Embed Size (px)
Citation preview


General Discussion
133
General discussion
Cardiovascular disease is a leading cause of morbidity and mortality in the western world. Acute
myocardial infarction (AMI) is an important contributor to this cause since it induces apoptosis, necrosis
and inflammation in the myocardium, resulting in a loss of cardiomyocytes.[1,2] As spontaneous
regeneration of cardiomyocytes is limited, injured myocardium is replaced by non-contractile scar tissue,
which leads to adverse remodelling of the left ventricle and can result in heart failure.[2,3] Therapy for
AMI focuses on restoration of reperfusion and on prevention of remodeling and secondary
cardiovascular events via specific drug therapy.[4,5] Although these strategies significantly reduce
mortality,[6,7] they do not replace lost cardiomyocytes. The ideal AMI therapy would therefore be to
stimulate revascularization of the ischemic region, to minimize the loss of cardiomyocytes, thus limiting
scar tissue formation, and to replace lost cardiomyocytes.[5] New therapies for AMI, focusing on
inflammatory inhibitors and stem cell therapy using adipose derived stem cells, are the subject of this
thesis.
Inflammatory inhibitors
The major goal of current AMI treatment is restoration of reperfusion in the ischemic myocardium.
Reperfusion, however, results in the induction of inflammation in the heart (figure 1). Through the
binding of inflammatory mediators on injured and dead cardiomyocytes, inflammatory cells, especially
neutrophilic granulocytes and macrophages are attracted, subsequently clearing injured cardiomyocytes
from the heart. However, this inflammatory reaction induces additional damage to the myocardium.[8-
10] After AMI, also non-lethal damaged cardiomyocytes lose their membrane phospholipid
asymmetry.[11] As a result, the anionic phospholipids phosphatidylserine and phosphatidyl-
ethanolamine, which are normally kept within the inner plasma membrane leaflet, are then exposed in
the outer membrane leaflet, a process known as `flip-flop’.[12] It has shown that this flip-flop can be a
reversible phenomenon.[13,14] However, during reperfusion also these flip-flopped/reversibly damaged
cells are targeted by inflammatory mediators, resulting in cell death of these cardiomyocytes, thereby
increasing infarct size in time. A crucial mediator herein is the acute phase protein type IIA secretory-
type phospholipase A2 (sPLA2-IIA), which was shown to bind to and to induce cell death of these
reversibly damaged cardiomyocytes.[13,15,16] As depicted in figure 1, sPLA2-IIA can not only induce cell
death via CRP and complement binding/activation, but also via a direct cytotoxic effect, independent of
other inflammatory mediators.[13] Inhibition of this inflammation cascade might therefore form a
potent therapy to reduce cell death of cardiomyocytes after AMI. To this purpose we analyzed the effect
of the sPLA2-IIA inhibitor PX-18 on cell death after AMI.We then found in a rat model of AMI, that
treatment with PX-18 significantly reduced infarct size and mortality, without impairing wound
healing.[17] The latest is of utmost importance since the inflammation response itself is also essential for
the healing process of the heart after myocardial infarction.[15] When using PX-18, CRP may still bind to
necrotic cardiomyocytes independent of sPLA2-IIA, meaning that necrotic tissue can be cleared via the
CRP-complement dependent cascade (Figure 1).[9,13] Interestingly, we also found in vitro that PX-18
reduced membrane flip-flop and apoptosis in cardiomyocytes subjected to metabolic inhibition in the

Chapter 8
134
absence of sPLA2-IIA, suggesting an additional cell protective effect of PX-18 independent of sPLA2-
IIA.[17] Finally we found that after treatment with PX-18, the numbers of so called late type
macrophages in the heart were significantly increased after AMI.[17] The function of macrophages in
wound healing after AMI is quite diverse, including digestion of damaged tissue, but also inhibition of
inflammation and promotion of regeneration.[18,19] It has been shown that early type macrophages
(type 1) digest damaged tissue, while late type macrophages (type 2) promote healing.[19,20] Therefore
the finding that these late type macrophages were increased after treatment with PX-18 suggests
another mechanism through which PX-18 exerts a protective effect (Figure 1). Taken together, PX-18 can
exert a cardiac protective effect on at least 4 different levels: (1) PX-18 inhibits membrane flip-flop, and
thus prevents the pro-inflammatory state of the plasma membrane of cardiomyocytes. (2) PX-18 inhibits
sPLA2-IIA activity, and thus prohibits sPLA2-IIA mediated cell death of at that moment reversibly
damaged cardiomyocytes. (3) PX-18 directly inhibits apoptosis of cardiomyocytes. (4) PX-18 attracts late
type macrophages which improve wound healing. (Figure 1)
Another inflammatory inhibitor we have studied in this thesis was clusterin. Clusterin is not only
an inhibitor of complement, but also protects cardiomyocytes after ischemia in vitro, independent of
complement.[21] We have now shown in a rat model of AMI, that treatment with clusterin significantly
reduced infarct size and mortality, as compared to untreated rats, again without impairing wound
healing.[22] The exact mechanism through which clusterin protects cardiomyocytes after AMI, next to
complement inhibition, is however not known. It has been shown that during oxidative stress clusterin
exerts a protective effect in H9c2 cells (a cardiomyoblast cell-line) via the Akt/GSK-3β signaling
pathway.[28] It also has been suggested that clusterin plays a role in membrane recycling of
cardiomyocytes in response to ischemia,[21,23-25] and that it functions as a secreted heat-shock protein
or chaperone molecule. As such it has an active role in the removal of damaged cells or toxic molecules
derived from (cardiac) cells.[25-27] The cytoprotective effect of clusterin has been related to its receptor
megalin.[29,30] Although we have shown that megalin is expressed in cardiomyocytes of the human
heart, the cell protective effect of clusterin was independent of megalin, at least in vitro after metabolic
inhibition, which is in line with findings of Jun et al.[22,28] We now have also shown that after treatment
with clusterin, the number of late type macrophages was significantly increased after AMI. Taken
together, also clusterin forms a potential therapeutic agent in the treatment of AMI.
Figure 1. In vivo mechanisms of cell death after AMI: the effect of PX-18 and clusterin hereon. Ischemia induces
reversible and irreversible membrane flip-flop in cardiomyocytes. During the subsequent inflammatory reaction, sPLA2-IIA
binds to flip-flopped membranes and induces cell-death, resulting in necrosis of both reversibly as irreversibly damaged
cardiomyocytes. PX-18 (red) can prevent cell death of reversibly flip-flopped cardiomyocytes by inhibiting flip-flop, by
inhibiting direct cytotoxic effects of sPLA2-IIA, and also by inhibiting sPLA2-IIA-facilitated binding of CRP and complement.
