Embed Size (px)
Citation preview

The Downstream Targets of Complex I Dysfunction in Bipolar Disorder
Helena Kyunghee Kim
A thesis submitted in conformity with the requirements for the degree of PhD
Graduate Department of Pharmacology and Toxicology
University of Toronto
© Copyright by Helena Kyunghee Kim (2015)

ii
Title: The Downstream Targets of Complex I Dysfunction in Bipolar Disorder
Name: Helena Kyunghee Kim, Doctor of Philosophy
Department of Pharmacology and Toxicology, University of Toronto 2015
ABSTRACT
Mitochondrial complex I dysfunction is consistently reported in bipolar disorder (BD),
implicating its role in increased oxidative stress. Therefore, the overall aim of my PhD was to
examine potential downstream targets of complex I dysfunction in BD. First, microarray studies
examining complex I subunits in patients with BD or schizophrenia were re-evaluated to
examine if complex I defect was specific to BD. Results revealed that complex I subunits that are
involved in the electron transfer process are decreased only in patients with BD, suggesting that
oxidative stress from complex I dysfunction may be specific to BD. Next, using post-mortem
brain, I confirmed lower levels of complex I and its subunit, NDUFS7, in the frontal cortex of
patients with BD, supporting the role of complex I dysfunction in the pathophysiology of BD. As
an extension of a post-mortem brain study from our group demonstrating the mitochondria and
the synapse as targets of oxidative stress, I first examined downstream targets of complex I
dysfunction in the post-mortem brain, focusing on the mitochondria and the synapse. To
demonstrate the mitochondria as a target of complex I dysfunction, I measured the activation of
the NLRP3-inflammasome, which is a marker of mitochondrial oxidative stress. Results revealed
that increased activation of the NLRP3-inflamamsome may be specific to BD, suggesting that
mitochondria may be a target of complex I dysfunction. To examine the synapse as a target of
complex I defect, I measured oxidative modifications to the dopaminergic synapse as increased
dopamine signaling is known to underlie mania. Oxidative modifications to dopaminergic

iii
proteins, the dopamine transporter and tyrosine hydroxylase, were altered in the post-mortem
prefrontal cortex of patients with BD, demonstrating the synapse as another potential target of
complex I dysfunction in BD. Downstream targets of complex I dysfunction were further
explored in cell models, allowing us to directly inhibit the electron transfer process of complex I
using rotenone. The effect of lithium was also tested as it is uniquely used for the treatment of
BD and therefore allows us to assess if the observed alterations may be relevant to BD
pathology. Inhibiting complex I with rotenone increased protein oxidation and nitration, and
caused an increase in methylation and hydroxymethylation of DNA. Moreover, lithium pre-
treatment was able to decrease these alterations, suggesting that they may be downstream targets
of complex I defect in BD. Together, these studies suggest that complex I dysfunction may be
specific to BD, and that it may play an important role in the pathophysiology of this disease.

iv
ACKNOWLEDGEMENTS
First and foremost, I would like to thank my two supervisors, Dr. L. Trevor Young and Dr. Ana
C. Andreazza for their continuous support. In addition to the technical knowledge and the
incredible training opportunities I received under their supervision, I also learned to appreciate
the process that goes into producing high quality data, telling a compelling story, and taking
ownership of my work. Their enthusiasm for research and passion that they poured into their
work were also contagious, and I am certain that I will not be as passionate about research today
if it was not for their guidance.
I would also like to thank my supervisory committee members, Dr. Stephen Kish and Dr.
Rebecca Laposa, for their impeccable advice and support throughout my PhD. Their instruction
helped shape my presentation and writing style, and truly allowed me to grow as a researcher. I
would also like to express my gratitude for their immense help with this thesis.
Furthermore, I would like to thank my collaborators from Brazil who I am very grateful to have
had a chance to work with, Camila Nascimento and Gustavo Scola. The learning experience I
shared with them are memories I will always treasure, and I want to thank them for all the new
things they introduced me to inside and outside the lab. I would also like to thank my amazing
students, Karina Mendonca, Cameron Isaacs-Trepanier, Nika Elmi, Wenjun Chen, and David
(Pok Yik) Yeung, who were some of the most intelligent, persistent, patient and diligent
individuals I have had the good fortune to meet.
I would also like to thank my mother (Eunja Yun), father (Jaegeun Kim), and my brother (Steven
Kim) for their encouragement and unyielding support. They had faith in me even in times I did

v
not, and I will not be here today without them. Lastly, I would like to thank my friend, Jackie
Liu, for her support and friendship.

vi
TABLE OF CONTENTS
CHAPTER 1. Introduction…………………………………………………………...…..………..1
1.1 The pathophysiology of bipolar disorder………………………………………….....………..1
1.1.1 Mitochondrial dysfunction and oxidative stress in bipolar disorder…………....………...4
1.1.2 Inflammation in bipolar disorder……………………………………………....…..……...7
1.1.3 Dopamine dysregulation in bipolar disorder………………………………..…....………11
1.2 Lithium…………………………………………………………………………………..…...14
1.3 Downstream effects of complex I dysfunction in bipolar disorder………………….....……17
1.3.1 Complex I dysfunction and the NLRP3-inflammasome in bipolar disorder………..…..18
1.3.2 Complex I dysfunction and the dopamine system in bipolar disorder…………………...20
1.4 Scope and Hypotheses…………………………………………………………………..…...22
1.4.1 Scope…...………………………………………………………………………………...22
1.4.2 Aims and Hypotheses…………………………………………………………………....24
CHAPTER 2. A Fresh Look at Complex I in Microarray Data: Clues to Understanding Disease-
Specific Mitochondrial Alterations in Bipolar Disorder…………………………………….…...27
- Statement of significance and impact …………………………………………………...34
CHAPTER 3. Nod-like Receptor Pyrin Containing 3 in the Post-mortem Frontal Cortex from
Patients with Bipolar Disorder: a Potential Mediator between Mitochondria and Inflammation 37
- Statement of significance and impact……………………………………………………58
CHAPTER 4. Oxidation and Nitration in Dopaminergic Areas Prefrontal Cortex from Patients
with Bipolar Disorder and Schizophrenia………………………………………………………..60
- Statement of significance and impact……………………………………………………86

vii
CHAPTER 5a. Glutathione-mediated Effects of Lithium in Decreasing Protein Oxidation
Induced by Mitochondrial Complex I Dysfunction……………………………………………..88
CHAPTER 5b. Lithium Reduces the Effects of Rotenone-induced Complex I Dysfunction on
DNA Methylation and Hydroxymethylation in Rat Cortical Primary Neurons………………..102
- Statement of significance and impact (Chapter 5a&b)…………………………………127
CHAPTER 6. Summary and General Discussion………………………………………………129
CHAPTER 7. Conclusions……………………………………………………………………..139
References………………………..……………………………………………………………..142
Copyright Releases……………………………………………………………………………..163

viii
LIST OF TABLES
CHAPTER 3
Table 1. Subject information……………………………………………………………………52
CHAPTER 4
Table 1. Subject information by group…………………………………………………………83

ix
LIST OF FIGURES
CHAPTER 2
Figure 1. Summary of mitochondrial complex I gene alterations in bipolar disorder and
schizophrenia……………………………………………………………………………………33
CHAPTER 3
Figure 1. Levels of electron transport chain components complex I, II, III, IV, V and
nicotinamide nucleotide transhydrogenase (NNT) in the post-mortem frontal cortex of patients
with BD, SCZ and non-psychiatric controls (CTL)……………………………………………53
Figure 2. Levels of NLRP3-inflammasome components NLRP3 (A), ASC (B) and caspase 1 (C)
in the brain homogenate (homogenate), cytoplasmic and crude mitochondrial fractions
(mitochondrial) in the post-mortem frontal cortex of patients with BD, SCZ and non-psychiatric
controls (CTL)…………………………………………………………………………………54
Figure 3. Levels of inflammatory factors downstream of NLRP3-inflammasome activation in the
IL-1β pathway, IL-1β, IL-6, TNF-α, and in the IL-18 pathway, IFNγ & IL-10 in the post-mortem
frontal cortex of patients with BD, schizophrenia (SCZ) and non-psychiatric controls
(CTL)………………………………………………………………………………………….55
CHAPTER 4
Figure 1. Immunohistochemistry……………………………………………………………...79
Figure 2. CPM intensity and 3NT intensity…………………………………………………...80
Figure 3. FRET between DAT and CPM, and TH and 3NT…………………………………..81

x
Figure 4.Correlations…………………………………...………………………………………82
CHAPTER 5a
Figure 1. Lithium prevents decrease in complex I activity and cell viability induced by
mitochondrial complex I dysfunction……………………………………………………………97
Figure 2. Glutathione is important for lithium’s ability to decrease protein carbonylation, but not
nitration…..………………………………………………………………………………………98
CHAPTER 5b
Figure 1. Complex I activity measurement in rat cortical primary neurons with lithium pre-
treatment (0.75mM) and rotenone treatment at different concentrations (5nM, 10nM, and
50nM)…………………………………………………………………………………………..116
Figure 2. The effect of lithium pre-treatment (0.75mM) and rotenone treatment at different
concentrations (5nM, 10nM, and 50nM) on ATP levels in primary cortical neurons…………117
Figure 3. The effect of lithium pre-treatment (0.75mM) and rotenone treatment at different
concentrations (5nM, 10nM, and 50nM) on cell mortality in primary cortical neurons measured
by the MTT assay………………………………………………………………………………118
Figure 4. The effect of lithium pre-treatment (0.75mM) and rotenone treatment at different
concentrations (5nM, 10nM, and 50nM) on apoptosis and necrosis in primary cortical
neurons………………………………………………………………………………………….119
Figure 5. Changes in methylation and hydroxymethylation levels with 7 days of lithium pre-
treatment (0.75mM) and 30 minutes of rotenone treatment (5nM, 10nM, and 50nM) in primary
cortical neurons…………………………………………………………………………………120

xi
Figure 6. Changes in methylation and hydroxymethylation levels of 30 minutes of H2O2
treatment (5µM, 10µM or 50µM) in primary cortical neurons………………………………121
Schema 1. Mitochondrial complex I dysfunction induced by rotenone and its effects on cell
viability and DNA methylation and hydroxymethylation: the role of lithium………………122
Online resource 1. Cross-reactivity for antibodies was assayed by adding different combinations
of the primary and secondary antibodies for assessing resultant fluorescence intensity……123
Online resource 2. Changes in methylation and hydroxymethylation levels with 7 days of lithium
pre-treatment followed by rotenone treatment in primary cortical neurons…………………124
Online resource 3. Changes in methylation and hydroxymethylation levels with H2O2 treatment
in primary cortical neurons…………………………………………………………………..124

1
1. INTRODUCTION
Bipolar disorder (BD) is a chronic mood disorder characterized by alternating episodes of mania
and depression. According to WHO, it is the sixth leading cause of disability worldwide, where
approximately 50% of the patients attempt suicide during their lifetime. Complex I dysfunction in
the electron transfer process and consequent generation of oxidative stress, which occurs when the
production of reactive oxygen or nitrogen species surpasses the cell’s capacity to maintain them at
an optimal level, is one of the most consistent findings in patients with BD. Therefore, the primary
aim of my PhD was to examine potential downstream targets of complex I dysfunction in BD.
1.1 The pathophysiology of bipolar disorder
Bipolar disorder is a mood disorder characterized by alternating episodes of mania/hypomania,
depression and/or mixed episodes. Mania, which is a defining feature of BD, is a sustained period in
which patients experience elevated mood and/or irritability, rapid speech, and impulsive behaviors
(Barnett & Smoller, 2009). Patients in the depressive phase exhibit persistent loss of interest in
pleasurable activities and/or depressed mood. Lastly, mixed episodes feature symptoms of both
depression and mania (Anderson, Haddad, & Scott, 2012). There are two subtypes of BD, BD I and
BD II. Patients with BD I exhibit one or more episodes of mania or mixed episodes with at least one
episode of depression, and patients with BD II have one or more episodes of depression with
hypomania (Muller-Oerlinghausen, Berghofer, & Bauer, 2002). In addition to the burden of the
symptoms of the disease, patients may require social welfare due to failure to maintain a job,
intervention from the justice system, and/or assistance from family members (Kilbourne et al.,
2004). BD is also associated with mild to moderate impairments in cognitive function, selective
attention and memory, which are present regardless of the state of the patient (Anderson et al., 2012;
Rajkowska, 2000). BD is also hypothesized to be a progressive disorder, where responsiveness to
therapy and cognitive ability declines with increasing number of recurrences (Kupfer, 2005). Indeed,

2
due to the chronic nature of the disorder, where greater than 90% of patients experience recurrent
episodes (Muller-Oerlinghausen et al., 2002), development of novel and more efficacious treatments
is of great interest.
BD is a highly complex disorder and multiple pathological pathways have been reported in brain
areas associated with emotional control, cognition and memory, such as the prefrontal cortex,
anterior cingulate cortex, hippocampus and the amygdala (Chen, Suckling, Lennox, Ooi, &
Bullmore, 2011). For example, reduced brain volume has been reported in the prefrontal cortex
(Almeida et al., 2009), hippocampus, amygdala, anterior cingulated gyrus and medial and inferior
frontal gyri (Lyoo et al., 2004; Malhi, Tanious, Das, Coulston, & Berk, 2013). In agreement with
these findings, decreased neuronal density was also reported in the prefrontal cortex (Rajkowska,
2000). Some other findings include increased protein and mRNA levels of pro-apoptotic factors
(Rao, Harry, Rapoport, & Kim, 2010), lower levels of brain derived neurotrophic factor (BDNF)
(Cunha et al., 2006), and increased glutamatergic and dopaminergic signaling in patients with BD
(Berk et al., 2007; Messiha, Agallianos, & Clower, 1970; Rao et al., 2010).
Genetic alterations, such as Val66Met polymorphism of the BDNF gene (Lohoff et al., 2005),
and genetic variations at the calcium channel CACNA1C have also been reported in BD (Ferreira et
al., 2008). In addition to genetic polymorphisms, recent studies have focused on epigenetic
modifications in BD, with a strong focus on methylation (Abdolmaleky et al., 2006; Connor &
Akbarian, 2008; D'Addario et al., 2012; Huzayyin et al., 2013). DNA methylation occurs when DNA
methyltransferase transfers a methyl group to the cytosine residue at the 5’ carbon position (Cheng,
Cahill, Kasai, Nishimura, & Loeb, 1992), and methylation of regulatory regions prevents the binding
of transcription factors to the DNA, inhibiting transcription (Bestor, 2000; Bird, 2002;
Lertratanangkoon, Wu, Savaraj, & Thomas, 1997; Reik, Dean, & Walter, 2001). Methylation is also
the most common epigenetic alteration in mammals, and is known to be affected by a large number

3
of different factors, such as nicotine, environmental toxins, and psychostimulants (Bird, 2002;
Connor & Akbarian, 2008; Reik et al., 2001). Alterations in methylation levels were reported in
patients with BD, where BDNF promoter region was found to be hypermethylated (D'Addario et al.,
2012), and promoter of the catechol-o-methyltransferase gene was found to be hypomethylated
(Abdolmaleky et al., 2006). A related alteration is hydroxylation of methylcytosine by the ten eleven
translocation enzyme (Tahiliani et al., 2009). Formation of 5-hydroxymethylcytosine is thought to
promote the demethylation of DNA, and also to inhibit the binding of 5-methylctyosine recognizing
proteins, limiting their ability to repress transcription (Jin, Kadam, & Pfeifer, 2010). The ten eleven
translocation enzyme requires molecular oxygen (Jin et al., 2010). Since the mitochondria are
primary consumers of oxygen in the cell, mitochondrial dysfunction may also contribute to different
hydroxymethylation patterns. Moreover, studies have shown that reactive oxygen species can cause
various DNA damage including both hypermethylation and hypomethylation using cancer cells
(Chestnut et al., 2011; H. J. Kang et al., 2012; Kang, Zhang, Kim, Bae, & Hyun, 2012), implicating
methylation and hydroxymethylation as possible downstream targets of mitochondrial dysfunction
and oxidative stress.
Indeed, mitochondrial complex I dysfunction has been strongly implicated in BD (Iwamoto,
Bundo, & Kato, 2005; Kato, 2005; Kato & Kato, 2000; Konradi, Sillivan, & Clay, 2012). Complex I
is involved in many different pathways in the brain, including the production of reactive oxygen and
nitrogen species (Halliwell, 2007). Production of these molecules can have multiple downstream
effects, as they can cause direct modifications to transporters and enzymes, changing their function,
and alter levels of genetic expression as mentioned above (Campos et al., 2007; K. A. Kang et al.,
2012), or act as signaling molecules to affect downstream pathways (Halliwell & Gutteridge, 2007;
H. K. Kim, Andreazza, Yeung, Isaacs-Trepanier, & Young, 2014). The following sections will
begin with a discussion of the role of mitochondrial complex I dysfunction in BD. I will then discuss

4
two potential downstream targets of complex I dysfunction in BD: dopaminergic dysfunction and
inflammation. Lithium, which is uniquely used for the treatment of BD, is also discussed as it
improves complex I activity and decrease oxidative stress, making it a good proxy for understanding
complex I dysfunction in BD.
1.1.1 Mitochondrial dysfunction and oxidative stress in bipolar disorder
Mitochondria are primarily responsible for oxidative phosphorylation, which produces adenosine
triphosphate (ATP) by relaying electrons extracted from nicotinamide adenine dinucleotide (NADH)
and flavin adenine dinucleotide (FADH2) through the electron transport chain (ETC) consisting of 5
multi-protein complexes located at the inner mitochondrial membrane. The process of electron
transfer is coupled to proton pumping into the intermembrane space of the mitochondria. This
process creates a proton gradient, which is used by the ATP-synthase (complex V) to phosphorylate
ATP. Molecular oxygen is the final acceptor of electrons, becoming water (Fariss, Chan, Patel, Van
Houten, & Orrenius, 2005). Studies have reported lower levels of high energy phosphates in the
brain of patients with BD since the 1990s using magnetic resonance spectroscopy (Deicken, Weiner,
& Fein, 1995; Kato et al., 1998; Kato, Takahashi, Shioiri, & Inubushi, 1993), suggesting decreased
efficiency of the mitochondria. Patients with BD were also found to have lower mRNA levels of
genes encoding mitochondrial proteins related to oxidative phosphorylation, suggesting simpairment
in this process (Konradi et al., 2004; Sun, Wang, Tseng, & Young, 2006). Decreased efficiency of
the ETC can compromise the process of electron transfer, resulting in increased production of
superoxide anions from the reaction between leaked electrons and oxygen (Fariss et al., 2005).
Complex I of the ETC consists of 45 or 46 subunits arranged into 2 functional arms, which are
the hydrophilic arm and the hydrophobic arm. The hydrophilic arm is largely responsible for the
transfer of electrons and contains subunits containing iron-sulfur clusters, while the hydrophobic arm
is responsible for proton pumping (Green & Kroemer, 2004; Lenaz, 2001). Interestingly, patients

5
with BD were found to have lower mRNA and protein levels of NADH dehydrogenase Fe-S protein
7 (NDUFS7), which is a subunit of complex I that contains the last iron-sulfur cluster N2 responsible
for the reduction of ubiquinone to ubiquinol (Andreazza, Shao, Wang, & Young, 2010), and levels
of this subunit were found to correlate with complex I activity (Andreazza et al., 2010). Because
complex I is the main site of electron leakage (Halliwell & Gutteridge, 2007; Jeong & Seol, 2008;
Sherer et al., 2003), these studies suggest that patients with BD may be more susceptible to the
production of superoxide anions. This is important because while products of oxidative stress are
found in both schizophrenia and BD (Ng, Berk, Dean, & Bush, 2008), the contributing sources may
be different for the two disorders. More specifically, while complex I dysfunction may be a major
contributor to oxidative stress production BD, other pathways may be involved for schizophrenia
(Ng et al., 2008).
Superoxide anions produced during this process is converted to hydrogen peroxide by the
superoxide dismutase. Hydrogen peroxide, which is relatively unreactive, can go through the Fenton
reaction in the presence of iron ions to produce hydroxyl radicals, which are highly reactive
(Halliwell, 1992). Indeed, hydroxyl radicals can cause lipid peroxidation, converting membrane
lipids to lipid hydroperoxides and releasing reactive unsaturated aldehydes (Halliwell, 1992). It can
also oxidize DNA, producing 8-hydroxydeoxyguanosine (8-OHdG), and oxidize proteins to cause
disulfide bridge formation or carbonylation (Halliwell, 2001). Superoxide anion can also react with
nitric oxide to produce peroxynitrite, a reactive nitrogen species (RNS) (Halliwell & Gutteridge,
1984; Jeong & Seol, 2008). Peroxynitrite can act similarly to the hydroxyl radical, oxidizing protein
sulfhydryls, lipids, and DNA (LaVoie & Hastings, 1999). On the other hand, peroxynitrite can also
produce a different type of oxidative damage called nitration, where it reacts with tyrosine residues
to produce 3-nitrotyrosine (Halliwell & Gutteridge, 2007). The central nervous system may be
particularly vulnerable to oxidative damage due to the richness of polyunsaturated fatty acid side

6
chains that are more sensitive to attack by free radicals, lower levels of antioxidant enzymes such as
superoxide dismutase and glutathione peroxidase, high oxygen consumption and high concentrations
of iron in certain brain areas (Halliwell, 1992).
Production of ROS and RNS is neutralized by antioxidant enzymes, including the
aforementioned superoxide dismutase, which converts the superoxide anion to hydrogen peroxide.
Hydrogen peroxide is then converted to water by catalase (Halliwell & Gutteridge, 2007). The main
antioxidant system in the brain is glutathione, which can neutralize both ROS and RNS by using its
sulfhydryl group (Gawryluk, Wang, Andreazza, Shao, & Young, 2011). Glutathione peroxidase
catalyzes the reaction between hydrogen peroxide and glutathione to produce water, and glutathione-
S-transferase conjugates glutathione to oxidized products, making them non-toxic (Dringen, 2000).
Oxidized glutathione groups are returned to their reduced form by glutathione reductase (Yao &
Keshavan, 2011). Oxidative stress can also occur due to the reduction in antioxidant defenses. Lower
levels of antioxidant enzymes including glutathione peroxidase, superoxide dismutase, catalase,
glutathione S-transferase and glutathione (Benes, Matzilevich, Burke, & Walsh, 2006) were found in
the post-mortem brain of patients with BD. Indeed, N-acetylcysteine, a cysteine derivative that
increases glutathione production, was found to decrease depression and improve global functioning
in patients with BD compared to placebo in a double-blind randomized study (Berk et al., 2008;
Magalhães et al., 2011).
While complex I dysfunction and increased oxidative stress are well-established in BD
(Andreazza et al., 2010; Andreazza, Wang, Salmasi, Shao, & Young, 2013; Scola, Kim, Young, &
Andreazza, 2013; Sun et al., 2006), the specific downstream targets have yet to be elucidated. To
answer this question, a recent study published from our group identified the mitochondria and the
synapse as two targets of oxidative stress in the frontal cortex of patients with BD (Andreazza et al.,
2013). To extend on these findings, the first part of my PhD aimed to examine the downstream

7
targets of complex I dysfunction in BD with a focus on the mitochondria and the synapse in the post-
mortem brain of patients with BD. In choosing a marker of mitochondrial oxidative damage, we
decided to examine the nod-like receptor, pyrin containing 3 (NLRP3)-inflammasome, a redox
sensor in the inflammatory system that was found to be downstream of mitochondrial ROS
production from complex I dysfunction in vitro (Zhou, Yazdi, Menu, & Tschopp, 2011). For a
marker of synaptic oxidative damage, we chose to examine oxidative modifications to the dopamine
system, which exists largely in the synapse and is known to underlie mania, a defining feature of BD
(Berk et al., 2007; Gerner, Post, & Bunney, 1976). The following sections will provide a review of
findings supporting inflammatory and dopaminergic abnormalities in BD.
1.1.2 Inflammation and bipolar disorder
Medical conditions associated with the activation of the inflammatory system are more
commonly diagnosed in patients with BD than in the normal population, particularly diabetes
mellitus, cardiovascular disease and autoimmune diseases (Barbosa et al., 2013). Chronic pain
conditions such as arthritis and headache are also more prevalent in BD (Goldstein, Fagiolini,
Houck, & Kupfer, 2009; Goldstein, Kemp, Soczynska, & McIntyre, 2009; Kilbourne et al., 2004),
which is in agreement with studies reporting a strong association between pain and inflammatory
cytokines IL-1β, IL-6, and TNF-α (J. Zhang et al., 2015). Patients with BD also show higher
monocyte phagocytic activity (Padmos et al., 2008). The first study demonstrating increased levels
of inflammatory cytokines in patients with BD was published in 1995 (Maes, Bosmans, Calabrese,
Smith, & Meltzer, 1995). Since then, an accumulating number of studies have demonstrated mild
and chronic activation of the inflammatory system in the brain and periphery of patients with BD
(Goldstein, Kemp, et al., 2009; Hope et al., 2011; Kauer-Sant'Anna et al., 2009; Leboyer et al., 2012;
Liu et al., 2004; Rao et al., 2010). Indeed, even after controlling for various demographic factors
such as age, sex, and even clinical variables, cytokine levels were found to be strongly associated

8
with BD (Goldstein, Kemp, et al., 2009). While the role of the inflammatory system in BD is
unclear, Barbosa et al (2014)’s hypotheses for increased monocyte activity in BD can also be applied
to the general inflammatory system (Barbosa et al., 2014). More specifically, presence of
inflammatory activation in BD can be explained by 1. The illness itself induces activation of the
inflammatory system, 2. Inflammatory activation may contribute to the development of BD, and/or
3. A factor may be responsible for both the development of BD and inflammatory conditions.
Studies examining genetic alterations in the inflammatory system reported polymorphisms in the
IL1B gene in patients with BD, and greater levels of haplotypic combinations in the IL1RA gene,
particularly in BD patients with a family history of BD, schizophrenia, or major depression (Papiol
et al., 2008). Moreover, expression levels of 19 inflammatory mRNAs were found to be altered, with
IL-6 being one of the strongest markers separating patients with BD from healthy controls (Padmos
et al., 2008). Interestingly, some studies reported increased activation of the inflammatory system
during mood episodes, where patients with mania were found to have greater levels of IL-6, TNF-α,
and soluble IL-2 receptor compared to patients with euthymia as well as controls (Barbosa et al.,
2013; Modabbernia, Taslimi, Brietzke, & Ashrafi, 2013; Munkholm, Braüner, Kessing, & Vinberg,
2013). However, findings are inconsistent and studies examining associations between treatment and
cytokine levels, as well as inflammation and symptom severity have largely reported negative
findings, suggesting that inflammatory activation may be a trait marker rather than a state marker in
BD (Goldstein, Kemp, et al., 2009). Another topic that has been garnering much attention is the
interaction between inflammatory cytokines in the periphery and in the central nervous system, as
inflammatory cytokines are larger and cannot easily pass through the blood-brain barrier (Barbosa et
al., 2013). Some methods of transport under physiological conditions include cytokine receptors,
endothelial activation and active transport (Barbosa et al., 2013). Cytokines can also pass through
the blood brain barrier when the blood brain barrier becomes leaky, which occurs with increased

9
inflammation in the brain, suggesting that neuroinflammation can exacerbate cytokine exchange
between the central nervous system and the periphery (Brietzke, Stabellini, Grassis-Oliveira, &
Lafer, 2011). Interestingly, cytokine activation profile differs between the central nervous system
and the periphery in BD, with increased levels of cytokines in the IL-2, TNF-α and IL-6 pathways in
the periphery, suggesting the activation of T cells (Kauer-Sant'Anna et al., 2009; Muller-
Oerlinghausen et al., 2002; Munkholm et al., 2013; O'Brien, Scully, Scott, & Dinan, 2006; Ortiz-
Domínguez et al., 2007; Rao et al., 2010), and greater levels of cytokines and receptors in the IL-1
pathway, including IL-1β, IL-1 receptor, NF-κB, glial fibrillary acidic protein, and cFOS in the
prefrontal cortex and cerebrospinal fluid of patients with BD (Rao et al., 2010; Söderlund et al.,
2011). Moreover, treatment with medications that are used in BD such as lithium and antipsychotics
was found to decrease levels of inflammatory cytokines, including IL-10, IL-6, IL-2 and IFN-γ,
suggesting that amelioration of inflammatory activation may partly contribute to their effect in BD
(Boufidou, Nikolaou, Alevizos, Liappas, & Christodoulou, 2004; Padmos et al., 2008).
Activation of the inflammatory system may have important implications for BD as inflammatory
cytokines can influence long-term potentiation, neuronal survival, astrocyte development, and
neurogenesis (Barbosa et al., 2013). Furthermore, inflammatory cytokines can contribute to changes
in neurotransmitter levels such as dopamine and cause cognitive decline, possibly contributing to the
behavioral symptoms of BD (Berk et al., 2013; Felger et al., 2013; Felger & Miller, 2012). As such,
anti-inflammatory agents have received attention as potential treatments for BD (Barbosa et al.,
2013). Celecoxib, a cyclooxygenase-2 inhibitor, was tested on patients with BD with mixed or
depressive episodes in a double-blind, randomized and placebo-controlled study as an adjunct (Nery
et al., 2008). The findings of this study revealed that patients treated with celecoxib experienced
faster reduction in their depressive symptoms compared to the control group.

10
The cause for inflammatory activation in BD appears to be multifactorial, with a few studies
demonstrating genetic alterations, and others suggesting that it may be occurring downstream of
other changes that occur in BD, such as excitatoxicity, apoptotic signaling, or activation of the
hypothalamic-pituitary-adrenal axis (Goldstein, Kemp, et al., 2009; Leboyer et al., 2012). Recent
studies have shown that mitochondrial complex I dysfunction and oxidative stress may contribute to
activation of the inflammatory system (López-Armada, Riveiro-Naveira, Vaamonde-García, &
Valcárcel-Ares, 2013; Zhou et al., 2011), suggesting that complex I dysfunction in BD may be a
contributing factor to inflammation as well. More specifically, signs of mitochondrial dysfunction,
such as release of mitochondrial DNA or ATP through the opening of permeability transition pores,
were found to correlate with increased levels of IL-1β, and inhibition of complex I using chemicals
such as rotenone was found to have the same effect (López-Armada et al., 2013; Zhou et al., 2011).
Indeed, drugs that decrease oxidative stress, such as N-acetyl cysteine were found to decrease the
release of inflammatory cytokines (Zhou et al., 2011). Lithium, which is the gold-standard of
treatment for the management of BD, was also shown to improve complex I activity, decrease
oxidative stress, and decrease pro-inflammatory cytokines (Burgess et al., 2001; Chuang, 2005;
Goldstein, Kemp, et al., 2009; Quiroz, Gould, & Manji, 2004), further suggesting the importance of
these pathways in BD. However, whether complex I dysfunction and subsequent release of
mitochondrial ROS underlies increased inflammation in BD has yet to be explored. Specific
mechanisms that may be involved in the link between complex I defect and inflammation in BD will
be further discussed in section 1.3.1, which examines potential downstream targets of complex I
dysfunction in BD.
In addition to activating the inflammatory system through generating mitochondrial oxidative
stress, complex I dysfunction can have other downstream effects (Andreazza et al., 2010; Naoi et al.,
2005; Watabe & Nakaki, 2007). As a continuation of a previous study by our group identifying the

11
synapse as a target of oxidative stress in BD (Andreazza et al., 2013), we decided to examine the
dopaminergic synapse as a potential downstream target of complex I dysfunction, since increased
dopamine signaling is strongly implicated in mania (Berk et al., 2007; Cousins, Butts, & Young,
2009). The next section will provide an overview of dopaminergic dysregulation in BD.
1.1.3 Dopamine dysregulation in bipolar disorder
Much attention has been given to the dopaminergic synapse as a site of damage in BD as
increased levels of dopamine is widely accepted to be one of the main mechanisms underlying mania
(Berk et al., 2007; Manji & Lenox, 2000; Salvadore et al., 2010). Dopamine is a neurotransmitter
that is involved in locomotion, reward, emotion and cognition (Giros et al., 1992) through four
pathways: the nigrostiratal pathway, which is involved in the motor system and includes substantia
nigra pars compacta and the striatum, the tuberinfundibular pathway, which is in the hypothalamus,
the mesolimbic pathway, which begins at the ventral tegmental area and project to the ventral
striatum, hippocampus, and septum, and finally the mescortical pathway, which also originates from
the ventral tegmental area and project to the frontal and temporal cortices (Ciliax et al., 1999;
Cousins et al., 2009). The mesocortical pathway has received much attention in BD as it contributes
to working memory, reward, impulsivity and emotional processing, characteristics often reported to
be altered in BD (Seamans & Yang, 2004).
Dopamine is regulated by many different enzymes, including tyrosine hydroxylase, which is the
rate-limiting enzyme for dopamine synthesis (Daubner, Le, & Wang, 2011). As the concentration of
its precursor, tyrosine, does not affect the rate in which dopamine is synthesized, the proper
functioning of tyrosine hydroxylase is critical for the dopamine system (Cousins et al., 2009). L-
DOPA produced by tyrosine hydroxylase is converted to dopamine by aromatic amino acid
decarboxylase. Dopamine’s actions are primarily terminated by the dopamine transporter, which
uptakes dopamine from the synapse (Cousins et al., 2009; Jaber, Jones, Giros, & Caron, 1997),

12
although it can also be oxidized by catechol-o-methyl transferase into homovanilic acid, or auto-
oxidized (Cousins et al., 2009). Once released into the synapse, dopamine exerts its effects through
two different families of dopamine receptors, D1 and D2. Activation of receptors of the D1 family,
consisting of D1 and D5, result in activation of the adenylyl cyclase pathway through its coupling
with Gα. Activation of D2 receptors, which consist of D2, D3 and D4, inhibits adenylyl cyclase
through Gαi/o (Elsworth & Roth, 1997).
First report of greater dopamine levels in patients with BD was in 1970, when Messiha et al.
reported higher levels of conjugated and free dopamine in the urine of patients in the manic phase
(Messiha et al., 1970). Administration of dopaminergic agonists such as amphetamine, which
reverses the direction of dopamine transport such that synaptic dopamine levels increase, and
pramipexole and bromocriptine (Cousins et al., 2009; Silverstone, 1984) leads to manic behavior as
measured by the Young Mania Scale in healthy individuals and in patients with BD (Anand et al.,
2000; Berk et al., 2007). The same is observed when L-DOPA, a dopamine precursor, is
administered (Cousins et al., 2009). Behavioral effects of dopaminergic agonists can be reversed by
medications used in BD, such as lithium, adding support to the dopamine theory of BD (Cousins et
al., 2009; Van Kammen & Murphy, 1975). Moreover, antagonizing the dopamine system with
antipsychotics and alpha-methyl-para-tyrosine, an inhibitor of dopamine synthesis, reduces manic
behavior (Berk et al., 2007; Cousins et al., 2009). Some studies also suggest that bipolar depression
may represent hypodopaminergic states, as bupropion, which inhibits dopamine removal from the
synapse, is effective in treating depression (Berk et al., 2007), and levels of homovanillic acid, a
metabolite of dopamine, in the cerebrospinal fluid of patients with BD in the depressive phase is
lower after drug washout (Cousins et al., 2009). Moreover, lower dopamine levels may be
underlying depression in Parkinson’s disease, which can be ameliorated by administering
dopaminergic agonists (Goldberg, Burdick, & Endick, 2004). The presence of cognitive dysfunction

13
and impaired working memory also suggests dopamine dysfunction in BD, as optimal levels of
dopamine is necessary for working memory, where too much or too little dopamine is thought to
impair performance (Berk et al., 2011). Indeed, both patients with mania or depression exhibit
lowered sustained attention, and lower scores on tests for executive function and working memory
(Clay, Sillivan, & Konradi, 2010; Cousins et al., 2009; H. W. Kim, Rapoport, & Rao, 2010;
Martinez-Aran et al., 2004). These findings lead to the dopamine dysregulation model of BD, which
states that increased dopamine transmission that underlies mania is followed by a subsequent
downregulation of elements of the dopamine system, resulting in the depressive phase (Berk et al.,
2007).
First-line treatment for acute mania is antipsychotics, which act by blocking dopamine D2
receptors (Anderson et al., 2012), prompting scientists to examine dopamine receptor alterations in
BD. Studies examining dopamine receptors by positron emission tomography have reported
inconsistent findings, with one study reporting alterations in D1-receptor binding (Suhara et al.,
1992), increased D2 receptor density (Pearlson et al., 1995; Wong et al., 1997), and other studies
reporting no alterations in D2 receptor density between BD and controls (Pearlson et al., 1995).
Differences in the results may be due to the different areas examined or the radioligand used
(Cousins et al., 2009). Genetic studies have also examined dopamine receptors, in which
predominantly negative results were reported for the D1 receptor (Mitchell et al., 1992; Nöthen et
al., 1992), D2 receptor (Cousins et al., 2009; Furlong et al., 1998; Nöthen et al., 1992), the D3
receptor (Elvidge et al., 2001; Savoye et al., 1998), D4 (Serretti et al., 1999) and the D5 receptor
genes (Muir et al., 2001). Coincidentally, genetic studies examining catechol-o-methyl transferase
also reported inconsistent findings (Craddock & Sklar, 2013), and mRNA levels of this enzyme was
not found to differ in the prefrontal cortex of patients with BD compared to controls (Tunbridge,
Burnet, Sodhi, & Harrison, 2004). As genetic studies suggest that dopaminergic abnormality in BD

14
may not occur due to alterations in gene expression, downstream pathways may be involved, such as
post-translational alterations. In fact, dopaminergic proteins are susceptible to oxidative
modifications due to their abundance in cysteine residues (Hastings, Lewis, & Zigmond, 1996;
Hastings & Zigmond, 1994), suggesting that complex I dysfunction and oxidative stress may
contribute to dopamine dysregulation in BD.
While antipsychotics are best recognized as decreasing dopamine signaling in mania, lithium
was also shown to decrease dopamine levels and increase dopamine uptake (Dunigan & Shamoo,
1995; Messiha et al., 1970). In addition to decreasing dopamine signaling, lithium also has a wide
range of effects, including decreasing glutamate levels, stabilizing calcium levels, decreasing
apoptosis, pro-inflammatory cytokines, and importantly, increasing complex I activity (Bearden et
al., 2007; Cordeiro, Gundersen, & Umbach, 2002; Cui, Shao, Young, & Wang, 2007; J. S. Lai,
Zhao, Warsh, & Li, 2006; Nascimento et al., 2014; Quiroz et al., 2004; Schäfer, Goodenough,
Moosmann, & Behl, 2004; Tan et al., 2011). Complex I dysfunction and generation of oxidative
stress can impact many systems due to the ability of ROS and RNS to directly modify proteins and
impact downstream pathways, suggesting that at least a part of lithium’s actions may be by
improving complex I function (Nascimento et al., 2014; Sun et al., 2006). Additionally, lithium is
uniquely used for the treatment of BD, making lithium a good proxy for studying complex I
dysfunction in BD. The next section further examines the use of lithium and its mechanisms of
action in BD.
1.2 Lithium
Lithium has been the first-line treatment for long-term management of BD since being
introduced 60 years ago, and is used uniquely for the treatment of BD (Malhi et al., 2013; Muller-
Oerlinghausen et al., 2002). While it is most recognized for its ability to stabilize mood and prevent
suicidal ideation during euthymia (Bauer, 2004; Burgess et al., 2001; Malhi et al., 2013), lithium is

