Embed Size (px)
Citation preview

Accepted Manuscript
Title: General approach for electrochemical functionalizationof glassy carbon surface by in situ generation of diazonium ionunder acidic and non-acidic condition with a cascade protocol
Author: Hamid Salehzadeh Davood Nematollahi VahidKhakyzadeh Banafsheh Mokhtari Luke C. Henderson
PII: S0013-4686(14)01350-4DOI: http://dx.doi.org/doi:10.1016/j.electacta.2014.06.134Reference: EA 22996
To appear in: Electrochimica Acta
Received date: 28-4-2014Revised date: 12-6-2014Accepted date: 13-6-2014
Please cite this article as: H. Salehzadeh, D. Nematollahi, V. Khakyzadeh, B.Mokhtari, L.C. Henderson, General approach for electrochemical functionalizationof glassy carbon surface by in situ generation of diazonium ion under acidicand non-acidic condition with a cascade protocol, Electrochimica Acta (2014),http://dx.doi.org/10.1016/j.electacta.2014.06.134
This is a PDF file of an unedited manuscript that has been accepted for publication.As a service to our customers we are providing this early version of the manuscript.The manuscript will undergo copyediting, typesetting, and review of the resulting proofbefore it is published in its final form. Please note that during the production processerrors may be discovered which could affect the content, and all legal disclaimers thatapply to the journal pertain.

Page 1 of 40
Accep
ted
Man
uscr
ipt
1
General approach for electrochemical functionalization of glassy carbon
surface by in situ generation of diazonium ion under acidic and non-acidic
condition with a cascade protocol
Hamid Salehzadeh,a Davood Nematollahi,*a Vahid Khakyzadeh,a Banafsheh Mokhtari,a Luke
C. Hendersonb,c
Faculty of Chemistry, Bu-Ali-Sina University, P. O. Box 65174, Hamedan, Iran.
Tel.: +98 811 8282807; Fax: +98 811 8257407. E-mail: [email protected].
bStrategic Research Centre for Chemistry and Biotechnology, Deakin University, Geelong,
Victoria, Australia , 3216
cInstitute for Frontier Materials, Deakin University, Geelong, Victoria, Australia , 3216
Abstract
Immobilization of catechol derivatives on GC electrode surfaces can be performed by
in situ generation and reduction of nitrocatechol. We present the oxidative nitration of
catechol in the presence of nitrous acid followed by electrochemically reduction of
the generated nitro aromatic group to the corresponding amine group and its
conversion to diazonium cation at the electrode surface to yield a surface covalently
modified with catechol. In this manner, some derivatives of catechol can be
immobilized on the electrode surface. Whole of the process is carried out in

Page 2 of 40
Accep
ted
Man
uscr
ipt
2
Triethylammonium acetate ionic liquid as an inert and neutral medium (pH~7.0).
Surface coverage can be easily controlled by the applied potential, time and
concentration of catechol. After modification, the electrochemical features of
modified surface have been studied. Also modified GC electrode exhibited
remarkable catalytic activity in the oxidation of NADH. The catalytic currents were
proportional to the concentration of NADH over the range 0.01-0.80 mM. This
condition can be used for modification of GC surfaces by various aromatic molecules
for different application such as design of sensors and biosensors.
Keywords: Electrochemical derivatization, Nitrocatechol, Diazonium ion, Triethylammonium
acetate.
1. Introduction
In recent year’s modification of surface with organic films have performed by several
methodologies, including chemisorption and physical adsorption [1]. Between them, one of
the most dominant procedures is the covalent attachment of aryl groups by electroreduction
of diazonium salts [1-4]. The functionalization of surfaces including carbon materials, metals,
electrodes and powders during the past decade has been widely performed by this procedure
[1-7]. Various substituted aryl groups can be immobilized onto a variety of surfaces in order
to change their properties for different applications [8-10], including bioelectrochemistry
[11], molecular electronics [12], immobilization of organometallics [13] and corrosion
protection [14], design of novel high-performance redox surfaces [10,15,16], (bio)sensors
[17,18], biocatalysts [19], and hybrid molecule-on-semiconductor nanoelectronic devices
[20].
Functionalization of carbon substrates by electroreduction of aryl diazonium salts is
affected, by multitude of factors such as: (i) the applied potential, (ii) the electrolysis time

Page 3 of 40
Accep
ted
Man
uscr
ipt
3
[21-23], (iii) concentration and electronic nature of the diazonium salt [24,25] (iv) the type of
carbon material used, and (v) the type of solvent [26,27]. Nonetheless, modification of
surfaces by this method suffers from high reactivity of aryl diazonium salts and so controlling
the extent of reaction can be challenging. It has been observed that surface grafted aryl
groups are attacked by aryl radicals which generally results in multiple attachments and
multilayer formation [28,29]. To resolve this problem, introduction of bulky substituents on
the ArN2+ moieties was proposed to limit the secondary radical reactions. Using this strategy,
ultrathin organic layers were obtained [25,30]. In other work, 2,2-diphenyl-1-picrylhydrazyl
was used as a radical scavenger to control, or prevent, the polymerization of the
electrogenerated aryl radicals on a carbon electrode [31]. In other strategies, to overcome the
high reactivity of diazonium ions, some precursors were used for in situ generation of these
ions. Recently, Tour's research group developed a convenient procedure for in situ
functionalizing of Si(100) and single-wall carbon nanotubes (SWNTs) in acidic aqueous
media using organic triazenes [32,33]. Also, a two-phase laminar flow procedure was applied
by Downard's group to form an aryldiazonium ion from an aryltriazene precursor at the
interface between the two streams including aryltriazene and acid [34]. In other work,
Cougnon et al. reported an organo-layer modification procedure based on stepwise reduction
of nitro precursors for in situ generation of diazonium ion [35-37]. In these works, the
modification could be carried out at room temperature and the problem of high reactivity of
aryldiazonium ions was reduced.
Nevertheless generation of diazonium ion in surface modification by precursors
(compounds containing amine, nitro or aryltriazene) requires the use of acidic environment
[32-37]. This condition may be a major limitation for the surface grafting of compounds
possessing acid-sensitive functional groups [38]. Thus there is a need for inert conditions to
overcome this limitation to expand the chemical space available by the methodology.

Page 4 of 40
Accep
ted
Man
uscr
ipt
4
In 2011, Daasbjerg and co-workers immobilized organic layers onto surface of a GCE by
electrochemical reduction of in situ generated diazonium ion from phenyltriazene
functionality in acetonitrile and non-acidic media by applying double-pulse potentials [39]. In
this work, the required protons for the conversion of phenyltriazene to the corresponding
diazonium ion is prepared from oxidation of N,N'-diphenylhydrazine by an oxidative pulse. A
reductive pulse converts diazonium ion to its reduced form.
In other work, reported by Cougnon et al., activated carbon was functionalized via the
spontaneous reduction of sulfonic acid functionalized phenyldiazonium salts generated in situ
in neutral water. In this elegant approach the proton source originate from sulfonic acid
substituent, thus requiring no additional acidic additive to promote reaction progression [40].
Spontaneous grafting of diazonium salts in a neutral aqueous medium was also reported [41].
In a different work, spontaneously reaction of diazoates, produced from reaction of hydroxyl
ions with diazonium salts in a basic condition, with Fe and Au surfaces was reported to
modification with aryl groups [42].
Function of more than 300 dehydrogenase based enzymes in living system is dependent
to the enzyme cofactor nicotinamide adenine dinucleotide (NAD+) [43]. NAD+ dependent
dehydrogenase biosensing device acts based on the quantification of enzymatically generated
NADH. The concentration of NADH which produced is enzymatically proportional to the
amount of available substrate in the sample. Though the redox potential of NAD+/NADH
couple is -0.56 V vs SCE at pH 7.0 [44], but due to slow electron transfer and dimerization of
intermediates, high positive potential, as large as 0.8-1 V, is required for the oxidation of
NADH on common electrodes [45]. Various methodologies have been used to improvement
of the electron transfer reaction and to decrease the overpotential of NADH oxidation [43].
The oxidation of NADH has been traditionally facilitated by the redox mediator based
electrodes [46-48].

Page 5 of 40
Accep
ted
Man
uscr
ipt
5
In the current work, we present a new general approach to covalently immobilize
catechol derivatives onto the GCE surface. We describe, in situ chemically generated
nitrocatechol, in the bulk solution, which is electrochemically reduced to aminocatechol
followed by immediate conversion to the corresponding diazonium ion that is
electrochemically reduced and immobilized onto the surface of GCE. The entire process is
done in aqueous triethylammonium acetate (TEAA) solution which is a neutral buffered
solution (pH ~ 7.0) and also is a source of protons for various steps of modification, e.g.
(generation of nitrous acid required for chemical oxidative nitration of catechol, etc). This
procedure can be applied to electro-grafting of other amino and nitro aryl groups to
appropriate applications. Finally, we will show that glassy carbon electrode modified with
catechol exhibit a good catalytic activity in the electrooxidation of NADH.
2. Experimental
2.1. Reagents and Apparatus
4-Nitrocatechol, catechol, 3-methoxycatechol, 3-methylcatechol, triethylammonium
acetate (TEAA) and sodium nitrite were obtained from Sigma-Aldrich. All other chemicals
used in this investigation were of analytical grade. Cyclic voltammetry, controlled-potential
coulometry and preparative electrolysis were performed using an Autolab model PGSTAT
302N potentiostat/galvanostat. The working electrode used in the voltammetry experiment
was a glassy carbon disc (1.8 mm diameter) and platinum wire was used as counter electrode.
The working electrode used in controlled-potential coulometry was an assembly of four
carbon rods (6 mm diameter and 4 cm length) and a large platinum gauze constitute the
counter electrode. The working electrode potentials were measured versus Ag/AgCl electrode
(saturated KCl) (all electrodes from AZAR electrode). The glassy carbon electrode was
polished using alumina slurry (from Iran Alumina Co.).