Importantly, irreversibly damaged cardiomyocytes can still be cleared due to binding of CRP to these cells independent of
sPLA2-IIA. Clusterin (blue) prevents cell death by inhibition of complement, and by inhibition of flip-flop independent of
the clusterin receptor megalin. In addition both PX-18 and clusterin induce macrophage infiltration, therewith improving
cardiac remodeling. C: complement, CRP: C-reactive protein, †: cell death, ⊥: inhibition, o: hydrophobic phospholipids
(phosphatidylcholine) that in normal cells composes most of the outer leaflet of plasma membranes, •: anionic
phospholipids phosphatidylserine (PS) and phosphatidylethanolamine (PE) that are normally kept within the inner leaflet
of the plasma membrane.

General Discussion
135
intr
acellu
lar
extr
a c
ellu
lar
no
rma
l
pla
sm
a m
em
bra
ne
intr
acellu
lar
extr
a c
ellu
lar
intr
acellu
lar
extr
a c
ellu
lar
extr
a c
ellu
lar
no
rma
l
pla
sm
a m
em
bra
ne
Ex
tra
ce
llu
lar In
tra
ce
llu
lar
†P
las
ma
me
mb
ran
e o
f
he
alt
hy c
ard
iom
yo
cyte
s
Ch
an
ge
s o
f th
e p
las
ma
me
mb
ran
e o
f c
ard
iom
yo
cyte
s
du
rin
g is
ch
em
ia/r
ep
erf
us
ion
in
du
ce
d i
nfl
am
ma
tio
n
Irre
vers
ibly
dam
ag
ed
Me
ga
lin
Fig
ure
1.
Revers
ibly
dam
ag
ed
Me
ga
lin
Macro
phag
e
infi
ltra
tio
n
++
xx
C CR
P
sP
LA
2sP
LA
2
Clu
ste
rin
PX
-18
+
Neutr
op
hili
c
gra
nulo
cyte
s
Macro
phag
e
infi
ltra
tio
n
Neutr
op
hili
c
gra
nulo
cyte
s
Macro
phag
e
infi
ltra
tio
n
†C C
RP
PX
-18
Clu
ste
rin
sP
LA
2sP
LA
2C C
RP
+M
acro
phag
e
infi
ltra
tio
n
++

Chapter 8
136
Next, an important topic is the optimal timeframe to apply these inflammatory inhibitors. We have
found that application of a single bolus of clusterin as well as PX-18 directly after reperfusion did not
result in a significant reduction of infarct size. Reduction was only found when the inflammatory
inhibitors were administered repeatedly during the first 3 days after infarct induction.[17,22] These
results therefore indicate that the acute inflammation period after AMI plays a major role in cell death
induction after AMI, in addition to ischaemia itself. In conclusion, inhibition of the inflammatory
response post AMI, by administration of PX-18 and clusterin, forms a new promising therapeutic
approach to decrease cell death after AMI, thus preventing the development of heart failure.
Adipose derived stem cells
Although the above described inflammatory inhibitors significantly reduced infarct size, they do not
replace lost cardiomyocytes. Stem cells, however, can replace lost cardiomyocytes, although results of
early clinical trials using stem cell therapy in AMI are disappointing. As such, stem cell therapy still needs
to be further optimized. This we have studied in the second part of this thesis, using the relatively new
and promising adipose derived stem cells (ASCs). ASCs are derived from the stromal vascular fraction
(SVF) of adipose tissue, and are suggested to have a high potential for the treatment of AMI.[18,32-34]
ASCs can be easily harvested and have properties similar to bone marrow mesenchymal stem cells
including the capacity to differentiate towards several cell types, amongst which cardiomyocytes.[18,32-
35] Even more, when compared to bone marrow, adipose tissue provides up to 100 times more
mesenchymal stem cells per gram tissue.[33,35] Meanwhile, several experimental studies have been
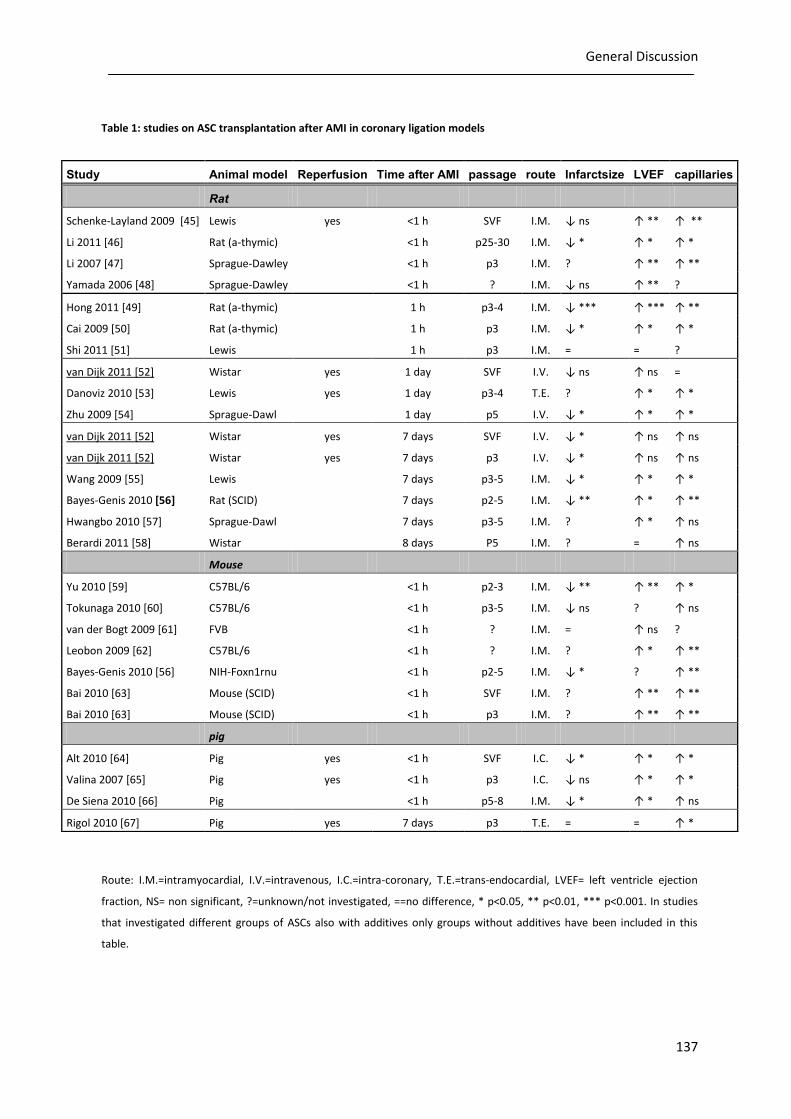
