Embed Size (px)
Citation preview

Hash Algorithm and Hash Algorithm and SSAHA SSAHA
ImplementationsImplementationsZemin NingZemin Ning
Production Software Group Production Software Group InformaticsInformatics

Outline of the Talk:
The need for fast search engine; SSAHA – Sequence Search and Alignment using
Hashing Algorithm; Hash table; Sequence search based on the hash table; Search speed; Memory requirement; How to use the package.

Algorithms and Software Tools Algorithms
- Dynamic programming; - Hash method; - Suffix tree; - …
Software tools - FASTA; - BLAST; - Cross_Match; - Mummer; - …
CPU vs Memory

Smith-Waterman Algorithm Only works effectively when
gap penalties are used In example shown
– match = +1– mismatch = -1/3– gap = -1+1/3k (k=extent of
gap) Start with all cell values = 0 Looks in subcolumn and
subrow shown and in direct diagonal for a score that is the highest when you take alignment score or gap penalty into account
C A G C C U C G C U U A GA 0.0 1.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 1.0 0.0A 0.0 1.0 0.7 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 1.0 0.7U 0.0 0.0 0.8 0.3 0.0 0.0 0.0 0.0 0.0 1.0 1.0 0.0 0.7G 0.0 0.0 1.0 0.3 0.0 0.0 0.7 1.0 0.0 0.0 0.7 0.7 1.0C 1.0 0.0 0.0 2.0 1.3 0.3 1.0 0.3 2.0 0.7 0.3 0.3 0.3C 1.0 0.7 0.0 1.0 3.0 1.7 ?AUUGACGG
Hij=max{Hi-1, j-1 +s(ai,bj), max{Hi-k,j -Wk}, max{Hi, j-l -Wl}, 0}
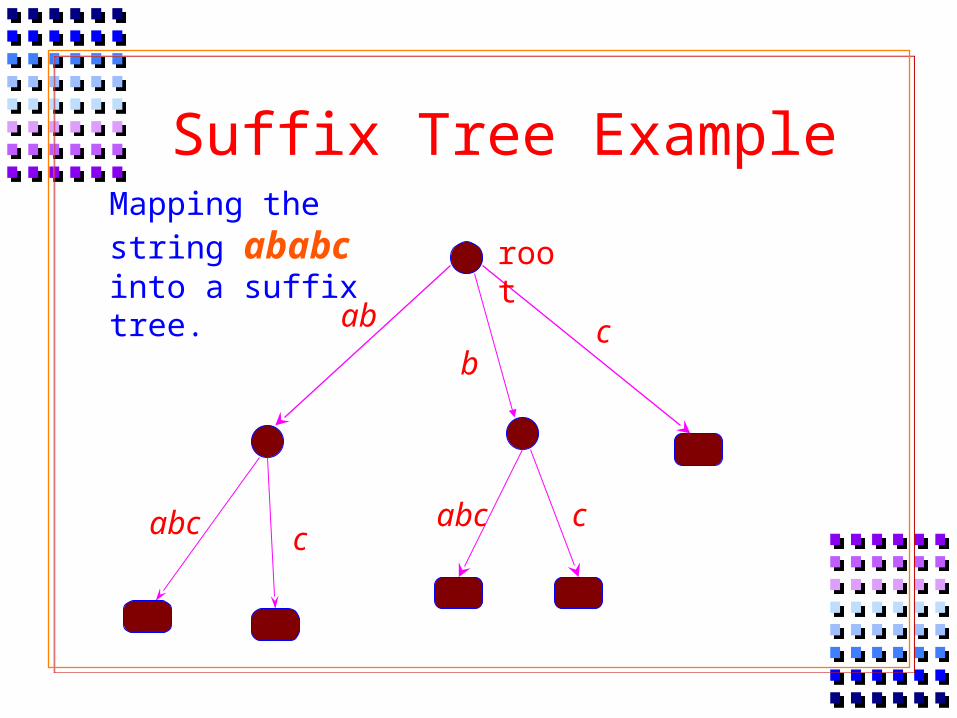
Mapping the string ababc into a suffix tree.
ab
abc c
bc
abc c
root
Suffix Tree Example

Motivation for sequence indexing
– faster (economy)– remove reliance on the external service
and network delays (user independence)– integrate fully with a database engine
(convenience)– exhaustive instead of heuristics (quality)– enable different statistics in sequence
evaluation (flexibility)

Objectives:
With SSAHA algorithm, we aim to achieve the following objectives:
(ii) To explore applications such as large scale sequence assembly and single nucleotide polymorphism (SNP) detection;
(i) To develop a sequence search engine to search genomic sequences with a fast speed and acceptable accuracy;
(iii)To provide possible tools for sequence analysis based on the search engine.

