tissue were used.136 When
Figure 6: MRI in patients with rabiesImages depict a conscious patient with furious rabies (A–C) and a comatose patient with paralytic rabies (D–F). (A) Coronal spin echo T2-weighted MRI of the brain showing diff use ill-defi ned mild hypersignal T2 change involving the cerebral white matter and brainstem (asterisks), hippocampi, and temporal lobes (white arrows). (B) Ill-defi ned hypersignal T2 change seen in spinal cord (white arrow). (C) Enhancing left brachial plexus (white arrow) between the anterior and middle/posterior scalene muscles of the bitten limb. (D) Marked diff use hypersignal T2 change involving the basal ganglia, hippocampi (asterisks), and cerebral white matter (green arrow). (E) Moderate ill-defi ned enhancing hypothalamus (green arrow), tectal plate and midbrain (white arrowhead), and the dorsal pons and medulla and anterior cervical cord (white arrows). (F) Moderate enhancement of the intrathecal ventral and dorsal nerve roots (white arrows). Reproduced from Laothamatas and colleagues,7 by permission of Elsevier.
CA
ED
B
F
**
*
*
*

www.thelancet.com/neurology Vol 12 May 2013 509
Review
previously positive clinical samples (saliva) from ten patients with rabies, and RABV-infected dog and human brain tissue, were tested, results with each technique were similar.136 However, the volume of tissue might be crucial for adequate sensitivity.136 Hemi-nested RT-PCR assay of nuchal skin biopsy specimens containing hair follicles (diameter roughly 4 mm; total volume 20 mm³) is almost 100% sensitive.130 Similar sensitivity was obtained when at least three serial saliva samples were examined or when three types of specimen (saliva, CSF, urine, or hair follicles) were assayed simultaneously.134,136 This result, however, might apply with certainty only to patients with furious rabies, since results were negative in half (three of six) of patients with the paralytic form.134 Negative results were higher in patients with paralytic rabies than in those with furious rabies: saliva samples (seven of nine (78%) vs eight of 53 [15%]), CSF (three of fi ve [60%] vs 14 of 25 [56%]), urine (fi ve of fi ve (100%) vs 20 of 36 [56%]).134 Hair follicle tests showed negative results in 12 of 25 (48%) patients with the furious form compared with one of one with the paralytic form.134
ManagementRecovery after rabies has been reported in four patients.15–18 Prediction of which patients are likely to recover is not possible, although most survivors with good functional recovery had bat RABV variant rabies.15–18 Patients infected with bat RABV variants (who died or survived) had clinical manifestations that diff ered in many respects from those who have survived classic forms of dog RABV variant rabies.3,4,13,23 Diff erences in cellular tropism37 or in the routes of spread, or both, might account for these discrepancies.28 Because only a few experimental studies are available of the propagation of bat RABV variants in vivo,37,137 it is unclear whether higher chances of survival in patients with bat RABV variant rabies might be related to diff erences in the modalities of propagation of bat versus dog RABV variants. Importantly, all four rabies survivors described in the scientifi c literature with good recovery, with or without treatment, had a vigorous and early immune response, with autosterilisation (ie, no detected virus or RNA in tissue or biological fl uids) and rabies antibodies detected in serum and CSF.13–18 Two received coma induction therapy,16,18 one had standard intensive care support,15 and another had presumptive abortive infection17 and did not receive any intensive support. Two patients, one of whom did not receive coma induction therapy, had non-neutralising antibodies, suggesting that other mechanisms played a part in eradication of the virus.17,18
The suff ering that patients with furious rabies go through cannot be readily explained by pain in the throat or phobic spasms. We have routinely used benzodiazepines, barbiturates, ketamine, or even intravenous morphine. However, deepening of consciousness by the use of sedatives to the extent that ventilatory support is needed should be avoided.
So far, no proven standard treatment for rabies exists. Combination therapy with rabies immunoglobulin (polyclonal or monoclonal) plus vaccination, ribavirin (antiviral drug), ketamine (with some NMDA receptor antagonistic eff ect), and interferon α has been advocated.138 Large doses of intravenous human rabies immunoglobulin (25 g for 4 consecutive days) were given to a patient with furious rabies. Anti-rabies antibody was not detected in the CSF after treatment, confi rming the intactness of the blood–brain barrier;125 however, this treatment was able to attenuate the autonomic symptoms.