performed analyzing the role of ASCs in AMI in both small and large animal models (table 1). Also the
first clinical trial using ASCs to treat AMI has started recently in the Netherlands.[36]
Although the results of these animal studies generally are promising, it was also found that due
to the harsh ischemic and inflammatory environment in which the cells are transplanted after AMI,
massive cell death of stem cells occurs, lowering the effectiveness of this therapy.[2,37] Several factors
play an important role herein, including the time point of administration, the environment of the
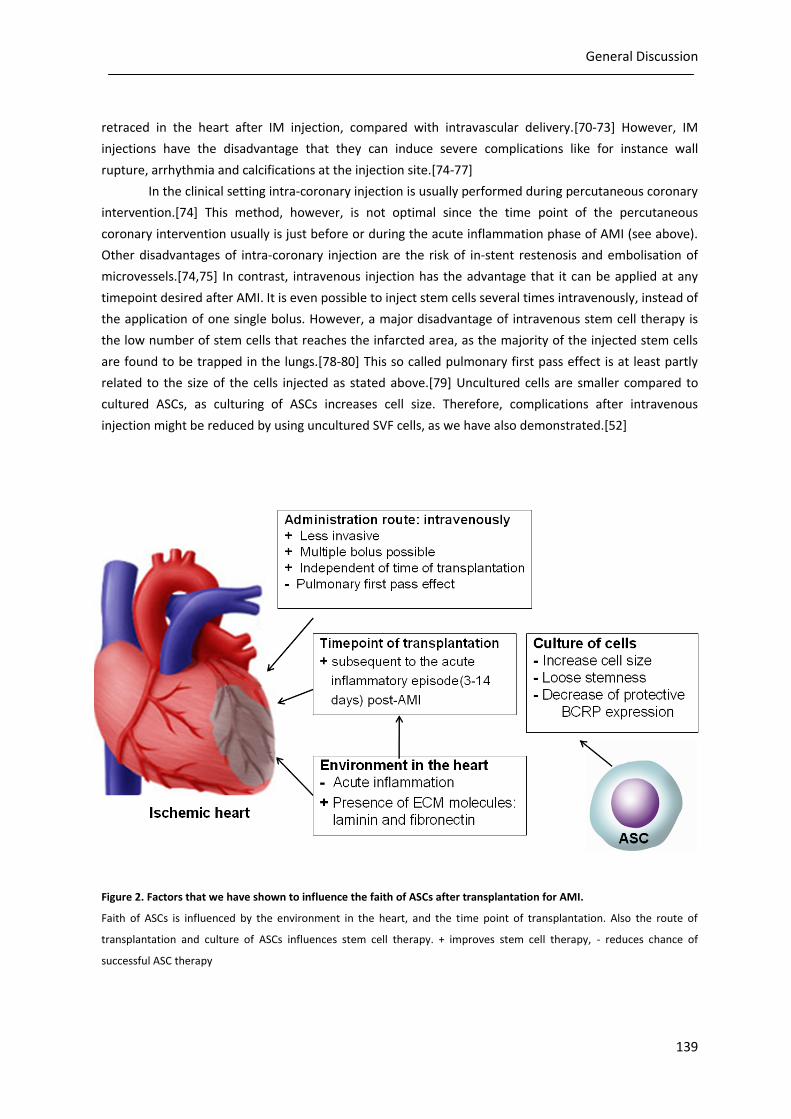
infarcted heart, the passage of ASCs used, and its administration route (figure 2).
Time point of administration
In most animal studies investigators applied stem cells during the same operational procedure as the
infarct induction itself (table 1). However, it is known that the environment in the heart changes
dramatically after AMI. Massive myocardial necrosis and leukocyte infiltration might then harm survival
of implanted stem cells, and therewith reduce the effectiveness of stem cell therapy.[2,38,39] Therefore,
the time point of administration might significantly affect stem cell survival after AMI. Several studies
have additionally investigated the time frame to apply stem cell therapy post AMI, although this was not
studied directly using ASCs.[38,40-42] These in vitro and in vivo studies suggested that the optimal timing
for stem cell therapy is subsequent to the acute inflammatory period (>3 days post AMI), but before two
weeks post AMI when scar tissue is formed and regeneration capacity is limited.[42] Interestingly we
found for ASCs that adhesion, proliferation and differentiation in vitro was improved by the extracellular
General Discussion
137
Table 1: studies on ASC transplantation after AMI in coronary ligation models
Route: I.M.=intramyocardial, I.V.=intravenous, I.C.=intra-coronary, T.E.=trans-endocardial, LVEF= left ventricle ejection
fraction, NS= non significant, ?=unknown/not investigated, ==no difference, * p<0.05, ** p<0.01, *** p<0.001. In studies
that investigated different groups of ASCs also with additives only groups without additives have been included in this
table.
Study Animal model Reperfusion Time after AMI passage route Infarctsize LVEF capillaries
Rat
Schenke-Layland 2009 [45] Lewis yes <1 h SVF I.M. ↓ ns ↑ ** ↑ **
Li 2011 [46] Rat (a-thymic) <1 h p25-30 I.M. ↓ * ↑ * ↑ *
Li 2007 [47] Sprague-Dawley <1 h p3 I.M. ? ↑ ** ↑ **
Yamada 2006 [48] Sprague-Dawley <1 h ? I.M. ↓ ns ↑ ** ?
Hong 2011 [49] Rat (a-thymic) 1 h p3-4 I.M. ↓ *** ↑ *** ↑ **
Cai 2009 [50] Rat (a-thymic) 1 h p3 I.M. ↓ * ↑ * ↑ *
Shi 2011 [51] Lewis 1 h p3 I.M. = = ?
van Dijk 2011 [52] Wistar yes 1 day SVF I.V. ↓ ns ↑ ns =
Danoviz 2010 [53] Lewis yes 1 day p3-4 T.E. ? ↑ * ↑ *
Zhu 2009 [54] Sprague-Dawl 1 day p5 I.V. ↓ * ↑ * ↑ *
van Dijk 2011 [52] Wistar yes 7 days SVF I.V. ↓ * ↑ ns ↑ ns
van Dijk 2011 [52] Wistar yes 7 days p3 I.V. ↓ * ↑ ns ↑ ns
Wang 2009 [55] Lewis 7 days p3-5 I.M. ↓ * ↑ * ↑ *
Bayes-Genis 2010 [56] Rat (SCID) 7 days p2-5 I.M. ↓ ** ↑ * ↑ **
Hwangbo 2010 [57] Sprague-Dawl 7 days p3-5 I.M. ? ↑ * ↑ ns
Berardi 2011 [58] Wistar 8 days P5 I.M. ? = ↑ ns
Mouse
Yu 2010 [59] C57BL/6 <1 h p2-3 I.M. ↓ ** ↑ ** ↑ *
Tokunaga 2010 [60] C57BL/6 <1 h p3-5 I.M. ↓ ns ? ↑ ns
van der Bogt 2009 [61] FVB <1 h ? I.M. = ↑ ns ?
