Embed Size (px)
Citation preview

Invited Review
Gender, sex hormones, and vascular tone
Julia M. Orshal and Raouf A. KhalilResearch and Development, Department of Veterans Affairs Medical Center, West Roxbury;and Department of Medicine, Harvard Medical School, Boston, Massachusetts 02132
Orshal, Julia M., and Raouf A. Khalil. Gender, sex hormones, and vasculartone.Am J Physiol Regul Integr Comp Physiol 286: R233–R249, 2004; 10.1152/ajpregu.00338.2003.—The greater incidence of hypertension and coronary arterydisease in men and postmenopausal women compared with premenopausal womenhas been related, in part, to gender differences in vascular tone and possiblevascular protective effects of the female sex hormones estrogen and progesterone.However, vascular effects of the male sex hormone testosterone have also beensuggested. Estrogen, progesterone, and testosterone receptors have been identifiedin blood vessels of human and other mammals and have been localized in theplasmalemma, cytosol, and nuclear compartments of various vascular cells, includ-ing the endothelium and the smooth muscle. The interaction of sex hormones withcytosolic/nuclear receptors triggers long-term genomic effects that could stimulateendothelial cell growth while inhibiting smooth muscle proliferation. Activation ofplasmalemmal sex hormone receptors may trigger acute nongenomic responses thatcould stimulate endothelium-dependent mechanisms of vascular relaxation such asthe nitric oxide-cGMP, prostacyclin-cAMP, and hyperpolarization pathways. Ad-ditional endothelium-independent effects of sex hormones may involve inhibitionof the signaling mechanisms of vascular smooth muscle contraction such asintracellular Ca2� concentration and protein kinase C. The sex hormone-inducedstimulation of the endothelium-dependent mechanisms of vascular relaxation andinhibition of the mechanisms of vascular smooth muscle contraction may contributeto the gender differences in vascular tone and may represent potential beneficialvascular effects of hormone replacement therapy during natural and surgicallyinduced deficiencies of gonadal hormones.
estrogen; progesterone; testosterone; endothelium; nitric oxide; vascular smoothmuscle; calcium
CARDIOVASCULAR DISEASES such as hypertension and coronaryartery disease are some of the most common and costlydiseases in the industrialized world. The incidence of cardio-vascular diseases is greater in men aged 30–50 yr comparedwith women of similar age (38, 43). Among women, theincidence of cardiovascular diseases is greater in postmeno-pausal compared with premenopausal women. Although re-ports from Heart and Estrogen-Progestin Replacement Study(HERS), HERS2, and Women’s Health Initiative (WHI) stud-ies do not support beneficial vascular effects of hormonereplacement therapy (HRT) particularly in elderly hypertensivewomen (37, 47, 74, 89, 117, 119), other studies have suggestedbeneficial effects of HRT in reducing the incidence of coronaryartery disease in postmenopausal women and have suggestedputative vascular protective effects of female sex hormones(43, 48, 108). Sex hormone receptors have been identified inthe cytosol and nuclear compartments of various cell typesincluding the endothelium and vascular smooth muscle (VSM)(Table 1). The interaction of sex hormones with cytosolic/nuclear receptors has long been known to stimulate a host ofgenomic effects that could affect vascular cell growth andproliferation. Recent evidence suggests that sex hormones mayalso interact with specific plasmalemmal receptors and induce
additional nongenomic vascular effects (38, 43). Several ex-cellent reviews have described the role of gender and femalesex hormones in modifying the incidence of cardiovasculardisease (38, 43, 48). Previous reviews have focused on possibleestrogen-induced beneficial effects such as modification ofcirculating lipoproteins, inhibition of lipoprotein oxidation(106), attenuation of atherosclerotic lesions, favorable modu-lation of homocysteine (134), changes in blood coagulation (4),and inhibition of intravascular accumulation of collagen (8)(Table 2). Also, several studies have suggested significanteffects of sex hormones on the renal control mechanisms of theblood pressure, particularly the renin-angiotensin system (104,105). For example, estradiol has been suggested to inhibit reninrelease and the angiotensin converting enzyme (126), whereastestosterone may increase the blood pressure by activating therenin-angiotensin system (105). Although alterations in vascu-lar tone play a major role in the control of blood pressure andthe coronary circulation and thereby the incidence of hyper-tension and coronary artery disease, little information is avail-able regarding the gender differences and the effects of sexhormones on vascular tone.
The purpose of this review is to provide an insight into thegender differences in vascular tone and the effects of sexhormones on vascular cells, namely the endothelium andsmooth muscle. We will first provide an overview on vasculartone and its modification with gender. The vascular sex hor-mone receptors, agonists, and antagonists will then be de-
Address for reprint requests and other correspondence: R. A. Khalil, Har-vard Medical School, VA Boston Healthcare-Research, 1400 VFW Parkway3/2B123, Boston, MA 02132 (E-mail: [email protected]).
Am J Physiol Regul Integr Comp Physiol 286: R233–R249, 2004;10.1152/ajpregu.00338.2003.
http://www.ajpregu.org R233
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from

scribed. We will follow with a description of the genomiceffects of sex hormones on endothelial as well as VSM cellgrowth and proliferation. The nongenomic effects of sex hor-mones on the endothelium-dependent mechanisms of vascularrelaxation will then be discussed. We will follow with adetailed description of the effects of sex hormones on the
signaling mechanisms of VSM contraction. The review willend with a perspective on potential areas for future investiga-tions to better understand the mechanisms underlying thegender differences and the effects of sex hormones on vasculartone and the possible clinical uses of HRT to reduce theincidence of cardiovascular disease.
Table 1. Examples of sex hormone receptor distribution, agonists, and antagonists in the endotheliumand vascular smooth muscle
Estrogen Progesterone Testosterone
Blood vessel distribution - Mouse aorta- Rat aorta, carotid, uterine, tail artery- Rabbit uterine artery- Bovine aortic endothelial cells- Primate coronary, carotid artery- Human coronary, uterine artery, umbilical
vein endothelial cells
- Mouse aorta- Rat aorta- Rabbit uterine artery- Bovine aortic endothelial cells- Primate aorta, coronary artery- Human aorta, internal carotid, coronary,
uterine artery
- Mouse aorta- Rat aorta- Rabbit aorta, coronary artery- Bovine aorta- Primate coronary artery- Human internal mammary artery
Subcellular distributionPlasma membrane � � �Cytosol � � �Nucleus � � �Mitochondria,
Endoplasmic� � undetermined
Reticulum, Golgi,Lysosomes
Agonists - 17�-Estradiol- Estradiol benzoate, cypionate, valerate- Ethinyl estradiol- Diethylstilbestrol- Idoxifene- Phytoestrogens: genistein, daidzein,
coumestrol, zearalenone, �-zearalanol,naringenin, taxifolin, biochanin A
- Progestins, progesterone- Medroxyprogesterone acetate- Norethisterone, norgestimate- 3-keto-desogestrel, Levonorgestrel- Gestodene- R5020
- Testosterone- Dihydrotestosterone- Dehyroepiandrosterone- Androstenedione- Cyproterone acetate
Antagonists - ICI-182780 (Fulvestrant), ICI-164384- Clomiphene citrate- RR-tetrahydrochrysene (ER-�)
- RU-486 (Mifepristone)- Onapristone- ZK-98299, ZK-98734
- Flutamide, Hydroxyflutamide- Casodex
Agonists/antagonists - Tamoxifen, 4-hydroxytamoxifen- Raloxifene, toremifene- 6-Carboxymethyl genistein- Enterodiol, enterolactone
Table 2. Beneficial vascular effects and possible clinical applications of sex hormones
Estrogen Progesterone Testosterone
Endothelium - Promotes proliferation/migration- Promotes release of EDRFs - Promotes release of EDRFs - Promotes release of EDRFs- Inhibits release of EDCFs - Inhibits release of EDCFs
Smooth muscle - Inhibits proliferation/migration - Inhibits proliferation/migration - Inhibits proliferation- VSM relaxation and vasodilation - Facilitates the vascular - Acute vascular relaxation
inhibitory effects of estrogen - Hyperpolarization- Acute vascular relaxation - Coronary vasodilation
Other vascular effects - Antiatherosclerotic - Antiatherosclerotic - Antiatherosclerotic- Antioxidant- Decreases low density lipoproteins,
increase high density lipoproteins
- Decreases low densitylipoproteins, increase highdensity lipoproteins
- Inhibition of lipoprotein oxidation- Decreases plasma homocysteine- Increases antiplatelet aggregation
factors, decrease platelet adhesion- Decreases vascular collagen
Clinical applications - Coronary artery disease - Coronary artery disease - Reduction of myocardial ischemia in- Postmenopausal hypertension - Thromboembolic events men with coronary artery disease- Thromboembolic events
EDRF, endothelium-derived relaxing factor; EDCF, endothelium-derived contracting factor; VSM, vascular smooth muscle.
Invited Review
R234 SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from

GENDER DIFFERENCES IN VASCULAR TONE
Vascular tone is defined as the degree of constriction of ablood vessel relative to its maximal diameter in the dilatedstate. Under basal conditions, most resistance and capacitancevessels exhibit some degree of smooth muscle contraction thatdetermines the diameter or tone of the vessel. Vascular tone isinfluenced by both the endothelium and VSM. The vasculartone is also determined by a multitude of vasoconstrictorfactors such as norepinephrine, ANG II, vasopressin, and5-hydroxytryptamine as well as vasodilator factors such asbradykinin and prostacyclin (PGI2). These factors can be di-vided into extrinsic factors that originate from outside theblood vessel and intrinsic factors that originate from the vesselitself. Extrinsic factors primarily serve the function of regulat-ing arterial pressure by altering systemic vascular resistance,whereas intrinsic mechanisms are concerned with regulation oflocal blood flow within an organ (30).
Gender differences in vascular tone have been described ina multitude of vascular beds in both human and experimentalanimals (123). For example, �-adrenergic agonists such asnorepinephrine cause less forearm vasoconstriction in womenthan in men (77). Also, oxidized low-density lipoprotein en-hances 5-hydroxytryptamine-induced contraction to a greaterextent in coronary arteries from male than female pigs (23). Inaddition, the contraction to norepinephrine or phenylephrine(Phe) is greater in the aorta of intact male than intact femalerats (25, 123). Interestingly, vasopressin-induced contraction inrat aorta exhibits sexual dimorphism, but the contraction infemales is almost twice that in males (123). The differencecould be related to possible tachyphylactic effects of vasopres-sin in isolated vessels, which are different from its effects invivo. This is supported by reports that the pressor response tovasopressin infusion in vivo is greater in male than female rats(123).
We recently found that the vascular contraction is notdifferent between castrated and intact male rats, but signifi-cantly enhanced in ovariectomized (OVX) females comparedwith intact females, suggesting that the gender differences invascular tone are less likely related to androgens and morelikely related to estrogens (25, 69). The data also suggest thatthe gender differences in vascular tone are due to directvascular effects of sex hormones, possibly through interactionwith specific hormone receptors in the vasculature.
SEX HORMONE RECEPTORS IN BLOOD VESSELS
Receptors for estrogen, progesterone, and testosterone areexpressed in varying numbers in both the endothelium andVSM of multiple vascular systems (75, 129). For instance, asignificant association between the number of estrogen recep-tors (ER) and normal endothelial cell function has been re-ported, and suggested that decreased number of endothelial ERmay represent a risk factor for cardiovascular diseases (109).Also, the sex hormone receptors appear to have differentsubtypes, tissue distribution, and subcellular location and canbe modulated by various agonists and antagonists (Table 1).
Two ER subtypes have been identified, ER-� and ER-� (84).Several variants of ER-�, such as ER-�A, ER-�C, ER-�E, andER-�F (79), as well as ER-�, such as ER-�1, ER-�2, ER-�4,and ER-�5, have been described (14, 115). Some studiessuggest that ER-� promotes the protective effects of estrogen
in response to vascular injury (98). However, ER-� is morewidely distributed in the body than ER-�. Also, ER-� is thereceptor form that is predominantly expressed in human VSM,particularly in women (84). Induction of ER-� mRNA expres-sion has also been demonstrated after balloon vascular injury tothe aorta of the male rat. Furthermore, experiments on trans-fected HeLa cells have shown that in response to 17�-estradiol(E2), ER-� is a stronger transactivator than ER-� at lowreceptor concentrations. However, at higher receptor concen-trations, ER-� activity self-squelches, and ER-� becomes thestronger transactivator. These data support a role for ER-� inthe direct vascular effects of estrogen and in the regulation ofvascular function (57).
ERs have been localized in the nucleus, and continuousshuttling of the receptor between the cytoplasm and the nucleushas been suggested (49). Sex steroids, such as estrogen, diffusethrough the plasma membrane and form complexes with spe-cific cytosolic and/or nuclear receptors, which then bind tochromatin and stimulate the transcription of a set of genes witha specific sex steroid-responsive regulatory element (60, 80)(Fig. 1). ER-mediated transcription requires coactivators toexert transcriptional activity. The steroid receptor coactivator-3(SRC-3) is highly expressed in VSM. SRC-3 interacts withestrogen-bound ERs and coactivates the transcription of targetgenes in cultured VSM cells, suggesting that SRC-3 facilitatesER-dependent vasoprotective effects during vascular injury(144). However, estrogen can also bind to the plasma mem-brane of various vascular cells and induce rapid cellular events,suggesting additional nongenomic action triggered by a signal-generating receptor on the cell surface (26, 38). Recent studiesalso suggest possible interactions of ER with signal-modulat-ing proteins or coactivators in the plasma membrane (103).
Another hormone receptor that has been located in theendothelium and VSM is the progesterone receptor (129, 135).Progesterone receptor-A and -B have been identified (97).Progesterone receptors, particularly the B isoform, appear tohave a direct role in the regulation of gene transcription andVSM cell proliferation (99).
Testosterone or androgen receptors have also been identifiedin endothelial cells and VSM. The expression of androgenreceptors in VSM appears to vary depending on the gender andthe status of the gonads. The androgen receptor protein, asdetected by Western blot in rat aortic VSM, is less in the cellsof females than those of males (55). In the smooth muscle ofprimates, androgen receptor mRNA levels are upregulated bycombined estradiol plus testosterone treatment, whereas estra-diol treatment alone had little or no effect, suggesting that acollaborative action of estradiol and testosterone enhancesandrogen receptor expression (142).
GENOMIC EFFECTS OF SEX HORMONES
The interaction of sex hormones with nuclear/cytosolic re-ceptors triggers a host of genomic effects leading to endothelialcell growth. The effects of sex hormones on endothelial cellgrowth appear to be mediated by activation of mitogen-acti-vated protein kinase (MAPK; Fig. 1). This is supported byreports that E2 induces the phosphorylation of p38 and p42/44MAPK as well as the migration and proliferation of porcineaortic endothelial cells (42).
Invited Review
R235SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from