Page 6 of 40
Accep
ted
Man
uscr
ipt
6
2.2. Controlled-potential coulometry
70 ml of aqueous phosphate buffer solution (c = 0.1 M, pH = 7.0) containing catechol
(0.25 mmol) and sodium nitrite (0.25 mmol) was subjected to electrolysis at 0.22 V versus
Ag/AgCl in a divided cell. The electrolysis was terminated when the decay of current became
more than 95%. At the end of electrolysis the yellow product was extracted in ethylacetate.
After drying of ethylacetate, the product was characterized by IR and cyclic voltammetry.
2.3. Chemical nitration of catechol
To a solution of catechol (0.5 mmol) in 30 ml of aqueous phosphate buffer (pH 3.0, c =
0.2 M), sodium nitrite (1.5 mmol) was added. The solution was stirred for approximately 45
min. Then, the reaction mixture was neutralized to pH = 7.0 by addition of sodium carbonate
and then product was extracted into ethylacetate. After drying, the product was characterized
by IR, 1HNMR and cyclic voltammetry.
2.3. Electrografting of Electrode Surface
Glassy carbon electrode was polished with alumina powder (0.05 μm) on a polishing
cloth and rinsed thoroughly with water. The polished electrode was then modified in situ
under potentiostatic condition at −0.60 V for catechol or by potential cycling over the range
of 0.0 to -0.9 V vs. Ag/AgCl in aqueous solution containing HCl (pH = 1.0) or TEAA (1/1
v:v water/ TEAA, pH ~ 7.0) as a proton source, catechol and NaNO2. Finally, the as prepared
catechol modified GCE was thoroughly rinsed with distilled water. In all cases, the electrode
modification was carried out at room temperature in an electrolytic mixture. The redox
activity of the catechol modified GCE was quantified by cyclic voltammetry in aqueous
buffered solutions.

Page 7 of 40
Accep
ted
Man
uscr
ipt
7
3. Results and discussion
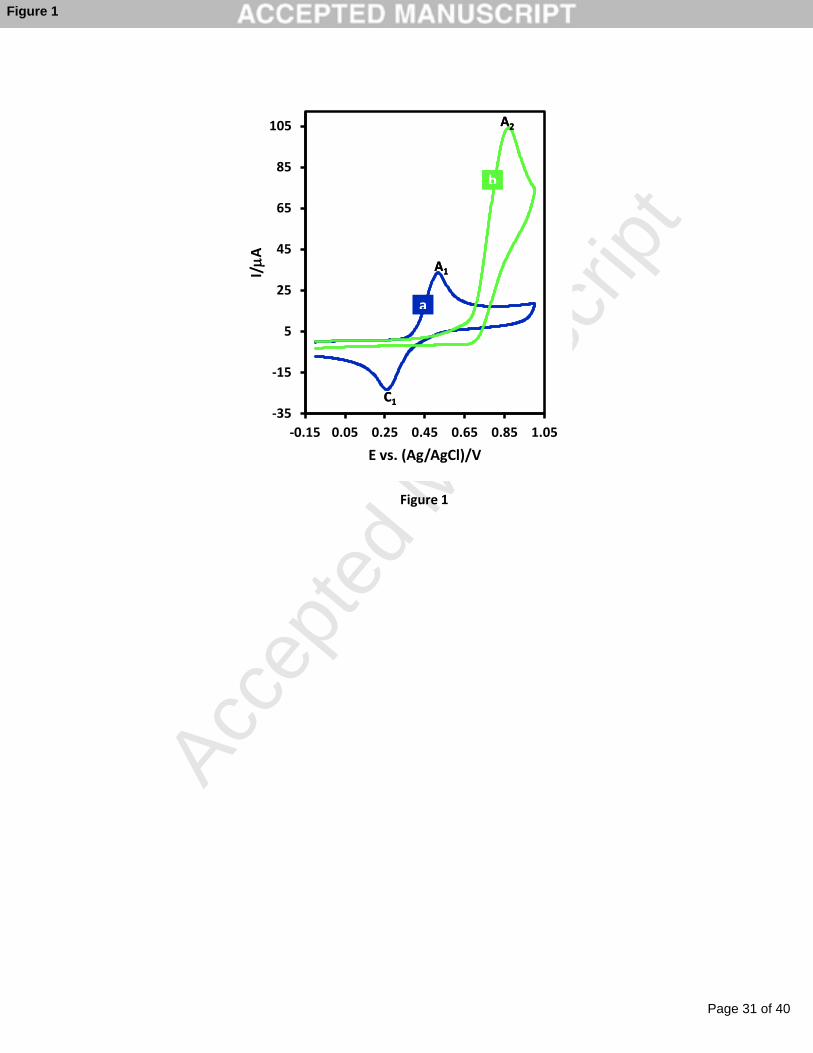
Cyclic voltammogram of catechol (Figure 1, curve a) shows one anodic peak (A1) and its
cathodic counterpart (peak C1) which correspond to the transformation of catechol to o-
benenzoquinone and vice-versa via a quasi-reversible two-electron process. In this figure,
cyclic voltammogram of nitrite ion (curve b) shows an irreversible oxidation peak situated at
0.85 V vs. Ag/AgCl which is attributed to the oxidation of nitrite to nitrate ion [49-51].
Figure 1
Reaction of nitrite ion with electrochemically generated o-benzoquinone was studied by
cyclic voltammetry and controlled potential coulometry. In the presence of nitrite ion, the
voltammogram of catechol shows a decrease in the cathodic peak C1 and the appearance of a
new anodic peak (A3) in the more positive potentials which is related to electrochemical
oxidation of produced 4-nitrocatechol [52]. More voltammetric studies were performed by
increasing of the potential scan rate. Our data shows that at high potential sweep rates, the
peak current ratio (IpC1/IpA1) is about one, while, the current of peak A3 (IpA3) is nearly zero.
These data confirm the reactivity of electrochemically generated o-benzoquinone toward
nitrite ion.
Controlled-potential coulometry of aqueous phosphate buffer solution (c = 0.2 M, pH =
7.0) containing catechol (0.25 mmol) and sodium nitrite (0.25 mmol) at 0.22 V versus
Ag/AgCl was also performed. Cyclic voltammetric analysis, carried out during the
electrolysis, shows the progressive formation of anodic peak A3, parallel to the disappearance
of the peak A1.
The plot of IpA1 vs. Q shows that IpA1 disappears when the charge consumption becomes
about 2e- per molecule of catechol. All of these analysis confirm the reaction of nitrite ion
with electrochemically produced o-benzoquinone and generation of 4-nitrocatechol (4-NC).

Page 8 of 40
Accep
ted
Man
uscr
ipt
8
As previously reported, nitrous acid (pKa = 3.27) [49,50] can act as an oxidizing agent
for compounds such as catechols and converts them to the corresponding o-benzoquinone via
a two-electron process [49,50]. The reaction pathway is shown in Scheme 1. The oxidation of
4-NC is more difficult than the oxidation of starting molecule by virtue of the presence of
electron-withdrawing nitro group on the catechol ring and therefore, its oxidation is
circumvented [52].
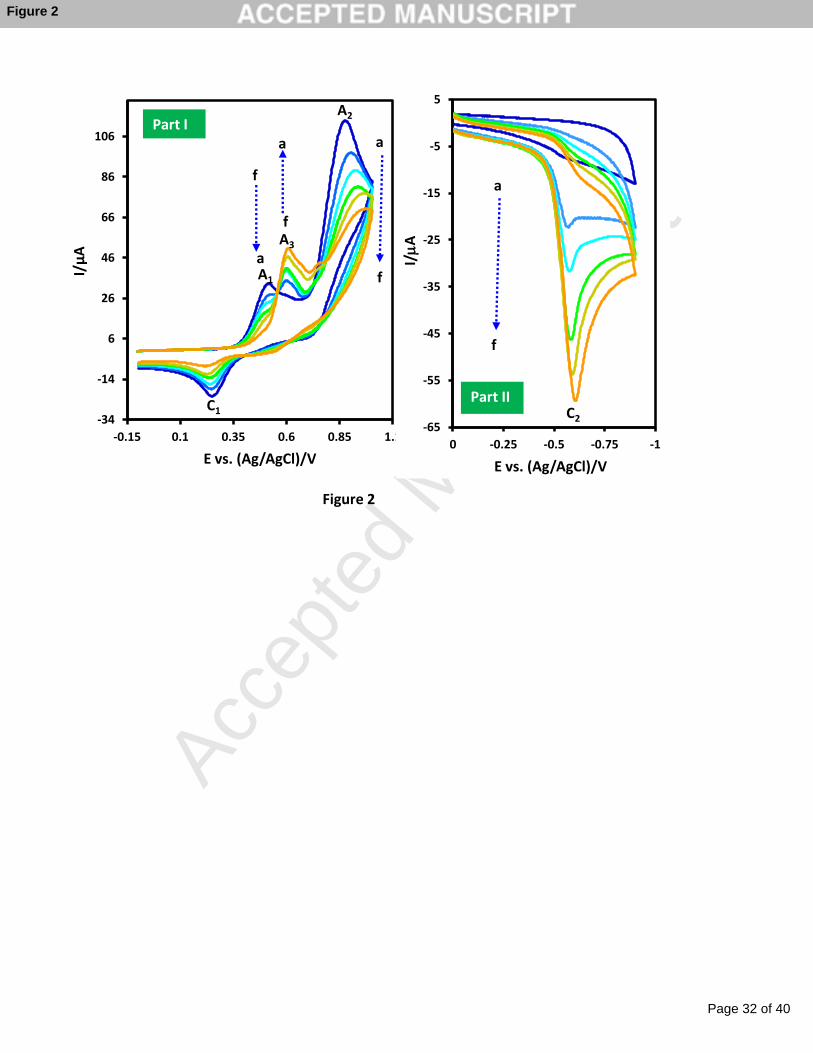
The progress of the chemical nitration of catechol with NO2- in acidic media was tracked
by cyclic voltammetry (Figure 2, Part I). As shown during the oxidation of catechol by
nitrous acid, anodic peak A1 and its cathodic counterpart (peak C1) decreases and disappears
after 40 minutes. In addition, anodic peak A2 (related to the oxidation of NO2-) decreases. On
the other hand, a new anodic peak (A3) which corresponds to the oxidation of chemically
produced 4-nitrocatechol appears at more positive potentials relative to the potential of peak
A1 [49-52]. Similar data was obtained for chemical nitration of catechol by NO2- in aqueous
solution of TEAA ionic liquid.
In addition, formation of 4-nitrocatechol during reaction of catechol by nitrous acid was
investigated by tracking of cathodic peak C2 The cyclic voltammetric curves show the
progressive increase of the peak C2 which is related to the reduction of chemically produced
4-nitrocatechol to the corresponding amine group (Scheme 1) (Figure 2, Part II).
Figure 2