15
also prescribed for acute mania, although its efficacy is limited as it takes 6-10 days to exert its
effects. Hence, lithium is often prescribed with antipsychotics (Malhi et al., 2013). Evidence
supporting lithium’s efficacy for bipolar depression is less consistent (Bhagwagar & Goodwin, 2002;
Fountoulakis, Gonda, Siamouli, & Rihmer, 2009), and recent clinical trials did not show differences
between lithium and placebo (Malhi et al., 2013). At a global level, patients who were prescribed
lithium were reported to have greater total gray matter volume compared to patients who were not
prescribed lithium and even healthy controls in areas implicated in BD, such as the anterior
cingulate, frontal cortex, amygdala, and hippocampus (Bearden et al., 2007; Hallahan et al., 2011;
Kempton, Geddes, Ettinger, Williams, & Grasby, 2008). Studies have also shown that lithium
attenuates dopamine release (Ferrie, Young, & McQuade, 2006) and decrease glutamate
transmission while increasing GABA release (Malhi et al., 2013), suggesting that it acts by
preventing alterations in neurotransmission. More specifically, lithium treated rats had lower levels
of dopamine (Gambarana et al., 1999), and lithium was shown to cause downregulation of the
NMDA receptor and increase glutamate reuptake (Dixon & Hokin, 1998), which could together be
contributing to decreasing manic symptoms. Furthermore, lithium was found to increase GABA
levels in the cerebrospinal fluid of patients (Ahluwalia, Grewaal, & Singhal, 1981), decreasing
glutamate-induced excitatoxicity.
Lithium was also shown to impact many signaling pathways through its ability to interfere with
magnesium binding (Malhi et al., 2013). For example, lithium inhibits inositol monophosphatase (Q.
Li et al., 2010; Malhi et al., 2013) and adenylyl cyclase (Quiroz et al., 2004), and minimize cAMP
fluctuations from stimulation, which may contribute to its ability to stabilize neurotransmission
(Bachmann, Schloesser, Gould, & Manji, 2005; El Khoury, Petterson, Kallner, Aberg-Wistedt, &
Stain-Malmgren, 2002; Quiroz et al., 2004). Lithium’s ability to regulate calcium levels are also well
documented, where it was shown to block calcium uptake, decrease calcium stores and its

16
intracellular levels (Dubovsky, Thomas, Hijazi, & Murphy, 1994; El Khoury et al., 2002; Perova,
Kwan, Li, & Warsh, 2010; Shalbuyeva, Brustovetsky, & Brustovetsky, 2007). Since calcium is an
important signaling molecule for many cellular processes, lithium’s ability to stabilize calcium levels
most likely plays an important role (Malhi et al., 2013). Lithium’s inhibition of glycogen synthase
kinase 3β is of interest, as it is a target of monoaminergic systems and produce hyperactivity in mice
(Prickaerts et al., 2006).
In addition, lithium is known to have neuroprotective effects, as it was shown to decrease pro-
apoptotic proteins, including Bax and p53, while increasing anti-apoptotic proteins, such bcl-2 (Q. Li
et al., 2010; Malhi et al., 2013). Lithium was also found to increase BDNF levels after 5 days of
treatment, which is very similar to how long lithium takes to have an effect in patients (6-10 days),
strongly implicating BDNF in lithium’s neuroprotective effects (Hashimoto et al., 2002).
Importantly, lithium was shown to protect against oxidative stress (Cui et al., 2007; Machado-Vieira,
Andreazza, et al., 2007; Shao, Cui, Young, & Wang, 2008; Sun et al., 2006). For example, lithium
was able to inhibit lipid peroxidation and protein oxidation in primary rat cortical cells (Frey et al.,
2006). Moreover, lithium was able to prevent other downstream effects of oxidative stress produced
by complex I inhibition or hydrogen peroxide, such as release of cytochrome c in hippocampal and
neuroblastoma cells (Cui et al., 2007; J. S. Lai et al., 2006). In addition, lithium was shown to
increase the activity of superoxide dismutase and glutathione peroxidase in the brain of rats, and
normalize superoxide dismutase to catalase ratios (Machado-Vieira, Andreazza, et al., 2007;
Machado-Vieira, Dietrich, et al., 2007), further demonstrating its ability to increase antioxidant
defenses. Lithium was also found to inhibit amphetamine-induced lipid peroxidation in rats,
suggesting that lithium can counteract oxidative stress produced by high levels of dopamine as well
(Frey et al., 2006). While the exact mechanism by which lithium exerts its antioxidant effects is
unknown, studies have demonstrated that lithium increases glutathione content (Shao et al., 2008),

17
expression levels of glutamate-cysteine ligase, and increase expression levels of glutathione S-
transferase (Shao et al., 2008). A number of studies have also shown that lithium improves complex
I function, which would decrease mitochondrial production of ROS (Bachmann et al., 2009; Quiroz,
Gray, Kato, & Manji, 2008). Also, patients with BD who were taking lithium were found to have
higher mRNA levels of complex I components compared to patients who were not (Sun et al., 2006).
These studies suggest that lithium may be able to prevent downstream effects of complex I
dysfunction and oxidative stress in BD.
1.3 Downstream effects complex I dysfunction in bipolar disorder
Complex I dysfunction in the electron transfer process can lead to increased leakage of electrons,
which can react with molecular oxygen to produce the superoxide anion (Halliwell & Gutteridge,
2007; Sherer et al., 2003). The superoxide anion can undergo a cascade of reactions to produce ROS
and RNS that can accumulate and lead to oxidative stress (Halliwell, 2007). Oxidative stress can
have many downstream effects as ROS and RNS can directly modify biomolecules such as proteins,
lipids and DNA, and also act as signaling molecules (Halliwell, 1992, 2006; Sies, 1991). Protein
oxidation and nitration products, such as 3-nitrotyrosine (Andreazza et al., 2009; Andreazza et al.,
2010), protein carbonyl groups (Andreazza et al., 2010) and disulfide bridges (H. K. Kim et al.,
2014) have also been found to be increased in the post-mortem brain of patients with BD,
particularly in the frontal cortex. Protein oxidation and nitration can produce aggregates and direct
alteration of protein function (Beal, 2002). Moreover, oxidative modifications can accumulate in
sufficient amounts to disrupt essential cellular processes when the production of oxidative stress
surpasses the cell’s antioxidant defense system (Halliwell, 2001), resulting in apoptosis or necrosis
depending on the degree of severity (Ng et al., 2008). Evidence of increased lipid peroxidation is one
of the most consistent findings in BD (Brown, Andreazza, & Young, 2014), where lipid peroxidation
products such as 4-hydroxynonenal, malondialdehyde, and thiobarbituric acid reactive substances

18
were reported in the brain and periphery of patients with BD (Andreazza et al., 2008; Brown et al.,
2014). Because products of lipid peroxidation can disturb cellular function by forming adducts,
increased levels of these products have functional implications (Kuloglu et al., 2002). RNA and
DNA are also targets of oxidative stress, as guanine residues are sensitive to oxidative attack, and
form 8-hydroxydeoxyguanosine in DNA (Barzilai & Yamamoto, 2004; Cheng et al., 1992).
Oxidative damage to DNA was also found to be increased in patients with BD, as higher 8-OHdG
levels were found in lymphocytes and the post-mortem hippocampus (Che, Wang, Shao, & Young,
2010; Soeiro-de-Souza et al., 2013).
Interestingly, a recent study by our group showed that complex I dysfunction and oxidative stress
can specifically target different subcellular fractions by examining the post-mortem frontal cortex of
patients with BD (Andreazza et al., 2013), suggesting that ROS and RNS may have specific
downstream targets rather than affecting all systems in the same direction. More specifically, the
myelin, synapse and mitochondrial fractions were found to be targets of oxidative modifications. In
the myelin fraction, 4-hydroxynonenal and 8-isoprostane levels were increased in patients with BD,
in agreement with previous studies supporting increased lipid peroxidation in patients with BD
(Andreazza et al., 2008; Brown et al., 2014; Wang, Shao, Sun, & Young, 2009). Lipid peroxidation
in patients with BD has been extensively studied by other members from our group, demonstrating
lipid peroxidation as one of the most consistent findings in BD and that it may contribute to
decreased white matter integrity (Andreazza et al., 2008; Brown et al., 2014; Versace et al., 2013).
These findings suggest that lipid peroxidation in the myelin fraction may be a downstream target of
complex I dysfunction in BD. Therefore, my PhD project aimed to extent on these findings and
examine specific downstream targets of complex I dysfunction in BD in the mitochondria and the
synapse. We first examined the mitochondria by using a marker of complex I dysfunction, NLRP3.
Not only does NLRP3 allow us to examine mitochondrial release of ROS through complex I defect,

19
its activation also causes the release of pro-inflammatory cytokines (Zhou et al., 2011). Increased
levels of inflammatory cytokines are consistently reported in the brain and peripheral samples of
patients with BD (Goldstein, Kemp, et al., 2009; Rao et al., 2010), making NLRP3 of particular
interest for the examination of complex I dysfunction in BD.
1.3.1 Complex I dysfunction and the NLRP3-inflammasome in bipolar disorder
A recent study demonstrated that complex I dysfunction and subsequent production of
mitochondrial ROS can cause the activation of a redox sensor, NLRP3 (Sorbara & Girardin, 2011;
Tschopp & Schroder, 2010; Zhou et al., 2011), suggesting that it can be used as a marker of
mitochondrial oxidative stress. More specifically, NLRP3 is a redox sensor in the inflammatory
system (Zhou et al., 2011) and a member of the NOD-like receptor family that are responsible for
activating IL-1β and IL-18 upon sensing a variety of different molecules including
lipopolysaccharide, ATP, and monosodium urate (Agostini et al., 2004; Bryant & Fitzgerald, 2009).
As it is highly unlikely that the ligand binding site of NLRP3 is flexible enough to allow it to
recognize such a large variety of triggers, it has been hypothesized that a common pathway must
exist for these molecules to activate NLRP3 (Schroder, Zhou, & Tschopp, 2010; Tschopp &
Schroder, 2010). NLRP3 is also activated by markers of mitochondrial oxidative damage, such as
opening of mitochondrial permeability transition pores and release of mitochondrial DNA and ATP,
demonstrating NLRP3 is a marker for mitochondrial oxidative stress (Schroder et al., 2010; Shimada
et al., 2012; Tschopp & Schroder, 2010; Zhong et al., 2013; Zhou et al., 2011). Indeed, superoxide
anions produced by complex I dysfunction can act as signaling molecules through redox sensors in
the cell that trigger downstream pathways by undergoing structural modifications upon reacting with
ROS or RNS (Jones, 2010). Studies have shown that treatment with rotenone, a complex I inhibitor,
causes activation of NLRP3 and that molecular triggers fail to activate NLRP3 when mitochondrial
ROS release is inhibited (N. Li et al., 2003; Zhou et al., 2011), suggesting that complex I

20
dysfunction and consequent release of superoxide anions may be the common pathway (Zhou et al.,
2011).
NLRP3 consists of three domains, a pyrin domain, a C-terminal leucine-rich domain, and a
central nucleotide binding domain (Agostini et al., 2004; Bryant & Fitzgerald, 2009). In its resting
state, NLRP3 is localized in the cytoplasm with its central nucleotide binding domain bound to the
leucine rich domain, which inhibits it from oligomerizing (Tschopp & Schroder, 2010). Upon its
activation, NLRP3 undergoes a conformational change exposing its oligomerization and pyrin
domain, allowing it to recruit another cytoplasmic protein, apoptosis-associated speck-like protein
containing a CARD (ASC) through pyrin-pyrin interaction (Zhou et al., 2011). The CARD domain
of ASC allows for the recruitment of procaspase-1, which completes the assembly of the NLRP3-
inflammasome. It was also found that inflammasomes co-localize with the mitochondrial membrane
using co-localization analysis and subcellular fractionation (Zhou et al., 2011). Assembly of the
inflammasome allows for the release of active caspase 1, which then cleaves IL-1β and IL-18 to their
active forms, triggering the inflammatory cascade. These pathways lead to the activation of MYD88
and NF-κB, resulting in increased expression of IL-6, TNFα, and prostaglandin E2, which have all
been shown to be increased in patients with BD (Eder, 2009; Sollberger, Strittmatter, Garstkiewicz,
Sand, & Beer, 2014).
The above studies suggest that complex I dysfunction may lead to the activation of the NLRP3-
inflammasome through the generation of mitochondrial ROS, making this system a marker of
mitochondrial oxidative damage. The aforementioned study has found the mitochondrial and the
synapse as targets of oxidative damage. While the synapse is a highly complex system consisting of
numerous transporters, enzymes, and receptors, we decided to focus on the dopamine system, as
increased activity of the dopamine system is known to underlie mania, a defining feature of BD.
1.3.2 Complex I dysfunction and the dopamine system in bipolar disorder

21
In vitro and animal studies have demonstrated that dopamine synthesis and transport are
influenced by oxidative stress and complex I inhibition, suggesting that the dopamine system may
also be a target of complex I dysfunction in BD (H. K. Kim & Andreazza, 2012; Maragos, Zhu,
Chesnut, & Dwoskin, 2002; Offen, Ziv, Sternin, Melamed, & Hochman, 1996; S. U. Park, Ferrer,
Javitch, & Kuhn, 2002; Watabe & Nakaki, 2008). In fact, bursts of dopamine synthesis and release
that occur during mania could make dopaminergic synaptic terminals particularly vulnerable to
oxidative damage, as dopamine and L-DOPA are highly susceptible to oxidation by ROS and RNS,
and produce more ROS upon being oxidized (H. K. Kim & Andreazza, 2012; Miyazaki et al., 2006;
S. U. Park et al., 2002). Moreover, oxidative metabolism of dopamine is increased in the presence of
ROS and RNS (Graham, 1978; Graham, Tiffany, Bell, & Gutknecht, 1978; LaVoie & Hastings,
1999; Tse, McCreery, & Adams, 1976), suggesting that complex I dysfunction could also be
contributing to oxidative damage in the dopaminergic system. Oxidation of dopamine results in the
formation of additional free radicals that are cytotoxic (Asanuma, Miyazaki, Diaz-Corrales, &
Ogawa, 2004), such as the superoxide anion, hydrogen peroxide and hydroxyl radicals, and
cytotoxic dopamine/DOPA-quinones which are electron poor and act similarly to ROS (Hastings,
2009; Hastings et al., 1996; Hastings & Zigmond, 1994). Quinones can produce functional
modifications to cysteine containing proteins by forming 5-cysteinyl dopamine (Maker, Weiss,
Silides, & Cohen, 1981; Tse et al., 1976). Indeed, protein-bound dopamine caused by intrastriatal
dopamine injections can be prevented by adding antioxidants (Hastings et al., 1996), and DA-
induced cytotoxicity can be prevented by adding glutathione and N-acetyl cysteine, which increases
glutathione levels (C. T. Lai & Yu, 1997), suggesting that dopamine-induced toxicity is through the
production of oxidative stress. Moreover, inducing mania-like states with d-amphetamine and
increasing synaptic levels of dopamine were shown to increase lipid peroxidation and protein
oxidation (Frey et al., 2006; Valvassori et al., 2010).

22
Two markers of dopamine-rich areas are tyrosine hydroxylase (Elsworth & Roth, 1997) and the
dopamine transporter (Akil et al., 1999). Areas immunoreactive for the dopamine transporter and
tyrosine hydroxylase overlap extensively in the human brain, suggesting that both are good markers
for the dopamine synapse (Akil et al., 1999; Lewis et al., 2001), These two proteins are particularly
interesting because they are also vulnerable to oxidative/nitrosative modifications (Ara et al., 1998;
Blanchard-Fillion et al., 2001; Fleckenstein, Metzger, Beyeler, Gibb, & Hanson, 1997; S. U. Park et
al., 2002). Tyrosine hydroxylase is a target of peroxynitrite-induced tyrosine nitration in vitro, and
nitration of a single tyrosine residue was found to be able to reduce its activity (Ara et al., 1998).
While tyrosine hydroxylase is particularly susceptible to nitration (Blanchard-Fillion et al., 2001),
dopamine transporter is susceptible to disulfide formation (S. U. Park et al., 2002). Indeed, the
dopamine transporter is rich in cysteine residues and is a target of products of dopamine oxidation,
resulting in decreased uptake of dopamine from the synapse and further increasing dopamine
signaling (S. B. Berman & Hastings, 1999; S. B. Berman, Zigmond, & Hastings, 1996; LaVoie &
Hastings, 1999; Miyazaki et al., 2006). Hence, since oxidation of dopamine makes dopaminergic
synapses more vulnerable to oxidative stress, and mitochondrial production of ROS/RNS from
complex I dysfunction would increase dopamine oxidation, subsequent oxidative damage to
dopaminergic proteins at the synapse may be contributing to dopamine dysregulation observed in
BD.
While complex I dysfunction may have many downstream effects such as the aforementioned
oxidation of dopaminergic proteins at the synapse and activation of the NLRP3-inflammasome in
animal and in vitro studies (Maragos et al., 2002; Sherer et al., 2003; Zhou et al., 2011), it remains to
be seen whether complex I dysfunction is underlying the alterations that are observed in BD.
Moreover, lithium, which was shown to improve complex I function, was also found to decrease
dopamine signaling and pro-inflammatory cytokines, suggesting that lithium may be exerting its

23
effects at least in part by ameliorating alterations that occur downstream of complex I defect and
oxidative stress.
1.4 Scope and Hypotheses
1.4.1 Scope
Bipolar disorder is a serious mood disorder with recurrent episodes, where symptom severity,
response to medications and cognitive ability decrease with recurrent episodes, bringing attention to
the urgent need for the development of novel and more efficacious treatments to prevent relapse in
patients with BD (Berk et al., 2011). Although studies have reported numerous alterations in the
brain and peripheral cells of patients with BD, one of the most consistent findings is alterations in
proteins of the mitochondrial electron transport chain, especially in complex I (Kato & Kato, 2000;
Konradi et al., 2004; Konradi et al., 2012; Sun et al., 2006). Complex I dysfunction can result in
increased generation of oxidative stress, which is also consistently reported in patients with BD
(Andreazza et al., 2008; Andreazza et al., 2013; Halliwell & Gutteridge, 2007). Indeed, increased
levels of carbonyl groups, 3-nitrotyrosine, lipid peroxidation products, and oxidized DNA and RNA
have been found in patients with BD (Andreazza et al., 2009; Andreazza et al., 2008; Andreazza et
al., 2010; Brown et al., 2014). This has important implications, as studies have demonstrated a wide
array of targets of oxidative stress in vitro and in animal models by structurally and functionally
modifying proteins that are susceptible to oxidative modifications (Halliwell, 1992, 2001) or by
acting as signaling molecules through redox sensors. What is unknown is whether complex I
dysfunction and oxidative stress underlie the changes that are observed in BD.
Complex I dysfunction leads to increased generation of ROS when subunits that are involved in
the electron transfer process are dysfunctional, allowing for electrons to escape to react with
molecular oxygen, producing the superoxide anion (Halliwell & Gutteridge, 2007). To examine if

24
complex I dysfunction and susceptibility to increased generation of ROS is specific to BD, we re-
examined microarray studies that measured complex I subunits in patients with BD or schizophrenia.
I also aimed to confirm complex I dysfunction in the post-mortem brain of patients with BD, which
was reported by two independent studies from our group (Andreazza et al., 2010; Andreazza et al.,
2013).
A recent study from our group identified the mitochondria and the synapse as two targets of
oxidative stress in the post-mortem brain of patients with BD (Andreazza et al., 2013). Therefore, I
aimed to extend on these findings by studying downstream targets of complex I dysfunction in the
post-mortem brain of patients with BD, focusing on the mitochondria and the synapse. To study the
mitochondria as a downstream target of complex I defect, I examined activation of the NLRP3-
inflammasome, which is a marker of mitochondrial oxidative stress (Zhou et al., 2011), in the post-
mortem frontal cortex. Activation of the NLRP3-inflammasome also results in the release of
inflammatory cytokines (Schroder et al., 2010), further suggesting that this marker may be relevant
for BD as increased inflammation is consistently found in patients with BD (Berk et al., 2011; Rao et
al., 2010). To examine the synapse as a target of complex I dysfunction, I examined oxidative
modifications to the dopamine system, as dopaminergic proteins largely reside in the synapse and
increased levels of synaptic dopamine is known to underlie manic symptoms (Berk et al., 2007;
Cousins et al., 2009).
To further understand the downstream targets of complex I in BD, we used cell-models, allowing
for direct inhibition of the electron transfer process in complex I using rotenone (Maragos et al.,
2002; Naoi et al., 2005; Sherer et al., 2003). Also, lithium was used to examine if the observed
alterations may be relevant to BD pathology, as lithium is uniquely used for the treatment of BD (J.
S. Lai et al., 2006; Maurer, Schippel, & Volz, 2009; Sun et al., 2006; Washizuka, Iwamoto,
Kakiuchi, Bundo, & Kato, 2009).

25
Examining post-mortem brains allows us to confirm complex I dysfunction in BD, and to
directly observe alterations that may be occurring downstream of complex I defect and oxidative
stress in patients. On the other hand, experiments using cell models provide us with the opportunity
to directly manipulate complex I functioning and observe its consequences. In examining the
downstream targets of complex I dysfunction using post-mortem brains and cell models, we hoped
to benefit from the advantages provided by both approaches in elucidating the role of complex I
dysfunction in the pathophysiology of BD.
1.4.2 Aims and hypotheses
The overall aim of my PhD was to examine downstream targets of complex I dysfunction in BD,
since complex I dysfunction may have specific targets through the production of oxidative stress as
demonstrated by a recent study from our group (Andreazza et al., 2013).
1. Re-examination of microarray studies:
a. Aim 1: To review microarray studies examining the post-mortem brain of patients
with BD or schizophrenia to determine if complex I dysfunction in the electron
transfer process is specific to patients with BD, making patients with BD more
susceptible to having increased generation of oxidative stress.
Hypothesis: Decreased levels of complex I subunits involved in the electron transfer
process will be unique to patients with BD, suggesting increased susceptibility to the
production of ROS in these patients.
2. Post-mortem brain studies: Studies in post-mortem brain aimed to confirm complex I
dysfunction in patients with BD and extend on a previous study from our group identifying
the mitochondria and the synapse as two targets of oxidative stress in the post-mortem frontal
cortex of patients with BD.

26
a. Aim 2: To confirm complex I dysfunction in the post-mortem brain of patients with
BD.
Hypothesis: Patients with BD will have decreased levels of complex I and its subunit,
NDUFS7, compared to non-psychiatric controls in the post-mortem frontal cortex.
b. Aim 3: To examine the mitochondria as a target of complex I dysfunction in BD by
measuring the activation of the NLRP3-inflammasome, a marker of mitochondrial
oxidative stress.
Hypothesis: Mitochondria will be found to be a potential downstream target of
complex I dysfunction in patients with BD, as shown by increased activation of the
NLRP3-inflamamsome in the post-mortem frontal cortex.
c. Aim 4: To examine the synapse as a target of complex I dysfunction in BD by
measuring oxidative modifications to the dopaminergic synapse, since dopaminergic
dysregulation underlies mania.
Hypothesis: The synapse will be found to be a potential downstream target of
complex I dysfunction in patients with BD, as shown by increased oxidative
modifications to the dopaminergic synapse in the post-mortem prefrontal cortex.
3. Cell model studies: Studies in cell models aimed to further examine downstream targets of
complex I dysfunction in a more controlled environment, allowing for the direct inhibition of
the electron transfer process in complex I using rotenone. The effect of lithium was also
examined, allowing us to see if resulting alterations from complex I inhibition may be
relevant to BD pathology.
a. Aim 5: To examine potential downstream targets of complex I dysfunction by using
rotenone to block the process of electron transfer in complex I, and to examine the
relevance of alterations occurring from rotenone treatment to BD by testing if lithium

27
is able to decrease these changes. Protein oxidation, nitration, and changes in
methylation and hydroxymethylation of DNA were measured as potential
downstream targets of complex I defect.
Hypothesis: Inhibition of the electron transfer process of complex I using rotenone
will result in increased generation of oxidative damage, as measured by increased
protein oxidation, nitration, and methylation and hydroxymethylation of DNA.
Lithium pre-treatment will be able to decrease these alterations produced by rotenone
treatment.
By testing these hypotheses in post-mortem brain and cell models, I aim to elucidate the
potential downstream targets of complex I dysfunction in BD, which will lead to a better
understanding of the pathways that are involved in the pathophysiology of this highly complex
disease.

28
CHAPTER 2
Title: A Fresh Look at Complex I in Microarray Data: Clues to Understanding Disease-Specific
Mitochondrial Alterations in Bipolar Disorder
Authors: Gustavo Scola*, Helena Kyunghee Kim*, L. Trevor Young, Ana Cristina Andreazza
*Authors GS and HKK contributed equally to this letter.
Journal: Biological Psychiatry
Volume: 2
Pages: e4-5
Work performed by the student:
The student performed all work included in this chapter, including literature review, and writing of
the manuscript.

29
A fresh look at complex I in microarray data: clues to understanding disease-specific
involvement of mitochondrial alterations in bipolar disorder
Scola G1*, Kim HK2*, Young LT1,2, Andreazza AC1,2
1. Departments of Psychiatry and Pharmacology, University of Toronto, ON, Canada
2. Centre for Addiction and Mental Health, Toronto, ON, Canada

30
To the Editor:
Mitochondrial dysfunction and the consequent generation of oxidative stress damage have
been consistently reported in bipolar disorder (BD) and schizophrenia (SCZ). Microarray studies are
a major source of evidence for this mechanism, where the expression of many electron transport
chain (ETC) genes from complexes I-V was found to be decreased in patients with BD in the frontal
cortex (Choi et al., 2011; Jurata et al., 2004; Sun et al., 2006; Washizuka et al., 2009; Washizuka et
al., 2003) and hippocampus (Konradi et al., 2004). Mitochondrial ETC complex I is one of the
major sites for the generation of reactive oxygen species (ROS) (Fariss et al., 2005). Recently,
volume 71, issue 11 of the Biological Psychiatry journal addressed the implications of using
antioxidants for the maintenance of redox balance in psychiatric disorders (O. M. Dean, Bush, &
Berk, 2012; Hardan et al., 2012; Shungu, 2012). Considering the complexity of mitochondrial
complex I and its significance for psychiatric disorders, we re-examined the reported microarray
findings and grouped the altered complex I subunits by their relevance for ROS generation.
Complex I is a large complex consisting of 45-46 subunits, each of which plays a unique role
for the structure or activity of this complex, or ROS generation (Green & Kroemer, 2004; Lenaz,
2001). Subunits of complex I are arranged in four main subcomplexes, which are λ, α-λ, γ, and β.
The hydrophilic arm, which is responsible for the transfer of electrons, contains subcomplexes λ and
α-λ, and the hydrophobic arm, which is largely responsible for the pumping of protons, consists of γ
and β. Upon reviewing the literature, we identified 34 microarray studies examining BD and/or SCZ.
Of these studies, we have selected the microarray studies that included probes for complex I genes.
Finally, 10 studies were selected to be included in this report, as they reported alterations in complex
I genes in BD or SCZ. Microarrays enable the examination of the expression pattern of a significant
portion of the human genome encompassing multiple systems, making it ideal for the exploration of
the pathophysiology of complex disorders such as BD and SCZ (Mirnics, Levitt, & Lewis, 2006). In

31
total, 18 genes were found to be reduced or increased in their levels of expression in BD or SCZ,
where 8 were found to be altered in BD, 6 in SCZ, and surprisingly, only 4 genes were found to be
altered in both BD and SCZ (Figure 1). Combined findings of these studies revealed that in BD,
expression of iron-sulfur cluster-containing subunits within the hydrophilic arm were reduced,
suggesting that patients with BD may be more prone to having a dysfunction in the electron transfer
process. For instance, NDUFV1, which contains FMN and N3, and initiates the electron transfer
process from NADH to the iron-sulfur clusters, was found to be downregulated in BD. Also,
NDUFS1, which contains N1b, N4, N5, and N7, and NDUFS8, which contains N6a and N6b, were
also found to be downregulated. Finally, NDUFS7, which contains N2, and is responsible for the
reduction of ubiquinone to ubiquinol, was also found to be reduced in BD (Janssen, Nijtmans, van
den Heuvel, & Smeitink, 2006) (Figure 1). Although 4 subunits were found to be down-regulated in
the α-ʎ subcomplex in BD, these subunits are non-catalytic and we therefore will focus on the ʎ
subunit. In contrast, gene expression alterations in SCZ were found to be scattered throughout
complex I, with up and downregulation, and did not include alterations in subunits directly involved
in electron transfer. These data may suggest that while both patients with BD and SCZ may have a
reduction in complex I functionality, patients with BD may be more prone to deficiencies in the
electron transfer process, which could increase the probability of electrons escaping the electron
transport chain to react with molecular oxygen, causing a cascade of reactions to increase the
production of ROS such as the hydroxyl radical (Fariss et al., 2005). In fact, gene expression levels
of NDUSF7 was found to be reduced in two microarray studies in BD (Iwamoto et al., 2005; Sun et
al., 2006), and this finding was supported by a separate study, where protein levels of NDUFS7 was
found to be reduced in patients with BD (Andreazza et al., 2010). Importantly, this reduction in
NDUFS7 levels was found to be correlated with decreased complex I activity (Andreazza et al.,

32
2010). This indicates that altered gene expression identified in microarray studies could have
functional implications for complex I activity and ROS generation.
There are several caveats to these data. For example, different platforms were used for data
acquisition and analysis (Konradi et al., 2004; Mirnics et al., 2006), which could generate variability
between studies. Although post-mortem brain allows for direct investigation of human pathology,
there are inherent limitations involved in the use of samples acquired post-mortem, such as pH, post-
mortem interval, and small sample size (Andreazza et al., 2010; Iwamoto et al., 2005).
In summary, the data from the microarray studies discussed here suggest important
differences in the expression of complex I genes between BD and SCZ. In patients with BD, there is
down-regulation specifically in genes involved in electron transfer in complex I. On the other hand,
altered genes in SCZ were found to be scattered through complex I and include increased as well as
decreased expression levels. The findings reported here suggest that in bipolar disorder, emphasis
must be placed on the electron transfer chain, which may contribute to the elucidation of the
pathogenesis of this condition.
Acknowledgements
The authors would like to acknowledge CNPq/Brazil (Gustavo Scola) and CIHR as sources
of funding in support of this report.

33
Figures
Figure 1. Summary of mitochondrial complex I gene alterations in bipolar disorder and
schizophrenia. Gene alterations and direction of alteration (arrows) in bipolar disorder (BD;
highlighted in grey) and schizophrenia (SCZ) are divided into different brain areas and
subcomplexes where the alterations were found. Downregulations are shown in blue, and
upregulations are shown in red. Mitochondrial complex I subunits NDUFV1, NDUFS1, NDUFS8,
and NDUFS7 within the λ subcomplex were found to be downregulated in BD. These subunits
contain iron-sulfur clusters that are directly involved in the electron transfer process from NADH to
ubiquinone.

34
Statement of significance and impact:
BD and SCZ contain many pathological similarities, including calcium alterations, decreased
numbers of neurons, neurotransmitter imbalance, and inflammation (Clay et al., 2010). Many studies
have reported alterations in energy metabolism and mitochondrial proteins in BD and SCZ, in
particular for those belonging to oxidative phosphorylation (Clay et al., 2010; Iwamoto, Kakiuchi,
Bundo, Ikeda, & Kato, 2004). However, a study by our group reported that only patients with BD
have lower protein levels of NDUFS7, a complex I subunit that is critical for the proper transfer of
electrons to ubiquinone, and may contribute to electron leakage when it is dysfunctional (Andreazza
et al., 2010). Hence, this review addressed Aim 1, which was to identify if complex I dysfunction in
the electron transfer process is specific to patients with BD. This review of microarray studies
examining complex I subunits in BD and SCZ revealed that patients with BD have lower levels of
complex I subunits that are involved specifically in the process of electron transfer, while patients
with SCZ have increases or decreases in subunits that are responsible for different functions, such as
proton pumping or structural support. It should be noted, however, that decreased ability to pump
protons may also contribute to production of mitochondrial ROS indirectly, such as by decreasing
ATP production and producing stress in the cell (Schütt, Aretz, Auffarth, & Kopitz, 2012). On the
other hand, inhibition in the electron transfer process leads to the direct production of superoxide
anions, and rotenone, which interferes with electron transferring subunits in complex I, was
consistently shown to increase mitochondrial ROS (Bashkatova, Alam, Vanin, & Schmidt, 2004;
Boveris & Chance, 1973; N. Li et al., 2003; Sherer et al., 2003; Sorbara & Girardin, 2011). These
findings suggest that patients with BD may be particularly vulnerable to having increased generation
of mitochondrial ROS.
Mitochondrial ROS can directly modify enzymes and transporters, altering their function,
and activate downstream pathways by acting as signaling molecules (Haddad, 2002; Lenaz, 2001),

35
suggesting that it may be contributing to other alterations observed in BD. A recent study from our
group demonstrated that mitochondria and the synapse are targets of oxidative stress in the post-
mortem frontal cortex of patients with BD (Andreazza et al., 2013). To extend on these findings, I
decided to first examine the post-mortem brain to confirm complex I dysfunction, and then to
examine a marker of mitochondrial oxidative stress, the NLRP3-inflammasome (Zhou et al., 2011).
Once the NLRP3-inflammasome is activated following complex I dysfunction and release of
mitochondrial ROS, it releases pro-inflammatory cytokines, making this pathway particularly
interesting as inflammation is consistently reported in patients with BD (Goldstein, Kemp, et al.,
2009; Leboyer et al., 2012; Liu et al., 2004).

36
CHAPTER 3
Title: Nod-like receptor pyrin containing 3 (NLRP3) in the postmortem frontal cortex from patients
with bipolar disorder: a potential mediator between mitochondria and inflammation
Authors: Helena Kyunghee Kim, Ana Cristina Andreazza, Wenjun Chen, L. Trevor Young
Work performed by the student:
The student performed all work included in this chapter, including planning and performing the
experiments, data analysis, and preparation of the manuscript.

37
Nod-like receptor pyrin containing 3 (NLRP3) in the postmortem frontal cortex from patients
with bipolar disorder: a potential mediator between mitochondria and inflammation
Helena Kyunghee Kim1, Ana Cristina Andreazza1,2, Wenjun Chen1, L. Trevor Young1,2
1Departments of Psychiatry and Pharmacology, University of Toronto, Canada
2Campbell Family Mental Health Research Institute, Centre for Addiction and Mental Health

38
Abstract
Mitochondrial complex I dysfunction and inflammation are consistently reported in bipolar disorder
(BD). Mitochondrial production of reactive oxygen species (ROS), which occurs when complex I is
working ineffectively, was recently linked to activation of a redox sensor in the inflammatory
system, the nod-like receptor family pyrin domain-containing 3 (NLRP3). Upon its activation,
NLRP3 recruits apoptosis-associated speck-like protein (ASC) and caspase-1 to form the NLRP3-
inflamamsome, activating IL-1β and IL-18. This study aimed to examine if inflammation may be a
downstream target of complex I dysfunction through the NLRP3-inflamamsome in BD. Post-mortem
frontal cortex from patients with BD (N=9), schizophrenia (N=10), and non-psychiatric controls
(N=9) were donated from the Harvard Brain Tissue Resource Center. Levels of NLRP3, ASC and
caspase-1 were measured by western blotting and ELISA, and levels of downstream cytokines were
measured using Luminex. While we found no effects of age, sex or post-mortem delay, lower levels
of complex I (F2,25=3.46, p<0.05) and NDUFS7, a subunit of complex I (F2,25= 4.13, p<0.05), were
found in patients with BD compared with healthy controls. Mitochondrial NLRP3 (F2,25=3.86,
p<0.05) and ASC (F2,25=4.61, p<0.05) levels were higher in patients with BD. However, levels of
caspase 1 (F2,25=4.13, p<0.05 for both), IL-1β (F2,25=7.05, p<0.01), IL-6 (F2,25=5.48, p<0.05), TNFα
(F2,25=7.14, p<0.01) and IL-10 (F2,25=5.02, p<0.05) were found to be increased in both BD and
schizophrenia. These findings suggest that inflammation in the frontal cortex may occur both in
patients with BD and schizophrenia, while complex I dysfunction and NLRP3-inflammasome
activation may be more specific to BD.

39
Evidence supporting mitochondrial dysfunction, such as decreased mitochondrial respiration,
metabolism (Kato et al., 1993) and mitochondrial DNA polymorphisms (Kato, 2008) are
consistently reported in BD (Clay et al., 2010), a debilitating disorder affecting 1-2% of the
population worldwide (Merikangas et al., 2011). In particular, decreased mRNA and protein levels
of subunits that are involved in the transfer of electrons in complex I, which is a member of the
electron transport chain, may be a finding that is specific to BD (Iwamoto et al., 2005; Konradi et al.,
2012; Scola, Kim, Young, & Andreazza, 2012). In microarray studies, for example, NDUFS7, the
final iron-sulfur cluster-containing subunit involved in electron transfer within complex I, was found
to be lower in patients with BD (Iwamoto et al., 2005; Scola et al., 2013; Sun et al., 2006). Protein
levels of NDUFS7 were also found to be lower in two separate studies examining post-mortem
brains from patients with BD (Andreazza et al., 2010; Andreazza et al., 2013). Decreased efficiency
of the electron transfer process within complex I has important implications, as it can result in
increased leakage of electrons that react with molecular oxygen to form the superoxide anion
(Halliwell & Gutteridge, 2007). The superoxide anion can undergo further reactions to form
powerful ROS (Halliwell & Gutteridge, 2007). In support of these findings, increased oxidative
damage to proteins, lipids and DNA are consistently reported in the brain and periphery of patients
with BD (Andreazza et al., 2010; Andreazza et al., 2013; Clay et al., 2010).
ROS also serve as important signaling molecules through redox sensors. Recent studies have
suggested that ROS produced by the mitochondria may be a potent activator of the inflammatory
system (López-Armada et al., 2013). Increasing levels of mitochondrial ROS production by
inhibiting complex I, for instance, was found to result in greater levels of inflammatory factors such
as NF-κB and IL-1β (N. Li et al., 2003). These findings have important implications for BD, as
increased levels of inflammatory cytokines have been found in the brain and periphery of patients
with BD (B. Dean et al., 2013; Kauer-Sant'Anna et al., 2009; Y. K. Kim, Jung, Myint, Kim, & Park,

40
2007; Leboyer et al., 2012; Munkholm et al., 2013; O'Brien et al., 2006; Rao, Kellom, Reese,
Rapoport, & Kim, 2012). More specifically, IL-6 and TNF-α were reported to be increased in studies
examining peripheral samples from patients with BD (Y. K. Kim et al., 2007; Munkholm et al.,
2013), and studies examining the central nervous system have reported increases in cytokines and
receptors involved in the IL-1 pathway (B. Dean et al., 2013; Rao et al., 2010; Söderlund et al.,
2011).
Nod-like receptor pyrin containing 3 (NLRP3) is a redox sensor in the inflammatory system
that has been implicated as a potential link between mitochondrial ROS production and
inflammation (Tschopp & Schroder, 2010; Zhou et al., 2011). NLRP3, which normally resides in the
cytoplasm, undergoes a structural change upon the release of mitochondrial ROS and migrates to the
mitochondria, allowing it to be close to the site of damage (Zhou et al., 2011). This structural change
allows NLRP3 to recruit two other cytoplasmic proteins - apoptosis-associated speck-like protein
containing a CARD (ASC) and caspase-1 which form the NLRP3-inflammasome (Schroder et al.,
2010; Shimada et al., 2012; Tschopp & Schroder, 2010; Zhou et al., 2011). The assembly of the
inflammasome allows for the cleavage of caspase-1 into its active form, releasing it into the
cytoplasm to cleave pro-IL-1β and pro-IL-18 to their biologically active forms (Perregaux & Gabel,
1994). IL-1β and IL-18 are then released from the cells to activate downstream pathways involving
MYD88 and NF-κB (Eder, 2009), leading to increased expression of other inflammatory molecules
such as IL-6 and TNF-α (Bryan et al., 2010; A. Weber, Wasiliew, & Kracht, 2010). What remains
unknown, however, is whether inflammation is a downstream target of mitochondrial production of
ROS in BD through the NLRP3-inflamamsome. Therefore, in this study, we examined
mitochondrial dysfunction, localization and levels of NLRP3, ASC, and caspase-1, and levels of
downstream cytokines of the inflammasome pathway in frontal cortex of patients with BD compared
to healthy controls.

