Embed Size (px)
Citation preview

C H A P T E R 37Biomarkers in Heart Failure, 545Markers of Myocardial Stretch, 545Markers of Inflammation, 549Markers of Myocardial Cell Death, 551Markers of Renal Function, 552
Multimarker Approach and Future Direction, 556
Conclusion, 557
The Use of Biomarkers in the Evaluation of Heart FailureLeo Slavin, Lori B. Daniels, and Alan S. Maisel
5
nfusythgadcotomddo
B
Mclabvevmerchpald
MN
BBeftemstvbcin
st(pbumbTcyC
The approach to the diagnosis of acute heart failure is complex and challenging because it has heterogeneous manifestations and
onspecific signs and symptoms.1 Classically, students are taught that a care-l history in patients presenting with congestive heart failure (CHF) elicits mptoms of dyspnea, orthopnea, and paroxysmal nocturnal dyspnea, whereas e physical examination reveals elevated jugular venous pressure, rales, an S3 llop, and pitting peripheral edema (see also Chapter 35). However, it is well
ocumented that in practice, the clinical manifestation of heart failure, even in mbination with chest radiographs, electrocardiograms, and standard labora-ry assessments, frequently does not clinch the diagnosis. Often clinicians ust consider other causes of dyspnea, such as chronic obstructive pulmonary isease or pneumonia, which can delay necessary treatment. The growth and evelopment of various classes of biomarkers are improving the understanding f the pathogenesis, diagnosis, and prognosis in heart failure.
IOMARKERS IN HEART FAILURE
orrow and de Lemos2 proposed three criteria required for a biomarker to be inically useful. First, the assay should be precise, accurate, and rapidly avail-le to the clinician at relatively low cost. Second, the biomarker should pro-
ide additional information that is not surmised from findings of the clinical aluation. Third, the absolute measured value should help in clinical decision aking.2 Currently, few biomarkers can fulfill all these requirements. Biomark-s that do meet these criteria reflect the different mechanisms (such as biome-anical stretch, inflammation, and myocyte injury) that are involved in the
athophysiology and natural history of heart failure. These markers individu-ly and jointly provide important information for assessing the progression of isease, diagnosing acute exacerbations, and establishing prognosis.
arkers of Myocardial Stretchatriuretic Peptides
iology. Three major natriuretic peptides—atrial natriuretic peptide (ANP), -type natriuretic peptide (BNP), and C-type natriuretic peptide—counter the fects of volume overload or adrenergic activation of the cardiovascular sys-m. ANP is synthesized primarily in the atria, stored in granules, and, under inor triggers such as exercise, released into the circulation.3 BNP has minimal orage in granules and is synthesized and secreted in bursts primarily by the entricles.3 C-type natriuretic peptide is a product of endothelial cells and may e protective in post–myocardial infarction remodeling.4 Upon release into the rculation, ANP and BNP bind to various tissues and induce vasodilation, atriuresis, and diuresis.5
Left ventricular pressure or volume overload results in myocardial wall ress that initiates the synthesis of precursor pro–B-type natriuretic peptide re-proBNP). Pre-proBNP is initially cleaved to proBNP and then to BNP, the
iologically active form, and the inactive N-terminal fragment, NT-proBNP (Fig-re 37-1). The mechanism of action of natriuretic peptides is mediated through embrane-bound natriuretic peptide receptors (NPRs). NPR-A preferentially
inds ANP and BNP, and NPR-B primarily binds C-type natriuretic peptide. he natriuretic peptide–receptor interaction activates the enzyme guanylyl clase, which leads to the production of cyclic guanosine monophosphate.
learance of natriuretic peptides is mediated through NPR-C, degradation by
54
neutral endopeptidase, and by direct renal clearance (Table 37-1).
Patients with heart failure are in a state of BNP insufficiency that results from a deficiency of the active BNP form plus molecular resistance to its effects.6 Studies have demonstrated that the BNP detected in acute heart failure is primar-ily the high–molecular-weight proBNP rather than the biologically active form.7 Some authors have suggested that abnor-mal cellular processing of BNP is a factor in the relative BNP-deficiency state in heart failure.8 In addition, upregulation of phos-phodiesterases leads to rapid clearance of the secondary messenger, cyclic guanosine monophosphate, despite high activation of the NPRs by natriuretic peptides.9
What constitutes a normal natriuretic peptide level depends on the specific clini-cal setting. Two studies revealed that BNP levels in normal adults without cardiovas-cular disease increase with age and appear to be higher in women; NT-proBNP levels show greater age dependence than do BNP levels.10,11 According to general guide-lines, 90% of young, healthy adults have a BNP level of less than 25 pg/mL and an NT-proBNP level of 70 pg/mL or lower.12 In patients presenting with acute dyspnea, a BNP cutoff level of less than 100 pg/mL and an NT-proBNP cutoff level of less than 300 pg/mL should be used to rule out heart failure.13,14
Natriuretic Peptides in Acute Heart Failure. BNP and NT-proBNP levels have become powerful diagnostic tools in the evaluation of patients presenting with dys-pnea in a variety of clinical settings. In the Breathing Not Properly Multinational Study, BNP levels were measured in 1586 patients with shortness of breath upon arrival in the emergency department (ED). Use of this information resulted in a higher diagnostic accuracy, with an area under the receiver-operating characteristic curve (AUC) of 0.91.13 A BNP cutoff level of 100 pg/mL was 90% sensitive and 76% specific for the diag-nosis of heart failure as the cause of dyspnea (Figure 37-2). The data were similar for NT-proBNP levels in the ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) study, in which NT-proBNP level was measured in 600 emergency department

546
CH 37
Less active
Inactive
proBNP(108 amino acids)
NT-proBNP(76 amino acids)
Active
BNP(32 amino acids)
1 76
Corin
77 108
FIGURE 37–1 Synthesis of B-type natriuretic peptide (BNP) and amino terminal B-type natriuretic peptide (NT-proBNP).
patients with dyspnea; its sensitivity and specificity were demonstrated in the diagnosis of CHF (AUC = 0.94).14 A NT-proBNP cutoff level of less than 300 pg/mL was proposed to rule out heart failure as the cause of dyspnea.
The use of natriuretic peptides has also proved to be sig-nificantly cost effective. In the BNP for Acute Shortness of breath EvaLuation (BASEL) study, 452 patients in the emer-gency department with dyspnea were randomly assigned to conditions involving a single measurement of BNP level ver-sus no such measurement. The researchers reported a 10% reduction in the rate of admissions and a 3-day decrease in the median length of stay with the use of BNP level, which amounted to a total cost savings of $1800 per patient with-out an increase in mortality or repeated hospitalization.15 These results were also obtained with NT-proBNP level in the Improved Management of Patients with Congestive Heart Failure (IMPROVE-CHF) study.16
Elevated Natriuretic Peptide Levels in Other Clinical Set-tings. For optimal use of natriuretic peptides, it is important to be aware of clinical scenarios other than acute heart failure that can cause natriuretic peptide levels to rise. Patients with a history of heart failure but without an acute exacerbation
TABLE 37–1 Biochemical Properties of BNP versus NT-proBNP
Property BNP NT-proBNP
Amino acids 32 76
Molecular weight (kDa) 3.5 8.5
Half-life (min) 22 60-120
Clearance
ll Primary Neutral endopeptidase Renal
ll C-R NPR-C Renal
ll Hemodialysis No No
Point-of-care Yes Pending
Correlation with GFR Moderate Strong
Biologically active Yes No
Clinical range (pg/mL) 0–5,000 0–35,000
Adapted from Daniels LB, Maisel AS. Natriuretic peptides. J Am Coll Cardiol 2007;50:2357-2368.BNP, B-type natriuretic peptide; C-R, clearance receptor; GFR, glomerular filtration rate; NPR, natriuretic peptide receptor; NT-proBNP, N-terminal pro–B-type natriuretic peptide.
can have intermediate BNP levels, as shown in the Breathing Not Properly trial.13 Acute coronary syndrome is also associ-ated with elevated levels of natriuretic peptides: Acute ische-mia causes transient diastolic dysfunction, which results in increased left ventricular end-diastolic pressure, a rise in wall stress, and increased synthesis of BNP.17 The role of BNP is perhaps most useful in differentiating between cardiac and pulmonary causes of shortness of breath. Although natri-uretic peptide values can be elevated to intermediate levels in patients with underlying lung disease, they tend to remain significantly lower than in patients presenting with CHF.18 The Breathing Not Properly trial demonstrated that natri-uretic peptide levels were useful in diagnosing heart failure, which was present in 87 of the 417 patients with a history of chronic obstructive pulmonary disease or asthma (BNP lev-els, 587 vs. 109 pg/mL, P < .0001).13 In addition, right-sided heart dysfunction from hemodynamically significant pulmo-nary embolism, severe lung disease, and pulmonary hyper-tension can lead to elevated levels of natriuretic peptides.19-22 Therefore, when levels of natriuretic peptide levels are inter-mediate in an acutely dyspneic patient, it is important to con-sider other life-threatening causes of dyspnea.
Hyperdynamic states of sepsis, cirrhosis, and hyperthy-roidism can also be associated with elevated levels of natri-uretic peptides.23-25 These levels are also increased in the setting of atrial fibrillation.26 The Breathing Not Properly trial demonstrated that BNP still performed well in patients with atrial fibrillation; the AUC was 0.84, in comparison with 0.91 for the entire cohort. Increasing the cutoff to 200 pg/mL for these patients improved specificity and the positive predic-tive value for diagnosing heart failure.27
Caveats. In addition to wall stress, other factors have been associated with elevated natriuretic peptide levels. Aging appears to be associated with elevated levels of BNP, inde-pendent of the degree of diastolic dysfunction.10,11 Possible mechanisms may include altered renal function, changes in the biosynthesis and processing of natriuretic peptides on a cellular level, or a reduction in the clearance receptor NPR-C.28 Women appear to have higher natriuretic peptide levels than do men the same age.10,11 Although the reasons for this sex difference are unclear, some authors have proposed that differences in estrogen or testosterone may be responsible.10,29
The association between natriuretic peptide levels and renal function is complex. In the setting of renal dysfunc-tion, increased concentrations of natriuretic peptides may stem from elevation in atrial pressure, systemic pressure, or ventricular mass. Many patients with renal disease have hypertension that results in significant left ventricular hyper-trophy, and comorbid cardiac conditions are also common.

The Use of B
iomarkers in the Evaluation of H
eart Failure
547
CH 37
0.0
A B
0.2
0.4
0.6
0.8
1.0
0.0 0.2
BNP, 150 pg/mL
Area under the receiver-operating-characteristic curve,0.91 (95% confidence interval, 0.90-0.93)
BNP, 125 pg/mLBNP, 100 pg/mL
BNP, 80 pg/mL
BNP, 50 pg/mL
0.4 0.6 0.8 1.0
BNP Sensitivity Specificity
Positivepredictive
value
Negativepredictive
value Accuracy
50 97 (96-98) 62 (59-66) 71 (68-74) 96 (94-97) 7980 93 (91-95) 74 (70-77) 77 (75-80) 92 (89-94) 83100 90 (88-92) 76 (73-79) 79 (76-81) 89 (87-91) 83125 87 (85-90) 79 (76-82) 80 (78-83) 87 (84-89) 83150 85 (82-88) 83 (80-85) 83 (80-85) 85 (83-88) 84
(95% confidence interval)pg/mL
1-Specificity
Sen
sitiv
ity
0
200
400
600
800
1000
1200
1400
I(N = 18)
II(N = 152)
III(N = 351)
IV(N = 276)
New York heart association class
B-T
ype
natr
iure
tic p
eptid
e (p
g/m
l)
FIGURE 37–2 A, Receiver-operating-characteristic curve from the Breathing Not Properly Multinational Study for various cutoff levels of B-type natriuretic peptides in differentiating dyspnea caused by heart failure from dyspnea with other causes. B, Box plots showing the median levels of B-type natriuretic peptide among patients in different New York Heart Association classifications. (From Maisel AS, Krishnaswamy P, Nowak RM, et al. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002;347:161-167.)