Page 9 of 40
Accep
ted
Man
uscr
ipt
9
Working electrode
Reference electrodeCounter electrode TEAA
Catechol
H2O
NaNO2
NaNO2HOOH
NO2
HOOH
NH2
Step One: Catalytic Oxidation of Catechol
Step Two: Attacking of Nitro group and Reduction of it
+ 6 e
+ 6 H
NaNO2 TEAA
HOOH
N2
- N2
+1 e
HOOH Surface Functionalization
of the Working Electrode
Step Three: In situ generation of diazonium ion
Step Four: Surface functionalization of the working electrodeonto surface of the working electode
In solution
Nitrogen Purging
(II)
(III)
(IV)
(V)
OHOH
OHOHOH
OHOH
OH
OHOH
OHOHOH
OHOH
OH
OHOH
OHOHOH
OHOH
OH
OHOH
OHOHOH
OHOH
OH
Wor
king
Elec
trod
e
Working Electrode
2 NaNO2
NOHO N
HO ONO O H2O
O
O
(I)
N O
HOO
H
N O
OO
NO
H
NO H
[TEAA] -H
TEAA
Scheme 1. Modification of GCE by electroreduction of in situ generated 4-nitrocatechol
For more confirmation, chemically formation of 4-nitrocatechol in acidic condition was
also tracked by UV–vis absorption spectroscopy. Our data show that, catechol has an
absorption peak (P1) at 273 nm while 4-nitrocatechl has two absorption peaks (P0 and P2) at
233 and 333 nm, respectively. During the reaction of catechol with nitrite ion at pH 3.0, peak
P1 is decreased and peaks P0 and P2 are increased, which shows the formation of 4-
nitrocatechol during the chemical reaction.
Possible derivatization of GCE surface by reduction of diazonium salt has been shown
by Cougnon et al in which glassy carbon electrode was modified by a protected catechol
structure (1,2-bis-(tertbutyldimethylsilanoxy)-4-(4-nitrophenylethynyl)-benzene) [36,37].
With respect to these data, we coupled in situ nitration of catechol with electrochemical
reduction of produced 4-nitrocatechol for one-pot functionalization of GCE. All of stages

Page 10 of 40
Accep
ted
Man
uscr
ipt
10
were carried out initially in acidic aqueous solution (HCl, pH = 1.0) and then independently
in buffered aqueous solution by TEAA ionic liquid with pH ~ 7.0 and results of two
conditions were compared. Figure 3 parts I and II shows the successive cyclic
voltammograms of 4-nitrocatechol and catechol respectively obtained at GCE in aqueous
solution containing potassium nitrite and HCl (pH = 1.0). In both cases, upon the first scan of
the GCE to negative potentials, voltammogram shows a cathodic peak (C2) at −0.52 V versus
Ag/AgCl. As shown, following nine cycles over the -0.2V to -0.65 V range vs. Ag/AgCl at a
scan rate of 100 mV s-1, potential of this peak (C2) shifts negatively and its current totally
disappears. After thorough rinsing with acetonitrile and distilled water, the blocking effect of
the electrode response persists. Fig 3, part II shows that similar to 4-nitrocatechol, in situ
chemically generated 4-nitrocatechol is electrochemically reduced to the corresponding 4-
aminicatechol [35]. In a similar way, cougnone et.al are shown that multicyclic
voltammograms of 1,2-bis-(tertbutyldimethylsilanoxy)-4-(4-nitrophenylethynyl)-benzene
results to its grafting on to the GCE surface [37].
Figure 3
In the next step, we investigated the possibility of GCE modification at neutral pH. But
in these conditions, the dissociation equilibrium of nitrous acid lies predominantly to the right
and thus is mainly in its nitrite ion form. Thus the nitrous acid concentration, as an oxidizing
agent, is not sufficient for the oxidation of catechol. So, in order to perform GCE surface
modification in neutral conditions, TEEA (an ionic liquid with pH ~ 7.0) was used.
Repetitive cyclic voltammograms of 4-nitrocatechol (Figure 4, part I) and catechol (Figure 4,
part II) in TEEA aqueous solution (pH ~ 7.0) containing sodium nitrite are shown. As seen,
similar to HCl solution, a cathodic peak C2 related to the reduction of nitro aromatic group
appears and its potential shifts to the negative right. In addition, its current decreases under
subsequent cycles and finally disappears following the twentieth cycle. The results, obtained

Page 11 of 40
Accep
ted
Man
uscr
ipt
11
in both acidic and neutral conditions, indicate that similar to 4-nitrocatechol, in situ produced
4-nitrocatechol (Scheme 1), is reduced to the corresponding 4-aminocatechol during the
cathodic peak C2 (Scheme 1, step III). In the presence of nitrous acid, cathodically generated
4-aminocatechol rapidly reacts with HNO2 to produce corresponding diazonium ion (Scheme
1, step IV). After this, the generated diazonium ion is further reduced on the surface of glassy
carbon electrode, resulting to the formation of aryl radicals. Reaction of these radicals, with
unsaturated carbon atoms of the GCE, yields catechol layer covalently attached to the GCE
surface (Scheme 1, step V). The formation of this layer prevents subsequent reduction, and
consequently cathodic peak shifts to the negative potentials and its current decreases and
finally disappears.
Figure 4
3.1. Modified Electrode
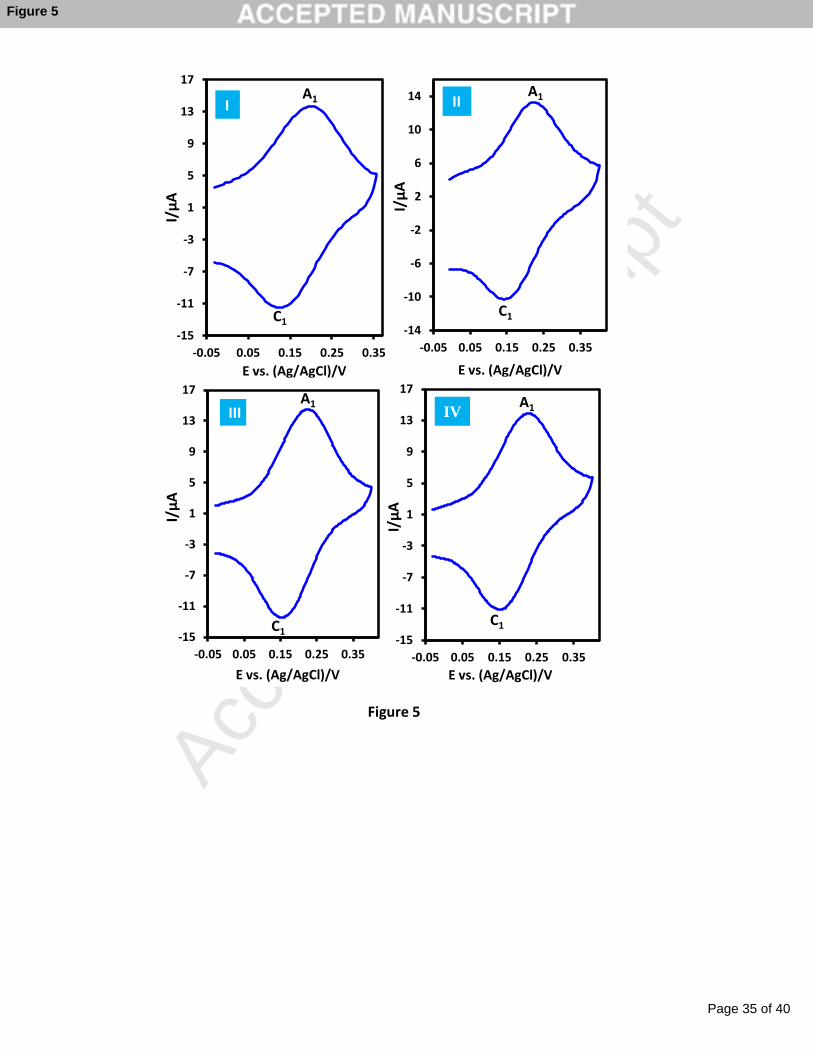
In Figure 5, CVs related to the catechol electrografted glassy carbon electrode using
discussed procedures are presented. In all of them, the modified electrode shows a reversible
peak at 0.19 V in which anodic peak A1 is related to the oxidation of catechol to the
corresponding o-benzoquinone and cathodic peak C1 corresponds to the revers reaction. In
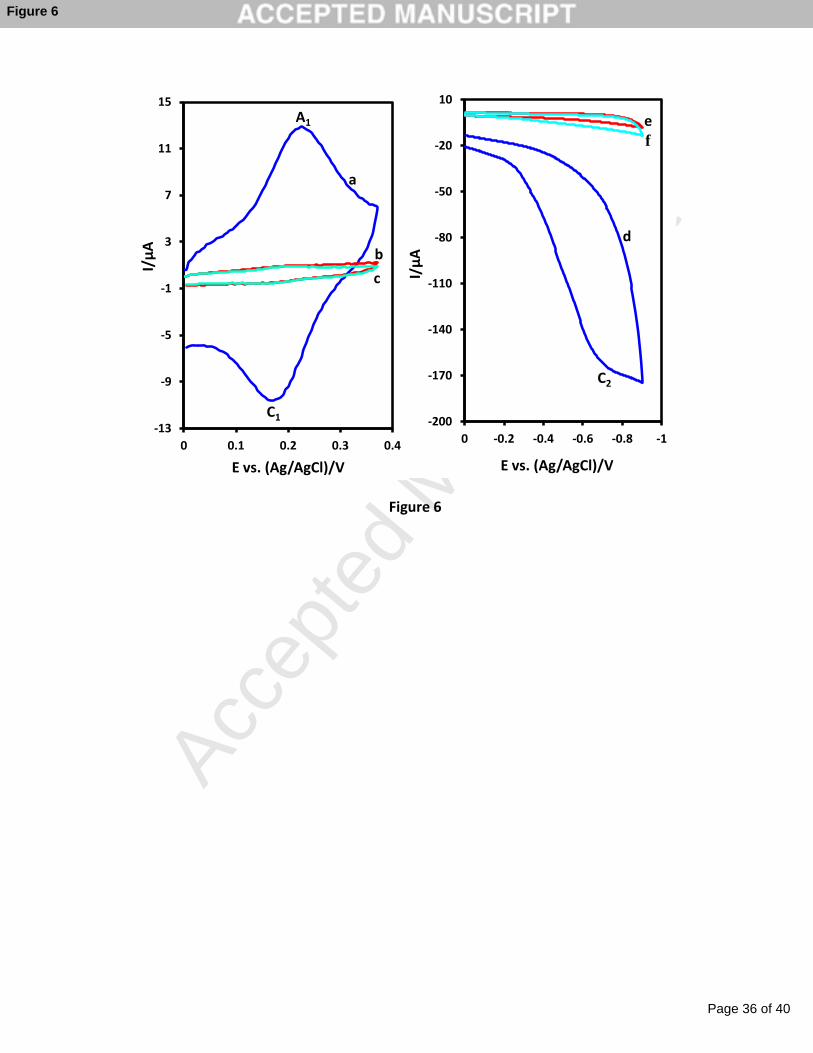
Figure 6, cyclic voltammograms correspond to the modification of GC electrode in the
presence and absence of NaNO2 or proton source are presented. Curve a is related to the
modification in solution containing catechol, NaNO2 and HCl. Modification condition of
cyclic voltammogram b is as same as a but in the absence of NaNO2 and modification
condition of cylic voltammogram c is as same as a but in the absence of HCl. As seen, only in
the presence of all reagents required for modification (catechol, NaNO2 and proton source)
catechol can be immobilized on to the GCE (Figure 6, curve a). CVs corresponding to the

