Embed Size (px)
Citation preview

Hipertensión pulmonar (HP)
JAVIER GAUDÓ NAVARRO MERCEDES GARCÍA-SALMONES MARTÍN
Monografías NEUMOMADRID
CO
D.
ES
DC
RE
0019
R E S P I R A T O R I O
VOLUMEN XIV / 2010
VO
LUM
EN
XIV
/ 20
10H
IPE
RT
EN
SIÓ
N P
ULM
ON
AR
(H
P)
Mon
ogra
fías
NE
UM
OM
AD
RID
Fec
ha d
e el
abor
ació
n: m
arzo
201
0
Cubierta NM 2010 18/2/10 16:06 Página 1

Monografías NEUMOMADRID
Javier Gaudó Navarro Mercedes García-Salmones Martín
VOLUMEN XIV / 2010
HIPERTENSIÓNPULMONAR (HP)
Monografía HAP NM 208 pag 18/2/10 17:08 Página 1

“La información contenida en este documento no debe considerarse como recomendación deuso de los productos farmacéuticos y sus indicaciones. Por favor, antes de prescribir cualquiermedicamento, consulte la Ficha Técnica vigente”
Reservados todos los derechos. Ni la totalidad ni parte de este libro pueden reproducirse o transmitirse por ningún procedimiento electrónico o mecánico, incluyendo fotocopias, grabación magnética o cualquier almacenamiento de información y sistema de recuperación, sin el previo permiso escrito del editor.
© NEUMOMADRID. C/ CEA BERMÚDEZ 46-1 derecha. 28003 Madrid
Edita: ERGON. C/ Arboleda, 1. 28221 Majadahonda (Madrid).
ISBN: 978-84-8473-829-9 Depósito Legal: M-9349-2010
Monografía HAP NM 208 pag 18/2/10 17:08 Página 2

HIPERTENSIÓN PULMONAR (HP)
Monografías de la Sociedad Madrileñade Neumología y Cirugía Torácica
Junta Directiva
Presidente: Dr. José M. Rodríguez González-Moro
Vicepresidente Neumólogo: Dr. Javier Flandes Aldeyturriaga
Vicepresidente Cirujano Torácico: Dr. Prudencio Díaz-Agero Álvarez
Secretaria: Dra. Belén López-Muñiz Ballesteros
Tesorero: Dr. Juan Luis Rodríguez Hermosa
Vocal Congresos: Dra. Sagrario Mayoralas Alises
Vocal Científico: Dra. Dolores Álvaro Álvarez
Vocal Grupos de Trabajo: Dr. Ricardo García Luján
Vocal Pediatría: Dra. Mª Isabel Barrio Gómez de AgüeroVocal M.I.R.: Dra. Celia Zamarro García
Expresidente en ejercicio: Dr. Rodolfo Álvarez-Sala Walther
Comité Científico
Presidente: Dra. Dolores Álvaro ÁlvarezVocales: Dra. Eva Arias AriasDra. Mercedes García-Salmones MartínDra. Ana Mª Gómez MartínezDra. Eva Mañas BaenaDra. Mª Antonia Gómez MendietaDra. Mª Teresa Río RamírezDr. Felipe Villar Álvarez
VOLUMEN XIV / 2010
Javier Gaudó Navarro Mercedes García-Salmones Martín
Monografía HAP NM 208 pag 24/2/10 12:44 Página 3

Monografía HAP NM 208 pag 18/2/10 17:08 Página 4

PrólogoJavier Gaudó Navarro, Mercedes García-Salmones Martín . . . . . . . . . . . . . . . . . . . . . . . . . 7
BLOQUE I. FISIOPATOLOGÍA, PATOGENIA, EPIDEMIOLOGÍA Y GENERALIDADESDIAGNÓSTICAS Y TERAPÉUTICAS EN HAP
Fisiopatología e histopatología de la hipertensión arterial pulmonar (HAP)María Jesús Rodríguez Nieto, Felipe Villar Álvarez . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .9
Patogenia de la hipertensión arterial pulmonar (HAP)Juan Manuel Díez Piña . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
Definición, clasificación, epidemiología y generalidades de la hipertensión arterial pulmonarJosé Javier Jareño Esteban, José Ignacio de Granda Orive, José Francisco Villegas Fernández . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
Generalidades diagnósticas en la hipertensión pulmonarFernando Pedraza Serrano, Javier de Miguel Díez, Gema Sánchez Muñoz . . . . . . . . . . . . . . . 37
Generalidades terapéuticas en la hipertensión arterial pulmonar (HAP)Julio Flores Segovia, Olga Navarrete Isidoro, Silvia Sánchez González . . . . . . . . . . . . . . . . . 47
BLOQUE II. ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS SEGÚN GRUPOS DECLASIFICACIÓN ETIOLÓGICA
Grupo I. Clasificación de Dana PointHipertensión arterial pulmonar idiopática y resto. Aspectos diagnósticos y terapéuticosCarlos Pindado Rodríguez, Marta de Riva Silva, Miguel Ángel Gómez Sánchez . . . . . . . . . . 55
Grupo II. Clasificación de Dana PointAspectos diagnósticos y terapéuticosSergio Alcolea Batres, Juan José Ríos Blanco . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71
Grupo III. Clasificación de Dana PointAspectos diagnósticos y terapéuticosMaría Asunción Nieto Barbero, Beatriz Morales Chacón, José Luis Álvarez-Sala Walther . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
Grupo IV. Clasificación de Dana PointHipertensión pulmonar tromboembólica crónica (HPTEC)Javier Gaudó Navarro, Antonio Sueiro Bendito, David Jiménez Castro . . . . . . . . . . . . . . . . . 99
Grupo V. Clasificación de Dana PointHipertensión pulmonar con mecanismos multifactoriales no aclaradosMercedes García-Salmones Martín, María Jesús Linares Asensio, Ángela Ramos Pinedo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 111
Índice de capítulos
Monografía HAP NM 208 pag 24/2/10 12:26 Página 5

BLOQUE III. OTROS ASPECTOS TERAPÉUTICOS EN LA HP
Trasplante pulmonar, cardiopulmonar y septostomía en la hipertensión pulmonar (HP)Cristina López García-Gallo, Rosalía Laporta Hernández, Piedad Ussetti Gil . . . . . . . . . . . . 121
Enfermería e hipertensión arterial pulmonar (HAP) Fulgencio González Garrido . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 129
Seguimiento extrahospitalario de los pacientes con hipertensión pulmonar (HP):programas de educación al paciente y familiares Asunción Perpiñá Ferri, Pilar Alba García-Baquero,Inmaculada Fernández Rozas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 139
Hipertensión pulmonar en cardiopatías congénitas del adultoVerónica Hernández Jiménez, Mª Teresa Río Ramírez, Mª Antonia Juretschke Moragues . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 147
BLOQUE IV. AVANCES EN HIPERTENSIÓN PULMONAR
Genética e hipertensión arterial pulmonar Adolfo Baloira Villar, Diana Valverde Pérez . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161
Futuras líneas de investigación en la hipertensión arterial pulmonarCarlos Almonacid Sánchez . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 173
Recursos en internet sobre hipertensión pulmonar (HP)Francisco Javier García Pérez, Carlos Disdier Vicente, Claudia Valenzuela . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 185
Lista de abreviaturas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 199
Índice de autores . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203
Índice de materias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .205
Monografía HAP NM 208 pag 24/2/10 11:33 Página 6

La hipertensión pulmonar (HP) es un estado hemodinámico y patofisiológico, que puedehallarse en diversas enfermedades clínicas, las cuales han sido clasificadas en seis grupos clí-nicos, con características definidas, de acuerdo a la última clasificación de Dana Point 2008, yque han sido desarrolladas extensamente en la reciente “Guía de práctica clínica para el diag-nóstico y tratamiento de la hipertensión pulmonar ESC/ERS 2009”.
La hipertensión arterial pulmonar (HAP) representa a la enfermedad que comprende a lasentidades agrupadas en el grupo clínico I –compartiendo todas ellas un cuadro clínico similar,y unas alteraciones patológicas idénticas a las presentes en la microcirculación pulmonar–. Espor tanto este grupo I en el que se ha centrado la mayoría de las publicaciones al respecto, debi-do a la disponibilidad de nuevos tratamientos específicos, que incrementan la supervivenciade estos pacientes con HAP en un 20%, como objetiva el reciente metaanálisis realizado de Galiéy cols. Por otra parte las técnicas quirúrgicas (trasplante pulmonar/cardiopulmonar/septostomía)también se han venido perfeccionando, e incluso algunas de ellas –como la tromboendarte-rectomía– pueden resultar potencialmente curativas en formas seleccionadas de HP trombo-embólica.
Este impacto descrito en la supervivencia resulta pues de crucial importancia, al enfren-tarnos ante una enfermedad rara (se estima una prevalencia de 15 casos por millón de habi-tantes), de fatal pronóstico –con una mediana de supervivencia inferior a los 5 años en laHAP idiopática–. Además sigue siendo diagnosticada todavía en estadíos avanzados (clasesfuncionales III y IV de la OMS), como demuestran los diversos registros –incluyendo el espa-ñol REHAP–, y para la que no existían fármacos realmente eficaces, exceptuando el epo-prostenol y los bloqueantes de los canales de calcio, en los escasos pacientes respondedo-res a éstos.
Esta nueva monografía sobre HP pretende concienciar sobre varios aspectos, entre los quedestacan la importancia de realizar un diagnóstico correcto –tanto de la HP como de su etiolo-gía– y lo más precozmente posible. También resulta clave el abordaje multidisciplinario de estoscomplejos pacientes, así como la necesidad de derivarlos a Unidades de Referencia especiali-zadas, cuando no se acrediten convenientemente los requisitos y resultados que establecen lasGuías y Normativas al respecto.
7
PRÓLOGO
Monografía HAP NM 208 pag 18/2/10 17:08 Página 7

Para tratar de conseguir este objetivo, hemos contado con la colaboración de especialistascon diversa dedicación en el ámbito de la HP, y que representan mayoritariamente a los Hos-pitales que integran a nuestra Sociedad. A todos ellos agradecemos sus aportaciones, haciendoespecial mención a la labor desempeñada por el Dr. Sueiro Bendito. Confiamos que entre todoshayamos conseguido despertar su interés ante una enfermedad que puede simular simplicidad,pero tremendamente compleja, ya que supone un reto al clínico que se enfrenta a su certerodiagnóstico, manejo, y todavía elevada mortalidad.
Javier Gaudó NavarroServicio de Neumología. Unidad de HipertensiónPulmonar. Hospital Ramón y Cajal. Madrid
Mercedes García-Salmones MartínUnidad de Neumología. Hospital UniversitarioFundación Alcorcón. Madrid
8
Monografía HAP NM 208 pag 18/2/10 17:08 Página 8

9
RESUMEN La circulación pulmonar es un circuito de alto
flujo y baja resistencia. Una de las característi-cas diferenciales del sistema vascular pulmonares su respuesta a la hipoxia. La vasoconstricciónhipóxica es el mecanismo regulador más impor-tante de la distribución del flujo pulmonar. Losvasos que confieren la resistencia al sistema sonlas arterias pulmonares musculares, que sonpequeños vasos que discurren en el interior delos lóbulos y acompañan a los bronquíolos. Haydistintos factores que, activa o pasivamente, cam-bian el diámetro de estos vasos, aumentando laresistencia vascular pulmonar.
La fisiopatología de la hipertensión arterialpulmonar es compleja y multifactorial. Ésta seproduce por una disfunción endotelial que con-duce a un desequilibrio de agentes vasoacti-vos (endotelina-1, óxido nítrico, prostaciclina,etc.), con predominio de la vasoconstricción.La vasoconstricción crónica genera cambiosmorfológicos en la pared de las arterias pul-monares, con remodelado de la pared vascu-lar. La lesión característica del remodelado delos vasos pulmonares es la hipertrofia de lascélulas musculares lisas de la capa media. Esteremodelado, finalmente termina con unaumento de la presión en la arteria pulmonar.
Por tanto, la hipertensión pulmonar tieneuna fisiopatología compleja, con mecanismosimplicados heterogéneos, que suponen un retopara encontrar un tratamiento eficaz.
CIRCULACIÓN PULMONAR
Anatomía de la circulación pulmonarEl pulmón dispone de un doble sistema
arterial: el sistema de las arterias bronquia-
les (destinado a la nutrición de los tejidos desostén), que se origina en la aorta descendentey drena al sistema venoso a través de las venasbronquiales (ácigos en el lado derecho, yhemiácigos y mediastínicas en el izquierdo),y el sistema de las arterias y venas pulmona-res, destinadas al intercambio alveolocapilarde gases. Nos centraremos en este segundosistema que constituye la irrigación funcio-nal del pulmón.
El tronco arterial se origina en el ventrícu-lo derecho. Los vasos se van ramificando enel interior de los pulmones, acompañando deforma paralela a los bronquios. Dentro de lasunidades respiratorias, las arterias y arteriolaspulmonares se localizan en el centro y danlugar a arteriolas precapilares, de las cualessurgen los capilares pulmonares como una redque cubre las paredes alveolares. Estos capi-lares alveolares se unen en la periferia de losacinos y drenan las vénulas localizadas en losseptos interlobulares, que en este caso estánalejadas de las ramificaciones bronquiales.Al final, convergen en dos grandes troncosvenosos en cada pulmón que drenan directa-mente en la aurícula izquierda.
Arterias pulmonaresSe distinguen dos tipos básicos de arterias
pulmonares en función de la estructura de sucapa media: arterias elásticas y musculares. Lasarterias elásticas tienen un diámetro superiora 1 mm y están formadas por distintas capasde tejido elástico, recubiertas por células mus-culares. Son vasos de conducción, altamentedistensibles a bajas presiones. A medida quevan disminuyendo de calibre, va desapare-
FISIOPATOLOGÍA E HISTOPATOLOGÍA DE LA HIPERTENSIÓN ARTERIALPULMONAR (HAP)María Jesús Rodríguez Nieto, Felipe Villar Álvarez
BLOQUE I. FISIOPATOLOGÍA, PATOGENIA, EPIDEMIOLOGÍA Y GENERALIDADESDIAGNÓSTICAS Y TERAPÉUTICAS EN HAP
Monografía HAP NM 208 pag 18/2/10 17:08 Página 9

ciendo la capa elástica y aumenta el músculoliso hasta que, en los vasos con diámetro de100 a 500 mm, se pierde el tejido elástico dela capa media y las arterias se vuelven mus-culares. Son los vasos que discurren en el inte-rior de los lóbulos y acompañan a los bron-quiolos. Las arteriolas son vasos de menos de100 mm que constituyen la parte distal del sis-tema arterial pulmonar y que se insertan en lared capilar pulmonar de la unidad respiratoria.La transición entre arterias musculares y arte-riolas no es brusca sino que, a medida que sehacen más distales, pierden progresivamentela capa muscular y quedan constituidas por unafina capa íntima y una única lámina elástica.Los capilares pulmonares están revestidos poruna capa continua de endotelio que descansasobre la membrana basal y están conectadoscon los neumocitos, localizados por debajo dela membrana basal.
La circulación pulmonar es un sistema dealto flujo (todo el gasto cardiaco derecho), bajaresistencia y gran capacidad de reserva. Susamplios vasos de pared fina permiten estascaracterísticas a diferencia de los vasos sisté-micos. Los vasos que confieren resistencia alsistema son las arterias musculares.
Regulación del tono vascular y flujosanguíneo pulmonar
El lecho vascular pulmonar tiene una resis-tencia al flujo 10 veces menor que el lecho vas-
cular sistémico. La resistencia vascular expre-sa la relación entre presión y flujo sanguíneo.La resistencia vascular pulmonar (RVP), quedepende tanto del diámetro de los vasos comode la viscosidad de la sangre, es proporcionalal gradiente de presión entre la arteria pulmo-nar y la aurícula izquierda e inversamente pro-porcional al flujo sanguíneo pulmonar, que encondiciones normales equivale prácticamenteal gasto cardiaco. La RVP se modifica por todosaquellos factores que activa o pasivamente cam-bian el diámetro de los vasos, y de forma máslimitada a las variaciones en el hematocrito.
Efecto de la insuflación pulmonarEn la figura 1 observamos de forma esque-
mática lo que ocurre considerando dos tiposde vasos: intraalveolares (sometidos a la pre-sión alveolar) y extraalveolares (sometidos alas presiones pleurales negativas durante lainspiración). Así, durante la insuflación pul-monar, los vasos intraalveolares se colapsanmientras que los extraalveolares aumentan sudiámetro. El efecto sobre la resistencia vascu-lar global es que ésta es máxima en ambosextremos del volumen pulmonar (volumen resi-dual y capacidad pulmonar total), y mínima acapacidad residual funcional.
Efecto de la gravedadDebido al efecto de la gravedad, en un suje-
to en posición erecto, la presión en el inte-
M.J. RODRÍGUEZ NIETO, F. VILLAR ÁLVAREZ
10
FIGURA 1. Cambios en la resistencia vas-cular pulmonar en función del volumen pul-monar. La línea roja muestra el efecto totalde los vasos intra y extraalveolares sobrela resistencia vascular global, que es máxi-ma a volumen residual y capacidad pul-monar total, y mínima a capacidad residualfuncional.
Resistencia vascularpulmonar
CPTVR
Volumen pulmonar
CRF
ExtraalveolarAlveolarTotal
Monografía HAP NM 208 pag 18/2/10 17:08 Página 10

rior del vaso pulmonar es mayor en las basesque en el vértice y, en consecuencia, se incre-menta el diámetro de los vasos con lo que laresistencia disminuye progresivamente endirección caudal. Como la presión de perfu-sión es igual en todas las zonas, el flujo san-guíneo es mayor en las bases y va decrecien-do hacia el vértice.
Efecto de los cambios en el flujo sanguíneopulmonar
La circulación pulmonar es capaz de aco-modar grandes aumentos de flujo con sólo lige-ros incrementos en la presión arterial. Así, amedida que aumenta el flujo, disminuye laresistencia vascular pulmonar a través de dosmecanismos: la apertura o el reclutamiento devasos que permanecían cerrados y el aumen-to en el diámetro de los vasos que ya estabanabiertos.
Efecto de los cambios de la composicióndel gas alveolar
El efecto del oxígeno sobre la vasculaturapulmonar es la diferencia más notable sobrela circulación sistémica. La hipoxia alveolarproduce vasoconstricción pulmonar, que secaracteriza por ser proporcional al grado dehipoxia y estar estrictamente limitada al áreapulmonar que padece la hipoxia.
Mecanismos que regulan la circulaciónpulmonar
El control de la circulación pulmonar serealiza por tres tipos de mecanismos: • Control nervioso (adrenégico): los vasos
pulmonares expresan receptores adrenér-gicos alfa y beta que ayudan a regular eltono vascular, produciendo vasoconstric-ción y vasodilatación, respectivamente. Laestimulación excesiva de receptores alfa-1adrenérgicos produce contracción, proli-feración y crecimiento del músculo liso.Sustancias como la cocaína, anorexígenoso norepinefrina, incrementan los recepto-res alfa-1, aumentan la contracción y pro-liferación del músculo liso.
• Control humoral (mediadores vasculares): lasecuencia de mecanismos de activación delos factores de crecimiento de las célulasmusculares lisas de la vasculatura pulmonar,están involucrados como mediadores vas-culares. Comprende las prostaglandinas (laprostaciclina – PGI2, potente vasodilatadorsintetizado por células endoteliares que, ade-más, inhibe la agregación plaquetaria), elóxido nítrico (vasodilatador pulmonar sin-tetizado en el endotelio vascular con efectoantitrombótico al inhibir la activación pla-quetaria y que inhibe el crecimiento de lascélulas musculares lisas vasculares), la sero-tonina (liberada por las plaquetas, es unpotente vasoconstrictor que, además, pro-mueve la hipertrofia e hiperplasia de las célu-las del músculo liso vascular), angiotensinaII (generado a partir de la angiotensina I,potente vasoconstrictor pulmonar, que indu-ce proliferación celular), endotelina 1 (sin-tetizada también en el endotelio, es un poten-te vasoconstrictor y mitógeno).
• Control local (vasoconstricción hipóxica):La vasoconstricción hipóxica es el meca-nismo regulador más importante de la dis-tribución del flujo pulmonar. Desempeñaun papel esencial en mantener una rela-ción ventilación/perfusión adecuadas. Aun-que los efectos agudos son beneficiosos(redistribuye el flujo sanguíneo a las zonasbien ventiladas), a la larga la hipoxia cró-nica puede producir una elevación soste-nida de la presión arterial pulmonar, remo-delado vascular y desarrollo de hipertensiónpulmonar. El mecanismo de la vasocons-tricción hipóxica está mediado por cam-bios en la permeabilidad de la membranacelular a distintos iones en las células de lamusculatura lisa de los vasos pulmonares.Los canales de potasio, calcio y cloro tie-nen un papel importante en mantener eltono vascular pulmonar y están alteradospor los cambios en la presión local de oxí-geno en la circulación pulmonar(1). Los cam-bios agudos en el tono vascular pulmonarpor la hipoxia son reversibles, pero la hipo-
FISIOPATOLOGÍA E HISTOPATOLOGÍA DE LA HIPERTENSIÓN ARTERIAL PULMONAR (HAP)
11
Monografía HAP NM 208 pag 18/2/10 17:08 Página 11

xia crónica conduce a remodelado vascu-lar. Los sujetos que viven a altas altitudestienen distinto grado de hipertensión pul-monar, reflejo de la susceptibilidad perso-nal a la vasoconstricción por la hipoxia cró-nica. Algunos grupos étnicos, como lostibetanos, están mejor adaptados a la viday el trabajo en altitud, quizá por factoresgenéticos(2).
FISIOPATOLOGÍA DE LA HIPERTENSIÓNARTERIAL PULMONAR
La fisiopatología de la hipertensión arterialpulmonar es compleja y multifactorial. Comose resume en la figura 2, la hipertensión pul-monar se produce por una alteración de la fun-ción endotelial, a veces relacionada con unasusceptibilidad genética o con la exposición adistintos agentes desencadenantes (anorexí-genos, aceite de colza, virus, hipoxia, etc.). Estadisfunción endotelial conduce a un desequili-brio de agentes vasoactivos con predominiode la vasoconstricción(3,4). La vasoconstriccióncrónica genera cambios morfológicos en lapared de las arterias pulmonares, con remo-delado de la pared vascular. Otros contribu-yentes de la respuesta son la inflamación ytrombosis in situ. Todos estos mecanismos
determinan los cambios obliterativos típicosobservados en el árbol vascular pulmonar, queincrementan las resistencias vasculares pul-monares. Finalmente, este aumento de lasresistencias produce una carga de trabajo exce-siva sobre el ventrículo derecho, que terminafracasando y puede ocasionar la muerte delpaciente.
Se ha estudiado la genética en la hiper-tensión pulmonar familiar y sigue un patrónde transmisión de herencia autonómica domi-nante, con expresión variable y penetranciaincompleta, es decir, que puede saltar gene-raciones y simular casos esporádicos(5). Se haidentificado la mutación responsable demuchos casos, en el gen asociado a un tipo dereceptor de la superfamilia de los TGF-beta, elBMPR2(6). Los TGF-beta son factores de creci-miento que regulan la diferenciación y apop-tosis de muchas líneas celulares, entre ellas elmúsculo liso vascular, fundamentalmente inhi-biendo su crecimiento y proliferación. Tam-bién se han descrito mutaciones en el gen ALK-1, el cual codifica el receptor tipo I del TGF-betaen pacientes con hipertensión pulmonar ytelangiectasia hemorrágica hereditaria(7).
La disfunción endotelial se expresa comoun desequilibrio entre agentes vasodilatado-
12
FIGURA 2. Esquema que muestra la fisiopatología de la hipertensión pulmonar.
FACTORES GENÉTICOS(Mutaciones BMPR-II, ALK-1)
FACTORES DESENCADENANTES(Hipoxia crónica, colagenosis,tóxicos, VIH…)
DISFUNCIÓNENDOTELIAL
VASOCONSTRICCIÓN PROLIFERACIÓN
OBLITERACIÓN VASCULARAUMENTO DE LAS RVPFRACASO VD
TrombosisInflamación
M.J. RODRÍGUEZ NIETO, F. VILLAR ÁLVAREZ
Monografía HAP NM 208 pag 18/2/10 17:08 Página 12

res y vasoconstrictores. La endotelina 1, poten-te vasoconstrictor producido por el endotelio,está aumentada en pacientes con hipertensiónpulmonar(8) y se correlaciona con la gravedadde la enfermedad. Además de su efecto vaso-constrictor, también se ha relacionado con lainducción del remodelado vascular, ya queaumenta la expresión de receptores para laserotonina en las células musculares lisas vas-culares, un potente estimulador del crecimientode estas células. El óxido nítrico, implicado enel mantenimiento de la baja resistencia del cir-cuito vascular pulmonar por su efecto vaso-dilatador, está disminuido en pacientes conhipertensión pulmonar(9). Otro vasomoduladorafectado por la disfunción endotelial es la pros-taciclina. Ésta tiene efectos vasodilatadores yantiagregantes plaquetarios. Además, dismi-nuye su síntesis en las arteriolas pulmonaresy se produce vasoconstricción y tendencia ala coagulación dentro de estos vasos en pacien-tes con hipertensión pulmonar(10). Otro cam-po que se ha estudiado es la alteración en loscanales de potasio de la célula muscular lisavascular. Esta alteración produciría una mayorrespuesta a las señales vasoconstrictoras quellegan del endotelio, porque se genera unpotencial de membrana más alto y una mayorconcentración del calcio en las células, el cualtiene un papel central en la vasoconstriccióny es, posiblemente, un estímulo para la hiper-plasia e hipertrofia del musculo liso(11).
La inflamación puede desempeñar unpapel importante en la patogénesis de algunostipos de hipertensión pulmonar(12). Es conoci-da la asociación de hipertensión pulmonar endistintas enfermedades autoinmunes o con lainfección por el virus de la inmunodeficienciahumana. Incluso en algunos casos de lupus sis-témico con hipertensión pulmonar severa, seha comunicado la mejoría de la hipertensióncon el tratamiento inmunosupresor. Tambiénse han descrito infiltrados inflamatorios alre-dedor de las lesiones plexiformes de pacien-tes con hipertensión arterial pulmonar.
Por tanto, la fisiopatología de la hiperten-sión pulmonar es compleja, con mecanismos
implicados heterogéneos que suponen un retopara encontrar un tratamiento eficaz.
HISTOPATOLOGÍA DE LA HIPERTENSIÓNARTERIAL PULMONAR
Como hemos visto, la disfunción endote-lial, producida por agentes conocidos o des-conocidos, conduce a un remodelado de losvasos pulmonares cuya lesión característica esla hipertrofia de las células musculares lisas dela capa media. Este remodelado termina final-mente con un aumento de la presión en la arte-ria pulmonar. La clasificación clínica actual dela hipertensión arterial (Dana Point, 2008, Tabla1), se basa en la etiología, datos funcionales ypresentación clínica. Pero, además, los estu-dios histológicos muestran diferencias en ladistribución y prevalencia de los cambios vas-culares entre los distintos grupos clínicos(13).Los avances en distintos mecanismos genéti-cos y moleculares, junto con las descripcionesde los hallazgos histológicos en la hipertensiónpulmonar de diferentes etiologías, pueden con-tribuir a un conocimiento mayor de las rela-ciones entre los distintos grupos, las implica-ciones pronosticas y las posibles opcionesterapéuticas. A continuación describiremos lasdistintas formas de enfermedad vascular hiper-tensiva pulmonar. • Arteriopatía plexogénica pulmonar. Son las
lesiones vasculares que aparecen en lamayoría de las enfermedades que formanparte del Grupo 1. Inicialmente hay hiper-trofia de la capa media y fibrosis laminarconcéntrica de la íntima y, cuando éstasprogresan, aparecen las lesiones específi-cas y patognomónicas: las lesiones ple-xiformes. Su falta no excluye el diagnós-tico, ya que la enfermedad podría estar enuna fase incipiente(14). La lesión plexifor-me es una pequeña lesión arterial, simi-lar a una dilatación, que consiste en unplexo de canalículos, intercalados con teji-do conectivo que contiene fibras muscu-lares y miofibroblastos, cuyas luces estántapizadas por células epiteliales. Además,es frecuente encontrar trombos en los
FISIOPATOLOGÍA E HISTOPATOLOGÍA DE LA HIPERTENSIÓN ARTERIAL PULMONAR (HAP)
13
Monografía HAP NM 208 pag 18/2/10 17:08 Página 13

M.J. RODRÍGUEZ NIETO, F. VILLAR ÁLVAREZ
14
espacios vasculares de la lesión plexifor-me. La lesión avanza con una marcada pro-liferación miofibroblástica, conformandola fibrosis intimal laminar concéntrica. Alfinal, la fibrosis domina el proceso y lasarterias afectadas se ocluyen casi por com-pleto. En la figura 3 podemos ver una arte-ria pulmonar muscular ocluida, con unasintensas hipertrofia y proliferación de lacapa media y fibrosis laminar concéntri-ca de la íntima.
• Enfermedad venooclusiva pulmonar yhemangiomatosis capilar pulmonar. Son laslesiones vasculares que aparecen en lasenfermedades raras que forman el Grupo1’. La enfermedad venooclusiva pulmonarse caracteriza por fibrosis obliterante devenas y vénulas pulmonares, acompaña-da de congestión del parénquima pulmo-nar circundante. Éstas son lesiones focalesque alternan con zonas sanas(15). La heman-giomatosis capilar pulmonar histológica-mente corresponde a zonas de exuberan-te proliferación de las células endoteliales,que forman conglomerados de vasos depared fina, similares a los capilares, queinfiltran los septos alveolares y el tejidointersticial de los ejes broncovasculares. Amenudo se asocian a hemosiderosis. Conmarcadores de remodelado vascular se havisto que las células endotelialres son dife-rentes a las lesiones plexiformes(16). Ambasentidades comparten la distribución focal,la presencia de fibrosis y hemosiderosis, yla combinación de obliteración de las vénu-las y proliferación capilar(14).
• Hipertensión pulmonar persistente del reciénnacido. Al nacer se ponen en marcha dis-tintos mecanismos de adaptación para con-vertir el sistema pulmonar en un circuito
FIGURA 3. Imagen de una arteria pulmonar mus-cular obstruida completamente por unas intensashipertrofia y proliferación de la capa media, y fibro-sis laminar concéntrica de la íntima, en un pacien-te afecto de hipertensión pulmonar. Imagen cedi-da por la Dra. Renedo, del Departamento deAnatomía Patológica de la Fundación Jiménez Díaz.
TABLA 1. Clasificación de la hipertensiónpulmonar (Dana Point, 2008)
1. Hipertensión arterial pulmonar- Idiopática- Hereditaria: BMPR2, ALK-1, desconocida- Inducida por fármacos o tóxicos- Asociada: enfermedades tejido conectivo,
infección por VIH, hipertensión portal, cor-tocircuito sistémico-pulmonar, esquisto-somiasis, anemia hemolítica crónica
- Hipertensión pulmonar persistente delrecién nacido
1’. Enfermedad venooclusiva pulmonar y/ohemangiomatosis capilar pulmonar
2. Hipertensión pulmonar por cardiopatía dellado izquierdo
3. Hipertensión pulmonar asociada a enferme-dad pulmonar y/o hipoxemia
4. Hipertensión pulmonar tromboembólica cró-nica
5. Hipertensión pulmonar de mecanismo incier-to o multifactorial- Trastornos hematológicos: mieloprolifera-
tivos, esplenectomía- Trastornos sistémicos: vasculitis, sarcoido-
sis, histiocitosis X, linfangioleiomatosis, neu-rofibromatosis
- Trastornos metabólicos: enfermedad deGaucher, enfermedades tiroideas, etc.
- Cardiopatías congénitas distintas del shuntsistémico-pulmonar
- Otras (obstrucción tumoral, mediastinitisfibrosante, etc.)
Monografía HAP NM 208 pag 18/2/10 17:08 Página 14

de baja resistencia. En esta rara enferme-dad, estos mecanismos fallan y las paredesarteriales pulmonares presentan hiperpla-sia e hipertrofia de las células musculareslisas, así como un aumento de la matrizextracelular. Finalmente, el proceso acabaen fibrosis intensa. En estos niños se handetectado concentraciones bajas de óxidonítrico(17), que podría explicar la patoge-nia de la enfermedad.
• Vasculopatía congestiva pulmonar. Apareceen las enfermedades del grupo 2 (insufi-ciencia cardiaca izquierda) y en aquellasenfermedades que producen obstrucciónde venas centrales (adenopatías o tumo-res mediastínicos, mediastinitis fibrosan-te: Grupo 5). La obstrucción crónica del flu-jo venoso ocasiona cambios en las venaspulmonares que se asemejan a arterias pul-monares musculares (“arterialización”).Hay congestión pulmonar, edema intersti-cial y ensanchamiento de los vasos linfá-ticos. Al progresar aparecen la fibrosis y lahemosiderosis. Estas alteraciones a menu-do desaparecen cuando se soluciona laenfermedad cardiológica de base.
• Vasculopatía hipóxica pulmonar. Como yahemos visto, la respuesta a la hipoxia agu-da alveolar es una vasoconstricción de losvasos pulmonares que intenta evitar elcortocircuito de las áreas mal ventiladas.Cuando se produce una exposición cró-nica a la hipoxia, aparecen cambiosestructurales en las arterias pulmonarescon remodelado y reducción de la canti-dad total de arterias pulmonares perifé-ricas(18). La magnitud y las característicasde estos cambios dependen de la espe-cie, el sexo y el estadio de desarrollo delpulmón en el momento de la exposicióna la hipoxia. Además, los cambios queinduce la hipoxia crónica son diferentesen función de si se producen en vasosgrandes o periféricos. Por otra parte, laexposición a hipoxia crónica induce unarespuesta inflamatoria en la pared delvaso(19). Los hallazgos característicos de
la vasculopatía pulmonar hipóxica son lahiperplasia muscular de la capa media,sobre todo en las arterias periféricas, y laneomuscularización de las arteriolas pul-monares(20). En la EPOC, aproximada-mente un 10-30% de los pacientes tienenhipertensión pulmonar, generalmente,leve(21). La disfunción endotelial produci-da por el humo del cigarrillo o algunoselementos inflamatorios se consideran laalteración primaria que inicia la secuen-cia de la hipertensión pulmonar en estaenfermedad(22).
• Arteriopatía pulmonar postrombótica-trom-boembólica. Aproximadamente, el 4% delos pacientes que sufren un tromboem-bolismo pulmonar sintomático, desarro-llan hipertensión pulmonar por la persis-tencia de una arteriopatía pulmonarpostrombótica(23). La oclusión trombóticade una arteria pulmonar puede ser elresultado de un tromboembolismo o deuna trombosis in situ, siendo ambos his-tológicamente indistinguibles. La trom-bosis in situ, sobre todo de las arteriaspequeñas, puede aparecer en cualquierenfermedad vascular hipertensiva y enlos trastornos fibroinflamatorios del inters-ticio pulmonar. Histológicamente, lostrombos se organizan, constituyendo unamasa fibrótica que ocasiona un engrosa-miento fibrótico excéntrico de la íntima.El trombo se recanaliza produciendo unao más neoluces, formando tabiques fibro-sos finos en la luz vascular(24). Éstas sonlesiones focales que pueden afectar a arte-rias de diferentes tamaños. La alteraciónde los pequeños vasos periféricos se con-sidera la causa principal de la progresiónde la hipertensión pulmonar en estospacientes(25).
• Otras formas de enfermedad vascular hiper-tensiva. Éstas están formadas por diferen-tes trastornos vasculares desarrollados enfunción de la enfermedad de base, comola presencia de una vasculitis granuloma-tosa en casos de sarcoidosis.
FISIOPATOLOGÍA E HISTOPATOLOGÍA DE LA HIPERTENSIÓN ARTERIAL PULMONAR (HAP)
15
Monografía HAP NM 208 pag 18/2/10 17:08 Página 15

BIBLIOGRAFÍA 1. Weir EK, Aarcher SL. The mechanism of acu-
te hypoxic pulmonary vasoconstriction: thetale of two channels. Faseb J. 1995; 9: 183-9.
2. Wu T, Kaiser B. High altitude adaptation inTibetans. High Alt Med Biol. 2006; 7: 193-208.
3. Caraballo Fonseca JC, Martínez Balsano CD,Sánchez de León R. Disfunción endotelial enla hipertensión pulmonar. Arch Bronconeu-mol. 2005; 41: 389-92.
4. Budhiraja R, Tuder RM, Hassoun PM. Endote-lial dysfunction in pulmonary hypertension.Circulation. 2004; 109: 159-65.
5. Loyd JE, Primm RK, Newman JH. Familiar pri-mary pulmonary hypertension: clinical pat-terns. Am Rev Respir Dis. 1984; 129: 194-7.
6. Deng ZM, Morse JH, Sleger SL. Familial pri-mary pulmonary hypertension (gen PPH1) iscaused by mutations in the bone morphoge-netic protein receptor II gene. Am J HumGenet. 2000; 67: 637-44.
7. Trembath RC, Thomson JR, Machado RD, etal. Clinical and molecular genetic features ofpulmonary hypertension in patients with here-ditary hemorrhagic telangiectasia. N Engl JMed. 2001; 345: 325-34.
8. Giaid A, Yanagisawa M, Langleben D, et al.Expression of endothelin-1 in the lung ofpatients with pulmonary hypertension. N EnglJ Med. 1993; 328: 1732-39.
9. Giaid A, Saleh D. Reduced expression of endo-thelial nitric oxide synthase in the lungs ofpatients with pulmonary hypertension. N EnglJ Med. 1995; 333: 214-21.
10. Tuder R, Cool C, Geraci M, et al. Prostacyclinsynthase expression is decreased in lung frompatients with severe pulmonary hypertension.Am J Respir Crit Care Med. 1999; 159: 1925-32.
11. Yuan L, Aldinger A, Juhaszova M, et al.Dysfunctional voltage-gated K+ channels inpulmonary artery smooth muscle cells ofpatients with primary pulmonary hyperten-sion. Circulation. 1998; 98: 1400-06.
12. Dorfmüller P, Perros F, Balabanian K, et al.Inflammation in pulmonary arterial hyper-tension. Eur Respir J. 2003; 22: 358-63.
13. Stewart S, Rassl D. Advances in the unders-tanding and classification of pulmonary hyper-tension. Histopathology. 2009; 54: 104-16.
14. Mooi WJ, Grünberg K. Histopathology of pul-monary hypertensive diseases. Curr DiagnostPathol. 2006; 12: 429-40.
15. Mandel J, Mark EJ, Hales CA. Pulmonary veno-occlusive disease Am J Respir Crit Care Med.2000; 162: 1964-73.
16. Sullivan A, Chmura K, Cool CD, et al. Pulmo-nary capillary hemangiomatosis: an inmuno-histochemical analysis of vascular remodeling.Eur J Med Res. 2006; 11: 187-93.
17. Dakshinamurti S. Pathophysiologic mecha-nisms of persistent pulmonary hypertensionof the newborn. Pediatr Pulmonol. 2005; 39:492-503.
18. Weir EK, López-Barneo J, Buckler KJ, et al. Acu-te oxygen-sensing mechanisms. N Engl J Med2005; 353; 2042-55.
19. Stenmark KR, Fagan KA, Frid MG. Hipoxia-induced pulmonary vascular remodeling: cellu-lar and molecular mechanisms. Circ Res. 2006;99: 675-91.
20. Mathai SC, Girgis RE. Pulmonary hypertensionrelated to respiratory diseases. Clin pulm Med.2007; 14: 286-95.
21. Elwing J, Panos RJ. Pulmonary hypertensionassociated to COPD. Int J Chron Obstruct Pul-mon Dis. 2008; 3: 55-70.
22. Peinado VI, Pizarro S, Barberà JA. Pulmonaryvascular involvement in COPD. Chest. 2008;134: 808-14.
23. Pengo V, Lesing AW, Prins MH, et al. Throm-boembolic Pulmonary Hypertension StudyGroup. Incidence of chronic thromboembo-lic pulmonary hypertension after pulmonaryembolism. N Engl J Med. 2004; 350: 2257-64.
24. Hoeper MM, Mayer E, Simmoneau G, et al.Chronic thromboembolic pulmonary hyper-tension. Circulation. 2006; 113: 2011-20.
25. Galiè N, Kim NH. Pulmonary microvasculardisease in chronic thromboembolic pulmo-nary hypertension. Proc Am Thorac Soc. 2006;3: 571-6.
M.J. RODRÍGUEZ NIETO, F. VILLAR ÁLVAREZ
16
Monografía HAP NM 208 pag 18/2/10 17:08 Página 16

17
RESUMENLa hipertensión arterial pulmonar (HAP)
tradicionalmente ha sido considerada comoun proceso de vasoconstricción y en los últi-mos años se han ido implicando factores decrecimiento y alteraciones en mecanismos deapoptosis. Esta enfermedad tiene una biopa-tología multifactorial que incluye varios tiposde células y varios procesos bioquímicos dis-tintos (Tabla 1). Aún no se conocen bien lasinteracciones entre estos mecanismos quedesencadenan y hacen progresar los procesospatológicos. La clásica explicación ha sido lainteracción entre predisposición genética y fac-tores de riesgo que pueden inducir cambiosen diferentes células (células musculares lisas,endoteliales, inflamatorias y plaquetas) y en lamatriz extracelular de la microcirculación pul-
monar. El desequilibrio entre factores trom-bogénicos, mitogénicos, proinflamatorios yvasoconstrictores frente a los mecanismos anti-coagulantes, antimitóticos y vasodilatadorespuede iniciar y, posteriormente, perpetuar, pro-cesos como la vasoconstricción, la prolifera-ción, la trombosis y la inflamación de la micro-circulación pulmonar(1). Estos mecanismos sonlos que causan el inicio y progresión de loscambios patológicos obstructivos típicos de laHAP. Como consecuencia, aumenta la resis-tencia vascular pulmonar (RVP), que conducea una sobrecarga del ventrículo derecho y, final-mente, a la insuficiencia cardiaca y muerte.
Vamos a repasar en este capítulo los fac-tores implicados en la patogenia de la hiper-tensión arterial pulmonar, revisando los dife-rentes mecanismos celulares y moleculares,
PATOGENIA DE LA HIPERTENSIÓNARTERIAL PULMONAR (HAP)
Juan Manuel Díez Piña
FIGURA 1.Biopatología multifactorial de la HAP.
Factores trombogénicos,mitogénicos, proinflamatorios
y vasoconstrictores
Mecanismosanticoagulantes, antimitóticos
y vasodilatadores
CambiosCélulas músculo lisoCélulas endotelialesPlaquetasMatriz extracelular
Factores genéticos y ambientales HAP
Monografía HAP NM 208 pag 18/2/10 17:08 Página 17

y los sustratos genéticos conocidos hasta aho-ra(2).
VASOCONSTRICCIÓNLa vasoconstricción es uno de los prime-
ros componentes de la hipertensión pulmo-nar, y guarda relación con disfunción del endo-telio y con función anormal de los canales depotasio en las células del músculo liso.
Efectos vascularesEl endotelio vascular detecta variaciones
del flujo de la luz vascular y genera estímulosque afectan al tono de las células del músculoliso. Si estos estímulos se mantienen en el tiem-po, pueden originar el remodelado de dichascélulas(3).
Sustancias vasodilatadorasEl óxido nítrico (NO) es un potente vaso-
dilatador pulmonar, un inhibidor de la activa-ción de las plaquetas y de la proliferación delas células del músculo liso. Su síntesis estámediada por la familia de las enzimas de la
NOS (e-NOS). La isoforma endotelial eNOS estáregulada por multitud de factores vasoacti-vos y por estímulos fisiológicos, como la hipo-xia, inflamación y estrés oxidativo. Los pacien-tes con HAP tienen una disminución de laexpresión de la enzima óxido nítrico-sintasa,lo que conduce a vasoconstricción y prolife-ración celular, así como a un incremento en laapoptosis de células endoteliales(4).
La prostaciclina (PGI2) activa la vía inde-pendiente de la adenosina monofosfato cicla-sa (cAMP), y actúa como vasodilatador, factorantiproliferativo del músculo liso vascular einhibidor de la activación y agregación de lasplaquetas. Los niveles de prostaciclina y la acti-vidad de la prostaciclina sintetasa están dis-minuidos en pacientes con HAP(5).
Otros factores vasodilatadores que se hanimplicado en la patogenia de la HAP cuandoson deficitarios son: el péptido intestinal vaso-activo (VIP), vasodilatador pulmonar que tam-bién inhibe la agregación plaquetaria y la pro-liferación de células musculares lisasvasculares(6,7); el monóxido de carbono (CO),
J.M. DÍEZ PIÑA
18
TABLA 1. Factores promotores, mecanismos celulares y moleculares implicadosen la patogenia de la HAP
Factores Factores Sustancias Remodelado Sustanciasambientales genéticos vasoactivas vascular inflamatorias Coagulación
VD VC
Hipoxia BMPR2 NO ET-1 Proliferación IL-1 Alteración cascada endotelial coagulación
Anorexígenos ALK-1 PGI-2 TxA2 Inhibición IL-6 Disfunciónapoptosis plaquetaria
Aceite tóxico 5-HT VIP 5-HT Aumento OPGmatriz extracelular
VIH Canales K+ VEGF Otros
Otros
VIH: virus de la inmunodeficiencia humana; BMPR-2: gen del receptor tipo 2 de las proteínas morfogenéticas delhueso; ALK-1: gen de la activina-cinasa “receptor-like” tipo 1; 5-HT: serotonina; NO: óxido nítrico; PGI-2: prosta-ciclina; VIP: péptido intestinal vasoactivo; ET-1: endotelina 1; TxA2: tromboxano A2; K+: potasio; VEGF: factorde crecimiento vascular del endotelio; IL-1: interleucina 1; IL-6: interleukina 6; OPG: osteoprotegerina.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 18

cuya enzima de producción hemooxigenasa 1(HO-1) está relacionada con el desarrollo deHAP cuando es deficitaria; y el sulfito de hidró-geno (H2S), que es también inhibidor de la pro-liferación vascular.
Sustancias vasoconstrictorasLa endotelina 1 (ET-1) se expresa en las célu-
las endoteliales del pulmón y es un mediadorvascular en la HAP, ya que es un potente vaso-constrictor y un agente mitogénico de célulasmusculares lisas vasculares. Esta acción mito-génica se produce a través de los receptores Acomo de los receptores B, siendo el primeroel responsable de la mitogénesis en arterias prin-cipales, mientras que en las arterias de resis-tencia están ambos implicados. Esta vasocons-tricción, la mitogénesis y el remodelado vascularresultante, provocan cambios hemodinámicosimportantes en la circulación pulmonar. En plas-ma de animales y humanos con HAP se hanpodido detectar niveles elevados de ET-1(8).
El tromboxano A2 (TxA2) es un derivadoaraquidónico que aumenta la vasoconstriccióny activa las plaquetas. En orina de pacientescon HAP se han visto niveles bajos de un pro-ducto de la degradación de la prostaciclina (6-keto-prostaciclina F2 alfa), así como un aumen-to de las concentraciones de un metabolito delTxA2, tromboxano B2(5).
La serotonina (5-HT) se codifica en el gendel cromosoma 17q11.2 y se expresa en variostipos de células, como neuronas, plaquetas ycélulas endoteliales y de músculo liso de arte-ria pulmonar. Los niveles de expresión en pul-món son mucho mayores que en cerebro, locual sugiere que modificaciones en su expre-sión pueden tener consecuencias directas enla función de las células musculares lisas de laarteria pulmonar.
Existen estudios que han comprobadocómo la serotonina promueve el desarrollo deHAP hipóxica mediante el estímulo de creci-miento de células musculares lisas de la arte-ria pulmonar(9).
También se ha publicado que la expresióndel gen de la serotonina está incrementada
en arterias pulmonares remodeladas de ani-males con HAP secundaria a exposición cró-nica a hipoxia(10). Además, se ha visto queratones con modificaciones en el gen de laserotonina desarrollan HAP hipóxica menossevera que aquellos a los que no se les modi-fica(11).
Eddahibi y cols. encontraron que las célu-las del músculo liso de arteria pulmonar cre-cen más rápido cuando son estimuladas conserotonina y que estos efectos eran debidos ala mayor expresión del transportador de sero-tonina. En este mismo estudio se comprue-ba que este efecto se reduce cuando se usaninhibidores de la serotonina. La expresión deserotonina estaba incrementada en cultivos decélulas del músculo liso de la arteria pulmo-nar, en plaquetas y pulmones de pacientes conHAP, sobre todo en la media de arterias pul-monares engrosadas y en las lesiones “en bul-bo de cebolla”(12).
Canales de potasioEl potencial transmembrana de reposo del
músculo liso vascular se regula a través delpotasio mediante canales voltaje-dependien-tes. La despolarización de estos canales pro-voca una despolarización celular, incremen-tando el calcio citoplasmático por apertura delos canales del calcio, lo que conlleva a vaso-constricción pulmonar, proliferación celulare inhibición de la apoptosis(13).
Un defecto en los canales de potasio encélulas del músculo liso puede ser el meca-nismo desencadenante y de mantenimientoen la vasoconstricción pulmonar de pacientescon HAP primaria. Yuan y cols. demostraronque la función de los canales de potasio vol-taje-dependientes en las células del músculoliso de las arterias pulmonares de pacientescon HAP primaria está inhibida en compara-ción con los pacientes con HAP secundaria.Esta inhibición origina una despolarización dela membrana y un incremento del calcio cito-plasmático, con las consecuencias referidasanteriormente(14). Además, este incrementodel calcio intracitoplasmático se relaciona con
PATOGENIA DE LA HIPERTENSIÓN ARTERIAL PULMONAR (HAP)
19
Monografía HAP NM 208 pag 18/2/10 17:08 Página 19

20
el desarrollo y mantenimiento de la HAP, yaque se ha visto que el 26% de los pacientescon HAP primaria presentan reducciones enla presión de la arteria pulmonar y de la resis-tencia vascular en respuesta a bloqueantes delos canales del calcio(15).
Se han identificado subtipos de canales deK+ en células musculares de la arteria pulmo-nar que se inhiben ante el estímulo hipóxico,lo cual podría estar en el origen de la HAP dealgunas enfermedades(16).
También en la HAP secundaria a hipoxiacrónica se ha implicado a los canales de K+,como se ha visto en células musculares lisas dearteria pulmonar cultivadas en las que existíauna disminución del ARNm y de la expresiónde las subunidades α de los canales, lo que pro-vocaba una reducción en el flujo de iones(17).
Se ha demostrado igualmente implicaciónde los canales de K+ en la HAP secundaria afármacos anorexígenos, ya que existe una
menor expresión de los mismos en las arte-rias pulmonares de estos pacientes(18).
Disfunción endotelialLa disfunción endotelial juega un papel fun-
damental en los cambios estructurales que seproducen en la vasculatura pulmonar. Un endo-telio dañado provoca una exposición del teji-do vascular subyacente a factores circulantesque pueden originar mayores cambios pato-lógicos. La disfunción endotelial también pue-de tener consecuencias adversas para lahemostasia pulmonar, alterando la producciónde factores anticoagulantes (Fig. 2)(19).
La permeabilidad de las células endote-liales puede verse alterada por daño direc-to, por excesiva producción del factor de cre-cimiento del endotelio vascular por el epitelioalveolar en respuesta a hipoxia, o por media-dores inflamatorios, como citocinas y oxi-dantes.
FIGURA 2. Mecanismos de disfunción endotelial y remodelado vascular en HAP. Modificado de Budhi-raja et al(19).
J.M. DÍEZ PIÑA
Mutaciones genéticas
Desequilibriohumoral
Proliferaciónendotelial
Incremento dela coagulación
Citocinas/factoresdel crecimiento
Exposición delsubendotelio a factores del crecimiento
Estrés mecánico Inflamación Autoinmunidad
Vasoconstricción, proliferación e hipertrofia de células músculo liso vascular y de la adventicia
Disfunción endotelial
Oclusión luz vascular
Monografía HAP NM 208 pag 18/2/10 17:08 Página 20

El endotelio vascular contribuye al man-tenimiento de la coagulabilidad mediante laelaboración de sustancias como factores humo-rales, heparina, trombomodulina, activadortisular del plasminógeno, activador del plas-minógeno tipo urocinasa, y el factor de vonWillebrand. Por tanto, una disfunción del mis-mo contribuye a la formación de procesostrombóticos.
Determinados estímulos, como pequeñasgrietas en el endotelio debidas a un flujo san-guíneo pulmonar aumentado, hipoxia alveo-lar, mutaciones genéticas (BMPR-2, ALK-1) ofenómenos inflamatorios, pueden producir unaactivación endotelial. Esta activación origina-ría una proliferación de células endoteliales,un incremento de la coagulabilidad, liberaciónde factores de crecimiento y disbalance entreéstos y mediadores vasoactivos, y una expo-sición del subendotelio a factores de creci-miento solubles. Como consecuencia, se pro-duce una obliteración de la luz vascular queorigina una vasoconstricción, hipertrofia y pro-liferación de la adventicia y del músculo lisoarterial.
El reflejo patológico de la disfunción endo-telial es la lesión plexiforme, una proliferaciónde células endoteliales que aparece en hiper-tensión pulmonar idiopática (HAPI) y que pare-ce tener un origen monoclonal. Estas lesiones,no sólo se ven en la HAPI, sino que tambiénse pueden ver en HAP secundarias a malfor-maciones cardiacas o secundarias a colage-nopatías. En estas lesiones se pueden apreciarmarcadores de angiogénesis, como el factorde crecimiento endotelial (VEGF)(20).
REMODELADO VASCULAR
Capa íntimaEl remodelado de la capa íntima puede ser
el resultado de daño endotelial y posterior acti-vación de la migración de células musculareslisas, depósito de matriz extracelular y proli-feración endotelial.
El factor de crecimiento endotelial (VEGF)es un factor mitogénico específico de la célu-
la endotelial, y un factor angiogénico que enla circulación pulmonar se une a las célulasendoteliales mediante dos tipos de receptores,el VEGF-R1 (FLT) y VEGF-R2 (KDR). En la HAPestán aumentados tanto la expresión del VEGFcomo los receptores VEGF-R1 en el endoteliopulmonar, y VEGF-R2 en las lesiones plexifor-mes(20). Estos hallazgos también se han obser-vado en pulmones sometidos a hipoxias agu-da y crónica(21).
En la HAPI, la progresión de las células endo-teliales hacia lesiones plexiformes y concéntri-cas puede deberse a mutaciones en genes supre-sores, como el gen del receptor 2 de la proteínamorfogénica ósea (BMPR2) (ver apartado: “Muta-ciones genéticas”), que también se expresa enlesiones plexiformes y en arterias pulmonaresremodeladas de pacientes con HAPI(22).
El VEGF, la ET-1 y la survivina están pre-sentes en las lesiones plexiformes y podríaninfluir aumentando la proliferación de célu-las endoteliales y musculares lisas, o dismi-nuyendo su apoptosis. Además, en estas lesio-nes están disminuidos factores como la NOS,la PGI2-sintasa y proteínas de supresión tumo-ral, como la caveolina 1 (CAV-1).
Capa mediaEl remodelado de la capa media incluye:
presencia de hiperplasia e hipertrofia de lacapa muscular; inhibición de la apoptosis celu-lar de la capa muscular y acúmulo de la matrizextracelular.
La hipoxia causa HAP al inducir hipertrofiay metaplasia de células precursoras potenciales,como los pericitos, apareciendo así músculo lisoen arterias precapilares que habitualmente notienen capa muscular(23).
Los déficits del receptor gamma activadodel peroxisoma (PPAR-γ) y de la apoproteínaE (apo-E) se han relacionado con la resisten-cia a la insulina, y esta resistencia puede serun factor que predisponga a HAP. En pacien-tes con HAPI se ha comprobado una menorexpresión de PPAR-γ. También existen evi-dencias que relacionan al factor de crecimientoderivado de las plaquetas (PDGF) con la pato-
PATOGENIA DE LA HIPERTENSIÓN ARTERIAL PULMONAR (HAP)
21
Monografía HAP NM 208 pag 18/2/10 17:08 Página 21

genia de la HAP. Este factor produce prolife-ración y migración de células musculares lisasvasculares. Se ha visto también que el recep-tor de PDGF está sobre-expresado en el tejidopulmonar de pacientes con HAP(24).
Adventicia y matriz extracelularLos fibroblastos de la adventicia son capa-
ces de diferenciarse en células musculares lisasante determinados estímulos de estrés, comola hipoxia, monocrotalina y otros, y así migrarhacia la capa media remodelada. Además, tam-bién pueden secretar factores de crecimientoque activan la proliferación de células muscu-lares lisas, permiten el reclutamiento de célu-las progenitoras de la médula ósea y crean unnicho favorable para la expansión de los nue-vos vasos(24).
El aumento de depósito de proteínas deltejido conectivo (colágeno y elastina) se harelacionado con el aumento de la capa mediade las arterias pulmonares musculares, cau-sante de una reducción de la luz vascular(24).
CÉLULAS INFLAMATORIAS Y PLAQUETASDeterminadas células inflamatorias, como
macrófagos y linfocitos, están aumentadas enlesiones plexiformes de los vasos de HAP. Unimportante porcentaje de pacientes con HAPpresentan niveles elevados de anticuerpos anti-nucleares, de citocinas proinflamatorias (IL-1y -6) y una mayor expresión pulmonar del fac-tor de crecimiento plaquetario y de la proteí-na 1-α de macrófagos. Esto proporciona unaevidencia de un componente de autoinmuni-dad y/o inflamación activa(25,26).
Existe en la bibliografía evidencia de uncomponente inflamatorio sistémico en la HAP,tal y como se puede ver en el estudio de Bala-banian y cols., que encontraron niveles signi-ficativamente elevados de varios marcadoresinflamatorios (factor de vonWillebrand, P-selec-tina, CD25, etc.) en pacientes con HAP gravefrente a controles(27).
La osteoprotegerina (OPG) es una gluco-proteína de 60 kDa que pertenece a la super-familia del factor de necrosis tumoral alfa (TNF-
α). Se expresa y se produce en varios tejidosque incluyen el corazón y el pulmón. Esta pro-teína está modulada por proteínas morfogené-ticas de hueso (BMP), serotonina e interleucina-1. En un reciente estudio, Lawrie y cols. hanrealizado estudios inmunohistoquímicos de lesio-nes de HAP y han demostrado un incrementoen la expresión de la osteoprotegerina, así comoniveles aumentados de la misma en plasma depacientes con HAP idiopática. En este mismotrabajo se demuestra que: la OPG recombinanteestimula la proliferación y migración de célulasdel músculo liso de la arteria pulmonar in vitro;la disminución de la función de BMP-R2 aumen-ta la secreción de OPG; y que la serotonina e IL-1 aumentan la secreción de OPG. Así demues-tran que la OPG está aumentada en HAP y quepuede regular la proliferación y migración decélulas musculares lisas de la arteria pulmonar,jugando así un papel importante en la patogé-nesis de la HAP(28).
ANOMALÍAS PROTROMBÓTICASLas alteraciones de la coagulación de la HAP
pueden ser debidas a la disfunción endotelial,a alteraciones en la cascada de la coagulacióny a una función plaquetaria alterada. Las lesio-nes trombóticas son frecuentes en la HAP, apa-reciendo entre un 20 y un 56% de pacientescon HAP esporádica o hereditaria, pudiendoencontrarse en cualquier tipo de HAP grave.Existe suficiente evidencia de que, en pacien-tes con HAP, se producen fenómenos de coa-gulación intravascular, detectándose niveles plas-máticos elevados de fibrinopéptido A ydímero-D. De la misma forma, la actividad pro-coagulante y fibrinolítica del endotelio pulmo-nar está alterada en la HAP, como se compruebaal detectar niveles altos del factor de von Wille-brand y del inhibidor tipo 1 del activador delplasminógeno. Tanto la HAP como otras formasde hipertensión pulmonar comparten la diáte-sis protrombótica descrita ampliamente en laliteratura(29). Este estado protrombótico puedecontribuir a la progresión de la HAP.
Cada vez está más aceptado que las anor-malidades vasculares de la HAP provocan una
J.M. DÍEZ PIÑA
22
Monografía HAP NM 208 pag 18/2/10 17:08 Página 22

liberación de factores procoagulantes, factoresvasoactivos (tromboxano A2, el factor activa-dor de plaquetas, serotonina) y mediadoresmitogénicos (factor del crecimiento derivadode las plaquetas, TGF-β y VEGF) por parte delas plaquetas. No obstante, no acaba de que-dar claro si la disfunción plaquetaria y la trom-bosis son realmente causa o consecuencia dela enfermedad(30).
Los pacientes con HAPI grave pueden pre-sentar trombocitopenia, lo cual se ha relacio-nado con la formación de agregados plaque-tarios secundarios a la activación de lasplaquetas por el endotelio(31). La plaquetope-nia suele asociarse a hemólisis microangiopá-tica que se produce por depósitos de fibrinaen las lesiones plexiformes.
MUTACIONES GENÉTICASLa mutación del gen que codifica el recep-
tor tipo 2 de las proteínas morfogenéticas delhueso (BMPR2) constituye el factor con mayorpredisposición genética a padecer HAP. Este genpertenece a la superfamilia del factor-β trans-formador de crecimiento y está involucrado enel control de la proliferación vascular celular.Mutaciones en este gen son responsables dealteraciones en la transducción de las proteínasmorfogenéticas del hueso, que conllevan a laproliferación de células musculares lisas en lasarterias pulmonares. No obstante, la penetran-cia es baja (en torno al 20%) y se desconocenaún los factores implicados en el inicio de laenfermedad y en el mecanismo molecular, quejustifica la responsabilidad de la mutación deBMPR2 en el desarrollo de la misma. En la hiper-tensión pulmonar arterial familiar se detectanmutaciones de este gen en al menos el 70% delos casos, habiéndose descrito ya hasta 140mutaciones(32,33).
Estas mutaciones también pueden serdetectadas en casos esporádicos en un por-centaje que varía entre el 10 y 25%. El estu-dio más amplio es el que incluía a 99 pacien-tes, de los cuales 11 eran portadores de lamutación(34). En porcentajes menores se hanvisto en HAP en pacientes con enfermedad
congénita cardiaca (6%)(35) y casos asociadosa fármacos anorexígenos(36). Por contra, no sehan visto mutaciones en pacientes con HAPasociada a esclerodermia(37) o SIDA(38).
Otras alteraciones genéticas descritas enla literatura son las del gen de la activina-kina-sa “receptor-like” tipo 1 (ALK-1), que se encuen-tra en el cromosoma 12q13, cromosoma enel que se encuentran también las alteracionesgenéticas de pacientes con telangiectasiahemorrágica hereditaria. Estas mutacionesgenéticas serían responsables de la dilataciónvascular característica de la telangiectasia y dela obstrucción de arterias pulmonares depequeño calibre de la HAP(39).
La variante alélica-L del gen promotor deserotonina se asocia con sobreexpresión deserotonina y mayor crecimiento de células demúsculo liso de arterias pulmonares. Además,esta mutación estaba presente en la formahomocigótica en el 65% de los pacientes, ysólo en 27% de controles. Por lo tanto, estoshallazgos sugieren que la serotonina tiene unpapel activo en la patogénesis de HAP prima-ria y que los polimorfismos de la misma le pro-porcionan susceptibilidad a padecerla(11).
CONCLUSIONESExisten múltiples mecanismos celulares,
moleculares y genéticos, implicados en el pro-ceso de instauración y mantenimiento de laHAP, sin que se conozca aún bien la interacciónentre ellos. Estos mecanismos originan un dese-quilibrio entre factores trombogénicos, mito-génicos, proinflamatorios y vasoconstrictores,por un lado, frente a mecanismos anticoagu-lantes, antimitóticos y vasodilatadores, por otro.
Es preciso conocer mejor estos mecanis-mos para desarrollar moléculas que actúendirectamente sobre ellos y controlar la pro-gresión de la enfermedad.
BIBLIOGRAFÍA1. Galiè N, Torbicki A, Barst R, et al. Guidelines
on diagnosis and treatment of pulmonary arte-rial hypertension. The Task Force on Diagno-sis and Treatment of Pulmonary Arterial Hyper-
PATOGENIA DE LA HIPERTENSIÓN ARTERIAL PULMONAR (HAP)
23
Monografía HAP NM 208 pag 18/2/10 17:08 Página 23

J.M. DÍEZ PIÑA
24
tension of the European Society of Cardiology.Eur Heart J. 2004; 25: 2243-78.
2. McLaughlin VV, McGoon MD. Pulmonary arte-rial hypertension. Circulation. 2006; 114: 1417-31.
3. Voelkel NF, Tuder RM. Hypoxia-induced pul-monary vascular remodeling: a model for whathuman disease? J Clin Invest 2000; 106: 733-8.
4. Giaid A, Saleh D. Reduced expression of endo-thelial nitric oxide synthase in the lungs ofpatients with pulmonary hypertension. N EnglJ Med. 1995; 333: 214-21.
5. Christman BW, McPherson CD, Newman JH,et al. An imbalance between the excretion ofthromboxane and prostacyclin metabolites inpulmonary hypertension. N Engl J Med. 1992;327: 70-5
6. Cox CP, Linden J, Said SI. VIP elevates plate-let cyclic AMP (AMPc) levels and inhibits invitro platelet activation induced by platelet-activating factor (PAF). Peptides. 1984; 5: 325-8.
7. Petkov V, Mosgoeller W, Zieske R, et al. Vaso-active intestinal peptide as a new drug for tre-atment of primary pulmonary hypertension.J Clin Invest. 2003; 111:1339-46.
8. Giaid A, Yanagisawa M, Langleben D, et al.Expression of endothelin-1 in the lungs ofpatients with pulmonary hypertension. N EnglJ Med. 1993; 328: 1732-9.
9. Eddahibi S, Raffestin B, Pham I, et al. Treat-ment with 5-HT potentiates development ofpulmonary hypertension in chronically hypo-xic rats. Am J Physiol. 1997; 272: 1173-81.
10. Eddahibi S, Fabre V, Boni C, et al. Induction ofserotonin transporter by hypoxia in pulmo-nary vascular smooth muscle cells. Relations-hip with the mitogenic action of serotonin. CircRes. 1999; 84: 329-36.
11. Eddahibi S, Hanoun N, Lanfumey L, et al. Atte-nuated hypoxic pulmonary hypertension inmice lacking the 5-hydroxytryptamine trans-porter gene. J Clin Invest. 2000; 105: 1555-62.
12. Eddahibi S, Humbert M, Fadel E, et al. Sero-tonin transporter overexpression is responsi-ble for pulmonary artery smooth musclehyperplasia in primary pulmonary hyperten-sion. J Clin Invest. 2001;108:1141-50.
13. Yuan XJ. Voltage-gated K+ currents regulateresting membrane potential and [Ca2+]i inpulmonary arterial myocytes. Circ Res. 1995;77: 370-8.
14. Yuan JX, Aldinger AM, Juhaszova M, et al.Dysfunctional voltage-gated K+ channels inpulmonary artery smooth muscle cells ofpatients with primary pulmonary hyperten-sion. Circulation. 1998; 98: 1400-6.
15. Rich S, Kaufmann E, Levy PS. The effect ofhigh doses of calcium-channel blockers on sur-vival in primary pulmonary hypertension. NEngl J Med. 1992; 327: 76-81.
16. Archer SL, Weir EK, Reeve HL, et al. Mole-cular identification of O2 sensors and O2-sensitive potassium channels in the pulmo-nary circulation. Adv Exp Med Biol. 2000;475: 219-40.
17. Takimoto K, Fomina AF, Gealy R, et al. Dexa-methasone rapidly induces Kv1.5 K+ channelgene transcription and expression in clonalpituitary cells. Neuron. 1993; 11: 359-69.
18. Wang J, Juhaszova M, Conte JV Jr, et al. Actionof fenfluramine on voltage-gated K+ channelsin human pulmonary-artery smooth-musclecells. Lancet. 1998; 352: 290.
19. Budhiraja R, Tuder RM, Hassoun PM. Endo-thelial dysfunction in pulmonary hyperten-sion. Circulation. 2004; 109: 159-65.
20. Tuder RM, Chacon M, Alger L, et al. Expres-sion of angiogenesis-related molecules in ple-xiform lesions in severe pulmonary hyper-tension: evidence for a process of disorderedangiogenesis. J Pathol. 2001; 195: 367-74.
21. Tuder RM, Flook BE, Voelkel NF. Increased geneexpression for VEGF and the VEGF receptorsKDR/Flk and Flt in lungs exposed to acute orto chronic hypoxia. Modulation of gene expres-sion by nitric oxide. J Clin Invest. 1995; 95:1798-807.
22. Atkinson C, Stewart S, Upton PD, et al. Pri-mary pulmonary hypertension is associatedwith reduced pulmonary vascular expressionof type II bone morphogenetic protein recep-tor. Circulation. 2002; 105: 1672-8.
23. Meyrick B, Reid L. Hypoxia and incorporationof 3H-thymidine by cells of the rat pulmonaryarteries and alveolar wall. Am J Pathol. 1979;96: 51-70.
24. Sala E, Palou A, Carrera M. Mecanismos pato-génicos en la hipertensión pulmonar. En: Suei-ro A, Gaudó J, ed. Tratado de hipertensión pul-monar. Barcelona: Ars Medica; 2009. p. 37-54.
25. Yamaguchi K, Kinosaki M, Goto M, et al. Cha-racterization of structural domains of humanosteoclastogenesis inhibitory factor. J BiolChem. 1998; 273: 5117-23.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 24

26. Dorfmüller P, Perros F, Balabanian K, et al.Inflammation in pulmonary arterial hyper-tension. Eur Respir J. 2003; 22: 358-63.
27. Balabanian K, Foussat A, Dorfmüller P, et al.CX(3)C chemokine fractalkine in pulmonaryarterial hypertension. Am J Respir Crit CareMed 2002; 165: 1419-25.
28. Lawrie A, Waterman E, Southwood M, et al.Evidence of a role for osteoprotegerin in thepathogenesis of pulmonary arterial hyperten-sion. Am J Pathol. 2008; 172: 256-64.
29. Humbert M, Morrell NW, Archer SL, et al. Cellu-lar and molecular pathobiology of pulmonaryarterial hypertension. J Am Coll Cardiol. 2004;43: 13S-24S.
30. Herve P, Humbert M, Sitbon O, et al. Patho-biology of pulmonary hypertension. The roleof platelets and thrombosis. Clin Chest Med.200; 22: 451-8.
31. Lopes AA, Maeda NY, Almeida A, et al. Circu-lating platelet aggregates indicative of in vivoplatelet activation in pulmonary hypertension.Angiology. 1993; 44: 701-6
32. Machado RD, Aldred MA, James V, et al. Muta-tions of the TGF-beta type II receptor BMPR2in pulmonary arterial hypertension. HumMutat. 2006; 27: 121-32.
33. Cogan JD, Pauciulo MW, Batchman AP, et al.High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial
hypertension. Am J Respir Crit Care Med.2006; 174: 590-8.
34. Koehler R, Grünig E, Pauciulo MW, et al. Lowfrequency of BMPR2 mutations in a Germancohort of patients with sporadic idiopathic pul-monary arterial hypertension. J Med Genet.2004; 41: 127.
35. Roberts KE, McElroy JJ, Wong WP, et al. BMPR2mutations in pulmonary arterial hypertensionwith congenital heart disease. Eur Respir J.2004; 24: 371-4.
36. Humbert M, Deng Z, Simonneau G, et al.BMPR2 germline mutations in pulmonaryhypertension associated with fenfluraminederivatives. Eur Respir J. 2002; 20: 518-23.
37. Morse J, Barst R, Horn E, et al. Pulmonaryhypertension in scleroderma spectrum of dise-ase: lack of bone morphogenetic protein recep-tor 2 mutations. J Rheumatol. 2002; 29: 2379-81.
38. Nunes H, Humbert M, Sitbon O, et al. Prog-nostic factors for survival in human immuno-deficiency virus-associated pulmonary arterialhypertension. Am J Respir Crit Care Med.2003; 167: 1433-9
39.- Trembath RC, Thomson JR, Machado RD, et al.Clinical and molecular genetic features of pul-monary hypertension in patients with here-ditary hemorrhagic telangiectasia. N Engl JMed. 2001; 345: 325-34.
PATOGENIA DE LA HIPERTENSIÓN ARTERIAL PULMONAR (HAP)
25
Monografía HAP NM 208 pag 18/2/10 17:08 Página 25

Monografía HAP NM 208 pag 18/2/10 17:08 Página 26

27
RESUMENLa hipertensión arterial pulmonar (HAP) es
una enfermedad grave, poco frecuente, de bajaprevalencia e incidencia, según lo han confir-mado los registros y estudios epidemiológicosrealizados en diferentes países: EE.UU., Fran-cia, Escocia (RU), China, etc. Desde su descrip-ción por Dresdale en 1954, conocemos quees una enfermedad con cierta tendencia fami-liar y actualmente disponemos de informacióny de las bases genéticas de la enfermedad. Laclínica es inespecífica y su diagnóstico se reali-za mediante procedimientos hemodinámicos,encontrándose, en general, los pacientes en unafase avanzada de la enfermedad. La HAP pre-domina en mujeres y con base familiar, estan-do el 75% de los pacientes diagnosticados enclase funcional III-IV de la OMS. Los pacientescon HAP presentan un mal pronóstico a corto-medio plazo si no se realiza una intervenciónterapéutica. La HAP idiopática es la entidad másfrecuente, seguidas de procesos asociados comola HAP de origen tromboembólico, asociadas aconectivopatías, hipertensión portal, HIV, tóxi-cos, etc. El reciente IV Simposio Internacionalde HP de Dana-Point (California), febrero de2008, ha establecido una nueva clasificación dela HAP, así como modificaciones en la estrate-gia del tratamiento, que recoge los avances tera-péuticos en esta enfermedad, basado en nive-les de evidencia científica.
INTRODUCCIÓNLa circulación pulmonar presenta unas
características muy diferentes a la circulación
sistémica, siendo un sistema de baja resistenciay baja presión, con gran distensibilidad vas-cular y escaso control vasomotor. La resisten-cia vascular pulmonar están muy disminuidasy representan casí un sexto de la resistenciavascular sistémica, la presión arterial mediapulmonar en un sujeto sano es de 13 mmHg,con intervalo entre 22/8 mmHg.
La hipertensión arterial pulmonar (HAP) esuna enfermedad infrecuente, progresiva, aso-ciada a mal pronóstico y elevada mortalidad.La supervivencia media tras el diagnóstico enlos pacientes no tratados se sitúa en una mediade edad inferior a los tres años. La introduc-ción de nuevos tratamientos antihipertensivospulmonares desde la década de los años noven-ta, ha mejorado la supervivencia y la esperanzade vida en estos pacientes, pero su pronósti-co a largo plazo conlleva una elevada morta-lidad próxima al 30% a los tres años.
La aparición de HAP se asocia a un incre-mento progresivo de las resistencias vascularespulmonares (RVP), con elevación de la presiónvascular en la circulación pulmonar y fallo delcorazón derecho y muerte del paciente. La HAPse caracteriza por importantes cambios en lavascularización pulmonar que conducen a unremodelado progresivo de la pared de las arte-rias pulmonares de mediano y pequeño calibres,apreciándose: fibrosis concéntrica de la íntima,hipertrofia de la capa media, engrosamiento dela adventicia, incremento de la muscularizaciónarteriolar y desarrollo de fenómenos trombóti-cos locales, con degeneración fibrinoide y reduc-ción del lecho vascular pulmonar(1,2).
DEFINICION, CLASIFICACION,EPIDEMIOLOGÍA Y GENERALIDADES DELA HIPERTENSION ARTERIAL PULMONAR
José Javier Jareño Esteban, José Ignacio de Granda Orive, José Francisco Villegas Fernández
Monografía HAP NM 208 pag 18/2/10 17:08 Página 27
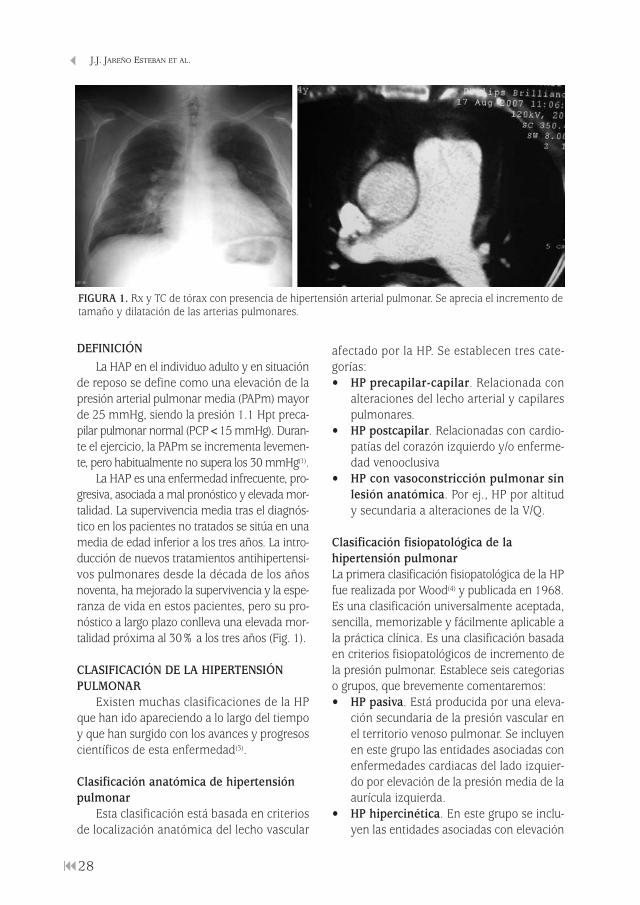
DEFINICIÓN La HAP en el individuo adulto y en situación
de reposo se define como una elevación de lapresión arterial pulmonar media (PAPm) mayorde 25 mmHg, siendo la presión 1.1 Hpt preca-pilar pulmonar normal (PCP<15 mmHg). Duran-te el ejercicio, la PAPm se incrementa levemen-te, pero habitualmente no supera los 30 mmHg(1).
La HAP es una enfermedad infrecuente, pro-gresiva, asociada a mal pronóstico y elevada mor-talidad. La supervivencia media tras el diagnós-tico en los pacientes no tratados se sitúa en unamedia de edad inferior a los tres años. La intro-ducción de nuevos tratamientos antihipertensi-vos pulmonares desde la década de los añosnoventa, ha mejorado la supervivencia y la espe-ranza de vida en estos pacientes, pero su pro-nóstico a largo plazo conlleva una elevada mor-talidad próxima al 30% a los tres años (Fig. 1).
CLASIFICACIÓN DE LA HIPERTENSIÓNPULMONAR
Existen muchas clasificaciones de la HPque han ido apareciendo a lo largo del tiempoy que han surgido con los avances y progresoscientíficos de esta enfermedad(3).
Clasificación anatómica de hipertensiónpulmonar
Esta clasificación está basada en criteriosde localización anatómica del lecho vascular
afectado por la HP. Se establecen tres cate-gorías:• HP precapilar-capilar. Relacionada con
alteraciones del lecho arterial y capilarespulmonares.
• HP postcapilar. Relacionadas con cardio-patías del corazón izquierdo y/o enferme-dad venooclusiva
• HP con vasoconstricción pulmonar sinlesión anatómica. Por ej., HP por altitudy secundaria a alteraciones de la V/Q.
Clasificación fisiopatológica de lahipertensión pulmonarLa primera clasificación fisiopatológica de la HPfue realizada por Wood(4) y publicada en 1968.Es una clasificación universalmente aceptada,sencilla, memorizable y fácilmente aplicable ala práctica clínica. Es una clasificación basadaen criterios fisiopatológicos de incremento dela presión pulmonar. Establece seis categoriaso grupos, que brevemente comentaremos: • HP pasiva. Está producida por una eleva-
ción secundaria de la presión vascular enel territorio venoso pulmonar. Se incluyenen este grupo las entidades asociadas conenfermedades cardiacas del lado izquier-do por elevación de la presión media de laaurícula izquierda.
• HP hipercinética. En este grupo se inclu-yen las entidades asociadas con elevación
J.J. JAREÑO ESTEBAN ET AL.
28
FIGURA 1. Rx y TC de tórax con presencia de hipertensión arterial pulmonar. Se aprecia el incremento detamaño y dilatación de las arterias pulmonares.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 28

de la presión pulmonar por aumento delflujo pulmonar, como los cortocircuitos sis-témico-pulmonares.
• HP obstructiva-obliterativa. Existe enestos casos una obstrucción del lecho vas-cular y reducción efectiva de la luz. Sonejemplos de este grupo la HP trom-boembólica.
• HP vasoconstrictiva. El mecanismo funda-mental es la vasoconstricción vascular rever-sible, demostrada mediante estudio hemo-dinámico y test de vasodilatador. Ejemplosde este grupo son la HP asociada a la altitudy secundaria a trastornos de ventilación-per-fusión.
• HP reactiva. Producida en situaciones enque la resistencia vascular pulmonar se ele-va de forma fija y constante, observándoseen situaciones de HP pasiva, hipercinética ovasoconstrictiva.
• HP primaria o idiopática. Situaciones deHP sin identificar la causa o tras la exclu-sión de otras entidades responsables.Con posterioridad a esta clasificación, algu-nos autores han introducido otra entidadconocida como –HP poligénica– que englo-baría las entidades en las cuales hay variosmecanismos fisiopatológicos implicados(5).
Clasificación diagnóstica de la hipertensiónpulmonar
Desde una visión diagnóstica y a lo largode los últimos cuarenta años se han realiza-do varias clasificaciones de la HP, que respondea la necesidad de incorporar a la misma, losavances cientificos que se han ido producien-do en los últimos años. Como resumen de estaevolución, seguidamente comentaremos lasdiferentes clasificaciones y los cambios expe-rimentados en las mismas.
Clasificación de la OMS (Ginebra-Suiza)La primera clasificación de la HP fue publi-
cada por la OMS en 1973. En la década de lossesenta y a raíz de la epidemia de casos con HAPque aconteció en Europa y Norteamérica por laadministración del anorexígeno-aminorex, cre-
ció el interés por el estudio de la HP, realizán-dose el primer consenso auspiciado por la Orga-nización Mundial de la Salud (OMS) en Gine-bra (Suiza), estableciéndose las bases de unanueva clasificación. Las principales conclusio-nes obtenidas del consenso OMS estuvieron rela-cionadas con la necesidad de establecer una cla-sificación clínica nueva, diferenciada de laclasificación histológica.
Respecto a esta última, se consensuó esta-blecer tres patrones histopatológicos: la arte-riopatía plexogénica, la tromboembólica y lavenooclusiva. Finalmente se decidió fomentary crear registros internacionales para el estu-dio de la HP, dada la baja prevalencia e inci-dencia de esta enfermedad, así surgió el Regis-tro Nacional Americano de HAP idiopática, quecaracterizaría a esta enfermedad (Fig. 2).
Clasificación de HP de Evian (Francia)De características muy similares a lo que
aconteció con la primera reunión de HAP, sedescribió en los países occidentales un aumen-to de la incidencia de HAP relacionada con laingesta de anorexígenos (fenfluramina y dex-fenfluramina). Asimismo, es de destacar la epi-demia de HAP que se relacionó en nuestro paíscon la ingesta del aceite de colza desnaturali-zado (síndrome tóxico)(1981)(6,7). Todo ellomotivó la celebración de un nueva reunióninternacional de HP, coordinada por el Dr Richy patrocinada por la OMS, cuyo lugar de reu-nión fue Evian (Francia) en 1998.
El documento de consenso en HP elabo-rado en la II Conferencia Internacional de HAP,supuso un punto de inflexión en la clasifica-ción y manejo de la de la HP. Se estableció porel grupo de expertos una nueva clasificaciónque comprende los aspectos anatomopatoló-gicos, diagnósticos y funcionales, de la HP. Seestablecieron cinco grandes grupos para la cla-sificación de la HP: 1. HP primaria; 2. HP veno-sa pulmonar; 3. HP asociada a enfermedadesrespiratorias y/o hipoxemia; 4. HP debida atromboembolismo crónico y/o enfemedadesembólicas; 5. HP debido a enfermedades queafectan a la vasculatura pulmonar(8).
DEFINICIÓN, CLASIFICACIÓN, EPIDEMIOLOGÍA Y GENERALIDADES DE LA HIPERTENSIÓN ARTERIAL PULMONAR
29
Monografía HAP NM 208 pag 18/2/10 17:08 Página 29