This interplay between the heart and kidneys in patients with reduced renal function accounts for one component of the increase in natriuretic peptide levels, reflecting elevated “true” physiological natriuretic peptide levels.30 The Breath-ing Not Properly study revealed a weak but significant corre-lation between glomerular filtration rate (GFR) and BNP level, and the researchers suggested higher cutoff levels for patients with a GFR of less than 60 mL/min/1.7 m2.31
Interpretation of NT-proBNP levels in the setting of renal dysfunction is more challenging because clearance is not mediated by NPR-C or neutral endopeptidase and is more dependent on renal function. GFR seems to be more strongly correlated with NT-proBNP (r = −0.55) than with BNP, although the discrepancy may be somewhat less prominent in patients with acute CHF (r = −0.33 for NT-proBNP level, and r = −0.18 for BNP level).32,33 An analysis from the PRIDE study demonstrated that NT-proBNP levels in patients with GFR of less than 60 mL/min/1.7 m2 were still the strongest predic-tors of outcome, and the researchers suggested a higher cutoff level, of more than 1200 pg/mL, for the diagnosis of heart fail-ure.33 In contrast, there is no significant relationship between NT-proBNP levels and GFR in relatively healthy patients with mild renal insufficiency.34 Despite the relationship between natriuretic peptide levels and GFR, both BNP and NT-proBNP levels still provide important diagnostic and prognostic infor-mation in patients with renal dysfunction.
Several studies have demonstrated an inverse relationship between body mass index and BNP levels.11,35-37 The reasons for this are not fully elucidated, although some authors have postulated a theory of increased clearance mediated though elevated levels of NPR-C in adipocytes38; data for this are con-flicting.34,39 A study of patients who had undergone bariat-ric surgery showed an increase in both BNP and NT-proBNP levels after the surgery, which suggests that downregula-tion of natriuretic peptide production in obesity, rather than increased clearance, may be responsible for the lower levels of natriuretic peptides in the obese population.40 Despite lower circulating levels, natriuretic peptide values retain
their diagnostic capability in obese patients, albeit at lower cutoff levels.37
Flash pulmonary edema, CHF with causes upstream from the left ventricle (i.e., acute mitral regurgitation and mitral stenosis), and pericardial disease do not lead to substan-tial elevations in natriuretic peptides.30 In flash pulmonary edema, because of the small amounts of natriuretic peptides that are preformed and residing in secretory granules, and because of the delay between wall stress and upregulation of gene expression, the levels of natriuretic peptides are dis-proportionately low in comparison to symptoms.30 Patients with constrictive pericardial disease can present with symptoms of right-sided heart failure; however, because the stiff pericardium limits the myocardium’s stretching, natriuretic peptide levels are typically normal or minimally elevated.30,41
Prognosis. There has been a tremendous increase in data demonstrating the prognostic power of natriuretic peptides in a variety of clinical settings. Several researchers reported that natriuretic peptide levels in patients presenting to the emergency department with heart failure are predictive of future cardiovascular events. Every 100-pg/mL increase is associated with a 35% increase in the risk of death in heart failure.42 The prospective, multicenter Rapid ED Heart Fail-ure Outpatient Trial (REDHOT), involving 464 patients pre-senting to the emergency department with heart failure, showed that BNP level was predictive of future heart fail-ure–related events and of mortality and was superior to the emergency department physician’s assessment of severity of illness. In patients with chronic heart failure, BNP level provides powerful prognostic information regarding survival and deterioration of functional status.43 In more than 4300 outpatients with chronic heart failure in the Valsartan Heart Failure Trial (Val-HeFT), patients with the greatest rise in BNP level despite therapy had the highest rates of morbidity and mortality.44
In addition to heart failure, both BNP and NT-proBNP levels have strong prognostic value in patients with coronary

54
CH
8
37
artery disease, acute coronary syndrome, and valvular heart disease and in the prediction of sudden cardiac death.45-50
Monitoring TherapyInpatient. Several investigators evaluated the relationship between natriuretic peptide levels and pulmonary capillary wedge pressure (PCWP) derived from invasive hemody-namic catheters.51 Because in various clinical scenarios, as described above, there can be discordance between natri-uretic peptide levels and clinical manifestation, natriuretic peptide levels are not always correlated with PCWP.52 How-ever, in patients with decompensated heart failure caused by volume overload, a treatment-induced drop in PCWP usually leads to a rapid decrease in BNP levels (35 to 50 pg/mL/hour), especially in the first 24 hours of therapy, if adequate urine output is maintained.53
In the management of hospitalized patients with heart failure, it is probably not necessary to measure daily levels of natriuretic peptides. Instead, measurements obtained at admission and before discharge can be useful in tailoring the intensity of therapy and has been shown to have strong prognostic implications.54,55 A third measurement of natri-uretic peptide level in the first 24 to 48 hours after admission often enhances the success of a treatment plan. In a study with 114 patients admitted with CHF, the rate of mortality or readmission at 6 months was 15 times higher among patients with a predischarge BNP level of more than 700 pg/mL than among patients with BNP levels of less than 350 pg/mL at discharge.56
Outpatient. Monitoring natriuretic peptide levels in the outpatient setting might help improve patient care. Changes in natriuretic peptide levels could potentially, help physi-cians titrate neurohormonal blockade agents and diuretics more aggressively and safely. Prospective studies in this area have yielded somewhat conflicting results. In the Systolic Heart Failure Treatment Supported by BNP (STARS-BNP) study, 220 patients with heart failure and left ventricular dys-function were randomly assigned to receive therapy guided by natriuretic peptide levels versus standard of care; natri-uretic peptide–guided treatment that targeted a BNP level of less than 100 pg/mL significantly reduced death and hospital stay for heart failure.57 The smaller (130-patient) Strategies for Tailoring Advanced Heart Failure Regimens in the Out-patient Settings: Brain NatrIuretic Peptides Versus the Clini-cal CongesTion ScorE (STARBRITE) study failed to show significant improvement in the primary outcome of nonhos-pital days alive in patients whose treatment was guided by BNP levels. However, STARBRITE enrolled patients with more severe disease and used a higher BNP cutoff level.58 In both studies, clinicians who adjusted medical therapy on the basis of natriuretic peptide levels prescribed higher doses of angiotensin converting enzyme (ACE) inhibitors and β-blockers.
In patients monitored in clinics, it is important to establish the “steady state” level of natriuretic peptide that corresponds to a patient’s optimized fluid status. Significant deviations from this baseline level allows for rapid diagnosis and institu-tion of therapy, with more aggressive diuresis and follow-up when levels rise substantially.59 Values that are considerably reduced from baseline can signal overdiuresis and impending prerenal azotemia. In view of significant intraindividual vari-ability in natriuretic peptide levels, a change of at least 50% in the steady-state natriuretic peptide level might be a reason-able impetus for a more aggressive evaluation and modifica-tion of treatment.30
Adrenomedullin
Biology. Adrenomedullin level, like natriuretic peptide lev-els, is elevated in the setting of myocardial stress. Adreno-medullin is a vasoactive peptide consisting of 52 amino acids that shares homology with calcitonin gene–related peptide.60
The downstream actions of adrenomedullin are mediated by an increase in cyclic adenosine monophosphate levels and nitric oxide, which lead to potent vasodilation, an increase in cardiac output, and diuresis and natriuresis.61-65 This biologi-cally active modulator is produced from the precursor pre-proadrenomedullin, which is produced by the heart, adrenal medulla, lungs, kidneys, and vascular endothelium.66,67 Adre-nomedullin levels are increased in endothelial dysfunction, which is common in patients with heart failure and indicates a poor prognosis. In patients with heart failure, the magnitude of increase in adrenomedullin corresponds with the sever-ity of heart failure and is inversely related to left ventricular function.66,68 Adrenomedullin levels increase with worsen-ing symptoms of heart failure and elevated PCWP and are reduced with appropriate therapy.66 Because adrenomedullin combines with complement factor H and is rapidly cleared from the circulation, direct measurements are difficult; how-ever, midregional proadrenomedullin (MR-proADM), another product of the precursor, appears to be more clinically stable and can be readily measured.69,70
Adrenomedullin and Heart Failure. In one of the first studies involving MR-proADM, Khan and colleagues71 evalu-ated its prognostic capability in 983 patients after myocar-dial infarction. The data showed that MR-proADM level was increased in patients who died or developed heart failure (median, 1.19 nmol/L [interquartile range, 0.09 to 5.39 nmol/L]), in comparison with survivors (median, 0.71 nmol/L [interquartile range, 0.25 to 6.66 nmol/L], P < .0001). The AUCs for MR-proADM, NT-proBNP, and the combination of both markers for prediction of death or heart failure were 0.77, 0.79, and 0.84, respectively,71 which demonstrate that MR-proADM level is a powerful predictor of adverse outcome in patients after myocardial infarction and provides additive information with natriuretic peptides.
The prognostic implications of elevated MR-proADM levels in outpatients with both ischemic and nonischemic causes of CHF were evaluated by Adlbrecht and associates.72 In 786 patients admitted with CHF, they demonstrated that MR-proADM level was a significant predictor of death (hazard ratio = 1.77, P < .001) at 24 months.72 Adrenomedullin also has pre-dictive capability in asymptomatic patients with risk factors for coronary artery disease. In a study of 121 patients, Nishida and colleagues73 reported that patients with elevated adrenomedul-lin levels had a higher risk of future cardiovascular events.
MR-proADM has also been evaluated in patients with acute decompensated heart failure. Gegenhuber and associ-ates74 evaluated the utility of natriuretic peptides, MR-pro-ADM, and other biomarkers in predicting mortality in 137 patients with acute decompensated heart failure. The AUCs for the prediction of 1-year mortality were similar for BNP level (0.716; 95% confidence interval [CI], 0.633 to 0.790), midregional pro–A-type natriuretic peptide (MR-proANP) (0.725; 95% CI, 0.642 to 0.798), MR-proADM (0.708; 95% CI, 0.624 to 0.782), and copeptin (0.688; 95% CI, 0.603 to 0.764).74 In the Biomarkers in the Assessment of Congestive Heart failure (BACH) study, MR-proADM level was com-pared with BNP and NT-proBNP levels in predicting mortal-ity at 90 days in patients hospitalized for decompensated heart failure.75 Of 1641 patients recruited, 568 (approxi-mately one third) ultimately received a diagnosis of heart failure. The data showed that MR-proADM level was more accurate than BNP or NT-proBNP level at predicting out-come at 90 days.75
Summary of MR-proADM. Adrenomedullin is a rela-tively new biomarker that reflects biomechanical stretch and increases with pressure or volume overload. Although experi-ence with adrenomedullin is limited, available data indicate that its levels are correlated with natriuretic peptide levels and may provide superior prognostic information in patients with acute and chronic heart failure.