Page 12 of 40
Accep
ted
Man
uscr
ipt
12
electrografted glassy carbon electrode in these conditions are also shown in Figure 6, curves
d-f. These data also confirm the results obtained from the comparison of curves a-c.
Figure 5
Figure 6
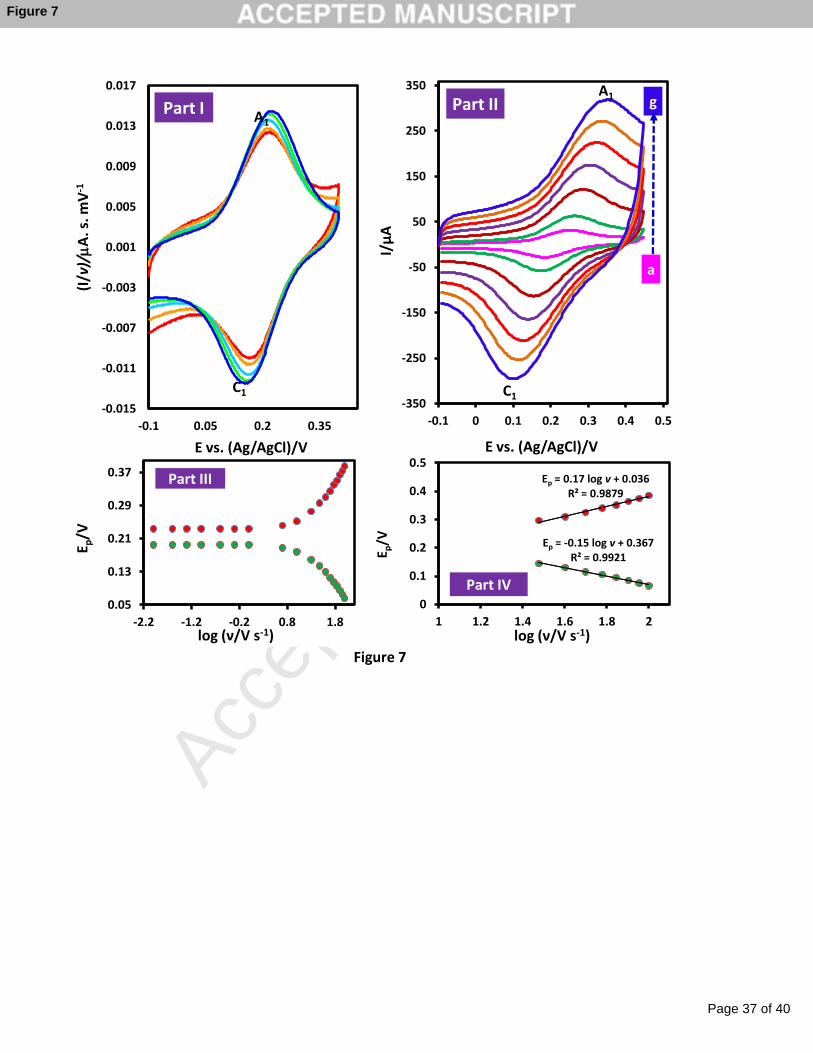
Figure 7 (part I) represents the normalized cyclic voltammograms of immobilized
catechol at different scan rates (these voltammograms were obtained by dividing the current
by the scan rate, I/ν). As shown, IpA1/ν and IPC1/ν remain nearly constant, upon increasing the
potential sweep rate, implying that electron transfer process is related to the electrografted
species. In addition, the cathodic to anodic peak current ratio (IpC/IpA) is about one at given
scan rates, indicating a reversible electron transfer process. A fast electron transfer at the
electrode surface is indicated by small peak potential separation (although not zero) and
independence of peak shape from scan rate. The formal potential of voltammograms is also,
independent of the potential scan rate so that the transfer coefficient (α) was anticipated to be
equal to 0.5 [48]. By plotting of log IpA1 vs. log v, a linear relationship with equation log IpA1
= 1.06 log v -2.34 (R2 = 0.9993) was obtained. It was reported that when the slope of log Ip
against log v is 0.5, the electrochemical reaction is a diffusion controlled process, while when
the slope equals to 1.0, the electrochemical reaction occurs via an adsorption-controlled
process [53,54].
Contrary to the low potential sweep rates (Figure 7, part I), at high potential sweep rates,
∆Ep values increased significantly with increasing sweep rate, suggesting a limitation in the
kinetics of charge transfer (Figure 7, part II). When ∆Ep > 200/n (at high potential scan rates),
the heterogeneous electron transfer rate constant (ks), (assuming that a monolayer of catechol
is adsorbed onto the surface of electrode) can be calculated by plotting Ep vs. log ν (see Table
1) [55]. Heterogeneous electron transfer rate constant of GC and Au electrodes modified by

Page 13 of 40
Accep
ted
Man
uscr
ipt
13
catechol derivatives was also calculated previously [56,57]. Our results are very close to the
values reported in Ref. 56 and are higher than Ref. 57.
Figure 7
Modification of the electrode surface has been influenced by the electrolysis potential,
time and concentration of catechol. Maximum surface coverage (Γ) was found to be at the
potential of -0.60 V, time of 250 s and in 1.0 mM of catechol or 4-nirocatechol. The GCE
was also modified with 3-methylcatechol and 3-methoxycatechol by the same method
described for catechol (time 250 s and c = 1.0 mM). The optimum potential of modification
of 3-methylcatechol and 3-methoxycatechol are -0.64 and -0.70 V vs. Ag/AgCl, respectively.
Parameters related to the catechol derivatives modified electrodes including surface coverage
(Γ), anodic and cathodic electron transfer rate constants ( sA and ks
C) and E1/2 are given in
Table 1. As shown, with respect to substituent of 3-methylcatechol and 3-methoxycatechol,
surface coverage for these derivatives is lower than that for catechol.
Table 1
As shown in Table 1, the calculated surface coverage (Γ) was found to vary in the order
catechol > 3-methylcatechol > 3-methoxycatechol. The observed trend is expected, since the
amount of surface coverage (Γ) is dependent on the size of the substituted group on the
catechol ring. The presence of methyl or methoxy groups on catechol ring causes a decrease
in Γ. Furthermore, the heterogeneous rate conxstant (ks) of the immobilized catechol derivatives
for both anodic and cathodic peaks, varies in the order catechol > 3-methoxycatechol > 3-
methylcatechol. The observed trend, confirms that catechol itself is more suitable mediator
than 3-methylcatechol and 3-methoxycatechol for the catalytic oxidation of NADH.
Because of the participation of proton(s) in the oxidation of catechols, the response of
catechol modified electrode would be pH dependent. The voltammetric response of the
catechol-bonded electrode was studied in buffer solutions with pH values from 1.0 to 8.0. It

