Embed Size (px)
Citation preview

1
Overview: Scheme of a Genomic SELEX experiment
Outcome: RNA pool enriched in RNA targets for the protein of interest, ready for sequencing
Questions to be answered: What are the direct RNA binding partners for your protein of interest?
What are the RNA binding motifs of the protein?
Bannikova and Barta, Genomic Selex

2
Comments have the following color codeComments by StefanGlossaryRefereal to other chapters, please do not changeList of vendorsReagents added to the database
look at the book chapters at the eurasnet site:http://www.eurasnet.info/noe/bookstefanstamm galadriel
Genomic SELEX to identify RNA targets of plant RNA binding proteins
Olga Bannikova and Andrea Barta
Max F. Perutz Laboratories, Medical University of Vienna, Dr. Bohrgasse 9/3, A-1030 Wien, Austria
Address correspondence to: Andrea Barta or Olga Bannikova, Max F. Perutz Laboratories, Medical
University of Vienna, Dr. Bohrgasse 9/3, A-1030 Wien, Austria ; email:
[email protected]; [email protected];
1. Abstract
Systematic evolution of ligands by exponential enrichment (SELEX) is an elegant technique
and allows the isolation of RNA and DNA sequences which directly interact with a protein of
interest. Genomic SELEX is an expression level independent selection method which is
useful when multiple RNA targets are expected. These RNAs might be expressed in different
conditions, or are differentially localized, or have diverse expression levels. Therefore, this
method allows the determination of RNA sequences within a particular genome which are
potential binding partners of a particular protein. At first a DNA library is constructed by
random priming of sheared Arabidopsis DNA with a direct and reverse primer and selection
of fragments with a desired length of 200-300 nucleotides. The RNA library is constructed by
transcribing the DNA fragments with T7 polymerase. Several rounds of selection with a
protein of choice yield a highly specific pool of potential RNA targets. This pool is best
sequenced by a deep sequencing method such as 454 sequencing technology and
sequences of the selected library are analyzed by bioinformatics methods. The result in a
two month time experiment of the experiment is a collection of RNAs which are binding
Bannikova and Barta, Genomic Selex

3
targets for the protein used for selection. A typical experiment takes about two months time.
These RNAs usually allow the determination of binding motifs.
Keywords: RNA selection, RNA binding proteins,
2. Introduction
Systematic evolution of ligands by exponential enrichment (SELEX) is a combination
of a combinatorial chemistry approach and experimental molecular biology techniques
allowing the isolation of high affinity binding partners to a given molecular target [1].(see
chapter 17a, singh) The first SELEX experiments used randomized artificial RNA-aptamer
libraries from which strong binders for RNAs that strongly bind to proteins or small molecules
were selected. Typically, the initial aptamer library contains around 1015 to 1016 DNA
oligonucleotides with a randomized central part and fixed flanking regions [2]. Such a pool
can be easily converted to RNA by in-vitro transcription and then, after the selection step,
back to DNA via RT-PCR. The selection is based on incubation of the library with the target
molecule followed by separation of the unbound fraction from the formed complex which is
usually performed on nitrocellulose filter [3, 4]. The nucleic acids of the selected complex are
isolated and then amplified. Such cycles are repeated several times to select for high affinity
binders for the target molecule. The aim of this selection is the isolation of RNA or DNA
oligonucleotides which have the strongest binding affinity to the target of interest. These tight
binding partners are often used in diagnostic and therapeutic applications [5].
Several problems can emerge throughout the selection procedure. One of the
possible troubles is losses of the bound oligonucleotides or enrichment of unspecific binding
targets due do you mean during the specificity selection procedure. To overcome that
problem few several other methods for the separation step were described [for review see 6].
As the described SELEX method is optimized to select the winners for best binding to a
particular molecule, they have their benefits if such artificial aptamers are used in clinical
studies. Can you tune down the statement, because in chapter 17a we show an application
of selex to map binding sites Applications range from inhibition of a particular protein with
this aptamers to using them as substitutions for antibodies. However, these selected aptamer
Bannikova and Barta, Genomic Selex