The Milwaukee protocol initially aimed to induce coma with an electroencephalographic stage of burst suppression.16 Various sedatives (midazolam, bar-biturates, ketamine), amantadine, which is supposed to reduce brain excitotoxicity,139 and ribavirin were given to a patient who then recovered with minimal sequelae.16 However, following the protocol did not save more than two dozen fully alert, previously healthy, young, or middle-aged patients with symptomatic rabies.12,14,129,140–142 The role of excitotoxic mechanisms and the benefi t of ketamine in in-vitro and in-vivo experiments are not supported by the data and scientifi c evidence.12,14,140,143,144 The current Milwaukee protocol consists of ketamine and midazolam, similar to what is used by physicians in dog RABV variant endemic countries to relieve suff ering and dysautonomia. Coma induction is no longer recommended in the protocol. Nimodipine has also been added to the protocol to relieve vasospasm.145,146 However, data have been confl icting; neuroimaging and post-mortem examination did not reveal fi ndings compatible with territorial spasm in patients with dog RABV variant rabies.6,7,147 Whether territorial spasm might be specifi cally related to bat RABV variants is not known. In view of autonomic disturbances in patients with rabies, nimodipine should be administered with extreme caution because it potentially poses the risk of severe hypotension and shock.3
Conclusions and future directionsRabies is unique because it carries the highest fatality rate among all viral encephalitides. The mechanisms that
Search strategy and selection criteria
We searched PubMed for English language articles published from 1967, to Dec 1, 2012, containing the terms “rabies” in conjunction with other key terms, including “encephalitis”, “human”, “virus”, “pathophysiology”, “pathology”, “treatment”, “propagation”, and “transneuronal”. Data for this review also came from references on neural connectivity and those contained within older relevant review articles. Most of the review articles that we have cited contain clinical data of many individual cases or case series, as well as many original references of basic science studies and experiments in animal models.

510 www.thelancet.com/neurology Vol 12 May 2013
Review
allow this virus to invade and partly hide from the host’s immune defences, sometimes for extensive periods of time, before overwhelming the host are fascinating for scientists and clinicians.
Improved understanding of the mechanisms underlying rabies neuropathogenesis in man and animal models is necessary for the development of new therapeutic approaches. Temporary blood–brain barrier disruption by the use of ultrasound and microbubbles, and therapeutic miRNAs or nanoengineered molecules, together with generalised or regional brain cooling methods are prospective treatment options.14,45,148,149 A live-attenuated triple-glycoprotein RABV variant is a promising vaccine candidate for both pre-exposure and postexposure prophylaxis of rabies because it induces immune mechanisms capable of containing experimental CNS infection with pathogenic RABV.150,151 Any new drugs or treatment protocol should be proven not to pose any potentially harmful risk to already seriously ill patients.
Physicians and neurologists need to promote awareness of the absolute requirement of prompt administration of evidence-based prophylactic treatment to all individuals exposed to RABV. This approach is still defi cient or even non-existent in many parts of the world. Equally, if not more importantly, is the need to control this disease in its main vector, the dog. The knowledge and means to do so are available, but the will and appropriate support from societies and governments are often not.
ContributorsAll authors contributed to the scientifi c literature search and the writing
and revision of the paper.
Confl icts of interestWe declare that we have no confl icts of interest.
AcknowledgmentsResearch by the authors described in this Review article was sponsored
by the Thailand Research Fund (DBG5180026, RDG5420089), the Higher
Education Research Promotion and National Research University Project
of Thailand, Offi ce of the Higher Education Commission (HR1160A-55),
the Thai Red Cross Society, the US Naval Health Research Center BAA-
10-93 under cooperative agreement number W911NF-11-2-004, the Centre
National de la Recherche Scientifi que (CNRS), France, and the European
Union (QLRT-2001-00151, EUROKINESIS, and BIO4-CT98-0546,
TransVirus). The views and conclusions contained in this document are
those of the authors.
References1 Wilde H, Hemachudha T, Wacharapluesadee S, Lumlertdacha B,
Tepsumethanon V. Rabies in Asia: the classical zoonosis. Curr Top Microbiol Immunol 2012; published online June 8. DOI:10.1007/82_2012_228.
2 Hemachudha T, Wacharapluesadee S, Mitrabhakdi E, Wilde H, Morimoto K, Lewis RA. Pathophysiology of human paralytic rabies. J Neurovirol 2005; 11: 93–100.
3 Hemachudha T, Laothamatas J, Rupprecht CE. Human rabies: a disease of complex neuropathogenetic mechanisms and diagnostic challenges. Lancet Neurol 2002; 1: 101–09.
4 Hemachudha T, Phuapradit P. Rabies. Curr Opin Neurol 1997; 10: 260–67.
5 Mitrabhakdi E, Shuangshoti S, Wannakrairot P, et al. Diff erence in neuropathogenetic mechanisms in human furious and paralytic rabies. J Neurol Sci 2005; 238: 3–10.
6 Laothamatas J, Hemachudha T, Mitrabhakdi E, Wannakrairot P, Tulayadaechanont S. MR imaging in human rabies. AJNR Am J Neuroradiol 2003; 24: 1102–09.
7 Laothamatas J, Sungkarat W, Hemachudha T. Neuroimaging in rabies. Adv Virus Res 2011; 79: 309–27.
8 Laothamatas J, Wacharapluesadee S, Lumlertdacha B, et al. Furious and paralytic rabies of canine origin: neuroimaging with virological and cytokine studies. J Neurovirol 2008; 14: 119–29.