41
Materials and Methods
Subjects
Frozen post-mortem frontal cortex (BA9,10,24) tissues were generously donated from the Harvard
Brain Tissue Resource Centre (Table 1). These well-characterized samples have been in previous
publications by our lab and others (Andreazza et al., 2013; Rao et al., 2012). Briefly, subject groups
consisted of patients with BD (N=9), schizophrenia (SCZ; N=10) and non-psychiatric controls
(N=9). There were no between-group differences for sex (F2,25 = 0.023, p = 0.98), age (F2,25 = 0.18, p
= 0.83) and post-mortem interval (F2,25 = 0.14, p = 0.87). Diagnoses were established according to
DSM-IV criteria. Samples were coded numerically, and the experimenters were blind to the codes
until all the experiments and data analyses were completed.
Measurement of mitochondrial ETC proteins in the post-mortem frontal cortex
Levels of mitochondrial ETC components complex I (NADH-ubiquinone oxidoreductase), II
(succinate ubiquinone oxidoreductase), III (ubiquinone cytochrome c oxidoreductase), IV
(cytochrome c oxidase) and V (ATP synthase) and nicotinamide nucleotide transhydrogenase (NNT)
were measured using the Human oxidative phosphorylation magnetic bead panel kit (EMD
Millipore, #H0XPSMAG-16K) according to manufacturer’s instructions. Briefly, tissue extracts
were prepared with the lysis buffer provided in the kit followed by centrifugation at 14,000xg for 20
minutes. Samples (5ug total protein) were incubated with antibody-coated magnetic beads followed
by detection antibodies and streptavidin-phycoerythrin. Luminex Magpix (EMD Millipore) was used
to read the plates. Analysis was performed with the xPONENT software, with results expressed as
median fluorescence intensity (MFI). NDUFS7 levels were measured by immunoblotting using a
previously published protocol (Andreazza et al., 2013). First, brain homogenate was prepared by

42
sonication in a mannitol-sucrose buffer (225mM mannitol, 75mM sucrose, 30mM Tris-HCL at
pH7.4) (Wieckowski, Giorgi, Lebiedzinska, Duszynski, & Pinton, 2009). 15ug of proteins were
loaded onto 12% polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF)
membranes. Blotting was performed using a primary antibody against NDUFS7 (Santa Cruz, Dallas,
TX, USA, sc-98644; 2h; 1:1000) followed by a secondary goat anti-rabbit IgG horseradish
peroxidase antibody (Abcam Inc, Cambridge, MA, USA, #ab97051; 1h; 1:2000). For loading
control, mouse monoclonal anti-beta actin (Abcam Inc, #ab8226, 1hr; 1:1000; secondary antibody –
1:3000, 30min) was used. Membranes were visualized by incubating with ECL reagents and
analyzed using Versa Doc from Bio-Rad (Bio-Rad Laboratories Ltd., Mississauga, Canada).
Measurement of NLRP3 in the post-mortem frontal cortex
First, post-mortem brain was homogenized by sonication and applied to a sugar gradient (mannitol +
sucrose) to isolate the crude mitochondrial fraction and the cytosolic fraction using a previously
reported method that preserves the association between mitochondrial membranes and NLRP3-
inflammasome (Wieckowski et al., 2009; Zhou et al., 2011). Samples were first homogenized as
described above, and centrifuged twice at 740g for 5min to collect the supernatant. The supernatant
was then centrifuged at 9000g for 10 minutes. The resulting supernatant was collected as the
cytosolic fraction, and the pellet containing the crude mitochondrial fraction was resuspended and
centrifuged twice at 10,000g for 10 minutes. The resulting pellet was then collected and resuspended
in mitochondrial resuspending buffer (250mM mannitol, 5mM HEPES, 0.5mM EGTA at pH7.4).
The following procedures were performed on the brain homogenate as well as the cytoplasmic and
crude mitochondrial fractions. To measure NLRP3 levels, we performed western blotting according
to a previously published protocol (Andreazza et al., 2013). Samples (10ug for brain homogenate,
20ug for cytoplasmic fraction, and 35ug for mitochondrial fraction) were loaded on to 10%

43
polyacrylamide gels, and transferred to PVDF membranes. The membranes were stained with
Memcode reversible stain (Pierce BCA protein Assay kit, 23227) according to manufacturer’s
instructions and blocked (1h, 5% BSA). Incubation with primary anti-NLRP3 polyclonal antibody
(Millipore Co, Billerica, MA, USA, #ABF23; 2h; 1:500 for mitochondria, 1:2000 for cytoplasmic,
1:3000 for homogenate) was followed by a secondary goat anti-rabbit IgG horseradish peroxidase
antibody (Abcam Inc; 1h; 1:2000 for mitochondrial, 1:3000 for cytoplasmic, 1:5000 for whole
brain). Membranes were incubated with ECL reagents and analyzed using Versa Doc from Bio-Rad
(Bio-Rad Laboratories Ltd., Mississauga, Canada). For loading control, Memcode was used for the
crude mitochondrial fraction, and β-actin was used for cytoplasmic fraction and brain homogenate.
Measurement of ASC and caspase-1 in the post-mortem frontal cortex
ASC and caspase-1 levels were measured in the brain homogenate, cytoplasmic and mitochondrial
fractions using the ELISA technique. For ASC, samples were diluted to 20μg/ml for the brain
homogenate, cytoplasmic and mitochondrial fractions. For caspase 1, samples were diluted to
10ug/mL for the brain homogenate and cytoplasmic fraction, and 20ug/mL for the mitochondrial
fraction. 100μl/well of the samples were loaded onto 96w microlon ELISA plates (Gibco Inc,
Sainte-Marie, Quebec, Canada). ASC peptide (Abcam Inc, #ab22875) was used to create a curve
ranging from 10ng - 0pg to allow for quantification of ASC in the samples. THP-1 cell lysate was
used as a positive control for caspase-1(Chanput, Mes, & Wichers, 2014). Samples were incubated
overnight at 4°C on a shaker followed by blocking using 1% BSA (1hr, 37°C). Then, samples were
incubated with either primary anti-ASC antibody (Abcam, #ab111852; 2h; 1:32000 for homogenate,
1:20000 for cytoplasmic and mitochondrial fractions) or the primary anti-caspase-1 antibody
(Abcam, #ab62698; 2h; 1:5000 for all). Samples were incubated with horse radish peroxidase-
conjugated secondary antibody (Abcam; 1h; #ab6885; 1:4000 RT for ASC, #ab97051; 1:2000 RT

44
for caspase-1), and visualized by adding 100uL/well of TMB solution followed by 100uL/well of
stopping solution (1.2M HCl). Synergy H1 hybrid reader (BioTekR, Winooski, VT, USA) was used
to read the optical density at 450nm using the Gen 5 1.11 software.
Measurement of downstream cytokines of the NLRP3-inflammasome pathway in the post-mortem
frontal cortex
Levels of IL-1β, IFN-γ, IL-10, IL-6, and TNF-α in the brain homogenate were measured using the
human cytokine magnetic bead panel Milliplex Map kit (EMD Millipore, #HCYTOMAG-60K)
according to manufacturer’s instructions. Briefly, samples (20ug total protein) were incubated with
antibody-coated magnetic beads followed by detection antibodies and streptavidin-phycoerythrin.
Luminex Magpix (EMD Millipore) was used to read the plates. Analysis was performed with the
xPONENT software, where the amount of cytokines in the samples was quantified using a standard
curve provided by the kit.
Statistical analysis
Statistical analysis was performed using SPSS ver 21. Kolmogorov-Smirnov test was used to
determine normal distribution of data. For parametrically distributed data, one-way ANOVA was
used to examine between-group differences followed by Least Significant Difference post-hoc test to
examine the effect of diagnosis. For non-parametrically distributed data, Kruskal-Wallis test was
used to examine between-group differences followed by Mann-Whitney U test to examine the effect
of diagnosis. Pearson’s correlation test was used to measure any associations between age, sex,
postmortem interval (PMI) and measured variables. If the correlation was significant, ANCOVA was
performed with age/sex/PMI as a covariate. Pearson’s correlation test was also used to examine
correlations. Data are presented as mean±standard error of the mean (SEM).

45
Results
Complex I and NDUSF7
Levels of complex I, complex II, complex III, complex IV, complex V and nicotinamide nucleotide
transhydrogenase were measured using the Luminex system. Age, sex and PMI did not correlate
with any of the variables. We found a significant group difference for complex I (F2,25 = 3.46, p <
0.05), where lower levels of complex I was found in patients with BD (Fig. 1) compared to non-
psychiatric controls (p < 0.05). Complex II, III, IV, V and nicotinamide nucleotide transhydrogenase
did not differ between groups. Since the decrease in BD was specific to complex I, and because
previous studies reported decreased levels of NDUFS7, a complex I subunit, in patients with
BD(Andreazza et al., 2010; Andreazza et al., 2013), levels of NDUFS7 were measured using
western blotting. We found a significant group difference, (F2,25=4.13, p < 0.05), where patients with
BD were found to have lower levels of NDUFS7 (Fig. 1G) than patients with SCZ (p < 0.05) and
non-psychiatric controls (p < 0.05).
Components of the NLRP3-inflammasome in different subcellular fractions
Levels of NLRP3, ASC and caspase-1 were measured in the brain homogenate, cytosolic and crude
mitochondrial fractions. Western blotting was used to examine NLRP3, and the ELISA technique
was used to measure levels of ASC and caspase-1. Age, sex, and PMI did not correlate with any of
the measured variables. Between-group difference for NLRP3 was found in the crude mitochondrial
fraction (F2,25 = 3.83, p < 0.05; Figure 2C), with higher levels in patients with BD compared to
patients with SCZ (p < 0.05) and non-psychiatric controls (p < 0.05). There were no differences in
either the brain homogenate or the cytoplasmic fraction. Levels of ASC were also found to differ
between groups in the crude mitochondrial fraction (F2,25 = 4.61, p < 0.05; Figure 2F), with higher

46
levels in patients with BD compared to patients with SCZ (p < 0.05) and non-psychiatric controls (p
< 0.01). Caspase-1 was found to differ between groups in the crude mitochondrial fraction (F2,25
=4.13, p < 0.05; Figure 2I), with higher levels in patients with BD (p < 0.05) or SCZ (p < 0.05)
compared to non-psychiatric controls.
Cytokines downstream of the NLRP3-inflammasome
Levels of cytokines involved in the IL-1β pathway, including IL-1β, IL-6, TNF-α, and cytokines
involved in the IL-18 pathway, including IFN-γ and IL-10, were measured using the Luminex
system. Age, sex and PMI did not correlate with any of the variables. For the IL-1β pathway, a
between-group difference was found for IL-1β (F2,25 = 7.05 p < 0.01; Figure 3A), with higher levels
in patients with BD or SCZ (p < 0.01 for both) compared to non-psychiatric controls. IL-6 also
significantly differed between groups (F2,25 = 5.48, p < 0.05; Figure 3B), such that patients with BD
and SCZ (p < 0.01 for both) had higher levels than non-psychiatric controls. Similar results were
found for TNF-α (F2,25 = 7.14, p < 0.01, p < 0.01 for both BD and SCZ compared to non-psychiatric
controls; Figure 3C). For the IL-18 pathway, group difference was found in IL-10 (F2,25 = 5.02, p <
0.05; Figure 3F), with higher levels in patients with BD or SCZ compared to non-psychiatric
controls (p < 0.05 for both). IFN-γ did not differ between groups.
Discussion
In this study, we examined activation of the NLRP3-inflammasome in BD using post-
mortem frontal cortex from patients with BD, SCZ and non-psychiatric controls. Our results showed
decreased levels of complex I and NDUFS7 in patients with BD, suggesting complex I dysfunction,
which could in turn lead to greater production of mitochondrial ROS. Consistent with these findings,
we also found increased levels of NLRP3 and ASC in the crude mitochondrial fraction of patients

47
with BD, suggesting increased activation of the inflammasome as a result of complex I dysfunction
in these patients. On the other hand, we found that levels of caspase-1 and downstream cytokines are
increased in both patients with BD or SCZ, demonstrating activation of the inflammatory system in
both disorders. Together, these findings suggest that complex I dysfunction and subsequent
activation of the NLRP3-inflammasome in the frontal cortex may be specific to BD. Moreover,
while the NLRP3-inflammasome may contribute to increased levels of inflammatory cytokines in
patients with BD, inflammation in SCZ may occur due to other causes.
When we examined components of the electron transport chain, we only found a group
difference in complex I, where lower levels were found in patients with BD than non-psychiatric
controls. Complex I extracts electrons from NADH to transfer it to ubiquinone, reducing it to
ubiquinol. This is done by relaying electrons through the hydrophilic arm of complex I, which
contains iron-sulfur cluster containing subunits (Halliwell & Gutteridge, 2007). When this process is
disturbed, electrons can escape the system to react with molecular oxygen, producing the superoxide
anion (Halliwell, 2007). Because of this, complex I has been strongly implicated in mitochondrial
production of ROS, and disruption of complex I using chemicals such as rotenone, has been linked
to increased oxidative damage in cells and in animals (Bashkatova et al., 2004; Nascimento et al.,
2014; Sorbara & Girardin, 2011). Interestingly, a review of microarray studies revealed that patients
with BD have lower mRNA levels of complex I subunits that are directly involved in the electron
transfer process, while patients with SCZ show changes in subunits that are involved in other
functions of complex I, such as structural support or proton pumping (Scola et al., 2012). This
suggests that patients with BD may be particularly vulnerable to increased production of
mitochondrial ROS from complex I dysfunction. As complex I is a multi-protein structure, we
wanted to explore this further and examine NDUFS7, which contains the last iron-sulfur cluster
responsible for transferring electrons to ubiquinone(Janssen et al., 2006). Importantly, levels of

48
NDUFS7 were found to correlate with complex I activity (Andreazza et al., 2010), suggesting that it
is most likely important for its functioning. Our findings showed lower levels of NDUFS7 in patients
with BD compared to patients with SCZ and non-psychiatric controls. These findings are in
agreement with two independent studies that examined this subunit in post-mortem brain (Andreazza
et al., 2010; Andreazza et al., 2013), suggesting that it is a very consistent finding for BD.
Our findings having suggested complex I dysfunction in BD, we examined evidence of
NLRP3-inflammsome activation as mitochondrial ROS production is an important precursor for
inflammasome assembly (Zhou et al., 2011). We examined the three components of the NLRP3-
inflammasome, NLRP3, ASC and caspase-1, in the brain homogenate, cytoplasmic and crude
mitochondrial fractions of the post-mortem frontal cortex. Our findings showed that levels of
NLRP3 and ASC are increased only in the mitochondrial fraction from patients with BD, while no
group differences were found in the brain homogenate and the cytoplasmic fraction. A previous
study reported that activating the NLRP3-inflammasome caused an increase in co-localization
between the mitochondria and inflammasome components using microscopy (Zhou et al., 2011).
This finding was confirmed using subcellular fractionation studies in which NLRP3 and ASC were
found to be increased in the mitochondrial fraction and mitochondria-associated membranes, both of
which are part of the crude mitochondrial fraction (Wieckowski et al., 2009; Zhou et al., 2011).
Moreover, adding a complex I inhibitor, rotenone, caused an increase in IL-1β secretion (N. Li et al.,
2003), while the addition of a mitochondrial ETC inhibitor did not result in IL-1β secretion in Nlrp3
KO mice (Zhou et al., 2011). Importantly, activation of the inflammasome was completely blocked
by adding an ROS inhibitor, ammonium pyrrolidine dithiocarbamate, suggesting that NLRP3-
inflammsome assembly at the mitochondria is triggered by mitochondrial release of ROS (Zhou et
al., 2011). Thus, our finding of increased NLRP3 and ASC only in the mitochondrial fraction of
patients with BD suggests increased activation of the inflammsome in these patients. This is in

49
agreement with our results showing decreased levels of complex I and NDUFS7, as decreased levels
of complex I and resulting increase in mitochondrial ROS production may lead to increased
assembly of the NLRP3-inflammasome complex at the mitochondria (Zhou et al., 2011). As our
results suggested complex I dysfunction and increased activation of the NLRP3-inflammasome in
patients with BD, we correlated levels of complex I with NLRP3, ASC and caspase 1 levels in the
brain homogenate, cytoplasmic fraction and crude mitochondrial fraction (Figure 4). While we did
not find significant correlations, it is difficult to conclude that this suggests a lack of association
between complex I levels and the NLRP3-inflammasome due to the small number of subjects (N=9
in BD group).
Inflammation has been hypothesized to play a role in psychiatric disorders since the 1970s.
Since that time, evidence demonstrating cytokine alterations in BD and SCZ has accumulated
(Libíková et al., 1979; Maes et al., 1995; Munkholm et al., 2013; Müller, Myint, & Schwarz, 2012).
In order to examine if the NLRP3-inflammsome contributes to inflammation in BD, we measured
levels of cytokines that are activated downstream of the NLRP3-inflammasome in our samples.
Caspase-1 cleaves and activates two cytokines, IL-1β and IL-18, both of which are released outside
of the cells to activate different signaling pathways (Sollberger et al., 2014). We examined IL-1β,
IL-6, and TNF-α for the IL-1β pathway. For cytokines activated downstream of IL-18, also known
as IFN-γ inducing factor, we examined IFN-γ and IL-10 (Gracie, Robertson, & McInnes, 2003; A.
Weber et al., 2010). Our results indicated that IL-1β, IL-6, TNF-α and IL-10 are increased in patients
with BD and SCZ, suggesting an activation of the IL-1β pathway in these individuals, but not the IL-
18 pathway, since IFN-γ is a direct product of IL-18 activation. A previous study demonstrated that
the IL-18 pathway is unaffected by the NLRP3-inflammasome in microglial cells (Hanamsagar,
Torres, & Kielian, 2011), which are the primary immune cells of the brain (Dheen, Kaur, & Ling,
2007). This may be why we did not see group differences for IFN-γ despite evidence supporting

50
activation of the NLRP3-inflammasome in the BD group. It is also worth noting that while evidence
supporting activation of the NLRP3-inflammasome was stronger for patients with BD than SCZ,
higher levels of cytokines were found in both BD and SCZ. This is in agreement with previous
studies demonstrating inflammation in the brain and periphery of patients with BD or SCZ (B. Dean
et al., 2013; Kauer-Sant'Anna et al., 2009; Leboyer et al., 2012; Müller et al., 2012; Rao et al., 2010).
Indeed, altered levels of cytokines in the IL-2, TNF-α, IL-6 and IL-1 pathways are consistently
reported in the central nervous system and periphery of patients with BD or SCZ (Barbosa et al.,
2011; Bresee & Rapaport, 2009; Fineberg & Ellman, 2013; B. J. Miller, Buckley, Seabolt, Mellor, &
Kirkpatrick, 2011; Munkholm et al., 2013; Müller et al., 2012; Rao et al., 2010; Söderlund et al.,
2011). Similar findings in cytokine alterations in BD and SCZ may be due to symptoms that exist in
both of these disorders, such as psychosis (B. J. Miller et al., 2011). Moreover, it is important to note
that expression of cytokines is influenced by many different factors in addition to the inflammasome,
such as apoptosis, neurotransmitter imbalance (Berk et al., 2013; Dheen et al., 2007; Felger et al.,
2013; Felger & Miller, 2012) and other inflammatory pathways such as those involving
phospholipase A2 (Weerasinghe, Rapoport, & Bosetti, 2004), all of which have been implicated in
both BD and SCZ (Berk et al., 2007; Jarskog, 2006; H. W. Kim, Rapoport, & Rao, 2011; Rao et al.,
2012). Hence, while activation of the NLRP3-inflammasome may be contributing to the activation
of theses cytokines in patients with BD, the aforementioned factors could be playing a much larger
role in the activation of cytokines in patients with SCZ. These studies, in addition to our findings,
demonstrate the complex involvement of the inflammatory system in both of these conditions,
indicating that extensive future studies are required to further elucidate this interaction.
Limitations of this study include small sample size, which limits generalizability. Repeating
these experiments in a larger sample will be useful in validating the findings reported in this study.
Also, pre and postmortem factors such as agonal states and storage conditions may have influenced

51
the results (Chandana, Mythri, Mahadevan, Shankar, & Srinivas Bharath, 2009; Vawter et al., 2006).
To verify the effect of such factors, we correlated our findings with PMI, sex and age. Also, we did
not directly measure the activity of the NLRP3-inflammasome. While it has been demonstrated in
vitro that components of the NLRP3-inflammasome associate with the mitochondria and can be
found in the mitochondrial fraction when the inflammasome is activated (Zhou et al., 2011), it
should be noted that immunocontent does not always represent activation. In addition, as patients
may have been taking medications that could have influenced the results, it would be important to
verify these findings in a large sample with medication information available to examine their effect
on complex I and the NLRP3-inflamamsome. Moreover, we only examined the frontal cortex.
Examination of other areas implicated in psychiatric disorders may yield interesting results.
While chronic inflammation of the brain has been consistently demonstrated in BD, its
causes remain largely unknown. (Leboyer et al., 2012). This study is the first to demonstrate
activation of the NLRP3-inflammasome in the frontal cortex of patients with BD, implicating
inflammation as a downstream target of complex I dysfunction in BD and linking two of the most
commonly reported alterations in this disorder - mitochondrial abnormalities and inflammation
(Andreazza et al., 2010; Berk et al., 2011; Clay et al., 2010). Future studies are required to further
elucidate this pathway, and the extent of its contribution to inflammation in brain tissue in patients
with BD. For example, since BD is conceptualized as progressive (Berk et al., 2011), repeating
these experiments in a larger sample that allows for examination of the extent of NLRP3-
inflammasome activation in relation to age or number of episodes may yield interesting findings.
The effects of past or current treatment will also be of great interest. Furthermore, given that
identification of biomarkers of BD is of great interest, measuring the NLRP3-inflammasome in
peripheral samples to identify its potential to be used as a biomarker may be beneficial. Lastly,

52
examination of the specific molecules that are involved in the complex I-NLRP3-inflammasome
pathway may contribute to a better understanding of the pathophysiology of BD.

53
Table 1. Subject information
Control (N=9) BD (N=9) SCZ (N=10)
Age: mean (range) 79.0 (65-91) 76.1 (58-92) 76.8 (55-91)
PMI: mean (range); hr 21.4 (13.0-30.1) 20.9 (10.0-32.9) 19.4 (8.0-43.5)
Male 4 4 4
Female 5 5 6

54
Figures
Figure 1. Levels of electron transport chain components complex I, II, III, IV and V, and
nicotinamide nucleotide transhydrogenase (NNT) in the post-mortem frontal cortex of patients with
BD, SCZ and non-psychiatric controls (CTL). Results for complexes I-V and NNT are expressed in
median fluorescence intensity (MFI). Inset illustrates levels of NDUFS7 measured by western
blotting and the blot for NDUFS7. NDUFS7 was quantified using β-actin as loading control. This
image was cropped to improve clarity. Differences between groups were analyzed using one-way
ANOVA followed by Least Significant Difference test (*p < 0.05).

55
Figure 2. Levels of NLRP3-inflammasome components NLRP3 (A), ASC (B) and caspase 1 (C) in
the brain homogenate (homogenate), cytoplasmic and crude mitochondrial fractions (mitochondrial)
in the post-mortem frontal cortex of patients with BD, SCZ and non-psychiatric controls (CTL).
Insets in panel A illustrate blots for NLRP3. The images were cropped to improve clarity. Arrows
indicate the NLRP3 band at 110kDa, which was quantified using β-actin as a loading control for the
brain homogenate and cytoplasmic fraction, and total protein (memcode) as loading control for the
crude mitochondrial fraction. Loading controls are shown in red squares below the blots with arrows.
Differences between groups were analyzed using one-way ANOVA followed by Least Significant
Difference test (*p < 0.05; **p < 0.01).

56
Figure 3. Levels of inflammatory factors downstream of NLRP3-inflammasome activation in the
IL-1β pathway, IL-1β, IL-6, & TNF-α, and in the IL-18 pathway, IFNγ & IL-10 in the post-mortem
frontal cortex of patients with BD, schizophrenia (SCZ) and non-psychiatric controls (CTL).
Differences between groups were analyzed using one-way ANOVA followed by Least Significant
Difference test (*p < 0.05; ** p < 0.01).

57
Figure 4. Correlation between NLRP3 inflammasome components in the brain homogenate (whole),
cytoplasmic fraction (cytoplasm) and the crude mitochondrial fraction (mitochondria) against
complex I levels in the post-mortem frontal cortex of patients with bipolar disorder.

58
Statement of significance and impact:
Mitochondrial complex I dysfunction and subsequent production of ROS can have important
consequences when ROS act as signaling molecules to activate downstream pathways through redox
sensors. This study first addressed Aim 2, which was to confirm complex I dysfunction in the post-
mortem brain of patients with BD. By examining levels of all components of the electron transport
chain, this study was able to show that only complex I had between-group differences, where
patients with BD had lower levels of complex I than non-psychiatric controls, suggesting that
complex I dysfunction is specific to BD. Also, this study confirmed the findings of 2 different
studies from our group demonstrating lower levels of NDUFS7, a complex I subunit that is critical
for the efficient transfer of electrons to ubiquinone (Halliwell, 2001). This study also addressed Aim
3, which was to examine the mitochondria as a target of complex I dysfunction by measuring the
activation of the NLRP3-inflammasome, a marker of mitochondrial oxidative stress (Zhou et al.,
2011). Indeed, NLRP3 is activated upon release of mitochondrial ROS from complex I inhibition,
making it a suitable marker for oxidative stress in the mitochondria. To measure the activation of the
NLRP3-inflammasome and not just the immunocontent, I measured the levels of NLRP3, ASC, and
caspase-1 in the crude mitochondrial fraction as well as in the brain homogenate, as the components
of the NLRP3-inflamamsome only associate with the mitochondria when the inflamamsome is
activated. The findings of this study suggested activation of the NLRP3-inflammasome in the frontal
cortex of patients with BD. Since activation of the NLRP3-inflammasome complex is strongly
influenced by mitochondrial production of ROS, and occur as a direct result of complex I inhibition
(Zhou et al., 2011), these results demonstrate that complex I dysfunction may be contributing at least
in part to the activation of the inflammatory system in patients with BD. It is also interesting to note
that inflammatory factors that are less specific to the NLRP3-inflamamsome, such as caspase-1 and
inflammatory cytokines, were found to be increased in both BD and schizophrenia, although

59
evidence supporting NLRP3-inflammasome activation was stronger for BD. These findings suggest
that mitochondria may be a downstream target of complex I dysfunction specifically in BD, as
shown in the activation of the NLRP3-inflammasome, while inflammation may be less specific to
BD.
The next experiment aimed to study the synapse as a target of complex I dysfunction, which
was shown to be a target of oxidative stress in the same study demonstrating oxidative damage in the
mitochondria (Andreazza et al., 2013). We focused on the dopaminergic synapse, as increased
dopamine signaling is known to underlie mania, the defining feature of BD (Berk et al., 2007; H. K.
Kim & Andreazza, 2012; Post, Jimerson, Bunney, & Goodwin, 1980). Hence, the next study
examined the dopamine system as a target of complex I dysfunction and oxidative stress by
measuring oxidation and nitration of two dopaminergic proteins that were shown to be targets of
oxidative modification in in vitro and animal studies, the dopamine transporter and the tyrosine
hydroxylase (Blanchard-Fillion et al., 2001; S. U. Park et al., 2002).

60
CHAPTER 4
Title: Oxidation and nitration in dopaminergic areas of the prefrontal cortex from patients with
bipolar disorder and schizophrenia
Authors: Helena Kyunghee Kim, Ana Cristina Andreazza, Pok Yik Yeung, Cameron Isaacs-
Trepanier, L. Trevor Young
Journal: Journal of Psychiatry & Neuroscience
Volume: 39
Pages: 276-285
Work performed by the student:
The student performed all work included in this chapter, including planning and performing the
experiments, image acquisition, data analysis, and preparation and submission of the manuscript.

61
Oxidation and nitration in dopaminergic areas of the prefrontal cortex from patients with
bipolar disorder and schizophrenia
Helena Kyunghee Kim BSc1, Ana Cristina Andreazza Pharm, PhD1,2, Pok Yik Yeung BSc1,
Cameron Isaacs-Trepanier2, L. Trevor Young MD, PhD1,2
1Departments of Psychiatry and Pharmacology, University of Toronto, Canada
2Campbell Family Mental Health Research Institute, Centre for Addiction and Mental Health
Corresponding author: L. Trevor Young
Center for Addiction and Mental Health, Clarke Site
250 College Street, Rm. 835
Toronto, ON
M5T 1R8
Telephone: 416-979-6948
FAX: 416-979-6928
Email: [email protected]

62
Abstract
Background: Increased oxidative stress is strongly implicated in bipolar disorder, where protein
oxidation, lipid peroxidation, and oxidative damage to DNA have been consistently reported. High
levels of dopamine in mania are also well-recognized in bipolar disorder, and dopamine produces
reactive oxygen species and electron-deficient quinones that can oxidize proteins when it is
metabolized.
Methods: Using immunohistochemistry and acceptor photobleaching FRET, we examined oxidation
and nitration of areas immunoreactive for the dopamine transporter and tyrosine hydroxylase in the
postmortem prefrontal cortex from patients with bipolar disorder, schizophrenia, major depression,
and non-psychiatric controls.
Results: We found increased oxidation of dopamine transporter-immunoreactive regions in patients
with bipolar disorder (F3,50=7.23, P<0.01; P<0.01), while decreased nitration of tyrosine
hydroxylase-immunoreactive regions was found in both patients with bipolar disorder (F3,46 = 3.59,
P<0.05; P<0.01) and schizophrenia (P<0.05) . On the other hand, global levels of oxidation were
found to be increased in patients with bipolar disorder (F3,46=5.91, P<0.01; P<0.01) and
schizophrenia (P<0.05), although nitration levels did not differ between groups (F3,46 = 1.75;
P=0.17).
Limitations: Limitations of this study include the use of postmortem brain sections, which may be
affected by factors such as postmortem interval and antemortem agonal states, although
demographic factors and postmortem interval were accounted for in statistical analysis.
Conclusion: These findings suggest alterations in levels of protein oxidation and nitration in
dopamine-rich regions of the prefrontal cortex in patients with bipolar disorder and schizophrenia,
but more markedly in bipolar disorder.

63
Introduction
Bipolar disorder (BD) is characterized by recurrent episodes of mania/hypomania and
depression, affects 1.5% of the population, and is associated with high morbidity and mortality
(Kupfer, 2005). The pathophysiology of BD is linked to a number of factors, including
neurotransmitter imbalance, oxidative stress and genetic causes among others (Salvadore et al.,
2010). Increased oxidative stress, which could result in oxidative and nitrosative damage to
biomolecules (Halliwell, 1992), is a consistent finding in BD. For example, mitochondrial
dysfunction and decreased expression of genes of the electron transport chain, particularly that of
complex I, are reported in patients with BD (Andreazza et al., 2010; Scola et al., 2013). Decreased
efficiency of the electron transport chain could result in increased production of reactive oxygen
species (ROS) (Halliwell, 1992). Moreover, increased levels of carbonyl groups, 3-nitrotyrosine and
decreased levels of antioxidants, such as glutathione, are all reported in BD (Andreazza et al., 2009;
Andreazza et al., 2008; Gawryluk et al., 2011).
It is widely held that dysregulation of the dopamine (DA) system is important in this disorder,
where high levels of DA is thought to underlie mania, which is a defining feature of BD (Berk et al.,
2007). Indeed, increasing synaptic DA levels with amphetamine and L-DOPA produces mania-like
behaviour (Berk et al., 2007), and antipsychotics, which act in part by blocking DA receptors, are
among the most effective treatments for acute mania (Cipriani et al., 2011). High levels of DA can
be cytotoxic in part through the generation of ROS and electron-deficient quinones during oxidation
of DA (Hastings et al., 1996). In fact, amphetamine increases markers of oxidative stress such as
lipid peroxidation and protein oxidation in animals (Frey et al., 2006; Valvassori et al., 2010).
Therefore, in this study, we examined oxidative and nitrosative damage in dopamine-rich
regions of the prefrontal cortex (PFC) using acceptor photobleaching forster resonance energy

64
transfer (apFRET). We examined the PFC, as it was previously shown to have increased oxidative
stress in patients with BD (Andreazza et al., 2010). In order to examine dopamine-rich areas, we
labelled the dopamine transporter (DAT), which is a transmembrane protein responsible for the
uptake of synaptic DA into the presynaptic terminal (Akil et al., 1999; Ciliax et al., 1999), and
tyrosine hydroxylase (TH), which converts L-tyrosine to L-DOPA, and is the rate-limiting enzyme
in DA synthesis. Previous studies used TH and DAT labeling with immunohistochemistry
techniques to visualize dopaminergic axons in the PFC, and reported extensive co-localization
between TH and DAT immunoreactive axons (Akil et al., 1999; Ciliax et al., 1999). Here, we report
evidence of oxidative and nitrosative damage in DA-rich regions of the post-mortem PFC of patients
with BD and schizophrenia (SCZ).
Methods
Post-mortem brain samples
Frozen post-mortem PFC (BA9/46) sections (14µm) were from the Stanley Foundation
Neuropathology Consortium (Table 1). The details of these samples have been published elsewhere
(Torrey, Webster, Knable, Johnston, & Yolken, 2000). Briefly, subject groups consisted of patients
with BD, major depressive disorder (MDD), SCZ, and non-psychiatric controls. There were no
differences for sex (F3,55 = 0.11, p = 1.00), age (F3,55 = 0.53, p = 0.67), post-mortem interval (F3,55 =
1.70, p = 0.18; PMI) and pH (F3,55 = 0.63, p = 0.60) between groups (N=15 per group). Diagnoses
were established using DSM-IV criteria. Sections with good structural integrity were used. Samples
were randomly coded numerically, and the experimenters were kept blind to the codes until all the
experiments were completed.
Immunohistochemistry

65
Sections were nissl stained and labelled for NeuN to examine tissue quality prior to
immunohistochemistry experiments (methods in supplementary information; Figure 1A&B). Four
adjacent sections per subject were processed for immunohistochemistry, where 2 sections were
labelled for DAT and free thiols, and the other 2 were labelled for TH and 3NT. FRET analysis and
intensity analysis were performed simultaneously on the same sections. 7-Diethylamino-3-
(4’maleimidylphenyl)-4-methylcoumarin (CPM; Invitrogen, Burlington, ON; D346) is a specific
label for free thiols (Mastroberardino, Orr, Hu, Na, & Greenamyre, 2008). CPM labeling was
performed using a previously published method by Mastroberardino et al. (Mastroberardino et al.,
2008) with slight modifications. We used CPM to directly label free thiols without prior
modification of the tissue to minimize alterations to the sections and introduction of modifications
from the experimental procedure. The indirect method by Mastroberardino et al., in which free thiols
are blocked and disulfide linkages are reduced to be labelled with CPM, was used to increase
sensitivity (Mastroberardino et al., 2008). For our samples, direct labeling method was sufficient to
detect between-individual differences. In the methods described here, lower CPM labeling is
indicative of greater oxidation of thiols. Sections fixed with 4% paraformaldehyde (10min) were
incubated in 0.5mM CPM in Tris-HCl (0.1M pH 6.8) for 1h. Sections were washed extensively and
immunohistochemistry (IHC) was carried out using previously published techniques (Waldvogel,
Curtis, Baer, Rees, & Faull, 2006). Sections were blocked in 4% goat serum for 1h, and incubated
with the DAT antibody (Millipore, Temacula, CA; MAB369; 1:250; 60h at 4°C) in 0.05% Triton-X.
The specificity of this antibody was demonstrated in previous studies (G. W. Miller et al., 1997;
Wills et al., 2010). Sections were then washed, incubated with Alexa Fluor®488 anti-rat (Molecular
Probes, Burlington, ON, Canada A11006) in 0.05% Triton-X for 3h, and mounted in Fluoromount™
(Cedarlane, Burlington, ON, Canada). To create a negative control for CPM labeling,
paraformaldehyde-fixed sections were alkylated with 100mM N-ethylmaleimide (NEM; Bioshop,

66
Burlington, ON; ETM222) and 100mM iodoacetamide (IAA; Sigma-Aldrich, Oakville, ON; I6125)
in Tris-0.1 M, pH 6.8 (15min) prior to CPM labeling. Oxidation of thiol groups with NEM and IAA
diminished CPM labeling, demonstrating specificity of CPM for free thiols. Hence, decreased CPM
labeling is indicative of greater thiol oxidation. These sections were used as controls to account for
non-specific labeling of CPM (Mastroberardino et al., 2008). For 3-nitrotyrosine and TH double
labeling, acetone-fixed sections were blocked with 10% goat serum in PBS-0.3% Triton X-100
(PBS-T) for 30min. Sections were then incubated with primary antibodies in PBS-T (48h, 4˚C): TH
(Millipore; AB152; 1:250) and 3NT (abcam, Cambridge, MA, USA; ab61392; 1:200). Sections were
washed, incubated with secondary antibodies (2h, RT) in PBS-T (1:300): Alexa Fluor®488 anti-
rabbit IgG (Molecular probes®; A11008) and Alexa Fluor®350 anti-mouse IgG (Molecular probes®;
A11045), and mounted in FluoromountTM (Cedarlane). Specificity of the anti-TH antibody was
demonstrated elsewhere (O'Connell, Matthews, Ryan, & Hofmann, 2010). The anti-3NT antibody
was previously used by our laboratory (Andreazza et al., 2009). To test the specificity of the anti-
3NT antibody, sections were treated with peroxynitrite (Millipore, 20-107; 1:100), which is used as a
positive control for 3NT labeling, or degraded peroxynitrite (Millipore, 20-247; 1:100) using a
previously published method (P. Zhang, Wang, Kagan, & Bonner, 2000). An increase in 3NT
labeling was observed only with peroxynitrite, demonstrating the specificity of the anti-3NT
antibody (Figure S1). To minimize the risk of antibody cross-reactivity, different combinations of
primary and secondary antibodies were tested, where only minor and non-specific binding was found,
indicating minimal cross-reactivity (Figure 1C&D).
Intensity Analysis
Intensity analysis was performed using a previously published method from our group (Che et al.,
2010). 60X water immersion objective and a confocal laser scanning microscope (CLSM; Olympus