The Use of B
iomarkers in the Evaluation of H
eart Failure
549
CH 37
ST2 Receptor
Biology. ST2 is a member of the interleukin (IL)–1 recep-tor family and has two primary isoforms: a transmembrane receptor form (ST2L) and a soluble receptor form (soluble ST2), each regulated by different promoters.76-78 Microarray analysis demonstrates marked upregulation of the transcript for both forms of ST2; the soluble form displays more robust expression in mechanically stimulated cardiomyocytes.78,79 In response to mechanical strain, there is also an increase in the transcription and translation of the ligand, IL-33.80 The IL-33/ST2 signaling cascade is thought to play a criti-cal role in the regulation of myocardial response to pressure overload, which inhibits development of fibrosis.81-83 Genetic knockouts of ST2 in mouse models demonstrated significant cardiac hypertrophy, interstitial fibrosis, and cavity dilation in response to transverse aortic constriction.78,84 A similar phenotype is generated by infusion of soluble ST2, which is believed to serve as a decoy receptor, thereby decreasing the beneficial IL-33 interaction with ST2L. In this way, the ratio of soluble ST2 to IL-33 may regulate the IL-33/ST2L system.78
ST2 and Acute Coronary Syndrome. Clinically, ST2 lev-els increase early in the course of acute myocardial infarc-tion and are inversely correlated with systolic function.79 When ST2 levels were analyzed in more than 800 patients presenting to the hospital with an acute ST wave–elevation myocardial infarction (STEMI), the concentration of ST2 at the time of presentation was independently associated with in-hospital and 30-day mortality, after adjustments for age, hemodynamics, infarct territory, Killip class, and time from symptom onset to treatment.85 Sabatine and colleagues86 confirmed the utility of ST2 as a prognostic biomarker in 1239 patients presenting with STEMI in the Clopidogrel as Adjunctive Reperfusion Therapy—Thrombolysis in Myocar-dial Infarction (CLARITY-TIMI 28) trial. After adjusting for baseline characteristics and NT-proBNP level, the researchers showed that a concentration of ST2 above the median was a powerful predictor of cardiovascular death and heart failure, independent of baseline characteristics. The combination of ST2 and NT-proBNP levels significantly improved risk strati-fication in this patient population.86
ST2 and Heart Failure. In 161 patients with severe chronic New York Heart Association (NYHA) class III or IV heart fail-ure, Weinberg and associates87 demonstrated that change in ST2 levels over a 2-week period was an independent predic-tor of subsequent mortality or need for transplantation. ST2 has also proved to be an important biomarker in patients pre-senting with acute decompensated heart failure. In the PRIDE study,88 ST2 concentrations were measured in 593 dyspneic patients in the emergency department and were found to be higher among patients with acute heart failure than in patients with other causes of dyspnea (0.50 vs. 0.15 ng/mL; P < .001). An ST2 level of 0.20 ng/mL or higher was strongly predictive of death at 1 year in the entire cohort (hazard ratio = 5.6; 95% CI, 2.2 to 14.2; P < .001). The 1-year mortality rate was less than 4.5% among patients in the lowest decile of ST2 levels but 45% among those in the highest decile (Figure 37-3).88 In another study, researchers investigated change in ST2 concentrations during the course of a hospitalization in 150 patients with acute heart failure.89 Multiple biomarkers were measured, including ST2, BNP, NT-proBNP, and blood urea nitrogen levels six times between admission and discharge. Among patients whose ST2 values decreased by 15.5% or more during the study period, the incidence of death was 7%, whereas those whose ST2 levels failed to decrease by 15.5% had a 33% chance of dying within 90 days.89 Rehman and coworkers further examined the patient-specific char-acteristics of ST2 in 346 patients hospitalized with acute heart failure, demonstrating that, even after they controlled for established and clinical characteristics, ST2 remained
a predictor of mortality (hazard ratio = 2.04; 95% CI, 1.3 to 3.24; P = .003)90 (see Figure 37-3). The highest mortality rate was observed in the subgroup that had elevated concentra-tions of both natriuretic peptides and ST2 (see Figure 37-3).90
Summary of ST2. ST2, induced by myocardial response to pressure or volume overload, has important clinical impli-cations for patients with acute and chronic heart failure and acute coronary syndrome. ST2 level is a powerful prognosti-cator of future cardiovascular morbidity and mortality, pro-viding incremental information to natriuretic peptides.91
Markers of InflammationAn abnormal inflammatory response in patients with heart failure is a mechanism for disease progression and deterio-ration (see also Chapter 11).92 Ischemia and other biological insults to the heart trigger an innate immune response that leads to the upregulation of proinflammatory cytokines.91 The activation of these mediators results in apoptosis, hypertro-phy, and dilation, which in turn accelerate disease progression ( Figure 37-4).91 Many features of heart failure, including hemodynamic compromise and vascular abnormalities, can be explained through the effects of proinflammatory cyto-kines, including C-reactive protein (CRP), tumor necrosis factor–α (TNF-α), IL-6, and IL-18.93-96
C-Reactive Protein
Biology. CRP is synthesized by hepatocytes in response mainly to IL-6.97 CRP is an acute-phase reactant, its levels becoming significantly elevated shortly within the onset of an inflammatory process. It is, however, a nonspecific marker; its levels are commonly increased in the settings of infection, inflammatory diseases, acute coronary syndrome, smoking, and neoplastic processes.98 CRP reduces the release of nitric oxide and increases expression of endothelin-1 and of endo-thelial adhesion molecules.99 These features are suggestive of mechanisms by which CRP plays an important role in vascu-lar diseases.
hsCRP and Future Development of Heart Failure. In four major population-based investigations (the Cardiovascular Health Study; the Framingham Heart Study; the Health, Aging and Body Composition [Health ABC] Study; and the Rotter-dam Study), researchers have studied the development of heart failure in asymptomatic populations. In a total of 15,282 elderly patients with a follow-up period of 3.6 to 6.2 years, there was a significant association between elevated levels of high-sensitivity C-reactive protein (hsCRP) and development of heart failure (Table 37-2).98,100-103 Subsets of patients with the highest levels of hsCRP had double the risk of developing heart failure. In the Health ABC study, high levels of other inflammatory cytokines such as TNF-α and IL-6 were also pre-dictive of the development of heart failure.102 The Rotterdam Study demonstrated that the association between hsCRP and heart failure was weaker in women and did not persist after the adjustment for traditional cardiovascular risk factors.103
hsCRP and Patients at High Risk. Large volumes of data have been collected to examine the relationship between hsCRP and the development of heart failure in high-risk patients, particularly those admitted with acute coronary syndrome (Table 37-3).98,104-112 The initial study, in which Pietilä and colleagues104 monitored 188 patients with acute myocardial infarction for 2 years, revealed that patients with higher peak CRP levels within the period immediately after myocardial infarction had increased risk of death and of development of heart failure. Subsequent analysis of more than 10,000 patients confirmed that elevated levels of CRP or hsCRP within 24 hours of acute myocardial infarction were predictive of short- and long-term development of heart failure. One of the largest studies of this association was a substudy from the Thrombolysis in Myocardial Infarction

550
CH 37
0.0
A
0.1
0.2
0.3
0.4
0.5
0.6 ST2↑/NT-proBNP↑(n=118)ST2↑/NT-proBNP↓(n=57)ST2↓/NT-proBNP↑(n=59)ST2↓/NT-proBNP↓(n=114)
NT-proBNPNo discrimination
BNPCRP [mg/L]ST2 (ng/mL)
ST2↑/BNP↑(n=100)ST2↑/BNP↓(n=63)ST2↓/BNP↑(n=66)ST2↓/BNP↓(n=106)
0 73 146
P < 0.001
P < 0.001
P < 0.001 for trend
219 292 365
Days from enrollment
Cum
ulat
ive
haza
rd
0.0
C
0.1
0.3
0.5
0.7
0.9
0.2
0.4
0.6
0.8
1.0
0 0.2 0.4 0.6 0.8 1
1-Specificity (false positives)
Sen
sitiv
ity (
true
pos
itive
s)
0.0
B
0.1
0.2
0.3
0.4
0.5
0.6
0 73 146 219 292 365
Days from enrollment
Cum
ulat
ive
haza
rd
0
D
10
20
30
40
50
60
10987654321
ST2 decile
One
yea
r m
orta
lity
(%)
FIGURE 37–3 Mortality rates among patients hospitalized with acute heart failure as a function of ST2 and amino-terminal B-type natriuretic peptide (NT-proBNP) concentration (A) or B-type natriuretic peptide (BNP) concentration (B). C, Receiver-operator characteristic curve analysis, in which ST2 level was compared with BNP, NT-proBNP and C-reactive protein levels for predicting death at 1 year after hospitalization for acute heart failure. D, Frequency of mor-tality at 1 year as a function of ST2 decile. (From Rehman SU, Mueller T, Januzzi JL Jr. Characteristics of the novel interleukin family biomarker ST2 in patients with acute heart failure. J Am Coll Cardiol 2008;52:1458-1465.)
(TIMI) trials, in which increased concentrations of hsCRP within 48 hours of symptoms were strongly associated with development of heart failure.105 More recently, analysis of data from more than 4000 patients with STEMI and non-STEMI revealed higher event rates for cardiovascular endpoints (car-diovascular death, recurrent myocardial infarction, stroke, and heart failure) with increasing quartile of hsCRP.106 Patients with chronic stable angina have a similar association between hsCRP levels and the development of heart failure.113,114
hsCRP and Prognosis in Heart Failure. The prognostic implications of hsCRP on patients with chronic heart failure has been evaluated in multiple studies with a combined total of more than 6600 patients (Table 37-4).98,115-123 There is a signifi-cant association between increasing level of hsCRP and worse cardiovascular outcome, including mortality, poorer left ventric-ular systolic function, higher prevalence of NYHA class III or IV symptoms, and poorer quality of life. This association appears to translate to both ischemic and nonischemic causes of heart failure.98,115-123
In contrast, there is a paucity of data regarding the prognostic significance of hsCRP in acute heart failure.
Alonso-Martínez and associates124 prospectively stud-ied the role of hsCRP in 76 patients admitted with acute heart failure. The data showed that elevated hsCRP levels were associated with increased rates of readmission at 18 months. Subsequently, Mueller and colleagues125 evalu-ated 214 patients with acute heart failure to determine the impact of CRP on rates of all-cause mortality at 24 months. Patients in the highest tertile of CRP had significantly higher in-hospital and all-cause mortality rates at 24 months. In a study in Finland, researchers evaluated clinical and bio-chemical predictors of 1-year mortality in patients hospital-ized for acute heart failure. The data showed that elevated CRP was independently predictive of mortality at 1 year.126 Villacorta and coworkers127 confirmed that finding in 119 patients hospitalized in Brazil for acute heart failure: CRP proved to be the most important prognosticator of mortal-ity at 1 year. Although the role of CRP as a prognostic bio-marker in patients hospitalized with heart failure is yet to be firmly established, limited data suggest that it may comple-ment other biomarkers in predicting mortality in this clini-cal setting.

The Use of B
iomarkers in the Evaluation of H
eart Failure
551
CH 37
Stimulation
Sympatheticnervoussystem
Cytokinesimpairing myocardial
function
Reducedcardiac output
Activatedmonocyte
Activatedmonocyte
Damagedmyocardium
Proinflammatorycytokines
Cytokines impairingmyocardial function
Natriureticpeptides
Hypoperfusedskeletal muscle
Release intobloodstream
Cytokines
Cytokines
FIGURE 37–4 Cytokine hypothesis of heart failure. Injury to the myocardium leads to increased production of proinflammatory cytokines (tumor necrosisfactor α, interleukin-6, and interleukin-18), leading to depressed cardiac function. Decreased cardiac output causes hypoperfusion of the skeletal muscle, whichactivates monocytes to produce proinflammatory cytokines, further impairing cardiac function. (From Braunwald E. Biomarkers in heart failure. N Engl J Med2008;358:2148-2159.)
Summary of CRP. CRP is perhaps the most important marker of inflammation and is analyzed routinely in clinical practice. It has validated utility in predicting the development of heart failure in populations at low and high risk, as well as prognostic value in patients with chronic heart failure. In practice, interpretation of CRP levels is somewhat hampered by the fact that different cutoff levels were used in different studies, which makes it difficult to interpret individual CRP levels. Determining the real-world clinical utility of hsCRP necessitates further investigation with uniform studies that use similar assays and cutoff levels.
Markers of Myocardial Cell DeathTroponin
Biology. Myocardial cell death occurs in heart failure and may represent the common final pathway to refractory fail-ure. Although cardiac troponin levels are well-validated markers of myocyte injury during myocardial infarction, the pathophysiological mechanisms underlying troponin eleva-tions in heart failure are probably distinct from those under-lying acute coronary syndrome.128 Multiple mechanisms, including inflammation, interstitial fibrosis, increased wall stress, oxidative stress, and neurohormonal activation, are responsible for adverse cardiac remodeling that leads to pro-gressive heart failure.129-132 Troponin is a cardiac structural protein that is part of the troponin-tropomyosin complex, with a small amount present in the cytosol.128 Mild elevations in troponin levels have been documented in a number of car-diovascular diseases, including heart failure. Hypotheses for why troponin levels may be elevated in the setting of heart failure, even in the absence of overt ischemia, include myo-cardial injury, loss of cell membrane integrity, excessive wall
tension that leads to subendocardial ischemia, apoptosis, or some combination.91,133,134
Troponin and Acute Heart Failure. Several small stud-ies have reported elevated cardiac troponin levels in patients with decompensated heart failure, in the absence of acute coronary syndrome, and found associations between elevated levels of troponin and poor prognosis.135-138 La Vecchia and associates evaluated the clinical associations and prognos-tic implications of detectable cardiac troponin I (cTnI) in 34 patients with severe heart failure.139 Modest elevations of cTnI (0.7 ± 0.3 ng/mL) were associated with lower left ven-tricular ejection fraction, and there was a trend toward higher pulmonary artery pressures. In patients who evinced clinical improvement after admission, cTnI levels became undetect-able after a few days.139 However, in patients with refractory heart failure who ultimately died in the hospital, detectable levels of cTnI persisted throughout the observation period.139 In a larger subsequent study, cTnI’s role in predicting adverse outcomes was evaluated in 238 patients with advanced heart failure who were referred for cardiac transplantation.128 Patients with detectable cTnI levels of 0.04 ng/mL or higher had higher levels of BNP, impaired hemodynamics (indicated by lower cardiac index and higher PCWP), progression of left ventricular dysfunction, and increased mortality (relative risk, 2.05; 95% CI, 1.22 to 3.43).128
Researchers for the Acute Decompensated Heart Fail-ure National Registry (ADHERE) evaluated the association between cardiac troponin level and adverse events in 84,872 patients with acute decompensated heart failure.140 Patients with positive troponin levels (6.2%) had lower systolic blood pressure on admission, a lower ejection fraction, and higher rates of in-hospital mortality (8.0% vs. 2.7%; P < .001) than did those with undetectable troponin levels (odds ratio for

55
CH 3
2
7
TABLE 37–2 Association Between hsCRP and Future Development of Heart Failure in Population-Based Studies
Reference Study Title (country) N Age (% Male)
Mean Follow-up Period (Years)
Incidence of Heart Failure
hsCRP Comparisons*
Hazard Ratio (95% CI) Adjusted for
Gottdiener et al100
Predictors of congestive heart failure in the elderly. The Cardiovascu-lar Health Study (United States)
5888 73 ± 5 years (42%)
5.5 597 (19.3%) ≥7.40 mg/L vs. ≤0.95 mg/L (5th vs. 1st quintile)
1.91 (1.43–2.57)
Age, sex, CHD or stroke at baseline, diabe-tes, SBP, LVEF ankle-arm index, internal carotid thickness, FEV1, creatinine
Vasan et al101 Inflammatory markers and risk of HF in elderly subjects without prior myocardial infarction. The Framingham Heart Study (United States)
732 78 ± 4 years (33%)
5.2 56 (7.6%) ≥5 mg/L vs. <5 mg/L (optimal cutoff)
2.81 (1.22–6.50)
Age, sex, smoking, diabetes, valve disease, AF, previous cardio-vascular disease, LV hypertrophy, total choles-terol/HDL
Cesari et al102 Inflammatory markers and onset of cardio-vascular events. Results from the Health ABC Study (United States)
2225 70–79 years (45%)
3.6 92 (4.1%) ≥2.51 mg/L vs. ≤1.15 mg/L (3rd vs. 1st tertile)
2.60 (1.45–4.67)
Age, sex, race, smoking, diabe-tes, hyperten-sion, BMI, HDL cholesterol, triglycerides, albumin
Kardys et al103 CRP and risk of heart failure. The Rotterdam Study (The Netherlands)
6437 ≥55 years; 69 ± 9 years (40%)
6.5 551 (8.6%) ≥3.5 mg/L vs. ≤0.9 mg/L (4th vs. 1st quartile)
2.08 (1.58–2.74)
Age, sex, smoking, hypertension, diabetes, BMI, HDL cholesterol
*hsCRP method in all studies except the Framingham Heart Study (standard method).AF, atrial fibrillation; BMI, body mass index; CI, confidence interval; FEV1, forced expiratory volume in 1 second; HDL, high-density lipoprotein; HF, heart failure; hsCRP, high-sensitivity C-reactive protein; LV, left ventricular; LVEF, left ventricular ejection fraction; SBP, systolic blood pressure.Modified from Araújo JP, Lourenço P, Azevedo A, et al. Prognostic value of high-sensitivity C-reactive protein in heart failure: a systematic review. J Card Fail 2009;15:256-266.
