Embed Size (px)
Citation preview

GENETICALLY MODIFIED MYOGLOBIN AS A MIMIC FOR HEME ENZYMES
by
SUBHASH CHAND
Presented to the Faculty of the Graduate School of
The University of Texas at Arlington in Partial Fulfillment
of the Requirements
for the Degree of
DOCTOR OF PHILOSOPHY
THE UNIVERSITY OF TEXAS AT ARLINGTON
May 2013

Copyright © by Subhash Chand 2013
All Rights Reserved

iii
ACKNOWLEDGEMENTS
I would like to take this opportunity to express my deepest gratitude to my advisor, Dr.
Roshan Perera, for his excellent guidance, support, understanding, patience, inspiration and
constant encouragement which have been instrumental in providing my graduate research
every success. I would also like to thank the members of my advisory committee, Dr.
Subhrangsu S. Mandal and Dr. Jongyun Heo, for their many invaluable opinions towards my
research. I would like to express my deep gratitude to Dr. Krishnan Rajeshwar, and Dr. Daniel
W. Armstrong for being extremely generous with their precious advice and collaborative
research and guidance that kept my research focused. In addition, I would like to thank Dr. Brad
Pierce and Dr. Kayunta Johnson-Winters for allowing me to use the various facilities for my
research and assisting me to learn some very significant techniques.
I am very much indebted to Dr. Brian Edwards, Dr. Norma Tacconi and Dr. Muhammed
Yousufuddin, for sharing their copious knowledge and helping me with many of the
instrumentation parts used in my research. I would like to express my utmost thankfulness to
the Department of Chemistry and Biochemistry and the Graduate school of The University of
Texas at Arlington for supporting my graduate studies here.
It was a pleasure to share my doctoral studies with lab-mates like Dr. Sriparna Ray, Dr.
Sridev Mohapatra, Leticia Loredo, Yanbo Zhang, Sriyani Liyanage, Nam Tran, Brian Stamos,
Tuan Phan, Chris Wilson, Jimmy Saing and Buuh Yousuf. I owe my sincere appreciation to Dr.
Sriparna Ray for her continuous efforts in checking thesis, which required many hours.
Last but not least, I would like to thank my family, my father, Mr. Keshaw Prasad Yadav,
my mother, Mrs. Urmila Devi, my brothers Shrikant Yadav, Sandeep Yadav, Shashikant Yadav
and Manish Yadav, my cousins Mr. Ram Chander Yadav and Sunil Yadav and my friends of
UTA and beyond - Prasad, Piyush, Bibhu, Chetan, Swanand, Megha, Prashant, Aakash, Amit,

iv
Soumyava, Saket, Kirti, Karthik, Irene, Valay, Vaishali, Ajoy, Mrunmayi, Prudhvi and Ajinkya for
their support during my stay in UTA as well as in my research.
I would like to make a special mention of my uncle, Mr. Harendra Kumar Yadav who
has showered me with unconditional love and endless support necessary to succeed in life.
April 16, 2013

v
ABSTRACT
GENETICALLY MODIFIED MYOGLOBIN AS A MIMIC FOR HEME ENZYMES
Subhash Chand, PhD
The University of Texas at Arlington, 2013
Supervising Professor: Roshan Perera
This study addresses the mechanistic relationships between formation of reactive
oxygen species (ROS) and their catalytic oxidation functions in oxygenation and peroxygenation
reactions in heme enzymes. Even though both cytochrome P450s (CYP P450s) and
peroxidases have different catalytic activities, the involvement of common ROS (Compound 0
and Compound I) have been proposed. Therefore, to understand the generation and activation
of peroxide to form ROS, genetically-engineered myoglobin (Mb) mutants were created by
incorporating redox-sensitive 3-amino-L-tyrosine (NH2Tyr) or L-3, 4-dihydroxyphenylalanine
(DOPA) into its active site. Distal His 64 replaced with redox amino acids mutant Mb showed
excellent turnover rates for thioanisole and benzaldehyde oxidation, compared to the wild-type
protein. A 9-fold and 81-fold increase in activity, respectively, was observed in the presence of
hydrogen peroxide (H2O2). The presence of a redox unnatural amino acid in the active site
enhances the rate of compound I formation and stabilizes it to form one extra H-bond as
compared to the wild type (WT) Mb. This increased oxidation activity in mutants offer insights
into the role of the distal active site residues which are involved in acid-base catalysis and distal
charge relay “pull” effect in peroxide activation and formation of ROS in heme proteins.

vi
Furthermore, cyclic voltammetry (CV) and atomic force microscopy (AFM) were used to
investigate the importance of active site orientation of an immobilized protein for direct electron
transfer (DET) and electrocatalysis. While the bioconjugated wild-type myoglobin (WT Mb) was
immobilized on the modified gold electrode surface in a random multilayered fashion, the Ser 3
replaced with NH2Tyr in Mb mutant, was immobilized via a Diels-Alder reaction specific to the
NH2Tyr residue to form a homogeneous monolayer. Electrochemical calculations for the
number of surface exposed redox-sensitive molecules on the electrode surface (Γ) and
heterogeneous rate constant for DET were 1.29 × 10–10
mol cm–2
; 2.3 sec–1
for the WT Mb and
1.54 × 10–10
mol cm–2
; 1.3 sec–1
for the S3NH2Tyr Mb mutant, respectively. Electro-catalytic
conversion of thioanisole to sulfoxide products showed similar turnover frequencies (TOF)
around 1.9 × 103 sec
–1 (with 87% conversion) for the WT Mb, and 1.5 × 10
3 sec
–1 for the mutant
Ser 3-amino-L-tyrosine (S3NH2Tyr) Mb (with 81% conversion). These results indicate that site-
directed single monolayer immobilization affords almost the same number of surface exposed
Mb active sites as the random multilayer immobilization strategy, though the latter contains a
greater number of protein molecules on the electrode surface. The microarray concept
development provides novelty to study protein-protein interactions, drug discovery, and
biomedical and proteomic research.
Another aspect of this research was that the importance and significance of electron
rich functional groups on the electronic nature of heme center had been extensively explored. It
has been found that the axial His attached to the heme center plays a decisive role in dictating
the electron cloud near the heme center. When the axial heme is replaced by a residue with an
electron rich functional group like pNO2 L-Phenyl alanine (pNO2Phe), the electron density was
higher near the heme center. The oxidation state of the metal center and the nature of the
ligand play important role in the determination of back-bonding and direction of charge transfer.

vii
TABLE OF CONTENTS
ACKNOWLEDGEMENTS.............................................................................................................iii
ABSTRACT...................................................................................................................................v
LIST OF ILLUSTRATIONS..........................................................................................................xii
LIST OF FIGURES......................................................................................................................xii
LIST OF TABLES........................................................................................................................xv
LIST OF SCHEMES....................................................................................................................xvi
PREFACE..................................................................................................................................xvii
Chapter Page
1. INTRODUCTION……………………………………………………………………………1
1.1 Heme proteins……………………………………………………………………1
1.1.1 Myoglobin ……………………………………………………………. 5
1.2 Incorporation of unnatural (noncanonical) amino acids into proteins……… 7
1.3 Direct electron transfer and bioelectrocatalysis……………..…………….... 10
1.4 Dissertation overview ............................................................................…..12
2. MECHANISTIC INSIGHTS AND IMPROVED OXIDATION ACTIVITY USING
GENETICALLY-ENGINEERED MYOGLOBIN........................................................... 14
2.1 Introduction..................................................................................................14
2.2 Experimental section...................................................................................17
2.2.1 Chemicals....................................................................................17
2.2.2 Preparation of WT Mb and H64NH2Tyr Mb mutant
construct...…………………………………………………………...17
2.2.3 WT Mb and H64NH2Tyr Mb protein purification………….………18
2.2.4 WT Mb and H64NH2Tyr Mb protein analysis.....………….………18
2.2.5 Spectroscopy………………………………………………..……….20

viii
2.2.6 Preparation of oxyferrous complexes……………………………..20
2.2.7 Effect of hydrogen peroxide………………………………………...21
2.2.8 Catalytic Sulfoxidation of Thioanisole and Oxidation
of Benzaldehyde………………………………………………………21
2.2.9 Electrochemical Instrumentation…………………………………...21
2.3 Results and Discussion…………………………………………………………25
2.3.1 Genetic design rationale and characterization…………………...25
2.3.2 Catalytic activity……………………………………………………...28
2.3.3 ABTS Peroxidase assay…………………………………………….29
2.3.4 Electrochemical characterization…………………………………..30
2.3.5 Ligand binding study of WT and H64NH2Tyr Mb mutant…….….33
2.3.6 Mechanistic insights………………………………….......…………36
2.4 Conclusions…………………………………………………………………...…37
3. INTRODUCTION OF PEROXIDASE ACTIVITY IN MYOGLOBIN BY INCORPORATING AN UNNATURAL AMINO ACID AT THE DISTAL HISTIDINE POSITION..............................................................................................38
3.1 Introduction……………….………………………………………………….…38
3.2 Experimental procedure..............................................................................40
3.2.1 Chemicals………………………………………..............................40
3.2.2 Preparation of WT Mb and H64DOPA Mb mutant
constructs.......................................................................................40
3.2.3 WT Mb and H64DOPA Mb protein purification and analysis ......40
3.2.4 Spectroscopy...…………………………………………..................42
3.2.5 Effect of hydrogen peroxide........................................................42
3.2.6 Preparation of oxyferrous complex..............................................42
3.2.7 Preparation of CO, NO, cyanide, and azide adduct
samples..........................................................................................42

ix
3.2.8 Catalytic sulfoxidation of thioanisole and oxidation
of benzaldehyde..........................................................................43
3.2.9 Electrochemical instrumentation................................................43
3.3 Results and discussion.............................................................................43
3.3.1 UV-visible spectroscopic characterization of WT and
H64DOPA mutant…....................................................................43
3.3.2 Deoxyferrous and oxyferrous species of WT Mb and
H64DOPA Mb............................................................................46
3.3.3 CO, NO, cyanide, and azide adducts of WT and
H64DOPA Mb..........................................................................47
3.3.4 Redox potentials of WT and mutant Mb....................................49
3.3.5 Thioanisole sulfoxidation and benzaldehyde oxidation..............51
3.4 Conclusion..............................................................................................55
4. FAVOURABLE BINDING SITE ORIENTATION IN MYOGLOBIN FOR DIRECT ELECTRON TRANSFER, ELECTROCATALYSIS, AND MICROARRAY INVESTIGATION...........................................................................56 4.1 Introduction…………................................................................................56
4.2 Experimental Section……........................................................................60
4.2.1 Chemicals................................................................................60
4.2.2 Preparation of WT Mb and S3NH2Tyr mutant Mb construct......60
4.2.3 WT Mb and S3NH2Tyr Mb protein purification..........................60
4.2.4 UV-vis spectroscopy................................................................63
4.2.5 AFM measurements…………………………..............................63
4.2.6 Ligands adduct formation...............................................…......64
4.2.7 Preparation of microarray........................................................64
4.2.8 Electrochemical instrumentation and procedures………...........65
4.2.9 Electrode modification procédures…………………………… …65
4.2.9.1 Au/L-Cys/WT Mb………………………………………..65
4.2.9.2 Au/L-Cys/TEGDA/ S3NH2Tyr Mb…….………...........66

x
4.2.9.3 Electrocatalysis..........................................................66
4.3. Results and discussion……………………………………………….....……..66
4.3.1 Monolayer vs. multilayer covalent immobilization of Mb on Au………………………………………………………….....….66 4.3.2 Electrochemical characterization................................................73
4.3.3 Catalytic studies.........................................................................82
4.3.4 Microarray studies......................................................................83
4.3.4.1 Preparation of functional protein microarrays...........................83
4.3.5 Ligand binding studies................................................................88
4.4. Conclusion………………………………………………………………………88
5. ELECTRONIC NATURE INVESTIGATION OF HEME CENTER BY
MODIFYING PROXIMAL AND DISTAL BINDING SITE ENVIRONMENT OF
SPERM WHALE MYOGLOBIN WITH ELECTRON RICH RESIDUES.......................90
5.1 Introduction.................................................................................................90
5.2 Experimental Section..................................................................................92
5.2.1 Chemicals...................................................................................92
5.2.2 Preparation of WT, H64pNO2Phe and H93pNO2Phe
Mb mutant constructs.................................................................92
5.2.3 WT, H64pNO2Phe and H93pNO2Phe Mb purification and
analysis..........................................................................................93
5.2.4 Electronic absorption spectroscopy............................................94
5.2.5 Preparation of deoxyferrous Mb..................................................96
5.2.6 Preparation of oxyferrous complex.............................................96
5.2.7 CO and NO complex preparation................................................96
5.2.8 Azide and cyanide complex preparation.....................................96
5.3 Results and discussion.............................................................................96

xi
5.3.1 Ferrous-CO, ferrous-NO and ferrous O2 complexes of WT
and H64pNO2Phe and H93pNO2Phe mutants of myoglobin.........98
5.3.2 Ferric-cyanide and ferric-azide complexes of WT
and H64pNO2Phe and H93pNO2Phe mutants of Mb...................105
5.4 Conclusion.................................................................................................110
REFERENCES..........................................................................................................................111
BIOGRAPHICAL INFORMATION.............................................................................................128

xii
LIST OF ILLUSTRATIONS
Figures Page
1.1 Structures of heme a and heme b........................................................................................2 1.2 Structures of naturally occurring iron porphyrines…............................................................3
1.3 Overall structure of sperm whale Mb.......…………………..........................................….....6
1.4 A general method for genetically encoding unnatural amino acids in live cells…................8
1.5 The list of unnatural amino acids that have been introduced to the genetic codes of E. coli, yeast and mammalian cells.......................................................................9
1.6 Schematic representations of bioelectrocatalysis involving direct electron transfer in an enzyme........…..............................................................................…….......12
2.1 Mechanistic pathway for generation of the highly reactive Compound I from the ferric resting form in heme-proteins………………………………………………………..15
2.2 Schematic representation of the oxyferrous complex of WT Mb..…………………………..17
2.3 Coomassie-stained SDS-PAGE analysis of expression of WT Mb and H64NH2Tyr Mb.............................................…………………………………….………....….19
2.4 The mass spectrum (MALDI-TOF) of the H64NH2Tyr Mb protein…………………...……20
2.5 GC analysis of oxidation products of thioanisole and benzaldehyde ………………...…….24
2.6 Electronic absorption spectra of the H64NH2Tyr Mb.........................................................27
2.7 Effect of H2O2 on WT ans mutant Mb…………………………………………………………..28
2.8 Deoxyferrous and oxyferrous spectra of WT and mutant Mb…………………………….....28
2.9 Differential pulse voltammetry (DPV) of WT and mutant Mb in presence and absence of H2O2.…..........................................................................................................32
2.10 Characterization of WT and H64NH2 Mb ligand complexes…......................................35
2.11 Mechanism of oxidation of 3-amino-L-tyrosine which releases 2-electrons and two protons to produce quinone product in the presence of an oxidant……….……37
3.1 Schematic representation of the formation of compound I and compound II

xiii
in Mb and HRP..…............................................................................................................39
3.2 Analysis of WT and H64DOPA mutant Mb by SDS-PAGE and MALDI-TOF.........……....41
3.3 High-valent heme complexes of WT and H64DOPA Mb..................................................44
3.4 Stability of WT and H64DOPA Mb in presence of H2O2...........................................…………….......45
3.5 Deoxyferrous and oxyferrous spectra of WT and H64DOPA mutant Mb………….…....…46
3.6 Study of WT and H64DOPA Mb ligand complexes....…………………………….……........50
3.7 Reduction potential of WT and H64DOPA Mb in presence and absence of H2O2 at pH 7.0 and at pH 10.0................................................................................…....50
3.8 Gas-chromatography data and MS data of thioanisol and benzaldehyde oxidation with WT and H64DOPA....................................................................................52
3.9 Proposed acid-base catalytic mechanism for compound I formation in H64DOPA mutant Mb in presence of H2O2 .................................................................55
4.1 Graphical representation of WT and S3NH2Tyr mutant Mb attachment on modified gold electrodes................................................................................................................59
4.2 Coomassie-stained sodium SDS-PAGE analysis of S3NH2Tyr Mb and WT Mb..............61
4.3 Electrospray ionization-time of flight-mass spectrometry (ESI-TOF-MS) analysis of the incorporation of S3NH2Tyr into S3(TAG) Mb …………………..…….....…62
4.4 Tapping mode AFM topographic images for chemically modified Au surface of immobilized S3NH2Tyr Mb mutant and with WT Mb...................................................72
4.5 CV response of L-Cys-modified, WT Mb and S3NH2Tyr Mb mutant immobilized gold electrode in nitrogen-purged 100 mM phosphate buffer…...………………….….....73
4.6 CV and peak current response of WT and mutant Mb.....................................…...........74
4.7 Plot of peak potentials vs pH for WT Mb and S3NH2Tyr Mb..........................................78
4.8 Comparision of immobilized Mb with hemin on modified gold electrode.........................81
4.9 A microarray of five proteins on a single slide displays...................................................85
4.10 Spectroscopic characterization of high spin, low spin and ligand complexes of WT and mutant Mb..........................................................………....................…........87
5.1 Showing structure of heme b and heme c.......................................................................91
5.2 Schematic representations of WT and mutants Mb.........................................................92
5.3 Coomassie-stained SDS-PAGE analysis of expression of WT Mb, H64pNO2Phe and H93pNO2Phe mutants Mb.......................................................................................94

xiv
5.4 Characterization of ferric WT and mutants Mb.................................................................97
5.5 Deoxyferrous electronic absorption spectra of WT, H64pNO2Phe and H93pNO2Phe mutant Mb..................................................................................................98
5.6 The heme carbonyl complex showing dπ–pπ* backbonding…........................................99
5.7 Showing presence of electron rich functional group (NO2
-) at distal position……………...99
5.8 Comparative studies of carbonyl complexes of WT, H64pNO2Phe, and H93pNO2Phe ……………………………………......………………………………..…...102
5.9 Comparative studies of NO complexes of WT, H64pNO2Phe, And H93pNO2Phe...........................................................................................................108
.
5.10 Characterization of oxyferrous complexes of WT, H64pNO2Phe, and H93pNO2Phe Mb.................................................................................................104 5.11 Proposed structure of Fe (II) O2, Fe (II) CO and Fe (II) NO..........................................105 5.12 Comparative studies of cyanide complexes of WT, H64pNO2Phe, and H93pNO2Phe Mb....................................................................................................107 5.13 Characterization of azide oxyferrous complexes of WT, H64pNO2Phe, and H93pNO2Phe Mb....................................................................................................108 5.14 Two canonical forms of azide bound to heme iron of WT Mb and H64pNO2Phe Mb mutant.............................................................................................109
5.15 Two canonical forms of cyanide bound to heme iron of WT Mb and H64pNO2Phe Mb mutant ............................................................................................109

xv
LIST OF TABLES
Table Page
1.1 Biological functions of heme proteins…………………………………………………….........…4
2.1 Rate of sulfoxidation of thioanisole and benzaldehyde oxidation by WT Mb and mutant H64NH2Tyr Mb...………………………………………………………..……………29
2.2 ABTS oxidation reaction assays of Mb and its mutant in the presence of 5 mM H2O2. Kinetic values are based on the average of at least 2 determinations……..….30
2.3 Reduction potentials were measured at pH 7.0 and 10.0 in the absence and presence of 5 mM H2O2………………………………………………………………….….……33
3.1 Reduction potential of WT Mb, H64DOPA Mb mutant and DOPA only at pH 7.0 and pH 10.0 in presence and absence of H2O2 ……………...........…………………..………49
3.2 Rate of sulfoxidation of thioanisole and benzaldehyde oxidation by WT Mb and H64DOPA mutant Mb..………....……..…….………………………………………….....…..53
3.3 ABTS oxidation reaction assays of Mb and its H64 DOPA mutant Mb in the presence of 5 mM H2O2.......................................................................................................54
4.1 Comparison of heterogeneous electron transfer rates of myoglobin on gold electrodes…..77
4.2 Comparison of direct electrochemistry of Mb on various electrode systems………………..79
4.3 Electrocatalytic conversion of thioanisole to its oxidized form and turnover frequency (TOF) using bare, multilayered WT Mb-immobilized and monolayered S3NH2Tyr Mb……..82
5.1 The electronic absorption spectral features of the oxy, carbonyl, NO, cyanide and azide complexes with H64pNO2Phe Mb, H93pNO2Phe Mb mutants and WT Mb………….100

xvi
LIST OF SCHEMES
Scheme Page
4.1 Schematic representation of Au electrode modification for monolayered immobilization of S3NH2Tyr Mb mutant on the Au surface; see text for details………...…..68
4.2 Schematic representation of multilayered WT Mb immobilization on the modified gold electrode surface using EDC catalyzed bioconjugation reaction...................................70

xvii
PREFACE
This study “Genetically modified myoglobin as a mimic for heme enzymes” is submitted by
Subhash Chand to The University of Texas at Arlington for the degree of Doctor of Philosophy
(Ph.D.), and has the following kind contributions. Chapter 4 was published as “S. Ray, S.
Chand, Y. Zhang, S. Nussbaum, R. Krishnan, R. Perera, Immobilization of Myoglobin on the
Gold Electrode to Promote Direct Electron Transfer, Electrochimica. Acta. 2013, 99, 85-93”. In
this study Dr. Ray has carried out electrochemistry studies. Figures (4.1, 4.5, 4.6, 4.7, and 4.8)
and schemes (4.1 and 4.2) were kindly contributed by her. The microarrays studies in chapter 4
have been publisted as “B. Stamos, L. Loredo, S. Chand, T. Phan, Y. Zhang, S. Mohapatra, R.
Perera, Biosynthetic Approach for Functional Protein Microarrays, Anal. Biochem. 2012, 424,
114-23”.