Clasificación de HAP de Venecia 2003(Italia)
En 2003 tuvo lugar la penúltima y III Con-ferencia de HP en Venecia 2003. Era necesa-ria una nueva reunión de expertos que reco-giera los avances científicos que se habíanproducido en los últimos años (genéticos, diag-nósticos, terapéuticos, etc.).
La primera modificación consistió en reco-ger los avances y descubrimientos en la gené-tica, con el decubrimiento en las mutacionesdel gen que codifica el receptor tipo II de laproteína morfogenética ósea (BMPR2), que
ocurre hasta en el 50% de los HP familiares yen el 20% de las HP esporádicas(9,10). Sinembargo, los expertos también quisieron queno se podía establecer una clasificación por elmomento en base a las determinaciones gené-ticas.
Se produjo el abandono del término HPprimaria acuñado 50 años antes por Dresda-le, por el término idiopática, englobando enesta entidad y grupo los casos de HP sin unacausa etiológica conocida.
También se produjeron cambios en el agru-pamiento de entidades como la enfermedad
J.J. JAREÑO ESTEBAN ET AL.
30
TABLA 1. Clasificación de la HP (Venecia-2003)
1. Hipertensión arterial pulmonar
• Idiopática
• Familiar
• Asociada a ETC, CC, HPT portal, fármacos, toxinas, etc.
• HAP con alteración significativa venosa y/capilar
• HAP persistente del recién nacido
2. Hipertensión pulmonar con enfermedad del corazón izquierdo
• Enfermedad cardiaca de la aurícula o ventrículo izquierdo
• Enfermedad valvular (mitral y aórtica)
3. Enfermedad pulmonar asociada a enfermedad pulmonar y/o hipoxemia
• EPOC
• Enfermedad pulmonar intersticial difusa (EPID)
• Trastornos respiratorios durante el sueño: SAHS, hipoventilación alveolar, exposición a elevadasalturas
• Anomalías del desarrollo
4. HP asociada a enfermedad trombótica y/o embólica
• Obstrucción tromboembólica de las arterias pulmonares proximales
• Obstrucción tromboembólica de las arterias pulmonares distales
• Embolismo pulmonar asociado (tumor, parásitos, material extraño)
5. Miscelánea
• Sarcoidosis, Histiocitoxis X, linfangioleiomiomatosis, compresión de los vasos pulmonares (ade-nopatías, tumores, mediastinitis fibrosante, etc.)
Abreviaturas: ETC: enfermedad del tejido conectivo; EPOC: enfermedad pulmonar obstructiva crónica; EPID: enfer-medad pulmonar intersticial difusa; VIH: virus de la inmunodeficiencia humana; RN: recién nacido; CC: cardiopa-tía congénita.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 30

venooclusiva pulmonar y de la hemangioma-tosis capilar pulmonar, al conocerse avancescientíficos que las relacionan en la etiopato-genia e histopatología.
El grupo de cardiopatías congénitas aso-ciadas a shunt sistémico-pulmonar sufrió modi-ficaciones, que permitiesen una mejor iden-tificación de los grupos de máximo riesgo dedesarrollo de HP y que, a su vez, permitieraestablecer un pronóstico y una terapéutica ade-cuada(11).
Clasificación de HP en Dana-Point 2008 CA (EE.UU.)
Finalmente y como todos conocemos, enfebrero de 2008 se celebró la IV ConferenciaInternacional de HP en Dana-Point (California)(EE.UU)(12) (Tabla II).
En la misma se han introducido algunoscambios que es necesario comentar.
El Grupo I se caracteriza por presentar cin-co grandes divisiones: idiopática; hereditaria yésta, a su vez, subdividida según la base gené-tica (BMPR2, ALK-1, desconocida); asociada afármacos y tóxicos; asociada a entidades con(conectivopatías, VIH, hipertensión portal, cor-tocircuito shunt sistémico pulmonar, esquisto-somiasis, anemia hemolítica crónica); e HP delrecién nacido.
Se establece un subgrupo especial de HPpor enfermedad venooclusiva y/o hemangio-matosis capilar.
El Grupo II, HP pulmonar secundaria a car-diopatía izquierda se ha realizado un subdivi-sión en tres entidades: disfunción sistólica, dis-función diastolica y enfermedad valvular. Estoes fruto del incremento de la frecuencia deobservación en clínica de disfunción diastóli-ca observada predominantemente en pacien-tes de tercera edad y con riesgo cardiovascu-lar (HTA, diabetes mellitus, etc.)(13).
El Grupo IV, encuadrado en la HP de ori-gen tromboembólico crónico, se modifica sinreferirse a la localización de la lesión pulmo-nar: proximal, distal, mixta, etc.
El Grupo V, HP de mecanismo incierto omultifactorial, engloba a un conjunto mucho
más amplio de procesos y entidades, difíci-les de encuadrar en otros grupos y se incre-menta respecto a la clasificación de HPT enVenecia.
EpidemiologíaEn este apartado vamos hacer referencia
a los principales estudios epidemiológicosactualmente publicados en HP.
Registro NIH (EE.UU.)La historia natural de la HAP idiopática
(HAPI) es bien conocida, desde la realizaciónpor el Instituto Nacional de la Salud de losEE.UU. entre 1981-1987 del Registro Nacionalde pacientes con HAP primaria (NIH). El estu-dio realizado entre 32 centros reclutó a 187pacientes, con una media de edad de 36 años,a los que se realizó un seguimiento de cincoaños. Un 6,4% de los pacientes presentabanHAP familiar, siendo la relación mujer/varónde 1,7:1, esta relación aumentaba hasta alcan-zar 4:1 entre la población de raza negra. Lamayor prevalencia parecía ocurrir en la 3ª y4ª décadas de la vida, aunque un (9%) de loscasos se presentan en edades más avanzadas.La media de la supervivencia de los pacientesincluidos fue de 2,8 años: (1º año: 68%; 2ºaño: 48%; 3º año: 34%). Desde la publica-ción de estos resultados conocemos cómo laHAPI es un enfermedad infrecuente, con unaincidencia de 1-2 casos millón de habitantes,de mal pronóstico, asociada a una baja super-vivencia sin tratamiento, establecida en 2,8años. Posteriormente otros registros realiza-dos han refrendado la validez de estos resul-tados(9,10,14) (Fig. 2).
Registro de HAP en Francia El registro de HAP realizado en Francia
constituye actualmente para los investigado-res y científicos la referencia epidemiológicaen HAP. Se realizó, de forma prospectiva y mul-ticentrica, en 17 hospitales universitarios deFrancia, con unidad de HAP y durante los años2002 y 2003. Se incluyeron 674 pacientes, conuna edad media de 50 ±15 años, rango (18-
DEFINICIÓN, CLASIFICACIÓN, EPIDEMIOLOGÍA Y GENERALIDADES DE LA HIPERTENSIÓN ARTERIAL PULMONAR
31
Monografía HAP NM 208 pag 18/2/10 17:08 Página 31

J.J. JAREÑO ESTEBAN ET AL.
32
TABLA 2. IV. Clasificación Internacional de Hipertensión Pulmonar (HP)
1. Hipertensión arterial pulmonar (HAP)
• Idiopática
• Hereditaria: BMPR2
ALK-1
Desconocida
• Inducida por fármacos o tóxicos
• Asociada:
- Enfermedades del tejido conectivo (ETC)
- Infección por VIH
- Hipertensión portal
- Cortocircuito (shunt) sistémico-pulmonar
- Esquistosomiasis
- Anemia hemolitica crónica
- Hipertensión pulmonar persistente del recién nacido
1’. Enfermedad venooclusiva pulmonar y/o hemangiomatosis capilar pulmonar
2. Hipertensión pulmonar debida a cardiopatía izquierda
• Disfunción sistólica
• Disfunción diastólica
• Enfermedad valvular
3. Hipertensión asociada a enfermedad pulmonar y/o hipoxemia
• Enfermedad pulmonar obstructiva crónica (EPOC)
• Neumopatias intersticiales
• Otras neumopatías
• Trastornos respiratorios durante el sueño
• Exposición crónica a grandes alturas
• Anomalías del desarrollo
4. Hipertensión pulmonar tromboembólica crónica
5. Hipertensión pulmonar de mecanismo incierto o multifactorial
• Trastornos hematológicos (trastornos mieloproliferativos, esplenectomia)
• Trastornos sistémicos (vasculitis, sarcoidosis, histiocitoxis pulmonar-enfermedad de Langerhans,linfangioleomiomatosis, neurofibromatosis)
• Trastornos metabólicos (enfermedad por almacenamiento del glucógeno, enfermedad de Gau-cher, enfermedades tiroideas)
• Cardiopatías congénitas (distintas de cortocircuito sistémico-pulmonar)
• Otras entidades (obstrucción tumoral, mediastinitos fibrosante, insuficiencia renal crónica/diáli-sis, otras)
Dana Point-2008 (Simmoneau et al.J Am Coll Cardiol. 2009;54: S43–54).
Monografía HAP NM 208 pag 18/2/10 17:08 Página 32

85 años) a los que se realizó un seguimientode 3 años. Los pacientes diagnósticados deHAP correspondían a las siguientes etiologías:idiopática (39,2%); familiar (3,9%); anorexí-genos (9,5%); conectivopatías (15,3%); car-diopatías congénitas (11,3%); hipertensión por-tal (10,4%; asociada a HIV(6,2%).
El 75% de los pacientes se encontrabanen clases funcionales III y IV (NYHA). El testde marcha de 6 minutos fue: 329±109 metros.Todos los pacientes fueron diagnósticadosmediante estudio hemodinámico: la presiónarterial media (PAP) fue de 55±15 mmHg;el índice cardiaco (IC), de 2,5±0,8 L/min/m2
y, finalmente, la resistencia vascular pulmonar(RVP) de 20,5±10,2 mmHg/L/min/m2. El regis-tro permitió establecer las caracteristicas epi-demiológicas de la HAP en Francia. La preva-lencia de HAP estimada en la población fue de15 casos/millón de habitantes adultos y la inci-dencia de 2,4 casos/millón de habitantes adul-tos. La supervivencia a un año desde el diag-nóstico de los pacientes incluidos fue del88%(15).
Registro de HAP en Escocia (RU)El registro de HAP realizado en Escocia (RU)
y publicado en 2007, está basado en los datosregistrados de pacientes diagnosticados deHAP en la Unidad Vascular Pulmonar Esco-cesa, Hospital Western Infirmary (Glasgow),centro de referencia para el estudio de HAP enEscocia. El registro incluye a pacientes entrelos 16-65 años, durante 1997-2006. Se regis-
traron 374 pacientes, la incidencia anual fuede 7.6 casos por millón de habitantes y la pre-valencia entre 26 casos por millón de habi-tantes. La HAPI fue la entidad más frecuente,seguida por la HAP asociada a conectivopatí-as, cardiopatías congénitas, portopulmonar,tóxicos y drogas, asociada a HIV(16).
Registro de HAP en ChinaEn el año 2007, se han publicado los resul-
tados de un estudio epidemiológico realizadoen China, cuyo objetivo fue conocer las mani-festaciones clínicas y la supervivencia enpacientes con HAPI y familiar. El estudio serealizó entre 1994 y 2004. Se incluyeron 72pacientes (51 mujeres; 21 varones, con unarelación mujer-varón de 2.4:1. La media deedad fue de 35,9±12 años, rango (9,7 y 73,8años). La incidencia de HAP familiar se regis-tró en 4 casos (5,6%). La clase pronóstico seestableció según la clasificación de la OMS: I(2 %); II (37%); III (44%); IV (17%).
Todos los pacientes fueron diagnósticadosmediante estudio hemodinámico. La PAPmedia fue de: clase funcional I/II (59,1±14)mmHg y para los pacientes en clase funcionalIII/IV (69±18,8) mmHg. El seguimiento clí-nico se realizó entre 40±20 meses. La super-vivencia registrada fue: 1er año (68%); 2º año(56%); 3er año (38,9%) y 5º año (20,8%). Seapreció una diferencia estadísticamente sig-nificativa entre los pacientes en clase funcio-nal I/II y los pacientes en clase funcional III/IV(p=0,02) (log rank test). Los autores, una vez
DEFINICIÓN, CLASIFICACIÓN, EPIDEMIOLOGÍA Y GENERALIDADES DE LA HIPERTENSIÓN ARTERIAL PULMONAR
33
FIGURA 2. Curva de supervivenciaen pacientes con HAP. Registro Ame-ricano 1987(9).
5,54,54,03,53,02,52,01,51,00,50
20
40
60
80
100Kaplan-Meier estimate of survival
Year of follow-up
Monografía HAP NM 208 pag 18/2/10 17:08 Página 33

analizados los datos obtenidos en el registro,aconsejan el establecimiento de una estrate-gia terapéutica en pacientes con HAPI y fami-liar en China(17).
REHAPEn España y, a iniciativa de la Sociedad
Española de Cardiología (SEC) y de Neumo-logía (SEPAR), disponemos de un RegistroNacional de pacientes con HAP, es el registroREHAP (www.rehap.org), que incluye a másde 1.000 pacientes y cuyo objetivo es conocerla incidencia, diagnóstico y tratamiento de laHAP en nuestro país. La distribución de loscasos registrados corresponde por frecuencias:idiopáticos (39%), (18%) asociadas a ETC,(15%) cardiopatías congénitas, (12%) trom-boembólica y, con menor frecuencia, otras enti-dades (HIV, hipertensión portal, drogas y acei-te tóxico, etc.). A finales de julio 2008, laincidencia de HAP en dicho registro era de 3,3casos/millón/año y la prevalencia, de 15,3casos/millón/año(18).
REVEALEn EE.UU. disponen del registro REVEAL
(Registry to Evaluate Early and Long –term PAHDisease Management), registro prospectivo ymúlticentrico realizado en 56 centros y con3.500 pacientes incluidos y que pretende dara conocer las tendencias de la HAP, procedi-mientos diagnósticos, terapéuticos, etc., cuyafinalización está prevista para 2012.
QUERIRecientemente, se han comunicado en la
ATS 2009 (EE.UU.), los resultados prelimina-res del estudio PAH-QUERI (Quality Enhance-ment Research Initiative). Sus objetivos con-sisten en conocer las prácticas clínicas, calidadde vida e intervención diagnóstica y terapéu-tica en pacientes con HAP. El estudio incluyó786 pacientes con HAP, reclutados entre losaños 2006 y marzo del 2009. El 37% presen-taban HAP idiopática; 3,7%, origen familiar;30% asociada ETC; 3,7%, cardiopatias con-génitas; 7%, drogas; 3,8%, HAP de origen por-
tal, 3,9%, asociada a HIV y otras causas, 8%.El 9% se encontraban en clase funcional I,38% clase II, 47% clase III y 5% clase IV. Laedad media de los pacientes fue de 55 años,con un índice de masa corporal de 27 kg/m2,siendo el 77% mujeres. El estudio hemodiná-mico reveló un test de vasorreactividad posi-tivo del 11%(19).
BIBLIOGRAFÍA1. Sinmonneau G, Galié N, Rubin LJ. Clinical clas-
sification of pulmonary hypertension. J AmColl Cardiol. 2004; 43 (Suppl. 12): 5S-12S.
2. Humbert M. More pressure on pulmonaryhypertension. ERR. 2009; 18 (111): 1-3.
3. Rubin LJ. Primary pulmonary hypertensionACCP Consensus Statement. Chest. 1993; 104:236-50.
4. Wood P. Diseases of the heart and circulation.Philadelphia: JB Lippincot; 1968. p. 976-83.
5. D’ Alonzo GR, Dantzker DR. Diagnosing pri-mary pulmonary hypertension. En: Rubin LJ,Rich S, editors. Primary pulmonary hyper-tension. New York: Marcel Decker, Inc., 1997.p. 227-52.
6. Gómez-Sánchez MA. Hipertensión arterial pul-monar. Revista Española de Cardiología 2002;Vol 2: 1-2.
7. Sáenz de la Calzada C, Sanz Calvo J, PindadoC, et al. Concepto y clasificación de la hiper-tensión pulmonar. Revista Española de Car-diología. 2002; 2: 3-8.
8. Rich S. Executive summary from the Worldsymposiun on primary pulmonary hiperten-sion 1998. World Health Organization websi-te. Disponible en: www,who.in/ cvd/pph. html.
9. Rich S, Dantzker DR, Ayres S, et al. Primarypulmonary Hyprtension. A National Prospec-tive Study. Ann Intern Med. 1987; 107: 216-23.
10. D`Alonzo GE, Bart RJ, Ayres SM, et al. Survi-val in patients with primary pulmonary hyper-tension. Ann Intern Med. 1991; 115: 343-9.
11. Jiménez C, Escribano P, Sáenz de la CalzadaC. Epidemiología y clasificación de la hiper-tensión arterial pulmonar. En: Hipertensiónpulmonar. M.A. Gómez Sánchez. Madrid: Edi-torial Ergon; 2008. p. 11-26.
12 . 4th World Symposium o Pulmonary Hiperten-sion; 2008 Feb 11-14; Dana Point CA (EE.UU)(En prensa). J Am Coll Cardiol. 2009; 54: S43-54.
J.J. JAREÑO ESTEBAN ET AL.
34
Monografía HAP NM 208 pag 18/2/10 17:08 Página 34

13. Sueiro A, Gaudó J. Tratado de HipertensiónArterial Pulmonar. Barcelona: Editorial Ars XXI;2009.
14. Mc Laughlin V, MD McGoon MD. PulmonaryArterial Hypertension. Circulation. 2008:141:7-31.
15. Humbert M, Sitbon O, Chaouat A, et al. Pulmo-nary Arterial Hypertension in France. Am J Res-pir Critical Care Medicine 2006; 173: 1023-30.
16 . Peacock AJ, Murphy NF, Mc Murray JV, et al. Anepidemiological study o pulmonary arterialhypertension. Eur Respir J. 2007; 30: 104-9.
17. Zhi Cheng Ping, Xi-Qi Xu, Zhi-Yan Han, et al.Registry and survival study in Chinesepatients with idiopathic and familial pulmo-nary arterial hypertension. Chest. 2007; 132:373-9.
18. Escribano P, Morales P. Registro de hiperten-sión pulmonar en España. www. rehap.org.
19. Mc Lauglin V, Langer A, Dragomir A, et al. Tre-atment of pulmonary aterial hypertension(PAH). Dat from the Quality Enhacement Rese-arch Initiative. ATS International Conference.San Diego (California) EE.UU. 2009.
DEFINICIÓN, CLASIFICACIÓN, EPIDEMIOLOGÍA Y GENERALIDADES DE LA HIPERTENSIÓN ARTERIAL PULMONAR
35
Monografía HAP NM 208 pag 18/2/10 17:08 Página 35

Monografía HAP NM 208 pag 18/2/10 17:08 Página 36

37
GENERALIDADES DIAGNÓSTICAS EN LA HIPERTENSIÓN PULMONAR
Fernando Pedraza Serrano, Javier de Miguel Díez, Gema Sánchez Muñoz
RESUMENEn los últimos años, gracias al desarrollo de
nuevos fármacos para el tratamiento de la hiper-tensión pulmonar (HP), se han publicado variasguías de práctica clínica donde se estandarizanlos métodos a seguir para llegar al diagnósticode este proceso. También, debido a ello se haconseguido mejorar la calidad de vida de muchospacientes con HP y prolongar la esperanza devida en las fases más graves de la enfermedad.Pero, la mayoría de las veces, estos tratamientosrequieren formas de administración complejasy presentan, en muchas ocasiones, efectos secun-darios que pueden llegar a ser graves. Por todasestas razones se sigue recomendando efectuarel tratamiento y el seguimiento de los pacientescon HP en unidades de referencia donde hayaprofesionales expertos en la materia.
Aunque es recomendable realizar el mane-jo de estos enfermos en unidades de referencia,en los centros menos especializados puede ydebe hacerse un diagnostico correcto. En estecapítulo se profundiza en este aspecto, propor-cionando pautas y algoritmos diagnósticos quepueden ser útiles para realizar un estudio com-pleto de la enfermedad y, una vez completadoel diagnostico, plantear el tratamiento a seguir.
ESTRATEGIA DIAGNÓSTICA EN LAHIPERTENSIÓN ARTERIAL PULMONAR
El esquema a seguir para llegar al diagnós-tico de la hipertensión pulmonar (HP) consta de4 fases. A lo largo de este capítulo se desarro-llan, una por una, cada una de ellas (Fig. 1)(1-4).
Sospecha clínicaEs el primer aspecto a tener en cuenta ya
que, si no se sospecha la enfermedad, nuncase podrá llegar a diagnosticar. Es imprescin-
dible incluir esta entidad en nuestro esquemamental de diagnostico diferencial. Además, setrata de una enfermedad que, en sus fases ini-ciales, presenta muy pocos síntomas y la explo-ración física del paciente suele ser bastanteanodina, por lo que es necesario estar atentosa pequeños detalles para sospecharla. En otrasocasiones la sospecha clínica aparece antealgún hallazgo electrocardiográfico o en laradiografía de tórax de un paciente que se harealizado estas pruebas por algún motivo aje-no a la sospecha clínica de HP como, por ejem-plo, un estudio preoperatorio.
Muchos pacientes consultan inicialmentepor disnea de esfuerzo. Otros, aunque conmenos frecuencia, por síncopes o presíncopes,cansancio, debilidad o dolor torácico. No obs-tante, en muchas ocasiones no suelen encon-trarse signos claros de enfermedad cardiaca opulmonar.
En la exploración física del paciente rara vezaparecen síntomas que orienten al diagnóstico,pero, en algunas ocasiones, pueden detectarselos siguientes: un segundo ruido aumentado enla auscultación cardiaca, un pulso en el bordeparaesternal izquierdo que puede correspondera la arteria pulmonar, un soplo sistólico de regur-gitación tricúspide, uno diastólico de insufi-ciencia pulmonar o un tercer o cuarto tono car-diaco. Si la enfermedad se encuentra en fasesmás avanzadas pueden objetivarse signos deinsuficiencia cardiaca derecha como edemasen miembros inferiores, hepatomegalia, ascitis,ingurgitación yugular o cianosis. La auscultaciónpulmonar es normal en la mayoría de las oca-siones. Cuando la enfermedad está más avan-zada pueden detectarse crepitantes bibasales.
Debido a que los síntomas y los signos sonescasos en la mayoría de las ocasiones, a la
Monografía HAP NM 208 pag 18/2/10 17:08 Página 37

vez que poco específicos, debe mantenerse unalto índice de sospecha en aquellas enferme-dades que pueden provocar HP secundaria(Tabla 1)(4).
Detección de la hipertensión arterialpulmonar
Los tres estudios iniciales que deben soli-citarse para llegar a detectar una HP son el
F. PEDRAZA SERRANO ET AL.
38
FIGURA 1. Algoritmo de diagnóstico en la hipertensión pulmonar(4).
HP: hipertensión pulmonar; VIH: virus de inmunodeficiencia humana; EVO: enfermedad venooclusiva; HCP: heman-giomatosis capilar pulmonar; CCD: cateterismo cardiaco derecho. TACAR: tomografía axial computarizada de altaresolución; OMS: Organización Mundial de la Salud.
Arteriografía y CCD
Patología cardiaca izquierdaCardiopatía congénita
TACAR y CCD
Población de riesgo para HPSíntomas clínicos y signos físicos Sospecha clínica de HPElectrocardiogramaRadiografía de tórax
Completar estudio etiológico
Grado de disnea (clase funcional de la OMS)Test de la marcha de 6 minutos
Ergometría con consumo de oxígeno
- Analítica (incluyendo hormonas tiroi-deas, pruebas de autoinmunidad, estu-dio de hipercoagulabilidad y serologíapara VIH y virus B y C)
- Pruebas de función respiratoria- Gammagrafía V/Q- Ecografía abdominal- TACAR en algunos casos
Sospecha deEVO o HCP
HTP secundaria apatología pulmonar
Si se sospecha HPtromboembólica
Angio-TAC
Ecocardiograma
Sospecha de HP secundaria y formas asociadas
Confirmar la HP con CCD (incluyendotest vasodilatador)
Valoración final
Monografía HAP NM 208 pag 18/2/10 17:08 Página 38

electrocardiograma (ECG), la radiografía detórax y el ecocardiograma transtorácico (ETT),siendo este último el más importante de todos.
Electrocardiograma El electrocardiograma puede orientar
hacia el diagnóstico de sospecha de HP, perotiene baja sensibilidad y especificidad (del 55al 70% en ambos casos)(5). De esta manera,no puede considerarse como una herramientaóptima para la detección de una HP signifi-cativa. En esta exploración podemos obser-var signos de sobrecarga del ventrículo dere-cho con crecimiento de la aurícula derecha,desviación del eje del QRS hacia la derecha,ondas R altas o patrón qR en las derivacio-nes precordiales derechas, junto con altera-ciones de la repolarización en dichas precor-diales.
Radiografía de tóraxEn algunos casos de HP, la radiografía
simple de tórax puede ser normal o mostrarescasas alteraciones. Sin embargo, en el 90%de los pacientes con HP idiopática esta explo-
ración es anormal en el momento del diag-nóstico(6). Al igual que en el caso del ECG, lainformación que nos aporta esta prueba esorientativa y sirve como sospecha. Entre lasimágenes que pueden apreciarse en estaprueba se encuentran las siguientes: aumen-to del índice cardiotorácico, dilatación delventrículo derecho (la superficie de contac-to entre el borde cardiaco anterior y el ester-nón supera el tercio de la longitud total deéste en el plano radiológico lateral) y dilata-ción de la arteria pulmonar central.
Ecocardiograma transtorácicoEs una herramienta diagnóstica funda-
mental e imprescindible en la actualidad parallegar al diagnóstico de HP o, al menos, poner-nos sobre la pista de ella. Al no ser invasiva,es una prueba excelente para detectar inicial-mente una HP. Con el ecocardiograma puedeestudiarse la presión sistólica en la arteria pul-monar (PSP). Para ello, el explorador debeencontrar el flujo de regurgitación de la válvulatricúspide, es decir, es necesario que existainsuficiencia tricúspide para poder calculardicha presión. Aplicando el Doppler del ecó-grafo se puede estimar la presión en la arteriapulmonar. Se considera que puede existir HPsi la PSP es mayor de 36 mmHg, aunque seconsidera significativa si es superior a 45-50mmHg, lo que correspondería a una velocidadde regurgitación tricúspide de 2,8-3,4 m/seg(7).En la mayor parte de los estudios se ha obser-vado que hay una buena correlación entre estevalor, calculado mediante el Doppler del eco-cardiógrafo y el obtenido mediante un estudioinvasivo(8,9). Sin embargo, hay que tener encuenta también las limitaciones de la ecogra-fía. Éstas son, sobre todo, que la presión cal-culada aumenta con la edad del paciente y conel índice de masa corporal, lo que puede darlugar a la existencia de falsos positivos. Porconsiguiente, para llegar al diagnóstico defi-nitivo de HP, es necesario realizar un estudiohemodinámico en todos los pacientes sinto-máticos. Por el contario, el Doppler puede tenertambién falsos negativos por lo que, sólo con
GENERALIDADES DIAGNÓSTICAS EN LA HIPERTENSIÓN PULMONAR
39
TABLA 1. Patologías que asocianhipertensión pulmonar con másfrecuencia
• Cardiopatías congénitas con cortocircuito sistémico-pulmonares.
• Hipertensión arterial pulmonar familiar.• Infección por el virus de la inmunodeficien-
cia humana.• Hipertensión portal/cirrosis hepática.• Enfermedades del tejido conectivo.• Exposición a fármacos y tóxicos:
- Aminorex- Femfluramina- Defemfluramina- Aceite de colza desnaturalizado- Anfetaminas- Metaanfetaminas- Quimiterápicos- Cocaína
Monografía HAP NM 208 pag 18/2/10 17:08 Página 39

esta prueba, no podría excluir una HP si la sos-pecha clínica es elevada(10).
El estudio de la ecocardiografía, no sóloinforma sobre la existencia de una HP de unmodo bastante fiable, sino que también pue-de proporcionar información sobre su origen,en el caso de que se aprecien miocardiopatías,enfermedades valvulares del corazón izquier-do o anomalías congénitas, entre otras altera-ciones. Por lo tanto, dentro del estudio eco-cardiográfico, además de la velocidad deregurgitación de la válvula tricúspide, debenvalorarse otros parámetros, como la dimen-sión y la función de los ventrículos derecho eizquierdo, la presencia de anormalidades val-vulares, la eyección del ventrículo derecho, lascaracterísticas de llenado del ventrículo izquier-do, el tamaño de la vena cava inferior y la posi-ble existencia de un derrame pericárdico(11,12).
En ocasiones, la información que aporta elecocardiograma transtorácico se puede mejo-rar o potenciar utilizando suero salino agitadointravenoso (a modo de contraste). Esta téc-nica puede ayudar a identificar una comuni-cación interauricular tipo seno venoso depequeño tamaño, o un foramen oval permea-ble que podrían haber pasado desapercibidoscon el ecocardiograma convencional. Si, apesar del suero salino agitado, persisten dudasacerca de la existencia de una malformacióncongénita, habría que recurrir al ecocardio-grama transesofágico.
Identificación del tipo de HPUna vez que con las pruebas anteriores,
sobre todo con el ecocardiograma, se ha detec-tado la presencia de HP, hay que intentar cla-sificarla y englobarla en uno de los cinco gru-pos de la nueva clasificación de Danna Point.Para ello debería realizarse una batería de prue-bas diagnósticas que irán orientando haciacada uno de ellos (Tabla 2).
Se debe realizar una analítica sanguíneacon hematología, bioquímica sanguínea y estu-dio de la función tiroidea. También es conve-niente estudiar el potencial trombótico,mediante la determinación de los anticuerpos
antifosfolípidos (anticoagulante lúdico y anti-cardiolipina). Para descartar la existencia deenfermedades del tejido conectivo (ETC) sedebería contar, además de con diversos cri-terios clínicos, con algunas pruebas de labo-ratorio, como anticuerpos antinucleares (ANA),anticuerpos anticentrómero, anti-SCL70 y RNP.Asimismo, conviene solicitar también serolo-gía para el virus de la inmunodeficiencia huma-na (VIH) y para los virus de las hepatitis B y C.
El estudio de las pruebas de función respi-ratoria (espirometría forzada, volúmenes pul-monares, test de difusión del monóxido de car-bono), incluyendo la gasometría arterial, permitedescartar una enfermedad respiratoria comocausa de HP. Si se sospecha un síndrome deapnea/hipopnea del sueño (SAHS) se deberá rea-lizar una poligrafía o una polisomnografia, yaque esta enfermedad puede causar también HP.
Con la gammagrafía de ventilación-perfu-sión (V/Q) pulmonar puede diagnosticarse unaHP secundaria a un tromboembolismo cróni-co(13). Esta prueba tiene una gran sensibilidady especificidad para distinguir entre una HPidiopática y una HP secundaria a un embo-lismo crónico. Debe siempre completarse conuna tomografía axial computarizada (TAC) heli-coidal con contraste, para así poder detectarposibles trombos en arterias pulmonares. Enotras ocasiones se apreciarán defectos de lle-nado excéntricos compatibles con trombos,signos de recanalización o la presencia de este-nosis o redes vasculares(14,15).
La arteriografía pulmonar, a pesar de seruna técnica invasiva, debe realizarse a lospacientes con sospecha de HP tromboembó-lica crónica, sobre todo si pueden beneficiar-se de una tromboendarterectomía(13). Esta prue-ba es más precisa que la TAC helicoidal concontraste para identificar obstrucciones dista-les de las arterias pulmonares. También per-mite cuantificar mejor el tamaño de los trom-bos y la extensión de los mismos. Aspectosútiles en relación con la técnica de realizaciónde esta prueba son la inyección selectiva delcontraste en las ramas principales izquierda yderecha y la práctica de múltiples angulacio-
F. PEDRAZA SERRANO ET AL.
40
Monografía HAP NM 208 pag 18/2/10 17:08 Página 40

nes y proyecciones. En cualquier caso, siem-pre es recomendable realizarla en el centro dereferencia donde se vaya a efectuar la inter-vención quirúrgica, en el caso de que final-mente se precise ésta.
La TAC de alta resolución (TACAR) es otraprueba a realizar si se quiere valorar adecuada-mente el parénquima pulmonar, para así poderdetectar la existencia de una enfermedad pul-monar intersticial difusa. También permite apre-ciar signos de hipertensión venosa pulmonar,como el engrosamiento de los septos, lo que pue-de sugerir la presencia de una enfermedad veno-oclusiva pulmonar. Si, además, se acompaña deopacidades centroacinares, puede tratarse deuna hemangiomatosis capilar pulmonar(16).
Con la ecografía abdominal puede detec-tarse una cirrosis hepática y/o una hiperten-sión portal. Aplicando en ella el Doppler, se
puede distinguir también la hipertensión por-tal pasiva o secundaria a un fallo cardiaco dere-cho de la hipertensión portal, que tiene su ori-gen en el aumento del gradiente venosotranshepático asociado con la cirrosis(17,18).
Evaluación funcional y hemodinámica
Pruebas de tolerancia al ejercicioIntentar cuantificar de forma objetiva la
capacidad de ejercicio de los pacientes con HPes un aspecto muy importante dentro de estaenfermedad. Con ello puede valorarse, no sólola gravedad de la misma(19,20), sino también laeficacia del tratamiento(21,22). Entre las pruebasque pueden emplearse se encuentran el testde la marcha de 6 minutos y la prueba deesfuerzo cardiopulmonar. A continuación sedescriben cada una de ellas por separado.
GENERALIDADES DIAGNÓSTICAS EN LA HIPERTENSIÓN PULMONAR
41
TABLA 2. Pruebas complementarias útiles para el estudio del tipo de hipertensiónpulmonar
1. Analítica:
• Hemograma y bioquímica básicas
• Determinación de hormonas tiroideas
• Serologías: VIH, herpes virus tipo 8 y virus de las hepatitis B y C
• Inmunología: anticuerpos antinucleares (ANA), incluyendo anticuerpos anticentrómero, anti-SCL70y RNP
• Estudio de hipercoagulabilidad: anticuerpos antifosfolípido
2. Pruebas de función respiratoria:
• Espirometría forzada
• Pletismografía para medir volúmenes pulmonares
• Test de difusión del monóxido de carbono
• Gasometría arterial
3. Ecografía abdominal y Doppler de vena porta
4. Gammagrafía de ventilación/perfusión. Si se sospecha HP tromboembólica, además:
• Angio-TAC para visualizar bien las arterias pulmonares
• Arteriografía pulmonar
• Estudio de hipercoagulabilidad
5. TAC de alta resolución en algunos casos:
• En pacientes con colagenosis para valorar si existe fibrosis pulmonar
Monografía HAP NM 208 pag 18/2/10 17:08 Página 41

a) Prueba de marcha de los 6 minutos: con ellase intenta reflejar la capacidad que tieneel paciente de realizar actividades de lavida diaria. Se trata de una prueba muysencilla, poco agresiva, de bajo coste y fácil-mente reproducible. Permite efectuar unaaproximación a la supervivencia del pacien-te que se encuentra en fases avanzadas dela enfermedad y valorar la eficacia del tra-tamiento. Sin embargo, esta prueba cuen-ta con importantes limitaciones (Tabla 3),ya que existen múltiples circunstancias quepueden modificar la distancia recorrida(23).Algunas de ellas son inherentes al propiopaciente e imposibles de evitar, tales comola edad, el peso o el sexo del paciente. Sinembargo, existen factores independientesde las condiciones del paciente que pue-den producir variaciones en los resultados,entre las que se encuentran la longitud delpasillo donde se realiza la prueba o el per-sonal técnico encargado de la supervisiónde la misma. Son estos factores los que sedeben intentar controlar a la hora de rea-lizar el test. Entre los factores que puedenreducir la distancia total recorrida puedencitarse la edad avanzada, el sobrepeso, elsexo femenino, la escasa longitud del pasi-llo y las enfermedades cardiopulmonareso musculoesqueléticas. Por otra parte, seríaaconsejable la realización de al menos dos
pruebas, tomando como válida aquella enla que el paciente haya caminado una dis-tancia mayor(24). Además, es recomenda-ble que transcurra un período de tiempoprudencial de descanso entre una pruebay otra.
b) Prueba de esfuerzo cardiopulmonar: aportainformación más precisa que la anteriorsobre la tolerancia al ejercicio, principal-mente en las clases funcionales iniciales,es decir, en los pacientes con HP con pocossíntomas, donde la prueba de marcha delos 6 minutos puede ser normal. Con laprueba de esfuerzo cardiopulmonar pue-de medirse la ventilación y el intercambiode gases durante el ejercicio. Estudia deforma no invasiva la fisiopatología de lossistemas respiratorios y cardiovascularesen condiciones de estrés físico, evaluandoobjetivamente el grado de limitación fun-cional y su mecanismo. Esta prueba es bas-tante más compleja de realizar que el testde los 6 minutos y requiere un análisis pre-ciso del pico del consumo de oxígeno (VO2),el consumo máximo del oxígeno (VO2
max), la producción de dióxido de carbo-no (VCO2), la medición del pulso de oxí-geno y el umbral de anaerobiosis, entreotros parámetros. Para su realización espreciso disponer, por lo tanto, de personalentrenado en dicha técnica y con expe-riencia a la hora de interpretarla.
Estudio hemodinámicoSi la ecografía transtorácica es, como se ha
explicado anteriormente, la prueba funda-mental para el diagnóstico de sospecha de laHP, el estudio hemodinánico mediante cate-terismo cardiaco derecho (CCD) es la pruebaque confirma definitivamente esa sospecha,ya que determina directamente de forma inva-siva el valor de la presión arterial pulmonar(3,25).Además de confirmar el diagnóstico, esta prue-ba permite valorar la severidad de la enfer-medad y ayuda a establecer su pronóstico.
Las mediciones que se realizan duranteeste estudio hemodinámico son:
F. PEDRAZA SERRANO ET AL.
42
TABLA 3. Limitaciones de la pruebade marcha de 6 minutos
• Variables antropométricas: edad, sexo, talla
• No se puede identificar el elemento fisiopa-tológico de base donde radica la limitación alejercicio
• “Efecto aprendizaje” en los pacientes que hanrealizado varias veces esta prueba, lo que pue-de sesgar los valores obtenidos
• “Efecto techo” que impide detectar variacio-nes en aquellos pacientes que caminan másde 450 metros
Monografía HAP NM 208 pag 18/2/10 17:08 Página 42

GENERALIDADES DIAGNÓSTICAS EN LA HIPERTENSIÓN PULMONAR
43
a) Presión arterial pulmonar (PAP).b) Presión en la aurícula derecha (PAD).c) Presión de enclavamiento pulmonar (PCP).
Este parámetro es muy útil porque puededistinguir entre un origen arterial o veno-so de la HP en los pacientes con patologíadel corazón izquierdo. Si la PCP es normalse deberá a una elevación de las resisten-cias vasculares pulmonares y, si está ele-vada, se trataría de una hipertensión veno-sa pulmonar. En este último caso, cuandola PCP está elevada, se debería realizar tam-bién un cateterismo cardiaco izquierdopara medir la presión telediastólica del ven-trículo izquierdo y con ella poder excluir laexistencia de disfunción del ventrículoizquierdo, estenosis mitral o enfermedadvenooclusiva.
d) Gasto cardiaco (GC). Se puede medirmediante termodilución, inyectando a tra-vés del catéter suero frío y midiendo lasdiferencias de temperatura en función deltiempo. Con este método se debe tener encuenta siempre la cantidad exacta de sue-ro administrado y la temperatura de dichosuero. Este método de medida por termo-dilución puede aportar datos inexactoscuando el GC es muy alto o muy bajo. Tam-poco es útil en presencia de cortocircuitosintracardiacos o si la regurgitación valvularde la tricúspide o de la pulmonar es exce-siva. Para estos casos el GC se debe calcu-lar mediante la fórmula de Fick (Tabla 4).
e) Resistencias vasculares pulmonares (RVP).RVP = PAP media - PCP media/GC).
f) Saturación venosa mixta (SatO2AP), que esla saturación de oxígeno en la arteria pul-monar.
Durante todo el procedimiento del cate-terismo cardiaco el paciente debe estar moni-torizado. Se debe registrar y controlar la fre-cuencia cardiaca, la tensión arterial sistémicay la saturación de oxígeno.
En cuanto a las indicaciones, habría querealizar un estudio hemodinámico a todos lospacientes del grupo 1 y 4 de la clasificación de
Dana Point: aquellos con hipertensión arterialpulmonar (HAP) y los pertenecientes al grupode la hipertensión pulmonar tromboembólicacrónica (HPTEC). En los casos en los que seobserve una PSP menor de 50 mmHg esti-mada por ecocardiografía, se debe individua-lizar la decisión de realizar el estudio invasivohemodinámico en función de la edad, lacomorbilidad y el fundamento de la sospechaclínica.
A los pacientes con hipertensión pulmo-nar asociada a enfermedad cardiaca izquier-da (grupo 2 de Dana Point), no es necesariorealizarles un cateterismo cardiaco derecho,salvo que existan dudas sobre si el origen dela HP es pre o postcapilar. En los del grupo 3con enfermedades respiratorias asociadas, tam-poco es necesario efectuarles un estudio hemo-dinámico invasivo, salvo en aquellos casos enlos que el valor de la PSP estimado por eco-cardiograma sea desproporcionadamente ele-vado (por encima de 55 mmHg) en relacióncon su enfermedad respiratoria, ya que la HPpuede ser un proceso asociado y podría bene-ficiarse de un tratamiento específico.
Test vasodilatador agudoUna vez confirmada la presencia de HP y
obtenido un valor objetivo de ésta, debería rea-lizarse una medición de la reversibilidad de lamisma, ya que el resultado puede influir en elmanejo y el pronóstico de estos pacientes. Con
TABLA 4. Ecuación de Fick para elcálculo del gasto cardiaco (GC)
VO2 = GC (CaO2-CvO2)
De ahí que el GC sea igual a:
GC = VO2/ (CaO2-CvO2)
Cálculo de CaO2:
CaO2 = (1,34 x Hb x Sat O2) + (PaO2 x 0.0031)
Abreviaturas. GC: gasto cardiaco; CaO2: contenidoarterial de oxígeno; CvO2: contenido de oxígeno ensangre venosa mezclada; Hb: concentración de hemo-globina; Sat O2: saturación de oxígeno.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 43

este test se intenta identificar a los individuosque van a poder beneficiarse de un tratamientoprolongado con bloqueadores de los canalesde calcio (BCC).
La prueba de la vasodilatación debe reali-zarse exclusivamente con vasodilatadores pul-monares de acción rápida, en el mismo pro-ceso del cateterismo del lado derecho en elque se ha obtenido el valor de la hipertensión.Los fármacos usados en este test son adeno-sina intravenosa, prostaglandina intravenosay óxido nítrico inhalado, siendo estos dos últi-mos los más empleados en la actualidad. Enla tabla 5 se describen las dosis, la vida media,la vía de administración y los efectos adversosde estos fármacos.
Para considerar la prueba como positivase necesitan tres requisitos(2,26,27): a) Disminución de la presión arterial pulmo-
nar media igual o mayor a 10 mmHg.b) Que la presión arterial pulmonar media
alcance un valor absoluto igual o menora 40 mmHg.
c) Que el gasto cardiaco no se modifique oque aumente.
Sólo un 10-15% de los pacientes con HPidiopática o los relacionados con la toma de ano-rexígenos van a cumplir estos criterios(26,28). Ade-más, de ese pequeño porcentaje, sólo la mitadvan a mostrar una respuesta mantenida al tra-tamiento prolongado con dosis elevadas de BCC.
Por lo tanto, se ha protocolizado la necesidadde reevaluar la eficacia del tratamiento con estosfármacos a los 3 y 6 meses de iniciar la medi-cación en aquellos pacientes con un test posi-tivo inicial(26). Por otra parte, hay que tener encuenta que el tratamiento empírico con BCC alos pacientes a los que no se les haya hecho pre-viamente el test vasodilatador está totalmentedesaconsejado, debido a los posibles efectosadversos graves de estos fármacos.
En otras enfermedades que cursan con HP,como las asociadas a enfermedades del tejidoconectivo o las secundarias a malformacionescongénitas, la utilidad del test vasodilatadoragudo es menos clara que en la HP idiopática.Aun así, los expertos siguen recomendandorealizarlo y, en los casos en los que sea positi-vo, buscar una respuesta a largo plazo de losBCC en los pacientes adecuados.
Biopsia pulmonarLa biopsia pulmonar abierta entraña ries-
gos importantes de morbilidad y mortalidad,por lo que se desaconseja realizarla de formasistemática a los pacientes con HP. Sí se reco-mienda efectuarla en aquellos casos en los quese requiere una confirmación histológica(29),para establecer el diagnóstico de vasculitis pul-monar activa, enfermedad intersticial pulmo-nar difusa, enfermedad venooclusiva o heman-giomatosis capilar pulmonar, siempre quehayan fracasado otras técnicas diagnósticas
F. PEDRAZA SERRANO ET AL.
44
TABLA 5. Vasodilatadores más usados en el test vasodilatador agudo
Epoprostenol Adenosina Óxido nítrico
Vía de administración Intravenosa Intravenosa Inhalada
Vida media 3 minutos 5-10 segundos 15-30 segundos
Rango de dosis 2-12 ng/kg/min 50-350 µg/kg/min 10-80 ppm
Incremento de dosis 2 ng/kg/min cada 50 µg/kg/min 10-80 ppm durante 10 min cada 2 min 5 min
Duración 1 minuto 2 minutos 5 minutos
Efectos secundarios Cefalea, rubor facial, Disnea, opresiónnáuseas torácica Escasos
Monografía HAP NM 208 pag 18/2/10 17:08 Página 44

de menor riesgo para el paciente y exista unalto grado de sospecha clínica.
BIBLIOGRAFÍA1. Estándares asistenciales en hipertensión pul-
monar. Documento de consenso elaboradopor la SEPAR y la SEC. Rev Esp Cardiol. 2008;61:170-84.
2. Barst RJ, McGoon M, Torbicki A, et al. Diag-nosis and differential assesment of pulmonaryarterial hypertension. J Am Coll Cardiol. 2004;43: S40-7.
3. Galie N, Torbicki A, Barst R, et al. Guías depráctica clínica sobre el diagnóstico y trata-miento de la hipertensión arterial pulmonar.Rev Esp Cardiol. 2005; 58: 523-66.
4. Gómez Sánchez MA, Escribano Subías P. Pro-tocolos de actuación en hipertensión arterialpulmonar. Madrid: Editores Médicos SA; 2008.
5. Ahearn GS, Tapson VF, Rebeiz A, et al. Elec-trocardiography to define clinical status in pri-mary pulmonary hypertension and pulmonaryarterial hypertension secondary to collagenvascular disease. Chest 2002:122: 524-7.
6. Rich S, Dantzker DR, Ayres SM, et al. Primarypulmonary hypertension. A national prospec-tive study. Ann Intern Med. 1987; 107: 216-23.
7. Brennan JM, Blair JE, Goonewardena S, et al.Reappraisal of the use of inferior vena cavafor estimating right atrial pressure. J Am SocEchocardiography. 2007; 20: 857-61.
8. Denton CP, Cailes JB, Phillips GD, et al. Com-parison of Doppler echocardiography and rightheart catheterization to assess pulmonaryhypertension in systemic sclerosis. Br J Rheu-matol. 1997; 36: 239-43.
9. Vachiery JL, Brimioulle S, Crasset V, et al. Fal-se-positive diagnosis of pulmonary hyperten-sion by Doppler echocardiography. Eur RespirJ. 1998; 12: 1476-8.
10. Mukerjee D, St George D, Knight C, et al. Echo-cardiography and pulmonary function as scre-ening test for pulmonary arterial hypertensionin systemic sclerosis. Rheumatology. 2004; 43:461-6.
11. Hinderliter AL, Willis PW, Barst RJ, et al. Effectsof long-term infusion of prostacyclin (epo-prostenol) on echocardiografic measures ofright ventricular structure and function in pri-mary pulmonary hypertension. Primary Pul-monary Hypertension Study Group. Circula-tion. 1997; 95: 1479-86.
12. Galie N, Hinderliter AN, Torbiki A, et al. Effectof the oral endotelin-receptor antagonistbosentan on echocardiografic and dopplermeasures in patientes with pulmonary arte-rial hypertension. J Am Coll Cardiol. 2003;41:1380-6.
13. Fedullo PF, Auger WR, Kerr KM, et al. Chronicthromboembolic pulmonary hypertension.N Engl J Med. 2001; 345:1465-72.
14. Ley S, Kreitner KF, Fink C, et al. Assessmentof pulmonary hypertension by CT and MR ima-ging. Eur Radiol. 2004; 14: 359-68.
15. Dartevelle P, Fadel E, Mussot S, et al. Chro-nic thromboembolic pulmonary hypertension.Eur Respir J. 2004; 23: 637-48.
16. Resten A, Maitre S, Capron F, et al. Pulmo-nary hipertensión: CT finding in pulmonaryveno-oclusive disease. J Radiol. 2003; 84:1739-45.
17. Albrecht T, Blomley MJ, Cosgrove DO, et al.Non-invasive diagnosis of hepatic cirrhosis bytransit-time analysis of an ultrasound contrastagent. Lancet. 1999; 353:1579-83.
18. Naeije R. Hepatopulmonary syndrome andportopulmonary hipertensión. Swiss MedicalWeekly. 2003; 133: 163-9.
19. Miyamoto S, Nagaya N, Satoh T, et al. Clinicalcorrelates and prognostic significance of six-minute walk test in patients with primary pul-monary hypertension. Comparison with car-diopulmonary exercise testing. Am J RespireCrit Care Med. 2000; 161: 487-92.
20. Wensel R, Opitz CF, Anker SD, et al. Assess-ment of survival in patients with primary pul-monary hypertension: importance of pulmo-nary exercise testing. Circulation. 2002;106:319-24.
21. Galie N, Manes A, Branzi A. The new clinicaltrials on pharmacological tratment in pulmo-nary arterial hypertension. Eur Respir J. 2002;20: 1037-49.
22. Peacock A, Naeije R, Galie N, et al. End-pointsfor clinical trials in pulmnary arterial hyper-tension: The way forward. Eur Respir. J 2004;23: 947-53.
23. ATS statement: guidelines for the six-minutewalk test. ATS committee on proficiency stan-dards for clinical pulmonary function labora-tories. Am J Respire Crit Care Med. 2002;166:111-17.
24. Guyyat GH, Pugsley SO, Sullivan MJ, et al. Effectof encouragement of walking test permorfance.Thorax. 1984; 39: 818-22.
GENERALIDADES DIAGNÓSTICAS EN LA HIPERTENSIÓN PULMONAR
45
Monografía HAP NM 208 pag 18/2/10 17:08 Página 45

25. Guillinta P, Peterson KL, Ben-Yehuda O. Car-diac catheterization techniques in pulmonaryhypertension. Cardiol Clin. 2004; 22: 401-15.
26. Sitbon O, Humbert M, Jaïs X, Ioos V,HamidAM, Provencher S, et al.Long-term responseto calcium channel blockers in idiopathic pul-monary arterial hypertension. Circulation.2005; 111: 3105-11.
27. Galie N. Seeger W, Naeije R, et al. Comparati-ve analysis of clinical trials and evidence-based
treatment algorithm in pulmonary arterial hyper-tension. J Am Coll Cardiol. 2004; 43: S81-8.
28. Galie N, Ussia G, Passarelli P, et al. Role of phar-macologic test in the treatment of primary pul-monary hypertension. Am J Cardiol. 1995: 75:A55-62.
29. McGoon M, Gutterman D, Steen V, et al. Scree-ning, early detection and diagnosis of pulmo-nary arterial hypertension. ACCP Evidence.BasedClinical Guideliness. Chest. 2004; 126: S14-34.
F. PEDRAZA SERRANO ET AL.
46
Monografía HAP NM 208 pag 18/2/10 17:08 Página 46

47
RESUMENEl tratamiento de soporte o conservador
de la HAP incluye la oxigenoterapia, diuréti-cos, digoxina, anticoagulación, calcioantago-nistas (CA) y rehabilitación. Las indicacionesde oxigenoterapia en pacientes con HAP sonlas mismas que las aplicadas a pacientes conEPOC, y se extrapolan a partir de los estudiosrealizados en pacientes con EPOC hace ya casitreinta años. La utilización de diuréticos, comofurosemida y espironolactona, tienen comometa el tratamiento sintomático del fracasoventricular derecho, fundamentalmente losedemas periféricos. La digoxina en pacientescon HAP tiene indicación en caso de insufi-ciencia cardiaca derecha y/o taquiarrítmia auri-cular. La utilización de anticoagulación se apo-ya en que la HAP se considera un estadoprotrombótico. Los anticoagulantes orales (ace-nocumarol y warfarina) son los más utilizados,considerándose indicados en pacientes conHAP y presión en arteria pulmonar (PAP) >60mmHg. En cuanto a los CA (nifedipino, dil-tiazem y amlodipino) están indicados ante untest de vasorreactividad positivo favorable (caí-da de la PAP media >10 mmHg y valor de laPAP media <40 mmHg con aumento o no dis-minución del gasto cardiaco). De cualquiermanera, un test de vasorreactividad positivoocurre en una baja proporción en pacientescon HAP, que se sitúa en torno al 10%.
Respecto a la rehabilitación pulmonar,incluyendo entrenamiento físico y respirato-rio, comienza a considerarse su beneficio entérminos de calidad de vida y capacidad de
ejercicio en pacientes con HAP, incluso en for-mas severas.
INTRODUCCIÓNEl último Simposio Internacional de Dana
Point clasifica la hipertensión pulmonar (HP)en cinco grupos, basados en su etiología(1). Enel grupo I incluye a pacientes que tienen hiper-tensión arterial pulmonar idiopática y otras. Aeste grupo I le denominaremos HAP, y es alque se hará especialmente referencia en el pre-sente capítulo.
El manejo terapéutico de la HAP es difícil.Se puede decir que hay dos grupos de terapias:1. Tratamiento general, de soporte o con-
vencional, común a todos los grupos deHP que incluye: oxigenoterapia, diuréticos,digitálicos, calcioantagonistas (CA) y anti-coagulantes. Todas estas terapias preten-den tratar la HAP y el fracaso ventriculardel lado derecho cuando existe.
2. Tratamiento avanzado o específico, deaparición en las últimas 2 décadas, dondese incluyen prostanoides, antogonistas dela endotelina e inhibidores de la fosfo-diesterasa tipo 5. Todos estos agentes tie-nen efecto vasodilatador pulmonar y pro-piedades antiproliferantes.
En el presente capítulo se comentaránexclusivamente aspectos relacionados con eltratamiento general o de soporte de la HAP,y no analizará el tratamiento avanzado de laHAP, que se discutirá específicamente en otroscapítulos de esta monografía.
GENERALIDADES TERAPÉUTICAS EN LA HIPERTENSIÓN ARTERIAL PULMONAR(HAP)
Julio Flores Segovia, Olga Navarrete Isidoro, Silvia Sánchez González
Monografía HAP NM 208 pag 18/2/10 17:08 Página 47

OXIGENOTERAPIA
El uso de la oxigenoterapia en pacientescon HP se extrapola de los datos existentespara el tratamiento de la enfermedad pulmo-nar obstructiva crónica (EPOC). Existen dosestudios prospectivos(2,3), clásicos, que handemostrado una marcada mejoría en la super-vivencia a largo plazo, con el tratamiento deloxígeno suplementario en pacientes conEPOC. En uno de los estudios mencionados(Medical Research Council Party, 1981), se estu-diaron 87 pacientes con EPOC e historia defallo del ventrículo derecho, con PaO2< 60 mHg. Los pacientes fueron randomizados a reci-bir oxígeno suplementario durante 15horas/día o no; en el grupo tratado con oxí-geno, la mortalidad a 5 años fue del 46%,frente al 67% en el grupo que no recibió dichotratamiento. Este estudio también mostró unaumento en los valores de resistencia vascu-lar pulmonar (RVP) en el grupo que no reci-bió oxígeno suplementario, mientras que laresistencia vascular no aumentó en el grupotratado; no obstante, en el estudio no se pudoevaluar correctamente este parámetro. El otroestudio mencionado (Nocturnal Oxygen The-rapy Trial Group, 1980), incluyó a 203 pacien-tes con EPOC e hipoxemia crónica; los resul-tados concluyeron que, en el grupo depacientes que seguía terapia con oxígeno deforma continua (al menos 19 horas/día), lamortalidad era inferior respecto a los pacien-tes que seguían tratamiento sólo durante 12horas/día, un 22% frente a un 42%, respec-tivamente. La mejora de la supervivencia resul-tó ser más significativa en los pacientes conHAP menos severa (PAP media <27 mmHg).La terapia con oxígeno de forma continua, portanto, mejoró la supervivencia a largo plazoen los pacientes con EPOC con PaO2<55mmHg en reposo, incluso aunque no exis-tiera una mejora significativa en los paráme-tros hemodinámicos pulmonares. En cuantoa los efectos en la RVP, hubo un beneficio mássignificativo en cuanto a la mortalidad en elgrupo de pacientes que tenían una RVP basalinferior, con la terapia continua frente al gru-
po de pacientes con una RVP basal más alta.Al realizar un cateterismo cardiaco del ladoderecho después de 6 meses de tratamientocon oxígeno de forma continua, no hubo bene-ficio en mortalidad en el grupo que presentómás descenso de la RVP, por lo que este datosugiere que el beneficio observado en cuantoa mortalidad en los pacientes con EPOC ehipoxemia, en tratamiento con oxigenotera-pia de forma continua, no es derivado de unaclara disminución en la RVP. Por tanto, segúnlos datos de los estudios anteriores, existe con-senso generalizado en que todos los pacien-tes con EPOC, con hipoxemia significativa enreposo o con el ejercicio, o signos de hipo-xemia crónica, deberían recibir oxígeno suple-mentario(4). No obstante, si esto es claramen-te aplicable a los pacientes con HAP, no estádel todo claro, si bien su indicación actual pre-senta un nivel de recomendación I-C.
La hipoxemia en los pacientes con HAP sepresenta tanto en reposo como con el ejerci-cio o durante el sueño, y está causada posi-blemente por la baja saturación de la sangrevenosa, el bajo gasto cardiaco y, en menormedida, los trastornos de la relación ventila-ción-perfusión o alteración de la difusión. Enla mayoría de los pacientes, a excepción deaquellos con enfermedad más avanzada, lahipoxemia en reposo y durante el sueño pue-de ser corregida con flujos de oxígeno entre 2-6 litros/minuto, administrado por cánula nasal,siendo necesario aumentar este flujo durantela actividad y el ejercicio. Las principales socie-dades americanas y europeas recomiendanque todos los pacientes con HAP y conPaO2<55 mmHg o saturación de oxihemo-globina<88%, en reposo, durante el sueño oen actividad, deben recibir terapia con oxí-geno para mantener una saturación de oxihe-moglobina mediante pulsioximetría >90%durante todo el tiempo. En los pacientes consignos clínicos de fallo de ventrículo derechoo datos sugestivos del mismo en el electro-cardiograma o ecocardiografía Doppler, o enaquellos con hematocrito >55%, la oxigeno-terapia debe ser iniciada cuando la PaO2<60
J. FLORES SEGOVIA ET AL.
48
Monografía HAP NM 208 pag 18/2/10 17:08 Página 48

mmHg. Los pacientes con disminución seve-ra de la capacidad de difusión (<60% del valorpredicho) en reposo deben ser evaluados paravalorar desaturación de la oxihemoglobina conla actividad o durante el sueño(5).
Otras entidades patológicas cardiacas pre-sentan hipoxemia resistente a la administra-ción de oxígeno, por presentar un cortocircuito(shunt) derecha-izquierda. El uso de la oxige-noterapia en las cardiopatías congénitas y enel síndrome de Eisenmenger es objeto de con-troversia. Un estudio realizado con 15 pacien-tes en edad pediátrica con HAP asociada a car-diopatía congénita, con hipoxemia, objetivóbeneficio en la mortalidad con la terapia conoxígeno durante 12 horas/día durante 5 años(6).Otro estudio bien diseñado, prospectivo, ran-domizado y controlado, con 23 pacientes adul-tos con síndrome de Eisenmenger e hipoxe-mia, objetivó que el uso de la oxigenoterapianocturna no tenía efecto alguno ni en la super-vivencia, ni en la calidad de vida ni en la mejo-ra de los valores del hematocrito ni en el testde la marcha de 6 minutos en estos pacien-tes(7). No obstante, existen otros estudios condatos favorables respecto al uso de terapia conoxígeno en estos pacientes, algunos objetivanque puede reducir la necesidad de flebotomíay puede reducir las complicaciones neuroló-gicas(8), otros han demostrado una disminu-ción en la tasa de progresión de policitemiaasí como mejora de los síntomas(9).
Pese a que la oxigenoterapia en ciertamanera supone cierta limitación en la movi-lidad y estilo de vida del paciente, para sucorrecto cumplimiento en general se aceptasu indicación en pacientes con hipoxemia enreposo o desaturación con el ejercicio habitual.
DIURÉTICOSLos diuréticos no tienen efecto vasodilata-
dor pulmonar y no modifican directamente laPAP ni la RVP(8). Sin embargo, resultan útiles paradisminuir el edema periférico, la congestiónvenosa sistémica y la congestión hepática. Suuso en pacientes con HAP y edemas periféricossecundarios a la disfunción del ventrículo dere-
cho está amplia y universalmente aceptado (Nivelde recomendación I-C)(10,29). No obstante, se des-conoce si su uso se asocia a una alteración de lamorbimortalidad en estos pacientes. Así, los diu-réticos están indicados en pacientes con evi-dencia de insuficiencia cardiaca derecha dadoque el mantenimiento de un volumen intravas-cular “casi” normal con ayuda de diuréticos jun-to a una restricción dietética cuidadosa en laingesta de líquidos y de sal es considerado unpunto muy importante en el manejo a largo pla-zo de los pacientes con HAP. No obstante, hayque tener en cuenta que una diuresis rápida yexcesiva puede dar lugar a hipotensión sisté-mica, insuficiencia renal y síncope, motivo porel que los niveles de electrólitos y la determina-ción de la función renal se deben monitorizarestrechamente en todos aquellos pacientes entratamiento diurético.
Los diuréticos de asa, como la furosemi-da, se utilizan en aquellos pacientes en los quepredominan signos y síntomas de congestión,aunque evitando una brusca disminución de laprecarga, con la subsiguiente caída del volumenminuto cardiaco y deterioro de la funciónrenal(11). No se han evaluado los efectos a largoplazo que la mayoría de los diuréticos ejercensobre la morbimortalidad en la insuficienca car-diaca izquierda o derecha. La espironolacto-na, un antagonista aldosterónico utilizado comodiurético ahorrador de potasio, disminuyó lamortalidad, en un ensayo a largo plazo, pros-pectivo, randomizado y controlado con place-bo en pacientes con una hospitalización recien-te por insuficiencia cardiaca izquierda con clasefuncional III o IV de la NYHA(12). Ningún estu-dio ha evaluado su papel en pacientes con HAPy disfunción ventricular derecha, sin embargo,la espironolactona es ampliamente utilizada enpacientes que tienen HAP y disfunción ventri-cular derecha junto a los diuréticos de asa. Eluso de espironolactona, por sus efectos diuré-ticos y moduladores del sistema neurohumo-ral, puede acompañarse de mejorías en el remo-delamiento ventricular y del lecho vascularpulmonar y, probablemente, deba ser utilizadodesde las etapas tempranas del diagnóstico de
GENERALIDADES TERAPÉUTICAS EN LA HIPERTENSIÓN ARTERIAL PULMONAR (HAP)
49
Monografía HAP NM 208 pag 18/2/10 17:08 Página 49

la HP. Su uso, igualmente, debe realizarse consumo cuidado, dado que la brusca caída de laprecarga ventricular se puede acompañar deuna caída del volumen minuto cardiaco y deldeterioro de la función renal. No debe utilizar-se en pacientes con cifras de creatinina >2,5mg/dl o potasio > 5 mEqu/L o en pacientes conhistoria de hiperpotasemia refractaria severa.También se debe tener precaución en pacien-tes con función renal alterada o diabetes, enancianos y pacientes que utilizan de forma con-junta IECAs o AINEs. Por último, análogos delamiloride (un diurético ahorrador de potasio)parece inhibir el desarrollo de hipoxia inducidapor la hipertensión pulmonar en modelos ani-males, aunque no hay estudios clínicos enpacientes con HAP que apoyen el significadoclínico de estas observaciones. Se debe tenercuidado en el ajuste de dosis de diuréticos. Noexiste evidencia o razones científicas para la eli-minación de grandes volúmenes de líquido encortos períodos de tiempo, excepto cuando exis-te un edema pulmonar cardiogénico o un empe-oramiento agudo de la hipoxemia arterial. Elajuste de dosis en pacientes con HAP y disfun-ción ventricular derecha debe perseguir un con-trol óptimo en el mantenimiento del gasto car-diaco, minimizar la hipoxemia arterial ycontrolar los signos de perfusión tisular. En elcaso de disfunción severa del ventrículo dere-cho, se pueden requerir altas dosis de diuréti-cos para conseguir una diuresis efectiva (hasta600 mg/d de furosemida); no obstante, enmuchas ocasiones en etapas avanzadas de laenfermedad debe permitirse cierto grado decongestión, siendo necesario un adecuado con-trol de la misma, siempre que no conlleve undeterioro progresivo de la función hepática(13).
DIGOXINAGlucósidos cardiacos, como la digoxina,
incrementan la contractibilidad miocárdicamediante el bloqueo de la bomba ATPasa Na-K localizada en la membrana celular de lascélulas miocárdicas. La utilidad de la digoxinaen pacientes con HAP no está definida, peroestudios a niveles de experimentación sobre
su utilidad en la sobrecarga sistólica del ven-trículo derecho muestran que su administra-ción ayuda a prevenir la reducción de la con-tractilidad. Además, la digoxina ha demostradoen pacientes con HAP incrementar el gastocardiaco y disminuir los niveles de noradre-nalina cuando es administrada de forma agu-da(14). Sin embargo, no existen datos sobre laseguridad, eficacia y el impacto sobre la mor-bilidad o mortalidad del tratamiento con digo-xina a largo plazo en pacientes con HAP y dis-función ventricular derecha. La digoxina seutiliza sobre todo en aquellos pacientes consignos y síntomas de insuficiencia cardiacaderecha. La disminución de la función sistóli-ca ventricular derecha es uno de los puntosfundamentales en la progresión de los signosy síntomas de la HAP. La digoxina, a pesar deposeer un efecto inotrópico positivo débil, pare-ce poseer cierta utilidad en pacientes con HAPcon deterioro de la función ventricular dere-cha. Su uso puede compensar el efecto inotró-pico negativo que produce el tratamiento vaso-dilatador con antagonistas del calcio. Tambiénse utiliza en pacientes con HAP que tienen alte-raciones del ritmo auricular (taquicardia auri-cular multifocal, flutter auricular o fibrilaciónauricular) (nivel de recomendación IIb-C).
Los glucósidos cardiacos se deben usar conprecaución, ya que su toxicidad puede verseaumentada por las alteraciones electrolíticasinducidas por los diuréticos. Además, la vidamedia de la digoxina está prolongada en casode deterioro de la función renal. La digoxinano debería utilizarse en caso de sospecha deisquemia coronaria o en pacientes con un sín-drome coronario agudo reciente debido almayor riesgo de muerte por arritmias o infar-to. No hay que olvidar que el uso concurrentede fármacos, como la amiodarona, claritro-micina, itroconazol, ciclosporina y otrosmuchos, puede incrementar la dosis de digo-xina y, por tanto, su potencial toxicidad.
ANTICOAGULACIÓNNo existen estudios prospectivos, rando-
mizados, controlados con placebo, que pro-
J. FLORES SEGOVIA ET AL.
50
Monografía HAP NM 208 pag 18/2/10 17:08 Página 50

porcionen evidencia científica a favor o en con-tra de la utilización de los anticoagulantes enel tratamiento de la HAP. Sin embargo, la uti-lización de anticoagulantes en pacientes conHAP está ampliamente aceptada siempre queno existan contraindicaciones (nivel de reco-mendación IIa-C)(10,29).
La base teórica para la utilización de la anti-coagulación en la HAP se apoya en que diver-sos estudios han demostrado que la HAP es unestado protrombótico(15,16). Se ha observadoque este estado protrombótico es más mar-cado en la HAP y la HP asociadas a trombo-embolismo crónico. Además, esa predisposi-ción a la trombosis revierte con tratamientoespecífico para la HAP, como la prostaciclina(17)
o bosentan(18).Estudios retrospectivos no controlados no
randomizados en pacientes con HAP parecensugerir que los anticoagulantes aumentan lasupervivencia en pacientes con HAP(19).
En otras formas de HP (excepto la secun-daria a embolismo crónico), la evidencia cien-tífica o clínica a favor o en contra de la anti-coagulación es muy escasa. De cualquiermanera, debido a la similitud de la HAP conotras formas de HP, el uso de anticoagulantesse ha extendido a estas últimas, aunque sinolvidar que, si no hay una indicación clara,como puede ser embolismo crónico, fibrilaciónauricular, prótesis valvular, lupus eritematososistémico o enfermedad de Rendu-Osler, no sedebe aplicar dicho tratamiento dado el riesgono despreciable de hemorragia(20).
En cuanto al tipo de anticoagulante a uti-lizar, no se han observado diferencias entre losdistintos tipos de anticoagulantes a utilizar,como son la heparina no fraccionada, hepari-na de bajo peso molecular o anticoagulantesorales (ACO), aunque en pacientes con HAPlos ACO (acenocumarol y warfarina) son losmás utilizados. Existen estudios sobre anima-les señalando un posible efecto de la hepari-na sobre la vasculatura pulmonar que no seproduce con los ACO. En este sentido, la hepa-rina produciría una inhibición del crecimien-to de las células musculares de las arterias pul-
monares(21). De cualquier manera, esto toda-vía no ha sido demostrado en humanos y enla práctica clínica el agente más utilizado sonlos ACO.
Se aconseja mantener el INR (Target Inter-national, Normalized Ratio) entre 1,5 -2,5 aun-que algunos proponen entre 2 y 3.
El papel de los antiagregantes, como laaspirina y el clopidogrel, no ha sido evaluadoen profundidad.
La indicación de anticoagulación empíri-ca debe ser considerada si existe sospechamoderada o alta de HAP y la ecocardiografíamuestra o estima una PAP media > 60mmHg(10,29).
CALCIOANTAGONISTASLos calcioantagonistas (CA) han sido utili-
zados desde hace más de veinte años en laHAP(8). Algunos pacientes (<10% según lasseries) que tienen un test de vasorreactividadpositivo y reciben tratamiento con CA, puedenllegar a tener una supervivencia más prolon-gada, mejoría funcional mantenida y mejo-ría hemodinámica; estos hechos han sidodemostrados en un estudio observacional de64 pacientes con HAP idiopática que compa-ró a pacientes no vasorreactivos con un gru-po de pacientes con test de vasorreactividadpositivo tratados con CA(22). La supervivenciaa 5 años fue mayor entre los pacientes querecibieron CA frente a aquellos no tratados (55vs. 95%, respectivamente).
Un reciente estudio, también observa-cional, observó que sólo el 54% de los pacien-tes vasorreactivos con HAP que reciben CAmantienen su estado funcional después deun año de tratamiento(23); aquellos que teníanmejor respuesta eran los que tuvieron un testde vasorreactividad más positivo (disminu-ción mayor de la PAP media durante el testde vasorreactividad) y una menor severidadde la enfermedad en el momento de iniciodel estudio.
De cualquier manera, la evidencia quesugiere el efecto beneficioso de los CA es limi-tada ya que no existen estudios randomiza-
GENERALIDADES TERAPÉUTICAS EN LA HIPERTENSIÓN ARTERIAL PULMONAR (HAP)
51
Monografía HAP NM 208 pag 18/2/10 17:08 Página 51

dos que comparen el tratamiento con CA ensujetos con HAP y test de vasorreactividadpositivos frente a sujetos con HAP no trata-dos con CA y test de vasorreactividad posi-tivo. Por este motivo, su actual nivel de reco-mendación es I-C.
La proporción de los pacientes con HAPque tienen un test de vasorreactividad positi-vo o son considerados “respondedores” des-graciadamente es pequeña, situándose en tor-no al 10%(24,25). En general, existe consensopara considerar a un paciente con HAP como“respondedor” favorable (test de vasorreacti-vidad positivo) cuando se cumplen los siguien-tes criterios(10,25): 1. Caída de la PAP media > de 10 mmHg y
valor de PAP media < 40 mmHg. 2. Con aumento o no disminución del gasto
cardiaco.3. Con reducción mínima o sin cambios en
la tensión arterial sistémica.
En los pacientes con test de vasorreactivi-dad positivo el tratamiento con CA puede rea-lizarse con nifedipino (30 mg/día) o diltiazem(120 mg/día), incrementado hasta la dosismáxima tolerada(26). Se monitorizarán TA sis-témica, frecuencia cardiaca y saturación deoxígeno durante el tratamiento.
La elección del CA puede realizarse de unaforma práctica de la siguiente manera(10,29):1. Nifedipino si la frecuencia cardiaca es
<100/min y diltiazem si es >100/min.2. Existen preparaciones de liberación sos-
tenida para ambos fármacos que minimi-zan sus efectos adversos.
3. Amlodipino puede utilizarse si hay efectosadversos importantes con nifedipino o dil-tiazem (edema en miembros inferiores,taquicardia significativa, bradicardia o hipo-tensión).
4. No se aconseja la utilización de verapami-lo debido a su efecto cardiodepresor.
5. Cuando se requieren altas dosis de CA serecomienda repartir sus dosis: (nifedipino>240 mg/día o diltiazen >720 mg/día,ambos se pueden repartir en 3 dosis/día;
amlodipino >5 mg/día, repartir en dosdosis/día).
6. Sólo se debe utilizar la administración oral.
Los pacientes con respuesta positiva, defi-nido como aquellos asintomáticos o mínima-mente sintomáticos –clase funcional I o II, conPSP cercana a valores normales– deberán serrevisados cada 3-6 meses(26). De no alcanzar-se esos objetivos, se comenzará tratamientoya con fármacos específicos.
MEDIDAS GENERALESEl tratamiento general o convencional
para cualquier modalidad de hipertensiónpulmonar (HP) incluye una dieta hiposódi-ca, evitar la realización de sobreesfuerzos ygrandes altitudes, vacunaciones antigri-pal/antineumocócica y el control de facto-res agravantes, como la anemia, infeccionesrespiratorias o el hipertiroidismo (Nivel derecomendación I-C).
También se aconseja evitar el embarazo porla elevada tasa de morbimortalidad que con-lleva –30 a 50%–(27) especialmente en el pos-parto inmediato (nivel de recomendación I-C).
Un estudio prospectivo reciente en 30pacientes ha demostrado una mejoría de laclase funcional, capacidad de ejercicio –100metros de incremento promedio– y consumopico de oxígeno en pacientes con HAP some-tidos a ejercicio respecto de pacientes con HAPsedentarios, aunque no se observó mejoría enlos parámetros hemodinámicos tras 15 sema-nas de ejercicio(28).
Parece, por tanto, que el ejercicio aeró-bico suave y progresivo, con una frecuen-cia de 4-5 días a la semana, resulta reco-mendable en estos pacientes (nivel derecomendación IIa-B)(29).
BIBLIOGRAFÍA1. Simonneau G, Galie N, Rubin LJ, et al. Clinical
classification of pulmonary hypertension. J AmColl Cardiol. 2004; 43: 10S.
2. Long term domiciliary oxygen therapy in chro-nic hypoxic cor pulmonale complicating chro-
J. FLORES SEGOVIA ET AL.
52
Monografía HAP NM 208 pag 18/2/10 17:08 Página 52

nic bronchitis and emphysema: report of theMedical Research Council Working Party. Lan-cet. 1981; 1 (8222):681-6
3. Nocturnal Oxygen Therapy Trial Group. Con-tinuous or nocturnal oxygen therapy in hypo-xemic chronic obstructive lung disease: a cli-nical trial. Nocturnal Oxygen Therapy TrialGroup. Ann Intern Med. 1980; 93 (3): 391-8.
4. Global strategy for the diagnosis, managementand prevention of chronic obstructive pulmo-nary disease: NHLBI/WHO workshop report.En: NHLBI/WHO. Global initiative for chronicobstructive pulmonary lung disease. Bethes-da, MD: National Heart, Lung and Blood Ins-titute;2005. Disponible en: http://www.gold-copd.org/
5. Owens GR, Rogers RM, Pennock BE, et al. Thediffusing capacity as a predictor of arterial oxy-gen desaturation during exercise in patientswith chronic obstructive pulmonary disease.N Engl J Med. 1984; 310 (19): 1218-21.
6. Bowyer JJ, Busst CM, Denison DM, et al. Effectof long term oxygen treatment at home in chil-dren with pulmonary vascular disease. Br HeartJ. 1986; 55 (4): 385-90.
7. Sandoval J, Aguirre JS, Pulido T, et al. Noctur-nal oxygen therapy in patients with the Eisen-menger syndrome. Am J Respir Crit Care Med.2001; 164 (9): 1682-7.
8. Badesch DB, Abman SH, Ahearn GS, et al.Medical therapy for pulmonary arterial hyper-tension: ACCP evidence-based clinical practi-ce guidelines. Chest. 2004; 126 (Suppl. 1): 35S-62S.
9. Widlitz A, Barst RJ. Pulmonary arterial hyper-tension in children. Eur Respir J. 2003; 21 (1):155-76.
10. Alam S, Palevsky HI. Standard therapies forpulmonary arterial hypertension. Clin ChestMed. 2007; 28 (1): 91-115, viii.
11. Eshaghian S, Horwich TB, Fonarow GC. Rela-tion of loop diuretic dose to mortality in advan-ced heart failure. Am J Cardiol. 2006; 97 (12):1759-64
12. Pitt B, Zannad F, Remme WJ, et al. The effectof spironolactone on morbidity and morta-lity in patients with severe heart failure: ran-domized Aldactone Evaluation Study Inves-tigators. N Engl J Med. 1999; 341 (10,29):709-17.
13. Galié N, Palazzini M, Negro L, et al. Pharma-cological impact on right ventricullar remo-delling in pulmonary arterial hypertensión.
European Heart Journal suppl. 2007; 9 (HSuppl): H68-H74.
14. Rich S, Seidlitz M, Dodin, et al. The short-termeffects of digoxin in patients with right ven-tricular dysfunction from pulmonary hiper-tension. Chest. 1998; 114 (3): 787-92.
15. Cacoub P, Karmochkine M, Dorent R, et al.Plasma levels of thrombomodulin in pulmo-nary hypertension. Am J Med. 1996; 101(2):160-4.
16. Hoeper MM, Sosada M, Fabel H. Plasma coa-gulation profiles in patients with severe pri-mary pulmonary hypertension. Eur Respir J.1998; 12 (6): 1446-9.
17. Friedman R, Mears JG, Barst RJ. Continuousinfusion of prostacyclin normalizes plasmamarkers of endothelial cell injury and plateletaggregation in primary pulmonary hyperten-sion. Circulation. 1997; 96 (9): 2782-4.
18. Girgis RE, Champion HC, Diette GB, et al.Decreased exhaled nitric oxide in pulmonaryarterial hypertension response to bosentantherapy. Am J Respir Crit Care Med. 2005; 172(3): 352-7.
19. Frank H, Miczoch J, Huber K, et al. The effectof anticoagulant therapy in primary and ano-retic drug-induced pulmonary hypertension.Chest. 1997; 112 (3); 714-21.
20. Ansell J, Hirsh J, Oller L, et al. The pharma-cology and management of the vitamin K anta-gonists: seventh ACCP conference on antith-rombotic and thrombolytic therapy: Chest.2004; 126 (3 suppl.): 204S-233S.
21. Yu L, Quinn DA, Garg HG, et al. Gene expres-sion of cyclin-dependent kinase inhibitors andeffect of heparin on their expression in micewith hypoxia-induced pulmonary hyperten-sion. Biochem Biophys Res Commun. 2006;345 (4): 1565-72.
22. Rich S, Kaufmann E, Levy PS, et al. The effectof high doses of calcium Channel blockers onsurvival in primary pulmonary hypertension.N Engl J Med. 1992; 327: 76-81.
23. Sitbon O, Humbert MJ, et al. Long-term res-ponse to calcium channel blockers in idiopa-thic pulmonary hypertension. Circulation.2005; 111; 3105-11.
24. Chin KM, Rubin LJ. Pulmonary arterial hyper-tension. J Am Coll Cardiol. 2008; 51 (16): 1527-38.
25. Badesch DB, Abman SH, Simonneau G, et al.Medical Therapy for pulmonary arterial hyper-tensión: Updated ACCP Evidence-Based Cli-
GENERALIDADES TERAPÉUTICAS EN LA HIPERTENSIÓN ARTERIAL PULMONAR (HAP)
53
Monografía HAP NM 208 pag 18/2/10 17:08 Página 53

nical Practice Guidelines. Chest. 2007; 131 (6):1917-28.
26. Hopkins W, Rubin LJ. Treatment of pulmonaryhypertension. Uptodate, enero 2009: Dispo-nible en www.uptodate.com
27. Sánchez Román J, Gracia Hernández FJ, Cas-tillo Palma MJ, et al. Diagnóstico y tratamien-to de la hipertensión pulmonar. Rev Clin Esp.2008; 208 (3): 142-55.
28. Mereles D, Ehlken N, Kreuscher S, et al. Exer-cise and respiratory training improve exerci-se capacity y quality of life in patients withsevere chronic pulmonary hypertension. Cir-culation. 2006; 114 (14): 1482-9.
29. Galiè N, Torbicki A, Barst R, et al. ESC Guide-lines. Guidelines on diagnosis and treatmentof pulmonary arterial hypertension. Eur HeartJ. 2009; 30: 2493-537.
J. FLORES SEGOVIA ET AL.
54
Monografía HAP NM 208 pag 18/2/10 17:08 Página 54

55
RESUMENEl diagnóstico de hipertensión arterial pul-
monar se basa en la realización del cateteris-mo del lado derecho, tras un proceso diag-nóstico secuencial. Debe realizarse unavaloración pronóstica inicial que evalúe la seve-ridad y estabilidad del paciente. Los objeti-vos terapéuticos se basan en alcanzar la situa-ción clínica estable y de buen pronóstico. Eltratamiento consiste en la adopción de medi-das generales, inicio de la terapia de soportey la remisión del paciente a un centro de refe-rencia. La prueba aguda de vasorreactividaddebería realizarse en todos los pacientes delgrupo 1. En aquellos pacientes candidatos a laterapia con calcio-antagonistas deberá confir-marse el mantenimiento de la respuesta a los3-4 meses de haber iniciado el tratamiento.Los pacientes no respondedores agudos en cla-se funcional II (OMS) deberían tratarse con unantagonista de la endotelina (ARE) o un inhi-bidor de la fosfodiesterasa tipo 5 (IPD5). Losno respondedores agudos, los respondedoresagudos que persisten en clase funcional III(OMS) o que progresan a ella serían candida-tos a tratamiento con un ARE, IPD5, o un pros-tanoide. En clase funcional IV (OMS) se empleala prostaciclina intravenosa y también debeconsiderarse el tratamiento combinado de ini-cio. En el caso de obtener una respuesta inade-cuada, se planteará terapia de combinaciónsecuencial. Las asociaciones más comunes sonARE con IPD5, o un prostanoide con ARE o
IPD5. En muchas ocasiones se debe llegar autilizar triple terapia. Si, a pesar de una tera-pia óptima, o en ausencia de tratamiento médi-co, la respuesta es inadecuada, se indicará sep-tostomía con balón o trasplante.
INTRODUCCIÓNLa hipertensión arterial pulmonar (HAP)
pertenece al grupo I de la clasificación de DanaPoint(1) Es el grupo en donde se han realizadomayores avances terapéuticos en los últimosaños. El valor pronóstico, demostrado enpacientes con HAP de etiología idiopática(HAPI), de tres parámetros hemodinámicos clá-sicos: presión de aurícula derecha, índice car-diaco y presión arterial pulmonar media, con-firma la importancia de la progresión del fracasoventricular derecho en la evolución de estospacientes. La modificación de este componentehemodinámico y el pronóstico del pacienteestán relacionados con una compleja relaciónfisiopatológica entre la progresión de los cam-bios obstructivos en la microcirculación pul-monar y el comportamiento de un ventrículoderecho sobrecargado, lo que a su vez pudie-ra estar determinado genéticamente.
ASPECTOS DIAGNÓSTICOSLa realización de un cateterismo del lado
derecho es necesaria para confirmar el diag-nóstico y valorar la severidad de la afectaciónhemodinámica. La hipertensión pulmonar (HP)se ha definido tradicionalmente como una pre-
GRUPO I. CLASIFICACIÓN DE DANA POINT
HIPERTENSIÓN ARTERIAL PULMONARIDIOPÁTICA Y RESTO. ASPECTOSDIAGNÓSTICOS Y TERAPÉUTICOSCarlos Pindado Rodríguez, Marta de Riva Silva, Miguel Ángel Gómez Sánchez
BLOQUE II. ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS SEGÚN GRUPOS DE CLASIFICACIÓN ETIOLÓGICA
Monografía HAP NM 208 pag 18/2/10 17:08 Página 55

sión arterial pulmonar media (PAPm) en repo-so > 25 mmHg, o con el ejercicio superior a30 mmHg(2). El grupo I de la clasificación (HAP)añade el criterio de que la presión arterial pul-monar de enclavamiento debe ser ≤ 15 mmHg.La presión arterial pulmonar media (PAPm) nor-mal en reposo es de aproximadamente 14 ±3,3 mmHg, y el límite superior de la normali-dad, de aproximadamente 20,6 mmHg(3). Seha abandonado el criterio de HP en el ejerci-cio, pues existe mucha variabilidad en su defi-nición incluyendo, por ejemplo, el hecho deque el nivel, el tipo y la postura del ejercicio nohan sido especificados. Además, resulta cono-cido cómo la presión arterial pulmonar en elejercicio varía con la edad. Durante el ejercicioligero la PAPm fue 32 (supino) y 30 mmHg(bipedestación); y durante el ejercicio máximofue 37 (supino) y 35 mmHg (vertical)(4).
La hipertensión pulmonar en la insuficien-cia cardiaca con fracción de eyección normal ydisfunción diastólica puede presentar valores depresión capilar pulmonar (PCP) levemente ele-vados o normales. En ocasiones, esta entidadplantea un problema diagnóstico diferencial conla HAP, al estar la PCP en rango de normalidad.Con frecuencia se requiere realización de ejer-cicio o sobrecarga de volumen para demostrar
un aumento desproporcionado de la PCP, aun-que estas maniobras diagnósticas no se encuen-tran suficientemente estandarizadas (Fig. 1).
La prueba aguda de vasorreactividad serealiza en el momento del cateterismo dere-cho diagnóstico, con objeto de identificar apacientes con respuesta mantenida al trata-miento con antagonistas del calcio, ofrecien-do información pronóstica adicional. Única-mente debe realizarse con fármacos de accióncorta, seguros, fáciles de administrar y conlimitados efectos sistémicos. En el momentoactual, el agente más usado es el óxido nítri-co por vía inhalada. También se ha utilizadoepoprostenol y adenosina por vía intraveno-sa(5). El iloprost inhalado y el sildenafilo oralpueden presentar marcados efectos vasodila-tadores; recientemente el iloprost inhalado hademostrado capacidad para predecir la res-puesta al tratamiento con antagonistas del cal-cio(6). Los propios calcioantagonistas están con-traindicados, para demostrar vasorreactividadde forma aguda, habiéndose descrito resulta-dos fatales con este uso. La respuesta positivase define como una reducción en la PAP media≥ 10 mmHg, alcanzando un valor absoluto ≤40 mmHg con un gasto cardiaco mantenido oaumentado. Menos de un 15% de los pacien-
C. PINDADO RODRÍGUEZ ET AL.
56
FIGURA 1. Algoritmo diagnós-tico para distinguir la HAP dela HP causada por disfuncióndiastólica.
Cateterismoderecho
PCP ≥ 15RVP < 3
PCP ≥ 15RVP ≥ 3
PCP < 15RVP ≥ 3
PCP <15RVP ≥ 3Tratamiento
Probable ICD Dudosa ICD Improbable ICD
PCP < 15RVP<3
PCP < 15RVP ≥ 3
Nitroprusiato o NTG Volumen o ejercicio
ICDHAP I
ICD: insuficiencia cardiaca diastólica (con fracción de eyección normal); PCP:presión capilar pulmnar media mmHg; RVP: resistencias vasculares pulmo-nares U. Wood; NTG: nitroglicerina; HAP: hipertensión arterial pulmonar.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 56