Sequence Representation
Sequence S: (s1s2, …, si, …, sm) i =1,2, …, m
K-tuple: (sisi+1...si+k-1)
Using two binary digits for each base, we may have the following representations:
“A” =00; “C” = 01; “G” = 10; “T” = 11
For any of the m/k no-overlapping k-tuples in the sequence, an integer may be used to represent the k-tuple in a unique way
k
i
ii EE
2
1
2kmax
1 1-2 with 2
where i = 0 or 1, depending on the value of the sequence base and Emax is the maximum value of the possible E values.
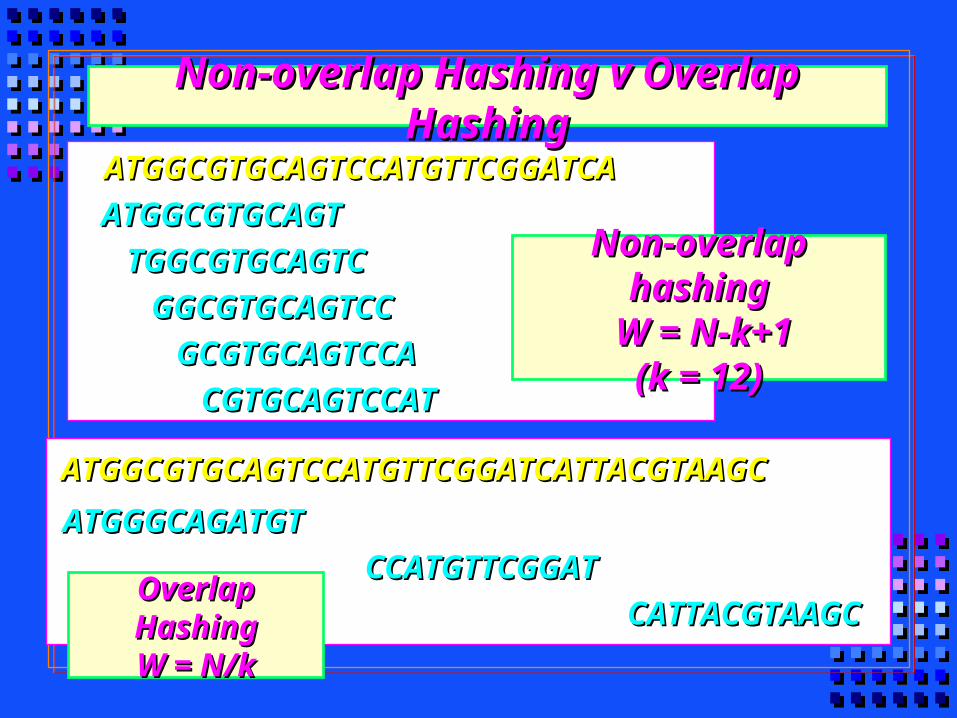
Overlap HashingOverlap HashingW = N/kW = N/k
ATGGGCAGATGTATGGGCAGATGT
CCATGTTCGGATCCATGTTCGGAT
CATTACGTAAGCCATTACGTAAGC
ATGGCGTGCAGTCCATGTTCGGATCATTACGTAAGCATGGCGTGCAGTCCATGTTCGGATCATTACGTAAGC
ATGGCGTGCAGTATGGCGTGCAGT
TGGCGTGCAGTCTGGCGTGCAGTC
GGCGTGCAGTCCGGCGTGCAGTCC
GCGTGCAGTCCAGCGTGCAGTCCA
CGTGCAGTCCATCGTGCAGTCCAT
ATGGCGTGCAGTCCATGTTCGGATCAATGGCGTGCAGTCCATGTTCGGATCA
Non-overlap hashingNon-overlap hashing W = N-k+1W = N-k+1
(k = 12)(k = 12)
Non-overlap Hashing v Overlap HashingNon-overlap Hashing v Overlap Hashing