4
sequences might not be present in the genome of the organism as such strong binders might
be detrimental for the function of the protein used in the selection. Biological procedures
necessitate a certain dynamic equilibrium as proteins have to bind specifically in one
situation but often have to be released again in order to continue the biological process. The
selction of splice site is governed by the formation of transient protein complexes, as
described in chapter 4/Hertel. Therefore, using a randomized aptamer library for the SELEX
procedure is not optimal for finding natural binding sites for your favorite RNA binding
protein, as the best binder might not corresponded to real binders occurring in vivo in the
cell.
To overcome some of the problems in the field of RNA –protein interactions, another
type of SELEX was developed termed Genomic SELEX. In contrast to a randomized library,
only sequences occurring in the genome of a specific organism are used for library
construction allowing the search for real-existing DNAs or RNAs from the same organism of
interest.
The first DNA library for Genomic SELEX was developed in 1997 by B.S. Singer [7] for
E.coli, S.cerevisiae and human genomes. Subsequently, several proteins have been used to
find in vivo RNA targets via Genomic SELEX experiments [8, 9]. These experiments showed
the ability of Genomic SELEX not only to find known targets for these proteins, but detected
many new targets which then were proven in in vivo studies.
The selection procedure does not vary significantly from a randomized aptamer
SELEX. The only difference lies in the library construction where short genomic sequences
are prepared with a DNA polymerase Klenow-fragment dependent addition of adapters to the
DNA. These adaptors are chosen to permit the construction of an RNA library which now
contains most genomic DNA sequences of an organism in form of RNA sequences. This
allows now to select for protein binding regions independent of their level of transcription.
Unsurprisingly, many problems which have to be dealt with are similar in Genomic SELEX as
in randomized aptamer-based selection.
A critical point for this genomic approach is the loss of weak but biologically
significant binders during selection. Consequently the stringency of selection conditions
especially in the first rounds of SELEX should be decreased which boosts the diversity of
RNA targets in comparison to selecting only a few winners. Other interference in such
experiments is the possibility of forming secondary structures between the middle part and
the adapter sequences in a library which could lead to unspecific selection of targets. An
approach to handle this issue was suggested by Wen and Grey [10] which developed a
primer-free Genomic SELEX. Finally, genomic SELEX as any other SELEX procedure
remains an in vitro technique and results of the selection procedure have to be proven by
Bannikova and Barta, Genomic Selex

5
other in vitro methods like a gel shift assay and by experiments to show binding and activity
in vivo (CLIP or ChIP-chip technology) [1], see chapter 22/Ule.
Here we present our variant of Genomic SELEX which was developed for an
Arabidopsis thaliana, (as well as an S. pombe) RNA-library where we tried to avoid possible
problems inherently residing in SELEX techniques. This protocol can be performed in two
months and results in a highly-specific RNA-pool saturated with targets sequences for the
protein of interest. These RNAs are suitable for further sequencing and bioinformatic
analysis.
3. ProtocolProtocol 1: Genomic library construction
3.1 DNA preparation
3.1.1 Genomic DNA isolation
Genomic DNA was isolated from 2-weeks old seedling of wild-type Arabidopsis
thaliana (Columbia) using the DNAeasy plant mini kit (Qiagen) by following the manufactures
instructions. The optimal amount for 30 µg of pure genomic DNA is about 2 gram plant tissue
grounded in liquid nitrogen. DNA should be in TE (10 mM Tris/HCl pH 8.0, 1 mM EDTA)
buffer, its concentration quantified by measuring OD260nm and DNA purity should be checked
on a 1% agarose gel (what is your criterion for intact DNA?). If DNA is still contaminated with
RNA an additional RNase treatment is required following the standard protocol.
3.1.2. DNA fragmentation
30 µg of pure genomic DNA was placed in a 13 ml round-bottom falcon tube and
fragmented by ultrasound treatment using a Bandelin Sonoplus UW2070 device with a MS73
microtip. Vendor? DNA was sonicated 8 times with 10 pulses for 10 seconds at 70% power.
Sonication should produce fragments from 100 bp to 4 kb in length, which should be checked
by agarose gel electrophoresis and compared with unshared control DNA (Figure 1A).Then
sheared DNA was precipitated overnight in the presence of 1/10 volume 3 M NaOAc pH 5.4,
and 3 volumes of absolute EtOH. The pellet was resuspended in 100 μl of TE buffer.
3.2 Library development
3.2.1 Primers design
Two pairs of primers were used in the protocol. The first pair was needed for the
introduction of adaptor sequences to the sheared DNA. The second pair introduced the T7
promoter sequence for creating the RNA library. The adapters should meet several
Bannikova and Barta, Genomic Selex