GRUPO I. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN ARTERIAL PULMONAR IDIOPÁTICA Y RESTO…
57
tes de este grupo presentan esta respuesta. Delos pacientes respondedores agudos con HAPde etiología idiopática (HAPI), alrededor dela mitad mantienen la respuesta a largo pla-zo con el tratamiento oral con antagonistas delcalcio(7). Sólo en estos casos está indicado con-tinuar con este tratamiento en monoterapia.Esta prueba ha demostrado ser útil en la etio-logía idiopática. En otros tipos de HAP, comola hereditaria, la asociada a conectivopatíasy la asociada a VIH, la utilidad es menos cla-ra, aunque expertos recomiendan la continui-dad de su práctica. No hay datos de su utili-dad en la HAP asociada a cardiopatíascongénitas donde su práctica es controverti-da. No se recomienda su realización en los gru-pos clínicos 2, 3, 4 y 5 de la clasificación deDana Point.
El proceso diagnóstico comienza con laidentificación de los grupos clínicos más comu-nes de hipertensión pulmonar, la enfermedadde corazón izquierdo y las patologías pulmo-nares (grupos 2 y 3, respectivamente). A con-tinuación se descarta la enfermedad trombo-embólica crónica (grupo 4) y, finalmente, sereconocen los diferentes tipos que integran elgrupo 1 y las raras condiciones del grupo 5. Laetiología idiopática es un diagnóstico por exclu-sión(5) (ver Fig. 2)
El ecocardiograma bidimensional Doppleres de gran ayuda en el proceso diagnóstico deHP. Con objeto de evitar falsos diagnósticos,se recomienda un punto de corte por encimade la presión sistólica en la arteria pulmonarmayor de 50 mmHg como dato especifico paraencontrar HP.
El screening ecocardiográfico se reco-mienda para la detección de hipertensión pul-monar, en pacientes sintomáticos con enfer-medades del espectro esclerodermia (Gradode recomendación y nivel de evidencia: IB),pacientes sintomáticos con el resto de conec-tivopatías (IC) y en todos los casos donde sesospeche HAP asociada a conectivopatías (IC).Por el mismo motivo se realiza en pacientessintomáticos con enfermedad hepática y/o can-didatos a trasplante hepático (IB). También se
indica en pacientes con infección por VIH ycon disnea no explicada, para detectar com-plicaciones cardiovasculares asociadas al VIH(IC)(5).
VALORACIÓN DE LA SEVERIDADEl pronóstico depende significativamente
de la etiología. En términos generales, la HAPasociada a enfermedades del tejido conectivocursa con peor pronóstico que la HAP idiopá-tica. A su vez esta última, en ausencia de tra-tamiento específico, tiene peor pronóstico quela HAP asociada a cardiopatías congénitas(8).
La capacidad funcional es un fuerte pre-dictor de supervivencia. Las edades extremas(<14 y >65 años), la reducción de la capa-cidad de ejercicio, la presencia de síncope,hemoptisis y signos de fallo derecho, conlle-van un pronóstico pobre en la etiología idio-pática. Determinados parámetros ecocardio-gráficos han demostrado su valor pronóstico:presencia de derrame pericárdico, área inde-xada de la aurícula derecha, índice de excen-tricidad del ventrículo izquierdo, índice Dop-pler del ventrículo derecho (Tei) y la excursióndel anillo tricúspide (TAPSE)(9,10). Algunos estu-dios sugieren que la saturación arterial pul-monar de oxígeno reducida, la baja presiónarterial sistólica y una frecuencia cardiaca ele-vada también se asocian a peor pronóstico. Elíndice cardiaco, la presión de la AD y la pre-sión media de la arteria pulmonar fueron incor-porados en una fórmula para predecir en pro-nóstico de la HAP idiopática previa a laaparición de terapia específica para esta pato-logía. No está claro que pueda aplicarse en lapráctica clínica actual.
La valoración de la capacidad de ejerciciose realiza con el test de la marcha de seis minu-tos. Una distancia recorrida inferior a 332 my la desaturación >10% indican peor pro-nóstico(11). El incremento de la distancia reco-rrida es un objetivo primario en muchos ensa-yos clínicos controlados de HAP. Esta pruebano está suficientemente validada en todos lossubgrupos de HAP y se ve influenciada porla edad, peso, talla, género y motivación del
Monografía HAP NM 208 pag 18/2/10 17:08 Página 57
C. PINDADO RODRÍGUEZ ET AL.
58
FIGURA 2. Algoritmo diagnóstico de la HP.
ECG: electrocardiograma; Rx: radiografía de torax; TAC AR: tomografía axial computarizada de alta resolución; PFR:pruebas de función respiratoria; HPTC: hipertensión pulmonar tromboembólica crónica; APm: presión arterial pul-monar media mmHg; PCP: presión capilar pulmonar media mmHg; EPVO: enfermedad pulmonar venooclusiva;ANA: anticuerpos antinucleares; HIV: virus inmunodeficiencia humana; ETT: ecocardiografía transtorácica; ETE; eco-cardiografía transesofágica; RMN: resonancia magnética nuclear; HTPP: hipertensión portopulmonar; BMPR2: recep-tor 2 de proteína morfogenética del hueso; ALK1: activina tipo cinasa tipo 1.
Drogas colagenopatías
EsquistosomiasisOtros del Grupo 5
Hemólisiscrónica
CC HTPP
Idiopática o hereditaria BMPR2, ALK1, etc.
Grupo 1
Enfermedad cardiaca izquierdaGrupo 2
Enfermedad pulmonar crónica/hipoxiaGrupo 3
EPVOHemangiomatosis
Exámenes específicos
Signos, síntomasANA, TAC AR
Historialab. específico
Serologíap/HIV
ETT-ETE-RMN
Ev. funciónhepática
HistoriaEx. específico
HistoriaLab. especifico
Considere otrascausas
Considere Grupo 4HPTC
ConsidereEVOP-Hemangiomatosis
Sí
No
No
Sí
Sí Sí
APm ≥ 25 mmHgPCP ≤ 15
HIV
Cateterismo derecho
Considere otras causas poco comunes
Defecto segmentario
HP proporcional a la severidad HP desproporcionada a la severidad
Gammagrafía V/Q
Diagnóstico confirmado: Grupo 2 y 3
ECG-Rx-TAC AR-PFR
Considerar causas comunes de HP
Evaluación no invasiva
Signos y síntomas sugestivos de HP
Monografía HAP NM 208 pag 24/2/10 11:41 Página 58

paciente. En el momento actual, debido a lafalta de estandarización en la realización de laprueba de ejercicio cardiopulmonar, el test deseis minutos caminando sigue siendo la prue-ba aceptada para la valoración de los efectosdel tratamiento en la HAP. Por otro lado, en laergoespirometría, el consumo pico de oxíge-no, el pulso de O2, la presión sistólica pico conel esfuerzo y la valoración de la eficiencia ven-tilatoria mediante la pendiente ventilaciónminuto/producción de CO2 proporciona infor-mación importante acerca de la función delventrículo derecho durante el ejercicio(12).
Biomarcadores como el BNP o el NT-proBNP son de utilización recomendada pararealizar una estratificación de riesgo inicial ypueden ser utilizados para monitorizar el efec-to del tratamiento(13). En la práctica, el valorpronóstico de la determinación de niveles deácido úrico es muy limitado por la interferen-cia del tratamiento habitual del paciente, que
a menudo requiere diuréticos y control de lahiperuricemia que éstos inducen con alopu-rinol. El valor de la monitorización de la tro-ponina T aún requiere confirmarse en nuevosestudios.
Un conjunto de parámetros nos permi-te separar, grupos de pacientes con mejor ypeor pronóstico (Tabla 1). Sin embargo, noexiste un umbral claramente definido paracada parámetro, dejando un grupo interme-dio cuyo pronóstico es más difícil de definir.En estos casos, además de tales parámetros,también debemos considerar conjuntamen-te otros factores como: edad, etiología ycomorbilidad(5). Es importante no fiarse úni-camente de un parámetro, pues con fre-cuencia múltiples mediciones arrojan resul-tados divergentes. No es preciso valorar latotalidad de los parámetros en cada visita. Enla tabla 2 se propone la relación de pruebasy temporalidad de las mismas en el segui-
TABLA 1. Parámetros que deben ser evaluados en el seguimiento de los pacientescon HAP
Mejor pronóstico Parámetros evaluados Peor pronóstico
No Presencia de insuficiencia Sícardiaca derecha
Lenta Velocidad de progresión Rápida
No Síncope Sí
I, II Clase funcional (OMS) IV
Larga (> 500 m)* T6MC Corta (< 300 m)
Normal o casi-normal Niveles plasmáticos Muy elevados y en aumentode BNP/NT-proBNP
No derrame pericárdico Hallazgos ecocardiográficos† Derrame pericárdicoTAPSE > 2.0 cm TAPSE < 1,5 cm
PAD < 8 mmHg PAD > 15 mmHg oe IC ≥ 2,5 L/min/m2 Hemodinámica IC ≤ 2,0 L/min/m2
*Varía con la edad y es preciso adaptar los protocolos.† TAPSE y el derrame pericárdico se han seleccionado porque pueden ser medidos en la mayoría de los pacientes.OMS: Organización Mundial de la Salud; T6MC: test de 6 minutos caminando; BNP/NT-proBNP: péptido natriuréticocerebral; TAPSE: excursión sistólica posterior del anillo tricuspídeo; PAD: presión aurícula derecha, mmHg; IC: índicecardiaco, L/min/m2.
GRUPO I. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN ARTERIAL PULMONAR IDIOPÁTICA Y RESTO…
59
Monografía HAP NM 208 pag 18/2/10 17:08 Página 59

C. PINDADO RODRÍGUEZ ET AL.
60
miento de pacientes con HAP. Con esta infor-mación podemos clasificar la condición delpaciente como: estable y satisfactoria, cuan-do cumple la mayoría de los parámetros de“mejor pronóstico”. Estable y no satisfactoria,en la que no se cumplen algunos de los cri-terios del grupo de “mejor pronóstico” y amenudo se requiere reevaluación y plantearcambio o incorporación de nuevos trata-mientos seguido de una valoración comple-ta en un centro de referencia de esta patolo-gía. Inestable y deteriorándose, cuando sereúna la mayoría de los criterios de “peor pro-nóstico”. El incremento de edemas, necesi-dad de diuréticos, frecuencia e intensidad deangina y frecuencia de síncopes son sínto-mas ominosos, como expresión de bajo gas-to cardiaco, y requieren atención inmediata.Las arritmias supraventriculares son relati-vamente frecuentes en esta condición y con-tribuyen a un mayor deterioro clínico. La res-tauración del ritmo sinusal en pacientes conHAP supone una mejor supervivencia a lar-go plazo, mientras que la persistencia en fibri-lación auricular se asocia a una mortalidadpor encima del 80% a los 2 años. Las arrit-mias ventriculares son raras en pacientes conHAP(14).
TRATAMIENTO
Medidas generalesEl esquema general del tratamiento de la
HAP se refleja en la figura 3. La HAP es una enfermedad crónica para
la que no existe, en el momento actual, trata-miento médico curativo. La única opción decuración es el trasplante pulmonar o cardio-pulmonar, cuando es aceptable asumir lacomorbilidad que conlleva. El tratamientomédico moderno permite mejorar la sinto-matología de los pacientes y frenar su dete-rioro clínico. Los casos más avanzados requie-ren terapias médicas e intervencionistasinvasivas y con efectos adversos significativos.
Los pacientes requieren consejos prácticosen relación a las actividades de la vida diaria ynecesitan adaptarse a la incertidumbre queacompaña a padecer una enfermedad crónicamuy grave. El aislamiento social es frecuente.El apoyo ofrecido por grupos de ayuda parapacientes y familiares suele ser positivo. Laansiedad y la depresión que acompañan aldetrimento en calidad de vida puede requerirdel tratamiento psicológico o psiquiátrico.
Recientemente se ha demostrado que elentrenamiento realizado dentro de un pro-
TABLA 2. Estrategia sugerida para el seguimiento de pacientes con HAP
3 meses Basal (previo Cada 3 Cada 6-12 Empeoramiento después de cambios
al trat.) meses* meses* clínico* en el tratamiento
Evaluación clínica/clase funtional (OMS) X X X X
6 min caminando X X X X
NT-proBNP X X X X
Pulsioximetría X X X X X
Ecocardiograma X X X X
Cateterismo derecho X X† X† X†
*Dependiendo de la severidad y estabilidad del paciente. † Varía con la experiencia del centro.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 60

grama controlado de rehabilitación corrige lapérdida de masa muscular periférica y mejo-ra la capacidad de ejercicio, en la misma medi-da que algunos tratamientos médicos. Sinembargo, la actividad física excesiva puededesencadenar síntomas alarmantes, por lo quedebe evitarse.
El embarazo está contraindicado, pues seasocia con una mortalidad del 30-50% en estospacientes, debiendo utilizarse una combina-ción de dos métodos anticonceptivos. Bosen-tan puede reducir la eficacia de los anticon-ceptivos orales. Llegado el caso, debe plantearse
la interrupción del embarazo tras informar a lapaciente de elevado riesgo en caso de conti-nuar con el mismo(15).
La cirugía electiva supone un riesgo aumen-tado en pacientes con HAP. Aunque no estáclaro qué modalidad de anestesia es preferi-ble, probablemente la epidural será mejor tole-rada que la general. Los pacientes que seencuentren recibiendo tratamiento oral pue-den requerir conversión temporal a tratamientointravenoso o nebulizado.
Debe considerarse la administración deoxigenoterapia durante el vuelo a pacientes en
GRUPO I. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN ARTERIAL PULMONAR IDIOPÁTICA Y RESTO…
61
FIGURA 3. Tratamiento de los pacientes con hipertensión arterial pulmonar, basado en la evidencia.
Continuarcalcioantagonista
Sí No
Vasorreactivo No vasorreactivo
*Mantener oxígeno al 92%
BASB y/o trasplantepulmonar
ProstanoidesiPDE-5 ARE
Anticoagulantes orales HAPIDiuréticosOxígeno*DigoxinaRehabilitación supervisada
Evitar ejercicio físico excesivoControl natalidadSoporte psicosocialPrevención infecciones
Tratamiento de soporte ymedidas generales
Referir a un experto
Clase III OMSARE (A) o iPDE-5Iloprost inhaladoTreprostinil subc.Treprostinil inhalado.Epoprostenol IVIloprost IVTreprostinil IVBeraprost
Test agudo de vasorreactividad
Respuesta mantenida (OMS I-III)
Clase I-IV OMSCalcioantago-nistas orales
Clase II OMSARE o iPDE-5
Clase III OMSEpoprostenol IVIloprost IVTreprostinil IVIloprost inhaladoTreprostinil subc.Treprostinil inhaladoAREiPDE-5Tto. inicial combinado
Respuesta clínica inadecuada
Respuesta clínicainadecuadaTratamiento combinado
Monografía HAP NM 208 pag 18/2/10 17:08 Página 61

clase funcional (CF-OMS) III-IV y aquellos conPO2 persistentemente por debajo de 60mmHg. Un flujo de 2 L/min elevará la presiónde oxígeno inspirada a un nivel equivalente alexistente a nivel del mar.
Los pacientes con HAP son propensos apadecer neumonía, la cual es la causa de muer-te en un 7% de los casos.(16). Aunque no se dis-pone de ensayos controlados, se recomienda lavacunación antigripal y la antineumocócica.
Terapia de soporteLa evidencia a favor de la anticoagulación
oral se limita a la etiología idiopática, heredi-taria y asociada a la ingesta de anorexígenos.Es generalmente retrospectiva y se basa en laexperiencia de un único centro. Debe valorarsela relación riesgo/beneficio, especialmentecuando el riesgo de sangrado es elevado, comoen la hipertensión portopulmonar o en la aso-ciada a cardiopatías congénitas. En esta últi-ma se indica en presencia de trombosis dearteria pulmonar y en ausencia de hemopti-sis, o con leve hemoptisis. Generalmente lospacientes que reciben tratamiento a largo pla-zo con prostaglandinas por vía intravenosa sonanticoagulados, en ausencia de contraindica-ciones, debido en parte al riesgo añadido detrombosis del catéter.
Aunque no hay ensayos clínicos controla-dos, los diuréticos han demostrado un clarobeneficio sintomático en pacientes con sobre-carga de volumen. Debe considerarse la adi-ción de antagonistas de la aldosterona. Esimportante la monitorización de la funciónrenal, debiendo evitarse la hipopotasemia y lahipovolemia, lo que podría derivar en fallo pre-rrenal.
No hay estudios randomizados que sugie-ran que la oxigenoterapia a largo plazo es bene-ficiosa. No se ha demostrado que prolonguela supervivencia, al menos administrada sólopor la noche, pero puede mejorar la sintoma-tología en determinados casos. En la indica-ción se suele seguir la evidencia basada enEPOC: cuando la PO2 es consistentementemenor de 60 mmHg se instaura, para alcan-
zar una PO2 >60 mmHg, durante al menos15 h al día.
En la HAP asociada a cardiopatías congé-nitas cianógenas debe evitarse la flebotomíarutinaria. Estará indicada si aparecen síntomasde hiperviscosidad y el hematocrito es >65%.Debe realizarse con reposición de volumen. Ladeficiencia de hierro debe corregirse.
La digital puede ser utilizada para el con-trol de frecuencia cardiaca en pacientes conHAP que desarrollan taquiarritmias auricula-res. Fármacos antiarrítmicos sin efecto inotró-pico negativo, como la amiodarona, debenconsiderarse para intentar restaurar el ritmosinusal. La información disponible sobre su efi-cacia en este contexto es escasa y deben valo-rarse las posibles interacciones con otros fár-macos.
La terapia inmunosupresora combinandoglucocorticoides y ciclofosfamida puede pro-ducir mejoría clínica en pacientes con HAPasociada a lupus eritematoso o enfermedadmixta del tejido conectivo.
Terapia específica
Antagonistas del calcioSólo un pequeño número de pacientes con
HAP que demostraron respuesta favorable enel test agudo vasodilatador mantienen el bene-ficio a largo plazo. Aunque otras etiologías delgrupo 1 pueden responder positivamente enla prueba aguda, la respuesta mantenida seasocia, casi exclusivamente, a la etiología idio-pática. Los antagonistas del calcio más utili-zados en los estudios realizados han sido nife-dipino y diltiazem seguidos de amlodipino.Dependiendo de la frecuencia cardiaca en repo-so del paciente se basaba la elección de lamolécula. La bradicardia favorecía el uso denifedipino o amlodipino y la taquicardia rela-tiva favorecía el uso de diltiazem. Se desa-conseja el uso de verapamilo, en este contex-to, debido a su efecto inotropo negativo. Lasdosis diarias utilizadas son relativamente altas:120-240 mg de nifedipino, 240-720 de diltia-zem y hasta 20 mg de amlodipino. Se debe
C. PINDADO RODRÍGUEZ ET AL.
62
Monografía HAP NM 208 pag 18/2/10 17:08 Página 62

comenzar con dosis bajas de estos fármacos:30 mg de nifedipino de liberación retardadados veces al día, 60 mg de diltiazem 3 vecesal día o 2,5 mg de amlodipino una vez al día.Debe aumentarse la dosis lentamente, bajo unseguimiento estrecho, hasta alcanzar la máxi-ma dosis tolerada. Los factores limitantes sue-len ser la hipotensión y el edema periférico enmiembros inferiores; este último puede mejo-rar parcialmente con el tratamiento diurético.Se debe realizar un seguimiento cercano deestos pacientes y reevaluar con cateterismodel lado derecho a los 3-4 meses de haber ini-ciado el tratamiento(7).
ProstanoidesEl uso clínico de la prostaciclina en pacien-
tes con HAP se ha extendido gracias a la sín-tesis de análogos estables, que poseen dife-rentes propiedades farmacocinéticas perocomparten cualitativamente similares efectosfarmacodinámicos.
Epoprostenol tiene una vida media corta(3-5 min) y es estable a temperatura ambien-te durante 8 horas. Debe ser administrado enbomba de infusión continua y mediante uncatéter permanente (Hickman). Su eficacia haquedado demostrada en ensayos clínicos con-trolados en pacientes con HAPI y en HAP aso-ciada a esclerodermia. Mejora la sintomatolo-gía, la capacidad de ejercicio y los parámetroshemodinámicos. Es el único tratamiento queha demostrado prolongar la supervivencia enun estudio randomizado(17). Se ha observadola persistencia de su eficacia a largo plazo enHAPI, en HAP asociada a diferentes condicio-nes, incluyendo la HAP asociada a enferme-dad tromboembólica crónica no susceptiblede cirugía. La dosis de inicio es de 2-4ng/kg/min y la dosis óptima suele ser alrede-dor de 20-40 ng/kg/min. Efectos secundarios,como el rubor, la cefalea, la diarrea y el doloren miembros inferiores limitan la escalada dedosis. Los eventos adversos de mayor grave-dad se deben al sistema de administración:malfuncionamiento de la bomba, infecciónlocal, obstrucción del catéter y sepsis. La inte-
rrupción brusca de la infusión puede producirun rebote de la hipertensión pulmonar condeterioro sintomático e incluso la muerte.
Iloprost es un análogo estable disponiblepor vías intravenosa, oral e inhalada. La tera-pia inhalada tiene la ventaja teórica de la selec-tividad por la circulación pulmonar. Se admi-nistran en dosis de 2,5-5 µg/inhalación, 6-9veces al día. Es generalmente bien tolerado,los efectos secundarios más frecuentes son enel rubor y el dolor mandibular. Ha demostra-do mejorar la sintomatología, la capacidad deejercicio, las resistencias vasculares pulmona-res y los eventos clínicos(18). En administraciónintravenosa continua parece ser tan efectivocomo el epoprostenol. No se han valorado susefectos por vía oral en HAP.
Teprostinil es un análogo del epoproste-nol con suficiente estabilidad como para seradministrado a temperatura ambiente. Puedeadministrarse por vía subcutánea. Se inicia auna dosis de 1-2 ng/kg/min(19). La dosis ópti-ma varía entre 20-80 ng/kg/min. El dolor en lazona de infusión es el efecto adverso más fre-cuente. Conlleva la interrupción del tratamientoen un 8% de los casos y limita el ascenso dedosis en otro porcentaje adicional de casos. Elrubor y la cefalea también limitan la escala-da de dosis. Mejora la sintomatología, la capa-cidad de ejercicio, los parámetros hemodiná-micos y parece prolongar la supervivencia(20,21).El mayor beneficio, en cuanto a mejora de lacapacidad de ejercicio, lo obtienen los pacien-tes con estado basal más comprometido y quetoleran dosis más elevadas. Por vía intraveno-sa, el efecto parece ser comparable al del epo-prostenol pero a una dosis entre dos y tresveces mayor. El reservorio puede ser cambia-do cada 48 h en lugar de cada 12 h, comosucede con el epoprostenol. Se están realizandoensayos clínicos controlados con esta drogaadministrada por vía oral y recientemente seha comprobado su eficacia por via inhalada.
Beraprost es el primer análogo estable yactivo por vía oral de la prostaciclina. Hademostrado mejorar la capacidad de ejerciciopero el beneficio no se mantiene trascurri-
GRUPO I. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN ARTERIAL PULMONAR IDIOPÁTICA Y RESTO…
63
Monografía HAP NM 208 pag 18/2/10 17:08 Página 63

dos 3-6 meses. No se han obtenido beneficioshemodinámicos con este fármaco.
Antagonistas del receptor de la endotelinaLa eficacia en la HAP de los antagonistas
de endotelina-1 con efecto sobre las dos iso-formas del receptor (A y B) parece similar a losque presentan selectividad por el receptor A.
Bosentan, antagonista oral de la endoteli-na (ARE) no selectivo, fue la primera molé-cula sintetizada. Ha sido evaluado en variosensayos clínicos controlados (en HAPI, HAPasociada a conectivopatías y síndrome deEisenmerger) demostrando mejoría en la capa-cidad de ejercicio, clase funcional, tiempo has-ta el deterioro clínico, variables hemodiná-micas y ecocardiográficas. Ha sido aprobadopara su uso en CF (OMS) II y en pacientes concircuitos sistémico-pulmonares congénitos ysíndrome de Eisenmenger(22-24). El tratamien-to se inicia a dosis de 62,5 mg dos veces al díay se incrementa hasta 125 mg dos veces al día,trascurridas 4 semanas. La duración de su efec-to a largo plazo ha quedado demostrada. Un10% de los pacientes presentan una elevaciónde transaminasas. Este efecto es dosis depen-diente y reversible tras la reducción de dosiso interrupción del tratamiento. Por este moti-vo, la función hepática debe monitorizarsemensualmente en pacientes que recibenbosentan. Se ha observado la disminución enlos niveles de hemoglobina y el deterioro deespermatogénesis con este fármaco.
Sitaxentan, antagonista oral selectivo parael receptor de endotelina-A, ha sido evaluadoen ensayos clínicos controlados en pacientescon clase funcional (OMS) II y III (en HAPI, HAPasociada a conectivopatías y síndrome de Eisen-merger). Ha demostrado mejorar la capacidadde ejercicio y la durabilidad de sus efectos a lolargo del tiempo (1 año)(25,26). Con la dosis apro-bada de 100 mg una vez al día, la incidencia dealteración en la función hepática es del 3-5%.La función hepática debe monitorizarse men-sualmente. Interacciona con dicumarínicos.
Ambrisentan, antagonista oral selectivopara el receptor de endotelina-A, evaluado en
dos grandes ensayos clínicos controlados(27,28).Ha demostrando eficacia en la mejoría de lossíntomas, capacidad de ejercicio, parámetroshemodinámicos y tiempo hasta el deterioroclínico en la etiología idiopática y HAP aso-ciada a conectivopatías e infección por VIH.También ha demostrado la durabilidad de susefectos durante al menos un año. Aprobadopara su uso en CF (OMS II y III) a una dosis de5 mg una vez al día. Si se tolera la dosis pue-de incrementarse hasta 10 mg una vez al día.La alteración del perfil hepático ocurre en un0,8-3% de los casos. La función hepática debemonitorizarse mensualmente. Se ha descritoun aumento de incidencia de edema periféri-co con su uso.
Inhibidores de la fosfodiesterasa tipo 5Los inhibidores de la fosfodiesterasa tipo 5
(IPD5) producen vasodilatación mediante la inhi-bición de la degradación del GMPc y presentanadicionalmente efectos antiproliferativos.
Sildenafilo, potente inhibidor selectivo dela PD5 activo por vía oral, ha demostrado bene-ficio, en un ensayo clínico controlado, en lacapacidad de ejercicio, síntomas y parámetroshemodinámicos(29). Se ha estudiado en la HAPI,HAP asociada a enfermedades del tejido conec-tivo, cardiopatías congénitas y enfermedadtromboembólica crónica. La dosis aprobadaes de 20 mg pero la durabilidad del efecto has-ta un año se ha demostrado sólo con la dosisde 80 mg. En la práctica clínica, con frecuen-cia es necesario escalar hasta dosis de 40-80mg. La mayoría de los efectos adversos sonleves o moderados y relacionados con la vaso-dilatación: cefalea, rubor y epíxtasis.
Tadalafilo, inhibidor selectivo de la PD5,se dispensa una vez al día. En un ensayo clí-nico controlado, con dosis de 5-40 mg al día,ha demostrado eficacia en la mejoría de lossíntomas, capacidad de ejercicio, parámetroshemodinámicos y tiempo hasta el deterioroclínico. También ha demostrado la durabilidadde su efecto. La dosis eficaz es de 40 mg al día.Los efectos secundarios son similares a los delsildenafilo(30).
C. PINDADO RODRÍGUEZ ET AL.
64
Monografía HAP NM 208 pag 18/2/10 17:08 Página 64

GRUPO I. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN ARTERIAL PULMONAR IDIOPÁTICA Y RESTO…
65
Fármacos experimentales y estrategiasmédicas alternativas
A pesar de los progresos realizados en eltratamiento de la HAP, el grado de limitaciónfuncional y el mal pronóstico asociado hacenque los recursos terapéuticos actuales seaninsuficientes. Es por esta razón que se estándesarrollando diferentes estrategias farmaco-lógicas, que se adapten a los cambios obser-vados en la biopatología de esta enfermedad.Existen estudios en marcha, actualmente enfases II y III con: estimuladores independien-tes del óxido nítrico, estimuladores del GMPc,vasopéptido intestinal activo, agonistas no pros-tanoides de los receptores de prostaciclina,antagonistas de los receptores tisulares deendotelina, inhibidores de la tirosín-cinasa ycon antagonistas serotoninérgicos. Otros com-puestos se encuentran en estadios precocesde desarrollo como: inhibidores de la R- cina-sa, inhibidores de los receptores del factor decrecimiento vascular endotelial, inhibidores dela angiopoyetina 1 y los inhibidores de la elas-tasa. El tratamiento con células madre ha sidoeficaz en el modelo animal (ratas con mono-crotalina) y se está ensayando en pacientescon HAP(5).
Tratamiento combinadoLa terapia de combinación se ha conver-
tido en un estándar de tratamiento en muchoscentros de HAP, a pesar de que su seguridady eficacia no ha sido completamente estudia-da. Numerosas series de casos sugieren quecombinaciones de varios fármacos parecenser efectivas y seguras. Diversos ensayos clí-nicos controlados realizados hasta el momen-to demuestran el efecto aditivo de la super-posición de fármacos, fundamentalmente enrelación con la mejora de la capacidad de ejer-cicio y, en mayor a menor grado, dependien-do de la combinación elegida. Muchas cues-tiones quedan por resolver: la elección de losagentes a combinar, sí debe empezar de ini-cio con terapia combinada o introducir secuen-cialmente los fármacos según respuesta a lasya instauradas, cuándo cambiar y cuándo aso-
ciar un nuevo fármaco. En general, se reco-mienda la terapia combinada, con fármacosya establecidos, para pacientes que no res-ponden adecuadamente con la monoterapia.Este tratamiento debería ser instaurado en cen-tros experimentados y dentro de ensayos clí-nicos o registros, siempre que sea posible.Deben considerarse las principales interac-ciones farmacológicas entre estos agentes, yde éstos con otros fármacos recogidos en latabla 3. En esta tabla se muestran las inter-acciones más importantes, pero no se inclu-yen interacciones teóricamente posibles nocomprobadas que, sin embargo, podrían serclínicamente relevantes.
TrasplanteEl trasplante es la alternativa final al tra-
tamiento de la HAP. Esta técnica está limitadapor la escasez de donantes. No obstante, elnúmero de pacientes en lista de espera ha dis-minuido por el efecto positivo de la terapiaespecífica actualmente disponible. Se calculaque, una cuarta parte de los pacientes con tra-tamiento intensivo, evolucionan mal, siendocandidatos a trasplante. Existen recomenda-ciones para referir a los pacientes a un pro-grama de trasplante y cuándo deben entrar enlista de espera. En cualquier caso, cuando lospacientes se encuentran inestables y con datosde mal pronóstico bajo tratamiento intensivomáximo, deben ser evaluados para trasplan-te. El peor pronóstico se observa para la enfer-medad veno-oclusiva pulmonar y la heman-giomatosis capilar pulmonar, dada la falta deeficacia de la medicación específica. Estospacientes deberían remitirse para inclusión enun programa de trasplante en el momento deldiagnóstico. En cuanto a la técnica quirúrgica,la opción más habitual es el doble trasplantepulmonar dado que la función del ventrículoderecho suele presentar recuperación posto-peratoria y que, tras el trasplante uni-pulmo-nar, la presencia de cualquier complicación seasocia a una hipoxemia severa. Los pacien-tes con síndrome de Eisenmenger por car-diopatías complejas deben ser considerados
Monografía HAP NM 208 pag 18/2/10 17:08 Página 65
C. PINDADO RODRÍGUEZ ET AL.
TABLA 3. Interacciones farmacológicas potencialmente significativas con laterapia específica para HAP
Terapia Droga depara HAP interacción Modo de interacción
Bosentan Sildenafilo Los niveles de sildenafilo se reducen a la mitad. Los de bosentanse duplican. Podría no requerirse el ajuste de dosis de cada fár-maco
Ciclosporina Los niveles de ciclosporina se reducen a la mitad. Los de bosen-tan se elevan por cuatro. Combinación contraindicada
Eritromicina Los niveles de bosentan se elevan. Podría no requerir ajuste dedosis durante un ciclo corto
Ketoconazol Los niveles de bosentan se duplican
Glibencamida Aumenta la incidencia de elevación de transaminasas. Potencialreducción del efecto hipoglucemiante de la glibencamida. Combi-nación contraindicada
Fluconazol, Los niveles de bosentan se elevan considerablemente.Amiodarona Combinación potencialmente contraindicada
Rifampicina, Los niveles de bosentan se reducen un 58%. Podría ser necesarioFenitoína ajustar la dosis
Estatinas Los niveles de simvastatina se reducen a la mitad. Efecto similarsobre la atorvastatina. Los niveles de colesterol deben ser moni-torizados
Dicumarínicos Aumenta el metabolismo de los dicumarínicos, pudiendo sernecesario ajustar la dosis del anticoagulante. Se recomienda unamonitorización cercana tras el inicio del tratamiento
Anticonceptivos Los niveles hormonales descienden y el efecto anticonceptivo es hormonales poco fiable
Sitaxentan Dicumarínicos Inhibe el metabolismo de dicumarínicos. La dosis de dicumaríni-cos debe reducirse en un 80% cuando se inicia sitaxentan y debeintensificarse la monitorización de INR
Ciclosporina Incrementa los niveles de sitaxentan. Combinación contraindicada
Ambrisentan Ciclosporina, La co-administracción de estos fármacos con ambrisentan debe ketoconazol realizarse con cautela
Sildenafilo Bosentan Los niveles de sildenafilo se reducen a la mitad. Los de bosentanse duplican. Podría no requerirse el ajuste de dosis de cada fár-maco
Estatinas Puede incrementar los niveles de simvastatina/atorvastatina porcompetición en el metabolismo. Los niveles de sildenafilo pue-den elevarse. Posible incremento en el riesgo de rabdomiólisis.
Inhibidores de Ritonavir y saquinovir aumentan marcadamente los niveles de la proteasa VIH sildenafilo. Suele ser necesario ajustar la dosis de sildenafilo
…/…
66
Monografía HAP NM 208 pag 18/2/10 17:08 Página 66

para trasplante cardiopulmonar. La supervi-vencia global a los 5 años postrasplante es del45-50%, con evidencia de buena calidad devida(31,32). Es importante mencionar que el tras-plante para pacientes con enfermedad vascu-lar pulmonar avanzada se realiza actualmen-te bajo tratamiento de fármacos vasoactivosy/o con soporte circulatorio (ECMO) y, comoconsecuencia, el grado de prioridad para estetipo de trasplantes es elevado. La infección porVIH se considera generalmente un criterio deexclusión para el trasplante; sin embargo, enalgunos centros se ha implementado un pro-grama específico.
Septostomía auricularLa septostomía auricular con balón debe
evitarse en pacientes con una presión mediade AD >20 mmHg o una saturación en repo-so con aire ambiente <80%. Los pacientesdeben encontrase bajo tratamiento óptimo,incluyendo un preacondicionamiento coninotropos intravenosos, antes de considerar laseptostomía(33). La evidencia sugiere un bene-ficio para pacientes en clase IV con insufi-
ciencia cardiaca derecha refractaria a trata-miento médico o con sintomatología sincopalsevera. También puede considerarse como unpuente al trasplante cuando la terapia médicano está disponible. Su impacto en la supervi-vencia a largo plazo no se ha establecido enensayos clínicos controlados. Debe ser consi-derado como un procedimiento paliativo ydebe ser realizado únicamente por centros conexperiencia(34,35).
ENFERMEDAD VENO-OCLUSIVA YHEMANGIOMATOSIS CAPILAR PULMONAR(GRUPO 1’)
Parecen representar diferentes manifes-taciones fenotípicas de la misma enfermedad.A menudo sólo el estudio anatomopatológicopuede distinguir las dos condiciones. Su diag-nóstico puede establecerse con alta probabili-dad a partir de la sospecha clínica, examenfísico, broncoscopia y hallazgos radiológicos.La radiografía torácica puede revelar líneas Bde Kerley e infiltrado intersticial periférico, ade-más de otros signos típicos de la HP. El méto-do a seguir es el escáner de TC de alta reso-
GRUPO I. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN ARTERIAL PULMONAR IDIOPÁTICA Y RESTO…
67
TABLA 3. Interacciones farmacológicas potencialmente significativas con laterapia específica para HAP (continuación)
Terapia Droga depara HAP interacción Modo de interacción
Sildenafilo Fenitoína Los niveles de sildenafilo pueden descender
Eritromicina Los niveles de sildenafilo se elevan. Podría no requerir ajuste dedosis durante un ciclo corto
Ketoconazol Los niveles de sildenafilo se elevan. Podría no requerir ajuste dedosis
Cimetidina Los niveles de sildenafilo se elevan. Podría no requerir ajuste dedosis
Nitratos, Profunda hipotensión sistémica. Combinación contraindicadaNicorandil
Tadalafilo Bosentan Los niveles de tadalafilo se reducen en un 42%, sin cambios sig-nificativos en los niveles de bosentan. Puede no requerir ajustede dosis
Nitratos, Profunda hipotensión sistémica. Combinación contraindicadaNicorandil
Monografía HAP NM 208 pag 18/2/10 17:08 Página 67

lución. Los típicos hallazgos que indican unaEVOP son la presencia de líneas septales sub-pleurales más espesas, opacidades centrolo-bulares en vidrio esmerilado (que contrastancon la distribución panlobular hallada en laHAPI) y linfadenopatía mediastínica(36). La aso-ciación de estos tres hallazgos resultó ser el100% precisa para la EVOP en casos de HAP,con una sensibilidad del 66%. Además, su pre-sencia se correlaciona de manera estrecha conel riesgo de edema pulmonar a causa de laterapia con epoprostenol(37).
Esto puede evitar la realización de biopsiapulmonar (técnica gold standard). Se ha infor-mado de mejoría clínica sostenida en algunospacientes con este tratamiento. No hay datosacerca del uso de ARE e IPD5 en este grupo.La septostomía auricular puede ser utilizadaaunque con frecuencia está contraindicada porla hipoxemia, más acentuada en estos pacien-tes. El único tratamiento curativo es el tras-plante. No hay datos de recurrencia de la enfer-medad tras el trasplante. En el momento, quese establece el diagnóstico el paciente debeser remitido a un centro con Unidad de Tras-plante.
BIBLIOGRAFÍA1. Simonneau G, Robbins I, Beghetti M, et al.
Updated clinical classification of pulmonaryhypertension. J Am Coll Cardiol. 2009; 54: S43-54.
2. Simonneau G, Galiè N, Rubin LJ, et al. Clini-cal classification of pulmonary hypertension.J Am Coll Cardiol. 2004; 43: 5S-12S.
3. Badesch BD, Champion HC, Gómez-SánchezMA, et al. Diagnosis and assessment of pul-monary arterial hypertension. J Am Coll Car-diol. 2009; 54: S55-6.
4. Kovacs G, Berghold A, Scheidl S, et al. Pul-monary arterial pressure during rest and exer-cise in healthy control subjects: a systematicreview. Eur Respir J. 2009; 34: 888-94.
5. Galié N, Hoeper MM, Humbert M, et al. Gui-delines for the diagnosis and treatment of pul-monary hypertension: The Task Force for theDiagnosis and Treatment of Pulmonary Hyper-tension of the European Society of Cardiology(ESC) and the European Respiratory Society
(ERS), endorsed by the International Societyof Heart and Lung Transplantation (ISHLT). EurHeart J. 2009; 30: 2493-537.
6. Jing ZC, Jiang X, Han ZY, et al. Iloprost for pul-monary vasodilator testing in idiopathic pul-monary arterial hypertension. Eur Respir J.2009; 33: 1354-60.
7. Sitbon O, Humbert M, Jais X, et al. Long-termresponse to calcium channel blockers in idio-pathic pulmonary arterial hypertension. Cir-culation. 2005; 111: 3105-11.
8. McLaughlin VV, Presberg KW, Doyle RL, et al.Prognosis of pulmonary arterial hypertensionACCP evidence-based clinical practice guide-lines. Chest. 2004; 126: 78S-92S.
9. Yeo TC, Dujardin KS, Tei C, et al. Value of aDoppler-derived index combining systolic anddiastolic time intervals in predicting outcomein primary pulmonary hypertension. Am J Car-diol. 1998; 81: 1157-61.
10. Forfia PR, Fisher MR, Mathai SC, et al. Tricus-pid annular displacement predicts survivalin pulmonary hypertension. Am J Respir CritCare Med. 2006; 174: 1034-41.
11. Miyamoto S, Nagaya N, Satoh T, et al. Clinicalcorrelates and prognostic significance of six-minute walk test in patients with primary pul-monary hypertension. Comparison with car-diopulmonary exercise testing. Am J RespirCrit Care Med. 2000; 161: 487-92.
12. Wensel R, Opitz CF, Anker SD, et al. Assess-ment of survival in patients with primary pul-monary hypertension: importance of cardio-pulmonary exercise testing. Circulation. 2002;106: 319-24.
13. Fijalkowska A, Kurzyna M, Torbicki A, et al.Serum N-terminal brain natriuretic peptide asa prognostic parameter in patients with pul-monary hypertension. Chest. 2006; 129: 1313-21.
14. Tongers J, Schwerdtfeger B, Klein G, et al. Inci-dence and clinical relevance of supraventri-cular tachyarrhythmias in pulmonary hyper-tension. Am Heart J. 2007; 153: 127-32.
15. Bedard E, Dimopoulos K, Gatzoulis MA. Hasthere been any progress made on pregnancyoutcomes among women with pulmonary arte-rial hypertension? Eur Heart J. 2009; 30: 256-65.
16. Rich S, Dantzker DR, Ayres SM, et al. Primarypulmonary hypertension. A national prospec-tive study. Ann Intern Med. 1987; 107: 216-23.
C. PINDADO RODRÍGUEZ ET AL.
68
Monografía HAP NM 208 pag 18/2/10 17:08 Página 68

17. Barst RJ, Rubin LJ, Long WA, et al. A compa-rison of continuous intravenous epoprostenol(prostacyclin) with conventional therapy forprimary pulmonary hypertension. The PrimaryPulmonary Hypertension Study Group. N EnglJ Med. 1996; 334: 296-302.
18. Olschewski H, Simonneau G, Galiè N, et al, forthe AIR Study Group. Inhaled iloprost in seve-re pulmonary hypertension. N Engl J Med.2002; 347: 322-9.
19. Simonneau G, Barst RJ, Galiè N, et al. Conti-nuous subcutaneous infusion of treprostinil, aprostacyclin analogue, in patients with pul-monary arterialhypertension. A double-blind,randomized, placebocontrolled trial. Am J Res-pir Crit Care Med. 2002; 165: 800-4.
20. Barst RJ, Galiè N, Naeije R, et al. Longterm out-come in pulmonary arterial hypertensionpatients treated with subcutaneous treprosti-nil. Eur Respir J. 2006; 28: 1195-203.
21. Lang I, Gómez-Sánchez M, Kneussl M, et al.Efficacy of long-term subcutaneous treprosti-nil sodium therapy in pulmonary hyperten-sion. Chest. 2006; 129: 1636-43.
22. Rubin LJ, Badesch DB, Barst RJ, et al. Bosen-tan therapy for pulmonary arterial hyperten-sion. N Engl J Med. 2002; 346: 896-903.
23. Galiè N, Rubin LJ, Hoeper M, et al. Treatmentof patients with mildly symptomatic pul-monary arterial hypertension with bosen-tan (EARLY study): a double-blind, rando-mized controlled trial. Lancet. 2008; 371:2093-100.
24. Galiè N, Beghetti M, Gatzoulis MA, et al, for theBosentan Randomized Trial of Endothelin Anta-gonist Therapy. Bosentan therapy in patientswith Eisenmenger syndrome: a multicenter,doubleblind, randomized, placebo-controlledstudy. Circulation. 2006; 114: 48-54.
25. Barst RJ, Langleben D, Badesch D, et al. Tre-atment of pulmonary arterial hypertensionwith the selective endothelin-A receptor anta-gonist sitaxsentan. J Am Coll Cardiol. 2006;47: 2049-56.
26. Benza RL, Barst RJ, Galiè N, et al. Sitaxsentanfor the treatment of pulmonary arterial hyper-tension: a one year, prospective, open label,observation of outcome and survival. Chest.2008; 134: 775-82.
27. Galiè N, Badesch BD, Oudiz R, et al. Ambri-sentan therapy for pulmonary arterial hyper-tension. J Am Coll Cardiol. 2005; 46: 529-35.
28. Galiè N, Olschewski H, Oudiz RJ, et al. Ambri-sentan for the treatment of pulmonary arte-rial hypertension. Results of the ambrisen-tan in pulmonary arterial hypertension,randomized, doubleblind, placebo-controlled,multicenter, efficacy (ARIES) study 1 and 2.Circulation. 2008; 117: 3010-9.
29. Galiè N, Ghofrani HA, Torbicki A, et al, the Sil-denafil Use in Pulmonary ArterialHyperten-sion (SUPER) Study Group. Sildenafil citratetherapy for pulmonary arterial hypertension.N Engl J Med. 2005; 353: 2148-57.
30. Galiè N, Brundage B, Ghofrani A, et al. Tada-lafil therapy for pulmonary arterial hyperten-sion. Circulation. 2009; 119: 2894-903.
31. Orens JB, Estenne M, Arcasoy S, et al. Pul-monary Scientific Council of the InternationalSociety for Heart and Lung Transplantation.International guidelines for the selection oflung transplant candidates: 2006 update-a con-sensus report from the Pulmonary ScientificCouncil of the International Society for Heartand Lung Transplantation. J Heart Lung Trans-plant. 2006; 25: 745-55.
32. Trulock EP, Edwards LB, Taylor DO, et al; Inter-national Society for Heart and Lung Trans-plantation. Registry of the International Societyfor Heart and Lung Transplantation: twentythird official adult lung and heart lung trans-plantation report-2006. J Heart Lung Trans-plant. 2006; 25: 880-92.
33. Sandoval J, Gaspar J, Pulido T, et al. Gradedballoon dilation atrial septostomy in severeprimary pulmonary hypertension. A thera-peutic alternative for patients nonresponsiveto vasodilator treatment. J Am Coll Cardiol.1998; 32: 297-304.
34. Kurzyna M, Dabrowski M, Bielecki D, et al.Atrial septostomy in treatmentof end-stageright heart failure in patients with pulmonaryhypertension. Chest. 2007; 131: 977-83.
35. Althoff TF, Knebel F, Panda A, et al. Long-termfollow-up of a fenestrated Amplatzer atrial-septal occlude in pulmonary arterial hyper-tension. Chest. 2008; 133: 283-5.
36 Resten A, Maitre S, Humbert M, et al. Pulmo-nary hypertension: CT of the chest in pulmo-nary venoocclusive disease. AJR Am J Roent-genol. 2004; 183: 65-70.
37. Montani D, Price LC, Dorfmuller P, et al. Pul-monary veno-occlusive disease. Eur Respir J.2009; 33: 189-200.
GRUPO I. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN ARTERIAL PULMONAR IDIOPÁTICA Y RESTO…
69
Monografía HAP NM 208 pag 18/2/10 17:08 Página 69

Monografía HAP NM 208 pag 18/2/10 17:08 Página 70

71
INTRODUCCIÓNLa hipertensión pulmonar (HP) es un hallaz-
go relativamente común en los pacientes conpatología cardiaca izquierda. Su presencia, gene-ralmente, se asocia a una elevada morbilidad(1).El porcentaje de pacientes con enfermedad car-diaca izquierda que desarrollan hipertensiónpulmonar es desconocido, sin embargo, endeterminados subgrupos, como aquellos conestenosis aórtica sintomática, se ha descritouna prevalencia de hasta un 65%(2). Además,el desarrollo de hipertensión pulmonar tras unepisodio de infarto de miocardio(3), miocardio-patía dilatada(4) o post-trasplante cardiaco(5), esun importante predictor de mortalidad.
DEFINICIÓN Y CLASIFICACIÓNLa hipertensión arterial pulmonar (HAP) se
define hemodinámicamente por la presenciade una cifra de presión media (PAPm) mayora 25 mmHg, medida mediante cateterismocardiaco derecho.
Con la ecocardiografía Doppler, se puedeestimar la presión arterial pulmonar sistólica(PAPs) de una forma no invasiva, a través delregistro de la velocidad máxima de regurgita-ción tricúspide. Se considera que existe HAPcuando, en ausencia de estenosis tricúspide,la velocidad máxima del flujo regurgitante esmayor o igual a 2,8 m/seg. Adicionalmente, laecocardiografía puede aportar información deotras patologías cardiacas, sospechadas o no,que ayuden en el proceso diagnóstico de la HP.
Desde el punto de vista de su clasificación,en un intento de unificar procesos con pato-
genia, histopatología y clínica similares, en el3º World Symposium on Pulmonary ArterialHypertension se categorizó la hipertensión pul-monar en cinco grupos(6). Esta clasificacióncon algunas pequeñas modificaciones fuerefrendada en el 4º World Symposium, cele-brado en Dana Point en 2008 (Tabla 1).
Este capítulo tratará exclusivamente delsegundo grupo: HP secundaria a cardiopatíaizquierda, probablemente el más frecuente detodos, en parte debido a la alta prevalencia depatologías cardiacas y al progresivo envejeci-miento de la población. Desde un punto de vis-ta funcional, se le puede definir como aquellahipertensión pulmonar secundaria a hiper-tensión venosa producida por enfermedadesdel lado izquierdo del corazón.
FISIOPATOLOGÍAEn ausencia de cortocircuitos derecha-
izquierda, el sistema vascular pulmonar reci-be todo el volumen minuto cardiaco. Al serun sistema de gran capacitancia, ese pasose realiza con presiones bajas, incluso duran-te el ejercicio, pudiendo acomodar hasta 5veces el volumen minuto basal sin que las pre-siones varíen significativamente, hecho dife-renciador fundamental con la circulación sis-témica. Esta característica, además, permiteregular el llenado de las cavidades izquierdas,manteniendo un gradiente transpulmonarcapilar medio (GTP) dentro de valores nor-males (7 mmHg):
GTP = PAPm – PCPsiendo PCP la presión capilar media.
GRUPO II. CLASIFICACIÓN DE DANA POINT
ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS
Sergio Alcolea Batres, Juan José Ríos Blanco
Monografía HAP NM 208 pag 18/2/10 17:08 Página 71

En la tabla 2 se enumeran diversas pato-logías cardiacas izquierdas que potencialmentepueden ser causantes de hipertensión pulmo-nar. En estos pacientes, la HP resulta del incre-mento de la presión de llenado de las cavida-des izquierdas, hecho que se correlaciona conla evidencia ecocardiográfica de disfuncióndiastólica(7). En pacientes con disfunción sis-tólica ventricular izquierda, la presencia de HPno se correlaciona únicamente con la presen-cia de disfunción diastólica, sino que tambiénestá influenciada por el grado de regurgitaciónmitral presente(8).
En las cardiopatías izquierdas que tienenun aumento de las presiones de llenado, se pro-duce un aumento de la PCP (> 15 mmHg). De
esta forma, para mantener un GTP adecuado,se ocasiona un incremento de la PAPm que, enun principio, es “pasivo” y reversible, si se nor-maliza la PCP. Sin embargo, la prolongación deesta situación hemodinámica provoca la apa-rición de una HP “reactiva”, que se suma alcomponente pasivo, aumentando el GTP.
El componente reactivo, a su vez, se sub-divide en un elemento dinámico o funcional,mediado por elementos vasoconstrictores queactúan sobre las arterias musculares y que esmodificable por fármacos vasodilatadores, yen un elemento fijo u obliterativo, reflejo delremodelado a nivel de la arteria muscular pul-monar, fundamentalmente hipertrofia de lamedia y, en menor grado, fibrosis de la ínti-
S. ALCOLEA BATRES, J.J. RÍOS BLANCO
72
TABLA 1. Clasificación de la HP. Dana Point 2008
Grupo I. Hipertensión arterial pulmonar • Idiopática• Heredada (BMPR2, ALK2, desconocido)• Drogas y tóxicos• Asociada: conectivopatías, HIV, portal, shunts, esquistosomiasis, anemia falciforme
Grupo I’. Hipertensión venoclusiva y hemangiomatosis capilar pulmonar
Grupo II. Consecuencia de enfermedad cardiaca izquierda• Disfunción sistólica• Disfunción diastólica• Valvulopatías
Grupo III. Consecuencia de la enfermedad pulmonar/hipoxia• Enfermedad pulmonar obstructiva crónica• Enfermedades pulmonares intersticiales• Síndrome apnea del sueño • Síndrome hipoventilación-obesidad• Exposición crónica altitud
Grupo IV. Hipertensión pulmonar tromboembólica crónica
Grupo V. Origen multifactorial o no aclarado:• Hematológico: Sd mieloproliferativo, esplenectomía• Sistémicas: vasculitis, sarcoidosis, histiocitosis X,…• Metabólicas: Gaucher, hipertiroidismo• Congénita cardiaco diferente de shunts• Otros: tumores, fibrosis mediastínica, insuficiencia renal crónica en diálisis…
Monografía HAP NM 208 pag 18/2/10 17:08 Página 72

ma, dilatación linfática y arterialización de lasvenas y vénulas, elemento que no es reversi-ble y sobre el que no actúan los fármacos vaso-dilatadores(9,10).
Desde el punto de vista hemodinámico,podemos diferenciar entre hipertensión pul-monar pasiva y reactiva, estimando el gradientetranspulmonar, siendo ≤12 mmHg en la pri-mera y >12 mmHg en la segunda (Fig. 1).
La HP reactiva finalmente produce dis-función del ventrículo derecho, ya que la pre-
sión pulmonar es el principal determinante desu postcarga, y, al final, insuficiencia cardiacaderecha y disminución del volumen minuto.
El desarrollo de HP fija, en presencia dehipertensión venosa pulmonar crónica, se hapostulado como un mecanismo protector fren-te al edema pulmonar, junto con el aumentodel drenaje linfático y el engrosamiento de lainterfase alvéolo-intersticio. A su vez, la HP reac-tiva, por vasculopatía pulmonar conlleva unareducción significativa del gasto cardiaco del
GRUPO II. CLASIFICACIÓN DE DANA POINT: ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS
73
TABLA 2. Cardiopatías izquierdas causantes de hipertensión pulmonar
1. Disfunción sistólica ventricular izquierda1.1 Isquémica1.2 No isquémica
1.2.1 Miocardiopatía dilatada familiar/idiopática1.2.2 Valvular
1.2.2.1 Insuficiencia mitral1.2.2.2 Insuficiencia aórtica1.2.2.3 Estenosis mitral1.2.2.4 Estenosis aórtica
2. Disfunción diastólica ventricular izquierda2.1 Miocardiopatías restrictivas y constrictivas2.2 Miocardiopatía hipertensiva2.3 Disfunción diastólica de la senectud/idiopática
3. Mixoma o trombo auricular izquierdo4. Cor triatriatum
FIGURA 1. Esquema de la circula-ción pulmonar, reflejando hiper-tensión pulmonar pasiva (A) e hiper-tensión pulmonar reactiva (B).Nótese que en todo caso la PCPdebe estar aumentada.
AP CP VP Aorta
AortaAP CP VP
Cava
Cava
VD
VD VI
VI
PAP90/35
50
PAP48/25
35
PCP25
PCP30
GC: 3,5 lpmGTP: 20
GC: 4 lpmGTP: 10
A
B
VD: ventrículo derecho; AP: arteria pulmonar; CP: capilar pulmonar; VP: venapulmonar; VI: ventrículo izquierdo; PAP: presión arterial pulmonar; PCP: pre-sión capilar pulmonar; GC: gasto cardiaco; GTP: gradiente transpulmonar.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 73

ventrículo derecho por aumento de su post-carga(11), hecho que reduce el aporte sanguíneoal lecho capilar pulmonar, siendo un meca-nismo igualmente protector frente al edema.
Evolución de HP pasiva a HP reactivaMientras que en la HP idiopática el daño
vascular inicial provoca secundariamente unremodelado e hipertensión pulmonar en susestadios finales, en la HP secundaria a car-diopatías izquierdas el mecanismo es al con-trario: el mecanismo inicial es la hipertensiónpulmonar pasiva, que produce un daño vas-cular, el cual inicia el proceso de remodelado,convirtiéndose en estadios finales en HP fija.
En este proceso, el endotelio desempeñaun papel fundamental, regulando el tono vas-cular, a través de la liberación de sustanciasvasoconstrictoras (endotelina 1) y vasodilata-doras (óxido nítrico).
El óxido nítrico (NO) es generado en la célu-la endotelial a partir de L-arginina. Una vez sin-tetizado, su acción se efectúa en las célulasmusculares lisas, activando la guanilil-ciclasa yaumentando los niveles de GMPc, producien-do relajación de la célula muscular lisa arte-riolar, e inhibiendo su proliferación e hipertro-fia. Además, posee efectos antiagregantesplaquetarios y antitrombóticos(12). Estudiosexperimentales y en sujetos con insuficienciacardiaca(13,14), sugieren que la vasodilataciónpulmonar mediada por NO en estos pacientesse encuentra alterada, sugiriendo un estadobasal vasoconstrictor por ausencia de produc-ción de NO a nivel endotelial, contribuyendoal mantenimiento y progresión de la HP.
La otra sustancia segregada por las célulasendoteliales, con participación en la regulacióndel tono vascular, es la endotelina. Existen dostipos de receptores: ETA y ETB. Los primerosse encuentran localizados en la célula muscu-lar lisa, donde median vasoconstricción y cre-cimiento celular. Los segundos, se localizanfundamentalmente en la membrana de la célu-la endotelial, donde estimulan vasodilatación,por estimular la liberación de NO y prostaci-clina, y también por participar en el aclarado
de la propia endotelina. Sin embargo, dadoque la relación de receptores ETA/ETB en ellecho vascular pulmonar es de 9/1, el efectopredominante es vasoconstrictor(15). En pacien-tes afectos de insuficiencia cardiaca, se handemostrado niveles elevados de endotelina,que se correlacionan con las presiones y lasresistencias vasculares pulmonares(16). Esteaumento puede estar mediado por determi-nadas citocinas (TNF-α estimula la producciónde endotelina)(17), así como por la infraexpre-sión de los receptores ETB
(18).La combinación de ambos factores, son los
mediadores conocidos que, a través de vaso-constricción, proliferación de células muscu-lares lisas y producción de colágeno, inician elremodelado vascular y la transición de HP pasi-va a reactiva.
APROXIMACIÓN DIAGNÓSTICACuando existe la sospecha de HP, la eco-
cardiografía transtorácica es el método descreening más utilizado, siendo además la prue-ba de elección según las guías diagnósticas(19).La ecocardiografía ofrece la posibilidad de eva-luar de una forma no invasiva la presión arte-rial pulmonar, teniendo la ventaja adicional deidentificar patología cardiaca izquierda con-comitante. Además de poder valorar la funciónsistólica izquierda, morfología y función val-vular, este método nos puede servir para ave-riguar la presencia de disfunción diastólica delventrículo izquierdo, condición clínica cadavez más prevalente, especialmente en aque-llos pacientes de mayor edad(20).
Sin embargo, para realizar un diagnósti-co definitivo en todo caso de hipertensión pul-monar, se hace necesaria la práctica de un ca-teterismo cardiaco derecho. Esto es debido aque la estimación de la PAP mediante eco-cardiografía muestra una sensibilidad muy ele-vada, sin embargo la especificada es baja,presentando una alta variabilidad interobser-vador(21). Además, mediante el cateterismocardiaco, se pueden realizar la medición de lapresión capilar pulmonar, parámetro no deter-minado por métodos no invasivos.
S. ALCOLEA BATRES, J.J. RÍOS BLANCO
74
Monografía HAP NM 208 pag 18/2/10 17:08 Página 74

Para realizar el cálculo de GTP en pacien-tes con HP, el cateterismo cardiaco derecho esmandatorio. En los casos en los que se sos-peche enfermedad cardiaca izquierda asocia-da, bien sea por historia clínica, hallazgosexploratorios o ecocardiografía, probablementesea también necesaria la práctica de un cate-terismo cardiaco izquierdo. Sin embargo, noexiste un protocolo de consenso en cuanto asu realización. En algunos centros, durante elcateterismo se realiza, al igual que en lospacientes con HAP idiopática o del grupo I dela clasificación de Dana Point, una prueba vaso-dilatadora(22), con el fin de definir la respuestaa largo plazo y las posibilidades terapéuticas.
Otra prueba diagnóstica utilizada en pacien-tes con disfunción sistólica y diastólica ventri-culares izquierdas es la prueba de esfuerzo car-dio-respiratoria(23). Este método no es invasivoy permite caracterizar la naturaleza de la limi-tación al ejercicio y proporciona informaciónpronóstica(24), pudiendo ser útil tanto en la eva-luación inicial, evolución, así como en el trata-miento, estimando el momento óptimo paratrasplante cardiaco/cardio-pulmonar(24).
La evaluación de la función ventricularderecha es igualmente importante en pacien-tes con patología izquierda. En un estudio lle-vado a cabo por Ghio y cols.(25), sobre 379pacientes con fallo ventricular izquierdo gra-ve (fracción de eyección del ventrículo izquier-do < 35%), observó que la fracción de eyec-ción del ventrículo derecho era un predictorde mortalidad independientemente de la pre-sencia de hipertensión pulmonar, sugiriendoque la función del ventrículo derecho, más quela PAP, determina el riesgo de muerte o la nece-sidad de trasplante cardiaco en estos pacien-tes. Similares hallazgos han sido obtenidos enotros estudios, con pacientes con fraccionesde eyección mayores (35-45%)(26).
TRATAMIENTOEn la mayor parte de los pacientes con HP
relacionada con cardiopatía izquierda, la basedel tratamiento es el de la propia enfermedadcardiaca. Teóricamente, en fases precoces (HP
pasiva), si conseguimos reducir la presión veno-sa, reducimos igualmente las cifras de hiper-tensión pulmonar(27). Sin embargo, cuandoexiste un componente reactivo, la “reversibi-lidad” ya no es factible, no respondiendo altratamiento de la enfermedad subyacente.
En los últimos años han aparecido diver-sos fármacos para el tratamiento de la HP idio-pática y del grupo I, mejorando parámetroshemodinámicos, capacidad de esfuerzo, clasefuncional y supervivencia. Se han generadograndes expectativas en su uso en pacientesdel grupo II, sin embargo, los ensayos clínicosrealizados han arrojado resultados contradic-torios y en la mayoría de los casos desalenta-dores (Tabla 3). Es por ello que la Agencia Ame-ricana del Medicamento (FDA, US Food andDrug Administration), así como la Europea(EMEA, European Medicines Angency), no tie-nen ningún fármaco aprobado para su usoespecífico en pacientes con HP asociada a car-diopatía izquierda.
En este apartado se van a revisar los dife-rentes fármacos utilizados habitualmente enHAP, así como su evidencia científica paraaquellos pacientes del grupo II. En cualquiercaso, cabe resaltar que un efecto fundamen-tal de todos los fármacos utilizados es el de lavasodilatación, cuyo uso puede ser poten-cialmente perjudicial en pacientes con pato-logía cardiaca izquierda, especialmente este-nosis valvulares y/o disfunción diastólica, yaque pueden provocar un aumento del retor-no venoso a cavidades izquierdas, sin modi-ficar sustancialmente las presiones de llena-do, condicionando una propensión al edemapulmonar por incremento de la PCP. En aque-llos pacientes con disfunción sistólica, su usocontinúa siendo incierto, si bien podrían jugarun papel beneficioso en pacientes seleccio-nados (PCP discretamente elevadas < 20mmHg, con GTP > 12 mmHg), disminuyen-do las cifras de presión en la arteria pulmo-nar y, por tanto, una mejoría de la postcargadel ventrículo derecho (como se ha comenta-do previamente, marcador pronóstico inde-pendiente de la HAP)(25).
GRUPO II. CLASIFICACIÓN DE DANA POINT: ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS
75
Monografía HAP NM 208 pag 18/2/10 17:08 Página 75

ProstanoidesLa prostaciclina es un potente vasodilata-
dor vascular, al inducir relajación de las célu-las musculares mediada por AMPc. Tiene tam-bién efectos antiproliferativos, antifibróticos yantiagregante plaquetario.
Existen estudios muy limitados que eva-lúen la eficacia de esta molécula en pacientescon HP y fallo ventricular izquierdo. Entre
ellos, cabe destacar un trabajo realizado porBrown y cols.(28), en pacientes con fallo ven-tricular agudo, a los que se les administrabailoprost inhalado para reducir las resistenciaspulmonares pre-trasplante, con resultadossatisfactorios, pero similares a los obtenidoscon óxido nítrico(29).
Otros estudios realizados a corto plazo eva-luando el análogo de la prostaciclina, epo-
S. ALCOLEA BATRES, J.J. RÍOS BLANCO
76
TABLA 3. Principales estudios de HP en patología cardiaca izquierda
Variable Referencia Fármaco Tipo principal Resultados Comentarios en texto
Epoprostenol Análogo Fracción Sin resultadosprostaciclina eyección VI Mejoría aguda a largo plazo 30
Epoprostenol Análogo — Mejoría hemo- Sin resultados 31prostaciclina dinámica aguda a largo plazo
Epoprostenol Análogo Supervivencia Mayor mortalidad Suspensión 35prostaciclina en subgrupo de prematura
tratamiento
Epoprostenol Análogos Isquemia Inductores de — 33Iloprost prostaciclina miocárdica isquemia
miocárdica
Iloprost Análogo Vasorreac- Fármaco seguro — 28prostaciclina tividad y efectivo
Óxido nítrico Vasodilatador Vasorreac- Fármaco seguro — 29tividad y efectivo
Sildenafilo Inhibidor Capacidad de Mejoría — 36PDE-5 esfuerzo significativa
Sildenafilo Inhibidor — Mejoría en Sin resultados 37PDE-5 _ variables a largo plazo
hemodinámicas
Tezosentan Antagonista Saturación a Sin cambios Empeoramiento 44endotelina la 1ª hora a dosis crecientesETA ETB
Bosentan Antagonista Cambio clínico Sin cambios Suspensión 45endotelina a los 6 meses prematura ETA ETB
Darusentan Antagonista Volumen tele- Sin cambios — 46endotelina sistólico delETA ventrículo izqdo.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 76

prostenol, han demostrado aumentar la frac-ción de eyección del ventrículo izquierdo(30),así como diversos cambios hemodinámicos(31),con reducción en las presiones de llenado auri-culares y disminución de las resistencias pul-monares y sistémicas(32). Aunque los resulta-dos de estos estudios fueron prometedores,los presuntos efectos adversos de las prosta-ciclinas sobre la circulación coronaria(33), asícomo la posibilidad de producir edema pul-monar(34), han hecho que se cuestione suempleo.
Entre los estudios a largo plazo realizados,cabe mencionar el FIRST (Flolan InternationalRandomized Survival Trial)(35), en el cual se inclu-yeron un total de 471 pacientes con insufi-ciencia cardiaca por disfunción sistólica delventrículo izquierdo en clase funcional III y IV.El estudio se suspendió prematuramente porun incremento en la mortalidad en el subgru-po tratado con epoprostenol, por progresiónde la enfermedad cardiaca. Sin embargo, seobservaron mejorías significativas en las resis-tencias pulmonares, presión arterial pulmonar,presión capilar pulmonar e índice cardiaco.
Inhibidores de la fosfodiesterasa 5 (PDE-5)El uso del inhibidor de la PDE-5, sildena-
filo, en la HP secundaria a cardiopatía izquier-da ha ganado recientemente adeptos. En diver-sos estudios, como el realizado por Bocchi ycols., se ha demostrado que disminuye la fre-cuencia cardiaca en reposo y mejora diferen-tes parámetros hemodinámicos en compara-ción con placebo(36). Guazzi y cols. estudiaronlos efectos de la administración aguda de sil-denafilo en 16 pacientes con patología car-diaca izquierda, encontrando un descenso sig-nificativo en el umbral anaeróbico, mejorandocon ello la capacidad de esfuerzo(37).
Sin embargo, y aunque los resultados deestos estudios son positivos, queda por deter-minar el efecto de sildenafilo a largo plazo,sobre todo por las malas experiencias previascon otros inhibidores de la fosfodiesterasa3, como la milrinona en la insuficiencia car-diaca(38).
En cualquier caso, los inhibidores de laPDE-5 podrían tener su campo en aquellospacientes con insuficiencia cardiaca y con-traindicación para trasplante. A este respectose han publicado resultados con mejoría sig-nificativa tras 1-2 meses de tratamiento, res-catándose a los pacientes para trasplante y rea-lizándose sin complicaciones(39,40).
Antagonistas de los receptores de laendotelina
Estudios preliminares realizados con losantagonistas de la endotelina, tezosentan(41),bosentan(42) y darusentan(43), han demostradouna mejoría significativa hemodinámica, perosin claro beneficio clínico.
Con respecto a tezosentan, se han realiza-do diversos estudios en pacientes con insufi-ciencia cardiaca aguda, síndrome coronarioagudo y edema agudo de pulmón, sin bene-ficio alguno, excepto en uno de ellos(44).
Los estudios realizados con bosentan,REACH (Randomised Endothelin Antagonism inChronic Heart Failure), suspendido prematu-ramente por toxicidad hepática, aunque concierta tendencia no significativa a la dismi-nución de la morbi-mortalidad, y el estudioENABLE, con dosis menores de bosentan, perosin diferencias significativas frente a placebo,no arrojaron datos concluyentes(45).
Con respecto a darusentan, antagonistaselectivo de los receptores ETA, el estudioEARTH (Endothelin A Receptor antogonist Trialin Heart failure) no demostró diferencias sig-nificativas hemodinámicas ni en la morbi-mor-talidad(46).
En conclusión, y a pesar de los datos alen-tadores de los estudios preliminares, los anta-gonistas de los receptores de la endotelina nohan demostrado ser efectivos en los pacientescon HP y fallo cardiaco izquierdo.
BIBLIOGRAFÍA1. Costanzo MR, Augustine S, Bourge R, et al.
Selection and treatment of candidates for hearttransplantation: a statement for health pro-fessionals from the Committee on Heart Fai-
GRUPO II. CLASIFICACIÓN DE DANA POINT: ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS
77
Monografía HAP NM 208 pag 18/2/10 17:08 Página 77

lure and Cardiac Transplantation of the Coun-cil on Clinical Cardiology, American Heart Asso-ciation. Circulation. 1995; 92: 3593-612.
2. Faggiano P, Antonini-Canterin F, Ribichin F, etal. Pulmonary artery hypertension in adultpatients with symptomatic valvular aortic ste-nosis. Am J Cardiol. 2000; 85: 204-8.
3. Moller JE, Hillis GS, Oh JK, et al. Prognosticimportance of secondary pulmonary hyper-tension after acute myocardial infarction. AmJ Cardiol. 2005; 96:199-203.
4. Abramson SV, Burke JF, Kelly JJ, et al. Pulmo-nary hypertension predicts mortality and mor-bidity in patients with dilated cardiomyopathy.Ann Intern Med. 1992; 116: 888-95.
5. Hosenpund JD, Bennett Le, Keck BM, et al. TheRegistry of the International Society of Heartand Lung Transplantation: seventeenth officialreport-2000. J Heart Lung Transplant. 2000;19: 909-31.
6. Simonneau G, Galie n, Rubin LJ, et al. Clinicalclassification of pulmonary hypertension. J AmColl Cardiol. 2004; 43(S): 5-12.
7. Lipkin DP, Poole-Wilson PA. Symptoms limi-ting exercise in chronic heart failure. Br MedJ. 1986; 292: 1030-1.
8. Enriquez-Sarano M, Rossi A, Seward JB, et al.Determinants of pulmonary hypertension inleft ventricular disfunction. J Am Coll Cardiol.1997; 29: 153-9.
9. Delgado JF, Sáenz de la Calzada C, Sánchez V,et al. Hipertensión pulmonar asociada a car-diopatías izquierdas. En: Gómez Sánchez MA(ed). Hipertensión pulmonar. Madrid: Ergon;2008. p. 133-46.
10. Delgado JF, Conde E, Sánchez V, et al. Pul-monary vascular remodeling in pulmonaryhypertension due to chronic heart failure. EurJ Heart Fail. 2005; 7: 1011-6.
11. Gelbach BK, Geppert E. The pulmonary mani-festations of left heart failure. Chest. 2004;125: 669-82.
12. Moraes DL, Colucci WS, Givertz MM. Secon-dary pulmonary hypertension in chronic heartfailure: the role of the endothelium inpathophysiology and management. Circula-tion. 2000; 102: 1718-23.
13. Ontkean M, Gay R, Greenberg B. Diminishedendothelium-derived relaxing factor activityin an experimental model of chronic failure.Circ Res. 1991; 69: 1088-96.
14. Cooper CJ, Landzber MJ, Anderson TJ, et al.role of nitric oxide in the local regulation of
pulmonary vascular resistance in humans. Cir-culation. 1996; 93: 266-71.
15. Fukuroda T, Kobayashi M, Ozaki S, et al. Endo-thelin receptor subtypes in human versus rab-bit pulmonary arteries. J Appl Physiol. 1994;76: 1976-82.
16. Cody TJ, Haas GJ, Binkley PF, et al. Plasmaendothelin correlates with the extent of pul-monary hypertension in patients with chroniccongestive heart failure. Circulation. 1992; 85:504-9.
17. Klemm P, Warner TD, Hohlfed T, et al. Endo-thelin 1 mediates ex vivo coronary vasocons-triction caused by exogenous and endogenouscytokines. Proc Natl Acad Sci EE.UU. 1995; 92:2691-5.
18. Zolk O, Quattek J, Sitzler G, et al. Expressionof endothelin-1, endothelin-converting enzy-me, and endothelin receptors in chronic heartfailure. Circulation. 1999; 99: 2118-23.
19. Mc Goon M, Gutterman D, Steen V, et al. Scre-ening, early detection, and diagnosis of pul-monary arterial hypertension: ACCP eviden-ce-based clinical practice guidelines. Chest.2004; 126(S): 14-34.
20. Aurigemma GP, Gaasch WH. Clinical practice:diastolic heart failure. N Engl J Med. 2004; 351:1097-105.
21. Rich S, D’Alonzo GE, Dantzker DR, et al. Mag-nitude and implications of spontaneoushemodynamic variability in primary pulmonaryhypertension. Am J Cardiol. 1985; 55: 159-63.
22. Sitbon O, Humbert M, Jais X, et al. Long-termresponse to calcium channel blockers in idio-pathic pulmonary arterial hypertension. Cir-culation. 2005; 111: 3105-11.
23. Markowitz DH, Systrom DM. Diagnosis of pul-monary vascular limit to exercise by cardio-pulmonary exercise testing. J Heart Lung Trans-plant. 2004; 23: 88-95.
24. Mancini DM, Eisen H, Kussmaul W, et al. Valueof peak oxygen consumption for optimaltiming of cardiac transplantation in ambula-tory patients with heart failure. Circula-tion.1991; 83: 778-86.
25. Ghio S, Gavazzi A, Campana C, et al. Inde-pendent and additive prognostic value or fightventricular systolic function and pulmonaryartery pressure in patients with chronic heartfailure. J Am Coll Cardiol. 2001; 37: 183-8.
26. De Groote P, Millaire A, Foucher-Hossein C, etal. Right ventricular ejection fraction is an inde-pendent predictor of survival in patients with
S. ALCOLEA BATRES, J.J. RÍOS BLANCO
78
Monografía HAP NM 208 pag 18/2/10 17:08 Página 78

moderate heart failure. J Am Coll Cardiol. 1998;32: 948-54.
27. Silke B. Hemodynamic impact of diuretic the-rapy in chronic heart failure. Cardiology. 1994;84(S): 115-23.
28. Brown S, Schrotter H, Schmeisser A, et al. Eva-luation of pulmonary vascular response toinhaled iloprost in heart transplant candida-tes with pulmonary venous hypertension. IntJ Cardiol. 2007; 115: 67-72.
29. Fojon S, Fernández-González C, Sánchez-Andrade J, et al. Inhaled nitric oxide througha noninvasive ventilation device to assessreversibility of pulmonary hypertension inselecting recipients for heart transplant. Trans-plant Proc. 2005; 37: 4028-30.
30. Virginoli I, Auinger C, Weissel M, et al. Incre-ase in left ventricular ejection fraction (LVEF)induced by PGE1 and PGI2. Prog Clin Biol Res.1989; 301: 463-7.
31. Yui Y, Nakajima H, Kawai C, et al. Prostacyclintherapy in patients with congestive heart fai-lure. Am J Cardiol. 1982; 50: 320-4.
32. Dzau VJ, Swartz SL, Creager MA. The role ofprostaglandins in the pathophysiology of andtherapy for congestive heart failure. Heart Fail.1986; 2: 6-13.
33. Bugiardini R, Galvani M, Ferrini D, et al. Myo-cardial ischemia during intravenous prostacy-clin administration: hemodynamic findingsand precautionary measures. Am Heart J.1987; 113: 234-40.
34. Haywood GA, Sneddon JF, Bashir Y, et al. Ade-nosine infusion for the reversal of pulmonaryvasoconstriction in biventricular failure. A goodtest but a poor therapy. Circulation. 1992; 86:896-902.
35. Califf RM, Adams KF, McKenna WJ, et al. Arandomized controlled trial of epoprostenoltherapy for severe congestive heart failure: theFlolan International Randomized Survival Trial(FIRST). Am Heart J. 1997;134: 44-54.
36. Bocchi EA, Guimaraes G, Mocelin A, et al. Sil-denafil effects on exercise, neurohormonalactivation, and erectile dysfunction in con-gestive heart failure. A double blind, place-bo-controlled, randomized study followed bya prospective treatment for erectile dysfunc-tion. Circulation. 2002; 106: 1097-103.
37. Guazzi M, Tumminello G, Di Marco F, et al.The effects of phosphodiesterase-5 inhibition
with sildenafil on pulmonary hemodynamicsand diffusion capacity, exercise ventilatoryefficiency, and oxygen uptake kinetics in chro-nic heart failure. J Am Coll Cardiol. 2004; 44:2339-48.
38. Packer M, Carver JR, Rodeheffer RJ, et al.Effect of oral milrinone on mortality in seve-re chronic heart failure. The PROMISE StudyResearch Group. N Engl J Med. 1991; 325:1468-75.
39. Jabbour A, Keogh A, Hayward C, Mc DonaldP. Chronic sildenafil lowers transpulmonarygradient and improves cardiac output allowingsuccessful heart transplantation. Eur J HeartFail. 2007; 9: 674-7.
40. Gómez-Moreno S, Lage E, Hernández A, et al.Use of oral sildenafil in patients with irrever-sible pulmonary hypertension not eligible forheart transplantation. Transplant Proc. 2005;37: 1550-1.
41. Torre-Amione G, Durand JB, Nagueh S, et al.A pilot safety trial of prolonged (48h) infusionof the dual endothelin-receptor antagonist tezo-sentan in patients with advanced heart failu-re. Chest. 2001; 120-460-466.
42. Kiowski W, Sutsch G, Hunziker P, et al. Evi-dence for endothelin-1-mediated vasocons-triction in severe chronic heart failure. Lancet.1995; 346: 732-36.
43. Spieker LE, Mitrovic V, Noll G, et al. Acutehemodynamic and neurohomonal effects ofselective ET(A) receptor blockade in patientswith congestive heart failure. ET 003 Investi-gators. J Am Coll Cardiol. 2000; 35: 1745-52.
44. Kaluski E, Kobrin I, Zimlichman R, et al. RITZ-5: randomized intravenous TeZosentan (anendothelin A/B antagonist) for the treatmentof pulmonary edema: a prospective, multi-center, double-blind, placebo-controlled study.J Am Coll Cardiol. 2003; 41: 204-10.
45. Packer M, Mc Murray J, Massie BM, et al. Clli-nical effects of endothelin receptor antogo-nism with bosentan in patients with severechronic heart failure: results of a pilot study.J Card Fail. 2005; 11: 12-20.
46. Anand I, Mc Murray J, Cohn JN, et al. Long-term effects of darusentan on left-ventricularremodelling and clinical outcomes in the Endo-thelin A Receptor Antagonist Trial in Heart Fai-lure (EARTH): randomised, double-blind, pla-cebo-controlled trial. Lancet. 2004; 364:347-54.
GRUPO II. CLASIFICACIÓN DE DANA POINT: ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS
79
Monografía HAP NM 208 pag 18/2/10 17:08 Página 79

Monografía HAP NM 208 pag 18/2/10 17:08 Página 80

81
RESUMENEn este capítulo se aborda la hipertensión
pulmonar debida a las enfermedades respira-torias. En estas enfermedades siempre se haconsiderado a la hipoxia alveolar crónica comoel mecanismo patogénico fundamental, capazde producir tanto una vasoconstricción comocambios estructurales y funcionales en el árbolvascular pulmonar. No obstante, también sonposibles otros mecanismos patogénicos, queen algunos casos, como en el de las neumo-patías intersticiales difusas, son similares a losque se encuentran en la hipertensión arterialpulmonar. Pese a que la prevalencia de la hiper-tensión pulmonar en estas enfermedades nose conoce bien, se aportan los datos más actua-les al respecto. Se insiste, asimismo, en la con-veniencia de establecer adecuadamente el diag-nóstico de la hipertensión pulmonar en todasestas situaciones, diferenciando aquéllas en lasque ésta debe confirmarse mediante un cate-terismo cardiaco derecho. Por último, se repa-san los aspectos terapéuticos más relevantespara cada una de las enfermedades respirato-rias que cursan con una hipertensión pulmo-nar. Se hace hincapié en que, hasta el momen-to actual, no hay evidencia suficiente como paraindicar el llamado tratamiento vasodilatador“específico” en estos casos.
INTRODUCCIÓNLa hipertensión arterial pulmonar se carac-
teriza por la existencia de un remodelado enlos vasos pulmonares, un aumento de presiónen el árbol vascular y una hipertrofia del ven-
trículo derecho. La hipoxia alveolar se ha con-siderado como un factor de riesgo importan-te en la patogenia de la hipertensión pulmonar,por ejemplo en las neumopatías obstructivasy en las restrictivas y en los trastornos asocia-dos con la altitud. La hipoxia alveolar agudacausa una vasoconstricción selectiva en las arte-riolas pulmonares y un aumento de la presiónvascular pulmonar. Sin embargo, la hipoxiaalveolar crónica induce cambios estructuralesy funcionales en el sistema vascular(1,2), lo queincluye la proliferación y la migración de lascélulas de músculo liso de la pared de los vasos,así como el aumento y el acúmulo de matrizextracelular en la pared de dichos vasos.
El “4th World Symposium on PulmonaryHypertension”, celebrado en febrero del 2008en Dana Point (California, Estados Unidos), hasido el primero en establecer un grupo de tra-bajo específico para estudiar las formas de hiper-tensión pulmonar denominadas como “no arte-riales”(3). Se han incluido aquí, entre otrasformas, a la hipertensión pulmonar secundariaa la enfermedad pulmonar obstructiva cróni-ca (EPOC) y a la asociada con las neumopatí-as intersticiales difusas (EPID). La introducciónde esta novedad se ha justificado por el hechode que se trata de formas de hipertensión pul-monar que, a pesar de ser más frecuentes quelas arteriales, se han estudiado mucho menos.Además, esa circunstancia no ha sido incon-veniente para que en ellas, pese a la ausenciade ensayos clínicos que lo avalen, haya aumen-tado el uso de fármacos específicos para las for-mas arteriales.
GRUPO III. CLASIFICACIÓN DE DANA POINT
ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS
María Asunción Nieto Barbero, Beatriz Morales Chacón, José Luis Álvarez-Sala Walther
Monografía HAP NM 208 pag 18/2/10 17:08 Página 81

