Embed Size (px)
Citation preview

This is an electronic reprint of the original article.This reprint may differ from the original in pagination and typographic detail.
Powered by TCPDF (www.tcpdf.org)
This material is protected by copyright and other intellectual property rights, and duplication or sale of all or part of any of the repository collections is not permitted, except that material may be duplicated by you for your research use or educational purposes in electronic or print form. You must obtain permission for any other use. Electronic or print copies may not be offered, whether for sale or otherwise to anyone who is not an authorised user.
Fraxedas, J.; Lee, Y.-J.; Jimenez, I.; Gago, R.; Nieminen, Risto; Ordejon, P.; Canadell, E.
Characterization of the unoccupied and partially occupied states of TTF-TCNQ byXANES and first-principles calculations
Published in:Physical Review B
DOI:10.1103/PhysRevB.68.195115
Published: 01/01/2003
Document VersionPublisher's PDF, also known as Version of record
Please cite the original version:Fraxedas, J., Lee, Y-J., Jimenez, I., Gago, R., Nieminen, R. M., Ordejon, P., & Canadell, E. (2003).Characterization of the unoccupied and partially occupied states of TTF-TCNQ by XANES and first-principlescalculations. Physical Review B, 68(19), 1-11. [195115]. DOI: 10.1103/PhysRevB.68.195115

PHYSICAL REVIEW B 68, 195115 ~2003!
Characterization of the unoccupied and partially occupied states of TTF-TCNQby XANES and first-principles calculations
J. Fraxedas,1 Y. J. Lee,2 I. Jimenez,3 R. Gago,4 R. M. Nieminen,2 P. Ordejon,1 and E. Canadell1
1Institut de Ciencia de Materials de Barcelona (CSIC), Campus de la U.A.B., 08193 Bellaterra, Spain2COMP/Laboratory of Physics, Helsinki University of Technology, P.O.Box 1100, FIN-02015 HUT, Finland
3Instituto de Ciencia y Tecnologı´a de Polı´meros (CSIC), 28006 Madrid, Spain4Forschungszentrum Rossendorf e.V. PF-510119, 01314 Dresden, Germany
~Received 17 July 2003; published 21 November 2003!
We report a combined experimental and theoretical study of the unoccupied electronic states of the neutralmolecular organic materials TTF~tetrathiafulvalene! and TCNQ~7,7,8,8-tetracyano-p-quinodimethane! and ofthe one-dimensional metallic charge transfer salt TTF-TCNQ. The experimental density of states~DOS! isobtained by x-ray absorption near edge spectroscopy~XANES! with synchrotron light and the predicted DOSby means of first-principles density functional theory calculations. Most of the experimentally derived element-specific XANES features can be associated to molecular orbitals of defined symmetry. Because of the planargeometry of the TTF and TCNQ molecules and the polarization of the synchrotron light, the energy dependents or p character of the orbitals can be inferred from angular dependent XANES measurements. The presentwork represents the state of the art analysis of the XANES spectra of this type of materials and points out theneed for additional work in order to elucidate the governing selection rules in the excitation process.
DOI: 10.1103/PhysRevB.68.195115 PACS number~s!: 71.20.Rv, 71.15.Mb, 61.10.Ht
eno-
-in
-ly
taicQ
-ouoola-eT.io
icecrergtio
den-enpy
eryin-ho-
e
I. INTRODUCTION
The successful marriage between the tetrathiafulval~TTF, C6S4H4) donor and the 7,7,8,8-tetracyanp-quinodimethane~TCNQ, C12N4H4) acceptor, which led tothe first truly molecular metal,1 opened a vast area of research which thirty years later remains as lively and excitas ever.2 TTF-TCNQ exhibits a monoclinic crystal structure3
built up from parallel segregated stacks of donors~TTF! andacceptors~TCNQ! ~see Fig. 1!. Due to thep-p interactionsalong the stacks (b direction! and the weak interactions between molecular chains, the electrical conductivity is highanisotropic with typical values ofsb;600–900V21 cm21
and sb /sa;103 at room temperature, wheresa and sb
stand for the conductivity along thea and b axes, respec-tively, increasing to a maximum value close to 63103 near60 K.4 TTF-TCNQ is thus a quasi-one-dimensional meand exhibits a series of transitions at 54, 49, and 38 K, whsuccessively destroy the metallic conductivity of the TCNand TTF chains.5 The discovery of TTF-TCNQ and its remarkable low-temperature behavior triggered an enorminterest on the physics of molecular conductors which sled to the discovery of superconductivity in the molecusolid (TMTSF)2PF6 ~TMTSF: tetramethyltetraselenenafulvalene!.6 Since then molecular conductors have provida seemingly endless series of exciting discoveries and TTCNQ has remained as one of the emblems of the fieldfact, it is one of the materials best suited for the exploratof the physics of one-dimensional~1D! materials.5
Despite the continued attention devoted to the physcharacterization of TTF-TCNQ, some aspects of its eltronic structure still remain ambiguous. Although somecent works on the experimental determination of the enedispersion of the occupied states by means of high resoluangle resolved photoemission spectroscopy~ARPES! have
0163-1829/2003/68~19!/195115~11!/$20.00 68 1951
e
g
lh
snr
dF-Inn
al--yn
been published,7–9 little is known concerning the unoccupielevels region of the band structure. Information on the dsity of unoccupied electronic states of TTF-TCNQ has beobtained with inelastic electron scattering spectrosco~IESS! in transmission mode with 300 keV electrons.10 Thesurfaces of molecular organic materials are in general vsensitive to irradiation, so that impinging electrons mayduce irreversible damage. Results derived from inverse ptoemission spectroscopy~IPS! might be also questionabl
FIG. 1. Crystal structure of TTF-TCNQ:~a! view along thebaxis and~b! view along the direction perpendicular to thebc plane.
©2003 The American Physical Society15-1

tiosspeis
annc
ingcac
ar
-otha
soedorni
ter-
re
c-
(
bniobinsico
rer-
g-o
roa
sly
eri-n
in
ex-l ofsre-icrheex-f,
f 5he
aro-ki-d iny.
F-0,fQ,ap-n-
heionstheect
-
hesfac-
veau-el.to
onsce
J. FRAXEDASet al. PHYSICAL REVIEW B 68, 195115 ~2003!
unless low energy electrons~around 10–20 eV! and lowbeam currents are used as in low energy electron diffrac~LEED! experiments.11 Since molecular materials are lesensitive to x-ray photons, x-ray absorption near edge stroscopy~XANES! represents an interesting alternative. Thtechnique is based on the induction of intra-atomic interbtransitions from core levels to empty states caused by ident x rays of sufficient energy.12 Therefore, the partial~ele-ment selective! density of unoccupied states correspondto the electronic excited state with one core hole, whichbe used in a first approximation as the image of the unocpied states in the electronic ground state, is obtained.13 Aclosely related technique, which would give complementinformation, is angle resolved constant initial state~ARCIS!spectroscopy.14 Here we report a XANES study of the density of unoccupied states of TTF-TCNQ salt as well asneutral TTF and TCNQ. These results are analyzed onbasis of a comparison with first-principles density functiontheory~DFT! calculations for the three materials, which alallow us to discuss in detail the nature of the partially fillbands of TTF-TCNQ. Thus, we believe that the present wprovides the most complete description of the electrostructure of these materials reported so far.