E k-tuple Ni Indices and Offsets0 AA 1 2, 19 1 AC 3 1, 9 2, 5 2, 11 2 AG 2 1, 15 2, 35 3 AT 2 2, 13 3, 3 4 CA 7 2, 3 2, 9 2, 21 2, 27 2, 33 3, 21 3, 235 CC 4 1, 21 2, 31 3, 5 3, 7 6 CG 1 1, 5 7 CT 6 1, 23 2, 39 2, 43 3, 13 3, 15 3, 17 8 GA 4 1, 3 1, 17 2, 15 2, 25 9 GC 0
10 GG 5 1, 25 1, 31 2, 17 2, 29 3, 1 11 GT 6 1, 1 1, 27 1, 29 2, 1 2, 37 3, 19 12 TA 1 3, 25 13 TC 6 1, 7 1, 11 1, 19 2, 23 2, 41 3, 11 14 TG 3 1, 13 2, 7 3, 9 15 TT
S1=(GTGACGTCACTCTGAGGATCCCCTGGGTGTGG) S2=(GTCAACTGCAACATGAGGAACATCGACAGGCCCAAGGTCTTCCT)S3=(GGATCCCCTGTCCTCTCTGTCACATA)
Hash Table: A 2-tuple hashing table of S1, S2 and S3

Query sequence: Sq = (TGCAACAT)
E k-tuple Ni Indices and Offsets0 AA 1 2, 19 1 AC 3 1, 9 2, 5 2, 11 2 AG 2 1, 15 2, 35 3 AT 2 2, 13 3, 3 4 CA 7 2, 3 2, 9 2, 21 2, 27 2, 33 3, 21 3, 235 CC 4 1, 21 2, 31 3, 5 3, 7 6 CG 1 1, 5 7 CT 6 1, 23 2, 39 2, 43 3, 13 3, 15 3, 17 8 GA 4 1, 3 1, 17 2, 15 2, 25 9 GC 0
10 GG 5 1, 25 1, 31 2, 17 2, 29 3, 1 11 GT 6 1, 1 1, 27 1, 29 2, 1 2, 37 3, 19 12 TA 1 3, 25 13 TC 6 1, 7 1, 11 1, 19 2, 23 2, 41 3, 11 14 TG 3 1, 13 2, 7 3, 9 15 TT
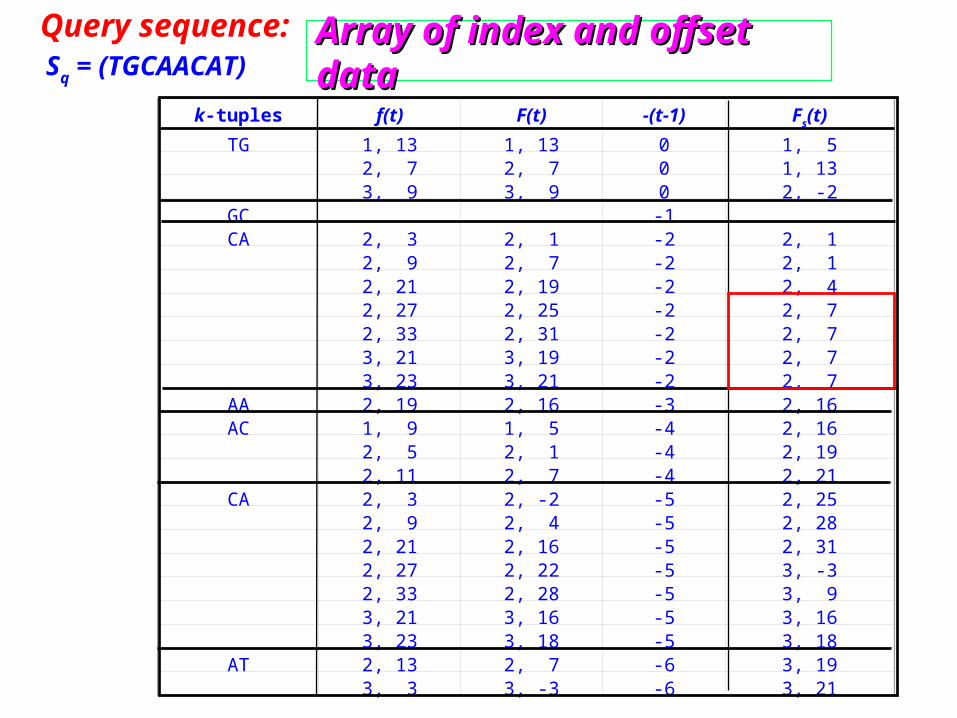
k-tuples f(t) F(t) -(t-1) Fs(t)
TG 1, 13 1, 13 0 1, 5 2, 7 2, 7 0 1, 13 3, 9 3, 9 0 2, -2
GC -1 CA 2, 3 2, 1 -2 2, 1 2, 9 2, 7 -2 2, 1 2, 21 2, 19 -2 2, 4 2, 27 2, 25 -2 2, 7 2, 33 2, 31 -2 2, 7 3, 21 3, 19 -2 2, 7 3, 23 3, 21 -2 2, 7
AA 2, 19 2, 16 -3 2, 16AC 1, 9 1, 5 -4 2, 16 2, 5 2, 1 -4 2, 19 2, 11 2, 7 -4 2, 21
CA 2, 3 2, -2 -5 2, 25 2, 9 2, 4 -5 2, 28 2, 21 2, 16 -5 2, 31 2, 27 2, 22 -5 3, -3 2, 33 2, 28 -5 3, 9 3, 21 3, 16 -5 3, 16 3, 23 3, 18 -5 3, 18
AT 2, 13 2, 7 -6 3, 19 3, 3 3, -3 -6 3, 21
Array of index and offset dataArray of index and offset dataSq = (TGCAACAT)
Query sequence:

Query sequence: Sq = (TGCAACAT)
E k-tuple Ni Indices and Offsets0 AA 1 2, 19 1 AC 3 1, 9 2, 5 2, 11 2 AG 2 1, 15 2, 35 3 AT 2 2, 13 3, 3 4 CA 7 2, 3 2, 9 2, 21 2, 27 2, 33 3, 21 3, 235 CC 4 1, 21 2, 31 3, 5 3, 7 6 CG 1 1, 5 7 CT 6 1, 23 2, 39 2, 43 3, 13 3, 15 3, 17 8 GA 4 1, 3 1, 17 2, 15 2, 25 9 GC 0
10 GG 5 1, 25 1, 31 2, 17 2, 29 3, 1 11 GT 6 1, 1 1, 27 1, 29 2, 1 2, 37 3, 19 12 TA 1 3, 25 13 TC 6 1, 7 1, 11 1, 19 2, 23 2, 41 3, 11 14 TG 3 1, 13 2, 7 3, 9 15 TT
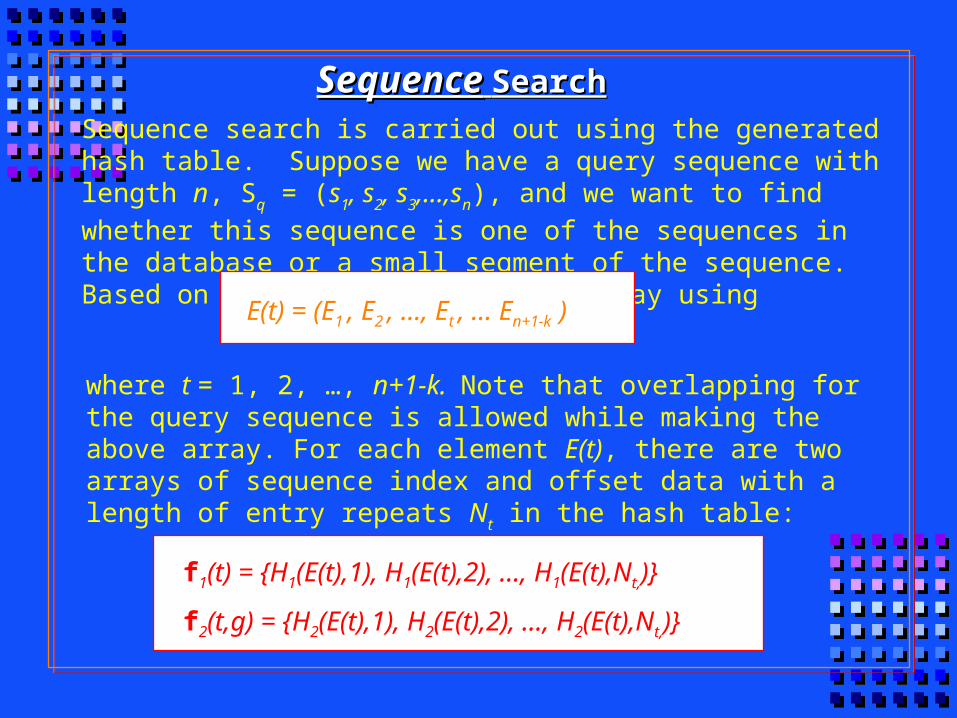
SequenceSequence SearchSearch
Sequence search is carried out using the generated hash table. Suppose we have a query sequence with length n, Sq = (s1, s2, s3,...,sn), and we want to
find whether this sequence is one of the sequences in the database or a small segment of the sequence. Based on Sq, we have an integer array using
where t = 1, 2, …, n+1-k. Note that overlapping for the query sequence is allowed while making the above array. For each element E(t), there are two arrays of sequence index and offset data with a length of entry repeats Nt in
the hash table:
E(t) = (E1 , E2 , …, Et , … En+1-k )
f1(t) = {H1(E(t),1), H1(E(t),2), …, H1(E(t),Nt,)}
f2(t,g) = {H2(E(t),1), H2(E(t),2), …, H2(E(t),Nt,)}