Although estradiol activates signaling pathways that stim-ulate endothelial cell proliferation, the steroid appears toinhibit cell growth and to induce antiproliferative effects inVSM (33). For example, the rate of growth in VSM offemale aorta is slower than that in male aorta (3). Theinhibitory effects of estrogen on VSM growth may beenhanced by its interaction with steroid receptor coactiva-tors such as SRC-3 (144). Estrogen has also been shown toinhibit MAPK activity in VSM, and this effect is blocked bythe estrogen antagonist ICI-182780, suggesting that inhibi-tion of the MAPK pathway via ERs contributes to theinhibitory effects of estrogen on VSM growth (33). Estrogenmay also antagonize the growth-promoting effect of ANG IIon VSM via the induction and activation of protein phos-phatases through genomic as well as nongenomic mecha-nisms (126). Interestingly, nitric oxide (NO) has beenshown to inhibit VSM growth and proliferation. Whether anestrogen-induced increase in NO production plays a role asa possible mediator of estrogen-induced inhibition of VSM
growth remains to be clarified. Furthermore, estradiol maystimulate cAMP production and the cAMP-derived adeno-sine may regulate VSM growth via adenosine receptors,suggesting that the cAMP/adenosine pathway may contrib-ute to the antiproliferative effects of estradiol (34).
Progesterone also inhibits VSM proliferation and migrationand may facilitate the inhibitory effects of estrogen. Proges-terone appears to mediate its inhibitory effects on VSM growthby reducing MAPK activity (93).
Some studies have suggested that androgens accelerate vas-cular growth by stimulating the proliferation of VSM, whereasother studies show androgen-induced inhibition of growth andproliferation (121, 142). The discrepancy in these reports maybe related to the concentration of androgens used. Androgensmay control the proliferation of their target cells by firstincreasing cell proliferation and later by inhibiting the prolif-eration of the same cells. For example, dihydrotestosteronemodulates human umbilical VSM cell proliferation in a dose-dependent manner, with low concentrations (3 nM) stimulating
Fig. 1. Estrogen-stimulated endothelium-dependent mechanisms of vascular smooth muscle (VSM) relaxation. In the genomicpathway, estrogen binds to endothelial cytosolic/nuclear estrogen receptors (ER), leading to activation of mitogen-activated proteinkinase (MAPK), increased gene transcription, endothelial cell proliferation, and increased endothelial nitric oxide synthase (eNOS)production. In the nongenomic pathway, estrogen binds to endothelial surface membrane ERs, which are coupled to increased Ca2�
release from the endoplasmic reticulum and stimulation of MAPK/Akt pathway, leading to activation of eNOS and increased nitricoxide (NO) production. NO diffuses into the VSM cells, binds to guanylate cyclase (GC), and increases cGMP. cGMP causes VSMrelaxation by decreasing [Ca2�]i and the myofilament sensitivity to Ca2�. ER may also inhibit the production of NADPH, therebypreventing the inactivation of NO and the formation of peroxynitrites (ONOO�). Endothelial ER may also activate cyclooxyge-nases (COX) and increase PGI2 production. PGI2 activates prostacyclin receptors in VSM, activates adenylate cyclase (AC), andincreases the formation of cAMP. cAMP causes VSM relaxation by mechanisms similar to those activated by cGMP. ER may alsoincrease the production of endothelium-derived hyperpolarizing factor (EDHF), which activates K� channels and causeshyperpolarization and inhibition of Ca2� influx via Ca2� channels leading to VSM relaxation. Interrupted arrows indicateinhibition. L-arg, L-arginine; L-cit, L-citrulline; AA, arachidonic acid.
Invited Review
R236 SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from
[3H]thymidine incorporation, and high concentrations (300nM) inhibiting [3H]thymidine incorporation (121, 142).
We should note that the genomic effects of sex hormonesmay alter the expression of a multitude of regulatory andsignaling proteins in endothelial cells and VSM. To avoidrepetition, these genomic effects will be described with thenongenomic effects of sex hormones on the endothelium andVSM as described below.
NONGENOMIC EFFECTS OF SEX HORMONES
The interaction of sex hormones with plasmalemmal recep-tors in the endothelium and VSM may initiate additionalnongenomic vascular effects. For example, estrogen may in-duce acute inhibition of vascular contraction (24, 129). Also,progestins may have direct vascular effects or modify theeffects of estrogen on vascular contraction (24). Interestingly,direct vascular effects of testosterone have also been described(142, 145). For example, testosterone induces pulmonary andcoronary artery dilation (24, 145). The acute nongenomicvasodilator effects of sex hormones appear to have both endo-thelium-dependent as well as endothelium-independent mech-anisms involving direct effects on VSM.
SEX HORMONES AND THE ENDOTHELIUM
The vascular endothelium plays an important role in medi-ating the gender-related and the estrogen-induced vasodilation(72). Physiological levels of E2 potentiate endothelium-depen-dent flow-mediated vasodilation in postmenopausal women(45). Also, endothelium-dependent relaxation of isolated aorta
is greater in female than in male spontaneously hypertensiverats (SHR; 67, 72). Similar to estrogen, progesterone maypromote endothelium-dependent vasodilation in porcine coro-nary artery (92). Also, some studies have shown that testos-terone induces endothelium-dependent vascular relaxation (20,22). The vascular endothelium is known to release relaxingfactors such as NO, prostacyclin (PGI2), and endothelium-derived hyperpolarizing factor (EDHF), as well as contractingfactors such as endothelin (ET-1) and thromboxane A2, and thesex hormones appear to induce vascular relaxation by modi-fying the synthesis/release/bioactivity of one or more of thesefactors.
SEX HORMONES AND NO
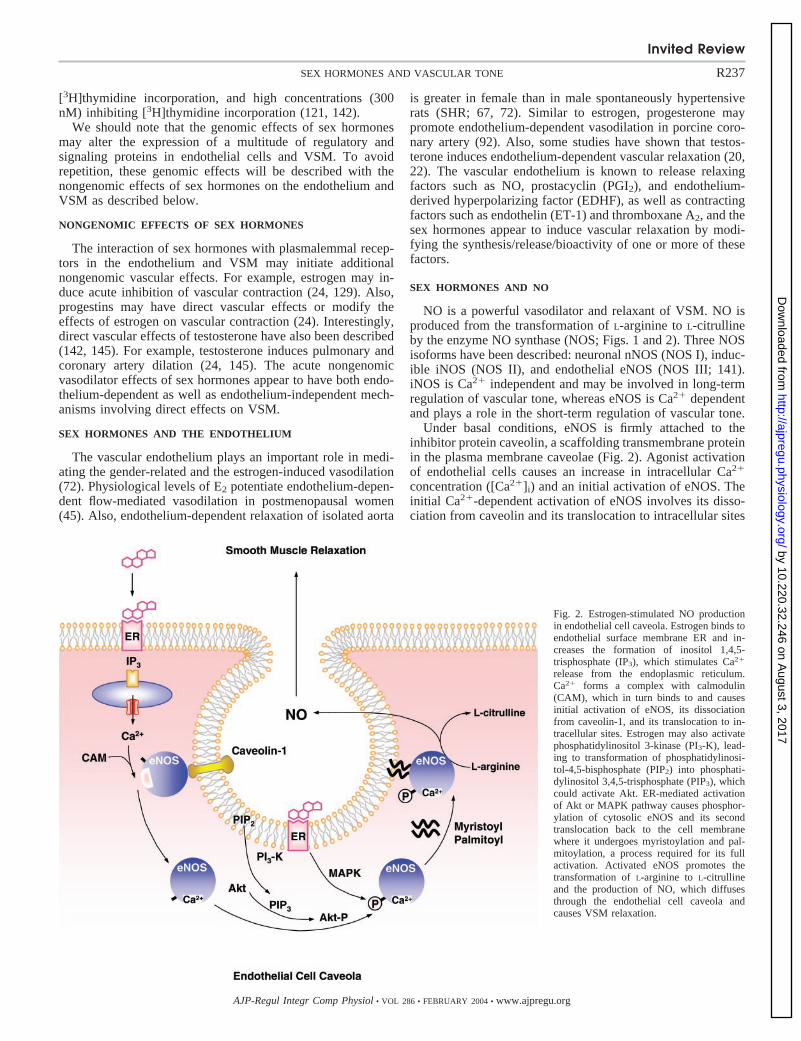
NO is a powerful vasodilator and relaxant of VSM. NO isproduced from the transformation of L-arginine to L-citrullineby the enzyme NO synthase (NOS; Figs. 1 and 2). Three NOSisoforms have been described: neuronal nNOS (NOS I), induc-ible iNOS (NOS II), and endothelial eNOS (NOS III; 141).iNOS is Ca2� independent and may be involved in long-termregulation of vascular tone, whereas eNOS is Ca2� dependentand plays a role in the short-term regulation of vascular tone.
Under basal conditions, eNOS is firmly attached to theinhibitor protein caveolin, a scaffolding transmembrane proteinin the plasma membrane caveolae (Fig. 2). Agonist activationof endothelial cells causes an increase in intracellular Ca2�
concentration ([Ca2�]i) and an initial activation of eNOS. Theinitial Ca2�-dependent activation of eNOS involves its disso-ciation from caveolin and its translocation to intracellular sites
Fig. 2. Estrogen-stimulated NO productionin endothelial cell caveola. Estrogen binds toendothelial surface membrane ER and in-creases the formation of inositol 1,4,5-trisphosphate (IP3), which stimulates Ca2�
release from the endoplasmic reticulum.Ca2� forms a complex with calmodulin(CAM), which in turn binds to and causesinitial activation of eNOS, its dissociationfrom caveolin-1, and its translocation to in-tracellular sites. Estrogen may also activatephosphatidylinositol 3-kinase (PI3-K), lead-ing to transformation of phosphatidylinosi-tol-4,5-bisphosphate (PIP2) into phosphati-dylinositol 3,4,5-trisphosphate (PIP3), whichcould activate Akt. ER-mediated activationof Akt or MAPK pathway causes phosphor-ylation of cytosolic eNOS and its secondtranslocation back to the cell membranewhere it undergoes myristoylation and pal-mitoylation, a process required for its fullactivation. Activated eNOS promotes thetransformation of L-arginine to L-citrullineand the production of NO, which diffusesthrough the endothelial cell caveola andcauses VSM relaxation.
Invited Review
R237SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from