II. EXPERIMENTAL
Thin TTF-TCNQ films~thickness;1 mm) have been ob-tained by thermal sublimation in high vacuum(;1026 mbar) of recrystallized TTF-TCNQ powder.Ex situcleaved KCl~001! surfaces were used as substrates, heldroom temperature during deposition. The films are orienwith their molecularab planes parallel to the substrate suface and consist of highly oriented and strongly texturectangular-shaped microcrystals, with theira and b axesparallel to both@110# and@ 110# substrate directions, respetively, due to the cubic symmetry of the substrate.15 Themean length of the microcrystals along their long axisbdirection! is around 1.5mm. Thin films of neutral TTF andTCNQ were prepared from their microcrystalline powderssuspension in ethanol and by stirring and deposition osilicon wafer immersed in the suspension. The informaton the spatial distribution of empty states is obtainedperforming XANES measurements at different angles ofcidence of the x-ray light, taking advantage of the intrinlinear polarization of synchrotron radiation. The examplegraphite is illuminating, since from initials statess ~i.e.,symmetric with respect to the molecular plane! or p ~i.e.,antisymmetric with respect to the molecular plane! finalstates are selected when the light polarization vectorE liesparallel or perpendicular to the basal plane, respectively.16 Ingeneral, intramolecular transitions, i.e., 1s→p* and 1s→s* , in molecules containing C, N, and O atoms astrongly polarized.17 The XANES measurements were peformed at the SACEMOR endstation~beamline SA72! of theLaboratoire pour l’ Utilisation du Rayonnement Electromanetique~LURE! in Orsay, France and at the beamline 8.2the Stanford Synchrotron Radiation Laboratory~SSRL! inStanford, CA. The data were acquired in the total electyield mode by measuring with an ammeter the current dr
19511
n
c-
di-
nu-
y
fel
kc
atd
d
yany-
f
f
nin
to ground. The signal was normalized to the simultaneourecorded photocurrent from a gold-covered grid.
III. COMPUTATIONAL DETAILS
The present calculations were carried out using a numcal atomic orbitals DFT~Ref. 18! approach, which has beerecently developed and designed for efficient calculationslarge systems and implemented in theSIESTA code.19 Wehave used the generalized gradient approximation to thechange correlation energy and, in particular, the functionaPerdew, Burke, and Ernzerhof.20 Only the valence electronare considered in the calculation, with the core beingplaced by norm-conserving scalar relativistpseudopotentials21 factorized in the Kleinman-Bylandeform.22 For the nonlinear core-valence interaction, tpartial-core correction scheme was used for all elementscept for hydrogen.23 We have used the following type obasis set: triple-z for the s orbitals of S, N, and C atomstriple-z plus single polarization functions for thes orbital ofH, double-z plus single polarization functions for thep or-bitals of S, N, and C, as obtained with an energy shift omeV.24 The integrals of the self-consistent terms of tKohn-Sham Hamiltonian are obtained with the help ofregular real space grid in which the electron density is pjected. The grid spacing is determined by the maximumnetic energy of the plane waves that can be representethat grid. In the present work, we used a cutoff of 250 RThe Brillouin zone~BZ! was sampled using grids of 33932, 43732, and 23331 k points25 for the determinationof density matrices and partial densities of states of TTTCNQ, TTF, and TCNQ, respectively. Sets of 23 0024 000, and 10 000k points were used for determination othe total densities of states of TTF-TCNQ, TTF, and TCNrespectively. The above-mentioned methods have beenplied to the electronic structure study of other organic coductors, and proved to give highly accurate results.26
IV. RESULTS AND DISCUSSION
A. Theoretical band structure close to the Fermi level
Before looking at the unoccupied levels region of tband structure let us consider how the present calculatdescribe the region around the Fermi level. Because ofmolecular nature and the relatively large unit cell, the corrdescription of the electronic structure of TTF-TCNQ~asmost of the molecular conductors! is still a challenge fromthe computational viewpoint. Initial first-principles calculations by Starikov27 as well as Kasowskiet al.28 are in con-tradiction with some of the symmetry requirements of tcrystal structure and thus cannot be considered as satitory. Very recently, Ishibashiet al.29 as well as Claessenet al.8,30 have reported band calculations by plane-wapseudopotential methods for TTF-TCNQ, although thesethors only consider in detail the region near the Fermi levBefore studying the XANES spectra it is thus interestingconsider how the present atomic orbitals band calculaticompare with them for this region as well as its significanin discussing very recent photoemission studies.
5-2
th
.n
dstrype
dQe
iptreform
.6Ont
wsioO0.o-a
thteont i
xi-bonter
italheforthee
l to
cal-
iceer-
lec-hus,
turemsthe
s-
ut aomveryar-a-
in-aintlyer
er
rgeat
m
t
to
CHARACTERIZATION OF THE UNOCCUPIED AND . . . PHYSICAL REVIEW B68, 195115 ~2003!
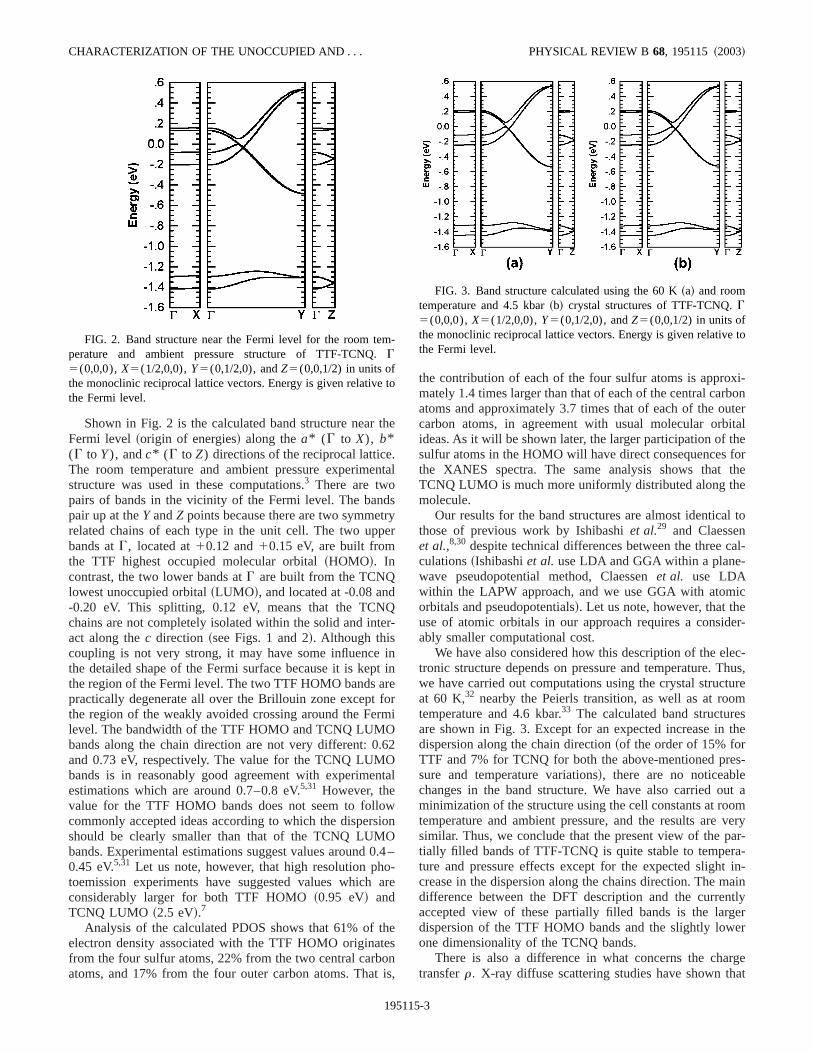
Shown in Fig. 2 is the calculated band structure nearFermi level~origin of energies! along thea* (G to X), b*(G to Y), andc* (G to Z) directions of the reciprocal latticeThe room temperature and ambient pressure experimestructure was used in these computations.3 There are twopairs of bands in the vicinity of the Fermi level. The banpair up at theY andZ points because there are two symmerelated chains of each type in the unit cell. The two upbands atG, located at10.12 and10.15 eV, are built fromthe TTF highest occupied molecular orbital~HOMO!. Incontrast, the two lower bands atG are built from the TCNQlowest unoccupied orbital~LUMO!, and located at -0.08 an-0.20 eV. This splitting, 0.12 eV, means that the TCNchains are not completely isolated within the solid and intact along thec direction ~see Figs. 1 and 2!. Although thiscoupling is not very strong, it may have some influencethe detailed shape of the Fermi surface because it is kethe region of the Fermi level. The two TTF HOMO bands apractically degenerate all over the Brillouin zone exceptthe region of the weakly avoided crossing around the Felevel. The bandwidth of the TTF HOMO and TCNQ LUMObands along the chain direction are not very different: 0and 0.73 eV, respectively. The value for the TCNQ LUMbands is in reasonably good agreement with experimeestimations which are around 0.7–0.8 eV.5,31 However, thevalue for the TTF HOMO bands does not seem to follocommonly accepted ideas according to which the dispershould be clearly smaller than that of the TCNQ LUMbands. Experimental estimations suggest values around0.45 eV.5,31 Let us note, however, that high resolution phtoemission experiments have suggested values whichconsiderably larger for both TTF HOMO~0.95 eV! andTCNQ LUMO ~2.5 eV!.7
Analysis of the calculated PDOS shows that 61% ofelectron density associated with the TTF HOMO originafrom the four sulfur atoms, 22% from the two central carbatoms, and 17% from the four outer carbon atoms. Tha
FIG. 2. Band structure near the Fermi level for the room teperature and ambient pressure structure of TTF-TCNQ.G5(0,0,0),X5(1/2,0,0),Y5(0,1/2,0), andZ5(0,0,1/2) in units ofthe monoclinic reciprocal lattice vectors. Energy is given relativethe Fermi level.