Page 14 of 40
Accep
ted
Man
uscr
ipt
14
was found that, both reduction and oxidation peak potentials shifted to the negative potentials
by increasing pH and half-wave potential showed a linear dependence with pH, with a slope
very close to the 59 mV/pH unit which is expected for a two-proton, two-electron process
according to Nernstian behavior. The electrochemical response and surface coverage of the
modified electrode remained nearly constant in all pHs, showing the stability of the catechol
bonded electrode over the pH range 1.0-8.0 [48].
3.2. Electrocatalysis
One of the aims of the present work is to examine the electrocatalytic oxidation of
NADH by the surface modified catechol. It is previously reported that quinones (specially, o-
quinones compared to the p-quinones) are an excellent mediator for the oxidation of NADH
[41]. Cyclic voltammetric response of the catechol bonded electrode was obtained in pH =
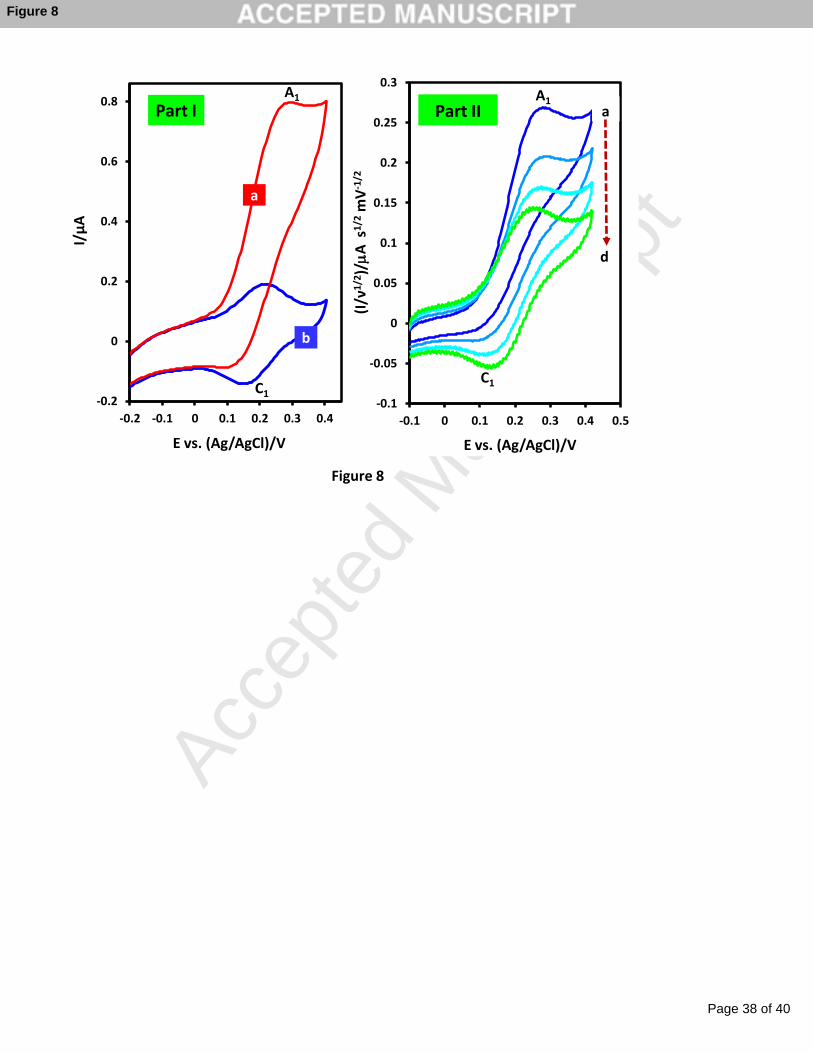
7.0 in the absence and in the presence of NADH (1.0 mM) in order to the study of its
electrocatalytic activity toward NADH oxidation (Figure 8). In the absence of NADH, a well-
defined voltammogram for the catechol on the electrode could be obtained at the formal
potential of 0.19 V vs. Ag/AgCl. The occurrence of a catalytic reaction in the presence of
NADH is supported by the increase of IpA1 and decrease of IpC1, which could indicate that
electro-catalytically active o-quinone formed at the surface of the electrode reacted with
NADH to form NAD+ and catechol. The catechol, which is regenerated at the electrode
surface subsequently reoxidized and again is accessible for another catalytic cycle (Scheme
2).
Figure 8
Scan rate-dependence studies further support the creation of catalytic current in the
oxidation of NADH by modified electrode. Normalized voltammograms (I/ν1/2) of the
modified electrode in the presence of NADH at different scan rates are shown in Figure 8,

Page 15 of 40
Accep
ted
Man
uscr
ipt
15
part II. By decreasing the potential scan rate (longer time scale), the reaction between the
electro-generated o-quinone on the surface of electrode and NADH via the EC' mechanism
has sufficient time to occur (Scheme 2). At shorter time scale (higher scan rates), the anodic
response for the mediated oxidation of NADH by catechol is not readily apparent. These
observations indicate that in longer time scale the catalytic reaction can be carried further and
thus lead to the higher catalytic signal.
OHHO
OO
2NADH 2NAD+
-2e- -2H+
Scheme 2. Catalytic oxidation of NADH by surface confined catechol.
It was also found that the catalytic current of surface modified electrode in the presence
of NADH depends on the solution’s pH. The anodic peak current (IpA1) increases firstly with
increasing pH, reaches to a maximum value at pH 7.0, and then decreases. Thus we choose
pH = 7.0 as an optimum pH for catalytic oxidation of NADH.
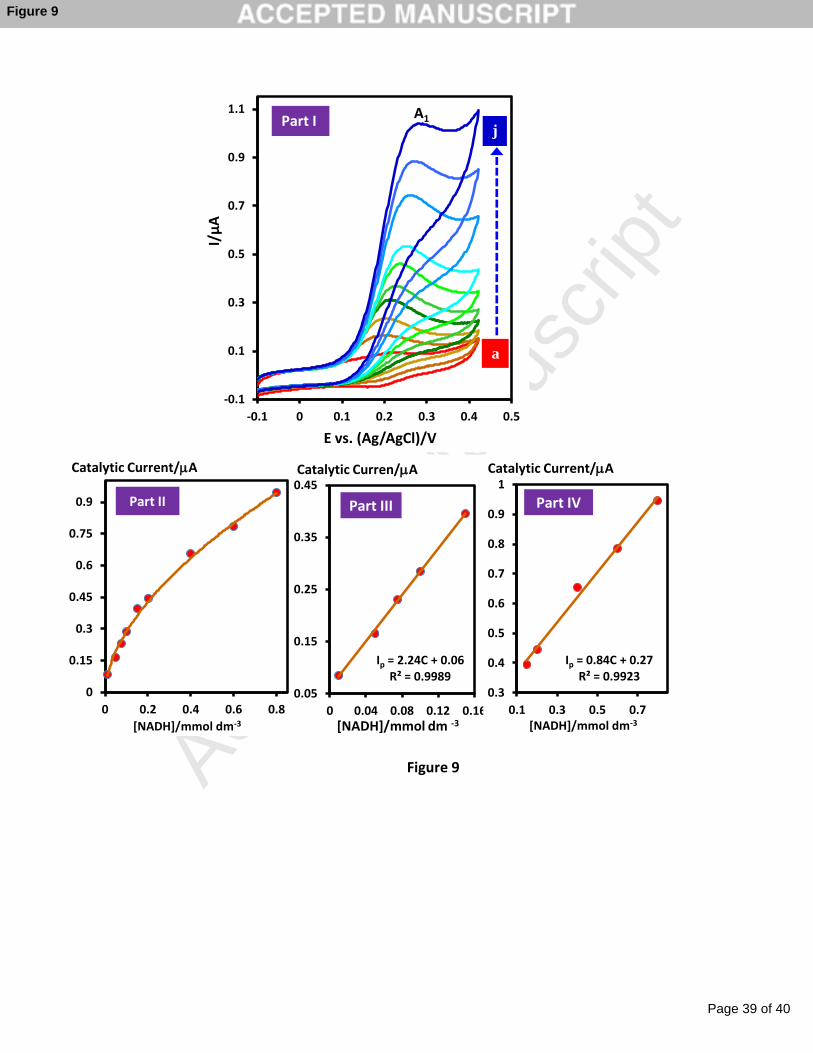
Figure 9 shows the electrocatalytic activity of surface bounded catechol toward the
oxidation of different NADH concentrations. The current of the GC-modified electrode
linearly responds to the NADH concentration over the two linear ranges consisting of 0.01-
0.15 and 0.15-0.80 mM, which covers values of a great relevance in biosensor design and
applications. In addition, for the modified electrode, detection limit of 6.0 μM was estimated.
As anticipated for a Michaelis- Menten-type reaction, the catalytic signal was leveled off at a

Page 16 of 40
Accep
ted
Man
uscr
ipt
16
high concentration (>0.8 mM) which was ascribed to kinetic limitations. The linear range of
this method is consistent with that of method reported previously [56], while, the detection
limit of this method is significantly improved.
Figure 9
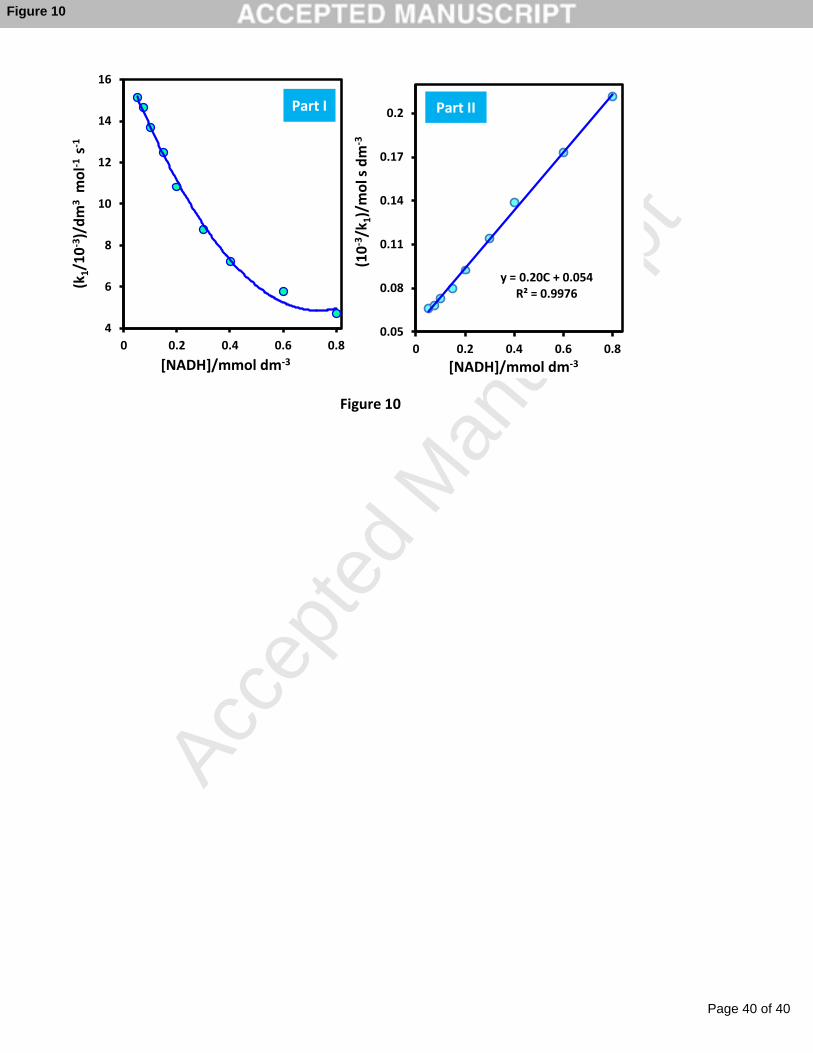
We have also investigated the reaction mechanism at pH = 7.0. By considering the
catalytic reaction as Eqs. (2) and (3), the reaction current, Ip, can be obtained using Eq. (4):
Mred Mox + 2e- + 2H+ (2)
NADH + Mox NAD+ + Mredk1 (3)
Ip = nFAΓk1C* (4)
Here Γ represents the surface coverage of the mediator and C* is the bulk concentration
of NADH. The catalytic reaction proceeds via an intermediate complex (Eq. 5) [57].
NADH + Mox NAD+ + Mredk+1
k-1[NADH-M]
k+2 (5)
According to the Michaelis–Menten type reaction model, and using the Lineweaver–
Burk type equation for the reaction dynamics:
k1 =k+2
Km + C*(6)
where Km is Michaelis parameter. The plots of k1 and k -11 vs. NADH concentration (C*) are
shown in Figure 10. Plot of k -11 vs. NADH concentration gives a straight line indicating that
in the catalytic oxidation of NADH by modified electrode an intermediate complex is
involved. The reaction parameters including, Km and k+2 can also be determined from the
slope and the intercept of the plots; they were 0.26 mM and 5.02 s−1, respectively. These
values are consistent with that of values reported previously [57-59].
Figure 10