6
requirements: 1. they should be able to prime at any sequence in genome, using randomized
8 nucleotides. 2. Have fixed sequence for further manipulation. 3. Sequences of the primers
should not be present in the genome.
Based on the features above, forward and reversed primers for the Klenow reaction were:
Fran: AGGGGAATTCGGAGCGGGGCAGCNNNNNNNNN
Rran: CGGGATCCTCGGGGCTGGGATGNNNNNNNNN
The second pair of primers should be complementary to the fixed part of the first primer pair.
In addition, the forward primer must have a T7 promoter sequence for the in vitro
transcription.
Fclcf: CCAAGTAATACGACTCACTATAGGGGAATTCGGAGCGGG
Rclcr: CGGGATCCTCGGGGCTG
3.2.2 Primer labeling
In order to visualize the incorporation of the randomized primer-adaptors to the
genome, a part of the adaptors was radioactively labeled at their 5’ end with [γ32P] ATP and
T4 polynucleotide kinase using standard protocols. Unincorporated nucleotides were
separated by a G-50 column from GEHealthcare following the manufacturer’s instructions.
3.2.3 Introducing adaptor sequences to the genomic DNA by Klenow reaction
The starting material was about 25 µg of sheared and purified DNA (3.1.2.) at a
concentration of 1mM. The concentration of the primers should be enough to allow annealing
once every 40 nucleotides as described in [8]. To monitor the reaction process, part of the
forward and the reverse primer (containing the randomized sequence) were end-lableled with
32P. kinased. Then the reaction mixture was split into two tubes to control the introduction of
the forward and reverse primer separately. The DNA solution was mixed with 255 μM primer
Rran to a final concentration of 12 μM and one tube was supplemented with 2 µM radioactive
Rran. Both tubes were incubated for 3 min at 93oC, and then placed on ice and further treated
in parallel. After addition of 10x Klenow buffer (composition, vendor) and
deoxyribonucleotides to a final concentration of 1 mM, the reaction was started with 67 U of
Klenow exo-minus enzyme (Fermentas) and incubated for 5 min on ice. Incubation was for
25 min at room temperature and then 5 min at 50oC. The reaction was inactivated by adding
EDTA (final conc. 15 mM) and heating for 10 min at 75oC. The reaction mixture was cleaned
up from low molecular weight substances with YM-30 Millipore columns. At this point the
efficiency of incorporation could be monitored in the radioactive sample by a denaturing 8%
polyacrylamide gel with 7 M urea (Figure 1B) normally people use 8 M urea, any reason for
Bannikova and Barta, Genomic Selex