19511
e
tal
r
r-
nin
ri
2
al
n
4–
re
es
s,
the contribution of each of the four sulfur atoms is appromately 1.4 times larger than that of each of the central caratoms and approximately 3.7 times that of each of the oucarbon atoms, in agreement with usual molecular orbideas. As it will be shown later, the larger participation of tsulfur atoms in the HOMO will have direct consequencesthe XANES spectra. The same analysis shows thatTCNQ LUMO is much more uniformly distributed along thmolecule.
Our results for the band structures are almost identicathose of previous work by Ishibashiet al.29 and Claessenet al.,8,30 despite technical differences between the threeculations~Ishibashiet al. use LDA and GGA within a plane-wave pseudopotential method, Claessenet al. use LDAwithin the LAPW approach, and we use GGA with atomorbitals and pseudopotentials!. Let us note, however, that thuse of atomic orbitals in our approach requires a considably smaller computational cost.
We have also considered how this description of the etronic structure depends on pressure and temperature. Twe have carried out computations using the crystal strucat 60 K,32 nearby the Peierls transition, as well as at rootemperature and 4.6 kbar.33 The calculated band structureare shown in Fig. 3. Except for an expected increase indispersion along the chain direction~of the order of 15% forTTF and 7% for TCNQ for both the above-mentioned presure and temperature variations!, there are no noticeablechanges in the band structure. We have also carried ominimization of the structure using the cell constants at rotemperature and ambient pressure, and the results aresimilar. Thus, we conclude that the present view of the ptially filled bands of TTF-TCNQ is quite stable to temperture and pressure effects except for the expected slightcrease in the dispersion along the chains direction. The mdifference between the DFT description and the currenaccepted view of these partially filled bands is the largdispersion of the TTF HOMO bands and the slightly lowone dimensionality of the TCNQ bands.
There is also a difference in what concerns the chatransferr. X-ray diffuse scattering studies have shown th
-
o
FIG. 3. Band structure calculated using the 60 K~a! and roomtemperature and 4.5 kbar~b! crystal structures of TTF-TCNQ.G5(0,0,0),X5(1/2,0,0),Y5(0,1/2,0), andZ5(0,0,1/2) in units ofthe monoclinic reciprocal lattice vectors. Energy is given relativethe Fermi level.
5-3

9
eue
r-hadssll
TF5Q
uin-erh
TTegoisa
duom
eeinm
eu-eth
unlly
ig-
ngsteerFe
hend.not
heeNQ
altlev-alt
aresalt
-theeof
ex-
inndtraally
lcu-thefor
-.
lS
ofO
J. FRAXEDASet al. PHYSICAL REVIEW B 68, 195115 ~2003!
r50.55 electrons/molecule at room temperature and 0.5low temperature near the Peierls transition.34 In contrast, thecalculatedr at room temperature is 0.74 and practically donot change with pressure or temperature. Similar val~0.70–0.72! were reported by Ishibashiet al.29 It is not clearif this overestimation ofr and the larger calculated dispesion for the TTF HOMO bands are related. Let us note t4kf (kf is the Fermi wave vector! fluctuations associatewith the TTF chains have been observed in x-ray diffuscattering experiments34 suggesting that electron correlationare important for TTF but less for TCNQ. Thus it may webe that both LDA and GGA approaches to DFT lead toslightly better description of the TCNQ chains than the Tones. However, let us note that just a shift of the order ofmeV of the TTF HOMO bands with respect to the TCNLUMO ones would lead to the correctr so that for the cur-rent purposes the disagreement is not really significant.
Finally, we also considered the possible effect of the sface structural relaxation on the band structure by carryout a model calculation for theab-plane relaxation. A geometry optimization of the outer layer while keeping the undlying ones fixed at the bulk geometry was carried out. Tgeometrical changes are not large and affect more thethan the TCNQ. They both change their inclination with rspect to theab plane but none of the two molecules underany rotational motion along the long molecular axChanges in the band dispersion along the chain directionsmall, of the same order but slightly smaller than thoseto thermal contraction or pressure reported above. The cponent of the Fermi wave vector alongb* practically doesnot change. Consequently, at least in what concerns thabplane, no noticeable changes inr are expected at the surfacdue to the structural relaxation, in line with recent scanntunneling microscopy results performed at cryogenic teperatures under ultrahigh vacuum.35
B. Electronic structure of unoccupied states
The crystal structure of neutral TTF (a57.36 Å, b54.023 Å, c513.922 Å,b5101.42°)~Ref. 36! and TCNQ(a58.906 Å, b57.060 Å, c516.395 Å, b598.54°) ~Ref.37! are shown in Figs. 4~a! and 4~b! and Figs. 5~a! and 5~b!,respectively. Note that there are two molecules per repunit of the crystal structure for neutral TTF but four for netral TCNQ. TTF contains one-dimensional stacks along thbdirection where every successive molecule slides alonglong molecular axis. These stacks are similar to those foin TTF-TCNQ. In contrast, neutral TCNQ does not reacontain one-dimensional stacks as in TTF-TCNQ~see Fig. 1!but two-dimensional ones in theab planes@see Figs. 5~a! and5~b!#. The calculated band structures are reported in F4~c! and 5~c! for TTF and TCNQ, respectively. The calculated band structure for TTF-TCNQ in the same energy rais shown in Fig. 6~a!. Note that the two TTF HOMO bandas well as the four TCNQ LUMO bands are well separafrom the bands above and this leads to a well defined engap between the partially filled and empty bands of the TTTCNQ. Whereas the TTF HOMO bands are practically d
19511
at
ss
t
e
a
0
r-g
-eF
-
.ree-
g-
at
ed
s.
e
dgy--
generate in TTF-TCNQ, they are not in neutral TTF, i.e., tinterchain interactions are larger in the neutral compouThe dispersion of these bands along the chain direction isvery different in TTF and TTF-TCNQ.