death, 2.55; 95% CI, 2.24 to 2.89; P < .001). The study con-firmed the powerful prognostic utility of elevated troponin levels in predicting mortality in patients hospitalized with decompensated heart failure (Figure 37-5).140
Troponin and Chronic Heart Failure. The role of myocyte injury in prognosis of stable outpatients with heart failure was evaluated by Hudson and colleagues.141 The authors assessed the association of elevated cardiac troponin T (cTnT) levels with the severity, cause, and prognosis of heart failure in 136 stable, ambulatory patients. Patients with elevated cTnT lev-els had an increased risk of death from or hospitalization for heart failure (relative risk, 2.7; 95% CI, 1.7 to 4.3; P = .001) and of death alone (relative risk, 4.2; 95% CI, 1.8 to 9.5, P = .001) at 14 months.141 In a larger outpatient data set from the Val-HeFT study, Latini and associates142 measured levels of cTnT and high-sensitive troponin T (hsTnT) in 4053 patients with chronic stable heart failure. Levels of cTnT were detect-able in 10.4% of the population with the cTnT assay (lower limit of detection, 0.01 ng/mL), in comparison with levels of htTNT in 92.0% with the new hsTnT assay (lower limit of detection, 0.001 ng/mL). Patients in whom cTnT level was elevated or in whom hsTnT level was above the median (0.012 ng/mL) had more severe heart failure and worse prognoses.142 Increased concentration of cTnT (cTnT level > 0.01 ng/mL) was associated with an increased risk of death (hazard ratio, 2.08; 95% CI, 1.72 to 2.52; P < .0001) and increased risk of first hospitalization for heart failure (hazard ratio, 1.55; 95% CI, 1.25 to 1.93; P < .0001) in multivariable models. For each 0.01-ng/mL increase in hsTnT level, risk of death increased 5% (95% CI, 4% to 7%).142
Troponin Summary. Cardiac troponin level, an indicator of myocardial necrosis, is an established biomarker in the evaluation and management of acute coronary syndrome. There is clear evidence that even low levels of detectable troponin in patients with heart failure have significant prog-nostic implications in terms of morbidity and mortality. Such data hold true for patients with both acute decompensated and chronic heart failure.
Markers of Renal Function (see also Chapter 18)Neutrophil Gelatinase–Associated Lipocalin
In patients with heart failure who develop concurrent renal dysfunction, management is challenging, requiring a careful balance between diuretic and vasodilator therapy. The car-diorenal syndrome is a well-documented phenomenon and is common among patients with acute heart failure; approxi-mately 60% of these patients manifest at least a 0.1-mg/dL rise in creatinine level during hospitalization.143 However, nephrotoxic insult, as manifested by an increase in serum creatinine level, usually takes at least 24 hours to become evi-dent. A biomarker that rapidly increases with acute kidney injury could prove to be very powerful in guiding therapy in these complex clinical scenarios.
Biology. Neutrophil gelatinase–associated lipocalin (NGAL) is a small (25-kDa) molecule that belongs to a super-family of lipocalins and is normally secreted in low amounts in the lungs, kidneys, trachea, stomach, and colon.144,145 Levels of NGAL are elevated in various pathological states, including acute or chronic kidney injury, neoplasia, chronic

The Use of B
iomarkers in the Evaluation of H
eart Failure
53
H 37
5
C
TABLE 37–3 Association Between hsCRP and Future Development of Heart Failure in High-Risk Population
Reference Study (Country) N Age (% Male)Follow-up Period
Incidence of Heart Failure
hsCRP Comparisons
Hazard Ratio (95% CI) Adjusted for
Berton et al107 C-reactive protein in acute myocardial infarction: asso-ciation with heart failure (Italy)
220 66.7 years (mean) (74%)
1 year 86 (39.1%) ≥15 mg/L vs. <15 mg/L (optimal cutoff)
4.3 (–) Age, gender, hypertension, diabetes, BMI, CK, previous MI or angor, thrombolytic therapy, LVEF
Suleiman et al108 Admission C-reactive protein levels and 30-day mortality in acute myocardial infarc-tion (Israel)
448 60 ± 12 years (78%)
30 days 121 (27.0%) >23.3 mg/L vs. <6.9 mg/L (3rd vs. 1st tertile)
2.60 (1.50–4.60)
Age, gender, smoking, diabe-tes, hyperten-sion, Killip class, BP, creatinine, aspirin, statin, anterior and ST-elevation MI, thrombolysis vs. primary angioplasty, wall motion score index
Suleiman et al109 Early inflammation and risk of long- term development of heart failure and mortality in survivors of acute myocardial infarc-tion: predictive role of C-reactive protein (Israel)
1044 60 ± 13 years (89%)
23 months (median)
112 (10.7%) ≥37.9 mg/L vs. ≤5.0 mg/L (4th vs. 1st quartile)
2.80 (1.40–5.90)
Age, gender, smoking, hypertension, diabetes, BB, previous HF, Killip class, anterior MI, BP, heart rate, peak CK, LVEF
Scirica et al105 Clinical application of C-reactive protein across the spectrum of acute coronary syndromes (United States)
1992 61.1 years (mean) (70%)
30 days and 10 months
— >25.4 mg/L vs. <3.4 mg/L (4th vs. 1st quartile)
30 days: 8.20 (–)10 months: 2.60 (–)
Age, gender, smoking, diabetes, prior MI, peripheral arterial or cerebrovascular disease, hyper-cholesterolemia BMI, Killip class, statin, aspirin, treatment with orbofiban
Bursi et al110 C-reactive protein and heart failure after myocardial infarction in the community (United States)
329 69 ± 16 years (52%)
1 year 92 (28.0%) > 15 mg/L vs. < 3 mg/L (3rd vs. 1st tertile)
2.83 (1.27–4.82)
Age, sex, comorbid conditions, previous MI, Killip class, troponin T
Kavsak et al111 Elevated C-reactive protein in acute coronary syndrome presentation is an independent predictor of long-term mortality and heart failure (United States)
446 62 ± 14 years (59%)
2 and 8 years
— >7.44 mg/L vs. <3 mg/L (arbi-trary cutoff)
2 years: 4.14 (–) 8 years: 3.69 (–)
Age, sex, presentation, and peak troponin I
Hartford et al112 C-reactive protein, interleukin-6, secre-tory phospholipase A2 group IIA and intercellular adhe-sion molecule–1 in the prediction of late outcome events after acute coronary syndromes (Sweden)
757 65 years (73%) 2 years for morbidity (median)
76 (10.0%) >16 mg/L vs. <2 mg/L (4th vs. 1st quartile)
1.40 (1.10–1.90)
Age, sex, smok-ing, diabetes, hypertension, hypercholester-olemia, obesity, previous MI, Killip class, cre-atinine, aspirin, statin, BB, ACE inhibitor
(Continued)
554
CH 37
Reference Study (Country) N Age (% Male)Follow-up Period
Incidence of Heart Failure
hsCRP Comparisons
Hazard Ratio (95% CI) Adjusted for
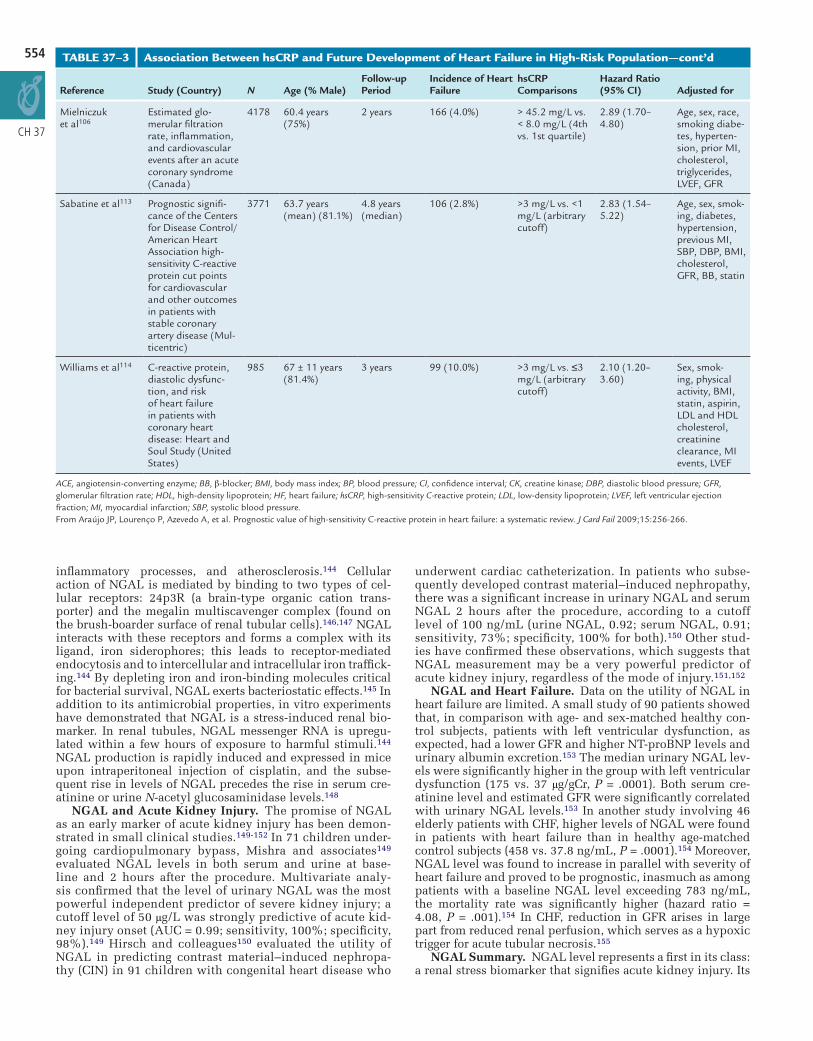
Mielniczuk et al106
Estimated glo-merular filtration rate, inflammation, and cardiovascular events after an acute coronary syndrome (Canada)
4178 60.4 years (75%)
2 years 166 (4.0%) > 45.2 mg/L vs. < 8.0 mg/L (4th vs. 1st quartile)
2.89 (1.70–4.80)
Age, sex, race, smoking diabe-tes, hyperten-sion, prior MI, cholesterol, triglycerides, LVEF, GFR
Sabatine et al113 Prognostic signifi-cance of the Centers for Disease Control/American Heart Association high-sensitivity C-reactive protein cut points for cardiovascular and other outcomes in patients with stable coronary artery disease (Mul-ticentric)
3771 63.7 years (mean) (81.1%)
4.8 years (median)
106 (2.8%) >3 mg/L vs. <1 mg/L (arbitrary cutoff)
2.83 (1.54–5.22)
Age, sex, smok-ing, diabetes, hypertension, previous MI, SBP, DBP, BMI, cholesterol, GFR, BB, statin
Williams et al114 C-reactive protein, diastolic dysfunc-tion, and risk of heart failure in patients with coronary heart disease: Heart and Soul Study (United States)
985 67 ± 11 years (81.4%)
3 years 99 (10.0%) >3 mg/L vs. ≤3 mg/L (arbitrary cutoff)
2.10 (1.20–3.60)
Sex, smok-ing, physical activity, BMI, statin, aspirin, LDL and HDL cholesterol, creatinine clearance, MI events, LVEF
ACE, angiotensin-converting enzyme; BB, β-blocker; BMI, body mass index; BP, blood pressure; CI, confidence interval; CK, creatine kinase; DBP, diastolic blood pressure; GFR, glomerular filtration rate; HDL, high-density lipoprotein; HF, heart failure; hsCRP, high-sensitivity C-reactive protein; LDL, low-density lipoprotein; LVEF, left ventricular ejection fraction; MI, myocardial infarction; SBP, systolic blood pressure.From Araújo JP, Lourenço P, Azevedo A, et al. Prognostic value of high-sensitivity C-reactive protein in heart failure: a systematic review. J Card Fail 2009;15:256-266.