M.A. NIETO BARBERO ET AL.
82
En este capítulo se hace referencia al gru-po 3 de la clasificación clínica del 4th WorldSymposium on Pulmonary Hypertension, en elque se incluye la hipertensión pulmonar debi-da a la hipoxia alveolar secundaria a enfer-medades pulmonares, a alteraciones en la regu-lación de la ventilación y a la exposicióncrónica a grandes alturas sobre el nivel delmar(4). El título del encabezamiento se ha modi-ficado y al grupo se le ha denominado “Hiper-tensión pulmonar debida a las enfermeda-des pulmonares y/o a la hipoxia”. Sin embargo,la principal modificación ha sido la de incor-porar a este grupo a las enfermedades pul-monares definidas por un patrón ventilatoriomixto (obstructivo y restrictivo), dentro del cualestán las bronquiectasias, la fibrosis quística yel síndrome recientemente descrito caracteri-zado por la combinación de una fibrosis pul-monar y un enfisema (Tabla 1).
HIPERTENSIÓN PULMONAR DEBIDA A ENFERMEDADES PULMONARESINTERSTICIALES DIFUSAS
Las EPID son un conjunto heterogéneo deprocesos caracterizados por la infiltración, celu-
lar y no celular, de las estructuras alveolo-inters-ticiales pulmonares. Estas enfermedades afec-tan al pulmón de una manera difusa y tienenen común rasgos fisiopatológicos, clínicos yradiológicos(5). Sin embargo, la inflamación, laproliferación fibrosa, la formación de granu-lomas y la destrucción del parénquima pul-monar normal varía considerablemente de unosprocesos a otros. El desarrollo de una hiper-tensión pulmonar en la evolución de una EPIDes una circunstancia conocida y supone unimpacto negativo en el pronóstico de la enfer-medad. No obstante, aún no se ha establecidocúal es la prevalencia real de la hipertensiónpulmonar en estos casos. La sarcoidosis, la his-tiocitosis de células de Langerhans y la linfan-gioleimiomatosis no se consideran que estén,por sus especiales peculiaridades, entre otraslas de índole patogénico, en el grupo 3 de lanueva clasificación de Dana Point, sino en ungrupo distinto (Grupo 5) dedicado a las “enfer-medades sistémicas”. Pese a ello, en este capí-tulo se hace mención a las tres puesto que, enrealidad, forman parte de las EPID.
PatogeniaAlgunas nuevas teorías sobre la patogenia
de una de las EPID más frecuentes y graves, lafibrosis pulmonar idiopática (FPI), compartenla implicación de algunos aspectos de los meca-nismos patogénicos propios de la hipertensiónpulmonar(6,7). Al respecto cabe citar, por unaparte, a las interrelaciones entre la fibroproli-feración y la vasculatura pulmonar y, por otrolado, al impacto de la biología vascular y laangiogénesis en la fibrosis pulmonar. Esta nue-va teoría patogénica de la FPI también podríaexplicar parte del desarrollo de otras EPID,como las asociadas a las conectivopatías o alas granulomatosis, como es el caso en la sar-coidosis(6,8,9).
Así, por ejemplo, la existencia de un des-equilibrio en el estrés oxidativo se ha postu-lado tanto para comprender la fibroprolifera-ción como la génesis de la hipertensiónpulmonar, al suponerse un efecto anti-vasodi-latador y proproliferativo(10,11). La falta del antio-
TABLA 1. Grupo 3 de la clasificaciónclínica de la hipertensión pulmonar(actualización del “4th WorldSymposium on PulmonaryHypertension” celebrado en DanaPoint en febrero del 2008)
Grupo 3. Hipertensión pulmonar debida a lasenfermedades pulmonares y/o a la hipoxia
3.1. Enfermedad pulmonar obstructiva crónica
3.2. Enfermedad pulmonar intersticial difusa
3.3. Otras enfermedades pulmonares con patrónmixto restrictivo y obstructivo
3.4. Trastornos respiratorios del sueño
3.5. Trastornos de hipoventilación alveolar
3.6. Exposición crónica a las grandes alturas
3.7. Anomalías del desarrollo broncopulmonar
Monografía HAP NM 208 pag 18/2/10 17:08 Página 82

xidante glutatión en presencia de los oxidan-tes pulmonares favorece la apoptosis de lascélulas epiteliales alveolares, la proliferaciónde los fibroblastos y la síntesis de la matrizextracelular, derivando todo ello en una fibro-sis pulmonar. La guanilato-ciclasa soluble (GCs)es esencial en la vía de la vasodilatación pro-ducida por el óxido nítrico (NO). El NO acti-va a la GCs, que sintetiza al segundo mensa-jero, la guanosina-monofosfato cíclica (GMPc),que a su vez activa a la GMP-cinasa, finalmenteresponsable de la vasodilatación; finalmentela GCs se inactiva por el estrés oxidativo y sereconstituye con antioxidantes(12).
Otro mecanismo patogénico común es elrelacionado con el sistema de la endotelina. Laendotelina 1 (ET-1) es un potente vasocons-trictor pulmonar, y también un gran factor decrecimiento para las células endoteliales y losmiofibroblastos. La ET-1 se ha encontradoaumentada en la hipertensión pulmonar arte-rial, pero también en conectivopatías, como laesclerosis sistémica progresiva (esclerodermia),en la FPI y en la sarcoidosis, tanto si se asociancomo si no con una hipertensión pulmonar.Además, la ET-1 parece que también favorecela fibrogénesis interactuando con las metalo-proteinasas de la matriz e iniciando la trans-formación de la célula epitelial hacia el miofi-broblasto, a través de la inducción del factor decrecimiento tisular beta-1 (TGF-β1 o transfor-ming growth factor β1)(13,14). Según esta teoríase producirían diversas interacciones patogé-nicas que serían mediadas por factores de cre-cimiento circulantes y locales. En realidad, todosestos eventuales mecanismos patogénicos hansuscitado gran interés por su posible relevan-cia terapéutica, como se discute en el epígrafededicado al tratamiento.
Además de los mecanismos que son comu-nes para el remodelado de los vasos y el delparénquima pulmonar, también hay otros queson únicos o específicos de cada proceso. Así,por ejemplo, en la esclerodermia se ha des-crito una relación con algunos polimorfismosdel factor de crecimiento del tejido conectivoy con la activación del factor de crecimiento
derivado de las plaquetas (PDGF o platelet-deri-ved growth factor) a través de autoanticuer-pos(15,16). Estos aspectos también pueden tenerimplicaciones terapéuticas.
EpidemiologíaLa prevalencia de la hipertensión pulmo-
nar en las EPID varía según el tipo de enfer-medad intersticial y la gravedad de la lesiónpulmonar. Las EPID que con más frecuencia seasocian a una hipertensión pulmonar y de lasque, por ello, se dispone de más informaciónson la FPI, la fibrosis pulmonar asociada a cola-genopatías (sobre todo, a la esclerosis sistémi-ca progresiva), la sarcoidosis, la histiocitosis decélulas de Langerhans y la linfangioleimioma-tosis(17). En todos estos procesos la hiperten-sión pulmonar no siempre se acompaña de unaalteración ventilatoria restrictiva, pero sí con lanecesidad de oxigenoterapia, la intolerancia alejercicio o la disminución del factor de trans-ferencia para el monóxido de carbono (“capa-cidad de difusión”).
Varios estudios retrospectivos indican quela hipertensión pulmonar puede ser frecuen-te en la FPI(18,19), con cifras que oscilan entreel 23 y el 84%. Sin embargo, estos datos noestán exentos de sesgos por ser retrospectivosu obtenidos de muestras seleccionadas, comopor ejemplo a partir de pacientes inscritos enuna lista de espera para un trasplante pulmo-nar(19,20). En realidad, la prevalencia de la hiper-tensión pulmonar en la FPI no se conoce conexactitud(3).
Según los resultados de dos trabajos recien-tes(21,22) la esclerodermia puede asociarse tan-to a una hipertensión pulmonar aislada (19-60% de los casos) como a una EPID aislada(22-21% de los casos) como a ambas simul-táneamente (18-22% de los casos). De cual-quier modo, la existencia de una hipertensiónpulmonar conlleva un peor pronóstico en cuan-to a la supervivencia de estos enfermos.
La hipertensión pulmonar también pue-de aparecer en los individuos que padecen unasarcoidosis, aunque apenas tengan alteraciónalguna en el parénquima pulmonar. Por ello,
GRUPO III. CLASIFICACIÓN DE DANA POINT: ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS
83
Monografía HAP NM 208 pag 18/2/10 17:08 Página 83

es muy probable que la vasoconstricción hipó-xica no sea el único mecanismo patogénicoen estos enfermos. El aumento de la ET-1 enel suero y en el lavado broncoalveolar, la infla-mación granulomatosa de las propias arterias,así como la compresión de la pared arterialpor adenopatías(23), son mecanimos que se hanimplicado en la patogenia de la hipertensiónpulmonar en estos pacientes. Se estima que laprevalencia de la hipertensión pulmonar varíaen estos casos entre un 5,7%(24) y un 74% (enlos enfermos en una lista de espera de tras-plante)(25,26). Su existencia se ha asociado, asi-mismo, con una mayor mortalidad.
La hipertensión pulmonar es muy frecuenteen el estadio final de la histiocitosis de célulasde Langerhans y tiende a ser más grave quela que aparece en otras EPID. Por otro lado,para que se desarrolle dicha hipertensión, noes necesario que exista una destrucción delparénquima pulmonar(17). De hecho, no haycorrelación alguna entre la presión arterial pul-monar media (PAPm) y la gravedad de las alte-raciones espirométricas(17,27,28).
Aproximación diagnósticaLa disnea y la limitación al flujo aéreo son
manifestaciones habituales tanto en las EPIDcomo en la hipertensión pulmonar, por lo laexistencia de esta última puede no sospecharseinicialmente. Ha de pensarse en ella cuandola intensidad de los síntomas sea mayor de laesperada y desproporcionada en atención a lagravedad de la EPID. Los signos que en laexploración física pueden sugerir una hiper-tensión pulmonar aparecen en fases avanza-das, por lo que no suelen ser útiles para esta-blecer la sospecha diagnóstica en los primerosestadios de la enfermedad.
En la radiografía de tórax puede observarseque las arterias pulmonares o el ventrículoderecho están aumentados de tamaño, peroestos hallazgos tienen una sensibilidad baja.En la tomografía computarizada (TC) de tóraxes posible encontrar, si la hipertensión pul-monar es moderada o grave, que la arteria pul-monar supera los 29 mm de diámetro o mues-
tra un aumento en comparación con la aor-ta(29). Sin embargo, no se ha podido evidenciarque exista una asociación entre el diámetro delas arterias pulmonares medidas mediante unaTC torácica y la PAPm(30).
El ecocardiograma transtorácico es el méto-do no invasivo más adecuado para detectaruna hipertensión pulmonar. En general se acep-ta que una presión sistólica ventricular dere-cha mayor de 50 mmHg es indicativa de unahipertensión pulmonar significativa en estospacientes. Valores entre 50 y 35 mmHg se con-sidera que son “borderline” y los que están pordebajo de de 35 mmHg se estiman como nor-males(31). Se ha señalado que hasta en la mitadde los individuos con una EPID no se puededeterminar la regurgitación tricuspídea porproblemas técnicos, por lo que los valores pre-dictivos positivos y negativos son menores delo real cuando se comparan con los resultadosque derivan de un cateterismo cardiaco dere-cho. Por este motivo es importante tener encuenta otras medidas, como las dimensionesde la aurícula y del ventrículo derechos, el índi-ce de excentricidad y el desplazamiento delplano anular tricuspídeo durante la sístole.
Algunos estudios han encontrado que laconcentración sérica del péptido natriuréticocerebral (BNP) y la de su porción N-terminal(NT-proBNP) aumentan en los enfermos quepadecen una fibrosis intersticial, de la causaque sea, y una hipertensión pulmonar con dis-función ventricular derecha(32). Además, esteaumento se asocia con un peor pronóstico. Sinembargo, estas sustancias no permiten efec-tuar un diagnóstico precoz, entre otras razo-nes porque en la hipertensión pulmonar leveo controlada sus niveles séricos pueden sernormales. Por el contrario, esta normalidad sípuede tener utilidad clínica, ya que es un mar-cador de mejor pronóstico.
El cateterismo cardiaco derecho sigue sien-do la “prueba de oro” para evaluar la hemo-dinámica pulmonar. Debe realizarse siem-pre para confirmar el diagnóstico de unahipertensión pulmonar cuya existencia se hasospechado por métodos no invasivos(3,33). Esta
M.A. NIETO BARBERO ET AL.
84
Monografía HAP NM 208 pag 18/2/10 17:08 Página 84

técnica también es necesaria para estratificarla gravedad de la hipertensión pulmonar, paradetectar una posible disfunción del ventrícu-lo derecho y para excluir otras posibles cau-sas de la hipertensión. Entre estas últimas,sobre todo una disfunción diastólica del ven-trículo izquierdo, que puede ser difícil de reco-nocer en la ecocardiografía. Si bien la pruebade vasodilatación está indicada y se reco-mienda en la mayoría de los pacientes quesufren una hipertensión pulmonar arterial, suutilidad no está indicada en los individuos quetienen una EPID(33). Además, los bloquean-tes de los canales de calcio a dosis altas nosirven en el tratamiento de estos enfermos,en los que pueden resultar incluso deletéreos.Según las recomendaciones del 4th WorldSymposium on Pulmonary Hypertension el cate-terismo cardiaco derecho sólo está indicadocuando se espera que sus resultados van aafectar al tratamiento final del paciente y, tam-bién, en ensayos clínicos en los que se quie-re identificar las subpoblaciones de sujetosque pueden beneficiarse más de los trata-mientos específicos de la hipertensión pul-monar(3).
El estudio de la función respiratoria no con-tribuye de manera esencial en la valoración dela hipertensión pulmonar que se asocia a lasEPID. Así, parece que la capacidad vital forza-da (FVC) tiene poco poder predictivo. Sinembargo, varios trabajos han puesto de mani-fiesto que un valor de la capacidad de trans-ferencia para el monóxido de carbono (DLCO)menor del 40% del estimado y la necesidad deuna oxigenoterapia son factores que predicenla existencia de una hipertensión pulmonar enlos individuos que tienen una FPI o una sar-coidosis(3,33). También debe hacer pensar endicha hipertensión la observación de una des-proporción entre la reducción de la DLCO y lade la FVC, con un cociente entre ambas ≥ 1,4.
En cuanto al estudio histológico, pareceque su realización no es necesaria en estoscasos para llegar al diagnóstico. De hecho, lapresencia de una hipertensión pulmonar incre-menta la morbimortalidad de las biopsias qui-
rúrgicas que se llevan a cabo en la valoraciónde los enfermos que tienen una EPID(34).
La actuación diagnóstica que puede poner-se en marcha en el estudio de la hipertensiónpulmonar de una EPID se expone, en formade algoritmo, en la figura 1.
TratamientoComo en las demás formas de hipertensión
pulmonar secundarias a una enfermedad res-piratoria, el tratamiento principal en ésta tam-bién es el de la neumopatía de base, lo que enlas EPID requiere el correcto diagnóstico de lasdistintas entidades histopatológicas. Para ello,a veces tiene que recurrirse a una biopsia qui-rúrgica(5). En muchas EPID se ha utilizado comotratamiento fundamental una combinación deesteroides sistémicos y de fármacos inmuno-supresores, como la azatioprina o la ciclofos-famida(35-37). Sin embargo, hay muy pocos datos,en ocasiones incluso contradictorios, sobre laacción terapéutica de los inmunosupresores enla hipertensión pulmonar de las EPID(38,39).
En realidad, a diferencia de lo que ocurreen la hipertensión arterial pulmonar del gru-po 1 de la clasificación clínica(3), no hay nor-mas claras sobre cómo debe tratarse la hiper-tensión pulmonar asociada a las EPID. Si sehace referencia a la anticoagulación, hay pocaspruebas en cuanto a sus eventuales efectosbeneficiosos y, sin embargo, es una posibili-dad no exenta de inconvenientes, como lahemorragia. Podría considerarse justificada enlos individuos con una EPID que están bieninformados o que tienen una hipertensión pul-monar grave unida a una disfunción del ven-trículo derecho, si bien no existen evidenciassuficientes al respecto.
El valor de los fármacos denominadosespecíficos tampoco se conoce bien en estoscasos, por lo que para ninguno de ellos se haaprobado esta indicación. No obstante, si serecuerda, por un lado, que existen factorespatogénicos comunes entre la hipertensiónpulmonar y la FPI y, por otra parte, que no haymedidas satisfactorias para esta última, se com-prende que se haya especulado sobre la posi-
GRUPO III. CLASIFICACIÓN DE DANA POINT: ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS
85
Monografía HAP NM 208 pag 18/2/10 17:08 Página 85

ble utilidad de estas sustancias. Se dispone deresultados procedentes de trabajos observa-cionales no comparativos que sugieren que,en estos enfermos, el sildenafilo oral aumen-ta la distancia recorrida en la prueba de 6minutos marcha (T6M), de manera similar acómo lo hace en los individuos con una hiper-tensión pulmonar arterial del grupo 1(40). Delmismo modo, teniendo en cuenta la impor-tancia de la ET-1 en la patogenia de la hiper-tensión pulmonar y de la FPI, también debenconsiderarse los datos de un ensayo clínico ale-atorizado que ha valorado el efecto antifibro-génico del bosentán en la FPI sin HP 2ª(41). Sibien no se encontró mejoría alguna en el obje-tivo primario del estudio (la distancia recorri-da en el T6M), sí se hallaron diferencias en unsegundo análisis, lo que ha llevado a continuarla investigación en este mismo sentido. Losresultados de ese nuevo trabajo aún no seconocen. También se han publicado datosobservacionales sobre la eficacia del bosentán
en otras EPID, como la sarcoidosis(42). Se hainsistido, asimismo, en la mejoría que se con-sigue con el iloprost nebulizado y actualmen-te está en marcha un estudio multicéntricopara analizar los beneficios reales que puedenesperarse de este fármaco en estos enfermos.Cabe destacar que en ninguno de los casosantes mencionados se produjo un deteriorodel intercambio gaseoso en los individuos tra-tados con estos fármacos, como consecuenciade su aparente selectividad pulmonar.
A pesar de todo lo señalado, aún no se dis-pone de pruebas suficientes como para reco-mendar el tratamiento especifico de la hiper-tensión pulmonar en las EPID. Se necesitanmás datos y así se recomienda en las norma-tivas nacionales e internacionales reciente-mente publicadas(3,43,44). A este respecto, cabeindicar que en este momento se están llevan-do a cabo distintos ensayos clínicos con ima-tinib, un inhibidor del PDGF, así como con inhi-bidores del TGF-β1.
M.A. NIETO BARBERO ET AL.
86
FIGURA 1. Algoritmo diagnóstico de la hipertensión pulmonar en las enfermedades pulmonares.
Disnea moderada o graveSignos en el ECG y la RX de tórax de HPDLCO < 50% del valor teóricoDesaturación de oxígeno con el ejercicioAumento de la BNP sérica
Sí Ecocardiograma
Prueba sin problemasde interpretaciónPSP < 40 mmHgVentrículo derecho normal
Prueba de dudosainterpretación
PSP 40-50 mmHgVentrículo derechodilatadoPSP > 50 mmHg
No
HP poco probable
Considerar una RMo un cateterismo cardiaco
derecho
No HP significativa:seguir la evolución
Descartar otras causas de HPOptimizar el tratamiento de la EPOC
Si persiste una HP grave
Considerar el cateterismo derecho si:- Se considera un tratamiento específico- Se valora el tratamiento quirúrgico- Se sospecha una HP desproporcionada para la alteración funcional existente
Abreviaturas: EPOC enfermedad pulmonarobstructiva crónica; ECG electrocardiograma;RX radiografía; RM resonancia magnéticanuclear; PSP presión sistólica pulmonar; HPhipertensión pulmonar; DLCO transferen-cia alveolo-capilar para el monóxido de car-bono; BNP péptido natriurético cerebral.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 86

En todo caso, puesto que la hipertensiónpulmonar es un factor de mal pronóstico enlas EPID, su existencia debe considerarse comouna indicación para la inclusión de estospacientes, en cuanto sea posible, en la lista deespera de un trasplante pulmonar, siempre ycuando se cumplan los demás criterios paraello(45). En tal sentido, conviene mencionar queactualmente existen datos contradictorios sobrecuál es el tipo de trasplante pulmonar (bilate-ral o unilateral) o cardiopulmonar más ade-cuado en estos casos.
HIPERTENSIÓN PULMONAR DEBIDA A LAENFERMEDAD PULMONAR OBSTRUCTIVACRÓNICA
EpidemiologíaLa prevalencia exacta de la hipertensión
pulmonar secundaria a la EPOC no se conocecon exactitud, en parte por la dificultad inhe-rente a hacer estudios poblacionales emple-ando una prueba diagnóstica invasiva comoes el cateterismo cardiaco derecho. La mayo-ría de los datos procede de trabajos en los quese ha utilizado la ecocardiografía, que es unatécnica no exenta de errores(46). La informa-ción disponible proviene de varias series decasos. En 120 pacientes con un enfisema gra-ve evaluados para participar en el estudio NETT(“National emphysema treatment trial”) se obser-vó que la media (± la desviación estándar) dela muestra tenía una PAPm de 26,3 (± 5,2)mmHg. Es decir, aunque se trataba de indivi-duos con una EPOC grave, la hipertensión pul-monar era leve o moderada, pero muy pre-valente, pues en un 91% de los sujetos la PAPmsuperaba los 20 mmHg(47). Vizza y cols.(48), enuna serie constituida por 168 enfermos estu-diados para valorar la indicación de un posi-ble trasplante pulmonar, encontraron resulta-dos similares, con un intervalo de confianzadel 95% para la PAPm que estaba entre 24,1y 25,9 mmHg. Thabut y cols.(49), en un grupode 215 individuos remitidos para considerarla posibilidad de una cirugía de resección devolumen pulmonar o de un trasplante pulmo-
nar, confirmaron que la mitad de los pacien-tes tenía una PAPm mayor de 25 mmHg.
En diversos trabajos se ha señalado coninsistencia que, pese a lo dicho, existe un sub-grupo de enfermos en los que la hipertensiónpulmonar es grave o “desproporcionada”, aun-que la obstrucción al flujo aéreo sea modera-da. Chaouat y cols.(50) hallaron que 27 de 998sujetos diagnosticados de EPOC tenían unaPAPm mayor o igual a 40 mmHg. En este gru-po, la DLCO fue muy baja y predominaba elenfisema en la TC de tórax de alta resolución.En la serie francesa de Thabut y cols.(49) seobservaron resultados muy similares. En amboscasos los pacientes tenían una hipoxemia gra-ve sin hipercapnia, estaban en situación clíni-ca estable y recibían un tratamiento óptimo.
Impacto de la hipertensión pulmonar enel pronóstico de la enfermedad pulmonarobstructiva crónica
En la EPOC la hipertensión pulmonar es,por lo general, leve o moderada, lo que no impi-de para que influya, de forma determinante,en la supervivencia de los enfermos. El grupofrancés de Strasbourg(51) comunicó una tasa desupervivencia a los cinco años del 36% en losindividuos que tenían una PAPm mayor de 25mmHg, comparada con el 62% que se obser-vó en los sujetos que no tenían hipertensiónpulmonar. En la serie de Chaouat y cols.(50), enla que los pacientes tenían una PAPm mayor oigual a 40 mmHg, la tasa de supervivencia alos cinco años fue menor del 20%.
PatogeniaEn la patogenia de la hipertensión pulmonar
de la EPOC se incluyen tres mecanismos poten-ciales, que deben estudiarse específicamente: elremodelado vascular, la disminución del núme-ro de vasos pulmonares debida a la destrucciónparenquimatosa y la trombosis pulmonar.
Remodelado vascularLa hipoxia alveolar crónica induce la “neo-
muscularizacion” de las arteriolas pulmona-res, previamente no “muscularizadas”, y la
GRUPO III. CLASIFICACIÓN DE DANA POINT: ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS
87
Monografía HAP NM 208 pag 18/2/10 17:08 Página 87

hipertrofia de la túnica media de las arteriolasmusculares. En la EPOC grave se produce unengrosamiento de la capa íntima como con-secuencia de una infiltración por células mus-culares lisas y por el depósito de fibras de colá-geno y de elastina. La “neomuscularización”se debe a la hipertrofia, la proliferación y latransformación de las células contráctiles deno-minadas pericitos, precursoras de los miocitosde la capa media. Cambios similares se hanencontrado también en los pacientes que tie-nen una EPOC leve y que son normoxémicosy no tienen una hipertensión pulmonar, asícomo en las personas fumadoras asintomáti-cas(52). Habitualmente se aprecia también lamencionada hipertrofia de la túnica media(53).El engrosamiento de la íntima de las arteriaspulmonares musculares y de las arteriolas seha hallado, asimismo, en la EPOC leve y en lasestadios finales de la enfermedad. Sin embar-go, no se han evidenciado anomalías com-plejas, como las lesiones plexiformes o lasangiomatoides que aparecen en la hiperten-sión arterial pulmonar. Sí se han detectado aveces, por el contrario, trombos o émbolosintravasculares.
Mecanismos patogénicos del remodeladovascular
Muchos de los mecanismos patogénicosdescritos a propósito de la hipertensión arte-rial pulmonar se observan también en la hiper-tensión pulmonar asociada a las enfermeda-des respiratorias.
En los pacientes con una EPOC y una hiper-tensión pulmonar se ha encontrado “in vitro”una alteración de la vasodilatación dependientedel endotelio. Esta disfunción endotelial de lasarterias pulmonares produce modificacionesen la expresión o en la liberación de los media-dores vasoactivos, perdiéndose así el equilibrionormalmente existente entre las sustanciasvasodilatadoras, como el NO o la prostaciclina,y las vasoconstrictoras, como la ET-1 o la angio-tensina(54). La expresión de la sintasa del NO(eNOS) en el endotelio de la arteria pulmonarestá disminuida en los individuos que tienen
una enfermedad avanzada y en los fumadoresasintomáticos. Esto parece indicar que la reduc-ción en la síntesis del NO podría contribuir alos cambios estructurales y de la función endo-telial de los vasos pulmonares que aparecen enlas enfermedades debidas al tabaco(55). Tam-bién se ha señalado que en los pacientes conuna EPOC desciende el NO exhalado, lo quepodría tener su origen en una menor liberaciónendotelial(56). En los enfermos con un enfise-ma grave se ha hallado, asimismo, una dis-minución en la expresión de la prostaciclina-sintasa en las arterias pulmonares(57). En cuantoa los niveles séricos circulantes de la ET-1, haydatos contradictorios. En cambio, sí se hademostrado que su expresión vascular pulmo-nar está aumentada en los individuos con unahipertensión pulmonar, incluidos los que tie-nen una EPOC(58).
En los sujetos fumadores con una EPOCmoderada se ha señalado que aumenta la expre-sión del factor de crecimiento endotelial en elmúsculo liso de los vasos, lo que se correlacio-na con el engrosamiento muscular de las arte-rias pulmonares(59). También se ha resaltado elpapel de la serotonina (5-HT) y de su transpor-tador (5-HTT) en el origen de la hiperplasia mus-cular propia de la hipertensión pulmonar.
Importante parece también la relación exis-tente entre la inflamación y el remodelado vas-cular. Este último se asocia con una infiltraciónen la vía aérea por células inflamatorias, fun-damentalmente linfocitos T activados, con pre-dominio de los de tipo CD8(60). Este infiltradose ha encontrado, igualmente, en los pacien-tes que tienen una EPOC leve y en las perso-nas fumadoras que mantienen una funciónpulmonar normal. Esto ha sugerido la hipó-tesis de que el tabaco podría tener un efectodirecto sobre la musculatura vascular. Del mis-mo modo, también se ha referido una posiblerelación en la EPOC entre los marcadores cir-culantes de inflamación sistémica y la hiper-tensión pulmonar(61). En algunos trabajos seha observado que los niveles séricos de la pro-teína C reactiva y del factor de necrosis tumo-ral alfa (TNF-α) eran significativamente mayo-
M.A. NIETO BARBERO ET AL.
88
Monografía HAP NM 208 pag 18/2/10 17:08 Página 88

res en los individuos con una EPOC que te-nían, además, una hipertensión pulmonar res-pecto a los que no la tenían.
La hipoventilación alveolar y la hipercap-nia producen una vasoconstricción sinérgicacon la hipoxia y también parece que influyenen el remodelado vascular. Quizás el factor cla-ve al respecto sea la concentración de los hidro-geniones(62).
Factores mecánicosDesde hace años se sugiere que la pérdi-
da de capilares intersticiales, secundaria a ladestrucción parenquimatosa de las paredesalveolares que es propia del enfisema, podíaser un mecanismo importante en la patogeniade la hipertensión pulmonar que se asocia conla EPOC. Sin embargo, esta posibilidad no hapodido demostrarse en modelos experimen-tales.
La PAPm es la suma de la presión de encla-vamiento de la arteria pulmonar, equivalentea la presión capilar pulmonar (PCP), y del resul-tado del producto del gasto cardiaco (GC) porla resistencia vascular pulmonar (RVP) total,de acuerdo con la siguiente ecuación: PAPm=PCP + (GC x RVP). De esta forma, la PAPmpuede elevarse si aumenta cualquiera de esastres variables. La PCP suele ser alta, incluso enreposo, en los pacientes que tienen unaEPOC(63). Este hecho puede ser el reflejo tantode una disfunción diastólica del ventrículoizquierdo, debida a alguna comorbilidad car-diovascular, como de un incremento de la pre-sión intratorácica, como el que aparece en elenfisema por la hiperinsuflación existente(64).
Una obstrucción al flujo aéreo grave tam-bién puede hacer que se incremente la pre-sión intraalveolar, lo que ocasiona una com-presión extrínseca de los vasos yuxtaalveolares.Conviene recordar que en la circulación pul-monar los capilares dan cuenta de más de lamitad de la RVP total. Además, la hiperinsu-flación distendería los vasos, originándose asíun estrechamiento vascular aún mayor. Enresumen, la RVP puede elevarse en la EPOCpor mecanismos muy diversos.
Hipoxia alveolarEl aumento de la PAPm y de la RVP que se
produce como respuesta a una hipoxia alveolaraguda se denomina reflejo de vasoconstricciónpulmonar hipóxica(41). Este reflejo afecta fun-damentalmente a las arterias pulmonares quetienen un calibre menor de 500 µm. La vaso-constricción pulmonar hipóxica es menos enér-gica en los individuos que ya muestran altera-ciones estructurales en las arteriolas pulmonares,como ocurre en los enfermos que tienen unaEPOC. Sin embargo, aún puede ser de granimportancia en algunas situaciones concretas,como las agudizaciones, el sueño o el ejercicio.
Las consecuencias de la hipoxia crónica enla circulación pulmonar se conocen a travésde modelos animales y de estudios in vitro(44),en los que se han puesto de manifiesto loscambios que se suceden en todas las célulasy en la matriz extravascular de la pared de losvasos pulmonares. Surge así un desequilibrioa favor del incremento del tono vascular y delestado proliferativo celular, debido a un aumen-to de la síntesis de ET-1 y a una disminucióndel NO y de la prostaciclina(65).
Los mecanismos patogénicos expuestos seintegran para articular la hipótesis de la pato-biología de la hipertensión pulmonar de laEPOC, tal y como se expone en la figura 2.
Aproximación diagnósticaEl reconocimiento de la existencia de una
hipertensión pulmonar en un paciente con unaEPOC es importante por varias razones. Porun lado, permite identificar, en su caso, otrasposibles causas de hipertensión pulmonar yrealizar, entonces, el tratamiento adecuado.Por otra parte, da información sobre el pro-nóstico del enfermo. Finalmente, condicionadecisiones terapéuticas importantes, como eltrasplante pulmonar o la oxigenoterapia.
Los síntomas habituales de la hipertensiónpulmonar, como la disnea o la intolerancia alejercicio, pueden estar enmascarados por losde la propia EPOC. Los signos característi-cos en la exploración física tampoco suelenestar presentes, salvo en algunas situaciones,
GRUPO III. CLASIFICACIÓN DE DANA POINT: ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS
89
Monografía HAP NM 208 pag 18/2/10 17:08 Página 89

como las agudizaciones graves. Los edemasperiféricos pueden aparecer en la EPOC y nodeberse a una insuficiencia ventricular dere-cha ocasionada por la hipertensión pulmo-nar(66). Además, la hipercapnia puede originaruna retención de agua y sodio por un meca-nismo renal, lo que también puede determi-nar la aparición de edemas en las extremi-dades.
El electrocardiograma puede mostrar lossignos propios de un crecimiento ventriculoderecho. Esta alteración tiene una buena espe-cificidad, mayor del 85%, pero una mala sen-sibilidad, menor del 40%, cuando la hiperten-sión pulmonar es leve. Algo similar ocurre conlos hallazgos que ofrece la radiografía de tórax.
Los datos que proporciona la espirometría,como la capacidad vital (VC), el volumen espi-ratorio forzado en el primer segundo (FEV1)y la relación FEV1/VC, no son útiles para pre-decir el valor de la PAPm. La presión arterialde oxígeno (PaO2) y la saturación transcutáneade oxígeno (StcO2) sólo podrían explicar un25% de la modificación de la PAPm, según lasecuaciones de predicción propuestas recien-temente(67). En cualquier caso, la prediccióndel valor de la PAPm con estas variables esmuy imprecisa. La DLCO está disminuida enla hipertensión pulmonar grave, pero esto tam-bién sucede en el enfisema, por lo que este
dato no es útil para predecir la existencia deuna hipertensión pulmonar.
Dos estudios recientes han insistido en lautilidad clínica de un patrón funcional respi-ratorio normalmente poco frecuente, pero queaparece en la mayoría de los enfermos que tie-nen una hipertensión pulmonar despropor-cionada. Consiste en la combinación de unapreservación de la mecánica pulmonar, conuna gran disminución de la PaO2, un aumen-to importante del gradiente alvéolo-arterial deoxígeno y una cierta tendencia a la hipocap-nia(49,50). Además, en uno de estos dos traba-jos el valor de la DLCO había descendido másde lo que cabría esperar por la gravedad delenfisema existente. La asociación de estepatrón funcional con una disnea grave debellevar a la sospecha de una hipertensión pul-monar desproporcionada.
La precisión diagnóstica del ecocardiogra-ma es más baja en la hipertensión pulmonarde la EPOC que en la hipertensión arterial pul-monar(46). Muchas veces es imposible estimarla presión sistólica del ventrículo derecho porimpedimentos anatómicos, fundamentalmentepor la hiperinsuflación pulmonar, y cuandosí es posible hacerlo suele existir una diferen-cia variable, hasta de 20 mmHg, con la medi-da obtenida mediante un cateterismo cardia-co derecho. Sin embargo, la visualización del
M.A. NIETO BARBERO ET AL.
90
FIGURA 2. Mecanismo patogénico de la hipertensión pulmonar en la enfermedad pulmonar obstructivacrónica.
Humo del tabaco y otros agentes + factores genéticos
Enfisema: reducción del lechopulmonar capilar
Remodelado vascular pulmonar:- Proliferación celular- Aumento de la matriz extracelular
Vasoconstricción arterialpulmonar
Hipertensión pulmonar
Inflamación Hipoxia
Monografía HAP NM 208 pag 18/2/10 17:08 Página 90

ventrículo derecho sí se consigue en la mayo-ría de los casos. La ausencia de alteracionesen este ventrículo tiene un buen valor predic-tivo negativo, que alcanza hasta un 90%(46). Adiferencia de lo que ocurre en la hipertensiónarterial pulmonar, en el momento actual sedesconoce si las medidas ecocardiográficas dela disfunción del ventrículo derecho, como elíndice de excentricidad o el desplazamientodel plano anular tricuspídeo durante la sísto-le, tienen o no un valor pronóstico en lospacientes que tienen una EPOC. A pesar detodas las dificultades técnicas, la ecocardio-grafía con doppler es el mejor procedimientono invasivo para estudiar la hipertensión pul-monar en la EPOC y complementa la infor-mación que se obtiene con el cateterismo car-diaco derecho(68).
La resonancia magnética cardiaca permi-te la medida de los volúmenes y de los flujossanguíneos que se producen en el tórax. Actual-mente se considera como el mejor métodopara medir la fracción de eyección y la masadel ventrículo derecho(69), por lo que podría seruna técnica no invasiva ideal para valorar lacirculación pulmonar. Lo cierto es, sin embar-go, que hasta la fecha no se ha evaluado suposible utilidad en el diagnóstico de la hiper-tensión pulmonar de la EPOC.
Los niveles séricos del BNP también pue-den ser útiles en el estudio de la hipertensiónpulmonar de la EPOC. Un trabajo realizado conpacientes afectos de enfermedades respirato-rias muy heterogéneas permitió establecer queun punto de corte de 33 pg/ml tenía capaci-dad para identificar una PAPm mayor de 35mmHg con una sensibilidad del 87% y unaespecificidad del 81%(32). Este mismo puntode corte se mostró útil, además, para prede-cir la mortalidad de los individuos que tení-an una EPOC y una hipertensión pulmonar.Sin embargo, es obvio que se requieren másestudios para confirmar la utilidad del BNPsérico en estos pacientes.
En los individuos que tienen una EPOC yuna hipertensión pulmonar es frecuente quese produzcan desaturaciones de oxígeno con
el esfuerzo, lo que se ha relacionado con unamenor supervivencia del enfermo. La distan-cia recorrida en el T6M es menos sensible alos cambios clínicos en estos casos que en lainsuficiencia cardiaca o en la hipertensión pul-monar arterial, probablemente porque la tole-rancia al ejercicio en la EPOC con hipertensiónarterial no sólo depende del rendimiento car-diaco.
El cateterismo cardiaco derecho es nece-sario para confirmar el diagnóstico de la hiper-tensión pulmonar, sobre todo cuando las téc-nicas no invasivas muestran resultados noconcluyentes. Sin embargo, este procedimientoes invasivo y no está exento de riesgos y moles-tias para el paciente. Además, su realizaciónpuede suponer un coste no despreciable. Porestos motivos no se recomienda llevarlo a cabode forma sistemática, sino únicamente cuan-do los hallazgos que puedan obtenerse condi-cionen el tratamiento posterior del enfermo,por ejemplo, en la valoración preoperatoria deun candidato a un trasplante pulmonar o a unacirugía de reducción de volumen(70). La prue-ba vasodilatadora carece de interés prácticoen este caso, y no está indicada por sus posi-bles efectos deletéreos sobre el intercambiogaseoso.
En resumen, en los pacientes que tienenuna EPOC y una hipertensión pulmonar hayque recorrer el mismo camino diagnóstico queel que se ha propuesto para cualquier indivi-duo que padece una hipertensión pulmonarde otro origen. La actuación diagnóstica quepuede ponerse en marcha en estos casos seexpone, en forma de algoritmo, en la figura 1.
TratamientoEste apartado se refiere al tratamiento de
la hipertensión pulmonar de la EPOC, no aldel cor pulmonale ni al de la EPOC en sí mis-ma. Sin embargo, es obvio que estos dos últi-mos deben optimizarse en todos los pacien-tes que tienen una hipertensión pulmonarasociada a una EPOC.
Clásicamente el tratamiento de la hiper-tensión pulmonar de la EPOC se ha basado,
GRUPO III. CLASIFICACIÓN DE DANA POINT: ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS
91
Monografía HAP NM 208 pag 18/2/10 17:08 Página 91

sobre todo, en la oxigenoterapia continuadomiciliaria (OCD), al considerarse a la hipo-xia alveolar como la causa fundamental en elaumento de la RVP y de la PAPm. Los traba-jos del Nocturnal oxygen therapy trial (NOTT)(71)
y del Medical Research Council (MRC)(72) valo-raron la utilidad de la OCD en los enfermosque padecían una insuficiencia respiratoriacrónica secundaria a una EPOC. En el estudiodel MRC los datos obtenidos del seguimientode 42 individuos durante más de un año mos-traron que los sujetos tratados con oxígenoa largo plazo tenían una PAPm estable, mien-tras que ésta aumentaba significativamenteen los pacientes no tratados. En el estudioNOTT se dispuso de los datos hemodinámi-cos de 117 enfermos(73). En las personas tra-tadas con oxígeno durante, al menos, 18 horasal día se consiguió disminuir significativa-mente la RVP y la PAPm en reposo y en ejer-cicio, objetivo que no se logró en los queemplearon el oxígeno sólo durante las horasde sueño. En el momento actual existe evi-dencia suficiente como para asegurar que laOCD produce una mejoría hemodinámica enlos pacientes que tienen una EPOC. Al res-pecto, los mejores resultados se observancuando la OCD se mantiene un mínimo de 17ó 18 horas al día(74).
Algunos pacientes con EPOC no tienen unainsuficiencia respiratoria diurna, sino sólo unahipoxemia moderada, pero experimentan unaimportante hipoxemia durante el sueño, conunos valores de la SaO2, que caen por debajodel 90% o, incluso, del 88%. Los estudios quehan analizado la utilidad de la OCD nocturnaen estos casos son contradictorios y no hanpodido confirmar que con ella mejore la evo-lución de la hemodinámica pulmonar(75). En elmomento actual, por tanto, no se recomiendala OCD nocturna en estos enfermos.
Tampoco se dispone de evidencia que jus-tifique el tratamiento farmacológico específi-co de la hipertensión pulmonar de la EPOC.Por el contrario, sí hay datos que sugieren queeste tratamiento puede ser prejudicial(70), tal ycomo se analiza a continuación.
Los fármacos vasodilatadores pulmonaresatenúan la vasoconstricción de los alvéolos malventilados, por lo que empeoran la relación, yade por sí alterada, de la ventilación con la per-fusión en el pulmón (cociente V/Q) y originan,por tanto, una mayor hipoxemia. Tal ocurre,por ejemplo, con los prostanoides, tanto admi-nistrados por vía intravenosa como en nebu-lización. El NO es un potente y selectivo vaso-dilatador, que puede mejorar la hemodinámicapulmonar y la hipoxemia en los pacientes quetienen una EPOC. Así, en los enfermos trata-dos con NO se obtiene una mejoría significa-tiva en la PAPm, la RVP y el GC. Sin embargo,la administración nebulizada del NO tieneimportantes problemas técnicos, además deocasionar efectos tóxicos que imposibilitan, porel momento, su empleo. En esta línea, el sil-denafilo, que es un inhibidor de la fosfodies-terasa V y que incrementa la producción de NOa través de la vía del GMPc, se ha utilizado conresultados favorables en algunos pacientes conuna EPOC asociada a una hipertensión arterial.No obstante, aún no se dispone de ensayos clí-nicos aleatorizados que demuestren fehacien-temente su eficacia. Por ello en estos casos,al igual que en la hipertensión pulmonar de lasEPID, no se aconseja el uso de los tratamien-tos específicos fuera del contexto de un ensa-yo clínico(3,43,44).
Recientemente, se han publicado los datosde un ensayo clínico aleatorizado diseñado paravalorar la acción de un inhibidor de la ET-1(bosentán) en los individuos con una EPOC gra-ve y una hipertensión pulmonar evaluadamediante ecocardiografía. El tratamiento nooriginó cambio alguno en la hemodinámica nien las funciones pulmonares. Por el contra-rio, sí se produjeron efectos perjudiciales en losenfermos tratados, tales como un aumento dela hipoxemia y del gradiente alvéolo-arterial deoxígeno y un deterioro de la calidad de vida(76).
En general, los pacientes con una EPOCy una hipertensión pulmonar grave no se con-sideran como candidatos a una cirugía dereducción de volumen pulmonar. Por otro lado,en al menos un estudio se ha puesto de mani-
M.A. NIETO BARBERO ET AL.
92
Monografía HAP NM 208 pag 18/2/10 17:08 Página 92

fiesto que los sujetos con una EPOC y unahipertensión pulmonar normalizan su hemo-dinámica pulmonar después de someterse aun trasplante pulmonar unilateral(77). En cual-quier caso, el trasplante pulmonar debe tener-se en cuenta como una posibilidad en cual-quier persona menor de 65 años de edad quepadece una EPOC y una hipertensión pulmo-nar grave, y que no tiene comorbilidad aso-ciada alguna.
HIPERTENSIÓN PULMONAR DEBIDA A LAASOCIACIÓN DE UNA FIBROSIS Y UNENFISEMA PULMONARES
La clasificación clínica del 4th World Sympo-sium on Pulmonary Hypertension ha incorpo-rado en el grupo 3 a las enfermedades pul-monares que tienen un patrón ventilatoriomixto (restrictivo y obstructivo). Entre ellas seincluyen las bronquiectasias, la fibrosis quísti-ca y una entidad recientemente descrita, quese caracteriza por la combinación de un enfi-sema localizado preferentemente en los lóbu-los superiores y una neumopatía intersticial,de afectación predominante en los inferiores.En lo que se refiere a este síndrome, en la seriemás extensa publicada hasta el momento, queprocede de un centro de referencia en enfer-medades pulmonares raras(78), se señala quela hipertensión pulmonar es muy prevalenteen estos pacientes y más grave que la que seobserva en cualquiera de las dos enfermeda-des por separado. El síndrome se caracterizatambién por cursar con unos volúmenes pul-monares bastantes conservados y con una dis-minución de la DLCO desproporcionada paralo que cabría esperar en atención a la altera-ción ventilatoria existente.
HIPERTENSIÓN PULMONAR DEBIDA A LOSTRASTORNOS RESPIRATORIOS DEL SUEÑO
El trastorno respiratorio del sueño más fre-cuente es el síndrome de apneas e hipopneasdel sueño (SAHS). Las consecuencias inme-diatas de las apneas del sueño son múltiplesy entre ellas se incluye el aumento de la pre-sión arterial pulmonar. Este aumento proba-
blemente no sólo se debe a la hipoxia alveo-lar, sino también a los grandes cambios de lapresión intratorácica que ocasionan las apne-as, que incrementan el retorno venoso y sobre-cargan el ventrículo izquierdo(79). El estímulohipóxico secundario a las apneas produciríauna vasoconstricción pulmonar y, posterior-mente, un remodelado vascular(80). Otros meca-nismos patogénicos invocados para explicarla hipertensión pulmonar que se observa enel SAHS son todos los que se sabe que elevanel tono del sistema vascular periférico, a saber,el sistema nervioso autónomo(81), los media-dores inflamatorios(82) y las especies reactivasde oxígeno resultantes del proceso de oxida-ción(83). También se ha observado la existen-cia de una reducción en la liberación del NO,que rápidamente se normaliza cuando se ins-taura el tratamiento con un equipo de presiónpositiva continua en la vía aérea (CPAP)(84). Porúltimo, como en otras formas secundarias dehipertensión pulmonar, también es posible queexista una susceptibilidad mediada genética-mente, capaz de favorecer el desarrollo de unahipertención pulmonar como respuesta a unahipoxemia crónica e intermitente.
Las normativas más recientes del Ameri-can College of Chest Physicians(85) recomiendanno investigar sistemáticamente la existenciade una hipertensión pulmonar en los enfer-mos que padecen un SAHS (grado de reco-mendación I), exceptuando una alta sospechaclínica sobre el mismo. Reconocen, sin embar-go, que las pruebas que permiten hacer estaafirmación son escasas, aunque se basan enla revisión de 12 artículos que analizaron laprevalencia de la hipertensión pulmonar en elSAHS(86). En diez de ellos el diagnóstico se esta-bleció con un cateterismo cardiaco y en losotros dos se estimó mediante un ecocardio-grama. En general, en estos estudios se obser-va que el valor medio de la PAPm era menorde 30 mmHg y en la mayoría menor de 25mmHg. Es decir, la hipertensión pulmonar delSAHS, cuando existe, es leve.
La prevalencia de la hipertensión pulmo-nar en el SAHS varía entre un 17 y un 53%.
GRUPO III. CLASIFICACIÓN DE DANA POINT: ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS
93
Monografía HAP NM 208 pag 18/2/10 17:08 Página 93

En realidad, si se tienen en cuenta los resul-tados de los trabajos que buscaron posiblesfactores acompañantes en los enfermos estu-diados, es muy posible que la hipertensión pul-monar de estos pacientes se deba no sólo alSAHS, sino también a la comorbilidad aso-ciada. El mayor índice de masa corporal y lasdesaturaciones de oxígeno más graves se rela-cionan mejor con la hipertensión pulmonarque con el índice de apneas e hipopneas(87,88).Del mismo modo, los individuos que tienenun SAHS y una hipertensión pulmonar tienenuna alteración funcional ventilatoria y del inter-cambio gaseoso mayor que la de los que notienen una hipertensión pulmonar. Y esto esasí en lo que se refiere a la FVC, al FEV1, a laPaO2 y a la PaCO2
(88-90). Cuando se valoró laexistencia de una hipertensión pulmonar enun grupo de 181 pacientes con un SAHS puro,sin comorbilidades acompañantes, se eviden-ció que la PAPm era de 15 mmHg y que sóloun 9% de los sujetos tenía una hipertensiónpulmonar (PAPm mayor de 20 mmHg)(86).
Otra comorbilidad que probablemente con-tribuye al desarrollo de la hipertensión pul-monar del SAHS es la enfermedad del ventrí-culo izquierdo(91). Por un lado, el SAHS es unfactor de riesgo conocido para la hipertensiónarterial sistémica(92) y esta última puede oca-sionar una disfunción diastólica del ventrícu-lo izquierdo. A su vez, el SAHS es un factor deriesgo independiente para el desarrollo dedicha insuficiencia(93). En efecto, los resultadosdel trabajo de Arias y cols.(94) apoyan la exis-tencia de una relación entre la hipertensiónpulmonar del SAHS y la disfunción diastólicadel ventrículo izquierdo. En su serie, nueve decada diez enfermos con un SAHS y una hiper-tensión arterial sin ningún otro factor de ries-go cardiovascular padecían una disfunción dias-tólica del ventrículo izquierdo. Sin embargo,esto sólo ocurrió en cuatro de los trece pacien-tes en los que el SAHS no se asociaba con unahipertensión pulmonar.
De acuerdo con todo lo anterior puede con-cluirse que la hipertensión pulmonar, aunquees frecuente en el SAHS, suele ser leve y pro-
bablemente dependiente, en su patogenia, decomorbilidades asociadas, como las altera-ciones diurnas de la función pulmonar y delintercambio gaseoso, y la disfunción diastóli-ca del ventrículo izquierdo(86). Si bien se dis-pone de pocos datos, los trabajos que han estu-diado estos aspectos muestran que la CPAPdisminuye la PAPm en comparación con laexistente al inicio del tratamiento(95-97).
BIBLIOGRAFÍA1. Naeije R. Pulmonary hypertensión and right
heart failure in chronic obstructive pulmonarydisease. Proc Am Thorac Soc. 2005; 2: 20-2.
2. Hopkins N, McLoughlin P. The structural basisof pulmonary hypertension in chronic lungdisease: remodelling, rarefaction or angioge-nesis? J Anat. 2002; 201: 335-48.
3. Hoeper MH, Barberà A, Channick RN, et al.Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension. J Am CollCardiol. 2009; 54: S85-S96.
4. Simonneau G, Robbins IM, Beghetti M, et al.Updated clinical classification of pulmonaryhypertension. J Am Coll Cardiol. 2009; 54: S43-S54.
5. Sánchez-Alarcos JMF, Nieto Barbero MA, Gar-cía de Castro AB. Enfermedades pulmonaresintersticiales, abordaje diagnóstico. MonogrNeumomadrid. 2008; 12: 25-38.
6. Gross T, Hunninghake GW. Ididopathic pul-monary fibrosis. N Engl J Med. 2001; 345: 517-25.
7. Ryu J, Colby T, Hartman TE.Idiopathic pul-monary fibrosis: current concepts. Mayo ClinProc. 1998; 73: 1085-101.
8. Thannickal VJ, Toews GB, White ES, et al.Mechanisms of pulmonary fibrosis. Annu RevMed. 2004; 55: 395-417.
9. Selman M, King TE, Pardo A. Idiopathic pul-monary fibrosis: prevailing and envolvinghypotheses about its pathogenesis and impli-cation for therapy. Ann Intern Med. 2001; 134:136-151.
10. Kamezaki F, Tasaki H, Yamashita K, et al. Genetransfer of extracellular superoxide dismuta-se ameliorates pulmonary hypertension in rats.Am J Respir Crit Care Med. 2008; 177: 219-26.
11. Dumitrascu R, Weissmann N, Ghofrani HA, etal. Activation of soluble guanylate cyclase rever-
M.A. NIETO BARBERO ET AL.
94
Monografía HAP NM 208 pag 18/2/10 17:08 Página 94

ses experimental pulmonary hypertension andvascular remodeling. Circulation. 2006; 113:286-95.
12. Evgenov OV, Pacher P, Schmidt PM, et al. NO-independent stimulators and activtaors of solu-ble guanylate cyclase: discovery and thera-peutic potential. Nat Rev Drug Discov. 2006;5: 755-68.
13. Abraham D, Ponticos M, Nagase H. Connecti-ve tissue remodelling: cross-talk between endo-thelins and matrix metalloproteinases. CurrVasc Pharmacol. 2005; 3: 369-79.
14. Jain R, Shaul W, Borok Z, et al. Endothelin-1induces alveolar epithelial-mesenchymal tran-sition through endothelin type A receptor-mediated production of TGF-β1. Am J RespirCell Mol Biol. 2007; 37: 38-47.
15. Fonseca C, Lindahl GE, Ponticos M, et al. Apolymorphism in the CTGF promoter regionassociated with systemic sclerosis. N Engl JMed. 2007; 357: 1210-20.
16. Baroni SS, Santillo M, Bevilacqua F, et al. Sti-mulatory autoantibodies to the PDGF recep-tor in systemic sclerosis. N Engl J Med. 2006;354: 2667-76.
17. Ryu JH, Krowka J, Pellikka PA, et al. Pulmonaryhypertension in patients with interstitial lungdiseases. Mayo Clin Proc. 2007; 82: 342-50.
18. Nadrous HF, Pellikka PA, Krowka MJ, et al. Theimpact of pulmonary hypertension on survi-val in patients with idiopathic pulmonary fibro-sis. Chest. 2005; 128 (Suppl. 6): 616S-7S.
19. Lettieri CJ, Nathan SD, Barnett SD, et al. Pre-valence and outcomes of pulmonary arterialhypertension in advanced idiopathic pulmo-nary fibrosis. Chest. 2006; 129: 746-52.
20. Shorr AF, Wainright JL, Cors CS, et al. Pulmo-nary hypertension in patients with pulmonaryfibrosis awaiting lung transplant. Eur Respir J.2007; 30: 715-21.
21. Chang B, Wigley FM, White B, et al. Sclero-derma patients with combined pulmonaryhypertension and interstitial lung disease. JRheumatol. 2003; 30: 2398-405.
22. Trad S, Amoura Z, Beiglman C, et al. Pulmo-nary arterial hypertension is a major morta-lity factor in diffuse systemic sclerosis, inde-pendent of interstitial lung disease. ArthritisRheum. 2006; 54: 184-191.
23. Nunes H, Humbert M, Capron F, et al. Pulmo-nary hypertension associated with sarcoido-sis: mechanisms, haemodynamics and prog-nosis. Thorax. 2006; 61: 68-74.
24. Handa T, Nagai S, Miki S, et al. Incidence ofpulmonary hypertension and its clinical rele-vance in patients with sarcoidosis. Chest. 2006;129: 1246-52.
25. Shorr AF, Helman DL, Davies DB, et al. Pul-monary hypertension in advanced sarcoido-sis: epidemiology and clinical characteristics.Eur Respir. J 2005; 25: 783-8.
26. Shorr AF, Davies DB, Nathan SD. Predictingmortality in patients with sarcoidosis awaitinglung transplantation. Chest. 2003; 124: 922-8.
27. Chaowalit N, Pellikka PA, Decker PA, et al.Echocardiographic and clinic characteristicsof pulmonary hypertension complicating pul-monary Langerhans cell histiocytosis. MayoClin Proc. 2004; 79: 1269-75.
28. Fartoukh M, Humbert M, Capron F, et al. Seve-re pulmonary hypertension in histiocytosisX. Am J Respir Crit Care Med. 2000;161: 216-23.
29. Ng CS, Wells AU, Padley SP. A CT sign of chro-nic pulmonary arterial hypertension: the ratioof main pulmonary artery to aortic diameter.J Thorac Imaging. 1999; 14: 270-8.
30. Zisman DA, Karlamangla AS, Ross DJ, et al.High-resolution CT findings do not predict thepresence of pulmonary hypertension in advan-ced idiopathic pulmonary fibrosis. Chest. 2007;132: 773-9.
31. Barst R, McGoon M, Torbicki A, et al. Diag-nosis and differential assessment of pulmo-nary arterial hypertension. J Am Coll Cardiol.2004; 43: 40-7.
32. Leuchte H, Baumgartner R, Elnounou M, et al.Brain natriuretic peptide is a prognostic para-meter in chronic lung disease. Am J Respir CritCare Med. 2006; 173: 744-50.
33. Behr J, Ryu JH. Pulmonary hypertension ininterstitial lung disease. Eur Respir J. 2008; 31:1357-67.
34. McGoon M, Gutterman D, Steen V, et al. Scre-ening, early detection, and diagnosis of pul-monary arterial hypertension: ACCP eviden-ce-based clinical practice guidelines. Chest.2004; 126: 14S-34S.
35. American Thoracic Society. Statement on sar-coidosis. Am J Respir Crit Care Med. 1999;160: 736-55.
36. American Thoracic Society (ATS) and the Euro-pean Respiratory Society (ERS). Idiopathic pul-monary fibrosis: diagnosis and treatment.International consensus statement. Am J Res-pir Crit Care Med. 2000; 161: 646-64.
GRUPO III. CLASIFICACIÓN DE DANA POINT: ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS
95
Monografía HAP NM 208 pag 18/2/10 17:08 Página 95

37. Vassallo R, Thomas CF. Advances in the treat-ment of rheumatic interstitial lung disease.Curr Opin Rheumatol. 2004; 16: 186-91.
38. Rodman D, Lindenfeld J. Successful treatmentof sarcoidosis-associated pulmonary hyper-tension with corticosteroids. Chest. 1990; 97:500-2.
39. Sánchez O, Sitbon O, Jais X, et al. Immuno-suppressive therapy in connective tissue dise-ases-associated pulmonary arterial hyperten-sion. Chest. 2006; 130: 182-9.
40. Collard HR, Anstrom KJ, Schwarz MI, et al. Sil-denafil improves walk distance in idiopathicpulmonary fibrosis. Chest. 2007; 131: 897-9.
41. King TE, Behr J, Brown KK, et al. BUILD-1: arandomized placebo-controlled trial of bosen-tan in idiopathic pulmonary fibrosis. Am J Res-pir Crit Care Med. 2008; 177: 75-81.
42. Foley RJ, Metersky ML. Successful treatmentof sarcoidosis-associated pulmonary hyper-tension with bosentan. Respiration. 2008; 75:211-4.
43. Galie N, Hoeper MM, Humbert M, et al. Gui-delines for the diagnosis and treatment of pul-monary hypertension. Eur Respir J. 2009; 30:2493-537.
44. Barberà JA, Escribano P, Morales P, et al. Están-dares asistenciales en hipertensión pulmonar.Arch Bronconeumol. 2008; 44: 87-99.
45. Orens JB, Estenne M, Arcasoy S, et al. Inter-national guidelines for the selection of lungtransplant candidates: 2006 update. A con-sensus report from the Pulmonary ScientificCouncil of the International Society for Heartand Lung Transplantation. J Heart Lung Trans-plant. 2006; 25: 745-55.
46. Arcasoy SM, Christie JD, Ferrari VA, et al. Echo-cardiographic assessment of pulmonary hyper-tension in patients with advanced lung disea-se. Am J Respir Crit Care Med. 2003; 167:735-40.
47. Scharf SM, Iqbal M, Keller C, et al. Hemody-namic characterization of patients with seve-re emphysema. Am J Respir Crit Care Med.2002; 166: 314-22.
48. Vizza CD, Lynch JP, Ochoa LL, et al. Right andleft ventricular dysfunction in patients withsevere pulmonary disease. Chest. 1998; 113:576-83.
49. Thabut G, Dauriat G, Stern JB, et al. Pulmonaryhemodynamics in advanced COPD candidatesfor lung volume reduction surgery or lung trans-plantation. Chest. 2005; 127: 1531-6.
50. Chaouat A, Bugnet AS, Kadaoui N, et al. Seve-re pulmonary hypertension and chronic obs-tructive pulmonary disease. Am J Respir CritCare Med. 2005; 172: 189-94.
51. Oswald-Mammosser M, Weitzenblum E, QuoixE, et al. Prognostic factors in COPD patientsreceiving long-term oxygen therapy. Impor-tance of pulmonary artery pressure. Chest.1995; 107: 1193-8.
52. Santos S, Peinado VI, Ramírez J, et al. Cha-racterization of pulmonary vascular remode-ling in smokers and patients with mild COPD.Eur Respir J. 2002; 19: 632-8.
53. MacNee W. Pathophysiology of cor pulmonalein chronic obstructive pulmonary disease. Partone. Am J Respir Crit Care Med. 1994; 150:833-52.
54. Peinado VI, Pizarro S, Barberà JA. Pulmonaryvascular involvement in COPD. Chest. 2008;134: 808-14.
55. Barberà JA, Peinado VI, Santos S,et al. Redu-ced expression of endothelial nitric oxidesynthase in pulmonary arteries of smokers.Am J Respir Crit Care Med. 2001; 164: 709-13.
56. Clini E, Cremona G, Campana M, et al. Pro-duction of endogenous nitric oxide in chronicobstructive pulmonary disease and patientswith cor pulmonale. Correlates with echo-dop-pler assessment. Am J Respir Crit Care Med.2000; 162: 446-50.
57. Nana-Sinkam SP, Lee JD, Sotto-Santiago S, etal. Prostacyclin prevents pulmonary endothe-lial cell apoptosis induced by cigarette smoke.Am J Respir Crit Care Med. 2007; 175: 676-85.
58. Giaid A, Yanagisawa M, Langleben D, et al.Expression of endothelin-1 in the lungs ofpatients with pulmonary hypertension. N EnglJ Med. 1993; 328: 1732-9.
59. Santos S, Peinado VI, Ramírez J, et al. Enhan-ced expression of vascular endothelial growthfactor in pulmonary arteries of smokers andpatients with moderate chronic obstructivepulmonary disease. Am J Respir Crit Care Med.2003; 167: 1250-6.
60. Peinado VI, Barberà JA, Abate P, et al. Inflam-matory reaction in pulmonary muscular arte-ries of patients with mild chronic obstructivepulmonary disease. Am J Respir Crit Care Med.1999; 159: 1605-11.
61. Joppa P, Petrasova D, Stancak B, et al. Syste-mic inflammation in patients with COPD and
M.A. NIETO BARBERO ET AL.
96
Monografía HAP NM 208 pag 18/2/10 17:08 Página 96

pulmonary hypertension. Chest. 2006; 130:326-33.
62. Weitzenblum E. Chronic cor pulmonale. Heart.2003; 89: 225-30.
63. Kessler R, Faller M, Weitzenblum E, et al. Natu-ral history of pulmonary hypertension in aseries of 131 patients with chronic obstructi-ve lung disease. Am J Respir Crit Care Med.2001; 164: 219-24.
64. Butler J, Schrijen F, Henríquez A, et al. Causeof the raised wedge pressure on exercise inchronic obstructive pulmonary disease. AmRev Respir Dis. 1988; 138: 350-4.
65. Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellu-lar and molecular mechanisms. Circ Res. 2006;99: 675-91.
66. Anand IS, Chandrashekhar Y, Ferrari R, et al.Pathogenesis of congestive state in chronic obs-tructive pulmonary disease. Studies of bodywater and sodium, renal function, hemodyna-mics, and plasma hormones during edema andafter recovery. Circulation. 1992; 86: 12-21.
67. Bishop JM, Cross KW. Use of other physiolo-gical variables to predict pulmonary arterialpressure in patients with chronic respiratorydisease. Multicentre study. Eur Heart J. 1981;2: 509-17.
68. Naeije R, Torbicki A. More on the noninvasi-ve diagnosis of pulmonary hypertension: dop-pler echocardiography revisited. Eur Respir J.1995; 8: 1445-9.
69. Vonk-Noordegraaf A, Marcus JT, Holverda S, etal. Early changes of cardiac structure and func-tion in COPD patients with mild hypoxemia.Chest. 2005; 127: 1898-903.
70. Chaouat A, Naeije E, Weitzenblum E. Pulmo-nary hypertension in COPD. Eur Respir J.2008; 32: 1371-85.
71. Nocturnal oxygen therapy trial group. Conti-nuous or nocturnal oxygen therapy in hypo-xemic chronic obstructive lung disease: a cli-nical trial. Ann Intern Med. 1980; 93: 391-8.
72. Medical Research Council working party. Longterm domiciliary oxygen therapy in chronichypoxic cor pulmonale complicating chronicbronchitis and emphysema. Report of theMedical Research Council working party. Lan-cet. 1981; 1: 681-6.
73. Timms RM, Khaja FU, Williams GW. Hemody-namic response to oxygen therapy in chronicobstructive pulmonary disease. Ann InternMed. 1985; 102: 29-36.
74. Zielinski J, Tobiasz M, Hawrylkiewicz I, et al.Effects of long-term oxygen therapy on pulmo-nary hemodynamics in COPD patients: a 6-yearprospective study. Chest. 1998; 113: 65-70.
75. Chaouat A, Weitzenblum E, Kessler R, et al. Arandomized trial of nocturnal oxygen therapyin chronic obstructive pulmonary diseasepatients. Eur Respir J. 1999; 14: 1002-8.
76. Stolz D, Rasch H, Linka A, et al. A randomi-sed, controlled trial of bosentan in severeCOPD. Eur. Respir J. 2008; 32: 619-28.
77. Bjortuft O, Simonsen S, Geiran OR, et al. Pul-monary haemodynamics after single-lung trans-plantation for end-stage pulmonary parenchy-mal disease. Eur Respir J. 1996; 9: 2007-11.
78. Cottin V, Nunes H, Brillet PY, et al. Combinedpulmonary fibrosis and emphysema: a distinctunderrecognised entity. Eur Respir J. 2205; 26:586-93.
79. Schafer H, Hasper E, Ewig S, et al. Pulmonaryhaemodynamics in obstructive sleep apnoea:time course and associated factors. Eur Res-pir J. 1998; 12: 679-84.
80. Presberg K, Dincer IIE. Pathophysiology of pul-monary hypertension due to lung disease. CurrOpin Pulm Med. 2003; 9: 131-8.
81. Imadojemu VA, Gleeson K, Gray KS, et al. Obs-tructive apnea during sleep is associated withperipheral vasoconstriction. Am J Respir CritCare Med. 2002; 165: 61-6.
82. Ohga E, Tomita T, Wada H, et al. Effects of obs-tructive sleep apnea on circulating ICAM-1, IL-8, and MCP-1. J Appl Physiol. 2003; 94: 179-84.
83. Dyugoskaya L, Lavie P, Lavie L. Increased adhe-sión molecules expresión and production ofreactive oxygen species in leukocytes of sle-ep apnea patients. Am J Respir Crit Care Med.2002; 165: 934-9.
84. Ip MS, Lam B, Chan LY, et al. Circulating nitricoxide is suppressed in obstructive sleep apneaand is reversed by nasal continuous positiveairway pressure. Am J Respir Crit Care Med.2000; 162: 2166-71.
85. Atwood CW, McCrory D, García JGN, et al. Pul-monary artery hypertension and sleep disor-dered breathing. Chest. 2004; 126: 72S-7S.
86. Giris RE, Mathai SC. Pulmonary hypertensionassociated with chronic respiratory disease.Clin Chest Med. 2007; 28: 219-32.
87. Bady E, Achkar A, Pascal S, et al. Pulmonaryarterial hypertension in patients with sleepapnoea syndrome. Thorax. 2000; 55: 934-9.
GRUPO III. CLASIFICACIÓN DE DANA POINT: ASPECTOS DIAGNÓSTICOS Y TERAPÉUTICOS
97
Monografía HAP NM 208 pag 18/2/10 17:08 Página 97

88. Chaouat A, Weitzenblum E, Krieger J, et al.Pulmonary hemodynamics in the obstructivesleep apnea syndrome. Results in 220 con-secutive patients. Chest. 1996; 109: 380-6.
89. Sajov D, Wang T, Saunders N, et al. Dayti-me pulmonary hemodynamics in patientswith obstructive sleep apnea without lungdisease. Am J Respir Crit Care Med. 1999;159: 1518-26.
90. Sanner BM, Doberauer C, Konermann M, etal. Pulmonary hypertension in patients withobstructive sleep apnea syndrome. Arch InternMed. 1997; 157: 2483-7.
91. Shamsuzzaman AS, Gersh BJ, Somers VK. Obs-tructive sleep apnea: implications for cardiacand vascular disease. JAMA. 2003; 290: 1906-14.
92. Peppard PE, Young T, Palta M, et al. Prospec-tive study of the association between sleep-disordered breathing and hypertension. N EnglJ Med. 2000; 342: 1378-84.
93. Arias MA, García Río F, Alonso Fernández A,et al. Obstructive sleep apnea syndrome affectsleft ventricular diastolic function: effects of
nasal continuous positive airway pressure inmen. Circulation. 2005; 112: 375-83.
94. Arias MA, García Río F, Alonso Fernández A, etal. Pulmonary hypertension in obstructive sle-ep apnoea: effects of continuous positive air-way pressure. A randomized controlled cross-over study. Eur Heart J. 2006; 27: 1106-13.
95. Alchanatis M, Tourkohoriti G, Kakouros S, etal. Daytime pulmonary hypertension inpatients with obstructive sleep apnea: theeffect of continuous positive airway pressureon pulmonary hemodynamics. Respiration.2001; 68: 566-72.
96. Sajkov D, Wang T, Saunders NA, et al. Conti-nuous positive airway pressure treatmentimproves pulmonary hemodynamics inpatients with obstructive sleep apnea. Am JRespir Crit Care Med. 2002; 165: 152-8.
97. Shivalkar B, Heyning CD, Kerremans M, et al.Obstructive sleep apnea syndrome moreinsights on structural and functional cardiacalterations, and the effects of treatment withcontinuous positive airway pressure. J Am CollCardiol. 2006; 47: 1433-9.
M.A. NIETO BARBERO ET AL.
98
Monografía HAP NM 208 pag 18/2/10 17:08 Página 98

99
RESUMENLa hipertensión pulmonar tromboembó-
lica crónica (HPTEC) es un trastorno comple-jo y prevalente, cuya constatación precisa deun cierto grado de sospecha clínica. Se tratade un proceso de remodelado vascular de losgrandes vasos no suficientemente aclarado,consecuencia de un embolismo pulmonar –sin-tomático o asintomático–, y de una arterio-patía pulmonar clásica, que produce un aumen-to de la resistencia vascular pulmonar (RVP),hipertensión pulmonar (HP) progresiva e insu-ficiencia cardiaca derecha.
Algunos pacientes pueden estar condicio-nados genéticamente para desarrollar laHPTEC, pero sólo se han descrito algunas muta-ciones del fibrinógeno, y un aumento de la fre-cuencia de polimorfismos de los antígenos leu-cocitarios humanos (HLA). Se han descritotambién diferentes factores clínicos predispo-nentes, que aumentan el riesgo de HPTEC,como la esplenectomía, las derivaciones ven-triculares, las enfermedades inflamatorias cró-nicas, la terapia tiroidea sustitutiva y el cáncer.Resulta trascendental, por lo tanto, reconoceréstos –y otros nuevos factores predisponen-tes– para facilitar un diagnóstico precoz y apli-car el tratamiento más efectivo posible en cen-tros experimentados –mediante la realizaciónde una tromboendarterectomía pulmonar enpacientes debidamente seleccionados– que,hoy por hoy, puede resultar potencialmentecurativa, al restablecer la hemodinámica pul-monar. Por otra parte, comienzan a surgir enlos pacientes inoperables –que representanhasta el 50% en algunas series–, ensayos clí-
nicos aleatorizados controlados con nuevosfármacos vasodilatadores pulmonares, paralos que se precisan nuevos estudios que acla-ren su eficacia futura y nivel de evidencia.
Palabras clave: Hipertensión pulmonartromboembólica crónica; Embolia pulmonar;Factores predisponentes; Tromboendarte-rectomía pulmonar; Vasodilatadores pulmo-nares.
INTRODUCCIÓN Y EPIDEMIOLOGÍALa hipertensión pulmonar tromboembó-
lica crónica (HPTEC) es una entidad produci-da por una oclusión de los vasos pulmonarespor coágulos sanguíneos organizados. LaHPTEC se encuadra en el grupo IV de la Cla-sificación de Dana Point, como una de las for-mas de hipertensión pulmonar más progresi-vamente prevalentes, y donde ya se abandonala clásica distinción previa de “formas proxi-males y distales”, refiriéndose desde ahora auna única categoría como HPTEC, ante laausencia de criterios bien establecidos en sudiferenciación(1).
La HPTEC es una complicación a largo pla-zo de la embolia pulmonar sintomática, conuna incidencia acumulada del 0,5-2%(2-5), sibien un trabajo prospectivo reciente en 223pacientes establece una prevalencia de hastaun 3,8% en los 2 años siguientes al episodio(6).Además, alrededor del 50% de los casos deHPTEC tienen su orígen en un tromboembo-lismo venoso asintomático.
En Estados Unidos se presentan entre 500a 2.500 nuevos casos al año, y se calcula quela prevalencia es de unos 3 casos de HPTEC
GRUPO IV. CLASIFICACIÓN DE DANA POINT
HIPERTENSIÓN PULMONARTROMBOEMBÓLICA CRÓNICA (HPTEC)
Javier Gaudó Navarro, Antonio Sueiro Bendito, David Jiménez Castro
Monografía HAP NM 208 pag 18/2/10 17:08 Página 99

de cada 100 casos de embolia pulmonar (unos20 por millón)(4,5). En España, se calcula quela tasa anual de pacientes con HPTEC podríaalcanzar la cifra de 1,4 casos por 100.000 habi-tantes al año.
El sexo masculino no representa general-mente un factor de riesgo para la HPTEC y, dehecho, en algunos países, como Japón, hay unpredominio del sexo femenino(7), y donde ade-más se constata una asociación a los HLAB5201 y DPB10202(8).
Por otra parte, no se ha identificado unabase genética de la HPTEC. A diferencia dela hipertensión arterial pulmonar (HAP) queafecta a vasos de menos de 300 µm, la HPTECafecta principalmente a vasos grandes y, poreste motivo, puede ser subsidiaria de un tra-tamiento quirúrgico en casos seleccionados(9,10).
PATOGENIALa patogenia de la HPTEC no está sufi-
cientemente aclarada, y no se conocen toda-vía de manera precisa los factores predispo-nentes para su aparición. Aunque la HPTEC seconsidera un trastorno secundario a trombo-embolismo venoso (TEV), no se dan los facto-res de riesgo plasmáticos tromboembólicostradicionalmente conocidos, si bien estudiosde trombofilia para TEV recurrente muestranun factor VIII elevado en el 39% de los pacien-tes con HPTEC(11), presencia de anticoagulan-tes lúpicos en un 10%, y hasta un 20% sonportadores de antifosfolípido, anticoagulantelúpico o ambos(12-14,16).
Las lesiones anatomopatológicas objetiva-das en la HPTEC se caracterizan por la pre-sencia de trombos organizados, fuertementeunidos a la capa media en las arterias elásticaspulmonares, que sustituyen a la íntima normal,conduciendo a una hipertrofia de la media y auna fibrosis de la íntima. Estos procesos pue-den ocluir completamente la luz, o bien pro-ducir diferentes grados de estenosis. Por otraparte, en la áreas no ocluídas, se puede desa-rrollar una arteriopatía pulmonar, indistingui-ble de la presente en la HAP (pudiendo existirtambién las típicas lesiones plexiformes)(2,3).
Clásicamente se ha considerado que laHPTEC es la consecuencia de uno o varios epi-sodios de TEP (“hipótesis embólica” como inter-pretación mecánica). La completa resolución delos episodios de TEP no es un hecho tan comúncomo se creía. Al realizar gammagrafías de per-fusión de control seriadas, hasta el 66% de lospacientes presentan defectos de perfusión resi-duales, varios meses después del evento inicial.De no resolverse completamente estos episo-dios previos, se produce la obstrucción del árbolvascular pulmonar; secundariamente aumen-tan las fuerzas de cizallamiento de los vasos noocluídos; se desarrolla una arteriopatía de lospequeños vasos de la circulación pulmonar noocluida –con una mayor incidencia si existentrastornos médicos asociados–(14,15) aumentanlas resistencias vasculares pulmonares y apa-rece la hipertensión pulmonar. Sin embargo,esta teoría no es capaz de explicar convincen-temente todos los casos de HPTEC.
Como en más de la mitad de los pacientescon HPTEC no hay antecedentes de eventostrombóticos venosos, se han desarrollado otrashipótesis alternativas –como la “teoría trom-bótica”–, para explicar parcialmente la patoge-nia de esta entidad. Dicha teoría postula que enestos pacientes existiría una arteriopatía pri-maria, alteraciones plaquetarias y una disfun-ción endotelial con remodelado vascular –conliberación de mediadores inflamatorios y vas-culotrópicos–, que darían lugar a una trombo-sis local, responsable en última instancia de lahipertensión pulmonar. Este hecho guardaríarelación sobre todo en pacientes con coexis-tencia de infecciones, fenómenos inmunitarios,tratamiento sustitutivo tiroideo y neoplasias(16,17).Sin embargo, esta hipótesis tampoco explicaríaplenamente la mayoría de los pacientes conHPTEC. Por lo tanto, se considera actualmen-te que, en la mayoría de los casos, coexistenambos procesos, y que la trombosis in situ jue-ga un papel facilitador de la progresión, másque propiamente iniciador de la enfermedad.
Por otra parte, no hay mutaciones genéti-cas específicas relacionadas con el desarrollode HPTEC.
J. GAUDÓ NAVARRO ET AL.
100
Monografía HAP NM 208 pag 18/2/10 17:08 Página 100

FACTORES PREDISPONENTES
Algunos pacientes pueden estar condicio-nados genéticamente para desarrollar laHPTEC, pero sólo se han descrito algunas muta-ciones del fibrinógeno y un aumento de la fre-cuencia de polimorfismos de los antígenos leu-cocitarios humanos (HLA)(8). En los pacientescon HPTEC, los defectos hereditarios de la coa-gulación no son más prevalentes que en loscontroles, con la excepción del factor VIII y delos anticuerpos antifosfolípido, como ya se hacomentado previamente en la patogenia de laenfermedad. Algunos estudios han analizadodiferencias en la expresión del inhibidor delactivador tisular del plasminógeno tipo 1 enel trombo de los pacientes con HPTEC (com-parado con los trombos de la tromboembo-lia pulmonar [TEP] aguda), y sugieren que latrombosis in situ puede contribuir a la persis-tencia del coágulo y a la progresión de la enfer-medad. Finalmente, no hay anomalías de lafibrinólisis identificadas en este proceso(12,16).
Algunas características del TEP agudo sehan asociado a su progresión a HPTEC, entrela que figurarían: el TEP idiopático, los defec-tos de perfusión grandes, el TEP masivo, elTEP recurrente, o la hipertensión pulmonarpersistente tras 5 semanas del episodio trom-bótico. Se han descrito, asimismo, diferentesfactores clínicos que aumentan el riesgo deHPTEC –asociándose a una mayor inciden-cia y a un peor pronóstico–, como la esple-nectomía, las derivaciones ventriculares, lasenfermedades inflamatorias crónicas y la oste-omielitis crónica(17) (Tabla 1).
En una base de datos retrospectiva recien-te, en la que participaron tres centros euro-peos de referencia en tromboendarterectomíapulmonar –entre 1996 y 2007–, se compara-ron los datos de 687 pacientes con HPTEC enel momento del diagnóstico, versus los pacien-tes evaluados con hipertensión arterial pul-monar precapilar no tromboembólica dedichos centros. Se observó una asociación másfrecuente con la HPTEC en los casos de deri-vaciones ventriculoauriculares y de marcapa-sos infectados –odds ratio (OR) (intervalo de
confianza [IC] del 95%), 76,40 (7,67-10.351);p < 0,001–, esplenectomía (OR = 17,87[1,56-2.438]; p = 0,017), TEV previo (OR =4,52 [2,35-9,12]; p < 0,001), TEV recurrente(OR = 14,49 [5,40-43,08]; p < 0,001), gru-pos sanguíneos distintos de 0 (OR = 2,09[1,12-3,94]; p = 0,019) y anticuerpos anti-coagulantes/antifosfolípidos del lupus (OR =4,20 [1,56-12,21]; p = 0,004). Por otra par-
GRUPO IV. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN PULMONAR TROMBOEMBÓLICA CRÓNICA (HPTEC)
101
TABLA 1. Factores predisponentespara hipertensión pulmonartromboembólica crónica (HPTEC)(19)
Factores genéticos
• Polimorfismos del fibrinógeno
• Polimorfismos de HLA7
Marcadores biológicos
• Factor VIII
• Factor de von Willebrand
• Anticuerpos antifosfolípido/anticoagulante
lúpico
• Grupo sanguíneo distinto al 0
• Lipoproteína A
• Alteraciones estructurales de la fibrina
• Dimetilarginina asimétrica
• BNP
Factores clínicos
• Edad > 70 años
• Edad joven
• Sexo: mujer
• PAP > 50 mmHg
• TEP masiva
• TEP submasiva
• TEP recurrente
• TEP idiopática
• Defectos de perfusión grandes
• Defectos de perfusión persistentes
• Esplenectomía
• Derivaciones ventrículo peritoneales
• Enfermedades inflamatorias crónicas
• Tratamiento tiroideo sustitutivo
• Neoplasia
Monografía HAP NM 208 pag 18/2/10 17:08 Página 101

te, surgieron como nuevos factores de riesgopara la HPTEC el tratamiento sustitutivo tiroi-deo (OR = 6,10 [2,73-15,05]; p < 0,001) ylos antecedentes de neoplasias (OR = 3,76;p = 0,005). Se ha postulado que el trata-miento con levotiroxina parece aumentar lasconcentraciones de factor de von Willebrandy acortaría la formación del trombo plaque-tario in vitro –medida con el tiempo de oclu-sión inducida por colágeno-adrenalina–, conlo que explicaría posiblemente el aumento detrombogenicidad(18) (Tabla 1).
No obstante, también se ha publicadorecientemente un registro multicéntrico pros-pectivo de casos incidentes, que no ha confir-mado la influencia pronóstica negativa de laesplenectomía aislada en los resultados, inde-pendientemente de la operabilidad del pacien-te, si bien el número de pacientes con ante-cedentes de esplenectomía era tan sólo de un6,7%, y no se incluyeron otros trastornos aso-ciados en el análisis. Las supervivencias a 1y 3 años tras del diagnóstico fueron del 82 yel 70% para los pacientes con una enferme-dad no quirúrgica, y del 88 y el 76% para lospacientes tratados quirúrgicamente (p =0,023). Se objetivó una mejoría funcional ini-cial en los pacientes con una enfermedad noquirúrgica, pero este efecto no persistió a los2 años. Sin embargo, sí se observaron mejo-rías funcionales y hemodinámicas significati-vas en los pacientes tratados quirúrgicamen-te, con un aumento de la distancia recorridaen el test de 6 minutos de 105 m (p < 0,001)a los 3 meses(19,20).
En la actualidad se está llevando a cabo unregistro multicéntrico prospectivo europeo deHPTEC creado en 2007, que permitirá defi-nir la verdadera incidencia, el adecuado diag-nóstico, nuevos factores asociados, los trata-mientos más eficaces y la evolución pronósticade los pacientes con HPTEC(21).
DIAGNÓSTICOCualquier paciente con HP de causa no acla-
rada debe ser evaluado para descartar la pre-sencia de HPTEC, con la máxima sospecha clí-
nica si ha existido un proceso embólico previoasociado a datos ecocardiográficos de disfun-ción ventricular derecha(22). Los síntomas deHPTEC son intermitentes, y se producen cuan-do resulta afectado más del 50-60% del terri-torio vascular pulmonar. La disnea y la intole-rancia al ejercicio son síntomas frecuentes, juntoal dolor torácico y al síncope. En general, el cur-so evolutivo se considera que es más benignoque el de la hipertensión arterial pulmonar(23).
La espirometría en los pacientes conHPTEC sin antecedentes de neumopatía cró-nica no resulta relevante, pudiendo mostrar unpatrón restrictivo leve hasta en el 20% de loscasos.
Gasométricamente, la diferencia alvéolo-arterial de oxígeno puede aumentar en unafase temprana de la enfermedad para poste-riormente, disminuir la presión parcial de oxí-geno y aumentar la presión parcial de dióxidode carbono, especialmente en relación conel esfuerzo(24).
Técnicas de imagen radiológicasLa gammagrafía de ventilación/perfusión
pulmonar (V/Q) ha de realizarse siempre, yaque la muestra de un defecto segmentarioimportante en ella conduce al diagnóstico deHPTEC, y su normalidad la excluye, con unasensibilidad del 90-100% y una especificidaddel 94-100%. En los casos en los que la gam-magrafía resulte indeterminada, o bien reveledefectos de perfusión, se hace también pre-ceptiva la realización de una tomografía com-puterizada helicoidal/angio TAC, que puedevalorar adicionalmente el estado del parén-quima pulmonar. No hay suficiente eviden-cia todavía para sugerir que una tomografíacomputarizada helicoidal/angioTAC normalexcluya la presencia de HPTEC operable, y hoyen día su realización permite prácticamenteexcluir la realización de una angiografía pul-monar tradicional(25).
Las bandas, redes y recesos vasculares,con incrementos y cambios bruscos del tama-ño vascular junto a las oclusiones vascula-res, son los signos angiográficos característi-
J. GAUDÓ NAVARRO ET AL.
102
Monografía HAP NM 208 pag 18/2/10 17:08 Página 102

cos que comúnmente se hallan, además devasos centrales dilatados, con un adelgaza-miento irregular de la circulación periférica yde la oclusión vascular segmentaria comple-ta mencionada(26,27).
Por otra parte puede estar además tambiénindicada la realización de una angiografía coro-naria en aquellos candidatos a una trombo-endarterectomía pulmonar y que, además, pre-senten factores de riesgo para enfermedadarterial coronaria.
Ecocardiografía y cateterismo cardiacoLa ecocardiografía transtorácica (ETT) es
una herramienta diagnóstica clave en la detec-ción de HPTEC, y debe realizarse tras 3 a 6meses del episodio agudo, para confirmar odescartar la presencia de HP no resuelta, quepueda estar ya presente desde las fases ini-ciales(28,29).
Como técnica diagnóstica gold standard,siempre ha de realizarse un cateterismo car-diaco antes de realizar cualquier intervenciónterapéutica –incluyendo la intervención qui-rúrgica–, y ha de procurarse que sea practica-do en el mismo centro de referencia –junto alas técnicas de imagen radiológicas mencio-nadas previamente–, por el elevado riesgo quela repetición de estas pruebas puede entrañaren este tipo de pacientes(30-32).
TRATAMIENTOLa HPTEC tiene un mal pronóstico, y más
de la mitad de los pacientes con una PAPmedia (PAPm) superior a 50 mmHg presentansupervivencias inferiores a 1 año tras el diag-nóstico, alcanzando una mortalidad del 90%a los 5 años. La insuficiencia cardiaca derechay la muerte son consecuencias de la HPTEC silos pacientes no reciben tratamiento(33). Pare-ce que la hemodinámica pulmonar y la PVRson factores predictivos pronósticos cruciales,ya que su reducción significativa tras la ciru-gía se asocia a un aumento de la superviven-cia, y los valores preoperatorios elevados aso-cian un riesgo significativo de morbimortalidadquirúrgica(34-36).
La tromboendarterectomía pulmonar esuna alternativa terapéutica curativa, con unamortalidad perioperatoria que oscila entreun 5-24%, proporcionando una excelentecalidad de vida y, en los casos favorables,una normalización de la capacidad de ejer-cicio –y, frecuentemente, también de losparámetros hemodinámicos en reposo–(37-
39). El riesgo de esta técnica puede ser mini-mizado, siempre que se cumplan unas indi-caciones estrictas, entre las que se incluyen(Fig. 1):1. Presencia de hipertensión pulmonar sin-
tomática confirmada hemodinámica-mente, con presión arterial pulmonarmedia (PAPm), con un valor superior a 25mmHg.
2. Reevaluación diagnóstica tras al menos 3meses de anticoagulación oral efectiva.
3. Signos indicativos de la presencia detrombos accesibles quirúrgicamentesegún la angiografía pulmonar y/o angio-TAC de última generación, u oclusión uni-lateral completa de la arteria pulmonarprincipal.
4. Una relación de la resistencia vascular pul-monar (< 1.200 din x cm x s-5) y una masaesperada del trombo que predigan unareducción > 50% de la resistencia vas-cular pulmonar (RVP) postquirúrgica.
5. Perfil de riesgo preoperatorio favorable, quepermita la operación:• RVP < 1.000 din x cm x s-5.• Ausencia de comorbilidades y trastor-
nos médicos asociados graves.• Suficiente parénquima pulmonar fun-
cional.• Disminución de la PAPm de al menos
un 10% tras la administración de NOinhalado durante un cateterismo car-diaco derecho diagnóstico(29).
• Perfil favorable de biomarcadores (pro-teína transportadora de ácidos grasoslibres de tipo cardiaco < 3 ng/ml, dime-tilarginina asimétrica < 0,6 mol/L).
• Cociente de escotadura < 1 (interva-lo desde el inicio del flujo sistólico arte-
GRUPO IV. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN PULMONAR TROMBOEMBÓLICA CRÓNICA (HPTEC)
103
Monografía HAP NM 208 pag 18/2/10 17:08 Página 103

rial pulmonar hasta la desaceleraciónmáxima del flujo sistólico [t1] dividi-do por el intervalo desde la desacele-ración máxima del flujo sistólico has-ta el final del flujo sistólico arterialpulmonar [t2]).
Técnicas quirúrgicas y resultadosAlgunos autores defienden una “clasifica-
ción quirúrgica” de la HPTEC, que podría tenercierta significación pronóstica(40), en funciónde 4 tipos establecidos: tipo I, presencia de untrombo central (mortalidad quirúrgica, 2,1%);tipo II, engrosamiento de la íntima, red fibro-sa y bandas dentro de las arterias lobulares(mortalidad quirúrgica, 5,3%); tipo III, oclu-siones en las ramas segmentarias y subseg-mentarias (mortalidad quirúrgica, 5%), o tipo
IV, trombos muy distales (mortalidad quirúr-gica, 25%).
La función cardiopulmonar en pacientescon HPTEC puede normalizarse mediante unatromboendarterectomía, como se ha mencio-nado. La intervención consiste en la reseccióndel tejido obstructivo fibroso organizado adhe-rido a las arterias pulmonares, bajo circulaciónextracorpórea y períodos de parada circulato-ria en hipotermia profunda. Tras la incisión enla arteria pulmonar intrapericárdica proximal,se establece el plano correcto para la trombo-endarterectomía y se continúa circularmente,hasta las ramas arteriales lobulares, segmen-tarias y subsegmentarias de cada lóbulo. Lamorbilidad perioperatoria se relaciona princi-palmente con el edema de reperfusión, la infec-ción y la hemorragia(41-43).
J. GAUDÓ NAVARRO ET AL.
104
FIGURA 1. Algoritmo de actuación terapéutica en la HPTEC(23,24).
Considerar trasplante pulmonar Tromboendarterectomía Considerar terapia médica
Sintomatología persistente
No Sí Sí No
Comorbilidades severaspre-cirugía
Comorbilidades severaspre-cirugía
Obliteración quirúrgicamente accesiblede las arterias pulmonares centrales.Reducción estimada postquirúrgica
de la RVP > 50%
Afectación extensa de pequeños vasos.RVP elevada desproporcionadamente.
Reducción estimada postquirúrgica de la RVP < 50%
Anticoagulatción
Diagnóstico de HPTEC con limitación funcionaly/o signos de insuficiencia cardiaca derecha
Monografía HAP NM 208 pag 18/2/10 17:08 Página 104