In contrast, the structural origin of the spreading of tTCNQ LUMO levels is very different in the neutral and thsalt compounds. Consequently, the appearance of the TCLUMO bands is very different in the neutral and the scompounds. However, note that the total spread of theseels in the neutral is only one third smaller than in the sand, as far as the gross features of the density of statesconcerned, the differences between the neutral and theare not very significant~see below!. Similar observations apply for the upper bands and make directly comparableXANES results for TTF-TCNQ and those for TCNQ. Thcalculated total DOS and PDOS for the relevant atomsTTF-TCNQ, TTF, and TCNQ are reported in Figs. 6~b!, 4~d!,and 5~d!, respectively, and compared to the appropriateperimental XANES data. The S2p, C1s, and N1s XANESspectra for TTF-TCNQ, TTF, and TCNQ are also shownFig. 7 where the relevant molecular orbitals of TTF aTCNQ associated with the different features of the specare indicated. These molecular orbitals are schematicshown in Figs. 8~TTF! and 9 ~TCNQ!. The energy valuesassociated with the orbitals of Figs. 8 and 9 are those calated for a single molecule with the same geometry as forneutral solid. The symmetry labels are those appropriate
FIG. 4. View along theb axis ~a! and along the direction perpendicular to thebc plane~b! of the crystal structure of neutral TTF~c! Band structure of TTF whereG5(0,0,0), X5(1/2,0,0), Y5(0,1/2,0), andZ5(0,0,1/2) in units of the monoclinic reciprocalattice vectors.~d! Total DOS calculated for neutral TTF and PDOfor the sulfur and carbon atoms~black lines!. The S2p and C1sXANES spectra of TTF~gray lines! are superposed to the PDOSS and C, respectively. The energy is given relative to the HOMmaximum.
5-4

ra
h
heo-
tri-
red
gedthem-trauree inThein-
re
lS
iven
-l
l
ive
CHARACTERIZATION OF THE UNOCCUPIED AND . . . PHYSICAL REVIEW B68, 195115 ~2003!
idealizedD2h symmetry. For simplicity, the sulfurd orbitalcontributions have not been shown in these schematic dings.
1. TTF-derived spectral features
Let us start considering neutral and charged TTF. Texperimental spectra for S2p corresponding to neutral TTF
FIG. 5. View along theb axis ~a! and along the direction perpendicular to thebc plane ~b! of the crystal structure of neutraTCNQ. ~c! Band structure of TCNQ whereG5(0,0,0), X5(1/2,0,0), Y5(0,1/2,0), andZ5(0,0,1/2) in units of the mono-clinic reciprocal lattice vectors.~d! Total DOS calculated for neutraTCNQ and PDOS for the nitrogen and carbon atoms~black lines!.The N1s and C1s XANES spectra of TCNQ~gray lines! are super-posed to the PDOS of N and C, respectively. The energy is grelative to the LUMO minimum.
19511
w-
e
is not quite structured whereas that for charged TTF is. TS2p lines are composed of two spin-orbit splitted compnents 2p3/2 and 2p1/2 separated by 1.1 eV,38 so that the S2pXANES spectra consists of the superposition of both conbutions. The opposite trend is observed for N1s in TCNQ,with narrower spectral lines in the neutral state as compato the TTF-TCNQ salt. On the other hand C1s XANES spec-tra show well-defined features for both neutral and charTTF and TCNQ. The explanation indeed stems fromcharge transferred upon formation of the TTF-TCNQ copound from the donor TTF to the acceptor TCNQ. The excharge that is allocated within the TCNQ electronic structbroadens its electronic levels, whereas the loss of chargTTF narrows the line shape of the corresponding states.narrowing of the TTF spectral width corresponds to an
FIG. 6. ~a! Band structure calculated for the room temperatustructure of TTF-TCNQ whereG5(0,0,0), X5(1/2,0,0), Y5(0,1/2,0), andZ5(0,0,1/2) in units of the monoclinic reciprocalattice vectors.~b! Total DOS calculated for TTF-TCNQ and PDOfor the sulfur, carbon, and nitrogen atoms~black lines!. The S2p,N1s and C1s XANES spectra of TTF-TCNQ~gray lines! are su-perposed to the PDOS of S, N, and C, respectively. Energy is grelative to the Fermi level.
n
Q
FIG. 7. S2p, C1s, and N1s XANES spectrafor the charge transfer compound TTF-TCNand for the neutral TTF and TCNQ.
5-5

othly
too
.cargth
r.
th
t
TF.S
r
eetys
C-
-
wothelderS.
ala-
e
er
ite
eso
cu-of
iate
cuo
ia
J. FRAXEDASet al. PHYSICAL REVIEW B 68, 195115 ~2003!
crease of the core-hole lifetime in the excited state thatcurs when charge is transferred to form the salt. Sincecharge is transferred from the TTF HOMO, which mainderives from the sulfur atoms as mentioned before@see Fig.4~d!#, the line narrowing is specially important in the S2pXANES. This is thus a rather direct but qualitative methodexperimentally determine the influence of charge transferthe unoccupied electronic states close to the Fermi level
The calculated DOS and the C and S PDOS of TTFbe quite simply understood. Let us note that for the eneregion of interest here, i.e., up to approximately 5–6 eV,C PDOS is clearly dominated by thep-type contributionswith the s- and d-type contributions being much smalleHowever, for the S PDOS the weight of thed-type contribu-tions become considerably larger. In fact, thed contributionsextend over all the energy range of interest so that even ifp contributions dominate, there is always somed contribu-tion of the appropriate symmetry and the levels are goingbe visible in some degree in the S2p spectra.
The first unoccupied peak in the total DOS of neutral T@see Fig. 4~d!# lies around 2 eV of the TTF HOMO peakAnalysis of the PDOS shows that it is about two thirdsbased and one third C based@note the different scales foeach PDOS in Fig. 4~d!#. This peak originates from theagorbital ~see Fig. 8!, a s-type orbital, which is essentiallyantibonding for all C-S bonds and to a lesser extent betwall C-H bonds. The reason why it is the lowest lying emporbital is that it mixes in C-C bonding contributions thumaking the level less antibonding. The main character isantibonding and this orbital will be labeled ass* (ag) and isnoted in Fig. 7~a! for charged TTF, where it is clearly observed. This feature is not observed in the C1s XANES
FIG. 8. Schematic representation of the relevant TTF unocpied molecular orbitals for the analysis of the XANES spectraTTF and TTF-TCNQ. The symmetry labels are those approprfor theD2h symmetry. The energy values given~in eV! are relativeto those of the HOMO of TTF in the isolated TTF molecule.
19511
c-e
n
nye
e
o
n
S
spectrum of TTF@dashed line in Fig. 7~b!# as if transitionsfrom an initials-state to a finals* (ag)-state were forbidden~see more about this later!.
The next peak in the DOS of TTF@see Fig. 4~d!#, which isthe most prominent one, is really the superposition of tcontributions, a larger one at around 2.5 eV aboveHOMO maximum and a smaller one, appearing as a shouslightly below. This is also the case for the C and S PDOThe two contributions originate fromp-type orbitals of TTF.The lower contribution originates from theb3g orbital lyingat 2.58 eV from the TTF HOMO~see Fig. 8!. The large peakslightly above originates from the pair ofau andb2g orbitalsin Fig. 8, which in the molecule are very near in energy~i.e.,at 2.94 and 3.07 eV from the HOMO! and thus appear assingle peak in the DOS. The associated feature will bebeled asp* (b3g ,au ,b2g), and it can be observed in thXANES curves for both S2p ~since it has a significantdcharacter in the S atoms! and C1s (s→p* transitions in Care allowed! spectra.
The next three peaks in the DOS@see Fig. 4~d!# ~seen astwo peaks and a shoulder! are due tos-type orbitals. Thefirst one is due to the lowerb2u orbital of Fig. 8, which liesat 3.26 eV from the HOMO in the molecule and is anothantibonding C-S level which will be labeleds* (b2u). Thetwo next peaks originate from two orbitals which are qusimilar in energy in the molecule: theb3u and upperb2uorbitals of Fig. 8, which lie at 3.61 and 3.86 eV from thHOMO in the molecule. These two orbitals are al
FIG. 9. Schematic representation of the relevant TCNQ unocpied molecular orbitals for the analysis of the XANES spectraTCNQ and TTF-TCNQ. The symmetry labels are those approprfor D2h symmetry. The energy values given~in eV! are relative tothose of the LUMO of TCNQ in the isolated TCNQ molecule.