TABLE 37–3 Association Between hsCRP and Future Development of Heart Failure in High-Risk Population—cont’d
inflammatory processes, and atherosclerosis.144 Cellular action of NGAL is mediated by binding to two types of cel-lular receptors: 24p3R (a brain-type organic cation trans-porter) and the megalin multiscavenger complex (found on the brush-boarder surface of renal tubular cells).146,147 NGAL interacts with these receptors and forms a complex with its ligand, iron siderophores; this leads to receptor-mediated endocytosis and to intercellular and intracellular iron traffick-ing.144 By depleting iron and iron-binding molecules critical for bacterial survival, NGAL exerts bacteriostatic effects.145 In addition to its antimicrobial properties, in vitro experiments have demonstrated that NGAL is a stress-induced renal bio-marker. In renal tubules, NGAL messenger RNA is upregu-lated within a few hours of exposure to harmful stimuli.144 NGAL production is rapidly induced and expressed in mice upon intraperitoneal injection of cisplatin, and the subse-quent rise in levels of NGAL precedes the rise in serum cre-atinine or urine N-acetyl glucosaminidase levels.148
NGAL and Acute Kidney Injury. The promise of NGAL as an early marker of acute kidney injury has been demon-strated in small clinical studies.149-152 In 71 children under-going cardiopulmonary bypass, Mishra and associates149 evaluated NGAL levels in both serum and urine at base-line and 2 hours after the procedure. Multivariate analy-sis confirmed that the level of urinary NGAL was the most powerful independent predictor of severe kidney injury; a cutoff level of 50 μg/L was strongly predictive of acute kid-ney injury onset (AUC = 0.99; sensitivity, 100%; specificity, 98%).149 Hirsch and colleagues150 evaluated the utility of NGAL in predicting contrast material–induced nephropa-thy (CIN) in 91 children with congenital heart disease who
underwent cardiac catheterization. In patients who subse-quently developed contrast material–induced nephropathy, there was a significant increase in urinary NGAL and serum NGAL 2 hours after the procedure, according to a cutoff level of 100 ng/mL (urine NGAL, 0.92; serum NGAL, 0.91; sensitivity, 73%; specificity, 100% for both).150 Other stud-ies have confirmed these observations, which suggests that NGAL measurement may be a very powerful predictor of acute kidney injury, regardless of the mode of injury.151,152
NGAL and Heart Failure. Data on the utility of NGAL in heart failure are limited. A small study of 90 patients showed that, in comparison with age- and sex-matched healthy con-trol subjects, patients with left ventricular dysfunction, as expected, had a lower GFR and higher NT-proBNP levels and urinary albumin excretion.153 The median urinary NGAL lev-els were significantly higher in the group with left ventricular dysfunction (175 vs. 37 μg/gCr, P = .0001). Both serum cre-atinine level and estimated GFR were significantly correlated with urinary NGAL levels.153 In another study involving 46 elderly patients with CHF, higher levels of NGAL were found in patients with heart failure than in healthy age-matched control subjects (458 vs. 37.8 ng/mL, P = .0001).154 Moreover, NGAL level was found to increase in parallel with severity of heart failure and proved to be prognostic, inasmuch as among patients with a baseline NGAL level exceeding 783 ng/mL, the mortality rate was significantly higher (hazard ratio = 4.08, P = .001).154 In CHF, reduction in GFR arises in large part from reduced renal perfusion, which serves as a hypoxic trigger for acute tubular necrosis.155
NGAL Summary. NGAL level represents a first in its class: a renal stress biomarker that signifies acute kidney injury. Its

5
37
The Use of B
iomarkers in the Evaluation of H
eart Failure
55
CH
TABLE 37–4 Prognostic Value of hsCRP in Heart Failure
Reference Study (Country) N Age (% Male)Follow-up Period
Readmission for Worsening Heart Failure or Death
hsCRP Comparisons
Hazard Ratio (95% CI) Adjusted for
Anand et al159 C-reactive protein in heart failure. Prognostic value and the effect of valsartan (Val-HeFT: Multi-centric)
4202 63 ± 11 years (80%)
36 months 1255 (29.9%) ≥7.3 mg/L vs. <1.4 mg/L (4th vs. 1st quartile)
1.53 (1.28–1.84)
Age, sex, CHD, NHYA class, LVEF, hemo-globin, GFR, uric acid, BNP, aldosterone, renin, nor-epinephrine, statin, aspirin, BB, valsartan vs. placebo
Yin et al115 Independent prog-nostic value of ele-vated high-sensitivity C-reactive protein in chronic heart failure (Taiwan)
108 62 ± 16 years (66%)
403 days (median)
35 (32.4%) >2.97 mg/L vs. <2.97 mg/L (optimal cutoff)
3.05 (1.15–8.05)
Age, sex, CHD, LVEF, left ventricular filling pressure, TNF-α, statin
Xue et al116 Prognostic value of high-sensitivity C-reactive protein in patients with chronic heart failure (China)
128 62 ± 15 years (79%)
378 days (mean)
42 (32.8%) >3.2 mg/L vs. <3.2 mg/L (optimal cutoff)
3.81 (2.14–9.35)
Age, sex, CHD, LVEF, troponin T, statin
Windram et al117
Relationship of high-sensitivity C-reactive protein to prognosis and other prognostic markers in outpatients with heart failure (United Kingdom)
957 71 ± 10 years (71%)
36 months 163 (17.0%; only death)
>11 mg/L vs. <2.8 mg/L (4th vs. 1st quartile)
3.00 (2.1–4.1) Age, sex, smok-ing diabetes, white blood cell count, BB, statin, NSAID
Yin et al118 Multimarker approach to risk stratification among patients with advanced chronic heart failure (Taiwan)
152 56 ± 14 years (77%)
186 days (median)
63 (41.4%) >4.92 mg/L vs. <4.92 mg/L (median)
2.16 (1.17–3.99)
Age, sex, etiol-ogy, SBP, LVEF, sodium, creati-nine clearance, troponin I, NT-proBNP
Tang et al119 Usefulness of C-reac-tive protein and left ventricular diastolic performance for prognosis in patients with left ventricular systolic heart failure (United States)
136 57 ± 14 years (76%)
33 months (mean)
36 (26.5%) >5.96 mg/L vs. <5.96 mg/L (optimal cutoff)
2.26 (1.11–4.63)
LVEF, echo-cardiographic indexes of diastolic dys-function, BNP
Lamblin et al120 High-sensitivity C-reactive protein: potential adjunct for risk stratification in patients with stable heart failure (France)
546 56 ± 12 years (82%)
972 days (median)
113 (20.7%) >3 mg/L vs. <3 mg/L (optimal cutoff)
1.78 (1.17–2.72)
Age, sex, cause, hypertension, diabetes, BMI, NYHA class, LVEF, BNP
Chirinos et al121
Usefulness of C-reactive protein as an independent predictor of death in patients with ische-mic cardiomyopathy (United States)
123 64 ± 10 years (100%)
3 years — For each 10-mg/L increase
1.26 (1.02–1.55)
Age, number of vessels involved with coro-nary disease, LVEF, sodium, creatinine, hematocrit, ACE inhibitor, BB
(Continued)

556
CH 37
Reference Study (Country) N Age (% Male)Follow-up Period
Readmission for Worsening Heart Failure or Death
hsCRP Comparisons
Hazard Ratio (95% CI) Adjusted for
Ronnow et al122
C-reactive protein predicts death in patients with non-ischemic cardio-myopathy (United States)
203 62 ± 13 years (59%)
2.4 years (mean)
40 (19.7%) >23.3 mg/L vs. <23.3 mg/L (3rd tertile vs. 1st and 2nd tertiles)
20.0 0 (1.04–3.80)
Age, sex, smoking, family history, hypertension, hyperlipidemia, diabetes, renal failure, BMI, LVEF
Ishikawa et al123
Prediction of mortal-ity by high-sensitivity C-reactive protein and brain natriuretic peptide in patients with dilated cardio-myopathy (Japan)
84 56 ± 2 years (81%)
42 months (mean)
— >1 mg/L vs. <1 mg/L (arbitrary cutoff)
3.30 (1.2–9.0) Age, sex, NYHA class, LVEF, cardiac index, hemoglobin, creatinine, BNP, IL-6, endothelin, norepinephrine
ACE, angiotensin-converting enzyme; BB, β-blocker; BMI, body mass index; BNP, B-type natriuretic peptide; CHD, coronary heart disease; CI, confidence interval; CRP, C-reactive protein; GFR, glomerular filtration rate; HF, heart failure; hsCRP, high-sensitivity C-reactive protein; IL-6, interleukin-6; LVEF, left ventricular ejection fraction; NSAID, nonsteroidal antiinflammatory drug; NT-proBNP, N-terminal pro–B-type natriuretic peptide; NYHA, New York Heart Association; SBP, systolic blood pressure; TNF-α, tumor necrosis factor–α.From Araújo JP, Lourenço P, Azevedo A, et al. Prognostic value of high-sensitivity C-reactive protein in heart failure: a systematic review. J Card Fail 2009;15:256-266.
TABLE 37–4 Prognostic Value of hsCRP in Heart Failure—cont’d
0
A
2
4
6
8
>0.20>0.10-0.20>0.04-0.10�0.04
>0.06>0.02-0.06>0.01-0.02�0.01
5.3
3.42.7
2.0
6.3
3.32.8
1.7
9534932310,36711,090
Troponin I quartile
No. of patients
In-h
ospi
tal m
orta
lity
(%)
0
B
2
4
6
8
111911385021773
Troponin T quartile
No. of patients
In-h
ospi
tal m
orta
lity
(%)
FIGURE 37–5 In-hospital mortality rates in comparison with troponin I (A) or troponin T (B) quartiles in patients admitted with decompensated heart failure. (From Peacock WF 4th, De Marco T, Fonarow GC, et al. Cardiac troponin and outcome in acute heart failure. N Engl J Med 2008;358:2117-2126.)
utility in predicting the development of renal insufficiency with good sensitivity and specificity has been shown in small studies involving cardiopulmonary bypass and angiography. Future studies may further define the role of NGAL level in assisting clinicians to achieve a proper balance between diuretics, vasodilators, and inotropes when treating patients with acute heart failure.
MULTIMARKER APPROACH AND FUTURE DIRECTION
The evolution of numerous biomarkers has created valuable assets for the classification and management of heart failure. There is growing interest in the use of multimarker strategies to stratify patients with heart failure, similar to acute coro-nary syndromes. The interaction in heart failure between bio-mechanical strain, inflammation, and cellular injury allows a variety of biomarkers to jointly lead to better understanding of the pathogenesis, clinical diagnosis, and prognosis of heart failure.91 These markers are complementary to the clinical data and enhance prediction of morbidity and mortality.