7
7M?. The same protocol was applied for the forward primer reaction and now the radioactive
Fran primer was added to the non-radioactive sample. Note, that the specific activity of the
primers was low, so no high incorporation of radioactivity was expected. But, typically a
smear of labeled DNA above the radioactive primer should be visible (Figure 1B).
3.2.4. Gel-purification and size selection of DNA fragments
At this point the DNA has to be separated from unincorporated primers. The two
samples from the previous steps were combined and fractionated for about 2h at 100V on a
preparative 8% denaturing polyacrylamide (7 M urea) gel with labeled size markers.
Then DNA of 100 to 700 bp was extracted from the gel which was divided into small pieces;
frozen to -80oC for 15 min, and then eluted with buffer (10 mM Tris-HCl pH=8.0, 2 mM EDTA
pH=8.0, 0.3 M NaOAc pH=5.4). The mixture was heated for 5 min at 95oC and left overnight
at 25oC shaking (900 rpm). Alternatively shaking could be at 65oC for 3 hours.
Next, the gel mixture was filtrated though a 0.22 µm nitrocellulose filter (Millipore) and
precipitated with 2 volume of EtOH for at least 3 hours.
3.2.5. Introduction of the T7 promoter
For the introduction of the T7 promoter sequence the second pair of primers (Fclcf and
Rclcr) were used for the PCR reaction. The number of PCR cycles shouldn’t be more then 10
to avoid artificial byproducts. One of the available proof-reading DNA polymerases (we
usually use Phusion polymerase from Finnzyme) should be used to prevent mutations.
Typically, PCR was performed: 40 s denaturation at 95oC, 40 s annealing at 55oC (-3oC
below the Tm of primer), 20 s elongation at 72oC. The PCR reaction was cleaned via
phenol/chloroform extraction followed by PCR-clean up kit which has a low cut off to leave
small DNA fragments in the library (e.g. Nucleospin extract II (Macherey-Nagel).
3.2.6. Library cleaning and verification
At his point the DNA library was almost ready, but still can contain some undesirable
features, such as fragments containing the same primer (forward or reverse) at both ends.
To get rid of such products, the library was subjected to an in vitro transcription reaction
followed by a reverse transcription and PCR reaction (RT-PCR).
In order to produce a lot of RNA (usually around 50 µg) from a limited amount of
DNA, a High Yield Transcription kit (e.g. from Fermentas) is strongly recommended. in your
hands, was the fermentas kit bettern than Ambion? Transcribed RNA was extracted with
Bannikova and Barta, Genomic Selex

8
phenol/chloroform and precipitated with 2 volumes of EtOH overnight + 1/10 vol naac?. For
the precise quantification of RNA, it was crucial to purify the RNA with any clean-up kit (e.g.
MegaClear RNA clean-up kit from Ambion). After that the quality of the RNA was checked by
agarose gel electrophoreses.
For reverse transcription of RNA and a one-step RT-PCR kit from Qiagen was used,
because it contained two types of reverse transcriptases which allowed reverse transcription
of low and high abundant transcripts from the mixture. In case of using this kit, the number of
PCR cycles should be kept to 7-9 to decrease formation of unspecific products. After
checking the concentration of the DNA library, it could be further amplified using one of the
proof-reading polymerases. Now, the DNA library is ready for a selection procedure and can
be store at -20oC for at least 6 months or for a longer period at -80oC.
It is important to check the quality and comprehensiveness of the library by choosing 5 - 15
single copy gene primers for amplification of an appropriate size product from the library by
standard PCR [7].
Protocol 2: Affinity selection of RNA targets
3.3. In vitro transcription
In each cycle of the SELEX procedure, the same amount of starting DNA from the
previous cycle was used. Optimally, about 1 µg of DNA should be utilized as starting material
for in vitro transcription. Transcription should be performed as described in section 3.2.6.
3.4. Selection of RNAs bound to the protein of choice
The protein of interest was usually a recombinant protein purified from a prokaryotic
or eukaryotic protein expression system. In our case, a recombinant GST-tagged protein (the
RRM and Zn-knuckle domain from atCyp59 [11]) expressed in E.coli was used for selection.
The protein should be dialyzed into binding buffer with a 1:1000 excess.
There were several important points to take care off in this step. First, at each cycle 10µg
clean RNA (without remaining DNA and nucleotides) was used. Second, each RNA-protein
complex behaved differently; meaning that in each case information about binding conditions
(such as buffer, working pH and binding temperature) should be empirically assembled. If
nothing is known about the protein of interest or of a similar protein, PBS buffer (0.135 M
NaCl, 27 mM KCl, 8mM Na2HPO4, 2 mM NaH2P04) with 10 mM Mg2+ could be used. Third, for
binding and washing siliconized eppendorf tubes were recommended to prevent loss of
glutathione beads. Do you buy siliconized tubes or do you siliconize yourself, if so how?
Bannikova and Barta, Genomic Selex