1
CHAPTER 1
INTRODUCTION
1.1 Heme proteins
Heme proteins are the big family of proteins and are widely distributed in nature. Some
of these proteins act as oxygen storage (e.g., myoglobin) and some as an oxygen carrier (e.g.,
hemoglobin). While other heme proteins play important roles either in mediating electron
transfers (e.g., Cyt b5, Cyt c), or in catalysis (e.g., cytochrome P450, peroxidase, and catalase)
(1, 2). Along with heme, many enzymes contain other cofactors such as molybdopterin, copper,
flavin, and iron-sulfur cluster (1, 2). In these proteins (e.g., flavocytochrome b2, flavocytochrome
c3, p-cresolmethyl hydroxylase, sulfite oxidase, fructose dehydrogenase, alcohol
dehydrogenase, and cytochrome c oxidase), charge transfer is the main function of heme
domain. Iron porphyrin prosthetic groups are the most common among all heme proteins (1-3).
Different varieties of porphyrins such as heme a, heme b, heme c, heme d, heme d1, heme
P460, heme o, siroheme, and chloroheme are present in heme proteins (Figures 1.1 and 1.2,
and Table 1.1). These porphyrins have a common basic skeleton, but due to substitution at
various positions, they differ in their structural details. In heme proteins, nitrogen from the
porphyrin ring occupy four coordination sites out of six of the heme iron, leaving only two
positions available for further ligand interactions which strongly influence the reactivity and
redox potential of the heme protein (1-3).
In many heme and globin enzymes, penta-coordinated heme iron is found (1, 3). In
globins, the proximal histidine (His) residue act as fifth ligand while axial heme position remains
open for O2 coordination, while the sixth coordination position is used for substrate binding in
some heme enzymes (1, 3). The redox potential of the heme center depends on the variety of
proximal ligands while distal ligands are responsible for the catalytic reaction in general (2, 3).

2
The spin state of the heme center is governed by the nature of axial ligands (1, 2). If a
water molecule is present as the sixth ligand, the center is in a high spin state, while presence
of carbon monoxide (CO), nitric oxide (NO), O2 azide (N3–),
and cyanide (CN
–) as the sixth
ligand makes it in a low spin state (2, 3).
Heme type b is the simplest form of all known hemes and contains methyl group at 1, 3,
5, and 8 positions. At the same time 6 and 7 positions have two propionate groups and also 2
and 4 positions have vinyl groups (1). Energetically favored orientation of heme is the coplanar
structure, where vinyl group and the aromatic heme plane have the capability to interact with the
amino acids scaffold in the proteins (1, 2).
Heme a Heme b
Figure1.1 Structures of heme a and heme b.

3
Heme c Heme d
Heme d1 Heme o
Figure1.2 Structures of naturally occurring iron porphyrines.

4
Table 1.1 Listing biological functions, type, axial ligation and oxidation state of heme proteins (2).
Protein family
Protein function Type of heme Axial ligation Oxidation/spin state
Cytochromes c Electron transfer c His Fe(II) (S = 0)
Fe(III) (S =1/2)
Mb Hemoglobins O2 storage
O2 transport
b
b
His
Deoxy Fe (II) (S = 2)
Oxy Fe(II) (S = 0)
Peroxidases A-H2+H2O2 → A+2H2O b His Fe(III) (S = 5/2)
Catalases H2O2+ H2O2 → O2 + 2H2O b Tyr Fe(III) (S = 5/2)
P450 proteins RH +2e– + 2H
+ + O2 → ROH + H2O b Cys Fe(III) (S = 5/2)
Cytochrome bo quinol
oxidases
O2 + 4 Fe(II)cyt c + 4e– + 4H
+ → 4
Fe(III)cyt c + 2H2O o His Fe(III) (S = 5/2)
Hydroxylamine
oxidoreductase H2O + NH2OH → NO2
– + 4e
– + 5H
+ P 460 His
Fe(III) (S = 5/2)
Or (S = 3/2)
Nitric reductases (cyt d1) 2H
+ + NO2
– + e
– → NO + 2H2O
4H+ + O2
– + 4e
– → 2H2O
d1 His
His+ Tyr
Fe(III) (S = 1/2)
Sulfate Reductase 8H+ + NO2
– + 6e
– → NH4
+ + 2H2O Siroheme Cys Fe(II) (S =1 or 2)

5
1.1.1 Myoglobin
Myoglobin (Mb) is a relatively small heme protein (17 kDa) that is less affected by
environmental conditions when compared to most of the other heme proteins. Mb is a single-
chain globular protein of 153 amino acids, as found in the sperm whale, and serves as oxygen
storage (4, 5). It consists of eight separate right-handed α helices in its apoprotein (Figure 1.3).
Oxygen can bind at the distal site of proteins, directly to the iron atom of the heme. The axial
His (His 93) links protein to the heme center through a bond between the iron atom and the His
(2, 4). As shown in figure 1.2, a wide variety of amino acid residues are present near the binding
site of Mb. These residues play a critical role in determining the electronic nature of the heme
center and catalytic nature of the heme proteins (4-6). Studies shows that in Mb, the positions
such as 64, 68, and 93 are the most crucial residues (4). Due to its ability to prepare mixed
ligand complexes with different valences (ferric, ferrous, and ferryl species), Mb is often used as
a model to mimic other heme proteins such as cytochromes, peroxidases, catalases,
chloroperoxidases, and many more (5-10).
The role of the proximal ligand in controlling the reactivity and stabilizing the heme has
been a matter of interest for many years. In Mb, the role of proximal and distal His has been
assessed by replacing it with different residues (11-15) .
Mb is an oxygen storage heme protein; however, in presence of H2O2, it catalyzes a
variety of oxidative reactions such as sulfoxidation and epoxidation. Since Mb does not form a
stable Mb compound I in presence of H2O2, the rate of epoxidation and sulfoxidation are
significantly slow in Mb as compared to those of peroxidases (16-19). Watanabe and coworkers
have created mutations at 64, 43, and 29 positions of Mb, replacing respective amino acids with
Asp, His, and Trp, as well as with Leu and His in double mutants. They have been able to
introduce catalase monooxygenase and peroxidase activity in Mb by generating compound I in
different Mb mutants (16-27).

6
Figure 1.3 Overall structure of sperm whale Mb. (A) WT Mb showing heme center and His at 64 and 93 positions and (B) schematic representation of binding site of WT Mb.
A
B
His 93
His 64
Heme center
Heme
Leu 29
Val 68
Ile 107 His 64
Leu 89 His 97
His 93
Ser 92
Phe 43

7
Dawson and coworkers have created an H93G Mb cavity mutant and explored it as an
excellent structural model for His-ligated and other heme containing proteins (15, 28-32). They
have successfully compared and used Mb mutants as a model for the study of cytochrome f,
nitrite reductases, peroxidases, cytochrome P450, and many other heme proteins (15, 29, 33-
43). They have also extensively studied the ligand binding properties of heme proteins (6, 35,
43).
1.2 Incorporation of unnatural (noncanonical) amino acids into proteins
There are two strategies for the incorporation of noncanonical amino acids/unnatural
amino acids (UAA) into proteins: (1) site-specific and (2) residue-specific (44, 45). The site-
specific approach involves replacement of a single amino acid residue (Figure 1.4) while the
residue-specific method allows replacement of all or a fraction of one of the natural amino acids
by UAA (44-52). In the site-specific method, to deliver the analog in response to a stop or four-
bases codon, a heterologous tRNA/aminoacyl-tRNA synthetase pair is used (53). Amber codon
suppression in mammalian cells has been used to incorporate unnatural amino acids by using
the E. coli tRNA/aminoacyl-tRNA synthetase pair; none of the suppressor tRNA was charged by
any of the mammalian aminoacyl-tRNA synthetases (54). For the site-specific incorporation of
UAA, amber codon suppression is the most frequently used method (Figure 1.3) and to date
more than 50 UAA have been introduced in recombinant proteins using these methods (Figure
1.4) (44, 52). For the incorporation of structurally, chemically and spectroscopically amino acids,
Schultz and coworkers have produced orthogonal suppressor tRNA/aminoacyl-tRNA synthetase
pairs and used it to incorporate many UAA in recombinant proteins (55-60). By using both four
base codon and nonsense codon suppression, Dougherty and coworkers have successfully
incorporated two UAA into an ion channel protein (61-63). Four bases codons, commonly
known as frameshift suppression, have also been used for the site-specific insertion of UAA into
fluorophore proteins and in many more recombinant proteins (50, 63-65).

8
Figure 1.4 A general method for genetically encoded unnatural amino acids in live cells.

9
Figure 1.5 The list of unnatural amino acids that have been introduced to the genetic codes of E. coli, yeast, and mammalian cells.

10
Furthermore, for the site-specific incorporation of UAA, analogous five bases codon and
reassignment of sense codons can also be used, but fidelity of the method is much lower than
that of anti-sense or frameshift suppression methods (66-68). It is possible in the genetic code
that one amino acid can be coded by more than one codon, as there are 61 codons for 20
natural amino acids; this makes the genetic code highly degenerate. In E.coli, many amino
acids are coded by more than one codon. Phe is coded by UUU and UUC. Only one tRNA has
been assigned for both codons. The tRNA uses base pairing and wobble interaction to decode
UUC and UUU, respectively. tRNA synthetases from different organisms have been used for
the reassignment of UUU into host E.coli. The discriminating capability of synthetases between
unnatural and natural amino acids plays a crucial role in the incorporation of UAA and yield of
recombinant proteins.
Similar to post-translational modification, chemical methods for protein modification are
equally important and they have been used to enhance the performance of therapeutic proteins
and labeling with fluorescent dyes (69, 70). Highly reactive Cys and Lys residues have been
most commonly used in the process of protein modifications (70).
1.3 Direct electron transfer and bioelectrocatalysis
Electrochemical methods are very useful in determining the redox properties of proteins
and kinetics of electron transfer (71). Electrochemistry can be used as a powerful tool in the
biocatalytic study of a protein and in biosensors and biofuel cell production (72). In general, to
facilitate the electron communication in between electrode and redox-sensitive protein, small
soluble mediators were used (72, 73). Due to the possibility of their unspecific side reaction,
these mediators lead to erroneous results; to overcome this problem, direct electron transfer
(DET) methods were developed. For the electron communication, the DET methods do not
require a mediator between the enzymes and the electrodes. These methods at times have

11
their own limitation such as denaturation of protein upon adsorption on electrodes, which can be
solved by using different varieties of linkers and insulators.
The acceleration of the electrochemical reactions by a biological enzyme comes under
the realm of bioelectrocatalysis, and electrons can be transferred between an enzyme active
site and the electrode without any mediator. Electrodes for fuel cells and for the biosensors are
the two most common applications of bioelectrocatalysis. It was essential to achieve DET in
redox enzymes on solid electrode for its further development. In addition, to achieve the most
effective electron transfer, the importance of protein orientation on the solid electrode surface
has been investigated (71-73).
In principles, bioelectrocatalysis involves acceleration of the electrochemical reactions
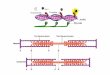
by biological catalysis (72). Figure 1.6 indicates the involvement of the enzymes in direct
bioelectrocatalysis. The communication of electrons with the electrode was provided by enzyme
catalyzing redox reactions. Oxidation and reduction reactions are the two halves of redox
reactions and one of these half reactions can be substituted in bioelectrocatalysis (72).
Heme is among those prosthetic groups which can communicate directly with the
electrode. Heme can be used both in formation of the enzyme active center as well as for the
communication with the redox mediator, thereby leading to communication with the electrode.
This makes heme one of the most important constituents of direct bioelectrocatalysis systems
and hence heme proteins are frequently used in bioelectrocatalysis (74, 75).

12
Figure 1.6 Schematic representations of bioelectrocatalysis involving direct electron transfer in an enzyme. One of the two coupled half-reactions is substituted by the electrochemical reaction.
1.4 Dissertation overview
Creation of genetically engineered Mb mutants by incorporating a non-cannonical
redox-active NH2Tyr into its active site for the mechanistic insight and oxidative activity has
been extensively explored in chapter 2. This study proposed the importance of the distal binding
site residues in acid-base catalysis and formation of reactive oxygen species (ROS) in heme
proteins.
In chapter 3, I reported the incorporation of another unnatural amino acid DOPA to a
specific position in the binding site of Mb. The mutant Mb protein could carry out the oxidation of
thioanisole and benzaldehyde and support the idea that the distal His is important for the
formation of compound I to exhibit peroxygenase activity in Mb.
In chapter 4, I reported monolayer and multilayer immobilization of Mb using two unique
immobilization techniques, onto a chemically-modified gold electrode surface. Further, using

13
DET method bioelectrocatalysis of thioanisole oxidation was carried out successfully with both
the protein immobilized electrodes. The results described in chapter 4 suggest the importance
of proper orientation of recombinant heme proteins containing UAA in bioelectrocatalysis and
effective electron transfer as comparison to the random multilayered WT Mb case. I have also
analyzed the protein-protein interactions of five different proteins which have been covalently
attached onto a solid support. Functional protein microarray concept development was
confirmed by catalytic activity assay using spectroscopic characterization, which will be of huge
importance in the field of drug discovery, and biomedical and proteomic research.
In chapter 5, I have summarized the importance and significance of electron rich
residues on the electronic nature of heme center. I have observed that the axial His attached to
the heme center plays a more crucial role in dictating the electron cloud near the heme center
and hence in formation of reactive species. When axial heme residue was replaced by electron
rich residue like pNO2Phe, it contributed to higher energy and red shift of the spectra due to the
higher electron density near the heme center. This study also explored the oxidation state of the
metal center, the nature of the ligand, the type of residues near the heme center that play an
important role in determining of backbonding, the direction of charge transfer, and the possibility
of hydrogen bonding. I also underlined that the size of UAA residue plays a crucial role in the
stability of ligand adducts.
Overall in this study I have used Mb as a model for the heme protein to study their
monooxygenase and peroxidase activity. No prior studies were reported using mutant protein
that utilizes its UAA acid residue to immobilize site specifically onto the electrode surface. I also
explored the DET and bioelctrocatalysis and importance of the proper orientation of protein
using Mb mutants as a model for heme proteins. This study also offers a similar orientation
concept while dealing with the protein microarray problems. The Mb mutants have also been
used to explore the role and effect of different varieties of residues near the heme center and
their effect in ligand binding spectra.

14
CHAPTER 2
MECHANISTIC INSIGHTS AND IMPROVED OXIDATION ACTIVITY USING GENETICALLY
ENGINEERED MYOGLOBIN
2.1 Introduction
Peroxidase enzymes couple substrate oxidation to H2O2 reduction (as generically
represented by Reaction 1). They are widely distributed throughout the plant and animal
kingdoms and are frequently isolated from bacteria, mold, and microorganisms (76-78). Their
main biological function is to act as antioxidants by protecting cells, tissues, and organs against
the oxidation of a variety of organic and inorganic compounds by H2O2, organic hydroperoxides,
peracids, or inorganic oxysalts, such as periodate or chlorate (79-85).
AH2 + H2O2 A + 2H2O (1)
Most heme-containing peroxidases (e.g., horseradish peroxidase, HRP) have a
proximal His and a polar distal pocket, with a non-ligated His as an H+ acceptor/donor and a
cationic arginine to stabilize developing negative charges during O-O bond cleavage.
Peroxidases transform the oxidizing power of H2O2 into high-valent, protein-bound oxidants. The
general peroxidase pathway (Figure 2.1) involves addition of H2O2 or other oxygen atom donors
([O]) to the ferric resting state (1) to form Compound I (5), a ferryl (FeIV
) radical species where
the π-cation radical is usually centered on the porphyrin (86). Compound I typically oxidizes
substrates by one electron to yield an organic radical and Compound II (6), a ferryl heme
complex (79, 87). Compound II then oxidizes another substrate molecule and returns to the
ferric state (1). A ferryl state-like peroxidase, Compound I, is thought to be the active species in
the catalytic cycle (88, 89). Involvement of a hydroperoxo-ferric intermediate (Compound 0) has
also been proposed (6, 82). Alternatively, addition of H2O2 to ferric-heme protein can generate
Compound 0 (4) via a peroxide “shunt” pathway. Heme-containing peroxidases such as HRP

15
form Compound I from Compound 0 using distal catalytic arginine amino acids to “pull” the O-O
bond apart heterolytically.
Figure 2.1 Mechanistic pathway for generation of the highly reactive Compound I (5) from the ferric resting form (1) in heme-proteins.
Lacking this distal machinery, WT Mb undergoes nearly equal amounts of homo- and
heterolytic cleavage upon H2O2 addition. Compound II, the stable product of homolytic cleavage
of the O-O bond of (6), is the prime product of Mb but has no catalytic activity. Previous
research shows that site-directed mutagenesis is a versatile technique to change the functional
properties of Mb. Thus the distal His (H64) has been replaced by amino acids such as Ala, Ser,
Leu and Asp, and the corresponding effect on the activity of Mb has been probed (90). Recent

16
reports evaluated the His93Gly (H93G) “cavity” mutant of Mb as a versatile scaffold for
modeling heme states (29, 35, 36). The difference in accessibility of the two sides of the heme
have frequently made it possible to prepare mixed ligand adducts in ferrous, ferric, as well as
ferryl, oxidation states with this mutant. In addition, the protection provided to the heme by the
protein environment allows for the preparation of stable oxyferrous and oxo-iron (IV) complexes
at near-ambient temperatures with sperm whale Mb. Similar studies have been carried out to
obtain spectroscopic characterization of homogeneous oxyferrous complexes of the cytochrome
P450 BM3 (CYP102) holoenzyme and heme domain (BMP) at –55 °C in presence of 70/30 (v/v)
glycerol/buffer cryosolvent (91).
Taking the above facts into account, I can effectively model the heme protein active site
using a natural probe. Here in this study, Mb mimic of peroxidase enzymes using unnatural
amino acids was used to characterize the active species in the catalytic cycle, investigate the
catalysis mechanism, and to model the heme protein active sites (44, 92). To this end, Mb, an
oxygen storage and carrier protein, was converted into a catalytically active peroxygenase using
a genetically incorporated redox-active NH2Tyr into its active site (Figure 2.2) (44, 93-95). The
mutant H64NH2Tyr Mb was then employed to investigate the structure-function relationship in
heme proteins (based on acid-base catalysis and distal charge relay effect).
A Mb mutant in which His 64 was replaced with NH2Tyr (H64NH2Tyr) showed high
turnover rates for thioanisole and benzaldehyde oxidation (9-fold and 81-fold), when compared
to WT, in the presence of H2O2. Our results indicate a possible situation where acid-base
catalysis of NH2Tyr coupled with distal “pull” effect via the hydrogen bonding network, play an
important mechanistic role to mimic peroxidase. Furthermore, peroxygenase activity of this
mutant exhibited remarkable stability against heme bleaching with the generation of active
catalytic species.

17
Figure 2.2 Schematic representation of the oxyferrous complex of Mb. (A) binding site of WT Mb. (B) active site of H64NH2Tyr, (PDB number 1MBO).
2.2 Experimental procedure
2.2.1 Chemicals
The gases (O2 and N2) were purchased from Air Liquide USA while NH2Tyr, sodium
dithionite (Na2S2O4), thioanisole, benzaldehyde, 30% H2O2, and 2,2’-azino-bis(3-
ethylbenzothiazoline-6-sulphonic acid) (ABTS) were purchased from Sigma-Aldrich USA, and
used without any further purification. All chemicals were at least analytical grade.
2.2.2 Preparation of WT Mb and H64NH2Tyr Mb mutant constructs
The WT Mb expression constructs, H64NH2Tyr Mb gene expression vector and
aminoacyl tRNA synthetase (aatRNA S) plasmids were gifts from Dr. Peter Schultz (The
Scripps Research Institute, La Jolla, CA USA). The H64NH2Tyr Mb expression vector and tRNA
synthetase plasmid were co-transformed in DH10B E. coli bacteria cells. The double antibiotic
resistant colonies were picked, grown, and the cell stocks were stored at –80 °C prior to protein
expression and purification.

18
2.2.3 WT Mb and H64NH2Tyr Mb protein purification
The H64NH2Tyr Mb was expressed in E. coli grown in glycerol minimal media (GMML)
containing 25% Luria Broth (LB) media and suitable antibiotics (ampicillin and tetracycline).
Both the WT Mb and mutated (H64NH2Tyr) Mb proteins were induced by adding 0.02%
arabinose to bacterial cultures when optical density at 600 nm (O.D600) was 0.5. Cells were
harvested by centrifugation at 13000 g and lysed in lysis buffer (Tris HCl buffer at pH 8.0) by
addition of RNase, Dnase, and lysozyme to the cell contents, followed by three freeze (liquid
N2) thaw (37 °C) cycles and 10 rounds of 40 sec sonications on ice. The crude lysate was
mixed thoroughly with Ni-NTA resin and the protein-bound nickel-nitrilotriacetic acid (Ni-NTA)
was collected in a column. Purified proteins were eluted from the column using an elute buffer
(300 mM NaCl, 250 mM imidazole, 50 mM phosphate buffer, pH 7.0). The purified WT Mb and
mutant proteins were stored at –80 °C until further use.
2.2.4 WT Mb and H64NH2Tyr Mb protein analysis
The sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis
were shown in figure 2.3. The mass of mutant H64NH2Tyr Mb protein was analyzed by matrix-
assisted laser desorption/ionization-time of flight (MALDI-TOF) and was observed to be 18397
Da, (Figure 2.4) which matched with the theoretically calculated mass.

19
Figure 2.3 Coomassie-stained SDS-PAGE analysis of expression of WT Mb and H64NH2Tyr Mb. (A) Lane 1, Molecular weight standards as indicated in kiloDalton (kDa); Lane 2, expressed WT Mb; Lane 3, expressed H64NH2Tyr Mb, (B) Lane 1, Molecular weight standards as indicated in kDa; Lane 2, expressed H64NH2Tyr mutant Mb in the absence (–) of 1 mM NH2Tyr; Lane 3, expressed H64NH2Tyr Mb in the presence (+) of NH2Tyr. No Mb protein was found by SDS−PAGE analysis in the absence of NH2Tyr.