Page 17 of 40
Accep
ted
Man
uscr
ipt
17
4. Conclusions
In summary, we have demonstrated that catechols can be successfully electrografted onto
GC surfaces via their chemical nitration and then electrochemical reduction. This method
involves the in situ chemical generation of 4-nitrocatechol from catechol and NO2- followed
by electrografting of diazonium ion produced from 4-aminocatechol which itself generated
from electrochemical reduction of created 4-nitrocatechol. Required protons (for conversion
of nitrite ion to nitrous acid and also for diazotization reaction) can be originated from HCl
(pH = 1.0) or TEAA (pH ~ 7.0) aqueous solutions. By electrochemical reduction of nitro
aromatic to amino aromatic group, diazonium salt is generated exactly in the adjustment of
electrode surface to be modified and also diazotization reaction of produced amine occurs in
non-acidic media resulted from TEAA aqueous solution. As a result, directly anchoring of
molecules containing functionalities that may be capable of reacting with the diazonium
group is possible. Finally, as an application modified electrode surface was used for the
catalytic oxidation of NADH at less positive potential. The catalytic currents are proportional
to the concentration of NADH in solution. Obtained results were very promising and could be
effectively applied in the field of electrocatalysis and electrochemical biosensors.
Acknowledgments
We acknowledge the Bu-Ali Sina University Research Council and the Center of
Excellence in Development of Environmentally Friendly Methods for Chemical Synthesis
(CEDEFMCS) for their support of this work. Also, the institute for frontier materials and the
strategic research center for biotechnology and chemistry are thanked for their continual
support of LH.

Page 18 of 40
Accep
ted
Man
uscr
ipt
18
Supplementary materials
Supplementary materials associated with this article can be found, in the online version, at
http://dx.doi.org...
References
[1] F. Barriére, A.J. Downard, Covalent modification of graphitic carbon substrates by non-
electrochemical methods, J. Solid State Electrochem. 12 (2008) 1231 and references
therein.
[2] K. Malmos, J. Iruthayaraj, S.U. Pedersen, K. Daasbjerg, General approach for monolayer
formation of covalently attached aryl groups through electrografting of arylhydrazines, J.
Am. Chem. Soc. 131 (2009) 13926.
[3] J. Pinson, F. Podvorica, Attachment of organic layers to conductive or semiconductive
surfaces by reduction of diazonium salts, Chem. Soc. Rev. 34 (2005) 429.
[4] M. Müri, B. Gotsmann, Y. Leroux, M. Trouwborst, E. Lörtscher, H. Riel, M. Mayor,
Modular functionalization of electrodes by cross-coupling reactions at their surfaces, Adv.
Funct. Mater. 21 (2011) 3706.
[5] P. Allongue, M. Delamar, B. Desbat, O. Fagebaume, R. Hitmi, J. Pinson, J.M. Savéant,
Covalent modification of carbon surfaces by aryl radicals generated from the
electrochemical reduction of diazonium salts, J. Am. Chem. Soc. 119 (1997) 201.
[6] M. Delamar, G. Désarmot, O. Fagebaume, R. Hitmi, J. Pinson, J.M. Savéant,
Modification of carbon fiber surfaces by electrochemical reduction of aryl diazonium
salts: application to carbon epoxy composites, Carbon 35 (1997) 801.
[7] S. Baranton, D. Bélanger, Electrochemical derivatization of carbon surface by reduction
of in situ generated diazonium cations, J. Phys. Chem. B 109 (2005) 24401.

Page 19 of 40
Accep
ted
Man
uscr
ipt
19
[8] J. Marwan, T. Addou, D. Bélanger, Fanctionaliation of glassy carbon electrodes with
metal-based species, Chem. Mater. 17 (2005) 2395.
[9] B.D. Bath, H.B. Martin, R.M. Wightman, M.R. Anderson, Dopamine adsorption at
surface modified carbon-fiber electrodes, Langmuir 17 (2001)7032.
[10] O. Ghodbane, G. Chamoulaud, D. Bélanger, Chemical reactivity of 4-bromophenyl
modified glassy carbon electrode, Electrochem. Commun. 6 (2004) 254.
[11] R. Polsky, J.C. Harper, S.M. Dirk, D.C. Arango, D.R. Wheeler, S.M. Brozik,
Diazonium-functionalized horseradish peroxidase immobilized via addressable
electrodeposition: direct electron transfer and electrochemical detection, Langmuir 23
(2007) 364.
[12] R.L. McCreery, U. Viswanathan, R.P. Kalakodimi, A.M. Nowak,
Carbon/molecule/metal molecular electronic junctions: the importance of ‘‘contacts’’,
Faraday Discuss. 131 (2006) 33.
[13] J.C. Swarts, D. Laws, W.E. Geiger, Organometallic electrode based on covalent
attachment of the cobaltocenium group to carbon, Organometallics 24 (2005) 341.
[14] T. Matrab, M.M. Chehimi, C. Perruchot, A. Adenier, A. Guillez, M. Save, B. Charleux,
E. Cabet-Deliry, J. Pinson, Novel approach for metallic surface-initiated atom transfer
radical polymerization using electrografted initiators based on aryl diazonium salts,
Langmuir 21 (2005) 4686.
[15] C. Saby, B. Ortiz, G.Y. Champagne, D. Bélanger, Electrochemical modification of
glassy carbon electrode using aromatic diazonium salts. 1. Blocking effect of 4-
nitrophenyl and 4-carboxyphenyl groups, Langmuir, 13 (1997) 6805.
[16] J. Agnès, J.C. Arnault, F. Omnés, B. Jousselme, M. Billon, G. Bidan, P. Mailley, XPS
study of ruthenium tris-bipyridine electrografted from diazonium salt derivative on
microcrystalline boron doped diamond, Phys. Chem. Chem. Phys. 11 (2009) 11647.

Page 20 of 40
Accep
ted
Man
uscr
ipt
20
[17] J. Wang, J.A. Carlisle, Covalent immobilization of glucose oxidase on conducting
ultrananocrystalline diamond thin films, Diamond Relat. Mater. 15 (2006) 279.
[18] A.E. Radi, X. Muñoz-Berbel, V. Lates, J.L. Marty, Label-free impedimetric
immunosensor for sensitive detection of ochratoxin A, Biosens. Bioelectron. 24 (2009)
1888.
[19] M. Pellissier, F. Barriére, A.J. Downard, D. Leech, Improved stability of redox enzyme
layers on glassy carbon electrodes via covalent grafting, Electrochem. Commun. 10 (2008)
835.
[20] M. Lu, W.M. Nolte, T. He, D.A. Corley, J.M. Tour, Direct covalent grafting of
polyoxometalates onto si surfaces, Chem. Mater. 21 (2009) 442.
[21] P.A. Brooksby, A.J. Downard, Multilayer nitroazobenzene films covalently attached to
carbon. An AFM electrochemical study, J. Phys. Chem. B 109 (2005) 8791.
[22] F. Anariba, U. Viswanathan, D.F. Bocian, R.L. McCreery, Determination of the
structure and orientation of organic molecules tethered to flat graphitic carbon by ATR-
FT-IR and Raman spectroscopy, Anal. Chem. 78 (2006) 3104.
[23] A. Ricci, C. Bonazzola, E. Calvo, An FT-IRRAS study of nitrophenyl mono- and
multilayers electro-deposited on gold by reduction of the diazonium salt, J. Phys. Chem.
Chem. Phys. 8 (2006) 4297.
[24] K. Malmos, M. Dong, S. Pillai, P. Kingshott, F. Besenbacher, S.U. Pedersen, K.
Daasbjerg, Using a hydrazone-protected benzenediazonium salt to introduce a near-
monolayer of benzaldehyde on glassy carbon surfaces, J. Am. Chem. Soc. 131 (2009)
4928.
[25] C. Combellas, F. Kanoufi, J. Pinson, F.I. Podvorica, Sterically hindered diazonium salts
for the grafting of a monolayer on metals, J. Am. Chem. Soc. 130 (2008) 8576.