9
Before performing the binding reaction of the protein to the GST- beads, an aliquot of
40 µl 50% 4B glutathione sepharose (which is sufficient to bind up to 8 µg protein) was
blocked with 0.5 ml tRNA 200 µg/ml (Sigma) and incubated 30 min at 4oC by slow rotation
and then washed three times with 10 volumes of binding buffer (1xPBS with 10mM MgCl2). In
parallel, the protein-RNA binding reaction was performed in solution (100 µl reaction mixture
contained 10 µg of RNA in binding buffer, supplemented with protease inhibitor cocktail
(Roche) and RNAse inhibitor (Invitrogen)). This mixture was heated for 5 min at 70oC and
then left for 10 min at 25oC (refolding process). Then the dialyzed protein was added in a 3:1
molar excess of RNA over protein in the first 3 cycles. Then the stringency was increased
and a ratio of 10:1 was used in later cycles. The binding reaction was incubated for 30 min
at 4oC (or another temperature appropriate for the protein of interest). Then, the blocked and
washed GST-beads were added to the binding reaction and incubated for another 30 min.
Then the beads were washed 3 times with 10 volumes of binding buffer and then eluted
twice with 100 µl of reduced glutathione elution buffer according to the manufactures
instructions (GEHealthcare). When performing the control – anti-GST selection, purified
GST-tag protein at the 10:1 molar ratio was used instead of the protein of interest. To release
RNA from the RNA-protein complex, 400 µl FES buffer (20 mM citric buffer pH 5.0, 7 M urea,
1mM EDTA pH 8.0) and 400 µl of Phenol pH 6.0 was added and vigorously shaken for 10
min at 900 rpm. Then 200 µl of H2O was added and the mixture extracted with an equal
volume of phenol/chloroform/isoamylalcohol : 25/24/1. The RNA was precipitated overnight
by adding 40 μg of glycogen (vendor, glycoblue?), 1/10 volume of 3 M NaOAc pH 5.4, and 2
volumes of EtOH. The precipitate was dissolved in 20 µl water, cleaned from residuals of
phenol with Megaclear RNA clean up kit and then the yield of the selected RNA was
measured.
3.5. RT-PCR, amplification.
In each step the selected RNA was reverse transcribed as described in section 3.2.3
and then the cycle of in vitro transcription and selection was repeated up to 7-12 times. The
number of cycles depends on the affinity to the protein and the diversity of the RNA targets.
Generally, selection may be stopped when half of the selected RNA pool binds to the protein.
For example, in a 10:1 excess of RNA over protein in the binding reaction, half of the
possible yield is 5% of RNA binding to the protein (Figure 2).
4. Example from of an experiment
Bannikova and Barta, Genomic Selex