67
Fluoview FV1000; Olympus America INC, Melville, NY, USA) were used to capture images from 5
fields of each section, which were chosen by dividing the section into 5 equally sized rectangles, and
imaging the middle region of each of the 5 rectangles. For CPM and DAT labelled sections, CPM
was excited with the 405nm laser (15% intensity) and Alexa488 was excited with the 488nm laser
(18% intensity). For 3NT and TH labelled sections, Alexa488 was excited with a 488nm laser (15%
laser intensity) and Alexa350 was excited with a 405nm laser (5% intensity). Detection wavelength
for Alexa488 was 500-600nm, and for Alexa350/CPM was 425-475nm. Confocal aperture diameter
was 110μm, and line-by-line sequential detection mode was used. Scanning parameters were kept
consistent for all experiments. Image analysis was performed using the FV10 – ASW 2.1 software
(Olympus). For analyzing DAT, TH, and 3NT intensity, first, background was subtracted from the
image by selecting 5 regions per field without specific immunoreactivity as background and
measuring the average intensity of the regions. This value was subtracted from the overall intensity
of the field. Then the average intensity of the 5 fields was calculated to represent the intensity for the
section. Therefore, DAT/TH/3NT intensity = (Average intensity of 5 fields from the section) –
(Average background intensity of 5 fields from the section). CPM intensity was determined using
the IAA and NEM-treated sections (negative control) to correct for non-specific labeling
(Mastroberardino et al., 2008) which had significantly decreased CPM labeling, demonstrating its
specificity for free thiols. Therefore, CPM intensity = (Average intensity of 5 fields from the section)
– (Average intensity of 5 fields from negative control).
Forster Resonance Energy Transfer (FRET)
Acceptor photobleaching FRET measures non-radiative energy transfer from a donor fluorophore to
an acceptor fluorophore by observing the fluorescence of the donor in the presence and absence
(photobleaching) of the acceptor (König et al., 2006). apFRET has been successfully used in

68
previous studies with human samples (Hynd, Lewohl, Scott, & Dodd, 2003; Keese et al., 2005;
Sharma et al., 2001). The energy transfer between a donor and an acceptor fluorophore only occurs
when the donor and the acceptor are within ~30-50nm of each other using indirect
immunohistochemistry, which allows us to use this technique to detect the proximity of the
molecules (König et al., 2006). For DAT and CPM FRET, Alexa488 (DAT) was the acceptor
fluorophore, and CPM was the donor fluorophore. For 3NT and TH FRET, Alexa488 (TH) was the
acceptor fluorophore, and Alexa350 (3NT) was the donor fluorophore. Acceptor photobleaching
FRET was performed with a CLSM (Olympus) using previously published techniques with small
modifications (König et al., 2006; Mastroberardino et al., 2008). 60X water immersion objective was
used. Image acquisition parameters were identical to those used for intensity analysis. Intensity of
the donor fluorophore before and after the photobleaching of the acceptor fluorophore was measured.
Photobleaching of the acceptor fluorophore was at 100% laser intensity (10μs/pixel) using the
488nm laser in a single bleaching step (1.5s for TH&3NT, 60ms for DAT&CPM). Scanning
parameters were kept consistent during all experiments. Change in fluorescence was represented as
increase in fluorescence (ΔIF), where ΔIF = intensity of donor after photobleaching – intensity of
donor before photobleaching. To account for non-specific binding of CPM, negative controls were
performed where sections were treated with IAA and NEM prior to CPM staining and
immunohistochemistry as detailed above. Then, ΔIF of the negative controls was subtracted from the
ΔIF determined for the samples to calculate the final ΔIF (Mastroberardino et al., 2008). ΔIF
between CPM and the secondary antibody (Alexa488) without the primary antibody against DAT
was undetectable. To test the effect of antibody cross-reactivity on FRET measurements for 3NT and
TH FRET, sections were labelled with only the primary antibody for TH and both secondary
antibodies, then FRET was measured. A very low level of FRET was detected (Average ΔIF = 12.5),
indicating negligible cross-reactivity. Five regions of interest (ROI) per section were chosen as

69
described in intensity analysis and examined. Therefore, ΔIF = (Average ΔIF of 5 ROIs in section) –
(Average ΔIF of 5 ROIs in negative control). We used ΔIF to measure energy transfer, which was
demonstrated to be more appropriate with tissue sections than FRET efficiency due to its lower
sensitivity to non-specific binding of fluorophores (König et al., 2006). For DAT&CPM FRET,
lower ∆IF indicates lower levels of free thiols in DAT-immunoreactive (IR) regions, and thus likely
increased oxidative stress status in DAT-IR regions. For TH&3NT FRET, higher ∆IF indicates
greater 3NT levels in TH-immunoreactive regions. All calculations were performed using FV10 –
ASW 2.1.
Mouse brain study for the effect of post-mortem interval on oxidation of neurons
Adult male C57/BL6 mice (8-12 weeks) were used to examine the effect of PMI on thiol oxidation
and 3-nitrotyrosine formation in neurons. Use of animals was approved by the University Animal
Care Committee of the University of Toronto (Protocol number: 20009477), and care of animals was
performed according to animal research guidelines of the University of Toronto. Animals were
euthanized by asphyxiation and kept at 4°C until an appropriate PMI was reached (6, 24, 48, 72h) to
mimic the storage of cadavers (Hynd et al., 2003) The brain was then extracted, sagittally bisected,
flash frozen in isopentane and dry ice, and stored at -80°C (Torrey et al., 2000). Each bisected brain
was processed and analyzed independently. 14µm sagittal sections were made using a cryostat, and
using standard immunohistochemistry techniques, acetone and methanol fixed sections were labeled
with NeuN (Millipore, Temacula, CA; MAB377B) and CPM, or 3NT (abcam, Cambridge, MA;
ab61392) and NeuN. The secondary antibodies were streptavidin-Alexa Fluor®568 (Molecular
probes®, Burlington, ON; S11226) and Alexa Fluor®350 anti-mouse IgG (Molecular probes®;
A11045). Controls for CPM and 3NT labeling done as described above. Sections were then used to
obtain images of the frontal cortex with a CLSM (Olympus) using a 60X water immersion objective.

70
CPM and Alexa350 were excited with the 405nm laser (32% intensity) and Alexa568 was excited
with the 543nm laser (38% intensity). Detection wavelength for Alexa568 was 555-655nm, and for
CPM and Alexa350 was 425-475nm. The images were analyzed using co-localization analysis
(Pearson’s correlation coefficient) between NeuN and CPM (free thiols in neurons) or between
NeuN and 3NT (protein nitration in neurons) with FV10-ASW2.1.
Statistical Analysis
Statistical analysis was performed using SPSS version 20. Normal distribution of data was
determined using Kolmogorov-Smirnov test. As the data were normally distributed, parametric tests
were used for further analysis. Effect of covariates (age, gender, PMI, and pH) was assessed using
Pearson’s correlation test. As age was the only variable that significantly correlated with one of our
dependent variables (DAT intensity; Pearson r = 0.34, P<0.05), ANCOVA model was used with age
as a covariate, and Dunnett’s post-hoc test were used to examine the effect of diagnosis. Data are
presented as mean±standard error of the mean (SEM).
Results
Subject characteristics
Subjects consisted of patients with BD (n=15), SCZ (n=15), and MDD (n=15), and non-psychiatric
controls (n=15). Demographic details can be found in Table 1. Age, post-mortem interval and brain
pH did not significantly correlate with CPM intensity, 3NT immunoreactivity, TH immunoreactivity,
∆IF between DAT&CPM, and ∆IF between 3NT&TH (Figure 4). DAT immunoreactivity did not
correlate with PMI or pH, but positively correlated with age (Pearson r = 0.34, P<0.05).
Furthermore, psychiatric patients with a history of substance abuse had lower DAT intensity
(F2,46=6.74; P<0.01) compared to healthy controls (P<0.01). We nissl stained the sections to examine

71
their quality. Sections were found to have good histological quality without large holes or tears
(Figure 1A). Also, the average glia to neuron ratio (1.37) was comparable to what was published
before in previous studies examining the same area (Selemon, Rajkowska, & Goldman-Rakic, 1998,
2004).
DAT and TH Immunoreactivity
We examined DAT immunoreactivity and TH immunoreactivity in the sections by measuring
fluorescence intensity. The groups did not differ from each other for DAT immunoreactivity
(F3,44=1.48, P=0.23; Figure 1C). TH immunoreactivity (F3,46 = 0.86, P=0.47; Figure 1D) also did not
differ between groups.
CPM Intensity and 3-nitrotyrosne (3NT) immunoreactivity
Intensity of CPM was quantified to assess the amount of free thiols in the sections. There was a
significant difference between groups (F3,44=6.74, P<0.01; Figure 2A), where patients with BD
(P<0.01) and SCZ (P<0.05) had lower CPM intensity in the PFC compared to the control group.
Age, gender, PMI, and pH did not correlate with CPM or 3NT intensity. To explore the potential
effect of psychotropic drugs, we compared psychiatric patients who were prescribed lithium,
antipsychotics, or antidepressants at the time of death with patients who were not prescribed drugs
and also with healthy controls. Patients who were prescribed antipsychotics at the time of death were
found to have lower CPM intensity (F2,44=7.69, P<0.01) compared to healthy controls (P<0.01). On
the other hand, patients who were not prescribed antidepressants had lower CPM intensity
(F2,44=8.31, P<0.01) compared to controls (P<0.01). There were no other effects of drug treatment on
either CPM intensity or 3NT immunoreactivity. 3NT immunoreactivity was also assessed by
measuring fluorescence intensity. In contrast to CPM intensity, 3NT immunoreactivity did not differ

72
between groups (F3,45 = 1.80, P=0.16; Figure 2B). Because protein stability may be affected by
longer post-mortem intervals, we decided to examine the effect of longer PMIs on the frontal cortex
using mouse brain. We chose to examine neurons as they have lower antioxidant capacity than glial
cells 3. CPM (F3,12=1.30, P=0.32) and 3NT labeling (F3,12=0.82, P=0.82) in neurons did not change
with increasing PMI up to 72h.
Acceptor photobleaching FRET analysis
Initially, our aim was to examine oxidation and nitration in both TH and DAT immunoreactive
regions using FRET analysis. However, FRET between CPM and Alexa488 labeling TH was not
detectable in our sections. Moreover, FRET between Alexa350 labeling 3NT and Alexa488 labeling
DAT was not detectable. Therefore, we examined free thiols in DAT-immunoreative regions
(DAT&CPM FRET), and nitration of TH-immunoreactive regions (TH&3NT FRET). There was a
significant difference between groups for DAT&CPM FRET (F3,48=6.76, P<0.01), where patients
with BD (P<0.01) were found to have lower energy transfer between CPM and DAT(Alexa488) than
the control group (Figure 3A). Also, subject groups were found to significantly differ from each
other for TH&3NT FRET (F3,45 = 3.10, P<0.05; Figure 3B), where patients with BD (P<0.01) and
SCZ (P<0.05) were found to have lower levels of nitration than the control group. Similarly, to
explore the potential effect of psychotropic drugs, we compared patients who were prescribed
antipsychotics, antidepressants, or lithium with those who were not prescribed the drugs and also
with healthy controls. The only significant effect was that patients who were not prescribed
antidepressants had lower TH&3NT FRET (F2,42=3.26; P<0.05) than healthy controls (P<0.05),
although this effect was not found comparing those who were prescribed antidepressants.
Discussion

73
In this study, we examined the relationship between oxidative stress and dopamine in BD by
measuring oxidative and nitrosative damage in DA-rich areas of the PFC. Using FRET, we found
increased oxidation of DAT-immunoreactive areas in patients with BD, while nitration of TH-
immunoreactive areas was found to be decreased in patients with BD and SCZ. When we examined
global levels of oxidative and nitrosative damage, however, we did not find between-group
differences in levels of nitration, but found thiol oxidation to be increased in both patients with BD
and SCZ. These results suggest alterations in oxidation and nitration of DA-rich areas in the PFC
that are particularly marked in BD, but also marked in SCZ.
When oxidation and nitration of DAT and TH immunoreactive (IR) regions were examined
using FRET, we found increased oxidation in DAT-IR areas in patients with BD while nitration in
TH-IR areas was found to be decreased in both patients with BD and SCZ. These results suggest
oxidative and nitrosative modifications to DA-rich areas in both BD and SCZ, which could be
contributing to the dysregulation of the DA system hypothesized to occur in both of these disorders.
More specifically, hyperactivity of the DA system in mania is well-recognized, where
antipsychotics, which act in part by blocking DA signaling, is one of the most effective treatments
for acute mania (Cipriani et al., 2011), and dopaminergic agonists were found to produce mania-like
behavior in humans and in animals (Berk et al., 2007; H. K. Kim & Andreazza, 2012). The DA
hypothesis for SCZ is more complex, however, as the direction of DA dysregulation may differ
depending on symptoms (positive vs. negative) and brain region (cortical vs. subcortical) (Howes &
Kapur, 2009). However, due to the limitations of using postmortem brain sections, future studies
examining the consequences of these alterations to DA signaling and behavior are required.
Furthermore, it is interesting to note that patients with BD and SCZ were found to have similar
alterations in our study, which is consistent with previous studies showing extensive overlap

74
between these two disorders including cognitive deficits (Schretlen et al., 2007), genetic alterations
(Potash & Bienvenu, 2009), changes in brain morphology (Maier, Zobel, & Wagner, 2006), and
oxidative stress (Ng et al., 2008). Our results also showed that patients who are not prescribed
antidepressants have lower TH&3NT FRET than healthy controls. This effect is difficult to interpret.
While antidepressants have been shown to have neuroprotective effects through other mechanisms
than oxidative stress including regulation of neurotrophin levels (Saarelainen et al., 2003), it is not
known whether the reduction in oxidative stress in patients treated with antidepressants reflect their
neuroprotective effects. A larger issue relates to the difficulty in disentangling the effects of
medication and diagnosis since antidepressants in most cases were prescribed to patients with
depression but not to those with BD or SCZ.
Acceptor photobleaching FRET has a resolution of ~30-50nm, and the effectiveness of FRET
increases as the distance between the fluorophores decreases (König et al., 2006), suggesting that
oxidation and nitration in DAT and TH immunoreactive areas as measured by FRET may in large
part include direct oxidation of the DAT, and nitration of the TH. Indeed, previous studies have
demonstrated that DAT is a target of thiol oxidation but not nitration (S. U. Park et al., 2002), which
may have contributed to why we could not measure FRET between the fluorophores labeling DAT
and 3NT (ΔIF undetectable). As DAT oxidation decreases its ability to uptake DA (H. K. Kim &
Andreazza, 2012; S. U. Park et al., 2002), increased DAT oxidation found in patients with BD may
contribute to increased levels of synaptic DA. Whether TH is primarily a target of nitration or
cysteine oxidation is controversial, with some studies showing evidence for both (S. Park, Geddes,
Javitch, & Kuhn, 2003) and other studies reporting TH nitration and oxidation only when the protein
is unfolded (Ara et al., 1998; Blanchard-Fillion et al., 2001). In our study, we could not detect FRET
between CPM labeling free thiols and TH. Since it is physiologically unlikely that all the cysteine
residues of TH were in the oxidized state, we believe that CPM may not have been able to access the

75
thiol groups of the TH protein as they are highly shielded from the surrounding environment
(Daubner et al., 2011). Decrease in 3NT levels in TH-IR regions was an unexpected finding, as
increased oxidative stress is strongly implicated in BD (Andreazza et al., 2009). This finding may be
specific to the TH protein or to L-DOPA-rich areas, since DA, L-DOPA and DOPAC are favorable
substrates for reactive nitrogen species, and may protect proteins in their proximity that are less
favorable substrates for nitration (S. U. Park et al., 2002). Future studies are required to elucidate
this further. Also, because TH nitration was shown to decrease its ability to produce L-DOPA
(Blanchard-Fillion et al., 2001; S. U. Park et al., 2002), decreased nitration of TH in BD and SCZ
suggest that nitration of TH may be present in normal conditions to limit the activity of this enzyme.
Consequently, decreased nitration of TH in BD and SCZ could act to ‘disinhibit’ TH and result in
higher levels of presynaptic DA. Also, while TH and DAT immunoreactive fibers co-localize in the
human prefrontal cortex (Ciliax et al., 1999), TH is also present in noradrenergic neurons, suggesting
that decreased nitration in TH-IR regions may be occurring in noradrenergic cells as well.
Interestingly, dysregulation of noradrenergic signaling has also been shown in BD (Manji & Lenox,
2000), suggesting a potential role for decreased TH nitration in the noradrenergic system.
Furthermore, while the focus of this study was the DA system, such changes could also occur in
other neuronal populations. Future studies examining these alterations in different neuronal cells
may increase our understanding of the pathophysiology of BD and SCZ.
Alterations in the prefrontal cortex (PFC) is consistently implicated in BD and other
psychiatric disorders, where studies have reported decreased metabolism, morphological changes
(Almeida et al., 2009), and increased oxidative damage (Andreazza et al., 2010). Moreover, the PFC
has a high relative density of dopaminergic axons compared to other cortical regions (Ciliax et al.,
1999), and DA is involved in the regulation of working memory by the PFC (D'Ardenne et al.,
2012), which is impaired in patients with BD and SCZ (Glahn et al., 2006; Thompson et al., 2007).

76
These studies suggest that dysregulation of the DA system in the PFC may be involved in the
pathophysiology of these disorders. In agreement with previous studies, we found that TH and DAT
labeling reveals fibers, which may be dopaminergic axons. We also saw punctate structures that
were occasionally connected by fibers, which may be axonal varicosities and terminals (Akil et al.,
1999; Ciliax et al., 1999) (Figure 1C&D). DAT and TH immunoreactivity examined by measuring
fluorescence intensity did not differ between groups, which is in agreement with previous studies
examining the same samples as those used in our study (data can be found in the Stanley
Neuropathology Consortium Integrative Database: http://sncid.stanleyresearch.org). We also found
that DAT intensity was lower in patients with a history of substance abuse compared to healthy
controls. Although we do not have details on the substances that were used by the patients, this
finding suggests that drug abuse may influence the dopaminergic system in the prefrontal cortex. It
should, however, be noted that fluorescence intensity is not a truly quantitative measure of
dopaminergic innervation. As our purpose was to visualize DA-rich areas of the PFC and our
sections (frozen, 14µm thick) were not amenable to stereological analysis (West, 2012), we did not
pursue this further using more quantitative methods (Akil et al., 1999; West, 2012).
Global levels of free thiols and 3-nitrotyrosine were examined by measuring fluorescence
intensity. We used CPM to label free thiols using a method validated by Mastroberardino et al.
(Mastroberardino et al., 2008). We chose to use thiol oxidation as a marker of protein oxidation as it
occurs more readily than formation of protein carbonyls, and hence may be a more sensitive measure
(Mastroberardino et al., 2008). 3-nitrotyrosine is a marker of protein nitration that has been used by
other studies examining oxidative stress (Andreazza et al., 2008; Andreazza et al., 2010; Machado-
Vieira, Andreazza, et al., 2007). We found increased thiol oxidation in patients with BD and SCZ,
while we did not find between-group differences in protein nitration. Previous studies have

77
consistently reported increased markers of oxidative stress for both SCZ and BD, where increased
levels of protein carbonyl groups and lipid peroxidation have been found (Andreazza et al., 2008).
Oxidation of thiol groups can alter protein structure and function (Winther & Thorpe, 2013), and
may be a contributing factor in the pathophysiology of these disorders. Increased thiol oxidation in
BD and SCZ may occur due to a number of different reasons, including lower levels of glutathione
(Gawryluk et al., 2011; Kulak et al., 2013) and mitochondrial dysfunction (Scola et al., 2013),
particularly that of complex I, which could increase the production of ROS (Halliwell, 1992) that
can oxidize proteins. Indeed, decreased levels of expression of subunits of the mitochondrial electron
transport chain were reported by a number of studies for both BD and SCZ (Scola et al., 2013).
Since cell loss and morphological alterations are found in the prefrontal cortex in patients with BD
and SCZ (Almeida et al., 2009; K. F. Berman, Doran, Pickar, & Weinberger, 1993; Frey et al., 2007;
Lopez-Larson, DelBello, Zimmerman, Schwiers, & Strakowski, 2002; Rajkowska, Halaris, &
Selemon, 2001), cellular dysfunction produced by protein oxidation may be contributing to these
alterations. This may lead to a decline in the proper functioning of the PFC. Indeed, as mentioned
previously, decreased working memory is consistently reported in BD (Glahn et al., 2006;
Thompson et al., 2007), suggesting that cellular dysfunction produced by oxidative damage could be
contributing to this as well. Furthermore, we found an effect of prescribed medications, where
patients who were prescribed antipsychotics had greater oxidative damage than the control group.
Since antipsychotics are D2 receptor antagonists, the effect of these drugs on oxidative damage in
the dopamine system is of potential interest (H. K. Kim & Andreazza, 2012). Also, patients who
were not prescribed antidepressants had greater global oxidative damage, which is consistent with a
potential protective effect of these drugs as described above for TH&3NT FRET results. 3-
nitrotyrosine, which is formed when reactive nitrogen species, such as peroxynitrite, react with
tyrosine residues (Greenacre & Ischiropoulos, 2001), was used as a marker of protein nitration. We

78
chose to label 3-nitrotyrosine to examine protein nitration as it has been successfully used by our
group as well as others to examine oxidative stress in post-mortem brain (Andreazza et al., 2007;
Andreazza et al., 2008; Andreazza et al., 2010). While we did not find global 3NT levels to differ
between groups, a previous study showed increased 3NT levels in the PFC of patients with BD and
SCZ (Andreazza et al., 2010). The difference between these findings may be due to the use of
different techniques, as Andreazza et al. measured 3NT levels using the ELISA technique from
whole tissue homogenates while we used immunohistochemistry to label cells in tissue sections.
Moreover, Andreazza et al. examined BA10 while we examined BA9/46. BA9 and BA10 have
different cytoarchitecture, suggesting that they may differ in the type and abundance of cells
(Semendeferi, Armstrong, Schleicher, Zilles, & Van Hoesen, 2001). Since different cells have
different vulnerabilities to oxidative stress, this may have contributed to the difference between these
two areas (Halliwell, 1992). Nonetheless, the differences in overall 3-NT levels between diagnostic
groups were in the same direction as those reported earlier by our group (Andreazza et al., 2010).
Also, while global 3NT levels did not differ between groups, nitration of TH-IR regions measured
by FRET was decreased in BD and SCZ. This suggests that nitration may be a more specific
phenomenon where increase or decrease in nitration of certain proteins does not always correspond
with global levels of oxidative stress (Greenacre & Ischiropoulos, 2001).
Limitations and Conclusion
The findings of this study must be interpreted in light of their limitations. First, we had a
small sample size and used multiple comparisons for statistical analysis. Second, we measured
fluorescence intensity in order to compare DAT, TH, and 3NT immunoreactivity between groups.
Fluorescence intensity obtained from tissue sections labelled with antibodies is a relative measure
and is limited by factors such as photobleaching and tissue quality. In order to minimize the effect of

79
these factors on our results, we kept the scanning parameters consistent, subtracted background
intensity, and only used sections with good histological quality. Also, because we used human
postmortem brain samples, pre and postmortem factors such as antemortem agonal states may have
affected our results (Chandana et al., 2009; Vawter et al., 2006). To account for this, we examined
the effect of demographic factors and post-mortem alterations in data analysis by correlating our
findings with PMI, pH, and age. Furthermore, in agreement with previous studies that examined
PMIs up to 48h (Chandana et al., 2009; Harish et al., 2011), thiol oxidation and nitration in the
mouse frontal cortex did not change up to 72h of PMI. Although we did find an effect of the history
of substance abuse on one measure, DAT intensity, we do not have detailed information on the
history of substance abuse or dependence to further explore how these factors could have influenced
our results. In addition, while the effects of drug treatment on CPM intensity (antipsychotics and
antidepressants) and TH&3NT FRET (antidepressants) are of potential interest, there are several
important limitations. These include small sample size of subjects, the differences in prescribing
patterns across diagnosis and no information of whether the patients were taking the medications
which were prescribed. Other approaches not solely relying on post-mortem brain may be more
helpful to determine drug treatment effects on these measures. We also only examined the prefrontal
cortex. Examining different areas of the brain may help further elucidate the relationship between
DA and oxidative stress in BD and SCZ. Finally, use of other techniques such as mass spectrometry
to examine oxidative and nitrosative modifications to DAT and TH in postmortem brain and
functional assays to examine the cause and consequences of such modifications may aid in further
elucidating the relationship between oxidative stress and DA in BD and SCZ.
In conclusion, our results demonstrate increased protein oxidation in the PFC of patients with
BD and SCZ. We also found increased thiol oxidation in DAT-IR regions of patients with BD and

80
dysregulation of protein nitration in TH-IR regions of patients with BD and SCZ, suggesting
oxidative and nitrosative modifications to DA-rich areas in both of these disorders. Therefore, our
findings suggest that the interaction between oxidative stress and DA may be important for the
pathophysiology of BD and SCZ, and that future studies examining the role of oxidative
modifications on dopaminergic proteins may contribute to a better understanding of how
dysregulation of the DA system occur in these disorders.
Acknowledgements / Conflict of interest disclosure
We acknowledge Ginnie Ng for her contributions in cell counting of the nissl stained sections, and
Dr. Dennis Grant for donating the mice for the post-mortem interval experiment. The authors also
declare CIHR, Brain and Behaviour Research Foundation (NARSAD), and OGS as sources of
funding, and Stanley Foundation Neuropathology Consortium as source of human post-mortem brain
sections. The authors declare no conflict of interest.

81
Figures
Figure 1. A. Example of a nissl-stained section taken with Nikon Eclipse 80i Microscope with good
section quality. Scale = 44µm. B. Example of a section labelled for NeuN (green). Image was taken
with a Nikon Eclipse 80i Microscope. Scale = 58μm. C. Image is an example of a section labelled
for DAT (green). Image was taken with a confocal laser scanning microscope (CLSM). The inset is
an example of a section labelled with only the secondary antibody, demonstrating minimal labeling,
and therefore specificity of the secondary antibody. D. Image is an example of a section labeled with
the TH antibody (green) taken with a CLSM. The inset is an example of a section labelled with the
primary antibody for 3NT, and secondary antibody for TH. Minimal labeling was observed,
indicating specificity of the secondary antibody. E. Image is an example of a FRET experiment for
DAT&CPM FRET. Red circle indicates the area that has been photobleached. Increase in the
intensity of the donor fluorophore (blue; CPM) can be observed after bleaching the acceptor

82
fluorophore (Alexa488; green), indicating the presence of FRET. F. Image is an example of a FRET
experiment for TH&3NT FRET. Red circle indicates the photobleached area. Increase in the
intensity of the donor fluorophore (Alexa350; blue) can be observed after photobleaching the
acceptor fluorophore (Alexa488; green), indicating presence of FRET.
Figure 2. A. Intensity of CPM labeling after correction for non-specific CPM binding with negative
control (IAA&NEM treated sections). Results are expressed as mean±SEM. * P<0.05; ** P<0.01.
N=50 (Control=13, BD=13, SCZ=11, MDD=13). B. 3NT immunoreactivity after correction for non-
specific signals as measured by fluorescence intensity. Results are expressed as mean±SEM. N=50
(Control=14, BD=10, SCZ=13, MDD=13). All analyses were performed on non-processed, original
images in Original Imaging Format (OIF).

83
Figure 3. A. ΔIF between Alexa488 labeling DAT and CPM labeling free thiols. Higher values
indicate greater amounts of free thiols in DAT-IR regions after correction for background ΔIF
(FRET in oxidized sections). Results are expressed as mean±SEM. *P<0.05 **P<0.01. N=54
(Control=14, BD=14, SCZ=12, MDD=14). B. ΔIF between Alexa488 labeling TH, and Alexa350
labeling 3NT. Greater ΔIF indicates greater nitration. Results are expressed as mean±SEM. *P<0.05;
**P<0.01. N=50 (Control=11, BD=11, SCZ=14, MDD=14). All analyses were performed on non-
processed, original images in Original Imaging Format (OIF).

84
Figure 4. A. Correlation between ∆IF for DAT&CPM and brain pH, age, and PMI. B. Correlation
between ∆IF for TH&3NT and brain pH, age, and PMI. Results were assessed using Pearson’s
correlation test. The correlations were not found to be significant.

85
Table 1. Subject information by group
Control BD SCZ MDD
Age: mean (range) 48 (29-68) 42 (25-61) 44.2 (25-62) 46.4 (30-65)
PMI: mean (range); hr 24 (8-42) 33 (13-62) 34 (12-61) 28 (7-47)
pH: mean (range) 6.3 (5.8-6.6) 6.2 (5.8-6.5) 6.1 (5.8-6.6) 6.2 (5.6-6.5)
Sex 9M & 6F 9M & 6F 9M & 6F 9M & 6F
Lithium 0 4 2 2
Antipsychotics 0 8 12 0
Antidepressants 0 8 5 10

86
Statement of significance and impact:
Our group has recently demonstrated that the synapse is a target of oxidative modifications in
the frontal cortex of patients with BD (Andreazza et al., 2013). As the dopamine system largely
exists in the synapse, and dopamine dysregulation has long been known to contribute to mania, a
defining feature of BD (Cousins et al., 2009), I decided to examine oxidative damage to the
dopaminergic system to examine if the synapse may be a downstream target of complex I
dysfunction in BD, addressing Aim 4. More specifically, as in vitro studies have shown the
dopamine transporter and tyrosine hydroxylase, two proteins critical for the regulation of dopamine
levels, as targets of oxidative modifications (Ara et al., 1998; S. U. Park et al., 2002), oxidation of
the dopamine transporter and nitration of tyrosine hydroxylase were measured. Findings of this study
demonstrated oxidative and nitrosative modifications to dopamine rich areas in the post-mortem
prefrontal cortex of patients with BD, where dopamine transporter-immunoreactive regions were
found to have increased thiol oxidation, and tyrosine hydroxylase-immunoreactive regions were
found to have decreased nitration. These findings suggest that oxidative modifications to
dopaminergic proteins could be contributing to dopamine dysregulation underlying BD,
demonstrating the dopamine system as a possible downstream target of complex I dysfunction.
Oxidation of the DAT would lead to decreased ability to uptake dopamine (S. U. Park et al., 2002),
and since DAT is the primary regulator of synaptic levels of dopamine, this would lead to increased
levels of synaptic dopamine levels. Furthermore, nitration of tyrosine hydroxylase was found to
decrease its ability to synthesize L-DOPA, resulting in lower levels of dopamine (Blanchard-Fillion
et al., 2001). This suggests that decreased nitration of tyrosine hydroxylase could “disinhibit” the
enzyme, causing increased synthesis of L-DOPA, although this has not been demonstrated directly.
Therefore, oxidation of DAT and decreased nitration of tyrosine hydroxylase could be contributing
to increased dopamine signaling in mania. Importantly, while global levels of nitration did not differ

87
between groups, patients with bipolar disorder and schizophrenia had lower levels of nitration in
tyrosine hydroxylase-immunoreactive regions. This suggests that oxidative stress can have specific
targets that are affected differently from global levels of oxidative/ nitrosative damage depending on
the target and surrounding conditions (i.e. presence of dopamine). To summarize, my studies
examining the post-mortem brain extended on previous findings from our group identifying the
mitochondria and synapse as targets of oxidative stress (Andreazza et al., 2013) by demonstrating
the activation of the NLRP3-inflammasome in patients of BD, a marker of complex I dysfunction
and mitochondrial ROS production, and by demonstrating oxidative modifications to the dopamine
transporter and tyrosine hydroxylase, which are markers of the dopaminergic synapse (Akil et al.,
1999).
The next set of studies aimed to further examine complex I dysfunction using cell models,
which allows us to directly inhibit the electron transfer process in complex I by using rotenone
(Turrens & Boveris, 1980). We also examined the effect of lithium because it is uniquely used for
the treatment of BD and was shown to improve complex I function and increase its mRNA levels
(Maurer et al., 2009; Sun et al., 2006), making it a good proxy for studying complex I dysfunction in
BD.

88
CHAPTER 5a
Title: Glutathione-mediated effects of lithium in decreasing protein oxidation induced by
mitochondrial complex I dysfunction
Authors: Helena Kyunghee Kim*, Camila Fernandes Nascimento*, L. Trevor Young, Karina
Martinez Mendonca, Beny Lafer, Lea Tenholz Grinberg, Ana Cristina Andreazza
• *HKK and CFN share first authorship
Journal: Journal of Neural Transmission
Volume: Epub ahead of print
Pages: Epub ahead of print
Work performed by the student:
The student performed all work included in this chapter, including planning and performing the
experiments, image acquisition, data analysis, and preparation and submission of the manuscript.

89
Title: Glutathione-mediated effects of lithium in decreasing protein oxidation induced by
mitochondrial complex I dysfunction
Category: Short communication
Authors:
#Camila Fernandes Nascimento1, #Helena Kyunghee Kim2, L. Trevor Young1,2,3, Karina Martinez
Mendonça5, Lea Tenenholz Grinberg1,4, Beny Lafer1, *Ana Cristina Andreazza2,3
#The first two authors (CFN and HKK) share first authorship.
1University of Sao Paulo Medical School, Sao Paulo, Brazil
2Departments of Pharmacology and Psychiatry, University of Toronto, Ontario, Canada
3Centre of Addiction and Mental Health, Ontario, Canada
4Memory and Aging Center, University of California, San Francisco, USA
5Department of Pharmacy, Federal University of Sao Paulo
Addresses:
CFN and BL: Rua Dr. Ovidio Pires de Campos, 785 - 1º andar - Sala 1C015, Cerqueira César,
05403-903 - Sao Paulo, SP – Brasil
HKK, LTY, KMM, and ACA: RM4204, 1 King’s College Circle Toronto, ON Canada
LTG: Sandler Neurosciences Center, 675 Nelson Rising Lane, Suite 190, San Francisco, California,
USA

90
Corresponding author:
Ana Cristina Andreazza
Address: RM4204, 1 King’s College Circle Toronto, ON Canada M5S 1A8
Phone number: 416-946-5722
E-mail: [email protected]

91
Abstract
The aim of this study was to elucidate whether glutathione is involved in lithium’s ability to decrease
carbonylation and nitration produced by complex I inhibition, which is consistently found in BD.
Neuroblastoma cells were treated with rotenone, a complex I inhibitor. Our results suggest that
glutathione is essential for lithium’s ability to ameliorate rotenone-induced protein carbonylation,
but not nitration.
Key words: 3-nitrotyrosine, carbonylation, lithium, mitochondrial complex I, rotenone

92
1. Introduction
Oxidative stress occurs when the production of reactive oxygen (ROS) or nitrogen (RNS)
species overwhelms the body’s antioxidant defense system (Halliwell, 2001). Mitochondrial
complex I is a major source of ROS and RNS, which form through the reaction between molecular
oxygen and leaked electrons during the process of adenosine triphosphate (ATP) production (Jeong
& Seol, 2008). Superoxide anions that form during this process can become hydroxyl radicals to
produce protein carbonyl groups, or react with nitric oxide to produce peroxynitrite, which can cause
protein nitration (Jeong & Seol, 2008). Both protein carbonylation and nitration can disrupt the
proper functioning of a variety of different proteins (Naoi et al., 2005; Ray, Huang, & Tsuji, 2012).
Glutathione, which is an endogenous antioxidant, can prevent such damages by directly reacting
with oxidizing substances and neutralizing them (Dringen, 2000). Importantly, glutathione is the
main antioxidant of the brain and was found to be decreased in the prefrontal cortex of patients with
BD (Dringen, 2000; Gawryluk et al., 2011). Glutathione peroxidase, which catalyzes the
consumption of free glutathione to reduce reactive oxygen species, was also found to be greater in
relatives of BD patients, suggesting dysfunction of the glutathione system in BD (Huzayyin et al.,
2014). Moreover, the production of oxidative stress through mitochondrial complex I aberrations
including decreased mRNA expression, activity, and protein levels has been strongly implicated in
bipolar disorder (BD) (Andreazza et al., 2010; Konradi et al., 2004; Scola et al., 2013).
Of significance, lithium was demonstrated to have antioxidant properties (Cui et al., 2007),
while whether lithium can prevent oxidative and nitrosative damage to proteins through modulation
of the glutathione system remains to be explored. Elucidating the exact pathway by which lithium
exerts its antioxidant effects could lead to the discovery of novel targets that can be used to develop
drugs for decreasing oxidative stress in BD. Therefore, the goal of this study was to investigate
whether lithium prevents mitochondrial complex I dysfunction-induced protein oxidation and

93
nitration by increasing glutathione levels. We hypothesized that mitochondrial complex I
dysfunction induced by rotenone will increase protein oxidation and nitration, and that lithium will
prevent both modifications by increasing glutathione levels. We tested our hypothesis in the
neuroblastoma cell line (SH-SY5Y).
2. Methods
2.1 Cell culture
Human neuroblastoma cells (SH-SY5Y) were grown in Dulbecco Modified Eagle’s Media (DMEM)
containing 10% fetal bovine serum (FBS) at 5% CO2, 37oC. To induce mitochondrial complex I
impairment, SH-SY5Y cells were treated with different concentrations of rotenone (16h; 5nM, 10nM,
15nM, 20nM and 30nM; Sigma-Aldrich, Oakville, ON). Rotenone was chosen as it has been
successfully used to model complex I dysfunction in neuroblastoma cells (Watabe & Nakaki, 2004).
From here on, rotenone-induced effects will be described as being due to mitochondrial complex I
dysfunction. The effect of lithium was assessed by pre-treating SH-SY5Y cells with a therapeutic
dose of lithium carbonate (72h; 0.75mM; Li; Bioshop, Burlington, ON) prior to rotenone treatment.
Buthionine sulfoximine (BSO; 1mM; Bioshop) was used to deplete glutathione and analyze its role
in the effect of lithium. For this, the cells were pre-treated with lithium (72 hours), followed by BSO
treatment (12 hours) and rotenone treatment (5nM, 15nM, 30nM only; 16 hours). The three different
series of treatments described so far will be referred throughout the text as rotenone alone,
Li+rotenone and Li+BSO+rotenone. All the experiments were performed in starving media
(DMEM+0.5% FBS) for all conditions as suggested by previous studies (Krämer, Bouzakri,
Holmqvist, Al-Khalili, & Krook, 2005; van der Valk et al., 2010; X. Zheng et al., 2006). Starving
media was not shown to decrease mitochondrial activity within 1 week in fibroblasts (Takeda et al.,
2002). In our experiments, mitochondrial function was shown to be preserved in cells treated with

94
starving media as observed by positive MitoTracker Red CMXRos staining, which only accumulates
in actively respiring mitochondria (Takeda et al., 2002).
2.2 Complex I activity
Activity of complex I was analyzed using a colorimetric kit (Abcam, Cambridge, UK) according to
manufacturer’s protocol.
2.3 Cell viability
Cell viability was measured using the CellTiter-blue assay (Promega, Madison, WI, USA) according
to manufacturer’s protocol.
2.4 Immunocytochemistry
Immunocytochemistry was carried out using a previously published method with slight
modifications (Watabe & Nakaki, 2007) to observe 3-nitrotyrosine (3NT). Briefly, cells were
incubated with MitoTracker Red CMXRos for 20min at 37oC (100nM; Life Technologies,
Burlington, ON). Cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton-
X for 5min. Following blocking (4% goat serum, 30min), cells were incubated with anti-3-
nitrotyrosine antibody (16h, 4 oC, 1:200; Abcam, Toronto, ON), followed by Alexa488 anti-mouse
(1h, Life Technologies), and mounted with ProLong Gold antifade reagent with DAPI (Life
Technologies). Carbonylation was visualized by using the OxyICC Oxidized Protein Detection Kit
(Millipore, Temecula, CA) according to manufacturer’s instructions. Mitochondria were visualized
using MitoTracker Red and the cells were mounted in ProLong Gold antifade reagent with DAPI. A
confocal laser scanning microscope (CLSM; Olympus FV1000; Olympus, Center Valley, PA) and a
60X objective were used to image the cells. Alexa488 was observed using a 488nm laser,

95
MitoTracker Red was observed using a 543nm laser, and DAPI was observed using a 405nm laser.
3NT and carbonyl fluorescence intensities were measured from five regions of interest (ROIs) per N
using the FV10-ASW2.1 software (Olympus), and the average value of the ROIs was determined.
All experiments were performed in triplicates.
2.5 Data analyses
Statistical analyses were performed using IBM SPSS 20 software (Chicago, USA). Differences
between the treatment groups (rotenone alone vs Li+rotenone or Li+rotenone vs Li+BSO+rotenone)
were assessed by two-way analysis of variance (ANOVA). Differences between rotenone treatments
against the appropriate control were assessed using one-way ANOVA followed by Dunnett post-hoc
analysis.
3. Results
3.1 Lithium prevents decrease of complex I activity and cell viability induced by mitochondrial
complex I dysfunction
Treating neuroblastoma cells with rotenone produced a decrease in complex I activity (F5,18 = 3.77,
P<0.05; P<0.05 for 30nM; Figure 1A). Lithium pre-treatment was able to prevent this decrease, such
that rotenone no longer produced a decrease in its activity. Dysfunction of mitochondrial complex I
induced by rotenone also demonstrated a dose-dependent effect in decreasing cell viability (F5,24 =
4.09, P<0.01, P<0.05 for 30nM), and lithium pre-treatment prevented this decrease. The addition of
BSO inhibited the ability of lithium to prevent decreased cell viability induced by mitochondrial
complex I dysfunction (F3,16 = 20.64, P<0.01, P<0.01 for all concentrations of rotenone; Figure 1B).
3.2 Lithium’s ability to decrease rotenone-induced protein carbonylation is dependent on glutathione.