close to the nucleus. During maintained endothelial cell stim-ulation with an agonist, activation of MAPK and/or the proteinkinase B/Akt pathway and subsequent phosphorylation ofeNOS causes a second translocation of cytosolic eNOS back tothe cell membrane where it undergoes myristoylation andpalmitoylation, a process required for its full activation (Fig.2). These rapid receptor-mediated effects on the NO pathwayare seen not only for “classic” eNOS agonists, such as ACh andbradykinin, but also for estradiol (86; Figs. 1 and 2).
Total NO production is greater in premenopausal womenthan in men (39). The cellular origin of the increased NO inwomen is not entirely clear, but differences in vascular endo-thelial NO production may underlie the gender differences invascular tone (39). NO release from the endothelium is in-creased in arteries of females compared with males (71, 78,139). This is supported by reports that the inhibitory effect ofthe NOS inhibitor N�-L-arginine methyl ester (L-NAME) onACh-induced relaxation is more pronounced in the mesentericartery of female than male rats (67). Estrogen appears to beresponsible for the gender differences in endothelial NO re-lease (28, 41, 78, 85). E2 replacement in OVX female guineapigs enhances the sensitivity to vasodilators in the coronarymicrocirculation through increased endothelial NO production(130). Also, prolonged treatment of human coronary endothe-lial cells with E2 increases the basal, adenosine triphosphate-,and A23187-induced NO release (143).
Estrogen may influence NO production by increasing NOSexpression (41, 78). Activation of genomic ERs may causeupregulation of eNOS (71; Fig. 1). It has been shown that ER-�gene transfer into bovine aortic endothelial cells induces eNOSgene expression (128). Also, estrogen increases the level ofeNOS mRNA in ovine fetal pulmonary artery endothelial cells(83). Estrogen may also prevent destabilization of eNOSmRNA induced by tumor necrosis factor-� through an ER-mediated mechanism (125).
In addition to the genomic effects of estrogen on eNOSexpression, estrogen may regulate NOS activity and therebyNO production and vascular tone by interacting with specificERs in the endothelial cell plasma membrane and activation ofrapid nongenomic signaling pathways (41, 78). Membrane-impermeant forms of estrogen bind to specific ERs at the cellsurface and stimulate NO release from human endothelial cells(112). Also, in bovine aortic endothelial cells, E2 causestransient translocation of eNOS from the plasma membrane tointracellular sites close to the nucleus, although during pro-longed exposure to E2, most of the eNOS returns to the plasmamembrane for its full activation (86).
The acute effect of E2 on eNOS activity and NO release hasbeen suggested to occur via activation of ER-� in COS-7 cells(19) and mouse aorta (27). However, studies have shown thatoverexpression of ER-� in COS-7 cells enhances rapid eNOSactivation by E2 and the ER-� protein association to the plasmamembrane caveolae and that these events occur independent ofER-�. These findings indicate that endogenous ER-� alsoplays a prominent role in the nongenomic effects of E2 oneNOS activity (17).
The acute effects of E2 on eNOS activity and NO releaseappear to be dependent on [Ca2�]i. Gender differences in theregulation of endothelial [Ca2�]i have been related to direct orindirect effects of estrogen on the Ca2� handling mechanismsin the vascular endothelium (78, 102, 139). Estrogen activation
of cell surface ERs has been suggested to be coupled toincreases in [Ca2�]i and acute stimulation of NO release fromhuman endothelial cells (124). Studies have also suggestedpossible gender differences in the eNOS sensitivity to [Ca2�]i.Experiments on pressurized coronary arteries of rats haveshown that increases in [Ca2�]i cause activation of eNOS witha similar slope and half-activation constant for both female andmale arteries. However, at [Ca2�]i � 100 nM, eNOS activity ishigher in females compared with males (78). Interestingly, E2
may promote the association of heat shock protein 90 witheNOS and thereby reduce the Ca2� requirement for eNOSactivation (112). However, E2 may stimulate eNOS activityand increase NO release from human endothelial cells, inde-pendent of cytosolic Ca2� mobilization (16), perhaps througheNOS phosphorylation via a mechanism involving MAPK orAkt (56, 112). In studies of ovine endothelial cells, E2 causedacute activation of eNOS as well as rapid activation of MAPK,and inhibition of MAPK kinase prevented the activation ofeNOS by E2. These data suggest that the acute vascular effectsof estrogen are mediated by ER functioning in a nongenomicmanner to activate eNOS via MAPK-dependent mechanisms(19). Also, E2 rapidly induces phosphorylation and activationof eNOS through the phosphatidylinositol-3 (PI3)-kinase-Aktpathway and thereby reduces its [Ca2�]i requirement for acti-vation (52; Fig. 2).
The effects of estrogen on the NO pathway may also berelated to its antioxidant effects. It has been shown that theincrease in arterial pressure in OVX female rats is associatedwith lower plasma antioxidant levels, reduced thiol groups, andincreased plasma lipoperoxides and vascular free radicals, andthat estrogen replacement prevents the increase in free radicalsand the decrease in plasma levels of nitrites/nitrates (54). Also,E2 inhibits NADPH oxidase expression and the generation ofreactive oxygen species and peroxynitrite (ONOO�) in humanumbilical vein endothelial cells (136; Fig. 1). ANG II stimu-lation of endothelial cells has been shown to increase theexpression of NADPH oxidase, which may contribute to oxi-dative stress, as evidenced by ONOO� formation. E2 appearsto inhibit ANG II-induced increases in oxidative-stress effects,possibly through reduced ANG II type 1 receptor expression(46). Also, measurements of superoxide anion (O2
�) in theisolated aorta have shown greater amounts in male than infemale rats (12). Furthermore, estrogen decreases the genera-tion of O2
� from cultured bovine aortic endothelial cells andthereby enhances NO bioactivity and decreases ONOO� re-lease (5). These data suggest that the decrease in vascular toneand arterial pressure with estrogen administration may berelated to preventing oxidative stress and improving endothe-lial function.
Although gender and estrogen treatment may affect NOproduction/bioactivity, their influence on factors downstreamin the NO signaling pathway is unclear. NO produced from theendothelium is known to activate guanylate cyclase in thesmooth muscle leading to increased cGMP and stimulation ofcGMP-dependent protein kinase (PKG; 140). PKG may de-crease [Ca2�]i by stimulating Ca2� extrusion pumps in theplasma membrane and Ca2� uptake pumps in the sarcoplasmicreticulum membrane and/or decrease the sensitivity of thecontractile myofilaments to [Ca2�]i and thereby promote VSMrelaxation (Fig. 1). It has been shown that the relaxation ofmesenteric arterial rings by the exogenous NO donor sodium
Invited Review
R238 SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from

nitroprusside is greater in female than male SHR (67), sug-gesting gender differences in smooth muscle reactivity to NOand possible hormonal regulation of PKG. However, in aorticrings from male and female SHR, sodium nitroprusside-in-duced relaxation is similar, making the possibility of genderdifferences in smooth muscle reactivity to NO and the hor-monal regulation of PKG rather less likely (72).
Carbon monoxide is formed by heme oxygenase-2 in thevascular endothelium and has been found to activate solubleguanylate cyclase and dilate blood vessels independently fromNO. Because of the parallels between NO and carbon monox-ide, it has been suggested that estrogen might affect carbonmonoxide production in vascular endothelium. Studies in hu-man umbilical vein and uterine artery endothelial cells haveshown that treatment with E2 causes significant increases inintracellular carbon monoxide production, heme oxygenase-2protein levels, and cellular cGMP, suggesting a potential rolefor carbon monoxide as a biological messenger molecule inestrogen-mediated regulation of vascular tone (133).
The effects of progesterone on the vascular endothelium areless clear. Experimental studies on canine coronary arterieshave suggested that progesterone may counteract the stimula-tory effects of estrogen on endothelium-dependent NO produc-tion and vascular relaxation (88). However, studies in post-menopausal women have shown that progesterone does notappreciably attenuate estradiol-induced endothelium-depen-dent vasodilation of the brachial artery (44). Other studies haveshown that progesterone may stimulate NO production in rataortic strips (116) and induce endothelium-dependent NO-mediated relaxation in rat resistance mesenteric arteries (18).Also, intravenous infusion of progesterone in pigs has beenshown to induce endothelium-dependent coronary vasodilationvia a mechanism involving the release of NO (92). Further-more, chronic administration of progesterone has been shownto increase the expression of eNOS in the endothelium of ovineuterine artery (111).
In regard to testosterone, acute intracoronary administrationof the hormone in canine coronary epicardial and resistancevessels has been shown to induce vasodilation that is mediatedin part by NO (20). Testosterone may also modulate the effectsof other agonists on endothelial NO production. Bradykinin, aknown activator of endothelium-dependent NO pathway, hasbeen shown to increase intracellular inositol 1,4,5-trisphos-phate (IP3) and stimulate rapid release of Ca2� from theendoplasmic reticulum in cultured endothelial cells of malerats. Treatment of the cells with testosterone blocks bradyki-nin-induced increases in [Ca2�]i and thereby NO production inendothelial cells, perhaps through an effect of testosterone onmembrane-bound bradykinin receptors or on bradykinin-in-duced Ca2� release mechanism (110). Recent studies haveshown that treatment of human endothelial cells with andro-gens such as dehydroepiandrosterone (DHEA) triggers NOsynthesis by enhancing the expression and stabilization ofeNOS. DHEA appears to activate eNOS through an MAPK-dependent mechanism, but not the PIP3 kinase/Akt pathway(120). Also, administration of DHEA in ovariectomized femaleWistar rats has been shown to restore aortic eNOS levels andeNOS activity (120). Interestingly, androgen antagonists suchas flutamide may also affect the NO pathway. It has beenshown that flutamide produces direct vasodilation by inducingNO release from the endothelium and subsequent activation of
guanylyl cyclase in rat aortic smooth muscle; however, thesevascular effects of flutamide do not appear to be mediated viaandrogen receptors (61). Additionally, administration of flut-amide reduces blood pressure in hypertensive transgenicTGR(mREN2)27 rats, which have an overactive renin-angio-tensin system. Furthermore, flutamide reduces the blood pres-sure in TGR(mREN2)27 rats with an additional testicularfeminizing mutation (tfm), suggesting that the vascular effectsof flutamide may be caused by androgen receptor-independentmechanisms (61).
SEX HORMONES AND PGI2
PGI2 is an endothelium-derived relaxing factor that is pro-duced from the metabolism of arachidonic acid by the enzymecyclooxygenase (COX; Fig. 1). COX has two isoforms, COX-1and COX-2. COX inhibitors such as indomethacin inhibit asignificant component of endothelium-dependent vascular re-laxation, and gender differences in the indomethacin-sensitivecomponent of vascular relaxation have been attributed to dif-ferences in the COX products (6). Estrogen may augment theproduction of COX products such as PGI2 (41). Physiologicallevels of E2 cause upregulation of COX-1 expression and PGI2
synthesis in ovine fetal pulmonary artery and human umbilicalvein endothelial cells (66). Also, E2 causes rapid ER-�-medi-ated stimulation of PGI2 synthesis in ovine fetal pulmonaryartery endothelial cells via a Ca2�-dependent, but MAPK-independent, pathway (118). It has also been suggested that theCOX-2 pathway plays a specific role in the rapid E2-inducedpotentiation of cholinergic vasodilation in postmenopausalwomen (13). However, other studies have reported that indo-methacin does not affect the E2-induced relaxation in endothe-lium-intact coronary artery, suggesting that the release ofvasodilator prostanoids may not be involved in the E2-inducedcoronary relaxation (62). It has also been suggested that estro-gen may modulate cross-talk between the NO synthase andCOX pathways of vasodilation and that estrogen-induced in-crease in the NO component of endothelium-dependent dila-tion may be associated with a decrease in the COX component(15).
In regard to other sex hormones, some studies have shownthat concomitant administration of progesterone with estrogenprevents the stimulatory effects of estrogen on PGI2 productionin cultured human umbilical vein endothelial cells (87). How-ever, other studies have shown that progesterone may exert adirect nongenomic effect on rat aorta, which involves COXactivation and increased PGI2 production (116). On the otherhand, experiments on the aorta of female rat have shown thattreatment with testosterone is associated with a decrease inPGI2 synthesis (137).
SEX HORMONES AND EDHF
The endothelium may release other relaxing factors evenduring complete inhibition of the NO-cGMP and the PGI2-cAMP pathways. Such factors have been shown to activateCa2�-activated K� channels (BKCa) and to cause hyperpolar-ization and relaxation of the smooth muscle and therebydesignated EDHF.
The greater endothelium-mediated relaxation in femalescompared with males may be related to differences in theendothelium-dependent hyperpolarization of VSM (67). It has
Invited Review
R239SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from