9 Shuangshoti S, Thepa N, Phukpattaranont P, et al. Reduced viral burden in paralytic compared to furious canine rabies is associated with prominent infl ammation at the brainstem level. BMC Vet Res 2013; 9: 31.
10 Lafon M. Evasive strategies in rabies virus infection. Adv Virus Res 2011; 79: 33–53.
11 Srinivasan A, Burton EC, Kuehnert MJ, et al. Transmission of rabies virus from an organ donor to four transplant recipients. N Engl J Med 2005; 352: 1103–11.
12 Maier T, Schwarting A, Mauer D, et al. Management and outcomes after multiple corneal and solid organ transplantations from a donor infected with rabies virus. Clin Infect Dis 2010; 50: 1112–19.
13 Feder HM Jr, Petersen BW, Robertson KL, Rupprecht CE. Rabies: still a uniformly fatal disease? Historical occurrence, epidemiological trends, and paradigm shifts. Curr Infect Dis Rep 2012; 14: 408–22.
14 Jackson AC. Therapy of human rabies. Adv Virus Res 2011; 79: 365–75.
15 Hattwick MA, Weis TT, Stechschulte CJ, Baer GM, Gregg MB. Recovery from rabies. A case report. Ann Intern Med 1972; 76: 931–42.
16 Willoughby RE Jr, Tieves KS, Hoff man GM, et al. Survival after treatment of rabies with induction of coma. N Engl J Med 2005; 352: 2508–14.
17 Presumptive abortive human rabies—Texas, 2009. MMWR Morb Mortal Wkly Rep 2010; 59: 185–90.
18 Centers for Disease Control and Prevention (CDC). Recovery of a patient from clinical rabies—California, 2011. MMWR Morb Mortal Wkly Rep 2012; 61: 61–65.
19 Hemachudha T, Phanuphak P, Sriwanthana B, et al. Immunologic study of human encephalitic and paralytic rabies. Preliminary report of 16 patients. Am J Med 1988; 84: 673–77.
20 Gadre G, Satishchandra P, Mahadevan A, et al. Rabies viral encephalitis: clinical determinants in diagnosis with special reference to paralytic form. J Neurol Neurosurg Psychiatry 2010; 81: 812–20.
21 Solanki A, Radotra BD, Vasishta RK. Correlation of cytokine expression with rabies virus distribution in rabies encephalitis. J Neuroimmunol 2009; 217: 85–89.
22 Mader EC Jr, Maury JS, Santana-Gould L, et al. Human rabies with initial manifestations that mimic acute brachial neuritis and Guillain-Barré syndrome. Clin Med Insights Case Rep 2012; 5: 49–55.
23 H emachudha T, Wacharapluesadee S, Laothamatas J, Wilde H. Rabies. Curr Neurol Neurosci Rep 2006; 6: 460–68.
24 H uman rabies—Minnesota, 2007. MMWR Morb Mortal Wkly Rep 2008; 57: 460–62.
25 S hantavasinkul P, Tantawichien T, Wacharapluesadee S, et al. Failure of rabies postexposure prophylaxis in patients presenting with unusual manifestations. Clin Infect Dis 2010; 50: 77–79.
26 L ewis P, Fu Y, Lentz TL. Rabies virus entry at the neuromuscular junction in nerve-muscle cocultures. Muscle Nerve 2000; 23: 720–30.
27 L entz TL, Burrage TG, Smith AL, Crick J, Tignor GH. Is the acetylcholine receptor a rabies virus receptor? Science 1982; 215: 182–84.
28 U golini G. Rabies virus as a transneuronal tracer of neuronal connections. Adv Virus Res 2011; 79: 165–202.
29 T ang Y, Rampin O, Giuliano F, Ugolini G. Spinal and brain circuits to motoneurons of the bulbospongiosus muscle: retrograde transneuronal tracing with rabies virus. J Comp Neurol 1999; 414: 167–92.
30 G raf W, Gerrits N, Yatim-Dhiba N, Ugolini G. Mapping the oculomotor system: the power of transneuronal labelling with rabies virus. Eur J Neurosci 2002; 15: 1557–62.
31 U golini G, Klam F, Doldan Dans M, et al. Horizontal eye movement networks in primates as revealed by retrograde transneuronal transfer of rabies virus: diff erences in monosynaptic input to “slow” and “fast” abducens motoneurons. J Comp Neurol 2006; 498: 762–85.

www.thelancet.com/neurology Vol 12 May 2013 511
Review
32 R athelot JA, Strick PL. Muscle representation in the macaque motor cortex: an anatomical perspective. Proc Natl Acad Sci USA 2006; 103: 8257–62.