20
Figure 2.4 The mass spectrum (MALDI-TOF) of the H64NH2Tyr Mb protein. A mass of 18397 Da matches the calculated mass for the Mb mutant. No WT Mb protein (predicted mass, 18356 Da) was observed.
2.2.5 Spectroscopy
UV-visible electronic absorption spectra of WT Mb and mutant Mb proteins were
acquired on a Varian Cary 50 Bio UV-visible spectrophotometer. The concentration of WT Mb
was determined using pyridine hemochromogen method (96) .
2.2.6 Preparation of oxyferrous complexes
The oxyferrous complexes were prepared in a chest freezer at –35 to –45 °C. The
protein sample was taken in 100 mM potassium phosphate buffer (pH 7.0) which contained
65% glycerol (v/v). The ferric protein was first degassed with N2 gas for 2 hr Then it was
reduced to a deoxyferrous species by addition of Na2S2O4 (20 mg/mL stock) under N2

21
atmosphere in a sealed cuvette at 4 °C. Pre-cooled O2 gas was bubbled into the cuvette for 60
s and the UV-visible spectra were recorded (Figure 2.8) as indicated in the previous study (32).
2.2.7 Effect of H2O2
The effect of H2O2 on ferric proteins was observed in the presence of 5 mM H2O2. The
protein concentration was 5 μM and heme bleaching of proteins was observed at 20 °C (Figures
2.6 and 2.7).
2.2.8 Catalytic sulfoxidation of thioanisole and oxidation of benzaldehyde
In the total volume of 500 μL, 100 mM potassium phosphate buffer pH 7.0, the 5 μM
protein (WT Mb or H64NH2Tyr mutant Mb) was added. To that 1 mM substrate, either
thioanisole or benzaldehyde was added. To initiate the reaction, 5 mM H2O2 was added to a
final volume of 500 µL. The reaction mixture was incubated for 1 hr After incubation the organic
products were extracted in dichloromethane. The solvent was reduced and the organic products
were analyzed by gas chromatography-mass spectrometry (GC-MS) (Figure 2.5).
2.2.9 Electrochemical instrumentation
Differential pulse voltammetry (DPV) was carried out using a single-compartment,
three-electrode cell at 22 °C on a CHI720C electrochemical analyzer (CH Instruments, Austin,
TX). The working electrode was polycrystalline gold, the counter electrode was a platinum wire,
and the reference electrode was Ag/AgCl/3.5 M KCl (BAS, West Lafayette, IN). All potential
values below are reported with respect to the Ag/AgCl reference electrode. The working
electrode surface was first polished on microcloth with alumina slurry suspension (0.05 μm),
then sonicated in ethanol, and finally rinsed thoroughly with Milli-Q water. The electrolyte
solution was 0.1 M K2HPO4/KH2PO4 buffer at a pH of 7.0. The solutions were deoxygenated by
bubbling nitrogen prior to each experiment. The concentration of H2O2 used was 5 mM. The

22
DPV measurements were recorded with pulse amplitude of 30 mV, step size set at 4 mV, pulse
width at 0.05 sec, and pulse period at 0.20 sec (Figure 2.9).

23

24
Figure 2.5 GC-MS analysis of oxidation products of thioanisole ans benzaldehyde. Conversion of thioanisole (a) to its sulfoxidation product (b) by 5 mM H2O2. (A), GC data of WT Mb; (B), GC data of H64NH2Tyr mutant Mb, (C), mass spectrum of thioanisole sulfoxide product, (D) mass spectrum of benzoic acid product, (E) and (F) show GC and MS data of oxidation of benzaldehyde (a) to benzoic acid (b) by 5 µM Mb and H2O2 for WT Mb and H64 H64NH2Tyr respectively. The reaction was carried out by adding 5 mM H2O2 to 5 µM WT or mutant Mb in 0.5 mL of 100 mM potassium phosphate buffer (pH 7.0) with 1 mM thioanisole or benzaldehyde at 25 °C.

25
2.3 Results and Discussion
2.3.1 Genetic design rationale and characterization
In the heme-containing peroxidases, the generation of active Compound I species
depends on precise delivery of two protons at the distal oxygen of the peroxo-ferric intermediate
(4). Another essential feature is the presence of the distal His and the Arg in the active site
pocket of the peroxidases (8, 21, 97). In HRP, the distal Arg influences the distal His to react
with H2O2 and form the active Compound I through an acid-base mechanism. But in WT Mb, the
distal His is not affected by any other distal amino acid and cannot induce the formation of
Compound I efficiently. Therefore, I decided to incorporate a redox-sensitive NH2Tyr amino acid
in place of the distal His. In the presence of an oxidant (e.g., H2O2), the redox-active NH2Tyr
amino acid should be able to facilitate an oxidation reaction which releases two electrons and
two protons (Figure 2.2) as well as to induce the formation of Compound I. Therefore, I
expected the H64NH2Tyr mutant Mb to behave as a considerably more potent peroxygenase
catalyst due to the increased oxygenase activity around the heme environment.
To facilitate the de novo design of Mb with unique peroxidase functionality, I have
genetically encoded NH2Tyr into Mb, replacing His 64 with the amber nonsense codon TAG into
a pBAD expression vector, using a previously reported technique. The method includes the
genetic incorporation of non-canonical amino acids into proteins using orthogonal
tRNACUA/aminoacyl-tRNA synthetase pairs in bacteria. The expressed H64NH2Tyr Mb showed
were analyzed by SDS- PAGE (Figure 2.3). Mass spectral analysis (MALDI-TOF) showed a
parent ion mass of 18397 Da as expected for the H64NH2Tyr Mb mutant (Figure 2.4).
The electronic absorption spectra of purified H64NH2Tyr mutant displayed a sharp
Soret absorption peak at 410 nm (visible peaks around 539 nm, 579 nm and 624 nm),
consistent with the aquo-ferric complex. Surprisingly, addition of 5 mM H2O2 did not show
significant Soret shift (λmax = 410 nm) or heme-bleaching for the NH2Tyr mutant Mb (Figure 2.6).

26
Figure 2.6 Electronic absorption spectra of the WT and mutant Mb. H64NH2Tyr Mb in the presence of 5 mM H2O2 (black solid line), in absence of H2O2 (blue dashed dot dot line) and WT Mb in the presence of 5 mM H2O2 (green dot line) and in absence of H2O2 (red dashed line). ε = Molar extinction coefficient.
However, there were isosbestic points in the spectra between the peaks in the
presence and absence of H2O2 with the mutant to indicate that a novel catalytic species had
been generated (Figure 2.6). As expected, the WT protein formed Compound II (the Soret
around 418 nm) with 5 mM H2O2 with significant heme loss within the first 15 min of the reaction
(Figure 2.7) (98).

27
Figure 2.7 Effect of H2O2 on WT and mutant Mb. Absorbance changes monitored over a 30 min period at λmax = 418 nm for WT Mb (blue dashed dot dot line) and λmax = 410 nm for H64NH2Tyr mutant Mb (black solid line) in the presence of 5 mM H2O2 and control (red dashed line). The spectra were measured in 100 mM potassium phosphate buffer, pH 7.0 at 20 °C with 10 μM protein.
In addition, the oxyferrous and deoxyferrous complexes of the H64NH2Tyr mutant and
the WT Mb were prepared at –35 to –45 °C. The UV-visible spectra were recorded (Figure 2.8),
which exhibited that the inherent characteristics of the native heme were intact in both the WT
and the H64NH2Tyr mutant Mb.

28
Figure 2.8 Deoxyferrous and oxyferrous spectra of WT and mutant Mb. The spectra of WT Mb deoxyferrous (blue dashed line), H64NH2Tyr Mb deoxyferrous (red dashed line), WT Mb oxyferrous (green dotted line) and H64NH2Tyr Mb oxyferrous (black solid line) species. The spectra were taken at –35 to –45 °C and the samples were examined in 60% glycerol, 100mM phosphate buffer at pH 7.0.
2.3.2 Catalytic activity
A number of substrates including thioanisole and benzaldehyde were successfully
oxidized to their products by the mutant. Sulfoxidation of thioanisole (Reaction 2) and oxidation
of benzaldehyde to benzoic acid (Reaction 3) by 5mM H2O2 for H64NH2Tyr mutant Mb showed
2.4 min–1
(9-fold higher activity compared to WT Mb) and 4.05 min–1
(81-fold higher activity than
WT Mb) turnover rates, respectively (Table 2.1). However, under the same conditions WT Mb
shows only 0.25 min–1
for sulfoxidation and 0.05 min–1
for the oxidation of benzaldehyde.

29
Table 2.1 Rate of sulfoxidation of thioanisole (Reaction 2) and benzaldehyde oxidation
(Reaction 3) by WT Mb and mutant H64NH2Tyr Mb. Kinetic values are based on the average of at least 2 determinations and the unit for rate is turnover per min.
Protein Km (µM) Vmax (µM min–1
)
WT Mb 19.3 25.6 [a]
H64NH2Tyr Mb 50.25 69.93
[a] This value is in agreement with data previously reported (26)
2.3.3 ABTS peroxidase assay
2,2’-Azino-bis (3-ethylbenzothiazoline-6-sulphonic acid) or ABTS has often been used
to estimate the reaction kinetics of enzymes like peroxidases. In the presence of H2O2 and
peroxidase.enzyme, it is converted to its radical cation. This blue colored radical cation is
utilized to indirectly identify the formation of the ferryl radical species in peroxidases (26, 99). In
order to detect the development of the reactive species in the WT and mutant H64NH2Tyr Mb,
the ABTS peroxidases assay was performed. The results are contained in Table 2.2. The

30
reaction mixture contained 1 µM protein, 5 mM H2O2 and ABTS. The concentration of ABTS
was varied from 0.02 mM to 2 mM (97, 100, 101).
Table 2.2 Peroxidase assay of WT and mutant Mb with ABTS in presence of H2O2. ABTS
oxidation reaction assays of Mb and its mutant in the presence of 5 mM H2O2. Kinetic values are based on the average of at least 2 determinations and the unit for rate is turnover per min.
Reaction has been done at pH 7.0 and at 20 °C.
Protein Km (µM) Vmax (µM min–1
)
WT Mb 19.3 [a]
25.6 [a]
H64NH2Tyr Mb 50.25 69.93
[a] This value is in consistent with data previously reported (99).
The ABTS radical cation formation was monitored at 730 nm. The Km and Vmax values
were calculated from Linewearver-Burk plots. The detection of the ABTS radical cation, in turn,
corroborated the peroxidase activity of the WT and mutant H64NH2Tyr Mb (101).
2.3.4 Electrochemical characterization
To understand the effect of the distal proton and electron delivery network via NH2Tyr
residue in Mb, we measured the reduction potentials (E°) for Mb and its mutant in the presence
of H2O2 at pH 7 as well as at pH 10 (Table 2.3) using differential pulse voltammetry (DPV). The
E° values confirmed that the primary amine and hydroxyl groups in NH2Tyr amino acid influence
the electronic nature of the iron at the heme active site. In the presence of H2O2 at pH 7.0, the
mutant (–360 mV) exhibited a 40 mV shift from the WT Mb (–320 mV), which is more than the
difference observed (27 mV) at pH 10.0 (Figure 2.9 and Table 2.3). Since a single hydrogen
bond results in a 25-60 mV change in the E° values (102, 103), the observed potential

31
difference between the mutant and the WT Mb is consistent with the presence of an additional
hydrogen bond for H64NH2Tyr mutant than the His-64 in WT Mb.

32
32
Figure 2.9 DPV of WT and mutant Mb in presence and absence of H2O2. The DPV response of 5 µM WT and mutant H64NH2Tyr Mb in nitrogen saturated 100 mM phosphate buffer and 10 mM KCl solutions (A and C: at pH 7.0; B and D: at pH 10.0) in absence (A and B) and in presence (C and D) of H2O2 at room temperature conditions. Pulse amplitude = 30 mV, step size = 4 mV, pulse duration = 0.05 sec and pulse period = 0.20 sec.

33
Table 2.3 Reduction potential of WT and mutant Mb. Reduction potentials were measured at pH 7.0 and 10.0 in the absence and presence of 5 mM H2O2. All reduction potentials are reported with respect to Ag/AgCl (3.5 M KCl) reference electrode.
Reduction Potential, E (V)
Protein
Without H2O2
With 5 mM H2O2
pH 7.0
pH 10.0
pH 7.0
pH 10.0
WT Mb
–0.232
–0.256
–0.260
–0.292
H64NH2Tyr Mb
–0.244
–0.256
–0.352
–0.320
A possible reason for the 27 mV shift with H2O2 at pH 10 (than the 40 mV at pH 7) is
that the redox-sensitive amino acid (NH2Tyr) can be more easily oxidized (pKa is ~10 for
hydroxyl group in NH2Tyr) to o-imino-quinone by releasing two electrons and two protons, which
would preferentially promote the formation of the heme high valent (ferryl) species in the Mb
mutant.
2.3.5 Ligand binding study of WT and H64NH2Tyr Mb mutant adducts
The replacement of distal His with redox residue like NH2Tyr gave us an important
insight of the nature of electronic absorption spectra. From Figure 3.6, I observed that after
addition of neutral ligands such as CO, NO and O2 to WT and mutant Mb, the Soret became
sharper at respective places. The CO adduct is linear and both of its π orbitals are empty,
which allows dπ-π orbitals to overlap in a perpendicular direction resulting in greater
overlapping. It provides the perfect condition for π backbonding, while under similar conditions
NO and O2 adducts have less π backbonding because they have one or two electrons in their
orbitals (104). Since mutant Mb form an extra hydrogen bond with the ligands, the adduct with

34
mutant Mb were stronger. The bigger size residue like NH2Tyr, if present near the active site,
also prohibits the release of ligands from the heme center and hence increases the stability.
CN– and N3
–, which are rich in electrons, also show the similar effect with lesser intensity as
compared to neutral ligands (Figure 2.10).

35
Figure 2.10 Characterization of WT and H64NH2 Mb ligand complexes. Electronic absorption spectra of (A) CO (B) NO, (C) cyanoferric and (D) ferric N3
– complexes of WT Mb (red solid line) and H64NH2Tyr (blue dashed line) Mb. The spectra were taken at 4 °C and the
samples were examined in 100 mM phosphate buffer at pH 7.0.

36
2.3.6 Proposed mechanical feature
The above spectroscopic and electrochemical characterizations demonstrate the role of
the redox-sensitive NH2Tyr in the introduction of peroxidase activity in Mb (Figure 2.11). When
H2O2 is present in the active site of the mutant Mb, the amine functional group of the NH2Tyr
can abstract a proton from the H2O2, thereby forming the Fe-O-OH moiety (Compound 0). This
in turn loses one molecule of H2O to form the active ferryl radical cation (Compound I). The
presence of the hydroxyl and the amine groups on the NH2Tyr is absolutely essential for this
step as it involves an acid-base catalysis mechanism. The distal “pull” effect via the hydrogen
bonding network facilitates heterolytic cleavage of the hydroperoxide-ferric O-O bond in
Compound 0. The substrate can now approach the ferryl (FeIV
) radical species become
oxidized, thereby regenerating the resting ferric state in the mutant Mb.
This indicates that the redox-properties of NH2Tyr can be exploited to generate the
elusive ferryl radical cation (Compound I) in the mutant Mb. Hence, H64NH2Tyr Mb
peroxygenase activities as well as isolation of catalytically active species in this mutant will
significantly increase our understanding of how nature utilizes the heme iron cofactor and the
protein scaffold around the active site in essential biological transformations involving peroxides
and O2.

37
Figure 2.11 Mechanism of oxidation of NH2Tyr which releases 2 electrons and two protons to produce quinone product in the presence of an oxidant [O], such as H2O2.
2.4 Conclusions
In this chapter, I report the creation of a genetically engineered Mb mutant by
incorporating a non-canonical redox-active NH2Tyr into its active site. The replacement of His in
the distal His 64 with NH2Tyr mutant Mb provided an insight into the role of the distal binding
site residues in acid-base catalysis and distal charge relay “pull” effect in peroxide activation
and formation of ROS in heme proteins. The H64NH2Tyr mutant Mb showed high turnover rates
for thioanisole oxidation (9 times) and benzaldehyde oxidation (81 times) when compared with
the WT Mb, in the presence of H2O2. The ligand binding studies also explored the importance of
redox residues (e.g., NH2Tyr) near the heme center.

38
CHAPTER 3
INTRODUCTION OF PEROXIDASE ACTIVITY IN MYOGLOBIN BY INCORPORATING
UNNATURAL AMINO ACIDS AT THE DISTAL HISTIDINE POSITION
3.1 Introduction
Among the various heme containing metalloproteins, Mb is found in the muscle tissue
of most vertebrates and all mammals (2). It is an oxygen binding single chain globular protein
with 153 amino acids (sperm whale Mb) (1, 2, 5). The binding site of Mb contains iron-heme as
the prosthetic group (105). Since Mb has been extensively characterized, it is frequently used
as a model to study the structure and function of the heme proteins. X-ray crystallography,
NMR, and EPR, have been successfully used to study the structure and function of the Mb
protein (106-108). The Mb gene of many organisms, including human and sperm whale, have
been cloned, and heterologous expression of recombinant protein in E. coli has been carried
out effectively (10, 109-112).
While studying other heme-proteins, like horseradish peroxidase (HRP) and
cytochrome c peroxidase (CcP), it has been revealed that the presence of distal His is very
critical for the formation of compound I and peroxidase activity (Figure 3.1) (106-108, 113-115).
The successful oxidation of thioanisole and benzaldehyde by HRP and CcP has demonstrated
their peroxidase property. In contrast, the rate of formation of compound I in Mb is quite slow
and the myoglobin compound I (Mb Cpd I) is also very unstable, hence the WT Mb cannot
catalyze sulfoxidation of thioanisole or benzaldehyde oxidation significantly. To carry out these
reactions, it is essential to increase the rate of compound I formation and provide a suitable
environment at the active site to prohibit its decay to compound II. The compound II is a

39
catalytically inactive and stable iron-oxygen species, which effectively stores the oxygen in the
tissues.
A Binding site of WT Mb
B Active site of HRP
Figure 3.1 Schematic representation of the formation of compound I and compound II in Mb and HRP. (A) Formation of compound II by homolytic cleavage of O-O bond in Mb; (B) Compound I formation in HRP by heterolytic cleavage.
In order to increase the rate of formation of compound I and to stabilize it, we have
incorporated an unnatural amino acid, DOPA, replacing the distal His in the binding site of Mb.
This H64DOPA Mb mutant was constructed on the basis of accommodating a redox-sensitive
amino acid in the binding site of Mb. Oxidation of thioanisole and benzaldehyde were carried

40
out in the presence of WT and mutant Mb to monitor their peroxidase property. Herein, we
report that the H64DOPA Mb mutant exhibited significantly high peroxidase activity as
compared to the WT Mb.
Furthermore, I have studied the electron rich and neutral ligand adducts of mutant and
WT Mb to understand the effect of redox amino acids such as DOPA, which is capable of giving
electrons to heme center and to ligand when present near the active site. It also was interesting
to explore the role played by hydrogen bonding favoring residues on the stability of ligand
adducts.
3.2 Experimental procedures
3.2.1 Chemicals
The gasses (O2, NO, CO and N2) were purchased from Air Liquide while DOPA,
Na2S2O4, sodium azide, potassium cyanide, thioanisole, benzaldehyde, and 30% H2O2 were
purchased from Sigma-Aldrich USA, and have been used without any further purification. All
chemicals were of analytical grade.
3.2.2 Preparation of WT Mb and H64DOPA Mb mutant constructs
The WT Mb and H64DOPA Mb expression constructs were made by the following
previous method (30). The H64DOPA Mb expression vector and tRNA synthetase plasmid have
been co-transformed in DH10B E.coli. The double antibiotic resistant colonies were picked,
grown, and the cell stocks were stored at –80 °C prior to protein purification.
3.2.3 WT Mb and H64DOPA Mb protein purification and analysis
The H64DOPA Mb was expressed in E. coli that was grown in GMML containing 25%
LB media and suitable antibiotics (ampicillin and tetracycline). Both the WT and mutated

41
(H64DOPA) Mb proteins were expressed, purified and analyzed as described in chapter 2
(Figure 3.2).
Figure 3.2 Analysis of WT and H64DOPA mutant Mb by SDS-PAGE and MALDI-TOF. (A) Lane 1, expressed WT Mb; Lane 2, expressed H64DOPA Mb; Lane 3, Molecular weight standards as indicated in kDa (B) MALDI-TOF of the H64DOPA mutant Mb protein. A mass of 18397 Da matches the calculated mass for the Mb mutant. No WT protein (predicted mass, 18356 Da) was observed.

42
3.2.4 Spectroscopy
UV-visible electronic absorption spectra of the WT Mb and mutant Mb proteins were
taken using Varian Cary 50 Bio UV-visible spectrophotometer. The concentration of WT and
mutant Mb was determined by heme chromatogen method (96).
3.2.5 Effect of H2O2
The effect of H2O2 on ferric proteins has been observed in the presence of 5 mM H2O2.
The protein concentration was 5 μM and heme bleaching of proteins was observed at 20 °C.
3.2.6 Preparation of oxyferrous complex
The oxyferrous complexes were prepared in a chest freezer at –45 to –55 °C. The
protein sample was taken in 100 mM potassium phosphate buffer (pH 7.0) which containing
65% glycerol (v/v). The ferric protein was first degassed with N2 gas for 2 hr Then, it was
reduced to deoxyferrous by addition of Na2S2O4 (20 mg/mL stock) under N2 atmosphere in a
sealed cuvette at 4 °C. Pre-cooled O2 gas was bubbled into the cuvette for 60 sec and the UV-
visible spectra were recorded.
3.2.7 Preparation of CO, NO, CN– and N3
– adduct samples
The ferrous-CO adducts were generated by gentle bubbling of CO in deoxyferrous
enzymes for 30 sec at 4 °C in 0.1 M potassium phosphate buffer pH 7.0. To prepare the
ferrous-NO adducts, minimal amount of buffer saturated with NO gas was added to the
deoxyferrous enzymes under N2 at 4 °C in 0.1 M potassium phosphate buffer pH 7.0. The ferric
N3– and cyanoferric adducts were formed by addition of minimal amounts of NaN3 and KCN
from their respective stocks (40 mM NaN3 and 1 M KCN) to the degassed ferric proteins. The
ferric proteins were in 100 mM potassium phosphate buffer pH 7.0 and the temperature was 4

43
°C. The deoxyferric enzymes were prepared by bubbling of N2 gas in the ferric enzymes for at
least 30 min.
3.2.8 Catalytic sulfoxidation of thioanisole and oxidation of benzaldehyde
The reactions were performed and the products were analyzed as described in chapter
2 and the data were shown in figure 3.8 and table 3.2.
3.2.9 Electrochemical instrumentation
DPV was carried out following the method as described in chapter 2 and the data were
shown in figure 3.7.
3.3 Results and discussion
The inherent property of Mb is to store and transport oxygen. The WT Mb forms a
stable and catalytically inert oxy-complex. It has been reported that the His 64 stabilizes the
sixth ligand water molecule through hydrogen bonding (110, 116, 117).
3.3.1 UV-visible spectroscopic characterization of WT and H64DOPA mutant Mb
The mutant protein and WT Mb exhibited similar electronic spectra as observed for the
ferric heme species. The maximum absorbance in Soret band was found to be 409 and 411 nm
for the WT and mutant Mb, respectively (Figure 3.3). In the case of WT Mb, small peaks were
observed at 632 nm, 580 nm, 544 nm, and 507 nm, while for H64DOPA Mb, only two peaks
were observed at 632 nm and 530 nm in addition to the 411 nm peak. His 64 has been replaced
by DOPA and the unnatural amino acid was expected to stabilize it further by forming an extra
hydrogen bond. Therefore, I have genetically encoded DOPA into Mb, replacing His 64 with the
amber nonsense codon TAG into a pBAD expression vector, using a previously reported
technique. The method includes the genetic incorporation of non-canonical amino acids into

44
proteins using orthogonal tRNACUA/aminoacyl-tRNA synthetase pairs in bacteria. Mass spectral
analysis (MALDI-TOF) showed a parent ion mass of 18597 Da as expected for the H64DOPA
Mb mutant (Figure 3.2 and Figure 3.3).
Furthermore, the H64DOPA Mb mutant showed remarkable tolerance to H2O2. Even at
25 mM concentration of H2O2, heme bleaching was not observed to a significant extent (Figure
3.4). The UV-visible absorption spectrum of H64DOPA Mb mutant in presence of H2O2 also
exhibited a Soret band at 411 nm similar to that of ferric resting species. However, the
isosbestic points present in the spectra indicate that an active catalytic species had been
formed (Figure 3.3). Under similar conditions, when H2O2 was added to the WT Mb, hemin was
bleached out and it formed an inactive compound II species as expected. This fact was verified
by the presence of a Soret at 418 nm (Figure 3.4) in the absorption spectrum of the WT Mb.
Figure 3.3 High-valent heme complexes of WT and H64DOPA Mb. Electronic absorption spectra of the H64DOPA Mb in presence (black solid line), in absence (red dashed line) and WT Mb in presence (green dotted line) and in absence (blue dashed dot line) of 5 mM H2O2.