F1(t) = f1(t)
F2(t) = {H2’(E(t),1), H2
’(E(t),2), …, H2’(E(t),Nt)}
with H2
’(E(t),i) = H2(E(t),2)-(t-1) i = 1,2,…, Nt
The above calculation to adjust offsets should be done for every element in the array.
Frequency ArrayFrequency Array
SubjectSubject
QueryQuery
t-1
Match Start Match Start
Reference Point
t-1
Match Start Match Start
Reference Point

In order to carry out search quickly and effectively, it would be helpful in the computer code to combine these two integer arrays into a single long integer array. We are targeting implementations on 64 bit machines. The long integer array can be expressed as
F (t) = {H (E(t),1), H (E(t),2),…, H (E(t),Nt)} with
H(E(t),i) = 232 H1(E(t),i) + H2’(E(t),i) i = 1,2,…, Nt
64 Bit Machines64 Bit Machines
It is seen from the above equation that the offset value takes the low bits while the index part takes high orders of bits in the long integer.
Index Offset

For the query sequence, there are n+1-k arrays in total and it is necessary that we combine all the arrays into one single arrays and
F = {F (1), F(2),…, F(t),…, F(n+1-k)}
Finally when the array is sorted into an ascending order, i.e.
F -> Fs
with Fs,1 < Fs,2 < … < Fs,i < …
the search results can be determined by the number of the data repeats in the array. In a section within the Fs array, if the found repeat level is higher than
a given threshold level, this means that there is a match between the query sequence and sequences in the database.
Array SortingArray Sorting

Power Law: CPU time v query lengthPower Law: CPU time v query length
y = 9E-05x1.3197
R2 = 0.9948
0
0.2
0.4
0.6
0.8
1
0 200 400 600 800 1000 1200
Number of k-tuples in query (n/k)
Nor
mal
ized
CPU
tim
e
Fig. 1 Normalized CPU time plotted against the number of k-tuples in query (k=12) using Quicksort.
iF Nknmkn
N ˆ)1()1(
Averaged length of frequency array:
where Ni is the average length of the entruy repeats.
^
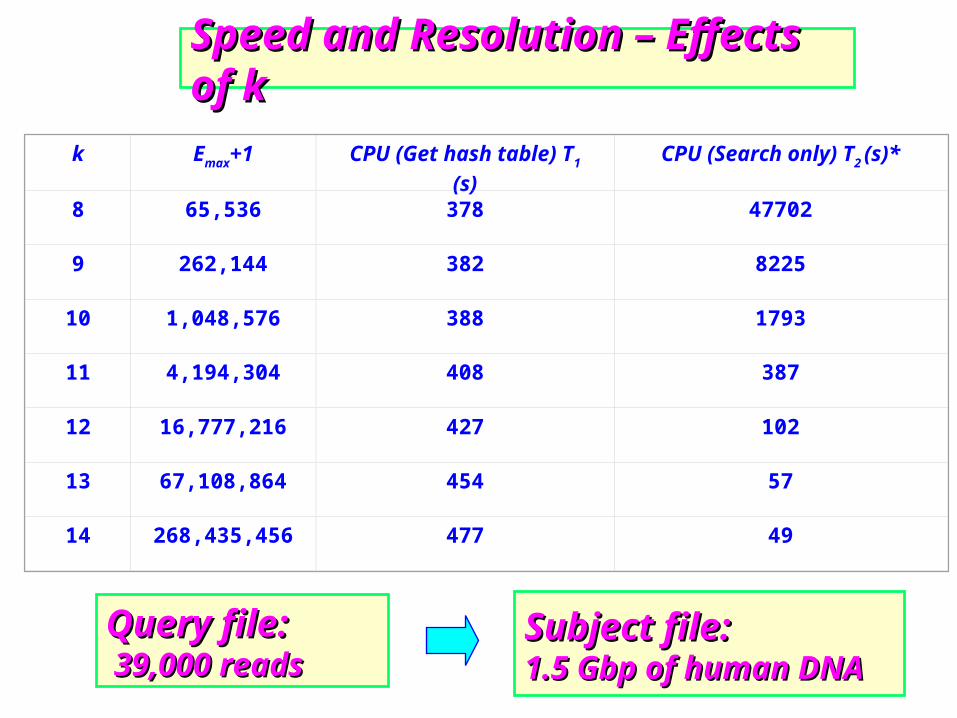
Query file:Query file: 39,000 reads39,000 reads
Speed and Resolution – Effects of kSpeed and Resolution – Effects of k
Subject file:Subject file:1.5 Gbp of human DNA1.5 Gbp of human DNA
k Emax+1 CPU (Get hash table) T1 (s) CPU (Search only) T2 (s)*
8 65,536 378 47702
9 262,144 382 8225
10 1,048,576 388 1793
11 4,194,304 408 387
12 16,777,216 427 102
13 67,108,864 454 57
14 268,435,456 477 49