-fte
5-6
ra
ene
CHARACTERIZATION OF THE UNOCCUPIED AND . . . PHYSICAL REVIEW B68, 195115 ~2003!
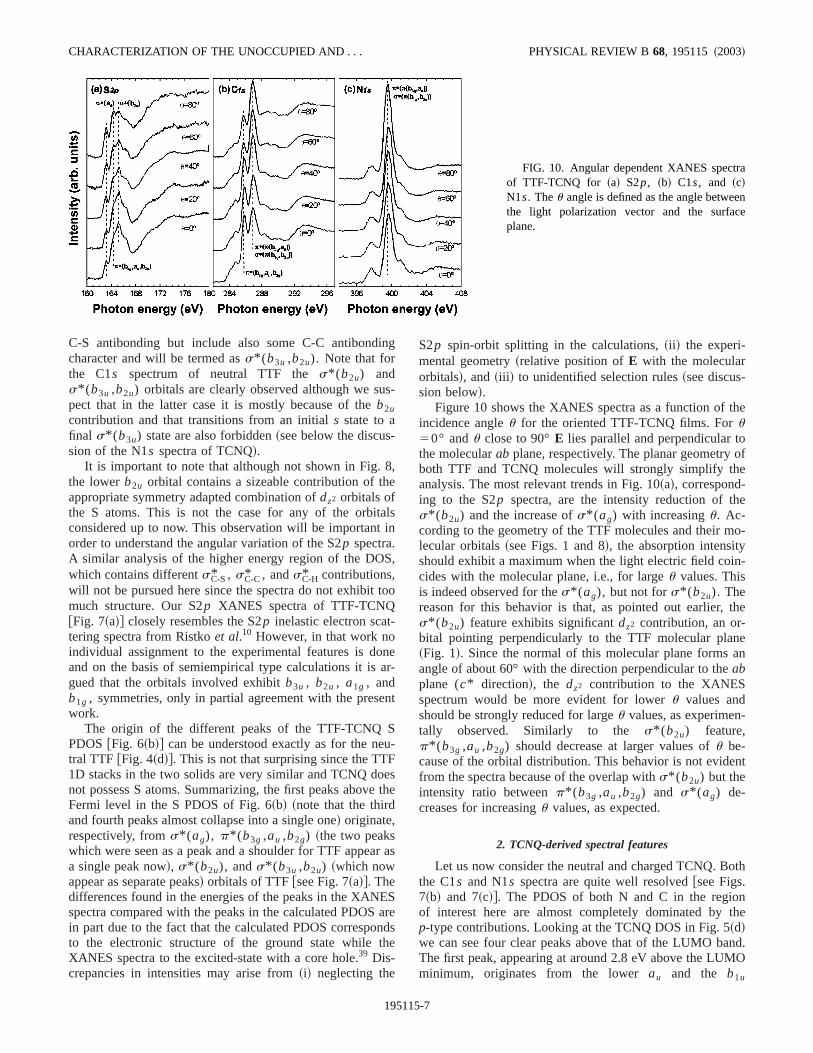
FIG. 10. Angular dependent XANES spectof TTF-TCNQ for ~a! S2p, ~b! C1s, and ~c!N1s. Theu angle is defined as the angle betwethe light polarization vector and the surfacplane.
in
s
-
8e
ain
S
to
-
ona
en
SuFe
e
ar
Ea
nth
the
oofe
e
o-
n-
the
ean
-
nt
oth
nthe
nd.O
C-S antibonding but include also some C-C antibondcharacter and will be termed ass* (b3u ,b2u). Note that forthe C1s spectrum of neutral TTF thes* (b2u) ands* (b3u ,b2u) orbitals are clearly observed although we supect that in the latter case it is mostly because of theb2ucontribution and that transitions from an initials state to afinal s* (b3u) state are also forbidden~see below the discussion of the N1s spectra of TCNQ!.
It is important to note that although not shown in Fig.the lowerb2u orbital contains a sizeable contribution of thappropriate symmetry adapted combination ofdz2 orbitals ofthe S atoms. This is not the case for any of the orbitconsidered up to now. This observation will be importantorder to understand the angular variation of the S2p spectra.A similar analysis of the higher energy region of the DOwhich contains differentsC-S* , sC-C* , andsC-H* contributions,will not be pursued here since the spectra do not exhibitmuch structure. Our S2p XANES spectra of TTF-TCNQ@Fig. 7~a!# closely resembles the S2p inelastic electron scattering spectra from Ristkoet al.10 However, in that work noindividual assignment to the experimental features is dand on the basis of semiempirical type calculations it isgued that the orbitals involved exhibitb3u , b2u , a1g , andb1g , symmetries, only in partial agreement with the preswork.
The origin of the different peaks of the TTF-TCNQPDOS@Fig. 6~b!# can be understood exactly as for the netral TTF @Fig. 4~d!#. This is not that surprising since the TT1D stacks in the two solids are very similar and TCNQ donot possess S atoms. Summarizing, the first peaks abovFermi level in the S PDOS of Fig. 6~b! ~note that the thirdand fourth peaks almost collapse into a single one! originate,respectively, froms* (ag), p* (b3g ,au ,b2g) ~the two peakswhich were seen as a peak and a shoulder for TTF appea single peak now!, s* (b2u), ands* (b3u ,b2u) ~which nowappear as separate peaks! orbitals of TTF@see Fig. 7~a!#. Thedifferences found in the energies of the peaks in the XANspectra compared with the peaks in the calculated PDOSin part due to the fact that the calculated PDOS correspoto the electronic structure of the ground state whileXANES spectra to the excited-state with a core hole.39 Dis-crepancies in intensities may arise from~i! neglecting the
19511
g
-
,
ls
,
o
er-
t
-
sthe
as
Sre
dse
S2p spin-orbit splitting in the calculations,~ii ! the experi-mental geometry~relative position ofE with the molecularorbitals!, and~iii ! to unidentified selection rules~see discus-sion below!.
Figure 10 shows the XANES spectra as a function ofincidence angleu for the oriented TTF-TCNQ films. Foru50° andu close to 90°E lies parallel and perpendicular tthe molecularab plane, respectively. The planar geometryboth TTF and TCNQ molecules will strongly simplify thanalysis. The most relevant trends in Fig. 10~a!, correspond-ing to the S2p spectra, are the intensity reduction of ths* (b2u) and the increase ofs* (ag) with increasingu. Ac-cording to the geometry of the TTF molecules and their mlecular orbitals~see Figs. 1 and 8!, the absorption intensityshould exhibit a maximum when the light electric field coicides with the molecular plane, i.e., for largeu values. Thisis indeed observed for thes* (ag), but not fors* (b2u). Thereason for this behavior is that, as pointed out earlier,s* (b2u) feature exhibits significantdz2 contribution, an or-bital pointing perpendicularly to the TTF molecular plan~Fig. 1!. Since the normal of this molecular plane formsangle of about 60° with the direction perpendicular to theabplane (c* direction!, the dz2 contribution to the XANESspectrum would be more evident for loweru values andshould be strongly reduced for largeu values, as experimentally observed. Similarly to the s* (b2u) feature,p* (b3g ,au ,b2g) should decrease at larger values ofu be-cause of the orbital distribution. This behavior is not evidefrom the spectra because of the overlap withs* (b2u) but theintensity ratio betweenp* (b3g ,au ,b2g) and s* (ag) de-creases for increasingu values, as expected.