33 M orcuende S, Delgado-Garcia JM, Ugolini G. Neuronal premotor networks involved in eyelid responses: retrograde transneuronal tracing with rabies virus from the orbicularis oculi muscle in the rat. J Neurosci 2002; 22: 8808–18.
34 B aer GM, Shaddock JH, Quirion R, Dam TV, Lentz TL. Rabies susceptibility and acetylcholine receptor. Lancet 1990; 335: 664–65.
35 L afon M. Rabies virus receptors. J Neurovirol 2005; 11: 82–87.
36 R eaves EJ, Salmon-Mulanovich G, Guevara C, et al. Susceptibility and lack of evidence for a viremic state of rabies in the night owl monkey, Aotus nancymaae. Virol J 2012; 9: 95.
37 M orimoto K, Patel M, Corisdeo S, et al. Characterization of a unique variant of bat rabies virus responsible for newly emerging human cases in North America. Proc Natl Acad Sci USA 1996; 93: 5653–58.
38 C harlton KM, Nadin-Davis S, Casey GA, Wandeler AI. The long incubation period in rabies: delayed progression of infection in muscle at the site of exposure. Acta Neuropathol 1997; 94: 73–77.
39 B ecker Y. Immunological and regulatory functions of uninfected and virus infected immature and mature subtypes of dendritic cells—a review. Virus Genes 2003; 26: 119–30.
40 L i J, McGettigan JP, Faber M, Schnell MJ, Dietzschold B. Infection of monocytes or immature dendritic cells (DCs) with an attenuated rabies virus results in DC maturation and a strong activation of the NFkappaB signaling pathway. Vaccine 2008; 26: 419–26.
41 S armento L, Li XQ, Howerth E, Jackson AC, Fu ZF. Glycoprotein-mediated induction of apoptosis limits the spread of attenuated rabies viruses in the central nervous system of mice. J Neurovirol 2005; 11: 571–81.
42 K uang Y, Lackay SN, Zhao L, Fu ZF. Role of chemokines in the enhancement of BBB permeability and infl ammatory infi ltration after rabies virus infection. Virus Res 2009; 144: 18–26.
43 B artel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–97.
44 C hen JF, Mandel EM, Thomson JM, et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and diff erentiation. Nat Genet 2006; 38: 228–33.
45 I srasena N, Mahavihakanont A, Hemachudha T. Rabies virus infection and microRNAs. Adv Virus Res 2011; 79: 329–44.
46 U golini G. Specifi city of rabies virus as a transneuronal tracer of motor networks: transfer from hypoglossal motoneurons to connected second-order and higher order central nervous system cell groups. J Comp Neurol 1995; 356: 457–80.
47 U golini G. Use of rabies virus as a transneuronal tracer of neuronal connections: implications for the understanding of rabies pathogenesis. Dev Biol (Basel) 2008; 131: 493–506.
48 Wacharapluesadee S, Hemachudha T. Nucleic-acid sequence based amplifi cation in the rapid diagnosis of rabies. Lancet 2001; 358: 892–93.
49 Ugolini G. Advances in viral transneuronal tracing. J Neurosci Methods 2010; 194: 2–20.
50 Murphy FA, Bauer SP. Early street rabies virus infection in striated muscle and later progression to the central nervous system. Intervirology 1974; 3: 256–68.
51 Murphy FA, Bauer SP, Harrison AK, Winn WC Jr. Comparative pathogenesis of rabies and rabies-like viruses. Viral infection and transit from inoculation site to the central nervous system. Lab Invest 1973; 28: 361–76.
52 Murphy FA, Harrison AK, Winn WC, Bauer SP. Comparative pathogenesis of rabies and rabies-like viruses: infection of the central nervous system and centrifugal spread of virus to peripheral tissues. Lab Invest 1973; 29: 1–16.
53 Charlton KM, Casey GA, Wandeler AI, Nadin-Davis S. Early events in rabies virus infection of the central nervous system in skunks (Mephitis mephitis). Acta Neuropathol 1996; 91: 89–98.
54 Stepien AE, Tripodi M, Arber S. Monosynaptic rabies virus reveals premotor network organization and synaptic specifi city of cholinergic partition cells. Neuron 2010; 68: 456–72.
55 Coulon P, Bras H, Vinay L. Characterization of last-order premotor interneurons by transneuronal tracing with rabies virus in the neonatal mouse spinal cord. J Comp Neurol 2011; 519: 3470–87.
56 Jankowska E. Interneuronal relay in spinal pathways from proprioceptors. Prog Neurobiol 1992; 38: 335–78.
57 Jankowska E, Edgley SA. Functional subdivision of feline spinal interneurons in refl ex pathways from group Ib and II muscle aff erents; an update. Eur J Neurosci 2010; 32: 881–93.
58 Kuypers HGJM. Anatomy of the descending pathways. In: Brooks V, ed. Handbook of physiology, section 1: neurophysiology, vol 2, part 1. Bethesda, MD: American Physiological Society, 1981: 579–666.