45
Figure 3.4 Stability of WT and H64DOPA Mb in presence of H2O2. Absorbance changes monitored over a 30 min period at λmax = 418 nm for WT Mb (blue dashed), λmax = 410 nm for H64DOPA mutant Mb (black dash dot line) in the presence of 5 mM H2O2.and control protein without H2O2 (red solid line) The spectra were measured in 100 mM potassium phosphate buffer, pH 7.0 at 20 °C with 10 μM protein.

46
3.3.2 Deoxyferrous and oxyferrous species of WT Mb and H64DOPA Mb
The substrate free ferric (low spin) of WT Mb and H64DOPA Mb have been reduced to
ferrous (high spin) with the Na2S2O4 in 65% (v/v) glycerol under N2 at 4 °C. The deoxyferrous
protein shows a shift in the Soret from 409 to 430 nm and from 411 to 432 nm in WT Mb and
mutant Mb respectively (Figure 3.5). In the visible region, a peak at 558 nm was observed in the
deoxyferrous WT Mb, while in mutant H64DOPA Mb, peaks were observed at 560 nm and 515
nm (118).
Figure 3.5 Deoxyferrous and oxyferrous spectra of WT and H64DOPA mutant Mb. The absorption spectra of WT Mb deoxyferrous (blue dashed dot line), H64DOPA Mb deoxyferrous (red dashed line), WT Mb oxyferrous (green dotted line) and H64DOPA Mb oxyferrous (black solid line) species. The spectra were taken at –35 to –45 °C and the samples were examined in 65% glycerol, 100 mM phosphate buffer at pH 7.0.
In the oxyferous WT Mb and mutant H64DOPA Mb, Soret bands at 416 nm and 418 nm
were observed, respectively. In the visible region, peaks at 581 nm and 540 nm for the WT Mb
and at 580 nm and 536 nm for the mutant H64DOPA have been recorded. Since DOPA can

47
give electrons to the heme and can also participate in hydrogen bonding, the Soret (λmax = 418)
has been observed to be sharper in the mutant protein as compared to WT mb. (Figure 3.5)
3.3.3 CO, NO, CN– and N3
– adducts of WT and H64DOPA Mb
Figure 3.6 clearly indicates that when CO is bound to the enzyme, the Soret (λmax =
422) became sharper for the ferrous enzyme bonded with CO. As Fe-CO is a linear adduct and
both the π orbitals are empty, it allows dπ-π orbitals to overlap in a perpendicular direction,
making the sharper Soret (104). Under similar conditions addition of NO and O2, (because of
having one or two electrons in their orbitals) reduce the chances of backbonding. These
adducts were stronger in the case of mutant protein because of the formation of extra hydrogen
bonding. The electron rich ligands such as CN– and N3
– have similar effects with less intensity
as π backbonding was observed less in these cases (32, 119).

48

49
Figure 3.6 Study of WT and H64DOPA Mb ligand complexes. The electronic absorption spectra of (A) CO (B) NO, (C) cyanoferric and (D) N3
– complexes of WT Mb and H64DOPA Mb. The spectra were taken at 4 °C and the samples were examined in 100mM phosphate
buffer at pH 7.0.

50
3.3.4. Redox potentials of WT and mutant Mb
Both the WT and the mutant H64DOPA Mb were further analyzed to obtain their redox
potential. The electrochemistry of the protein solutions were probed by differential pulse
voltammetry. The redox potential of WT and mutant H64DOPA Mb protein in the absence of
H2O2 WT were found to be the same, i.e., –232 mV, while in the presence of H2O2 it was
observed to be –260 mV and –300 mV, respectively (Figure 3.7 and Table 3.1). This
observation was expected as H2O2 was bonded to the heme center (120). The binding of H2O2
to the heme center replaced the H2O molecule, which was the sixth ligand, and a high spin
ferric state was formed. The difference in the reduction potential also removed the energy
barrier of electron transfer to the heme center (79, 120). Upon addition of H2O2 in the mutant
H64DOPA Mb protein, the reduction potential had decreased, which clearly indicates that an
extra hydrogen bond is formed at the active site of the mutant protein when compared to the
WT Mb binding site (121).
Table 3.1 Reduction potential of WT Mb and H64DOPA Mb mutant at pH 7.0 and pH 10.0 in presence and absence of H2O2 as obtained from DPV. All protein concentrations were 5 µM and the potentials were recorded vs. Ag/AgCl (3.5 M KCl).
Reduction Potential, E (V)
Protein
Without H2O2
With 5mM H2O2
pH 7
pH 10
pH 7
pH 10
WT Mb
–0.232
–0.256
–0.260
–0.292
H64DOPA Mb
–0.232
–0.244
–0.300
–0.268

51
Figure 3.7 Reduction potential of WT and H64DOPA Mb in presence and absence of H2O2 at pH 7.0 and at pH 10.0. The DPV response of 5 µM WT and mutant H64DOPA Mb in nitrogen saturated 100 mM phosphate buffer and 10 mM KCl solutions (A and C: at pH 7.0; B

52
and D: at pH 10.0) in presence (A and B) and absence (C and D) of H2O2 at room temperature conditions. Pulse amplitude = 30 mV, step size = 4 mV, pulse duration = 0.05 sec and pulse period = 0.20 sec.

53
3.3.5 Thioanisole sulfoxidation and benzaldehyde oxidation
As reported earlier (98, 122), WT Mb reacts with H2O2 and forms ferryl heme (FeIV
=O)
equivalent to compound II. Compound I has never been isolated, as it is highly unstable, which
makes peroxidase activity of the WT Mb very low. It was observed that, in the presence of H2O2,
WT Mb converts only 14% (Figure 3.8) thioanisole to sulfoxide with the turnover rate of 0.25
min–1
which matches with the literature reports (Table 3.2). But under identical conditions,
H64DOPA Mb converts 97% (Figure 3.8) thioanisole to its sulfoxide with a turnover rate of 2.5
min–1
. Thus the turnover rate of sulfoxidation is 10 times higher in the case of the mutant
H64DOPA Mb than that of WT Mb.

54
Figure 3.8 GC analysis of thioanisol and benzaldehyde oxidation. Conversion of thioanisole (a) to its sulfoxidation product (b), (A) WT Mb, (B) H64DOPA Mb. (C) and (D) show oxidation of benzaldehyde (a) to benzoic acid (b) by WT Mb and H64DOPA Mb respectively. The reaction mixture contained 5 µM Mb and 5Mm H2O2.

55
Furthermore, H64DOPA Mb converts 80% (Figure 3.8) benzaldehyde to benzoic acid
with 2.5 min–1
turnover rate (Table 3.2) while under similar conditions WT Mb converts 2%
(Figure 3.8) benzaldehyde to benzoic acid with 0.01 min–1
turnover rate (Table 3.2). The
turnover rate of benzaldehyde to benzoic acid was 54 times higher by mutant H64DOPA Mb as
compared to the WT Mb.
Table 3.2 Rate of sulfoxidation of thioanisole reaction (Rxn 1) and benzaldehyde oxidation reaction (Rxn 2) by WT and H64DOPA mutant Mb.
Protein
Rates (min–1
)
Rxn 1
Rxn 2
WT Mb
0.25
0.05
H64DOPA Mb
2.5
2.67
In addition to the above experiments, ABTS oxidation reaction assays of Mb and its
H64DOPA mutant Mb in the presence of 5 mM H2O2 were also carried out at pH 7.0 and at 4
°C. The kinetic values and turnover factor for the WT and the mutant H64DOPA Mb were
calculated based on the data obtained. The H64DOPA mutant Mb exhibited a Km of 0.67 ×103
µM, while that for the WT Mb was 19.3 µM, which corresponded to the literature value. The Vmax
for the WT and the mutant was observed to be 25.6 and 15.2 µM min–1
, respectively (Table 3.3)
(100).

56
Table 3.3 Peroxidase assay of WT and H64DOPA mutant Mb with ABTS in presence of H2O2. ABTS oxidation reaction with WT Mb and its H64DOPA mutant Mb in the presence of 5 mM H2O2. Kinetic values are based on the average of at least 2 determinations and the unit for rate is turnover per min. Reaction has been done at pH 7.0 and at 4 °C.
[a] This value is in agreement with data previously reported
Significantly enough, under similar conditions, epoxidation of styrene did not form
substantial amounts of the epoxide product (result not shown). In order to explain the observed
catalytic reactions and the non-formation of epoxides, we propose that the formation of
compound I was achieved through general acid base catalysis. The hydroxyl groups of the
DOPA facilitate the mechanism (Figure 3.9) as they are closer to the heme center and can form
a hydrogen bond with H2O2. The extra hydrogen bond is instrumental in stabilizing the
compound I species in the mutant H64DOPA Mb, which further generates the peroxidase
activity in the mutant Mb.
Protein
Km (µM)
Vmax (µM min–1
)
WT Mb
19.3 [a]
25.6 [a]
H64DOPA Mb 0.67 × 103 15.2

57
Figure 3.9 Proposed acid-base catalytic mechanism for compound I formation in H64DOPA mutant Mb in presence of H2O2.
3.4 Conclusion
In this chapter we report the incorporation of an unnatural amino acid to a specific place
in the binding site of Mb. The mutant H64DOPA Mb is an active and stable protein. It could
carry out the oxidation of two substrates, namely, thioanisole and benzaldehyde with 20 to 40
fold higher catalytic rate in mutant Mb as compared to WT Mb. In addition, our observations
support the idea that the distal His is important for the formation of compound I. In order to
exhibit peroxygenase activity in Mb, the mutation of the distal His with an unnatural amino acid
assisted in substrate binding at the active site and enhanced the compound I formation. In
addition, the ligand binding studies demonstrate electron deficient ligands exhibit sharper Soret
than the electron rich ones for the mutant protein.

58

59
CHAPTER 4
FAVOURABLE BINDING SITE ORIENTATION IN MYOGLOBIN FOR DIRECT ELECTRON
TRANSFER, ELECTROCATALYSIS AND MICROARRAY INVESTIGATION
4.1. Introduction
Much attention has been focused in recent years on the immobilization of
proteins on electrode surfaces (123-126). Such studies are driven by possible applications in a
broad range of chemical, biological, and medical technologies spanning bioelectrocatalysis,
biomaterials, and biosensors (127-129). Further, immobilized protein assemblies provide a
versatile platform for designing mechanistic studies on heterogeneous electron transfer
between a cofactor and an active center of a protein. However, direct electron transfer (DET)
between proteins and the electrode surface, which can provide new mechanistic insights, is
difficult to achieve, mainly due to the lack of good immobilization techniques. Most of the current
immobilization techniques show denaturation of the protein on adsorption on the electrode
surface. Unfavorable active site orientation of the protein during random adsorption process on
the electrode also hinders DET. Strategies to circumvent these difficulties include the use of
organic mediators (e.g., dye molecules, conducting polymers, etc) and protein films which again
can lead to denaturing of the proteins (130-133). In the past, various immobilization methods
have been employed to immobilize proteins on the electrode surface. For example, non-
covalent attachment of proteins (134, 135), entrapment in porous matrix (136-138),
nanoparticles (139, 140), and sol gels (141-143) have been extensively studied. There are also
some reports of covalent immobilization of proteins on the surface of the electrodes, where
either chemically modified protein (which often leads to protein denaturation due to harsh
chemical treatment) or the functional groups (such as Cys, Arg, Lys, Asp, Glu, etc.) available on

60
the surface of the proteins have been utilized to covalently attach the protein onto the suitably
modified electrode surface (123, 144-146).
Mb is a water soluble heme containing protein, which is present in all mammals and
vertebrates, and has been used extensively for investigating electron transfer and
electrocatalysis with the protein immobilized on an electrode surface. Its main function is to
store oxygen and enhance diffusion of oxygen in the muscle. This globular protein has a single
153 or 154 amino acid chain as in sperm whale, with a molecular weight of ~17 kDa (147). It is
a versatile protein with high tolerance for chemical and mechanical environments and can be
easily expressed and purified in large quantities in E. coli. Another important feature is that it
can accommodate various mutations without any adverse effects on its conformational and
functional properties (19, 24, 148). Mb contains a single iron-porphyrin center (heme b) that can
accommodate ferrous, ferric or ferryl oxidation states within this heme active site moiety. For
these reasons Mb has become an accepted natural model for exploring redox functional
properties and electron transfer reactions in electrochemistry (149-152). For example, co-
adsorption of Mb with surfactants like sodium dodecyl sulfate or cetyltrimethylammonium
bromide on various electrodes such as glassy carbon, pyrolytic graphite, and platinum was
reported (150). These protein films were utilized for the electrocatalytic reduction of 1,2-
dibromocyclohexane and trichloroacetic acid (150). In the case of immobilization of Mb, a self-
assembled monolayer using L-cysteine (L-Cys) on the modified electrode surface exhibited DET
between the protein active site and the electrode as well as good electrocatalytic activity toward
ascorbic acid oxidation (151).
Even though a wide range of reports are available for immobilization of proteins for
carrying out DET and electrocatalysis with electrodes, a direct comparison involving active site
orientation of monolayer and multilayer covalent attachment of proteins is absent. This is mainly
due to the difficulty to achieve a site-directed covalent tagging of the native conformation of Mb
on the surface with the full control over active site orientation. Furthermore, formation of both a

61
monolayer as well as a multilayer on the electrode surface is a challenging task. Herein we use
a unique biochemical technique based on the incorporation of an unnatural amino acid in vivo to
generate a native Mb protein with additional functional groups (92, 153). NH2Tyr was genetically
encoded with the amber nonsense codon, TAG, by means of a previously evolved orthogonal
tRNACUA/aminoacyl-tRNA synthetase pair that is specific for NH2Tyr (154). The S3NH2Tyr
mutant Mb was immobilized by forming a benzoxazine ring through a Diels-Alder reaction in
buffer (155). This provides an effective strategy for covalent immobilization of a single
monolayer of Mb mutant on a gold electrode surface without impairing its electroactivity (Figure
4.1A). The random multiple covalent attachment of the WT Mb was achieved through 1-ethyl-3-
(dimethylamino-propyl) carbodiimide (EDC)-catalyzed bioconjugation reaction. The DET
characteristics and electrocatalytic behavior of such random multilayered assembly of
covalently-bound WT Mb counterpart (Figure 4.1B) are compared in this study with a site-
directed monolayer protein on the gold surface.
Due to its ability to perform total genomic analysis, DNA microarray has revolutionized
the genomic biotechnology. Likewise, protein microarray has the potential to generate advanced
bioinformatics platforms for proteomics and it could transform the field of drug discovery,
medical diagnostics, and biochemical analysis. Development of functional protein microarray is
more difficult than DNA, peptide, and antibody microarrays. Strong attachment of proteins on
surface, native protein conformation, homogeneous protein monolayer, control over active site
orientation, and retention of protein activity have been the major problem while dealing with
protein microarray.
In order to understand the role of the amino acid residues which are located far from the
active site such Ser 3 in ligand adduct formation, we have extensively formed and studied
ligand adducts with various ligands such as CO, NO, O2, CN–, and N3
– in this chapter.

62
Figure 4.1 Graphical representation of WT and S3NH2Tyr mutant Mb attachment on modified gold electrodes. (A) Ser 3 replaced with NH2Tyr Mb, with its heme active site; which leads to site-directed covalent attachment, with full control over active site orientation, of S3NH2Tyr Mb mutant on the modified gold electrode; (B) WT Mb (PDB: 1MBO) with all surface-exposed amino acids are shown, aspartate and glutamate in red color (with acidic side-chains) and arginine and lysine in blue color (with basic side-chains) as points of immobilization for the bioconjugated crosslinking of the WT Mb proteins. Orange ellipses represent random active site orientation (with limited substrate accessibility) of bioconjugated protein cluster.

63
4.2 Experimental procedures
4.2.1 Chemicals
L-Cys, tetraethyleneglycol diacrylate (TEGDA), sodium periodate (NaIO4) and
N,N-di-iso-propylethyl amine (DIPEA) were obtained from Sigma-Aldrich USA and were used
without further purification. All chemicals were of analytical grade.
4.2.2 Preparation of WT Mb and S3NH2Tyr mutant Mb constructs
The WT Mb expression construct (pBAD with ampicillin resistant marker), S3NH2Tyr Mb
gene expression vector (pBAD with kanamycin resistant marker) and aminoacyl tRNA
synthetase (aatRNA S) with tetracycline resistant marker vector (PAC) were a gift from Dr.
Peter Schultz (The Scripps Research Institute, La Jolla, CA USA). The S3NH2Tyr Mb
expression vector and tRNA synthetase plasmid were co-transformed in DH10B E. coli cells.
The double antibiotic resistant colonies were picked, grown, and the cell stocks were stored at –
80 °C prior to protein expression.
4.2.3 WT Mb and S3NH2Tyr Mb protein purification and analysis
The S3NH2Tyr Mb was expressed in DH10B and was grown in GMML containing
ampicillin and tetracycline antibiotics and the unnatural amino acid, NH2Tyr. The WT and
S3NH2Tyr Mb mutated proteins were expressed, purified, and analyzed as described in chapter
2 (Figure 4.2). The mass of mutant S3NH2Tyr Mb protein mass was analyzed by MALDI-TOF
and was observed to be 18446 Da (Figure 4.3), which matched with the theoretically calculated
mass.

64
1 2 3
Figure 4.2 Coomassie-stained sodium SDS-PAGE analysis of S3NH2Tyr Mb and WT Mb proteins. Lane 1, expressed WT Mb; Lane 2, expressed S3NH2Tyr Mb mutant in presence of 1 mM NH2Tyr; Lane 3, molecular weight standards as indicated in kDa unit.
15 kDa
20 kDa
150 kDa

65

66
Figure 4.3 Electrospray ionization-time of flight-mass spectrometry (ESI-TOF-MS) analysis of the incorporation of S3NH2Tyr into S3 (TAG) Mb mutant showing a mass of 18446 Da (calculated mass, 18446 Da) for S3NH2Tyr Mb.

67
4.2.4 UV-vis spectroscopy
UV-visible spectra were acquired on a Cary 50 Bio UV-visible spectrophotometer. The
concentrations of the WT and mutant Mb were determined from the absorption spectrum of the
protein, using an extinction coefficient of 170 mM–1
cm–1
at 410 nm.
4.2.5 AFM measurements
The experiments were performed with a Veeco MultiMode V SPM instrument. The
cantilevers used in this experiment were made of P-doped n-type Si. The cantilever was
oscillated in the constant excitation mode. Tapping mode Atomic force microscopy (AFM)
images were acquired in the constant frequency shift mode using frequency modulation (FM)
detection. Data analysis was done with the diNanoscope software 7.0.

68
4.2.6 Ligands adduct formation
The protein samples containing 65% glycerol (v/v) were taken in airtight cuvettes
containing 0.1 M potassium phosphate buffer pH 7.0 and were degassed with the N2 gas for ~2
hr at 4 °C and then reduced with minimal amount of Na2S2O4 (in µL) from the stock of 20
mg/mL. In a chest freezer at –35 to –45 °C containing the above sample, pre-cooled O2 gas
was bubbled for 60 s and the UV-visible spectra were recorded. The ferrous-CO adducts were
generated by gentle bubbling of CO in the deoxyferrous enzymes under N2 for 30 s at 4 °C in
0.1 M potassium phosphate buffer pH 7.0 while ferrous-NO adducts were generated by µL
addition of buffer saturated with NO gas. Ferric azide and cyanoferric adducts were prepared by
addition of minimal amount of NaN3 and potassium cyanide solution from their respective stocks
4.2.7 Preparation of microarray
Clean glass slides were silanized, extended, and acryloyl functionalized. To obtain a
pegylated linker, extended acryloyl glass slides were obtained from amine-coated glass slide by
treating with 1 mM diisopropylethylamine followed by reaction with 1 mM tetra (ethylene glycol)
diacrylate in dry dimethylformamide (DMF). In the presence of 100 µM NaIO4, at room
temperature, the mutant protein (3NH2Tyr incorporated) was attached to the pegylated acryloyl
glass slide, for 2 hr The slides were washed in 100 mM potassium phosphate and water for 2
hr. At room temperature, carbonic anhydrase (CA) catalytic assay was carried out using p-
nitrophenyl acetate (0.1 mM) in 15 mM Tris buffer (pH 7.6). Using Veeco Multimode V SPM
instrument, AFM measurements were recorded for glass chip-bound proteins. To prepare
microarrays, proteins were mixed with 0.1 mM NaIO4 for 10 min prior to printing in a solution of
100 mM potassium phosphate buffer (pH 6.0). BioRobotics MicroGrid II 600 with an internal
chamber humidity of 70% was used for printing. After printing, before being transferred to
phosphate buffered saline (PBS) buffer (pH 7.5) for 48 hr of washing, proteins were allowed to
bind for 4 hr at room temperature. With a 1:500 dilution in PBS buffer containing 1% bovine

69
serum albumin (BSA) and 0.1% Tween 20 for 1 hr, binding of HiLyte 647 fluorophoretagged
anti-6xHis tag rabbit Immunoglobulin G (IgG) was performed, followed by washing in PBS buffer
containing 0.1% Tween 20 for 5 min twice. Using Coomassie bromophenol blue staining
solution, Coomassie staining was done for 10 min, followed by 24 hr of washing in water.
4.2.8 Electrochemical instrumentation and procedures
Cyclic voltammetry (CV) was carried out using a three-electrode cell at 22 °C on a
CHI720C electrochemical analyzer (CH Instruments, Austin, TX). The working electrode was
polycrystalline gold (1 × 1 cm2 in area, Alfa Aesar), the counter electrode was a platinum wire,
and the reference electrode was Ag/AgCl (3.5 M KCl). The counter electrode and reference
electrode were obtained from CH Instruments. All potential values below are reported with
respect to the Ag/AgCl (3.5 M KCl) reference electrode. The working electrode surface was first
polished on microcloth (Buehler No. 40-7212) with alumina slurry suspension (0.05 μm), then
sonicated in ethanol, and finally rinsed thoroughly with Milli-Q water. The electrolyte solution
was 0.1 M potassium phosphate buffer with 10 mM KCl solutions at pH 7.0. The solutions were
deoxygenated by bubbling nitrogen prior to each experiment. The CV measurements were
recorded at potential scan rates ranging from 0.05 Vsec–1
to 0.5 Vsec–1
.
4.2.9 Electrode modification procédures
4.2.9.1 Au/L-Cys/WT Mb
The freshly polished and clean gold electrode was immersed in a 2 mM ethanol
solution of L-Cys for 2 hr. It was then washed thoroughly and immersed in the 5 µM WT Mb
solution in MES buffer at pH 5.0. To this solution, a freshly prepared aqueous solution of EDC
was added and the electrode was incubated in this protein medium (2 mL) overnight at 4 °C.
The electrode was then removed, rinsed thoroughly with Milli-Q water, and allowed to dry under
nitrogen flow before use.