96
Mitochondrial complex I inhibition through rotenone treatment produced an increase in protein
carbonylation (F5, 24 = 15.38, P<0.01; P<0.01 for all concentrations; Figure 2). Although lithium pre-
treatment did not result in complete prevention of this increase in carbonylation (F5,24 = 28, P<0.01;
P<0.01 for all concentrations), it produced a decrease in carbonylation compared to cells treated with
rotenone without lithium pre-treatment (F1,48 = 22.23, P<0.01). The addition of BSO inhibited the
ability of lithium to decrease carbonylation such that carbonylation levels became similar or higher
than cells treated with rotenone alone (F3,16 = 25.10, P<0.01; P<0.01 for all).
3.3 Lithium prevents protein nitration induced by mitochondrial complex I dysfunction
independently of glutathione.
Protein nitration was increased by mitochondrial complex I inhibition with rotenone treatment (F5,36
= 9.58, P<0.01; P<0.01 for all rotenone concentrations; Figure 2). Lithium pre-treatment was able to
completely prevent this increase in nitration such that rotenone no longer produced an increase. The
ability of lithium to decrease nitration did not change with the addition of BSO.
4. Discussion
Mitochondrial dysfunction and increased oxidative stress are two of the most consistent
alterations reported in patients with BD (Andreazza et al., 2010; Clay et al., 2010; Scola et al., 2013)
and lithium, which is considered a gold standard of mood stabilizers (Goodwin & Malhi, 2007), was
shown to have antioxidant-like effects (Cui et al., 2007). Yet, the mechanisms underlying these
effects are poorly understood. In agreement with previous studies, we found that lithium prevented
rotenone-induced cell death and decreased complex I activity (Allagui et al., 2009; Maurer et al.,
2009). The findings of our study are the first to demonstrate that mitochondrial complex I inhibition
is able to increase protein oxidation and nitration and that lithium can ameliorate both alterations in

97
SH-SY5Y cells. Furthermore, our findings suggest that while the ability of lithium to reduce
carbonylation may be mediated by glutathione, its ability to prevent nitration may not be. This
demonstrates that multiple systems are involved in lithium’s antioxidant effects, and suggests that
elucidation of such pathways could contribute to the development of specific drug interventions to
target oxidative stress in BD.
In agreement with our results, many studies have demonstrated that lithium has
neuroprotective effects (Bachmann et al., 2009) such as decreasing apoptotic signaling (Q. Li et al.,
2010), improving energy metabolism (Bachmann et al., 2009; Maurer et al., 2009), and decreasing
oxidative damage (Cui et al., 2007; Shao et al., 2008). Although the mechanisms by which lithium
exerts its antioxidant effects are not clear, it has been hypothesized to work in part through the
glutathione system (Shao et al., 2008) and by improving mitochondrial functioning (Bachmann et al.,
2009). Since complex I inhibition can lead to increased leakage of electrons, resulting in the
production of the superoxide anion that can produce protein carbonylation and nitration (Kamat,
2006), lithium may be ameliorating these alterations by improving complex I functioning.
Glutathione, which is the most abundant antioxidant in the brain, acts by directly reacting
with ROS/RNS to neutralize them (Halliwell, 2001). In our study, glutathione depletion with BSO
increased carbonylation in the absence of rotenone as well, suggesting that glutathione alone is
important for decreasing the formation of carbonyl groups. BSO reduces the levels of glutathione,
thus our experiments suggest that lithium requires glutathione to prevent formation of carbonyl
groups. Indeed, glutathione is important to decrease levels of H2O2, which could alone contribute to
the formation of carbonyl groups, again suggesting the relevance of glutathione for prevention of
carbonyl groups (Dringen, 2000). Previous studies have shown that lithium can increase levels of
glutathione (Cui et al., 2007) and glutathione transferase, which conjugates glutathione to radicals
(Shao et al., 2008). While our results support the involvement of glutathione in the ability of lithium

98
to decrease carbonylation, the effect of lithium on nitration may occur through a different pathway,
such as improving complex I activity (N. Li et al., 2003). The effect of lithium on nitric oxide
production is controversial (Karimollah, Ghasemi, Ghahremani, & Dehpour, 2009; Wegener et al.,
2004). Further studies are required to examine the mechanism by which lithium prevents protein
nitration.
Limitations of this study include the use of a single technique, immunocytochemistry (ICC),
to quantify protein nitration and oxidation. Although measures were taken to minimize bias, such as
background subtraction and normalization by control, ICC is limited by factors such as
photobleaching. Furthermore, because we only used SH-SY5Y cells, which are well-characterized
neuronal cells (Cui et al., 2007; Watabe & Nakaki, 2004, 2007), using animal models may help to
expand the generalizability of these findings.
Acknowledgements
The authors have no conflict of interest to disclose. The authors thank Brain & Behavior Research
Foundation (Formerly NARSAD, ACA, BL), Coordenação de Aperfeiçoamento de Pessoal de Nível
Superior (CAPES-DFAIT) Program /Brazil and Canada (BL, LTG and CFN) and Canadian
Institutes of Health Research (CIHR) as sources of funding in support of this report (LTY and ACA).

99
Figures
Fig. 1 Lithium prevents decrease in complex I activity and cell viability induced by mitochondrial
complex I dysfunction. A. Complex I activity. Rotenone treatment (F5,36 = 3.67, P<0.01, *P<0.05
against DMSO control; green line); lithium pre-treatment (F1,36 = 4.68, P<0.05, red line). N =4. B.
Cell viability. Rotenone treatment (F5,44 = 3.83, P<0.01, *P<0.05 against DMSO control; green line);
lithium pre-treatment (F1,44=32.50, P<0.01, red line); BSO following lithium pre-treatment (F3,16 =
20.64, P<0.01; ++P<0.01 against lithium+BSO alone; blue line). N = 5. Statistics shown in brackets
are results of two-way ANOVA followed by Dunnett post-hoc analysis. Symbols represent Dunnett
post-hoc analysis against appropriate control
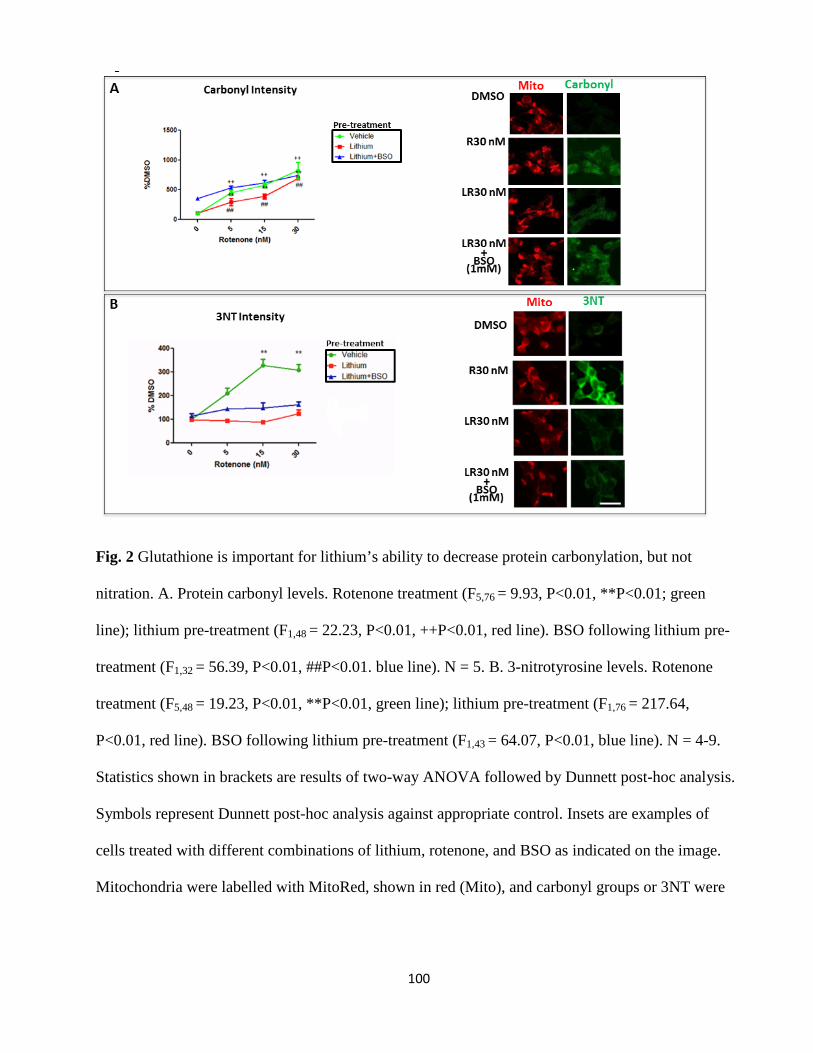
100
Fig. 2 Glutathione is important for lithium’s ability to decrease protein carbonylation, but not
nitration. A. Protein carbonyl levels. Rotenone treatment (F5,76 = 9.93, P<0.01, **P<0.01; green
line); lithium pre-treatment (F1,48 = 22.23, P<0.01, ++P<0.01, red line). BSO following lithium pre-
treatment (F1,32 = 56.39, P<0.01, ##P<0.01. blue line). N = 5. B. 3-nitrotyrosine levels. Rotenone
treatment (F5,48 = 19.23, P<0.01, **P<0.01, green line); lithium pre-treatment (F1,76 = 217.64,
P<0.01, red line). BSO following lithium pre-treatment (F1,43 = 64.07, P<0.01, blue line). N = 4-9.
Statistics shown in brackets are results of two-way ANOVA followed by Dunnett post-hoc analysis.
Symbols represent Dunnett post-hoc analysis against appropriate control. Insets are examples of
cells treated with different combinations of lithium, rotenone, and BSO as indicated on the image.
Mitochondria were labelled with MitoRed, shown in red (Mito), and carbonyl groups or 3NT were

101
labelled with Alexa488 (green). Images were taken with a 60X objective and a confocal laser
scanning microscope (Olympus). Scale bar = 20um

102
CHAPTER 5b
Title: Lithium reduces the effects of rotenone-induced complex I dysfunction on DNA methylation
and hydroxymethylation in rat cortical neurons.
Authors: Helena K. Kim*, Gustavo Scola*, L. Trevor Young, Mirian Salvador, Ana C. Andreazza
• HKK and GS share first authorship
Journal: Psychopharmacology
Volume: 21
Pages: 4189-4198
Work performed by the student:
The student performed all work included in this chapter, including planning and performing the
experiments, image acquisition, data analysis, and preparation and submission of the manuscript.

103
Title: Lithium reduces the effects of rotenone-induced complex I dysfunction on DNA
methylation and hydroxymethylation in rat cortical primary neurons.
Gustavo Scola1,3,4*, Helena K. Kim2,3*, L. Trevor Young1,2,3 Mirian Salvador4, Ana C.
Andreazza1,2,3#
1Departments of Psychiatry and 2Pharmacology, University of Toronto, ON, Canada
3Centre for Addiction and Mental Health, Toronto, ON, Canada
4Institute of Biotechnology, University of Caxias do Sul, RS, Brazil
*both authors contribute equally for this study
#Corresponding Author
Ana Andreazza, Pharm, PhD
University of Toronto
Medical Science Building, Room 4204.
1 king’s College Circle
Toronto, ON, M5S 1A8
Phone: 416-978-6042
e-mail: [email protected].

104
Abstract
Rationale Mitochondrial complex I dysfunction and alterations in DNA methylation levels are
consistently reported in bipolar disorder (BD) and are regulated by lithium. One of the mechanisms
by which lithium may exert its effects in BD is by improving mitochondrial complex I function.
Therefore, we examined whether complex I dysfunction induces methylation and
hydroxymethylation of DNA and whether lithium alters these effects in rat primary cortical neurons.
Methods Rotenone was used to induce mitochondrial complex I dysfunction. Cell viability was
measured by MTT assay and ATP levels were assessed by Cell-Titer-Glo®. Complex I activity was
measured using an ELISA based assay. Apoptosis, DNA methylation and hydroxymethylation levels
were measured by immunocytochemistry.
Results Rotenone decreased complex I activity and ATP production, but increased cell death and
apoptosis. Rotenone treatment increased levels of 5-methylcytosine (5mc) and
hydroxymethylcytosine (5hmc), suggesting a possible association between complex I dysfunction
and DNA alterations. Lithium prevented rotenone-induced changes in mitochondrial complex I
function, cell death and changes to DNA methylation and hydroxymethylation.
Conclusions These findings suggest that decreased mitochondrial complex I activity may increase
DNA methylation and hydroxymethylation in rat primary cortical neurons, and that lithium may
prevent these effects.
Keywords: lithium, complex I, methylation, hydroxymethylation, bipolar disorder.

105
Introduction
Mitochondrial complex I dysfunction and increased oxidative stress have been consistently
demonstrated in bipolar disorder (BD) (Clay et al., 2010; Scola et al., 2013). Complex I dysfunction
has a number of consequences, including production of reactive oxygen species and subsequent
oxidative damage to proteins, lipids, and DNA (Andreazza et al., 2008; Halliwell, 1992). Indeed,
increased 8-hydroxy-2-deoxyguanosine (8-OhdG) has been reported in lymphocytes (Che et al.,
2010; Soeiro-de-Souza et al., 2013) and post-mortem hippocampus (Che et al., 2010; Soeiro-de-
Souza et al., 2013) from patients with BD, suggesting that oxidative modifications to DNA may be a
factor in this disorder. Of significance, recent studies have shown that oxidative stress can modulate
the levels of DNA methylation in cancer cells (Campos et al., 2007; Chestnut et al., 2011; K. A.
Kang et al., 2012; Lim et al., 2008; R. Zhang et al., 2013); however, the role of mitochondrial
complex I dysfunction in the regulation of DNA methylation levels, especially in neurons, is still
unclear.
There is growing interest in the role of epigenetic modifications in BD (Abdolmaleky et al.,
2006; D'Addario et al., 2012; Nohesara et al., 2011; Petronis, 2003). For example, increased levels
of 5-methylcytosine (5mc) have been reported at the BDNF promoter and the COMT gene in
patients with BD (Abdolmaleky et al., 2006; D'Addario et al., 2012). 5mc plays an important role in
cell dynamics, such as regulation of gene expression, X-chromosome inactivation, and maintenance
of epigenetic memory, where increase in methylation levels at promoter regions will decrease gene
expression (Bird, 2002). 5mc can be oxidized by the ten-eleven translocation (TET; EC 1.14.11.n2)
enzyme, producing 5-hydroxymethylcytosine (5hmc) (Jin et al., 2010; Wu et al., 2010). Although
the role of 5hmc in gene expression is unclear, it was shown to increase transcription of methylated
DNA and has been suggested to be an important factor in regulating the balance between
methylation and demethylation (Grayson & Guidotti, 2013; Jin et al., 2010; Wu et al., 2010). Of

106
significance, increased levels of 5hmc were found in the inferior parietal lobule of patients with
psychosis when compared to healthy controls (Dong, Gavin, Chen, & Davis, 2012).
Lithium, which is the most commonly prescribed drug for the treatment of BD (Kupfer,
2005), has a number of specific cellular targets including: activation of the Wnt signaling pathway,
inhibition of glycogen synthase kinase 3β, changes in cytoplasmic β-catenin (Boyadjieva &
Varadinova, 2012; Malhi et al., 2013), among others in addition to an effect on mitochondrial
function. More recently, lithium was reported to increase the antioxidant capacity of the cell by
increasing glutathione content (Cui et al., 2007; Shao et al., 2008) and improving mitochondrial
function, particularly that of complex I (Bachmann et al., 2009; Quiroz et al., 2008; Sun et al., 2006).
Another pathway by which lithium could be exerting its effects in BD is through the regulation of
DNA oxidation and methylation, as shown in in vitro studies and in patients with BD (Boyadjieva &
Varadinova, 2012; D'Addario et al., 2012). For example, decreased global levels of 5mc were
reported in patients with BD who respond to lithium (Huzayyin et al., 2013).
The aforementioned studies are in support of the critical roles that mitochondrial complex I
dysfunction and alterations in methylation levels play in BD. Lithium, the most prescribed
medication for patients with BD, has also been demonstrated to regulate antioxidant activity, cell
survival response and to induce changes in methylation patterns in patients with BD. However, it is
still unclear whether neuronal mitochondrial complex I dysfunction can directly induce changes in
DNA methylation and hydroxymethylation. To study this phenomenon, we treated rat cortical
primary neurons with rotenone, a direct mitochondrial complex I inhibitor (Halliwell 2001). We
also tested whether alterations in DNA methylation and hydroxymethylation levels require
mitochondrial dysfunction or can also occur in a high oxidative stress environment, such as that
produced by hydrogen peroxide (H2O2) treatment. Lastly, we examined the ability of lithium to
prevent mitochondrial complex I dysfunction, and any alterations to global 5mc and 5hmc levels

107
which may occur. Here, we report that rotenone decreases complex I activity leading to increased
levels of DNA methylation and hydroxymethylation. Treatment with H2O2 decreased 5mc levels, but
did not change the levels of 5hmc, suggesting that oxidative stress alone is insufficient to change
DNA hydroxymethylation levels. Lithium treatment was found to prevent mitochondrial complex I
dysfunction and consequent changes in 5mc and 5hmc levels. These results suggest that the effect of
lithium to regulate 5mc and 5hmc levels in rat primary cortical neurons may be through its ability to
enhance mitochondrial complex I function.
Methods
Cell culture
Rat E18 cortical neurons (PC35102 Neuromics, US) were purchased from Neuromics and
cultured according to instructions from Neuromics in Neurobasal® medium (Gibco, Burlington,
ON) supplemented with B27® (Gibco, Burlington, ON) and 0.5 mM glutamine. Cells were cultured
in plates/coverslips coated with poly-D-lysine (Sigma, St. Louis, MO) for 7 days and treated with
0.75mM lithium (BioShop, Burlington, ON) for 7 more days. Next, cells were treated with different
concentrations of rotenone (5nM, 10nM, and 50nM; Sigma, St. Louis, MO) or H2O2 (5µM, 10µM
and 50µM; Sigma, St. Louis, MO) for 30 minutes. After washout, cells were cultured additionally
for 24 hours in the same conditions.
Complex I activity assay
Cells (3 x 105 per well in 6 well plate) were treated with lithium, and then exposed to
different concentrations of rotenone (5, 10, 50nM) for 30 minutes. After 24 hours in media without
B27, cells were assayed for complex I activity using the Complex I Enzyme Activity Microplate

108
Assay Kit (Abcam, Cambridge, MA, USA) according to manufacturer’s instructions. Data are
presented as percent of non-lithium treated DMSO control.
ATP measurement assay
Cells (4 x 104 per well in 96 well plate) were treated with lithium, followed by exposure to
different concentrations of rotenone (5, 10, and 50nM) for 30 minutes. Cells were grown in media
without B27 for 24 hours and assayed for ATP levels using Cell-Titer-Glo® assay (Promega,
Madison, WI) according to manufacturer’s instructions. Data are presented as percent of DMSO
treated control cells.
Annexin V assay
Annexin V labels phosphatidylserine that has been exposed at the outer leaflet of the plasma
membrane during early apoptosis, and propidium iodide (PI) enters the cell when membrane
integrity is lost during necrosis or late stage apoptosis. Therefore, labeling the cells simultaneously
with Annexin V and PI allows for the discrimination of cells in the early apoptotic stages with the
integrity of the membrane still intact (annexin V+ only) and cells in the late apoptotic stage with
exposure of phosphatidylserine and compromised membrane integrity (positive for both annexin V
and PI) (Schutte, Nuydens, Geerts, & Ramaekers, 1998). ApoDETECT Annexin V-FITC kit
(Invitrogen) was used according to manufacturer’s instructions. Briefly, cells were seeded in 24
wells (2 x 105 cells per well) and pre-treated with media or lithium followed by treatment with
different concentrations of rotenone as described above. Cells were viewed using a confocal laser
scanning microscope. Number of annexin V only positive cells (AV+ cells), cells positive for both
Annexin V and PI (AVPI+ cells), and total number of cells were counted by two examiners based on
previously published methods (Farinacci, 2007; Schutte et al., 1998). Cells positive only for Annexin

109
V were considered early apoptotic, cells positive for AV and PI were considered late apoptotic (B.
Weber et al., 2004).
Cell mortality
The MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay (Sigma, St.
Louis, MO) was used to determine cell mortality. Cells (4 x 104 per well in 96 well plate) were
treated with lithium, and then exposed to different concentrations of rotenone (5, 10, and 50nM) for
30 minutes. After 24 hours, 1mg/mL of MTT solution was added per well and the cells were
cultured for another 2 h. Following incubation, supernatant fluid was discarded, and precipitates
were dissolved in 100 µl of DMSO per well. After 20 min of agitation, optical density of the
resultant solution was measured with a microplate reader (BioTek® Instruments, Inc.) at 517 nm.
The increase or decrease in mortality of cells in each well was expressed as the percentage of non-
lithium treated DMSO control.
Immunocytochemistry
For immunocytochemistry, cells (2 x 105) were seeded in 12mm coverslips, pre-treated with
lithium or media, and exposed to different concentrations of rotenone (5, 10, and 50nM) and H2O2
(5, 10, and 50µM) for 30 minutes. After the removal of treatment media and 24 hours in media
without B27, cells were prepared for immunocytochemistry procedures as previously described (Ficz
et al., 2011; Guo, Su, Zhong, Ming, & Song, 2011; Wossidlo et al., 2011) with slight modifications.
Briefly, cells were fixed in pre-cooled methanol for 10 min at -20°C, washed 3 times with
Dulbecco's Phosphate Buffered Saline (DPBS), and permeabilized with 0.2% Triton X-100. Next,
samples were treated with 2N HCl for 30 min at RT, neutralized with 0.1M Tris-HCl (pH 8) for 10
min, and washed in DPBS 3 times. Cells were blocked with 1% bovine serum albumin for 1 hour

110
and washed 2 times in DPBS. Cells were then incubated with primary antibodies for 24 hours at 4°C
diluted in DPBS-0.4% Triton-X: anti-5-hydroxymethylcytosine (Millipore, Temecula, CA;
MABE176; 1:100) and anti-5-methylcytosine (Millipore, Temecula, CA; MABE146; 1:200). Slides
were washed 3 times and incubated with secondary antibodies for 1 hour at RT in DPBS-0.4%
Triton-X: Alexa Fluor®488 goat-anti rat IgG (Molecular Probes, Burlington, ON; A11006; 1:200)
and Alexa Fluor®568 goat-anti mouse IgG (Molecular Probes, Burlington, ON; A11031; 1:200).
Next, slides were washed 3 times and incubated in 300nM 4',6-diamidino-2-phenylindole (DAPI)
(Molecular Probes, Burlington, ON; D21490) in DPBS for 10 min. Finally, the cells were mounted
in Fluoromount™ (Cedarlane, Burlington, ON; CLSG80116). Cross-reactivity for antibodies was
assayed by adding different combinations of the primary and secondary antibodies for assessing
resultant fluorescence intensity (Online Resource 1).
Image Analysis
All images were taken using a confocal laser scanning microscope (Olympus Fluoview1000).
A 40X objective was used for the Annexin V assay, and Annexin V and PI were visualized using
excitation with a 488nm laser at 28% laser intensity. Detection wavelength was 500-530nm for
Annexin V, and 560-660nm for PI. At least two hundred cells were counted in each experiment per
condition. A 60X water-immersion objective was used for visualizing 5mc and 5hmc. Alexa568
labeling 5mc was excited with the 543nm laser at 34% laser intensity, Alexa488 labeling 5hmc was
excited with the 488nm laser at 33% laser intensity, and DAPI was excited with the 405nm laser at
20% laser intensity. Detection wavelength was 560-660nm for Alexa568, 500nm-530nm for
Alexa488, and 425-475nm for DAPI. Scanning parameters were kept consistent for all images.
Image analysis was done using the FV10-ASW2.1 software connected to the microscope with
images in Original Imaging Format (OIF). For measuring fluorescence intensity, individual cells

111
were selected using DAPI labeling as individual regions of interest (ROI) in order to analyze 5mc
and 5hmc levels within the nucleus. At least 100 cells were included in each experiment per
condition.
Statistical Analysis
Statistical analysis was performed using SPSS version 20 (Illinois, USA). Parametric
distribution of data was assessed using the Kolmogorov-Smirnov test. As majority of the data was
found to be normally distributed, parametric tests were used for subsequent analyses. One-way
ANOVA was used to examine the effect of different concentrations of rotenone, and the effect of
lithium in combination with different concentrations of rotenone. LSD post-hoc analysis was used to
examine the difference between treatment groups against the appropriate control. Independent t-test
was used to identify differences between lithium pre-treatment and rotenone at each concentration
(5, 10, 50nM).
Results
Lithium reduces rotenone-induced complex I dysfunction, decreased ATP production and
cellular mortality in rat cortical primary neurons
Rotenone produced a dose-dependent decrease in complex I activity (F3,20 = 38.75; p < 0.01;
Figure 1). Rotenone treatment at all concentrations (5nM, 10nM, 50nM) decreased complex I
activity when compared to DMSO (F3,10 = 34.33; p < 0.01; LSD post-hoc p< 0.01 for all). Lithium
pre-treatment decreased the extent of rotenone-induced decline on complex I activity such that the
different concentrations of rotenone were no longer significantly different from control (Figure 1).
Furthermore, rotenone decreased the production of ATP levels in a dose dependent manner (Figure

112
2; F3,37 = 2083.62; p < 0.01), which was prevented by lithium (Figure 2; R5 t=-21.95; p<0.001; R10 t
=-13.49; p<0.001; R50 t =-4.75; p<0.001). Rotenone treatment significantly increased cell mortality
(F3,20 = 393.47; p < 0.01) at all concentrations when compared to DMSO (Figure 3; p < 0.01 for all).
Lithium pre-treatment was able to decrease cell mortality, where rotenone at 5nM did not
statistically differ from control (R5 t =0.88; p=0.401; R10 t =-13.70; p<0.001; R50 t =-25.18;
p<0.001).
Lithium decreases apoptosis induced by complex I dysfunction in rat cortical primary neurons
Cells in early stage of apoptosis are represented by Annexin V+ and those in late stage of
apoptosis are represented by Annexin V and PI positive staining (AVPI+; Figure 4). Rotenone
treatment increased Annexin V+ cells at all concentrations when compared to DMSO (Figure 4A;
F3,18 = 17.07; p < 0.01; LSD p < 0.05 for 5 and 10nM; p < 0.01 for 50nM). Lithium pre-treatment
was able to decrease the levels of Annexin V+ cells in comparison to rotenone treatment alone
(Figure 4A; R5 t =3.20; p=0.013; R10 t =4.21; p=0.002; R50 t =5.11; p=0.001). Mitochondrial
complex I dysfunction induced by rotenone increased the presence of AVPI+ cells at all
concentrations (Figure 4B; F3,18 = 4.52; p < 0.05; LSD p < 0.05 for 5 and 10nM; p < 0.01 for 50nM).
However, lithium pre-treatment did not change rotenone-induced increase in AVPI+ cells (Figure
4B).
Lithium prevents increased DNA methylation and hydroxymethylation levels induced by
mitochondrial complex I dysfunction in rat cortical primary neurons
Rotenone treatment increased DNA methylation levels at concentrations of 10nM and 50nM
in comparison to DMSO (Figure 5A; F3,16 = 4.19; p < 0.05; LSD p < 0.05 for both). A similar pattern
was observed for DNA hydroxymethylation (Figure 5B; F3,16 = 5.87; p < 0.01), where rotenone at

113
10nM (p < 0.01) and 50nM (p < 0.05) significantly increased hydroxymethylation levels compared
to DMSO. Lithium pre-treatment decreased rotenone-induced DNA methylation (Figure 5A; R5 t
=7.50; p<0.001; R10 t =5.65; p<0.001; R50 t =16.48; p<0.001) and hydroxymethylation (Figure 5A;
R5 t =4.49; p<0.001; R10 t =2.51; p<0.013; R50 t =9.46; p<0.001) at all concentrations.
Hydrogen peroxide decreases methylation of DNA in rat cortical primary neurons
In order to examine if the effect of rotenone is specific to mitochondrial impairment or due to
the general induction of oxidative stress, we treated the cells with H2O2. A different pattern was
obtained when cells were treated with H2O2, where decreased levels of DNA methylation (Figure
6A; F3,16 = 8.95; p < 0.01) at all concentrations (LSD 5µM p < 0.05, 10µM p < 0.01, and 50µM p <
0.01) were observed. However, a lack of difference in DNA hydroxymethylation levels showed that
the groups do not significantly differ from each other (Figure 6B; F3,16 = 1.27; p = 0.32).
Discussion
In rat primary cortical neurons, rotenone treatment decreased complex I activity while
increasing DNA methylation and hydroxymethylation. Notably, treatment of neurons with H2O2
decreased 5mc levels without changing the levels of 5hmc. Lithium pre-treatment was able to
prevent the effects of rotenone on complex I activity and simultaneously decreased rotenone-induced
changes in global methylation and hydroxymethylation levels. Decreased expression of complex I
subunits and its activity are consistent findings in BD, while lithium is one of the most commonly
prescribed and effective drugs to treat this disease. Our findings suggest that mitochondrial complex
I dysfunction may lead to changes in DNA methylation and hydroxymethylation levels.
Furthermore, these results suggest that lithium, through regulation of mitochondrial complex I
activity, may normalize the levels of DNA methylation and hydroxymethylation. Since DNA

114
methylation and hydroxymethylation may be critical processes involved in the pathophysiology of
BD and other major psychiatric disorders (Grayson & Guidotti, 2013), the present results may
suggest another role of mitochondrial dysfunction in BD and its implications for the development of
novel treatments.
Mitochondrial electron transport chain is the main source of ATP for cells (Brandt, 2006;
Scola et al., 2013). Importantly, decreased mRNA (Sun et al., 2006) and protein levels (Andreazza et
al., 2010; Andreazza et al., 2013) of mitochondrial complex I, particularly that of NDUFS7, have
been described in patients with BD (Brandt, 2006). In this study, we chose to use rotenone because it
inhibits complex I activity by obstructing the transfer of electrons to ubiquinone, resulting in
decreased efficiency of the electron transport chain, which could potentially decrease ATP
production (Eckert et al., 2003; N. Li et al., 2003). Here, we demonstrated that rotenone decreases
complex I activity and ATP levels, while lithium pre-treatment prevents these changes, suggesting
that lithium may improve complex I function and consequently other rotenone-induced alterations.
These findings are in support of previous studies reporting that lithium increases complex I activity
in post-mortem brain (Maurer et al., 2009), protects against mitochondrial damage produced by high
calcium concentrations (Shalbuyeva et al., 2007) and enhances mitochondrial function (Bachmann et
al., 2009).
Using the Annexin V assay, we demonstrated that rotenone produces an increase in the
number of cells in the early and late apoptotic stages. Annexin V binds to phosphatidylserine that
has been exposed to the outer leaflet of the membrane during apoptosis, which is a calcium-
dependent process (Schutte et al., 1998). Our results are in agreement with previous studies that have
consistently shown that rotenone increases apoptosis by a number of different mechanisms,
including disruption of ATP levels and elevation of intracellular free calcium levels that activate the
apoptotic pathway. In addition, lithium prevented rotenone-induced increase in early apoptosis.

115
Indeed, studies have shown that lithium is able to decrease pro-apoptotic factors such as p53 and bax
(Quiroz et al., 2008), normalize intracellular calcium levels, and regulate ATP production, which
could combat the activation of apoptotic machinery (Q. Li et al., 2010).
In this study, decreasing mitochondrial complex I activity by rotenone increased DNA
methylation in primary cortical neurons. DNA methylation occurs when DNA methyltransferase
catalyzes the methylation of cytosine, resulting in the formation of 5mC (Branco, Ficz, & Reik,
2012; K. A. Kang et al., 2012). Consistent with these results, a previous study reported increased
DNA methylation in healthy adult subjects with mitochondrial J haplotype and lower levels of ATP
(Bellizzi, D'Aquila, Giordano, Montesanto, & Passarino, 2012). In our study, we also demonstrated
an increase in hydroxymethylation, as measured by an increase in 5hmc levels. 5hmc formation has
been suggested as possible compensatory response to overcome high levels of methylation (Lister et
al., 2013; Wu et al., 2010). Studies have shown that high levels of both 5mc and 5hmc in promoters
lead to increased gene transcription (Ficz et al., 2011; Jin et al., 2010; Wu et al., 2010). Furthermore,
we examined if the effect of rotenone on DNA methylation/hydroxymethylation was specific to
mitochondrial inhibition or was a result of general increase in oxidative stress using H2O2 (Halliwell
& Gutteridge, 2007). H2O2 treatment decreased methylation while not altering 5hmc levels,
suggesting that the effect of rotenone on DNA methylation/hydroxymethylation may differ from that
of a general increase in oxidative stress induced by H2O2. This suggests that while decreases in
mitochondrial complex I activity may increase both DNA methylation and hydroxymethylation,
general oxidative stress may only decrease methylation levels. Furthermore, lithium was found to
prevent rotenone-induced increase in 5mc and 5hmc levels. These findings are in support of previous
studies suggesting that lithium promotes hypomethylation in embryonic stem cells and in neural
stem cells (Guo, Ma, et al., 2011), and patients with BD who were taking lithium were shown to
have reduced methylation of the BDNF promoter compared to BD subjects who were not prescribed

116
lithium (D'Addario et al., 2012). Moreover, induction of apoptosis in neurons was found to increase
methylation levels (Chestnut et al., 2011; Hernandez & Singleton, 2012) in vitro, and lithium was
consistently shown in previous studies to prevent apoptotic processes, possibly through the
inhibition of GSK3β (King, Bijur, & Jope, 2001; Quiroz et al., 2008).
The findings of the present study should be interpreted in light of its limitations. First, this
study only examined a limited number of factors that are affected by or involved in the modulation
of 5mc and 5hmc levels. Examining a broader range of factors may help further elucidate the role of
mitochondrial dysfunction in epigenetic modifications. Moreover, we used primary neurons, which
do not allow for the examination of the effect of cell division on 5mc and 5hmc levels (Ito et al.,
2010; Tahiliani et al., 2009). Examining the effect of rotenone and lithium using dividing cells such
as embryonic stem cells may aid in furthering our understanding of the interaction between
mitochondrial dysfunction and epigenetic aberrations. Third, we used immunocytochemistry to
measure the amount of 5mc and 5hmc in cells. Although we used previously published methods and
tested for cross-reactivity, immunofluorescence is limited in that it only allows for the assessment of
global levels. Using more specific techniques such as MeDIP-Seq and hMeDIP-Seq in future studies
to verify the findings of the present study may aid in further elucidating the effect of rotenone and
lithium on methylation and hydroxymethylation (Matarese, Carrillo-de Santa Pau, & Stunnenberg,
2011).
In conclusion, the findings of our study are the first to demonstrate the ability of
mitochondrial complex I dysfunction induced by rotenone to increase methylation and
hydroxymethylation in primary neurons, and to show that lithium is able to ameliorate these
alterations. Moreover, our findings showed that in contrast to rotenone, H2O2 decreased 5mc levels
without changing the levels of 5hmc. This suggests that changes in methylation and
hydroxymethylation levels following rotenone-induced suppression of complex I activity may not be

117
due to a general increase in oxidative stress. Although preliminary, these results suggest a potential
mechanism by which lithium reverts aberrations in methylation and hydroxymethylation levels in
patients with BD, and shed new light for further studies investigating the role of methylation/
hydroxymethylation pattern in BD.
Acknowledgement
The authors have no conflict of interest to disclose. The authors thank Brain & Behavior Research
Foundation (Formerly NARSAD, ACA), CNPq/Brazil (GS) and Canadian Institutes of Health
Research (CIHR) as sources of funding in support of this report (LTY and ACA).

118
Figures
Figure 1. Complex I activity measurement in rat cortical primary neurons with lithium pre-treatment
(0.75mM) and rotenone treatment at different concentrations (5nM, 10nM, and 50nM). Li(-)
indicates pre-treatment with media alone, and Li(+) indicates pre-treatment with lithium. Values are
expressed as % DMSO control, mean±SEM. **p < 0.01 against DMSO control; #p < 0.05 lithium
versus rotenone. N=4.