59 Jackson AC, Ye H, Phelan CC, et al. Extraneural organ involvement in human rabies. Lab Invest 1999; 79: 945–51.
60 Christianson JA, Davis BM. The role of visceral aff erents in disease. In: Kruger L, Light AR, eds. Translational pain research: from mouse to man. Boca Raton, FL: CRC Press, 2010.
61 McNeill DL, Burden HW. Convergence of sensory processes from the heart and left ulnar nerve onto a single aff erent perikaryon: a neuroanatomical study in the rat employing fl uorescent tracers. Anat Rec 1986; 214: 441–44, 396–97.
62 Sung JH, Hayano M, Mastri AR, Okagaki T. A case of human rabies and ultrastructure of the Negri body. J Neuropathol Exp Neurol 1976; 35: 541–59.
63 Vural SA, Alcigir G, Berkin S. Immunohistochemical and histopathological studies of fi xed rabies virus in goats. Onderstepoort J Vet Res 2001; 68: 83–89.
64 Mignini F, Streccioni V, Amenta F. Autonomic innervation of immune organs and neuroimmune modulation. Auton Autacoid Pharmacol 2003; 23: 1–25.
65 Irwin DJ, Wunner WH, Ertl HC, Jackson AC. Basis of rabies virus neurovirulence in mice: expression of major histocompatibility complex class I and class II mRNAs. J Neurovirol 1999; 5: 485–94.
66 Bousso P. T-cell activation by dendritic cells in the lymph node: lessons from the movies. Nat Rev Immunol 2008; 8: 675–84.
67 Charlton KM, Casey GA, Campbell JB. Experimental rabies in skunks: mechanisms of infection of the salivary glands. Can J Comp Med 1983; 47: 363–69.
68 Sheikh KA, Ramos-Alvarez M, Jackson AC, Li CY, Asbury AK, Griffi n JW. Overlap of pathology in paralytic rabies and axonal Guillain-Barré syndrome. Ann Neurol 2005; 57: 768–72.
6 9 Hemachudha T, Panpanich T, Phanuphak P, Manatsathit S, Wilde H. Immune activation in human rabies. Trans R Soc Trop Med Hyg 1993; 87: 106–08.
7 0 Held JR, Tierkel ES, Steele JH. Rabies in man and animals in the United States, 1946–65. Public Health Rep 1967; 82: 1009–18.
7 1 Willoughby RE Jr. “Early death” and the contraindication of vaccine during treatment of rabies. Vaccine 2009; 27: 7173–77.
7 2 WHO. WHO Expert Committee on Rabies. World Health Organ Tech Rep Ser 1992; 824: 1–84.
7 3 Schnell MJ, McGettigan JP, Wirblich C, Papaneri A. The cell biology of rabies virus: using stealth to reach the brain. Nat Rev Microbiol 2010; 8: 51–61.
7 4 Faber M, Faber ML, Papaneri A, et al. A single amino acid change in rabies virus glycoprotein increases virus spread and enhances virus pathogenicity. J Virol 2005; 79: 14141–48.
7 5 Langevin C, Tuff ereau C. Mutations conferring resistance to neutralization by a soluble form of the neurotrophin receptor (p75NTR) map outside of the known antigenic sites of the rabies virus glycoprotein. J Virol 2002; 76: 10756–65.
7 6 Masatani T, Ito N, Shimizu K, et al. Amino acids at positions 273 and 394 in rabies virus nucleoprotein are important for both evasion of host RIG-I-mediated antiviral response and pathogenicity. Virus Res 2011; 155: 168–74.
7 7 Hemachudha T, Wacharapluesadee S, Lumlertdaecha B, et al. Sequence analysis of rabies virus in humans exhibiting encephalitic or paralytic rabies. J Infect Dis 2003; 188: 960–66.
7 8 Prehaud C, Wolff N, Terrien E, et al. Attenuation of rabies virulence: takeover by the cytoplasmic domain of its envelope protein. Sci Signal 2010; 3: ra5.
7 9 Mita T, Shimizu K, Ito N, et al. Amino acid at position 95 of the matrix protein is a cytopathic determinant of rabies virus. Virus Res 2008; 137: 33–39.
8 0 Larrous F, Gholami A, Mouhamad S, Estaquier J, Bourhy H. Two overlapping domains of a lyssavirus matrix protein that acts on diff erent cell death pathways. J Virol 2010; 84: 9897–906.

512 www.thelancet.com/neurology Vol 12 May 2013
Review
1 05 Baloul L, Camelo S, Lafon M. Up-regulation of Fas ligand (FasL) in the central nervous system: a mechanism of immune evasion by rabies virus. J Neurovirol 2004; 10: 372–82.
1 06 Hooper DC, Phares TW, Fabis MJ, Roy A. The production of antibody by invading B cells is required for the clearance of rabies virus from the central nervous system. PLoS Negl Trop Dis 2009; 3: e535.