Los datos hemodinámicos pre y postqui-rúrgicos definen en buena medida el pro-nóstico de la técnica quirúrgica. Una RVP pre-operatoria superior a 1.000 din x cm x s-5
aumenta significativamente el riesgo opera-torio. Además, una RVP postoperatoria >500 din x cm x s-5 tiene valor predictivo paraidentificar mal pronóstico a largo plazo(41-43).
Finalmente, como establecen las guíasrecientemente, un centro experto en trom-boendarterectomía se considera aquel que rea-liza al menos 20 intervenciones al año, acom-pañado de una tasa de mortalidad asumiblede en torno a un 10%(44). El trasplante pul-monar bilateral es una opción a considerar enlos casos severos contraindicados a la realiza-ción de la tromboendarterectomía, y será abor-dado en otro capítulo.
Tratamiento médicoDe acuerdo con las publicaciones de los
centros expertos en tromboendarterectomía,una proporción significativa de los pacientesremitidos no resultan idóneos para dicha téc-nica –pueden resultar inoperables hasta un50% de los mismos–, o bien muestran una fal-ta de mejoría o recurrencia de la hipertensiónpulmonar tras la cirugía –entre un 15 a un 25%de los pacientes–(45). La enfermedad de peque-ños vasos concomitante en la HPTEC es unfactor predictivo pronóstico, e hipotéticamen-te constituye una diana terapéutica para utili-zar el mismo tratamiento farmacológico queel empleado en la hipertensión arterial pul-monar idiopática (HAPI) ya que, además,ambas comparten patogenia y cambios vas-culares similares.
Con los escasos datos disponibles actual-mente, podría valorarse un tratamiento médi-co individualizado de la HPTEC –se recomiendadentro de ensayos clínicos, cuando ello resul-te posible– con prostanoides, antagonistas delos receptores de endotelina o inhibidores dela fosfodiesterasa-5, en las siguientes situa-ciones debidamente seleccionadas –dada lafalta de pruebas sólidas para apoyar el uso deestos fármacos–:
• Enfermedad tromboembólica crónica dis-tal inoperable.
• Comorbilidades de alto riesgo para la ciru-gía.
• Como puente para la tromboendarterec-tomía o al trasplante pulmonar, en pacien-tes de elevado riesgo.
• Hipertensión pulmonar residual o persis-tente tras la tromboendarterectomía.
Los resultados de los ensayos clínicos uti-lizando terapias farmacológicas para la HPTECen pacientes inoperables no resultan conclu-yentes hasta el momento. Todavía no exis-ten medicamentos aprobados para esta indi-cación como se ha hecho mención, ya queexiste la necesidad de nuevos estudios que per-mitan aclarar su eficacia. No obstante, resultareseñable resaltar cómo las tasas de supervi-vencia de los pacientes que recibieron trata-mientos médicos –con o sin cirugía– parecehaber mejorado en los últimos años, como harevelado un estudio de 469 pacientes conHPTEC, de los cuales 148 (32%) tenían unaafectación distal no quirúrgica.
Las tasas de supervivencia a los 1 y 3 añosdesde el diagnóstico fueron del 82 y 70% parael grupo no quirúrgico y del 88 y 76% para lospacientes tratados quirúrgicamente, respectiva-mente. La mayoría de los pacientes no quirúr-gicos en este estudio recibieron tratamientos far-macológicos para su HP fundamentalmente conprostanoides hasta el año 2004 y, posterior-mente, con antagonistas de los receptores deendotelina e inhibidores de la fosfodiesterasa-5,tras la aparición de los mismos. Además, el estu-dio también mostraba tendencias cambiantesen relación con la aplicación farmacológica pre-quirúrgica de estas terapias: antes de 2004, el29% de los pacientes recibieron estos fármacos,frente al 65% a partir de 2004(45).
Hasta la fecha se han publicado 3 estudiosrandomizados y aleatorizados en HPTEC(Tablas 2 y 3), utilizando iloprost inhalado (estu-dio AIR, Aerosolised Iloprost Randomised(46), sil-denafilo(47) y bosentan. Este último ya habíasido empleado previamente en estudios no
GRUPO IV. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN PULMONAR TROMBOEMBÓLICA CRÓNICA (HPTEC)
105
Monografía HAP NM 208 pag 18/2/10 17:08 Página 105

controlados en pacientes con HPTEC, mejo-rando la capacidad de ejercicio, la hemodi-námica y su papel en modelos animales deHPTEC, dado el reconocido protagonismo delremodelado vascular en este proceso media-do por la endotelina(48-50).
El estudio BENEFIT (Bosentan Effects iniNopErable Forms of chronIc Thromboembolic
pulmonary hipertension(51) ha contado con unapotencia estadística suficiente para detectaruna diferencia significativa entre los pacientestratados y el grupo control. Incluyó a 157pacientes con HPTEC inoperable o hiperten-sión pulmonar persistente/recurrente tras másde 6 meses después de una tromboendarte-rectomía, distribuyéndose aleatoriamente en
J. GAUDÓ NAVARRO ET AL.
106
TABLA 2. Principales estudios controlados y aleatorizados que incluyen apacientes con HPTEC
Autor Estudio Nº pacientes Fármaco Comparación Duración Etiologías
Olschewski AIR 203 Iloprost Placebo 12 Semanas HPTEC (n 57)N Engl J Med. HAPI (n 51)2002; 347:322-329
Suntharalingam 19 Sildenafilo Placebo 12 Semanas HPTECChest. 2008; 134: 229-36
JaïsJ Am Coll BENEFIT 157 Bosentan Placebo 16 Semanas HPTECCardiol. 2008; 52: 2127-34
TABLA 3. Resultados principales estudios controlados y aleatorizados queincluyen a pacientes con HPTEC
HemodinámicaAutor RVP T6M CF (NYHA/OMS) Otros
Olschewski Reducción Mejoría NS Mejoría Iloprost inhaladoN Engl J Med. (p =0,007) (p=0,007) es un tto. efectivo2002; 347: para pacientes con322 -9 HP severa
Suntharalingam Reducción Sin cambios Mejoría Estudio pilotoChest. 2008; 134: (p=0,044) (p=0,025) Efectos beneficiosos229-36 del sildenafilo
JaïsJ Am Coll Cardiol. Reducción Sin cambios Previene deterioro Mejora la calidad 2008; 52: (p<0.0001) clínico de vida y previene2127-34 el deterioro clínico
Monografía HAP NM 208 pag 18/2/10 17:08 Página 106

77 pacientes con medicación (dosis de 62,5mg cada 12 horas, aumentando a 125 mg cada12 horas tras 4 semanas) versus placebo (n:80), con los siguientes criterios de inclusión:edad: 18 - 80 años; clase funcional (CF) II-IVOMS (Organización Mundial de la Salud); testde 6 minutos marcha (T6M) < 450 metros;PAP media >25 mmHg, PCP < 15 mmHg yresistencia vascular pulmonar (RVP) >300dyn/sec/cm–5.
Las variables de valoración coprincipalesindependientes fueron el cambio obtenidoen la RVP (expresado en porcentaje respec-to al valor basal) y el cambio de la distan-cia recorrida en 6 minutos respecto a la situa-ción basal, tras 16 semanas de tratamiento.Las variables de valoración secundarias fue-ron el cambio de la clase funcional de laOMS, y de otros parámetros hemodinámicosrespecto a la situación basal [resistencia pul-monar total (RPT), índice cardiaco medio (IC),presión media de aurícula derecha (PAD),saturación de oxígeno venosa mixta (SVO2)],y tiempo hasta deterioro clínico (TDC). Sedemostró un efecto estadísticamente signi-ficativo del tratamiento con bosentan res-pecto a placebo, en cuanto a la RVP, con unadisminución del 24,1% respecto al valorbasal (intervalo de confianza del 95%, -31,5a -16; p < 0,0001). La resistencia pulmonartotal RPT (efecto del tratamiento, -193 din xcm x s-5; intervalo de confianza del 95%, -283 a -104; p < 0,0001) y el índice cardia-co IC (efecto del tratamiento, 0,3 l x min-1
x m-2; Intervalo de confianza del 95%, 0,14-0,46; p = 0,0007), mostraron también mejo-rías. El efecto medio del tratamiento en eltest de 6 min marcha fue de tan sólo +2,2m (intervalo de confianza del 95%, -22,5 a26,8; p = 0,5449). Por otra parte, el bosen-tan también mejoró la PAD y la SVO2, redu-jo significativamente el pro-BNP, menornúmero de pacientes deterioraron su CF, yuna mayor proporción mejoraron tanto suíndice de Borg (44,7 vs. 31,7%) como el TDC(variable combinada que incluía: muerte, hos-pitalización, interrupción del tratamiento o
inicio de epoprostenol i.v.). Con todo ello, eltratamiento con bosentan fue bien tolerado.La tasa de eventos adversos (EA) de bosen-tan vs. placebo fue del 67,5 y 52,5%, res-pectivamente, presentándose principalmen-te edemas periféricos (13%) y alteracioneshepáticas (7,8%). Un 9,1% de los pacientesque recibio bosentan (12,5% placebo) tuvoEA graves, mientras que el 2,6% debió inte-rrumpirlo (5% placebo). Por lo tanto, esteestudio demuestra un efecto positivo del fár-maco sobre la hemodinámica en esta pobla-ción de pacientes, no así en la capacidad deejercicio. Para valorar sus posibles efectosa largo plazo y, en particular, la tolerancia alesfuerzo, se está llevando a cabo en la actua-lidad una extensión abierta del estudio, conun período de seguimiento de 2 años.
En el plano de los biomarcadores, elaumento de los niveles plasmáticos de NT-proBNP durante la evolución de estos pacien-tes con HPTEC tratados farmacológicamentese ha relacionado con un peor pronóstico. Ade-más, se ha observado también que elevacio-nes de troponina T cardiaca constituye un pre-dictor –órgano independiente– de desenlacefatal durante el seguimiento de 51 pacientescon HAP, y cinco con CTEPH(44).
Recordar, por último, que los pacientes conHPTEC deben recibir anticoagulación indefi-nida con antagonistas de la vitamina K, ajus-tados a un INR entre 2,0 y 3,0(44).
CONCLUSIONESLa HPTEC representa un trastorno vas-
cular pulmonar «dual», con un remodeladovascular consecuente con la organizacióndel trombo en los vasos principales, –quepuede abordarse curativamente mediante larealización de una tromboendarterectomía–junto a una arteriopatía pulmonar de peque-ños vasos –que puede constituir la diana delos nuevos tratamientos vasodilatadores–.Sin embargo, serán necesarios más ensayosque definan el papel y pronóstico de estosnuevos fármacos en esta enfermedad(44,52)
(Tabla 4).
GRUPO IV. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN PULMONAR TROMBOEMBÓLICA CRÓNICA (HPTEC)
107
Monografía HAP NM 208 pag 18/2/10 17:08 Página 107
BIBLIOGRAFÍA1. McLaughlin VV, Archer SL, Badesch DB, et al.
ACCF/AHA 2009 Expert Consensus Documenton Pulmonary Hypertension: A Report of theAmerican College of Cardiology FoundationTask Force on Expert Consensus Documentsand the American Heart Association Develo-ped in Collaboration With the American Colle-ge of Chest Physicians; American ThoracicSociety, Inc.; and the Pulmonary Hyperten-sion Association. J Am Coll Cardiol. 2009; 53:1573-619.
2. Dartevelle P, Fadel E, Mussot S, et al. Chro-nic thromboembolic pulmonary hypertension.Eur Respir J. 2004; 23: 637-48.
3. Fedullo PF, Auger WR, Kerr KM, et al. Chronicthromboembolic pulmonary hypertension.N Engl J Med. 2001; 345: 1465-72.
4. Lang IM. Chronic thromboembolic pulmonaryhypertension -not so rare after all. N Engl JMed. 2004; 350: 2236-8.
5. Becattini C, Agnelli G, Pesavento R, et al. Inci-dence of chronic thromboembolic pulmonaryhypertension after a first episode of pulmo-nary embolism. Chest. 2006; 130: 172-5.
6. Pengo V, Lensing AW, Prins MH, et al. Incidenceof chronic thromboembolic pulmonary hyper-tension after pulmonary embolism. N Engl JMed. 2004; 350: 2257-64.
7. Shigeta A, Tanabe N, Shimizu H, et al. GenderDifferences in Chronic Thromboembolic Pul-monary Hypertension in Japan. Circ J. 2008;72: 2069-74.
8. Tanabe N, Kimura A, Amano S, et al. Asso-ciation of clinical features with HLA in chro-nic pulmonary thromboembolism. Eur RespirJ. 2005; 25: 131-8.
J. GAUDÓ NAVARRO ET AL.
108
TABLA 4. Grado de recomendaciones ERS/ESC para la HPTEC(44)
Clase de Nivel derecomendación evidencia
El diagnóstico de HPTEC se basa en la presencia de HP I Cprecapilar (PAP media ≥ 25 mmHg, PCP ≤ 15 mmHg, PVR > 2 unidades Wood) en pacientes con múltiples trombos/ émbolos oclusivos crónicos organizados en las arterias pulmonares elásticas (principal, lobar, segmentaria, subsegmentarios)
En los pacientes con HPTEC está indicada la I Canticoagulación permanente (INR 2-3)
La tromboendarterectomía pulmonar es el tratamiento I Crecomendado para pacientes con HPTEC
Una vez que la gammagrafía de perfusión y/o angioTAC IIa Cmuestran signos compatibles con HPTEC, el paciente debe ser remitido a un centro con experiencia en trombo-endarterectomía pulmonar quirúrgica
La selección de los pacientes para la cirugía debe basarse IIa Cen la extensión y localización de la trombos organizados, en el grado de HP confirmado hemodinámicamente, y en la presencia de comorbilidades
La terapia dirigida con medicamentos específicos para la HAP IIb Cpuede estar indicada en pacientes con HTPEC seleccionados, tales como no candidatos para la cirugía, o bien en pacientes con HP residual tras la realización de una tromboendarterectomía pulmonar
Monografía HAP NM 208 pag 18/2/10 17:08 Página 108

9. Tapson VF. Acute Pulmonary Embolism. N EnglJ Med. 2008; 358: 1037-52.
10. Moser KM, Auger WR, Fedullo PF. Chronicmajor-vessel thromboembolic pulmonaryhypertension. Circulation. 1990; 81: 1735-43.
11. Bonderman D, Turecek PL, Jakowitsch J, et al.High prevalence of elevated clotting factor VIIIin chronic thromboembolic pulmonary hyper-tension. Thromb Haemost. 2003; 90: 372-6.
12. Suntharalingam J, Goldsmith K, Van Marion V,et al. Fibrinogen A{alpha} Thr312Ala poly-morphism is associated with chronic throm-boembolic pulmonary hypertension. Eur Res-pir J. 2008; 31: 736-41.
13. Skoro-Sajer N, Mittermayer F, Panzenboeck A,et al. Asymmetric dimethylarginine is incre-ased in chronic thromboembolic pulmonaryhypertension. Am J Respir Crit Care Med. 2007;176: 1154-60.
14. Lankeit M, Dellas C, Panzenbock A, et al. Heart-type fatty acid-binding protein for risk assess-ment of chronic thromboembolic pulmonaryhypertension. Eur Respir J. 2008; 31: 1024-9.
15. Lewczuk J, Piszko P, Jagas J, et al. Prognosticfactors in medically treated patients with chro-nic pulmonary embolism. Chest. 2001; 119:818-23.
16. Wolf M, Boyer-Neumann C, Parent F,et al.Thrombotic risk factors in pulmonary hyper-tension. Eur Respir J 2000;15:395-9.
17. Bonderman D, Jakowitsch J, Adlbrecht C, et al.Medical conditions increasing the risk of chro-nic thromboembolic pulmonary hypertension.Thromb Haemost. 2005; 93: 512-6.
18. Bonderman D, Wilkens H, Wakounig S, et al.Risk factors for chronic thromboembolic pul-monary hypertension. Eur Respir J. 2009; 33:325-31.
19. Jiménez D, Gaudó J, Sueiro A. Factores de ries-go en la hipertensión pulmonar tromboem-bólica crónica. Arch Bronconeumol. 2009; 45(Supl. 6): 11-14.
20. Condliffe R, Kiely DG, Gibbs JSR, et al. Impro-ved Outcomes in Medically and Surgically Tre-ated Chronic Thromboembolic PulmonaryHypertension. Am J Respir Crit Care Med.2008; 177: 1122-7.
21. Simonneau G, Delcroix M, Mayer E, et al. Firstinternational registry on chronic thrombo-embolic pulmonary hypertension (CTEPH).European Respiratory Society Annual Congress2008. Poster P1045. Disponible en: http:www.
ersnet. org/learning_resources_player/abs-tract_print_08/ main_frameset.htm
22. Zhu T, Martínez I, Emmerich J. Venous Throm-boembolism: Risk Factors for Recurrence.Arterioscler Thromb Vasc Bio. 2009; 29: 298-310.
23. Barberá JA, Escribano P, Morales P, et al. Están-dares asistenciales en hipertensión pulmonar.Documento de consenso elaborado por laSociedad Española de Neumología y CirugíaTorácica (SEPAR) y la Sociedad Española deCardiología (SEC). Arch Bronconeumol 2008;44: 87-99.
24. Hoeper MM, Mayer E, Simonneau G, et al.Chronic thromboembolic pulmonary hyper-tension. Circulation. 2006; 113: 2011-20.
25. Castaner E, Gallardo X, Ballesteros E, et al. CTDiagnosis of Chronic Pulmonary Thrombo-embolism. RadioGraphics. 2009; 29: 31-50.
26. Auger WR, Fedullo PF, Moser KM, et al. Chro-nic major-vessel thromboembolic pulmonaryartery obstruction: appearance at angiography.Radiology. 1992;182: 393-8.
27. Moser KM, Bloor CM. Pulmonary vascularlesions occurring in patients with chronic majorvessel thromboembolic pulmonary hyperten-sion. Chest. 1993; 103: 685-92.
28. Ribeiro A, Lindmarker P, Johnsson H, et al. Pul-monary embolism: one-year follow-up withechocardiography doppler and five-year sur-vival analysis. Circulation. 1999; 99: 1325-30.
29. Kline JA, Steuerwald MT, Marchick MR, etal.Prospective evaluation of right ventricularfunction and functional status 6 months afteracute submassive pulmonary embolism: fre-quency of persistent or subsequent elevationin estimated pulmonary artery pressure. Chest.2009; 136: 1202-10.
30. Skoro-Sajer N, Hack N, Sadushi R, et al. Pul-monary vascular reactivity and prognosis inpatients with chronic thromboembolic pul-monary hypertension. Circulation. 2007; 116:503-4.
31. Skoro-Sajer N, Hack N, Sadushi-Kolici R, et al.Pulmonary Vascular Reactivity and Prognosisin Patients With Chronic Thromboembolic Pul-monary Hypertension: A Pilot Study. Circula-tion 2009; 119: 298-305.
32. National Pulmonary Hypertension Centres ofthe UK. Consensus statement on the mana-gement of pulmonary hypertension in clinicalpractice in the UK and Ireland. Heart. 2008;94: i1-i41.
GRUPO IV. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN PULMONAR TROMBOEMBÓLICA CRÓNICA (HPTEC)
109
Monografía HAP NM 208 pag 18/2/10 17:08 Página 109

J. GAUDÓ NAVARRO ET AL.
110
33. Kunieda T, Nakanishi N, Satoh T, et al. Prog-noses of primary pulmonary hypertensionand chronic majorvessel thromboembolic pul-monary hypertension determined from cumu-lative survival curves. Intern Med. 1999; 38:543-6.
34. Riedel M, Stanek V, Widimsky J, et al. Long-term follow-up of patients with pulmonarythromboembolism. Late prognosis and evolu-tion of hemodynamic and respiratory data.Chest. 1982; 81: 151-8.
35. Hardziyenka M, Reesink HJ, Bouma BJ, et al.A novel echocardiographic predictor of in-hos-pital mortality and mid-term haemodynamicimprovement after pulmonary endarterectomyfor chronic thromboembolic pulmonary hyper-tension. Eur Heart J. 2007; 28: 842-9.
36. Kim NH, Fesler P, Channick RN, et al. Preo-perative partitioning of pulmonary vascularresistance correlates with early outcome afterthromboendarterectomy for chronic throm-boembolic pulmonary hypertension. Circula-tion 2004; 109: 18-22.
37. Bonderman D, Skoro-Sajer N, Jakowitsch J, etal. Predictors of outcome in chronic throm-boembolic pulmonary hypertension. Circula-tion. 2007; 115: 2153-8.
38. Toshner M, Suntharalingam J, Goldsmith K, etal. Current differences in referral patterns forpulmonary endarterectomy in the UK. Eur Res-pir J. 2008; 32: 660-3.
39. Jamieson SW. Pulmonary thromboendarte-rectomy. Heart. 1998; 79: 118-20.
40. Thistlethwaite PA, Kaneko K, Madani MM, etal. Technique and outcomes of pulmonaryendarterectomy surgery. Ann Thorac Cardio-vasc Surg. 2008;14: 274-82.
41. Corsico AG, D'Armini AM, Cerveri I, et al. Long-term outcome after pulmonary endarterec-tomy. Am J Respir Crit Care Med. 2008; 178:419-24.
42. Jamieson SW, Kapelanski DP, Sakakibara N, etal. Pulmonary endarterectomy: experienceand lessons learned in 1,500 cases. Ann Tho-rac Surg. 2003; 76: 1457-62.
43. Couturaud F, Frachon I, Leroyer C. Chronicthromboembolic pulmonary hypertension: atribute to pulmonary endarterectomy. Eur Res-pir J. 2009; 33: 230-2.
44. Galié N, Hoeper MM, Humbert M, et al. Gui-delines for the diagnosis and treatment of pul-monary hypertension: The Task Force for theDiagnosis and Treatment of Pulmonary Hyper-tension of the European Society of Cardiology(ESC) and the European Respiratory Society(ERS), endorsed by the International Societyof Heart and Lung Transplantation (ISHLT). EurHeart J. 2009; 30: 2493-537.
45. Hill N S, Preston I R, Roberts K E. InoperableChronic Thromboembolic Pulmonary Hyper-tension: Treatable With Medical Therapy. Chest.2008; 134: 221-3.
46. Olschewski H, Simonneau G, Galiè N, et al.Inhaled iloprost for severe pulmonary hyper-tension. N Engl J Med. 2002; 347: 322-9.
47. Suntharalingam J, Treacy CM, Doughty NJ, etal. Long-term use of sildenafil in inoperablechronic thromboembolic pulmonary hyper-tension. Chest. 2008; 134: 229-36.
48. Hoeper MM, Kramm T, Wilkens H, et al. Bosen-tan therapy for inoperable chronic thrombo-embolic pulmonary hypertension. Chest. 2005;128: 2363-7.
49. Hughes RJ, Jais X, Bonderman D, et al. The effi-cacy of bosentan in inoperable chronic throm-boembolic pulmonary hypertension: a 1-yearfollow-up study. Eur Respir J. 2006; 28: 138-43.
50. Bonderman D, Nowotny R, Skoro-Sajer N, etal. Bosentan therapy for inoperable chronicthromboembolic pulmonary hypertension.Chest. 2005; 128: 2599-603.
51. Jaïs X, D'Armini AM, Jansa P, et al. Bosentanfor treatment of inoperable chronic thrombo-embolic pulmonary hypertension: BENEFiT(Bosentan Effects in iNopErable Forms of chro-nIc Thromboembolic pulmonary hyperten-sion), a randomized, placebo-controlled trial.J Am Coll Cardiol. 2008; 52: 2127-34.
52. Lang IM. Managing chronic thromboembolicpulmonary hypertension: pharmacologicaltreatment options. ERR. 2009;18: 24-8.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 110

111
RESUMENLa clasificación de la hipertensión pulmo-
nar ha sufrido una serie de cambios duranteel 4º Simposio Mundial que se desarrolló en2008 en Dana Point, California. El grupo V dedicha clasificación incluye varias entidades, lamayoría poco frecuentes, que se han asocia-do a hipertensión pulmonar, pero en las quela etiología de la hipertensión es poco clara omultifactorial. Este grupo se ha visto modifi-cado y mejor descrito en esta última clasifi-cación.
Sin embargo, debido a la heterogeneidady rareza de muchas de estas entidades, haypocos datos disponibles sobre respuesta a tra-tamientos específicos para la hipertensión pul-monar, sin existir ensayos clínicos aleatoriza-dos, por lo que no es posible emitir unarecomendación, y en todos estos procesos eltratamiento debe individualizarse.
El propósito de este capítulo es profundi-zar en los pocos aspectos conocidos hasta aho-ra de la relación entre la hipertensión pulmo-nar y el variado grupo de enfermedadesincluidas en el grupo V.
INTRODUCCIÓNEl grupo V de la clasificación de la hiper-
tensión pulmonar ha sufrido una serie de cam-bios durante el 4º Simposo Mundial que sedesarrolló en 2008 en Dana Point, California(1).
Este grupo incluye varias entidades en lasque la etiología de la hipertensión es poco cla-
ra o multifactorial y se ha visto engrosado ymejor descrito en esta ultima clasificación,incluyendo cuatro subgrupos, como se refle-ja en la tabla 1:• En el primero se incluyen enfermedades
hematológicas: se ha descrito hipertensiónpulmonar en enfermedades mieloprolife-rativas crónicas, como la policitemia vera,trombocitemia esencial y la leucemia mie-loide crónica.
• El segundo subgrupo comprende enfer-medades sistémicas con riego aumentadode producir hipertensión pulmonar, talescomo la sarcoidosis, histiocitosis, linfan-gioleiomiomatosis y neurofibromatosis tipo1 o enfermedad de von Recklinghausen.
• El tercer subgrupo agrupa diversos tras-tornos metabólicos, tales como la enfer-medad de Gaucher, enfermedad de depó-sito de glucógeno tipo I a y enfermedadesdel tiroides (hipotiroidismo e hipertiroi-dismo, tiroiditis de Hashimoto).
• Por último, el cuarto subgrupo es una mis-celánea de enfermedades: obstruccióntumoral de arterias pulmonares, embolis-mos por micro- metástasis tumorales, fibro-sis mediastínica y enfermedad renal enhemodiálisis.
El 4º Simposio Mundial ha sido la prime-ra reunión internacional en profundizar, nosólo en la hipertensión pulmonar arterial, sinotambién en las formas de hipertensión pul-
GRUPO V. CLASIFICACIÓN DE DANA POINT
HIPERTENSIÓN PULMONAR CONMECANISMOS MULTIFACTORIALES NO ACLARADOS
Mercedes García-Salmones Martín, María Jesús Linares Asensio,Ángela Ramos Pinedo
Monografía HAP NM 208 pag 18/2/10 17:08 Página 111

monar no arterial (que incluye el grupo V, obje-to de este capítulo, así como los grupos II,III y IV, de la clasificación mencionada). Sinembargo, se centra en el diagnóstico y trata-miento de la hipertensión pulmonar en lostrastornos más comunes [EPOC, enfermedadintersticial pulmonar difusa (EPID), trombo-embolismo crónico y enfermedad cardiacaizquierda](2).
Repasando la también la recientementepublicada Guía para el diagnóstico y tratamientode la hipertensión pulmonar 2009, de la ESC(Sociedad Europea de Cardiología)/ERS (Socie-dad Europea de Respiratorio) deja también delado la problemática del grupo V debido a laheterogeneidad y rareza de las condicionesincluidas en él(3).
El propósito de este capítulo es profundi-zar en los pocos aspectos conocidos hasta aho-ra de la relación entre la hipertensión pulmo-nar y el variado grupo de enfermedadesincluidas en el grupo V, centrándonos espe-cialmente en aquellas entidades más fre-cuentemente tratadas por los neumólogos,como la sarcoidosis, y en enfermedades habi-tuales, como la insuficiencia renal en diálisis.
SUBGRUPO 1: ENFERMEDADESHEMATOLÓGICAS
En este subgrupo se incluyen enfermeda-des hematológicas severas (Tabla 2).
Se ha descrito hipertensión pulmonar aso-ciada a enfermedades crónicas mieloprolife-rativas tales como la policitemia vera, la trom-bocitemia esencial y la leucemia mieloidecrónica(4).
Dichos trastornos pueden producir hiper-tensión pulmonar por varios mecanismos. Sehan descrito, entre ellos:• Obstrucción de las arterias pulmonares por
megacariocitos circulantes(5).• Asociación con tromboembolismo pulmo-
nar crónico(6).• Elevación de la presión en la circulación
portal originando hipertensión portopul-monar.
• Coexistencia de insuficiencia cardiaca con-gestiva.
• Esplenectomía.
La esplenectomia es frecuente en estospacientes, bien sea autoasplenia funcional obien quirúrgica como resultado de un trau-
M. GARCÍA-SALMONES MARTÍN ET AL.
112
TABLA 1. Clasificación de hipertensión pulmonar (Dana Point, 2008). Grupo V: hipertensión pulmonar con mecanismos multifactoriales no aclarados
5.1. Enfermedades hematológicas: trastornos mieloproliferativos, esplenectomía5.2. Enfermedades sistémicas: sarcoidosis, histiocitosis, linfangioleiomiomatosis, neurofibromatosis5.3. Trastornos metabólicos: enfermedades de depósito de glucógeno, Gaucher, enfermedades tiroideas5.4. Otros: obstrucción tumoral, mediastinitis fibrosante, insuficiencia renal crónica en diálisis
TABLA 2. Enfermedades hematológicas que se asocian a hipertensión pulmonar
Grupo I: Hipertensión pulmonar arterial (HAP)1.4.6. Anemia hemolítica crónica
Grupo V: Hipertensión pulmonar de mecanismo incierto multifactorial5.1. Enfermedades hematológicas:
- Síndromes mieloproliferativos crónicos: policitemia vera, leucemia mieloide crónica, trombocitemia esencial
- Esplenectomía
Dana Point, 2008.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 112

matismo o como parte del tratamiento. Se hadescrito en esplenectomizados, por una par-te, mayor riesgo de desarrollar tromboembo-lismo pulmonar crónico y, por otra, casos dehipertensión pulmonar arterial con hipertro-fia de la media, fibrosis interna y lesiones ple-xiformes en la vasculatura pulmonar(7).
Las anemias hemolíticas crónicas adquiri-das o hereditarias, incluyendo la talasemia y laesferocitosis entre otros procesos, se asociancon el desarrollo de hipertensión pulmonar arte-rial. Anteriormente situadas en el grupo de otrascondiciones asociadas con hipertensión pul-monar, en la clasificación de Dana Point seincluyen en el grupo I, y se describen en otrocapítulo de esta monografía.
SUBGRUPO II: ENFERMEDADESSISTÉMICAS
Este subgrupo incluye enfermedades sis-témicas que se han asociado con un riesgoaumentado de desarrollar hipertensión pul-monar.
Sarcoidosis Es una frecuente enfermedad inflamatoria
granulomatosa sistémica de origen descono-cido caracterizada por la presencia de granu-lomas no caseificantes. La hipertensión pul-monar es una complicación frecuente que sepresenta a menudo como causa de disnea inex-plicable y limitación al ejercicio. La hiperten-sión pulmonar es una complicación cada vezreconocida con más frecuencia, con una pre-valencia que varía del 1 al 28% según lasseries(8,9).
Se ha encontrado, además, en un 47% delos pacientes con disnea desproporcionada asu alteración funcional respiratoria y en un74% de los pacientes en lista de trasplante pul-monar, produciendo mayor mortalidad en estegrupo.
La hipertensión pulmonar podría produ-cirse por varios mecanismos. A menudo seatribuye a la destrucción del lecho capilar porel proceso fibrótico y/o a la hipoxemia cróni-ca(10). La necesidad de oxígeno se correlacio-
na con la hipertensión pulmonar y la mayoríade los casos tienen enfermedad estadio radio-lógico IV.
Sin embargo, la severidad de la hiperten-sión pulmonar no siempre se correlaciona biencon el grado de enfermedad del parénquimay las anomalías del intercambio gaseoso, locual sugiere que hay otros mecanismos quepueden contribuir al desarrollo de hipertensiónpulmonar. De hecho se ha descrito hiperten-sión pulmonar severa en ausencia de fibrosispulmonar. En este sentido, tales mecanismospodrían incluir la presencia de una vasculo-patía intrínseca sarcoidea, compresión extrín-seca de grandes arterias pulmonares por ade-nopatías hiliares y mediastínicas, e infiltracióngranulomatosa directa de la vasculatura pul-monar, especialmente las venas pulmonares,lo que simula una enfermedad pulmonar veno-oclusiva. Con menos frecuencia puede produ-cirse hipertensión portopulmonar secundariaa afectación hepática. Por otra parte, la afec-tación cardiaca izquierda por la propia sarcoi-dosis debería descartarse, con presión pul-monar capilar elevada en un 11% de loscasos(11).
El diagnóstico de hipertensión pulmonarasociada a sarcoidosis puede ser problemáti-co. Algunos autores han encontrado diferen-cias con tomografía de de alta resolución(TACAR) entre pacientes con y sin hipertensiónpulmonar, con compresión extrínseca por lin-fadenopatias pulmonares en un 21,4% decasos con hipertensión pulmonar y mayor por-centaje de fibrosis(12).
Sulica y cols.(12) han mostrado las caracte-rísticas clínicas de 54 pacientes con hiperten-sión pulmonar, con hallazgos ecocardiografi-cos, consistentes en elevación de la presiónsistólica del ventrículo derecho, aumento delventrículo derecho y disfunción ventricularderecha. El cateterismo derecho, sigue siendoel gold standard del diagnóstico en estos pacien-tes con sospecha de HP.
La sarcoidosis generalmente tiene muybuen pronóstico y hasta dos tercios de lospacientes presentan remisión espontánea. Sin
GRUPO V. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN PULMONAR CON MECANISMOS MULTIFACTORIALES NO ACLARADOS
113
Monografía HAP NM 208 pag 18/2/10 17:08 Página 113

embargo, de un 1 al 5% fallece por insufi-ciencia respiratoria, enfermedad del sistemanervioso central o afectación miocárdica. Lapresencia de hipertensión pulmonar asociadaproduce disnea refractaria y capacidad de ejer-cicio disminuida, y condiciona un peor pro-nóstico con alta mortalidad, por lo que debeconsiderarse su tratamiento, si bien la falta deestudios específicos mantiene incertidumbreen este campo.
El tratamiento incluye el tratamiento de lapropia sarcoidosis con corticoesteroides, asícomo otros antiinflamatorios sistémicos, anti-coagulantes y oxígeno suplementario, pero sueficacia es poco clara.
Se han realizado algunos estudios retros-pectivos en escasos pacientes con vasodila-tadores pulmonares, con un tiempo de segui-miento de al menos 4 meses (6 pacientes conbosentan y/o epoprostenol, 6 pacientes conepoprostenol continuo intravenoso y 12 pacien-tes con sildenafilo), que han demostrado algu-na mejoría en la hemodinámica pulmonar,pero con morbimortalidad significativa, inclu-yendo un caso de muerte súbita y otro de ede-ma agudo de pulmón(14,15).
Otro estudio retrospectivo de 22 pacientescon sarcoidosis tratados con distintas terapiasespecíficas para la hipertensión pulmonar yseguidos durante una media de 11 meses, hademostrado incremento en la distancia reco-rrida en el test de 6 minutos, en especial enaquellos pacientes con defectos restrictivosmenos severos, así como reducción de la pre-sión de la arteria pulmonar y de la resistenciapulmonar vascular. Sin embargo, la supervi-vencia sin trasplante a los 3 años fue sólo del74%. En este estudio, decisiones tales comotratamiento inmunosupresor, o qué vasodila-tador utilizar, fueron totalmente arbitrarias, porlo que no aporta información sobre qué pacien-tes tratar o qué terapia es mejor(16).
En la actualidad a falta de estudios con-trolados, se podría sugerir que pacientes cui-dadosamente seleccionados con hipertensiónpulmonar asociada a sarcoidosis, que hayanrecibido previamente tratamiento antiinfla-
matorio, podrían beneficiarse de un ensayo conterapia dirigida al tratamiento específico de lahipertensión pulmonar, posiblemente pacien-tes con una FVC mayor del 50% de su teórico.Deberían siempre descartarse otras causas dehipertensión pulmonar asociadas, especial-mente disfunción diastólica o sistólica por afec-tación cardiaca de la propia sarcoidosis.
El pronóstico de los pacientes con sarcoi-dosis e hipertensión pulmonar es pobre, porlo que estos pacientes deberían evaluarse paratrasplante pulmonar.
Histiocitosis pulmonar por células de Langerhans
Es una enfermedad infiltrativa pulmonarpoco frecuente asociada con cambios des-tructivos en el parénquima pulmonar. Enpacientes con enfermedad muy avanzada, esfrecuente encontrar hipertensión pulmonaren probable relación con hipoxemia cróni-ca(17),si bien encontramos casos sin relacióncon afectación parenquimatosa pulmonar,especialmente aquellos con hipertensión pul-monar más severa, en relación con vasculo-patía severa difusa con hipertrofia de la mediay fibrosis de la íntima, que afecta funda-mentalmente a las venas pulmonares intra-lobares. Fartoukh y cols. encontraron unaPAPm de 59 mmHg en 21 pacientes conse-cutivos con esta enfermedad (todos estabanpor encima de 40 mmHg) y que habían sidoremitidos para valoración de trasplante. Enseis de ellos se realizó biopsia pulmonar endos momentos de la evolución, observandoun empeoramiento de la vasculopatía conestabilización de las lesiones parenquimato-sas y bronquiolares(18).
LinfangioleiomiomatosisEs una rara enfermedad que afecta fun-
damentalmente a mujeres, caracterizada pordestrucción pulmonar con formación de quis-tes, anomalías linfáticas y tumores abdomi-nales.
La alteración funcional más frecuente esuna alteración del intercambio gaseoso, con
M. GARCÍA-SALMONES MARTÍN ET AL.
114
Monografía HAP NM 208 pag 18/2/10 17:08 Página 114

descenso de la difusión pulmonar y obstruc-ción al flujo aéreo. La alteración de la difusiónse correlaciona bien con los hallazgos histo-lógicos de severidad de la enfermedad, y esel mejor predictor de hipoxemia inducida porel ejercicio. Sin embargo, esta hipoxemia pue-de ocurrir en pacientes con difusión pulmo-nar, probablemente en relación con apariciónde hipertensión pulmonar. En un estudio rea-lizado con ecocardiograma en 95 pacientescon linfangioleiomiomatosis, se encuentrahipertensión pulmonar en un 8%, de los cua-les aproximadamente un tercio tenían oxige-noterapia. Parece por tanto que el desarrollode hipertensión pulmonar es bastante infre-cuente y se produce probablemente por hipo-xemia crónica y por destrucción de los capi-lares pulmonares debido a las lesionesquísticas(19).
Neurofibromatosis tipo I o enfermedad devon Recklinghausen
Es una enfermedad autonómica dominan-te secundaria a mutaciones en el gen supresorNF1 con expresión muy variable, con una inci-dencia de 1 de cada 4.000 nacidos vivos, y quese caracteriza por la aparición de fibromas cutá-neos, manchas cutáneas café con leche, nódu-los de Lish en el iris y líneas axilares. Otrasmanifestaciones clínicas comprenden anoma-lías cardiovasculares, gastrointestinales, rena-les y endocrinas, déficit cognitivo y neoplasiasdel sistema nervioso periférico y sistema ner-vioso central. Puede complicarse ocasional-mente con vasculopatía sistémica, que es hete-rogénea y afecta a variedad de arterias dedistinto tamaño en todo el cuerpo. Se han pro-puesto distintos mecanismos para explicar supatogenia, incluyendo alteración de la histo-génesis vascular, alteración del mantenimien-to y reparación vascular, mutaciones y factoresambientales. Lie clasifica la vasculopatía aso-ciada a neurofibromatosis como: 1) prolifera-ción de las células musculares de la intima vas-cular en arterias grandes; 2) proliferación decélulas musculares de la intima vascular confibrosis y neoangiogénesis en arterias de media-
no tamaño, y 3) proliferación plexiforme de laíntima en pequeñas arteras y arteriolas(20).
Se han descrito unos pocos casos de hiper-tensión pulmonar de mecanismo no aclara-do(21,22). En estos casos la hipertensión pul-monar parece deberse a afectación vascularsegún los escasos estudios histológicos quesuele correlacionarse con un patrón radioló-gico en mosaico en la TAC.
Algunos autores sobre la base de estoshallazgos han considerado la hipertensión pul-monar asociada a neurofibromatosis tipo I unaentidad bien caracterizada, que debería con-siderarse dentro del grupo 1 de la clasificaciónde hipertensión pulmonar (hipertensión pul-monar arterial asociada)(23).
La fibrosis pulmonar y el tromboembolis-mo crónico podrían tener también un papel enla aparición de hipertensión pulmonar (Fig. 1).
El pronóstico es infausto, si bien se ha des-crito alguna mejoría en hemodinámica pul-monar y síntomas en los casos descritos conel tratamiento con epoprostenol, bosentan, sil-denafilo y beraprost.
SUBGRUPO III: ENFERMEDADES METABÓLICAS
Enfermedad de depósito de glucógeno tipoIa o enfermedad de Pompe
Se trata de una rara enfermedad autosó-mica recesiva causada por un déficit de la glu-
GRUPO V. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN PULMONAR CON MECANISMOS MULTIFACTORIALES NO ACLARADOS
115
FIGURA 1. Neurofibromatosis con extensa afec-tación pulmonar.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 115

cosa-6-fosfatasa(24,25). El mecanismo de hiper-tensión pulmonar es incierto y se ha asociadoa shunts portocava, defectos del septo atrial oalteración restrictiva severa de la función pul-monar. También se han descrito fenómenostrombóticos asociados que podrían tener algúnpapel. En un caso se han descrito lesiones ple-xiformes.
Enfermedad de Gaucher Se caracteriza por una deficiencia de la glu-
cosidasa lisosomal B, lo cual produce un acú-mulo de glucocerebrósido en las células reti-culoendoteliales. Las manifestaciones típicasincluyen hepatoesplenomegalia e infiltraciónde la médula ósea. En un estudio con ecocar-diografia de 134 pacientes con enfermedadde Gaucher, la aparición de hipertensión pul-monar fue frecuente(26). Se han propuesto diver-sos mecanismos, entre ellos la aparición deenfermedad pulmonar intersticial, hipoxemiacrónica, alteración capilar y esplenectomía,e incluso se ha descrito un caso de hallazgoshistológicos similares a hipertensión pulmo-nar idiopática(27).
Enfermedades tiroideasSe ha encontrado hipertensión pulmonar
mediante ecocardiograma en más de un 40%de pacientes con enfermedad tiroidea (hipo ohipertiroidismo)(28). Además, en un estudio pros-pectivo de 63 adultos con hipertensión pul-monar arterial, se ha encontrado una inciden-cia de enfermedad tiroidea autoinmune del49%, sugiriendo una asociación entre ambasenfermedades, mientras que en otro estudiose encontraba un 30% de pacientes con hiper-tensión pulmonar arterial con anticuerpos anti-tiroglobulina(29). Asimismo, se ha descrito uncaso de enfermedad pulmonar venooclusivaconfirmada histológicamente en un pacientecon tiroiditis de Hashimoto(30). En un estudiocon 356 pacientes con hipertensión pulmonarencuentran un 24% de enfermedad tiroideafrente a un 15% en grupo control, fundamen-talmente hipotiroidismo, lo que sugiere quedebería probarse la función tiroidea en todos
los pacientes con hipertensión pulmonar(31). Lamayoría de los datos existentes en la literatu-ra sugiere que el tratamiento de la alteracióntiroidea mejora la hipertensión pulmonar, fun-damentalmente en paciente con tirotoxicosis.El mecanismo de unión entre ambas perma-nece sin aclarar pero se ha propuesto la exis-tencia de un mecanismo autoinmune común.
SUBGRUPO IV: OTRAS ENFERMEDADES
Neoplasias En algunos raros casos de sarcomas de las
arterias pulmonares puede producirse un cre-cimiento tumoral dentro de las arterias pulmo-nares, produciendo obstrucción progresiva delas mismas e hipertensión pulmonar arterial.Tales casos son de difícil diagnóstico diferencialcon hipertensión por tromboembolismo pul-monar crónico, generalmente, mediante TAC yangiografía, y su curso es rápidamente fatal(32,33).
Otra rara causa de hipertensión pulmonarrápidamente progresiva es la obstrucción de lamicrovasculatura por émbolos metastásicostumorales. La mayoría de casos se han asocia-do con tumores del pulmón, mama o carcino-mas gástricos. Suelen presentarse con hipoxe-mia, generalmente, severa. La TAC no muestratrombos proximales (si acaso, engrosamientode los septos), mientras que la gammagrafía deventilación/perfusión suele mostrar múltiplesdefectos subsegmentarios de perfusión(34,35).
Fibrosis mediastínicaLos pacientes con fibrosis mediastínica pue-
den desarrollar hipertensión pulmonar severadebido a la compresión de arterias y venas pul-monares. Esto puede producirse principal-mente en algunas entidades, como la histo-plasmosis, la tuberculosis, sarcoidosis yenfermedades por otros hongos. El diagnósti-co diferencial con tromboembolismo puederesultar difícil(36,37).
Enfermedad renal terminalEn pacientes con enfermedad renal ter-
minal en programa de hemodiálisis prolonga-
M. GARCÍA-SALMONES MARTÍN ET AL.
116
Monografía HAP NM 208 pag 18/2/10 17:08 Página 116

GRUPO V. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN PULMONAR CON MECANISMOS MULTIFACTORIALES NO ACLARADOS
117
do se ha descrito la aparición de hipertensiónpulmonar inexplicada hasta en un 40% (basa-do en estudios ecocardiográficos). La mortali-dad en el grupo de pacientes con hipertensiónpulmonar fue significativamente mayor queen el grupo sin hipertensión (30% frente al3%) La causa de la hipertensión pulmonar enestos pacientes es multifactorial (Tabla 3).
Se ha postulado que la vasoconstricciónde la vasculatura pulmonar debido a altera-ciones metabólicas y hormonales propias dela insuficiencia renal produciría un incremen-to de la resistencia vascular pulmonar.
Por otra parte, la anemia crónica que pre-sentan, el propio acceso mediante fístula arte-riovenosa y la disfunción ventricular sistólicay diastólica, que aparece frecuentemente enestos pacientes, jugarían también un papel.
A pesar de casi 4 décadas de aplicaciónde la hemodiálisis, no se ha estudiado sufi-cientemente el efecto a largo plazo sobre lacirculación pulmonar de la hemodiálisis a tra-vés de un acceso arteriovenoso creado qui-rúrgicamente. Se han comparado variables clí-nicas hemodinámicas y metabólicas entrepacientes en hemodiálisis con y sin hiperten-sión pulmonar. Los pacientes con hipertensiónpulmonar tenían significativamente mayor gas-to cardiaco, niveles de hematocrito y hemo-globina inferiores, y tenían menor duracióndel tratamiento con hemodiálisis, lo que se atri-buyó a menor supervivencia o a realización deun trasplante renal(38). Aunque la anemia seasocia con aumento compensatorio del gastocardiaco, la anemia más pronunciada en los
pacientes con hipertensión pulmonar no expli-ca todas las diferencias en el gasto cardiaco.Parece que otros factores tales como el tama-ño del acceso arteriovenoso, están involucra-dos en el aumento del gasto cardiaco. Sinembargo, debido a la gran capacidad de lamicrocirculación pulmonar, el aumento del gas-to cardiaco, por sí mismo, no puede causarhipertensión pulmonar, a no ser que haya unaumento de la resistencia vascular pulmonaro una disminución de la respuesta vasodilata-dora al incremento del gasto cardiaco.
La disminución de las respuesta vasodila-tadora podría deberse a cambios anatómicoso funcionales. Un estudio en perros con insu-ficiencia renal terminal mostraba calcificacio-nes y aumento de la resistencia vascular pul-monar así como hipertrofia ventricular derechasólo en perros con glándulas paratiroides intac-tas, por lo que se ha sugerido que la afectaciónpulmonar se debe al aumento de la actividadde la hormona paratiroidea(39). La endotelina1 está, por otra parte, aumentada en pacien-tes con insuficiencia renal crónica, siendo unpotente vasoconstrictor pulmonar que se harelacionado con la hipertensión pulmonar pri-maria y, secundaria.
En resumen, hay un gran número depacientes con insuficiencia renal terminal enprograma de hemodiálisis que desarrollanhipertensión pulmonar, lo que probablemen-te se asocia con una disminución de su super-vivencia. Debería por ello realizarse una esti-mación y seguimiento de la presión arterialpulmonar mediante ecocardiografía Doppler
TABLA 3. Etiología de la hipertensión pulmonar en la insuficiencia renal crónicaterminal
• Aumento de la endotelina 1 → Vasonconstricción de la musculatura pulmonar con aumento de lasresistencias vasculares pulmonares
• Hiperparatiroidismo → Disminución de la respuesta vasodilatadora• Anemia crónica → Aumento del gasto cardiaco• Fístula arterio-venosa → Aumento del gasto cardiaco• Disfunción ventricular sistólica y diastólica
Monografía HAP NM 208 pag 18/2/10 17:08 Página 117

a todos los pacientes con insuficiencia renalen hemodiálisis. La disminución o cierre dela fístula arteriovenosa en algunos casos pue-de disminuir la presión pulmonar. Por otraparte, ésta disminuye también con el tras-plante, por lo que quizá debería considerarsela presencia de hipertensión pulmonar comootro criterio adicional para indicar el trasplanterenal.
CONCLUSIÓNEl grupo V de la clasificación de la hiper-
tensión pulmonar incluye varias entidades enlas que la etiología de la hipertensión es pococlara o multifactorial. En todos estos proce-sos, la aparición de hipertensión pulmonarcondiciona un peor pronóstico y, en muchoscasos, suele conducir a un desenlace rápida-mente fatal. Es importante conocer la aso-ciación de los mismos con la hipertensiónpulmonar, sospecharla y tratar de diagnosti-carla. El tratamiento depende de la enfer-medad subyacente y en algunos casos se haintentado tratamiento específico de la hiper-tensión con éxito relativo. En muchos casosel diagnóstico de hipertensión pulmonar seve-ra va a condicionar la toma de decisiones fun-damentales, como remitir al paciente a tras-plante pulmonar (como en los casos desarcoidosis), o disminución o cierre de fís-tula arteriovenosa y remitir a trasplante renalen los casos de insuficiencia renal crónica.De cualquier modo, siempre hemos de estaratentos a la posibilidad de aparición de hiper-tensión pulmonar en estos procesos y su tra-tamiento ha de individualizarse.
BIBLIOGRAFÍA1. Simonneau G, Robbins IM, Beghetti M, et al.
Updated Clinical Classification of PulmonaryHypertension. J Am Coll Cardiol. 2009; 54:S43-54.
2. Hoeper MM, Barberá JA, Channick RN, Diag-nosis, Assessment and Treatment of Non-pul-monary Arterial Hypertension Pulmonary Hyper-tension. J Am Coll Cardiol. 2009; 54: S85-96.
3. The Task Force for de Diagnosis and Treatmentof Pulmonary Hypertension of the European
Society of Cardiology and the European Res-piratory Society endorsed by the Internatio-nal Society of Heart and Lung Transplantation.Guidelines for the diagnosis and treatment ofpulmonary hypertension. Eur Heart J. 2009;30: 2493-537.
4. Dingli D, Utz JP. Unexplained pulmonaryHypertension in chronic myeloproliferativedisorders. Chest. 2001; 120: 801-8.
5. Marvin KS, Spellberg RD. Pulmonary hyper-tension secondary to thrombocytosis in apatient with mieloide metaplasia. Chest. 1993;103: 642-4.
6. Nand S, Orfei E. Pulmonary hypertension inpolycythemia vera. Am J Hematol. 1994; 47:242-4.
7. Peacock AJ. Pulmonary hypertension after sple-nectomy: a consequence of loss of splenic fil-ter o is there something more? Thorax. 2005;60: 983-4.
8. Handa T, Nagai S, Miki S, et al. Incidence ofpulmonary hypertension and its clinical rele-vance in patients with sarcoidosis. Chest. 2006;129: 1246-52.
9. GlusKowski J, Hawrylkiewicz I, Zych D, et al.Pulmonary hemodynamic at rest and duringexercise in patients with sarcoidosis. Respira-tion. 1964; 46: 26-32.
10. Bourbonnais JM, Samavati L. Clinical predic-tors of pulmonary hypertension in sarcoido-sis. Eur Respir J. 2008; 32: 296-302.
11. Baughman RP, Engel PJ, Meyer CA, et al. Pul-monary hypertension in sarcoidosis. Sarcoi-dosis vasc Diffuse Lung Dis. 2006; 23: 108-16(Abstrat).
12. Nunes H, Humbert M, Capron F, et al. Pulmo-nary Hypertension associated with sarocido-sis: Mechanisms, haemodynamics and prog-nosis. Thorax. 2006; 61: 68-74.
13. Sulica R, Teirsten A, Kakarla S, et al. Distinc-tive clinical, radiographic and functional cha-racteristics of patients with sarcoidosis-rela-ted pulmonary hypertension. Chest 2005;128:1483-9.
14. Milman N, Burton CM, Iversen M, et al. Pul-monary hypertension in end-stage pulmo-nary sarocidosis: therapeutic effect of silde-nafil? J Heart Lung Transplant. 2008; 27:329-34.
15. Fisher KA, Serlin DM, Wilson KC, et al. Sar-coidosis- associated pulmonary hypertension:outcome with long- term epoprosternol treat-ment. Chest. 2006; 130: 1481-8.
M. GARCÍA-SALMONES MARTÍN ET AL.
118
Monografía HAP NM 208 pag 18/2/10 17:08 Página 118

GRUPO V. CLASIFICACIÓN DE DANA POINT: HIPERTENSIÓN PULMONAR CON MECANISMOS MULTIFACTORIALES NO ACLARADOS
119
16. Barnett CF, Bonura EJ, Nathan SD, et al. Tre-atment of sarcoidosis- associated pulmonaryhypertension: a two centres experience. Chet.2009; 135: 1455-61.
17. Dauriat G, Mal H, Thabut G, et al. Lung trans-plantation for pulmonary Langerhans’ cell his-tiocytosis: a multicenter analysis. Transplan-tation. 2006; 81: 746-50.
18. Fartoukh M, Humbert M, Capron F, et al. Seve-re pulmonary hypertension in histiocytosis X.Am J Respir Crit Care Med. 2000; 161: 216-23.
19. Taveira-DaSilva AM, Hathaway OM, SachdevV, et al. Pulmonary artery pressure in lymphan-gioleiomyomatosis:an echocardiographic study.Chest. 2007; 132: 1573-8.
20. Lie JT. Vasculopathies of neurofibromatosistype I. Cardiovasc Pathol. 1998; 7: 97-108.
21. Simeoni S, Puccetti A, Chilosi M, et al. Type 1neurofibromatosis complicated by pulmonaryartery hypertension: a case report. J MedInvest. 2007; 54: 354-8.
22. Engel PJ, Baughman RP, Menon SG, et al. Pul-monary hypertension in neurofibromatosis.Am J Cardiol. 2007; 99: 1177-8.
23. Stewart DR, Cogan JD, Kramer MR. Is pulmo-nary hypertension in neurofibromatosis TypeI Secondary to a Plexogenic Arteriopathy?Chest. 2007; 132: 798-808.
24. Hamaoka K, Nakagawa M, Furukawa N, et al.Pulmonary hypertension in type I glycogen sto-rage disease. Pediatr Cardiol. 1990; 11: 54-6.
25. Humbert M, Labrune P, Sitbon O, et al. Pul-monary arterial hypertension and type-I glyco-gen-storage disease: the serotonin hypothesis.Eur Respir J. 2002; 20: 59-65.
26. Elstein D, Klutstein MW, Lahad A, et al. Echo-cardiographic assessment of pulmonary hyper-tension in Gaucher’s disease. Lancet. 1998;351: 1544-6.
27. Theise ND, Ursell PC. Pulmonary hyperten-sion and Gaucher’s disease: logical associationor mere coincidence? Am J Pediatr HematolOncol. 1990; 12: 74-6.
28. Mercé J, Ferra´s S, Oltra C, et al. Cardiovascu-lar abnormalities in hyperthyroidism: a pros-pective Doppler echocardiographic study. AmJ Med. 2005; 118: 126-31.
29. Chu JW, Kao PN, Faul JL, et al. High prevalen-ce of autoimmune thyroid disease in pulmo-nary arterial hypertension. Chest. 2002; 122:1668-73.
30. Kokturk N, Demir N, Demircan S, et al. Pul-monary veno-occlusive disease in a patientwith a history of Hashimoto’s thyroiditis. IndianJ Chest Dis Allied Sci. 2005; 47: 289-92.
31. LI JH, Safford RE, Aduen JF, Heckman MG. Pul-monary Hypertension and Thyroid Disease.Chest. 2007; 132: 793-7.
32. Mayer E, Kriegsmann J, Gaumann A, et al.Surgical treatment of pulmonary artery sar-coma. J Thorac Cardiovasc Surg. 2001; 121:77-82.
33. Anderson MB, Kriett JM, Kapelanski DP, et al.Primary pulmonary artery sarcoma: a reportof six cases. Ann ThoracSurg. 1995; 59: 1487-90.
34. Roberts KE, Hamele-Bena D, Saqi A, et al. Pul-monary tumor embolism: a review of the lite-rature. Am J Med. 2003; 115: 228-32.
35. Dot JM, Sztrymf B, Yaici A, et al. [Pulmonaryarterial hypertension due to tumor emboli].Rev Mal Respir. 2007; 24: 359-66.
36. Loyd JE, Tillman BF, Atkinson JB, et al. Medias-tinalfibrosis complicating histoplasmosis. Medi-cine (Baltimore) 1988; 67: 295-310.
37. Goodwin RA, Nickell JA, Des Prez RM. Medias-tinal fibrosiscomplicating healed primary his-toplasmosis and tuberculosis. Medicine. (Bal-timore) 1972; 51: 227-46.
38. Yigla M, Nakhoul F, Sabag A, et al. Pulmonaryhypertension inpatients with end-stage renaldisease. Chest. 2003; 123: 1577-82.
39. Akmal M, Barnarr RR, Ansari AN, et al. ExcessPTH in CRF induces pulmonary calcification,pulmonary hypertension and right ventricularhypertrophy. Kidney INt. 1995; 47: 158-63.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 119

Monografía HAP NM 208 pag 18/2/10 17:08 Página 120

121
RESUMENEl trasplante pulmonar es una opción tera-
péutica a considerar en pacientes afectos deenfermedad pulmonar avanzada que no res-ponden a otros tratamientos.
En la hipertensión pulmonar severa, la mejo-ría en el tratamiento y control farmacológico(1)
ha supuesto un cambio en el control de la enfer-medad, aunque la cirugía sigue siendo el trata-miento de primera elección en los pacientes conhipertensión pulmonar tromboembólica cróni-ca(2) y tiene un papel relevante en la hiperten-sión pulmonar idiopática o asociada a otras con-diciones cuando fracasa el tratamiento médico(3).
Las alternativas quirúrgicas a consideraren la hipertensión pulmonar severa son(4): • El trasplante pulmonar, considerado como
la última alternativa terapéutica en pacien-tes con hipertensión pulmonar severa.
• La tromboendarterectomía, tratamiento deelección en pacientes con hipertensión pul-monar tromboembólica crónica. Cuandolas alteraciones vasculares son de locali-zación muy distal, o en presencia de hiper-tensión pulmonar muy severa, se debe con-siderar la opción de un trasplante pulmonar.
• La septostomía interauricular es una téc-nica quirúrgica que descomprime la aurí-cula y el ventrículo derecho a través de unacomunicación interauricular, lo que per-mite mejorar la presión arterial sistémicaa expensas de un descenso en la satura-ción arterial de oxígeno(5).
Cada una de estas técnicas quirúrgicas tie-ne sus indicaciones precisas y, en algunos
casos, pueden ser complementarias en un mis-mo paciente. En este sentido, la septostomíapuede ser utilizada como puente al trasplantepulmonar y, en algunos pacientes en los quefracasa la tromboendarterectomía, se puedeplantear el trasplante pulmonar.
TRASPLANTE PULMONAR Y CARDIOPULMONAR
IntroducciónTras aproximadamente 20 años de expe-
riencia y más de 25.000 procedimientos entodo el mundo, el trasplante pulmonar ha evo-lucionado considerablemente y, actualmente,constituye una opción terapéutica a conside-rar en pacientes con enfermedad pulmonaravanzada. No obstante, sus inicios fueron difí-ciles y, tras una fase experimental en perros,el primer trasplante pulmonar en humanos fuerealizado por Hardy en 1963(6). A partir de esafecha y hasta 1978, se realizaron 36 trasplan-tes pulmonares en todo el mundo, con muypoco éxito, ya que sólo dos receptores logra-ron sobrevivir durante más de 1 mes. La intro-ducción de la ciclosporina A como fármacoinmunosupresor de primera línea, en la déca-da de los 80, y las mejoras en las técnicas qui-rúrgicas, preservación del pulmón, y en elmanejo de la lesión isquemia-perfusión e infe-cciones han contribuido al progreso del tras-plante pulmonar. Así, en 1981, el grupo deStanford realizó el primer trasplante cardio-pulmonar con éxito a un paciente con enfer-medad pulmonar vascular(7). Dos años más tar-de se realizó el primer trasplante unilateral en
TRASPLANTE PULMONAR,CARDIOPULMONAR Y SEPTOSTOMÍA EN LA HIPERTENSIÓN PULMONAR (HP)Cristina López García-Gallo, Rosalía Laporta Hernández, Piedad Ussetti Gil
BLOQUE III. OTROS ASPECTOS TERAPÉUTICOS EN LA HP
Monografía HAP NM 208 pag 18/2/10 17:08 Página 121

un enfermo de 58 años afecto de fibrosis pul-monar, que sobrevivió a la intervención conbuena calidad de vida durante más de 5 años.En los años 90 se consolida el trasplante pul-monar como opción terapéutica y, según losdatos del Registro Internacional(8), el númerode trasplantes fue incrementándose progre-sivamente, hasta llegar a una meseta en losúltimos años en relación con la estabilizacióndel número de donantes, en unos 1.500 tras-plantes anuales. Sin embargo, la indicación detrasplante pulmonar por hipertensión pulmo-nar ha descendido desde el 17% en 1990 has-ta el 2% en el 2007. La disminución de las indi-caciones de trasplante por enfermedadvascular pulmonar se atribuye a la mejora enel tratamiento médico de estos pacientes(1).
En nuestro país, la actividad en trasplantepulmonar se inició en el año 1990(9), y has-ta finales del año 2008 se han realizado untotal de 2018 trasplantes, en los siete cen-tros acreditados, con un incremento progre-sivo en los primeros años, y últimamente conuna tendencia a la estabilización, de alre-dedor de 140 trasplantes por año. La indi-cación de trasplante pulmonar por hiper-tensión pulmonar en nuestro medio essimilar a la del Registro Internacional, y osci-la en alrededor del 2% anual.
En los últimos años el número de órganosdonantes disponibles es menor que el núme-ro de pacientes con enfermedad pulmonar ter-minal, que se pueden beneficiar de la cirugía.Por ello, es importante optimizar el uso de esterecurso y realizar una selección de los candi-datos a trasplante, que se espere que presen-ten mejores resultados a largo plazo(10). En estesentido, es de destacar las medidas que seestán tomando en nuestro país como son: elcompartir a un donante para dos receptorespulmonares(11,12), la reducción de volumen pul-monar para adaptar pulmones mayores dedonantes en receptores más pequeños, y lautilización de donantes pulmonares en para-da cardiaca, programa pionero de los hospi-tales Clínico San Carlos y Puerta de Hierro, deMadrid(13).
IndicacionesLos pacientes con hipertensión pulmonar
deben ser considerados para trasplante pul-monar cuando cumplen los requisitos gene-rales de indicaciones (Tabla 1) y contraindica-ciones (Tabla 2) de trasplante pulmonar. Lapresencia de contraindicaciones relativas debeser valorada de forma individual en cadapaciente en concreto (Tabla 2)(14).
El receptor de trasplante pulmonar debesuperar el procedimiento quirúrgico y tolerarla inmunosupresión posterior. La edad límitede contraindicación varía entre los 55 y los 65años, según se plantee el trasplante cardio-pulmonar en bloque o el trasplante exclusiva-mente pulmonar. Aunque no existe un acuer-do general sobre cuál es la mejor opción enestos pacientes, la posibilidades de consideraren la hipertensión pulmonar son el trasplantecardiopulmonar en bloque, el trasplante pul-monar bilateral y el unilateral. La elección deltipo de trasplante depende de la enfermedadsubyacente y de la situación hemodinámica.
Momento o ventana de trasplanteLa valoración clínica nos permite deter-
minar si la indicación de trasplante es ade-cuada, y si el paciente se encuentra en el“momento óptimo” para ser incluido en la lis-ta de espera. Se considera que se ha alcanza-do el “momento o ventana de trasplante” cuan-do la expectativa y la calidad de vida son losuficientemente malas como para precisar laintervención, pero el estado físico del pacien-
C. LÓPEZ GARCÍA-GALLO ET AL.
122
TABLA 1. Indicaciones del trasplantepulmonar
• Enfermedad pulmonar avanzada (clase fun-cional III/IV)
• Sin otras posibilidades terapéuticas• Adecuadamente informados • Edad < 65 años• Expectativa de vida < 2 años• Ausencia de contraindicaciones
(Modificado de Referencia 14).
Monografía HAP NM 208 pag 18/2/10 17:08 Página 122

te es lo suficientemente bueno como parasobrevivir a la cirugía.
Debemos diferenciar dos escenarios fun-damentales en relación con la decisión de tras-plante. En primer lugar hay que “remitir alpaciente a una unidad de trasplante” y, ensegundo lugar, hay que tomar la decisión de“incluir al paciente en lista de espera”. La deci-sión de “remitir a una unidad de trasplante”depende del médico encargado del cuidadodel paciente, que deberá conocer los requisi-tos necesarios para ser candidato a trasplan-te, e informar al paciente sobre esta posibili-dad terapéutica.
La decisión respecto a la aceptación delpaciente y su “inclusión en la lista de espe-ra” depende de la unidad de trasplante. Lastres variables fundamentales que entran enconsideración en la decisión de incluir a un
paciente en lista de espera son: la supervi-vencia con trasplante, el pronóstico de la enfer-medad de base y el tiempo medio en la listade espera.
Supervivencia con trasplanteSe considera indicado el trasplante, cuan-
do estimamos que la supervivencia sin tras-plante va a ser menor que con trasplante.
Según los datos del Registro Americanopublicados en 1991, la supervivencia mediade los pacientes con hipertensión pulmonarprimaria era de 2,8 años –con una supervi-vencia del 68% el primer año y del 48% a lostres años–(15). Series posteriores de Francia(16)
y Reino Unido(17) describen una supervivenciamedia similar: 3 años desde el momento deldiagnóstico. No obstante, el tratamiento convasodilatadores ha modificado estas cifras, ylos pacientes con respuesta a los antagonistasdel calcio tienen un buen pronóstico a los 5años(1). En los pacientes no respondedores,aunque el pronóstico es similar al previamen-te descrito, el tratamiento con epoprostenolproduce una mejora en la supervivencia y enlos síntomas(18). Los pacientes con enferme-dad pulmonar venooclusiva o hemangioma-tosis pulmonar no responden al tratamientomédico, y pueden desarrollar edema pulmo-nar con epoprostenol. Por ello, deben incluir-se en lista de espera de forma más precoz antela ausencia de la alternativa del tratamientomédico.
Según los datos del Registro Internacionalde Trasplante, la supervivencia general del tras-plante pulmonar es del 79% en el primer año,del 64% a los 3 años y del 52% a los 5 años(8).Por grupos de enfermedad, los pacientes conhipertensión pulmonar idiopática y síndromede Eisenmenger presentan una mayor morta-lidad perioperatoria y una menor superviven-cia precoz (0-30 días), que aquellos que pre-sentan otros procesos patológicos, comoenfisema, fibrosis quistica o fibrosis pulmo-nar(3). Esto es debido a la mayor complejidadquirúrgica, al mayor riesgo de sangrado, por lanecesidad constante de circulación extracor-
TRASPLANTE PULMONAR, CARDIOPULMONAR Y SEPTOSTOMÍA EN LA HIPERTENSIÓN PULMONAR (HP)
123
TABLA 2. Contraindicacionesabsolutas y relativas del trasplantepulmonar
Absolutas• Disfunción de otros órganos vitales• Neoplasia reciente (<5 años)• Serología para Ag HBs positiva• Serología para virus de la hepatitis C con
enfermedad hepática• Deformidades torácicas o espinales
significativas• Adicción a drogas (alcohol, tabaco) • Falta de adherencia a tratamientos previos• Enfermedad psiquiátrica no tratable• Ausencia de soporte social
Relativas• Edad >65 años• Severa limitación funcional • Obesidad (IMC >30)• Osteoporosis severa sintomática• Ventilación mecánica• Colonización por gérmenes panresistentes,
hongos o micobacterias
(Modificado de Referencia 14).
Monografía HAP NM 208 pag 18/2/10 17:08 Página 123

pórea, y a la inestabilidad hemodinámica pre-coz, como consecuencia de la disfunción tran-sitoria del ventrículo derecho. Los datos desupervivencia con trasplante pulmonar son glo-balmente peores que los del registro de hiper-tensión pulmonar. Por ello, es muy importan-te que estos pacientes sean tratados en unaunidad con experiencia, que deberá conside-rar el trasplante en los pacientes muy sinto-máticos sin respuesta al tratamiento médico(Tabla 3), teniendo en cuenta el tiempo de espe-ra y el elevado porcentaje de mortalidad en lis-ta de los pacientes con hipertensión pulmonar.
Pronóstico de la enfermedadLa predicción de la expectativa de vida de
un paciente en concreto es muy compleja, aun-que nos podemos aproximar a ella por gruposde enfermedades.
Las sociedades médicas implicadas en elprocedimiento de trasplante pulmonar publi-caron en 1998 las primeras guías para la selec-ción de los candidatos a trasplante pulmonarpor grupos de enfermedades, que han sidorecientemente renovadas(14) (Tabla 3).
En la hipertensión pulmonar, los test pro-nósticos más relevantes son las pruebas de
esfuerzo, siendo el más utilizado el test decaminar 6 minutos, que se correlaciona con elgrado funcional, el consumo de oxígeno y conla mortalidad de los pacientes con hiperten-sión pulmonar. La desaturación >10% en estetest incrementa la mortalidad 2,9 veces en lospacientes con hipertensión pulmonar idiopá-tica, y una distancia recorrida de menos de332 metros pronostica una mortalidad al añodel 40%(20). En el test de esfuerzo cardiopul-monar, un consumo máximo de oxígeno demenos de 10,4 ml/kg/min y una presión sis-tólica máxima de < 120 mmHg se ha asociadoa una mortalidad al año del 50 y 70%, res-pectivamente. Cuando coinciden estos dosvalores en un mismo paciente, la superviven-cia al año es del 23%(21). El cateterismo car-diaco también es predictivo de supervivencia,en función del índice cardiaco, resistencias pul-monares medias y presión de la arteria pul-monar media(15). La determinación de algunosneuropéptidos, como el BNP, ha demostradotambién correlacionarse con la disfunción ven-tricular y la mortalidad en la hipertensión pul-monar(22).
Tiempo en lista de esperaEl tiempo medio que los pacientes deben
permanecer en espera depende de la dispo-nibilidad de órganos, por lo que no es com-parable de unos países a otros, ni tampocodentro de las distintas unidades de un mismopaís.
La posición de los pacientes en lista de espe-ra en nuestro medio depende de la antigüedad.Cuando surge un donante, se selecciona elreceptor más antiguo en función del grupo san-guíneo y la capacidad pulmonar, que es unavariable directamente dependiente de la altu-ra. Por ello, otros factores que influyen en eltiempo de espera son el grupo sanguíneo y eltamaño del receptor. La desproporción entre elelevado número de candidatos y el limitadonúmero de donantes es la responsable de lamortalidad en lista de espera.
Los pacientes con fibrosis pulmonar idio-pática, fibrosis quistica o hipertensión pul-
C. LÓPEZ GARCÍA-GALLO ET AL.
124
TABLA 3. Criterios de trasplante en lahipertensión pulmonar
Criterios de derivación• Clase funcional III/IV (NYHA)• Enfermedad rápidamente progresiva
Criterios de inclusión en lista de espera• Clase funcional III/IV con máximo tratamien-
to médico• <380 metros en el T6M• Fracaso del epoprosterenol i.v.• Índice cardiaco <2 L/min/m2
• PAD >15 mmHg
(Modificado de Referencia 14)NYHA: New York Heart Association, T6M: test de lamarcha de los 6 minutos, PAD: presión de la aurícu-la derecha.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 124

monar idiopática tienen una menor supervi-vencia en lista de espera, en comparación conpacientes con enfisema o síndrome de Eisen-menger(23).
Tipo de trasplanteEn la HP idiopática se puede realizar tras-
plante unipulmonar, bipulmonar o cardiopul-monar (nivel de recomendación I-C). Según elregistro internacional, la HP supone un 3,3%de los trasplantes pulmonares realizados. Deellos, el 90% son bipulmonares. En cuanto altrasplante cardiopulmonar, la HP supone laindicación en el 27% de los casos(8).
La decisión de qué tipo de trasplante reali-zar es a veces difícil, y depende de cada cen-tro y de cada paciente. El trasplante unipul-monar ofrece la ventaja de que es técnicamentemás sencillo, con menos tiempo de isquemia,permitiendo además optimizar el número deórganos disponibles. El mayor problema de rea-lizar un trasplante unipulmonar es el manejopostoperatorio más complejo, ya que es fre-cuente la inestabilidad hemodinámica y el desa-rrollo de edema de reperfusión en el postope-ratorio inmediato, por la persistencia de ladisfunción ventricular en ese período, la pre-sencia de disbalance entre la ventilación y laperfusión pulmonar, y la diferencia de perfu-sión que existe entre el injerto y el pulmón nati-vo, donde persiste elevada la RVP(23,24).
Por el contrario, el trasplante bipulmonarpresenta menor disbalance ventilación/perfu-sión, lo que facilita el manejo postoperatorio.Además, algunos autores han publicado unamejoría en la función pulmonar y en la super-vivencia en estos pacientes(23). Sin embargo,otros estudios publicados por diferentes cen-tros no encontraron diferencias significativasen la supervivencia entre los dos tipos de tras-plante(25,26).
Por tanto, en la actualidad las recomen-daciones se inclinan a la realización de un tras-plante bilateral(27). En aquellos pacientes conHP secundaria a patología respiratoria, el tras-plante bipulmonar se considera de eleccióncuando la PAP > 40 mmHg
En cuanto al trasplante cardiopulmonar enla HP, inicialmente se asocia con un menornúmero de complicaciones postoperatorias.Sin embargo, en la actualidad, los buenos resul-tados obtenidos con el trasplante bipulmonary la escasez de órganos disponibles llevaríana la realización del trasplante cardiopulmonarsólo en aquellos pacientes con HP asociada adisfunción del ventrículo izquierdo, Síndromede Eissenmenger, o malformaciones cardiacasno reparables por cirugía(23).
Complicaciones postrasplante La principal diferencia en el manejo post-
operatorio del trasplante pulmonar por HPse debe a la mayor incidencia de disfunciónprecoz del injerto en estos pacientes. Se carac-teriza por la aparición de edema pulmonar,que se manifiesta como hipoxemia e infiltra-dos pulmonares, y es secundaria al daño deisquemia-reperfusión del injerto, que en el casode HP se ve además favorecida por la persis-tencia en algunos pacientes de disfunción ven-tricular derecha en el postrasplante inmedia-to, así como por el mayor flujo sanguíneo através del injerto en el caso del trasplante uni-lateral. Por ello hay que forzar la realizaciónde balance negativo, y utilizar ventilaciónmecánica con PEEP, la infusión de prosta-glandina o la administración de óxido nítricoinhalado(27).
El resto de las complicaciones postrasplanteson similares a las que se encuentran en otraspatologías(4). Las más frecuentes son infeccio-nes pulmonares, rechazo agudo o síndromede bronquiolitis obliterante (SBO). • Infecciones pulmonares. La infección más
frecuente en el postoperatorio inmediatoes la neumonía bacteriana y los gérmeneshabituales son la Pseudomonas aeruginosay el Staphylococcus aureus. A partir del pri-mer mes aumenta la incidencia de infec-ción por Citomegalovirus (CMV) que, ade-más, tiene efecto inmunomodulador(4).
• Rechazo agudo. Es otra complicación muyfrecuente, y un alto porcentaje de pacien-tes presentan al menos un episodio duran-
TRASPLANTE PULMONAR, CARDIOPULMONAR Y SEPTOSTOMÍA EN LA HIPERTENSIÓN PULMONAR (HP)
125
Monografía HAP NM 208 pag 18/2/10 17:08 Página 125

te el primer año. Aunque no suele ser cau-sa de mortalidad, se considera el factor deriesgo más importante para el desarrolloposterior de SBO.
• Síndrome de bronquiolitis obliterante(SBO). Es la complicación que más mor-bimortalidad ocasiona pasado el primeraño postrasplante. Se define como un des-censo del FEV1 > 20%, tras descartar otrascausas, como infecciones o alteraciones dela sutura bronquial. No se ha observadorelación entre su aparición y el tipo de tras-plante realizado. Para evitar estas complicaciones se debeiniciar desde el postoperatorio inmediatotratamiento inmunosupresor con tres fár-macos, tratamiento precoz de infecciones,así como profilaxis antibiótica en todos lospacientes.
Resultados• Supervivencia: según datos del Registro
Internacional de Trasplantes, la supervi-vencia del trasplante pulmonar en la HP esdel 78% a los tres meses, inferior a la obte-nida en la EPOC y en la FQ, probablementepor la mayor incidencia de complicacionesprecoces. Sin embargo, aquellos que sobre-viven el primer año tienen mayor super-vivencia a los diez años (45%) que los tras-plantados por EPOC (28%) y FPI (30%)(8),probablemente por ser pacientes más jóve-nes y con menor comorbilidad asociada.Al igual que en otras patologías, la mayorlimitación de la supervivencia a largo pla-zo se debe a al desarrollo de SBO.Por su parte, la supervivencia del trasplantecardiopulmonar por HP es del 72% a lostres meses, y del 64% al año. La esperan-za de vida media es de 9,2 años una vezsuperado el primer año.
• Parámetros hemodinámicos: tras la rea-lización, trasplante pulmonar, el pacientehabitualmente experimenta ya desde elpostrasplante inmediato, una reducción dela presión media de la arteria pulmonarque, en la mayoría de los casos, oscila entre
20-25 mmHg(28). Otros parámetros hemo-dinámicos que mejoran en el postopera-torio inmediato son el índice cardiaco, laRVP y la disfunción ventricular derecha.En los casos en que esto no ocurre, elpaciente presenta mayor riesgo de morta-lidad perioperatoria.
SEPTOSTOMÍALa septostomía consiste en crear un defec-
to interauricular que permita la descompre-sión del ventrículo derecho, para mejorar asíel gasto cardiaco. Se produce un shunt dere-cha-izquierda, que conduce a un descenso enla saturación arterial de oxígeno, pero incre-menta el gasto cardiaco y mejora el transpor-te de oxígeno a nivel sistémico(4,23).
Las principales indicaciones son el empe-oramiento de la función ventricular derecha–a pesar de tratamiento médico–, que produ-ce insuficiencia cardiaca congestiva y síncopede esfuerzo.
Además, puede utilizarse como puente altrasplante y cuando no existe otra alternati-va terapeútica eficaz (nivel de recomendaciónI-C).
Se realiza mediante cateterismo cardiacoderecho e izquierdo. Se perfora el septo inter-auricular, y posteriormente se dilata con balón.Durante la intervención se monitoriza la presióndiastólica del ventrículo izquierdo y la saturaciónde O2, hasta conseguir un descenso del 10%.
La mortalidad asociada al procedimientoes del 7% las primeras 24 horas, y del 15%durante el primer mes, por lo que se debe rea-lizar en centros con experiencia y con posi-bilidad de trasplante en los pacientes candi-datos al mismo(23).
Tras la intervención se produce mejoría dela función ventricular derecha, descenso de lapresión de aurícula derecha así como dismi-nución de los episodios sincopales y mejoríade la clase funcional.
CONCLUSIÓNLa hipertensión pulmonar es una enfer-
medad infrecuente y grave, sobre la que se está
C. LÓPEZ GARCÍA-GALLO ET AL.
126
Monografía HAP NM 208 pag 18/2/10 17:08 Página 126

investigando con nuevas modalidades terape-úticas. El trasplante pulmonar representa la últi-ma alternativa terapeútica a considerar enpacientes con hipertensión pulmonar severa.El trasplante bipulmonar es el procedimientode elección en la actualidad, aunque es nece-sario valorar cada paciente individualizada-mente, contando con la experiencia del gru-po trasplantador. Son necesarias, pues, unidadesde referencia, con suficiente experiencia en elmanejo de estos complejos pacientes, para dilu-cidar la mejor opción terapéutica en cada caso.
BIBLIOGRAFÍA1. Rich S, Kaufmann E, Levy PS. The effect of
high doses of calcium-channel blockers on sur-vival in primary pulmonary hypertension. NEngl J Med. 1992; 327 (2): 76-81.
2. Daily PO, Dembitsky WP, Jamieson SW.Theevolution and the current state of the art ofpulmonary thromboendarterectomy. SeminThorac Cardiovasc Surg. 1999; 11 (2): 152-63.
3. Trulock EP. Lung transplantation for primarypulmonary hypertension. Clin Chest Med.2001; 22 (3): 583-93. Review.
4. Ussetti P, Varela A. Alternativas quirúrgicas enel tratamiento de la hipertensión pulmonargrave. Arch Bronconeumol. 2002; 38 (supl 1):33-8.
5. Gaine SP, Rubin LJ. Medical and surgical tre-atment options for pulmonary hypertension.Am J Med Sci. 1998; 315 (3): 179-84. Review.
6. Hardy JD, Webb WR, DaltonML Jr, et al. Lunghomotransplantations in man. JAMA. 1963;186: 1065-74.
7. Reitz BA, Wallwork JL, Hunt SA, et al. N EnglJ Med. 1982; 306 (10): 557-64.
8. Christie JD, Edwards LB, Aurora P, et al. Theregistry of the international society for heartand lung transplantation: twenty-sixth officialadult lung and heart-lung transplantationReport-2009.J Heart Lung Transplant. 2009;28 (10): 1031-49.
9. Astudillo J, Morell F, Bravo C, et al. The lungtransplant. JANO 1992; 63: 121-2.
10. Organización Nacional de Trasplantes. Minis-terio de Sanidad y Consumo. Estadistica dedonación y trasplante.www.ont.es
11. Morales P, Borro JM, Sales G, et al. The firstsimultaneous double unilateral lung trans-
plantation in Spain: clinical course and results.Transplantation Group.Arch Bronconeumol.1999; 35 (2): 97-9.
12. Padilla J, Calvo V, Teixidor J, et al. Pulmonary"twinning" transplantation procedure.Trans-plant Proc. 2002; 34 (4): 1287-9.
13. Núñez JR, Varela A, del Río F, et al. Bipulmo-nary transplants with lungs obtained from twonon-heart-beating donors who died out of hos-pital. J Thorac Cardiovasc Surg. 2004; 127 (1):297-9
14. Orens JB, Estenne M, Arcasoy S, et al. Inter-nacional Guidelines for the selection of lungtransplant candidates: 2006 Update- A con-sensos Report From the Pulmonary ScientificCouncil of the Internacional Society for Heratand Lung Transplantation. J Heart Lung Trans-plant. 2006; 25: 745-55.
15. D'Alonzo GE, Barst RJ, Ayres SM, et al. Survi-val in patients with primary pulmonary hyper-tension. Results from a national prospectiveregistry.Ann Intern Med. 1991; 115 (5): 343-9.
16. Brenot F. Primary pulmonary hypertension.Case series from France. Chest. 1994; 105 (2Suppl): 33S-36S.
17. Oaklley CW. Primary pulmonary hypertension.Case series from United Kingdom. Chest.1994; 105: 29S.
18. Barst RJ, Rubin LJ, Long WA, et al. A compa-rison of continuous intravenous epoprostenol(prostacyclin) with conventional therapy forprimary pulmonary hypertension. The PrimaryPulmonary Hypertension Study Group. N EnglJ Med. 1996; 334 (5): 296-302.
19. Hosenpud JD, Bennett LE, Keck BM, et al. TheRegistry of the International Society for Heartand Lung Transplantation: eighteenth OfficialReport-2001. J Heart Lung Transplant. 2001;20 (8): 805-15.
20. Miyamoto S, Nagaya N, Satoh T, et al. Clinicalcorrelates and prognostic significance of six-minute walk test in patients with primary pul-monary hypertension. Comparison with car-diopulmonary exercise testing. Am J RespirCrit Care Med. 2000; 161 (2 Pt 1): 487-92.
21. Wensel R, Opitz CF, Anker SD, et al. Assess-ment of survival in patients with primary pul-monary hypertension: importance of cardio-pulmonary exercise testing. Circulation 2002;106 (3): 319-24.
22. Nagaya N, Nishikimi T, Uematsu M, et al. Plas-ma brain natriuretic peptide as a prognostic indi-cator in patients with primary pulmonary hyper-tension. Circulation. 2000; 102 (8): 865-70.
TRASPLANTE PULMONAR, CARDIOPULMONAR Y SEPTOSTOMÍA EN LA HIPERTENSIÓN PULMONAR (HP)
127
Monografía HAP NM 208 pag 18/2/10 17:08 Página 127

C. LÓPEZ GARCÍA-GALLO ET AL.
128
23. Corris PA. Atrial septostomy and transplanta-tion for patients with pulmonary arterial hiper-tensión. Seminars in respiratory and criticalcare medicine 2009; 4 (30): 493-501.
24. Galie N, Hoeper M, Humbert M, et al. Guide-lines for the diagnosis and treatment of pul-monary hypertension. Eur Heart J. 2009; 30:2493-537.
25. Conte JV, Borja MJ, Pate ChB, et al. Lungtransplantation for primary and secondaryhypertension. Ann Thorac Surg. 2001; 72:1673-80.
26. Pasque MK, Trulock EP, Cooper JD, et al. Sin-gle lung transplantation for pulmonary hyper-tension: single institution experience in 34patients. Circulation. 1995; 92: 2252-8.
27. Keogh AM, Mayer E, Benza RL, et al. Inter-ventional and Surgical Modalities of Treatmentin Pulmonary Hypertension. JACC. 2009; 54,(1 Suppl): S67-77.
28. Doyle RL, McCrory D, ChannicK RN, et al. Sur-gical Treatments/Interventions for PulmonaryArterial Hypertension. Chest. 2004; 126; 63S-71S.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 128