In evaluating the association between cTnI and mortality in advanced heart failure, Horwich and coworkers128 demon-strated that using both cTnI and BNP levels further improves prognostic value.128 Patients with both detectable cTnI level and a BNP level exceeding 485 pg/mL had a twelve-fold higher risk of death than did those with both undetectable cTnI level and a BNP level of less than 485 pg/mL. The prognostic value of this combination was confirmed with the hsTnT assay.142 Prediction was also enhanced with measurements of natri-uretic peptides and MR-proADM, ST2, or CRP.71,88,156 In 983 consecutive patients who had suffered myocardial infarction, Khan and associates showed that the combination of MR-proADM provided further risk stratification for patients with NT-proBNP levels above the median (P < .001).71 The PRIDE study verified the prognostic capability of the multimarker approach. Of the 184 patients with NT-proBNP level of 986 pg/mL or higher, 56 (30.4%) died. All but 1 of the decedents within this group had an ST2 level of 0.2 ng/mL or higher.88 In contrast, of the 28 patients with NT-proBNP levels of

The Use of B
iomarkers in the Evaluation of H
eart Failure
57
CH 37
986 pg/mL or higher and ST2 levels of less than 0.2 pg/mL, only 1 (3.5%) died at 1 year.88
Dunlay and colleagues157 used CRP, BNP, and cTnT levels in 593 patients with heart failure to evaluate the prognostic utility of a multimarker approach. Among patients with CRP (<11.8 mg/L), BNP (<350 pg/mL), and cTnT (≤0.01 ng/mL) levels all lower than the median, the 1-year mortality rate was low (3.3%), whereas among those with two or three biomark-ers higher than the median, the mortality rate was markedly increased (30.8% and 35.5%, respectively). A strategy involv-ing two biomarkers offered greater improvement in predicting 1-year mortality risk than did use of a single biomarker. In this study, however, the addition of a third biomarker con-ferred no added benefit over a two-biomarker approach.157
Short-term prognosis with a multimarker strategy was also evaluated in a study of 577 consecutive patients admitted with severe decompensated NYHA class III or IV low-output heart failure, to determine the combined prognostic value of admission BNP, cTnI, and hsCRP levels at 31 days.158 The total mortality rate in the group was 17.7%. Multivariate Cox analysis revealed that elevated levels of BNP, cTnI, and hsCRP were independent predictors of mortality.158 There was a sig-nificant, gradually increased risk of 31-day cardiac death with increased numbers of elevated biomarkers (P < .001).158
The multimarker approach provides powerful prognostic information in both compensated and decompensated heart failure. Combinations of biomarkers allow clinicians to bet-ter stratify risk for patients and perhaps tailor more aggres-sive therapy for those with a higher predicted risk. As new biomarkers are developed and implemented, more powerful multimarker approaches are likely to follow.
CONCLUSION
Biomarker use has skyrocketed since 2000. Natriuretic pep-tide levels, because of their low cost and rapid and accurate ability to provide additional information not surmised from clinical evaluation, are the standard biomarkers. But work with natriuretic peptide levels has also shown that they are not to be used as stand-alone tests; rather they are best used as adjuncts to other tests used by health care providers. There are many caveats to using natriuretic peptide levels, as there will be with all future biomarkers; thus, there is always much to learn about them. Finally, because complex pathological conditions are widespread, panels of multiple biomarkers will be needed in evaluation, risk stratification, and, ulti-mately, treatment initiation and follow-up.
REFERENCES 1. Stevenson, L. W., & Perloff, J. K. (1989). The limited reliability of physical signs for
establishing hemodynamics in chronic heart failure. JAMA, 261, 884–888. 2. Morrow, D. A., & de Lemos, J. A. (2007). Benchmarks for the assessment of novel car-
diovascular biomarkers. Circulation, 115, 949–952. 3. Yasue, H., Yoshimura, M., Sumida, H., et al. (1994). Localization and mechanism of
secretion of B-type natriuretic peptide in comparison with those A-type natriuretic peptide in normal subjects and patients with heart failure. Circulation, 90, 195–203.
4. Yoshimura, M., Yasue, H., Okumura, K., et al. (1993). Different secretion patterns of atrial natriuretic peptide and brain natriuretic peptide in patients with congestive heart failure. Circulation, 87, 464–469.
5. Nakao, K., Ogawa, Y., Suga, S., et al. (1992). Molecular biology and biochemistry of the natriuretic peptide system. II: natriuretic peptide receptors. J Hypertens, 10, 1111–1114.
6. Chen, H. H. (2007). Heart failure: a state of brain natriuretic peptide deficiency or resis-tance or both!. J Am Coll Cardiol, 49, 1089–1091.
7. Shimizu, H., Masuta, K., Aono, K., et al. (2002). Molecular forms of human brain natri-uretic peptide in plasma. Clin Chim Acta, 316, 129–135.
8. Lam, C. S., Burnett, J. C., Jr., Costello-Boerrigter, L., et al. (2007). Alternate circulat-ing pro–B-type natriuretic peptide and B-type natriuretic peptide forms in the general population. J Am Coll Cardiol, 49, 1193–1202.
9. Forfia, P. R., Lee, M., Tunin, R. S., et al. (2007). Acute phosphodiesterase 5 inhibition mimics hemodynamic effects of B-type natriuretic peptide effects in failing but not normal canine heart. J Am Coll Cardiol, 49, 1079–1088.
10. Redfield, M. M., Rodeheffer, R. J., Jacobsen, S. J., et al. (2002). Plasma brain natriuretic peptide concentration: impact of age and gender. J Am Coll Cardiol, 40, 976–982.
5 11. Wang, T. J., Larson, M. G., Levy, D., et al. (2002). Impact of age and sex on plasma natri-uretic peptide levels in healthy adults. Am J Cardiol, 90, 254–258.
12. Daniels, L. B., Allison, M. A., Clopton, P., et al. (2008). Use of natriuretic peptides in pre-participation screening of college athletes. Int J Cardiol, 124, 411–414.
13. Maisel, A. S., Krishnaswamy, P., Nowak, R. M., et al. (2002). Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med, 347, 161–167.
14. Januzzi, J. L., Jr., CamargoAnwaruddin, C. A. S., et al. (2005). The N-terminal Pro-BNP Investigation of Dyspnea in the Emergency Department (PRIDE) study. Am J Cardiol, 95, 948–954.
15. Mueller, C., Scholer, A., Laule-Kilian, K., et al. (2004). Use of B-type natriuretic peptide in the evaluation and management of acute dyspnea. N Engl J Med, 350, 647–654.
16. Moe, G. W., Howlett, J., Januzzi, J. L., et al. (2007). N-terminal pro–B-type natriuretic peptide testing improves the management of patients with suspected acute heart fail-ure: primary results of the Canadian prospective randomized multicenter IMPROVE-CHF study. Circulation, 115, 3103–3110.
17. Morita, E., Yasue, H., Yoshimura, M., et al. (1992). Increasing plasma levels of brain natriuretic peptide in patients with acute myocardial infarction. Circulation, 88, 82–91.
18. Morrison, L. K., Harrison, A., Krishnaswamy, P., et al. (2002). Utility of a rapid B-natri-uretic peptide assay in differentiating congestive heart failure from lung disease in patients presenting with dyspnea. J Am Coll Cardiol, 39, 202–209.
19. Kucher, N., Printzen, G., & Goldhaber, S. Z. (2003). Prognostic role of brain natriuretic peptide in acute pulmonary embolism. Circulation, 107, 2545–2547.
20. Nagaya, N., Nishikimi, T., Okano, Y., et al. (1998). Plasma brain natriuretic peptide lev-els increase in proportion to the extent of right ventricular dysfunction in pulmonary hypertension. J Am Coll Cardiol, 31, 202–208.
21. Bando, M., Ishhi, Y., Sugiyama, Y., et al. (1999). Elevated plasma brain natriuretic pep-tide levels in chronic respiratory failure with cor pulmonale. Respir Med, 93, 507–514.
22. Nagaya, N., Nishikimi, T., Uematsu, M., et al. (2000). Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circula-tion, 102, 865–870.
23. Rudiger, A., Gasser, S., Fischler, M., et al. (2006). Comparable increase of B-type natri-uretic peptide and amino-terminal pro–B-type natriuretic peptide levels in patients with severe sepsis, septic shock, and acute heart failure. Crit Care Med, 34, 2140–2144.
24. Schultz, M., Faber, J., Kistorp, C., et al. (2004). N-terminal–pro–B-type natriuretic peptide (NT-pro-BNP) in different thyroid function states. Clin Endocrinol (Oxf), 60, 54–59.
25. Yildiz, R., Yildirim, B., Karincaoglu, M., et al. (2005). Brain natriuretic peptide and severity of disease in non-alcoholic cirrhotic patients. J Gastroenterol Hepatol, 20, 1115–1120.
26. Ellinor, P. T., Low, A. F., Patton, K., et al. (2005). Discordant atrial natriuretic peptide and brain natriuretic peptide levels in lone atrial fibrillation. J Am Coll Cardiol, 45, 82–86.
27. Knudsen, C. W., Omland, T., Clopton, P., et al. (2005). Impact of atrial fibrillation on the diagnostic performance of B-type natriuretic peptide concentration in dyspneic patients: an analysis from the Breathing Not Properly Multinational Study. J Am Coll Cardiol, 46, 838–844.
28. Kawai, K., Hata, K., Tanaka, K., et al. (2004). Attenuation of biologic compensatory action of cardiac natriuretic peptide system with aging. Am J Cardiol, 93, 719–723.
29. Chang, A. Y., Abdullah, S. M., Jain, T., et al. (2007). Association among androgens, estrogens, and natriuretic peptides in young women: observations from the Dallas Heart Study. J Am Coll Cardiol, 49, 109–116.
30. Daniels, L. B., & Maisel, A. S. (2007). Natriuretic peptides. J Am Coll Cardiol, 50, 2357–2368.
31. McCullough P. A., Duc, P., Omland, T., et al. (2003). B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the Breathing Not Properly Multinational Study. Am J Kidney Dis, 41, 571–579.
32. Lamb, E. J., Vickery, S., & Price, C. P. (2006). Amino-terminal pro-brain natriuretic pep-tide to diagnose congestive heart failure in patients with impaired kidney function. J Am Coll Cardiol, 48, 1060–1061.
33. Anwaruddin, S., Lloyd-Jones, D. M., Baggish, A., et al. (2006). Renal function, con-gestive heart failure, and amino-terminal pro-brain natriuretic peptide measurement: results from the ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) study. J Am Coll Cardiol, 47, 91–97.
34. Costello-Boerrigter, L. C., Boerrigter, G., Redfield, M. M., et al. (2006). Amino-terminal pro–B-type natriuretic peptide and B-type natriuretic peptide in the general community: determinants and detection of left ventricular dysfunction. J Am Coll Cardiol, 47, 345–353.
35. Mehra, M. R., Uber, P. A., Park, M. H., et al. (2004). Obesity and suppressed B-type natriuretic peptide levels in heart failure. J Am Coll Cardiol, 43, 1590–1595.
36. Wang, T. J., Larson, M. G., Levy, D., et al. (2004). Impact of obesity on plasma natriuretic peptide levels. Circulation, 109, 594–600.
37. Daniels, L. B., Clopton, P., Bhalla, V., et al. (2006). How obesity affects the cut-points for B-type natriuretic peptide in the diagnosis of acute heart failure. Results from the Breathing Not Properly Multinational Study. Am Heart J, 151, 999–1005.
38. Sarzani, R., Dessi-Fulgheri, P., Paci, V. M., et al. (1996). Expression of natriuretic pep-tide receptors in human adipose and other tissues. J Endocrinol Invest, 19, 581–585.
39. Krauser, D. G., Lloyd-Jones, D. M., Chae, C. U., et al. (2005). Effect of body mass index on natriuretic peptide levels in patients with acute congestive heart failure: a ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) substudy. Am Heart J, 149, 744–750.
40. van Kimmenade, R., van Dielen, F., Bakker, J., et al. (2006). Is brain natriuretic peptide production decreased in obese subjects?. J Am Coll Cardiol, 47, 886–887.
41. Leya, F. S., Arab, D., Joyal, D., et al. (2005). The efficacy of brain natriuretic peptide levels in differentiating constrictive pericarditis from restrictive cardiomyopathy. J Am Coll Cardiol, 45, 1900–1902.

558
CH 37
42. Doust, J. A., Pietrzak, E., Dobson, A., et al. (2005). How well does B-type natriuretic peptide predict death and cardiac events in patients with heart failure: systematic review. BMJ, 330, 625.
43. Maisel, A., Hollander, J. E., Guss, D., et al. (2004). Primary results of the Rapid Emer-gency Department Heart Failure Outpatient Trial (REDHOT). A multicenter study of B-type natriuretic peptide levels, emergency department decision making, and out-comes in patients presenting with shortness of breath. J Am Coll Cardiol, 44, 1328–1333.
44. Anand, I. S., Fisher, L. D., Chiang, Y. T., et al. (2003). Changes in brain natriuretic pep-tide and norepinephrine over time and mortality and morbidity in the Valsartan Heart Failure Trial (Val-FeFT). Circulation, 107, 1278–1283.
45. Schnabel, R., Lubos, E., Rupprecht, H. J., et al. (2006). B-type natriuretic peptide and the risk of cardiovascular events and death in patients with stable angina: results from the AtheroGene study. J Am Coll Cardiol, 47, 552–558.