119
Figure 2. The effect of lithium pre-treatment (0.75mM) and rotenone treatment at different
concentrations (5nM, 10nM, 50nM) on ATP levels in primary cortical neurons. Li(-) indicates pre-
treatment with media alone, and Li(+) indicates pre-treatment with lithium. Values are expressed as
% DMSO control, mean±SEM. **p < 0.01 against DMSO control; ##p < 0.01 lithium versus
rotenone . N=4-8.

120
Figure 3. The effect of lithium pre-treatment (0.75mM) and rotenone treatment at different
concentrations (5nM, 10nM, 50nM) on cell mortality in primary cortical neurons measured by the
MTT assay. Li(-) indicates pre-treatment with media alone, and Li(+) indicates pre-treatment with
lithium. Values are expressed as % DMSO control, mean±SEM. **p < 0.01 against DMSO control;
##p < 0.01 lithium versus rotenone. N=5-8.
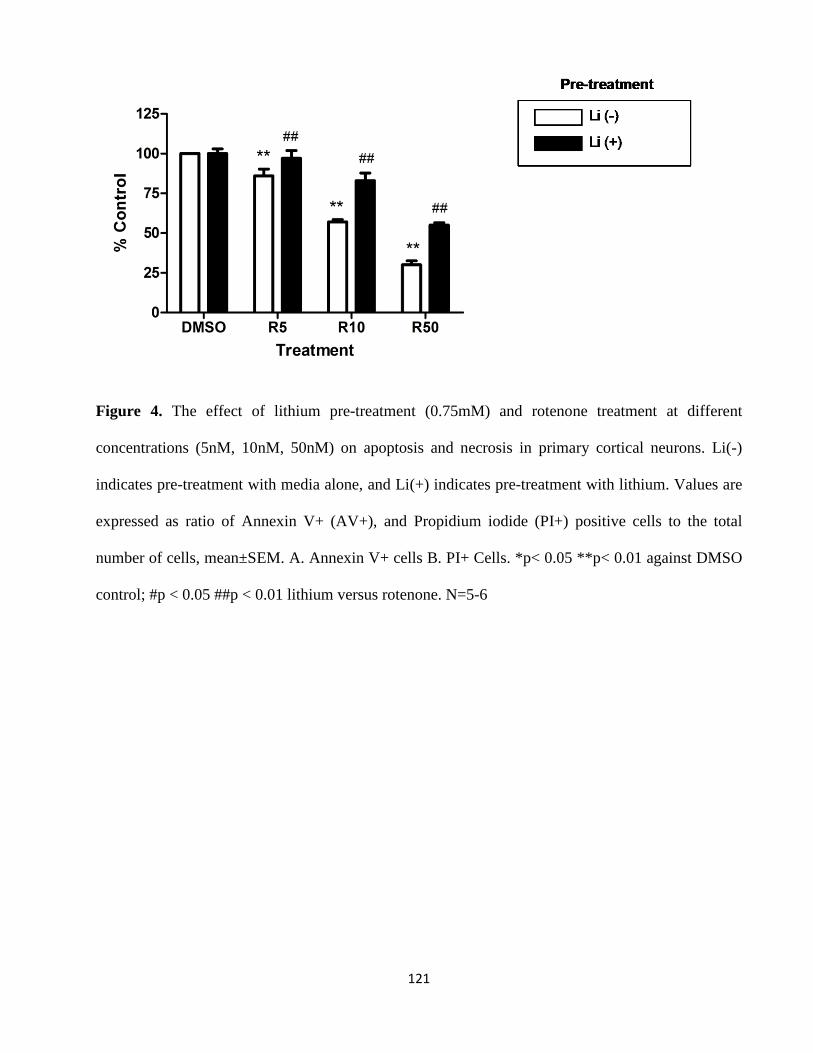
121
Figure 4. The effect of lithium pre-treatment (0.75mM) and rotenone treatment at different
concentrations (5nM, 10nM, 50nM) on apoptosis and necrosis in primary cortical neurons. Li(-)
indicates pre-treatment with media alone, and Li(+) indicates pre-treatment with lithium. Values are
expressed as ratio of Annexin V+ (AV+), and Propidium iodide (PI+) positive cells to the total
number of cells, mean±SEM. A. Annexin V+ cells B. PI+ Cells. *p< 0.05 **p< 0.01 against DMSO
control; #p < 0.05 ##p < 0.01 lithium versus rotenone. N=5-6

122
Figure 5. Changes in methylation and hydroxymethylation levels with 7 days of lithium pre-
treatment (0.75mM) and 30 minutes of rotenone treatment (5nM, 10nM, or 50nM) in primary
cortical neurons. Values are expressed as mean±SEM. A. Effect of lithium pre-treatment and
rotenone treatment on methylation levels. B. Effect of lithium pre-treatment and rotenone treatment
on hydroxymethylation levels. *p < 0.05 **p < 0.01 against DMSO control. N = 5. C. Images of
DAPI, 5-methylcytosine (5mc), and 5-hydroxymethylcytosine (5hmc) labeling with a confocal laser
scanning microscope and a 60X water immersion objective. DAPI is shown in blue, 5mc is shown in
red, and 5hmc is shown in green. Images are shown for DMSO control, rotenone treatment at 50nM,
and lithium pre-treatment (0.75mM) followed by rotenone treatment at 50nM. Images of 5mc and
5hmc for rotenone and lithium treatment in other doses are shown in Online Resource 2.

123
Figure 6. Changes in methylation and hydroxymethylation levels of 30 minutes of H2O2 treatment
(5µM, 10µM or 50µM) in primary cortical neurons. Values are expressed as mean±SEM. A. Effect
of H2O2 treatment on methylation levels. B. Effect of H2O2 treatment on hydroxymethylation levels.
*p < 0.05 **p < 0.01 against control. N = 5. C. Images of DAPI, 5-methylcytosine (5mc), and 5-
hydroxymethylcytosine (5hmc) labeling with a confocal laser scanning microscope and a 60X water
immersion objective. DAPI is shown in blue, 5mc is shown in red, and 5hmc is shown in green.
Images are shown for control, H2O2 treatment at 50µM. Images of 5mc and 5hmc for H2O2 treatment
in other doses are shown in Online Resource 3.

124
Schema 1. Mitochondrial complex I dysfunction induced by rotenone and it effect on cell viability
and DNA methylation and hydroxymethylation: the role of lithium.

125
Online Resource

126

127
Statement of significance and impact (Chapter 5a&b):
Downstream targets of complex I dysfunction was further examined in cell models, which
allow us to directly inhibit the electron transfer process in complex I using rotenone. Also, by
examining if lithium can decrease rotenone-induced alterations, we can observe if alterations
occurring from complex I defect are relevant to BD pathology. To examine downstream targets of
complex I dysfunction, we decided to measure protein oxidation and nitration, and methylation and
hydroxymethylation of DNA, addressing Aim 5. These factors were chosen because oxidative
damage to proteins and changes in methylation levels were reported in patients with BD (Andreazza
et al., 2010; D'Addario et al., 2012), and our lab has experience in measuring these factors using
different samples (Andreazza et al., 2010; Andreazza et al., 2013; Huzayyin et al., 2014). To
examine the effects of complex I dysfunction and lithium pre-treatment, we used one-way ANOVA.
We chose this statistical model because one-way ANOVA allows us to compare between the cells
that received the same pre-treatment (lithium vs. media only). For example, lithium was found to
increase cell viability on its own, which may lead to a significant main effect of lithium in a two-way
ANOVA model, even if lithium is unable to prevent the decrease in cell viability produced by an
assault (Allagui et al., 2009). By using one-way ANOVA, cells pre-treated with lithium followed by
rotenone will be compared against cells pre-treated with lithium only without rotenone treatment,
allowing us to specifically examine the protective effects of lithium. T-tests were used to compare
cells pre-treated with lithium against cells that received no pre-treatment.
First, we wanted to examine if complex I dysfunction is sufficient to produce oxidation and
nitration to proteins using SH-SY5Y cells. This study demonstrated that disrupting the electron
transfer process in complex I with rotenone increases oxidative and nitrosative modifications to
proteins. Previous studies have shown that lithium increases levels of complex I subunits and its
activity, suggesting that lithium may be able to prevent oxidative damage to proteins by improving

128
complex I function (Maurer et al., 2009; Sun et al., 2006). On the other hand, lithium was also
shown to increase glutathione levels as well as enzymes related to glutathione function, such as
glutathione peroxidase, suggesting that multiple factors are involved in lithium’s antioxidant
capabilities (Cui et al., 2007). The findings of this study demonstrated that lithium can decrease
carbonylation and nitration induced by rotenone treatment, suggesting that protein oxidation and
nitration from complex I dysfunction may be a part of BD pathology. Glutathione was found to be
critical in lithium’s ability to prevent cell death and carbonylation, suggesting that improvement of
complex I activity alone may not be sufficient to prevent against damages produced by complex I
inhibition.
The next cell study aimed to examine another potential downstream target of complex I
dysfunction, methylation and hydroxymethylation of DNA. To examine this, we pre-treated rat
cortical primary neurons with lithium followed by rotenone treatment, and measured levels of
methylation and hydroxymethylation using immunocytochemistry. The results demonstrated that
complex I inhibition with rotenone increases methylation as well as hydroxymethylation, suggesting
that methylation and hydroxymethylation may also be downstream targets of mitochondrial complex
I dysfunction. Surprisingly, it was also shown that complex I inhibition is most likely causing these
changes through a different pathway than oxidative stress, as hydrogen peroxide produced a different
pattern of alteration, where it decreased levels of methylation while levels of 5-
hydroxymethylcytosine were not affected. Complex I inhibition has other consequences in addition
to superoxide anion production, such as decreased ATP production and activation of apoptotic
mechanisms as demonstrated in this study, which could be contributing to alterations in 5-
methylcytosine and 5-hydroxymethylcytosine levels. Lithium was found to protect against the
effects of rotenone, suggesting that methylation and hydroxymethylation of DNA occurring as a
downstream effect of complex I dysfunction may have a role in BD pathology.

129
6. SUMMARY AND GENERAL DISCUSSION
Complex I dysfunction and oxidative stress are consistently reported in BD, where lower
protein and mRNA levels of complex I subunits, decreased complex I activity, and increased
oxidative modifications to proteins, lipids, and DNA have been found (Andreazza et al., 2010;
Andreazza et al., 2013; Sun et al., 2006). Complex I is the first member of the electron transport
chain that can produce oxidative stress through the leakage of electrons when dysfunctional
(Halliwell, 1992). Therefore, the overall aim of my PhD was to examine downstream targets of
complex I dysfunction in BD.
My first project aimed to examine if complex I dysfunction in the electron transport process
is specific to BD by reviewing microarray studies that measured expression levels of complex I
subunits in patients with BD or schizophrenia. Our findings revealed that the differentiating factor
between BD and schizophrenia is that complex I subunits that are specifically involved in the
electron transfer process are downregulated in BD, while those altered in schizophrenia are involved
in other functions such as proton pumping or structural support. Therefore, while other aspects of
mitochondrial dysfunction, such as lower production of ATP and activation of apoptotic pathways
may be common to BD and schizophrenia, mitochondrial generation of ROS through leakage of
electrons from complex I may be more specific to BD (Scola et al., 2013).
As complex I dysfunction and subsequent production of mitochondrial ROS is most likely
unique to this disorder and may be an important contributor to its pathophysiology, my first study in
post-mortem brain aimed to confirm complex I dysfunction in the frontal cortex. To do this, I
measured levels of all five complexes of the electron transport chain to test if decrease in complex I
is unique to this complex, or is a part a general downregulation of electron transport chain
components. The results showed that while complexes II, III, IV and V had no between-group
differences, complex I was lower in patients with BD compared to non-psychiatric controls,

130
suggesting that complex I dysfunction in BD is specific. To further examine the potential of this
complex I defect to lead to increased generation of ROS, I measured levels of NDUFS7, a complex I
subunit that is critical for the fidelity of the electron transfer process (Andreazza et al., 2010; Lebon
et al., 2007). NDUFS7 levels were lower in patients with BD compared to non-psychiatric controls
and patients with schizophrenia, adding support to the aforementioned review that complex I
dysfunction most likely contributes to oxidative stress in BD. Moreover, two previous studies from
our lab demonstrated lower NDUFS7 levels in patients with BD (Andreazza et al., 2010; Andreazza
et al., 2013), suggesting that this is a very consistent finding.
Following the confirmation of complex I dysfunction in the post-mortem brain of patients
with BD, I aimed to extend on the findings of a recent study from our group demonstrating the
mitochondria and the synapse as targets of oxidative stress in the frontal cortex of patients with BD
(Andreazza et al., 2013). For this, I aimed to examine the mitochondria and the synapse as potential
downstream targets of complex I dysfunction using post-mortem brains from patients with BD. First,
I examined the mitochondria as a downstream target by measuring the activation of the NLRP3-
inflamamsome, which is a marker of mitochondrial oxidative stress (Zhou et al., 2011). Furthermore,
activation of the NLRP3-inflammasome results in the production of inflammatory cytokines
(Schroder et al., 2010; Tschopp & Schroder, 2010; Zhong et al., 2013), making it an interesting
target to examine as increased inflammation is consistently found in patients with BD (Goldstein,
Kemp, et al., 2009; Leboyer et al., 2012; O'Brien et al., 2006; Rao et al., 2010). Using post-mortem
frontal cortex from patients with BD, schizophrenia and non-psychiatric controls, I found increased
levels of NLRP3 and ASC only in the crude mitochondrial fraction in patients with BD, suggesting
increased activation of the NLRP3-inflamamsome and identifying the mitochondria as a potential
target of complex I dysfunction in BD (Zhou et al., 2011). On the other hand, levels of caspase-1 and
cytokines IL-1β, IL-6, TNF-α, and IL-10 were increased in BD and schizophrenia, suggesting

131
increased inflammation in both disorders. This also suggests that while complex I dysfunction and
mitochondrial damage as a downstream target of complex I defect may be specific to BD,
inflammation may be common to both BD and schizophrenia.
The next study examined the synapse as a potential downstream target of complex I
dysfunction in BD, as the synapse was identified as another target of oxidative stress in the post-
mortem brain of patients with BD (Andreazza et al., 2013). Because increased dopamine signaling is
known to underlie mania (Berk et al., 2007) and the dopamine system mainly exists at the synapse
(Elsworth & Roth, 1997), we decided to examine oxidative and nitrosative modifications to areas
rich in dopamine as a marker of synaptic damage due to complex I defect. For this, we examined the
post-mortem prefrontal cortex of patients with BD, schizophrenia, major depression and non-
psychiatric individuals. Dopamine transporter (DAT) and tyrosine hydroxlase (TH) were chosen to
label dopamine rich areas because they have been used by previous studies to label dopaminergic
synapses along axons (Akil et al., 1999; Ciliax et al., 1999), and both have been demonstrated to be
vulnerable to oxidative modifications in vitro (Ara et al., 1998; Blanchard-Fillion et al., 2001; S. U.
Park et al., 2002). Findings of this study revealed that oxidation of DAT-immunoreactive regions are
increased patients with BD as measured by acceptor photobleaching FRET, which has a resolution
of 30-50nm with the immunohistochemistry technique that was used in this study (Kenworthy, 2001;
König et al., 2006), allowing us to examine oxidative modifications to the dopaminergic synapase.
Using the same technique, nitration in areas immunoreactive for TH was found to be decreased in
patients with BD and schizophrenia. This was contrary to my hypothesis, as complex I dysfunction
in BD was expected to contribute to greater levels of oxidation and nitration of dopaminergic
synapses. Patients with BD and schizophrenia can both exhibit psychosis, which is thought to be
caused in part by increased levels of dopamine at the synapse (Howes & Kapur, 2009; Pearlson et
al., 1995; Wong et al., 1997). Since dopamine and L-DOPA are favorable substrates for

132
peroxynitrite (S. Park et al., 2003), which is a strong nitrating agent (Greenacre & Ischiropoulos,
2001; Kamat, 2006), patients with BD and SCZ may have lower levels of nitration in dopamine-rich
areas as peroxynitrite may be consumed by its reaction with dopamine at the synaptic terminal.
Therefore, the findings of this study suggest that the synapse may be another potential downstream
target of complex I dysfunction in BD.
We also aimed to further examine downstream targets of complex I dysfunction by using cell
models, allowing us to directly inhibit the electron transfer process in complex I using rotenone
(Turrens & Boveris, 1980). Also, lithium, which is uniquely used for the treatment of BD and was
shown to improve complex I function (J. S. Lai et al., 2006; Maurer et al., 2009; Sun et al., 2006),
was used as a proxy to understand if downstream alterations caused by complex I inhibition may be
relevant to BD pathology. To examine the consequences of complex I dysfunction, we measured
protein oxidation and nitration, and methylation and hydroxymethylation of DNA as oxidative
modifications and methylation alterations were found in patients with BD, and our group has
examined these factors in patients with BD using different samples (Abdolmaleky et al., 2006;
Andreazza et al., 2010; Andreazza et al., 2013; D'Addario et al., 2012; Huzayyin et al., 2014). First,
using human neuroblastoma cells (SH-SY5Y), we examined if complex I inhibition produced by
rotenone is sufficient to cause protein oxidation and nitration, and if lithium can prevent these
changes by improving complex I activity or through the glutathione system, which is the most
important antioxidant system in the brain (Gawryluk et al., 2011). Rotenone, as expected, decreased
complex I activity, and increased protein carbonylation and nitration in a dose-dependent manner.
This suggests that complex I dysfunction in the electron transfer process is an important contributor
to oxidative stress, resulting in increased oxidation and nitration of proteins. Furthermore, lithium
pre-treatment was able to improve complex I activity, and decrease protein nitration and
carbonylation produced by complex I inhibition from rotenone. The results also suggested that

133
lithium may be doing this by improving complex I function to offset the inhibitory effects of
rotenone, as cells pre-treated with lithium showed higher complex I activity compared to cells pre-
treated with only media regardless of the presence of rotenone. This suggests that protein oxidation
and nitration from complex I dysfunction may be contributing to the pathology of BD (Maurer et al.,
2009). The ability of lithium to protect against rotenone induced cell death was abolished with the
addition of buthionine sulfoximine to deplete glutathione, again demonstrating that the main
mechanism of toxicity exerted by blocking electron transfer in complex I is through the generation
of oxidative stress. Also, lithium was no longer able to decrease protein carbonylation produced by
rotenone after blocking glutathione synthesis, suggesting that lithium’s ability to increase glutathione
levels are also important for its antioxidant effects. Surprisingly, buthionine sulfoximine did not
affect lithium’s ability to prevent nitration, suggesting that lithium’s ability to decrease nitration does
not significantly depend on glutathione. As superoxide anion is required for the formation of
peroxynitrite, a powerful nitrating reagent, lithium may be decreasing nitration by inhibiting
superoxide production from complex I. While this may be through decreasing nitric oxide
production, findings of previous studies examining lithium’s effect on nitric oxide synthase are
inconsistent (Karimollah et al., 2009; Nahman, Belmaker, & Azab, 2012; Wegener et al., 2004).
The next study further examined downstream effects of complex I dysfunction using cell
models by measuring methylation and hydroxymethylation of DNA following rotenone treatment.
Also, as our previous study demonstrated that lithium is able to protect against oxidative damage
produced by complex I inhibition, it was of interest to see if lithium could protect against another
downstream target of complex I dysfunction. Findings of our study showed that inhibiting complex I
with rotenone increased global levels of methylation and hydroxymethylation, suggesting that these
alterations may also be downstream targets of complex I dysfunction. In order to examine if these
changes were occurring as a consequence of oxidative stress produced by complex I inhibition, we

134
measured methylation and hydroxymethylation in cells treated with hydrogen peroxide. Surprisingly,
hydrogen peroxide produced a decrease in methylation and produced no changes in
hydroxymethylation, suggesting that changes that were observed with rotenone were due to other
effects of complex I inhibition other than generation of oxidative stress. Results from this study as
well as previous studies have shown that complex I dysfunction causes activation of apoptotic
pathways (Eckert et al., 2003), and inducing apoptosis in neurons was found to increase methylation
(Chestnut et al., 2011), suggesting that this could be the mechanism by which complex I dysfunction
is producing changes in methylation. Hence, lithium may be preventing rotenone-induced changes in
methylation and hydroxylation by improving complex I function, and therefore decreasing complex I
inhibition-induced apoptosis. In fact, lithium, in addition to improving complex I function, is also
known to decrease apoptosis (Chuang, 2005; Q. Li et al., 2010), possibly contributing to its ability to
prevent methylation changes. Mechanisms that underlie changes in hydroxymethylation patterns are
less well-known as it was discovered more recently (Song et al., 2011; Tahiliani et al., 2009).
Together, the findings of this study suggest that complex I dysfunction may cause alterations in
methylation and hydroxymethylation, and that this may be relevant to the pathophysiology of BD, as
lithium pre-treatment was able to prevent rotenone-induced alterations. Hence, by inducing complex
I dysfunction in the electron transfer process using rotenone and measuring its effects on methylation
and hydroxymethylation, this study extended the examination of downstream effects of complex I
dysfunction in BD.
Our findings suggest that complex I dysfunction may have an important role in the
pathophysiology of BD. For example, complex I dysfunction, by increasing the production of
oxidative stress, may contribute to oxidative modifications to enzymes and transporters, such as
those in the dopamine system. Dopamine dysregulation theory of BD states that increased dopamine
levels underlie mania, which is followed by a subsequent downregulation of dopaminergic signaling,

135
resulting in depression (Berk et al., 2007). Oxidative and nitrosative modifications to dopaminergic
proteins with cysteine or tyrosine residues, such as the dopamine transporter or tyrosine hydroxylase,
may contribute to this imbalance and to the cyclic nature of BD. For example, accumulating
oxidative modifications to dopaminergic proteins could result in increased production or decreased
uptake of dopamine from the synapse, resulting in increased dopamine signaling, which may
contribute to mania (H. K. Kim & Andreazza, 2012). According to Berk and colleagues (Berk et al.,
2007), excessive activation of the dopamine system can be followed by downregulation of
dopaminergic proteins, which may result in depression. Furthermore, according to kindling theory of
BD (Weiss et al., 2015), due to the increased vulnerability of the system from various neurological
defects including oxidative stress, the brain may be more vulnerable to experiencing a similar
hyperactivation upon facing a trigger, such as stress, resulting in another episode of mania and
contributing to the cyclical nature of BD. Dysregulation of the dopamine system in the prefrontal
and frontal cortices may also contribute to the cognitive decline in BD, as it is a part of the
mesocortical pathway critical for working memory, emotional processing, and attention, which have
been shown to be impaired in BD (Nikolaus, Antke, & Muller, 2009; Seamans & Yang, 2004).
Complex I dysfunction can also activate downstream signaling pathways through redox sensors,
such as the NLRP3-inflammasome (Zhou et al., 2011). Our study found increased activation of the
NLRP3-inflamamsome in patients with BD, suggesting that complex I dysfunction may be
contributing to increased inflammation in the frontal cortex of patients with BD as well.
Inflammation can lead to pyroptosis and apoptosis through the release of cytokines that activate the
NF-κB pathway, possibly contributing to neuronal and glial cell death and cognitive decline
(Shimada et al., 2012; Sollberger et al., 2014; Takuma, Baba, & Matsuda, 2004). Inflammation is
also hypothesized to play an important role in the progressive decline that is experienced by many
patients with BD (Berk et al., 2011). The strongest predictor of response to treatment is the number

136
of previous episodes, and cognitive performance in various tasks decreases with disease duration, all
suggesting that BD is a progressive disorder (Anderson et al., 2012; Berk et al., 2013; Kupfer, 2005).
One hypothesis states that since cells of monocytic origin, such as the microglia, react stronger and
faster to various assaults upon multiple activations due to immune memory, patients with BD may
become increasingly vulnerable to inflammatory activation with increasing number of episodes
(Barbosa et al., 2014), contributing to the progressive decline in this disease. Hence, these findings
suggest that complex I dysfunction may be contributing to the cyclical nature of BD by causing
dopamine dysregulation through oxidative modifications to dopaminergic proteins, and contributing
to the progressive nature of this disease by increasing the vulnerability of the brain through the
production of oxidative stress and inflammatory activation. Interestingly, while evidence suggesting
complex I dysfunction has been reported in other brain areas as well, such as the hippocampus
(Konradi et al., 2004), accumulating studies are suggesting that complex I defect and oxidative stress
are very consistent findings in the frontal and prefrontal cortices of patients with BD (Andreazza et
al., 2010; Andreazza et al., 2013; Frey et al., 2007; Gawryluk et al., 2011; Sun et al., 2006).
BD is a chronic and progressive disorder (Berk et al., 2011) where the development of more
efficacious treatments is of great interest (Aubry, Ferrero, & Schaad, 2007; Berk et al., 2011). Our
studies included in this thesis strongly suggest that complex I dysfunction may be an important
contributor to BD, suggesting that drugs that ameliorate complex I defect may be effective on their
own or as adjunct treatments. N-acetyl cysteine, which increases glutathione levels, was shown to
improve depression in patients with BD (Berk et al., 2008), suggesting that targeting oxidative stress,
which could occur as a result of complex I defect, may be useful for the treatment of BD as well.
Coenzyme Q10 or ubiquinone, which receives electrons from complex I – a process disturbed by
rotenone – was shown to decrease depressive symptoms in older adult patients with BD (Forester et
al., 2012), further suggesting that improving the electron transfer process in complex I may be

137
beneficial for BD. To my knowledge, while drugs commonly used to treat BD such as valproate or
lithium were shown to improve complex I activity and decrease oxidative stress (Cui et al., 2007;
Frey et al., 2006; Jornada et al., 2011; Sun et al., 2006), drugs that directly improve complex I
function have yet to be tested in patients with BD. Melatonin, which first received attention in BD to
correct irregular sleep patterns (Salvadore et al., 2010; Srinivasan et al., 2006), is well known for its
antioxidant properties and was shown to increase expression levels of complex I genes (Acuña-
Castroviejo, Escames, León, Carazo, & Khaldy, 2003; Lowes, Almawash, Webster, Reid, & Galley,
2011; Srinivasan et al., 2006; Srinivasan, Spence, Pandi-Perumal, Brown, & Cardinali, 2011). These
findings suggest that melatonin may be efficacious as an adjunct in BD by improving complex I
function and decreasing oxidative stress. In fact, patients with BD were found to have genetic
variations in the melatonin gene (Etain et al., 2012), further suggesting that melatonin may have
positive effects on the treatment of BD. Moreover, melatonin was found to decrease metabolic
effects of antipsychotic medications such as increased blood pressure or weight gain in patients with
BD, demonstrating that melatonin is safe to use as an adjunct for the treatment of BD and that it may
have multiple beneficial effects (Romo-Nava et al., 2014). Hence, a randomized clinical trial
examining the effect of melatonin as an adjunct on symptom severity, cognitive decline, complex I
dysfunction and oxidative stress may be beneficial for the development of better treatments for BD.
To summarize, I first re-examined microarray studies measuring mRNA levels of complex I
subunits in patients with BD or SCZ, and determined that patients with BD have lower levels of
subunits that are involved in the process of electron transfer, suggesting that they may be more
susceptible to increased leakage of electrons and greater generation of ROS. I also confirmed lower
levels of complex I and its subunit, NDUFS7, in the post-mortem brain of patients with BD. After
confirming that complex I dysfunction in BD is likely to contribute to increased oxidative stress, I
aimed to extend on a previous study identifying the mitochondria and the synapse as two targets of

138
oxidative stress in the frontal cortex of patients with BD. To examine the mitochondria as a target of
complex I dysfunction, I examined activation of the NLRP3-inflammasome, a marker of complex I
dysfunction and mitochondrial production of ROS, and found that the activation of the NLRP3-
inflammasome is increased in the post-mortem frontal cortex of patients with BD, demonstrating the
mitochondria as a potential downstream target of complex I dysfunction in BD. The next study
aimed to examine the second potential target, the synapse, by focusing on the dopaminergic synapse
as dopamine dysregulation is known to underlie mania. This study demonstrated the synapse as a
potential target of complex I dysfunction in BD, as oxidative modifications to the dopaminergic
synapse were found to be altered in the post-mortem prefrontal cortex from patients with BD.
Downstream targets of complex I dysfunction were further examined using cell models, allowing us
to directly inhibit the electron transfer process in complex I with rotenone. Using cell models, I
could also examine the effect of lithium, which is uniquely used for the treatment of BD and can be
used to examine if alterations produced by rotenone may be relevant to BD pathology. Complex I
dysfunction produced by rotenone increased protein nitration and oxidation, and increased
methylation and hydroxymethylation of DNA. These alterations were decreased with lithium pre-
treatment, suggesting that downstream effects of complex I dysfunction may have important
pathological roles in BD. While much remains to be explored in the role of complex I dysfunction in
the pathophysiology of BD, these studies suggest that examining complex I defect and its
downstream pathways may increase our understanding of the different systems that are involved in
BD, and potentially reveal novel targets that can be used to improve therapeutic interventions.

139
7. CONCLUSIONS
The overall aim of my PhD was to elucidate downstream targets of complex I dysfunction in
BD. Our review of microarray studies examining complex I subunits in psychiatric disorders
revealed that the differentiating factor between BD and schizophrenia is the downregulation of
complex I subunits specifically involved in the process of electron transfer in BD, suggesting that
patients with BD may be more vulnerable to the generation of oxidative stress. We then confirmed
complex I dysfunction in post-mortem brains of patients with BD, where patients with BD were
found to have lower levels of complex I, but not other members of the electron transport chain, and
also lower levels of NDUFS7, a complex I subunit that is critical for the electron transfer process.
After confirming complex I dysfunction in the frontal cortex of patients with BD, we aimed to
extend on a recent study published from our group, identifying the mitochondria and the synapse as
targets of oxidative stress in the post-mortem brain of patients with BD. I first examined the
mitochondria as a potential downstream target of complex I dysfunction in BD by measuring a
marker of mitochondrial oxidative stress, the NLRP3-inflamamsome, in the post-mortem frontal
cortex. Patients with BD were found to have increased activation of the NLRP3-inflammasome,
demonstrating the mitochondria as a potential downstream target of complex I dysfunction in BD. I
then examined the synapse as a downstream target of complex I defect by examining oxidative
modifications to the dopaminergic synapse, as dopamine dysregulation is known to underlie mania.
Results showed altered levels of oxidation and nitration in areas immunoreactive for markers of the
dopamine synapse, suggesting that complex I dysfunction may also target the synapse in BD. To
further examine downstream effects of complex I dysfunction, we used cell models, allowing us to
directly inhibit the electron transfer process in complex I using rotenone. We also examined the
effect of lithium, as it is uniquely used for the treatment of BD and can aid us in determining if
alterations occurring due to complex I inhibition with rotenone may be relevant to BD pathology.

140
Results revealed that complex I dysfunction produced by rotenone causes an increase in protein
oxidation and nitration and methylation and hydroxymethylation of DNA, suggesting that complex I
defect in the electron transfer process can directly cause potentially pathological alterations
downstream. Importantly, lithium pre-treatment was able to decrease these alterations caused by
rotenone, suggesting that consequences of complex I dysfunction may have pathological
implications in BD.
Our findings in post-mortem brain and cell models suggest that complex I dysfunction and its
downstream targets have important roles in the pathophysiology of BD. In vitro and animal studies
have shown that complex I dysfunction can lead to other downstream alterations as well (Beal, 2002;
Halliwell, 1992). Of interest, oxidative stress from complex I defect can increase intracellular
calcium levels (Persson et al., 2013; L. S. Zheng, Ishii, Zhao, Kondo, & Sasahara, 2013), which has
been consistently reported in patients with BD (Dubovsky et al., 1994; Perova et al., 2010; Perova,
Wasserman, Li, & Warsh, 2008), suggesting that complex I dysfunction may have multiple effects in
BD. Future directions include confirmation of these findings using different techniques. For
example, while DAT and TH immunoreactive regions were found to be targets of oxidative
modifications using FRET, most likely representing alterations of DAT and TH themselves,
confirming this using a biochemical technique, such as co-immunoprecipitation would help solidify
the dopaminergic synapse as a downstream target of complex I dysfunction. Furthermore, as
identification of biomarkers in BD is of great interest, examining whether markers of complex I
dysfunction, such as the NLRP3-inflammasome, can be measured in peripheral cells from patients
with BD may yield interesting results. This will also allow us to confirm our findings using a larger
sample, increasing the generalizability of the results. Also, using animal models to confirm the
findings of our studies using neuroblastoma cells and primary neurons would allow for the validation
of the identified mechanisms in a more complex system. Lastly, as lithium was demonstrated to be

141
effective in preventing downstream effects of complex I inhibition through its ability to improve
complex I functioning, examining the effect of other reagents that were shown to improve complex I
activity as an adjunct may contribute to developing better treatments for BD.

142
References
Abdolmaleky, H. M., Cheng, K. H., Faraone, S. V., Wilcox, M., Glatt, S. J., Gao, F., . . . Thiagalingam, S. (2006). Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum Mol Genet, 15(21), 3132-3145. doi: 10.1093/hmg/ddl253
Acuña-Castroviejo, D., Escames, G., León, J., Carazo, A., & Khaldy, H. (2003). Mitochondrial regulation by melatonin and its metabolites. Adv Exp Med Biol, 527, 549-557.
Agostini, L., Martinon, F., Burns, K., McDermott, M. F., Hawkins, P. N., & Tschopp, J. (2004). NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity, 20(3), 319-325.
Ahluwalia, P., Grewaal, D. S., & Singhal, R. L. (1981). Brain gabaergic and dopaminergic systems following lithium treatment and withdrawal. Prog Neuropsychopharmacol, 5(5-6), 527-530.
Akil, M., Pierri, J. N., Whitehead, R. E., Edgar, C. L., Mohila, C., Sampson, A. R., & Lewis, D. A. (1999). Lamina-specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am J Psychiatry, 156(10), 1580-1589.
Allagui, M. S., Nciri, R., Rouhaud, M. F., Murat, J. C., El Feki, A., Croute, F., & Vincent, C. (2009). Long-term exposure to low lithium concentrations stimulates proliferation, modifies stress protein expression pattern and enhances resistance to oxidative stress in SH-SY5Y cells. Neurochem Res, 34(3), 453-462. doi: 10.1007/s11064-008-9804-8
Almeida, J. R., Akkal, D., Hassel, S., Travis, M. J., Banihashemi, L., Kerr, N., . . . Phillips, M. L. (2009). Reduced gray matter volume in ventral prefrontal cortex but not amygdala in bipolar disorder: significant effects of gender and trait anxiety. Psychiatry Res, 171(1), 54-68.
Anand, A., Verhoeff, P., Seneca, N., Zoghbi, S. S., Seibyl, J. P., Charney, D. S., & Innis, R. B. (2000). Brain SPECT imaging of amphetamine-induced dopamine release in euthymic bipolar disorder patients. Am J Psychiatry, 157(7), 1108-1114.
Anderson, I. M., Haddad, P. M., & Scott, J. (2012). Bipolar disorder. BMJ, 345, e8508. Andreazza, A. C., Cassini, C., Rosa, A. R., Leite, M. C., de Almeida, L. M., Nardin, P., . . .
Goncalves, C. A. (2007). Serum S100B and antioxidant enzymes in bipolar patients. J Psychiatr Res, 41(6), 523-529.
Andreazza, A. C., Kapczinski, F., Kauer-Sant'Anna, M., Walz, J. C., Bond, D. J., Goncalves, C. A., . . . Yatham, L. N. (2009). 3-Nitrotyrosine and glutathione antioxidant system in patients in the early and late stages of bipolar disorder. J Psychiatry Neurosci, 34(4), 263-271.
Andreazza, A. C., Kauer-Sant'anna, M., Frey, B. N., Bond, D. J., Kapczinski, F., Young, L. T., & Yatham, L. N. (2008). Oxidative stress markers in bipolar disorder: a meta-analysis. J Affect Disord, 111(2-3), 135-144.
Andreazza, A. C., Shao, L., Wang, J. F., & Young, L. T. (2010). Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch Gen Psychiatry, 67(4), 360-368.
Andreazza, A. C., Wang, J. F., Salmasi, F., Shao, L., & Young, L. T. (2013). Specific subcellular changes in oxidative stress in prefrontal cortex from patients with bipolar disorder. J Neurochem. doi: 10.1111/jnc.12316
Ara, J., Przedborski, S., Naini, A. B., Jackson-Lewis, V., Trifiletti, R. R., Horwitz, J., & Ischiropoulos, H. (1998). Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Proc Natl Acad Sci U S A, 95(13), 7659-7663.