INTRODUCCIÓNAunque la hipertensión pulmonar (HP) ha
sido durante años una patología poco cono-cida, infradiagnosticada y con escasos recur-sos para su tratamiento, en los últimos añosse han realizado numerosos avances tanto enel conocimiento de su fisiopatología como ensu diagnóstico, que ha llevado a una mayorinvestigación en las opciones terapéuticas queha favorecido el desarrollo de nuevos fárma-cos y estrategias de tratamiento hasta hacealgunos años impensables. Sin embargo, estemismo avance ha hecho que se multiplique lacomplejidad de los métodos diagnósticos quehace necesaria una mayor especialización detodo el personal sanitario implicado en la asis-tencia a estos pacientes.
La complejidad de administración demuchos de los fármacos que actualmente seemplean en el tratamiento de esta enferme-dad hace que el personal encargado de elloprecise una formación específica tanto en elmétodo de aplicación como de los distintosefectos adversos que pueden producir, asícomo la capacidad y conocimientos para reac-cionar con prontitud y diligencia si éstos seproducen.
Por otra parte, el elevado coste tanto de losmétodos diagnósticos como de tratamientoempleados hace que sea necesaria una opti-mización de recursos sanitarios que sólo pue-de ser llevada a cabo por personal perfecta-mente cualificado.
Un buen número de especialistas puedenser necesarios para intervenir en el diagnosti-co y tratamiento de estos pacientes: neumó-logos, cardiólogos clínicos, hemodinamistas,cirujanos cardiovasculares, intensivistas…
Todo esto hace necesario que exista per-sonal cualificado que coordine y sirva de nexode unión del paciente con todos los profesio-nales implicados en su tratamiento y segui-miento mediante un programa perfectamenteorganizado y definido en el que cada vez tienemás peso la enfermería especializada, que coor-dine un oportuno y adecuado soporte asisten-cial, sirviendo de nexo de unión en el manejohospitalario así como en su relación con otrosniveles asistenciales, como la asistencia ambu-latoria y seguimiento domiciliario.
La educación del paciente e incluso de sufamilia es un proceso continuo que deberíaincorporar entrenamiento verbal, tanto por par-te del médico como del personal de enferme-ría así como materiales escritos, también comola opción para que pueda obtener informaciónfidedigna y la posibilidad de ponerse en con-tacto con otros pacientes que presenten su mis-ma patología e integrarse en asociaciones delos mismos.
Este capítulo pretende dar una visión glo-bal de la participación de la enfermería espe-cializada en este ya apasionante campo, asícomo ofrecer algunas pautas concretas demanejo a este nivel.
PRIMER ENCUENTRO-ACOGIDACuando nos encontramos por primera vez
ante un paciente es adecuado dirigirse a élsiempre por su nombre, esto produce tran-quilidad. Deberemos aquí iniciar una relaciónde confianza con el paciente, lo cual es útilpara poder realizar una valoración global desu estado y poder así ofrecer un mejor apoyoen todos los ámbitos. Es importante conocercómo se siente el paciente y cómo ve él su
129
ENFERMERÍA E HIPERTENSIÓN ARTERIALPULMONAR (HAP)
Fulgencio González Garrido
Monografía HAP NM 208 pag 18/2/10 17:08 Página 129

enfermedad, y también su familia más direc-ta, la que le servirá de apoyo durante todo elproceso, para poder conocer sus miedos e inse-guridades e intentar despejarlas.
Deberemos explicar al paciente con pala-bras claras, sencillas y serenas, pero tambiéncon sinceridad, cuál es su situación en esemomento y lo que se espera conseguir contodo lo que a partir de ese momento se va ainiciar; es importante que nos aseguremos deque el paciente comprenda lo que le estamoscomunicando. Se le expondrán al paciente laspruebas que se le pretende realizar y la expe-riencia que el equipo acumula en las mismaspara así tranquilizarle y hacer que pueda sen-tirse más seguro y con confianza.
DIAGNÓSTICOLa enfermería participa de manera activa
en un buen número de las pruebas diagnósti-cas que se van a realizar en los pacientes consospecha de HP, algunas de ellas podrán ser rea-lizadas por la enfermería con formación gené-rica pero otras precisarán de una formación alta-mente especializada. Entre la primeras seencuentran: analítica (microbiología, hemato-logía y bioquímica). Pruebas respiratorias (prue-bas funcionales respiratorias, gasometría), Prue-bas cardiacas (electrocardiograma).
La enfermería especializada participará enpruebas más específicas, como son: el test dela marcha y el cateterismo cardiaco.
Es posible que no se realicen todas estasexploraciones en todos los enfermos y oca-sionalmente se pueden añadir otras en fun-ción de las necesidades del paciente.
Antes del ingreso del paciente en la uni-dad, dispondremos de un plan con las prue-bas que se le realizarán dándole informacióndetallada y comprensible sobre ellas y asegu-rándole que nos encargaremos personalmen-te de su coordinación.
Test de la marcha Puede ser realizado por el el personal de
enfermería especializada contando con la asis-tencia del personal médico especializado.
Se realizará en un lugar de longitud ade-cuada, de alrededor de 30 m o mayor, con elpiso plano, duro y nivelado, que no presenteobstáculos con una temperatura y humedadagradables. Debemos disponer de cronóme-tro y medidor de la saturación de oxígeno por-tátil. Se indicará al paciente que use ropa cómo-da, calzado adecuado y evite comer en lashoras previas al estudio y no haga ejercicio enlos minutos previos. Se tomarán la frecuenciacardiaca, saturación de oxígeno y tensión arte-rial basal, antes de iniciar la marcha. Solicita-remos al paciente que nos comunique si tie-ne sensación de disnea, dolor torácico u otrossíntomas para detener la marcha si se produ-cen o si cae la saturación de oxígeno por deba-jo de los limites establecidos. Al finalizar laprueba se volverán a tomar al paciente de nue-vo las constantes tomadas a su inicio así comose deberá recoger la distancia recorrida y eltiempo empleado así como si ha tenido quedetener la marcha antes de su finalización esti-pulada por algún síntoma.
Cateterismo cardiacoEn esta prueba, de imprescindible realiza-
ción en todo paciente en el que se sospecheHTP, el papel de la enfermería es de sumaimportancia y en muchos programas de mane-jo de esta patología es la encargada de facilitaral paciente un entorno seguro y confortable,pues en este tipo de pruebas los pacientes sue-len presentar una gran carga de inseguridady ansiedad, manifestando un nerviosismo quela enfermera deberá intentar calmar, indicán-dole en qué consiste la prueba de forma claray sencilla aunque detallada, habiéndole ins-truido previamente sobre la necesidad de queesté en ayunas en el momento de la realiza-ción de la técnica. Indicará la duración apro-ximada de la prueba, el instrumental que seutilizará para que cuando se realice este ya algofamiliarizado con él, así como la necesidad deestar inmóvil y de colaborar con las instruc-ciones que le dé el médico que realiza la prue-ba. La enfermera le tranquilizará, asegurándo-le que estará en todo momento controlado por
F. GONZÁLEZ GARRIDO
130
Monografía HAP NM 208 pag 18/2/10 17:08 Página 130

ella misma, manteniendo una conversaciónsegura y serena con el paciente, cubriendo enmuchos casos un apoyo casi familiar que enese momento el paciente no tiene, ya que eneste tipo de pacientes habitualmente no estáindicada la administración de sedantes. Tam-bién será la encargada de procurar la vía veno-sa y preparar el contenido de la perfusión queadministrar al paciente en coordinación con elmédico que realiza el cateterismo. Se informaráal paciente de los posibles efectos de la medi-cación para que se lo comunique en caso deque aparezcan para ajustar así la infusión ydosis máxima que se administra. Por otra par-te, participará en una completa y exacta reco-gida de la datos obtenidos en la prueba de vaso-rreactividad y la custodia de los registrosrealizados para ser posteriormente analizados.Finalmente, una vez terminada la prueba, toma-rá al paciente las constantes vitales, compro-bando su estabilidad antes de abandonar la salade hemodinámica y posteriormente dará alpaciente las instrucciones que ayuden a evitarcomplicaciones por movilización en las siguien-tes horas a la misma.
TRATAMIENTO EN EL HOSPITAL
Antes de iniciar el tratamientoUna vez que se ha decidido el tratamien-
to que se va a instaurar la enfermería, en coor-dinación con el equipo médico, debería re-visar con el paciente la indicación deltratamiento así como comentar con él lasexpectativas reales de la mejoría de sus sín-tomas, los potenciales efectos adversos quese pueden producir, los criterios de notifica-ción de posibles problemas al equipo de tra-tamiento y la programación de las revisio-nes y pruebas a realizar para su control.También ofrecer al paciente la posibilidad decomunicar los problemas surgidos entre lasvisitas programadas mediante llamada tele-fónica y la posibilidad de solución por esta víao mediante una visita no programada o asis-tencia de la enfermera especializada a niveldel domicilio del paciente.
La enfermería juega un papel clave en elmanejo día a día de este tratamiento.
Asimismo, la enfermera deberá informaral paciente de que la suspensión repentina dela medicación puede provocar un empeora-miento significativo de los síntomas; por estemotivo deberá indicar al paciente que dispongade suficiente tratamiento para utilizar si la visi-ta programada sufre algún retraso no con-templado.
Tratamiento específicoLa mayoría de los tratamientos son caros
y complejos, lo que requiere un exhaustivoentrenamiento del paciente y su familia, estohabitualmente recae en la enfermería espe-cializada. No debería comenzarse hasta estarseguros de que el paciente y su entorno com-prenden y aceptan el tratamiento que se le haindicado.
Prostaciclina parenteralEn general, el tratamiento con prostaciclina
parenteral debería ser iniciado en el hospital.Una amplia información acerca del manejo delmedicamento y del sistema de administracióndebería darse antes, durante y después del ini-cio del tratamiento y puede ser dada por laenfermería especializada en coordinación conel equipo médico.
El tratamiento con prostaciclina i.v. requie-re un vía central y la enfermera, conjuntamentecon el paciente, debe identificar el sitio ade-cuado para ésta, así como, es importe la pre-sencia de la enfermera especializada en el cui-dado y control durante el reemplazamiento deesta vía y que la infusión se produce correc-tamente y no se interrumpe.
Como se ha descrito en otros capítulos,el epoprostenol es la prostaciclina de síntesispara utilización como tratamiento intraveno-so de la HP. Dada la complejidad y los posiblesefectos, tanto del tratamiento como de su posi-ble repentina suspensión, la actuación en sumanejo hace imprescindible el concurso deenfermería altamente especializada que debe-rá comprobar la adecuada monitorización del
ENFERMERÍA E HIPERTENSIÓN ARTERIAL PULMONAR (HAP)
131
Monografía HAP NM 208 pag 18/2/10 17:08 Página 131

paciente, que se encuentra relajado y tumba-do en decúbito supino y, posteriormente, pro-curar un acceso venoso por donde suministrarel fármaco que previamente ha preparado bajouna adecuada supervisión médica. Durante elproceso de infusión deberá comprobar lasconstantes del paciente, incluida la oximetríay la aparición de posibles efectos adversos quese pueden producir. Una vez iniciada la per-fusión deberá controlar las constantes vitalesde forma programada según se va incremen-tando el ritmo de administración, así comodisminuirla si aparecen efectos secundarios.Pasadas la primeras 24/48 horas, el pacientecomenzara a levantarse y deambular tran-quilamente y será función de la enfermeracomprobar nuevamente las constantes en estanueva situación comprobando asi que el tra-tamiento se podrá mantener de forma ambu-latoria, siendo en ese momento clave la correc-ta preparación de la bomba de infusión asícomo asegurarse de su correcto funciona-miento y tolerancia previamente al abandonodel paciente del hospital.
A partir de este momento comienza otrade las acciones de gran importancia de la enfer-mera especializada en HP como es el la for-mación del paciente y su soporte familiar paraun correcto uso del tratamiento, instruyendotanto al paciente como a su familia sobre losmecanismos del dispositivo y sus posibles defi-ciencias así como comprobar que el pacientecomprende perfectamente las enseñanzas reci-bidas, observando de forma práctica cómo pro-cede su manipulación antes de ir a su domi-cilio. No menos importante es el insistir en laabsoluta adherencia al tratamiento así comode las posibles consecuencias de su abandonosuministrándole, además de la formación, ins-trucciones escritas para que el paciente y sufamilia las puedan consultar en su domicilio.
El fármaco debe ser reconstituido antes deusarse. Se ha de adoptar un cuidado especialen la preparación y en el cálculo de la veloci-dad de perfusión. La reconstitución y dilucióndeberá realizarse en condiciones asépticas,inmediatamente antes de la utilización clínica.
Se debe utilizar únicamente la disolucióntampón estéril para la reconstitución. Se toma-rán unos 10 ml de la disolución tampón esté-ril con una jeringa también estéril e inyectar-los dentro del vial, que contiene 0,5 mg delproducto liofilizado, y agitar suavemente has-ta que se haya disuelto el polvo. Posterior-mente, rellenar la jeringa con la disoluciónresultante, volver a inyectarla con el volumenremante de la disolución tampón estéril y mez-clar concienzudamente. A esta disolución sele denomina disolución concentrada y contie-ne 10.000 ng por ml de epoprostenol. Sólo estadisolución concentrada será la adecuada paraposteriores diluciones antes de su uso.
Dilución: sólo el diluyente estéril suminis-trado puede ser utilizado en la dilución poste-rior de flolan reconstituido. La disolución sali-na fisiológica no deberá ser utilizada si se vaa administrar en el tratamiento de la HP y nodebe ser administrada con otras disolucioneso medicaciones parenterales.
Para diluir la solución concentrada, se debepasar ésta a una jeringa grande y luego aco-plar el filtro estéril suministrado a la jeringa.Dispersar la solución concentrada directamenteal diluyente estéril, utilizando una presión sufi-ciente pero no excesiva. Normalmente, se tar-dan unos 70 segundos para la filtración de 50ml de solución concentrada. Mezclarlo bien.La unidad de filtro deberá ser utilizada una solavez y luego, desechada.
Las concentraciones habitualmente utili-zadas en el tratamiento de la HP son lassiguiente: • 150.000 ng/ml-1,5 mg de epoprostenol
reconstituido y diluido a un volumen totalde 100 ml en diluyente estéril.
• 10.000 ng/ml- dos viales que contiene 500µg de epoprostenol reconstituido y diluidoa un volumen total de 100 ml.
• Cálculo de la velocidad de perfusión: lavelocidad de perfusión puede ser calcula-da a partir de la fórmula:Velocidad de perfusión (ml/min): dosis(ng/kg/min) x peso corporal (kg) /concen-tración de la disolución (ng/ml).
F. GONZÁLEZ GARRIDO
132
Monografía HAP NM 208 pag 18/2/10 17:08 Página 132
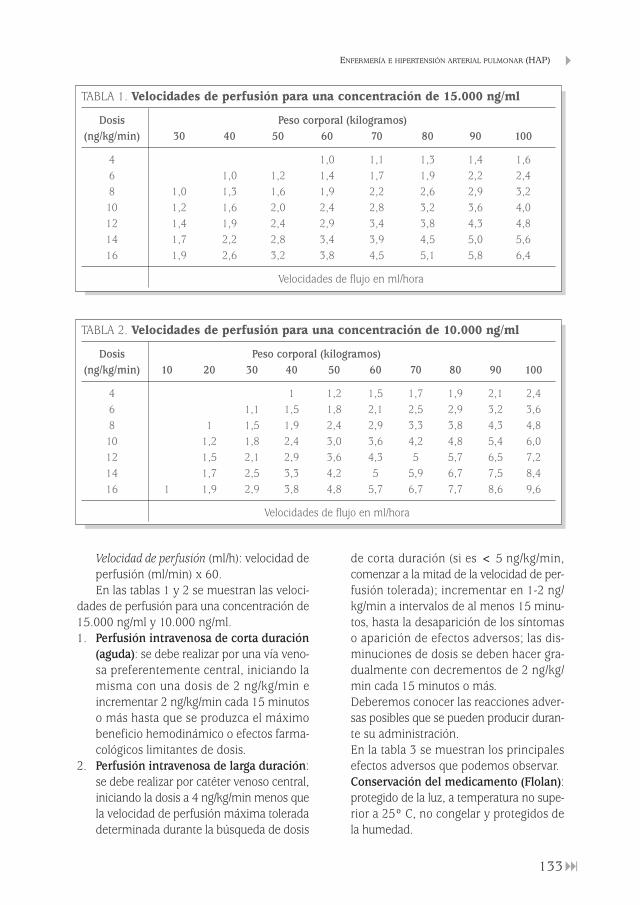
Velocidad de perfusión (ml/h): velocidad deperfusión (ml/min) x 60. En las tablas 1 y 2 se muestran las veloci-
dades de perfusión para una concentración de15.000 ng/ml y 10.000 ng/ml.1. Perfusión intravenosa de corta duración
(aguda): se debe realizar por una vía veno-sa preferentemente central, iniciando lamisma con una dosis de 2 ng/kg/min eincrementar 2 ng/kg/min cada 15 minutoso más hasta que se produzca el máximobeneficio hemodinámico o efectos farma-cológicos limitantes de dosis.
2. Perfusión intravenosa de larga duración:se debe realizar por catéter venoso central,iniciando la dosis a 4 ng/kg/min menos quela velocidad de perfusión máxima toleradadeterminada durante la búsqueda de dosis
de corta duración (si es < 5 ng/kg/min,comenzar a la mitad de la velocidad de per-fusión tolerada); incrementar en 1-2 ng/kg/min a intervalos de al menos 15 minu-tos, hasta la desaparición de los síntomaso aparición de efectos adversos; las dis-minuciones de dosis se deben hacer gra-dualmente con decrementos de 2 ng/kg/min cada 15 minutos o más.Deberemos conocer las reacciones adver-sas posibles que se pueden producir duran-te su administración.En la tabla 3 se muestran los principalesefectos adversos que podemos observar. Conservación del medicamento (Flolan):protegido de la luz, a temperatura no supe-rior a 25º C, no congelar y protegidos dela humedad.
ENFERMERÍA E HIPERTENSIÓN ARTERIAL PULMONAR (HAP)
133
TABLA 1. Velocidades de perfusión para una concentración de 15.000 ng/ml
Dosis Peso corporal (kilogramos)(ng/kg/min) 30 40 50 60 70 80 90 100
4 1,0 1,1 1,3 1,4 1,66 1,0 1,2 1,4 1,7 1,9 2,2 2,48 1,0 1,3 1,6 1,9 2,2 2,6 2,9 3,210 1,2 1,6 2,0 2,4 2,8 3,2 3,6 4,012 1,4 1,9 2,4 2,9 3,4 3,8 4,3 4,814 1,7 2,2 2,8 3,4 3,9 4,5 5,0 5,616 1,9 2,6 3,2 3,8 4,5 5,1 5,8 6,4
Velocidades de flujo en ml/hora
TABLA 2. Velocidades de perfusión para una concentración de 10.000 ng/ml
Dosis Peso corporal (kilogramos)(ng/kg/min) 10 20 30 40 50 60 70 80 90 100
4 1 1,2 1,5 1,7 1,9 2,1 2,46 1,1 1,5 1,8 2,1 2,5 2,9 3,2 3,68 1 1,5 1,9 2,4 2,9 3,3 3,8 4,3 4,810 1,2 1,8 2,4 3,0 3,6 4,2 4,8 5,4 6,012 1,5 2,1 2,9 3,6 4,3 5 5,7 6,5 7,214 1,7 2,5 3,3 4,2 5 5,9 6,7 7,5 8,416 1 1,9 2,9 3,8 4,8 5,7 6,7 7,7 8,6 9,6
Velocidades de flujo en ml/hora
Monografía HAP NM 208 pag 18/2/10 17:08 Página 133

Fármacos subcutáneosEl teprostinil es el medicamento que actual-
mente se administra por esta vía (aunque tam-bién se puede administrar por vía intraveno-sa); se presenta en frascos-ampolla de 20 mlcon diversas concentraciones (1,0; 2,5; 5 y 10mg/ml), pudiéndose administrar además depor vía subcutánea sin diluir como por infu-sión intravenosa diluido en agua estéril o solu-ción fisiológica al 0,9%.
La infusión se realiza utilizando una bom-ba diseñada para la administración subcutá-nea de medicamentos, que suelen ser depequeño tamaño y bajo peso, que dispone deuna jeringa conectada al sistema de infusión.Se puede programar el ritmo de infusión y sumanejo es lo suficientemente asequible comopara que pueda hacerse sin problemas.
En la administración subcutánea continuase realiza mediante un sistema de alargaderamás catéter subcutáneo; se deberá desinfec-tar previamente la zona donde se va a inser-tar, habiendo realizado previamente una correc-ta higiene de manos. Los materiales que seutilizan son de un solo uso y, por tanto, des-echable. Es frecuente la inflamación de la zona
donde se inserta y debe ser tratado de acuer-do con la seriedad del mismo, que irá desdetratamientos tópicos (frío, analgésicos…) has-ta la necesidad de analgésicos y antibióticossistémicos. Con frecuencia deberemos pasara administrar al paciente un tratamiento sub-cutáneo cuando el paciente se encuentra contratamiento iniciado a nivel intravenoso. En latabla 4 se ofrece un ejemplo de las dosis deprostaciclina durante en cambio de trata-miento.
Es preferible utilizar una dosis de infusiónfija para así poder cambiar el sistema de infu-sión a una hora concreta siempre. Se debetener preparada la siguiente dosis antes deinterrumpir la actual.
Cuando está preparada la infusión y pur-gada la alargadera se realiza el clampaje reti-rando la vía anterior y colocando la nueva, vol-viendo luego a desclampar.
La dosis se administra en función del pesodel paciente, la concentración y la dosis a admi-nistrar según la formula:
Velocidad de infusión subcutánea (ml/h)= [Dosis (ng/kg/min) x Peso (kg) x 0,00006*]
por ejemplo, si en una persona de 60 kg ladosis inicial fuese de 1,25 mg/kg/min utilizandouna ampolla con la concentración de 1 mg/mlla velocidad de infusión se calcularía:
[1,25 mg/kg/min x 60 x 0,00006] 1 mg/ml= 0,005 ml/hora
Pauta dosificación EE.UU.: inicio: 1,25ng/kg/min; incrementar dosis 1,25 ng porsemana hasta estabilización de síntomas; encaso de efectos secundarios severos, reducirdosis. Pauta dosificación actual: inicio: con 2-2,5 ng/kg/min.; incremento en 2,5 ng c/12 a24 horas según tolerancia; el objetivo es esta-bilizar al paciente en 10-15 ng/kg/min.
En la tabla 5 se expresan las principalesreacciones adversas a las que la enfermeradeberá estar atenta para su posible actuacióno comunicación al personal médico.
F. GONZÁLEZ GARRIDO
134
TABLA 3. Efectos adveros duranteadministración de epoprostenol
Rubor 58%Cefalea 49%Náuseas/vómitos 32%Hipotensión 16%Ansiedad, nerviosismo, agitación 11%Dolor torácico 11%Mareo 8%Bradicardia 5%Dolor abdominal 5%Dolor muscular 3%Disnea 2%Dolor de espalda 2%Sudoración 1%Dispepsia 1%Hipoestesia/parestesia 1%Taquicardia 1%
Monografía HAP NM 208 pag 18/2/10 17:08 Página 134

Fármacos inhaladosLos inconvenientes producidos por la medi-
cación parenteral favoreció la investigación connuevas formas de de administración del trata-miento más seguras y con menos efectos adver-sos pero con un potente efecto hipotensor, comoson los fármacos inhalados, esencialmente ilo-prost. Sin embargo, este tipo de administraciónde la medicación también plantea algunosinconvenientes en los que la participación dela enfermera experta es fundamental. Por una
parte, los equipos de inhalación pueden resul-tar complejos de entender y manipular por algu-nos pacientes y, aunque las empresas que lossuministran poseen personal que realiza estafunción, la enfermera especializada debe cono-cer el funcionamiento y manejo de los diversosdispositivos que en cada medio se pueden uti-lizar para poder solucionar los problemas quese le planteen a los pacientes con ellos y; ade-más, mantener al día la formación sobre el tra-tamiento que éstos necesitan. Por otra partela relativa complejidad de estos dispositivos pue-de hacer que la adherencia al tratamiento dis-minuya y seria también una razonable actua-ción de la enfermería el potenciarla en cadavisita o consulta que realicen.
La solución se presenta ya lista para usar,sin necesitad de preparación ni reconstituciónsi se emplea el Ventavis y se administramediante dispositivos adecuados para la inha-lación, nebulizadores. Se mantendrá el trata-miento previo, ajustándolo a las necesidadesindividuales. Los nebulizadores adecuados parala inhalación de iloprost deben administrar 2,5ó 5 microgramos de iloprost a través de laboquilla en un tiempo de unos 5 a 10 minu-tos. La mediana del diámetro aerodinámico demasa (Mass Median Aerodynamic Diameter,MMAD) del aerosol debe ser de 3-4 micróme-tros y el caudal total, de 0,05-0,4 mg/min.
Para reducir al mínimo la exposición acci-dental, se recomienda administrarlo con nebu-lizadores con un filtro o sistema estimuladopor inhalación, y mantener la habitación bienventilada.
ENFERMERÍA E HIPERTENSIÓN ARTERIAL PULMONAR (HAP)
135
TABLA 4. Dosis de prostaciclina durante en cambio de tratamiento
Basal (día 0) Día 1 Día 2 Día 3 Día 4 Día 5Caso Ep Tep Ep Tep Ep Tep Ep Tep Ep Tep Ep Tep
1 13 2,5 9 5 4 10 0 162 10 3,75 8,5 6,25 4 10 0 12,53 12 2,5 10 5 7 7,5 5 10 2 12,5 0 154 11 3,75 8 6,25 5 8,75 3 11,25 0 15
Ep: dosis de epoprostenol intravenoso (ng/kg/min); Tep: dosis de treprostinil subcutáneo (ng/kg/min)
TABLA 5. Principales reacciones adversasdurante la infusión de teprostinil
Remodulin PlaceboEfecto adverso N=236 N=233
Dolor en el sitio 85 27de infusión
Reacción en el 83 27sitio de infusión
Cefalea 27 23
Diarrea 25 16
Náuseas 22 18
Exantema 14 11
Dolor mandibular 13 5
Vasodilatación 11 5
Vértigo 9 8
Edema 9 3
Prurito 8 6
Hipotensión 4 2
Monografía HAP NM 208 pag 18/2/10 17:08 Página 135

Dosis recomendada• Adultos: cada sesión de inhalación debe
empezar con 2,5 microgramos de iloprost(liberados por la boquilla del inhalador). Ladosis se puede aumentar a 5,0 microgra-mos de iloprost, dependiendo de las nece-sidades y la tolerancia de cada paciente.La dosis de cada sesión de inhalación seadministrará de 6 a 9 veces al día, en fun-ción de las necesidades y la tolerancia indi-viduales.Dependiendo de la dosis que se desee enla boquilla y en el nebulizador, cada sesiónde inhalación dura de 5 a 10 minutos apro-ximadamente.En la tabla 6 se muestran las dosis y tiem-pos medios según el tipo de dispositivo.
• Pacientes con insuficiencia hepática: laeliminación de iloprost se reduce en lospacientes con disfunción hepática.Para evitar una acumulación indeseabledurante el transcurso del día, se debe ejer-cer especial precaución con estos pacien-tes durante la fase de titulación inicial dela dosis. Inicialmente se deben adminis-trar dosis de 2,5 microgramos con inter-valos de dosis de por lo menos 3 horas (locual corresponde a una administración demáximo 6 veces al día). Posteriormente,se pueden reducir los intervalos entre dosiscon precaución, teniendo en cuenta la tole-rancia individual. Si está indicado un
aumento adicional de la dosis hasta 5,0microgramos, nuevamente se deben ele-gir inicialmente intervalos de por lo menos3 horas, los cuales se acortarán de acuer-do con la tolerancia individual. No es pro-bable que se presente una acumulaciónindeseable adicional del medicamento des-pués de varios días de tratamiento, debi-do a la presencia de la pausa nocturna quese intercala durante la administración delmedicamento.
• Pacientes con insuficiencia renal: no haynecesidad de ajustar la dosis en pacientescon un aclaración de creatinina > 30ml/min. Con base en datos obtenidos coniloprost administrado por vía intravenosa,se sabe que la eliminación está reducidaen pacientes con insuficiencia renal querequieren diálisis.En la tabla 7 se muestran los principalesefectos adversos en la administración deiloprost.
TRATAMIENTO AMBULATORIOEl tratamiento ambulatorio del paciente con
HP tiene en esta patología especial importanciadado que la mayor parte del mismo se realiza-rá a este nivel, tanto si se realiza a nivel hospi-talario como a nivel mixto hospitalario y domi-ciliario en las unidades preparadas para ello.
No cabe ninguna duda de la importanciadel papel de enfermería en el manejo ambu-
F. GONZÁLEZ GARRIDO
136
TABLA 6. Dosis y tiempos medios de inhalación según el tipo de dispositivo
Tiempo de inhalaciónDispositivo Dosis de iloprost 15 respiraciones/minuto
HaloLite 2,5 microgramos 4-5 minutos5 microgramos 8-10 minutos
Prodose 2,5 microgramos 4-5 minutos5 microgramos 8-10 minutos
Venta-Neb 2,5 microgramos 4 minutos5 microgramos 8 minutos
I-Neb AAD 2,5 microgramos 3,2 minutos5 microgramos 6,5 minutos
Monografía HAP NM 208 pag 18/2/10 17:08 Página 136

lante hospitalario, pero donde esta figura cobrauna importancia que la hace imprescindiblees cuando se puede hacer, cosa que seria loideal según diversas entidades internaciona-les, a nivel domiciliario.
En el ámbito domiciliario la enfermeraespecialista en HP debería figurar como ele-mento clave para el control y seguimiento delpaciente entre las distintas visitas hospitala-rias programadas. En cada visita sería la encar-gada de realizar una evaluación de la situaciónclínica del paciente, observando y anotando laevolución de la sintomatología, así como consu evolución de forma objetiva verificandosu tensión arterial, frecuencia cardiaca, reali-zación de electrocardiograma y registro oxi-métrico, entre otras, según las característicasdel paciente.
En aquellos casos en que el tratamientosea administrado mediante dispositivos espe-ciales, como bombas de infusión, dispositivosde inhalación, entre otros, comprobará elcorrecto funcionamiento de los mismos asícomo la capacidad del paciente en su uso.Podría proceder a extracción de controles ana-líticos tanto si están programados como si lacondición del estado del paciente le indicansu pertinencia.
La enfermería experta estaría especial-mente entrenada en la detección de posiblesproblemas psicológicos a los que estos pacien-tes están especialmente expuestos, así como
valorar la necesidad de un apoyo por otro per-sonal especialista, como sicólogos o psiquia-tras.
Una vez finalizada la visita a domicilio, laenfermera recogerá todos los datos, tanto dela entrevista como del estado de los mecanis-mos de dispensación de la medicación y de losresultados de los test objetivos realizados paraque puedan ser incorporados al historial clíni-co del paciente y puedan ser evaluados por todoel equipo encargado de su asistencia.
Finalmente, si la situación del paciente asílo aconseja, podría establecer un adelanto delas visitas programadas y, si fuera necesario,contactar con el personal médico del hospi-tal para proceder a su ingreso si las circuns-tancias objetivadas así lo aconsejan.
Las visitas a domicilio del paciente po-drían ser útiles, además de disminuir en algu-nos casos las visitas hospitalarias, para apor-tar un mayor confort y seguridad del mismoya que serian programadas en función de lasituación del paciente, ofreciendo la suficienteflexibilidad para acordar visitas extraordina-rias si el contacto telefónico con el pacienteasí lo aconseja.
BIBLIOGRAFÍA1. Yates G, Saunders K. Pulmonary hypertension:
a review for nurses Can J Cardiovasc Nurs.2008; 18 (1): 7-14.
2. Cheever KH. An verview of pulmonary arte-rial hypertension: risks, pathogenesis, clinicalmanifestations, and management. J Cardio-vasc Nurs. 2005; 20 (2): 108-16; quiz, 117-8.
3. Badesch DB, Raskob GE, Elliott CG, et al. Pul-monary Arterial Hypertension: Baseline Cha-racteristics From the REVEAL Registry. Chest.2009; Oct 16.
4. Galie N, Hoeper MM, Humbert M, et al. Gui-delines for the diagnosis and treatment of pul-monary hypertension. The task force for thediagnosis and treatment of pulmonary hyper-tension of the European Society of Cardiology(ESC) and the European Respiratory Society(ERS), endorsed by the International Societyof Heart and Lung Transplantation (ISHLT). EurRespir J. 2009; Sep 24.
ENFERMERÍA E HIPERTENSIÓN ARTERIAL PULMONAR (HAP)
137
TABLA 7. Efectos adversos posibles deliloprost inhalado
Efecto Iloprost Placebo Valor de P
Aumento de tos 38,6% 25,5% 0,0513Cefalea 306% 19,6% 0,1054Vasodilatación 27% 8,8 % 0,0009Síndrome gripal 14% 9,8% nsEdema periférico 13% 15,7% nsNáuseas 13% 7,8% nsTrismus 12% 2,9% 0,0166Hipotensión 11% 5,9% 0,2163
Monografía HAP NM 208 pag 18/2/10 17:08 Página 137

F. GONZÁLEZ GARRIDO
138
5. Gaudó Navarro J, Suerio Bendito A. Aproxi-mación al diagnóstico de la hipertensión pul-monar. De la sospecha clínica a los procedi-mientos invasivos. Revista de PatologíaRespiratoria. 2007; 10 (supl. 2): 136-42.
6. Gardner A, Hase S, Gardner G, et al. From com-petence to capability: a study of nurse prac-titioners in clinical practice. J Clin Nurs. 2008;17 (2): 250-8.
7. Grantham G, McMillan V, Dunn SV, et al.Patient self-medication--a change in hospitalpractice. J Clin Nurs. 2006; 15 (8): 962-70.
8. Lluis Valera J, Melgosa MT, Barberá JA. Estu-dio hemodinámico pulmonary y prueba vaso-dilatadora aguda. En: Técnicas y procedi-
mientos en Hipertensión pulmonar. Manualde Procedimientos. SEPAR. 17-35.
9. Bueno N, Fernández A, Noguera S, et al. Admi-nistración de prostaciclina por via endoveno-sa. En: Técnicas y procedimientos en Hiper-tensión pulmonar. Manual de Procedimientos.SEPAR. 36-48.
10. Hernández Carcereny C. Control extrahospi-talario. En: Tecnicas y procedimientos enHipertensión pulmonar. Manual de Procedi-mientos. SEPAR. 69-76.
11. Escribano Subíasa P, Cea-Calvoa L, Tello deMenessesa R, et al. Transición de prostaciclinaintravenosa a subcutánea en la hipertensiónpulmonar. Rev Esp Cardiol. 2003; 56: 818-21.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 138

139
RESUMENUna vez diagnosticada la HP, el paciente
debe enfrentarse a una enfermedad que tieneun pronóstico sombrío, un protocolo de segui-miento y un tratamiento específico.
En el seguimiento hay que valorar la res-puesta al tratamiento, prevenir las complica-ciones, detectar precozmente el deterioro clí-nico y modificar la pauta de tratamiento segúnla evolución clínica. Por otro lado es precisoaportar información sobre la enfermedad yorientar a cerca de las fuentes de información,administrar consejos prácticos sobre las acti-vidades de la vida diaria y dar a conocer la exis-tencia de las asociaciones de enfermos. En oca-siones los pacientes presentan trastornospsicológicos y precisan soporte psicológico.
SEGUIMIENTO DE LOS PACIENTES CONHIPERTENSIÓN PULMONAR
En los últimos años, hemos asistido aimportantes avances en el tratamiento de lahipertensión arterial pulmonar, lo que hasupuesto el desarrollo de nuevas guías de actua-ción clínica centradas en el diagnóstico y tra-tamiento de estos pacientes. Sin embargo, estasguías de actuación no recogen aspectos másprácticos de organización asistencial o proto-colos de seguimiento para tales pacientes.
La Sociedad Española de Neumología yCirugía Torácica (SEPAR) y la Sociedad Espa-ñola de Cardiología (SEC) han publicado en el2008 un documento de consenso sobre los
estándares asistenciales en HP(1,19). En estedocumento se plantea un abordaje racional yescalonado del paciente con HP, estableciendouna vía clínica que va desde atención primariahasta las unidades especializadas de trasplan-te cardiopulmonar o cardiopatías congénitas,pasando por unidades clínicas locales y uni-dades de referencia en HP. Para cada escalónde la vía clínica se establecen unas competen-cias diferentes en el proceso diagnóstico, detratamiento y en el seguimiento que ahora nosocupa (Tabla 1).
Las unidades clínicas locales y las unida-des de referencia en HP van a ser los dos nive-
SEGUIMIENTO EXTRAHOSPITALARIO DE LOS PACIENTES CON HIPERTENSIÓNPULMONAR (HP): PROGRAMAS DEEDUCACIÓN AL PACIENTE Y FAMILIARES
Asunción Perpiñá Ferri, Pilar Alba García-Baquero, Inmaculada Fernández Rozas
TABLA 1. Niveles asistenciales en elmanejo de HP
Unidad clínica localSeguimiento regular• Control de función renal, hepática y equilibrio
hidroelectrolítico• Ajuste de tratamiento convencional• Cuidados de emergencia si hay complicaciones
Unidad de referenciaSeguimiento• Visitas cada 3-6 meses según situación clínica• Modificación del plan terapéutico según evo-
lución de éste• Asistencia al paciente si hay empeoramiento
o complicaciones• Indicación de procedimientos especiales
Monografía HAP NM 208 pag 18/2/10 17:08 Página 139

les implicados en el seguimiento del pacien-te con HP, aunque el objetivo va a ser comúnpara ambos, teniendo que existir una comu-nicación fluida y continua entre ambos.
El objetivo final siempre será mantener alpaciente en un perfil de bajo riesgo según semuestra en la tabla 2, a partir de lo que hoyse consideran indicadores pronósticos(2-4).
Todo el seguimiento se debe basar en: 1.valorar la respuesta al tratamiento; 2. prevenirlas complicaciones; 3. detectar precozmenteel deterioro clínico; 4. modificar la pauta detratamiento según la evolución clínica, y 5. lainformación sobre la enfermedad, consejosprácticos sobre las actividades de la vida dia-ria y soporte psicológico al paciente.
La SEC y la SEPAR recogen en el docu-mento de consenso unas recomendacionesrespecto a la frecuencia y contenido de las visi-tas de seguimiento, así como el nivel asisten-cial en el que se deben realizar éstas segúnmuestran las tablas 1 y 3(1,19).
Las herramientas básicas en estas visitasse pueden resumir en los siguientes cinco apar-tados:1. Valoración del grado funcional, síntomas
y signos de insuficiencia cardiaca derecha,considerándose datos de mal pronósti-co(3,5,6).• Clase funcional NYHA/OMS avanzada
III/IV.
• Signos de insuficiencia cardiaca derecha2. Determinaciones analíticas.
• Funciones hepática, renal y electrolitos,siendo la hiperuricemia un dato de malpronóstico.
• Determinación de péptidos natriuréti-cos. Se considera dato de mal pronós-tico la presencia de valores elevados.Valores de BNP >180 pg/ml se asociana una menor supervivencia(7).
3. La prueba de los 6 minutos de marcha. Seconsideran datos de mal pronóstico(2-4).• La distancia recorrida < 325 metros • Desaturación de oxígeno >10%.
4. Prueba de esfuerzo con estudio de inter-cambio gaseoso. Se consideran datos demal pronóstico(8).• Consumo máximo de oxígeno< 10,4
ml/kg/min.• Presión arterial sistólica < 120 mmHg.
5. Ecocardiograma Doppler transtorácico.El seguimiento ecocardiográfico permitevalorar la progresión de la enfermedad, elremodelado vascular y el pronóstico. Losparámetros que han demostrado más utili-dad clínica en el seguimiento, analizan lageometría y función del ventrículo derecho,la interdependencia entre ambos ventrícu-los, y la presencia de derrame pericárdico.• Valoración de las funciones sistólica y
diastólica de VD.
A. PERPIÑÁ FERRI ET AL.
140
TABLA 2. Perfil de riesgo de la evolución en el tratamiento de HP(1)
Bajo Determinante del riesgo Alto
No Evidencia clínica insuficiencia cardiaca derecha Sí
Estabilidad Progresión Rápida
> 500 m Distancia recorrida en la prueba de la marcha < 350 m
Valor normal cercano Péptidos natriuréticos Valor muy elevado
Disfunción leve de VD Ecocardiograma Derrame pericárdico. Disfunción grave de VD
Disfunción leve de VD Hemodinámica pulmonar PADm > 12 mmHgIC < 2 L/min/m2
SvO2 < 63%
Monografía HAP NM 208 pag 18/2/10 17:08 Página 140

- TAPSE, valora el acortamiento de lasfibras longitudinales de la pared libredel ventrículo derecho y se correlacio-na bien con la fracción de eyección iso-tópica o por resonancia cardiaca. Seconsidera factor pronóstico negativo unTAPSE <18 mm y, por cada descensode 1 mm de TAPSE, se eleva el riesgoen un 17%(9).
- El índice de Tei permite valorar la fun-ción ventricular del lado derecho inde-pendientemente de su geometría. Valo-ra la eficiencia del ventrículo derecho,y se establece que valores mayores de0,76 se asocian a mal pronóstico(10).
- Doppler tisular, se trata de valores quedependen poco de la precarga. Veloci-dades tisulares < 8,8 cm/seg, mues-tran una buena correlación con la dis-función ventricular derecha(11).
• Tamaño de la aurícula derecha. El índi-ce de área de la aurícula derecha >5cm2/m2 es factor predictivo de mortali-dad(12).
• Valoración de parámetros hemodiná-micos.
- Presiones en arteria pulmonar sistóli-ca, diastólica, media y presión de pul-so. Hay que recordar que la PSP comodato aislado no tiene valor pronósticoen el seguimiento.
- Índices de trabajo dP/dt.- Presión en aurícula derecha media,
valorando el colapso de la vena cavainferior.
- Resistencia vascular pulmonar, capa-citancia pulmonar e insuficiencias val-vulares.
• Presencia de derrame pericárdico. Supresencia, en ausencia de conectivopa-tía, se relaciona con la disfunción delventrículo derecho y se considera factorde mal pronóstico(12).
• Inter-independencia ventricular.- Ratio área del ventrículo derecho/
izquierdo > 1,72 se asocia a mal pro-nóstico(10).
- Índice de excentricidad, mide el gra-do de desviación septal y refleja larelación de presiones entra ambosventrículos > 1,8 se asocia a mal pro-nóstico(13).
SEGUIMIENTO EXTRAHOSPITALARIO DE LOS PACIENTES CON HIPERTENSIÓN PULMONAR (HP)…
141
TABLA 3. Seguimiento clínico(1)
Exploración Periodicidad
Síntomas (clase funcional) Variable. Primera a los 3 meses y, posteriormente a los 3-6 meses
Prueba de la marcha de 6 min Primera a los tres meses y, posteriormente, cada 6 meses
Radiografía de tórax 6 meses
Electrocardiograma 6 meses
Analítica 6 meses (excepto en pacientes tratados con antagonistas de los receptores de endotelina, control mensual de enzimashepáticas)
Ecocardiograma transtorácico 6-12 meses
Estudio hemodinámico pulmonar Según evolución, repetir si hay deterioro clínico o cambio terapéutico
Prueba de esfuerzo cardiopulmonar 6-12 meses (en pacientes en clases I-II o con distancia recorrida en 6 minutos>450 m o 80% del valor de referencia)
Péptidos natriuréticos 6 meses
Monografía HAP NM 208 pag 18/2/10 17:08 Página 141

- Función diastólica del ventrículoizquierdo. Su mejoría es indicador debuen pronóstico.
6. El estudio hemodinámico realizado a lostres meses del inicio del tratamiento tienevalor pronóstico, especialmente el cambioen el gasto cardiaco; sin embargo, al tra-tarse de un procedimiento invasivo, es pre-ferible reservarlo para pacientes con malaevolución. Se consideran datos de mal pro-nóstico(5):• Presión en aurícula derecha media> 20
mmHg.• Presión en arteria pulmonar media >
85 mmHg.• Índice cardiaco medio < 2 L/min/m2.
EL PACIENTE ANTE EL DIAGNÓSTICO DEHIPERTENSIÓN PULMONAR
Tras el diagnóstico de HP el paciente debeenfrentarse a una enfermedad que tiene unpronóstico sombrío, un protocolo de segui-miento y un tratamiento específico.
Afrontar el diagnósticoEn primer lugar ha de afrontar el diagnós-
tico, aceptar y conocer su enfermedad. Los pro-fesionales sanitarios debemos ayudar al pacien-te proporcionando información y programaseducativos. Podemos enseñarles a reorganizarsu actividad y a asumir sus limitaciones.
El paciente va a buscar información y, hoydía, Internet es la primera fuente de informa-ción(14). Nosotros podemos y debemos orien-tar acerca de las fuentes de información másveraces y dar a conocer la existencia de lasasociaciones de enfermos. En dichas asocia-ciones los pacientes, además de encontrarinformación sobre su patología, se relacionancon personas que padecen su misma enfer-medad y encuentran apoyo emocional paraellos y sus familias.
Reacción ante el diagnósticoCon la progresión de la enfermedad, el
paciente va a tener menos tolerancia al ejer-cicio, menor vitalidad y verá disminuida su
motilidad y su autonomía. Todo ello puede con-ducir a que, en ocasiones, los pacientes pre-senten trastornos psicológicos, como depresióno ansiedad. En un estudio realizado por Whitey cols. en pacientes con enfermedad avanza-da en clase funcional III-IV, se ha observadodepresión en un 26% y ansiedad en un 19%(15)
de los casos. En el mismo estudio se objetivóque en estos paciente existe un deterioro cog-nitivo en el 58% con pérdida de memoria, fal-ta de atención y lentitud en los procesos men-tales, relacionando todo ello con la hipoxemiay el fallo cardiaco derecho. Es probable que, enestadios más tempranos de la enfermedad,exista un menor índice de deterioro.
El paciente puede precisar y se le acon-sejará apoyo psicológico.
Calidad de vida en pacientes con HPy como mejorarla
Todo lo anteriormente expuesto repercuteen la calidad de vida de los pacientes con HP.Existen múltiples estudios que relacionan laHP con el descenso de la calidad de vida debi-do al deterioro funcional (el descenso serámayor cuanto más elevada sea la clase fun-cional) y a los efectos secundarios de la medi-cación. El descenso de la calidad de vida essimilar al que ocurre en otras enfermedadesdebilitantes y potencialmente fatales, siendopeor en la HP asociada a la esclerodermia yalgo mejor en la HP idiopática. No se relacio-na con los valores hemodinámicos, pero sí conlos resultados del test de la marcha(16).
Para mejorar la calidad de vida debemosllevar a cabo: • En primer lugar, un tratamiento y segui-
miento correctos.• Evaluar la capacidad del individuo para rea-
lizar las actividades de su vida diaria concuestionarios de calidad de vida y esca-las de disnea que, además, nos permitendemostrar la eficacia de los tratamientos,el control de los síntomas y la capacidadde ejercicio.
• Como parte del tratamiento puede incor-porarse la rehabilitación respiratoria.
A. PERPIÑÁ FERRI ET AL.
142
Monografía HAP NM 208 pag 18/2/10 17:08 Página 142

• Programas de educación. No sólo con lasindicaciones dadas por los profesionalessanitarios, sino a través de las asociacio-nes de pacientes, quienes administran pro-gramas de educación a pacientes y fami-liares con la oportunidad de aprenderdiferentes aspectos de la HP. También seorganizan reuniones donde se exponen,entre otros, los avances en investigación ytratamiento. Las asociaciones de pacien-tes, además de dar información sobre laenfermedad, apoyan a pacientes y fami-liares y establecen un punto de unión entreellos y con las autoridades sanitarias, laadministración y los laboratorios farma-céuticos.
PROGRAMAS DE EDUCACIÓN ALPACIENTE Y FAMILIARES
Sería recomendable que, desde el momen-to del diagnóstico, se realizara una evaluaciónglobal del paciente y de su familia para contarcon la participación activa de ambos en losprogramas de educación terapéutica. Éstos sepueden realizar individualmente o en grupo(17).Todo programa de educación terapéutica debeincluir un programa básico y otro específico.
En el programa básico debe existir mate-rial educativo que explique el concepto dehipertensión arterial pulmonar, los signos ysíntomas, cómo se diagnostica, opciones detratamiento disponibles y efectos secundarios,resultados esperados, actuación ante situacio-nes de emergencia y una serie de consejosprácticos sobre cómo enfrentarse a esta nue-va situación(17).
En el programa específico se deben incluirinformación sobre qué es y cómo funciona unnebulizador, una bomba de perfusión y un caté-ter de Hickman. Las diferentes vías de admi-nistración de los fármacos utilizados, así comolos cuidados básicos de los diferentes accesosy tratamientos(17).
Dentro del programa básico vamos a desa-rrollar las recomendaciones o consejos prácti-cos para vivir con hipertensión arterial pulmo-nar. El resto de apartados de ambos programas
serán tratados en otros capítulos de esta mono-grafía.
Actividad físicaEl ejercicio físico puede aumentar la pre-
sión arterial pulmonar media (PAPm) y pro-voca síntomas, como síncope o presíncope ydolor torácico. El ejercicio debe estar guiadosiempre por los síntomas. Por ello se reco-mienda una actividad física regular, como cami-nar. El entrenamiento debe ser suave, aeró-bico y gradual, para obtener una mejoría ensu capacidad física y calidad de vida, sin pro-ducir con ello deterioro hemodinámico. No serecomienda cargar o levantar objetos pesados.
Se ha evaluado recientemente el efecto delejercicio y entrenamiento respiratorio en un estu-dio aleatorizado prospectivo en 30 pacientes conhipertensión pulmonar severa sintomática.
A la semana, 15 pacientes del grupo aleato-rizado al entrenamiento tuvieron una mayormejoría en el test de 6 minutos marcha, con unadiferencia promedio de 100 metros, –de 65 a139 m; intervalo de confianza 95%; p =<0,001–. Mejoró también la calidad de vida eneste grupo, el consumo máximo de oxígeno y laclase funcional de la OMS, recalcándose por tan-to con este interesante estudio la importancia delentrenamiento físico y del respiratorio –añadidoal tratamiento médico–, en pacientes con HP(18).
NutriciónAunque existe poca bibliografía sobre este
aspecto, se observa un porcentaje de malnu-trición no desdeñable en estos pacientes. Tan-to la desnutrición como el sobrepeso debenser controlados en la HP. La malnutrición pro-voca una disminución de la fuerza de la mus-culatura respiratoria y una alteración de losmecanismos de defensa pulmonar. Se reco-mienda una dieta normo o hipocalórica, ricaen fibra y baja en sal. Evitar las comidas pre-cocinadas y controlar la ingesta de líquidos.
EmbarazoDurante la gestación, y también durante
el parto y el posparto, se producen una serie
SEGUIMIENTO EXTRAHOSPITALARIO DE LOS PACIENTES CON HIPERTENSIÓN PULMONAR (HP)…
143
Monografía HAP NM 208 pag 18/2/10 17:08 Página 143

de cambios hormonales y hemodinámicosque son fatales para pacientes con HP grave,considerando contraindicado el embarazo enlas mujeres con HP. La mortalidad materna eselevada, situándose en un 30-50%(19). Duran-te la edad fértil se deberán utilizar métodosanticonceptivos. Se desaconsejan los anti-conceptivos hormonales combinados por suposible efecto trombótico, siendo de elecciónlos métodos de barrera y los anticonceptivoshormonales sin estrógenos. La esterilizaciónquirúrgica y la implantación de dispositivosintrauterinos requieren seguimiento y anes-tesia especializada por la posibilidad de com-plicaciones (reacciones vasovagales) poten-cialmente fatales. En caso de embarazo, serecomienda su interrupción durante el primertrimestre.
En caso de que la paciente quiera conce-bir, se recomienda completar un año de tra-tamiento eficaz y la normalización de la fun-ción ventricular derecha antes de considerarla gestación(20).
CirugíaEs un factor de riesgo importante para los
pacientes con HP. Se debe valorar el riesgo-beneficio de la cirugía porque son particular-mente susceptibles a cuadros vasovagales,compromisos hemodinámico y ventilatorio.En aquellos pacientes con una PAP media >30 mmHg se detecta una mayor mortalidadpostoperatoria y una elevada tasa de compli-caciones tanto en cirugía cardiaca como encirugía no cardiaca. Las principales causas demuerte perioperatorias son el fracaso respira-torio y el fallo ventricular derecho.
El procedimiento anestésico parece quetiene poco que ver sobre la morbimortalidad.La mayoría de los anestésicos tienen poco efec-to sobre la circulación pulmonar, excepto laketamina y el óxido nitroso. Ambos producenun aumento de las resistencias vasculares pul-monares pero, al no acompañarse de otrasalteraciones hemodinámicas, el uso del óxidonitroso no está contraindicado. El isofluranopuede producir un efecto inotropo negativo,
que es perjudicial sobre el ventrículo derecho,por lo que es preferible el propofol(20).
AltitudEs conocido que los pacientes con HP pue-
den deteriorarse al respirar en ambientespobres de oxígeno. La hipoxia produce unaumento de la resistencia vascular pulmonar,lo que deriva en valores elevados de presiónarterial pulmonar (PAP). Este hecho ocurre enla mayoría de los individuos expuestos a alti-tudes por encima de los 3.000 m. Por ello seaconseja a los pacientes con HP, no viajar azonas o países que superen esta altitud y eltraslado de domicilio si viven en zonas de altamontaña.
Con respecto a los vuelos comerciales, noexisten consideraciones especiales para lospacientes con HP en la normativa sobre pato-logía respiratoria y vuelos en avión de laSEPAR(21). Se recomienda a los pacientes conoxigenoterapia domiciliaria, aumentar el flujode oxígeno durante el vuelo, en 1-2 litros. Parael resto de pacientes, sería conveniente esti-mar la hipoxemia durante el vuelo medianteecuaciones que calculan la PaO2 a una deter-minada altura a partir de la PaO2 a nivel delmar o bien realizar una prueba de simulaciónde altitud. En general se recomienda oxíge-no suplementario si se realiza un vuelo pro-longado (> 2 horas).
También sería recomendable iniciar el tra-tamiento con heparinas de bajo peso molecu-lar para prevenir la enfermedad tromboem-bólica venosa en aquellos pacientes que noestuvieran anticoagulados de forma crónica.
Otras recomendaciones(22)
Se aconseja la abstinencia tabáquica.Ya que las infecciones respiratorias son par-
ticularmente complicadas en estos pacientes,se recomienda la vacunación contra el virusde la gripe y contra el neumococo.
Deben evitar el agua excesivamente calien-te en las duchas y baños que sean prolonga-dos y la utilización de saunas por el peligro dela vasodilatación.
A. PERPIÑÁ FERRI ET AL.
144
Monografía HAP NM 208 pag 18/2/10 17:08 Página 144

SEGUIMIENTO EXTRAHOSPITALARIO DE LOS PACIENTES CON HIPERTENSIÓN PULMONAR (HP)…
145
Los descongestionantes nasales que con-tengan pseudoefedrina pueden empeorar lavasoconstricción.
Preparados que contengan ginseng puedenalterar la acción de los calcio-antagonistas, losniveles de digoxina y disminuir los efectos dela warfarina.
BIBLIOGRAFÍA1. Documento de consenso de la Sociedad Espa-
ñola de Neumología y Cirugía Torácica y laSociedad Española de Cardiologia. Revista EspCardiol. 2008; 61 (2): 170-84.
2. Hoeper MM, Markevych I, Spiekerkoetter E,et al. Goal-oriented treatment and combina-tion therapy for pulmonary arterial hyperten-sionEur Respir J. 2005; 26: 858-63.
3. Benza RL, Park MH, Keogh A, et al. Manage-ment of pulmonary arterial hypertension witha focus on combination therapies. J Heart LungTransplant. 2007; 26 (5): 437-46.
4. McLaughlin VV, McGoon MD. Pulmonary Arte-rial Hypertension. Circulation. 2006; 114:1417-31.
5. Sitbon O, Humbert M, Nunes H, et al. Long-term intravenous epoprostenol infusion in pri-mary pulmonary hypertension: prognostic fac-tors and survival. J Am Coll Cardiol 2002; 40(4): 780-8.
6. McLaughlin VV, Shillington A, Rich S. Survivalin primary pulmonary hipertension. Theimpact of epoprostenol therapy. Circulation2002;106:1477-82
7. Nagaya N, Nishikimi T, Uematsu M, et al. Plas-ma brain natriuretic peptide as a prognosticindicator in patients with primary pulmonaryhypertension. Circulation 2000; 102: 865-70.
8. Wensel R, Opitz CF, Anker SD, et al. Impor-tance of cardiopulmonary exercise testing. Cir-culation. 2002; 106: 319-24.
9. Forfia PR, Fisher MR, Mathai SC, et al. Tricus-pid Annular Displacement Predicts Survival inPulmonary Hypertension. Am J Respir CritCare Med. 2006; 174: 1034-41.
10. Tello Meneses R, Jiménez López-Guarch C, Chi-meno García J, et al. Echocardiographic deter-minants of functional capacity in pulmonaryarterial hypertension. Euro J Echocardiogr.2006; 7: S217.
11. Wang J, Prakasa K, Bomma C, et al. J Am SocEchocardiogr. 2007; 20 (9): 1058-64.
12. Raymond RJ, Hinderliter AL, Willis PW, et al.J Am Coll Cardiol. 2002; 39: 1214-9.
13. Raymond RJ, Hinderliter AL, Willis PW, et al.JACC. 2002; 39: 1214-9.
14. Flattery MP, Pinson JM, Savage L, et al. Livingwith pulmonary artery hypertension: Patiens’experiences. Heart Lung. 2005; 34: 99-107.
15. White J, Hopkins Ro, Glissmeyer EW, et al.Cognitive, emocional and quality of life out-comes in patients with pulmonary arterialhypertension. Respir Res. 2006; 7: 55.
16. Taichman D B, Shin J, Hud L, et al. Health-rela-ted quality of life in patients with pulmonaryarterial hypertension. Respir Res. 2005; 6: 92.
17. Barberá JA, Valera JL. Técnicas y procedi-mientos en hipertensión pulmonar. ManualSEPAR de Procedimientos 2006; 10: 69-81.
18. Mereles D, Ehlken N, Kreuscher S, et al. Exer-cise and respiratory training improve exerci-se capacity y quality of life in patients withsevere chronic pulmonary hypertension. Cir-culation. 2006; 114 (14): 1482-9.
19. Barberá JA, et al. Documento de consenso. Están-dares asistenciales en hipertensión pulmonar.Arch Bronconeumol. 2008; 44 (2): 87-99.
20. Casado Moreno I, González Gutiérrez MV. Reco-mendaciones generales para el paciente conhipertensión arterial pulmonar y sus familia-res. En: Sueiro y Gaudó eds. Tratado de hiper-tensión arterial pulmonar. 1ª edición. Barce-lona: Ars Medica; 2009. p. 306-8.
21. García Río F, et al. Normativa sobre patolo-gía respiratoria y vuelos en avión. Arch Bron-coneumol. 2007; 43 (2): 101-25.
22. Yates G, et al. Pulmonary Hipertension: ARewiew for Nurses. Can J Cardiovasc Nurs.2008; 18 (1): 7-14.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 145

Monografía HAP NM 208 pag 18/2/10 17:08 Página 146

147
RESUMENEl pronóstico y la supervivencia de las car-
diopatías congénitas han mejorado enorme-mente desde que la cirugía reparativa se haextendido y, además, con escasa mortalidad,de forma que el 85% de estos enfermos alcan-zan la edad adulta. Esto ha hecho necesario elestablecimiento de nuevas pautas de manejode los problemas que van a encontrar en estosenfermos los especialistas no pediátricos, enespecial el tratamiento de la hipertensión pul-monar (HAP). Esta situación va a aparecer fre-cuentemente en aquellos enfermos con cor-tocircuito crónico izquierda-derecha y laprobabilidad de que aparezca va a dependerdel tamaño y la localización del defecto. Conel aumento de las resistencias pulmonares,la dirección del flujo cambia, apareciendo elcuadro conocido como síndrome de Eisen-menger. Este síndrome tiene muchas caracte-rísticas propias, entre otras su relativo buenpronóstico, por lo que merece ser tratado deforma específica.
INTRODUCCIÓNHoy día, se estima que el 85% de los niños
nacidos con cardiopatías congénitas (CC) sobre-viven hasta la edad adulta. El espectro de las CCdel adulto se está modificando y casi todos lospacientes que llegan a la edad adulta presentanlesiones residuales, secuelas o complicacionesque pueden tener carácter evolutivo(1). Las CCdel adulto suponen un desafío para cardiólogos,neumólogos y para el resto del personal sani-tario implicados en el diagnóstico, seguimien-to y tratamiento de dichos pacientes.
La hipertensión arterial pulmonar (HAP) sedefine como la elevación de la presión arterial
pulmonar media > 25 mmHg en reposo, conresistencia vascular pulmonar (RVP) >3 uni-dades Wood y con presión capilar pulmonar<15 mmHg. La HAP es frecuente en lospacientes adultos con CC (aproximadamenteel 10%) y es un factor implicado en el pro-nóstico y la supervivencia de estos pacien-tes. Se sabe que la HAP en las CC tiene dife-rentes mecanismos patogénicos, objetivosterapéuticos y pronóstico respecto a la HAPidiopática (HAPI). La HAP asociada a las CCestá incluida en el grupo I de la clasificaciónactual de la DANA POINT de la HAP(2).
Las CC que pueden dar lugar a HAP pue-den ser simples o complejas (Tabla 1). Además,las CC asociadas a HAP se pueden agrupar endos categorías: CC que afectan al corazónizquierdo y producen una elevación de la pre-sión pulmonar postcapilar, y CC con cortocir-cuito izquierda-derecha intra o extracardia-co, que inducen elevación de la presiónprecapilar. En este capítulo nos vamos a cen-trar en esta última categoría.
La HAP en las CC suele ser debida a un flu-jo pulmonar excesivo en las primeras etapasde la vida a través de un cortocircuito sisté-mico-pulmonar preexistente de gran tamaño.Con el paso del tiempo, la presión pulmonaraumenta progresivamente por los cambiosestructurales y funcionales en la vasculaturapulmonar y, cuando alcanza los valores de lapresión arterial sistémica, tiene lugar la inver-sión de la dirección del cortocircuito (bidirec-cional o bien derecha-izquierda), con la apa-rición de hipoxemia y cianosis. Esta situaciónse denomina síndrome de Eisenmenger (SE)y se sitúa en el extremo del espectro de la HAP.El SE afecta a alrededor del 1-2% de los pacien-
HIPERTENSIÓN PULMONAR ENCARDIOPATÍAS CONGÉNITAS DEL ADULTO
Verónica Hernández Jiménez, Mª Teresa Río Ramírez, Mª Antonia Juretschke Moragues
Monografía HAP NM 208 pag 18/2/10 17:08 Página 147

tes con CC(3); aparece en casi todos los pacien-tes no intervenidos de Truncus arteriosus, enel 50% de las comunicaciones interventricu-lares (CIV) de > 1,5 cm y en el 10% de lospacientes con comunicación interauricular(CIA) (Tabla 2). En algunos casos, especialmenteen las CIA y las CIV de pequeño tamaño, quenunca han presentado sobrecarga de volumen,aparecen una HAP severa. En estos pacientes,la HAP debe interpretarse y manejarse comouna HAPI coincidente con un cortocircuito debajo flujo.
El desarrollo de enfermedad vascular obs-tructiva pulmonar depende de múltiples fac-tores: a) tamaño y localización del cortocir-cuito (pre o postricúspide); b) grado desobrecarga de presión y volumen en el lechovascular pulmonar; c) edad en el momentodel diagnóstico; d) cirugía o intervención pre-
via; e) edad a la que se realizó la cirugía; f)anomalías intra y extracardiacas asociadas yg) susceptibilidad individual y factores gené-ticos. En pacientes con CC pueden existir otrosfactores favorecedores del desarrollo de HAP:la presencia de enfermedad pulmonar res-trictiva, la hipoxemia, la policitemia, la hipo-ventilación, la exposición a grandes alturas,la predisposición genética, como la trisomía21. La HAP en pacientes con CC tiene mejorpronóstico que la HAPI.
FISIOPATOLOGÍAEl estado del lecho vascular pulmonar en
pacientes con HAP asociada a CC es a menu-do el principal determinante de las manifes-taciones clínicas, la evolución y la posibilidadde tratamiento quirúrgico(4). La enfermedadvascular pulmonar es una enfermedad de laspequeñas arteriolas pulmonares caracterizadapor una obliteración progresiva de la luz vas-cular que implica una creciente elevación delas RVP, lo que produce un incremento de lapostcarga del ventrículo derecho y, posterior-mente, disfunción VD y la aparición de insu-ficiencia cardiaca.
Las RVP caen tras el nacimiento debido alinicio de la ventilación y la oxigenación. Duran-te los tres primeros meses de vida, la capa mus-cular de las arteriolas pulmonares se adelga-za progresivamente para adaptarse a lascondiciones normales de la circulación pul-monar. En algunos niños con grandes comu-nicaciones sistémico-pulmonares, la presiónsistémica es transmitida al lecho pulmonar yno se produce el adelgazamiento de la mediade las arterias pulmonares, manteniéndose RVPelevadas después del nacimiento y, al llegar ala edad escolar o adulta, las RVP quedarán final-mente fijas por cambios obliterativos en el lechovascular pulmonar(5); este comportamiento esmás frecuente en los niños que viven en gran-des altitudes. Sin embargo, en la mayoría delos pacientes, las RVP disminuyen inicialmen-te, produciendo un incremento del cortocircuitoizquierda-derecha. El incremento del flujo pul-monar debido a un cortocircuito da lugar a un
V. HERNÁNDEZ JIMÉNEZ ET AL.
148
TABLA 1. Clasificación clínica de lahipertensión pulmonar en cardiopatíascongénitas
Cardiopatías congénitas que afectan al cora-zón izquierdo:• Estenosis mitral• Estenosis del tracto de salida del VI• Cor triatriatum• Estenosis de venas pulmonares
Cardiopatías congénitas con cortocircuitosistémico pulmonar:• Cardiopatías simples
- Comunicación interauricular- Comunicación interventricular- Ductus arterioso persistente- Drenaje venoso pulmonar anómalo sin
obstrucción de venas pulmonares• Cardiopatías complejas
- Canal auriculoventricular completo- Ventrículo único- Truncus arteriosus- Transposiciones complejas- Ventrículo derecho de doble salida- Otras cardiopatías complejas
Monografía HAP NM 208 pag 18/2/10 17:08 Página 148

aumento de presión cuando se supera la capa-citancia del lecho vascular pulmonar. La expo-sición crónica de la vasculatura pulmonar alincremento de presión y de flujo sanguíneo pro-voca un exceso del estrés tangencial o de ciza-llamiento sobre las paredes vasculares que des-encadena la disfunción endotelial y liberaciónde sustancias vasoactivas, produciendo cam-bios funcionales y estructurales (Tabla 3). Estoscambios son conocidos como arteriopatía pul-monar plexiforme.
La arteriopatía plexiforme es una formaespecífica de arteriopatía pulmonar que ocu-rre más frecuentemente en pacientes con CCcon un flujo pulmonar aumentado. Difiere deotras formas de arteriopatía pulmonar, comola arteriopatía hipóxica o la arteriopatía pul-
monar congestiva, no sólo por las lesiones vas-culares específicas, sino también por el cursoevolutivo de la enfermedad(6). En las fases ini-ciales de la arteriopatía plexiforme, los cam-bios estructurales son reversibles y desapare-cen una vez corregida la cardiopatía. Sinembargo, si no se corrige el cortocircuito y per-siste el hiperaflujo pulmonar, la enfermedadprogresará hasta un punto en el que los cam-bios se hagan irreversibles.
Llama la atención la variabilidad que pre-sentan pacientes con CC similares tanto enel momento de aparición como en la veloci-dad de progresión de la enfermedad vascularpulmonar. Se han implicado factores genéti-cos que podrían explicar esta diferente evolu-ción(7); varios estudios se han centrado en elgen BMPR2 y concluyen que la presencia demutaciones de este gen puede ser un factor deriesgo para el desarrollo de HAP. En un estu-dio realizado con pacientes con HAP de dife-rente etiología se detectó un 6% de mutacio-nes del gen BMPR2 en pacientes con HAPasociada a CC(8).
VALORACIÓN NO INVASIVA DE LA HAP ENLAS CC
La historia clínica y la exploración físicason los pilares básicos en la valoración de unpaciente con una CC. Si existe HAP asocia-da a un gran cortocircuito izquierda-derecha,los signos y síntomas que encontremos son
HIPERTENSIÓN PULMONAR EN CARDIOPATÍAS CONGÉNITAS DEL ADULTO
149
TABLA 2. Frecuencia del síndrome de Eisenmenger en las cardiopatías nocorregidas
Tipo de cardiopatía Frecuencia al nacer (%) Incidencia del S. Eisenmenger (%)
CIA 8 10-20CIV 23-34 15DAP 2,4-4,5 15Transposición con CIV o DAP 5-6 75Defecto septal AV 5-10 90Troncus arteriosus <1 100
CIA: comunicación interauricular; CIV: comunicación interventricular; DAP: ductus arterioso persistente; AV: aurí-culoventricular.
TABLA 3. Fisiopatología de la HAP enlas CC
Cortocircuito izquierda-derecha_
Hiperaflujo pulmonar–
Disfunción endotelial y remodelado vascular–
Aumento de las resistencias vasculares_
Inversión del cortocircuito _
Síndrome de Eisenmenger
Monografía HAP NM 208 pag 18/2/10 17:08 Página 149

reflejo de la malformación cardiaca respon-sable.
En la exploración física, aparece una ondaa prominente en el pulso venoso yugular porla contracción auricular derecha vigorosa debi-da a la distensibilidad reducida del ventrículoderecho; a veces existen grandes ondas c-v sis-tólicas, que sugieren insuficiencia tricúspide.En la auscultación, se oye un soplo sistólicoeyectivo suave pulmonar, precedido de un clicde apertura seguido del refuerzo del compo-nente pulmonar del segundo ruido; tambiénpuede oírse un soplo protodiastólico decre-ciente de insuficiencia pulmonar. Si existe dila-tación y disfunción ventricular derecha se aus-cultará un soplo sistólico de insuficienciatricúspide en el borde paraesternal izquierdoinferior y un 3er ruido del lado derecho.
Los síntomas están relacionados con la dis-minución del gasto cardiaco (fatiga e intole-rancia al esfuerzo), con la insuficiencia car-diaca congestiva, con el desarrollo de arritmiase hipoxemia. Los pacientes suelen presentaruna capacidad funcional adecuada hasta lasegunda década de la vida; posteriormentepresentan una reducción progresiva de la tole-rancia al esfuerzo y cianosis.
En pacientes con un gran cortocircuitoizquierda-derecha y RVP elevadas, el ECG sue-le mostrar ondas P picudas por sobrecarga auri-cular derecha, desviación a la derecha del ejedel QRS y diferentes grados de hipertrofia ven-tricular derecha. Pueden registrarse tambiénarritmias auriculares.
La radiografía de tórax refleja la magnitudde la derivación y el grado de hipertensión pul-monar. Si la derivación es moderada, aparececardiomegalia con crecimiento ventricularizquierdo y plétora pulmonar. Los pacientes conun gran cortocircuito, y aquellos en situaciónde Eisenmenger, desarrollarán dilatación dearterias pulmonares centrales con «recorte» dela vasculatura pulmonar periférica. La presen-cia de calcificación de la arteria pulmonar esuna señal inequívoca de HAP de larga evolu-ción. Si la causa del Eisenmenger es una CIV oun DAP, el índice cardiotorácico será normal o
ligeramente aumentado; si la cardiopatía sub-yacente es una CIA, el índice cardiotorácicoestará aumentado por dilatación auricular.
La ecocardiografía es una técnica impres-cindible para el diagnóstico de las cardiopa-tías congénitas y para la evaluación de la HAP.Proporciona el diagnóstico de la cardiopatíasubyacente en la mayoría de los pacientes (altasensibilidad); es una técnica diagnóstica fia-ble, ampliamente disponible y de bajo coste.Ofrece información sobre la severidad y el pro-nóstico de la HAP en estos pacientes. Las varia-bles habituales en el estudio ecocardiográfi-co de un paciente con HAP asociada a unacardiopatía son:• Presión sistólica de la arteria pulmonar: la
estimación de la presión por medio delDoppler se basa en el principio de con-servación de la energía y requiere para sucálculo un chorro de regurgitación tricús-pide. En general, la correlación con la pre-sión pulmonar calculada por hemodiná-mica es alta aunque el ecocardiogramatiende a sobreestimar sus valores. Si exis-te flujo de insuficiencia pulmonar, tambiénse pueden calcular la presión arterial pul-monar diastólica y la media.
• Estudio del flujo pulmonar que, en ausen-cia del flujo de regurgitación tricúspide,puede sugerir la presencia de HAP.
• Estudios morfológico y funcional del ven-trículo derecho.
• Estudios morfológico y funcional del ven-trículo izquierdo.
• Presencia de derrame pericárdico.
Las pruebas de función respiratoria en estosenfermos suelen demostrar una alteración res-trictiva leve, con aumento del volumen resi-dual; en ocasiones aparecen un defecto mix-to y disminución de la difusión(9). La afectacióndel intercambio gaseoso, fundamentalmentela hipoxemia, es en parte debida al cortocir-cuito y la consecuente mezcla de sangre nooxigenada, y en parte a las alteraciones de laventilación/perfusión (V/Q) secundarias a laplétora pulmonar y a los cambios estructura-
V. HERNÁNDEZ JIMÉNEZ ET AL.
150
Monografía HAP NM 208 pag 18/2/10 17:08 Página 150

les propios de la HAP. Esta hipoxemia empeo-ra en el decúbito, probablemente por altera-ciones V/Q(10) y, fundamentalmente, con el ejer-cicio, por aumento del flujo a través delcortocircuito.
Las pruebas de esfuerzo incluyen el testde la marcha y la ergoespirometría. El test dela marcha es preferida por su fácil aplicabili-dad y característicamente, en los enfermoscon SE, una desaturación de más de 10 pun-tos no es un factor de mal pronóstico ya queen ellos es habitual la desaturaración con elejercicio.
En la ergoespirometría se demuestra, enenfermos con cortocircuito izquierda-derecha,que el consumo máximo de oxígeno y elumbral anaeróbico están reducidos(11).La res-puesta ventilatoria está marcadamente ele-vada al inicio del ejercicio para compensar lacuantía de sangre que pasa por el cortocir-cuito y es un factor predictivo de mortalidad,sobre todo en enfermos no cianóticos(12). Estoproduce un descenso de la presión de dióxidode carbono al final de la espiración, con unaumento de la presión de oxígeno y un aumen-to del equivalente respiratorio que suele man-tenerse hasta iniciar la recuperación. La PaCO2
y el pH suelen permanecer inalterables, sugi-riendo que los mecanismos de control respi-ratorio no se afectan por las altas presionesexistentes en el lecho pulmonar.
Los adultos con SE consideran satisfacto-ria su capacidad para el ejercicio a pesar depresentar valores de consumo máximo de oxí-geno significativamente más bajos(13) que otrosenfermos portadores de CC. La capacidad deejercicio se correlaciona estrechamente conlos niveles de BNP y tiene valor pronóstico.
VALORACIÓN HEMODINÁMICA DE LA HAPEN LAS CC
Los adultos con HAP asociada a CC se pue-den agrupar en tres categorías:• Pacientes en situación de Eisenmenger: la
mayoría padecen defectos distales a la vál-vula tricúspide. No son susceptibles de nin-gún procedimiento de reparación.
• Pacientes con cortocircuito restrictivo, sinhiperaflujo pulmonar pero que sin embar-go han desarrollado HAP. Este grupo semaneja de forma similar a los pacientescon HAPI.Pacientes con un cortocircuito no restric-tivo, generalmente supratricuspídeo quedesarrollan HAP severa pero mantienen unflujo izquierda-derecha. Al llegar a la edadadulta, es frecuente que mantengan estasituación de HAP hiperquinética sin llegara la fisiología de Eisenmenger. Este gru-po es el más numeroso y su evolución natu-ral es peor que la del SE. En estos pacien-tes, la reparación de la cardiopatía seríaeficaz si disminuyesen las presiones pul-monares tras el cierre del cortocircuito.Si la presión pulmonar permaneciese ele-vada después de la reparación, la supervi-vencia disminuiría drásticamente y el pro-nóstico sería más sombrío que la HAPhipercinética no reparada. En este últimogrupo es fundamental el estudio de rever-sibilidad previo a la reparación del corto-circuito sistémico-pulmonar(14).
Peculiaridades del cateterismo derecho en las CC
Los hallazgos clínicos y electrocardiográfi-cos no distinguen entre elevaciones reversi-bles e irreversibles de las RVP. Por otra parte,la ecocardiografía permite diagnosticar la pre-sencia de HAP pero no proporciona un cálcu-lo del todo exacto de la presión pulmonar nide las RVP. El cateterismo cardiaco es la prue-ba fundamental para confirmar el diagnósticode HAP, determinar la severidad y el grado dereversibilidad(15). Permite el cálculo preciso de:presiones pulmonares y presiones sistémicas,presión capilar pulmonar, resistencias pulmo-nares y sistémicas y el gasto cardiaco. Enpacientes sin cortocircuitos, el flujo pulmonar(Qp) y el sistémico (Qs) son iguales y se pue-den calcular por el método de termodilucióno por el método de Fick. En el estudio hemo-dinámico de los pacientes con CC existen unaserie de particularidades(16):
HIPERTENSIÓN PULMONAR EN CARDIOPATÍAS CONGÉNITAS DEL ADULTO
151
Monografía HAP NM 208 pag 18/2/10 17:08 Página 151

1. Para estimar el gasto cardiaco en presen-cia de un cortocircuito sistémico-pulmonares recomendable el método de Fick, quees el método más exacto en pacientes conbajo gasto cardiaco y con regurgitación tri-cúspide. Según el método de Fick, el gas-to cardiaco es el resultado del cocienteentre el consumo de oxígeno (ml/min) y ladiferencia en el contenido de oxígeno dela sangre arterial y de la mezcla venosa. Elinconveniente de este método es la difi-cultad para obtener una medición preci-sa del consumo de oxígeno. La imprecisiónde las medidas del consumo de oxígenosupone hasta un 10% de variación en elgasto cardiaco calculado. El cálculo de ladiferencia en el contenido de oxígeno entrela sangre arterial y la mezcla venosa impli-ca también un grado de imprecisión; si elcortocircuito es infratricuspídeo se extraela sangre de la aurícula derecha, si el defec-to es supratricuspídeo hay que utilizar lamezcla de las cavas, algo problemático porel contenido variable de oxígeno de la cavainferior (la sangre venosa renal tiene unalto contenido en oxígeno y la hepática,muy bajo); para su cálculo se pueden uti-lizar varios métodos: la media aritméticade las dos cavas, la utilización de la cavasuperior o una fórmula matemática (3/4saturación de la cava superior + 1/4 satu-ración de la cava inferior).
2. Si existe estenosis de una rama de la arte-ria pulmonar o si el cortocircuito se diri-ge preferentemente a una de las ramas, lasRVP no pueden ser calculadas con fiabili-dad. También resultará complicado el cál-culo de las resistencias vasculares sisté-micas (RVS) en presencia de una coartaciónaórtica.
Estudio hemodinámico de reversibilidadEl estudio de reversibilidad de la HAP en
pacientes con cortocircuito sistémico-pulmo-nar con HAP hipercinética antes del cierre deldefecto es fundamental. En general, se con-sidera que, cuando la RVP es < 6 U Wood, la
HAP es reversible y subsidiaria de correcciónsi el Qp/Qs es > 1,5. Si la RVP es mayor, hayque hacer un estudio más minucioso de rever-sibilidad(17).
Test vasodilatadorUna respuesta positiva a los diferentes fár-
macos vasodilatadores indica que los cambiosestructurales en el lecho vascular están en fasesiniciales y, por tanto, reversibles. Para el pacien-te con HAP, se considera una respuesta posi-tiva al test vasodilatador si se obtiene una dis-minución de las RVP entre 20 y el 30%, unacaída de la presión arterial pulmonar mediade al menos 10 mmHg respecto al valor basal,con un gasto cardiaco mantenido o aumen-tado. En pacientes con HAP asociada a CC, seestá utilizando también la relación RVP/RVS,si se obtiene una relación RVP/RVS < 0,33 trasel test vasodilatador se puede proceder a lacirugía correctora(18). Los fármacos utilizadosen el test vasodilatador son fármacos de acciónrápida: oxígeno al 100%, oxído nítrico, iloprostinhalado, epoprostenol intravenoso, tolazoli-na y sildenafilo.
Además, la respuesta hemodinámica en eltest vasodilatador puede ayudar a seleccionarel fármaco más efectivo para manejar la HAPen el postoperatorio inmediato.
Oclusión temporal del defectoEl objetivo es valorar la respuesta hemodi-
námica tras la oclusión temporal del cortocir-cuito izquierda-derecha, habitualmente una CIAtipo ostium secundum. Se considera una res-puesta positiva, si la presión pulmonar dismi-nuye entre el 25-40%, sin disminución de lapresión sistémica ni aumento de la presión capi-lar pulmonar ni datos de claudicación del VD.
Las recomendaciones generales en la eva-luación del paciente con HAP asociada a CCse recogen en las guías del AHA/ACC sobre lasCC del adulto(19).
SÍNDROME DE EISENMENGERLa tríada cortocircuito sistémico-pulmonar,
enfermedad vascular pulmonar y cianosis se
V. HERNÁNDEZ JIMÉNEZ ET AL.
152
Monografía HAP NM 208 pag 18/2/10 17:08 Página 152

denomina SE. El desarrollo de enfermedadvascular pulmonar en el Eisenmenger es con-secuencia de un incremento del flujo pulmo-nar y para su diagnóstico se deben excluir otrascausas de HAP.
La clínica suele comenzar a partir de la 3ªdécada de la vida; los síntomas de insuficien-cia cardiaca aparecen más tardíamente que enla HAPI pero la cianosis lo hace más precoz-mente. La supervivencia de los pacientes conSE es mejor que la de los pacientes con HAPI.La supervivencia estimada a los 30 y los 50años de edad en pacientes con SE es del 75y del 55%, respectivamente(20).
La principal causa de muerte en estosenfermos es la muerte súbita de origen car-diaco. Otras causas de fallecimiento son la insu-ficiencia cardiaca congestiva y, en ocasiones,la hemoptisis masiva. El pronóstico dependetanto de la gravedad de la HAP como de la car-diopatía congénita subyacente. Son factorespredictores de mal pronóstico el inicio precozde los síntomas, una cardiopatía congénitacompleja de base, la capacidad funcional redu-cida, clínica de síncopes, el desarrollo de arrit-mias supraventriculares, la presión auricularderecha media elevada y el síndrome deDown(21). El pronóstico también depende delgrado de hipoxemia. La hipoxemia crónica pro-voca un aumento de la síntesis de eritropo-yetina y, secundariamente, aumenta el núme-ro de eritrocitos. La hipoxemia crónica y laeritrocitosis secundaria dan lugar a una seriede complicaciones en diferentes órganos y sis-temas:• Alteraciones hematológicas: eritrocitosis
secundaria; déficit de hierro (por sangríasde repetición y por desplazamiento de lacurva de disociación de la hemoglobinahacia la derecha). Clínicamente el pacien-te puede presentar un síndrome de hiper-viscosidad (somnolencia, cefalea, visiónborrosa, amaurosis fugaz, parestesias, mial-gias...). Entre las anomalías de la he-mostasia más frecuentes se encuentran:fibrinolisis acelerada, trombocitopenia,retracción anormal del coágulo, hipofibri-
nogenemia, prolongación del tiempo deprotrombina y prolongación del tiempoparcial de tromboplastina. El paciente pue-de sufrir tanto fenómenos hemorrágicos(hemorragias mayores y menores) comofenómenos trombóticos. Los trombos enlas arterias pulmonares proximales apare-cen en más del 50% de los pacientes conSE(22).
• Trastornos neurológicos: accidentes cere-brovasculares; se considera que la eritro-citosis secundaria, las flebotomías de repe-tición y la microcitosis podrían favorecersu aparición; abscesos cerebrales(17): sonmás frecuentes en pacientes con cortocir-cuitos derecha-izquierda y posiblementesean debidos al paso de la sangre venosaa la circulación sistémica sin pasar por elfiltro pulmonar.
• Alteraciones osteoarticulares: acropaquías(común en pacientes con hipoxemia deotras etiologías); cifoescoliosis que puedeagravar la función pulmonar de estospacientes; osteoartropatía hipertrófica, queconsiste en el engrosamiento del periostiocon formación de tejido óseo a lo largo dela metáfisis y diáfisis de los huesos largosy que se manifiesta con artralgias.
• Hiperuricemia y gota: la hiperuricemia esdebida a una disminución de la excreciónde ácido úrico y puede provocar crisis goto-sas, nefropatía litiásica por ácido úrico.
• Alteraciones renales: la hiperviscosidad san-guínea por la eritrocitosis y la vasocons-tricción arteriolar producen glomeruloes-clerosis, disminución del filtrado glomerulary aumento progresivo de las cifras de cre-atinina. El primer signo de disfunción renales la albuminuria; si la enfermedad evolu-ciona puede acabar en una insuficienciarenal establecida. El grado de disfunciónrenal depende de la severidad de la eri-trocitosis y del tiempo de evolución de lacianosis. Otra consecuencia de la cianosiscrónica a nivel renal es la acidosis tubu-lar renal, que suele aparecer en pacientesde mayor edad.
HIPERTENSIÓN PULMONAR EN CARDIOPATÍAS CONGÉNITAS DEL ADULTO
153
Monografía HAP NM 208 pag 18/2/10 17:08 Página 153