70
4.2.9.2 Au/L-Cys/TEGDA/ S3NH2Tyr Mb
The freshly polished and clean gold electrode was immediately immersed in a 2 mM
ethanolic solution of L-Cys for 2 hr. It was then washed thoroughly and immersed in 20 mM
N,N-di-iso-propyl-ethylamine in dimethylformamide (2 mL). To this solution, 10 mM
tetraethylene glycol diacrylate was added and the electrode was kept in it for 3 hr. The
S3NH2Tyr Mb was added in phosphate buffer solution and an aqueous solution of 100 µM
NaIO4 was added to it. The chemically-modified gold electrode was washed thoroughly to
remove any traces of unreacted molecules and incubated in the mutant protein solution
overnight at 4 °C. The electrode was then removed, rinsed with Milli-Q water thoroughly for 1 hr,
and allowed to dry under nitrogen flow before use.
4.2.9.3 Electrocatalysis
Electrocatalysis was carried out in 8 mL of potassium phosphate buffer (100 mM at pH
7.0) containing 5 mM thioanisole dissolved in 30% t-butanol. O2 was bubbled into the solution
throughout the reaction time. Bulk electrolysis was then performed for 2 hr at a constant
potential of –0.50 V. On completion of the catalytic cycle, the organic products were extracted in
dichloromethane (3 × 2 mL). The solution was concentrated, and the products were analyzed by
GC.
4.3. Results and discussion
4.3.1 Monolayer vs. multilayer covalent immobilization of Mb on Au
A central challenge in designing an Mb array for DET is that the electro-active heme
centers are randomly oriented within the protein matrix. Further, conversion of heme protein into
a protein film on the electrode surface may also lead to heme bleaching and protein denaturing
(156-158). To address these challenges, a mild and native immobilization strategy was

71
developed. In this study, site-directed mutagenesis was used to incorporate an unnatural amino
acid (Scheme 4.1) to immobilize Mb on the gold electrode as a monolayer using a unique Diels-
Alder reaction in water or buffer at room temperature or 4 °C (155). The Ser 3 group replaced
with an NH2Tyr of the Mb provides an ideal basis for this reaction as it is solvent exposed at that
position as well as it orients the active site away from the surface.

72
Scheme 4.1 Schematic representation of Au electrode modification for monolayered- immobilization of S3NH2Tyr Mb mutant on the Au surface; see text for details.

73
Incorporation of NH2Tyr into the protein was carried out by using orthogonal
tRNACUA/aminoacyl-tRNA synthetase pairs in bacteria. The mutant Mb was analyzed by SDS-
PAGE (Figure 4.2) and MALDI-TOF (Figure 4.3). Surface modification of the gold electrode was
initiated by modifying it first with an amine-functionalized thiol surface (Scheme 4.1). This
chemically-modified surface was reacted with tetraethyleneglycol diacrylate to obtain the
acroylyl moiety on the surface. Note that the tetraethyleneglycol portion serves to also provide a
lipid-like environment to the covalently attached protein, thus protecting its native conformation
on the surface. In the presence of mild oxidant NaIO4, the NH2Tyr on the surface of the
S3NH2Tyr Mb mutant was easily oxidized to form the o-iminoquinone. This intermediate, in turn,
underwent a Diels-Alder cyclo-addition reaction with suitable alkenes to form a benzoxazine
moiety. The entire process of the covalent attachment of mutant S3NH2Tyr Mb onto the acryloyl
derivatized gold electrode surface was carried out under ambient or 4 °C temperature. The
mutation at Ser 3 with the unnatural amino acid, NH2Tyr, enables full control of the orientation of
the active site of the protein, when covalently immobilized onto the modified gold electrode
(Figure 4.1A). Thus the catalytic and electro-activity of the heme can be easily achieved even
when the mutant protein is covalently attached to the surface of the electrode.
To immobilize the WT Mb as a multilayer, a different strategy had to be employed
(Figure 4.1B). The presence of carboxylic acid and amine groups on the protein surface can be
utilized to form covalent cross linking bonds between these groups and also with suitably
modified, i.e., amine-functionalized, gold surfaces. This bioconjugation reaction is facilitated by
the amino acids with carboxylate groups, such as aspartate (20, 27, 44, 122, 126, and 141
positions), glutamate (4, 6, 18, 38, 41, 52, 54, 59, 83, 136, and 148), and amine groups, such as
arginine (31, 45, 118, and 139) and lysine (16, 34, 42, 47, 50, 56, 62, 63, 77, 78, 79, 96, 98,
102, 133, 140, 145, and 147 positions). Compounds such as 1-ethyl-3-(3-dimethylaminopropyl)
carbodiimide possessing the carbodiimide functional group can be successfully used for
crosslinking these amino acids to form amide linkages (159, 160). The EDC reaction chemistry

74
(Scheme 4.2) was exploited in this study to covalently bind the WT Mb to the amine-
functionalized gold surface, as well as to bioconjugate the Mb as a polymer to generate Mb
multilayer with disoriented binding sites. The random orientation of the binding sites hinders
exposure of some of them to the solvent phase for catalytic or electrochemical activity.
Scheme 4.2 Schematic representation of multilayered WT Mb immobilization on the modified gold electrode surface using EDC catalyzed bioconjugation reaction. Inset: Graphical representation of WT Mb (PDB: 1MBO) with all surface-exposed amino acids. The aspartate and glutamate are represented in red color and arginine and lysine residues in blue color.

75
The surface topography of the covalently attached Mb assemblies were probed
by AFM. Figure 2 compares the AFM images for a chemically-modified amine-functionalized Au
surface before (Figure 4.2a) and after S3NH2Tyr Mb mutant (Figure 4.2b) or WT Mb (Figure
4.2c) covalent attachment. While the pristine surface before protein attachment was featureless
(Figure 4.2 a), an average feature height of ~5 nm was observed in the case of the mutant
protein monolayer, which correlated to the actual size of the protein based on the X-ray
crystallographic structure of Mb (Figure 4.2b). On the other hand, covalently-attached WT Mb
was noticeably rougher, with an average feature height of ~175 nm (Figure 4.2c). As a control
experiment, the WT Mb was also immobilized on the acryolyl-derivatized gold electrode surface
using NaIO4 under the same reaction conditions as the S3NH2Tyr Mb mutant. However, as
expected by the absence of the o-iminoquinone moiety, the WT Mb did not form a monolayer on
the modified surface (Figure 4.3) compared to the genetically modified S3NH2Tyr Mb mutant
(Figure 4.2b). Since the covalent linkage is formed only between the o-iminoquinone moiety of
the protein and the acryloyl group on the modified electrode surface by the Diels-Alder reaction,
the WT Mb does not get covalently immobilized on the electrode surface.

76
Figure 4.4 Tapping mode AFM topographic images for (a) chemically modified Au surface, (b) with immobilized S3NH2Tyr Mb mutant and (c) and with WT Mb on the surface.

77
4.3.2 Electrochemical characterization
Cyclic voltammetry (CV) was used to study the DET electrochemistry of immobilized Mb
on the Au electrodes. In this study, though L-Cys and cysteamine were used to modify the gold
electrode surface (Figure 4.1 and Table 4.1), cysteamine-bound Mb failed to display reversible
redox properties in CV measurements (data not shown). Both WT Mb and the S3NH2Tyr Mb
mutant were immobilized on L-Cys functionalized surface to obtain the CV data. The results
show well defined redox electrochemistry when examined by CV from scan rates 0.05 Vsec–1
to
0.5 Vsec–1
(Figure 4.4). Immobilized Mb on modified Au electrode (CV)
Potential /V vs. Ag/AgCl
-0.4-0.20.00.20.4
Cu
rren
t /A
-6e-5
-4e-5
-2e-5
0
2e-5
4e-5
Au-LCys
Au-LCys-WT Mb
Au-LCys-TEGDA-S3NH2Tyr Mb
Figure 4.5 CV response of L-Cys-modified, WT Mb and S3NH2Tyr Mb mutant immobilized gold electrode in nitrogen-purged 100 mM phosphate buffer with 10 mM KCl (pH 7.0) at room temperature at potential scan rate = 0.1 Vsec
–1.

78

79
Figure 4.6 CV and peak current response of WT and mutant Mb. Mutant (A) S3NH2Tyr Mb and (C) WT Mb immobilized gold electrode in nitrogen-purged 100 mM phosphate buffer with 10 mM KCl (pH 7.0) at room temperature. Potential scan rate ranges from 0.05 to 0.5 V/sec. Plot of peak current vs scan rate for mutant S3NH2Tyr Mb (B) and WT Mb (D) immobilized on L-Cys modified gold electrode in nitrogen-purged 100 mM phosphate buffer with 10 mM KCl (pH 7.0) at room temperature. Potential scan rate ranges from 0.05 to 0.50 Vsec
–1

80
It was observed that the peak currents were directly proportional to the scan rates,
which signals that the proteins were immobilized to the electrode surface (Figure 4.4). The peak
current, Ip for a surface confined reactant is given by Eq. 1 (161).
ART
FnI p
4
22
(Eq. 1)
where, n = number of electrons transferred, F = Faraday constant, R = universal gas
constant, T = absolute temperature in Kelvin (T = 295 K), A = surface area of the electrode, Γ =
surface coverage or the concentration of the redox-sensitive covalently immobilized Mb protein
in mol/cm2, υ = scan rate. From equation 1, the slopes of the plots were given by (n
2F
2/4RT) AΓ
and substituting the known values, such as the geometric area of the electrode surface (A = 2.0
cm2) and the other constants, the estimated values of the number of molecules of the enzyme
associated with the electrode surface (Γ) were 1.54 × 10–10
mol cm–2
for the S3NH2Tyr Mb
mutant protein immobilized on the modified gold electrode. Similarly, for the WT Mb protein, Γ
was 1.29 × 10–10
mol cm–2
on the modified gold electrode through the bioconjugation reaction. A
comparison of the surface coverage of the monolayered S3NH2Tyr Mb mutant and the
multilayered WT Mb indicated that random orientation of the binding sites in the multilayered
WT
Mb leads to lesser number of redox-sensitive proteins on the surface. In contrast, the
S3NH2Tyr Mb mutant allows proper orientation of the active site, thereby exhibiting
homogenously distributed redox-sensitive proteins on the electrode surface.
To investigate the influence of pH on the electron transfer between the active site of the
immobilized protein and the gold electrode, CV was carried out in solutions of different pH
ranging from 5 to 9. It was observed that with increasing pH, the reduction potential of the Fe-
center exhibited a linear negative shift (Figure 4.5). These data support the notion that
denaturation of the Mb protein did not occur within the pH range of 5 to 9.

81
Table 4.1 Comparison of heterogeneous electron transfer rates of Mb on gold electrodes.
Surface modification used Approximate
theoretical length of
anchoring tether (Å);
molecule used
Heterogeneous
electron transfer
rate, ks (s–1
)
Reference
5.35
Homocysteine
0.93 (162)
4.75
L-Cys 1.66 (151)
4.75
L-Cys 2.26 This work
26.66
L-Cys-linked with
tetraethylglycol
diacrylate
1.25 This work

82
Influence of pH on immobilized WT Mb and S3NH2Tyr Mb
pH
5 6 7 8 9
Po
ten
tia
l/ V
vs
Ag
/Ag
Cl
0.00
0.05
0.10
0.15
0.20
WT Mb
S3NH2Tyr Mb
Figure 4.7 Plot of peak potentials vs pH for WT Mb and S3NH2Tyr Mb. Proteins are immobilized on L-Cys-modified gold electrode in nitrogen purged 100 mM phosphate buffer with 10 mM KCl solutions (pH 7.0) at room temperature.
A wide variety of electrode systems have been used to explore the electrochemistry of
Mb. A comparison of some of these systems is tabulated below (Table 4.2). Similar cathodic
and anodic peak separation due to slow DET has also been reported in earlier literature. In this
study, when employing L-Cys for the gold electrode surface modification, the electrochemical
behaviour of the WT Mb and the S3NH2Tyr Mb mutant was significantly different. Since the
electrochemical reaction was quasi-reversible in nature, the formal reduction potential E°′ was
estimated from the midpoint potential E1/2 [obtained from (Epc + Epa)/2]. The WT Mb and the
immobilized S3NH2Tyr Mb mutant gave values of E1/2 between 0.080 V and 0.103 V with
respect to Ag/AgCl (3.5 M KCl) reference, respectively. These midpoint potentials of the heme
center of the WT Mb are in accord with reported values in the literature.(151, 162) The rather
large separation of the cathodic and anodic peaks indicated that DET to the protein was quite
slow.

83
Table 4.2 Comparison of direct electrochemistry of Mb on various electrode systems.
Electrode E1/2 values
(V)[a]
∆Ep
(V)
Reference
Gold/L-Cys-linked with
tetraethylglycol diacrylate and
S3NH2Tyr Mb mutant
0.103 ~0.150 This work
Gold/L-Cys-WT Mb 0.080 ~0.091 This work
Gold/L-Cys-WT Mb 0.086 (151)
Gold/Homocysteine-WT Mb -0.012 ~0.246 (162)
Gold nanopyramids-WT Mb 0.210 ~0.060 (163)
Gold/azide-terminated alkane thiols-
WT Mb
-0.335 (123)
NAF/Mb/IL/PtNPs/MWCNTs/GCE[b]
~0.140 (134)
Mb-CA/GCE[c]
-0.281 ~0.102 (164)
Mb/DDAB–HIMIMPF6[d]
-0.261 ~0.044 (165)
[a]All literature values for E1/2 have been converted to Ag/AgCl (3.5 M KCl) reference electrode,
for clarity; [b]
NAF/Mb/IL/PtNPs/MWCNTs/GCE = Glassy carbon electrode modified with multi-walled carbon nanotubes (MWCNTs), followed by platinum nanoparticles (PtNPs), ionic liquids (IL) and Nafion (NAF) with WT Mb;
[c]Mb-CA/GCE = Glassy carbon electrode modified with
calcium alginate and WT Mb; [d]
Mb/DDAB–HIMIMPF6 = Glassy carbon electrode modified with didodecyldimethylammonium bromide (DDAB), 1-hexyl-3-methylimidazolium hexafluorophosphate (HIMIMPF6) and WT Mb.
The rate of heterogeneous electron transfer between the electrode and the immobilized
protein was calculated using Laviron’s method (161). The value of the transfer coefficient ( )
was determined using Eq. 2:
ln)1(
)1(ln
)1(
)1(ln
)1(
lnlnln
00
00
nF
RT
RTk
nF
nF
RTE
RTk
nF
nF
RTEE
nF
RT
RTk
nF
nF
RTE
RTk
nF
nF
RTEE
ss
pa
ss
pc
(Eq. 2)
where Eº is the standard potential, ks is the heterogeneous electron transfer rate
constant, and n = 1. The potentials for the cathodic (Epc) and anodic (Epa) peaks were plotted
against ln (v) and on analyzing the ratio of the slopes of the plots, an average value of the

84
transfer coefficient, , was found to be 0.54 for the S3NH2Tyr Mb mutant, and 0.51 for the WT
Mb immobilized electrode. The heterogeneous electron transfer rate constants for immobilized
WT Mb and the S3NH2Tyr Mb mutant were determined from the following equation (Eq. 3) at
different scan rates:
log)1(
3.2logloglog)1()1log(
)1(
3.2
3.2
)1(loglog)1()1log(log
nF
RT
nF
RTk
nF
RTE
RT
EnF
nF
RTk
sp
p
s
(Eq. 3)
where ΔEp is the peak potential separation. Using the respective values of α and from
the plot of ΔEp vs log v (see Supporting Information Figure SI-6), ks was estimated to be 1.3
sec–1
and 2.3 sec–1
respectively for the immobilized S3NH2Tyr Mb mutant and WT Mb on the
chemically modified gold electrode. The S3NH2Tyr Mb mutant monolayer has slightly slower
electron transfer kinetics than the multilayered WT Mb presumably due to the varying length of
the anchoring tethers in the two cases (see Table 4.1).
Table 4.1 compares surface modification chemistries based on anchoring molecules
used for the immobilization of Mb on gold electrode surfaces and their associated ks values
reported in two previous studies (151, 162) with the results from this study. It is observed that
random immobilization of WT Mb leads to a decrease in the DET from 2.26 sec–1
to 0.93 sec–1
as the linker molecule length is increased from L-Cys (~4.75 Å) to homocysteine (~5.35 Å).
Though the theoretical length of the tether is longer (~26 Å) in the case of S3NH2Tyr Mb mutant,
it facilitates faster electron transfer compared to homocysteine. This could be due to the
tetraethylene glycol-linker providing a through-bond electron transfer environment from the
electrode to the redox-sensitive heme center of the Mb (164, 166).

85
Potential /V vs. Ag/AgCl
-0.4-0.20.00.20.4
Cu
rren
t /A
-2e-4
-2e-4
-1e-4
-5e-5
0
5e-5
Au-LCys
Au-LCys-WT Mb
Au-LCys-TEGDA-S3NH2Tyr Mb
Au-Lcys-Hemin
Hemin (5 M) in solution
Figure 4.8 Comparision of immobilized Mb with hemin on modified gold electrode. CV response of hemin in solution, L-Cys modified, hemin immobilized, WT Mb and S3NH2Tyr Mb mutant immobilized gold electrode in nitrogen-purged 100 mM phosphate buffer with 10 mM KCl (pH 7.0) at room temperature and at a potential scan rate of 0.1 Vsec
–1.
An important aspect of the heme bleaching from the protein immobilized electrode
suface was carefully investigated for both the S3NH2Tyr Mb mutant and the WT Mb protein. The
redox responses of the free hemin (5 µM) in solution and when immobilized on the amine-
functionalized gold electrode surface were observed to be quite different from those of the
S3NH2Tyr Mb and the WT Mb-bound surface (Figure 4.6). From these data, it can be inferred
that heme bleaching was absent from both the S3NH2Tyr Mb mutant and the WT Mb
immobilized electrodes under the above mentioned experimental conditions.

86
4.3.3 Catalytic studies
Mb has often been utilized to explore the monooxygenase activity of heme proteins in
presence of H2O2, in order to investigate the P450 catalytic shunt-pathway. A common
substrate used in such studies is thianisole, which can be catalytically oxidized to its sulfoxide
by high valent ferryl-porphyrin-π-cation radical species (also known as Compound I) of Mb.
Hence, electrocatalytic conversion of thioanisole to its oxidized form provides an ideal
comparison platform for the binding site orientation in these two modes of immobilization.
Dioxygen was bubbled into the solution and bulk electrolysis was carried out for 2 hr with the
covalently immobilized S3NH2Tyr Mb mutant and the WT Mb (Table 4.3).
Table 4.3 Electrocatalytic conversion of thioanisole to its oxidized form and turnover frequency
(TOF) using bare, multilayered WT Mb-immobilized and monolayered S3NH2Tyr Mb-immobilized Au electrodes.
Unreacted
thioanisole (%)
Thioanisole
sulfoxide
(%)
TOF for the oxidized
products (sec–1
)
Bare electrode 86 14
WT Mb 13 87 1.9 × 103
S3NH2Tyr Mb 19 81 1.5 × 103
The GC results demonstrated that 87% conversion of the thioanisole to its oxidized
form had taken place for the WT Mb immobilized electrode, while for the S3NH2Tyr Mb mutant,
81% conversion was obtained. A reduced reaction time (1 hr) did not yield sufficient product
formation in comparison to that of 2 hr catalytic reaction. A control experiment was also carried
out for the electrocatalytic oxidation of thioanisole in the presence of bare Au. Only 14%
conversion of the thioanisole to its sulfoxide was observed in this case after 2 hr. Considering

87
the amount of electro-active protein covalently attached onto the surface from Eq. 1, the
turnover frequency (TOF) for the WT Mb was 1.9 × 103 sec
–1. Similarly, the TOF for the
S3NH2Tyr Mb mutant was 1.5 × 103 sec
–1. Significantly, the comparable catalytic activity
observed for the multilayered WT Mb and the monolayered S3NH2Tyr Mb mutant reiterates the
importance of orientation of the active sites of both the proteins. The random orientation of
binding site in the multilayered protein leads to loss of catalytic activity (considering the greater
number of proteins on the surface), than the monolayered mutant S3NH2Tyr Mb. Proper
orientation of the heme active site in the monolayered S3NH2Tyr Mb mutant assisted the
catalytic and redox-activity of the protein, when covalently immobilized onto the modified gold
electrode surface.
4.3.4 Microarray studies
Functionalization of glass surface with polyethylene glycol (PEG) was done by using the
methods as previously described (167). To attach S3NH2Tyr Mb on PEG linker, Diels–Alder
reaction was used in mild conditions, such as in water (168). Since the protein will bind only
through its unnatural amino acid, we have control of the active site in mutants. The immobilized
protein on glass surface was analyzed by AFM, in which monomeric S3NH2Tyr Mb showed
expected dimensions of height and width (Figure 4.3) based on crystal structure.
4.3.4.1 Preparation of functional protein microarrays
A library of five proteins was printed on a single slide, to determine if our technique can
be used for microarray studies (Figure 4.7). In order to understand orientation of the protein
arrays and selective protein-protein interaction, we allowed HiLyte 647 fluorophore-conjugated
anti-6×His tag rabbit IgG to bind with the proteins that were spotted on the surface. S3NH2Tyr
Mb and T365 (3NH2Tyr P450 BM3) (Figure 4.7) were the only two proteins that show surface-
exposed 6×His-tag chains when we observed the site of immobilization. Thus these were the

88
two proteins that showed the direct and efficient binding with IgG. However, all other proteins
except the control were attached to the surface after the tagging reaction, as they were visible in
coomassie blue staining. This experiment confirmed the interaction between IgG and the protein
could be generated in a specific manner as well as control over the orientation of the protein.