SSAHA MemorySSAHA Memory
Memory for subjectMemory for subject: M: Mss = 4*N = 4*Nss/k+ 4*2/k+ 4*22k2k
Memory for queryMemory for query: M: Mqq = N = Nqq
House keepingHouse keeping: 10-20% total: 10-20% total
Total memoryTotal memory: M: Mss = 1.2*(M = 1.2*(Mss+M+Mqq) )
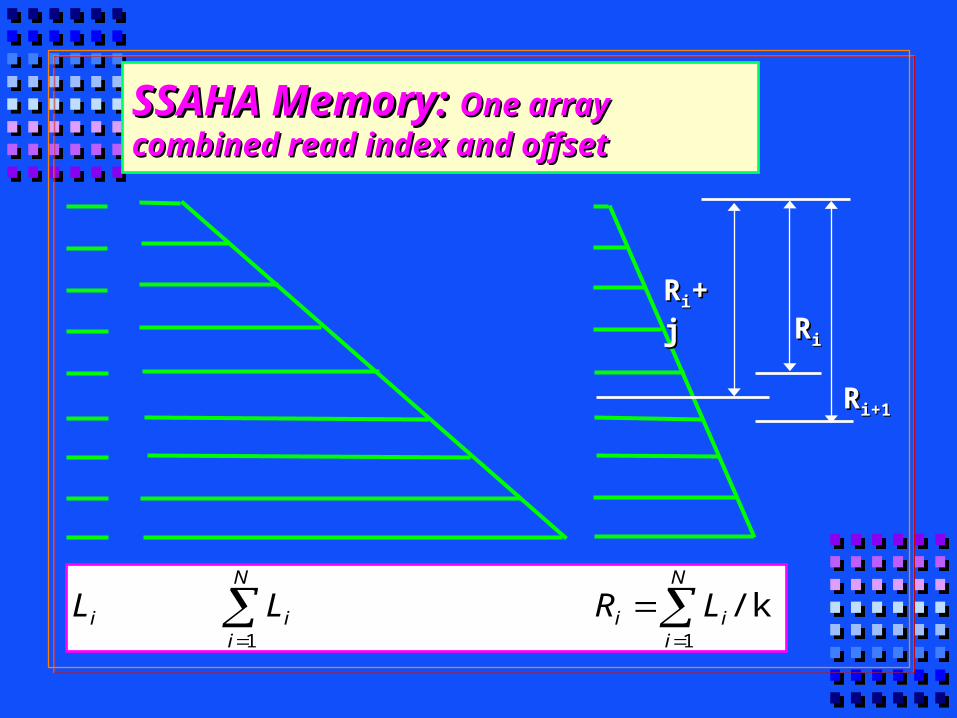
RRii+j+j
RRi+1i+1
RRii
N
iii
N
iii LRLL
11
/k
SSAHA Memory: SSAHA Memory: One array combined One array combined read index and offsetread index and offset
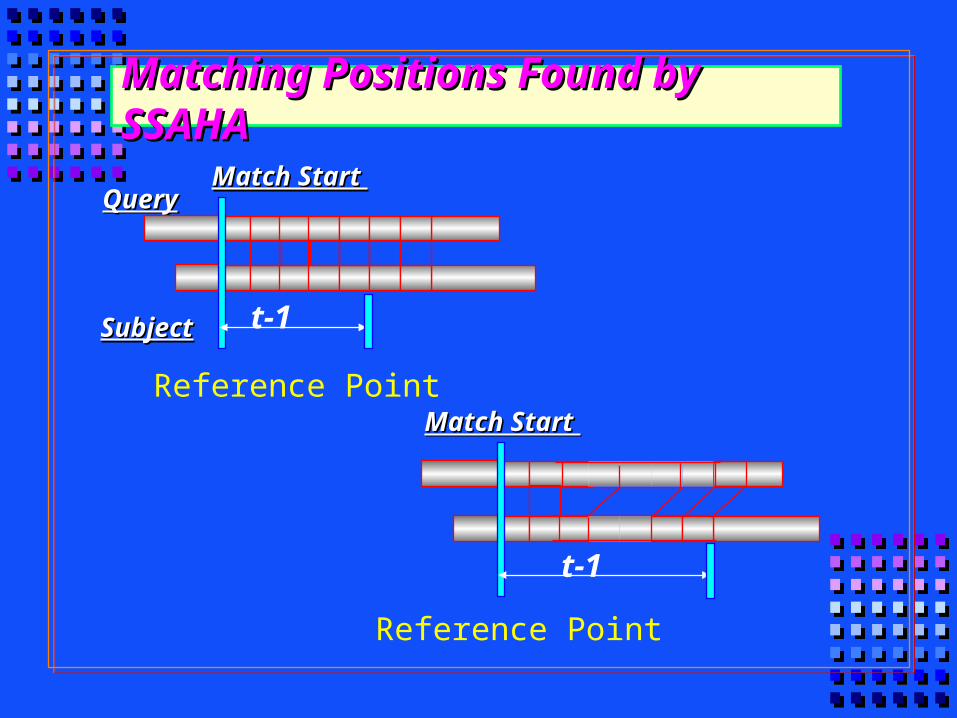
Matching Positions Found by SSAHAMatching Positions Found by SSAHA
SubjectSubject
QueryQuery
t-1
Match Start Match Start
Reference Point
t-1
Match Start Match Start
Reference Point