• Alteraciones dermatológicas: acné, lentacicatrización de las heridas cutáneas.
• Alteraciones en las vías biliares: litiasis biliarpor cálculos de bilirrubinato cálcico y epi-sodios de colecistitis.
MANEJO TERAPÉUTICO
Medidas generalesLos enfermos con SE presentan un frágil
equilibrio entre las presiones en el circuito pul-monar y circulación sistémica que condicionala cuantía del cortocircuito y con ello la canti-dad de sangre que se oxigena y el aporte desangre oxigenada a órganos vitales(23). Por tan-to, una de las primeras medidas a tomar esevitar toda intervención que pueda desestabi-lizar este equilibrio. En principio, hay que evi-tar y tratar enérgicamente todas las situacio-nes que conlleven una disminución de lapresión sistémica(18), como son episodios dedeshidratación, exposición al calor excesivo(saunas o duchas calientes), ejercicios mode-rado/severos y consumo importante de alco-hol; se deben evitar las situaciones que pue-dan aumentar las presiones pulmonares ydisminuir la presión inspiratoria de oxígenocomo, por ejemplo, la exposición crónica a lasalturas.
Parecen seguros los viajes en avión enenfermos no críticos y no se deben limitar(24).
Tampoco se recomiendan los bloqueantesde canales del calcio1(17). Se deben tratar enér-gicamente las arritmias y los procesos infec-ciosos.
Merecen una mención especial los siguien-tes apartados:• Insuficiencia respiratoria crónica: siempre
debe valorarse una desaturación que secorrige con oxígeno. En las demás situa-ciones, la oxigenoterapia no parece tenerun papel significativo puesto que la fisio-patología se basa en el cortocircuito másque en las alteraciones V/Q. Además, variosestudios realizados con oxígeno domici-liario no han conseguido demostrar dife-rencias significativas en los parámetros
hemodinámicos, capacidad de ejercicio, nicalidad de vida(25). Aun así, se suele indi-car la oxigenoterapia cuando el enfermocumple criterios de hipoxemia severa.
• Poliglobulia y síndrome de hiperviscosidad:su presencia determina un empeoramien-to de la calidad de vida y la muerte tem-prana de muchos de los enfermos con SE.Si desarrollan síntomas severos, la hiper-viscosidad sanguínea debe tratarse con fle-botomías cuidadosas y con reposiciónsimultánea de líquidos(26) y evitando el défi-cit de hierro y microcitosis(27).
• Embarazo: debe desaconsejarse totalmen-te y, en caso de producirse, es indicaciónabsoluta de aborto terapéutico debido alalto riesgo de muerte materna. La morta-lidad materna es debida a la incapacidadde las resistencias pulmonares a acomo-darse a los cambios hemodinámicos delembarazo, parto y postparto. Es en esteperíodo donde suelen acontecer la mayo-ría de las complicaciones. También el fetosufre un riesgo muy importante por el esca-so aporte de oxígeno de la sangre mater-na, que disminuye aún más durante la ges-tación al caer las resistencias sistémicas.Sólo un 15-25% de las gestaciones pro-gresan(13,17,28). Tampoco la contracepciónes fácil de plantear: la terapia hormonalestá contraindicada por aumento del ries-go de trombosis y la esterilización quirúr-gica conlleva el riesgo de una intervenciónen estos enfermos. Lo más recomendablees la esterilización por histeroscopia(29).
• Cirugía no cardiaca: el riesgo perioperato-rio es elevado (19%) y no es adscribiblea la naturaleza de la anestesia sino a laenvergadura de la cirugía(30) y la situacióncardiaca del enfermo. En general debenevitarse y sólo deben realizarse en centroscon experiencia en el manejo de estospacientes.
• Trombosis y anticoagulación: la frecuentepresencia de trombos en las arterias pul-monares, las alteraciones de la cascada dela coagulación y el riesgo de hemoptisis
V. HERNÁNDEZ JIMÉNEZ ET AL.
154
Monografía HAP NM 208 pag 18/2/10 17:08 Página 154

masivas hacen muy difícil la clara indica-ción de anticoagulación. Esta contraindi-cada en casos con episodios de hemopti-sis siginificativas(13,31).
Tratamiento específico de la HAP en las CC
La estrategia de tratamiento específico delos pacientes con HAP asociado a un cortocir-cuito sistémico-pulmonar y, en particular, lospacientes con SE, se basa principalmente enexperiencia clínica, más que en estudios ale-atorizados. De hecho, el único estudio con-trolado en pacientes con SE ha sido publicadorecientemente(32).
Como se ha descrito previamente, en lospacientes con HAP asociado a CC es necesa-rio valorar la respuesta vasodilatadora posi-tiva mediante el test agudo vasodilatador. Sinembargo, no hay evidencia de que el trata-miento crónico con calcioantagonistas tengaun papel destacado y favorable en estospacientes. En particular, en los pacientes conSE, el efecto vasodilatador sistémico e inotró-pico negativo puede dar lugar a un aumen-to del cortocircuito derecha-izquierda y a unasituación de insuficiencia cardiaca por bajogasto.
En los últimos años se han publicado múl-tiples estudios controlados que han incluidoprincipalmente a pacientes con HAPI o aso-ciada a conectivopatía. Sólo un escaso núme-ro de éstos han incluido a pacientes con CC.El uso de los diferentes fármacos específicostiene su base racional en que comparten simi-litudes patogénicas, biopatológicas e histoló-gicas con otras formas de HAP del grupo I dela clasificación de DANA POINT. Los fármacosdisponibles en la actualidad para su uso se des-criben a continuación.
Fármacos inhibidores de los receptores de la endotelina
La endotelina-1 (ET-1) juega un papel muyimportante en la patogénesis de la HAP, habién-dose descrito una elevación significativa enplasma en pacientes con SE(33).
El bosentan es un fármaco oral que bloquealos receptores ET-A y ET-B de la ET. Estudiospreliminares no controlados, con reducidonúmero de pacientes, mostraron un efectobeneficioso del bosentan en pacientes muy sin-tomáticos, mejorando la clase funcional, lacapacidad de ejercicio y el perfil hemodiná-mico, con un perfil de seguridad bueno tantoa corto como a largo plazos(34-36). Reciente-mente se ha publicado el estudio multicéntri-co, doble ciego, aleatorizado y controlado conplacebo BREATHE-5(32), en el que se evaluaronla eficacia y seguridad del bosentan en pacien-tes HP asociada a CC con fisiología de Eisen-menger. En dicho estudio, 54 pacientes en cla-se funcional III, fueron aleatorizados a bosentan(n=37) y placebo (n=17) durante 16 sema-nas. Los objetivos primarios fueron evaluarla seguridad del fármaco y los cambios en elperfil hemodinámico. Se observó que el gru-po de pacientes asignados a bosentan no dete-rioraron significativamente la saturación sis-témica de oxígeno, evidenciándose ademásuna mejoría significativa del índice de RVP, dela clase funcional y de la capacidad de ejerci-cio (>34 metros en el test de la marcha). Elperfil de seguridad fue bueno, y sólo un pacien-te del brazo de bosentan presentó una eleva-ción de las transaminasas 3 veces superior alvalor normal. En el estudio abierto de segui-miento, se mantuvo la respuesta favorable alas 24 semanas(37).
El sitaxsentan es un inhibidor selectivo delos receptores ET-A de la endotelina. En el estu-dio STRIDE-1(38), de los 178 pacientes inclui-dos en CF II-IV, el 24% presentaban HP aso-ciada a CC. Aunque no se han publicado losresultados del subgrupo de CC específicamente,tras las 12 semanas de seguimiento, global-mente se objetivó un efecto beneficioso enla clase funcional, la capacidad de ejercicio yel perfil hemodinámico.
Inhibidores de la PDE-5El sildenafilo es un potente inhibidor selec-
tivo de la fofodiesterasa tipo-5 ampliamentepresente en el lecho vascular pulmonar, que
HIPERTENSIÓN PULMONAR EN CARDIOPATÍAS CONGÉNITAS DEL ADULTO
155
Monografía HAP NM 208 pag 18/2/10 17:08 Página 155
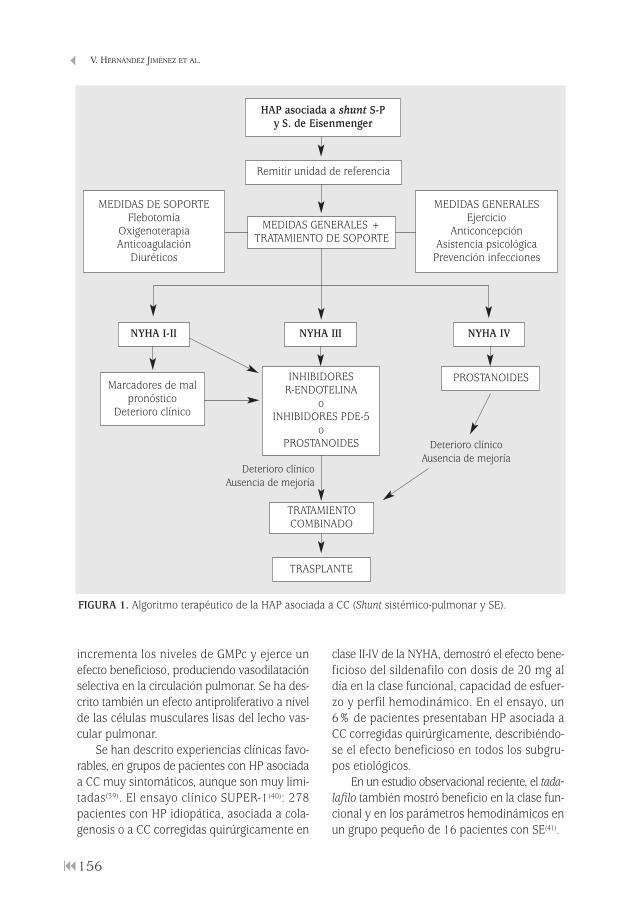
incrementa los niveles de GMPc y ejerce unefecto beneficioso, produciendo vasodilataciónselectiva en la circulación pulmonar. Se ha des-crito también un efecto antiproliferativo a nivelde las células musculares lisas del lecho vas-cular pulmonar.
Se han descrito experiencias clínicas favo-rables, en grupos de pacientes con HP asociadaa CC muy sintomáticos, aunque son muy limi-tadas(39). El ensayo clínico SUPER-1(40): 278pacientes con HP idiopática, asociada a cola-genosis o a CC corregidas quirúrgicamente en
clase II-IV de la NYHA, demostró el efecto bene-ficioso del sildenafilo con dosis de 20 mg aldía en la clase funcional, capacidad de esfuer-zo y perfil hemodinámico. En el ensayo, un6% de pacientes presentaban HP asociada aCC corregidas quirúrgicamente, describiéndo-se el efecto beneficioso en todos los subgru-pos etiológicos.
En un estudio observacional reciente, el tada-lafilo también mostró beneficio en la clase fun-cional y en los parámetros hemodinámicos enun grupo pequeño de 16 pacientes con SE(41).
V. HERNÁNDEZ JIMÉNEZ ET AL.
156
FIGURA 1. Algoritmo terapéutico de la HAP asociada a CC (Shunt sistémico-pulmonar y SE).
PROSTANOIDES
TRASPLANTE
Deterioro clínicoAusencia de mejoría
Deterioro clínicoAusencia de mejoría
Remitir unidad de referencia
HAP asociada a shunt S-P y S. de Eisenmenger
MEDIDAS GENERALES + TRATAMIENTO DE SOPORTE
TRATAMIENTO COMBINADO
MEDIDAS DE SOPORTEFlebotomía
OxigenoterapiaAnticoagulación
Diuréticos
MEDIDAS GENERALESEjercicio
AnticoncepciónAsistencia psicológica
Prevención infecciones
NYHA I-II NYHA IV
INHIBIDORES R-ENDOTELINA
oINHIBIDORES PDE-5
oPROSTANOIDES
NYHA III
Marcadores de mal pronóstico
Deterioro clínico
Monografía HAP NM 208 pag 18/2/10 17:08 Página 156

Prostaciclina y análogosEl epoprostenol se administra por vía intra-
venosa y ha demostrado su efecto beneficiosomejorando la clase funcional, capacidad deesfuerzo, los parámetros hemodinámicos y lasupervivencia en pacientes con HPI(42). Basadosen la experiencia clínica favorable, Rosenzweigy cols.(43) describieron el efecto beneficioso delepoprostenol en 20 pacientes con HP asociadaa CC, mejorando la clase funcional, la capacidadde esfuerzo y la severidad del perfil hemodiná-mico tras un año de seguimiento. Sin embargo,la administración de epoprostenol está limitadapor las serias complicaciones que se derivan delsistema de administración del fármaco, como lainfección, la trombosis o el desplazamiento delcatéter, todas ellas potencialmente fatales.
La eficacia del treprostinil administrado porvía subcutánea fue evaluada en el estudio lle-vado a cabo por Simonneau y cols.(44). En dichoestudio, el 25% de los pacientes presentaronHP asociada a CC. Aunque no se han publi-cado los datos en este subgrupo de los pacien-tes, se describió un efecto beneficioso en lapoblación global de estudio.
Existe una experiencia muy limitada con ilo-prost inhalado a largo plazo en los pacientes conCC, y su uso principal se ha dirigido al manejopostoperatorio de la HAP en la cirugía de las CC.
El beraprost es un análogo de la prostaci-clina que se administra por vía oral. En el estu-dio randomizado europeo(45) no se objetivóbeneficio clínico ni hemodinámico en el sub-grupo de pacientes con HP asociada a CC.Actualmente no está aprobado su uso por laAgencia Europea del Medicamento.
Tratamiento combinadoLa experiencia del tratamiento combinado
en pacientes con HP asociada a CC es muy limi-tada. Dada la ausencia de datos publicados,el tratamiento combinado debe valorarse indi-vidualmente, en centros con experiencia.
Trasplante pulmonar/cardiopulmonarEl trasplante pulmonar con reparación del
defecto cardiaco o el cardiopulmonar se reser-
va como opción terapéutica para aquellospacientes con indicadores de mal pronóstico(insuficiencia cardiaca derecha, clase funcio-nal IV, hipoxemia severa, síncope), refractariosal tratamiento médico. El momento de inclu-sión en lista de espera es controvertido, y deberealizarse teniendo en cuenta los limitadosresultados del trasplante en estos pacientes yla aceptable supervivencia a largo plazo.
El algoritmo terapéutico se muestra en lafigura 1.
BIBLIOGRAFÍA1. Oliver JM, Mateos M, Bret M. Evaluación de
las cardiopatías congénitas en el adulto. RevEsp Cardiol. 2003; 56 (6): 607-20.
2. Simonneau G, Robbins IM, Beghetti M, et al.Updated in clinical classication of pulmonaryHypertension. J Am Coll Cardiol 2009: 54;S43-54.
3. Bouzas B y Gatzoulis MA. Hipertensión arterialpulmonar en adultos con cardiopatía congé-nita. Rev Esp Cardiol. 2005; 58 (5): 465-9.
4. Heath D, Edwards JE. The pathology of hyper-tensive pulmonary vascular disease. Circula-tion. 1958; 18: 533.
5. Burchenal JEB, Loscalzo J. Endothelial dysfunc-tion and pulmonary hypertension. PrimaryCardiol. 1994; 20: 28.
6. Berger RMF. Possibilities and impossibilitiesin the evaluation of pulmonary vascular dise-ase in congenital heart defects. Eur Heart J.2000; 21 (1): 17-27.
7. Beghetti M. Congenital heart disease and pul-monary hypertension. Rev Por Cardiol. 2004;23: 273-81.
8. Roberts KE, McElroy JJ, Wong WPK. BMPR2mutations in pulmonary arterial hypertensionwith congenital heart disease. Eur Respir. 2002;24: 371-74.
9. MacArthur CG, Hunter D, Gibson GJ. Ventila-tory function in the Eisenmenger syndrome.Thorax. 1979; 34 (3): 348-53.
10. Sandoval J, Alvarado P, Martínez-Guerra ML, etal. Effect of body position on pulmonary gasexchange in Eisenmenmger´s Syndrome. AmJ Respir Crit Care Med. 1999; 159: 1070-3.
11. Wasserman K, Hansen JE, Sue DY, et al. Prin-ciples of Exercise Testing and Interpretation.Lippincott Willianms x Wilkins; 2005.
HIPERTENSIÓN PULMONAR EN CARDIOPATÍAS CONGÉNITAS DEL ADULTO
157
Monografía HAP NM 208 pag 18/2/10 17:08 Página 157

12. Dimopoulos k, Okonko DO, Diller GP, et al.Abnormal ventilatory response to exercise inadults with congenital heart disease relates tocyanosis and predicts survival. Circulation.2006; 113 (24): 2796-802.
13. Trojnarska O, Gwizdala A, Katarzynski S, et al.Evaluation of exercise capacity with cardio-pulmonary exercise test and B- type natriure-tic peptide in adults with congenital heart dise-ase. Cardiol J. 2009; 16 (2): 133-41.
14. Palenzuela H, Pérez H, Carballés F. Limita-ciones del cateterismo cardiaco como predic-tor de reversibilidad en la hipertensión pul-monar severa asociada a comunicacionesinterventriculares. Rev Peru Pediatr. 2008;61(3): 151-56.
15. Escribano P, Jiménez C. Hipertensión Pulmo-nar. Monocardio nº 1. 2007; Vol IX: 34-42.
16.- Steele PM, Fuster V, Cohen M, et al. Isolatedatrial septal defect with pulmonary vascularobstructive disease-long term follow up andprediction outcome after surgical correc-tion.Circulation. 1987; 76: 1037-42.
17. Haneda K, Sato N, Topo T, et al. Late resultsalter correction of ventricular septal defect withsevere pulmonary hypertension. Tohoku J ExpMed. 1994; 174: 41-8.
18. Daliento L, Somerville J, Presbitero P, et al. Eis-senmenger syndrome. Factors relating to dete-rioration and death. Eur Heart J. 1998; 19:1845-55.
19. Warners, et al ACC/AHA 2008 Guidelines forthe management of adults with congenitalHeart disease. JACC. 2008; 52 (23).
20. Diller GP, Dimopoulos K, Broberg C. Presen-tation, survival prospects, and predictors ofdeath in Eisenmenger syndrome: a combinedretrospective and case-control study. Eur HeartJ. 2006; 27 (14): 1737-42.
21. Cantor WJ, Harrison DA, Moussadaji JS, et al.Determinants of survival and length in survi-val in adults in Eisenmenger syndrome. AmJ Cardiol. 1998; 84: 677-81.
22. Perloff JK, Hart EM, Greaves SM, et al. Proxi-mal pulmonary arterial and intrapulmonaryradiologic features of Eisenmenger syndromeand primary pulmonary hypertension. Am JCardiol. 2003; 92: 182-7.
23. Vongpatanasian W, Brickner ME, Hillis LD, etal. The Eisenmenger syndrome in adults. AnnIntern Med. 1998; 128: 745-55.
24. Broberg CS, Uebing A, Cuomo L, et al. Adultpatients with Eisenberger syndrome report
flying safely on commercial airlines. Heart.2007; 93(12): 1599-603.
25. Sandoval J, Aguirre JS, Pulido T, et al. Noctur-nal oxigen therapy in patients with the Eisen-menger syndrome. Am J Respir Crit Care Med.2001; 164: 1682-87.
26. Perloff JK, Marelli AJ, Miner PD. Risk of stro-ke in adults with cyanotic congenital heart dise-ase. Circulation. 1993; 87: 1954-59.
27. DeFilippis AP, Law K, Curtis S, et al. Blood isthicker than water: the management ofhypervscosity in adults with cyanotic heartdisease. Cardiol Rev 2007; 15 (1): 31-4.
28. Makaryus AN, Forouzesh AS, Johnson M. Preg-nancy in the patient with Eisenmenger´ssyndrome. MT Sinai J Med. 2006; 73: 1033-36.
29. Andersson S, Erikson S, Mints M. Hysteros-copic female sterilization with Essure in anoutpatient setting. Acta Obstet Gynecol Scand.2009; 88: 743-6.
30. Martín JT, Tautz TJ, Antognini JF. Safety of regio-nal anesthesia in Eisenmenger´s syndrome.RegAnesth Pain Med.2002; 27: 509-13.
31. Broberg C, Ujita M, Babu-Narayan S, et al. Mas-sive pulmonary artery thrombosis with hae-moptysis in adults with Eisenmenger´s syndro-me: a clinical dilemma. Heart. 2004; 90: 1-4.
32. Galie N, Beghetti M, Gatzoulis MA, et al, forthe Bosentan Randomized trial of Endothe-lin Antagonist Therapy. Bosentan therapy inpatients with Eisenmenger´s Syndrome: a mul-ticenter, double-blind, randomized, placebocontrolled study. Circulation. 2006; 114; 48-54.
33. Giaid A, Yangisawa M, Langleben D, et al.Expression of endothelin-1 in the lungs ofpatients with pulmonary hypertension. N EnglJ Med. 1993; 328: 1732-9.
34. Apostolopoulou SC, Manginas A, Cokkinos DV,et al. Effect of the oral endothelin antagonistbosentan on the clinical, exercise, and hemody-namic status of patients with pulmonary arte-rial hypertension related to congenital heartdisease. Heart. 2005; 91: 1447-52.
35. Schulze-Neick I, Gilbert N, Ewert R, et al. Adultpatients with congenital heart disease and pul-monary arterial hypertension: first open pros-pective multicenter study of bosentan therapy.Am Heart J. 2005; 150: 716.
36. Diller GP, Dimopoulos K, Kaya MG, et al. Long-term safety, tolerability and efficacy of bosen-tan in adults with pulmonary arterial hyper-
V. HERNÁNDEZ JIMÉNEZ ET AL.
158
Monografía HAP NM 208 pag 18/2/10 17:08 Página 158

HIPERTENSIÓN PULMONAR EN CARDIOPATÍAS CONGÉNITAS DEL ADULTO
159
tension associated with congenital heart disea-se. Heart. 2007; 93: 974-6.
37. Gatzoulis MA, Begghetti M, Galie N, et al. Lon-ger-term bosentan therapy improves functio-nal capacity in Eisenmenger´s Syndrome:results of the BREATHE-5 open-label extensionstudy. Int J Cardiol. 2008; 127: 27-32
38. Langleben D, Hirsh, Shalit M, et al. SustainedSymptomatic,Functional, and HemodynamicBenefit With the Selective endothelin-a recep-tor antagonist, sitaxsentan, in patients withpulmonaryarterial hypertension A 1-YearFollow-up Study. Chest. 2004; 126: 1377-81.
39. Chau EMC, Fan KYY, Chow WH. Effects of chro-nic sildenafil in patients with Eisenmenger´ssyndrome versus idiopathic pulmonary arterialhypertension. Int J Cardiol. 2007; 120: 301-5.
40. Galiè N, Ghofrani HA, Torbicki A, et al, for theSildenafil Use in Pulmonary Arterial Hyper-tension (SUPER) Study Group. Sildenafil Citra-te Therapy for Pulmonary Arterial Hyperten-sion. N Engl J Med. 2005; 353: 2148-57.
41. Mukhopadhyay S, Sharma M, Rmakrshnan S,et al. Phosphodiesterase-5 inhibitor in Eisen-
menger´s syndrome: a preliminary observa-tional study. Circulation. 2006; 114: 1807-10.
42. Barst RJ, Rubin LJ, Lon WA, et al. A compari-son of continuous intravenous epoprostenolwith conventional therapy for primary pul-monary hypertension. The Primary Pulmo-nary Hypertension Study Group. N Eng J Med.1996; 334: 296-302.
43. Rosenzweig EB, Kerstein D, Barst R. Long TermProstacyclin for Pulmonary Hypertension withassociated Congenital Heart Defects. Circula-tion. 1999; 99: 1858-65.
44. Simonneau G, Barst RJ, Galie N, et al. Conti-nuous subcutaneous infusion of treprostinil, aprostacyclin analogue in patients with pul-monary arterial hypertension. A double-blind,randomized, placebo-controlled trial. Am J Res-pir Crit Care Med. 2002; 165: 800-4.
45. Galie N, Humbert M, Vachiery JL, et al. Effectsof beraprost sodium, an oral prostacyclin ana-logue, in patients with pulmonary arterialhypertension: a randomized, double blind pla-cebo controlled trial. J Am Coll Cardiol. 2002;39: 1496-502.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 159

Monografía HAP NM 208 pag 18/2/10 17:08 Página 160

161
RESUMENLa hipertensión arterial pulmonar (HAP)
es una rara enfermedad que en algunos casostiene un componente familiar. Tras cuatrodécadas de búsqueda, en el año 2000 se pudoidentificar el gen donde radicaba el origen deesta afección, y correspondía a un receptorde la familia del TGF-β, BMPR2. Actualmen-te se han descrito más de 280 mutaciones enese gen. En torno a un 80% de pacientes conHAP hereditaria son portadores de alguna deestas mutaciones que también se observanen un 20% de casos no hereditarios. Lasmutaciones fundamentalmente producen unadisminución de la cantidad final de proteína,es decir, actúan por haploinsuficiencia. Otrarara enfermedad, la telangiectasia hemorrá-gica hereditaria, cuando presenta una muta-ción en el gen ALK1, de la misma familia delTGF-β, se asocia a HAP.
La vía intracelular de BMPR2 es muycompleja, precisando formar un dímero conBMPR1 y fosforilarse, estimulando unaspequeñas proteínas llamadas Smad que, uni-das a cofactores o corepresores, regulan laexpresión genética. Cuando esta vía se alte-ra, es posible utilizar otras alternativas, perosu eficiencia en el control de la proliferacióncelular es claramente inferior. Recientementese han podido establecer relaciones entreesta vía y citocinas proinflamatorias, lo quecomplica algo más el proceso pero abre nue-vas expectativas. Se han estudiado otras víasen la patogenia de la HAP, sobre todo la sero-tonina, existiendo algunos polimorfismos conposible importancia en el desarrollo de laenfermedad.
INTRODUCCIÓNLa HAP es una enfermedad poco frecuen-
te, con una incidencia estimada en torno a 2-3 casos por millón de habitantes año(1). Ello difi-culta notablemente hacer estudios paraestablecer tendencias familiares. En el año1954, Dresdale fue el primero en reconocerque podría darse algún tipo de agrupamientofamiliar en pacientes con HAP inicialmente eti-quetada de primaria(2), al observar que unamadre, su hermana y su hijo presentabanhallazgos compatibles con la enfermedad. Entrelos años 1957-1974 se siguieron buscando fami-lias en las que más de un miembro padecierala enfermedad, publicándose varios casos(3,4).Con el fin de conocer más fondo, la incidenciay características de la HAP familiar, Jim Loyd yJohn Newman crearon una fundación para con-tactar con familiares de pacientes. Gracias aeste esfuerzo se pudieron conocer más fami-lias y, posteriormente, establecer el tipo deherencia. En 1984 publican los resultados desu trabajo donde demuestran que el patrón detransmisión es autosómico dominante(5). Tre-ce años después, Jane Morse y Bill Nichols loca-lizaron un marcador de HAP familiar en el cro-mosoma 2q31-32(6). Por último, en el año 2000dos grupos independientes, uno estadounidensey otro internacional, fundamentalmente eu-ropeo, localizan las mutaciones en el gen deBMPR2(7,8). Al año siguiente, otro grupo lide-rado por Richard Trembath demostró que lapresencia de mutaciones en el gen que codifi-ca ALK-1, otro receptor que pertenece a la fami-lia del factor transformante del crecimiento β(TGF-β), era responsable de la HAP asociadaa la telangiectasia hemorrágica hereditaria(9).
GENÉTICA E HIPERTENSIÓN ARTERIALPULMONAR
Adolfo Baloira Villar, Diana Valverde Pérez
BLOQUE IV. AVANCES EN HIPERTENSIÓN PULMONAR
Monografía HAP NM 208 pag 18/2/10 17:08 Página 161

Aunque inicialmente se estimó una fre-cuencia de mutaciones en BMPR2 en HAPhereditaria en torno al 50-60% de los casos,actualmente, tras exhaustiva investigación ycon nuevas técnicas de secuenciación de genes,esta cifra se aproxima al 80%. En torno a un20% de pacientes etiquetados como HAP idio-pática también tienen alguna murtación eneste gen. La estimación de portadores de estasmutaciones en la población general oscila entreel 0,001 al 0,01%(10), es decir, extremadamenteinfrecuentes.
HIPERTENSIÓN ARTERIAL PULMONARHEREDITARIA
La hipertensión arterial pulmonar heredi-taria (HAPH), anteriormente denominada fami-liar, muestra unas características histológicas,clínicas y pronósticas muy similares a lasencontradas en las formas esporádicas. Es muydifícil dar cifras sobre su incidencia y posible-mente, a medida que se conozcan mejor lasalteraciones genéticas de la enfermedad, elporcentaje de pacientes con HAPH se incre-mentará. En el primer registro realizado enEstados Unidos, un 6% del total de casos eranfamiliares(11) mientras que en el más reciente,realizado en Francia, donde se incluyeron mástipos de HAP, la cifra baja al 3,9%(12). No esfácil obtener datos fiables acerca de la trans-misión familiar de la HAP. Todavía es una enti-dad poco conocida y posiblemente existancasos sin diagnosticar. Un porcentaje muypequeño de pacientes portadores de mutacio-nes en BMPR2 refieren antecedentes familia-res, quizá por la baja penetrancia con expre-sión muy variable dentro de la misma familiao por la presencia de mutaciones espontáne-as. Pueden existir individuos portadores de lamutación sin ninguna expresión clínica duran-te varias generaciones. La presencia de muta-ciones en BMPR2 sólo supone en torno a un20% de posibilidades de desarrollar HAP. Elmayor registro de familias con al menos dosmiembros afectos de HAP pertenece a la Uni-versidad de Vanderbilt. Incluye a unas 100 fami-lias con 3.750 individuos estudiados, existiendo
más de 350 con criterios de HAP. Las seis fami-lias con mayor número de pacientes mostra-ron un 24,2% de posibilidades de padecer laenfermedad en caso de ser familiar en primergrado de alguien afecto (13). El inició de los sín-tomas sucede habitualmente en edades mástempranas que la HAP idiopática. Existe unarelación 1,7:1 a favor de las mujeres en laHAPH. Se ha postulado que posiblemente exis-ta una asociación ente el cromosoma X o fac-tores hormonales y la expresión clínica. Otrahipótesis con base experimental es que los fetosvarones que presentan la mutación podríantener defectos en el desarrollo embrionario quedarían lugar a mayor número de abortos pre-coces, de ahí las diferencias en la incidencia.Un estudio de Loyd y cols.(14) mostrando quenacen más niñas vivas portadoras obligadasde la mutación va a favor de la anterior hipó-tesis. Otro aspecto interesante es el de la anti-cipación genética, es decir, el comienzo másprecoz de la enfermedad en las sucesivas gene-raciones. En el registro estadounidense, la edadmedia de fallecimiento en la primera genera-ción fue de 45,6 años, en la siguiente 36,3 y,en la última, 24,2(5). Ello implica que debe exis-tir una base biológica para explicar este fenó-meno. Existen otras enfermedades en las quetambién se ha constatado anticipación genéti-ca, como la corea de Huntington, síndrome delcromosoma X frágil o la distrofia miotónica.Una repetición expandida de trinucleótidos esla base tanto de la anticipación genética comode la penetrancia incompleta en las dos últi-mas enfermedades(15). En los pacientes conHAPH no se ha encontrado, por el momento,esta alteración.
No hay diferencias tanto en la sintomato-logía como en el resto de hallazgos entre lasformas hereditarias y esporádicas de HAP. Com-parando, los 12 pacientes con HAPH con losotros 175 del estudio del NIH, se observó enlos primeros un diagnóstico más precoz desdeel inicio de los síntomas (0,68 años versus 2,56años)(11) con una supervivencia media similar.Un estudio postmortem de los pulmones de 23pacientes fallecidos por HAPH correspondien-
A. BALOIRA VILLAR, D. VALVERDE PÉREZ
162
Monografía HAP NM 208 pag 18/2/10 17:08 Página 162

tes a 13 familias mostró lesiones similares alas descritas en la HAP esporádica(16). En elregistro francés se incluyeron 233 pacientes enlos que se había estudiado la presencia de muta-ciones en BMPR2. Un total de 68 presentabanalguna. Existieron diferencias en cuanto a laedad de presentación (36 años en portadoresvs. 46 en los no portadores), los parámetroshemodinámicos eran más graves en los porta-dores de mutación y sólo un 1,5% de ellos res-pondieron al test vasodilatador comparado conel 10,3% en los no portadores(17). Es decir, unaenfermedad de aparición más temprana y concriterios de mayor gravedad. Es probable quepatogénicamente sean dos enfermedades lige-ramente diferentes.
TELANGIECTASIA HEMORRÁGICAHEREDITARIA
La telangiectasia hemorrágica hereditaria(THH) es una enfermedad autosómica domi-nante que se caracteriza por la aparición demúltiples telangiectasias mucocutáneas y mal-formaciones arteriovenosas pulmonares, hepá-ticas y cerebrales. A lo largo de la vida delpaciente se desarrollan de forma repetidahemorragias gastrointestinales, epistaxis y, enalgunos casos, HAP. En 1994 se publicó el pri-mer caso con una mutación en el gen que codi-fica la endoglina, otro miembro de la super-familia del TGF-β, situado en el cromosoma9(18). La endoglina facilita la unión del TGF-βa sus receptores tipos I y II en la membranacelular. Posteriormente se describieron 3 muta-ciones en cuatro familias con THH tipo II enotro gen de la familia del TGF-β, el que codi-fica el receptor similar a la cinasa 1 para la acti-vina (ALK1), localizado en el cromosoma 12(19).Las mutaciones en ALK1 parecen ser la causade la presencia de HAP en estos pacientes. Seha publicado el caso de un niño que a los 3meses fue diagnosticado de HAP y posterior-mente, a los 8 años, de THH, que presentabauna mutación en el gen de la endoglina. Secomprobó que esta mutación daba lugar a unaintensa disminución de la producción de estasustancia(20). Su padre, sin embargo, era por-
tador de la misma mutación pero no padecíala enfermedad.
No se ha podido encontrar hasta elmomento ninguna vía común entre BMPR2 yALK1 o endoglina, aunque es posible que laseñal intracelular que producen a través dela familia de coactivadores Smad pueda teneralgún tipo de interacción(21).
MUTACIONES EN BMPR2 EN HAP NO HEREDITARIA
Desde el descubrimiento de las mutacio-nes en el gen de BMPR2 en los pacientes conHAPH, se ha intentado conocer si la impor-tancia que podrían tener estas mutaciones enotros tipos de HAP. En el mismo año en que seconoció el gen responsable, se publicó un tra-bajo realizado en 50 pacientes con HAP espo-rádica no relacionados en donde se observóque 13 de ellos (26%) presentaban algunamutación(22). Uno de los estudios más ampliosfue realizado en Alemania(23) incluyendo a 99pacientes, de los cuales 11 presentaban muta-ciones (11%). Otro estudio hecho en Japón mos-tró, sobre un total de 30 pacientes, 12 casoscon mutaciones (40%)(24). Un trabajo realizadoen Finlandia, con una población genéticamentehomogénea, encontró sólo 3 casos sobre untotal de 26 pacientes analizados (12%)(25). Ennuestro país se ha publicado un trabajo con 8pacientes no relacionados, de los cuales 3 tuvie-ron mutaciones(26).
La HAP asociada a anorexígenos, sobretodo fenfluramina, es muy probable que pre-cise factores individuales para su desarrollodada la baja tasa de pacientes expuestos quepresentan la enfermedad (un caso por 10 mil).En un estudio francés se observó la presenciade mutaciones en BMPR2 en un 9% de pacien-tes(10). Un hecho destacable fue que el iniciode la enfermedad se produjo tras un períodode exposición a la fenfluramina significativa-mente más corto que en aquellos que no pre-sentaban mutaciones.
Apenas hay datos en cuanto a cardiopatí-as congénitas. Un estudio mostró sobre un totalde 106 niños y adultos con HAP asociada a
GENÉTICA E HIPERTENSIÓN ARTERIAL PULMONAR
163
Monografía HAP NM 208 pag 18/2/10 17:08 Página 163

este tipo de cardiopatías un 6% de casos conmutaciones en BMPR2(27). Tanto en HAP aso-ciada a esclerodermia, enfermedad trombo-embólica crónica como al virus de inmuno-deficiencia, no se han detectado mutaciones.Se desconoce lo que sucede con la hiperten-sión porto-pulmonar.
ESTRUCTURA DE BMPR2 Y SEÑAL DE TRANSDUCCIÓN
Los receptores tipo 2 de la familia TGF- βtienen cuatro dominios funcionales, la parteN-terminal de unión al ligando, una regióntransmembrana, una región serina/treoninacinasa y el dominio citoplasmático. En el casode BMPR2, la porción intracitoplasmática esexcepcionalmente larga, codificada por el exón12 del gen. Existe una segunda isoforma gene-rada por una división de este exón 12 que seexpresa de forma amplia a nivel del ARNmpero con una función, en lo que se refiere alproducto peptídico final, desconocida(28).
BMPR2 está codificado por un gen con 13exones y aproximadamente 4.000 pares debases que codifican 1.038 aminoácidos(29). Laparte extracelular, es decir, el ligando, está codi-ficada por los exones 1-3 (Fig. 1). Se hanencontrado un total de 298 mutaciones loca-lizadas en todos los exones incluido reciente-mente el 13. De ellas, 122 son pequeñas dele-ciones o inserciones en lugares con secuenciade baja complejidad o como secundarias a unatransición de citosina a timina (C>T) debido
probablemente a deaminación de citosina, algoque sucede de forma espontánea con ciertafrecuencia(30). Un total de 85 mutaciones (29%)fueron del tipo sin sentido; 73 (24%), del tipocambio de sentido; 26 (9%), por ruptura pre-matura de la secuencia y 19 (6%), duplica-ciones o deleciones. Aproximadamente lamitad de las mutaciones afectan a la porciónquinasa del gen (exones 6-11). Esta región estácompartimentada en 12 subdominios conimportancia variable en la unión al trifosfatode adenosina y la transferencia del grupo fos-fato. Un lugar frecuente de mutaciones es laposición 491, donde las mutaciones tipo pér-dida de sentido dan lugar a la sustitución delresiduo de arginina. La desaparición de la inter-acción entre este residuo y el de ácido glutá-mico en posición 386 convierte al receptorcinasa en inactivo, aboliendo prácticamentepor completo la señal vía Smad(31). Utilizandola técnica de ligazón múltiple se pudo demos-trar la presencia de mutaciones en un 28% defamilias con HAPH en las que con las técnicashabituales no se habían detectado(32). No todaslas mutaciones tienen la misma importancia.Mutaciones del tipo sin sentido en el dominiocitoplasmático no afectan a la activación de lavía Smad (Tabla 1). Como ya se comentó ante-riormente, los pacientes con HAP en relacióncon administración de fenfluramina sólo tie-nen mutaciones tipo pérdida de sentido encomparación con poco más del 30% en el casode la HAPH. El gen de BMPR2 es muy largo,
A. BALOIRA VILLAR, D. VALVERDE PÉREZ
164
FIGURA 1. Estructura del gen que codifica BMPR2.
1 2 3 4 5 6 7 8 9 10 11 12 13
Ligando Porción Porción extracelular transmembrana citoplásmica
Monografía HAP NM 208 pag 18/2/10 17:08 Página 164

con muchos posibles loci potencialmente afec-tos por mutaciones, incluso en zonas del recep-tor sin funcionalidad conocida. De manera ruti-naria no se estudian algunas de estas zonasque no participan en la traducción genética.El grupo de Machado ha encontrado en laregión no traducida 5´ una nueva mutaciónconsistente en una doble sustitución que con-tiene un codón de finalización prematura(33).Algunas mutaciones que aparecen en cardio-patías congénitas y asociadas a derivados dela fenfluramina han mostrado in vitro una capa-cidad de señalización prácticamente similar ala que tiene el receptor normal(34). Esto indi-ca la importancia de secuenciar las zonas nocodificantes del gen en todos los casos de HAPfen los que no se haya encontrado ningunamutación.
El mecanismo por el que las mutacionesen BMPR2 dan lugar a HAP es fundamental-mente haploinsuficiencia, es decir, síntesis dis-minuida de la proteína en cuestión, con lo quehay un déficit funcional. El grupo de Machadofue el que primero lo propuso, sugiriendo queun 60% de las mutaciones daban lugar a uncese prematuro de la traducción genética(35),confirmándose posteriormente en otros tra-bajos(36). La consecuencia de la mutación esuna proteína alterada, sin función, con lo queel número de receptores eficientes en la super-ficie celular disminuye. Se ha comprobado que
estas mutaciones impiden en parte el despla-zamiento del receptor hacia la superficie, acu-mulándose en el citoplasma sin posibilidad deunión a su ligando.
Un hecho importante es la baja penetran-cia de las mutaciones en BMPR2. Ello implicaque deben existir otros factores asociados aellas, ya sean genéticos o ambientales, paraque se desarrolle la enfermedad. Reciente-mente se ha observado que la penetranciaparece estar modulada por el grado de expre-sión de BMPR2 no mutado(37). Se ha intentadocomparar la HAP con una enfermedad tumo-ral. Las lesiones plexiformes tienen un desa-rrollo que no difiere mucho de las neoplasias,con proliferación de células musculares y endo-teliales en las arterias pulmonares y dismi-nución de la apoptosis. Algunos estudios hanmostrado una proliferación anormal mono-clonal de células endoteliales tanto en HAPidiopática como en la asociada a anorexíge-nos(38,39). En 7 pacientes con HAPH se estudióla posibilidad de que los alelos “normales” deBMPR2 fueran inactivados por las formas muta-das y ello diera lugar a expresión de la enfer-medad, siguiendo el modelo de la segundasupresión (second hit) de desarrollo tumoral(40).Sin embargo, no se pudo confirmar que la ines-tabilidad genética con pérdida de funcionali-dad en los alelos no mutados tuviera impac-to en el desarrollo de las lesiones.
GENÉTICA E HIPERTENSIÓN ARTERIAL PULMONAR
165
TABLA 1. Algunas de las mutaciones en BMPR2(30)
Localización Dominio Cambio de nucleótido Cambio de aminoácido
Exón 1 71C>A p. A24EExón 1 c28C>T p. T10WExón 2-3 Extracelular 77-?_418+? delExón 2 Extracelular c124C>T p. Q42XExón 2 Extracelular c247G>A p. G83RExón 3 Extracelular c248G>A p. G83EExón 3 Extracelular c295T>C p. C99RExón 6 Kinasa c794A>G p. E265GExón 8 Kinasa c1019T>C p. L340PExón 9 Kinasa c1171G>A p. A391TExón 12 Citoplásmico c2695C>T p. R899X
Monografía HAP NM 208 pag 18/2/10 17:08 Página 165

ESTUDIOS GENÉTICOS EN HAPActualmente es posible realizar estudios
genéticos en BMPR2, ALK1 y endoglina. Lomás habitual es iniciar el estudio con BMPR2salvo que el paciente tenga datos de THH. Laindicación sería cualquier individuo con ante-cedentes familiares de HAP o que padezca unaHAPi. Deberá informarse siempre al pacien-te de que, en caso de ser portador de algunamutación, la enfermedad podría aparecer enotros familiares. Es muy importante estudiarel gen completo. Una vez que está montada latécnica, el precio puede oscilar en torno a 300-500 euros. Es fundamental que se realice encentros con experiencia, dado que no es unapráctica clínica habitual. La realización del testimplica consejo genético posterior. Se ha dis-cutido mucho sobre el impacto que puedetener en el individuo. Dada la baja penetran-cia, es preciso valorar siempre los beneficiosde tener una información que puede no llegarnunca a tener consecuencias patológicas y cre-ar angustia innecesaria. Quizá es más impor-tante el estudio genético para descartar posi-bles candidatos a padecer la enfermedaddentro de familias con HAPH. En niños debetenerse todavía más cuidado dado que, por elmomento, no existe ninguna estrategia quepueda prevenir la enfermedad, lo que haríapoco útil conocer si son portadores de algu-na mutación
LA GRAN FAMILIA DEL TGF-βBMPR2 pertenece a la superfamilia del TGF-
β. Las proteínas morfogenéticas del hueso(BMP) son el grupo más numeroso dentro deesta superfamilia, recibiendo su nombre porimplicarse inicialmente en el crecimiento ydiferenciación de hueso y cartílago. Actual-mente sabemos que intervienen en el creci-miento y apoptosis de múltiples tipos celula-res tanto mesenquimales como epiteliales, enperíodos embrionarios y en tejidos maduros(41).BMPR2 es un receptor del tipo serina/treoni-na quinasa que precisa formar heterocomple-jos con uno de los receptores tipo 1 (ALK-3/BMPR1A, ALK-6/BMPR1B) para iniciar la
señal correspondiente una vez que se ha uni-do a sus ligandos específicos (BMP-2, BMP-4,BMP-6, BMP-7 y los factores de crecimientoy diferenciación 5 y 6)(42). Esta unión activael receptor tipo 1 mediante la fosforilación deuna porción proximal intracitoplasmática. Unavez activado el receptor, se inicia la señal cito-plásmica mediante la fosforilación del segun-do mensajero, que son las proteínas denomi-nadas Smad. Ellas son las responsables de laseñal de transducción de la superfamilia delTGF-β (Fig. 2). Existen tres clases funcionales:Smad receptor (R-Smad), Smad comediador(Co-Smad) y Smad inhibidor. Las que resultanfosforiladas son las R-Smad (Smad 1,5 y 8), lascuales deben formar un complejo con Co-Smady Smad 4 para traslocarse al núcleo e iniciarla transcripción genética(43). La unión de lasSmads al núcleo es débil, por lo que se requie-re la presencia de co-activadores o co-repre-sores, los cuales van a regular su función. Laseñal Smad puede reducirse mediante facto-res reguladores denominados Smurfs y por fos-fatasas(44). Las BMP pueden iniciar otras seña-les independientes de la vía Smad. Se hademostrado que regulan también la activaciónde las proteín-cinasas activadas por mitógeno(MAPK)(45), entre ellas p38MAPK, que tambiénparece tener un papel destacado en otras enfer-medades. Existen muchas variables que par-ticipan en la regulación de las BMP y un grannúmero de co-activadores o co-represores,
A. BALOIRA VILLAR, D. VALVERDE PÉREZ
166
FIGURA 2. Vía de la BMPR2.
BMP
BMPR1BMPR2
FosforilaciónR-Smad
Smad 4
p38 MAPK
Regulación de la expresión genética
Monografía HAP NM 208 pag 18/2/10 17:08 Página 166

entre ellos el tipo y densidad de los recepto-res 1 y 2 disponibles para formar dímeros. Posi-blemente, este sea el motivo de la baja pene-trancia de la enfermedad y de la especificidadtisular de las BMP. Sobre las células muscula-res lisas de las arterias pulmonares inducenapoptosis mientras que en las células endo-teliales pulmonares favorecen la superviven-cia inhibiendo la apoptosis(46). Es posible quelas mutaciones en BMPR2 produzcan un incre-mento de la apoptosis en las células endote-liales, lo cual favorecería la pérdida del lechocapilar pulmonar y obliteración de las áreasmás distales, es decir, el inicio de las lesionesque dan lugar a HAP. Se ha descrito reciente-mente otro mecanismo postranscripcional deregulación de BMPR2 al comprobar que lainterleucina-6, a través de la activación de STAT-3, disminuye intensamente la expresión delreceptor(47).
BMPR2 está ampliamente expresado. Lascélulas endoteliales de las arterias pulmona-res expresan de forma intensa receptores paralos diversos miembros de la familia TGF-β yproteínas Smad. En la capa media muscularla expresión es mucho menos intensa, inde-pendientemente de presentar o no HAP. Sinembargo, las células endoteliales del centro delas lesiones plexiformes apenas expresanreceptores para TGF-β o proteínas Smad(48),existan o no mutaciones en BMPR2. Es decir,la pérdida de la señal de las citocinas que for-man la superfamilia TGF-β parece tener unpapel muy importante en el desarrollo de HAPfavoreciendo la proliferación de células endo-teliales. Aunque la expresión de BMPR2 estádisminuida en todos los pacientes con HAP, esmás intensa en los que presentan mutaciones.Existen diferencias según el tipo de HAP, porejemplo, es más marcada en los casos con HAPidiopática que asociada a enfermedades deltejido conectivo(49).
Para conocer la importancia que tieneBMPR2 en la génesis de HAP, una posibilidadsería diseñar mediante ingeniería genética rato-nes knockout para este gen. Mediante una cepatransgénica se comprobó que tanto los homo-
cigotos como los heterocigotos presentabancifras de presión arterial pulmonar significati-vamente mayores que los controles a los 2 y7 meses de edad(50). Más del 50% de los homo-cigotos mostraban cifras por encima de 30mmHg.
Uno de los efectos de las mutaciones enBMPR2 es romper la vía BMP/Smad y poten-ciar la vía BMP/MAPK(51). Las mutaciones tipocambio de sentido pueden originar una sus-titución de los residuos de cisteína, tanto en lazona de unión al ligando como en el domi-nio cinasa, disminuyendo el tráfico de la pro-teína mutante hacia la superficie celular algoque, a su vez, interfiere con la unión al recep-tor BMP tipo 1. Por el contrario, las mutacio-nes en el dominio cinasa que no afectan a resi-duos de cisteína no son capaces de fosforilarBMPR1, con lo que no se activa la vía Smad.Si las mutaciones afectan a la parte citoplas-mática fuera del dominio cinasa se producendificultades en la señal de transducción víaSmad. El modelo experimental más habitualpara estudiar la HAP es mediante la inyecciónde monocrotalina. En este modelo se pudocomprobar una intensa reducción de la expre-sión de BMPR1b, BMPR2, y Smad 4, 5, 6 y 8en los pulmones aunque no en los riñones delas ratas estudiadas a las 4 semanas de la admi-nistración de monocrotalina(52). No se observódisminución en los niveles de fosforilación dep38MAPK. Cuando se induce HAP por exposi-ción a hipoxia también se observa una dis-minución significativa de la expresión deBMPR2(53). En este caso no se encontraroncambios en la fosforilación de Smads 1, 5 y8 y; sin embargo, sí en p38MAPK. Esto implicaque muy probablemente el mecanismo por elque la hipoxia y la monocrotalina inducen HAPsean diferentes, aunque en ambas están impli-cadas las BMP. Se ha encontrado otra proteí-na que interactúa con BMPR2, el receptor parala cinasa-C activada (RACK1)(54). Se trata de unaproteína de 31 kDa que se encuentra en el cito-plasma celular y se expresa de forma ubicua.Su misión es facilitar interacciones entre pro-teínas para mejorar las señales intracelulares.
GENÉTICA E HIPERTENSIÓN ARTERIAL PULMONAR
167
Monografía HAP NM 208 pag 18/2/10 17:08 Página 167

Fundamentalmente, disminuiye la prolifera-ción celular. En este estudio se indujeronmediante ingeniería genética cuatro mutacio-nes en BMPR2, observándose que, aunque nose impedía por completo la unión con RACK1,sí disminuía de forma significativa con lo cualse perdía en parte su capacidad anti-prolife-rativa. En HAP experimental inducida pormonocrotalina, tanto el ARNm como la con-centración de RACK1 en las células muscula-res lisas de las arterias pulmonares disminu-yeron de forma significativa. Estos resultadosapoyarían el posible papel de la interacciónBMPR2-RACK1 en la génesis de la HAP. Otrasproteínas importantes son las llamadas Id (inhi-bidores de la unión al ADN). Están distribui-das en múltiples células y tienen un impor-tante papel en la transcripción genética,impidiendo la unión de factores nucleares asus dianas. Los pacientes portadores de muta-ciones en BMPR2 presentan una disregulaciónde los genes Id en las células musculares lisasde las arterias pulmonares. Se ha comproba-do experimentalmente que esto significa unaalteración de de la supresión del crecimientocelular por parte de las BMP en las célulasmutantes(55). Las BMP también pueden unirsea receptores activina tipo II (ActRIIa) cuandoexiste deficiencia de BMPR2, aunque la señalmediada por esta unión no es comparable ala producida cuando se unen al receptor espe-cífico. Transitoriamente se estimulan Smad1/5/8 vía unión ActRIIa pero a largo plazo sólopersiste la señal vía BMPR2(56). En resumen, ladisfunción de la vía BMP/BMPR produce nota-bles alteraciones en la regulación de la proli-feración celular vascular pulmonar lo que, uni-do a factores apropiados, propiciaría laaparición de HAP.
LA VÍA DE LA SEROTONINALa serotonina es un jugador de primer
orden en la regulación del tono vascular pul-monar. Tiene un efecto importante como esti-mulante de proliferación de las células mus-culares lisas de las arterias bronquiales en unavía en la que es crucial el transportador de la
serotonina (TRSE)(57). La presencia de poli-morfismos, es decir, variaciones genéticas rela-tivamente frecuentes en la población general,tanto en el gen de la serotonina como en el delTRSE, podrían contribuir al desarrollo de HAP.En 1995 se describió el incremento de la con-centración de serotonina plasmática en 16pacientes con HAP idiopática y en un pacien-te con una enfermedad de depósito con acú-mulo excesivo de serotonina en las plaque-tas(58). A nivel experimental se demostró quelas células musculares de arteriolas pulmo-nares de pacientes con HAP en presencia deserotonina proliferaban de una forma signifi-cativamente más intensa que las de individuosnormales. Si se sometían a hipoxia simultá-neamente la proliferación era considerable-mente mayor. Se atribuyó este hecho funda-mentalmente al TRSE(59).
El gen del TRSE está localizado en el cro-mosoma 17 locus q11.1-12. Tiene 31 kilobasesy consta de 14 exones. La parte promotora delgen puede presentar una variante larga (L) ocorta (S) con diferencias notables en cuanto ala eficiencia transcripcional. En un estudio queincluyó a 89 pacientes con HAP grave y 84 con-troles sanos se observó que en el primer gru-po, un 65% presentaban de forma homoci-gota la variante larga (LL) mientras que estosólo sucedía en el 27% de los controles(60). Elgrupo de Machado ha estudiado el papel quelos polimorfismos en el gen del TRSE tienenen la predisposición y desarrollo de HAP en528 pacientes(61). De ellos, 133 tenían HAPH omutaciones en BMPR2. No se observaron dife-rencias en los alelos del gen entre los diferen-tes grupos lo que hace pensar que este gen notiene un papel importante en la expresión feno-típica de la enfermedad. Incluso en el sexo, laúnica variable clara en la incidencia de la enfer-medad, tampoco hubo diferencias en relacióncon los polimorfismos. Dado que no existierontampoco diferencias en relación con ser por-tador o no de una mutación en BMPR2, noparece que TRSE tenga algún papel en la pene-trancia de la HAPf. Otro estudio diseñado parainvestigar si el alelo L podría implicar un inicio
A. BALOIRA VILLAR, D. VALVERDE PÉREZ
168
Monografía HAP NM 208 pag 18/2/10 17:08 Página 168

GENÉTICA E HIPERTENSIÓN ARTERIAL PULMONAR
169
más precoz y una menor expectativa de vidacomparado con el alelo S, sólo encontró lo pri-mero en los pacientes con HAPf y alelo L. Estono sucedió en los casos con HAP esporádi-ca(62).Quizá ello implique algún tipo de relaciónentre BMPR2 y serotonina todavía no elucida-da La serotonina entra en el citoplasma mer-ced a la TRSE y allí activa la vía de las MAPK,la cual actúa como estimulante de la prolifera-ción celular. En células musculares lisas de arte-rias pulmonares se ha observado un potentepapel de la serotonina como activador de la víarho-cinasa, vía que está adquiriendo una granimportancia tanto en la patogenia de la HAPcomo en posible diana terapéutica(63). Este efec-to es opuesto al que realizan las BMPs. Un des-equilibrio hacia la vía serotonina-TRSE debidoa polimorfismos en este último o bien a la dis-minución de la señal BMP como consecuenciade mutaciones en BMPR2 podría iniciar laslesiones que darán lugar a HAP.
POLIMORFISMOS EN OTROS GENESSe han publicado algunos estudios aisla-
dos en general con escaso número de pacien-tes estudiando polimorfismos en otros genes.Por ejemplo, se comprobó que los pacientescon esclerodermia y HAP tenían de forma sig-nificativa diferencias en la presencia de dospolimorfismos de nucleótido único en posición1.026 y 277 así como en la repetición de unpentanucleótido (CCTTT) en el gen de la sin-tasa del óxido nítrico (NOs) comparados conlos pacientes con esclerodermia sin HAP o con-troles(64). La actividad transcripcional de la NOsen los fibroblastos a los que se les introdujoeste pentanucleótido fue claramente mayor.Nuestro grupo ha comprobado una diferenciasignificativa en el número de pentanucleóti-dos entre individuos sanos y pacientes conHAP (datos pendientes de publicación). Loscanales del potasio también han suscitado inte-rés. Un trabajo reciente ha mostrado cómo lospolimorfisrmos en la región promotora o tras-lacional del gen que codifica el canal del pota-sio Kv1.5 son más frecuentes en las célulasmusculares de las arterias pulmonares de los
pacientes con HAP(65). Un polimorfismo denucleótido único en una isoforma de los cana-les no voltaje dependientes del calcio, laTRPC6, está aumentado casi tres veces res-pecto a la población general en pacientes conHAP(66). Este canal tiene una importante rela-ción con el factor nuclear κB, fundamental enla transcripción de citocinas proinflamatorias.Se han estudiado diversos polimorfismos enel gen de la endotelina-1, alguno de los cua-les, como el G/T K198N, presenta asociacióncon hipertensión arterial sistémica, sobre todoen individuos obesos. Nuestro grupo ha encon-trado en una serie pequeña un posible aumen-to de la presencia de este polimorfismo en lospacientes con HAP asociada a fibrosis pulmo-nar (datos no publicados).
CONCLUSIONESAl igual que en otras enfermedades cróni-
cas, la genética juega un papel fundamental enla aparición de HAP. Si bien es necesaria la pre-sencia de otros factores exógenos, un terrenopreparado por una o varias mutaciones en algu-nos de los genes que regulan la proliferación delas células musculares de las arterias pulmona-res facilitan que se desarrolle la enfermedad yen muchos casos la hacen diferente. No hayduda de que la familia del TGF-β y, más con-cretamente, las BMP son uno de los pilares sobrelos que descansa buena parte de la patogeniade la HAP, pero también la vía de la serotoninay algunos de los canales del K+ y del Ca2
+ tie-nen un papel importante que posiblemente seirá conociendo mejor en los próximos años.Dada la variabilidad en la expresión clínica dealgunas de las mutaciones descritas, por elmomento es difícil dar consejos genéticos orecomendaciones sobre la realización de estu-dios a familiares de pacientes con la enferme-dad, debiendo ser muy cautos para no crearalarma innecesaria. Será preciso ir desgranan-do un poco más la intrincada vía de señales queimplica TGF-β para poder llegar a conocer mejorel riesgo potencial de desarrollo de HAP y, sobretodo, poder hacer algún tipo de prevención, algoque por ahora no es posible.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 169

BIBLIOGRAFÍA1. Gaine SP, Rubin LJ. Primary pulmonary hyper-
tension. Lancet. 1998; 352: 719-25.2. Dresdale DT, Michtom RJ, Schultz M. Recent
studies in primary pulmonary hypertension,including pharmacodynamic observations onpulmonary vascular resistence. Bull N Y AcadMed. 1954; 30 (3): 195-207.
3. Melmon KL, Braunwald E. Familial pulmonaryhypertension. N Eng J Med. 1963; 269: 770-5.
4. Tsagaris TJ, Tikoff G. Familial primary pulmo-nary hypertension. Am Rev Respir Dis. 1968;97: 127-30.
5. Loyd JE, Primm RK, Newman JH. Familial pri-mary pulmonary hypertension: clinical patterns.Am Rev Respir Dis. 1984; 129 (1):194-7.
6. Nicholls WC, Koller DL, Slovis B, et al. Locali-sation of the gene for familial primary pul-monary hypertension to chromosome2q31–32. Nat Genet. 1997; 15: 277-80.
7. Lane KB, Machado RD, Pauciulo MW, et al.Heterozygous germ-line mutations inBMPR2,encoding a TGF-β receptor, cause familial pri-mary pulmonary hypertension. The Interna-tional PPH Consortium. Nat Genet. 2000; 26:81-4.
8. Deng Z, Morse JH, Slager SL, et al. Familial pri-mary pulmonary hypertension (gene PPH1) iscaused by mutations in the bone morphoge-netic protein receptor-II gene. Am J HumGenet. 2000; 67: 737-44.
9. Trembath RC, Thomson JR, Machado RD, etal. Clinical and molecular genetic features ofpulmonary hypertension in patients with here-ditary hemorrhagic telangiectasia. N Engl JMed. 2001; 345 (5): 325-34.
10. Humbert M, Deng Z, Simonneau G, et al.BMPR2 germline mutations in pulmonaryhypertension associated with fenfluraminederivatives. Eur Respir J. 2002; 20: 518-23.
11. Rich S, Dantzker DR, Ayres SM, et al. Primarypulmonary hypertension: a national prospec-tive study. Ann Intern Med. 1987; 107: 216-30.
12. Humbert M, Sitbon O, Chaouat A, et al. Pul-monary arterial hypertension in France: resultsfrom a national registry. Am J Respir Crit CareMed. 2006; 173 (9):1023-30.
13. Austin ED, Loyd JE. Genetics and mediators inpulmonary hypertension. Clin Chest Med.2007; 28: 43-57.
14. Loyd JE, Butler MG, Foroud TM, et al. Gene-tic anticipation and abnormal gender ratio
at birth in familial primary pulmonary hyper-tension. Am J Respir Crit Care Med. 1995; 152(1): 92-7.
15. Ashley CT Jr, WarrenST. Trinucleotide repeatexpansion and human disease. Annu RevGenet. 1995; 29: 703-28.
16. Loyd JE, Atkinson JB, Pietra GG, et al. Hete-rogeneity of pathologic lesions in familial pri-mary pulmonary hypertension. Am Rev Res-pir Dis. 1988; 138: 952-7.
17. Sztrymf B, Coulet F, Girerd B, et al. Clinical out-comes of pulmonary arterial hypertension incarriers of BMPR2 mutation. Am J Respir CritCare Med. 2008; 177: 1377-83.
18. McAllister KA, Grogg KM, Johnson DW, et al.Endoglin, a TGF-β binding protein of endo-thelial cells, is the gene for hereditary hemor-rhagic telangiectasia type 1. Nat Genet. 1994;8: 345-51.
19. Johnson DW, Berg JN, Baldwin MA, et al. Muta-tions in the activin receptor-like kinase in here-ditary hemorrhagic telangiectasia type 2. NatGen. 1996; 13: 189-95.
20. Harrison RE, Berger R, Haworth SG, et al.Transforming growth factor-beta receptor muta-tions and pulmonary arterial hypertensionin childhood. Circulation. 2005; 111(4): 435-41.
21. Fernández LA, Sanz-Rodríguez F, Blanco FJ, etal. Hereditary hemorrhagic telangiectasia, avascular dysplasia affecting the TGF-beta sig-naling pathway. Clin Med Res. 2006; 4 (1):66-78.
22. Thomson JR, Machado RD, Pauciulo MW, et al.Sporadic primary pulmonary hypertension isassociated with germline mutations of the geneencoding BMPR-II, a receptor member of theTGF-α family. J Med Genet. 2000; 37: 741-5.
23. Koehler R, Grünig E, Pauciulo MW, et al. Lowfrequency of BMPR2 mutations in a Germancohort of patients with sporadic idiopathic pul-monary arterial hypertension. J Med Genet.2004; 41: e127.
24. Morisaki H, Nakanishi N, Kyotani S, et al.BMPR2 mutations found in Japanese patientswith familial and sporadic primary pulmonaryhypertension. Hum Mutat. 2004; 23: 632.
25. Sankelo M, Flanagan JA, Machado RD, et al.BMPR2 mutations have short lifetime expec-tancy in primary pulmonary hipertensión.Hum Mutat. 2005; 26:119-24.
26. Baloira A, Vilariño C, Leiro V, et al. Mutacio-nes en el gen que codifica BMPR2 en pacien-
A. BALOIRA VILLAR, D. VALVERDE PÉREZ
170
Monografía HAP NM 208 pag 18/2/10 17:08 Página 170

GENÉTICA E HIPERTENSIÓN ARTERIAL PULMONAR
171
tes con hipertensión pulmonar arterial idio-pática. Arch Bronconeumol. 2008; 44 (1): 29-34.
27. Roberts KE, McElroy JJ, Wong WPK, et al.BMPR2 mutations in pulmonary arterial hyper-tension with congenital heart disease. Eur Res-pir J. 2004; 24: 371-4.
28. Liu F, Ventura F, Doody J, Massagué J. Humantype II receptor for bone morphogenic pro-teins (BMPs): extension of the two-kinasereceptor model to the BMPs. Mol Cell Biol1995; 15: 3479-86.
29. Elliot CG. Genetics of pulmonary arterial hyper-tension: current and future implications. SemRespir Crit Care Med. 2005; 26(4): 365-71.
30. Machado RD Eickelberg O, Elliot CG, et al. Gene-tics and genomics of pulmonary arterial hyper-tension. J Am Coll Cardio. 2009; 54: s32-42.
31. Nishihara A, Watabe T, Imamura T, et al. Func-tional heterogeneity of bone morphogeneticprotein receptor type-II mutants founf inpatients with primary pulmonary hyperten-sion. Mol Biol Cell. 2002; 13: 3055-63.
32. Aldred MA, Vijayakrishnan J, James V, et al.BMPR2 gene rearrangements account for asignificant proportion of mutations in familialand idiopathic pulmonary arterial hyperten-sion. Hum Mutat. 2006; 27: 212-3.
33. Aldred MA, Machado RD, James V, et al. Cha-racterization of the BMPR2 5´-untranslatedregion and a novel mutation in pulmonaryhypertension. Am J Respir Crit Care Med. 2007;176: 819-24.
34. Nasim MT, Ghouri A, Patel B, et al. Stoichio-metric imbalance in the receptor complex con-tributes to dysfunctional BMPR-II mediatedsignalling in pulmonary arterial hypertension.Hum Mol Genet. 2008; 17: 1683-94.
35. Machado RD, Pauciulo MW, Thomson JR, et al.BMPR2 haploinsufficiency as the inherited mole-cular mechanism for primary pulmonary hyper-tension. Am J Hum Genet. 2001; 68: 92-102.
36. Cogan JD, Pauciulo MW, Batchman AP, et al.High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterialhypertension Am J Respir Crit Care Med. 2006;174: 590-8.
37. Hamid R, Cogan JD, Hedges LK, et al. Pene-trance of pulmonary arterial hypertension ismodulated by the expression of normalBMPR2 allele. Hum Mut. 2009; 30: 649-54.
38. Lee S-D, Shroyer KR, Markham NE, et al.Monoclonal endothelial cell proliferation is pre-
sent in primary but not secondary pulmonaryhypertension. J Clin Invest. 1998; 101: 927-34.
39. Tuder RM, Radisavljevic Z, Shroyer KR, et al.Monoclonal endothelial cells in appetite sup-pressant–associated pulmonary hypertension.Am J Respir Crit Care Med. 1998; 158:1999-2001.
40. Machado RD, James V, Southwood M, et al.Investigation of Second Genetic Hits at theBMPR2 Locus as a Modulator of Disease Pro-gression in Familial Pulmonary Arterial Hyper-tension. Circulation. 2005; 111: 607-13.
41. Miyazono K, Maeda S, Imamura T. BMP recep-tor signaling: transcriptional targets, regula-tion of signals, and signaling cross-talk. Cito-kine Growth Factor Rev. 2005; 16: 251-63.
42. Rosenzweig BL, Imamura T, Okadome T, et al.Cloning and characterization of a human typeII receptor for bone morphogenetic proteins.Pro Natl Acad Sci USA. 1995; 92: 7632-6.
43. Shi Y, Massagué J. Mechanisms of TGF-β sig-naling from cell membrane to the nucleus. Cell2003;113: 685-700.
44. Chen HB, Shen J, Ip YT, et al. Identification ofphosphatases for Smad in the BMP/DPP path-way. Genes Dev. 2006; 20: 648-53.
45. Massagué J, Chen Y-G. Controlling TGF-β sig-naling. Genes Dev. 2000; 14: 627-44.
46. Teichert-Kuliszewska K, Kutryk M, KuliszewskiMA, et al. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endo-thelial cell survival. Implications for loss-of-func-tion mutations in the pathogenesis of pulmonaryhypertension. Circ Res. 2006; 98: 209-17.
47. Brock M, Trenkmann M, Gay RE, et al. Inter-leukin-6 modulates the expression of the bonemorphogenic protein receptor typo II througha novel STAT3-microRNA cluster 17/92 path-way. Cir Res. 2009; 104: 1184-91.
48. Richter AM, Yeager ME, Zaiman A, et al. Impai-red transforming growth factor-β signaling inidiopathic pulmonary hypertension. Am J Res-pir Crit Care Med. 2004; 170: 1340-8.
49. Atkinson C, Stewart S, Upton PD, et al. Pri-mary pulmonary hypertension is associatedwith reduced pulmonary vascular expressionof type II bone morphogenetic protein recep-tor. Circulation. 2002; 105: 1672-8.
50. Hong K, Lee YJ, Lee E, et al. Genetic ablationof the BMPR2 gene in pulmonary endotheliumis sufficient to predispose to pulmonary hyper-tension. Circulation. 2008; 118: 722-30.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 171

A. BALOIRA VILLAR, D. VALVERDE PÉREZ
172
51. Nishihara A, Watabe T, Imamura T, Miyazo-no K. Functional heterogeneity of bone mor-phogenetic protein receptor-II mutants foundin patients with primary pulmonary hyper-tension. Mol Biol Cell 2002; 13: 3055-63.
52. Morty RE, Nejman B, Kwapiszewska G, et al.Dysregulated bone morphogenetic protein sig-nalin in monocrotaline-induced arterial hyper-tension. Arterioscler Thromb Vasc Biol. 2007;27: 1072-8.
53. Takahashi H, Goto N, Kojima Y, et al. Down-regulation of type II bone morphogenetic pro-tein receptor in hypoxic pulmonary hyper-tension. Am J Physiol Lung Cell Mol Physiol.2006; 290: L450–L458.
54. Zakrzewicz A, Hecker M, Marsh LM, et al.Receptor for activated C-Kinase 1, a novel inter-action partner of type II bone morphogeneticprotein receptor, regulates smooth muscle cellproliferation in pulmonary arterial hyperten-sion. Circulation. 2007; 115: 2957-68.
55. Yang J, Davies RJ, Southwood M, et al. Muta-tions in bone morphogenetic protein type IIreceptor cause dysregulation of Id gene expres-sion in pulmonary artery smooth muscle cells.Cir Res. 2008; 102: 1212-21.
56. Yu PB, Deng DY, Beppu H, et al. Bone mor-phogenetic protein (BMP) type II receptor isrequired for BMP-mediated growth arrest anddifferentiation in pulmonary artery smoothmuscle cells. J Biol Chem. 2008; 283: 3877-88.
57. Lee SL, Wang WW, Moore BJ, et al. Dual effectof serotonin on growth of bovine pulmonaryartery smooth muscle cells in culture. Cir Res.1991; 68 (5): 1362-8.
58. Herve P, Launay JM, Scrobohaci ML, et al. Incre-ased plasma serotonin in primary pulmonaryhypertension. Am J Med. 1995; 99: 249-54.
59. Eddahibi S, Raffestin B, HamonM, et al. Is theserotonin transporter involved in the patho-genesis of pulmonary hypertension? J Lab ClinMed. 2002; 139: 194-201.
60. Eddahibi S, Humbert M, Fadel E, et al. Sero-tonin transporter overexpression is responsi-ble for pulmonary artery smooth musclehyperplasia in primary pulmonary hyperten-sion. J Clin Invest. 2001; 108: 1141-50.
61. Machado RD, Koehler R, Glissmeyer E, et al.Genetic association of the serotonin transporterin pulmonary arterial hypetension. Am J Res-pir Crit Care Med. 2006; 173 (7): 793-7.
62. Willers ED, Newman JH, Loyd JE. Serotonintransporter polimorphisms in familial and idio-pathic pulmonary arterial hypertension. AmJ Respir Crit Care Med. 2006; 173 (7): 798-802.
63. Liu Y, Suzuki YJ, Day RM, et al. Rho kinase-induced nuclear translocation of ERK1/ERK2in smooth muscle cell mitogenesis caused byserotonin. Cir Res. 2004; 95 (6): 579-86.
64. Kawaguchi Y, Tochimoto A, Hara M, et al.NOS2 polymorphisms associated with the sus-ceptibility to pulmonary arterial hypertensionwith systemic sclerosis: contribution to thetranscriptional activity. Arthritis Res Ther. 2006;8 (4):1-10.
65. Remillard CV, Tigno DD, Platoshyn O, et al.Function of Kv1.5 channels and genetic varia-tions of KCNA5 in patients with idiopathic pul-monary arterial hypertension. Am J PhysiolCell Physiol 2007; 292: C1837-53.
66. Yu Y, Keller SH, Remillard CV, et al. A func-tional single-nucleotide polymorphism in theTRPC6 gene promoter associated with idio-pathic pulmonary hypertension. Circulation.2009; 119: 2313-22.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 172

173
RESUMENLa hipertensión arterial pulmonar (HAP) es
una patología en la que están involucradasnumerosas etiologías y en la que están impli-cados varios mecanismos. Durante los últimosaños se ha avanzado notablemente en el cono-cimiento de la fisiopatología de esta enferme-dad. En la actualidad el tratamiento se centraen el uso aislado o combinado de prostanoi-des, antagonistas de los receptores de la endo-telina o inhibidores de la fosfodiesterasa, sinolvidar como última opción el trasplante pul-monar. La estrategia terapéutica actual sigueasociándose a una tasa elevada de mortalidad,así como a un deterioro progresivo hemodi-námico y funcional en muchos de los pacien-tes con HAP. Los progresos obtenidos con lasnuevas terapias dirigidas siguen siendo insu-ficientes, por lo que se requiere un mayoresfuerzo de cara a investigar nuevas estrate-gias y opciones terapéuticas que mejoren losresultados obtenidos hasta la fecha. En estecapítulo vamos resumir las futuras líneas deinvestigación y posibles opciones terapéuticas.
INTRODUCCIÓNLa hipertensión arterial pulmonar (HAP) es
una patología en la que están involucradas nume-rosas etiologías y en la que están implicadosvarios mecanismos. Durante los últimos años seha avanzado notablemente en el conocimien-to de la fisiopatología de esta enfermedad, sien-do la vasoconstricción, la disfunción endotelialy la proliferación de las células musculares lisasde los vasos pulmonares (CMLV) los principa-les protagonistas de la HAP. Las células endote-liales (CE) juegan un papel fundamental en laetiopatogenia de esta enfermedad, participan-
do en la regulación del tono vascular, la home-ostasis, la respuesta inflamatoria y angiogénica,así como en los fenómenos de remodelado dela pared vascular. También se sabe que existe undesequilibrio entre los factores que predisponeno que protegen de la vasoconstricción, la divi-sión celular y la inflamación. Por otro lado, eldescubrimiento de mutaciones genéticas espe-cíficas, el reconocimiento de nuevas sustanciasvasoactivas y de factores del crecimiento haabierto nuevas vías de investigación de cara aldesarrollo de nuevas terapias(1).
En la actualidad el tratamiento se centra enel uso aislado o combinado de prostanoides,antagonistas de los receptores de la endotelina1 (ARE) o inhibidores de la fosfodiesterasa 5(IPDE-5), sin olvidar como última opción el tras-plante pulmonar. Las nuevas terapias handemostrado una mejoría de los síntomas, de lacalidad de vida, de los parámetros hemodiná-micos y de la capacidad de ejercicio. Sin embar-go, los logros obtenidos en relación al aumen-to de la supervivencia de los pacientes con HAPson más modestos. Un meta-análisis publicadorecientemente pone de manifiesto, que sí queexiste un beneficio en la supervivencia de aque-llos pacientes que sí recibieron tratamiento conprostanoides, antagonistas de los receptores dela ET-1 o inhibidores de la PDE-5(2). No obstan-te, estas conclusiones no están exentas de con-troversia, dado que dos años antes se publicóotro meta-análisis en el que no encontraronmejoría significativa en lo que se refiere a lasupervivencia(3).
La estrategia terapéutica actual sigue aso-ciándose a una tasa elevada de mortalidad, asícomo a un deterioro progresivo hemodinámicoy funcional en muchos de los pacientes con HAP.
FUTURAS LÍNEAS DE INVESTIGACIÓN ENLA HIPERTENSIÓN ARTERIAL PULMONAR
Carlos Almonacid Sánchez
Monografía HAP NM 208 pag 18/2/10 17:08 Página 173

Los progresos obtenidos con las nuevas terapiasdirigidas siguen siendo insuficientes, por lo quese requiere un mayor esfuerzo de cara a inves-tigar nuevas estrategias y opciones terapéuticas
que mejoren los resultados obtenidos hasta lafecha. Los nuevos ensayos clínicos deben mejo-rar aspectos en su diseño; por un lado, convie-ne hacer seguimientos a más largo plazo que losactuales y por otro, deben incorporar como obje-tivo principal el análisis de la supervivencia y nosólo centrarse en las mejoras en la capacidad deejercicio o de variables hemodinámicas(4).
En este capítulo vamos resumir las futuraslíneas de investigación y potenciales opcionesterapéuticas en la HAP a corto y a más largoplazo (Tabla 1).
NUEVOS PROSTANOIDESLos prostanoides siguen ocupando un papel
muy importante en la terapia de los pacientescon HAP y es muy posible que su importanciano cambie a corto plazo. El gran inconvenien-te de este tipo de tratamiento suele ser la vía deadministración y la vida media de este tipo defármacos. Por estos motivos se ha trabajadomucho en desarrollar prostanoides que puedanser administrados por vía oral y con un perío-do más largo de vida media. Hoy en día dis-ponemos de beraprost y treprostinil como pros-tanoides orales. Los beneficios obtenidos conberaprost no se mantuvieron a partir del nove-no mes de tratamiento y, en el caso del tre-prostinil oral, todavía no disponemos de losresultados del estudio FREEDOM(5,6). El princi-pal problema de estos fármacos son los efectossecundarios que son dosis dependientes, enespecial las cefaleas, dolor mandibular y alte-raciones digestivas. El NS-304 es un nuevo pros-tanoide con una vida media de diez horas yse espera que tenga menos efectos secundariosa nivel sistémico que los prostanoides oralesdisponibles en la actualidad(7).
NUEVOS ANTAGONISTAS DE LOSRECEPTORES DE LA ENDOTELINA 1
Como ya se ha mencionado con detalle enesta obra, la endotelina 1 (ET-1) es un poten-te vasoconstrictor y mitógeno de las CMLV. Has-ta la fecha se ha aprobado el uso de bosentan,sitaxentan y ambrisentan para el tratamientode la HAP, siendo estos dos últimos antago-
C. ALMONACID SÁNCHEZ
174
TABLA 1. Futuras líneas terapéuticas
Corto plazo• Nuevos prostaniodes orales:
- NS-304• Nuevos inhibidores de las fosfodiesterasas:
- Tadalafilo - 8MM-IBMX- Pumafentrina
• Nuevos antagonistas de los receptores de laendotelina:- Macitentan
Medio-largo plazo• Estimuladores de la guanilato ciclasa soluble:
- Riociguat (BAY 63-2521) • Activadores de la guanilato ciclasa soluble:
- YC-1- BAY 41-2272- BAY 58-2667- CFM-1571- A-350619- HMR-1766
• Inhibidores de la tirosin cinasa: - Imatinib- Sorafenib
• Análogos de la tetrahidrobiopterina: - Sapropterina
• Potenciadores de la transcripción de la eNOS: - AVE-3085 - AVE-9488
• Antagonistas de los receptores de serotonina: - Sarprogelato
• Estatinas: - Simvastatina- Atorvastatina
• Inhibidores de la Rho-cinasa: - Fasudil
• Inhibidores de las elastasas• Péptido intestinal vasoactivo (VIP)• Adrenomedulina• Otros
Monografía HAP NM 208 pag 18/2/10 17:08 Página 174

nistas selectivos del receptor A de la ET-1. Enla actualidad es difícil decidir si alguno de estosfármacos es más eficaz que el resto, pero loque sí aportan los ARE más selectivos son unamenor toxicidad y una menor interacción conotros fármacos. En la actualidad se está estu-diando en fase III el efecto sobre la morbi-mortalidad, como objetivo principal, de un nue-vo y potente ARE tisular –macitentan– (estudioSERAPHIN) en 700 pacientes(8,9).
NUEVOS INHIBIDORES DE LAFOSFODIESTERASA
Las fosfodiesterasas (PDE) son enzimasencargadas de inactivar el guanosín mono-fosfato cíclico (GMPc), que es una de las prin-cipales moléculas implicadas en la respuestacelular. Existen varios tipos de fosfodiestera-sas implicadas en la HAP, la más conocida esla PDE-5, y actualmente está aprobado el usode su inhibidor, el sildenafilo, como terapiapara los pacientes con HAP. En la actualidadestá en estudio el tadalafilo, que es otro inhi-bidor de la PDE de larga duración más poten-te. Recientemente se ha publicado con estefármaco un ensayo clínico controlado en 405pacientes con HP, mostrando su eficacia y segu-ridad a la dosis de 40 mg/día (PHIRST-1)(10).
Una nueva diana en estudio es la PDE-1,de la que existen tres isoformas que hidrolizantanto el AMPc como el GMPc. Las isoformas Ay B tienen una mayor afinidad por el GMPc.Estas enzimas, en especial las isoformas A y C,se han relacionado con el remodelado de lapared vascular. La utilización en ratas con HAPinducida por monocrotalina o por hipoxia deun inhibidor de la PDE-1C, el 8-metoximetil-isobutil-1-metilxantina (8MM-IBMX), ha demos-trado que es capaz de disminuir la resistenciavascular pulmonar (RVP) y la hipertrofia delventrículo derecho (HVD). El sildenafilo tam-bién actúa sobre la isoenzima de la PDE-1 perono el tadalafilo y se desconoce qué relevanciaclínica puede tener esta peculiaridad(11).
Pumafentrina es un inhibidor de la PDE3 y 4. Se ha observado que es eficaz en mode-los animales, disminuyendo HVD, el remo-
delado vascular y aumentando los niveles deAMPc. Sin embargo, los ensayos realizados coninhibidores de la PDE-3 en el tratamiento dela insuficiencia cardiaca aumenta de forma sig-nificativa la tasa de mortalidad, así que es pocoprobable que sean de utilidad en un futuro enla práctica clínica(12).
ESTIMULADORES O ACTIVADORES DE LAGUANILATO CICLASA SOLUBLE
La guanilato ciclasa soluble (GCs) es unadiana terapéutica que ha suscitado un graninterés en estos últimos años. Para que el óxi-do nítrico (NO) ejerza su función vasodilata-dora debe activar el GMPc mediante la GCs,que es una enzima formada por un grupohemo-Fe(II). La oxidación del grupo hemo deFe(II) a Fe(III) desactiva la GCs y, como resul-tado, el NO, al igual que ocurre con otrosvasodilatadores, como el péptido natriuréti-co, pierden su capacidad vasodilatadora(13).
Las investigaciones se centran en dosgrandes grupos, los estimuladores y los acti-vadores de la GCs (Fig. 1). Como comenta-mos previamente, en condiciones normalesel NO activa la GCs, uniéndose a su grupo elhemo-Fe(II), pero bajo condiciones de estrésoxidativo, los radicales O2 inactivan la GCs alreducir el grupo hemo de Fe(II) a Fe(III). Losestimuladores actúan preferentemente sobreel grupo hemo-Fe(II) no oxidado, mientrasque los activadores aumentan la producciónde GMPc actuando preferentemente sobre elgrupo hemo-Fe(III). Sobre esta base los esti-muladores requieren de la presencia de NOmientras que los activadores de la GCs actú-an con independencia del NO. Tanto los acti-vadores como los estimuladores de GCs handemostrado en modelos animales que soncapaces de revertir, en mayor o menor gra-do, el remodelado vascular(14).
Riociguat (BAY 63-2521) es un estimuladororal de la GCs que ha conseguido una reduc-ción del 30% de la RVP, aunque no sólo dis-minuye la presión de las arterias pulmonaressino que también actúa sobre la circulaciónsistémica, mejorando el gasto cardiaco. No
FUTURAS LÍNEAS DE INVESTIGACIÓN EN LA HIPERTENSIÓN ARTERIAL PULMONAR
175
Monografía HAP NM 208 pag 18/2/10 17:08 Página 175

obstante, el efecto sistémico desaparece trasvarios meses de tratamiento(15,16).
Próximos estudios con riociguat contro-lados con placebo incluyen el estudio CHEST-1, en el que participarán 270 pacientes conhipertensión pulmonar tromboembólica cró-nica (HPTC) durante 16 semanas evaluando lacapacidad de ejercicio. Por otro lado, el estu-dio PATENT-1 incluirá a 460 pacientes con HAP,evaluando el mismo objetivo principal ante-riormente mencionado tras 12 semanas detratamiento con riociguat.
El primero de los activadores que se desa-rrolló fue el YC-1 y, basándose en su estruc-tura, se han desarrollado nuevas moléculas
como el BAY 41-2272. Comparado con silde-nafilo BAY 41-2272 demostró que sus efectoseran más intensos y duraderos, disminuyen-do la PAP y la RVP. BAY 41-2272 también teníaun efecto sinérgico combinado con NO,aumentando y prolongando la respuesta vaso-dilatadora en las arterias pulmonares. BAY 58-2667 es otro activador que no necesita dela presencia del grupo hemo reducido paraactivar la GCs y, además, produce un efectosinérgico, más que aditivo, cuando se com-bina con NO. BAY 41-2272 y BAY 58-2667 handemostrado que pueden revertir la HAP indu-cida por monocrotalina en ratas, encontran-do que reducía la PAP media, la HVD y el
C. ALMONACID SÁNCHEZ
176
FIGURA 1. Vía de la guanilato ciclasa soluble (GCs). En condiciones normales el NO activa el GMPc median-te la GCs. Cuando la GCs se oxida por acción de radicales libres pierde su función. Existen activadoresde la GCs oxidada que activan el GMPc sin ayuda del NO. Los estimuladores sí necesitan del NO para quela GCs cumpla su función. La activación del GMPc conlleva una respuesta celular. Los PDE-5 degradan elGMPc a GMP, inactivándolo. Para que el GMP se convierta en GTP se requiere de energía en forma deATP y así de nuevo comenzar el ciclo. eNOS: óxido nítrico sintetasa. NO: óxido nítrico. O2- radicales super-óxido. GMPc: guanosín monofosfato cíclico. GMP: guanosín monofosfato. GTP: guanosín trifosfato. PDE 5:inhibidores de la fosfodiesterasa 5. ATP: adenosín trifosfato.
L-Arg
eNOS
NO
GCs
GCs
Heme Fe (II)
Heme Fe (III)
GMPc
GTPATP
GMP
PDE 5
–
+
ACTIVADORESYC-1BAY 412272BAY 582667
CFM-1571A-350619HMR-1766
Resouesta celularVASODILATACIÓN
Celula endotelialESTIMULADORES
BAY 632521
Célula muscular lisa vascular
O2-
VÍA GUANILATO CICLASA SOLUBLE
Monografía HAP NM 208 pag 18/2/10 17:08 Página 176

remodelado vascular. Una ventaja de estosnuevos fármacos es que pueden ser admi-nistrados por vía oral y con un vida media dehasta 12 horas. Los datos de que disponemosde los ensayos realizados hasta el momentoindican un adecuado perfil de seguridad yuna buena tolerancia por parte del pacien-te. Otros activadores en estudio son el CFM-1571, el A-350619 y el HMR-1766(17).
INHIBIDORES DE LA TIROSÍN-CINASAEl remodelado vascular, como ya se ha
mencionado, juega un papel muy importan-te en la fisiopatología de la HAP. Explicadode forma sencilla, el remodelado consiste enla proliferación incontrolada mediada por fac-tores de crecimiento de las CMLV, miofibro-blastos y CE. En la HAP, el factor de creci-miento derivado de plaquetas (PDGF) se harelacionado con la disfunción y proliferaciónde las CMLV y CE. Existen otras patologías enlas que también se observa un crecimientocelular incontrolado, como es el caso de lasneoplasias, en las que ya desde hace variosaños se vienen utilizando terapias dirigidascontra los receptores transmembrana tirosín-cinasa (TK)(11). El PDGF también ejerce su fun-ción a través de un receptor tipo TK y, tantoen modelos animales como en humanos conHAP, se ha visto una regulación al alza deestos receptores. Es por ello que los inhibi-dores de la tirosin-cinasa (TKI) se han posi-cionado como una de las más prometedorasopciones terapéuticas en el tratamiento de laHAP(18).
El imatinib es uno de los primeros TKI quecomenzó a utilizarse en los pacientes con leu-cemia mielogénica crónica ya que actúa sobreel receptor TK BCR-ABL, que está involucradoen la patogenia de de esta enfermedad. Ima-tinib también actúa sobre el receptor TK delPDGF, lo que le confiere un potencial efectobeneficioso sobre el remodelado vascular yjustifica su uso en los pacientes con HAP. Enmodelos animales de HTA inducida por mono-crotalina o hipoxia, imatinib demostró que eracapaz de revertir completamente la prolifera-
ción celular en los vasos pulmonares, reducirla HVD y la presión en el ventrículo derecho,aumentando el gasto cardiaco(19).
Imatinib se ha probado en casos aisladosde pacientes con HAP severa y ha demos-trado beneficios en la clase funcional, la capa-cidad de ejercicio y parámetros hemodiná-micos. No obstante, no todos los pacientesresponden de una forma adecuada a este fár-maco, habiéndose descrito casos de dete-rioro acelerado en pacientes con HAP refrac-taria a otros tratamientos y en los que seutilizó imatinib como uso compasivo(20,21).Por lo tanto, es necesario seguir investigan-do para comprobar la eficacia de este trata-miento como terapia adyuvante y el perfil deseguridad en este tipo de pacientes, ya queel uso prolongado de imatinib se ha relacio-nado con disfunción ventricular y fallo car-diaco, lo que puede limitar su uso en la prác-tica clínica.
Se están estudiando nuevos TKI que seanmenos cardiotóxicos. Sorafenib es un TKI múl-tiple que actúa sobre los receptores 1 y 3 delfactor de crecimiento endotelial vascular(VEGFR), el receptor β del PDGF y la cinasaRaf-1. Sorafenib ha demostrado que es capazde revertir la HAP inducida por hipoxia y/o lainhibición de VEGFR 1 y 2 en modelos ani-males de HAP. Estamos a la espera de los resul-tados de seguridad y eficacia de los ensayosclínicos que se están llevando a cabo(22).
TETRAHIDROBIOPTERINA Y ÓXIDONÍTRICO SINTETASA ENDOTELIAL
La tetrahidrobiopterina (BH4) es un cofactorde la óxido nítrico sintetasa endotelial (eNOS)que precisa para su síntesis de la GTP-ciclohi-drolasa-1 (GTP-CH1). En ratones con una activi-dad alterada de la GTP-CH1 se ha observado quepresentan niveles bajos de BH4 y terminan desa-rrollando HAP. Esto puede deberse a que sin laBH4 la eNOS no sintetiza NO sino aniones super-óxido y empeoran aún más la situación hemo-dinámica (Fig. 2). Por otro lado, la sobreexpre-sión de la GTP-CH1 en el endotelio vascularprotege a los ratones de desarrollar HAP(11).
FUTURAS LÍNEAS DE INVESTIGACIÓN EN LA HIPERTENSIÓN ARTERIAL PULMONAR
177
Monografía HAP NM 208 pag 18/2/10 17:08 Página 177