89

90
Figure 4.9 A microarray of five proteins on a single slide displays. (a) T365 (3NH2Tyr) P450 BM3. (b) P450 BM3 control without NaIO4 (control shows that printed proteins were detached from the surface after washing). (c) T73 (3NH2Tyr) CA. (d) D125 (3NH2Tyr) CA. (e) D133 (3NH2Tyr) EGFP. (f) S3 (3NH2Tyr) Mb. Column 2 displays an image of excitation at 649 nm and emission at 666 nm.

91

92
Figure 4.10 Spectroscopic characterization of high spin, low spin and ligand complexes of WT and mutant Mb. Electronic absorption spectra of (A) ferric and ferrous, (B) CO, (C) NO, (D) oxyferrous, (E) cyanoferric and (F) N3
– complexes of WT Mb and S3NH2Tyr Mb.
The spectra were taken at 4 °C and the samples were examined in 100mM phosphate buffer at pH 7.0.

93
4.3.5 Ligand binding studies
To understand the effect of different ligands upon replacement of residues which are
away from the active heme center of the protein, we have replaced the serine 3 (S3) with
3NH2Tyr, a redox amino acid. Figure 4.8 clearly indicates that S3NH2Tyr Mb protein behaves
more or less similar to that of WT Mb with respect to ligand-binding studies. Since position 3 is
quite distant from the heme center, it has almost no role in influencing the electronic density of
the Asheme center. As position 3 is quite away, it did not play a major role in determining the
ligand-metal charge transfer, metal-ligand charge transfer, and in π backbonding (104, 119).
We did not observe significant difference in the electron absorption spectra of mutant protein as
compared to WT Mb (15, 31).
4.4. Conclusion
Though, previous research groups have observed facile electron transfer using specific
organic molecules as promoters, no prior studies were reported using mutant protein that
utilizes its unnatural amino acid moiety to specifically immobilize onto the electrode surface. In
this chapter, I report monolayer and multilayer immobilization of Mb on to chemically-modified
gold electrode surfaces using two unique immobilization techniques as described above.
Further, electrocatalysis of thioanisole oxidation was carried out successfully with both the
protein immobilized electrode cases, albeit at variant facility. These results underline that the
S3NH2Tyr Mb mutant monolayer exhibits well-defined catalytic and electro-activity because of
proper orientation of its heme active site, in comparison to the random multilayered WT Mb
case.
To investigate protein-protein interactions, five separate proteins P450 BM3, Mb,
enhanced green fluorescent protein (EGFP), HRP, and CA have been covalently attached onto
a solid support and analyzed. Functional protein microarray concept development was
confirmed by a catalytic activity designed assay using spectroscopic characterization. This

94
concept developed microarray will bring novelty in the field of drug discovery and biomedical
and proteomic research. The ligand binding studies showed that the S3 position being quite
away from the active heme center, does not affect the metal-ligand binding to a greater extent.

95
Chapter 5
ELECTRONIC NATURE INVESTIGATION OF HEME CENTER BY MODIFYING PROXIMAL AND DISTAL BINDING SITE ENVIRONMENT OF SPERM WHALE MYOGLOBIN WITH
ELECTRON RICH RESIDUES
5.1 Introduction
The heme group is the most common prosthetic group in the metalloproteins (1). The
iron center, porphyrin, and axial ligand constitute the three covalently linked integral
components of a heme protein. In the heme proteins, a wide variety of porphyrines are present
such as heme a, heme b, heme c, heme d, heme d1, heme o, heme P460, chloroheme, and
siroheme. Among the globins and cytochrome, heme b and heme c are the most common
(Figure 5.1). A widely studied example is Mb (sperm whale), a small protein with 153 amino
acids and molecular weight of 17.5 kD, with oxygen storage as its main function in vertebrates
(2, 14, 169, 170). The presence of different varieties of amino acids around the binding site has
always been a matter of interest for the understanding of heme ligand interaction and its
catalytic activity (169, 170). With the tool of site directed mutagenesis divers, residues can be
introduced to the proteins, and complexes formed with different ligands can be investigated.
Among the heme protein, various axial ligands such as His, Cys, Met, Tyr, and Lys are found
and they play a crucial role in their properties (14, 22). His present at 64 position plays a major
role in stabilizing the ligand adduct by forming bonds to the ligands attached to the heme
moiety. It has been proposed that with open and closed confirmation distal His can act like a
gate for ligand adducts (1, 15, 29, 171, 172).
Figure 5.2 B shows the details of amino acid residues around the heme center in sperm
whale Mb. It is also believed that the size of certain residues also contributes significantly to
determining the heme ligand complex stability. But the role played by the electronic nature
around heme center is not clearly understood. The introduction of noncannonical amino acids
with electron rich functional groups such as pNO2Phe, will allow us to investigate the electronic
nature of heme center, and its effect on ligand (CO, NO, O2, CN–, and N3
–) binding to a greater

96
extent. For our further understanding of interaction of CO, NO, O2, N3–, and CN
− with Mb as a
model for heme protein, I have created mutations at its distal and proximal sites (15). By site
directed mutagenesis we have replaced the His with pNO2Phe at His 64 and His 93 positions
and compared the interaction of different ligands with these mutants, as well as to WT Mb (15,
88).
Figure 5.1 Structures of heme b and heme c.

97
Figure 5.2 Schematic representations of WT and mutants Mb. (A) Overall structure of WT Mb. (B) enlarged binding site of WT Mb, (C) structure of H64pNO2Phe and (D) structure of H93pNO2Phe mutants.
5.2 Experimental procedure
5.2.1 Chemicals
The gasses (O2, CO, and N2) have been purchased from Air Liquide, USA while
pNO2Phe, sodium azide, potassium cyanide, and Na2S2O4 were purchased from Sigma-Aldrich

98
USA. All chemicals were of analytical grade and have been used without any further purification
(173).
5.2.2 Preparation of WT, H64pNO2Phe and H93pNO2Phe Mb mutant constructs
The WT Mb expression constructs H64pNO2Phe and H93pNO2Phe Mb gene
expression vector and aminoacyl tRNA synthetase (aatRNA S) plasmids were donated by Dr.
Peter Schultz (The Scripps Research Institute, La Jolla, CA USA). The H64pNO2Phe and
H93pNO2Phe Mb expression vector were co-transformed with tRNA synthetase separately in
DH10B E.coli. The bacterial colonies which are double antibiotic (ampicillin and tetracycline)
resistant were picked, grown, and the cell stocks were stored at –80 °C prior to the start of
bacterial cell culture for protein purification (15).
5.2.3 Purification and analysis of WT Mb, H64pNO2Phe and H93pNO2Phe Mb
The H64pNO2Phe Mb and H93pNO2Phe Mb were expressed in E. coli bacteria which
were grown in GMML containing 5% LB media and suitable antibiotics (ampicillin and
tetracycline). Both the WT and mutated (H64pNO2Phe and H93pNO2Phe) Mb proteins were
induced, expressed, purified, and analyzed as described in chapter 2.

99
Figure 5.3 Coomassie-stained SDS-PAGE analysis of expression of WT, H64pNO2Phe and H93pNO2Phe mutants Mb. Lane 1 expressed WT Mb, Lane 2 H64pNO2Phe mutant Mb, Lane 3 Molecular weight standards and Lane 4 H93pNO2Phe mutant Mb. 5.2.4 Electronic absorption spectroscopy
UV-visible electronic absorption spectra of WT Mb proteins and the mutant H64pNO2
and H93pNO2 Mb proteins were taken using Varian Cary 50 Bio UV-visible spectrophotometer.
The concentrations of proteins were determined from the absorption spectrum of the protein,
using the molar extinction coefficient determined by the heme chromatogen method (96).

100
5.2.5 Preparation of deoxyferrous Mb
The WT Mb and mutants Mb proteins were taken in their respective airtight cuvettes
containing 0.1 M potassium phosphate buffer, pH 7.0. The N2 gas was passed for ~2 hr at 4 °C
to degas the sample. The minimal amount of Na2S2O4 (in µL) was added from the stock of 20
mg/mL and the electronic absorption spectra were recorded by Varian Cary 50 Bio UV-visible
spectrophotometer (15).
5.2.6 Preparation of oxyferrous complex
The oxyferrous complexes were prepared in a chest freezer at −35 to −45 °C. The
protein sample was placed in a 100 mM potassium phosphate buffer (pH 7.0), which contained
65% glycerol (v/v). The ferric protein was first degassed with N2 gas for 2 hrs. Then it was
reduced to deoxyferrous by addition of Na2S2O4 (20 mg/mL stock) under N2 atmosphere in a
sealed cuvette at 4 °C. Pre-cooled O2 gas was bubbled into the cuvette for 60 sec and the UV-
visible spectra were recorded (15, 31, 40).
5.2.7 CO and NO complex preparation
The ferrous-CO adducts were generated by gentle bubbling of CO in deoxyferrous
enzymes for 30 sec at 4°C in 0.1 M potassium phosphate buffer pH 7.0. The ferrous-NO
complexes were generated by micoliter addition of buffer saturated with NO gas in deoxyferrous
enzymes under N2 at 4 °C in 0.1 M potassium phosphate buffer pH 7.0. The saturated buffer
was prepared by gentle bubbling of NO gas in a sealed cuvette containing 0.1 M potassium
phosphate buffer pH 7.0 for 30 min at room temperature (15, 31, 32, 40).
5.2.8 N3– and CN
– complex preparation

101
WT Mb and mutants Mb N3– complexes were formed by µL addition of NaN3 solution
from 40 mM stock solution. The NaN3 stock solution and protein were in 100 mM potassium
phosphate buffer pH 7.0 and the temperature was kept at 4 °C. The cyanoferric adducts were
prepared from the deoxyferric enzymes by addition of minimal volumes (µL) of a 1 M KCN stock
solution, prepared in 100 mM potassium phosphate buffer pH 7.0. The deoxyferric enzymes
were prepared by bubbling of N2 gas in the ferric enzymes for at least 30 min.
5.3 Results and discussion
Here we studied the ferrous and ferric electronic absorption spectra of Mb and its
mutants (H64pNO2Phe Mb and H93pNO2Phe) in absence and presence of exogenous ligands
such as CO, NO, O2, N3–, and CN
– using UV-vis spectroscopy. The electronic absorption
spectra of the ferric form of the enzyme showed that there are significant differences in their
character, particularly at 550-650 nm (Figure 5.4). The peaks around 539, 578, and 628 nm in
ferric and around 662 nm in ferrous enzyme were not prominent in mutants when compared to
the WT Mb. Due to the presence of the electron rich functional group (pNO2Phe), the Sorets
around 409 nm in ferric and around 430 nm in ferrous were a little sharper in case of mutant
enzymes (Figures 5.4 and 5.5).

102
Figure 5.4 Characterization of ferric WT and mutants Mb. Electronic absorption spectra of the H64pNO2Phe (blue dashed-dot), H93pNO2Phe (black solid line) and WT (red dashed line) of ferric Mb enzymes. The spectra were recorded in 100 mM potassium phosphate buffer, pH 7.0 at 4 °C with 5 μM protein.

103
Figure 5.5 Deoxyferrous electronic absorption spectra of WT, H64pNO2Phe and H93pNO2Phe mutant Mb. WT Mb (red dashed line), H64pNO2Phe (blue dashed-dot) and H93pNO2Phe Mb mutant (black solid line). The spectra were taken at 4 °C and the samples were examined in 100mM phosphate buffer at pH 7.0.
5.3.1 Ferrous-CO, ferrous-NO and ferrous O2 complexes of WT, H64pNO2Phe and H93pNO2Phe Mb
To understand the effect of different ligands upon replacement of distal and proximal
His and electronic nature of the heme center, I have investigated the ferrous-XO (X= C, N, O)
complexes of Mb and its mutants. As XO molecules have vacant π orbitals which closely
matched with filled dπ orbitals of the Fe (II) ion of porphyrin, it makes the heme group
electronically more favorable to bind to CO, NO and O2. Furthermore, this kind of arrangement
was found to be optimal for the Fe-XO π-backbonding. The backbonding was responsible for
shortening (strengthening) the Fe-X bond and lengthening (weakening) of the X-O bond
(Figures 5.6 and 5.7) (40, 174-176).

104
Figure 5.6 The heme carbonyl complex showing dπ-pπ* backbonding.
Figure 5.7 Showing presence of electron rich functional group (NO2−) at distal position.

105
Table 5.1 The electronic absorption spectral features of the oxy, carbonyl, NO, CN−, and N3
− complexes with H64pNO2Phe Mb,
H93pNO2Phe Mb mutants and WT Mb.
H64pNO2Phe
Soret (nm)
Visible (nm)
H93pNO2Phe
Soret (nm)
Visible (nm)
WT Mb
Soret (nm)
Visible (nm) (References)
Ferric 408.9 Ferric 408.9 635 Ferric 409 505; 539; 578; 628; 675
(175) Deoxyerrous 430 558 Ferrous 433 635 Ferrous 431 556; 662
(175)
Fe3+
- CN– 422.9 537; 576 423 536.9 420 542; 642.2
(175) Fe
3+- N3
– 422 541; 576 421 536; 571 416 534; 583;
636; 661
Fe2+
-CO 424 539; 573 422.9 542; 577 422 540; 570; 649; 611 (10)
Fe2+
-O2 419.9 542; 580 416.9 541.5; 579.6
416 540; 581; 668 (15)
Fe2+
-NO 419 545, 577 419 546, 580 420 547, 581, (40)

106
Ferrous-CO complexes of the H64 and H93 pNO2Phe have shown higher intensity of
absorbance compared to ferrous-NO and ferrous–O2 complexes (Figure 5.8). This may be due
to Fe-CO being linear adduct and both of its π orbitals have empty shells. It allows dπ-π
orbitals to overlap in a perpendicular direction, which results in a prominent overlapping, and
more backbonding giving rise to sharper Soret (119). However under the similar conditions, NO
and O2, occupies one and two π electrons, respectively in their π orbitals. In order to
accommodate any π-antibonding interaction, the NO and O2 adducts have to bend, which
further lowers the possibility of overlapping and backbonding. This leads to less prominent Soret
in NO and O2 as compare to CO (Figure 5.8, 5.9 and 5.10). It is noteworthy that there were
significant differences within the XO adducts of different proteins’ electronic spectra (Table 5.1).
When axial His H93 attached to heme center, was replaced by pNO2Phe (an electron rich
functional group), the sharper Soret was recorded due to stretching one dπ-π backbonding
electron transition via push effect The pNO2Phe increases the electron density near the Fe (II)
center, which enhances the metal-ligand charge transfer and backbonding. Similar results were
not observed when the distal His 64 was replaced by pNO2Phe. The distal pNO2Phe probably
increases the electron density around the CO orbitals which favors ligand-metal charge transfer
and lowers the backbonding. Since O2 has two π electrons, reverse characteristics have been
observed in the oxyferrous complex (Figure 5.10). The different enzymes show different peaks
in 550-700 nm regions (Table 5.1), which explains that they also have diverse, features in
visible region (3, 176).

107
Figure 5.8 Comparative studies of carbonyl complexes of WT, H64pNO2Phe, and H93pNO2Phe. Electronic absorption spectra of CO complex of WT Mb (red dashed line), H64pNO2Phe (blue dashed-dot) and H93pNO2Phe Mb mutant (black solid line). The spectra were taken at 4 °C and the samples were examined in 100mM phosphate buffer at pH 7.0.

108
Figure 5.9 Comparative studies of NO complexes of WT, H64pNO2Phe, and H93pNO2Phe. Electronic absorption spectra of NO complex of WT Mb (red dashed line), H64pNO2Phe (blue dashed-dot) and H93pNO2Phe Mb mutant (black solid line). The spectra were taken at 4 °C and the samples were examined in 100mM phosphate buffer at pH 7.0.

109
Figure 5.10 Characterization of oxyferrous complexes of WT, H64pNO2Phe, and H93pNO2Phe Mb. Electronic absorption spectra of WT Mb oxyferrous (red dashed line), H64pNO2Phe mutant Mb oxyferrous (blue dashed-dot) and H93pNO2Phe mutant Mb oxyferrous (black solid line). The spectra were taken at −35 to −45 °C and the samples were examined in 60% glycerol, 100mM phosphate buffer at pH 7.0.
The stability of XO adducts are dictated by hydrogen bonding between the XO and
distal protein moiety (figure 5.11). In WT Mb at the 64 position, His is present, which is primarily
responsible for the H-bonding, hence the stability of XO adducts (176). When I replaced the His
with pNO2Phe, the possibility of H-bonding at distal pocket was reduced, which lowers stability
of complex. Furthermore, the electron rich oxygen allowed more electronic distribution, which
led to prominent ligand to metal charge transfer as seen in uv-vis spectrum. The replacement of
the proximal His 93 with pNO2Phe led to increased electron density, and backbonding was
more prominent around XO orbital; it helped in forming the H-bond with distal His. Thus I

110
conclude that when we replaced the axial His with the pNO2Phe, the XO adducts were more
stable than in other cases.
Figure 5.11 Proposed structure of (A) Fe (II) O2, (B) Fe (II) CO and (C) Fe (II) NO.
5.3.2 Ferric-cyanide and ferric-azide complexes of WT and H64pNO2Phe and H93pNO2Phe
mutants of Mb
Characterization of anionic ligand adducts such as CN– and N3
– to ferric WT Mb and
mutant Mb were important to understand how the electronic nature affects heme center. It gives
us the idea of the impact of protein environment on heme center. Ligand to metal charge
transfer are ideal for metals in high oxidation states such as Fe3+
that are bound to electron rich,
low electronegativity ligands such as N3– and CN
– (15, 175-177). The charge transfer from
ligand to metal will cause the increase in Soret, but due to lack of backbonding, it was not as

111
prominent as in the case of the CO adduct. The charge transfer from ligand to heme center
made it a low spin complex; the red shift was observed to high energy (Figures 5.12 and 5.13).
As CN– is a stronger field ligand, the Soret was less sharp in the ferrycyanide complex .
The introduction of electron rich oxygen in pNO2Phe at axial and distal positions has a
significant impact on the electronic density of the heme center. When we replaced distal His
with pNO2Phe in electron density around CN– and N3
– ligands, orbitals increased.
Consequently, there was more ligand-metal charge transfer, resulting in an increase in Soret
and red shift. While we replaced axial His with pNO2Phe, the Fe center was more electron rich,
which caused the red shift (λmax 423, 436.9, 437.4, and 576). There was less possibility of metal
to ligand charge transfer and backbonding, as the ligand was electron rich and its orbitals are
filled, and therefore no significant increase in Soret (119). The adducts also showed quite a
distinct feature in their 550-700 nm region (Table 5.1), which supports that the heme
environment has a significant role on heme proteins in the determination of their catalytic and
ligand binding properties.

112
Figure 5.12 Comparative studies of CN
– complexes of WT, H64pNO2Phe, and H93pNO2Phe
Mb. Electronic absorption spectra of ferric CN– complex of WT Mb (red dashed line),
H64pNO2Phe (blue dashed-dot) and H93pNO2Phe Mb mutant (black solid line). The spectra were taken at 4 °C and the samples were examined in 100mM phosphate buffer at pH 7.0.

113
Figure 5.13 Characterization of N3
– complexes of WT, H64pNO2Phe, and H93pNO2Phe Mb.
Electronic absorption spectra of ferric N3– complex of WT Mb (red solid line), H64pNO2Phe
(blue dashed-dot) and H93pNO2Phe Mb mutant (black solid line). The spectra were taken at 4 °C and the samples were examined in 100mM phosphate buffer at pH 7.0.
The H-bonding also plays a significant role in the stability of CN– and N3
– adducts
(Figures 5.14 and 5.15). Any residue that strengthens the H-bonding will make a stronger
adduct and vice versa. In this case, as pNO2Phe reduces the possibility of H-bonding at the
distal pocket, it leads to weaker adducts. But the size of the residues also plays a significant
role in holding the ligands by not allowing them to leave the binding site environment.

114
Figure 5.14 Two canonical forms of N3
– bound to heme iron of WT Mb and H64pNO2Phe Mb
mutant.
Figure 5.15 Two canonical forms of CN
– bound to heme iron of WT Mb and H64pNO2Phe Mb
mutant.

115
5.3 Conclusion
To summarize, the importance and significance of electron rich functional groups on the
electronic nature of heme center had been explored. It has been found that the axial His
attached to the heme center plays a crucial role in dictating the electron cloud near the heme
center. When the axial heme is replaced by an electron rich residue like pNO2Phe, the electron
density was higher near heme center, which contributes to higher energy and red shift of the
spectra. This study also concludes that the oxidation states of metal centers and the natures of
ligand (electron rich or electron deficient) play an important role in determining the backbonding
and direction of charge transfer (from metal to ligand or ligand to metal). The redox nature and
size of the distal residue also has a high impact on determining the stability of the ligand
adducts. It is well known that the H-bonding plays an important role in the stability of ligand
adducts, but what role the size of noncanonical (such as pNO2Phe) residues play in the stability
of ligand adducts is uncertain.