143
Asanuma, M., Miyazaki, I., Diaz-Corrales, F. J., & Ogawa, N. (2004). Quinone formation as dopaminergic neuron-specific oxidative stress in the pathogenesis of sporadic Parkinson's disease and neurotoxin-induced parkinsonism. Acta Med Okayama, 58(5), 221-233.
Aubry, J.-M., Ferrero, F.-P., & Schaad, N. (2007). Pharmacotherapy of bipolar disorders (pp. xiii, 277 p.). Retrieved from http://myaccess.library.utoronto.ca/login?url=http://books.scholarsportal.info/viewdoc.html?id=/ebooks/ebooks2/wiley/2011-12-13/1/9780470510421 Retrieved from http://myaccess.library.utoronto.ca/login?url=http://onlinelibrary.wiley.com/book/10.1002/9780470510421
Bachmann, R. F., Schloesser, R. J., Gould, T. D., & Manji, H. K. (2005). Mood stabilizers target cellular plasticity and resilience cascades: implications for the development of novel therapeutics. Mol Neurobiol, 32(2), 173-202.
Bachmann, R. F., Wang, Y., Yuan, P., Zhou, R., Li, X., Alesci, S., . . . Manji, H. K. (2009). Common effects of lithium and valproate on mitochondrial functions: protection against methamphetamine-induced mitochondrial damage. Int J Neuropsychopharmacol, 12(6), 805-822. doi: S1461145708009802 [pii]
10.1017/S1461145708009802
Barbosa, I. G., Huguet, R. B., Mendonça, V. A., Sousa, L. P., Neves, F. S., Bauer, M. E., & Teixeira, A. L. (2011). Increased plasma levels of soluble TNF receptor I in patients with bipolar disorder. Eur Arch Psychiatry Clin Neurosci, 261(2), 139-143. doi: 10.1007/s00406-010-0116-z
Barbosa, I. G., Morato, I. B., de Miranda, A. S., Bauer, M. E., Soares, J. C., & Teixeira, A. L. (2014). A preliminary report of increased plasma levels of IL-33 in bipolar disorder: further evidence of pro-inflammatory status. J Affect Disord, 157, 41-44. doi: 10.1016/j.jad.2013.12.042
Barbosa, I. G., Rocha, N. P., Bauer, M. E., de Miranda, A. S., Huguet, R. B., Reis, H. J., . . . Teixeira, A. L. (2013). Chemokines in bipolar disorder: trait or state? Eur Arch Psychiatry Clin Neurosci, 263(2), 159-165. doi: 10.1007/s00406-012-0327-6
Barnett, J. H., & Smoller, J. W. (2009). The genetics of bipolar disorder. Neuroscience, 164(1), 331-343. doi: S0306-4522(09)00576-4 [pii]
10.1016/j.neuroscience.2009.03.080
Barzilai, A., & Yamamoto, K. (2004). DNA damage responses to oxidative stress. DNA Repair (Amst), 3(8-9), 1109-1115.
Bashkatova, V., Alam, M., Vanin, A., & Schmidt, W. J. (2004). Chronic administration of rotenone increases levels of nitric oxide and lipid peroxidation products in rat brain. Exp Neurol, 186(2), 235-241. doi: 10.1016/j.expneurol.2003.12.005
Bauer, M. (2004). Review: lithium reduces relapse rates in people with bipolar disorder. Evid Based Ment Health, 7(3), 72.
Beal, M. F. (2002). Oxidatively modified proteins in aging and disease. Free Radic Biol Med, 32(9), 797-803.
Bearden, C. E., Thompson, P. M., Dalwani, M., Hayashi, K. M., Lee, A. D., Nicoletti, M., . . . Soares, J. C. (2007). Greater cortical gray matter density in lithium-treated patients with bipolar disorder. Biol Psychiatry, 62(1), 7-16. doi: 10.1016/j.biopsych.2006.10.027
Bellizzi, D., D'Aquila, P., Giordano, M., Montesanto, A., & Passarino, G. (2012). Global DNA methylation levels are modulated by mitochondrial DNA variants. Epigenomics, 4(1), 17-27. doi: 10.2217/epi.11.109

144
Benes, F. M., Matzilevich, D., Burke, R. E., & Walsh, J. (2006). The expression of proapoptosis genes is increased in bipolar disorder, but not in schizophrenia. Mol Psychiatry, 11(3), 241-251.
Berk, M., Berk, L., Dodd, S., Cotton, S., Macneil, C., Daglas, R., . . . Malhi, G. S. (2013). Stage managing bipolar disorder. Bipolar Disord. doi: 10.1111/bdi.12099
Berk, M., Copolov, D. L., Dean, O., Lu, K., Jeavons, S., Schapkaitz, I., . . . Bush, A. I. (2008). N-acetyl cysteine for depressive symptoms in bipolar disorder--a double-blind randomized placebo-controlled trial. Biol Psychiatry, 64(6), 468-475.
Berk, M., Dodd, S., Kauer-Sant'anna, M., Malhi, G. S., Bourin, M., Kapczinski, F., & Norman, T. (2007). Dopamine dysregulation syndrome: implications for a dopamine hypothesis of bipolar disorder. Acta Psychiatr Scand Suppl(434), 41-49.
Berk, M., Kapczinski, F., Andreazza, A. C., Dean, O. M., Giorlando, F., Maes, M., . . . Malhi, G. S. (2011). Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci Biobehav Rev, 35(3), 804-817. doi: 10.1016/j.neubiorev.2010.10.001
Berman, K. F., Doran, A. R., Pickar, D., & Weinberger, D. R. (1993). Is the mechanism of prefrontal hypofunction in depression the same as in schizophrenia? Regional cerebral blood flow during cognitive activation. Br J Psychiatry, 162, 183-192.
Berman, S. B., & Hastings, T. G. (1999). Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson's disease. J Neurochem, 73(3), 1127-1137.
Berman, S. B., Zigmond, M. J., & Hastings, T. G. (1996). Modification of dopamine transporter function: effect of reactive oxygen species and dopamine. J Neurochem, 67(2), 593-600.
Bestor, T. (2000). The DNA methyltransferases of mammals. Human Molecular Genetics, 9(16), 2395-2402. doi: 10.1093/hmg/9.16.2395
Bhagwagar, Z., & Goodwin, G. M. (2002). Role of mood stabilizers in bipolar disorder. Expert Rev Neurother, 2(2), 239-248. doi: 10.1586/14737175.2.2.239
Bird, A. (2002). DNA methylation patterns and epigenetic memory. Genes Dev, 16(1), 6-21. doi: 10.1101/gad.947102
Blanchard-Fillion, B., Souza, J. M., Friel, T., Jiang, G. C., Vrana, K., Sharov, V., . . . Ischiropoulos, H. (2001). Nitration and inactivation of tyrosine hydroxylase by peroxynitrite. J Biol Chem, 276(49), 46017-46023. doi: M105564200 [pii]
10.1074/jbc.M105564200
Boufidou, F., Nikolaou, C., Alevizos, B., Liappas, I. A., & Christodoulou, G. N. (2004). Cytokine production in bipolar affective disorder patients under lithium treatment. J Affect Disord, 82(2), 309-313. doi: 10.1016/j.jad.2004.01.007
Boveris, A., & Chance, B. (1973). The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J, 134(3), 707-716.
Boyadjieva, N., & Varadinova, M. (2012). Epigenetics of psychoactive drugs. J Pharm Pharmacol, 64(10), 1349-1358. doi: 10.1111/j.2042-7158.2012.01475.x
Branco, M. R., Ficz, G., & Reik, W. (2012). Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet, 13(1), 7-13. doi: 10.1038/nrg3080
Brandt, U. (2006). Energy converting NADH:quinone oxidoreductase (complex I). Annu Rev Biochem, 75, 69-92. doi: 10.1146/annurev.biochem.75.103004.142539
Bresee, C., & Rapaport, M. H. (2009). Persistently increased serum soluble interleukin-2 receptors in continuously ill patients with schizophrenia. Int J Neuropsychopharmacol, 12(6), 861-865. doi: 10.1017/S1461145709000315

145
Brietzke, E., Stabellini, R., Grassis-Oliveira, R., & Lafer, B. (2011). Cytokines in Bipolar Disorder: Recent Findings, Deleterious Effects But Promise for Future Therapeutics. CNS Spectr.
Brown, N. C., Andreazza, A. C., & Young, L. T. (2014). An updated meta-analysis of oxidative stress markers in bipolar disorder. Psychiatry Res, 218(1-2), 61-68. doi: 10.1016/j.psychres.2014.04.005
Bryan, N. B., Dorfleutner, A., Kramer, S. J., Yun, C., Rojanasakul, Y., & Stehlik, C. (2010). Differential splicing of the apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes. J Inflamm (Lond), 7, 23. doi: 10.1186/1476-9255-7-23
Bryant, C., & Fitzgerald, K. A. (2009). Molecular mechanisms involved in inflammasome activation. Trends Cell Biol, 19(9), 455-464. doi: 10.1016/j.tcb.2009.06.002
Burgess, S., Geddes, J., Hawton, K., Townsend, E., Jamison, K., & Goodwin, G. (2001). Lithium for maintenance treatment of mood disorders. Cochrane Database Syst Rev(3), CD003013. doi: 10.1002/14651858.CD003013
Campos, A. C., Molognoni, F., Melo, F. H., Galdieri, L. C., Carneiro, C. R., D'Almeida, V., . . . Jasiulionis, M. G. (2007). Oxidative stress modulates DNA methylation during melanocyte anchorage blockade associated with malignant transformation. Neoplasia, 9(12), 1111-1121.
Chandana, R., Mythri, R. B., Mahadevan, A., Shankar, S. K., & Srinivas Bharath, M. M. (2009). Biochemical analysis of protein stability in human brain collected at different post-mortem intervals. Indian J Med Res, 129(2), 189-199.
Chanput, W., Mes, J. J., & Wichers, H. J. (2014). THP-1 cell line: An in vitro cell model for immune modulation approach. Int Immunopharmacol. doi: 10.1016/j.intimp.2014.08.002
Che, Y., Wang, J. F., Shao, L., & Young, T. (2010). Oxidative damage to RNA but not DNA in the hippocampus of patients with major mental illness. J Psychiatry Neurosci, 35(5), 296-302.
Chen, C. H., Suckling, J., Lennox, B. R., Ooi, C., & Bullmore, E. T. (2011). A quantitative meta-analysis of fMRI studies in bipolar disorder. Bipolar Disord, 13(1), 1-15. doi: 10.1111/j.1399-5618.2011.00893.x
Cheng, K. C., Cahill, D. S., Kasai, H., Nishimura, S., & Loeb, L. A. (1992). 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G----T and A----C substitutions. J Biol Chem, 267(1), 166-172.
Chestnut, B. A., Chang, Q., Price, A., Lesuisse, C., Wong, M., & Martin, L. J. (2011). Epigenetic regulation of motor neuron cell death through DNA methylation. J Neurosci, 31(46), 16619-16636. doi: 10.1523/JNEUROSCI.1639-11.2011
Choi, K. H., Higgs, B. W., Wendland, J. R., Song, J., McMahon, F. J., & Webster, M. J. (2011). Gene expression and genetic variation data implicate PCLO in bipolar disorder. Biol Psychiatry, 69(4), 353-359. doi: 10.1016/j.biopsych.2010.09.042
Chuang, D. M. (2005). The antiapoptotic actions of mood stabilizers: molecular mechanisms and therapeutic potentials. Ann N Y Acad Sci, 1053, 195-204.
Ciliax, B. J., Drash, G. W., Staley, J. K., Haber, S., Mobley, C. J., Miller, G. W., . . . Levey, A. I. (1999). Immunocytochemical localization of the dopamine transporter in human brain. J Comp Neurol, 409(1), 38-56.
Cipriani, A., Barbui, C., Salanti, G., Rendell, J., Brown, R., Stockton, S., . . . Geddes, J. R. (2011). Comparative efficacy and acceptability of antimanic drugs in acute mania: a multiple-treatments meta-analysis. Lancet, 378(9799), 1306-1315. doi: 10.1016/S0140-6736(11)60873-8
Clay, H. B., Sillivan, S., & Konradi, C. (2010). Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int J Dev Neurosci, 29(3), 311-324.

146
Connor, C. M., & Akbarian, S. (2008). DNA methylation changes in schizophrenia and bipolar disorder. Epigenetics, 3(2), 55-58.
Cordeiro, M. L., Gundersen, C. B., & Umbach, J. A. (2002). Lithium ions modulate the expression of VMAT2 in rat brain. Brain Res, 953(1-2), 189-194.
Cousins, D. A., Butts, K., & Young, A. H. (2009). The role of dopamine in bipolar disorder. Bipolar Disord, 11(8), 787-806. doi: 10.1111/j.1399-5618.2009.00760.x
Craddock, N., & Sklar, P. (2013). Genetics of bipolar disorder. Lancet, 381(9878), 1654-1662. doi: 10.1016/S0140-6736(13)60855-7
Cui, J., Shao, L., Young, L. T., & Wang, J. F. (2007). Role of glutathione in neuroprotective effects of mood stabilizing drugs lithium and valproate. Neuroscience, 144(4), 1447-1453.
Cunha, A. B., Frey, B. N., Andreazza, A. C., Goi, J. D., Rosa, A. R., Gonçalves, C. A., . . . Kapczinski, F. (2006). Serum brain-derived neurotrophic factor is decreased in bipolar disorder during depressive and manic episodes. Neurosci Lett, 398(3), 215-219. doi: 10.1016/j.neulet.2005.12.085
D'Addario, C., Dell'Osso, B., Palazzo, M. C., Benatti, B., Lietti, L., Cattaneo, E., . . . Altamura, A. C. (2012). Selective DNA methylation of BDNF promoter in bipolar disorder: differences among patients with BDI and BDII. Neuropsychopharmacology, 37(7), 1647-1655. doi: 10.1038/npp.2012.10
D'Ardenne, K., Eshel, N., Luka, J., Lenartowicz, A., Nystrom, L. E., & Cohen, J. D. (2012). Role of prefrontal cortex and the midbrain dopamine system in working memory updating. Proc Natl Acad Sci U S A, 109(49), 19900-19909. doi: 10.1073/pnas.1116727109
Daubner, S. C., Le, T., & Wang, S. (2011). Tyrosine hydroxylase and regulation of dopamine synthesis. Arch Biochem Biophys, 508(1), 1-12. doi: S0003-9861(10)00514-X [pii]
10.1016/j.abb.2010.12.017
Dean, B., Gibbons, A. S., Tawadros, N., Brooks, L., Everall, I. P., & Scarr, E. (2013). Different changes in cortical tumor necrosis factor-α-related pathways in schizophrenia and mood disorders. Mol Psychiatry, 18(7), 767-773. doi: 10.1038/mp.2012.95
Dean, O. M., Bush, A. I., & Berk, M. (2012). Translating the rosetta stone of N-acetylcysteine. Biol Psychiatry, 71(11), 935-936. doi: 10.1016/j.biopsych.2012.04.001
Deicken, R. F., Weiner, M. W., & Fein, G. (1995). Decreased temporal lobe phosphomonoesters in bipolar disorder. J Affect Disord, 33(3), 195-199.
Dheen, S. T., Kaur, C., & Ling, E. A. (2007). Microglial activation and its implications in the brain diseases. Curr Med Chem, 14(11), 1189-1197.
Dixon, J. F., & Hokin, L. E. (1998). Lithium acutely inhibits and chronically up-regulates and stabilizes glutamate uptake by presynaptic nerve endings in mouse cerebral cortex. Proc Natl Acad Sci U S A, 95(14), 8363-8368.
Dong, E., Gavin, D. P., Chen, Y., & Davis, J. (2012). Upregulation of TET1 and downregulation of APOBEC3A and APOBEC3C in the parietal cortex of psychotic patients. Transl Psychiatry, 2, e159. doi: 10.1038/tp.2012.86
Dringen, R. (2000). Glutathione metabolism and oxidative stress in neurodegeneration. Eur J Biochem, 267(16), 4903.
Dubovsky, S. L., Thomas, M., Hijazi, A., & Murphy, J. (1994). Intracellular calcium signalling in peripheral cells of patients with bipolar affective disorder. Eur Arch Psychiatry Clin Neurosci, 243(5), 229-234.
Dunigan, C. D., & Shamoo, A. E. (1995). Li+ stimulates ATP-regulated dopamine uptake in PC12 cells. Neuroscience, 65(1), 1-4.

147
Eckert, A., Keil, U., Marques, C. A., Bonert, A., Frey, C., Schüssel, K., & Müller, W. E. (2003). Mitochondrial dysfunction, apoptotic cell death, and Alzheimer's disease. Biochem Pharmacol, 66(8), 1627-1634.
Eder, C. (2009). Mechanisms of interleukin-1beta release. Immunobiology, 214(7), 543-553. doi: 10.1016/j.imbio.2008.11.007
El Khoury, A., Petterson, U., Kallner, G., Aberg-Wistedt, A., & Stain-Malmgren, R. (2002). Calcium homeostasis in long-term lithium-treated women with bipolar affective disorder. Prog Neuropsychopharmacol Biol Psychiatry, 26(6), 1063-1069.
Elsworth, J. D., & Roth, R. H. (1997). Dopamine synthesis, uptake, metabolism, and receptors: relevance to gene therapy of Parkinson's disease. Exp Neurol, 144(1), 4-9. doi: 10.1006/exnr.1996.6379
Elvidge, G., Jones, I., McCandless, F., Asherson, P., Owen, M. J., & Craddock, N. (2001). Allelic variation of a BalI polymorphism in the DRD3 gene does not influence susceptibility to bipolar disorder: results of analysis and meta-analysis. Am J Med Genet, 105(4), 307-311.
Etain, B., Dumaine, A., Bellivier, F., Pagan, C., Francelle, L., Goubran-Botros, H., . . . Jamain, S. (2012). Genetic and functional abnormalities of the melatonin biosynthesis pathway in patients with bipolar disorder. Hum Mol Genet, 21(18), 4030-4037. doi: 10.1093/hmg/dds227
Farinacci, M. (2007). Improved apoptosis detection in ovine neutrophils by annexin V and carboxyfluorescein diacetate staining. Cytotechnology, 54(3), 149-155. doi: 10.1007/s10616-007-9086-z
Fariss, M. W., Chan, C. B., Patel, M., Van Houten, B., & Orrenius, S. (2005). Role of mitochondria in toxic oxidative stress. Mol Interv, 5(2), 94-111. doi: 10.1124/mi.5.2.7
Felger, J. C., Li, L., Marvar, P. J., Woolwine, B. J., Harrison, D. G., Raison, C. L., & Miller, A. H. (2013). Tyrosine metabolism during interferon-alpha administration: association with fatigue and CSF dopamine concentrations. Brain Behav Immun, 31, 153-160. doi: 10.1016/j.bbi.2012.10.010
Felger, J. C., & Miller, A. H. (2012). Cytokine effects on the basal ganglia and dopamine function: the subcortical source of inflammatory malaise. Front Neuroendocrinol, 33(3), 315-327. doi: 10.1016/j.yfrne.2012.09.003
Ferreira, M. A., O'Donovan, M. C., Meng, Y. A., Jones, I. R., Ruderfer, D. M., Jones, L., . . . Craddock, N. (2008). Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet, 40(9), 1056-1058.
Ferrie, L., Young, A. H., & McQuade, R. (2006). Effect of lithium and lithium withdrawal on potassium-evoked dopamine release and tyrosine hydroxylase expression in the rat. Int J Neuropsychopharmacol, 9(6), 729-735.
Ficz, G., Branco, M. R., Seisenberger, S., Santos, F., Krueger, F., Hore, T. A., . . . Reik, W. (2011). Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature, 473(7347), 398-402. doi: 10.1038/nature10008
Fineberg, A. M., & Ellman, L. M. (2013). Inflammatory cytokines and neurological and neurocognitive alterations in the course of schizophrenia. Biol Psychiatry, 73(10), 951-966. doi: 10.1016/j.biopsych.2013.01.001
Fleckenstein, A. E., Metzger, R. R., Beyeler, M. L., Gibb, J. W., & Hanson, G. R. (1997). Oxygen radicals diminish dopamine transporter function in rat striatum. Eur J Pharmacol, 334(1), 111-114.
Forester, B. P., Zuo, C. S., Ravichandran, C., Harper, D. G., Du, F., Kim, S., . . . Renshaw, P. F. (2012). Coenzyme Q10 effects on creatine kinase activity and mood in geriatric bipolar depression. J Geriatr Psychiatry Neurol, 25(1), 43-50. doi: 10.1177/0891988712436688

148
Fountoulakis, K. N., Gonda, X., Siamouli, M., & Rihmer, Z. (2009). Psychotherapeutic intervention and suicide risk reduction in bipolar disorder: a review of the evidence. J Affect Disord, 113(1-2), 21-29. doi: 10.1016/j.jad.2008.06.014
Frey, B. N., Stanley, J. A., Nery, F. G., Monkul, E. S., Nicoletti, M. A., Chen, H. H., . . . Soares, J. C. (2007). Abnormal cellular energy and phospholipid metabolism in the left dorsolateral prefrontal cortex of medication-free individuals with bipolar disorder: an in vivo 1H MRS study. Bipolar Disord, 9 Suppl 1, 119-127.
Frey, B. N., Valvassori, S. S., Réus, G. Z., Martins, M. R., Petronilho, F. C., Bardini, K., . . . Quevedo, J. (2006). Effects of lithium and valproate on amphetamine-induced oxidative stress generation in an animal model of mania. J Psychiatry Neurosci, 31(5), 326-332.
Furlong, R. A., Coleman, T. A., Ho, L., Rubinsztein, J. S., Walsh, C., Paykel, E. S., & Rubinsztein, D. C. (1998). No association of a functional polymorphism in the dopamine D2 receptor promoter region with bipolar or unipolar affective disorders. Am J Med Genet, 81(5), 385-387.
Gambarana, C., Ghiglieri, O., Masi, F., Scheggi, S., Tagliamonte, A., & De Montis, M. G. (1999). The effects of long-term administration of rubidium or lithium on reactivity to stress and on dopamine output in the nucleus accumbens in rats. Brain Res, 826(2), 200-209.
Gawryluk, J. W., Wang, J. F., Andreazza, A. C., Shao, L., & Young, L. T. (2011). Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int J Neuropsychopharmacol, 14(1), 123-130.
Gerner, R. H., Post, R. M., & Bunney, W. E., Jr. (1976). A dopaminergic mechanism in mania. Am J Psychiatry, 133(10), 1177-1180.
Giros, B., el Mestikawy, S., Godinot, N., Zheng, K., Han, H., Yang-Feng, T., & Caron, M. G. (1992). Cloning, pharmacological characterization, and chromosome assignment of the human dopamine transporter. Mol Pharmacol, 42(3), 383-390.
Glahn, D. C., Bearden, C. E., Cakir, S., Barrett, J. A., Najt, P., Serap Monkul, E., . . . Soares, J. C. (2006). Differential working memory impairment in bipolar disorder and schizophrenia: effects of lifetime history of psychosis. Bipolar Disord, 8(2), 117-123. doi: 10.1111/j.1399-5618.2006.00296.x
Goldberg, J. F., Burdick, K. E., & Endick, C. J. (2004). Preliminary randomized, double-blind, placebo-controlled trial of pramipexole added to mood stabilizers for treatment-resistant bipolar depression. Am J Psychiatry, 161(3), 564-566.
Goldstein, B. I., Fagiolini, A., Houck, P., & Kupfer, D. J. (2009). Cardiovascular disease and hypertension among adults with bipolar I disorder in the United States. Bipolar Disord, 11(6), 657-662.
Goldstein, B. I., Kemp, D. E., Soczynska, J. K., & McIntyre, R. S. (2009). Inflammation and the phenomenology, pathophysiology, comorbidity, and treatment of bipolar disorder: a systematic review of the literature. J Clin Psychiatry, 70(8), 1078-1090. doi: 10.4088/JCP.08r04505
Goodwin, G. M., & Malhi, G. S. (2007). What is a mood stabilizer? Psychol Med, 37(5), 609-614. doi: 10.1017/S0033291706009305
Gracie, J. A., Robertson, S. E., & McInnes, I. B. (2003). Interleukin-18. J Leukoc Biol, 73(2), 213-224.
Graham, D. G. (1978). Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol, 14(4), 633-643.
Graham, D. G., Tiffany, S. M., Bell, W. R., Jr., & Gutknecht, W. F. (1978). Autoxidation versus covalent binding of quinones as the mechanism of toxicity of dopamine, 6-

149
hydroxydopamine, and related compounds toward C1300 neuroblastoma cells in vitro. Mol Pharmacol, 14(4), 644-653.
Grayson, D. R., & Guidotti, A. (2013). The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology, 38(1), 138-166. doi: 10.1038/npp.2012.125
Green, D. R., & Kroemer, G. (2004). The pathophysiology of mitochondrial cell death. Science, 305(5684), 626-629.
Greenacre, S. A., & Ischiropoulos, H. (2001). Tyrosine nitration: localisation, quantification, consequences for protein function and signal transduction. Free Radic Res, 34(6), 541-581.
Guo, J. U., Ma, D. K., Mo, H., Ball, M. P., Jang, M. H., Bonaguidi, M. A., . . . Song, H. (2011). Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci, 14(10), 1345-1351.
Guo, J. U., Su, Y., Zhong, C., Ming, G. L., & Song, H. (2011). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell, 145(3), 423-434. doi: 10.1016/j.cell.2011.03.022
Haddad, J. (2002). Antioxidant and prooxidant mechanisms in the regulation of redox(y)-sensitive transcription factors. Cellular Signalling, 14(11), 879-897. doi: 10.1016/S0898-6568(02)00053-0
Hallahan, B., Newell, J., Soares, J. C., Brambilla, P., Strakowski, S. M., Fleck, D. E., . . . McDonald, C. (2011). Structural magnetic resonance imaging in bipolar disorder: an international collaborative mega-analysis of individual adult patient data. Biol Psychiatry, 69(4), 326-335. doi: 10.1016/j.biopsych.2010.08.029
Halliwell, B. (1992). Reactive oxygen species and the central nervous system. J Neurochem, 59(5), 1609-1623.
Halliwell, B. (2001). Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging, 18(9), 685-716.
Halliwell, B. (2006). Oxidative stress and neurodegeneration: where are we now? J Neurochem, 97(6), 1634-1658.
Halliwell, B. (2007). Biochemistry of oxidative stress. Biochem Soc Trans, 35(Pt 5), 1147-1150. Halliwell, B., & Gutteridge, J. M. (1984). Free radicals, lipid peroxidation, and cell damage. Lancet,
2(8411), 1095. Halliwell, B., & Gutteridge, J. M. C. (2007). Free Radicals in Biology and Medicine (4 ed.). Oxford:
Clarendon Press. Hanamsagar, R., Torres, V., & Kielian, T. (2011). Inflammasome activation and IL-1β/IL-18
processing are influenced by distinct pathways in microglia. J Neurochem, 119(4), 736-748. doi: 10.1111/j.1471-4159.2011.07481.x
Hardan, A. Y., Fung, L. K., Libove, R. A., Obukhanych, T. V., Nair, S., Herzenberg, L. A., . . . Tirouvanziam, R. (2012). A randomized controlled pilot trial of oral N-acetylcysteine in children with autism. Biol Psychiatry, 71(11), 956-961. doi: 10.1016/j.biopsych.2012.01.014
Harish, G., Venkateshappa, C., Mahadevan, A., Pruthi, N., Srinivas Bharath, M. M., & Shankar, S. K. (2011). Glutathione metabolism is modulated by postmortem interval, gender difference and agonal state in postmortem human brains. Neurochem Int, 59(7), 1029-1042. doi: 10.1016/j.neuint.2011.08.024
Hashimoto, R., Takei, N., Shimazu, K., Christ, L., Lu, B., & Chuang, D. M. (2002). Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: an essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology, 43(7), 1173-1179.

150
Hastings, T. G. (2009). The role of dopamine oxidation in mitochondrial dysfunction: implications for Parkinson's disease. J Bioenerg Biomembr, 41(6), 469-472.
Hastings, T. G., Lewis, D. A., & Zigmond, M. J. (1996). Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc Natl Acad Sci U S A, 93(5), 1956-1961.
Hastings, T. G., & Zigmond, M. J. (1994). Identification of catechol-protein conjugates in neostriatal slices incubated with [3H]dopamine: impact of ascorbic acid and glutathione. J Neurochem, 63(3), 1126-1132.
Hernandez, D. G., & Singleton, A. B. (2012). Using DNA methylation to understand biological consequences of genetic variability. Neurodegener Dis, 9(2), 53-59. doi: 10.1159/000333097
Hope, S., Dieset, I., Agartz, I., Steen, N. E., Ueland, T., Melle, I., . . . Andreassen, O. A. (2011). Affective symptoms are associated with markers of inflammation and immune activation in bipolar disorders but not in schizophrenia. J Psychiatr Res, 45(12), 1608-1616. doi: 10.1016/j.jpsychires.2011.08.003
Howes, O. D., & Kapur, S. (2009). The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophr Bull, 35(3), 549-562. doi: 10.1093/schbul/sbp006
Huzayyin, A. A., Andreazza, A. C., Turecki, G., Cruceanu, C., Rouleau, G. A., Alda, M., & Young, L. T. (2013). Decreased global methylation in patients with bipolar disorder who respond to lithium. Int J Neuropsychopharmacol, 1-9. doi: 10.1017/S1461145713001569
Huzayyin, A. A., Andreazza, A. C., Turecki, G., Cruceanu, C., Rouleau, G. A., Alda, M., & Young, L. T. (2014). Decreased global methylation in patients with bipolar disorder who respond to lithium. Int J Neuropsychopharmacol, 17(4), 561-569. doi: 10.1017/S1461145713001569
Hynd, M., Lewohl, J., Scott, H., & Dodd, P. (2003). Biochemical and molecular studies using human autopsy brain tissue. Journal of Neurochemistry, 85(3), 543-562. doi: 10.1046/j.1471-4159.2003.01747.x
Ito, S., D'Alessio, A., Taranova, O., Hong, K., Sowers, L., & Zhang, Y. (2010). Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature, 466(7310), 1129-U1151. doi: 10.1038/nature09303
Iwamoto, K., Bundo, M., & Kato, T. (2005). Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum Mol Genet, 14(2), 241-253.
Iwamoto, K., Kakiuchi, C., Bundo, M., Ikeda, K., & Kato, T. (2004). Molecular characterization of bipolar disorder by comparing gene expression profiles of postmortem brains of major mental disorders. Mol Psychiatry, 9(4), 406-416.
Jaber, M., Jones, S., Giros, B., & Caron, M. G. (1997). The dopamine transporter: a crucial component regulating dopamine transmission. Mov Disord, 12(5), 629-633. doi: 10.1002/mds.870120502
Janssen, R. J., Nijtmans, L. G., van den Heuvel, L. P., & Smeitink, J. A. (2006). Mitochondrial complex I: structure, function and pathology. J Inherit Metab Dis, 29(4), 499-515. doi: 10.1007/s10545-006-0362-4
Jarskog, L. F. (2006). Apoptosis in schizophrenia: pathophysiologic and therapeutic considerations. Curr Opin Psychiatry, 19(3), 307-312. doi: 10.1097/01.yco.0000218603.25346.8f
Jeong, S. Y., & Seol, D. W. (2008). The role of mitochondria in apoptosis. BMB Rep, 41(1), 11-22. Jin, S., Kadam, S., & Pfeifer, G. (2010). Examination of the specificity of DNA methylation
profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Research, 38(11). doi: 10.1093/nar/gkq223
Jones, D. P. (2010). Redox sensing: orthogonal control in cell cycle and apoptosis signalling. J Intern Med, 268(5), 432-448. doi: 10.1111/j.1365-2796.2010.02268.x

151
Jornada, L. K., Valvassori, S. S., Steckert, A. V., Moretti, M., Mina, F., Ferreira, C. L., . . . Quevedo, J. (2011). Lithium and valproate modulate antioxidant enzymes and prevent ouabain-induced oxidative damage in an animal model of mania. J Psychiatr Res, 45(2), 162-168. doi: S0022-3956(10)00163-9 [pii] 10.1016/j.jpsychires.2010.05.011
Jurata, L. W., Bukhman, Y. V., Charles, V., Capriglione, F., Bullard, J., Lemire, A. L., . . . Altar, C. A. (2004). Comparison of microarray-based mRNA profiling technologies for identification of psychiatric disease and drug signatures. J Neurosci Methods, 138(1-2), 173-188. doi: 10.1016/j.jneumeth.2004.04.002
Kamat, J. P. (2006). Peroxynitrite: a potent oxidizing and nitrating agent. Indian J Exp Biol, 44(6), 436-447.
Kang, H. J., Voleti, B., Hajszan, T., Rajkowska, G., Stockmeier, C. A., Licznerski, P., . . . Duman, R. S. (2012). Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med, 18(9), 1413-1417. doi: nm.2886 [pii]
10.1038/nm.2886
Kang, K. A., Zhang, R., Kim, G. Y., Bae, S. C., & Hyun, J. W. (2012). Epigenetic changes induced by oxidative stress in colorectal cancer cells: methylation of tumor suppressor RUNX3. Tumour Biol, 33(2), 403-412. doi: 10.1007/s13277-012-0322-6
Karimollah, A. R., Ghasemi, M., Ghahremani, M. H., & Dehpour, A. R. (2009). Inhibition by lithium of the nitrergic relaxation of rat anococcygeus muscle. Nitric Oxide, 20(1), 31-38. doi: 10.1016/j.niox.2008.08.007
Kato, T. (2005). [Mitochondrial dysfunction in bipolar disorder]. Nihon Shinkei Seishin Yakurigaku Zasshi, 25(2), 61-72.
Kato, T. (2008). Role of mitochondrial DNA in calcium signaling abnormality in bipolar disorder. Cell Calcium, 44(1), 92-102.
Kato, T., & Kato, N. (2000). Mitochondrial dysfunction in bipolar disorder. Bipolar Disord, 2(3 Pt 1), 180-190.
Kato, T., Murashita, J., Kamiya, A., Shioiri, T., Kato, N., & Inubushi, T. (1998). Decreased brain intracellular pH measured by 31P-MRS in bipolar disorder: a confirmation in drug-free patients and correlation with white matter hyperintensity. Eur Arch Psychiatry Clin Neurosci, 248(6), 301-306.
Kato, T., Takahashi, S., Shioiri, T., & Inubushi, T. (1993). Alterations in brain phosphorous metabolism in bipolar disorder detected by in vivo 31P and 7Li magnetic resonance spectroscopy. J Affect Disord, 27(1), 53-59.
Kauer-Sant'Anna, M., Kapczinski, F., Andreazza, A. C., Bond, D. J., Lam, R. W., Young, L. T., & Yatham, L. N. (2009). Brain-derived neurotrophic factor and inflammatory markers in patients with early- vs. late-stage bipolar disorder. Int J Neuropsychopharmacol, 12(4), 447-458. doi: 10.1017/S1461145708009310
Keese, M., Magdeburg, R. J., Herzog, T., Hasenberg, T., Offterdinger, M., Pepperkok, R., . . . Bastiaens, P. I. (2005). Imaging epidermal growth factor receptor phosphorylation in human colorectal cancer cells and human tissues. J Biol Chem, 280(30), 27826-27831. doi: M504485200 [pii]
10.1074/jbc.M504485200
Kempton, M. J., Geddes, J. R., Ettinger, U., Williams, S. C., & Grasby, P. M. (2008). Meta-analysis, database, and meta-regression of 98 structural imaging studies in bipolar disorder. Arch Gen Psychiatry, 65(9), 1017-1032. doi: 10.1001/archpsyc.65.9.1017
Kenworthy, A. K. (2001). Imaging protein-protein interactions using fluorescence resonance energy transfer microscopy. Methods, 24(3), 289-296. doi: S1046-2023(01)91189-2 [pii]

152
10.1006/meth.2001.1189
Kilbourne, A. M., Cornelius, J. R., Han, X., Pincus, H. A., Shad, M., Salloum, I., . . . Haas, G. L. (2004). Burden of general medical conditions among individuals with bipolar disorder. Bipolar Disord, 6(5), 368-373. doi: 10.1111/j.1399-5618.2004.00138.x
Kim, H. K., & Andreazza, A. C. (2012). The relationship between oxidative stress and post-translational modification of the dopamine transporter in bipolar disorder. Expert Rev Neurother, 12(7), 849-859. doi: 10.1586/ern.12.64
Kim, H. K., Andreazza, A. C., Yeung, P. Y., Isaacs-Trepanier, C., & Young, L. T. (2014). Oxidation and nitration in dopaminergic areas of the prefrontal cortex from patients with bipolar disorder and schizophrenia. J Psychiatry Neurosci, 39(4), 276-285.
Kim, H. W., Rapoport, S. I., & Rao, J. S. (2010). Altered expression of apoptotic factors and synaptic markers in postmortem brain from bipolar disorder patients. Neurobiol Dis, 37(3), 596-603.
Kim, H. W., Rapoport, S. I., & Rao, J. S. (2011). Altered arachidonic acid cascade enzymes in postmortem brain from bipolar disorder patients. Mol Psychiatry, 16(4), 419-428. doi: 10.1038/mp.2009.137
Kim, Y. K., Jung, H. G., Myint, A. M., Kim, H., & Park, S. H. (2007). Imbalance between pro-inflammatory and anti-inflammatory cytokines in bipolar disorder. J Affect Disord, 104(1-3), 91-95. doi: 10.1016/j.jad.2007.02.018
King, T. D., Bijur, G. N., & Jope, R. S. (2001). Caspase-3 activation induced by inhibition of mitochondrial complex I is facilitated by glycogen synthase kinase-3beta and attenuated by lithium. Brain Res, 919(1), 106-114.
Konradi, C., Eaton, M., MacDonald, M. L., Walsh, J., Benes, F. M., & Heckers, S. (2004). Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch Gen Psychiatry, 61(3), 300-308.
Konradi, C., Sillivan, S. E., & Clay, H. B. (2012). Mitochondria, oligodendrocytes and inflammation in bipolar disorder: evidence from transcriptome studies points to intriguing parallels with multiple sclerosis. Neurobiol Dis, 45(1), 37-47. doi: 10.1016/j.nbd.2011.01.025
Krämer, D. K., Bouzakri, K., Holmqvist, O., Al-Khalili, L., & Krook, A. (2005). Effect of serum replacement with plysate on cell growth and metabolismin primary cultures of human skeletal muscle. Cytotechnology, 48(1-3), 89-95. doi: 10.1007/s10616-005-4074-7
Kulak, A., Steullet, P., Cabungcal, J. H., Werge, T., Ingason, A., Cuenod, M., & Do, K. Q. (2013). Redox dysregulation in the pathophysiology of schizophrenia and bipolar disorder: insights from animal models. Antioxid Redox Signal, 18(12), 1428-1443. doi: 10.1089/ars.2012.4858
Kuloglu, M., Ustundag, B., Atmaca, M., Canatan, H., Tezcan, A. E., & Cinkilinc, N. (2002). Lipid peroxidation and antioxidant enzyme levels in patients with schizophrenia and bipolar disorder. Cell Biochem Funct, 20(2), 171-175.
Kupfer, D. J. (2005). The increasing medical burden in bipolar disorder. Jama, 293(20), 2528-2530. König, P., Krasteva, G., Tag, C., König, I. R., Arens, C., & Kummer, W. (2006). FRET-CLSM and
double-labeling indirect immunofluorescence to detect close association of proteins in tissue sections. Lab Invest, 86(8), 853-864. doi: 3700443 [pii]
10.1038/labinvest.3700443
Lai, C. T., & Yu, P. H. (1997). Dopamine- and L-beta-3,4-dihydroxyphenylalanine hydrochloride (L-Dopa)-induced cytotoxicity towards catecholaminergic neuroblastoma SH-SY5Y cells. Effects of oxidative stress and antioxidative factors. Biochem Pharmacol, 53(3), 363-372.