Leobon 2009 [62] C57BL/6 <1 h ? I.M. ? ↑ * ↑ **
Bayes-Genis 2010 [56] NIH-Foxn1rnu <1 h p2-5 I.M. ↓ * ? ↑ **
Bai 2010 [63] Mouse (SCID) <1 h SVF I.M. ? ↑ ** ↑ **
Bai 2010 [63] Mouse (SCID) <1 h p3 I.M. ? ↑ ** ↑ **
pig
Alt 2010 [64] Pig yes <1 h SVF I.C. ↓ * ↑ * ↑ *
Valina 2007 [65] Pig yes <1 h p3 I.C. ↓ ns ↑ * ↑ *
De Siena 2010 [66] Pig <1 h p5-8 I.M. ↓ * ↑ * ↑ ns
Rigol 2010 [67] Pig yes 7 days p3 T.E. = = ↑ *

Chapter 8
138
matrix molecules fibronectin and laminin, which are upregulated in the infarcted area after AMI
(fibronectin from 12 hours on and laminin from 3 days on). This indicates that in case ASCs are
transplanted subsequent to the acute inflammatory period, thus when fibronectin and laminin
depositions are high, these proteins might stimulate ASC attachment and differentiation.[43,44]
As depicted in table 1 infarct size reduction in rats indeed was more often non-significant after
early ASC injection (within 24 hours), compared with injection after the acute inflammatory period (1
week). We have now shown that application of stem cells 7 days post AMI, leads to a significant
reduction in scar size, which was not found when cells were injected at day 1 post AMI.[52] To the best
of our knowledge, no other studies have compared the effect of injection of ASCs at different timepoints
within one study. Taken together, after the acute inflammation period after AMI (>3 days), the
environment in the heart is more favourable for stem cell transplantation. In future studies we will
address whether this inflammatory environment can be modified, to improve stem cell therapy, for
example by adding inflammatory inhibitors prior to stem cell therapy.
Culturing of ASCs before transplantation
Another factor that might influence the fate of stem cells after AMI is the process of culturing these cells
before transplantation, which is usually done to achieve a high number of stem cells. Culturing, however,
is time consuming, expensive, increases cell size, and, probably most important, affects the functional
characteristics of cells.[33,35,52,68] As such it has been shown that already during early culture ASCs
loose stem cell markers like CD34.[69] Additionally, we have now found that ASCs express the multi drug
resistance protein BCRP, that protects ASCs against ischemia. Also the expression of this protective
factor decreases during culture. These data suggest that using the uncultured stromal vascular fraction
cells (SVF), which contains ASCs, is more favourable for clinical practice than using long cultured ASCs.
This is possible since high numbers of SVF-cells/ASCs can be harvested from a relatively small amount of
adipose tissue.[33,35]
The question then arises whether there is a difference in therapeutic effect between uncultured
SVF-cells and cultured ASCs. We therefore compared the potential of uncultured SVF-cells and passage 3
ASCs in a rat model of AMI with reperfusion. We have found that uncultured SVF cells reduced infarct
size and improved cardiac outcome comparable to cultured ASCs. However, injection with cultured ASCs
(p3) occasionally resulted in severe pulmonary complications (thrombo-embolism), which was not found
with SVF cells. Since the results in cardiac outcome between uncultured SVF cells and cultured ASCs were
comparable, the use of uncultured SVF cells theoretically would be clinically more favourable.
Administration route
The route of delivery of stem cells is another important determinant for the fate of the stem cells after
AMI. Commonly used administration routes are intra-myocardial (IM) injection (directly, trans-coronary
or trans-endocardial) and intravascular delivery (intravenously, or intra-coronary). Several studies have
compared the outcome for these different administration routes. Generally it was found that
independent of the administration route the majority of the cells is lost, but slightly more cells can be
General Discussion
139
retraced in the heart after IM injection, compared with intravascular delivery.[70-73] However, IM
injections have the disadvantage that they can induce severe complications like for instance wall
rupture, arrhythmia and calcifications at the injection site.[74-77]
In the clinical setting intra-coronary injection is usually performed during percutaneous coronary
intervention.[74] This method, however, is not optimal since the time point of the percutaneous
coronary intervention usually is just before or during the acute inflammation phase of AMI (see above).
Other disadvantages of intra-coronary injection are the risk of in-stent restenosis and embolisation of
microvessels.[74,75] In contrast, intravenous injection has the advantage that it can be applied at any
timepoint desired after AMI. It is even possible to inject stem cells several times intravenously, instead of
the application of one single bolus. However, a major disadvantage of intravenous stem cell therapy is
the low number of stem cells that reaches the infarcted area, as the majority of the injected stem cells
are found to be trapped in the lungs.[78-80] This so called pulmonary first pass effect is at least partly
related to the size of the cells injected as stated above.[79] Uncultured cells are smaller compared to
cultured ASCs, as culturing of ASCs increases cell size. Therefore, complications after intravenous
injection might be reduced by using uncultured SVF cells, as we have also demonstrated.[52]
Figure 2. Factors that we have shown to influence the faith of ASCs after transplantation for AMI.
Faith of ASCs is influenced by the environment in the heart, and the time point of transplantation. Also the route of
transplantation and culture of ASCs influences stem cell therapy. + improves stem cell therapy, - reduces chance of
successful ASC therapy

Chapter 8
140
In future studies we will further optimize the intravenous delivery method of ASCs/SVF cells, by
specifically targeting the cells to the infarction area using microbubbles. Microbubbles are gas-filled
bubbles with a diameter of 2-5µm, that originally were used as ultrasound contrast agents.[81] These
microbubbles will be labelled with an antibody against ASCs (within the SVF fraction) as well as against a
specific target in the infarction area. As such we not only intend to increase the retention time of ASCs
within the infarcted area, but also intend to facilitate the binding of this complex to the endothelium by
pushing the ASC-microbubble complexes to the vessel wall using the acoustic radiation force of
ultrasound.
Mechanisms through which ASCs can improve cardiac outcome
Although many studies now have shown that ASCs form a promising new therapy in myocardial
infarction, the mechanisms through which ASCs improve cardiac outcome after AMI are not fully
elucidated. Studies have postulated different mechanisms for this cardiac improvement, mainly via an
effect of ASCs on differentiation or via paracrine effects (figure 3). In vitro and in vivo studies have shown
differentiation of ASCs towards cardiomyocytes and endothelial cells, suggesting that ASCs could
improve cardiac outcome by differentiation into these cells.[43,52,63,65,82-88] However, the number of
differentiated endothelial cells and cardiomyocytes that could be retrieved in the heart in animal studies
was relatively low, suggesting that the improvement of cardiac outcome was not only related to
differentiation effects of ASCs.[45,50,52,63] In line with this, several in vivo studies have now shown that
after treatment with ASCs the number of vessels in the peri-infarction area was increased, albeit without
evidence of differentiation of ASCs into endothelial cells.[45,50,52,55]
It was therefore suggested that ASCs might also improve cardiac outcome by so called paracrine
effects, indicating that factors excreted by stem cells affect the surrounding cells. Growth factors,
cytokines and signalling molecules produced by the infused stem cells can improve vascularisation,
reduce inflammation, and favour the viability of cardiomyocytes by inhibition of apoptosis.[89] As such,
it has been shown that ASCs secreted significant amounts of vascular endothelial growth factor (VEGF),
insulin-like growth factor (IGF)-1, hepatic growth factor and transforming growth factor-β (TGF-
β).[90,91] Both the secreted VEGF and IGF-1 have an anti-apoptotic effect on cardiomyocytes subjected
to hypoxia in vitro,[91] while VEGF also promotes endothelial cell growth and reduced endothelial
apoptosis in vitro.[90] Finally, it has been suggested that ASCs might also improve cardiac outcome by
reducing inflammation or attract resident stem cells to the infarcted area, but this has not been
confirmed yet.[92] Also, we did not find a significant effect of ASC therapy on the number of
macrophages in the heart after AMI.[52] Taken together, ASCs can improve cardiac outcome after AMI
through both induction of differentiation into cardiomyocytes and endothelial cells and through
paracrine effects (figure 3).