116

117
REFERENCES
1. Montelanno, P. R. O. d., (Ed.) (2008) Cytochrome P450: Structure, Mechanism, and
Biochemistry, Third ed., Springer-Verlag New York, LLC, New York.
2. Bertini, I., Sigel, A., Sigel, H., (Ed.) (2001) Handbook on metalloproteins, Marcel
Dekker, Inc., New York.
3. Spiro, T. G., Soldatova, A. V., and Balakrishnan, G. (2013) CO, NO and O-2 as
vibrational probes of heme protein interactions, Coord. Chem. Rev. 257, 511-527.
4. Matsui, T., Ozaki, S., and Watanabe, Y. (1997) On the formation and reactivity of
compound I of the His-64 myoglobin mutants, J. Biol. Chem. 272, 32735-32738.
5. Bates, D. M., Popescu, C. V., Khoroshilova, N., Vogt, K., Beinert, H., Munck, E., and
Kiley, P. J. (2000) Substitution of leucine 28 with histidine in the Escherichia coli
transcription factor FNR results in increased stability of the [4Fe-4S](2+) cluster to
oxygen, J. Biol. Chem. 275, 6234-6240.
6. Denisov, I. G., Makris, T. M., and Sligar, S. G. (2002) Formation and decay of
hydroperoxo-ferric heme complex in horseradish peroxidase studied by cryoradiolysis,
J. Biol. Chem. 277, 42706-42710.
7. Tatoli, S., Zazza, C., Sanna, N., Palma, A., and Aschi, M. (2009) The role of Arginine 38
in horseradish peroxidase enzyme revisited: A computational investigation, Biophys.
Chem. 141, 87-93.
8. Coulter, E. D., Cheek, J., Ledbetter, A. P., Chang, C. K., and Dawson, J. H. (2000)
Preparation and initial characterization of the compound I, II, and III states of iron
methylchlorin-reconstituted horseradish peroxidase and myoglobin: models for key
intermediates in iron chlorin enzymes, Biochem. Biophys. Res. Commun. 279, 1011-
1015.
9. Rohlfs, R. J., Mathews, A. J., Carver, T. E., Olson, J. S., Springer, B. A., Egeberg, K.
D., and Sligar, S. G. (1990) The effects of amino acid substitution at position E7
(residue 64) on the kinetics of ligand binding to sperm whale myoglobin, J. Biol. Chem.
265, 3168-3176.
10. Adachi, S., Nagano, S., Ishimori, K., Watanabe, Y., Morishima, I., Egawa, T., Kitagawa,
T., and Makino, R. (1993) Roles of proximal ligand in heme proteins: replacement of
proximal histidine of human myoglobin with cysteine and tyrosine by site-directed

118
mutagenesis as models for P-450, chloroperoxidase, and catalase, Biochemistry 32,
241-252.
11. RodriguezLopez, J. N., Smith, A. T., and Thorneley, R. N. F. (1996) Recombinant
horseradish peroxidase isoenzyme C: The effect of distal haem cavity mutations
(His42->Leu and Arg38->Leu) on compound I formation and substrate binding, J. Biol.
Inorg. Chem. 1, 136-142.
12. Frauenfelder, H. (2010) Myoglobin as an example of protein complexity, Chem. Phys.
375, 612-615.
13. Cohen, F. E., and Sternberg, M. J. E. (1980) Use of Chemically Derived Distance
Constraints in the Prediction of Protein-Structure with Myoglobin as an Example, J. Mol.
Biol. 137, 9-22.
14. Brunori, M., Bourgeois, D., and Vallone, B. (2004) The structural dynamics of
myoglobin, J. Struct. Biol. 147, 223-234.
15. Du, J., Sono, M., and Dawson, J. H. (2011) The H93G myoglobin cavity mutant as a
versatile scaffold for modeling heme iron coordination structures in protein active sites
and their characterization with magnetic circular dichroism spectroscopy, Coord. Chem.
Rev. 255, 700-716.
16. Yang, H. J., Matsui, T., Ozaki, S., Kato, S., Ueno, T., Phillips, G. N., Fuku-Zumi, S., and
Watanabe, Y. (2008) Molecular engineering of myoglobin: Influence of residue 68 on
the rate and the enantioselectivity of oxidation reactions catalyzed by H64D/V68X
myoglobin (vol 42, pg 10174, 2003), Biochemistry 47, 2700-2700.
17. Pfister, T. D., Ohki, T., Ueno, T., Hara, I., Adachi, S., Makino, Y., Ueyama, N., Lu, Y.,
and Watanabe, Y. (2005) Monooxygenation of an aromatic ring by F43W/H64D/V68I
myoglobin mutant and hydrogen peroxide - Myoglobin mutants as a model for P450
hydroxylaation chemistry, J. Biol. Chem. 280, 12858-12866.
18. Sato, H., Hayashi, T., Ando, T., Hisaeda, Y., Ueno, T., and Watanabe, Y. (2004)
Hybridization of modified-heme reconstitution and distal histidine mutation to
functionalize sperm whale-myoglobin, J. Am. Chem. Soc. 126, 436-437.
19. Yang, H. J., Matsui, T., Ozaki, S., Kato, S., Ueno, T., Phillips, G. N., Fukuzumi, S., and
Watanabe, Y. (2003) Molecular engineering of myoglobin: Influence of residue 68 on
the rate and the enantioselectivity of oxidation reactions catalyzed by H64D/V68X
myoglobin, Biochemistry 42, 10174-10181.

119
20. Watanabe, Y., and Hayashi, T. (2005) Functionalization of Myoglobin, Prog. Inorg.
Chem. 54, 449-493.
21. Kato, S., Yang, H. J., Ueno, T., Ozaki, S., Phillips, G. N., Fukuzumi, S., and Watanabe,
Y. (2002) Asymmetric sulfoxidation and amine binding by H64D/V68A and H64D/V68S
Mb: Mechanistic insight into the chiral discrimination step, J. Am. Chem. Soc. 124,
8506-8507.
22. Hara, I., Ueno, T., Ozaki, S., Itoh, S., Lee, K., Ueyama, N., and Watanabe, Y. (2001)
Oxidative modification of tryptophan 43 in the heme vicinity of the F43W/H64L
myoglobin mutant, J. Biol. Chem. 276, 36067-36070.
23. Herold, S., Matsui, T., and Watanabe, Y. (2001) Peroxynitrite isomerization catalyzed
by His64 myoglobin mutants, J. Am. Chem. Soc. 123, 4085-4086.
24. Ozaki, S., Hara, I., Matsui, T., and Watanabe, Y. (2001) Molecular engineering of
myoglobin: The improvement of oxidation activity by replacing Phe-43 with tryptophan,
Biochemistry 40, 1044-1052.
25. Roach, M. P., Ozaki, S., and Watanabe, Y. (2000) Investigations of the myoglobin
cavity mutant H93G with unnatural imidazole proximal ligands as a modular peroxide O-
O bond cleavage model system, Biochemistry 39, 1446-1454.
26. Matsui, T., Ozaki, S., and Watanabe, Y. (1999) Formation and catalytic roles of
compound I in the hydrogen peroxide-dependent oxidations by His64 myoglobin
mutants, J. Am. Chem. Soc. 121, 9952-9957.
27. Tada, T., Watanabe, Y., Matsuoka, A., Ikeda- Saito, M., Imai, K., Yukio, N., and
Shikama, K. (1998) African elephant myoglobin with an unusual autoxidation behavior:
comparison with the H64Q mutant of sperm whale myoglobin, Biochim. Biophys. Acta.
M 1387, 165-176.
28. Dawson, J. H. (2011) His93Gly Myoglobin Cavity Mutant: A Versatile Scaffold for
Modeling Heme Protein Active Sites. Applications to Novel Heme Transport and Redox
Proteins, Biophys. J. 100, 379-379.
29. Du, J., Perera, R., and Dawson, J. H. (2011) Alkylamine-Ligated H93G Myoglobin
Cavity Mutant: A Model System for Endogenous Lysine and Terminal Amine Ligation in
Heme Proteins such as Nitrite Reductase and Cytochrome f, Inorg. Chem. 50, 1242-
1249.

120
30. Du, J., Sono, M., and Dawson, J. H. (2011) Ferric His93Gly myoglobin cavity mutant
and its complexes with thioether and selenolate as heme protein models, J. Porphyr.
Phthalocya. 15, 29-38.
31. Perera, R., Sono, M., Kinloch, R., Zhang, H. M., Tarasev, M., Im, S. C., Waskell, L., and
Dawson, J. H. (2011) Stabilization and spectroscopic characterization of the dioxygen
complex of wild-type cytochrome P4502B4 (CYP2B4) and its distal side E301Q, T302A
and proximal side F429H mutants at subzero temperatures, Biochim. Biophys. Acta.
1814, 69-75.
32. Perera, R., Sono, M., Voegtle, H. L., and Dawson, J. H. (2011) Molecular basis for the
inability of an oxygen atom donor ligand to replace the natural sulfur donor heme axial
ligand in cytochrome P450 catalysis. Spectroscopic characterization of the Cys436Ser
CYP2B4 mutant, Arch. Biochem. Biophys. 507, 119-125.
33. Huang, X., Wang, C. X., Celeste, L. R., Lovelace, L. L., Sun, S. F., Dawson, J. H., and
Lebioda, L. (2012) Complex of myoglobin with phenol bound in a proximal cavity, Acta.
Crystallogr. 68, 1465-1471.
34. Du, J., Sono, M., and Dawson, J. H. (2008) The proximal and distal pockets of the
H93G myoglobin cavity mutant bind identical ligands with different affinities:
Quantitative analysis of imidazole and pyridine binding, J.Spectroscopy 22, 123-141.
35. Qin, J., Perera, R., Lovelace, L. L., Dawson, J. H., and Lebioda, L. (2006) Structures of
thiolate- and carboxylate-ligated ferric H93G myoglobin: Models for cytochrome P450
and for oxyanion-bound heme proteins, Biochemistry 45, 3170-3177.
36. Perera, R., and Dawson, J. H. (2004) Modeling heme protein active sites with the
his93gly cavity mutant of sperm whale myoglobin: complexes with nitrogen-, oxygen-
and sulfur-donor proximal ligands, J. Porphyr. Phthalocya. 8, 246-254.
37. Perera, R., Sono, M., and Dawson, J. H. (2003) His93Gly cavity mutant of sperm whale
myoglobin as a template for ferric, ferrous, and ferryl heme states of mixed ligand
complexes., Biochemistry 42, 8649-8649.
38. Dawson, J. H., Pond, A. E., and Roach, M. P. (2002) H93G myoglobin cavity mutant as
versatile template for modeling heme proteins: Magnetic circular dichroism studies of
thiolate- and imidazole-ligated complexes, Biopolymers 67, 200-206.

121
39. Dawson, J. H., Pond, A. E., Roach, M. P., Perera, R., Dasgupta, A., Thomas, M. R.,
and Boxer, S. G. (2001) Modeling ferrous, ferric and ferryl heme states using His93Gly
myoglobin and related heme iron systems, J. Inorg. Biochem. 86, 39-39.
40. Pond, A. E., Roach, M. P., Thomas, M. R., Boxer, S. G., and Dawson, J. H. (2000) The
H93G myoglobin cavity mutant as a versatile template for modeling heme proteins:
Ferrous, ferric, and ferryl mixed-ligand complexes with imidazole in the cavity, Inorg.
Chem. 39, 6061-6066.
41. Pond, A. E., Roach, M. P., Sono, M., Rux, A. H., Franzen, S., Hu, R., Thomas, M. R.,
Wilks, A., Dou, Y., Ikeda-Saito, M., de Montelano, P. R. O., Woodruff, W. H., Boxer, S.
G., and Dawson, J. H. (1999) Assignment of the heme axial ligand(s) for the ferric
myoglobin (H93G) and heme oxygenase (H25A) cavity mutants as oxygen donors
using magnetic circular dichroism, Biochemistry 38, 7601-7608.
42. Das, T. K., Franzen, S., Pond, A., Dawson, J. H., and Rousseau, D. L. (1999)
Formation of a five-coordinate hydroxide-bound heme in the His93Gly mutant of sperm
whale myoglobin, Inorg. Chem. 38, 1952-1953.
43. Roach, M. P., Franzen, S., Pang, P. S. H., Boxer, S. G., Woodruff, W. H., and Dawson,
J. H. (1997) Thiolate adducts of cavity mutant myoglobin H93G as models for
cytochrome P450, J. Inorg. Biochem, 67. 134-134.
44. Wang, L., Brock, A., Herberich, B., and Schultz, P. G. (2001) Expanding the genetic
code of Escherichia coli, Science 292, 498-500.
45. Budisa, N. (2004) Prolegomena to future experimental efforts on genetic code
engineering by expanding its amino acid repertoire, Angew. Chem. Int. Ed. Engl. 43,
6426-6463.
46. Hendrickson, T. L., de Crecy-Lagard, V., and Schimmel, P. (2004) Incorporation of
nonnatural amino acids into proteins, Annu. Rev. Biochem. 73, 147-176.
47. Budisa, N., and Pal, P. P. (2004) Designing novel spectral classes of proteins with a
tryptophan-expanded genetic code, Biol. Chem. 385, 893-904.
48. Taki, M., Hohsaka, T., Murakami, H., Kuno, A., Hasegawa, T., Taira, K., and Sisido, M.
(2002) A novel fluorescent nonnatural amino acid that can be incorporated into a
specific position of streptavidin, Nucleic Acids Res. Suppl., 203-204.
49. Hohsaka, T., and Sisido, M. (2002) Incorporation of non-natural amino acids into
proteins, Curr. Opin. Chem. Biol. 6, 809-815.

122
50. Taki, M., Hohsaka, T., Murakami, H., Taira, K., and Sisido, M. (2002) Position-specific
incorporation of a fluorophore-quencher pair into a single streptavidin through
orthogonal four-base codon/anticodon pairs, J. Am. Chem. Soc. 124, 14586-14590.
51. Link, A. J., Mock, M. L., and Tirrell, D. A. (2003) Non-canonical amino acids in protein
engineering, Curr. Opin. Biotech. 14, 603-609.
52. Wang, L., and Schultz, P. G. (2005) Expanding the genetic code, Angew. Chem. Int.
Ed. Engl. 44, 34-66.
53. Furter, R. (1998) Expansion of the genetic code: Site-directed p-fluoro-phenylalanine
incorporation in Escherichia coli, Protein Sci. 7, 419-426.
54. Drabkin, H. J., Park, H. J., and RajBhandary, U. L. (1996) Amber suppression in
mammalian cells dependent upon expression of an Escherichia coli aminoacyl-tRNA
synthetase gene, Mol. Cell. Biol. 16, 907-913.
55. Anderson, J. C., Wu, N., Santoro, S. W., Lakshman, V., King, D. S., and Schultz, P. G.
(2004) An expanded genetic code with a functional quadruplet codon, Proc. Natl. Acad.
Sci. U.S.A. 101, 7566-7571.
56. Xie, J. M., and Schultz, P. G. (2005) Adding amino acids to the genetic repertoire, Curr.
Opin. Chem. Biol. 9, 548-554.
57. Anderson, J. C., and Schultz, P. G. (2003) Adaptation of an orthogonal archaeal leucyl-
tRNA and synthetase pair for four-base, amber, and opal suppression, Biochemistry 42,
9598-9608.
58. Santoro, S. W., Anderson, J. C., Lakshman, V., and Schultz, P. G. (2003) An
archaebacteria-derived glutamyl-tRNA synthetase and tRNA pair for unnatural amino
acid mutagenesis of proteins in Escherichia coli, Nuc. Acids Res. 31, 6700-6709.
59. Chin, J. W., Cropp, T. A., Anderson, J. C., Mukherji, M., Zhang, Z. W., and Schultz, P.
G. (2003) An expanded eukaryotic genetic code, Science 301, 964-967.
60. Chin, J. W., Cropp, T. A., Chu, S., Meggers, E., and Schultz, P. G. (2003) Progress
toward an expanded eukaryotic genetic code, Chem. Biol. 10, 511-519.
61. Beene, D. L., Dougherty, D. A., and Lester, H. A. (2003) Unnatural amino acid
mutagenesis in mapping ion channel function, Curr. Opin. Neurobiol. 13, 264-270.
62. Dougherty, D. A. (2000) Unnatural amino acids as probes of protein structure and
function, Curr. Opin. Chem. Biol. 4, 645-652.

123
63. Rodriguez, E. A., Lester, H. A., and Dougherty, D. A. (2006) In vivo incorporation of
multiple unnatural amino acids through nonsense and frameshift suppression, Proc.
Natl. Acad. Sci. U.S.A. 103, 8650-8655.
64. Taki, M., Hohsaka, T., Murakami, H., Taira, K., and Sisido, M. (2001) A non-natural
amino acid for efficient incorporation into proteins as a sensitive fluorescent probe,
FEBS Lett. 507, 35-38.
65. Hohsaka, T., Kajihara, D., Ashizuka, Y., Murakami, H., and Sisido, M. (1999) Efficient
incorporation of nonnatural amino acids with large aromatic groups into streptavidin in
in vitro protein synthesizing systems, J. Am. Chem. Soc. 121, 34-40.
66. Anderson, J. C., Magliery, T. J., and Schultz, P. G. (2002) Exploring the limits of codon
and anticodon size, Chem. Biol. 9, 237-244.
67. Hohsaka, T., Ashizuka, Y., Murakami, H., and Sisido, M. (2001) Five-base codons for
incorporation of nonnatural amino acids into proteins, Nuc. Acids Res. 29, 3646-3651.
68. Kwon, I., Kirshenbaum, K., and Tirrell, D. A. (2003) Breaking the degeneracy of the
genetic code, J. Am. Chem. Soc. 125, 7512-7513.
69. Ibba, M., and Soll, D. (2000) Aminoacyl-tRNA synthesis, Annual Review of
Biochemistry 69, 617-650.
70. Ibba, M., Kast, P., and Hennecke, H. (1994) Substrate-Specificity Is Determined by
Amino-Acid Binding Pocket Size in Escherichia-Coli Phenylalanyl-Transfer-Rna
Synthetase, Biochemistry 33, 7107-7112.
71. Wollenberger, U., Spricigo, R., Leimkuhler, S., and Schroder, K. (2008) Protein
electrodes with direct electrochemical communication, Adv. Biochem. Eng. Biotechnol.
109, 19-64.
72. Karyakin, A. A. (2012) Principles of direct (mediator free) bioelectrocatalysis,
Bioelectrochemistry 88, 70-75.
73. D'Souza, F., Rogers, L. M., O'Dell, E. S., Kochman, A., and Kutner, W. (2005)
Immobilization and electrochemical redox behavior of cytochrome c on fullerene film-
modified electrodes, Bioelectrochemistry 66, 35-40.
74. Betso, S. R., Klapper, M. H., and Anderson, L. B. (1972) Electrochemical studies of
heme proteins. Coulometric, polarographic, and combined spectroelectrochemical
methods for reduction of the heme prosthetic group in cytochrome c, J. Am. Chem. Soc.
94, 8197-8204.

124
75. Yeh, P., and Kuwana, T. (1977) Reversible Electrode-Reaction of Cytochrome-C,
Chem. Lett., 1145-1148.
76. Demontellano, P. R. O. (1992) Catalytic Sites of Hemoprotein Peroxidases, Annu. Rev.
Pharmacol. 32, 89-107.
77. Atamna, H., and Boyle, K. (2006) Amyloid-beta peptide binds with heme to form a
peroxidase: relationship to the cytopathologies of Alzheimer's disease, Proc. Natl. Acad.
Sci. U.S.A. 103, 3381-3386.
78. Karthikeyan, M., Jayakumar, V., Radhika, K., Bhaskaran, R., Velazhahan, R., and Alice,
D. (2005) Induction of resistance in host against the infection of leaf blight pathogen
(Alternaria palandui) in onion (Allium cepa var aggregatum), Indian J. Biochem.
Biophys. 42, 371-377.
79. Dawson, J. H. (1988) Probing structure-function relations in heme-containing
oxygenases and peroxidases, Science 240, 433-439.
80. Sono, M., Roach, M. P., Coulter, E. D., and Dawson, J. H. (1996) Heme-containing
oxygenases, Chem. Rev. 96, 2841-2887.
81. Schlichting, I., Berendzen, J., Chu, K., Stock, A. M., Maves, S. A., Benson, D. E.,
Sweet, B. M., Ringe, D., Petsko, G. A., and Sligar, S. G. (2000) The catalytic pathway
of cytochrome P450cam at atomic resolution, Science 287, 1615-1622.
82. Gerber, N. C., and Sligar, S. G. (1992) Catalytic Mechanism of Cytochrome-P-450 -
Evidence for a Distal Charge Relay, J. Am. Chem. Soc. 114, 8742-8743.
83. Gerber, N. C., and Sligar, S. G. (1994) A Role for Asp-251 in Cytochrome P-450cam
Oxygen Activation, J. Biol. Chem. 269, 4260-4266.
84. Vidakovic, M., Sligar, S. G., Li, H. Y., and Poulos, T. L. (1998) Understanding the role of
the essential Asp251 in cytochrome P450cam using site-directed mutagenesis,
crystallography, and kinetic solvent isotope effect, Biochemistry 37, 9211-9219.
85. Sitter, A. J., Reczek, C. M., and Terner, J. (1985) Heme-Linked Ionization of
Horseradish-Peroxidase Compound-Ii Monitored by the Resonance Raman Fe(Iv)=O
Stretching Vibration, J. Biol. Chem. 260, 7515-7522.
86. Gajhede, M., Schuller, D. J., Henriksen, A., Smith, A. T., and Poulos, T. L. (1997)
Crystal structure of horseradish peroxidase C at 2.15 angstrom resolution, Nat. Struct.
Biol. 4, 1032-1038.

125
87. Roach, M. P., Chen, Y. P., Woodin, S. A., Lincoln, D. E., Lovell, C. R., and Dawson, J.
H. (1997) Notomastus lobatus chloroperoxidase and Amphitrite ornata
dehaloperoxidase both contain histidine as their proximal heme iron ligand,
Biochemistry 36, 2197-2202.
88. Poulos, T. L. (1988) Heme Enzyme Crystal-Structures, Adv. Inorg. Biochem. 7, 1-36.
89. Poulos, T. L., Finzel, B. C., Gunsalus, I. C., Wagner, G. C., and Kraut, J. (1985) The
2.6-A crystal structure of Pseudomonas putida cytochrome P-450, J. Biol. Chem. 260,
16122-16130.
90. Watanabe, Y., Nakajima, H., and Ueno, T. (2007) Reactivities of oxo and peroxo
intermediates studied by hemoprotein mutants, Acc. Chem. Res. 40, 554-562.
91. Perera, R., Sono, M., Raner, G. M., and Dawson, J. H. (2005) Subzero-temperature
stabilization and spectroscopic characterization of homogeneous oxyferrous complexes
of the cytochrome P450BM3 (CYP102) oxygenase domain and holoenzyme, Biochem.
Bioph. Res. 338, 365-371.
92. Alfonta, L., Zhang, Z., Uryu, S., Loo, J. A., and Schultz, P. G. (2003) Site-specific
incorporation of a redox-active amino acid into proteins, J. Am. Chem. Soc. 125, 14662-
14663.
93. Magliery, T. J., Anderson, J. C., and Schultz, P. G. (2001) Expanding the genetic code:
Selection of efficient suppressors of four-base codons and identification of "shifty" four-
base codons with a library approach in Escherichia coli, J. Mol. Biol. 307, 755-769.
94. Park, H. S., Hohn, M. J., Umehara, T., Guo, L. T., Osborne, E. M., Benner, J., Noren, C.
J., Rinehart, J., and Soll, D. (2011) Expanding the Genetic Code of Escherichia coli with
Phosphoserine, Science 333, 1151-1154.
95. Seyedsayamdost, M. R., Xie, J., Chan, C. T. Y., Schultz, P. G., and Stubbe, J. (2007)
Site-specific insertion of 3-aminotyrosine into subunit alpha 2 of E-coli ribonucleotide
reductase: Direct evidence for involvement of Y-730 and Y-731 in radical propagation,
J. Am. Chem. Soc. 129, 15060-15071.
96. Paul, K. G., Theorell, H., Akeson, A. (1953) The Molar Light Absorption of Pyridine
Ferroprotoporphyrin, Acta. Chem. Scand. 1284-1287.
97. Ozaki, S., and Demontellano, P. R. O. (1995) Molecular Engineering of Horseradish-
Peroxidase - Thioether Sulfoxidation and Styrene Epoxidation by Phe-41 Leucine and
Threonine Mutants, J. Am. Chem. Soc. 117, 7056-7064.