Page 21 of 40
Accep
ted
Man
uscr
ipt
21
[26] J.K. Kariuki, M.T. McDermott, Formation of multilayers on glassy carbon electrodes via
the reduction of diazonium salts, Langmuir 17 (2001) 5947.
[27] C. Combellas, F. Kanoufi, J. Pinson, F.I. Podvorica, Time-of-flight secondary ion mass
spectroscopy characterization of the covalent bonding between a carbon surface and aryl
groups, Langmuir 21 (2005) 280.
[28] J.K. Kariuki, M.T. McDermott, Nucleation and growth of functionalized aryl films on
graphite electrodes, Langmuir 15 (1999) 6534.
[29] P.A. Brooksby, A.J. Downard, Electrochemical and atomic force microscopy study of
carbon surface modification via diazonium reduction in aqueous and acetonitrile solutions,
Langmuir, 20 (2004) 5038.
[30] Y.R. Leroux, H. Fei, J.M. Noël, C. Roux, P. Hapiot, Efficient covalent modification of a
carbon surface: use of a silyl protecting group to form an active monolayer, J. Am. Chem.
Soc. 132 (2010) 14039.
[31] T. Menanteau, E. Levillain, T. Breton, Electrografting via diazonium chemistry: from
multilayer to monolayer using radical scavenger, Chem. Mater. 25 (2013) 2905.
[32] J.L. Hudson, H.A. Jian, D. Leonard, J.J. Stephenson, J.M. Tour, Triazenes as a stable
diazonium source for use in functionalizing carbon nanotubes in aqueous suspensions,
Chem. Mater. 18 (2006) 2766.
[33] B. Chen, A.K. Flatt, H. Jian, J.L. Hudson, J.M. Tour, Molecular grafting to silicon
surfaces in air using organic triazenes as stable diazonium sources and HF as a constant
hydride-passivation source, Chem. Mater. 17 (2005) 4832.
[34] A.J. Gross, V. Nock, M.I.J. Polson, M.M. Alkaisi, A.J. Downard, Surface patterning
using two-phase laminar flow and in situ formation of aryldiazonium salts, Angew. Chem.
Int. Ed. 52 (2013) 10261.

Page 22 of 40
Accep
ted
Man
uscr
ipt
22
[35] C. Cougnon, F. Gohier, D. Bélanger, J. Mauzeroll, In situ formation of diazonium salts
from nitro precursors for scanning electrochemical microscopy patterning of surfaces,
Angew. Chem. Int. Ed. 48 (2009) 4006.
[36] C. Cougnon, J. Mauzeroll, D. Bélanger, Patterning of surfaces by oxidation of amine-
containing compounds using scanning electrochemical microscopy, Angew. Chem. Int.
Ed. 48 (2009) 7395.
[37] C. Cougnon, N.H. Nguyen, S. Dabos-Seignon, J. Mauzeroll, D. Bélanger, Carbon
surface derivatization by electrochemical reduction of a diazonium salt in situ produced
from the nitro precursor, J. Electroanal. Chem. 661 (2011) 13.
[38] V. Zimmermann, F. Avemaria, S. Brase, Chemoselective reduction of nitroarenes in the
presence of acid-sensitive functional groups:� solid-phase syntheses of amino aryl azides
and benzotriazoles, J. Comb. Chem. 9 (2007) 200.
[39] M. Kongsfelt, J. Vinther, K. Malmos, M. Ceccato, K. Torbensen, C.S. Knudsen, K.V.
Gothelf, S.U. Pedersen, K. Daasbjerg, Combining aryltriazenes and electrogenerated acids
to create well-defined aryl-tethered films and patterns on surfaces, J. Am. Chem. Soc. 133
(2011) 3788.
[40] E. Lebégue, L. Madec, T. Brousse, J. Gaubicher, E. Levillain, C. Cougnon, Modification
of activated carbons based on diazonium ions in situ produced from aminobenzene organic
acid without addition of other acid, J. Mater. Chem. 21 (2011) 12221.
[41] C. Combellas, M. Delamar, F. Kanoufi, J. Pinson, F.I. Podvorica, Spontaneous grafting
of iron surfaces by reduction of aryldiazonium salts in acidic or neutral aqueous solution.
application to the protection of iron against corrosion, Chem. Mater. 17 (2005) 3968.
[42] F.I. Podvorica, F. Kanoufi, J. Pinson, C. Combellas, Spontaneous grafting of diazoates
on metals, Electrochim. Acta 54 (2009) 2164.

Page 23 of 40
Accep
ted
Man
uscr
ipt
23
[43] L. Gorton, E. Dominguez, Electrochemistry of NAD(P)+/NAD–(P)H. In encyclopedia of
electrochemistry, bioelectrochemistry, G.S. Wilson, Ed.; Wiley-VCH, Weinheim,
Germany, 2002, Vol. 9, pp. 67-143, and the references cited therein.
[44] W.M. Clark, Oxidation-Reduction Potentials of Organic Synthesis; RE Krieger
Publishing, Huntington, NY, 1972.
[45] W.J. Blaedel, R.A. Jenkins, Electrochemical oxidation of reduced nicotinamide adenine
dinucleotide, Anal. Chem. 47 (1975) 1337.
[46] Q. Wu, M. Maskus, F. Pariente, F. Tobalina, V.M. Fernández, E. Lorenzo, H.D. Abruña,
Electrocatalytic oxidation of NADH at glassy carbon electrodes modified with transition
metal complexes containing 1,10-phenanthroline-5,6-dione ligands, Anal. Chem. 68
(1996) 3688.
[47] M. Wooten, W. Gorski, Facilitation of NADH electro-oxidation at treated carbon
nanotubes, Anal. Chem. 82 (2010) 1299.
[48] C.R. Raj, S. Behera, Electrochemically triggered Michael addition on the self-assembly
of 4-thiouracil: generation of surface-confined redox mediator and electrocatalysis,
Langmuir 23 (2007) 1600.
[49] L. Khalafi, M. Rafiee, Kinetic study of the oxidation and nitration of catechols in the
presence of nitrous acid ionization equilibria, J. Hazard. Mat. 174 (2010) 801.
[50] M. Rafiee, D. Nematollahi, H. Salehzadeh, CEC mechanism in electrochemical
oxidation of nitrocatechol–boric acid Complexes, Electrochim. Acta 56 (2011) 9946.
[51] D. Nematollahi, M. Rafiee, L. Fotouhi, Mechanistic study of homogeneous reactions
coupled with electrochemical oxidation of catechols, J. Iran. Chem. Soc. 6 (2009) 448.
[52] D. Nematollahi, A. Ariapad, M. Rafiee, Electrochemical nitration of catechols: Kinetic
study by digital simulation of cyclic voltammograms, J. Electroanal. Chem. 602 (2007) 37.

Page 24 of 40
Accep
ted
Man
uscr
ipt
24
[53] E.J. Laviron, L. Roullier, C. Degrand, A multilayer model for the study of space
distributed redox modified electrodes: Part I. Description and discussion of the model, J.
Electroanal. Chem. 112 (1980) 1.
[54] H. Beiginejad, D. Nematollahi, M. Bayat, Electrochemical oxidation of hematoxylin.
Part 1: Experimental and theoretical studies in an aqueous acidic medium, J. Electroanal.
Chem. 681 (2012) 76.
[55] E.J. Laviron, General expression of the linear potential sweep voltammogram in the case
of diffusionless electrochemical systems, Electroanal. Chem. 101 (1979) 19.
[56] F. Pariente, F. Tobalina, M. Darder, E. Lorenzo, H.D. Abruña, Electrodeposition of
redox-active films of dihydroxybenzaldehydes and related analogs and their
electrocatalytic activity toward NADH oxidation, Anal. Chem. 68 (1996) 3135.
[57] K. Nakanoa, K. Ohkubo, H. Taira, M. Takagi, Toshihiko Imato, Electrocatalytic
oxidation of dihydronicotineamide adenine dinucleotide on gold electrode modified with
catechol-terminated alkanethiol self-assembly, Anal. Chim. Acta 619 (2008) 30.
[58] H. Jaegfeldt, A.B.C. Torstensson, L.G.O. Gorton, G. Johansson, Catalytic oxidation of
reduced nicotinamide adenine dinucleotide by graphite electrodes modified with adsorbed
aromatics containing catechol functionalities, Anal. Chem. 53 (1981) 1979.
[59] F. Pariente, F. Tobalina, G. Moreno, L. Hernández, E. Lorenzo, H.D. Abruña,
Mechanistic studies of the electrocatalytic oxidation of NADH and ascorbate at glassy
carbon electrodes modified with electrodeposited films derived from 3,4-
dihydroxybenzaldehyde, Anal. Chem. 69 (1997) 4065.

Page 25 of 40
Accep
ted
Man
uscr
ipt
25
Figure Captions
Fig. 1. Cyclic voltammograms of (a) catechol (1.0 mM) and (b) sodium nitrite (3.0 mM) at
glassy carbon electrode, in phosphate buffer solution (pH 3.0, c = 0.2 M). Scan rate: 500
mV s-1. Temp. = 25 ± 1 oC.
Fig. 2. Cyclic voltammograms of catechol (1.0 mM) in the presence of sodium nitrite (3.0 mM)
during chemical reaction in aqueous phosphate buffer (pH = 3.0) at various times; reaction
times are 1, 5, 10, 15, 20, and 40 min from (a) to (f), respectively. Part I: Es = -0.1 and Ef =
1.0 V vs. Ag/AgCl. Part II: Es = 0.0 and Ef = -0.9 V vs. Ag/AgCl. Scan rate 500 mV s-1.
Temp. = 25±1 ◦C.
Fig. 3. a) First, b) second, c) third and d) ninth cycles of cyclic voltammograms of (I) 4-
nitrocatechol (1.0 mM) and (II) catechol (1.0 mM) at a bare glassy carbon electrode, in
water solution containing sodium nitrite (5 mM) and HCl (pH = 1.0). Scan rate: 100 mV
s−1. Temp. = 25±1 oC.
Fig. 4. a) First, b) Second, c) fifth, d) tenth and e) twentieth cycles of cyclic voltammograms
of 4-nitrocatechol (1.0 mM) (left) and catechol (1.0 mM) (right) at a bare glassy carbon
electrode, in aqueous/TEAA (1:1) solution containing sodium nitrite (5 mM) (pH = 7.0).
Scan rate: 100 mV s−1. Temp. = 25±1 oC.
Fig. 5. Cyclic voltammograms of electrografted catechol in various conditions (I) from 4-
nitrocatechol precursor in the presence of HCl (pH = 1.0) (II) from catechol precursor in
the presence of HCl (pH = 1.0) (III) from 4-nitrocatechol precursor in the presence of
TEAA (pH = 7.0) (IV) from catechol precursor in the presence of TEAA (pH = 7.0).
Concentration of NaNO2 = 5 mM, TEAA/water ratio is 1/1 v:v. Scan rate: 1000 mV s−1.