En el ser humano con HAP no se ha detec-tado una deficiencia o disfunción de la GTP-CH1, sin embargo, sí se ha visto que existenpolimorfismos de la GTP-CH1 que se asociancon la disponibilidad de NO. Los pacientes conHAP muestran un aumento de los marcadoresde estrés oxidativo y esto puede deberse a unareducción de la BH4 a dihidrobiopterina(18).
La sapropterina es una forma sintética deBH4 que ya se ha utilizado en pacientes confenilcetonuria. Los estudios demostraron unabuena tolerancia de este fármaco por parte delos pacientes, lo que facilita que se comiencenensayos clínicos con sapropterina en pacien-tes con HAP.
Los potenciadores de la transcripción de laeNOS, como AVE-3085 y AVE-9488, tambiénestán siendo estudiados. Este último ha demos-trado que incrementa el contenido vascular deBH4 y revierte el desacoplamiento de la eNOSen modelos animales. Está también por ver sipuede existir un sinergismo entre estos com-puestos y los suplementos de BH4 o activa-dores de la GCs.(23).
SEROTONINALa serotonina (5-hidroxitriptamina o 5-HT)
es una monoamina neurotransmisora sinteti-zada en las neuronas serotoninérgicas en elsistema nervioso central (SNC) y en las célu-
C. ALMONACID SÁNCHEZ
178
FIGURA 2. La GTP-CH1 estimula la síntesis de BH4, que es un cofactor necesario para que la eNOS llevea cabo su función y estimule la síntesis de NO. Los radicales libres (aniones superóxido) inhiben la activi-dad de la GTP-CH1 y, como consecuencia de ello, disminuye la síntesis de BH4, alterándose la funciónde la eNOS y favoreciendo que se sinteticen más aniones superóxido en vez de NO. Como consecuenciade estas alteraciones se favorecerían las alteraciones del tono vascular, proliferación celular e inflamaciónque se ven en los modelos animales de HAP. BH4: tetrahidrobiopterina. NO: óxido nítrico. eNOS: óxidonítrico sintetasa endotelial. GTP-CH1: guanosín trifosfato-ciclohidrolasa-1.
eNOS eNOSBH4 ↓ BH4
O2-
GTP-CH1GTP-CH1
+
+
O2-
–
–
–
L-ArgL-Arg
NO
NO HAP HAP
VasoconstricciónAdhesión leucocitaria
Agregación plaquetariaProliferación celular
VasoconstricciónAdhesión leucocitaria
Agregación plaquetariaProliferación celular
NO
Monografía HAP NM 208 pag 18/2/10 17:08 Página 178

las enterocromafines del tracto gastrointesti-nal de los animales y del ser humano. La hipó-tesis serotoninérgica surgió a partir de la apa-rición de casos de HAP en pacientes tratadoscon anorexígenos serotoninérgicos (amino-rex)(11,24). La serotonina estimula la vasocons-tricción y la proliferación de las células mus-culares lisas de las arterias pulmonares. Enmodelos animales se observó que la inhibi-ción del transportador de serotonina (5-HTT)y de los receptores de la serotonina podía inhi-bir la aparición de HAP. Durante los últimosaños se publicaron diversos estudios que rela-cionaban el sistema serotoninérgico con laaparición de hipertensión pulmonar funda-mentalmente a través de los receptores sero-toninérgicos, la triptófano hidroxilasa-1 y el5-HTT(25).
Los receptores serotoninérgicos se encuen-tran divididos en 7 familias que presentan dife-rencias estructurales entre sí. Los tipos 5-HT1A, 5-HT 2A y 5-HT 2B estarían involucradosen la biología de la hipertensión pulmonar. Enmodelos animales de HAP desencadenada porhipoxia la ausencia de receptores 5-HT 1B y2B demostró una mejoría de la HVD y unmenor grado de remodelado vascular. En ratascon HAP inducida por monocrotalina el blo-queo con sarprogelato del receptor 5-HT 2Ainhibía el desarrollo de HAP.
Los estudios en humanos sugieren que elreceptor relacionado con el desarrollo de HAPes fundamentalmente el 5-HT 1B. Esto justi-fica que la ketanserina, que es un antagonistafundamentalmente del receptor 5-HT 2A, hayatenido tan malos resultados en el tratamien-to de la HAP(26).
La expresión del gen de la triptófano hidro-xilasa-1 (TH1) se encuentra aumentada en lascélulas pulmonares y endoteliales de lospacientes con HAP. En ratones sin el gen de laTH1 y con niveles periféricos bajos de seroto-nina se observó una inhibición del remodela-do vascular pulmonar y la desaparición de lahipoxia inducida por la hipertensión pulmo-nar. Tanto la hipoxia como el estiramientomecánico de los vasos aumentan la expresión
de la TH1 y la liberación de serotonina, lo quesugiere que la hipoxia crónica induce la sínte-sis de la TH1 y la liberación de serotonina. Esteneurotransmisor actúa como mitógeno en lasarterias pulmonares, lo que contribuye a laaparición de remodelado vascular e hiperten-sión pulmonar(11).
Los estudios que analizan los polimorfis-mos del gen promotor del 5-HTT han encon-trado que la variante LL confiere susceptibili-dad a desarrollar HAP. No obstante, los datosen humanos que analizan el efecto de inhibirel 5-HTT son limitados y, en algunas ocasio-nes, contradictorios, por lo que se debe seguirinvestigando(18).
ESTATINASLa estatinas, además de bloquear la sín-
tesis de colesterol al inhibir la 3-hidroxi-metil-glutaril-CoA-reductasa (HMG-CoA), han demos-trado tener propiedades anti-inflamatorias alinhibir las moléculas de adhesión VCAM-1 eICAM1 y ejercer una regulación al alza de laGTP-CH en las células endoteliales, aumen-tando los niveles de BH4. La simvastatina, ade-más, ha demostrado que también es capaz deinhibir la actividad de las Rho-cinasas 1 y 2(ROCK 1 y 2)(27).
El uso de simvastatina en modelos ani-males de HAP desencadenada por hipoxia omonocrotalina se asoció a una reducción delremodelado vascular al disminuir la prolifera-ción y favorecer la apoptosis de las CMLV. Lamejoría del remodelado se acompañó de undescenso de la PAP media y del grado de HVD.También produce una regulación a la baja devarios genes inflamatorios y una regulación alalza de la eNOS, inhibidor del ciclo celular p27 kip 1 y el receptor morfogenético óseo tipo1a. Sin embargo no se han encontrado resul-tados positivos en todos los estudios que ana-lizan el uso de estatinas en la HAP, como es elcaso de los estudios que utilizaron atorvasta-tina. Esto sugiere que puedan existir diferen-cias entre las estatinas(28).
Estudios observacionales abiertos que hanutilizado simvastatina en pacientes con HAP
FUTURAS LÍNEAS DE INVESTIGACIÓN EN LA HIPERTENSIÓN ARTERIAL PULMONAR
179
Monografía HAP NM 208 pag 18/2/10 17:08 Página 179

que ya estaban en tratamiento con prostanoi-des han encontrado resultados positivos enparámetros hemodinámicos y en el test dela marcha. Los datos obtenidos, tanto in vitrocomo en animales y en humanos, apoyan quese siga investigando en esta línea pero conestudios aleatorizados y controlados con pla-cebo para valorar el valor real que las estati-nas puedan tener en el tratamiento de laHAP(18).
INHIBIDORES DE LA RHO-QUINASA En el citoesqueleto los receptores Rho acti-
van la proteína serina/treonína cinasa deno-minada Rho-cinasa. Se ha visto que la Rho ysu blanco, la Rho-cinasa, desempeñan un papelimportante en la regulación de la presión arte-rial y en la contracción de la musculatura lisavascular. Diversos agonistas de los receptoresacoplados a la proteína G de la membrana celu-lar (angiotensina II, fenilefrina) activan la Rho(RhoA), que activa la Rho-cinasa. Al activarsela Rho-cinasa por la RhoA se fosforila la fosfa-tasa de la cadena ligera de la miosina, favore-ciendo la contracción de las células muscula-res lisas vasculares, la formación de fibras deestrés y la migración celular(29).
La inhibición de la rho-cinasa disminuyela contracción de las CMLV, aumenta la expre-sión de la eNOS y disminuye la migración delas células inflamatorias. El fasudil es un inhi-bidor potente y selectivo de la rho-cinasa queen modelos animales demostró que era capazde disminuir la RVP e incluso revertía el remo-delado. En ratas, la combinación de la prosta-ciclina oral, beraprost y fasudil fue más efec-tiva que por separado en la reducción de losniveles de presión(30).
En estudios abiertos de pacientes con HAPgrave en los que se utilizó fasudil se observóun descenso significativo de la RVP y de la PAPmedia. También se vio que tenía efectos sobrela circulación sistémica que podrían evitarsemediante la administración inhalada del fár-maco(31).
La atorvastatina también es capaz de inhi-bir las rho-cinasas (ROCK) y el sildenafilo tam-
bién puede jugar un papel en la fosforilaciónde la RhoA y la prevención de su translocacióna la membrana plasmática.
INHIBIDORES DE LA ELASTASA VASCULARENDÓGENA
En la HAP se ha observado una fragmen-tación de la lámina elástica interna de la paredde los vasos como consecuencia de la activa-ción de las elastasa vascular endógena (EVE),quedando la barrera endotelial comprometi-da. Esta elastasa puede dirigir la degradaciónde la matriz en la enfermedad a través de laactivación de las metaloproteinasas de matriz(MMP), que son una familia de enzimas pro-teolíticas que se encargan del remodelado dela matriz extracelular y cuya presencia se hadocumentado en zonas en las que existe unremodelado vascular. Otros estudios handemostrado que, tanto la EVE como la elas-tasa leucocitaria, son capaces de liberar mitó-genos, como el factor de crecimiento fibro-blástico endógeno 2 (FGF-2) de las célulasmusculares lisas y la tenascina C, favorecien-do el remodelado vascular(32).
En modelos animales se ha visto que lainhibición directa de las MMP o de la EVE redu-ce los niveles de tenascina e induce la apop-tosis celular, pérdida de matriz extracelular yla regresión de la hipertrofia de las arterias pul-monares(33). Sin embargo, su uso en la prácti-ca clínica se ha visto retrasado dado el gradode toxicidad asociado, por ello se están inves-tigando otras vías, como aumentar la expre-sión de los inhibidores endógenos de las elas-tasas.
PÉPTIDO INTESTINAL VASOACTIVO (VIP)El péptido intestinal vasoactivo (VIP) es una
hormona polipeptídica producida por muchasestructuras del cuerpo humano. Esta hormo-na, además de intervenir en el metabolismodel agua y electrolitos, tiene propiedades anti-proliferativas, antiagregantes y vasodilatado-ras. Estos efectos son mediados a través de losreceptores unidos a la proteína G, VPAC1 yVPAC2. La estimulación de estos receptores
C. ALMONACID SÁNCHEZ
180
Monografía HAP NM 208 pag 18/2/10 17:08 Página 180

activa las vías de la adenilato ciclasa y la gua-nilato ciclasa. VIP también induce la síntesisde BH4 que, como ya mencionamos previa-mente en este capítulo, juega un papel muyimportante en el correcto funcionamiento dela eNOS(34).
La ausencia de VIP en ratones se ha aso-ciado a HAP grave, HVD y remodelado vas-cular que se logró atenuar tras administrar estahormona durante 4 semanas de tratamiento.Otras investigaciones han puesto de manifiestoque el VIP es un regulador clave de los genesque controlan el remodelado vascular.
Los estudios realizados en pacientes conHAP confirman que la concentración plasmá-tica del VIP está reducida y que la adminis-tración de 200 µg de VIP por vía inhalada seasocia con mejoría clínica y hemodinámica.No obstante, un problema que plantea el tra-tamiento con este péptido es que tiene unavida media muy corta debido a la rápida degra-dación por las proteasas endógenas. Actual-mente se está trabajando en un nuevo méto-do, basado en liposomas, que proporcionaríauna liberación mantenida del VIP, facilitandosu implantación en la práctica clínica. Estamosa la espera de los resultados de un ensayo clí-nico que se está llevando a cabo hoy en día yque estudia qué dosis es más efectiva, así comootros parámetros de seguridad y eficacia(35).
ADRENOMEDULINALa adrenomedulina (AM) es un péptido
hipotensor aislado del feocromocitoma. El gende la AM es expresado en numerosos tejidos,existiendo altas tasas de transcripción del geny síntesis del péptido en CMLV y CE(36).
La acción de la AM sobre el tono vasculary la proliferación celular es dependiente delAMPc aunque también puede existir una víamediada por el NO y el GMPc.
El efecto vasodilatador de la AM es mayoren los vasos pulmonares que en los sistémi-cos, por lo cual se piensa que tiene un papelen la regulación de la circulación pulmonar. Enmodelos animales de HAP por hipoxia seobservó que el uso de AM reducía la PAP
media, la HVD y además tenía propiedadesantiproliferativas(37).
Los estudios realizados en humanos conAM intravenosa demostraron que tenía efec-tos sobre la RVP, la PAP y la presión arterialsistémica. Los efectos sistémicos no se obser-varon cuando se administró el fármaco por víainhalada. Estos datos deben ser confirmadoscon nuevos estudios a largo plazo, aleatoriza-dos y controlados con placebo(38).
INFLAMACIÓN E INMUNIDADNumerosas evidencias sugieren que exis-
te una relación entre los mecanismos infla-matorios e inmunes en la etiopatogenia de laHAP. Esta hipótesis inflamatoria se apoya enla identificación de infiltrados inflamatoriosperivasculares en las lesiones plexiformescompuestos por macrófagos, linfocitos T y B.Estas células inflamatorias pueden ser el ori-gen de una fuente importante de citocinas,quimiocinas y factores del crecimiento quepodrían hacer diana en las células endotelia-les y contribuir al remodelado de la pared vas-cular(11).
Las células dendríticas también están impli-cadas en los fenómenos de inmunidad y se haencontrado un acúmulo de estas células en laslesiones vasculares de los pacientes con HAP.En los modelos animales de HAP en ratas indu-cida por monocrotalina se observa que la infil-tración por estas células precede los fenóme-nos de remodelado vascular y las alteracioneshemodinámicas. Se piensa que los TKI, comoimatinib, tiene un efecto modulador sobre estascélulas(18).
Los linfocitos T parece que también pue-den tener un papel en la patogenia de estaenfermedad y las investigaciones orientan aque es necesario que el sistema inmune estéintacto para un adecuado control de la prolife-ración vascular. En ratas sin linfocitos T trata-das con semaxinib (SU-5416), que es una anta-gonista del VEGF, se observó que desarrollabanHAP grave en condiciones normales de oxí-geno, mientras que las que tenían linfocitos Tprecisaban de una situación de hipoxia para
FUTURAS LÍNEAS DE INVESTIGACIÓN EN LA HIPERTENSIÓN ARTERIAL PULMONAR
181
Monografía HAP NM 208 pag 18/2/10 17:08 Página 181

C. ALMONACID SÁNCHEZ
182
desarrollar HAP. En pacientes con HAP se haobservado un aumento de los linfocitos T regu-ladores CD4+/CD25+ aunque el significadoen la patogenia de la HAP de estos hallazgosno está claro. Por otro lado, la estimulaciónconstante con antígenos solubles inhaladosinduce, a través de los linfocitos Th2, un aumen-to de la capa muscular de las arterias pulmo-nares de pequeño y mediano tamaños(39).
En la HAP, además de los infiltrados de lascélulas inflamatorias, se ha descrito a nivel delpulmón la expresión de quimiocinas entre lasque destacan la CCL2 (proteína quimioatrac-tante de monocitos MCP-1) que se ha rela-cionado con la migración de los monoci-tos/macrófagos y la proliferación de las CMLV.Los antagonistas de la CCL2 en modelos ani-males atenúan el remodelado cardiovasculary la infiltración por monocitos del pulmón,aminorando el desarrollo de HAP. Otras qui-miocinas que se ha visto que están implica-das la HAP son el CCL5 (RANTES) y CX3CL1(fractalcina)(40).
Varias líneas de investigación tambiénimplican como causa de los cambios vascu-lares a un proceso autoinmune. Se han des-crito la existencia de autoanticuerpos dirigidoscontra las células endoteliales, fibroblastos ycontra el receptor del PDGF en los casos deHAP asociado a esclerodermia. Por otro lado,en la HAP asociada a ciertas enfermedades deltejido conectivo o lupus eritematoso sistémi-co se pueden beneficiar de un tratamientoinmunosupresor(11).
Sin embargo, aunque la inflamación y losmecanismos autoinmunes juegan un papel enla patogenia de la HAP no queda claro hastaqué punto estos cambios son una consecuen-cia del desarrollo de la enfermedad.
OTRAS POTENCIALES DIANASOtras vías en investigación sobre poten-
ciales dianas terapéuticas se centran en la pro-teína morfogenética ósea (BMP) y su receptor(BMPR 2), en el receptor activado proliferadorde peroxisomas (PPAR-γ), en la apolipoproteí-na E (apoE), en el dicloroacetato, en los facto-
res nucleares de linfocitos T activados y en lascélulas endoteliales progenitoras(41).
CONCLUSIONES Es necesario conocer mejor los mecanis-
mos implicados en la patogenia de la HAP paraidentificar mejor las potenciales dianas tera-péuticas y facilitar de ese modo el desarrollode nuevos fármacos. Las opciones terapéuti-cas de la HAP son cada día más numerosas,habiéndose identificado en un corto períodode tiempo numerosos fármacos que puedenser efectivos sobre las nuevas dianas identifi-cadas. No obstante, es muy importante reali-zar un estudio detallado de estas nuevas molé-culas y que los ensayos clínicos que se diseñensean a largo plazo e incorporen los objetivosprincipales del estudio variables relaciona-das con la supervivencia de estos pacientes.Estos datos cobran cada vez más importancia,en especial dado el elevado coste económicodel tratamiento y que posiblemente el futuroimplique el uso de terapias combinadas múl-tiples.
BIBLIOGRAFÍA1. Barberà JA, Escribano P, Morales P, et al; Socie-
dad Española de Neumología y Cirugía Torá-cica; Sociedad Española de Cardiología [Stan-dards of care in pulmonary hypertension.Consensus statement of the Spanish Societyof Pulmonology and Thoracic Surgery (SEPAR)and the Spanish Society of Cardiology (SEC)].Rev Esp Cardiol. 2008; 61 (2): 170-84.
2. Galiè N, Manes A, Negro L, et al. A meta-analy-sis of randomized controlled trials in pulmo-nary arterial hypertension. Eur Heart J. 2009;30 (4): 394-403.
3. Macchia A, Marchioli R, Marfisi R, et al. A meta-analysis of trials of pulmonary hypertension:a clinical condition looking for drugs and rese-arch methodology. Am Heart J. 2007; 153 (6):1037-47.
4. McLaughlin VV, Archer SL, Badesch DB, et al;American College of Cardiology FoundationTask Force on Expert Consensus Documents;American Heart Association; American Colle-ge of Chest Physicians; American ThoracicSociety, Inc; Pulmonary Hypertension Asso-ciation. ACCF/AHA 2009 expert consensus
Monografía HAP NM 208 pag 18/2/10 17:08 Página 182

document on pulmonary hypertension a reportof the American College of Cardiology Foun-dation Task Force on Expert Consensus Docu-ments and the American Heart Associationdeveloped in collaboration with the AmericanCollege of Chest Physicians; American Thora-cic Society, Inc.; and the Pulmonary Hyper-tension Association. J Am Coll Cardiol. 2009;53 (17): 1573-619.
5. Galiè N, Humbert M, Vachiéry JL, et al; Arte-rial Pulmonary Hypertension and BeraprostEuropean (ALPHABET) Study Group. Effectsof beraprost sodium, an oral prostacyclin ana-logue, inpatients with pulmonary arterialhypertension: a randomized, double-blind,pla-cebo-controlled trial. J Am Coll Cardiol. 2002;39 (9): 1496-502.
6. Freedom M. Oral Treprostinil as Monotherapyfor the Treatment of Pulmonary Arterial hyper-tension (PAH) Study NCT00325403.
7. Kuwano K, Hashino A, Asaki T, et al. 2-[4-[(5,6-diphenylpyrazin-2-yl) (isopropyl)ami-no]butoxy]- N -(methylsulfonyl) acetamide(NS-304), an orally available and long-actingprostacyclin receptor agonist prodrug. J Phar-macol Exp Ther. 2007; 322 (3): 1181-8. Epub2007 Jun 1.
8. Long-Term Extension Study of the SERAPHINStudy, to Assess the Safety and Tolerabilityof ACT 064992 in Patients With SymptomaticPulmonary Arterial Hypertension (SERAPHINOL). Study NCT00667823.
9. Iglarz M, Binkert C, Morrison K, et al. Phar-macology of macitentan, an orally active tis-sue-targeting dual endothelin receptor anta-gonist. J Pharmacol Exp Ther. 2008; 327 (3):736-45. Epub 2008 Sep 9.
10. Galiè N, Brundage BH, Ghofrani HA, et al; Pul-monary Arterial Hypertension and Responseto Tadalafil (PHIRST) Study Group. Tadalafiltherapy for pulmonary arterial hypertension.Circulation. 2009; 119 (22): 2894-903.
11. Rhodes CJ, Davidson A, Gibbs JS, et al. The-rapeutic targets in pulmonary arterial hyper-tension. Pharmacol Ther. 2009; 121 (1): 69-88.
12. Dony E, Lai YJ, Dumitrascu R, et al. Partialreversal of experimental pulmonary hyper-tension by phosphodiesterase-3/4 inhibition.Eur Respir J. 2008; 31 (3): 599-610.
13. Schermuly RT, Stasch JP, Pullamsetti SS, et al.Expression and function of soluble guanyla-te cyclase in pulmonary arterial hypertension.Eur Respir J. 2008; 32 (4): 881-91.
14. Michelakis ED. Soluble guanylate cyclase sti-mulators as a potential therapy for PAH: enthu-siasm, pragmatism and concern. Eur RespirJ. 2009; 33 (4): 717-21.
15. Grimminger F, Weimann G, Frey R, et al. Firstacute haemodynamic study of soluble guany-late cyclase stimulator riociguat in pulmonaryhypertension. Eur Respir J. 2009; 33(4): 785-92.
16. Mittendorf J, Weigand S, Alonso-Alija C, et al.Discovery of riociguat (BAY 63-2521): a potent,oral stimulator of soluble guanylate cyclase forthe treatment of pulmonary hypertension.ChemMedChem. 2009; 4 (5): 853-65.
17. Stasch JP, Hobbs AJ. NO-independent, haem-dependent soluble guanylate cyclase timula-tors. Handb Exp Pharmacol. 2009; 191: 277-308.
18. Olsson KM, Hoeper MM. Novel approaches tothe pharmacotherapy of pulmonary arterialhypertension. Drug Discov Today. 2009;14 (5-6): 284-90.
19. Souza R, Sitbon O, Parent F, et al. Long termimatinib treatment in pulmonary arterialhypertension. Thorax. 2006; 61 (8): 736.
20. Patterson KC, Weissmann A, Ahmadi T, et al.Imatinib mesylate in thetreatment of refrac-tory idiopathic pulmonary arterial hyperten-sion. Ann Intern Med. 2006; 145 (2): 152-3.
21. García Hernández FJ, Castillo Palma MJ, Gon-zález León R, et al. [Experience with imatinibto treat pulmonary arterial hypertension]. ArchBronconeumol. 2008; 44 (12): 689-91.
22. Klein M, Schermuly RT, Ellinghaus P, et al.Combined tyrosine and serine/threonine kina-se inhibition by sorafenib prevents progres-sion of experimental pulmonary hypertensionand myocardial remodeling. Circulation. 2008;118 (20): 2081-90.
23. Katusic ZS, d'Uscio LV, Nath KA. Vascular pro-tection by tetrahydrobiopterin: progress andtherapeutic prospects. Trends Pharmacol Sci.2009; 30 (1): 48-54.
24. Fanburg BL, Lee SL. A role for the serotonintransporter in hypoxia-induced pulmonaryhypertension. J Clin Invest. 2000; 105 (11):1521-3.
25. Weir EK, Hong Z, Varghese A. The serotonintransporter: a vehicle to elucidate pulmonaryhypertension? Circ Res. 2004; 94 (9): 1152-4.
26. MacLean MR. Pulmonary hypertension andthe serotonin hypothesis: where are we now?Int J Clin Pract Suppl. 2007; 156: 27-31.
FUTURAS LÍNEAS DE INVESTIGACIÓN EN LA HIPERTENSIÓN ARTERIAL PULMONAR
183
Monografía HAP NM 208 pag 18/2/10 17:08 Página 183

C. ALMONACID SÁNCHEZ
184
27. Xing XQ, Gan Y, Wu SJ, et al. Statins may ame-liorate pulmonary hypertension via RhoA/Rho-kinase signaling pathway. Med Hypotheses.2007; 68 (5): 1108-13.
28. Hsu HH, Ko WJ, Hsu JY, et al. Simvastatin ame-liorates established pulmonary hypertensionthrough a heme oxygenase-1 dependent path-way in rats. Respir Res. 2009; 10: 32.
29. Do E Z, Fukumoto Y, Takaki A, et al. Evidencefor Rho-Kinase Activation in Patients With Pul-monary Arterial Hypertension. Circ J. 2009.
30. Tawara S, Fukumoto Y, Shimokawa H. Effectsof combined therapy with a Rho-kinase inhi-bitor and prostacyclin on monocrotaline-indu-ced pulmonary hypertension in rats. J Car-diovasc Pharmacol. 2007; 50 (2): 195-200.
31. Jalila J, Lavanderob S, Chiongb M, OcaranzaaMP. La vía de señalización Rho/Rho-cinasa enla enfermedad y el remodelado cardiovascu-lar. Rev Esp Cardiol. 2005; 58: 951-61.
32. Cowan KN, Jones PL, Ravinovitch M. Elastaseand Matrix Metaloproteinase Inhibitors Indu-ce Regression, and Tenascin-C Antisense Pre-vents Progression, of Vascular Disease. J ClinInvest. 2000; 105: 21-34.
33. Rabinovitch M. Elastase and the pathobilologyof unexplained pulmonary hypertension. Chest1998; 114 (3 suppl): 213s-224s.
34. Petkov V, Mosgoeller W, Ziesche R, et al. Vaso-active intestinal peptide as a new drug for tre-
atment of primary pulmonary hypertension.J Clin Invest. 2003;111(9):1339-46.
35. Hamidi SA, Prabhakar S, Said SI. Enhancementof pulmonary vascular remodeling and inflam-matory genes with VIP gene deletion. Eur Res-pir J. 2008; 31(1): 135-9.
36. Troughton RW, Lewis LK, Yandle TG, et al.Hemodynamic, hormone, and urinary effectsof adrenomedullin infusion in essential hyper-tension. Hypertension 2000; 36: 588-93.
37. Nagaya N, Nishikimi T, Uematsu M, et al. Hae-modynamic and hormonal effects of adreno-medullin in patients with pulmonary hyper-tension. Heart. 2000; 84: 653-58.
38. Murakami S, Kimura H, Kangawa K, et al.Physiological significance and therapeuticpotential of adrenomedullin in pulmonaryhypertension. Cardiovasc Hematol Disord DrugTargets. 2006; 6 (2): 125-32.
39. Hall SM, Brogan P, Haworth SG, et al. Contri-bution of inflammation to the pathology ofIdiopathic Pulmonary Arterial Hypertensionin children. Thorax; 2009.
40. Hassoun PM, Mouthon L, Barberà JA, et al.Inflammation, growth factors, and pulmonaryvascular remodeling. J Am Coll Cardiol. 2009;54 (1 Suppl): S10-9.
41. Newman JH, Phillips JA, Loyd JE. Narrativereview: the enigma of pulmonary arterialhypertension: new insights from genetic stu-dies. Ann Intern Med. 2008; 148 (4): 278-83.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 184

185
RESUMENLa poderosa irrupción, hace algunos años,
de Internet ha supuesto una verdadera revo-lución en el mundo de las comunicaciones.Sus mayores logros han consistido en la difu-sión global de la información y la democrati-zación del acceso a la misma, así como la posi-bilidad de interconexión entre indivíduos muyalejados geográficamente. La medicina, comootras disciplinas científicas, no ha permaneci-do ajena a este fenómeno y hoy se consideraimpensable un correcto ejercicio de la prácti-ca médica sin la consulta a Internet.
La hipertensión arterial pulmonar es consi-derada actualmente como una de las enferme-dades de moda, debido al gran volumen gene-rado de publicaciones, protocolos, normativasy ensayos clínicos. Los recursos disponiblesen Internet sobre esta patología son abundan-tes y crecen constantemente, y el propósitode este capítulo es revisarlos en sus aspectosmás destacados. Así, nos detendremos, entreotras, en aquellas páginas web que nos infor-men sobre buscadores generales y especializa-dos en bibliografía médica, sociedades y aso-ciaciones médicas interesadas en esta patología,hospitales y centros de referencia en el mane-jo de los enfermos, y direcciones de entidadesy fármacos específicos para su tratamiento.
INTRODUCCIÓNInternet surgió de un proyecto desarrolla-
do en Estados Unidos para apoyar a sus fuer-zas militares; tras su creación, empezó a serutilizado por el gobierno, universidades y otroscentros académicos.
Internet ha supuesto una revolución sinprecedentes en el mundo de la informática y
de las comunicaciones. Los inventos del telé-grafo, teléfono, radio y ordenador sentaron lasbases para esta integración de capacidadesnunca antes vivida. Internet es, a la vez, unaoportunidad de difusión global, un mecanis-mo de propagación de la información y unmedio de colaboración e interacción entre losindividuos y sus ordenadores independiente-mente de su localización geográfica. No obs-tante, la gran cantidad de información quesuministra puede convertirse en un serio pro-blema cuando no es analizada con espíritu crí-tico y clasificada con criterios rigurosos, porlo que, en ocasiones, la búsqueda de conte-nidos se convierte en un proceso largo, com-plejo y poco fiable.
La información necesaria para una correc-ta práctica médica crece progresivamente y elauge experimentado por Internet ha posibili-tado el acceso a toda la documentación médi-ca indispensable desde todos los rincones delmundo, especialmente aquéllos alejados delos grandes centros hospitalarios, universita-rios y de investigación.
La patología que nos ocupa, la hiperten-sión pulmonar (HP), es una entidad poco fre-cuente que ha experimentado un vertiginosodesarrollo en los últimos años en sus nivelesde conocimiento. Así, la navegación por la webpermite al personal sanitario, a los pacientesy a sus cuidadores, obtener información actua-lizada sobre el desarrollo de la enfermedad,su diagnóstico, las posibilidades terapéuticasy las líneas de investigación.
Mediante Internet podemos obtener infor-mación sobre “hipertensión pulmonar” a par-tir de buscadores públicos generales y de por-tales específicos sobre la enfermedad. La
RECURSOS EN INTERNET SOBREHIPERTENSIÓN PULMONAR (HP)
Francisco Javier García Pérez, Carlos Disdier Vicente, Claudia Valenzuela
Monografía HAP NM 208 pag 18/2/10 17:08 Página 185

búsqueda en las bases de datos médicas,como Medline o similares, permite seleccio-nar y adquirir artículos de revistas científi-cas o libros. El uso del correo electrónico posi-bilita recibir periódicamente bibliografíaactualizada, compartir información y reali-zar consultas entre médicos y pacientes. Através de chats, blogs, foros privados de dis-cusión o del mismo correo electrónico per-sonal se puede contactar con grupos de tra-bajo e investigación para intercambio deinquietudes.
También a través de Internet es factiblerecibir consejo y apoyo de asociaciones depacientes, acceder a la información de los cen-tros hospitalarios de referencia, concertar citascon médicos expertos e incluso recibir ase-soría jurídica gratuita en caso de sufrir, porejemplo, esta patología por la ingesta de ano-rexígenos.
Si introducimos actualmente en Googleel término pulmonary hypertension nos encon-tramos con 7.290.000 páginas; una búsquedasimilar con el epígrafe en castellano “hiper-tensión pulmonar” nos ofrece 211.000 pági-nas web.
RECURSOS EN INTERNET SOBREHIPERTENSIÓN PULMONAR
Los amplios recursos disponibles en Inter-net sobre la HP pueden ser clasificados en:• Buscadores generales.• Buscadores especializados en bibliografía
médica.• Actualización en la información bibliográ-
fica.• Asociaciones de pacientes con HP.• Páginas específicas sobre HP.• Sociedades médicas interesadas en esta
enfermedad.• Asociaciones de patologías relacionadas
con la HP.• Agencias farmacológicas y bases de fár-
macos.• Direcciones de fabricantes y fármacos
específicos para la HP.• Hospitales y centros de referencia.
Buscadores generalesUn motor de búsqueda es un sistema infor-
mático que indexa archivos almacenados enservidores web gracias a su spider. Un ejem-plo son los buscadores de Internet (algunos bus-can sólo en la web pero otros buscan, además,en noticias, servicios, etc.) cuando se pide infor-mación sobre algún tema. Las búsquedas sehacen con palabras clave o con árboles jerár-quicos por temas; el resultado de la búsquedaes un listado de direcciones web en los que semencionan temas relacionados con las palabrasclave buscadas. Se pueden clasificar en dos tipos:a) Índices temáticos: son sistemas de bús-
queda por temas o categorías jerarquiza-dos (aunque también suelen incluir siste-mas de búsqueda por palabras clave). Setrata de bases de datos de direcciones webelaboradas "manualmente", es decir, haypersonas que se encargan de asignar cadapágina web a una categoría o tema deter-minado.
b) Motores de búsqueda: son sistemas debúsqueda por palabras clave. Son bases dedatos que incorporan automáticamentepáginas web mediante "robots" de bús-queda en la red.
El mercado actual de los grandes busca-dores está dominado por Google, Yahoo y Micro-soft; el resto suelen ser portales que remitena los resultados de otros buscadores, ofreciendotambién otro tipo de contenidos. En la tabla 1se presentan los buscadores más conocidoscon sus correspondientes direcciones.• Google (www.google.es) es el motor de bús-
queda en Internet más grande, más famo-so y más utilizado. Permite encontrar infor-mación en la red de forma rápida ysencilla, con acceso a un índice de más de8.000 millones de páginas web y más de880 millones de imágenes.
• Yahoo search (www.yahoo.es) es unaempresa global de medios radicada enEE.UU., que posee un portal de Internet,un directorio web y una serie de servicios,incluido su popular correo electrónico
F.J. GARCÍA PÉREZ ET AL.
186
Monografía HAP NM 208 pag 18/2/10 17:08 Página 186
RECURSOS EN INTERNET SOBRE HIPERTENSIÓN PULMONAR (HP)
TABLA 1. Buscadores generales
Portales, directorios y buscadores internacionales generalesBuscador Alcance Idioma(s) Dirección
Global Inglés Google Buscador que enfoca sus resultados Español para cada país y a nivel internacional tanto en
castellano, catalán, gallego, euskara e inglés:http://www.google.com/
Global Inglés Altavista es un buscador en inglés y español,Español de grandes características, de la empresa Over-
ture Services, Inc. comprada, a su vez, porYahoo!: http://www.altavista.com/
Global Inglés Yahoo!: http://www.yahoo.com/Español
Global Inglés Excite: http://www.excite.com/ Español
Global Inglés HotBot: http://www.hotbot.com/
Global Inglés Infoseek: http://www.infoseek.com/
Global Inglés WebCrawler: http://www.webcrawler.com/
Global Inglés Lycos: http://www.lycos.com/Español
Global Inglés Netscape: http://www.netscape.com/
Global Inglés Aol: http://search.aol.com
Global Inglés MSN: http://search.msn.com/Español
Global Inglés Dmoz: http://www.dmoz.org/
Global Inglés NBCi: http://www.nbci.com/
Glonal Inglés All The Web: http://www.alltheweb.com/
Global Inglés DogPile: http://thunderstone.go2net.com/texis/websearch/
Global Inglés Hooting Owl: http://www.hootingowl.com/index.cfm?u=253&Dir=World/Espa%f1ol/
…/…
Monografía HAP NM 208 pag 18/2/10 17:08 Página 187

F.J. GARCÍA PÉREZ ET AL.
188
Yahoo! En 2004 lanzó su propio busca-dor basado en una combinación de tec-nologías de sus adquisiciones y propor-cionando un servicio en el que yaprevalecía la búsqueda de webs sobre eldirectorio.
• Live Search (www. live.com) de Microsoft(antes MSN Search) ofrece característicasinnovadoras, como la posibilidad de verresultados de búsqueda adicionales en lamisma página y la opción de ajustar de for-ma dinámica la cantidad de informaciónmostrada para cada resultado obtenido enla búsqueda. También permite al usuarioguardar las búsquedas y verlas actualiza-das automáticamente en live.com.
Buscadores especializados en bibliografíamédica
PubMed Medline: http://www.ncbi.nlm.nih.gov/sites/entrez
PubMed es unos de los servicios de Medli-ne más completos y rápidos. Es muy fiable yfácil de usar, con actualizaciones casi perma-nentes. Está desarrollado por la National Libraryof Medicine de EE.UU. con la colaboración deeditores médicos de todo el mundo, y permi-te el acceso a la indexación de Medline y dePre-Medline que incluyen 15 millones de artí-culos que se publican en más de 4.800 revis-tas médicas. Se pueden realizar búsquedas deforma intuitiva y potente en un sistema de bús-
TABLA 1. Buscadores generales (continuación)
Portales, directorios y buscadores internacionales generalesBuscador Alcance Idioma(s) Dirección
Global Inglés Go: http://www.go.com/
Global Inglés Celestina: http://www.celestina.com/USA
Global Inglés Looksmart: http://www.looksmart.com/ Indice Internacional
Global Inglés Ask Jeeves: http://www.askjeeves.com/Español Índice Internacional
Global Inglés Teoma: http://www.teoma.com/ Índice Internacional
Global Inglés Xupiter: http://www.xupiter.com/
Global Inglés Alexa: http://www.alexa.com/USA
Global Inglés MyWay: http://www.myway.com/ Resultados de: Google, Alta Vista, Ask Jeeves,AlltheWeb, LookSmart.
Global Inglés Search.com: http://www.search.com/
Global Inglés Search the web: http://www.lop.com/
celestina.com
XUPITERXUPITER
SSeeaarrcchh tthhee WWeebb!!You r sea rch ends he re
Monografía HAP NM 208 pag 18/2/10 17:08 Página 188

189
queda avanzada o por palabras, nombres deautores, título de revistas, abreviaturas válidaspara Medline o por el número de ISSN. Se pue-de acotar o ampliar la búsqueda mediante lostérminos AND, OR, NOT,*. Los resultados dela búsqueda aparecerán en inglés (Fig. 1).
Medscape: http://www.medscape.com/Surgió específicamente para Internet. Con-
tiene información abundante y de calidad paramédicos y pacientes, publicaciones y artículosen línea, noticias, etc. Para acceder de formagratuita a todos los servicios de Medscape esnecesario registrarse. El modo de búsquedaavanzada es también intuitivo y fácil de enten-der. Pueden encontrarse enlaces, bancos deimágenes y otros recursos con páginas espe-cíficas para hipertensión pulmonar: http://www.medscape.com/resource/pulmaryhypertension,así como presentaciones de congresos:h t tp : / /www.medscape.com/v iewpro -gram/3266?src=mp.
Centers for Disease Control andPrevention (CDC): http://www.cdc.gov/search.do
Es la base de datos de la Agencia del Depar-tamento de Prevención y Control de Enfer-medades. Incluye información sobre enfer-medades, factores de riesgo, directrices deprevención, estadísticas sobre salud, etc. Losresultados de la búsqueda aparecerán en inglés.Pueden introducirse palabras o frases, sepa-radas por comas. Se pueden filtrar las bús-quedas usando AND, OR, NOT o*.
Diseases Database: http://diseasesdatabase.com/
Esta base de datos de enfermedades es uníndice de referencias cruzadas de enfermeda-des humanas, signos, síntomas y medicaciones.Esta dirección funciona como el índice de unlibro de texto y un portal de búsquedas que inclu-ye enfermedades médicas, semiología, trastor-nos congénitos, microorganismos y fármacos.
RECURSOS EN INTERNET SOBRE HIPERTENSIÓN PULMONAR (HP)
FIGURA 1. Ejemplo de búsqueda en PubMed.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 189

Embase: http://www.embase.comProducida por Elsevier, contiene siete millo-
nes de citas. Cubre unas 3.500 revistas biomé-dicas de 110 países, especialmente de Europa.Su actualización es semanal, y es muy potenteen información sobre fármacos y toxicología.
Cochrane Library: http://www.cochrane.orgRevisiones sistemáticas basadas en ensa-
yos controlados y randomizados con meta-aná-lisis sobre intervenciones médicas. Mantenidopor la Cochrane Collaboration y actualizado cua-tro veces al año. Tiene un coste anual.
Novo|seek: http://www.novoseek.comDesarrollada por la empresa española Bioal-
ma y disponible de forma gratuita en Internet.Las búsquedas hay que hacerlas en inglés, igualque pasa en PubMed, pero es muy fácil de uti-lizar y por sus resultados merece la pena pro-barla.
Actualización en la informaciónbibliográfica
Amedeo: http://www.amedeo.com/medicine/phy.htm
Amedeo es una guía de literatura médicaque envía de forma gratuita a sus suscriptoresla actualización bibliográfica semanal de laspublicaciones seleccionadas por áreas temá-ticas a través del correo electrónico (www.ame-deo.com/email.php). A comienzos del 2007,Amedeo ya ofrecía información de más de 100temas (incluida la hipertensión pulmonar).Cuenta con 130.000 suscriptores y es una delas websites médicas más populares. Está sub-vencionada por numerosas compañías far-macéuticas. Su autor, Bernd Sebastian Kamps,es de nacionalidad alemana.
Asociaciones de pacientes conhipertensión arterial pulmonar
Entidades sin ánimo de lucro, promuevenla difusión y el conocimiento de la patología,promocionan la investigación, ofrecen educa-ción sanitaria y apoyo social e informan de las
terapias disponibles. A continuación se expo-nen las principales asociaciones españolas,europeas y americanas.
Asociación Nacional de Hipertensión Pulmonar:http://www.hipertensionpulmonar.es/
Esta asociación española, creada en juniode 2005 y declarada de utilidad pública ennoviembre de 2008, trabaja intensamente paradifundir conocimientos sobre la enfermedady sobre las mejores opciones terapéuticas. Rea-liza actividades periódicas, promueve la rela-ción entre los afectados (mediante foros deopinión) y les suministra atención psicológicay asesoría jurídica (Fig. 2).
PHA Europe: www. phaeurope.orgEsta asociación, fundada en 2003, pre-
tende aglutinar a todas las asociaciones nacio-nales europeas sobre hipertensión pulmonarpara servir de apoyo a las ya constituidas, asícomo ayudar al desarrollo de las nuevas, conel fin de promover un estándar europeo detratamiento y educación sanitaria en la enfer-medad. La PHA Europe facilita la comunica-ción e información entre miembros y ofrecesoporte social a enfermos y familiares. Otrosobjetivos que se plantean como asociaciónson conseguir que los fármacos específicostan costosos para esta patología estén dispo-nibles para todos los enfermos europeos y ser-vir de mediador entre los organismos políti-cos, médicos e industria farmacéutica; asícomo favorecer la investigación para mejorarla calidad de vida de los pacientes con estaenfermedad.
PHA Pulmonary Hypertension Association:http://www.phassociation.org
Organización sin ánimo de lucro que ofre-ce apoyo y educación sanitaria a los pacientescon hipertensión pulmonar y promueve lainvestigación médica en ese ámbito. Fue fun-dada gracias al estímulo de tres pacientes nor-teamericanos que querían conocer la situaciónde otros enfermos con la misma patología en1990, y cuenta en la actualidad con más de
F.J. GARCÍA PÉREZ ET AL.
190
Monografía HAP NM 208 pag 18/2/10 17:08 Página 190

191
6.000 miembros. Además de proporcionarinformación en forma de noticias de prensa(newsletters, http://www. phassociation.org/Newsletters/index.asp) y mensajes para todoslos miembros, tiene secciones para profesio-nales médicos y sanitarios interesados en com-partir información y avances en la enferme-dad, como la PH Doctor (http://www.phassociation.org/ PHDoctor/index.asp) y la PHResource Network (http://www.phassociation.org/ PHRN/ index.asp). Ofrece herramientas enlínea para la comunicación entre miembros(Messages boards, http://www.phassociation.org/Message_Boards), para ayudar en la infor-mación y educación de la enfermedad. La aso-ciación ha constituido una red de más de ciengrupos de apoyo (Support group network, http://www.phassociation.org/Support_Groups/index.asp) y receptores voluntarios de llamadas en línea(Helpline, http://www.phassociation.org/Con-tact_us/index.asp) para casos nuevos o enfer-mos con problemas. Estos voluntarios suelenser supervivientes a largo plazo de la enfer-medad. La PHA tiene contactos con otras orga-nizaciones (Allied organizations, http://www.phassociation. org/intl/ index.aspy) y realizaactividades de difusión de la enfermedad (PHAwareness Month, http://www. phassocia-tion.org/Awareness/Awareness_Month.asp).
Finalmente, también colabora promoviendo lainvestigación (PH research, http:// www.phas-sociation.org/support/ResearchFunding.asp).Esta asociación edita cada trimestre desde elaño 2002 la revista Advances in PulmonaryHypertension (revista oficial de la asociación dehipertensión pulmonar), de gran interés prác-tico: http://www. phassociation.org/ Medi-cal/Advances%5Fin%5FPH/article_list.asp
Otras asociaciones. En la tabla 2 se detallainformación sobre otras asociaciones interna-cionales.
Páginas especÍficas sobre hipertensiónarterial pulmonar
PH Central: http://www.Phcentral.org/Web dedicada a proporcionar información
actualizada sobre hipertensión pulmonar einvestigación farmacológica sobre la misma.No requiere suscripción ni ser asociado.
Phneighborhood: http://www.phneighborhood.com/
Página que ofrece información y apoyogeneral. Dispone de chats y ofrece informa-ción sobre aspectos médicos, ensayos clíni-cos, etc.
RECURSOS EN INTERNET SOBRE HIPERTENSIÓN PULMONAR (HP)
FIGURA 2. Página web de la asociación española de hipertensión pulmonar.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 191
F.J. GARCÍA PÉREZ ET AL.
192
TABL
A 2.
Nom
bres
y d
irec
cion
es d
e la
s as
ocia
cion
es d
e hi
pert
ensi
ón p
ulm
onar
nac
iona
les
y co
ntin
enta
les
Asoc
iaci
ónId
iom
aNo
mbr
eDi
recc
ión
web
Corr
eo
Alem
ania
Alem
ánPH
eV [P
ulm
onar
e Hy
perto
nie
e.V.]
ww
w.ph
ev.d
ein
fo@
phev
.de
(Ger
man
)
Arge
ntin
a Es
paño
lPH
A Ar
gent
ina
alab
ro@
ffava
loro
.org
mdi
ez@
ffava
loro
.org
,
Aust
ralia
Ingl
ésPH
A Au
stra
liaw
ww.
phaa
ustra
lia.c
om.a
u/ca
rmel
@ph
aaus
tralia
.com
.au
Aust
riaIn
glés
The
Child
ren
PH (P
ulm
onar
y w
ww.
phaa
ustri
a.or
gof
fice@
phaa
ustri
a.or
gHy
perte
nsio
n) R
esea
rch
Foun
datio
n
Bélg
icaHo
land
és
Belg
ium
PH
vzw
[Pul
mon
ale
ww
w.ph
-vzw
.be
hend
rik.ra
mak
er@
tele
net.b
eHy
perte
nsie
Pat
ient
enve
reni
ging
]
Bélg
icaFr
ancé
sHT
AP B
elgi
que
ww
w.ht
ap-b
elgi
que.b
eht
apbe
lgiq
ue@
hotm
ail.c
om
Bras
ilPo
rtugu
ésAB
RAF
- Ass
ocia
ção
Bras
ileira
de
ww
w.re
spira
revi
ver.o
rg.b
r/pa
ulaf
rmen
ezes
@ya
hoo.
com
.br
Amig
os e
Fam
iliar
es d
e Po
rtado
res
de H
iper
tens
ão A
rteria
l Pul
mon
ar
Cana
da -
Ingl
ésBC
PHS
[Brit
ish C
olum
bia
Pulm
onar
yw
ww.
bcph
s.org
lg
mcc
all@
shaw
.ca
Briti
sh C
olum
bia
Hype
rtens
ion
Socie
ty]
Cana
da -
Man
itoba
In
glés
The
Man
itoba
Pul
mon
ary
Hype
rtens
ion
http
://ca
.geo
citie
s.com
/mb_
ph_g
roup
/m
b_ph
_gro
up@
yaho
o.ca
Supp
ort G
roup
Cana
da-
Ingl
ésNe
w B
runs
wick
Pul
mon
ary
Hype
rtens
ion
Socie
tyw
ww.
nbph
s.org
jg
endr
on@
nb.sy
mpa
tico.
caNe
w B
runs
wick
Chec
a, R
epúb
lica
Sdru
zeni
Pac
ient
u s P
licni
Hyp
erte
nzi
jans
apav
el@
yaho
o.co
m
Chin
aCh
ino
PHA
Chin
aw
ww.
phac
hina
.com
phac
hina
@ya
hoo.
com
.cn
Espa
ñaEs
paño
lAs
ocia
ción
Nacio
nal d
e Hi
perte
nsió
n Pu
lmon
arw
ww.
hipe
rtens
ionp
ulm
onar
.es
info
rmac
ion@
hipe
rtens
ionp
ulm
onar
.es
Esta
dos U
nido
sIn
glés
PHA
Pulm
onar
y Hy
perte
nsio
n As
socia
tion
ww
w.ph
asso
ciatio
n.or
gw
eb@
phas
socia
tion.
org
Euro
paIn
glés
PHA
Euro
pe
ww
w.ph
aeur
ope.o
rgin
fo@
phae
urop
e.org
Fran
ciaFr
ancé
sHT
AP F
ranc
e [H
yper
tens
ion
Arte
rielle
w
ww.
htap
franc
e.com
secr
etar
iat@
htap
franc
e.com
Pulm
onai
re F
ranc
e]
…/…
Monografía HAP NM 208 pag 18/2/10 17:08 Página 192
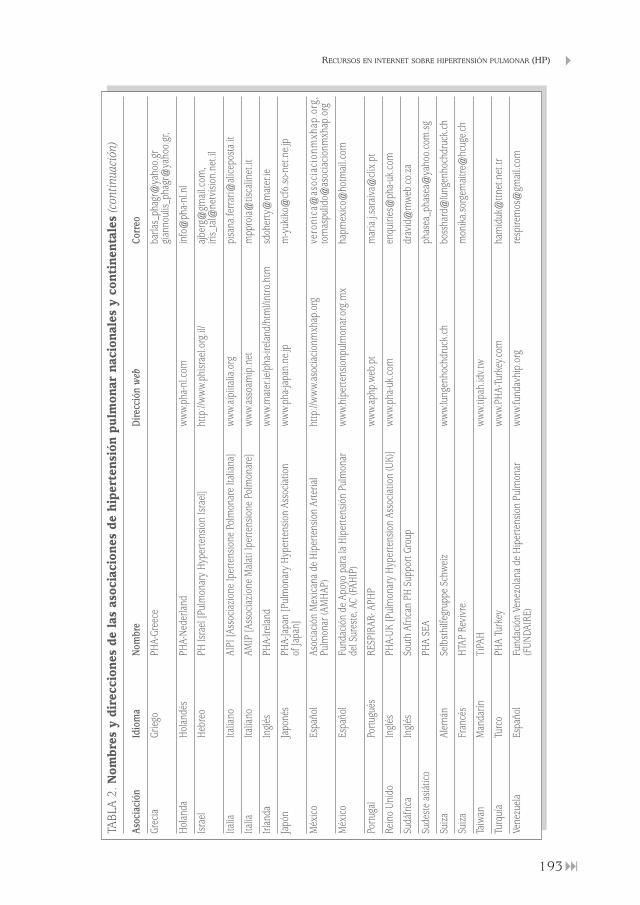
193
RECURSOS EN INTERNET SOBRE HIPERTENSIÓN PULMONAR (HP)
TABL
A 2.
Nom
bres
y d
irec
cion
es d
e la
s as
ocia
cion
es d
e hi
pert
ensi
ón p
ulm
onar
nac
iona
les
y co
ntin
enta
les
(con
tinua
ción
)
Asoc
iaci
ónId
iom
aNo
mbr
eDi
recc
ión
web
Corr
eo
Grec
iaGr
iego
PHA-
Gree
ce
barla
s_ph
agr@
yaho
o.gr
gian
noul
is_ph
agr@
yaho
o.gr
,
Hola
nda
Hola
ndés
PHA-
Nede
rland
w
ww.
pha-
nl.c
omin
fo@
pha-
nl.n
l
Israe
lHe
breo
PH Is
rael
[Pul
mon
ary
Hype
rtens
ion
Israe
l]ht
tp://
ww
w.ph
israe
l.org
.il/
ajbe
rg@
gmai
l.com
, iri
s_ta
l@ne
tvisi
on.n
et.il
Italia
Italia
noAI
PI [A
ssoc
iazio
ne Ip
erte
nsio
ne P
olm
onar
e Ita
liana
]w
ww.
aipi
italia
.org
pisa
na.fe
rrari@
alice
posta
.it
Italia
Italia
noAM
IP [A
ssoc
iazio
ne M
alat
i Ipe
rtens
ione
Pol
mon
are]
ww
w.as
soam
ip.n
etm
ppro
ia@
tisca
linet
.it
Irlan
daIn
glés
PHA-
Irela
nd
ww
w.m
ater
.ie/p
ha-ir
elan
d/ht
ml/i
ntro
.htm
sdoh
erty
@m
ater
.ie
Japó
nJa
poné
sPH
A-Ja
pan
[Pul
mon
ary
Hype
rtens
ion
Asso
ciatio
n w
ww.
pha-
japa
n.ne
.jpm
-yuk
iko@
cf6.
so-n
et.n
e.jp
of Ja
pan]
Méx
icoEs
paño
lAs
ocia
ción
Mex
icana
de
Hipe
rtens
ion
Arte
rial
http
://w
ww.
asoc
iacio
nmxh
ap.o
rg
vero
nica
@as
ocia
cion
mxh
ap.o
rg,
Pulm
onar
(AM
HAP)
tom
aspu
lido@
asoc
iacio
nmxh
ap.o
rg
Méx
icoEs
paño
lFu
ndac
ión
de A
poyo
par
a la
Hip
erte
nsió
n Pu
lmon
arw
ww.
hipe
rtens
ionp
ulm
onar
.org
.mx
hapm
exico
@ho
tmai
l.com
del S
ures
te, A
C (F
AHIP
)
Portu
gal
Portu
gués
RESP
IRAR
- APH
Pw
ww.
aphp
.web
.pt
mar
ia.j.
sara
iva@
clix.
pt
Rein
o Un
ido
Ingl
ésPH
A-UK
[Pul
mon
ary
Hype
rtens
ion
Asso
ciatio
n (U
K)]
ww
w.ph
a-uk
.com
enqu
iries
@ph
a-uk
.com
Sudá
frica
Ingl
ésSo
uth
Afric
an P
H Su
ppor
t Gro
updr
avid
@m
web
.co.
za
Sude
ste
asiá
tico
PHA
SEA
phas
ea_p
hase
a@ya
hoo.
com
.sg
Suiza
Alem
ánSe
lbst
hilfe
grup
pe S
chw
eiz
ww
w.lu
ngen
hoch
druc
k.ch
boss
hard
@lu
ngen
hoch
druc
k.ch
Suiza
Fran
cés
HTAP
Rev
ivre
mon
ika.
sorg
emai
tre@
hcug
e.ch
Taiw
anM
anda
rínTi
PAH
ww
w.tip
ah.id
v.tw
Turq
uía
Turc
oPH
A Tu
rkey
w
ww.
PHA-
Turk
ey.c
omha
mid
uk@
ttnet
.net
.tr
Vene
zuel
aEs
paño
lFu
ndac
ión
Vene
zola
na d
e Hi
perte
nsio
n Pu
lmon
ar
ww
w.fu
ndav
hip.
org
resp
irem
os@
gmai
l.com
(FUN
DAIR
E)
Monografía HAP NM 208 pag 18/2/10 17:08 Página 193

http://www.pah-info.com: http://www.pah-info.com/
Página patrocinada por Actelion, que ofre-ce información para familiares y pacientes.
Sociedades médicas interesadas en estaenfermedad
American Heart Association:http:// www.americanheart.org/
Asociación americana de cardiología, fun-dada en 1924, que tiene por objetivo reducir laincidencia de cardiopatía coronaria, enferme-dad cerebral vascular y los factores de riesgorelacionados con estas enfermedades. Realizainversiones en los ámbitos de la investigacióny la educación dirigida al público y profesio-nales sanitarios. Posee una sección en español.Cuenta con una página dedicada específica-mente a la hipertensión pulmonar: www.ame-ricanheart.org/presenter.jhtml?identifier=4.752
American Lung Association: http://www.lungusa.org/
Es la asociación de voluntariado sanitariamás antigua de Estados Unidos, fundada en1904 para luchar inicialmente contra la tuber-culosis. Su objetivo es la prevención de lasenfermedades pulmonares y la promoción dela salud respiratoria. Recibe subvenciones públi-cas y privadas.
American College of Chest Physicians:http://www.chestnet.org/
Órgano de la Sociedad Americana de Ciru-janos y Especialistas Médicos en neumología,cardiología, hipertensión, área cardiovascular,cirugía cardiotorácica y cuidados críticos.
National Heart, Lung, and Blood Institute:http://www.nhlbi.nih.gov/
El Instituto Nacional del Corazón, Pulmóny Sangre (The National Heart, Lung, and BloodInstitute –NHLBI–) lidera un programa nacio-nal americano sobre enfermedades cardiacas,vasculares, hematológicas, pulmonares y tras-tornos del sueño. Planifica, diseña y apoya pro-
gramas coordinados de investigación básica,investigación clínica y ensayos clínicos, asícomo estudios observacionales y proyectos deeducación sanitaria.
National Institutes of Health:http://www.nih.gov/
Agencia norteamericana del Departamen-to de la Salud y Servicios Humanos que ofre-ce recursos de información para el cuidado dela salud e investigación médica. Oferta el ser-vicio MedlinePlus del National Institutes ofHealth y U.S. National Library of Medicine. Tra-baja sobre búsquedas en Medline y permite elacceso a artículos médicos. Brinda informa-ción sobre fármacos, enciclopedia médica ilus-trada, tutoriales interactivos para pacientes ynoticias sobre salud (www.nlm.nih.gov/medli-neplus/pulmonaryhypertension.html). En lapágina http://clinicaltrials.gov/ se pueden bus-car los ensayos actuales registrados sobre hiper-tensión pulmonar.
Sociedad Española de Neumología y CirugíaTorácica (SEPAR): http://www.separ.es/
Web de la Sociedad Española de Neumo-logía y Cirugía Torácica. Esta sociedad estácompuesta por 10 áreas de trabajo. El área decirculación pulmonar de la SEPAR, junto conel área de circulación de la Sociedad Españo-la de Cardiología tienen un registro en líneaque requiere clave de acceso sobre hiperten-sión arterial pulmonar en España (http://www.rehap. org) (Fig. 3). Ambas sociedadeshan promovido, elaborado y publicado undocumento de consenso que establece en nues-tro medio los estándares de calidad adecua-dos para el diagnóstico y tratamiento de lahipertensión pulmonar en sus diversas formasde presentación. Además de abordar los aspec-tos más convencionales de esta patología, eldocumento introduce pautas de organizaciónasistencial de las unidades de referencia parael abordaje de la HP, incluyendo requisitosmínimos de personal y equipamiento, volu-men de actividad, sistemas de información,prestaciones y actividad investigadora. Todo
F.J. GARCÍA PÉREZ ET AL.
194
Monografía HAP NM 208 pag 18/2/10 17:08 Página 194

195
ello con el fin prioritario de intentar crear cir-cuitos asistenciales eficaces, rigurosos y pre-cisos que ayuden a mejorar la respuesta sani-taria ante esta dramática enfermedad.
World Health Organization: http://www.who.int/
Asociación de enfermedades raras: http://www.rarediseases.org/
En su registro, entre más de 1.150 enfer-medades raras, figuran la hipertensión pul-monar primaria y, secundaria, con bastanteinformación y enlaces de interés.
Asociaciones de trasplantes• Organización Nacional de Trasplantes
(ONT): http://www.ont.es/Organismo coordinador de carácter técni-co, perteneciente al Ministerio de Sanidady Consumo, encargado de desarrollar lasfunciones relacionadas con la obtención yutilización clínica de órganos, tejidos y célu-las. Su estructura se basa en una organi-zación reticular a tres niveles: coordinaciónnacional, coordinación autonómica y coor-dinación hospitalaria. Su principal objeti-vo es la promoción de la donación altruis-ta con el único fin de que el ciudadanoespañol que necesite un trasplante tenga
las mayores y mejores posibilidades deconseguirlo.
• American Society of Transplantation:http://www.a-s-t.org/ Fundada en 1982, engloba a más de 2.800profesionales dedicados a la investigación,educación, defensa y cuidado de pacien-tes trasplantados.
• Second Wind: http://www.2ndwind.org/Foro para pacientes trasplantados o en lis-ta de trasplante de pulmón u otros órga-nos.
Asociaciones de patologías relacionadascon la hipertensión pulmonar
Adult Congenital Heart Association (ACHA):http://www.achaheart.org/
Web dedicada a prestar educación, asis-tencia y apoyo a los enfermos y médicos quetratan la enfermedad congénita en el adulto.
Children´s Health Information Network:Congenital Heart Disease: http://www.tchin.org/
El objetivo de esta organización es ofrecerinformación y recursos a los familiares de niñoscon enfermedades cardiacas congénitas yadquiridas, adultos con cardiopatías congéni-tas y a los profesionales que trabajan con ellos.
RECURSOS EN INTERNET SOBRE HIPERTENSIÓN PULMONAR (HP)
FIGURA 3. Registro español de hipertensión arterial pulmonar (REHAP).
Monografía HAP NM 208 pag 18/2/10 17:08 Página 195

Scleroderma Foundation: http://www.srfcure.org/Un espacio para pacientes con esclero-
dermia, cuidadores y familiares dedicados alapoyo, educación e investigación en la enfer-medad. La dirección de la Fundación para lainvestigación en esclerodermia es http://www.srfcure.org/srf/home.htm
American Academy of Sleep Medicine:http://www.aasmnet.org/
American Liver Foundation: http://www.liverfoundation.org/
American Society of Hematology: http://www.hematology.org/
Hereditary Hemorragic Telangiectasia (HHT)Foundation: http://www.hht.org/
National Organization for Rare Disorders,Inc. NORD: http://www.rarediseases.org/
Genetics home reference: http://ghr.nlm.nih.gov/condition=pulmonaryarterialhypertension
Agencias farmacológicas y bases de fármacos
EMEA: http://www.emea.europa.eu/La Agencia Europea de Medicamentos
(EMEA) es un organismo de la Unión Europeacon sede en Londres cuya mayor responsabi-lidad es la protección y promoción de la saludmediante la evaluación y supervisión de medi-camentos de uso humano y veterinario.
FDA: http://www.fda.gov/La Food and Drug Administration (FDA) de
Estados Unidos es la agencia responsable de laprotección de la salud pública americana, encar-gada de certificar la eficacia y seguridad de losfármacos de uso humano y veterinario, pro-ductos biológicos, aparatos médicos, productoscosméticos y sustancias que emiten radiación.
Health Touch: Drug Information: http://www.healthtouch.com/level1/p_dri.htm
Esta web ofrece información sobre más de10.000 fármacos, con referencia a la prescrip-
ción, efectos adversos, vías de administración,etc. Debe introducirse el nombre genérico delfármaco de forma total o parcial en inglés.
Center Watch: http://www.centerwatch.com/index.html
Incluye un listado de ensayos clínicos y defármacos aprobados por la FDA.
ClinicalTrials.Gov: http://www.clinicaltrials.gov/ct/gui/c/b
Oferta una base de datos de ensayos clí-nicos (más de 74.400) desarrollada por losNational Institutes of Health, otras agenciasfederales e industrias privadas.
Direcciones de fabricantes y fármacosespecíficos para la hipertensión pulmonar
Actelion: http://www.actelion.com/• Bosentan (Tracleer®): http://www.tracle-
er.com/Pfizer: http://www.pfizer.es/• Sildenafilo (Revatio®): http://www.reva-
tio.com• Sitaxentan (Thelin®)GSK: http://www.gsk.es/• Ambrisentan (Volibris®): http://www.voli-
bris.eu/• Epoprostenol (Flolan®): http://www.flolan-
center.com/, http://www.flolanresource.com/,http://emc.medicines.org.uk/emc/assets/c/html/displaydoc.asp?documentid=7173,http://home.earthlink.net/~dtpchick/flo-fashions/
United Therapeutics y Grupo Ferrer:http://www.unither.com/, http://www.ferrergrupo.com/• Treprostinil (Remodulin®): http://www.
remodulin.com/Bayer Schering Pharma: http://www.bayerscheringpharma.es• Iloprost (Ventavis“): http://www.ventavis.
comBombas de infusión continua • Bombas para infusion subcutánea mini-
med (Treprostinil sc): www.minimed.com/
F.J. GARCÍA PÉREZ ET AL.
196
Monografía HAP NM 208 pag 18/2/10 17:08 Página 196

197
• Bombas CADD-Legacy: http://www.smiths-medical.com/catalog/ambulatory-pumps-sets/cadd-ambulatory-infusion-pumps/cadd-legacy/cadd-legacy-1-pump.html
Hospitales y centros de referencia• University of California San Francisco
(UCSF) Medical Center: http://www.ucs-fhealth.org/adult/medical_services/heart_care/hypertension/index.html
• Mayo Clinic: http://www.mayoclinic.com/health/heart-disease/HB99999
• Cleveland Clinic: http://cms.clevelandcli-nic.org/ccfpulmonary
• Unidad de Hipertensión Pulmonar delHospital 12 de octubre: http://www.uni-dadhp12.es/principal/index.htmEsta unidad de referencia nacional tienesu web ya operativas y un proyecto deconexión con centros asociados para con-sultas de pacientes por internet (iclinic).
RECURSOS EN INTERNET SOBRE HIPERTENSIÓN PULMONAR (HP)
Monografía HAP NM 208 pag 18/2/10 17:08 Página 197

Monografía HAP NM 208 pag 18/2/10 17:08 Página 198

199
AAF: anticuerpos antifosfolípido.AD: aurícula derecha.ADN: ácido desoxirribonucleico.AI: aurícula izquierda.ALK-1: pseudorreceptor de la activina-cinasade tipo 1 (de Activin receptor-like kinase).ANA: anticuerpos antinucleares.AP: arterias pulmonares.apoE: apoproteína E.AR: artritis reumatoide.ARE: antagonistas de receptores deendotelina.ARNM: ácido ribonucleico mensajero.BCC: bloqueantes de los canales de calcio. BMP: proteína morfogenética ósea (de bonemorphogenetic protein)BMPR1: receptor (de tipo) 1 de la proteínamorfogenética ósea.BMPR2: receptor (de tipo) 2 de la proteínamorfogenética ósea.BNP: péptido natriurético cerebral (de brainnatriuretic peptide).cAMP: monofosfato cíclico de adenosina (decyclic adenosine monophosphate).CAV-1: caveolina 1.CCD: cateterismo cardiaco derecho. cCMP: monofosfato cíclico de citidina (decyclic citidine monophosphate).CE: células endoteliales CF: clase funcional.CIA: comunicación interauricular.CIV: comunicación interventricular.CMLV: células del músculo liso vascular.CO: monóxido de carbonoDLCO: capacidad de difusión del monóxidode carbono (de carbon monoxide diffusingcapacity).EAV: escala analógica visual.
ECA: enzima de conversión de laangiotensina.EGF: factor de crecimiento epidérmico (deepidermic growth factor).EMEA: Agencia Europea de Medicamentos(de European Medicines Evaluation Agency).EMTC: enfermedad mixta del tejidoconectivo.eNOS: óxido nítrico-sintetasa endotelial.EPID: enfermedad pulmonar intersticialdifusa.EPOC: enfermedad pulmonar obstructivacrónicaES: esclerosis sistémica.EScd: esclerosis sistémica cutánea difusa.EScl: esclerosis sistémica cutánea limitada.ET-1: endotelina 1.ETA: receptor (de tipo) A de la endotelina.ETB: receptor (de tipo) B de la endotelina.ETC: enfermedad del tejido conectivo.ETE: ecocardiografía transesofágica.ETT: ecocardiografía transtorácica.EVO: enfermedad venooclusiva.EVOP: enfermedad venooclusiva pulmonar.FAP: factor activador de plaquetas.FDA: Food and Drug Administration.FDG: fluorodesoxiglucosa (18F).FEV1: volumen espiratorio forzado en elprimer segundo (de forced expiratory volumein one [in the first] second).FEVD: fracción de eyección del ventrículoderecho.FEVI: fracción de eyección del ventrículoizquierdo.FOP: foramen oval permeable.FPI: fibrosis pulmonar idiopática.FR: factor reumatoide.FRy: fenómeno de Raynaud.
LISTA DE ABREVIATURAS
Neumomadrid 2010
Monografía HAP NM 208 pag 18/2/10 17:08 Página 199

FVC: capacidad vital forzada (de forced vitalcapacity).GC: gasto cardiaco.GMPc: monofosfato cíclico de guanosina (decyclic guanosine monophosphate).GRAD-T: gradiente tricuspídeo.GTP: gradiente transpulmonar.HAP: hipertensión arterial pulmonar.HAPA: hipertensión arterial pulmonarrelacionada con factores de riesgo oenfermedades asociadas.HAPH: hipertensión arterial pulmonarhereditaria.HAPI: hipertensión arterial pulmonaridiopática.HCP: hemangiomatosis capilar pulmonar.HIF-1: factor inducible por hipoxia 1 (dehypoxia inducible factor).HO-1: hemooxigenasa 1.HP: hipertensión pulmonar.HPPRN: hipertensión pulmonar persistentedel recién nacido.HPTEC: hipertensión pulmonartromboembólica crónica.H2S: sulfito de hidrógeno.5.HT2b-R: receptor del transportador de laserotonina.HTO: hematocrito.5-HTT: gen transportador de la serotonina.IAo, IM, IP, IT: insuficiencia aórtica, mitral,pulmonar o tricuspídea.IC: índice cardiaco.ICC: insuficiencia cardiaca congestiva.IL-2: interleucina 2.INF-γ: interferón γ.IPDE-5: inhibidores de fosfodiesterasa tipo 5.IRVP: índice de resistencia vascularpulmonar.ISHLT: International Society for Heart andLung Transplantation.iv: intravenoso.Kv: canales de K+ regulados por cambios devoltaje.LES: lupus eritematoso sistémico.5-LO: 5-lipooxigenasa.MAPK: proteína-cinasa activada por mitógeno(de mitogen-activated protein kinase).
MMP: metaloproteasa de matriz (de matrixmetalloproteinases).NADPO: dinucleótido fosfato denicotinamida y adenina oxidasa (denicotinamide adenine dinucleotidephosphate oxidase).NIH: National Institutes of Health.NO: óxido nítrico.NOS: óxido nítrico-sintetasa.NT-proBNP: parte inactiva N terminal delpéptido natriurético cerebral.NYHA: New York Heart Association.OCD: oxigenoterapia crónica domiciliaria.OMS: Organización Mundial de la Salud.ONT: Organización Nacional de Trasplantes.PaCO2: presión parcial arterial de dióxido decarbono.PAD: presión auricular derecha. PAI: presión auricular izquierda.PAI-1: inhibidor del activador delplasminógeno de tipo 1 (de plasminogenactivator inhibitor-1). PaO2: presión parcial arterial de oxígeno.PAP: presión arterial pulmonar.PAPm: presión arterial pulmonar media.PAPd: presión arterial pulmonar diastólica.PAPs: presión arterial pulmonar sistólica.PAS: presión arterial sistémica.PAWP: presión arterial pulmonar en cuña(de pulmonary arterial wedge pressure).PCP: presión capilar pulmonar.PDE-5: fosfodiesterasa de tipo 5 (dephosphodiesterase 5).PDGF: factor de crecimiento derivado de lasplaquetas (de platelet-derived growth factor).PDGF-R: receptor del factor de crecimientoderivado de las plaquetas.PECP: prueba de esfuerzo cardiopulmonar.PFR: pruebas de la función respiratoria.PGI2: prostaciclina.PPARγ: receptor gamma activado delperoxisoma (de peroxisome proliferator-activated receptor γ).PSP: presión sistólica pulmonar.PSVD: presión sistólica del ventrículoderecho.PT: potencia transmembrana.
LISTA DE ABREVIATURAS
200
Monografía HAP NM 208 pag 18/2/10 17:08 Página 200

LISTA DE ABREVIATURAS
201
PVC: presión venosa central.PVD: presión ventricular derecha.PVP: presión venosa pulmonar.Q: gasto cardiaco.QP: flujo pulmonar.RACK1: receptor de la cinasa-C activada (dereceptor for activated C-kinase).RM: resonancia magnética. RN: recién nacido.RPT: resistencia pulmonar total.RVP: resistencia vascular pulmonar.RVS: resistencia vascular sistémica.SAHS: síndrome de apnea/hipoapnea delsueño.SaO2: saturación arterial de oxígeno.SAT: síndrome del aceite tóxico.SEC: Sociedad Española de CardiologíaSEPAR: Sociedad Española de Neumología yCirugía Torácica.T6M: prueba de 6 minutos de marcha.TC: tomografía axial computarizada.TCAR: tomografía computarizada de altaresolución. TGA: transposición de grandes arterias.TGF: factor de crecimiento transformador, d.t., factor de transformación del crecimiento(de transforming growth factor).TGF-RII: receptor 2 del factor de crecimientotransformador.
TGFβ2: factor de crecimiento transformadorβ2.THH: telangiectasia hemorrágica hereditaria.TIMP: inhibidor tisular de la metaloproteasade matriz.TLC: capacidad pulmonar total.TNF-α: factor de necrosis tumoral α (detumor necrosis factor-alpha).TxA2: tromboxano A2.TxCP: trasplante cardiopulmonar.TxP: trasplante pulmonar.UW: Unidades Word.VC: capacidad vital (de vital capacity)VD: ventrículo derecho.VEGF: factor de crecimiento del endoteliovascular (de vascular endothelial growthfactor).VHB/C: virus de la hepatitis B/C.VI: ventrículo izquierdo. VIH: virus de la inmunodeficiencia humana.VIP: péptido vasointestinal (de vasoactiveintestinal peptide).VM: volumen minuto.VO2: consumo máximo de oxígeno.VP: volúmenes pulmonares.VPH: vasoconstricción pulmonar hipóxica.V/Q: ventilación/perfusión.VTD: volumen telediastólico.VTS: volumen telesistólico.
Monografía HAP NM 208 pag 18/2/10 17:08 Página 201

Monografía HAP NM 208 pag 18/2/10 17:08 Página 202

203
Alba García-Baquero, PilarSección de Neumología. Hospital Severo Ochoa.Leganés, Madrid
Alcolea Batres, Sergio Servicio de Neumología. Hospital Universitario La Paz.Hospital General. Madrid
Almonacid Sánchez, CarlosServicio de Neumología. Hospital Universitario deGuadalajara
Álvarez-Sala Walther, José Luis Servicio de Neumología. Hospital Clínico San Carlos.Madrid
Baloira Villar, AdolfoCoordinador del Servicio de Neumología. ComplexoHospitalario de Pontevedra
de Granda Orive, José Ignacio Servicio de Neumología. Hospital Central de Defensa,Gómez Ulla. Madrid
de Miguel Díez, Javier Servicio de Neumología. Hospital General UniversitarioGregorio Marañón. Madrid
de Riva Silva, MartaServicio de Cardiología. Unidad de InsuficienciaCardiaca e Hipertensión Pulmonar. HospitalUniversitario 12 de Octubre. Madrid
Díez Piña, Juan ManuelServicio de Neumología. Hospital Universitario deMóstoles
Disdier Vicente, Carlos Sección de Neumología. Hospital San Pedro deAlcántara. Cáceres
Fernández Rozas, Inmaculada Servicio de Cardiología. Hospital Severo Ochoa.Leganés, Madrid
Flores Segovia, Julio Sección de Neumología. Hospital Príncipe de Asturias.Madrid
García Pérez, Francisco Javier Servicio de Neumología. Hospital Universitario de laPrincesa. Madrid
García-Salmones Martín, MercedesUnidad de Neumología. Hospital UniversitarioFundación Alcorcón. Madrid
Gaudó Navarro, Javier Servicio de Neumología. Unidad de HipertensiónPulmonar. Hospital Ramón y Cajal. Madrid
Gómez Sánchez, Miguel Ángel Servicio de Cardiología. Unidad de InsuficienciaCardiaca e Hipertensión Pulmonar. HospitalUniversitario 12 de Octubre. Madrid
González Garrido, Fulgencio Servicio de Neumología. Hospital 12 de Octubre. Madrid
Hernández Jiménez, Verónica Servicio de Cardiología. Hospital Universitario Getafe.Madrid
Jareño Esteban, José JavierServicio de Neumología. Hospital Central de Defensa,Gómez Ulla. Madrid
Jiménez Castro, David Servicio de Neumología. Hospital Ramón y Cajal.Madrid
Juretschke Moragues, María Antonia Servicio de Neumología. Hospital Universitario deGetafe. Madrid
Laporta Hernández, Rosalía Servicio Neumología. Hospital Universitario Puerta de Hierro. Majadahonda, Madrid
Linares Asensio, María Jesús Unidad de Neumología. Hospital UniversitarioFundación Alcorcón. Madrid
López García-Gallo, Cristina Servicio Neumología. Hospital Universitario Puerta de Hierro. Majadahonda, Madrid
Morales Chacón, Beatriz Servicio de Neumología. Hospital Clínico San Carlos.Madrid
Navarrete Isidoro, Olga Sección de Neumología. Hospital Príncipe de Asturias.Madrid
Nieto Barbero, María Asunción Servicio de Neumología. Hospital Clínico San Carlos.Madrid
Pedraza Serrano, Fernando Servicio de Neumología. Hospital General UniversitarioGregorio Marañón. Madrid
Índice de autores
Monografía HAP NM 208 pag 18/2/10 17:08 Página 203

Perpiñá Ferri, Asunción Sección de Neumología. Hospital Severo Ochoa.Leganés, Madrid
Pindado Rodríguez, CarlosServicio de Cardiología. Unidad de InsuficienciaCardiaca e Hipertensión Pulmonar. HospitalUniversitario 12 de Octubre. Madrid
Ramos Pinedo, ÁngelaUnidad de Neumología. Hospital UniversitarioFundación Alcorcón. Madrid
Río Ramírez, María TeresaServicio de Neumología. Hospital Infanta Cristina. Parla, Madrid
Ríos Blanco, Juan José Servicio de Neumología. Hospital Universitario La Paz.Hospital General. Madrid
Rodríguez Nieto, María Jesús Servicio de Neumología. Fundación Jiménez Díaz-Capio. CIBERes
Sánchez González, SilviaSección de Neumología. Hospital Príncipe de Asturias.Madrid
Sánchez Muñoz, Gema Servicio de Neumología. Hospital General UniversitarioGregorio Marañón. Madrid
Sueiro Bendito, Antonio Servicio de Neumología. Unidad de HipertensiónPulmonar. Hospital Ramón y Cajal. Madrid
Ussetti Gil, Piedad Servicio Neumología. Hospital Universitario Puerta de Hierro. Majadahonda, Madrid
Valenzuela, Claudia Servicio de Neumología. Hospital Universitario de La Princesa. Madrid
Valverde Pérez, DianaDepartamento de Genética. Facultad de Biología.Universidad de Vigo
Villar Álvarez, Felipe Servicio de Neumología. Fundación Jiménez Díaz-Capio. CIBERes
Villegas Fernández, José Francisco Servicio de Neumología. Hospital Central de Defensa,Gómez Ulla. Madrid
204
Monografía HAP NM 208 pag 18/2/10 17:08 Página 204

205
Índice de materias
Actividad física 61, 143Adrenomedulina 174, 181Altitud 12, 28, 29, 72, 81, 144Anomalías protrombóticas 22Antagonistas
del calcio 50, 56, 57, 62, 123del receptor de la endotelina 64
Anticoagulación 47, 50, 51, 62, 85, 103,107, 108, 154-156Arteriopatía
plexogénica pulmonar 13pulmonar postrombótica-tromboembólica15
Asociaciones de pacientes 143, 186, 190
Biopsia pulmonar 44, 68, 114
Calcioantagonistas 47, 51, 56, 61, 85, 155Calidad de vida 34, 37, 47, 49, 60, 67, 92,103, 122, 142, 143, 154, 173, 190Canales de potasio 11, 13, 18, 19Cardiopatías,
congénitas 6, 14, 31-34, 39, 49, 57, 62,64, 139, 147-151, 153, 155, 157, 159,163, 165, 195izquierdas 72, 73, 74, 78
Cateterismo,cardiaco 38, 42, 43, 48, 71, 74, 75, 84,85, 87, 90, 91, 93, 103, 124, 126, 130,151, 158, 199derecho 56, 58, 60, 113, 151
Células inflamatorias 22, 88, 180-182Circulación pulmonar 9-11, 19, 21, 27, 63, 73,89, 91, 100, 117, 144, 148, 156, 181, 194Cirugía 3, 61, 63, 87, 91, 92, 103, 105, 108,109, 121-123, 125, 139, 144, 145, 147, 148,152, 154, 157, 182, 194, 201
Clasificación,anatómica 28diagnóstica 29fisiopatológica 28
Digoxina 47, 50, 61, 145Disfunción endotelial 9, 12, 13, 15, 16, 20,21, 22, 88, 100, 149, 173Diuréticos 47, 49, 50, 59, 60-62, 156
Ecocardiografía transtorácica (ETT) 103Ecocardiograma transtorácico 39, 40, 84,141Electrocardiograma 38, 39, 48, 58, 86, 90,130, 137, 141Embarazo 52, 61, 143-154Enfermedad de depósito de glucógeno tipo Ia 115Enfermedad,
de Gaucher 14, 32, 111, 116de Pompe 115de von Recklinghausen 111, 115pulmonar obstructiva crónica (EPOC) 15,30, 32, 47, 48, 62, 72, 81, 82, 86-93, 112,126, 199 renal terminal 116veno-oclusiva 65, 67
pulmonar 14, 30, 32, 41, 199Enfermedades,
hematológicas 111, 112metabólicas 115pulmonares intersticiales difusas (EPID)30, 81-86, 92, 112, 199sistémicas 82, 111, 112, 113tiroideas 14, 32, 112, 116
Enfisema 82, 87-89, 90, 93, 123, 125
Monografía HAP NM 208 pag 18/2/10 17:08 Página 205

Estatinas 66, 174, 179, 180Estudio/s,
hemodinámico 29, 33, 34, 39, 42, 43,138, 141, 142, 151, 152
de reversibilidad 152genéticos 166
Fármacos,inhalados 135inhibidores de los receptores de la endote-lina 155subcutáneos 134
Fibrosis 13-15, 27, 41, 72, 82-84, 93-97,100, 111, 113-116, 122-124, 169, 199
mediastínica 72, 111, 116Flujo sanguíneo pulmonar 10, 11, 21Futuras líneas terapéuticas 174
Gammagrafía de ventilación/perfusión pul-monar (V/Q) 102Guanilato ciclasa soluble 174-176
Hemangiomatosis capilar pulmonar 14, 31,32, 38, 41, 44, 65, 67, 72, 200Hipertensión,
arterial pulmonar hereditaria 162, 200pulmonar hipercinética 28pulmonar obstructiva-obliterativa 29pulmonar pasiva 28, 29, 74, 75pulmonar primaria o idiopática 29pulmonar persistente del recién nacido14, 32, 200pulmonar tromboembólica crónica(HPTEC) 5, 99, 101, 103, 105, 107, 109pulmonar reactiva 29, 73, 74pulmonar vasoconstrictiva 29
Hipoxia alveolar 11, 21, 81, 82, 87, 89, 91, 93Histiocitosis pulmonar 114
Infecciones pulmonares 125Inhibidores,
de la Elastasa Vascular Endógena 180de la fosfodiesterasa 5 (PDE-5) 77
de la fosfodiesterasa tipo 5 47, 64de la PDE-5 77, 155, 173de la Rho-quinasa 180de la tirosín-cinasa 65, 177
Internet 6, 142, 185-187, 189-191, 193,195, 197
Linfangioleiomiomatosis 30, 111, 112, 114, 115
Mutaciones en BMPR2 162-165, 167-169genéticas 20, 21, 23, 100, 173
Neoplasias 100, 102, 115, 116, 165, 177Neurofibromatosis tipo I 115Nuevos,
antagonistas de los receptores de la endo-telina 1 174inhibidores de la fosfodiesterasa 175prostanoides 174
Nutrición 9, 143
Oclusión temporal del defecto 152Óxido nítrico sintetasa endotelial 177, 178Oxigenoterapia 47-49, 61, 62, 83, 85, 89,91, 115, 144, 154, 156, 200
Péptido intestinal vasoactivo (VIP) 174, 180Perfusión intravenosa
de corta duración 133de larga duración 133
Programas de educación al paciente y familiares 6, 139, 143Prostaciclina 9, 11, 13, 18, 19, 51, 55, 63,65, 74, 76, 88, 89, 131, 134, 135, 138, 157,180, 200
parenteral 131Prostanoides 47, 61, 63, 65, 76, 92, 105,156, 173, 174, 180Pruebas de tolerancia al ejercicio 41
Radiografía de tórax 37-39, 84, 90, 141,150
206
Monografía HAP NM 208 pag 18/2/10 17:08 Página 206

Rechazo agudo 125Recursos en internet 6, 185-187, 189, 191,193, 195, 197Remodelado vascular 11-14, 19-21, 74, 87-89, 93, 99, 100, 106, 107, 140, 149, 175,177, 179-181
Sarcoidosis 14, 15, 30, 32, 72, 82, 83, 85,86, 95, 96, 111-114, 116, 118, 119Septostomía auricular 67, 68Serotonina 11, 13, 18, 19, 22, 23, 88, 161,168, 169, 174, 178-200Síndrome
de bronquiolitis obliterante 125, 126de Eisenmenger 49, 64, 65, 123, 125,147, 149, 152
Sustancias,vasoconstrictoras 19, 74vasodilatadoras 18, 88
Técnicas quirúrgicas 104, 121Telangiectasia hemorrágica hereditaria 12,23, 161, 163, 201Test,
de la marcha 38, 41, 49, 57, 124, 130,142, 151, 155, 180vasodilatador 38, 43, 44, 152, 163agudo 43, 44
Tetrahidrobiopterina 174, 177, 178TGF-β 23, 161, 163, 166, 167, 169, 170, 171Tomografía computarizada helicoidal/angio 102Tono vascular 10, 11, 74, 89, 168, 173,178, 181Trasplante 6, 55, 57, 60, 61, 65, 67, 68, 71,75-77, 83, 84, 87, 89, 91, 92, 105, 113, 114,117, 118, 121-127, 139, 156, 157, 173, 195, 201
pulmonar 6, 60, 65, 83, 87, 89, 91, 92, 105, 113, 114, 118, 121-127, 157, 173, 201cardiopulmonar 6, 41, 42, 59, 60, 67,104, 121-127, 139, 141, 157, 200, 201
Tratamiento,ambulatorio 136combinado 55, 61, 65, 157
Valoración, de la severidad 57hemodinámica 151no invasiva 149
Vasculopatía,congestiva pulmonar 15hipóxica pulmonar 15
Vasoconstricción 9, 11-13, 15, 17-21, 28, 29,74, 81, 84, 89, 92, 93, 117, 145, 153, 173,179, 201
Monografía HAP NM 208 pag 18/2/10 17:08 Página 207





