General Discussion
141
Figure 3. Mechanisms through which ASCs can improve outcome after AMI. ASCs can directly improve cardiac outcome
after AMI through differentiation into cardiomyocytes or endothelial cells. In addition, ASCs secrete factors that promote
survival of ischaemic cardiomyocytes and that induce angiogenesis, thereby improving perfusion around the ischaemic
area. In addition, it has also been suggested that ASCs might improve clinical outcome after AMI in a paracrine manner by
reducing inflammation through modulating protease activity and scar formation and by recruitment of resident cardiac
stem cells. HGF= hepatocyte growth factor; IGF= insulin-like growth factor; VEGF= vascular endothelial growth factor.
Conclusions of this thesis
The aim of this thesis was to develop and improve new putative therapies for myocardial infarction. We
now have shown that related to the inflammation, the first three days post AMI play a major role in the
induction of additional damage to cardiomyocytes, next to ischaemia itself. Specific inflammatory
inhibitors, clusterin and PX-18, indeed significantly reduced infarct size, without impairing normal cardiac
wound healing, when applied during these first three days post AMI. Therefore these inflammation
inhibitors form a promising new therapy in patients with AMI, especially since they not only inhibit
inflammatory mediators, but also reduce the pro-inflammatory state of the cardiomyocyte membrane.
To replace lost cardiomyocytes post AMI human ASCs in the SVF fraction of adipose tissue form
a potent therapy. These cells are abundantly available and show a high potential of endothelial and
cardiomyocyte differentiation. We have now shown that uncultured SVF cells are more favourable over
cultured ASCs for therapy after AMI. Furthermore, our studies indicate that SVF-cells/ASCs should be
transplanted after the acute inflammation period (>3 days), as the acute inflammatory environment
after infarction is a harmful environment also for stem cells. Finally, we have shown that intravenous
injection is a save transplantation route for stem cells that can be used at any desired timepoint post
AMI. In conclusion, inflammatory inhibitors and ASCs or their combination, can further improve clinical
outcome after AMI, by preventing development of heart failure later on.

Chapter 8
142
References
1. Daher M. (2001) Overview of the World Health Report 2000 Health systems: improving performance.
J.Med.Liban. 49:22-24.
2. Wang QD, Sjoquist PO. (2006) Myocardial regeneration with stem cells: pharmacological possibilities for efficacy
enhancement. Pharmacol.Res. 53:331-340.
3. Smits AM, van Vliet P, Hassink RJ, Goumans MJ and Doevendans PA. (2005) The role of stem cells in cardiac
regeneration. J.Cell Mol.Med. 9:25-36.
4. Erhardt L, Herlitz J, Bossaert L, Halinen M, Keltai M, Koster R, Marcassa C, Quinn T and van WH. (2002) Task force
on the management of chest pain. Eur.Heart J. 23:1153-1176.
5. Fraser JK, Schreiber RE, Zuk PA and Hedrick MH. (2004) Adult stem cell therapy for the heart. Int.J.Biochem.Cell
Biol. 36:658-666.
6. Lange RA, Hillis LD. (2002) Reperfusion therapy in acute myocardial infarction. N.Engl.J.Med. 346:954-955.
7. Khalil ME, Basher AW, Brown EJ, Jr. and Alhaddad IA. (2001) A remarkable medical story: benefits of angiotensin-
converting enzyme inhibitors in cardiac patients. J.Am.Coll.Cardiol. 37:1757-1764.
8. Arumugam TV, Magnus T, Woodruff TM, Proctor LM, Shiels IA and Taylor SM. (2006) Complement mediators in
ischemia-reperfusion injury. Clin.Chim.Acta 374:33-45.
9. Krijnen PA, Meischl C, Nijmeijer R, Visser CA, Hack CE and Niessen HW. (2006) Inhibition of sPLA2-IIA, C-reactive
protein or complement: new therapy for patients with acute myocardial infarction?
Cardiovasc.Hematol.Disord.Drug Targets. 6:113-123.
10. Yellon DM, Hausenloy DJ. (2007) Myocardial reperfusion injury. N.Engl.J.Med. 357:1121-1135.
11. Post JA, Verkleij AJ and Langer GA. (1995) Organization and function of sarcolemmal phospholipids in control
and ischemic/reperfused cardiomyocytes. J.Mol.Cell Cardiol. 27:749-760.
12. Daleke DL. (2003) Regulation of transbilayer plasma membrane phospholipid asymmetry. J.Lipid Res. 44:233-
242.
13. Nijmeijer R, Willemsen M, Meijer CJ, Visser CA, Verheijen RH, Gottlieb RA, Hack CE and Niessen HW. (2003) Type
II secretory phospholipase A2 binds to ischemic flip-flopped cardiomyocytes and subsequently induces cell
death. Am.J.Physiol Heart Circ.Physiol 285:H2218-H2224.
14. Hammill AK, Uhr JW and Scheuermann RH. (1999) Annexin V staining due to loss of membrane asymmetry can
be reversible and precede commitment to apoptotic death. Exp.Cell Res. 251:16-21.
15. Frangogiannis NG, Smith CW and Entman ML. (2002) The inflammatory response in myocardial infarction.
Cardiovasc.Res. 53:31-47.
16. Nijmeijer R, Lagrand WK, Baidoshvili A, Lubbers YT, Hermens WT, Meijer CJ, Visser CA, Hack CE and Niessen HW.
(2002) Secretory type II phospholipase A(2) binds to ischemic myocardium during myocardial infarction in
humans. Cardiovasc.Res. 53:138-146.
17. van Dijk A, Krijnen PA, Vermond RA, Pronk A, Spreeuwenberg M, Visser FC, Berney R, Paulus WJ, Hack CE, van
Milligen FJ and Niessen HW. (2009) Inhibition of type 2A secretory phospholipase A2 reduces death of
cardiomyocytes in acute myocardial infarction. Apoptosis. 14:753-763.
18. Arras M, Ito WD, Scholz D, Winkler B, Schaper J and Schaper W. (1998) Monocyte activation in angiogenesis and
collateral growth in the rabbit hindlimb. J.Clin.Invest 101:40-50.
19. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R and Pittet
MJ. (2007) The healing myocardium sequentially mobilizes two monocyte subsets with divergent and
complementary functions. J.Exp.Med. 204:3037-3047.
20. Gordon S, Taylor PR. (2005) Monocyte and macrophage heterogeneity. Nat.Rev.Immunol. 5:953-964.
21. Krijnen PA, Cillessen SA, Manoe R, Muller A, Visser CA, Meijer CJ, Musters RJ, Hack CE, Aarden LA and Niessen
HW. (2005) Clusterin: a protective mediator for ischemic cardiomyocytes? Am.J.Physiol Heart Circ.Physiol
289:H2193-H2202.