126
98. King, N. K., and Winfield, M. E. (1963) The mechanism of metmyoglobin oxidation, J.
Biol. Chem. 238, 1520-1528.
99. Ozaki, S., and Ishikawa, Y. (2006) One- and two-electron oxidation by the GLY-65 to
threonine myoglobin mutant, React. Kinet. Catal. Lett. 89, 21-28.
100. Carlsen, C. U., and Skibsted, L. H. (2004) Myoglobin species with enhanced
prooxidative activity is formed during mild proteolysis by pepsin, J. Agric. Food. Chem.
52, 1675-1681.
101. Carlsen, C. U., Skovgaard, I. M., and Skibsted, L. H. (2003) Pseudoperoxidase activity
of myoglobin: kinetics and mechanism of the peroxidase cycle of myoglobin with H2O2
and 2,2-azino-bis(3-ethylbenzthiazoline-6-sulfonate) as substrates, J. Agric. Food.
Chem. 51, 5815-5823.
102. Iwagami, S. G., Creagh, A. L., Haynes, C. A., Borsari, M., Felli, I. C., Piccioli, M., and
Eltis, L. D. (1995) The Role of a Conserved Tyrosine Residue in High-Potential Iron-
Sulfur Proteins, Protein Sci. 4, 2562-2572.
103. Sarma, S., DiGate, R. J., Goodin, D. B., Miller, C. J., and Guiles, R. D. (1997) Effect of
axial ligand plane reorientation on electronic and electrochemical properties observed in
the A67V mutant of rat cytochrome b(5), Biochemistry 36, 5658-5668.
104. Zemojtel, T., Rini, M., Heyne, K., Dandekar, T., Nibbering, E. T. J., and Kozlowski, P.
M. (2004) NO-bound myoglobin: Structural diversity and dynamics of the NO ligand, J.
Am. Chem. Soc. 126, 1930-1931.
105. Schoenbo.Bp, Watson, H. C., and Kendrew, J. C. (1965) Binding of Xenon to Sperm
Whale Myoglobin, Nature 207, 28-&.
106. Sundaramoorthy, M., Kishi, K., Gold, M. H., and Poulos, T. L. (1994) The crystal
structure of manganese peroxidase from Phanerochaete chrysosporium at 2.06-A
resolution, J. Biol. Chem. 269, 32759-32767.
107. Kunishima, N., Amada, F., Fukuyama, K., Kawamoto, M., Matsunaga, T., and
Matsubara, H. (1996) Pentacoordination of the heme iron of Arthromyces ramosus
peroxidase shown by a 1.8 A resolution crystallographic study at pH 4.5, FEBS Lett
378, 291-294.
108. Finzel, B. C., Poulos, T. L., and Kraut, J. (1984) Crystal structure of yeast cytochrome c
peroxidase refined at 1.7-A resolution, J. Biol. Chem. 259, 13027-13036.

127
109. Springer, B. A., and Sligar, S. G. (1987) High-level expression of sperm whale
myoglobin in Escherichia coli, Proc. Natl. Acad. Sci. U. S. A. 84, 8961-8965.
110. Phillips, G. N., Jr., Arduini, R. M., Springer, B. A., and Sligar, S. G. (1990) Crystal
structure of myoglobin from a synthetic gene, Proteins 7, 358-365.
111. Quillin, M. L., Arduini, R. M., Olson, J. S., and Phillips, G. N., Jr. (1993) High-resolution
crystal structures of distal histidine mutants of sperm whale myoglobin, J. Mol. Biol.
234, 140-155.
112. Matsui, T., Nagano, S., Ishimori, K., Watanabe, Y., and Morishima, I. (1996)
Preparation and reactions of myoglobin mutants bearing both proximal cysteine ligand
and hydrophobic distal cavity: protein models for the active site of P-450, Biochemistry
35, 13118-13124.
113. Edwards, S. L., Raag, R., Wariishi, H., Gold, M. H., and Poulos, T. L. (1993) Crystal
structure of lignin peroxidase, Proc. Natl. Acad. Sci. U. S. A. 90, 750-754.
114. Poulos, T. L., Freer, S. T., Alden, R. A., Edwards, S. L., Skogland, U., Takio, K.,
Eriksson, B., Xuong, N., Yonetani, T., and Kraut, J. (1980) The crystal structure of
cytochrome c peroxidase, J. Biol. Chem. 255, 575-580.
115. Patterson, W. R., and Poulos, T. L. (1995) Crystal structure of recombinant pea
cytosolic ascorbate peroxidase, Biochemistry 34, 4331-4341.
116. Matsui, T., Ozaki, S., Liong, E., Phillips, G. N., Jr., and Watanabe, Y. (1999) Effects of
the location of distal histidine in the reaction of myoglobin with hydrogen peroxide, J.
Biol. Chem. 274, 2838-2844.
117. Takano, T. (1977) Structure of myoglobin refined at 2-0 A resolution. I. Crystallographic
refinement of metmyoglobin from sperm whale, J. Mol. Biol. 110, 537-568.
118. Sono, M., Perera, R., Jin, S., Makris, T. M., Sligar, S. G., Bryson, T. A., and Dawson, J.
H. (2005) The influence of substrate on the spectral properties of oxyferrous wild-type
and T252A cytochrome P450-CAM, Arch. Biochem. Biophys. 436, 40-49.
119. Montgomery, C. D. (2007) pi Backbonding in carbonyl complexes and carbon-oxygen
stretching frequencies: A molecular modeling exercise, J. Chem. Educ. 84, 102-105.
120. Kimmich, N., Das, A., Sevrioukova, I., Meharenna, Y., Sligar, S. G., and Poulos, T. L.
(2007) Electron transfer between cytochrome P450cin and its FMN-containing redox
partner, cindoxin, J. Biol. Chem. 282, 27006-27011.

128
121. Hiner, A. N. P., Raven, E. L., Thorneley, R. N. F., Garcia-Canovas, F., and Rodriguez-
Lopez, J. N. (2002) Mechanisms of compound I formation in heme peroxidases, J.
Inorg. Biochem. 91, 27-34.
122. Yonetani, T., and Schleyer, H. (1967) Studies on cytochrome c peroxidase. IX. The
reaction of ferrimyoglobin with hydroperoxides and a comparison of peroxide-induced
compounds of ferrimyoglobin and cytochrome c peroxidase, J. Biol. Chem. 242, 1974-
1979.
123. Mukherjee, S., Sengupta, K., Das, M. R., Jana, S. S., and Dey, A. (2012) Site-specific
covalent attachment of heme proteins on self-assembled monolayers, J. Biol. Inorg.
Chem. 17, 1009-1023.
124. Liu, S. Q., and Ju, H. X. (2003) Electrocatalysis via direct electrochemistry of myoglobin
immobilized on colloidal gold nanoparticles, Electroanalysis 15, 1488-1493.
125. Rusling, J. F. (1998) Enzyme bioelectrochemistry in cast biomembrane-like films, Acc.
Chem. Res. 31, 363-369.
126. Armstrong, F. A., Hill, H. A. O., and Walton, N. J. (1988) Direct Electrochemistry of
Redox Proteins, Acc. Chem. Res. 21, 407-413.
127. Yarman, A., Nagel, T., Gajovic-Eichelmann, N., Fischer, A., Wollenberger, U., and
Scheller, F. W. (2011) Bioelectrocatalysis by Microperoxidase-11 in a Multilayer
Architecture of Chitosan Embedded Gold Nanoparticles, Electroanalysis 23, 611-618.
128. Moyo, M., Okonkwo, J. O., and Agyei, N. M. (2012) Recent Advances in Polymeric
Materials Used as Electron Mediators and Immobilizing Matrices in Developing Enzyme
Electrodes, Sensors 12, 923-953.
129. Hill, H. A. O. (1983) The Exploitation of the Electrochemistry of Proteins, Biochem. Soc.
Trans. 11, 453-455.
130. Krzeminski, L., Cronin, S., Ndamba, L., Canters, G. W., Aartsma, T. J., Evans, S. D.,
and Jeuken, L. J. C. (2011) Orientational Control over Nitrite Reductase on Modified
Gold Electrode and Its Effects on the Interfacial Electron Transfer, J. Phys. Chem. B
115, 12607-12614.
131. Uygun, A., Oksuz, L., Chowdhury, S., and Bhethanabotla, V. (2010) Fluorescence study
of protein immobilization on poly(4-hydroxyphenyl thiophene-3-carboxylate)-coated
electrodes, Mater Sci Eng C Mater Biol Appl 30, 868-872.

129
132. Gates, A. J., Kemp, G. L., To, C. Y., Mann, J., Marritt, S. J., Mayes, A. G., Richardson,
D. J., and Butt, J. N. (2011) The relationship between redox enzyme activity and
electrochemical potential-cellular and mechanistic implications from protein film
electrochemistry, Phys. Chem. Chem. Phys. 13, 7720-7731.
133. Rusling, J. F., and Forster, R. J. (2003) Electrochemical catalysis with redox polymer
and polyion-protein films, J. Colloid Interface Sci. 262, 1-15.
134. Babaei, A., Garrett, D. J., and Downard, A. J. (2012) Electrochemical Investigations on
a Third Generation Biosensor for Determination of Hydrogen Peroxide Based on
Immobilization of Myoglobin on a Novel Platinum Nanoparticle/Carbon Nanotube/Ionic
Liquid/Nafion Composite, Int. J. Electrochem. Sc. 7, 3141-3154.
135. Loget, G., Chevance, S., Poriel, C., Simonneaux, G., Lagrost, C., and Rault-Berthelot,
J. (2011) Direct Electron Transfer of Hemoglobin and Myoglobin at the Bare Glassy
Carbon Electrode in an Aqueous BMI.BF4 Ionic-Liquid Mixture, Chemphyschem. 12,
411-418.
136. Choi, O., Kim, B. C., An, J. H., Min, K., Kim, Y. H., Um, Y., Oh, M. K., and Sang, B. I.
(2011) A biosensor based on the self-entrapment of glucose oxidase within biomimetic
silica nanoparticles induced by a fusion enzyme, Enzyme Microb. Technol. 49, 441-445.
137. You, C. P., Yan, X. W., Kong, J. L., Zhao, D. Y., and Liu, B. H. (2008) Direct
electrochemistry of myoglobin based on bicontinuous gyroidal mesoporous carbon
matrix, Electrochem. Commun. 10, 1864-1867.
138. Ferri, T., Poscia, A., and Santucci, R. (1998) Direct electrochemistry of membrane-
entrapped horseradish peroxidase. Part I. A voltammetric and spectroscopic study,
Bioelectroch. Bioener 44, 177-181.
139. Chen, L. F., Xie, H. Q., and Li, J. (2012) Electrochemical glucose biosensor based on
silver nanoparticles/multiwalled carbon nanotubes modified electrode, J. Solid State
Electrochem. 16, 3323-3329.
140. Zhang, F., Wu, J., and Zhang, H. B. (2012) Construction of hyaluronan-silver
nanoparticle-hemoglobin multilayer composite film and investigations on its
electrocatalytic properties, J. Solid State Electrochem. 16, 1683-1692.
141. Wang, X. H., Han, M., Bao, J. C., Tu, W. W., and Dai, Z. H. (2012) A superoxide anion
biosensor based on direct electron transfer of superoxide dismutase on sodium alginate

130
sol-gel film and its application to monitoring of living cells, Analytica. Chimica. Acta.
717, 61-66.
142. Nadzhafova, O., Etienne, M., and Walcarius, A. (2007) Direct electrochemistry of
hemoglobin and glucose oxidase in electrodeposited sol-gel silica thin films on glassy
carbon, Electrochem. Commun. 9, 1189-1195.
143. Chen, H. J., and Dong, S. J. (2007) Direct electrochemistry and electrocatalysis of
horseradish peroxidase immobilized in sol-gel-derived ceramic-carbon nanotube
nanocomposite film, Biosens. Bioelectron. 22, 1811-1815.
144. Ran, Q., Peng, R., Liang, C., Ye, S. Q., Xian, Y. Z., Zhang, W. J., and Jin, L. T. (2011)
Covalent immobilization of horseradish peroxidase via click chemistry and its direct
electrochemistry, Talanta 83, 1381-1385.
145. Mazzei, F., Favero, G., Frasconi, M., Tata, A., and Pepi, F. (2009) Electron-Transfer
Kinetics of Microperoxidase-11 Covalently Immobilised onto the Surface of Multi-Walled
Carbon Nanotubes by Reactive Landing of Mass-Selected Ions, Chem.Eur. J. 15, 7359-
7367.
146. Albrecht, T., Li, W. W., Ulstrup, J., Haehnel, W., and Hildebrandt, P. (2005)
Electrochemical and spectroscopic investigations of immobilized de novo designed
heme proteins on metal electrodes, Chemphyschem. 6, 961-970.
147. Berg, J. M., Tymoczko, J. L., and Stryer, L. (2006) in Biochemistry, 6th ed., pp 491-550,
Freeman, New York.
148. Ozaki, S., Matsui, T., and Watanabe, Y. (1996) Conversion of myoglobin into a highly
stereospecific peroxygenase by the L29H/H64L mutation, J. Am. Chem. Soc. 118,
9784-9785.
149. Lvov, Y. M., Lu, Z. Q., Schenkman, J. B., Zu, X. L., and Rusling, J. F. (1998) Direct
electrochemistry of myoglobin and cytochrome p450(cam) in alternate layer-by-layer
films with DNA and other polyions, J. Am. Chem. Soc. 120, 4073-4080.
150. Kamau, G. N., Guto, M. P., Munge, B., Panchagnula, V., and Rusling, J. F. (2003)
Myoglobin coadsorbed on electrodes from microemulsions provides reversible
electrochemistry and tunable electrochemical catalysis, Langmuir 19, 6976-6981.
151. Paulo, T. D., Diogenes, I. C. N., and Abruna, H. D. (2011) Direct Electrochemistry and
Electrocatalysis of Myoglobin Immobilized on L-Cysteine Self-Assembled Gold
Electrode, Langmuir 27, 2052-2057.

131
152. Zhang, J. D., and Oyama, M. (2005) Gold nanoparticle-attached ITO as a biocompatible
matrix for myoglobin immobilization: direct electrochemistry and catalysis to hydrogen
peroxide, J. Electroanal. Chem. 577, 273-279.
153. Schultz, K. C., Supekova, L., Ryu, Y., Xie, J., Perera, R., and Schultz, P. G. (2006) A
genetically encoded infrared probe, J. Am. Chem. Soc. 128, 13984-13985.
154. Seyedsayamdost, M. R., Xie, J., Chan, C. T., Schultz, P. G., and Stubbe, J. (2007) Site-
specific insertion of 3-aminotyrosine into subunit alpha2 of E. coli ribonucleotide
reductase: direct evidence for involvement of Y730 and Y731 in radical propagation, J.
Am. Chem. Soc. 129, 15060-15071.
155. Stamos, B., Loredo, L., Chand, S., Phan, T. V., Zhang, Y. B., Mohapatra, S.,
Rajeshwar, K., and Perera, R. (2012) Biosynthetic approach for functional protein
microarrays, Anal. Biochem. 424, 114-123.
156. de Groot, M. T., Merkx, M., and Koper, M. T. M. (2005) Heme release in myoglobin-
DDAB films and its role in electrochemical NO reduction, J. Am. Chem. Soc. 127,
16224-16232.
157. de Groot, M. T., Merkx, M., and Koper, M. T. M. (2006) Additional evidence for heme
release in myoglobin-DDAB films on pyrolitic graphite, Electrochem. Commun. 8, 999-
1004.
158. de Groot, M. T., Merkx, M., and Koper, M. T. M. (2007) Evidence for heme release in
layer-by-layer assemblies of myoglobin and polystyrenesulfonate on pyrolitic graphite,
J. Biol. Inorg. Chem. 12, 761-766.
159. Nakajima, N., and Ikada, Y. (1995) Mechanism of Amide Formation by Carbodiimide for
Bioconjugation in Aqueous-Media, Bioconjugate Chem. 6, 123-130.
160. Wang, L. W., Ran, Q., Tian, Y. A., Xu, J. J., Xian, Y. Z., Peng, R., and Jin, L. T. (2010)
Covalent immobilization of redox protein via click chemistry and carbodiimide reaction:
Direct electron transfer and biocatalysis, J. Colloid Interface Sci. 350, 544-550.
161. Bard, A. J., and Faulkner, L. R. (2001) Electrochemical Methods, Wiley, New York.
162. Zhang, H. M., and Li, N. Q. (2001) The direct electrochemistry of myoglobin at a DL-
homocysteine self-assembled gold electrode, Bioelectrochemistry 53, 97-101.
163. Xia, P. P., Liu, H. Q., and Tian, Y. (2009) Cathodic detection of H2O2 based on
nanopyramidal gold surface with enhanced electron transfer of myoglobin, Biosensors.
Bioelectron. 24, 2470-2474.

132
164. Chai, H. M., Liu, H. Y., Guo, X. H., Zheng, D., Kutes, Y., Huey, B. D., Rusling, J. F., and
Hu, N. F. (2012) Long Distance Electron Transfer Across > 100 nm Thick Au
Nanoparticle/Polyion Films to a Surface Redox Protein, Electroanalysis 24, 1129-1140.
165. Xu, Y. X., Hu, C. G., and Hu, S. S. (2010) Electrochemical behavior of biocatalytical
composite based on heme-proteins, didodecyldimethylammonium bromide and room-
temperature ionic liquid, Analytica Chimica Acta 663, 19-26.
166. Mayo, S. L., Ellis, W. R., Jr., Crutchley, R. J., and Gray, H. B. (1986) Long-range
electron transfer in heme proteins, Science 233, 948-952.
167. Beier, M., and Hoheisel, J. D. (2004) Derivatization of glass and polypropylene
surfaces, Curr Protoc Nucleic Acid Chem. 12, Unit 12.4.
168. Rideout, D. C., and Breslow, R. (1980) Hydrophobic Acceleration of Diels-Alder
Reactions, J. Am. Chem. Soc. 102, 7816-7817.
169. Ordway, G. A., and Garry, D. J. (2004) Myoglobin: an essential hemoprotein in striated
muscle, J. Exp. Biol. 207, 3441-3446.
170. Collman, J. P., Boulatov, R., Sunderland, C. J., and Fu, L. (2004) Functional analogues
of cytochrome c oxidase, myoglobin, and hemoglobin, Chem. Rev. 104, 561-588.
171. Loew, G. H., and Harris, D. L. (2000) Role of the heme active site and protein
environment in structure, spectra, and function of the cytochrome p450s, Chem. Rev.
100, 407-419.
172. Anderson, J. L., and Chapman, S. K. (2005) Ligand probes for heme proteins, Dalton
Trans, 13-24.
173. Coulter, E. D., Sono, M., Chang, C. K., Lopez, O., and Dawson, J. H. (1995) Electron
paramagnetic resonance spectroscopy as a probe of coordination structure in green
heme systems: Iron chlorins and iron formylporphyrins reconstituted into myoglobin,
Inorg. Chim. Acta 240, 603-608.
174. Solomon, E. I. (2006) Spectroscopic methods in bioinorganic chemistry: Blue to green
to red copper sites, Inorg. Chem. 45, 8012-8025.
175. Roach, M. P., Pond, A. E., Thomas, M. R., Boxer, S. G., and Dawson, J. H. (1999) The
role of the distal and proximal protein environments in controlling the ferric spin state
and in stabilizing thiolate ligation in heme systems: Thiolate adducts of the myoglobin
H93G cavity mutant, J. Am. Chem. Soc. 121, 12088-12093.

133
176. Olson, J. S., and Phillips, G. N. (1997) Myoglobin discriminates between O-2, NO, and
CO by electrostatic interactions with the bound ligand, J. Biol Inorg. Chem. 2, 544-552.
177. Maurus, R., Bogumil, R., Nguyen, N. T., Mauk, A. G., and Brayer, G. (1998) Structural
and spectroscopic studies of azide complexes of horse heart myoglobin and the His-64-
->Thr variant, Biochem. J. 332, 67-74.

134
BIOGRAPHICAL INFORMATION
Subhash Chand the elder son of Mr. Keshaw Prasad Yadav and Mrs. Urmila Devi
Yadav has obtained his high school education at Shri Shanker Ji Intermediate College,
Pushpanagar, Azamgarh. Ever since the initial years of education, his interests were focused in
subjects like Science and Mathematics. In his primary school days, he has been academically
oriented, always stood out for his scholastic performance and earned his way into the top rank
in his class. He received B.Sc. (Biology) degree from the Ewing Christian College, Allahabad,
India and then M.Sc. (Botany) degree from University of Allahabad, Allahabad, India. His
respectable educational credentials landed him in the Indian Institute of Technology, Kharagpur
(IIT Kharagpur), the prestigious science and technology institute of India, to pursue Master of
Technology in Applied Botany (Biotechnology). After receiving his Masters degree he joined
Meerut Institute of Engineering and Technology and served as Lecturer in Department of
Biotechnology and Applied Sciences. He soon realized that higher education plays an important
role in advancing one’s professional life and moved to USA to join University of Texas at
Arlington so as carry out doctoral research. He joined Dr. Roshan Perera’s group to obtain his
Doctoral degree in Biochemistry. There he conducted stimulating research to incorporate
unnatural amino acids in genetically modified myoglobin mutants so as to mimic the catalytic
activities of various heme proteins.