Page 26 of 40
Accep
ted
Man
uscr
ipt
26
Fig. 6. Cyclic voltammograms for modification of GCE (a) catechol (1.0 mM) in the
presence of sodium nitrite (5 mM) and HCl (pH = 1.0), (b) catechol (1.0 mM) in the
presence of HCl (pH = 1.0) and (c) catechol (1.0 mM) in the presence of sodium nitrite (5
mM). Corresponding cyclic voltammograms for modified electrode in aqueous solution
containing phosphate buffer (pH = 7.0). d, e and f are related to a, b and c respectively.
Scan rate 1000 mV s-1.
Fig. 7. Part I: Normalized cyclic voltammograms of the surface-confined catechol in
phosphate buffer solution (pH = 7.0, c = 0.10 M) at (low) potential scan rates: 0.05, 0.10,
0.25, 0.50 and 1.00 V s-1. Part II: Cyclic voltammograms of above electrode at (high)
potential scan rates: (a) 5, (b) 10, (c) 20, (d) 30, (e) 40, (f) 50 and (g) 60 V s-1. Part III:
Experimental variation of Ep vs. the log v for part II. Part IV: Magnification of the same
plot for high sweep rates.
Fig. 8. Part I: Cyclic voltammograms obtained for the surface-modified catechol in phosphate
buffer solution (c = 0.2M, pH = 7.0). (a) In the absence and (b) in the presence of NADH
(1 mM). Scan rate: 20 mV s-1. Part II: Normalized cyclic voltammograms (I/ν1/2) in the
presence of NADH (0.5 mM) at different potential sweep rates: (a) 10, (b) 25, (c) 50 and
(d) 75 mV s-1.
Fig. 9. Part I: Cyclic voltammograms of the GC-modified catechol electrode in the presence
of different concentration of NADH in phasphate buffer (pH 7.0, c = 0.20 M). [NADH]:
(a) 0.000 (b) 0.010, (c) 0.050, (d) 0.075, (e) 0.100, (f) 0.150, (g) 0.200, (h) 0.400, (i)
0.600, and (j) 0.800 mM. Scan rate: 20 mV s-1. Parts II, III and IV show the corresponding
calibration plots.

Page 27 of 40
Accep
ted
Man
uscr
ipt
27
Fig. 10. Plots of k1 and k -11 vs. NADH concentration for the CV responses of the modified
electrode. Experimental conditions are as same as Fig. 9.

Page 28 of 40
Accep
ted
Man
uscr
ipt
28
Table 1. Surface coverage (Γ), heterogeneous rate constants and E1/2 of derivatized GCE prepared by
reduction of diazonium cations in situ produced from nitrocatechol derivatives.
Redox mediator
OHHO
OHHO
OHHO O
Γ[a] 8.0 5.8 5.1
E1/2 0.214 0.138 0.130
ks (anodic peak)[b] 67.9 24.8 42.3
ks(cathodic peak)[b] 54.6 20.4 33.6
[a] Anodic surface coverage (in mol cm-2 × 1011) [b] Heterogeneous electron transfer rate constant (s−1)

Page 29 of 40
Accep
ted
Man
uscr
ipt
GRAPHICAL ABSTRACT
General approach for electrochemical functionalization of glassy carbon
surface by in situ generation of diazonium ion under acidic and non-acidic
condition with a cascade protocol
Hamid Salehzadeh, Vahid Khakyzadeh, Davood Nematollahi* Banafsheh Mokhtari and Luke
C. Henderson
Graphical Abstract (for review)

Page 30 of 40
Accep
ted
Man
uscr
ipt
Highlighted
Selective electrochemical determination of NADH.
Electrochemical functionalization of glassy carbon surface by in situ
generation of diazonium ion.
Oxidative nitration of catechols.
Electrografting of catechols onto the GC surface.
Research Highlights
Page 31 of 40
Accep
ted
Man
uscr
ipt
-35
-15
5
25
45
65
85
105
-0.15 0.05 0.25 0.45 0.65 0.85 1.05
I/m
A
E vs. (Ag/AgCl)/V
A1
C1
A2
a
b
A1
C1
A2
a
b
Figure 1
Figure 1
Page 32 of 40
Accep
ted
Man
uscr
ipt
-34
-14
6
26
46
66
86
106
-0.15 0.1 0.35 0.6 0.85 1.1
I/µ
A
E vs. (Ag/AgCl)/V
A1
C1
A3
A2
a
f a
f
f
a
Part I
-65
-55
-45
-35
-25
-15
-5
5
-1 -0.75 -0.5 -0.25 0
I/m
A
E vs. (Ag/AgCl)/V
C2
a
f
Part II
Figure 2
Figure 2

Page 33 of 40
Accep
ted
Man
uscr
ipt
-180
-160
-140
-120
-100
-80
-60
-40
-20
0
-0.75 -0.55 -0.35 -0.15
I/µ
A
E vs. (Ag/AgCl)/V
a
C2
Part I
d
-160
-140
-120
-100
-80
-60
-40
-20
0
-0.75 -0.55 -0.35 -0.15
I/µ
A
E vs. (Ag/AgCl)/V
a
d
C2
Part II
Figure 3
Figure 3

Page 34 of 40
Accep
ted
Man
uscr
ipt
-69
-59
-49
-39
-29
-19
-9
1
-1 -0.8 -0.6 -0.4 -0.2
I/µ
A
E vs. (Ag/AgCl)/V
C2
a
e
Part II
-62
-52
-42
-32
-22
-12
-2
-1 -0.8 -0.6 -0.4 -0.2
I/µ
A
E vs. (Ag/AgCl)/V
a
e
C2 I
Part I
Figure 4
Figure 4
Page 35 of 40
Accep
ted
Man
uscr
ipt
-15
-11
-7
-3
1
5
9
13
17
-0.05 0.05 0.15 0.25 0.35
I/µ
A
E vs. (Ag/AgCl)/V
A1
C1
I
-14
-10
-6
-2
2
6
10
14
-0.05 0.05 0.15 0.25 0.35
I/µ
A
E vs. (Ag/AgCl)/V
A1
C1
II
-15
-11
-7
-3
1
5
9
13
17
-0.05 0.05 0.15 0.25 0.35
I/µ
A
E vs. (Ag/AgCl)/V
C1
III
A1
-15
-11
-7
-3
1
5
9
13
17
-0.05 0.05 0.15 0.25 0.35
I/µ
A
E vs. (Ag/AgCl)/V
C1
A1 IV
Figure 5
Figure 5
Page 36 of 40
Accep
ted
Man
uscr
ipt
-200
-170
-140
-110
-80
-50
-20
10
-1 -0.8 -0.6 -0.4 -0.2 0
I/µ
A
E vs. (Ag/AgCl)/V
C2
d
e
f
-13
-9
-5
-1
3
7
11
15
0 0.1 0.2 0.3 0.4
I/µ
A
E vs. (Ag/AgCl)/V
A1
C1
a
b
c
Figure 6
Figure 6
Page 37 of 40
Accep
ted
Man
uscr
ipt
-350
-250
-150
-50
50
150
250
350
-0.1 0 0.1 0.2 0.3 0.4 0.5
I/µ
A
E vs. (Ag/AgCl)/V
a
g A1
C1
Part II
0.05
0.13
0.21
0.29
0.37
-2.2 -1.2 -0.2 0.8 1.8
E p/V
log (ν/V s-1)
Part III Ep = 0.17 log v + 0.036 R² = 0.9879
Ep = -0.15 log v + 0.367 R² = 0.9921
0
0.1
0.2
0.3
0.4
0.5
1 1.2 1.4 1.6 1.8 2
E p/V
log (ν/V s-1)
Part IV
Figure 7
-0.015
-0.011
-0.007
-0.003
0.001
0.005
0.009
0.013
0.017
-0.1 0.05 0.2 0.35
(I/v)/m
A. s
. mV
-1
E vs. (Ag/AgCl)/V
A1
C1
Part I
Figure 7
Page 38 of 40
Accep
ted
Man
uscr
ipt
-0.2
0
0.2
0.4
0.6
0.8
-0.2 -0.1 0 0.1 0.2 0.3 0.4
I/µ
A
E vs. (Ag/AgCl)/V
A1
C1
a
b
Part I
-0.1
-0.05
0
0.05
0.1
0.15
0.2
0.25
0.3
-0.1 0 0.1 0.2 0.3 0.4 0.5
(I/ν
1/2
)/m
A s
1/2
mV
-1/2
E vs. (Ag/AgCl)/V
A1
a
C1
d
Part II
Figure 8
Figure 8
Page 39 of 40
Accep
ted
Man
uscr
ipt
-0.1
0.1
0.3
0.5
0.7
0.9
1.1
-0.1 0 0.1 0.2 0.3 0.4 0.5
I/µ
A
E vs. (Ag/AgCl)/V
A1
a
j
Part I
0
0.15
0.3
0.45
0.6
0.75
0.9
0 0.2 0.4 0.6 0.8
[NADH]/mmol dm-3
Catalytic Current/mA
Part II
Ip = 2.24C + 0.06 R² = 0.9989
0.05
0.15
0.25
0.35
0.45
0 0.04 0.08 0.12 0.16 [NADH]/mmol dm -3
Catalytic Curren/mA
Part III
Ip = 0.84C + 0.27 R² = 0.9923
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
0.1 0.3 0.5 0.7 [NADH]/mmol dm-3
Catalytic Current/mA
Part IV
Figure 9
Figure 9
Page 40 of 40
Accep
ted
Man
uscr
ipt
4
6
8
10
12
14
16
0 0.2 0.4 0.6 0.8
(k1/1
0-3
)/d
m3 m
ol-1
s-1
[NADH]/mmol dm-3
Part I
y = 0.20C + 0.054 R² = 0.9976
0.05
0.08
0.11
0.14
0.17
0.2
0 0.2 0.4 0.6 0.8 (1
0-3
/k1)/
mo
l s d
m-3
[NADH]/mmol dm-3
Part II
Figure 10
Figure 10





