2. TCNQ-derived spectral features
Let us now consider the neutral and charged TCNQ. Bthe C1s and N1s spectra are quite well resolved@see Figs.7~b! and 7~c!#. The PDOS of both N and C in the regioof interest here are almost completely dominated byp-type contributions. Looking at the TCNQ DOS in Fig. 5~d!we can see four clear peaks above that of the LUMO baThe first peak, appearing at around 2.8 eV above the LUMminimum, originates from the lowerau and the b1u
5-7

ee
-thh
d
d
in
fomet
he
to
nbit
ap
o
try
djn
dle
and
apa
at
ha
th
zene
Nthe
tra,
-u-dbutatin
theenhe
f
eakhn
lar
ofste
ular
-
he
fr
2
on
J. FRAXEDASet al. PHYSICAL REVIEW B 68, 195115 ~2003!
orbitals of TCNQ~see Fig. 9!, which for the isolated mol-ecule lie at 2.55 and 2.65 eV from the LUMO. Thesep-typeorbitals p* (au ,b1u) are quite strongly concentrated on thbenzenic region~small contributions are not shown in thschematic representations of Fig. 9!. These orbitals originatefrom the degenerate pair of lowest emptyp orbitals (e2u) ofbenzene,40 which only slightly delocalize towards the substituent in TCNQ. The next two peaks are associated toCN groups. The first one contains the contribution of tbands based on theag and b3u orbitals of TCNQ lying at3.51 and 3.63 eV from the LUMO, and will be labeles* @p(ag ,b3u)# ~see the next paragraph for notation!, whilethe second peak contains the contribution of bands basefour orbitals of TCNQ lying at 4.55 (b3g) and 4.66 eV (au),labeled asp* @p(b3g ,au)#, and 4.77 (b1g) and 4.86 eV(b2u), labeled ass* @p(b1g ,b2u)#, from the LUMO ~seeFig. 9!. Essentially, thep* (ag ,b3u) andp* (b1g ,b2u) orbit-als are the four symmetry adapted combinations of theplanep* orbitals of the CN groups.
It is important to distinguish between the twop* or-thogonal orbitals of the CN group. They are degenerateCN itself because of the cylindrical symmetry but beconondegenerate in TCNQ. Because of the symmetry planthe molecule, the two formally degenerate orbitals leadone in-planep* orbital, denoted ass* (p) because it is anantibonding orbital which is symmetrical with respect to tmolecular plane even if locally it is ap-type orbital, and oneout-of-planep* orbital, denoted asp* (p) because it is anantibonding orbital which is antisymmetrical with respectthe molecular plane and is locally ap-type orbital. The fours* (p) orbitals lead to four symmetry adapted combinatiowhich are the main components of the four molecular orals. Two of them are lower in energys* @p(ag ,b3u)#, be-cause they are symmetrical with respect to theC2 axis alongthe long molecular axis and they exhibit positive overlbetween the C components of the two adjacents* (p) orbit-als. This lowers considerably the energy. The other twobitals are based on theb1g andb2u orbitalss* @p(b1g ,b2u)#~see Fig. 9!. These orbitals are the two remaining symmeadapted combinations of thes* (p) orbitals. By havingnegative overlap between the C component of the two acent s* (p) orbitals they lie now around 1 eV higher ienergy than theirag and b3u counterparts~see Fig. 9!. Thep* @p(b3g ,au)# orbitals are two of the symmetry adaptecombinations of thep* (p) orbitals. By being symmetricawith respect to theC2 axis along the long molecular axis, thp orbital of the central C atom of the C(CN)2 substituentcannot mix into these symmetry adapted combinationsthus thep* (p) orbitals practically do not delocalize towarthe benzene ring.
Before concluding let us note that the next peak, atproximately 5.6 eV from the LUMO minimum, arises fromp-type orbital delocalized all over the molecule but origining from the highestp-type orbital of benzene (b2g).40 Su-perposed to this contribution there is a second one, ofs typeand also localized on the benzene moiety, whose main cacter issC-H antibonding. However, since this orbital is ofagsymmetry, we suspect it does not lead to any feature in
19511
ee
on
-
reofo
s-
r-
a-
d
-
-
r-
e
experimental spectra~see below!. Thus, the first and forthpeaks are considerably more concentrated in the benring whereas all other peaks are strongly based onpCN* or-bitals. This is clear from the comparison of the C andPDOS. The higher energy region of the DOS containscontributions of several quite delocalizeds-type orbitalscontainingsC-C* , sC-H* , and sC-N* contributions, the lowestone of which leads to the next feature in the TCNQ specwhose analysis will not be pursued here.
The origin of the different peaks of the N PDOS of TTFTCNQ @Fig. 6~b!# can be understood exactly as for the netral TCNQ @Fig. 5~d!#. As noted in the discussion of the banstructure, there are really no structural reasons for this,the molecular nature of TCNQ and TTF-TCNQ makes ththe differences in the packing do not induce differencesthe band topology, which lead to noticeable changes inDOS. Again, small variations in energy differences betwesome of the peaks in TCNQ and TTF-TCNQ arise from tdifferential effect induced by the charge transfer~and to aminor degree from the structural differences!. The origin ofthe peaks for the C1s and N1s spectra of TCNQ is shown inFigs. 7~b! and 7~c!, respectively. Note that the contribution os* @p(ag ,b3u)#, which should appear@see the dotted line inFig. 7~c!# in between those of p* (au ,b1u) andp* @p(b3g ,au)#1s* @p(b1g ,b2u)#, both clearly visible inthe N1s spectra, does not appear at all. That, the larger pin the N1s spectra is partially associated wits* @p(b1g ,b2u)# is clear not only from the energy separatiofrom the first peak but, more importantly, from the angudependence of the peaks, as discussed next.
Figures 10~b! and 10~c! show the angular dependencethe C1s and N1s XANES spectra, respectively. The mosalient feature of Fig. 10~c! is the intensity increase of thp* @p(b3g ,au)#1s* @p(b1g ,b2u)# peak for increasinguvalues. However, the benzenic-typep* (au ,b1u) andp* (b2g) peaks remain nearly unchanged. For TCNQ theC2molecular axis forms an angle of about 36° with thec*direction. Since CN bonds form an angle of 60° with thisC2molecular axis within the molecular plane@see Fig. 1~a!#, theintensity associated tos* @p(b3g ,au)# should increase forlargeru. However, the intensity associated top* (p) orbit-als, the benzeniclike orbitals andp* @p(b3g ,au)#, shouldexhibit the opposite behavior because they are perpendicto the s* (p) orbitals. From Fig. 10~c! it is clear that thep* @p(b3g ,au)#1s* @p(b1g ,b2u)# peak increases with regard to the benzenic-type orbitals for increasingu valuesbecause of the increasings* (p) and decreasingp* (p) con-tributions. The same arguments apply for tp* @p(b3g ,au)#1s* @p(b1g ,b2u)# C1s peak @Fig. 10~b!#,which indeed increases for increasingu angles. In the case oTTF p* (b3g ,au ,b2g) @Fig. 10~a!#, its intensity decreases foincreasingu angles, which is again ascribed to thep char-acter of the bonds as pointed out earlier@see discussionabove for S2p XANES spectra and note that in Fig. 10~b!the intensity decrease is more readily patent than in the Spcase of Fig. 10~a! because of reduced overlap#.
In view of these observations we are quite confidentthe appropriateness of the assignations in the N1s spectra of
5-8

CHARACTERIZATION OF THE UNOCCUPIED AND . . . PHYSICAL REVIEW B68, 195115 ~2003!