been shown that ACh-induced hyperpolarization and relaxationof mesenteric arteries are reduced in OVX female and intactmale rats compared with intact female rats and that the differ-ences in the ACh responses in OVX female compared withintact female rats are eliminated in the presence of K� channelblockers such as apamin or charybdotoxin. Also, the hyperpo-larizing response to ACh is improved in OVX female ratstreated with E2. These data suggest that estrogen-deficientstates attenuate relaxation transduced by EDHF (81, 113).
Testosterone may also promote endothelium-mediated hy-perpolarization of VSM. In aortic rings of both Wistar-Kyoto(WKY) and SHR, testosterone induces concentration-depen-dent relaxation (58). Testosterone-induced relaxation is re-duced by denudation of endothelium in SHR, but not WKY.Indomethacin and L-NAME show little influence on testoster-one-induced relaxation in both WKY and SHR aortic rings.4-Aminopyridine, inhibitor of voltage-dependent K� channels,and tetraethylammonium, inhibitor of BKCa, reduce testoster-one-induced relaxation in SHR, but not WKY. On the otherhand, glibenclamide, inhibitor of ATP-sensitive K� channels,reduces testosterone-induced relaxation in both WKY and SHRaortic rings. These data suggest that in SHR aortic rings,testosterone may release endothelium-derived substances thatcause hyperpolarization of the cells by a mechanism thatinvolves voltage-dependent and BKCa channels. However, asignificant component of testosterone-induced vasorelaxationin both WKY and SHR appears to be endothelium-independentand may involve ATP-sensitive K� channels in aortic smoothmuscle (58).
SEX HORMONES AND ENDOTHELIUM-DERIVEDCONTRACTING FACTORS
The gender differences in vascular tone may be related todifferences in the release of or sensitivity to endothelium-derived contracting factors (EDCF) such as ET-1 and throm-boxane A2. ET-1 release from endothelial cells appears to bereduced in females and may explain the decreased vasculartone and blood pressure in female compared with male SHR(67). The gender difference in ET-1 production by endothelialcells may be related to the plasma levels of estrogen (1).
ET-1 is known to interact with ETA and ETB receptors. Theactivation of endothelial ETB causes the release of variousrelaxing factors that promote VSM relaxation. On the otherhand, the interaction of ET-1 with ETA and ETB receptors inVSM activates signaling mechanisms of smooth muscle con-traction. Gender differences in the vascular responses to ET-1have been reported in DOCA-salt hypertensive rats, with thearteries of males exhibiting marked contraction to ET-1 com-pared with those of females, and functional changes in ETB
receptors have been suggested as one possible mechanism(131). For example, in the mesenteric arteries of DOCA rats,the ETB agonist IRL-1620 induces mild vasoconstriction inintact females, but marked vasoconstriction in OVX females.Estradiol or estradiol/progesterone decreases IRL-1620-in-duced vasoconstriction in the OVX rats. Ovariectomy is alsoassociated with increases in ET-1 and ETB receptor mRNA inmesenteric arteries, and treatment with estradiol or estradiol/progesterone reverses these changes. These data suggest thatthe ovarian hormones attenuate ET-1/ETB receptor expressionand the vascular responses in DOCA-salt hypertension (29). It
has also been shown that prolonged treatment of culturedendothelial cells with estradiol inhibits basal and stimulatedET-1 expression and release in response to serum, tumornecrosis factor-�, transforming growth factor-�1, ANG II, andthrombin (9, 35, 93). Also, short-term intracoronary adminis-tration of E2 decreases ET-1 levels in coronary sinus plasma ofpostmenopausal women with coronary artery disease (138).
Similar to estrogen, progesterone inhibits serum- and ANGII-stimulated ET-1 synthesis and release from cultured bovineaortic endothelial cells (93). Androgens appear to have theopposite effect on ET-1 production. Studies on female-to-maletranssexuals receiving large doses of testosterone have shownhigh plasma levels of ET-1 (100). Whether testosterone-in-duced increase in ET-1 production could increase the risk ofhypertension in males is unclear. Although ETA-receptor an-tagonists prolong survival and improve renal status in maleSHR, they do not reduce the blood pressure significantly,suggesting little role of ET-1 in male SHR hypertension (132).
In addition to the gender differences in ET-1 responses, ithas been shown that the release of COX-derived constrictingfactors such as thromboxane A2 is more pronounced in malethan in female SHR (67).
SEX HORMONES AND VSM CONTRACTION
In addition to the nongenomic effects of sex hormones onthe endothelium, rapid nongenomic effects on VSM have beendescribed (24, 62, 64, 129; Fig. 3). For example, estrogencauses vasodilation in endothelium-denuded vessels, suggest-ing that the estrogen-induced inhibition of vascular tone has anendothelium-independent component that involves direct ac-tion on VSM (24, 38, 62, 64). Also, estrogen causes relaxationin endothelium-denuded rabbit, porcine, and human coronaryarteries precontracted by ET-1, PGF2�, and high KCl depolar-izing solution (24, 51, 64; Fig. 4). The vasodilator effects ofestrogen do not appear to be mediated by the classic cytosolic-nuclear ER or stimulation of protein synthesis, but ratherthrough a direct effect of estrogen on plasmalemmal receptorsin VSM (24, 38).
The acute vasodilator effects of estrogen may be influencedby gender, vessel type, estrous cycle, and previous exposure toestrogen. For example, in rat aorta, E2 causes greater relaxationin males than females (25). Among female rats, the largestE2-induced vasodilation is seen in the tail and mesentericarteries from females at the proestrous stage. However, themagnitude of relaxation in microvessels of estradiol-replacedOVX female rats is smaller than that of nonreplaced OVX rats,suggesting that chronic estradiol replacement may downregu-late the acute nongenomic vasorelaxation effects of estrogen insmall arteries of OVX rats (68).
The effects of progesterone on vascular reactivity are lessclear and range between no effect, inhibition of vasorelaxation,and potent vascular relaxation (24, 63, 129). Progesterone maycause endothelium-independent relaxation of VSM, although itis smaller than that induced by estrogen (24; Fig. 4). Proges-terone induces relaxation of primate, porcine, rabbit, and ratcoronary arteries (24, 36, 63, 90). The vasodilator effect ofprogesterone in isolated VSM suggests that its benefits inhormone replacement therapy may be related to its non-genomic relaxant effects on VSM.
Invited Review
R240 SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from

Some studies suggest that testosterone enhances vascularcontraction either by inhibiting endothelium-dependent relax-ation or by directly stimulating VSM contraction (142). Forexample, treatment of porcine coronary artery with nanomolarconcentrations of testosterone impairs bradykinin- andA23187-induced endothelium-dependent vascular relaxation.Also, testosterone enhances thromboxane A2-induced coronaryvasoconstriction in guinea pigs. Flutamide, a testosterone re-ceptor antagonist, has been shown to cause direct vasodilationin rat vessels and flutamide-induced vascular relaxation issmaller in females compared with males, suggesting that tes-tosterone may promote vascular contraction (2). However,other studies have shown that testosterone induces relaxationof rabbit coronary artery and aorta, rat aorta, and canine andporcine coronary artery (24, 145; Fig. 4). A significant portionof the testosterone-induced vascular relaxation appears to beendothelium independent because only small differences could
be observed between the relaxation in vessels with and withoutendothelium. Also, inhibition of endothelium-dependent relax-ation pathways such as NOS and COX may not abolish thevasorelaxing effect of testosterone (145), providing evidencethat a significant component of testosterone-induced relaxationis endothelium independent and involves direct action onVSM. The testosterone-induced vasorelaxation appears to be astructurally specific effect of the androgen molecule and isenhanced in more polar analogs that have a lower permeabilityto the VSM cell membrane (31).
We should note that although both the endogenous presenceand exogenous application of sex hormones may be associatedwith reduction in vascular contraction, the mechanisms ofhormone-induced relaxation in isolated vascular strips or cellsand the possible vasorelaxant effects of the hormone in vivomay not be identical. The acute effects of estrogen on vascularcontraction in vitro are often observed at micromolar concen-
Fig. 3. Genomic and nongenomic effects of estrogen in VSM. In the genomic pathway, estrogen binds to cytosolic/nuclear ER,leading to inhibition of growth factor (GF)-activated MAPK and gene transcription, and thereby inhibition of VSM growth andproliferation. In the nongenomic pathway, estrogen binds to plasma membrane ER, leading to inhibition of agonist-activatedmechanisms of VSM contraction. An agonist (A) activates a specific receptor (R), stimulates membrane phospholipase (PLC), andincreases the production of IP3 and diacylglycerol (DAG). IP3 stimulates Ca2� release from the sarcoplasmic reticulum (SR). Also,the agonist stimulates Ca2� entry through Ca2� channels. Ca2� binds CAM, activates myosin light chain (MLC) kinase, causesMLC phosphorylation, and initiates VSM contraction. DAG causes activation of protein kinase C (PKC). PKC could phosphorylatecalponin (CaP) and/or activate a protein kinase cascade involving Raf, MAPK kinase (MEK), and MAPK, leading to phosphor-ylation of caldesmon (CaD) and an increase in the myofilament force sensitivity to Ca2�. Possible effects of estrogen includeactivation of K� channels, leading to membrane hyperpolarization, inhibition of Ca2� entry through Ca2� channels, and therebyinhibition of the Ca2�-dependent MLC phosphorylation and inhibition of VSM contraction. Estrogen may also inhibit PKC and/orthe MAPK pathway and thereby further inhibit VSM contraction. SRC-3, steroid receptor coactivator-3; SMP, signal-modulatingprotein. Interrupted arrows indicate inhibition.
Invited Review
R241SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from

trations, which are several-fold higher than the physiologicalnanomolar concentrations observed in vivo. Although agenomic action of physiological concentrations of estrogenmay underlie the reduced cell contraction in VSM of intactfemales, it is less likely to account for the acute inhibitoryeffects of exogenous micromolar concentrations of E2 onvascular contraction. The acute vasorelaxant effects of exoge-nous estrogen may represent additional nongenomic effects ofestrogen on the mechanisms of VSM contraction (24).
We should also note that although most of the sex hormonestested appear to cause vascular relaxation and inhibit VSMcontraction, the vascular relaxant effects of estrogen signifi-cantly surpass those of progesterone or testosterone (Fig. 4).Inasmuch as the estrogen levels are greater in females com-pared with males, this could explain the gender differences invascular tone and the reduced vascular contraction in femalescompared with males (Table 3). However, because the expres-sion of sex hormone receptors in arterial smooth muscle mayvary depending on the gender and the status of the gonads(127), the gender differences in vascular contraction may berelated to the relative abundance of sex hormone receptors.This is supported by reports that females have higher levels of
ERs in their arteries than males (21). The gender differences invascular contraction could also be related to effects of sexhormones on the gene expression of the specific receptors ofvasoconstrictor agonists such as ANG II (Table 3). Westernblot analyses in VSM have revealed that estrogen induces adownregulation and progesterone an upregulation of the an-giotensin AT1 receptor protein. Also, E2 decreases the AT1
receptor mRNA half-life, whereas progesterone induces stabi-lization of AT1 receptor mRNA (96). Other studies have shownthat progesterone replacement in OVX monkeys decreasesthromboxane A2 receptors in coronary arteries (91). Neverthe-less, the gender differences in vascular contraction may also berelated to gender differences in the signaling mechanisms ofVSM contraction downstream from receptor activation.
SIGNALING MECHANISMS OF VSM CONTRACTION
It is widely accepted that VSM contraction is triggered byincreases in [Ca2�]i due to initial Ca2� release from thesarcoplasmic reticulum and maintained Ca2� entry from theextracellular space (73, 95). Also, activation of protein kinasessuch as myosin light chain (MLC) kinase, Rho kinase, andMAPK as well as inhibition of MLC phosphatase may con-tribute to smooth muscle contraction (59, 122; Fig. 3). Addi-tionally, the interaction of an �-adrenergic agonist such as Phewith its receptor is coupled to increased breakdown of plasmamembrane phospholipids and increased production of diacyl-glycerol (DAG), which activates protein kinase C (PKC; 70).PKC is mainly cytosolic under resting conditions and under-goes translocation from the cytosolic to the particulate fractionwhen it is activated by DAG or phorbol esters. PKC is nowknown to be a family of several isoforms that have differentenzyme properties, substrates, and functions and exhibit dif-ferent subcellular distributions in the same blood vessel fromdifferent species and in different vessels from the same species(69, 70).
Table 3. Potential causes of the gender differences invascular tone
MalePremenopausal
FemalePostmenopausal
Female
Plasma levelEstrogen, pg/ml 15–25 100–700 0–60Progesterone, pg/ml 25–100 50–700 200–500Testosterone, ng/ml 4–10 0.3–0.7 0.1–0.7
Vascular hormone receptorEstrogen � ��� ��Progesterone � ��� ��Testosterone ��� � �
EndotheliumeNOS � ��� �Ca2� � ��� �
Smooth muscleANG II Receptor ��� � ��TXA2 Receptor ��� � ��[Ca2�]i (nM)Basal �100 �60 �100Transient �400 �400 �400Maintained �200 �150 �200�-PKC ��� � ���
eNOS, endothelial nitric oxide synthase; [Ca2�]i, intracellular Ca2� concn;PKC, protein kinase C.
Fig. 4. Effect of sex hormones on PGF2�-induced contraction in porcinecoronary artery. Endothelium-denuded coronary artery strips were stimulatedwith PGF2� (10�5 M), then treated with the vehicle ethanol (A) or with 10�5
M 17�-estradiol (B), progesterone (C), or testosterone (D). [Modified from(24)].
Invited Review
R242 SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from