General Discussion
143
22. van Dijk A, Vermond RA, Krijnen PA, Juffermans LJ, Hahn NE, Makker SP, Aarden LA, Hack E, Spreeuwenberg M,
van Rossum BC, Meischl C, Paulus WJ, van Milligen FJ and Niessen HW. (2010) Intravenous clusterin
administration reduces myocardial infarct size in rats. Eur.J.Clin.Invest 40:893-902.
23. Vakeva A, Laurila P and Meri S. (1993) Co-deposition of clusterin with the complement membrane attack
complex in myocardial infarction. Immunology 80:177-182.
24. Partridge SR, Baker MS, Walker MJ and Wilson MR. (1996) Clusterin, a putative complement regulator, binds to
the cell surface of Staphylococcus aureus clinical isolates. Infect.Immun. 64:4324-4329.
25. Jones SE, Jomary C. (2002) Clusterin. Int.J.Biochem.Cell Biol. 34:427-431.
26. Wilson MR, Easterbrook-Smith SB. (2000) Clusterin is a secreted mammalian chaperone. Trends Biochem.Sci.
25:95-98.
27. Michel JB. (2003) Anoikis in the cardiovascular system: known and unknown extracellular mediators.
Arterioscler.Thromb.Vasc.Biol. 23:2146-2154.
28. Jun HO, Kim DH, Lee SW, Lee HS, Seo JH, Kim JH, Kim JH, Yu YS, Min BH and Kim KW. (2011) Clusterin protects
H9c2 cardiomyocytes from oxidative stress-induced apoptosis via Akt/GSK-3beta signaling pathway.
Exp.Mol.Med. 43:53-61.
29. Bartl MM, Luckenbach T, Bergner O, Ullrich O and Koch-Brandt C. (2001) Multiple receptors mediate apoJ-
dependent clearance of cellular debris into nonprofessional phagocytes. Exp.Cell Res. 271:130-141.
30. Morales CR, Igdoura SA, Wosu UA, Boman J and Argraves WS. (1996) Low density lipoprotein receptor-related
protein-2 expression in efferent duct and epididymal epithelia: evidence in rats for its in vivo role in endocytosis
of apolipoprotein J/clusterin. Biol.Reprod. 55:676-683.
31. Li Y, Qu J, Shelat H, Gao S, Wassler M and Geng YJ. (2010) Clusterin induces CXCR4 expression and migration of
cardiac progenitor cells. Exp.Cell Res. 316:3435-3442.
32. Rangappa S, Fen C, Lee EH, Bongso A and Sim EK. (2003) Transformation of adult mesenchymal stem cells
isolated from the fatty tissue into cardiomyocytes. Ann.Thorac.Surg. 75:775-779.
33. Zhu Y, Liu T, Song K, Fan X, Ma X and Cui Z. (2008) Adipose-derived stem cell: a better stem cell than BMSC. Cell
Biochem.Funct. 26:664-675.
34. Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, Mizuno H, Alfonso ZC, Fraser JK, Benhaim P and Hedrick MH.
(2002) Human adipose tissue is a source of multipotent stem cells. Mol.Biol.Cell 13:4279-4295.
35. Oedayrajsingh-Varma MJ, van Ham SM, Knippenberg M, Helder MN, Klein-Nulend J, Schouten TE, Ritt MJ and
van Milligen FJ. (2006) Adipose tissue-derived mesenchymal stem cell yield and growth characteristics are
affected by the tissue-harvesting procedure. Cytotherapy. 8:166-177.
36. Houtgraaf HJ, Den Dekker W, Swager S, Van Geuns W, Fernandez-Aviles F, Serruys PW, Duckers HJ. (2011) First-
in-man experience of adipose tissue-derived regenerative cell transplantation in the treatment of patients with
an acute ST-elevation myocardial infarction. Report No.: NR10-1154 (SS10/Duckers) .
37. Zhang M, Methot D, Poppa V, Fujio Y, Walsh K and Murry CE. (2001) Cardiomyocyte grafting for cardiac repair:
graft cell death and anti-death strategies. J.Mol.Cell Cardiol. 33:907-921.
38. Lu L, Zhang JQ, Ramires FJ and Sun Y. (2004) Molecular and cellular events at the site of myocardial infarction:
from the perspective of rebuilding myocardial tissue. Biochem.Biophys.Res.Commun. 320:907-913.
39. Malek S, Kaplan E, Wang JF, Ke Q, Rana JS, Chen Y, Rahim BG, Li M, Huang Q, Xiao YF, Verheugt FW, Morgan JP
and Min JY. (2006) Successful implantation of intravenously administered stem cells correlates with severity of
inflammation in murine myocarditis. Pflugers Arch. 452:268-275.
40. Ma J, Ge J, Zhang S, Sun A, Shen J, Chen L, Wang K and Zou Y. (2005) Time course of myocardial stromal cell-
derived factor 1 expression and beneficial effects of intravenously administered bone marrow stem cells in rats
with experimental myocardial infarction. Basic Res.Cardiol. 100:217-223.
41. Zhang S, Sun A, Liang Y, Chen Q, Zhang C, Wang K, Zou Y and Ge J. (2009) A role of myocardial stiffness in cell-
based cardiac repair: a hypothesis. J.Cell Mol.Med. 13:660-663.
42. Jiang CY, Gui C, He AN, Hu XY, Chen J, Jiang Y and Wang JA. (2008) Optimal time for mesenchymal stem cell
transplantation in rats with myocardial infarction. J.Zhejiang.Univ Sci.B 9:630-637.

Chapter 8
144
43. van Dijk A, Niessen HW, Zandieh Doulabi B, Visser FC and van Milligen FJ. (2008) Differentiation of human
adipose-derived stem cells towards cardiomyocytes is facilitated by laminin. Cell Tissue Res. 334:457-467.
44. van Dijk A, Niessen HW, Ursem W, Twisk JW, Visser FC and van Milligen FJ. (2008) Accumulation of fibronectin in
the heart after myocardial infarction: a putative stimulator of adhesion and proliferation of adipose-derived
stem cells. Cell Tissue Res. 332:289-298.
45. Schenke-Layland K, Strem BM, Jordan MC, Deemedio MT, Hedrick MH, Roos KP, Fraser JK and MacLellan WR.
(2009) Adipose tissue-derived cells improve cardiac function following myocardial infarction. J.Surg.Res.
153:217-223.
46. Ii M, Horii M, Yokoyama A, Shoji T, Mifune Y, Kawamoto A, Asahi M and Asahara T. (2011) Synergistic effect of
adipose-derived stem cell therapy and bone marrow progenitor recruitment in ischemic heart. Lab Invest
91:539-552.
47. Li B, Zeng Q, Wang H, Shao S, Mao X, Zhang F, Li S and Guo Z. (2007) Adipose tissue stromal cells transplantation
in rats of acute myocardial infarction. Coron.Artery Dis. 18:221-227.