FIG. 11. C1s XANES spectra of TTF-TCNQtaken at incidence angles~a! u50°, ~b! u540°, and~c! u570°. Black and gray lines cor-respond to 300 and 10 K, respectively.
b
et
--
inninisthet
mo
icrin
d
ta
-
rv
sire
an
NQ
F-glenst
rys-
mi
nallict israinb-
ndy a
yattherar-the
eennetion
d-m-faceesebye
at
Fig. 7~c!. This leads to the intriguing observation of the asence of thes* @p(ag ,b3u)# contribution in the N1sXANES spectra of both neutral TCNQ and TTF-TCNQ. Lus now note the absence of precisely theag andb3u contri-butions in the C1s spectra of TTF discussed above. However, the C1s XANES spectrum of TCNQ shows some intensity in thes* @p(ag ,b3u)# region „betweenp* (au ,b1u)andp* @p(b3g ,au)#1s* @p(b1g ,b2u)#…. This is due to thesignificant d contribution from carbon for neutral TCNQwhile nitrogen contributes negligibly. Thus, it seems thataddition to the intraatomic selection rules there are additiorestrictions behind the possibility of observation of certafeatures of the DOS in molecular materials like those dcussed here. We have no appropriate explanation forinteresting observation which certainly calls for both theorical and experimental additional work~for instance the de-tailed comparison of the N1s XANES spectra of appropriatemono- and di-cyanosubstituted systems could bring solight into this problem!. Our molecular orbital assignation tthe N1s XANES spectra differs from that of Ritskoet al.10
mainly because of their lower experimental resolution, whresults in a single peak in the inelastic electron scattespectra, which they assign to haveb3u , a1g , anda1u sym-metry. Additionally, the origin of the two lower energy C1sXANES features~at around 284 eV! for both neutral TCNQand TTF-TCNQ is also uncertain. No peaks are associatethe predicted PDOS in this region@see Figs. 5~d! and 6~b!#and no instrumental artifacts are expected~i.e., higher-orderlight from the monochromator!. Contribution from interfacialstates@at the TTF-TCNQ / KCl~100! interface#, which can beclearly distinguished in XANES spectra,41 can be ruled outbecause of the large film thickness~around 1mm). Thepresence of both peaks might be related to the excited snature of the absorption process but we are unable tomore precise on this point.
Figure 11 shows the C1s XANES spectra taken at different incidence angles(0°, 40°, and 70°) andmeasured at 10and 300 K, respectively. No relevant changes are obseupon undergoing the Peierls transition (;55 K), except forlarge values ofu, evidencing that XANES mainly explorethe molecular contribution to the electronic structure. Thisin line with XANES experiments on perylene crystals, whethe spectra look very similar for botha andb polymorphs,which exhibit differentiated structures.41 The conduction bar-rier energy or activation energyD of the films is D.30 meV as determined from resistivity measurements
19511
-
al
-is-
e
hg
in
tebe
ed
s
d
the Peierls transition has been only observed for TTF-TCfilms with D,25 meV. The reduction ofD is intimatelyrelated to the reduction of the grain boundary density.15
C. Electronic structure of occupied states
The electronic structure of the occupied states of TTTCNQ has been experimentally determined for sincrystals7–9 and thin films prepared under the same conditioas those reported here42 by means of ARPES performed asample temperatures below 100 K. In the case of single ctals dispersion along theG-Y direction is clearly observedtogether with extremely low spectral intensity at the Ferlevel, a common property of quasi-1D materials.43 Due to thesurface stability of oriented thin TTF-TCNQ films in air,exsitu grown films do also exhibit band dispersion. In additioto the 60 meV pseudogap already observed in the metphase in the single crystal samples about 50 meV shifobserved due to contamination and nonstoichiometry at gboundaries. In thin films the band dispersion is also oserved, even with the overlap of botha and b directions inthe films ~see Sec. II!, because of the shape of the basurface in reciprocal space, which may be approximated bsheet in E vs G-X, G-Y space with ]E/]ka* 50 and]E/]kb* Þ0, whereE represents the band energy andka*andkb* stand for the wave vectors alonga* andb* direc-tions, respectively.44 The x-ray photoelectron spectroscop~XPS! analysis of single crystals and films reveals thcharged and neutral TCNQ may coexist at the surface, eidynamically, if the conversion period is larger than the chacteristic time of the photoexcitation process, or static, incase of incomplete charge transfer or contamination.9,42 Thecoexistence of bulk charged and neutral species has bconfirmed for the quasi-1D organic conductor diperylehexafluorophosphate as determined by high-resolunuclear magnetic resonance~NMR!.45 In the case of thinfilms neutral TCNQ is probably allocated at grain bounaries. Band dispersion is only observed at cryogenic teperatures because of the considerable vibration of the surmolecules in ultrahigh vacuum at room temperature. Thsurface thermal vibrations, caused by a small surface Detemperature, smaller than 160 K, the bulk value,46 induce areduction ofr. According to our band calculations@see Fig.5~c!# neutral TCNQ should contribute with energy statesabout 0.8 eV binding energy~the Fermi level is taken half-
5-9

on
cotoaje
oussri
ensbc
rucuivro
muearaQveti
ntTF,edof
larleteup
hatpe-
u-stillas--s
in-
rn
eirlloedn-m
J. FRAXEDASet al. PHYSICAL REVIEW B 68, 195115 ~2003!
way of the HOMO-LUMO gap!. A band at this energy isfound for both TTF-TCNQ single crystals and thin films.
The ARPES spectra may be thus divided in three regi~i! 0<E<0.5 eV, ~ii ! E;0.8 eV, and~iii ! E;1.5 eV. Re-gions ~i! and ~iii ! agree with most of band calculations8,29,30
including the present work and discrepancies arise whensidering region~ii !. If the 0.8 eV peak could be ascribedthe presence of neutral TCNQ on the surface, the only mcontribution from the one dimensionality would be thstrong reduction of electronic states at the Fermi level. Inopinion the separation of spin-charge suggested by Claeet al.8 cannot be concluded only from the ARPES expements.
V. CONCLUSIONS
The electronic structure of the unoccupied states of ntral TTF and TCNQ as well as of the quasi-1D charge trafer salt TTF-TCNQ has been experimentally determinedmeans of XANES and compared to DFT calculations. Sinthe intermolecular interactions are weak the electronic stture of molecular solids is mainly determined by the molelar character, so that the energy levels are essentially gby the molecular orbital energies. The use of synchrotradiation allows the determination of the PDOS becausethe element selectivity of x-ray absorption and the deternation of the spatial distribution of the molecular orbitals dto the polarization of the synchrotron light. State of theDFT computations have been used to calculate the bstructure, DOS, and PDOS of TTF, TCNQ, and TTF-TCNThe partially filled bands of the 1D metal TTF-TCNQ habeen characterized and then compared with recent theore
m.ol
ta
ys
d
S.
hy
m-.
19511
s
n-
or
ren
-
u--yec--ennofi-
tnd.
cal
studies. An in depth discussion of the origin of the differefeatures of the unoccupied DOS of the three materials, TTCNQ, and TTF-TCNQ, has been possible. The combinexperiment-theory study has permitted the assignationmost of the experimentally obtained features to molecuorbitals of specific energy and symmetry. Thus, a compdescription of the nature of the states from the Fermi levelto approximately 6 eV above is obtained. It is observed tthe contributions associated with molecular orbitals of scific symmetry (ag andb3u in D2h) are not observed in theXANES spectra, despite being clearly visible in the calclated DOS. This suggests that there are selection rulesnot well understood for materials structurally as complexTTF, TCNQ, and TTF-TCNQ. More work on carefully chosen molecular materials~i.e., based on molecules with symmetry lower thanD2h) is clearly needed in order to progresalong these lines.
ACKNOWLEDGMENTS
The work at HUT was supported by the Academy of Fland through the Center of Excellence Program~2002-2005!.Work at Bellaterra was supported by DGI-Spain~ProjectsNo. MAT1999-1128 and BFM2000-1312-C02-01!, Generali-tat de Catalunya~2001 SGR 33!, and by grants for computetime from the CESCA-CEPBA. Regarding synchrotrowork, we are indebted to P. Parent and C. Laffon for thhelp with the measurements at LURE and to L. J. Terminefor helping with the SSRL experiments. The SSRL is fundby the US-DOE, Office of Basic Energy Science. The sychrotron work at LURE was financed by the TMR Prograof the European Union.
P.llo,
. BD.
-
D.