SEX HORMONES AND VSM [CA2�]I
Because [Ca2�]i is important for the initiation of smoothmuscle contraction, several studies have used isolated vascularstrips and smooth muscle cells from intact and gonadectomizedmale and female experimental animals to investigate the effectof gender and sex hormones on [Ca2�]i and the Ca2� mobili-zation mechanisms of smooth muscle contraction (25, 95, 146).Studies in isolated VSM cells have shown that the resting celllength is longer and the basal [Ca2�]i is smaller in intact femalecompared with intact male rats, suggesting gender differencesin the Ca2� handling mechanisms (95). The gender differencesin resting cell length and [Ca2�]i appear to be related toestrogen because the cell length and [Ca2�]i are greater inOVX females compared with intact females, but not differentbetween OVX females with E2 implants and intact females orbetween castrated and intact males (95; Fig. 5).
In cells incubated in the presence of external Ca2�, Phecauses an initial peak in [Ca2�]i mainly due to Ca2� releasefrom the intracellular stores, followed by a smaller but main-tained increase in [Ca2�]i due to Ca2� entry from the extra-
cellular space (95; Fig. 5). In Ca2�-free solution, Phe causes atransient increase in smooth muscle contraction and [Ca2�]i
that is not different between intact and gonadectomized maleand female rats, suggesting that the IP3-mediated Ca2� releaseis not involved in the gender differences in cell contraction and[Ca2�]i (95). Also, caffeine, which stimulates the Ca2�-in-duced Ca2� release mechanism, causes a small cell contractionand a transient increase in [Ca2�]i that are similar in magnitudein intact and gonadectomized male and female rats, suggestingthat the gender differences in cell contraction and [Ca2�]i arenot related to the Ca2�-induced Ca2� release mechanism (95).
On the other hand, the maintained Phe-induced [Ca2�]i inVSM cells incubated in the presence of external Ca2� is greaterin intact male than intact female rats, suggesting gender dif-ferences in the Ca2� entry mechanism of VSM contraction.The maintained Phe-induced [Ca2�]i is enhanced in OVXcompared with intact females, but not different between OVXfemales with estrogen implants and intact females, or betweencastrated and intact males, suggesting that the gender differ-ences are more likely related to estrogen than androgen (95;Fig. 5).
Membrane depolarization by high KCl mainly stimulatesCa2� entry from the extracellular space. The reports that theKCl-induced smooth muscle contraction, Ca2� influx, and[Ca2�]i are greater in intact males than intact females furthersupport gender differences in the Ca2� entry mechanisms (25,95). Also, the KCl-induced cell contraction and [Ca2�]i areenhanced in OVX females compared with intact females, butnot different between OVX females with estrogen implants andintact females, lending support to the contention that thegender differences are more likely related to endogenous es-trogen. The causes of the gender differences in the Ca2� entrymechanism are not clear, but may be related to the plasmale-mmal density and/or the permeability of the Ca2� channelsdepending on the presence or deficiency of endogenous estro-gen. This is supported by reports that the expression of theL-type Ca2� channels in cardiac muscle is substantially in-creased in ER-deficient mice and that the L-type Ca2� currentis significantly greater in coronary smooth muscle of malescompared with females (11, 65).
Inasmuch as VSM contraction and [Ca2�]i are often en-hanced in animal models of hypertension, any gender differ-ences in the Ca2� mobilization mechanisms of VSM contrac-tion are expected to be more apparent in hypertensive SHRthan normotensive WKY rats. Aortic strips of SHR showgreater vascular contraction and Ca2� entry than those ofWKY rats (25). Also, VSM cells of SHR show shorter restingcell length, greater basal [Ca2�]i, and greater maintained Phe-and KCl-induced contraction and [Ca2�]i than those of WKYrats (95; Fig. 5). Additionally, the reduction in vascular con-traction, Ca2� entry, and [Ca2�]i in intact females or OVXfemales with estrogen implants compared with intact males orOVX females is greater in SHR than WKY rats, suggestingpossible differences in the number of ERs or the number andpermeability of the plasma membrane Ca2� channels (25, 95).We should note that the gender differences in the mechanismsof Ca2� mobilization into VSM could be due to a multitude ofeffects of sex hormones in vivo. However, E2 causes relativelyrapid relaxation of isolated vascular strips of rabbit, porcine,and human coronary artery, suggesting that it may be mediatedby an effect on Ca2� mobilization and/or fluxes (24; Fig. 5).
Fig. 5. Effect of Phe (10�5 M) on [Ca2�]i in aortic smooth muscle cellsisolated from intact and gonadectomized, male and female Wistar-Kyoto(WKY; A) and spontaneously hypertensive rats (SHR; B), and ovariectomized(OVX) female rats with 17�-estradiol implants and incubated in Hank’ssolution (1 mM Ca2�). Dashed lines are drawn at the smallest basal andmaintained Phe-induced increase in [Ca2�]i in cells of intact females tofacilitate comparison with the other groups. [Modified from (95)].
Invited Review
R243SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from

Several studies have shown that estrogen does not inhibitcaffeine- or carbachol-induced smooth muscle contraction or[Ca2�]i in Ca2�-free solution, suggesting that it does notinhibit Ca2� release from the intracellular stores (24, 94).However, supraphysiological concentrations of estrogen mayinhibit thromboxane A2-induced Ca2� release in porcine cor-onary artery (50). On the other hand, estrogen inhibits themaintained Phe-, PGF2�-, and thromboxane A2-induced con-traction, Ca2� influx, and [Ca2�]i, suggesting inhibition ofCa2� entry from the extracellular space (24, 50, 94). Also,estrogen inhibits the high KCl-induced contraction, Ca2� in-flux, and [Ca2�]i, suggesting that it may act by inhibiting Ca2�
entry through voltage-gated channels (24, 40, 76, 94; Fig. 6).Estrogen may inhibit Ca2� entry by direct or indirect action
on plasmalemmal Ca2� channels. Some studies have shownthat estrogen blocks Ca2� channels in cultured A7r5 and aortic
smooth muscle cells (146). Other studies have shown thatestrogen activates BKCa channels in coronary smooth musclecells, which could lead to hyperpolarization and decreasedCa2� entry through voltage-gated channels (140). However,estrogen-induced vasorelaxation and inhibition of Ca2� influxinto VSM have been observed even in the absence of activationof K� efflux, lending support to direct effects of estrogen onCa2� channels (114).
Estrogen may also decrease [Ca2�]i by stimulating Ca2�
extrusion via plasmalemmal Ca2� pump (101). However, thismechanism seems less likely because the rate of decay ofcaffeine- and carbachol-induced contraction and [Ca2�]i tran-sients in smooth muscle incubated in Ca2�-free solution, whichare often used as a measure of Ca2� extrusion, are not affectedby estrogen (24, 94).
In contrast to estrogen, the effects of progesterone on[Ca2�]i have not been clearly established. However, severalstudies have shown that acute application of progesteronedecreases Ca2� influx and [Ca2�]i in rabbit and porcine coro-nary smooth muscle (24, 94; Fig. 6). There have also beeninconsistent reports regarding the effects of testosterone onVSM [Ca2�]i. However, the majority of studies suggests thattestosterone has a potent vasorelaxant effect in the rabbitcoronary artery and aorta and porcine coronary artery and thattestosterone decreases VSM [Ca2�]i by inhibiting Ca2� entryfrom the extracellular space (24, 94, 145; Fig. 6). It has beenshown that the relaxing effect of testosterone is attenuated byK� channel blockers, suggesting that stimulation of K� con-ductance through specific K� channels, e.g., voltage-depen-dent (delayed rectifier) K� channel may be involved in theinhibitory effects of testosterone on [Ca2�]i (145).
The progesterone- and testosterone-induced inhibition ofPGF2�-induced contraction is greater than the inhibition of theKCl-induced responses (24). These data suggest that proges-terone and testosterone not only inhibit Ca2� entry throughvoltage-gated channels, but may also inhibit additional VSMcontraction mechanisms activated by PGF2� such as PKC.
SEX HORMONES AND PKC
Recent studies have investigated whether the gender differ-ences in vascular contraction reflect differences in the expres-sion/activity of PKC isoforms in VSM. Phorbol esters, whichactivate PKC, produce greater contraction in isolated vessels ofintact male than intact female rats (69). The greater Phe- andphorbol ester-induced contraction and PKC activity in intactmale compared with intact female rats have suggested genderdifferences in the PKC-mediated pathway of VSM contraction(69), which may be related to differences in the amount of PKCexpressed in VSM and/or the sensitivity of the PKC pathway toendogenous sex hormones.
Immunoblot analysis in aortic smooth muscle of intact maleWKY rat has shown significant amounts of �-, �-, and -PKC(Fig. 7). In the same preparation, both Phe and phorbol estercause activation and redistribution of �- and �-PKC from thecytosolic to the particulate fraction. The amount of �-, �-, and-PKC, and the Phe- and phorbol ester-induced redistributionof �- and �-PKC are reduced in intact females compared withintact males, suggesting that the gender differences in vascularcontraction are related, in part, to underlying changes in theamount and activity of �-, �-, and -PKC (69; Fig. 7).
Fig. 6. Effect of sex hormones on KCl-stimulated [Ca2�]i in male porcinecoronary smooth muscle cells. Cells were stimulated with 51 mM KCl solutionfor 5 min. Cells were then treated with the vehicle ethanol (A) or with 10�7 M17�-estradiol (B), progesterone (C), or testosterone (D). [Modified from (94)].
Invited Review
R244 SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from

The Phe- and phorbol ester-induced contraction and PKCactivity are not different between castrated and intact male rats,but greater in OVX than intact females, suggesting that thegender differences in vascular contraction and PKC activity aremore likely related to estrogens than androgens. This is sup-ported by reports that inserting E2 implants in OVX female andcastrated male rats is associated with reduction in vascularcontraction and PKC activity (69).
Previous studies have shown that vascular PKC activity isaugmented in the SHR rat model of enhanced vascular con-traction. Also, the reduction in vascular contraction and PKCactivity in intact females compared with intact males is greaterin SHR than WKY. The greater reduction in vascular contrac-tion and PKC activity in intact female SHR compared withWKY could not be explained by differences in their plasmaestrogen levels and appear to be related to inherent differencesin the amount of PKC isoforms expressed in VSM (69; Fig. 7).
A genomic action of estrogen on the expression of PKCisoforms in VSM might well underlie the reduction in vascularcontraction and PKC activity observed in intact females com-pared with intact males. However, additional nongenomiceffects of estrogen on the PKC molecule or its lipid cofactorsor other protein kinases upstream from PKC cannot be ex-cluded. Although a direct effect of estrogen on PKC activityhas not been established, progesterone has been shown toinhibit phorbol ester-induced contraction and PKC transloca-tion in VSM, suggesting an effect of progesterone on PKCactivity (53). The progesterone-induced inhibition of PKC maybe mediated by increasing cAMP levels in VSM (53).
Perspectives
It is apparent that there is an array of factors contributing tothe greater incidence of cardiovascular disease in men andpostmenopausal women compared with premenopausalwomen. One contributing factor is gender differences in theregulation of vascular tone. Numerous studies have suggestedboth genomic and nongenomic effects of sex hormones on theendothelium and VSM, but there are yet many unansweredquestions.
One important question is related to the subtypes, distribu-tion, and function of sex hormone receptors in vascular cells. Inblood vessels of wild-type mice, estrogen attenuates vasocon-striction via an ER-�-mediated increase in iNOS expression.Initial studies in ER knockout mice have shown that deficiencyof ER-� renders the aortic wall supersensitive to relaxation byE2, but does not change the vascular wall morphology, sug-gesting that ER-� may not be involved in vascular structuredevelopment. Other studies have shown that ER-�-deficientmice develop hypertension as they age, and their blood vesselsshow abnormal ion channel functions (84, 147), supporting arole for ER-� in the regulation of vascular function and bloodpressure. However, complex tissue-specific effects of sex hor-mones may be mediated by the expression of heterogeneousforms of their cognate receptors. Variant estrogen, progester-one, and testosterone receptor transcripts are expressed inhuman vascular cells and may alter the physiological effectsof estrogen, progestins, and androgens on the endotheliumand VSM.
In addition to the nuclear ERs that mediate the classictranscriptional effects of estrogen, ERs may associate with thecell membrane, and a subpopulation of these membrane-boundERs may mediate the rapid effects of estrogen. However, littleis known regarding the pathways that regulate the distributionof ER between the nuclear and membrane fractions. Phosphor-ylation of transcription factors plays an important role inregulation of gene expression, and subcellular trafficking ofspecific transcription factors is regulated by phosphorylation/dephosphorylation. Steroid hormone receptors are phosphopro-teins, and mutations in phosphorylation sites may affect thetransactivation capacity of these transcription factors (10).Studies in human VSM cells transiently transfected with ER-�have shown translocation of ER-� from the membrane to thenucleus. Nuclear localization of ER-� was blocked by bothpharmacological and genetic inhibition of MAPK. Also, con-stitutive activation of MAPK resulted in nuclear translocationof ER-�. These studies suggest that MAPK-mediated phos-phorylation of ER-� induces its nuclear localization (82).
Another question relates to the effect of sex hormones oncell growth and proliferation. Why estrogen enhances theproliferation of endothelial cells but inhibits the proliferation ofVSM cells remains an enigma and should represent an impor-tant area for future studies.
The rapid vasodilator effects of estrogen have suggestedother mechanisms in addition to the classic genomic pathwayof steroid action, possibly involving effects on the cellularmechanisms of vascular relaxation and/or contraction. Recentevidence indicates that Ca2� and K� channels in VSM cellsplay an important role in mediating estrogen-induced relax-ation of many vascular beds; however, elucidation of the signaltransduction mechanisms coupling ER-� and ER-� activationto generation of second messengers and effector mechanismsremains an area of intense study.
Although the gender differences in vascular contraction maybe related to effects of sex hormones on vascular [Ca2�]i orPKC activity, other protein kinases such as MLC kinase, Rhokinase, and tyrosine kinase as well as MLC phosphatase couldregulate smooth muscle contraction. Whether the expressionand activity of smooth muscle protein kinases and phospha-tases differ with gender and by the presence or deficiency of
Fig. 7. Expression of �-PKC in aortic smooth muscle of intact and gonadec-tomized male and female WKY (open bars) and SHR rats (solid bars). Westernblot analysis was performed using whole tissue homogenate of rat aorta andanti �-PKC antibody. Position of the molecular mass marker is shown at right.Amount of �-PKC is expressed as optical density. *SHR significantly different(P 0.05) from WKY. #Significantly different from other WKY. †Signifi-cantly different from other SHR. [Modified from (69)].
Invited Review
R245SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from