46. Kragelund, C., Gronning, B., Kobel, L., et al. (2005). N-terminal pro–B-type natriuretic peptide and long-term mortality in stable coronary heart disease. N Engl J Med, 352, 666–675.
47. Bibbins-Domingo, K., Gupta, R., Na, B., et al. (2007). N-terminal fragment of the prohor-mone brain-type natriuretic peptide (NT-proBNP), cardiovascular events, and mortality in patients with stable coronary heart disease. JAMA, 297, 169–176.
48. Lindahl, B., Lindback, J., Jernberg, T., et al. (2005). Serial analyses of N-terminal pro–B-type natriuretic peptide in patients with non–ST-segment elevation acute coronary syndromes: a Fragmin and fast Revascularisation during In Stability in Coronary artery disease (FRISC)–II substudy. J Am Coll Cardiol, 45, 533–541.
49. Berger, R., Huelsman, M., Strecker, K., et al. (2002). B-type natriuretic peptide predicts sudden death in patients with chronic heart failure. Circulation, 105, 2392–2397.
50. Tapanainen, J. M., Lindgren, K. S., Makikallio, T. H., et al. (2004). Natriuretic peptides as predictors of non-sudden and sudden cardiac death after acute myocardial infarc-tion in the beta-blocking era. J Am Coll Cardiol, 43, 757–763.
51. Taub, P. R., Daniels, L. B., & Maisel, A. S. (2009). Usefulness of B-type natriuretic pep-tide levels in predicting hemodynamic and clinical decompensation. Heart Fail Clin, 5, 169–175.
52. Forfia, P. R., Watkins, S. P., Rame, J. E., et al. (2005). Relationship between B-type natri-uretic peptides and pulmonary capillary wedge pressure in the intensive care unit. J Am Coll Cardiol, 45, 1667–1671.
53. Kazanegra, R., Cheng, V., Garcia, A., et al. (2001). A rapid test for B-type natriuretic peptide correlates with falling wedge pressures in patients treated for decompensated heart failure: a pilot study. J Card Fail, 7, 21–29.
54. Cheng, V., Kazanagra, R., Garcia, A., et al. (2001). A rapid bedside test for B-type pep-tide predicts treatment outcomes in patients admitted for decompensated heart failure: a pilot study. J Am Coll Cardiol, 37, 386–391.
55. Bettencourt, P., Ferreira, S., Azevedo, A., et al. (2002). Preliminary data on the potential usefulness of B-type natriuretic peptide levels in predicting outcome after hospital discharge in patients with heart failure. Am J Med, 113, 215–219.
56. Logeart, D., Thabut, G., Jourdain, P., et al. (2004). Predischarge B-type natriuretic pep-tide assay for identifying patients at high risk of re-admission after decompensated heart failure. J Am Coll Cardiol, 43, 625–641.
57. Jourdain, P., Jondeau, G., Funck, F., et al. (2007). Plasma brain natriuretic peptide–guided therapy to improve outcome in heart failure: the STARS-BNP multicenter study. J Am Coll Cardiol, 49, 1733–1739.
58. Shah, M. R., Claise, K. A., Bowers, M. T., et al. (2005). Testing new targets of ther-apy in advanced heart failure: the design and rationale of the Strategies for Tailoring Advanced Heart Failure Regimens in the Outpatient Setting: BRain NatrIuretic Peptide Versus the Clinical CongesTion ScorE (STARBRITE) trial. Am Heart J, 150, 893–898.
59. Lee, S. C., Stevens, T. L., Sandberg, S. M., et al. (2002). The potential of brain natriuretic peptide as a biomarker for New York Heart Association class during the outpatient treatment of heart failure. J Card Fail, 8, 149–154.
60. Kitamura, K., Kangawa, K., Kawamoto, M., et al. (1993). Adrenomedullin: a novel hypotensive peptide isolated from human pheochromocytoma. Biochem Biophys Res Commun, 192, 553–560.
61. Takahashi, K., Satoh, F., Hara, E., et al. (1997). Production and secretion of adrenomedul-lin from glial cell tumors and its effects on cAMP production. Peptides, 18, 1117–1124.
62. Nakamura, M., Yoshida, H., Makita, S., et al. (1997). Potent and long-lasting vasodila-tory effects of adrenomedullin in humans. Comparisons between normal subjects and patients with chronic heart failure. Circulation, 95, 1214–1221.
63. Parkes, D. G., & May, C. N. (1995). ACTH-suppressive and vasodilator actions of adre-nomedullin in conscious sheep. J Neuroendocrinol, 7, 923–929.
64. Parkes, D. G., & May, C. N. (1997). Direct cardiac and vascular actions of adrenomedul-lin in conscious sheep. Br J Pharmacol, 120, 1179–1185.
65. Vari, R. C., Adkins, S. D., & Samson, W. K. (1996). Renal effects of adrenomedullin in the rat. Proc Soc Exp Biol Med, 211, 178–183.
66. Kato, J., Kobayashi, K., Etoh, T., et al. (1996). Plasma adrenomedullin concentration in patients with heart failure. J Clin Endocrinol Metab, 81, 180–183.
67. Kitamura, K., Sakata, J., Kangawa, K., et al. (1993). Cloning and characterization of cDNA encoding a precursor for human adrenomedullin. Biochem Biophys Res Com-mun, 194, 720–725.
68. Jougasaki, M., Rodeheffer, R. J., Redfield, M. M., et al. (1996). Cardiac secretion of adre-nomedullin in human heart failure. J Clin Invest, 97, 2370–2376.
69. Hinson, J. P., Kapas, S., & Smith, D. M. (2000). Adrenomedullin, a multifunctional regulatory peptide. Endocr Rev, 21, 138–167.
70. Struck, J., Tao, C., Morgenthaler, N. G., et al. (2004). Identification of an adrenomedul-lin precursor fragment in plasma of sepsis patients. Peptides, 25, 1369–1372.
71. Khan, S. Q., O’Brien, R. J., Struck, J., et al. (2007). Prognostic value of midregional pro-adrenomedullin in patients with acute myocardial infarction. The LAMP (Leicester Acute Myocardial Infarction Peptide) study. J Am Coll Cardiol, 49, 1525–1532.
72. Adlbrecht, C., Hülsmann, M., Strunk, G., et al. (2009). Prognostic value of plasma midregional pro-adrenomedullin and C-terminal–pro–endothelin-1 in chronic heart failure outpatients. Eur J Heart Fail, 11, 361–366.
73. Nishida, H., Horio, T., Suzuki, Y., et al. (2008). Plasma adrenomedullin as an indepen-dent predictor of future cardiovascular events in high-risk patients: comparison with C-reactive protein and adiponectin. Peptides, 29, 599–605.
74. Gegenhuber, A., Struck, J., Dieplinger, B., et al. (2007). Comparative evaluation of B-type natriuretic peptide, mid-regional pro–A-type natriuretic peptide, mid-regional pro-adrenomedullin, and Copeptin to predict 1-year mortality in patients with acute destabilized heart failure. J Card Fail, 13, 42–49.
75. Coletta, A. P., Clark, A. L., & Cleland, J. G. (2009). Clinical trials update from the Heart Failure Society of America and the American Heart Association meetings in 2008: SADHART-CHF, COMPARE, MOMENTUM, thyroid hormone analogue study, HF-ACTION, I-PRESERVE, beta-interferon study, BACH, and ATHENA. Eur J Heart Fail, 11, 214–219.
76. Tominaga, S. (1989). A putative protein of a growth specific cDNA from BALB/c-3T3 cells is highly similar to the extracellular portion of mouse interleukin 1 receptor. FEBS Lett, 258, 301–304.
77. Townsend, M. J., Fallon, P. G., Matthews, D. J., et al. (2000). T1/ST2-deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. J Exp Med, 191, 1069–1076.
78. Kakkar, R., & Lee, R. T. (2008). The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov, 7, 827–840.
79. Weinberg, E. O., Shimpo, M., De Keulenaer, G. W., et al. (2002). Expression and regula-tion of ST2, an interleukin-1 receptor family member, in cardiomyocytes and myocar-dial infarction. Circulation, 106, 2961–2966.
80. Schmitz, J., Owyang, A., Oldham, E., et al. (2005). IL-33, an interleukin-1–like cytokine that signals via the IL-1 receptor–related protein ST2 and induces T helper type 2–asso-ciated cytokines. Immunity, 23, 479–490.
81. Diez, J., Gonzalez, A., Lopez, B., et al. (2005). Mechanisms of disease: pathologic struc-tural remodeling is more than adaptive hypertrophy in hypertensive heart disease. Nat Clin Pract Cardiovasc Med, 2, 209–216.
82. Sadoshima, J., & Izumo, S. (1997). The cellular and molecular response of cardiac myo-cytes to mechanical stress. Annu Rev Physiol, 59, 551–571.
83. Manabe, I., Shindo, T., & Nagai, R. (2002). Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res, 91, 1103–1113.
84. Sanada, S., Hakuno, D., Higgins, L. J., et al. (2007). IL-33 and ST2 comprise a criti-cal biomechanically induced and cardioprotective signaling system. J Clin Invest, 117, 1538–1549.
85. Shimpo, M., Morrow, D. A., Weinberg, E. O., et al. (2004). Serum levels of the inter-leukin-1 receptor family member ST2 predict mortality and clinical outcome in acute myocardial infarction. Circulation, 109, 2186–2190.
86. Sabatine, M. S., Morrow, D. A., Higgins, L. J., et al. (2008). Complementary roles for bio-markers of biomechanical strain ST2 and N-terminal prohormone B-type natriuretic pep-tide in patients with ST-elevation myocardial infarction. Circulation, 117, 1936–1944.
87. Weinberg, E. O., Shimpo, M., Hurwitz, S., et al. (2003). Identification of serum soluble ST2 receptor as a novel heart failure biomarker. Circulation, 107, 721–726.
88. Januzzi, J. L., Jr., Peacock, W. F., Maisel, A. S., et al. (2007). Measurement of the inter-leukin family member ST2 in patients with acute dyspnea: results from the PRIDE (Pro-Brain Natriuretic Peptide Investigation of Dyspnea in the Emergency Department) study. J Am Coll Cardiol, 50, 607–613.
89. Boisot, S., Beede, J., Isakson, S., et al. (2008). Serial sampling of ST2 predicts 90-day mortality following destabilized heart failure. J Card Fail, 14, 732–738.
90. Rehman, S. U., Mueller, T., & Januzzi, J. L., Jr. (2008). Characteristics of the novel inter-leukin family biomarker ST2 in patients with acute heart failure. J Am Coll Cardiol, 52, 1458–1465.
91. Braunwald, E. (2008). Biomarkers in heart failure. N Engl J Med, 358, 2148–2159. 92. Levine, B., Kalman, J., Mayer, L., et al. (1990). Elevated circulating levels of tumor
necrosis factor in severe chronic heart failure. N Engl J Med, 323, 236–241. 93. Seta, Y., Shan, K., Bozkurt, B., et al. (1996). Basic mechanisms in heart failure: the
cytokine hypothesis. J Card Fail, 2, 243–249. 94. Bozkurt, B., Kribbs, S. B., Clubb, F. J., Jr., et al. (1998). Pathophysiologically relevant
concentrations of tumor necrosis factor–alpha promote progressive left ventricular dys-function and remodeling in rats. Circulation, 97, 1382–1391.
95. Steele, I. C., Nugent, A. M., Maguire, S., et al. (1996). Cytokine profile in chronic car-diac failure. Eur J Clin Invest, 26, 1018–1022.
96. Kubota, T., McTiernan, C. F., Frye, C. S., et al. (1997). Dilated cardiomyopathy in trans-genic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ Res, 81, 627–635.
97. Yamauchi-Takihara, K., Ihara, Y., & Ogata, A. (1995). Hypoxic stress induces cardiac myocyte–derived interleukin-6. Circulation, 91, 1520–1524.
98. Araújo, J. P., Lourenço, P., Azevedo, A., et al. (2009). Prognostic value of high-sensitiv-ity C-reactive protein in heart failure: a systematic review. J Card Fail, 15, 256–266.
99. Venugopal, S. K., Devaraj, S., & Jialal, I. (2005). Effect of C-reactive protein on vascular cells: evidence for a proinflammatory, proatherogenic role. Curr Opin Nephrol Hyper-tens, 14, 33–37.
100. Gottdiener, J. S., Arnold, A. M., Aurigemma, G. P., et al. (2000). Predictors of congestive heart failure in the elderly: the Cardiovascular Health Study. J Am Coll Cardiol, 35, 1628–1637.