SSAHA2 = SSAHA + Cross_MatchSSAHA2 = SSAHA + Cross_Match
SSAHA for matching seeds, cross_match forSSAHA for matching seeds, cross_match forsequence alignment.sequence alignment.
SSAHA seedsSSAHA seeds
Edge Edge lengthlength
Sequence for cross_matchSequence for cross_match
Edge Edge lengthlength
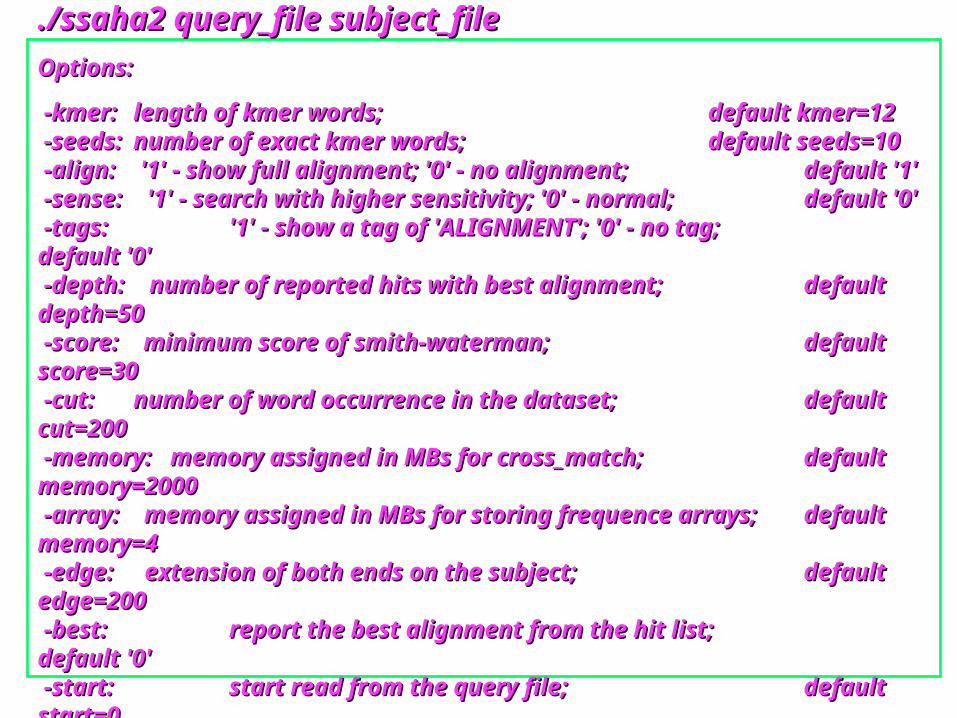
SSAHA2 Command LineSSAHA2 Command Line./ssaha2 query_file subject_file./ssaha2 query_file subject_file
Options:Options:
-kmer: -kmer: length of kmer words;length of kmer words; default kmer=12default kmer=12 -seeds: -seeds: number of exact kmer words;number of exact kmer words; default seeds=10default seeds=10 -align: '1' - show full alignment; '0' - no alignment; -align: '1' - show full alignment; '0' - no alignment; default '1'default '1' -sense: '1' - search with higher sensitivity; '0' - normal; -sense: '1' - search with higher sensitivity; '0' - normal; default '0'default '0' -tags: -tags: '1' - show a tag of 'ALIGNMENT'; '0' - no tag;'1' - show a tag of 'ALIGNMENT'; '0' - no tag; default '0'default '0' -depth: number of reported hits with best alignment; -depth: number of reported hits with best alignment; default depth=50default depth=50 -score: minimum score of smith-waterman; -score: minimum score of smith-waterman; default score=30default score=30 -cut: -cut: number of word occurrence in the dataset; number of word occurrence in the dataset; default cut=200default cut=200 -memory: memory assigned in MBs for cross_match; -memory: memory assigned in MBs for cross_match; default memory=2000default memory=2000 -array: memory assigned in MBs for storing frequence arrays; -array: memory assigned in MBs for storing frequence arrays; default memory=4default memory=4 -edge: extension of both ends on the subject; -edge: extension of both ends on the subject; default edge=200default edge=200 -best: -best: report the best alignment from the hit list;report the best alignment from the hit list; default '0'default '0' -start: -start: start read from the query file;start read from the query file; default start=0default start=0 -end: end read from the query file; -end: end read from the query file; default start= Total number of the reads in the query file;default start= Total number of the reads in the query file;