10
Figure 1b shows the size distribution of the radioactive labeled DNA library on a 8%
denaturing polyacrylamide gel after the ligation is ligation the right word here, as you do
klenow fill in , of the first pair of primer-adaptors.
Figure 2 shows a typical example of RNA recovery after each cycle in an SELEX experiment
using the RRM and Zn-knuckle domain of atCyp59 [11].
Table 1 shows a statistics analysis of the sequenced RNA pool after the 11th cycle, enriched
with binding targets for the AtCyp59 protein after a SELEX experiment.
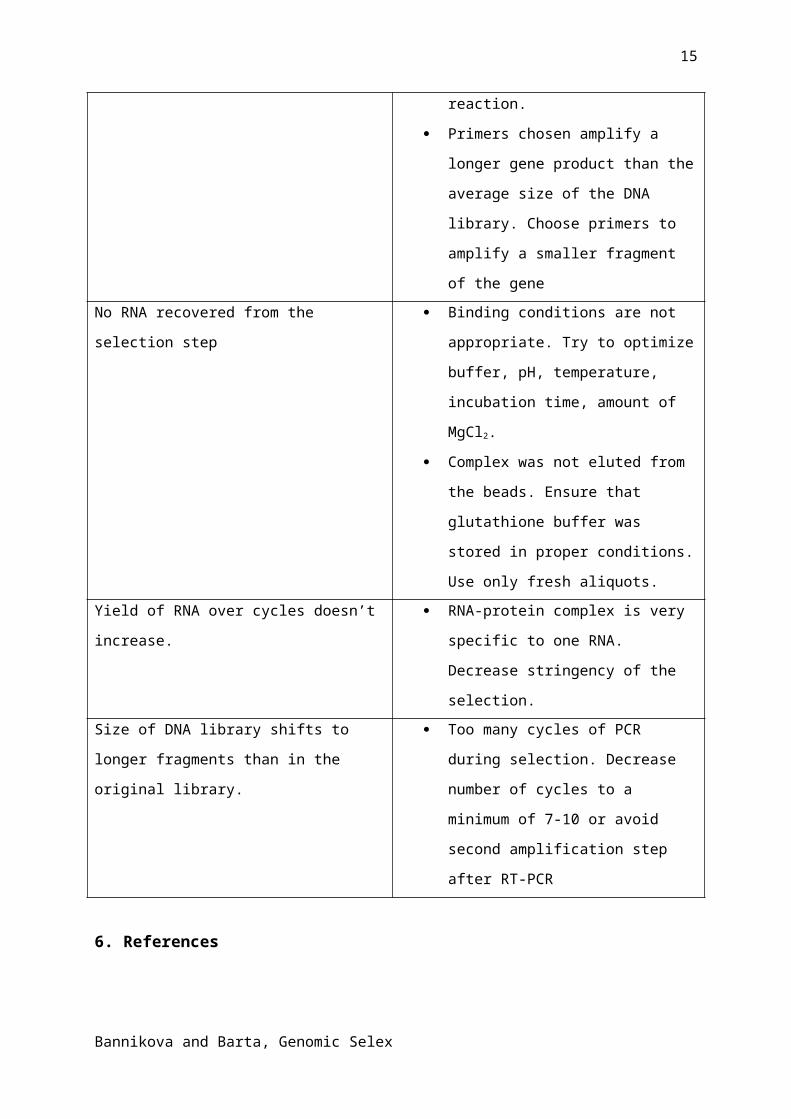
5. TroubleshootingProblem Reason + solution
No PCR product for a particular gene during
library verification step
Library is not representative. Check
the purity of genomic DNA and try to
increase the amount of the first pair of
primers during Klenow extension
reaction.
Primers chosen amplify a longer gene
product than the average size of the
DNA library. Choose primers to
amplify a smaller fragment of the gene
No RNA recovered from the selection step Binding conditions are not
appropriate. Try to optimize buffer,
pH, temperature, incubation time,
amount of MgCl2.
Complex was not eluted from the
beads. Ensure that glutathione buffer
was stored in proper conditions. Use
only fresh aliquots.
Yield of RNA over cycles doesn’t increase. RNA-protein complex is very specific
to one RNA. Decrease stringency of
the selection.
Size of DNA library shifts to longer
fragments than in the original library.
Too many cycles of PCR during
selection. Decrease number of cycles
to a minimum of 7-10 or avoid second
amplification step after RT-PCR
6. References
Bannikova and Barta, Genomic Selex