gonadal hormones is unclear and should be examined in futureinvestigations.
There is considerable evidence that both female and malesex hormones affect the mechanisms of vascular contraction;however, the vascular effects of sex hormones may not beuniform. Sex hormones have different sexual effects in bothsexes, and it is reasonable to believe that the vascular effects ofsex hormones are different in the two sexes. Preliminarystudies suggest gender differences in the effects of estrogen onthe mechanisms of vascular contraction (25), a research areathat should be more thoroughly examined.
Because the vascular effects of estrogen and progesteronemay involve modulation of the Ca2� channels, HRT mayrepresent a natural approach to decrease the severity of certainforms of hypertension that are responsive to Ca2� channelblockers. To use or not to use HRT in postmenopausal womenwith hypertension or coronary artery disease is still controver-sial. Reports from HERS, HERS2, and WHI studies do notappear to support beneficial vascular effects of HRT, particu-larly in elderly hypertensive women (37, 47, 74, 89, 117, 119).However, HRT may be beneficial in reducing the incidence ofcoronary artery disease in postmenopausal women (43, 48,108). Also, some studies demonstrated that postmenopausalHRT is accompanied with a reduction in arterial pressure whennatural hormones are used in a manner that avoids first-passliver effects and in doses that produce hormone levels similarto those in the premenopausal state (32). Furthermore, estradiolmetabolism may be an important determinant of its cardiovas-cular protective effects, and nonfeminizing estradiol metabo-lites may confer cardiovascular protection in both genders (7).Finally, compared with the role of estradiol in the regulation ofvascular tone, there are sparse data on the effects of androgensand androgen receptors on the vascular control mechanisms.Whether the recently discovered effects of testosterone on themechanisms of vascular relaxation/contraction justify its po-tential use in prevention of cardiovascular disease remains tobe explored (107).
GRANTS
This work was supported by grants from National Heart, Lung, and BloodInstitute (HL-52696, HL-65998, and HL-70659). R. A. Khalil is an EstablishedInvestigator of the American Heart Association.
REFERENCES
1. Akishita M, Kozaki K, Eto M, Yoshizumi M, Ishikawa M, Toba K,Orimo H, and Ouchi Y. Estrogen attenuates endothelin-1 production bybovine endothelial cells via estrogen receptor. Biochem Biophys ResCommun 251: 17–21, 1998.
2. Ba ZF, Wang P, Kuebler JF, Rue LW III, Bland KI, and ChaudryIH. Flutamide induces relaxation in large and small blood vessels. ArchSurg 137: 1180–1186, 2002.
3. Bacakova L and Kunes J. Gender differences in growth of vascularsmooth muscle cells isolated from hypertensive and normotensive rats.Clin Exp Hypertens 22: 33–44, 2000.
4. Bar J, Tepper R, Fuchs J, Pardo Y, Goldberger S, and Ovadia J. Theeffect of estrogen replacement therapy on platelet aggregation andadenosine triphosphate release in postmenopausal women. Obstet Gy-necol 81: 261–264, 1993.
5. Barbacanne MA, Rami J, Michel JB, Souchard JP, Philippe M,Besombes JP, Bayard F, and Arnal JF. Estradiol increases rat aortaendothelium-derived relaxing factor (EDRF) activity without changes inendothelial NO synthase gene expression: possible role of decreasedendothelium-derived superoxide anion production. Cardiovasc Res 41:672–681, 1999.
6. Barber DA and Miller VM. Gender differences in endothelium-depen-dent relaxations do not involve NO in porcine coronary arteries. Am JPhysiol Heart Circ Physiol 273: H2325–H2332, 1997.
7. Barchiesi F, Jackson EK, Gillespie DG, Zacharia LC, Fingerle J, andDubey RK. Methoxyestradiols mediate estradiol-induced antimitogen-esis in human aortic SMCs. Hypertension 39: 874–879, 2002.
8. Beldekas JC, Smith B, Gerstenfeld LC, Sonenshein GE, and Franz-blau C. Effects of 17�-estradiol on the biosynthesis of collagen incultured bovine aortic smooth muscle cells. Biochemistry 20: 2162–2167,1981.
9. Bilsel AS, Moini H, Tetik E, Aksungar F, Kaynak B, and Ozer A.17�-Estradiol modulates endothelin-1 expression and release in humanendothelial cells. Cardiovasc Res 46: 579–584, 2000.
10. Blok LJ, de Ruiter PE, and Brinkmann AO. Androgen receptorphosphorylation. Endocr Res 22: 197–219, 1996.
11. Bowles DK. Gender influences coronary L-type Ca2� current and adap-tation to exercise training in miniature swine. J Appl Physiol 91: 2503–2510, 2001.
12. Brandes RP and Mugge A. Gender differences in the generation ofsuperoxide anions in the rat aorta. Life Sci 60: 391–396, 1997.
13. Calkin AC, Sudhir K, Honisett S, Williams MR, Dawood T, andKomesaroff PA. Rapid potentiation of endothelium-dependent vasodi-lation by estradiol in postmenopausal women is mediated via cyclooxy-genase 2. J Clin Endocrinol Metab 87: 5072–5075, 2002.
14. Campbell-Thompson M, Lynch IJ, and Bhardwaj B. Expression ofestrogen receptor (ER) subtypes and ER� isoforms in colon cancer.Cancer Res 61: 632–640, 2001.
15. Case J and Davison CA. Estrogen alters relative contributions of nitricoxide and cyclooxygenase products to endothelium-dependent vasodila-tion. J Pharmacol Exp Ther 291: 524–530, 1999.
16. Caulin-Glaser T, Garcia-Cardena G, Sarrel P, Sessa WC, andBender JR. 17�-Estradiol regulation of human endothelial cell basalnitric oxide release, independent of cytosolic Ca2� mobilization. CircRes 81: 885–892, 1997.
17. Chambliss KL, Yuhanna IS, Anderson RG, Mendelsohn ME, andShaul PW. ER� has nongenomic action in caveolae. Mol Endocrinol 16:938–946, 2002.
18. Chan HY, Yao X, Tsang SY, Chan FL, Lau CW, and Huang Y.Different role of endothelium/nitric oxide in 17�-estradiol- and proges-terone-induced relaxation in rat arteries. Life Sci 69: 1609–1617, 2001.
19. Chen Z, Yuhanna IS, Galcheva-Gargova Z, Karas RH, MendelsohnME, and Shaul PW. Estrogen receptor � mediates the nongenomicactivation of endothelial nitric oxide synthase by estrogen. J Clin Invest103: 401–406, 1999.
20. Chou TM, Sudhir K, Hutchison SJ, Ko E, Amidon TM, Collins P,and Chatterjee K. Testosterone induces dilation of canine coronaryconductance and resistance arteries in vivo. Circulation 94: 2614–2619,1996.
21. Collins P, Rosano GM, Sarrel PM, Ulrich L, Adamopoulos S, BealeCM, McNeill JG, and Poole-Wilson PA. 17�-Estradiol attenuatesacetylcholine-induced coronary arterial constriction in women but notmen with coronary heart disease. Circulation 92: 24–30, 1995.
22. Costarella CE, Stallone JN, Rutecki GW, and Whittier FC. Testos-terone causes direct relaxation of rat thoracic aorta. J Pharmacol ExpTher 277: 34–39, 1996.
23. Cox DA and Cohen ML. Influence of gender on vasomotor effects ofoxidized low-density lipoprotein in porcine coronary arteries. Am JPhysiol Heart Circ Physiol 272: H2577–H2583, 1997.
24. Crews JK and Khalil RA. Antagonistic effects of 17�-estradiol, pro-gesterone and testosterone on Ca2� entry mechanisms of coronaryvasoconstriction. Arterioscler Thromb Vasc Biol 19: 1034–1040, 1999.
25. Crews JK and Khalil RA. Gender-specific inhibition of Ca2� entrymechanisms of arterial vasoconstriction by sex hormones. Clin ExpPharmacol Physiol 26: 707–715, 1999.
26. Dan P, Cheung JC, Scriven DR, and Moore ED. Epitope-dependentlocalization of estrogen receptor-�, but not -�, in en face arterialendothelium. Am J Physiol Heart Circ Physiol 284: H1295–H1306,2003.
27. Darblade B, Pendaries C, Krust A, Dupont S, Fouque MJ, Rami J,Chambon P, Bayard F, and Arnal JF. Estradiol alters nitric oxideproduction in the mouse aorta through the �-, but not �-, estrogenreceptor. Circ Res 90: 413–419, 2002.
Invited Review
R246 SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from