101. Vasan, R. S., Sullivan, L. M., Roubenoff, R., et al. (2003). Inflammatory markers and risk of heart failure in elderly subjects without prior myocardial infarction: the Framing-ham Heart Study. Circulation, 107, 1486–1491.
102. Cesari, M., Penninx, B. W., Newman, A. B., et al. (2003). Inflammatory markers and car-diovascular disease (The Health, Aging and Body Composition [Health ABC] Study). Am J Cardiol, 92, 522–528.
103. Kardys, I., Knetsch, A. M., Bleumink, G. S., et al. (2006). C-reactive protein and risk of heart failure. The Rotterdam Study. Am Heart J, 152, 514–520.
104. Pietilä, K. O., Harmoinen, A. P., Jokiniitty, J., et al. (1996). Serum C-reactive protein con-centration in acute myocardial infarction and its relationship to mortality during 24 months of follow-up in patients under thrombolytic treatment. Eur Heart J, 17, 1345–1349.

The Use of B
iomarkers in the Evaluation of H
eart Failure
559
CH 37
105. Scirica, B. M., Morrow, D. A., Cannon, C. P., et al. (2007). Clinical application of C-reactive protein across the spectrum of acute coronary syndromes. Clin Chem, 53, 1800–1807.
106. Mielniczuk, L. M., Pfeffer, M. A., & Lewis, E. F. (2008). Estimated glomerular filtra-tion rate, inflammation, and cardiovascular events after an acute coronary syndrome. Am Heart J, 155, 725–731.
107. Berton, G., Cordiano, R., Palmieri, R., et al. (2003). C-reactive protein in acute myocar-dial infarction: association with heart failure. Am Heart J, 145, 1094–1101.
108. Suleiman, M., Aronson, D., Reisner, S. A., et al. (2003). Admission C-reactive protein levels and 30-day mortality in patients with acute myocardial infarction. Am J Med, 115, 695–701.
109. Suleiman, M., Khatib, R., Agmon, Y., et al. (2006). Early inflammation and risk of long-term development of heart failure and mortality in survivors of acute myocardial infarction: predictive role of C-reactive protein. J Am Coll Cardiol, 47, 962–968.
110. Bursi, F., Weston, S. A., Killian, J. M., et al. (2007). C-reactive protein and heart failure after myocardial infarction in the community. Am J Med, 120, 616–622.
111. Kavsak, P. A., MacRae, A. R., Newman, A. M., et al. (2007). Elevated C-reactive pro-tein in acute coronary syndrome presentation is an independent predictor of long-term mortality and heart failure. Clin Biochem, 40, 326–329.
112. Hartford, M., Wiklund, O., Mattsson Hultén, L., et al. (2007). C-reactive protein, inter-leukin-6, secretory phospholipase A2 group IIA and intercellular adhesion molecule–1 in the prediction of late outcome events after acute coronary syndromes. J Intern Med, 262, 526–536.
113. Sabatine, M. S., Morrow, D. A., Jablonski, K. A., et al. (2007). Prognostic significance of the Centers for Disease Control/American Heart Association high-sensitivity C-reactive protein cut points for cardiovascular and other outcomes in patients with stable coro-nary artery disease. Circulation, 115, 1528–1536.
114. Williams, E. S., Shah, S. J., Ali, S., et al. (2008). C-reactive protein, diastolic dysfunc-tion, and risk of heart failure in patients with coronary disease: Heart and Soul Study. Eur J Heart Fail, 10, 63–69.
115. Yin, W. H., Chen, J. W., Jen, H. L., et al. (2004). Independent prognostic value of ele-vated high-sensitivity C-reactive protein in chronic heart failure. Am Heart J, 147, 931–938.
116. Xue, C., Feng, Y., Wo, J., et al. (2006). Prognostic value of high-sensitivity C-reactive protein in patients with chronic heart failure. N Z Med J, 119, U2314.
117. Windram, J. D., Loh, P. H., Rigby, A. S., et al. (2007). Relationship of high-sensitivity C-reactive protein to prognosis and other prognostic markers in outpatients with heart failure. Am Heart J, 153, 1048–1055.
118. Yin, W. H., Chen, J. W., Feng, A. N., et al. (2007). Multimarker approach to risk stratifi-cation among patients with advanced chronic heart failure. Clin Cardiol, 30, 397–402.
119. Tang, W. H., Shrestha, K., Van Lente, F., et al. (2008). Usefulness of C-reactive protein and left ventricular diastolic performance for prognosis in patients with left ventricular systolic heart failure. Am J Cardiol, 101, 370–373.
120. Lamblin, N., Mouquet, F., Hennache, B., et al. (2005). High-sensitivity C-reactive pro-tein: potential adjunct for risk stratification in patients with stable congestive heart failure. Eur Heart J, 26, 2245–2250.
121. Chirinos, J. A., Zambrano, J. P., Chakko, S., et al. (2005). Usefulness of C-reactive pro-tein as an independent predictor of death in patients with ischemic cardiomyopathy. Am J Cardiol, 95, 88–90.
122. Ronnow, B. S., Reyna, S. P., Muhlestein, J. B., et al. (2005). C-reactive protein predicts death in patients with non-ischemic cardiomyopathy. Cardiology, 104, 196–201.
123. Ishikawa, C., Tsutamoto, T., Fujii, M., et al. (2006). Prediction of mortality by high-sensitivity C-reactive protein and brain natriuretic peptide in patients with dilated cardiomyopathy. Circ J, 70, 857–863.
124. Alonso-Martínez, J. L., Llorente-Diez, B., Echegaray-Agara, M., et al. (2002). C-reactive protein as a predictor of improvement and readmission in heart failure. Eur J Heart Fail, 4, 331–336.
125. Mueller, C., Laule-Kilian, K., Christ, A., et al. (2006). Inflammation and long-term mor-tality in acute congestive heart failure. Am Heart J, 151, 845–850.
126. Siirilä-Waris, K., Lassus, J., Melin, J., et al. (2006). Characteristics, outcomes, and pre-dictors of 1-year mortality in patients hospitalized for acute heart failure. Eur Heart J, 27, 3011–3017.
127. Villacorta, H., Masetto, A. C., & Mesquita, E. T. (2007). C-reactive protein: an inflamma-tory marker with prognostic value in patients with decompensated heart failure. Arq Bras Cardiol, 88, 585–589.
128. Horwich, T. B., Patel, J., MacLellan, W. C., et al. (2003). Cardiac troponin I is associated with impaired hemodynamics, progressive left ventricular dysfunction, and increased mortality rates in advanced heart failure. Circulation, 108, 833–838.
129. Mann, D. L. (1999). Mechanisms and models in heart failure. Circulation, 100, 999–1008.
130. Olivetti, G., Abbi, R., Quaini, F., et al. (1997). Apoptosis in the failing human heart. N Engl J Med, 336, 1131–1141.
131. Logeart, D., Beyne, P., Cusson, C., et al. (2001). Evidence of cardiac myolysis in severe nonischemic heart failure and the potential role of increased wall strain. Am Heart J, 141, 247–253.
132. Anversa, P., & Kajstura, J. (1998). Myocyte cell death in the diseased heart. Circ Res, 82, 1231–1233.
133. Wu, A. H. B., & Ford, L. (1999). Release of cardiac troponin in acute coronary syn-dromes: ischemia or necrosis? Clin Chim Acta, 284, 161–174.
134. Ilva, T., Lassus, J., Siirilä-Waris, K., et al. (2008). Clinical significance of cardiac tropo-nins I and T in acute heart failure. Eur J Heart Fail, 10, 772–779.
135. Sato, Y., Yamada, T., Taniguchi, R., et al. (2001). Persistently increased serum concen-trations of cardiac troponin T in patients with idiopathic dilated cardiomyopathy are predictive of adverse outcomes. Circulation, 103, 369–374.
136. Missov, M., Calzolari, C., & Pau, B. (1997). Circulating cardiac troponin I in severe congestive heart failure. Circulation, 96, 2953–2958.
137. Setsuta, K., Seino, Y., Takahashi, N., et al. (1999). Clinical significance of elevated levels of cardiac troponin T in patients with chronic heart failure. Am J Cardiol, 84, 608–611.
138. Del Carlo, C. H., & O’Connor, C. M. (1999). Cardiac troponins in congestive heart fail-ure. Am Heart J, 138, 646–653.
139. La Vecchia, L. L., Mezzena, G., Zanolla, L., et al. (2000). Cardiac troponin I as a diagnos-tic and prognostic marker in severe heart failure. J Heart Lung Transplant, 19, 644–652.
140. Peacock, W. F., 4th, De Marco, T., Fonarow, G. C., et al. (2008). Cardiac troponin and outcome in acute heart failure. N Engl J Med, 358, 2117–2126.
141. Hudson, M. P., O’Connor, C. M., Gattis, W. A., et al. (2004). Implications of elevated cardiac troponin T in ambulatory patients with heart failure: a prospective analysis. Am Heart J, 147, 546–552.
142. Latini, R., Masson, S., Anand, I. S., et al. (2007). Prognostic value of very low plasma concentrations of troponin T in patients with stable chronic heart failure. Circulation, 116, 1242–1249.
143. Ronco, C., Haapio, M., House, A. A., et al. (2008). Cardiorenal syndrome. J Am Coll Cardiol, 52, 1527–1539.
144. Bolignano, D., Donato, V., Coppolino, G., et al. (2008). Neutrophil gelatinase–associ-ated lipocalin (NGAL) as a marker of kidney damage. Am J Kidney Dis, 52, 595–605.
145. Schmidt-Ott, K. M., Mori, K., Li, J. Y., et al. (2007). Dual action of neutrophil gelatin-ase–associated lipocalin. J Am Soc Nephrol, 18, 407–413.
146. Devireddy, L. R., Gazin, C., Zhu, X., et al. (2005). A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell, 123, 1293–1305.
147. Hvidberg, V., Jacobsen, C., Strong, R. K., et al. (2005). The endocytic receptor megalin binds the iron transporting neutrophil-gelatinase–associated lipocalin with high affin-ity and mediates its cellular uptake. FEBS Lett, 579, 773–777.
148. Mishra, J., Mori, K., Ma, Q., et al. (2004). Neutrophil gelatinase–associated lipocalin: a novel early urinary biomarker for cisplatin nephrotoxicity. Am J Nephrol, 24, 307–315.
149. Mishra, J., Dent, C., Tarabishi, R., et al. (2005). Neutrophil gelatinase–associated lipo-calin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet, 365, 1231–1238.
150. Hirsch, R., Dent, C., Pfriem, H., et al. (2007). NGAL is an early predictive biomarker of contrast-induced nephropathy in children. Pediatr Nephrol, 22, 2089–2095.
151. Wagener, G., Jan, M., Kim, M., et al. (2006). Association between increases in urinary neutrophil gelatinase–associated lipocalin and acute renal dysfunction after adult car-diac surgery. Anesthesiology, 105, 485–491.
152. Bachorzewska-Gajewska, H., Malyszko, J., Sitniewska, E., et al. (2006). Neutrophil-gelatinase–associated lipocalin and renal function after percutaneous coronary inter-ventions. Am J Nephrol, 26, 287–292.
153. Damman, K., van Veldhuisen, D. J., Navis, G., et al. (2008). Urinary neutrophil gelatin-ase associated lipocalin (NGAL), a marker of tubular damage, is increased in patients with chronic heart failure. Eur J Heart Fail, 10, 997–1000.
154. Bolignano, D., Basile, G., Parisi, P., et al. (2009). Increased plasma neutrophil gelatin-ase–associated lipocalin levels predict mortality in elderly patients with chronic heart failure. Rejuvenation Res, 12, 7–14.
155. Ljungman, S., Laragh, J. H., & Cody, R. J. (1990). Role of the kidney in congestive heart failure. Relationship of cardiac index to kidney function. Drugs, 39(Suppl 4), 10–21.
156. Ng, L. L., Pathik, B., Loke, I. W., et al. (2006). Myeloperoxidase and C-reactive protein augment the specificity of B-type natriuretic peptide in community screening for sys-tolic heart failure. Am Heart J, 152, 94–101.
157. Dunlay, S. M., Gerber, Y., Weston, S. A., et al. (2009). Prognostic value of biomarkers in heart failure: application of novel methods in the community. Circ Heart Fail, 2, 393–400.
158. Zairis, M. N., Tsiaousis, G. Z., Georgilas, A. T., et al. (2009 Jan 19). Multimarker strategy for the prediction of 31 days cardiac death in patients with acutely decompensated chronic heart failure. Int J Cardiol, [Epub ahead of print].
159. Anand, I. S., Latini, R., Florea, V. G., et al. (2005). In C-reactive protein in heart failure: prognostic value and the effect of valsartan. Circulation, 112, 428–434.





