48. Yamada Y, Wang XD, Yokoyama S, Fukuda N and Takakura N. (2006) Cardiac progenitor cells in brown adipose
tissue repaired damaged myocardium. Biochem.Biophys.Res.Commun. 342:662-670.
49. Hong SJ, Kihlken J, Choi SC, March KL and Lim DS. (2011) Intramyocardial transplantation of human adipose-
derived stromal cell and endothelial progenitor cell mixture was not superior to individual cell type
transplantation in improving left ventricular function in rats with myocardial infarction. Int.J.Cardiol.
50. Cai L, Johnstone BH, Cook TG, Tan J, Fishbein MC, Chen PS and March KL. (2009) IFATS collection: Human
adipose tissue-derived stem cells induce angiogenesis and nerve sprouting following myocardial infarction, in
conjunction with potent preservation of cardiac function. Stem Cells 27:230-237.
51. Shi CZ, Zhang XP, Lv ZW, Zhang HL, Xu JZ, Yin ZF, Yan YQ and Wang CQ. (2011) Adipose tissue-derived stem cells
embedded with eNOS restore cardiac function in acute myocardial infarction model. Int.J.Cardiol.
52. van Dijk A, Naaijkens BA, Jurgens WJFM, Nalliah K, Sairras S, van der Pijl RJ, Vo K, Vonk ABA, van Rossum AC,
Paulus WJ, van Milligen FJ and Niessen HWM. (2011) Reduction of infarct size by intravenous injection of
uncultured adipose derived stromal cells in a rat model is dependent on the time point of application. Stem Cell
Research 7:219-229.
53. Danoviz ME, Nakamuta JS, Marques FL, dos SL, Alvarenga EC, dos Santos AA, Antonio EL, Schettert IT, Tucci PJ
and Krieger JE. (2010) Rat adipose tissue-derived stem cells transplantation attenuates cardiac dysfunction post
infarction and biopolymers enhance cell retention. PLoS.One. 5:e12077.
54. Zhu XY, Zhang XZ, Xu L, Zhong XY, Ding Q and Chen YX. (2009) Transplantation of adipose-derived stem cells
overexpressing hHGF into cardiac tissue. Biochem.Biophys.Res.Commun. 379:1084-1090.
55. Wang L, Deng J, Tian W, Xiang B, Yang T, Li G, Wang J, Gruwel M, Kashour T, Rendell J, Glogowski M, Tomanek B,
Freed D, Deslauriers R, Arora RC and Tian G. (2009) Adipose-derived stem cells are an effective cell candidate for
treatment of heart failure: an MR imaging study of rat hearts. Am.J.Physiol Heart Circ.Physiol 297:H1020-H1031.
56. Bayes-Genis A, Roura S, Soler-Botija C, Farre J, Hove-Madsen L, Llach A and Cinca J. (2005) Identification of
cardiomyogenic lineage markers in untreated human bone marrow-derived mesenchymal stem cells.
Transplant.Proc. 37:4077-4079.
57. Hwangbo S, Kim J, Her S, Cho H and Lee J. (2010) Therapeutic potential of human adipose stem cells in a rat
myocardial infarction model. Yonsei Med.J. 51:69-76.
58. Berardi GR, Rebelatto CK, Tavares HF, Ingberman M, Shigunov P, Barchiki F, Aguiar AM, Miyague NI, Francisco JC,
Correa A, Senegaglia AC, Suss PH, Moutinho JA, Sotomaior VS, Nakao LS and Brofman PS. (2011) Transplantation
of SNAP-treated adipose tissue-derived stem cells improves cardiac function and induces neovascularization
after myocardium infarct in rats. Exp.Mol.Pathol. 90:149-156.
59. Yu LH, Kim MH, Park TH, Cha KS, Kim YD, Quan ML, Rho MS, Seo SY and Jung JS. (2010) Improvement of cardiac
function and remodeling by transplanting adipose tissue-derived stromal cells into a mouse model of acute
myocardial infarction. Int.J.Cardiol. 139:166-172.

General Discussion
145
60. Tokunaga M, Liu ML, Nagai T, Iwanaga K, Matsuura K, Takahashi T, Kanda M, Kondo N, Wang P, Naito AT and
Komuro I. (2010) Implantation of cardiac progenitor cells using self-assembling peptide improves cardiac
function after myocardial infarction. J.Mol.Cell Cardiol. 49:972-983.
61. van der Bogt KE, Schrepfer S, Yu J, Sheikh AY, Hoyt G, Govaert JA, Velotta JB, Contag CH, Robbins RC and Wu JC.
(2009) Comparison of transplantation of adipose tissue- and bone marrow-derived mesenchymal stem cells in
the infarcted heart. Transplantation 87:642-652.
62. Leobon B, Roncalli J, Joffre C, Mazo M, Boisson M, Barreau C, Calise D, Arnaud E, Andre M, Puceat M, Penicaud L,
Prosper F, Planat-Benard V and Casteilla L. (2009) Adipose-derived cardiomyogenic cells: in vitro expansion and
functional improvement in a mouse model of myocardial infarction. Cardiovasc.Res. 83:757-767.
63. Bai X, Yan Y, Song YH, Seidensticker M, Rabinovich B, Metzele R, Bankson JA, Vykoukal D and Alt E. (2010) Both
cultured and freshly isolated adipose tissue-derived stem cells enhance cardiac function after acute myocardial
infarction. Eur.Heart J. 31:489-501.
64. Alt E, Pinkernell K, Scharlau M, Coleman M, Fotuhi P, Nabzdyk C, Matthias N, Gehmert S and Song YH. (2010)
Effect of freshly isolated autologous tissue resident stromal cells on cardiac function and perfusion following
acute myocardial infarction. Int.J.Cardiol. 144:26-35.
65. Valina C, Pinkernell K, Song YH, Bai X, Sadat S, Campeau RJ, Le Jemtel TH and Alt E. (2007) Intracoronary
administration of autologous adipose tissue-derived stem cells improves left ventricular function, perfusion, and
remodelling after acute myocardial infarction. Eur.Heart J. 28:2667-2677.
66. De SR, Balducci L, Blasi A, Montanaro MG, Saldarelli M, Saponaro V, Martino C, Logrieco G, Soleti A, Fiobellot S,
Madeddu P, Rossi G, Ribatti D, Crovace A, Cristini S, Invernici G, Parati EA and Alessandri G. (2010) Omentum-
derived stromal cells improve myocardial regeneration in pig post-infarcted heart through a potent paracrine
mechanism. Exp.Cell Res. 316:1804-1815.
67. Rigol M, Solanes N, Farre J, Roura S, Roque M, Berruezo A, Bellera N, Novensa L, Tamborero D, Prat-Vidal C,
Huzman MA, Batlle M, Hoefsloot M, Sitges M, Ramirez J, Dantas AP, Merino A, Sanz G, Brugada J, Bayes-Genis A
and Heras M. (2010) Effects of adipose tissue-derived stem cell therapy after myocardial infarction: impact of
the route of administration. J.Card Fail. 16:357-366.