28. Darkow DJ, Lu L, and White RE. Estrogen relaxation of coronaryartery smooth muscle is mediated by nitric oxide and cGMP. Am JPhysiol Heart Circ Physiol 272: H2765–H2773, 1997.
29. David FL, Carvalho MH, Cobra AL, Nigro D, Fortes ZB, ReboucasNA, and Tostes RC. Ovarian hormones modulate endothelin-1 vascularreactivity and mRNA expression in DOCA-salt hypertensive rats. Hy-pertension 38: 692–696, 2001.
30. Davis MJ and Hill MA. Signaling mechanisms underlying the vascularmyogenic response. Physiol Rev 79: 387–423, 1999.
31. Ding AQ and Stallone JN. Testosterone-induced relaxation of rat aortais androgen structure specific and involves K� channel activation. J ApplPhysiol 91: 2742–2750, 2001.
32. Dourakis SP and Tolis G. Sex hormonal preparations and the liver. EurJ Contracept Reprod Health Care 3: 7–16, 1998.
33. Dubey RK, Gillespie DG, Imthurn B, Rosselli M, Jackson EK, andKeller PJ. Phytoestrogens inhibit growth and MAP kinase activity inhuman aortic smooth muscle cells. Hypertension 33: 177–182, 1999.
34. Dubey RK, Gillespie DG, Mi Z, Rosselli M, Keller PJ, and JacksonEK. Estradiol inhibits smooth muscle cell growth in part by activatingthe cAMP-adenosine pathway. Hypertension 35: 262–266, 2000.
35. Dubey RK, Jackson EK, Keller PJ, Imthurn B, and Rosselli M.Estradiol metabolites inhibit endothelin synthesis by an estrogen recep-tor-independent mechanism. Hypertension 37: 640–644, 2001.
36. English KM, Jones RD, Jones TH, Morice AH, and Channer KS.Gender differences in the vasomotor effects of different steroid hormonesin rat pulmonary and coronary arteries. Horm Metab Res 33: 645–652,2001.
37. Enstrom I, Lidfeldt J, Lindholm LH, Nerbrand C, Pennert K, andSamsioe G. Does blood pressure differ between users and non-users ofhormone replacement therapy? The Women’s Health In the Lund Area(WHILA) Study. Blood Press 11: 240–243, 2002.
38. Farhat MY, Lavigne MC, and Ramwell PW. The vascular protectiveeffects of estrogen. FASEB J 10: 615–624, 1996.
39. Forte P, Kneale BJ, Milne E, Chowienczyk PJ, Johnston A, Ben-jamin N, and Ritter JM. Evidence for a difference in nitric oxidebiosynthesis between healthy women and men. Hypertension 32: 730–734, 1998.
40. Freay AD, Curtis SW, Korach KS, and Rubanyi GM. Mechanism ofvascular smooth muscle relaxation by estrogen in depolarized rat andmouse aorta. Role of nuclear estrogen receptor and Ca2� uptake. CircRes 81: 242–248, 1997.
41. Geary GG, Krause DN, and Duckles SP. Estrogen reduces mousecerebral artery tone through endothelial NOS- and cyclooxygenase-dependent mechanisms. Am J Physiol Heart Circ Physiol 279: H511–H519, 2000.
42. Geraldes P, Sirois MG, Bernatchez PN, and Tanguay JF. Estrogenregulation of endothelial and smooth muscle cell migration and prolif-eration: role of p38 and p42/44 mitogen-activated protein kinase. Arte-rioscler Thromb Vasc Biol 22: 1585–1590, 2002.
43. Gerhard M and Ganz P. How do we explain the clinical benefits ofestrogen from bedside to bench. Circulation 92: 5–8, 1995.
44. Gerhard M, Walsh BW, Tawakol A, Haley EA, Creager SJ, SeelyEW, Ganz P, and Creager MA. Estradiol therapy combined withprogesterone and endothelium-dependent vasodilation in postmeno-pausal women. Circulation 98: 1158–1163, 1998.
45. Gilligan DM, Badar DM, Panza JA, Quyyumi AA, and Cannon ROIII. Acute vascular effects of estrogen in postmenopausal women. Cir-culation 90: 786–791, 1994.
46. Gragasin FS, Xu Y, Arenas IA, Kainth N, and Davidge ST. Estrogenreduces angiotensin II-induced nitric oxide synthase and NAD(P)Hoxidase expression in endothelial cells. Arterioscler Thromb Vasc Biol23: 38–44, 2003.
47. Grimes DA and Lobo RA. Perspectives on the Women’s HealthInitiative trial of hormone replacement therapy. Obstet Gynecol 100:1344–1353, 2002.
48. Grodstein F, Stampfer MJ, Manson JE, Colditz GA, Willett WC,Rosner B, Speizer FE, and Hennekens CH. Postmenopausal estrogenand progestin use and the risk of cardiovascular disease. N Engl J Med335: 453–461, 1996.
49. Guiochon-Mantel A. Regulation of the differentiation and proliferationof smooth muscle cells by the sex hormones. Rev Mal Respir 17:604–608, 2000.
50. Han SZ, Karaki H, Ouchi Y, Akishita M, and Orimo H. 17�-Estradiolinhibits Ca2� influx and Ca2� release induced by thromboxane A2 inporcine coronary artery. Circulation 91: 2619–2626, 1995.
51. Harder DR and Coulson PB. Estrogen receptors and effects on mem-brane electrical properties of coronary vascular smooth muscle. J CellPhysiol 100: 375–382, 1979.
52. Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, Sessa WC, and Bender JR. Membrane estrogen receptorengagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res 87: 677–682,2000.
53. Herkert O, Kuhl H, Busse R, and Schini-Kerth VB. The progestinlevonorgestrel induces endothelium-independent relaxation of rabbit jug-ular vein via inhibition of calcium entry and protein kinase C: role ofcyclic AMP. Br J Pharmacol 130: 1911–1918, 2000.
54. Hernandez I, Delgado JL, Diaz J, Quesada T, Teruel MJ, LlanosMC, and Carbonell LF. 17�-Estradiol prevents oxidative stress anddecreases blood pressure in ovariectomized rats. Am J Physiol RegulIntegr Comp Physiol 279: R1599–R1605, 2000.
55. Higashiura K, Mathur RS, and Halushka PV. Gender-related differ-ences in androgen regulation of thromboxane A2 receptors in rat aorticsmooth-muscle cells. J Cardiovasc Pharmacol 29: 311–315, 1997.
56. Hisamoto K, Ohmichi M, Kurachi H, Hayakawa J, Kanda Y, NishioY, Adachi K, Tasaka K, Miyoshi E, Fujiwara N, Taniguchi N, andMurata Y. Estrogen induces the Akt-dependent activation of endothelialnitric-oxide synthase in vascular endothelial cells. J Biol Chem 276:3459–3467, 2001.
57. Hodges YK, Tung L, Yan XD, Graham JD, Horwitz KB, andHorwitz LD. Estrogen receptors � and �: prevalence of estrogen recep-tor � mRNA in human vascular smooth muscle and transcriptionaleffects. Circulation 101: 1792–1798, 2000.
58. Honda H, Unemoto T, and Kogo H. Different mechanisms for testos-terone-induced relaxation of aorta between normotensive and spontane-ously hypertensive rats. Hypertension 34: 1232–1236, 1999.
59. Horowitz A, Menice CB, Laporte R, and Morgan KG. Mechanisms ofsmooth muscle contraction. Physiol Rev 76: 967–1003, 1996.
60. Horwitz KB and Horwitz LD. Canine vascular tissues are targets forandrogens, estrogens, progestins, and glucocorticoids. J Clin Invest 69:750–758, 1982.
61. Iliescu R, Campos LA, Schlegel WP, Morano I, Baltatu O, and BaderM. Androgen receptor independent cardiovascular action of the antian-drogen flutamide. J Mol Med 81: 420–427, 2003.
62. Jiang C, Sarrel PM, Lindsay DC, Poole-Wilson PA, and Collins P.Endothelium-independent relaxation of rabbit coronary artery by 17�-estradiol in vitro. Br J Pharmacol 104: 1033–1037, 1991.
63. Jiang C, Sarrel PM, Lindsay DC, Poole-Wilson PA, and Collins P.Progesterone induces endothelium-independent relaxation of rabbit cor-onary artery in vitro. Eur J Pharmacol 211: 163–167, 1992.
64. Jiang C, Sarrel PM, Poole-Wilson PA, and Collins P. Acute effect of17�-estradiol on rabbit coronary artery contractile responses to endothe-lin-1. Am J Physiol Heart Circ Physiol 263: H271–H275, 1992.
65. Johnson BD, Zheng W, Korach KS, Scheuer T, Catterall WA, andRubanyi GM. Increased expression of the cardiac L-type calciumchannel in estrogen receptor-deficient mice. J Gen Physiol 110: 135–140,1997.
66. Jun SS, Chen Z, Pace MC, and Shaul PW. Estrogen upregulatescyclooxygenase-1 gene expression in ovine fetal pulmonary artery en-dothelium. J Clin Invest 102: 176–183, 1998.
67. Kahonen M, Tolvanen JP, Sallinen K, Wu X, and Porsti I. Influenceof gender on control of arterial tone in experimental hypertension. Am JPhysiol Heart Circ Physiol 275: H15–H22, 1998.
68. Kakucs R, Varbiro S, Nadasy GL, Monos E, and Szekacs B. Acute,nongenomic vasodilatory action of estradiol is attenuated by chronicestradiol treatment. Exp Biol Med (Maywood) 226: 538–542, 2001.
69. Kanashiro CA and Khalil RA. Gender-related distinctions in proteinkinase C activity in rat vascular smooth muscle. Am J Physiol CellPhysiol 280: C34–C45, 2001.
70. Kanashiro CA and Khalil RA. Signal transduction by protein kinase Cin mammalian cells. Clin Exp Pharmacol Physiol 25: 974–985, 1998.
71. Kauser K and Rubanyi GM. Gender difference in bioassayable endo-thelium-derived nitric oxide from isolated rat aortae. Am J Physiol HeartCirc Physiol 267: H2311–H2317, 1994.
Invited Review
R247SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from

72. Kauser K and Rubanyi GM. Gender difference in endothelial dysfunc-tion in the aorta of spontaneously hypertensive rats. Hypertension 25:517–523, 1995.
73. Khalil RA and van Breemen C. Mechanisms of calcium mobilizationand homeostasis in vascular smooth muscle and their relevance tohypertension. In: Hypertension: Pathophysiology, Diagnosis, and Man-agement, edited by Laragh JH and Brenner BM. New York: Raven, 1995,p. 523–540.
74. Khan NS and Malhotra S. Effect of hormone replacement therapy oncardiovascular disease: current opinion. Exp Opin Pharmacother 4:667–674, 2003.
75. Kim-Schulze S, McGowan KA, Hubchak SC, Cid MC, Martin MB,Kleinman HK, Greene GL, and Schnaper HW. Expression of anestrogen receptor by human coronary artery and umbilical vein endothe-lial cells. Circulation 94: 1402–1407, 1996.
76. Kitazawa T, Hamada E, Kitazawa K, and Gaznabi AK. Non-genomicmechanism of 17�-oestradiol-induced inhibition of contraction in mam-malian vascular smooth muscle. J Physiol 499: 497–511, 1997.
77. Kneale BJ, Chowienczyk PJ, Brett SE, Coltart DJ, and Ritter JM.Gender differences in sensitivity to adrenergic agonists of forearmresistance vasculature. J Am Coll Cardiol 36: 1233–1238, 2000.
78. Knot HJ, Lounsbury KM, Brayden JE, and Nelson MT. Genderdifferences in coronary artery diameter reflect changes in both endothe-lial Ca2� and ecNOS activity. Am J Physiol Heart Circ Physiol 276:H961–H969, 1999.
79. Kos M, Denger S, Reid G, and Gannon F. Upstream open readingframes regulate the translation of the multiple mRNA variants of theestrogen receptor �. J Biol Chem 277: 37131–37138, 2002.
80. Landers JP and Spelsberg TC. New concepts in steroid hormoneaction: transcription factors, proto-oncogenes, and the cascade model forsteroid regulation of gene expression. Crit Rev Eukaryot Gene Expr 2:19–63, 1992.
81. Liu MY, Hattori Y, Fukao M, Sato A, Sakuma I, and Kanno M.Alterations in EDHF-mediated hyperpolarization and relaxation in mes-enteric arteries of female rats in long-term deficiency of oestrogen andduring oestrus cycle. Br J Pharmacol 132: 1035–1046, 2001.
82. Lu Q, Ebling H, Mittler J, Baur WE, and Karas RH. MAP kinasemediates growth factor-induced nuclear translocation of estrogen recep-tor �. FEBS Lett 516: 1–8, 2002.
83. MacRitchie AN, Jun SS, Chen Z, German Z, Yuhanna IS, ShermanTS, and Shaul PW. Estrogen upregulates endothelial nitric oxide syn-thase gene expression in fetal pulmonary artery endothelium. Circ Res81: 355–362, 1997.
84. Mendelsohn ME. Genomic and nongenomic effects of estrogen in thevasculature. Am J Cardiol 90: 3F–6F, 2002.
85. Meyer MC, Cummings K, and Osol G. Estrogen replacement attenu-ates resistance artery adrenergic sensitivity via endothelial vasodilators.Am J Physiol Heart Circ Physiol 272: H2264–H2270, 1997.
86. Michel T. Targeting and translocation of endothelial nitric oxide syn-thase. Braz J Med Biol Res 32: 1361–1366, 1999.
87. Mikkola T, Viinikka L, and Ylikorkala O. Administration of transder-mal estrogen without progestin increases the capacity of plasma andserum to stimulate prostacyclin production in human vascular endothelialcells. Fertil Steril 73: 72–74, 2000.
88. Miller VM and Vanhoutte PM. Progesterone and modulation of endo-thelium-dependent responses in canine coronary arteries. Am J PhysiolRegul Integr Comp Physiol 261: R1022–R1027, 1991.
89. Mills PJ, Farag NH, Matthews S, Nelesen RA, Berry CC, andDimsdale JE. Hormone replacement therapy does not affect 24-h am-bulatory blood pressure in healthy non-smoking postmenopausal women.Blood Press Monit 8: 57–61, 2003.
90. Minshall RD, Pavcnik D, Browne DL, and Hermsmeyer K. Non-genomic vasodilator action of progesterone on primate coronary arteries.J Appl Physiol 92: 701–708, 2002.
91. Minshall RD, Pavcnik D, Halushka PV, and Hermsmeyer K. Proges-terone regulation of vascular thromboxane A2 receptors in rhesus mon-keys. Am J Physiol Heart Circ Physiol 281: H1498–H1507, 2001.
92. Molinari C, Battaglia A, Grossini E, Mary DA, Stoker JB, Surico N,and Vacca G. The effect of progesterone on coronary blood flow inanaesthetized pigs. Exp Physiol 86: 101–108, 2001.
93. Morey AK, Razandi M, Pedram A, Hu RM, Prins BA, and Levin ER.Oestrogen and progesterone inhibit the stimulated production of endo-thelin-1. Biochem J 330: 1097–1105, 1998.
94. Murphy JG and Khalil RA. Decreased [Ca2�]i during inhibition ofcoronary smooth muscle contraction by 17�-estradiol, progesterone, andtestosterone. J Pharmacol Exp Ther 291: 44–52, 1999.
95. Murphy JG and Khalil RA. Gender-specific reduction in contractilityand [Ca2�]i in vascular smooth muscle cells of female rat. Am J PhysiolCell Physiol 278: C834–C844, 2000.
96. Nickenig G, Strehlow K, Wassmann S, Baumer AT, Albory K, SauerH, and Bohm M. Differential effects of estrogen and progesterone onAT1 receptor gene expression in vascular smooth muscle cells. Circula-tion 102: 1828–1833, 2000.
97. Nisolle M, Gillerot S, Casanas-Roux F, Squifflet J, Berliere M, andDonnez J. Immunohistochemical study of the proliferation index, oes-trogen receptors and progesterone receptors A and B in leiomyomata andnormal myometrium during the menstrual cycle and under gonadotro-phin-releasing hormone agonist therapy. Hum Reprod 14: 2844–2850,1999.
98. Pare G, Krust A, Karas RH, Dupont S, Aronovitz M, Chambon P,and Mendelsohn ME. Estrogen receptor-� mediates the protectiveeffects of estrogen against vascular injury. Circ Res 90: 1087–1092,2002.
99. Pieber D, Allport VC, Hills F, Johnson M, and Bennett PR. Interac-tions between progesterone receptor isoforms in myometrial cells inhuman labour. Mol Hum Reprod 7: 875–879, 2001.
100. Polderman KH, Stehouwer CD, van Kamp GJ, Dekker GA, Ver-heugt FW, and Gooren LJ. Influence of sex hormones on plasmaendothelin levels. Ann Intern Med 118: 429–432, 1993.
101. Prakash YS, Togaibayeva AA, Kannan MS, Miller VM, FitzpatrickLA, and Sieck GC. Estrogen increases Ca2� efflux from female porcinecoronary arterial smooth muscle. Am J Physiol Heart Circ Physiol 276:H926–H934, 1999.
102. Rahimian R, Wang X, and van Breemen C. Gender difference in thebasal intracellular Ca2� concentration in rat valvular endothelial cells.Biochem Biophys Res Commun 248: 916–919, 1998.
103. Razandi M, Oh P, Pedram A, Schnitzer J, and Levin ER. ERsassociate with and regulate the production of caveolin: implications forsignaling and cellular actions. Mol Endocrinol 16: 100–115, 2002.
104. Reckelhoff JF, Zhang H, and Srivastava K. Gender differences indevelopment of hypertension in spontaneously hypertensive rats: role ofthe renin-angiotensin system. Hypertension 35: 480–383, 2000.
105. Reckelhoff JF. Gender differences in the regulation of blood pressure.Hypertension 37: 1199–1208, 2001.
106. Rifici VA and Khachadurian AK. The inhibition of low-density li-poprotein oxidation by 17-� estradiol. Metabolism 41: 1110–1114, 1992.
107. Rosano GM, Leonardo F, Pagnotta P, Pelliccia F, Panina G, Cerqu-etani E, della Monica PL, Bonfigli B, Volpe M, and Chierchia SL.Acute anti-ischemic effect of testosterone in men with coronary arterydisease. Circulation 99: 1666–1670, 1999.
108. Rosano GM, Sarrel PM, Poole-Wilson PA, and Collins P. Beneficialeffect of oestrogen on exercise-induced myocardial ischaemia in womenwith coronary artery disease. Lancet 342: 133–136, 1993.
109. Rubanyi GM, Freay AD, Kauser K, Sukovich D, Burton G, LubahnDB, Couse JF, Curtis SW, and Korach KS. Vascular estrogen recep-tors and endothelium-derived nitric oxide production in the mouse aorta.Gender difference and effect of estrogen receptor gene disruption. J ClinInvest 99: 2429–2437, 1997.
110. Rubio-Gayosso I, Garcia-Ramirez O, Gutierrez-Serdan R, Guevara-Balcazar G, Munoz-Garcia O, Morato-Cartajena T, Zamora-GarzaM, and Ceballos-Reyes G. Testosterone inhibits bradykinin-inducedintracellular calcium kinetics in rat aortic endothelial cells in culture.Steroids 67: 393–397, 2002.
111. Rupnow HL, Phernetton TM, Shaw CE, Modrick ML, Bird IM, andMagness RR. Endothelial vasodilator production by uterine and sys-temic arteries. VII Estrogen and progesterone effects on eNOS. Am JPhysiol Heart Circ Physiol 280: H1699–H1705, 2001.
112. Russell KS, Haynes MP, Sinha D, Clerisme E, and Bender JR.Human vascular endothelial cells contain membrane binding sites forestradiol, which mediate rapid intracellular signaling. Proc Natl Acad SciUSA 97: 5930–5935, 2000.
113. Sakuma I, Liu MY, Sato A, Hayashi T, Iguchi A, Kitabatake A, andHattori Y. Endothelium-dependent hyperpolarization and relaxation inmesenteric arteries of middle-aged rats: influence of oestrogen. Br JPharmacol 135: 48–54, 2002.
Invited Review
R248 SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from