COOKBOOKCOOKBOOK
BACends placement - find the best hit in the database:BACends placement - find the best hit in the database: -seeds 14 -kmer 13 -align 0 -tags 1 -depth 5 -score 200 -cut 50000; -seeds 14 -kmer 13 -align 0 -tags 1 -depth 5 -score 200 -cut 50000;
EST/cDNA alignment - produce splice on the subject sequence:EST/cDNA alignment - produce splice on the subject sequence: -seeds 4 -kmer 13 -align 0 -tags 1 -depth 5 -score 20 -edge 20000; -seeds 4 -kmer 13 -align 0 -tags 1 -depth 5 -score 20 -edge 20000;
Primer/gene Marks alignment - find the matches of short motifs to the database:Primer/gene Marks alignment - find the matches of short motifs to the database: -seeds 1 -kmer 13 -tags 1 -score 12 -skip 1 -sense 1 -cut 50000; -seeds 1 -kmer 13 -tags 1 -score 12 -skip 1 -sense 1 -cut 50000;
Search with higher sensitivity:Search with higher sensitivity: -seeds 2 -kmer 13 -tags 1 -score 20 -sense 1 -cut 50000; -seeds 2 -kmer 13 -tags 1 -score 20 -sense 1 -cut 50000;
Both query and subject are large (q: 100Kb < query < 1MB; s: no limit):Both query and subject are large (q: 100Kb < query < 1MB; s: no limit): -seeds 50 -kmer 13 -tags 1 -score 2000 -array 40 -memory 10000; -seeds 50 -kmer 13 -tags 1 -score 2000 -array 40 -memory 10000;

Summary:
Speed - Fast enough to perform genomic scale searches between large genomes;
Memory – linear;
Sensitivity – not as good as BLAST, but applicable in assembly and SNP detection;





