1 07 Roy A, Hooper DC. Lethal silver-haired bat rabies virus infection can be prevented by opening the blood-brain barrier. J Virol 2007; 81: 7993–98.
1 08 Roy A, Phares TW, Koprowski H, Hooper DC. Failure to open the blood-brain barrier and deliver immune eff ectors to central nervous system tissues leads to the lethal outcome of silver-haired bat rabies virus infection. J Virol 2007; 81: 1110–18.
1 09 Bailey SL, Carpentier PA, McMahon EJ, Begolka WS, Miller SD. Innate and adaptive immune responses of the central nervous system. Crit Rev Immunol 2006; 26: 149–88.
1 10 Thanomsridetchai N, Singhto N, Tepsumethanon V, et al. Comprehensive proteome analysis of hippocampus, brainstem, and spinal cord from paralytic and furious dogs naturally infected with rabies. J Proteome Res 2011; 10: 4911–24.
1 11 Reinke SN, Resch L, Maingat F, et al. Metagenomic and metabolomic characterization of rabies encephalitis: new insights into the treatment of an ancient disease. J Infect Dis 2012; published online Aug 21. DOI:10.1093/infdis/jis479.
1 12 Dietzschold B, Morimoto K, Hooper DC. Mechanisms of virus-induced neuronal damage and the clearance of viruses from the CNS. Curr Top Microbiol Immunol 2001; 253: 145–55.
1 13 Fu ZF, Jackson AC. Neuronal dysfunction and death in rabies virus infection. J Neurovirol 2005; 11: 101–06.
1 14 Morimoto K, Hooper DC, Spitsin S, Koprowski H, Dietzschold B. Pathogenicity of diff erent rabies virus variants inversely correlates with apoptosis and rabies virus glycoprotein expression in infected primary neuron cultures. J Virol 1999; 73: 510–18.
1 15 Javier RT, Rice AP. Emerging theme: cellular PDZ proteins as common targets of pathogenic viruses. J Virol 2011; 85: 11544–56.
1 16 Guigoni C, Coulon P. Rabies virus is not cytolytic for rat spinal motoneurons in vitro. J Neurovirol 2002; 8: 306–17.
1 17 Dhingra V, Li X, Liu Y, Fu ZF. Proteomic profi ling reveals that rabies virus infection results in diff erential expression of host proteins involved in ion homeostasis and synaptic physiology in the central nervous system. J Neurovirol 2007; 13: 107–17.
1 18 Jackson AC, Kammouni W, Zherebitskaya E, Fernyhough P. Role of oxidative stress in rabies virus infection of adult mouse dorsal root ganglion neurons. J Virol 2010; 84: 4697–705.
119 Kammouni W, Hasan L, Saleh A, Wood H, Fernyhough P, Jackson AC. Role of nuclear factor-kappaB in oxidative stress associated with rabies virus infection of adult rat dorsal root ganglion neurons. J Virol 2012; 86: 8139–46.
1 20 Scott CA, Rossiter JP, Andrew RD, Jackson AC. Structural abnormalities in neurons are suffi cient to explain the clinical disease and fatal outcome of experimental rabies in yellow fl uorescent protein-expressing transgenic mice. J Virol 2008; 82: 513–21.
1 21 Jackson AC, Randle E, Lawrance G, Rossiter JP. Neuronal apoptosis does not play an important role in human rabies encephalitis. J Neurovirol 2008; 14: 368–75.
1 22 Juntrakul S, Ruangvejvorachai P, Shuangshoti S, Wacharapluesadee S, Hemachudha T. Mechanisms of escape phenomenon of spinal cord and brainstem in human rabies. BMC Infect Dis 2005; 5: 104.
1 23 Chopra JS, Banerjee AK, Murthy JM, Pal SR. Paralytic rabies: a clinico-pathological study. Brain 1980; 103: 789–802.
1 24 Hemachudha T, Sunsaneewitayakul B, Mitrabhakdi E, et al. Paralytic complications following intravenous rabies immune globulin treatment in a patient with furious rabies. Int J Infect Dis 2003; 7: 76–77.
1 25 Tangchai P, Vejjajiva A. Pathology of the peripheral nervous system in human rabies. A study of nine autopsy cases. Brain 1971; 94: 299–306.
1 26 Awasthi M, Parmar H, Patankar T, Castillo M. Imaging fi ndings in rabies encephalitis. AJNR Am J Neuroradiol 2001; 22: 677–80.
1 27 Desai RV, Jain V, Singh P, Singhi S, Radotra BD. Radiculomyelitic rabies: can MR imaging help? AJNR Am J Neuroradiol 2002; 23: 632–34.
8 1 Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 2010; 11: 373–84.
8 2 Yoneyama M, Fujita T. Recognition of viral nucleic acids in innate immunity. Rev Med Virol 2010; 20: 4–22.