11
1. Djordjevic, M. (2007). SELEX experiments: new prospects, application and data analysis
in inferring regulatory pathways. Biomol. Engineering 24, 179-189
2. Gold, L. (1995). Oligonucleotides as re search, diagnostic, and therapeutic agents. J. boil.
Chem. 270 (23), 13581-13584.
3. Tuerk, C., Gold, L. (1990). Systematic evolution of ligands by exponential enrichment:
RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505-510
4. Schneider, D., Gold, L., Platt, T., (1993). Selective enrichment of RNA species for tight-
binding to Escherichia-Coli Rho-factor. FASEB J. 7, 201-207
5. Bunka, D.H., Stockley, P.G., (2006) Aptamers come of age-at last. Nat. Rev. Microbiol.
4(8), 588-596.
6. Gopinath, S.C. (2007). Methods developed for SELEX. Anal. Bioanal. Chem. 387, 171-
182.
7. Singer, B.S., Shtatland,T., Brown, D., Gold, L., (1997). Libraries for genomic SELEX.
Nucleic Acid Research 25 (4), 781-786
8. Lorenz, C., Pelchrzim, F., Schroeder, R., (2006). Genomic systematic evolution of
ligandsby exponential enrichment (Genomic SELEX) for the identification of protein-binding
RNAs independent of their expression levels. Nat.Protocols 1(5), 2204-2212
9. Kim, S., Shi, H., Lee, d., Lis, J.T., (2003). Specific SR protein-dependent splicing
substrates identified through genomic SELEX. Nucleic Acids Research 31(7), 1955-1961
10. Wen, J.-D., Gray, D. M., (2004). Selection of genomic sequences that bind tighly to Ff
gene 5 protein: primer-free genomic SELEX. Nucleic Acids Research 32(22), 182-192
11. Gullerova, M., Barta, A., Lorkovic, Z., (2006). AtCyp59 is a multidomain cyclophilin from
Arabidopsis thaliana that interacts with SR proteins and the C-terminal domain of the RNA
polymerase II. RNA 12(4), 631-643
Acknowledgements:The authors are grateful to M. Kalyna for fruitful discussions. This work was funded by the
EU FP6 Programme Network of Excellence on Alternative Splicing (EURASNET) [LSHG-CT-
2005-518238]; the Austrian Science Foundation (FWF: SFB-F017/10/11; DK W1207, RNA
Biology) and the Austrian GEN-AU program (ncRNAs).
Figure legends
Figure1. Library development
A. Ethidium bromide stained 1.2 % agarose gel shows size distributions of genomic
Arabidopsis thaliana DNA after isolation (lane 1) and after fragmentation via ultrasound (lane
2). Lane M1 is a lamda HindIII marker, lane M2 is 100 bp DNA size ladder (Fermentas).
Bannikova and Barta, Genomic Selex

12
B. Example of an autoradiogram showing size distribution of the fragmented DNA after
ligation of primer-adaptors. Lane 2 and 4 are control lanes showing radioactively labeled
reverse (Rran) and forward (Fran) primers, respectively. Lanes 1 and 3 are DNA from the first
and second Klenow’ extension reaction, respectively. Lane M is a φX174 DNA/HinfI labeled
size marker (Promega).
Figure 2. Recovery of RNA during the selection cycles with atCyp59 as a bait
Example of a typical Genomic SELEX experiment is showing increasing recovery of selected
RNAs with increasing numbers of selection cycles. The first 4 cycles are carried out in
relaxed selection conditions with a molar excess of RNA over protein of 3:1. These
conditions allow selection of ligands with low affinity. Further cycles of selection are
performed with excess RNA over protein of 10:1. In the penultimate step, a counter selection
step is included with GST-tag protein alone to assure exclusion of binders to the GST-tag
from the pool.
Bannikova and Barta, Genomic Selex

13
Bannikova and Barta, Genomic Selex

14
Bannikova and Barta, Genomic Selex
15
can you please put in these vendors?
Columbia barta
GEHealthcare barta
Fermentas barta
Finnzyme barta
Macherey-Nagel barta
Can you explain this for the glossary?Genomic SELEX barta
Bannikova and Barta, Genomic Selex

