68. Rombouts WJ, Ploemacher RE. (2003) Primary murine MSC show highly efficient homing to the bone marrow
but lose homing ability following culture. Leukemia 17:160-170.
69. Mitchell JB, McIntosh K, Zvonic S, Garrett S, Floyd ZE, Kloster A, Di Halvorsen Y, Storms RW, Goh B, Kilroy G, Wu
X and Gimble JM. (2006) Immunophenotype of human adipose-derived cells: temporal changes in stromal-
associated and stem cell-associated markers. Stem Cells 24:376-385.
70. Hale SL, Dai W, Dow JS and Kloner RA. (2008) Mesenchymal stem cell administration at coronary artery
reperfusion in the rat by two delivery routes: a quantitative assessment. Life Sci. 83:511-515.
71. Hou D, Youssef EA, Brinton TJ, Zhang P, Rogers P, Price ET, Yeung AC, Johnstone BH, Yock PG and March KL.
(2005) Radiolabeled cell distribution after intramyocardial, intracoronary, and interstitial retrograde coronary
venous delivery: implications for current clinical trials. Circulation 112:I150-I156.
72. Perin EC, Silva GV, Assad JA, Vela D, Buja LM, Sousa AL, Litovsky S, Lin J, Vaughn WK, Coulter S, Fernandes MR
and Willerson JT. (2008) Comparison of intracoronary and transendocardial delivery of allogeneic mesenchymal
cells in a canine model of acute myocardial infarction. J.Mol.Cell Cardiol. 44:486-495.
73. Baklanov DV, Moodie KM, McCarthy FE, Mandrusov E, Chiu J, Aswonge G, Cheng J, Chow M, Simons M and de
Muinck ED. (2006) Comparison of transendocardial and retrograde coronary venous intramyocardial catheter
delivery systems in healthy and infarcted pigs. Catheter.Cardiovasc.Interv. 68:416-423.
74. van der Spoel TI, Lee JC, Vrijsen K, Sluijter JP, Cramer MJ, Doevendans PA, van BE and Chamuleau SA. (2011)
Non-surgical stem cell delivery strategies and in vivo cell tracking to injured myocardium.
Int.J.Cardiovasc.Imaging 27:367-383.
75. Wu KH, Han ZC, Mo XM and Zhou B. (2011) Cell delivery in cardiac regenerative therapy. Ageing Res.Rev.

Chapter 8
146
76. Fukushima S, Varela-Carver A, Coppen SR, Yamahara K, Felkin LE, Lee J, Barton PJ, Terracciano CM, Yacoub MH
and Suzuki K. (2007) Direct intramyocardial but not intracoronary injection of bone marrow cells induces
ventricular arrhythmias in a rat chronic ischemic heart failure model. Circulation 115:2254-2261.
77. Hwangbo S, Kim J, Her S, Cho H and Lee J. (2010) Therapeutic potential of human adipose stem cells in a rat
myocardial infarction model. Yonsei Med.J. 51:69-76.
78. Assis AC, Carvalho JL, Jacoby BA, Ferreira RL, Castanheira P, Diniz SO, Cardoso VN, Goes AM and Ferreira AJ.
(2010) Time-dependent migration of systemically delivered bone marrow mesenchymal stem cells to the
infarcted heart. Cell Transplant. 19:219-230.
79. Fischer UM, Harting MT, Jimenez F, Monzon-Posadas WO, Xue H, Savitz SI, Laine GA and Cox CS, Jr. (2009)
Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem
Cells Dev. 18:683-692.
80. Gao J, Dennis JE, Muzic RF, Lundberg M and Caplan AI. (2001) The dynamic in vivo distribution of bone marrow-
derived mesenchymal stem cells after infusion. Cells Tissues.Organs 169:12-20.
81. Dijkmans PA, Juffermans LJ, Musters RJ, van WA, ten Cate FJ, van GW, Visser CA, de JN and Kamp O. (2004)
Microbubbles and ultrasound: from diagnosis to therapy. Eur.J.Echocardiogr. 5:245-256.
82. Bai X, Pinkernell K, Song YH, Nabzdyk C, Reiser J and Alt E. (2007) Genetically selected stem cells from human
adipose tissue express cardiac markers. Biochem.Biophys.Res.Commun. 353:665-671.
83. Ning H, Liu G, Lin G, Yang R, Lue TF and Lin CS. (2009) Fibroblast growth factor 2 promotes endothelial
differentiation of adipose tissue-derived stem cells. J.Sex Med. 6:967-979.
84. Planat-Benard V, Menard C, Andre M, Puceat M, Perez A, Garcia-Verdugo JM, Penicaud L and Casteilla L. (2004)
Spontaneous cardiomyocyte differentiation from adipose tissue stroma cells. Circ.Res. 94:223-229.
85. Song YH, Gehmert S, Sadat S, Pinkernell K, Bai X, Matthias N and Alt E. (2007) VEGF is critical for spontaneous
differentiation of stem cells into cardiomyocytes. Biochem.Biophys.Res.Commun. 354:999-1003.
86. Strem BM, Zhu M, Alfonso Z, Daniels EJ, Schreiber R, Beygui R, MacLellan WR, Hedrick MH and Fraser JK. (2005)
Expression of cardiomyocytic markers on adipose tissue-derived cells in a murine model of acute myocardial
injury. Cytotherapy. 7:282-291.
87. Miyahara Y, Nagaya N, Kataoka M, Yanagawa B, Tanaka K, Hao H, Ishino K, Ishida H, Shimizu T, Kangawa K, Sano
S, Okano T, Kitamura S and Mori H. (2006) Monolayered mesenchymal stem cells repair scarred myocardium
after myocardial infarction. Nat.Med. 12:459-465.
88. Zhang DZ, Gai LY, Liu HW, Jin QH, Huang JH and Zhu XY. (2007) Transplantation of autologous adipose-derived
stem cells ameliorates cardiac function in rabbits with myocardial infarction. Chin Med.J.(Engl.) 120:300-307.
89. Gnecchi M, Zhang Z, Ni A and Dzau VJ. (2008) Paracrine mechanisms in adult stem cell signaling and therapy.
Circ.Res. 103:1204-1219.
90. Rehman J, Traktuev D, Li J, Merfeld-Clauss S, Temm-Grove CJ, Bovenkerk JE, Pell CL, Johnstone BH, Considine RV
and March KL. (2004) Secretion of angiogenic and antiapoptotic factors by human adipose stromal cells.
Circulation 109:1292-1298.
91. Sadat S, Gehmert S, Song YH, Yen Y, Bai X, Gaiser S, Klein H and Alt E. (2007) The cardioprotective effect of
mesenchymal stem cells is mediated by IGF-I and VEGF. Biochem.Biophys.Res.Commun. 363:674-679.
92. Bai X, Alt E. (2010) Myocardial regeneration potential of adipose tissue-derived stem cells.
Biochem.Biophys.Res.Commun. 401:321-326.





