8 3 Rieder M, Conzelmann KK. Interferon in rabies virus infection. Adv Virus Res 2011; 79: 91–114.
8 4 Jensen S, Thomsen AR. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol 2012; 86: 2900–10.
8 5 Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nature Rev Mol Cell Biol 2007; 8: 49–62.
8 6 Prehaud C, Megret F, Lafage M, Lafon M. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J Virol 2005; 79: 12893–904.
8 7 Jackson AC, Rossiter JP, Lafon M. Expression of Toll-like receptor 3 in the human cerebellar cortex in rabies, herpes simplex encephalitis, and other neurological diseases. J Neurovirol 2006; 12: 229–34.
8 8 Chiron D, Pellat-Deceunynck C, Amiot M, Bataille R, Jego G. TLR3 ligand induces NF-{kappa}B activation and various fates of multiple myeloma cells depending on IFN-{alpha} production. J Immunol 2009; 182: 4471–78.
8 9 Menager P, Roux P, Megret F, et al. Toll-like receptor 3 (TLR3) plays a major role in the formation of rabies virus Negri bodies. PLoS Pathog 2009; 5: e1000315.
9 0 Faul EJ, Wanjalla CN, Suthar MS, Gale M, Wirblich C, Schnell MJ. Rabies virus infection induces type I interferon production in an IPS-1 dependent manner while dendritic cell activation relies on IFNAR signaling. PLoS Pathog 2010; 6: e1001016.
9 1 Hornung V, Ellegast J, Kim S, et al. 5�-triphosphate RNA is the ligand for RIG-I. Science 2006; 314: 994–97.
9 2 Brzozka K, Finke S, Conzelmann KK. Identifi cation of the rabies virus alpha/beta interferon antagonist: phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3. J Virol 2005; 79: 7673–81.
9 3 Vidy A, Chelbi-Alix M, Blondel D. Rabies virus P protein interacts with STAT1 and inhibits interferon signal transduction pathways. J Virol 2005; 79: 14411–20.
9 4 Vidy A, El Bougrini J, Chelbi-Alix MK, Blondel D. The nucleocytoplasmic rabies virus P protein counteracts interferon signaling by inhibiting both nuclear accumulation and DNA binding of STAT1. J Virol 2007; 81: 4255–63.
9 5 Brzozka K, Finke S, Conzelmann KK. Inhibition of interferon signaling by rabies virus phosphoprotein P: activation-dependent binding of STAT1 and STAT2. J Virol 2006; 80: 2675–83.
9 6 Moseley GW, Lahaye X, Roth DM, et al. Dual modes of rabies P-protein association with microtubules: a novel strategy to suppress the antiviral response. J Cell Sci 2009; 122: 3652–62.
9 7 Lafon M. Immune evasion, a critical strategy for rabies virus. Dev Biol (Basel) 2008; 131: 413–19.
9 8 Vuaillat C, Varrin-Doyer M, Bernard A, et al. High CRMP2 expression in peripheral T lymphocytes is associated with recruitment to the brain during virus-induced neuroinfl ammation. J Neuroimmunol 2008; 193: 38–51.
99 Suja MS, Mahadevan A, Madhusudana SN, Shankar SK. Role of apoptosis in rabies viral encephalitis: a comparative study in mice, canine, and human brain with a review of literature. Patholog Res Int 2011; 2011: 374286.
1 00 Fernandes ER, de Andrade HF Jr, Lancellotti CL, et al. In situ apoptosis of adaptive immune cells and the cellular escape of rabies virus in CNS from patients with human rabies transmitted by Desmodus rotundus. Virus Res 2011; 156: 121–26.
1 01 Chopy D, Pothlichet J, Lafage M, et al. Ambivalent role of the innate immune response in rabies virus pathogenesis. J Virol 2011; 85: 6657–68.
1 02 Lafon M, Megret F, Meuth SG, et al. Detrimental contribution of the immuno-inhibitor B7-H1 to rabies virus encephalitis. J Immunol 2008; 180: 7506–15.
1 03 Lafon M, Prehaud C, Megret F, et al. Modulation of HLA-G expression in human neural cells after neurotropic viral infections. J Virol 2005; 79: 15226–37.
1 04 Megret F, Prehaud C, Lafage M, et al. Modulation of HLA-G and HLA-E expression in human neuronal cells after rabies virus or herpes virus simplex type 1 infections. Hum Immunol 2007; 68: 294–302.

www.thelancet.com/neurology Vol 12 May 2013 513
Review
1 28 Pleasure SJ, Fischbein NJ. Correlation of clinical and neuroimaging fi ndings in a case of rabies encephalitis. Arch Neurol 2000; 57: 1765–69.
1 29 van Thiel PP, de Bie RM, Eftimov F, et al. Fatal human rabies due to Duvenhage virus from a bat in Kenya: failure of treatment with coma-induction, ketamine, and antiviral drugs. PLoS Negl Trop Dis 2009; 3: e428.