114. Salom JB, Burguete MC, Perez-Asensio FJ, Torregrosa G, andAlborch E. Relaxant effects of 17�-estradiol in cerebral arteries throughCa2� entry inhibition. J Cereb Blood Flow Metab 21: 422–429, 2001.
115. Scobie GA, Macpherson S, Millar MR, Groome NP, Romana PG,and Saunders PT. Human oestrogen receptors: differential expression ofER alpha and beta and the identification of ER beta variants. Steroids 67:985–992, 2002.
116. Selles J, Polini N, Alvarez C, and Massheimer V. Nongenomic actionof progesterone in rat aorta: role of nitric oxide and prostaglandins. CellSignal 14: 431–436, 2002.
117. Shah SH and Alexander KP. Hormone replacement therapy for primaryand secondary prevention of heart disease. Curr Treat Options Cardio-vasc Med 5: 25–33, 2003.
118. Sherman TS, Chambliss KL, Gibson LL, Pace MC, Mendelsohn ME,Pfister SL, and Shaul PW. Estrogen acutely activates prostacyclinsynthesis in ovine fetal pulmonary artery endothelium. Am J Respir CellMol Biol 26: 610–616, 2002.
119. Simon JA, Hsia J, Cauley JA, Richards C, Harris F, Fong J,Barrett-Connor E, and Hulley SB. Postmenopausal hormone therapyand risk of stroke: The Heart and Estrogen-Progestin Replacement Study(HERS). Circulation 103: 638–642, 2001.
120. Simoncini T, Mannella P, Fornari L, Varone G, Caruso A, andGenazzani AR. Dehydroepiandrosterone modulates endothelial nitricoxide synthesis via direct genomic and nongenomic mechanisms. Endo-crinology 144: 3449–3455, 2003.
121. Somjen D, Kohen F, Jaffe A, Amir-Zaltsman Y, Knoll E, and SternN. Effects of gonadal steroids and their antagonists on DNA synthesis inhuman vascular cells. Hypertension 32: 39–45, 1998.
122. Somlyo AP and Somlyo AV. Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle my-osin II. J Physiol 522: 177–185, 2000.
123. Stallone JN, Crofton JT, and Share L. Sexual dimorphism in vaso-pressin-induced contraction of rat aorta. Am J Physiol Heart Circ Physiol260: H453–H458, 1991.
124. Stefano GB, Prevot V, Beauvillain JC, Cadet P, Fimiani C, WeltersI, Fricchione GL, Breton C, Lassalle P, Salzet M, and Bilfinger TV.Cell-surface estrogen receptors mediate calcium-dependent nitric oxiderelease in human endothelia. Circulation 101: 1594–1597, 2000.
125. Sumi D, Hayashi T, Jayachandran M, and Iguchi A. Estrogen pre-vents destabilization of endothelial nitric oxide synthase mRNA inducedby tumor necrosis factor � through estrogen receptor mediated system.Life Sci 69: 1651–1660, 2001.
126. Takeda-Matsubara Y, Nakagami H, Iwai M, Cui TX, Shiuchi T,Akishita M, Nahmias C, Ito M, and Horiuchi M. Estrogen activatesphosphatases and antagonizes growth-promoting effect of angiotensin II.Hypertension 39: 41–45, 2002.
127. Tamaya T, Wada K, Nakagawa M, Misao R, Itoh T, Imai A, andMori H. Sexual dimorphism of binding sites of testosterone and dihy-drotestosterone in rabbit model. Comp Biochem Physiol 105A: 745–749,1993.
128. Tan E, Gurjar MV, Sharma RV, and Bhalla RC. Estrogen receptor-�gene transfer into bovine aortic endothelial cells induces eNOS geneexpression and inhibits cell migration. Cardiovasc Res 43: 788–797,1999.
129. Thompson J and Khalil RA. Gender differences in the regulation ofvascular tone. Clin Exp Pharmacol Physiol 30: 90–105, 2003.
130. Thompson LP, Pinkas G, and Weiner CP. Chronic 17�-estradiolreplacement increases nitric oxide-mediated vasodilation of guinea pigcoronary microcirculation. Circulation 102: 445–451, 2000.
131. Tostes RC, David FL, Carvalho MH, Nigro D, Scivoletto R, andFortes ZB. Gender differences in vascular reactivity to endothelin-1 indeoxycorticosterone-salt hypertensive rats. J Cardiovasc Pharmacol 36:S99–S101, 2000.
132. Touyz RM, Turgeon A, and Schiffrin EL. Endothelin-A-receptorblockade improves renal function and doubles the lifespan of stroke-prone spontaneously hypertensive rats. J Cardiovasc Pharmacol 36:S300–S304, 2000.
133. Tschugguel W, Stonek F, Zhegu Z, Dietrich W, Schneeberger C,Stimpfl T, Waldhoer T, Vycudilik W, and Huber JC. Estrogenincreases endothelial carbon monoxide, heme oxygenase 2, and carbonmonoxide-derived cGMP by a receptor-mediated system. J Clin Endo-crinol Metab 86: 3833–3839, 2001.
134. Van der Mooren MJ, Mijatovic V, van Baal WM, and StehouwerCD. Hormone replacement therapy in postmenopausal women withspecific risk factors for coronary artery disease. Maturitas 30: 27–36,1998.
135. Vazquez F, Rodriguez-Manzaneque JC, Lydon JP, Edwards DP,O’Malley BW, and Iruela-Arispe ML. Progesterone regulates prolif-eration of endothelial cells. J Biol Chem 274: 2185–2192, 1999.
136. Wagner AH, Schroeter MR, and Hecker M. 17�-Estradiol inhibitionof NADPH oxidase expression in human endothelial cells. FASEB J 15:2121–2130, 2001.
137. Wakasugi M, Noguchi T, Kazama YI, Kanemaru Y, and Onaya T.The effects of sex hormones on the synthesis of prostacyclin (PGI2) byvascular tissues. Prostaglandins 37: 401–410, 1989.
138. Webb CM, Ghatei MA, McNeill JG, and Collins P. 17�-Estradioldecreases endothelin-1 levels in the coronary circulation of postmeno-pausal women with coronary artery disease. Circulation 102: 1617–1622,2000.
139. Wellman GC, Bonev AD, Nelson MT, and Brayden JE. Genderdifferences in coronary artery diameter involve estrogen, nitric oxide, andCa2�-dependent K� channels. Circ Res 79: 1024–1030, 1996.
140. White RE, Darkow DJ, and Lang JLF. Estrogen relaxes coronaryarteries by opening BKCa channels through a cGMP-dependent mecha-nism. Circ Res 77: 936–942, 1995.
141. Wilcox JN, Subramanian RR, Sundell CL, Tracey WR, Pollock JS,Harrison DG, and Marsden PA. Expression of multiple isoforms ofnitric oxide synthase in normal and atherosclerotic vessels. ArteriosclerThromb Vasc Biol 17: 2479–2488, 1997.
142. Wynne FL and Khalil RA. Testosterone and coronary vascular tone:implications in coronary artery disease. J Endocrinol Invest 26: 181–186,2003.
143. Yang S, Bae L, and Zhang L. Estrogen increases eNOS and NOxrelease in human coronary artery endothelium. J Cardiovasc Pharmacol36: 242–247, 2000.
144. Yuan Y, Liao L, Tulis DA, and Xu J. Steroid receptor coactivator-3 isrequired for inhibition of neointima formation by estrogen. Circulation105: 2653–2659, 2002.
145. Yue P, Chatterjee K, Beale C, Poole-Wilson PA, and Collins P.Testosterone relaxes rabbit coronary arteries and aorta. Circulation 91:1154–1160, 1995.
146. Zhang F, Ram JL, Standley PR, Sowers JR. 17�-Estradiol attenuatesvoltage-dependent Ca2� currents in A7r5 vascular smooth muscle cellline. Am J Physiol Cell Physiol 266: C975–C980, 1994.
147. Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, Hodgin J, Shaul PW,Thoren P, Smithies O, Gustafsson JA, and Mendelsohn ME. Abnor-mal vascular function and hypertension in mice deficient in estrogenreceptor �. Science 295: 505–508, 2002.
Invited Review
R249SEX HORMONES AND VASCULAR TONE
AJP-Regul Integr Comp Physiol • VOL 286 • FEBRUARY 2004 • www.ajpregu.org
by 10.220.32.246 on August 3, 2017
http://ajpregu.physiology.org/D
ownloaded from





