153
Lai, J. S., Zhao, C., Warsh, J. J., & Li, P. P. (2006). Cytoprotection by lithium and valproate varies between cell types and cellular stresses. Eur J Pharmacol, 539(1-2), 18-26. doi: 10.1016/j.ejphar.2006.03.076
LaVoie, M. J., & Hastings, T. G. (1999). Peroxynitrite- and nitrite-induced oxidation of dopamine: implications for nitric oxide in dopaminergic cell loss. J Neurochem, 73(6), 2546-2554.
Lebon, S., Minai, L., Chretien, D., Corcos, J., Serre, V., Kadhom, N., . . . Rotig, A. (2007). A novel mutation of the NDUFS7 gene leads to activation of a cryptic exon and impaired assembly of mitochondrial complex I in a patient with Leigh syndrome. Mol Genet Metab, 92(1-2), 104-108.
Leboyer, M., Soreca, I., Scott, J., Frye, M., Henry, C., Tamouza, R., & Kupfer, D. J. (2012). Can bipolar disorder be viewed as a multi-system inflammatory disease? J Affect Disord, 141(1), 1-10. doi: 10.1016/j.jad.2011.12.049
Lenaz, G. (2001). The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life, 52(3-5), 159-164.
Lertratanangkoon, K., Wu, C. J., Savaraj, N., & Thomas, M. L. (1997). Alterations of DNA methylation by glutathione depletion. Cancer Lett, 120(2), 149-156.
Lewis, D. A., Melchitzky, D. S., Sesack, S. R., Whitehead, R. E., Auh, S., & Sampson, A. (2001). Dopamine transporter immunoreactivity in monkey cerebral cortex: regional, laminar, and ultrastructural localization. J Comp Neurol, 432(1), 119-136.
Li, N., Ragheb, K., Lawler, G., Sturgis, J., Rajwa, B., Melendez, J. A., & Robinson, J. P. (2003). Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem, 278(10), 8516-8525. doi: 10.1074/jbc.M210432200
Li, Q., Li, H., Roughton, K., Wang, X., Kroemer, G., Blomgren, K., & Zhu, C. (2010). Lithium reduces apoptosis and autophagy after neonatal hypoxia-ischemia. Cell Death Dis, 1, e56. doi: 10.1038/cddis.2010.33
Libíková, H., Breier, S., Kocisová, M., Pogády, J., Stünzner, D., & Ujházyová, D. (1979). Assay of interferon and viral antibodies in the cerebrospinal fluid in clinical neurology and psychiatry. Acta Biol Med Ger, 38(5-6), 879-893.
Lim, S. O., Gu, J. M., Kim, M. S., Kim, H. S., Park, Y. N., Park, C. K., . . . Jung, G. (2008). Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: methylation of the E-cadherin promoter. Gastroenterology, 135(6), 2128-2140, 2140.e2121-2128. doi: 10.1053/j.gastro.2008.07.027
Lister, R., Mukamel, E. A., Nery, J. R., Urich, M., Puddifoot, C. A., Johnson, N. D., . . . Ecker, J. R. (2013). Global epigenomic reconfiguration during mammalian brain development. Science, 341(6146), 1237905. doi: 10.1126/science.1237905
Liu, H. C., Yang, Y. Y., Chou, Y. M., Chen, K. P., Shen, W. W., & Leu, S. J. (2004). Immunologic variables in acute mania of bipolar disorder. J Neuroimmunol, 150(1-2), 116-122. doi: 10.1016/j.jneuroim.2004.01.006
Lohoff, F. W., Sander, T., Ferraro, T. N., Dahl, J. P., Gallinat, J., & Berrettini, W. H. (2005). Confirmation of association between the Val66Met polymorphism in the brain-derived neurotrophic factor (BDNF) gene and bipolar I disorder. Am J Med Genet B Neuropsychiatr Genet, 139B(1), 51-53. doi: 10.1002/ajmg.b.30215
Lopez-Larson, M. P., DelBello, M. P., Zimmerman, M. E., Schwiers, M. L., & Strakowski, S. M. (2002). Regional prefrontal gray and white matter abnormalities in bipolar disorder. Biol Psychiatry, 52(2), 93-100.
Lowes, D. A., Almawash, A. M., Webster, N. R., Reid, V. L., & Galley, H. F. (2011). Melatonin and structurally similar compounds have differing effects on inflammation and mitochondrial

154
function in endothelial cells under conditions mimicking sepsis. Br J Anaesth, 107(2), 193-201. doi: 10.1093/bja/aer149
Lyoo, I. K., Kim, M. J., Stoll, A. L., Demopulos, C. M., Parow, A. M., Dager, S. R., . . . Renshaw, P. F. (2004). Frontal lobe gray matter density decreases in bipolar I disorder. Biol Psychiatry, 55(6), 648-651.
López-Armada, M. J., Riveiro-Naveira, R. R., Vaamonde-García, C., & Valcárcel-Ares, M. N. (2013). Mitochondrial dysfunction and the inflammatory response. Mitochondrion, 13(2), 106-118. doi: 10.1016/j.mito.2013.01.003
Machado-Vieira, R., Andreazza, A. C., Viale, C. I., Zanatto, V., Cereser, V., Jr., da Silva Vargas, R., . . . Gentil, V. (2007). Oxidative stress parameters in unmedicated and treated bipolar subjects during initial manic episode: a possible role for lithium antioxidant effects. Neurosci Lett, 421(1), 33-36.
Machado-Vieira, R., Dietrich, M. O., Leke, R., Cereser, V. H., Zanatto, V., Kapczinski, F., . . . Gentil, V. (2007). Decreased plasma brain derived neurotrophic factor levels in unmedicated bipolar patients during manic episode. Biol Psychiatry, 61(2), 142-144. doi: 10.1016/j.biopsych.2006.03.070
Maes, M., Bosmans, E., Calabrese, J., Smith, R., & Meltzer, H. Y. (1995). Interleukin-2 and interleukin-6 in schizophrenia and mania: effects of neuroleptics and mood stabilizers. J Psychiatr Res, 29(2), 141-152.
Magalhães, P. V., Dean, O. M., Bush, A. I., Copolov, D. L., Malhi, G. S., Kohlmann, K., . . . Berk, M. (2011). N-acetyl cysteine add-on treatment for bipolar II disorder: a subgroup analysis of a randomized placebo-controlled trial. J Affect Disord, 129(1-3), 317-320. doi: 10.1016/j.jad.2010.08.001
Maier, W., Zobel, A., & Wagner, M. (2006). Schizophrenia and bipolar disorder: differences and overlaps. Curr Opin Psychiatry, 19(2), 165-170. doi: 10.1097/01.yco.0000214342.52249.82
Maker, H. S., Weiss, C., Silides, D. J., & Cohen, G. (1981). Coupling of dopamine oxidation (monoamine oxidase activity) to glutathione oxidation via the generation of hydrogen peroxide in rat brain homogenates. J Neurochem, 36(2), 589-593.
Malhi, G. S., Tanious, M., Das, P., Coulston, C. M., & Berk, M. (2013). Potential mechanisms of action of lithium in bipolar disorder. Current understanding. CNS Drugs, 27(2), 135-153. doi: 10.1007/s40263-013-0039-0
Manji, H. K., & Lenox, R. H. (2000). The nature of bipolar disorder. J Clin Psychiatry, 61 Supp 13, 42-57.
Maragos, W. F., Zhu, J., Chesnut, M. D., & Dwoskin, L. P. (2002). Mitochondrial toxin inhibition of [(3)H]dopamine uptake into rat striatal synaptosomes. Biochem Pharmacol, 63(8), 1499-1505.
Martinez-Aran, A., Vieta, E., Reinares, M., Colom, F., Torrent, C., Sanchez-Moreno, J., . . . Salamero, M. (2004). Cognitive function across manic or hypomanic, depressed, and euthymic states in bipolar disorder. Am J Psychiatry, 161(2), 262-270.
Mastroberardino, P. G., Orr, A. L., Hu, X., Na, H. M., & Greenamyre, J. T. (2008). A FRET-based method to study protein thiol oxidation in histological preparations. Free Radic Biol Med, 45(7), 971-981.
Matarese, F., Carrillo-de Santa Pau, E., & Stunnenberg, H. G. (2011). 5-Hydroxymethylcytosine: a new kid on the epigenetic block? Mol Syst Biol, 7, 562. doi: 10.1038/msb.2011.95
Maurer, I. C., Schippel, P., & Volz, H. P. (2009). Lithium-induced enhancement of mitochondrial oxidative phosphorylation in human brain tissue. Bipolar Disord, 11(5), 515-522. doi: 10.1111/j.1399-5618.2009.00729.x

155
Merikangas, K. R., Jin, R., He, J. P., Kessler, R. C., Lee, S., Sampson, N. A., . . . Zarkov, Z. (2011). Prevalence and correlates of bipolar spectrum disorder in the world mental health survey initiative. Arch Gen Psychiatry, 68(3), 241-251.
Messiha, F. S., Agallianos, D., & Clower, C. (1970). Dopamine excretion in affective states and following Li2Co3 therapy. Nature, 225(5235), 868-869.
Miller, B. J., Buckley, P., Seabolt, W., Mellor, A., & Kirkpatrick, B. (2011). Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry, 70(7), 663-671. doi: 10.1016/j.biopsych.2011.04.013
Miller, G. W., Staley, J. K., Heilman, C. J., Perez, J. T., Mash, D. C., Rye, D. B., & Levey, A. I. (1997). Immunochemical analysis of dopamine transporter protein in Parkinson's disease. Ann Neurol, 41(4), 530-539. doi: 10.1002/ana.410410417
Mirnics, K., Levitt, P., & Lewis, D. A. (2006). Critical appraisal of DNA microarrays in psychiatric genomics. Biol Psychiatry, 60(2), 163-176. doi: 10.1016/j.biopsych.2006.02.003
Mitchell, P., Selbie, L., Waters, B., Donald, J., Vivero, C., Tully, M., & Shine, J. (1992). Exclusion of close linkage of bipolar disorder to dopamine D1 and D2 receptor gene markers. J Affect Disord, 25(1), 1-11.
Miyazaki, I., Asanuma, M., Diaz-Corrales, F., Fukuda, M., Kitaichi, K., Miyoshi, K., & Ogawa, N. (2006). Methamphetamine-induced dopaminergic neurotoxicity is regulated by quinone formation-related molecules. Faseb Journal, 20(1), 571-+. doi: 10.1096/fj.05-4996fje
Modabbernia, A., Taslimi, S., Brietzke, E., & Ashrafi, M. (2013). Cytokine alterations in bipolar disorder: a meta-analysis of 30 studies. Biol Psychiatry, 74(1), 15-25. doi: 10.1016/j.biopsych.2013.01.007
Muir, W. J., Thomson, M. L., McKeon, P., Mynett-Johnson, L., Whitton, C., Evans, K. L., . . . Blackwood, D. H. (2001). Markers close to the dopamine D5 receptor gene (DRD5) show significant association with schizophrenia but not bipolar disorder. Am J Med Genet, 105(2), 152-158.
Muller-Oerlinghausen, B., Berghofer, A., & Bauer, M. (2002). Bipolar disorder. Lancet, 359(9302), 241-247.
Munkholm, K., Braüner, J. V., Kessing, L. V., & Vinberg, M. (2013). Cytokines in bipolar disorder vs. healthy control subjects: a systematic review and meta-analysis. J Psychiatr Res, 47(9), 1119-1133. doi: 10.1016/j.jpsychires.2013.05.018
Müller, N., Myint, A. M., & Schwarz, M. J. (2012). Inflammation in schizophrenia. Adv Protein Chem Struct Biol, 88, 49-68. doi: 10.1016/B978-0-12-398314-5.00003-9
Nahman, S., Belmaker, R. H., & Azab, A. N. (2012). Effects of lithium on lipopolysaccharide-induced inflammation in rat primary glia cells. Innate Immun, 18(3), 447-458. doi: 10.1177/1753425911421512
Naoi, M., Maruyama, W., Shamoto-Nagai, M., Yi, H., Akao, Y., & Tanaka, M. (2005). Oxidative stress in mitochondria: decision to survival and death of neurons in neurodegenerative disorders. Mol Neurobiol, 31(1-3), 81-93.
Nascimento, C., Kim, H. K., Young, L. T., Mendonça, K. M., Grinberg, L. T., Lafer, B., & Andreazza, A. C. (2014). Glutathione-mediated effects of lithium in decreasing protein oxidation induced by mitochondrial complex I dysfunction. J Neural Transm. doi: 10.1007/s00702-014-1318-8
Nery, F. G., Monkul, E. S., Hatch, J. P., Fonseca, M., Zunta-Soares, G. B., Frey, B. N., . . . Soares, J. C. (2008). Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: a double-blind, randomized, placebo-controlled study. Hum Psychopharmacol, 23(2), 87-94. doi: 10.1002/hup.912

156
Ng, F., Berk, M., Dean, O., & Bush, A. I. (2008). Oxidative stress in psychiatric disorders: evidence base and therapeutic implications. Int J Neuropsychopharmacol, 11(6), 851-876.
Nikolaus, S., Antke, C., & Muller, H. W. (2009). In vivo imaging of synaptic function in the central nervous system: II. Mental and affective disorders. Behav Brain Res, 204(1), 32-66.
Nohesara, S., Ghadirivasfi, M., Mostafavi, S., Eskandari, M. R., Ahmadkhaniha, H., Thiagalingam, S., & Abdolmaleky, H. M. (2011). DNA hypomethylation of MB-COMT promoter in the DNA derived from saliva in schizophrenia and bipolar disorder. J Psychiatr Res, 45(11), 1432-1438.
Nöthen, M. M., Erdmann, J., Körner, J., Lanczik, M., Fritze, J., Fimmers, R., . . . Propping, P. (1992). Lack of association between dopamine D1 and D2 receptor genes and bipolar affective disorder. Am J Psychiatry, 149(2), 199-201.
O'Brien, S. M., Scully, P., Scott, L. V., & Dinan, T. G. (2006). Cytokine profiles in bipolar affective disorder: focus on acutely ill patients. J Affect Disord, 90(2-3), 263-267. doi: 10.1016/j.jad.2005.11.015
O'Connell, L. A., Matthews, B. J., Ryan, M. J., & Hofmann, H. A. (2010). Characterization of the dopamine system in the brain of the túngara frog, Physalaemus pustulosus. Brain Behav Evol, 76(3-4), 211-225. doi: 10.1159/000321715
Offen, D., Ziv, I., Sternin, H., Melamed, E., & Hochman, A. (1996). Prevention of dopamine-induced cell death by thiol antioxidants: possible implications for treatment of Parkinson's disease. Exp Neurol, 141(1), 32-39.
Ortiz-Domínguez, A., Hernández, M. E., Berlanga, C., Gutiérrez-Mora, D., Moreno, J., Heinze, G., & Pavón, L. (2007). Immune variations in bipolar disorder: phasic differences. Bipolar Disord, 9(6), 596-602. doi: 10.1111/j.1399-5618.2007.00493.x
Padmos, R. C., Hillegers, M. H., Knijff, E. M., Vonk, R., Bouvy, A., Staal, F. J., . . . Drexhage, H. A. (2008). A discriminating messenger RNA signature for bipolar disorder formed by an aberrant expression of inflammatory genes in monocytes. Arch Gen Psychiatry, 65(4), 395-407. doi: 10.1001/archpsyc.65.4.395
Papiol, S., Molina, V., Desco, M., Rosa, A., Reig, S., Sanz, J., . . . Fañanás, L. (2008). Gray matter deficits in bipolar disorder are associated with genetic variability at interleukin-1 beta gene (2q13). Genes Brain Behav, 7(7), 796-801. doi: 10.1111/j.1601-183X.2008.00421.x
Park, S., Geddes, T. J., Javitch, J. A., & Kuhn, D. M. (2003). Dopamine prevents nitration of tyrosine hydroxylase by peroxynitrite and nitrogen dioxide: is nitrotyrosine formation an early step in dopamine neuronal damage? J Biol Chem, 278(31), 28736-28742. doi: M304362200 [pii]
10.1074/jbc.M304362200
Park, S. U., Ferrer, J. V., Javitch, J. A., & Kuhn, D. M. (2002). Peroxynitrite inactivates the human dopamine transporter by modification of cysteine 342: potential mechanism of neurotoxicity in dopamine neurons. J Neurosci, 22(11), 4399-4405.
Pearlson, G. D., Wong, D. F., Tune, L. E., Ross, C. A., Chase, G. A., Links, J. M., . . . Wagner, H. N. (1995). In vivo D2 dopamine receptor density in psychotic and nonpsychotic patients with bipolar disorder. Arch Gen Psychiatry, 52(6), 471-477.
Perova, T., Kwan, M., Li, P. P., & Warsh, J. J. (2010). Differential modulation of intracellular Ca2+ responses in B lymphoblasts by mood stabilizers. Int J Neuropsychopharmacol, 13(6), 693-702.
Perova, T., Wasserman, M. J., Li, P. P., & Warsh, J. J. (2008). Hyperactive intracellular calcium dynamics in B lymphoblasts from patients with bipolar I disorder. Int J Neuropsychopharmacol, 11(2), 185-196.

157
Perregaux, D., & Gabel, C. A. (1994). Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem, 269(21), 15195-15203.
Persson, A. K., Kim, I., Zhao, P., Estacion, M., Black, J. A., & Waxman, S. G. (2013). Sodium channels contribute to degeneration of dorsal root ganglion neurites induced by mitochondrial dysfunction in an in vitro model of axonal injury. J Neurosci, 33(49), 19250-19261. doi: 10.1523/JNEUROSCI.2148-13.2013
Petronis, A. (2003). Epigenetics and bipolar disorder: new opportunities and challenges. Am J Med Genet C Semin Med Genet, 123C(1), 65-75. doi: 10.1002/ajmg.c.20015
Post, R. M., Jimerson, D. C., Bunney, W. E., Jr., & Goodwin, F. K. (1980). Dopamine and mania: behavioral and biochemical effects of the dopamine receptor blocker pimozide. Psychopharmacology (Berl), 67(3), 297-305.
Potash, J. B., & Bienvenu, O. J. (2009). Neuropsychiatric disorders: Shared genetics of bipolar disorder and schizophrenia. Nat Rev Neurol, 5(6), 299-300. doi: 10.1038/nrneurol.2009.71
Prickaerts, J., Moechars, D., Cryns, K., Lenaerts, I., van Craenendonck, H., Goris, I., . . . Steckler, T. (2006). Transgenic mice overexpressing glycogen synthase kinase 3beta: a putative model of hyperactivity and mania. J Neurosci, 26(35), 9022-9029. doi: 10.1523/JNEUROSCI.5216-05.2006
Quiroz, J. A., Gould, T. D., & Manji, H. K. (2004). Molecular effects of lithium. Mol Interv, 4(5), 259-272.
Quiroz, J. A., Gray, N. A., Kato, T., & Manji, H. K. (2008). Mitochondrially mediated plasticity in the pathophysiology and treatment of bipolar disorder. Neuropsychopharmacology, 33(11), 2551-2565.
Rajkowska, G. (2000). Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol Psychiatry, 48(8), 766-777.
Rajkowska, G., Halaris, A., & Selemon, L. D. (2001). Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder. Biol Psychiatry, 49(9), 741-752.
Rao, J. S., Harry, G. J., Rapoport, S. I., & Kim, H. W. (2010). Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol Psychiatry, 15(4), 384-392.
Rao, J. S., Kellom, M., Reese, E. A., Rapoport, S. I., & Kim, H. W. (2012). Dysregulated glutamate and dopamine transporters in postmortem frontal cortex from bipolar and schizophrenic patients. J Affect Disord, 136(1-2), 63-71. doi: S0165-0327(11)00489-7 [pii] 10.1016/j.jad.2011.08.017
Ray, P. D., Huang, B. W., & Tsuji, Y. (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal, 24(5), 981-990. doi: 10.1016/j.cellsig.2012.01.008
Reik, W., Dean, W., & Walter, J. (2001). Epigenetic reprogramming in mammalian development. Science, 293(5532), 1089-1093. doi: 10.1126/science.1063443
Romo-Nava, F., Alvarez-Icaza González, D., Fresán-Orellana, A., Saracco Alvarez, R., Becerra-Palars, C., Moreno, J., . . . Buijs, R. M. (2014). Melatonin attenuates antipsychotic metabolic effects: an eight-week randomized, double-blind, parallel-group, placebo-controlled clinical trial. Bipolar Disord, 16(4), 410-421. doi: 10.1111/bdi.12196
Saarelainen, T., Hendolin, P., Lucas, G., Koponen, E., Sairanen, M., MacDonald, E., . . . Castrén, E. (2003). Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci, 23(1), 349-357.

158
Salvadore, G., Quiroz, J. A., Machado-Vieira, R., Henter, I. D., Manji, H. K., & Zarate, C. A. (2010). The neurobiology of the switch process in bipolar disorder: a review. J Clin Psychiatry, 71(11), 1488-1501. doi: 10.4088/JCP.09r05259gre
Savoye, C., Laurent, C., Amadeo, S., Gheysen, F., Leboyer, M., Lejeune, J., . . . Mallet, J. (1998). No association between dopamine D1, D2, and D3 receptor genes and manic-depressive illness. Biol Psychiatry, 44(7), 644-647.
Schretlen, D. J., Cascella, N. G., Meyer, S. M., Kingery, L. R., Testa, S. M., Munro, C. A., . . . Pearlson, G. D. (2007). Neuropsychological functioning in bipolar disorder and schizophrenia. Biol Psychiatry, 62(2), 179-186. doi: 10.1016/j.biopsych.2006.09.025
Schroder, K., Zhou, R., & Tschopp, J. (2010). The NLRP3 inflammasome: a sensor for metabolic danger? Science, 327(5963), 296-300. doi: 10.1126/science.1184003
Schutte, B., Nuydens, R., Geerts, H., & Ramaekers, F. (1998). Annexin V binding assay as a tool to measure apoptosis in differentiated neuronal cells. J Neurosci Methods, 86(1), 63-69.
Schäfer, M., Goodenough, S., Moosmann, B., & Behl, C. (2004). Inhibition of glycogen synthase kinase 3 beta is involved in the resistance to oxidative stress in neuronal HT22 cells. Brain Res, 1005(1-2), 84-89. doi: S0006899304001544 [pii] 10.1016/j.brainres.2004.01.037
Schütt, F., Aretz, S., Auffarth, G. U., & Kopitz, J. (2012). Moderately reduced ATP levels promote oxidative stress and debilitate autophagic and phagocytic capacities in human RPE cells. Invest Ophthalmol Vis Sci, 53(9), 5354-5361. doi: 10.1167/iovs.12-9845
Scola, G., Kim, H. K., Young, L. T., & Andreazza, A. C. (2012). A Fresh Look at Complex I in Microarray Data: Clues to Understanding Disease-Specific Mitochondrial Alterations in Bipolar Disorder. Biol Psychiatry. doi: S0006-3223(12)00583-5 [pii]
10.1016/j.biopsych.2012.06.028
Scola, G., Kim, H. K., Young, L. T., & Andreazza, A. C. (2013). A fresh look at complex I in microarray data: clues to understanding disease-specific mitochondrial alterations in bipolar disorder. Biol Psychiatry, 73(2), e4-5. doi: 10.1016/j.biopsych.2012.06.028
Seamans, J. K., & Yang, C. R. (2004). The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol, 74(1), 1-58. doi: 10.1016/j.pneurobio.2004.05.006
Selemon, L. D., Rajkowska, G., & Goldman-Rakic, P. S. (1998). Elevated neuronal density in prefrontal area 46 in brains from schizophrenic patients: application of a three-dimensional, stereologic counting method. J Comp Neurol, 392(3), 402-412. doi: 10.1002/(SICI)1096-9861(19980316)392:3<402::AID-CNE9>3.0.CO;2-5 [pii]
Selemon, L. D., Rajkowska, G., & Goldman-Rakic, P. S. (2004). Evidence for progression in frontal cortical pathology in late-stage Huntington's disease. J Comp Neurol, 468(2), 190-204. doi: 10.1002/cne.10938
Semendeferi, K., Armstrong, E., Schleicher, A., Zilles, K., & Van Hoesen, G. W. (2001). Prefrontal cortex in humans and apes: a comparative study of area 10. Am J Phys Anthropol, 114(3), 224-241. doi: 10.1002/1096-8644(200103)114:3<224::AID-AJPA1022>3.0.CO;2-I
Serretti, A., Cusin, C., Lattuada, E., Lilli, R., Lorenzi, C., Di Bella, D., . . . Smeraldi, E. (1999). No interaction between serotonin transporter gene and dopamine receptor D4 gene in symptomatology of major psychoses. Am J Med Genet, 88(5), 481-485.
Shalbuyeva, N., Brustovetsky, T., & Brustovetsky, N. (2007). Lithium desensitizes brain mitochondria to calcium, antagonizes permeability transition, and diminishes cytochrome C release. J Biol Chem, 282(25), 18057-18068. doi: 10.1074/jbc.M702134200
Shao, L., Cui, J., Young, L. T., & Wang, J. F. (2008). The effect of mood stabilizer lithium on expression and activity of glutathione s-transferase isoenzymes. Neuroscience, 151(2), 518-524.

159
Sharma, N., Hewett, J., Ozelius, L. J., Ramesh, V., McLean, P. J., Breakefield, X. O., & Hyman, B. T. (2001). A close association of torsinA and alpha-synuclein in Lewy bodies: a fluorescence resonance energy transfer study. Am J Pathol, 159(1), 339-344. doi: S0002-9440(10)61700-2 [pii]
Sherer, T. B., Betarbet, R., Testa, C. M., Seo, B. B., Richardson, J. R., Kim, J. H., . . . Greenamyre, J. T. (2003). Mechanism of toxicity in rotenone models of Parkinson's disease. J Neurosci, 23(34), 10756-10764.
Shimada, K., Crother, T. R., Karlin, J., Dagvadorj, J., Chiba, N., Chen, S., . . . Arditi, M. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity, 36(3), 401-414. doi: 10.1016/j.immuni.2012.01.009
Shungu, D. C. (2012). N-acetylcysteine for the treatment of glutathione deficiency and oxidative stress in schizophrenia. Biol Psychiatry, 71(11), 937-938. doi: 10.1016/j.biopsych.2012.03.025
Sies, H. (1991). Oxidative stress: from basic research to clinical application. Am J Med, 91(3C), 31S-38S.
Silverstone, T. (1984). Response to bromocriptine distinguishes bipolar from unipolar depression. Lancet, 1(8382), 903-904.
Soeiro-de-Souza, M. G., Andreazza, A. C., Carvalho, A. F., Machado-Vieira, R., Young, L. T., & Moreno, R. A. (2013). Number of manic episodes is associated with elevated DNA oxidation in bipolar I disorder. Int J Neuropsychopharmacol, 1-8. doi: 10.1017/S1461145713000047
Sollberger, G., Strittmatter, G. E., Garstkiewicz, M., Sand, J., & Beer, H. D. (2014). Caspase-1: the inflammasome and beyond. Innate Immun, 20(2), 115-125. doi: 10.1177/1753425913484374
Song, C. X., Szulwach, K. E., Fu, Y., Dai, Q., Yi, C., Li, X., . . . He, C. (2011). Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol, 29(1), 68-72. doi: 10.1038/nbt.1732
Sorbara, M. T., & Girardin, S. E. (2011). Mitochondrial ROS fuel the inflammasome. Cell Res, 21(4), 558-560. doi: 10.1038/cr.2011.20
Srinivasan, V., Smits, M., Spence, W., Lowe, A. D., Kayumov, L., Pandi-Perumal, S. R., . . . Cardinali, D. P. (2006). Melatonin in mood disorders. World J Biol Psychiatry, 7(3), 138-151. doi: 10.1080/15622970600571822
Srinivasan, V., Spence, D. W., Pandi-Perumal, S. R., Brown, G. M., & Cardinali, D. P. (2011). Melatonin in mitochondrial dysfunction and related disorders. Int J Alzheimers Dis, 2011, 326320. doi: 10.4061/2011/326320
Suhara, T., Nakayama, K., Inoue, O., Fukuda, H., Shimizu, M., Mori, A., & Tateno, Y. (1992). D1 dopamine receptor binding in mood disorders measured by positron emission tomography. Psychopharmacology (Berl), 106(1), 14-18.
Sun, X., Wang, J. F., Tseng, M., & Young, L. T. (2006). Downregulation in components of the mitochondrial electron transport chain in the postmortem frontal cortex of subjects with bipolar disorder. J Psychiatry Neurosci, 31(3), 189-196.
Söderlund, J., Olsson, S. K., Samuelsson, M., Walther-Jallow, L., Johansson, C., Erhardt, S., . . . Engberg, G. (2011). Elevation of cerebrospinal fluid interleukin-1ß in bipolar disorder. J Psychiatry Neurosci, 36(2), 114-118. doi: 10.1503/jpn.100080
Tahiliani, M., Koh, K., Shen, Y., Pastor, W., Bandukwala, H., Brudno, Y., . . . Rao, A. (2009). Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science, 324(5929), 930-935. doi: 10.1126/science.1170116
Takeda, K., Akagi, S., Takahashi, S., Onishi, A., Hanada, H., & Pinkert, C. A. (2002). Mitochondrial activity in response to serum starvation in bovine (Bos taurus) cell culture. Cloning Stem Cells, 4(3), 223-229. doi: 10.1089/15362300260339502

160
Takuma, K., Baba, A., & Matsuda, T. (2004). Astrocyte apoptosis: implications for neuroprotection. Prog Neurobiol, 72(2), 111-127.
Tan, H., Young, L., Shao, L., Che, Y., Honer, W., & Wang, J. (2011). Mood stabilizer lithium inhibits amphetamine-increased 4-hydroxynonenal-protein adducts in rat frontal cortex. The Int J of Neuropsychopharmacol, 1-11.
Thompson, J. M., Gray, J. M., Hughes, J. H., Watson, S., Young, A. H., & Ferrier, I. N. (2007). Impaired working memory monitoring in euthymic bipolar patients. Bipolar Disord, 9(5), 478-489. doi: 10.1111/j.1399-5618.2007.00470.x
Torrey, E. F., Webster, M., Knable, M., Johnston, N., & Yolken, R. H. (2000). The stanley foundation brain collection and neuropathology consortium. Schizophr Res, 44(2), 151-155.
Tschopp, J., & Schroder, K. (2010). NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol, 10(3), 210-215. doi: 10.1038/nri2725
Tse, D. C., McCreery, R. L., & Adams, R. N. (1976). Potential oxidative pathways of brain catecholamines. J Med Chem, 19(1), 37-40.
Tunbridge, E., Burnet, P. W., Sodhi, M. S., & Harrison, P. J. (2004). Catechol-o-methyltransferase (COMT) and proline dehydrogenase (PRODH) mRNAs in the dorsolateral prefrontal cortex in schizophrenia, bipolar disorder, and major depression. Synapse, 51(2), 112-118. doi: 10.1002/syn.10286
Turrens, J. F., & Boveris, A. (1980). Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J, 191(2), 421-427.
Valvassori, S. S., Rezin, G. T., Ferreira, C. L., Moretti, M., Goncalves, C. L., Cardoso, M. R., . . . Quevedo, J. (2010). Effects of mood stabilizers on mitochondrial respiratory chain activity in brain of rats treated with d-amphetamine. J Psychiatr Res, 44(14), 903-909.
van der Valk, J., Brunner, D., De Smet, K., Fex Svenningsen, A., Honegger, P., Knudsen, L. E., . . . Gstraunthaler, G. (2010). Optimization of chemically defined cell culture media--replacing fetal bovine serum in mammalian in vitro methods. Toxicol In Vitro, 24(4), 1053-1063. doi: 10.1016/j.tiv.2010.03.016
Van Kammen, D. P., & Murphy, D. L. (1975). Attenuation of the euphoriant and activating effects of d- and l-amphetamine by lithium carbonate treatment. Psychopharmacologia, 44(3), 215-224.
Vawter, M. P., Tomita, H., Meng, F., Bolstad, B., Li, J., Evans, S., . . . Bunney, W. E. (2006). Mitochondrial-related gene expression changes are sensitive to agonal-pH state: implications for brain disorders. Mol Psychiatry, 11(7), 615, 663-679.
Versace, A., Andreazza, A. C., Young, L. T., Fournier, J. C., Almeida, J. R., Stiffler, R. S., . . . Phillips, M. L. (2013). Elevated serum measures of lipid peroxidation and abnormal prefrontal white matter in euthymic bipolar adults: toward peripheral biomarkers of bipolar disorder. Mol Psychiatry. doi: 10.1038/mp.2012.188
Waldvogel, H. J., Curtis, M. A., Baer, K., Rees, M. I., & Faull, R. L. (2006). Immunohistochemical staining of post-mortem adult human brain sections. Nat Protoc, 1(6), 2719-2732. doi: nprot.2006.354 [pii]
10.1038/nprot.2006.354
Wang, J. F., Shao, L., Sun, X., & Young, L. T. (2009). Increased oxidative stress in the anterior cingulate cortex of subjects with bipolar disorder and schizophrenia. Bipolar Disord, 11(5), 523-529.
Washizuka, S., Iwamoto, K., Kakiuchi, C., Bundo, M., & Kato, T. (2009). Expression of mitochondrial complex I subunit gene NDUFV2 in the lymphoblastoid cells derived from patients with bipolar disorder and schizophrenia. Neurosci Res, 63(3), 199-204.

161
Washizuka, S., Kakiuchi, C., Mori, K., Kunugi, H., Tajima, O., Akiyama, T., . . . Kato, T. (2003). Association of mitochondrial complex I subunit gene NDUFV2 at 18p11 with bipolar disorder. Am J Med Genet B Neuropsychiatr Genet, 120B(1), 72-78. doi: 10.1002/ajmg.b.20041
Watabe, M., & Nakaki, T. (2004). Rotenone induces apoptosis via activation of bad in human dopaminergic SH-SY5Y cells. J Pharmacol Exp Ther, 311(3), 948-953. doi: jpet.104.071381 [pii]
10.1124/jpet.104.071381
Watabe, M., & Nakaki, T. (2007). Mitochondrial complex I inhibitor rotenone-elicited dopamine redistribution from vesicles to cytosol in human dopaminergic SH-SY5Y cells. J Pharmacol Exp Ther, 323(2), 499-507. doi: jpet.107.127597 [pii]
10.1124/jpet.107.127597
Watabe, M., & Nakaki, T. (2008). Mitochondrial complex I inhibitor rotenone inhibits and redistributes vesicular monoamine transporter 2 via nitration in human dopaminergic SH-SY5Y cells. Mol Pharmacol, 74(4), 933-940. doi: mol.108.048546 [pii]
10.1124/mol.108.048546
Weber, A., Wasiliew, P., & Kracht, M. (2010). Interleukin-1beta (IL-1beta) processing pathway. Sci Signal, 3(105), cm2. doi: 10.1126/scisignal.3105cm2
Weber, B., Serafin, A., Michie, J., Van Rensburg, C., Swarts, J. C., & Bohm, L. (2004). Cytotoxicity and cell death pathways invoked by two new rhodium-ferrocene complexes in benign and malignant prostatic cell lines. Anticancer Res, 24(2B), 763-770.
Weerasinghe, G. R., Rapoport, S. I., & Bosetti, F. (2004). The effect of chronic lithium on arachidonic acid release and metabolism in rat brain does not involve secretory phospholipase A2 or lipoxygenase/cytochrome P450 pathways. Brain Res Bull, 63(6), 485-489. doi: 10.1016/j.brainresbull.2004.04.005
Wegener, G., Bandpey, Z., Heiberg, I. L., Volke, V., Trabace, L., Rosenberg, R., & Harvey, B. H. (2004). Combined chronic treatment with citalopram and lithium does not modify the regional neurochemistry of nitric oxide in rat brain. J Physiol Pharmacol, 55(3), 575-586.
Weiss, R. B., Stange, J. P., Boland, E. M., Black, S. K., LaBelle, D. R., Abramson, L. Y., & Alloy, L. B. (2015). Kindling of life stress in bipolar disorder: comparison of sensitization and autonomy models. J Abnorm Psychol, 124(1), 4-16. doi: 10.1037/abn0000014
West, M. J. (2012). Introduction to stereology. Cold Spring Harb Protoc, 2012(8). doi: 10.1101/pdb.top070623
Wieckowski, M. R., Giorgi, C., Lebiedzinska, M., Duszynski, J., & Pinton, P. (2009). Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat Protoc, 4(11), 1582-1590. doi: 10.1038/nprot.2009.151
Wills, J., Jones, J., Haggerty, T., Duka, V., Joyce, J. N., & Sidhu, A. (2010). Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson's disease brains with and without dementia. Exp Neurol, 225(1), 210-218. doi: 10.1016/j.expneurol.2010.06.017
Winther, J. R., & Thorpe, C. (2013). Quantification of thiols and disulfides. Biochim Biophys Acta. doi: 10.1016/j.bbagen.2013.03.031
Wong, D. F., Pearlson, G. D., Tune, L. E., Young, L. T., Meltzer, C. C., Dannals, R. F., . . . Gjedde, A. (1997). Quantification of neuroreceptors in the living human brain: IV. Effect of aging and elevations of D2-like receptors in schizophrenia and bipolar illness. J Cereb Blood Flow Metab, 17(3), 331-342. doi: 10.1097/00004647-199703000-00010

162
Wossidlo, M., Nakamura, T., Lepikhov, K., Marques, C. J., Zakhartchenko, V., Boiani, M., . . . Walter, J. (2011). 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun, 2, 241. doi: 10.1038/ncomms1240
Wu, H., Coskun, V., Tao, J., Xie, W., Ge, W., Yoshikawa, K., . . . Sun, Y. (2010). Dnmt3a-Dependent Nonpromoter DNA Methylation Facilitates Transcription of Neurogenic Genes. Science, 329(5990), 444-448. doi: 10.1126/science.1190485
Yao, J. K., & Keshavan, M. S. (2011). Antioxidants, redox signaling, and pathophysiology in schizophrenia: an integrative view. Antioxid Redox Signal, 15(7), 2011-2035.
Zhang, J., Echeverry, S., Lim, T. K., Lee, S. H., Shi, X. Q., & Huang, H. (2015). Can Modulating Inflammatory Response be a Good Strategy to Treat Neuropathic Pain? Curr Pharm Des, 21(7), 831-839.
Zhang, P., Wang, Y. Z., Kagan, E., & Bonner, J. C. (2000). Peroxynitrite targets the epidermal growth factor receptor, Raf-1, and MEK independently to activate MAPK. J Biol Chem, 275(29), 22479-22486. doi: 10.1074/jbc.M910425199
Zhang, R., Kang, K. A., Kim, K. C., Na, S. Y., Chang, W. Y., Kim, G. Y., . . . Hyun, J. W. (2013). Oxidative stress causes epigenetic alteration of CDX1 expression in colorectal cancer cells. Gene, 524(2), 214-219. doi: 10.1016/j.gene.2013.04.024
Zheng, L. S., Ishii, Y., Zhao, Q. L., Kondo, T., & Sasahara, M. (2013). PDGF suppresses oxidative stress induced Ca2+ overload and calpain activation in neurons. Oxid Med Cell Longev, 2013, 367206. doi: 10.1155/2013/367206
Zheng, X., Baker, H., Hancock, W. S., Fawaz, F., McCaman, M., & Pungor, E. (2006). Proteomic analysis for the assessment of different lots of fetal bovine serum as a raw material for cell culture. Part IV. Application of proteomics to the manufacture of biological drugs. Biotechnol Prog, 22(5), 1294-1300. doi: 10.1021/bp060121o
Zhong, Z., Zhai, Y., Liang, S., Mori, Y., Han, R., Sutterwala, F. S., & Qiao, L. (2013). TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun, 4, 1611. doi: 10.1038/ncomms2608
Zhou, R., Yazdi, A. S., Menu, P., & Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature, 469(7329), 221-225. doi: 10.1038/nature09663

163
Copyright releases

164

165
January 15, 2015
Helena K. Kim
Ph.D. Candidate
Department of Pharmacology and Toxicology
University of Toronto
SUBJECT: J Psychiatry Neurosci 2014; 39(4):276-85 DOI: 10.1503/jpn.130155
Oxidation and nitration in dopaminergic areas of the prefrontal cortex from patients with bipolar disorder and schizophrenia
Dear Helena,
I am pleased to confirm that the Canadian Medical Association as the copyright holder, grants you permission to reproduce this material as requested in your thesis. We do ask that you use the final, published text as opposed to the originally submitted manuscript.
The Royalty fee has been waived provided that this material will be used for research/education purposes only and that it will not be resold or used for commercial purposes in any way.
Please credit the original publication of the material as follows:
Oxidation and nitration in dopaminergic areas of the prefrontal cortex from patients with bipolar disorder and schizophrenia — Reprinted from, J Psychiatry Neurosci; 2014; 39(4):276-85 DOI: 10.1503/jpn.130155 by permission of the publisher.
© 2014 Canadian Medical Association
If you require further information or assistance, please do not hesitate to contact the undersigned.
Sincerely,
Janis Murrey Program Manager Journals
1867 Alta Vista Drive, Ottawa ON K1G 5W8

167

168






