1 30 Dacheux L, Reynes JM, Buchy P, et al. A reliable diagnosis of human rabies based on analysis of skin biopsy specimens. Clin Infect Dis 2008; 47: 1410–17.
1 31 Kasempimolporn S, Hemachudha T, Khawplod P, Manatsathit S. Human immune response to rabies nucleocapsid and glycoprotein antigens. Clin Exp Immunol 1991; 84: 195–99.
1 32 Wacharapluesadee S, Hemachudha T. Urine samples for rabies RNA detection in the diagnosis of rabies in humans. Clin Infect Dis 2002; 34: 874–75.
1 33 Hemachudha T, Wacharapluesadee S. Antemortem diagnosis of human rabies. Clin Infect Dis 2004; 39: 1085–86.
1 34 Wacharapluesadee S, Hemachudha T. Ante- and post-mortem diagnosis of rabies using nucleic acid-amplifi cation tests. Expert Rev Mol Diagn 2010; 10: 207–18.
1 35 Dacheux L, Wacharapluesadee S, Hemachudha T, et al. More accurate insight into the incidence of human rabies in developing countries through validated laboratory techniques. PLoS Negl Trop Dis 2010; 4: e765.
1 36 Wacharapluesadee S, Phumesin P, Supavonwong P, Khawplod P, Intarut N, Hemachudha T. Comparative detection of rabies RNA by NASBA, real-time PCR and conventional PCR. J Virol Methods 2011; 175: 278–82.
137 Preuss MA, Faber ML, Tan GS, et al. Intravenous inoculation of a bat-associated rabies virus causes lethal encephalopathy in mice through invasion of the brain via neurosecretory hypothalamic fi bers. PLoS Pathog 2009; 5: e1000485.
1 38 Jackson AC, Warrell MJ, Rupprecht CE, et al. Management of rabies in humans. Clin Infect Dis 2003; 36: 60–63.
1 39 Nigg AJ, Walker PL. Overview, prevention, and treatment of rabies. Pharmacotherapy 2009; 29: 1182–95.
1 40 Hemachudha T, Sunsaneewitayakul B, Desudchit T, et al. Failure of therapeutic coma and ketamine for therapy of human rabies. J Neurovirol 2006; 12: 407–09.
1 41 McDermid RC, Saxinger L, Lee B, et al. Human rabies encephalitis following bat exposure: failure of therapeutic coma. CMAJ 2008; 178: 557–61.
1 42 Hunter M, Johnson N, Hedderwick S, et al. Immunovirological correlates in human rabies treated with therapeutic coma. J Med Virol 2010; 82: 1255–65.
1 43 Weli SC, Scott CA, Ward CA, Jackson AC. Rabies virus infection of primary neuronal cultures and adult mice: failure to demonstrate evidence of excitotoxicity. J Virol 2006; 80: 10270–73.
1 44 Jackson AC, Kammouni W, Fernyhough P. Role of oxidative stress in rabies virus infection. Adv Virus Res 2011; 79: 127–38.
1 45 Willoughby RE, Roy-Burman A, Martin KW, et al. Generalised cranial artery spasm in human rabies. Dev Biol (Basel) 2008; 131: 367–75.
1 46 Sing TM, Soo MY. Imaging fi ndings in rabies. Australas Radiol 1996; 40: 338–41.
1 47 Tirawatnpong S, Hemachudha T, Manutsathit S, Shuangshoti S, Phanthumchinda K, Phanuphak P. Regional distribution of rabies viral antigen in central nervous system of human encephalitic and paralytic rabies. J Neurol Sci 1989; 92: 91–99.
1 48 McDannold N, Arvanitis CD, Vykhodtseva N, Livingstone MS. Temporary disruption of the blood-brain barrier by use of ultrasound and microbubbles: safety and effi cacy evaluation in rhesus macaques. Cancer Res 2012; 72: 3652–63.
1 49 Israsena N, Supavonwong P, Ratanasetyuth N, Khawplod P, Hemachudha T. Inhibition of rabies virus replication by multiple artifi cial microRNAs. Antiviral Res 2009; 84: 76–83.
1 50 Faber M, Li J, Kean RB, Hooper DC, Alugupalli KR, Dietzschold B. Eff ective preexposure and postexposure prophylaxis of rabies with a highly attenuated recombinant rabies virus. Proc Natl Acad Sci USA 2009; 106: 11300–05.
1 51 Li J, Ertel A, Portocarrero C, et al. Postexposure treatment with the live-attenuated rabies virus (RV) vaccine TriGAS triggers the clearance of wild-type RV from the central nervous system (CNS) through the rapid induction of genes relevant to adaptive immunity in CNS tissues. J Virol 2012; 86: 3200–10.





