1J. Ferraris, D.O. Cowan, V.V. Walatka, and J.H. Perlstein, J. AChem. Soc.95, 948 ~1973!; L.B. Coleman, M.J. Cohen, D.JSandman, F.G. Yamagishi, A.F. Garito, and A.J. Heeger, SState Commun.12, 1125~1973!.
2T. Ishiguro, K. Yamaji, and G. Saito,Organic Superconductors,2nd ed.~Springer-Verlag, Berlin, 1998!.
3T.J. Kistenmacher, T.E. Philips, and D.O. Cowan, Acta Cryslogr., Sect. B: Struct. Crystallogr. Cryst. Chem.30, 763 ~1974!.
4E.M. Conwell in Semiconductors and Semimetals, edited by E.Conwell ~Academic Press, New York, 1988!, Vol. 27, p. 215.
5D. Jerome and H. Schulz, Adv. Phys.31, 299 ~1982!.6D. Jerome, A. Mazaud, M. Ribault, and K. Bechgaard, J. Ph
~France! Lett. 41, L-95 ~1980!.7F. Zwick, D. Jerome, G. Margaritondo, M. Onellion, J. Voit, an
M. Grioni, Phys. Rev. Lett.81, 2974~1998!.8R. Claessen, M. Sing, U. Schwingenschlo¨gl, P. Blaha, M. Dressel,
and C.S. Jacobsen, Phys. Rev. Lett.88, 096402~2002!.9M. Sing, U. Schwingenschlo¨gl, R. Claessen, M. Dressel, and C.
Jacobsen, Phys. Rev. B67, 125402~2003!.10J.J. Ritsko, N.O. Lipari, P.C. Gibbons, and S.E. Schnatterly, P
Rev. Lett.37, 1068~1976!.11W. Gebauer, M. Bassler, R. Fink, M. Sokolowski, and E. U
bach, Chem. Phys. Lett.266, 177 ~1997!; J. Poppensieker, Ch
.
id
l-
.
s.
Rothig, and H. Fuchs, Adv. Funct. Mater.11, 188 ~2001!.12J. Stohr, NEXAFS Spectroscopy~Springer, Berlin, 1992!.13A. Curioni, W. Andreoni, R. Treusch, F.J. Himpsel, E. Haskal,
Seidler, C. Heske, S. Kakar, T. Van Buuren, and L.J. TermineAppl. Phys. Lett.72, 1575~1998!.
14A. Bianconi, S.B.M. Hagstrom, and R.Z. Bachrach, Phys. Rev16, 5543 ~1977!; J. Fraxedas, A. Stampfl, R.C.G. Leckey, J.Riley, and L. Ley, Phys. Rev. B42, 8966~1990!.
15J. Fraxedas, S. Molas, A. Figueras, I. Jime´nez, R. Gago, P. AubanSenzier, and M. Goffman, J. Solid State Chem.168, 384~2002!.
16R.A. Rosenberg, P.J. Love, and V. Rehn, Phys. Rev. B33, 4034~1986!.
17A.M. Bradshaw and N.V. Richardson, Pure Appl. Chem.68, 457~1996!.
18P. Hohenberg and W. Kohn, Phys. Rev.136, 864~1964!; W. Kohnand L.J. Sham,ibid. 140, 1133~1965!.
19P. Ordejon, E. Artacho, and J.M. Soler, Phys. Rev. B53, R10441~1996!; D. Sanchez-Portal, P. Ordejo´n, E. Artacho, and J.M.Soler, Int. J. Quantum Chem.65, 453 ~1997!; J.M. Soler, E.Artacho, J.D. Gale, A. Garcia, J. Junquera, P. Ordejon, andSanchez-Portal, J. Phys.: Condens. Matter14, 2745~2002!.
20J.P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett.77,3865 ~1996!.
5-10

v.
ev
m
cta
nd
gr.
sc.
m-
:
na,
H.
CHARACTERIZATION OF THE UNOCCUPIED AND . . . PHYSICAL REVIEW B68, 195115 ~2003!
21N. Trouiller and J.L. Martins, Phys. Rev. B43, 1993~1991!.22L. Kleinman and D.M. Bylander, Phys. Rev. Lett.48, 1425
~1982!.23S.G. Louie, S. Froyen, and M.L. Cohen, Phys. Rev. B26, 1738
~1982!.24E. Artacho, D. Sa´nchez-Portal, P. Ordejo´n, A. Garcia, and J.M.
Soler, Phys. Status Solidi B215, 809 ~1999!.25H.J. Monkhorst and J.D. Pack, Phys. Rev. B13, 5188~1976!.26Y.J. Lee, R.M. Nieminen, P. Ordejo´n, and E. Canadell, Phys. Re
B 67, 180505~R! ~2003!.27E.B. Starikov, Solid State Commun.91, 45 ~1994!.28R.V. Kasowski, M.-H. Tsai, S.T. Chui, and J.D. Dow, Phys. R
B 46, 10017~1992!.29S. Ishibashi and M. Kohyama, Phys. Rev. B62, 7839~2000!.30M. Sing, R. Claessen, Th. Finteis, S. Hao, S. Hu¨fner, and P.
Blaha, J. Electron Spectrosc. Relat. Phenom.114-116, 717~2001!.
31C.S. Jacobsen, Mat. Fys. Medd. K. Dan. Vidensk. Selsk.41, 251~1985!.
32A.J. Schultz, G.D. Stucky, R.H. Blessing, and P. Coppens, J. AChem. Soc.98, 3194~1976!.
33A. Filhol, G. Bravic, J. Gaultier, D. Chasseau, and C. Vettier, ACrystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem.37, 1225~1981!.
34J.P. Pouget, inSemiconductors and Semimetals, edited by E. Con-well ~Academic, New York, 1988!, Vol. 27, p. 88.
19511
.
.
35Z.Z. Wang, J.C. Girard, C. Pasquier, D. Je´rome, and K. Bech-gaard, Phys. Rev. B67, 121401~2003!.
36W.F. Cooper, N.C. Kenney, J.W. Edmons, A. Nagel, F. Wudl, aP. Coppens, Chem. Commun.~Cambridge! 1971, 889.
37R.E. Long, R.A. Sparks, and K.N. Trueblood, Acta Crystallo18, 932 ~1965!.
38A. Barrie, I.W. Drummond, and Q.C. Herd, J. Electron SpectroRelat. Phenom.5, 217 ~1974!.
39E.L. Shirley, Phys. Rev. Lett.80, 794 ~1998!.40T.A. Albright, J.K. Burdett, and M.-H. Whangbo,Orbital Inter-
actions in Chemistry~John Wiley & Sons, New York, 1985!.41J. Taborski, P. Vaterlein, H. Dietz, U. Zimmermann, and E. U
bach, J. Electron Spectrosc. Relat. Phenom.75, 129 ~1995!.42C. Rojas, J. Caro, M. Grioni, and J. Fraxedas, Surf. Sci.482-485,
546 ~2001!.43S.F. Lin, W.E. Spicer, and B.H. Schechtman, Phys. Rev. B12,
4184 ~1975!; C. Coluzza, H. Berger, P. Alme´ras, F. Gozzo, G.Margaritondo, G. Indlekofer, L. Forro, and Y. Hwu,ibid. 47,6625 ~1993!; K.E. Smith, Annu. Rep. Prog. Chem., Sect. CPhys. Chem.90, 115 ~1993!.
44J. Fraxedas, H.J. Trodahl, S. Gopalan, L. Ley, and M. CardoPhys. Rev. B41, 10068~1990!.
45G. Fischer and E. Dormann, Phys. Rev. B58, 7792~1998!.46K. Saito, Y. Yamamura, H. Akatsu, M. Takeda, H. Asaoka,
Nishikawa, I. Ikemoto, and M. Sorai, J. Phys. Soc. Jpn.68, 1277~1999!.
5-11





























